
Submitted:
23 September 2025
Posted:
25 September 2025
You are already at the latest version
Abstract
Background/Objectives: Vaccine elicitation of antibodies with high HIV-1-neutralization breadth is a long-standing goal. Recently, induction of such antibodies has been achieved at the fusion peptide-site of vulnerability; questions remain, however, as to whether their neutralization breadth and potency were sufficient to prevent HIV-1 infection. Methods: Here, we use yeast display coupled with deep-mutational screening, biochemical and structural analysis to study improvement of the best fusion-peptide-directed, vaccine-elicited antibody, DFPH_a.01, with initial 59% breadth. Results: Yeast display identified both single and double mutations that improved recognition of HIV-1-envelope trimers. We characterized two paratope-distal light chain (LC) mutations, S10R and S59P, which together increased breadth to 63%. Biochemical analysis demonstrated DFPH-a.01_10R59P-LC, and its component mutations, to have increased affinity and stability. Cryo-EM structural analysis revealed elbow-angle influencing by S10RLC and isosteric positioning by S59PLC as explanations for enhanced breadth, affinity, and stability. Conclusions: These results, along with another antibody with enhanced performance (DFPH-a.01_1G10A56K-LC with 64% breadth), suggest possible mutations that improve DFPH_a.01 are plentiful, an important vaccine insight.
Keywords:
antibody improvement
; broadly neutralizing antibody
; epitope
; fusion peptide
; HIV-1 vaccine
; paratope
1. Introduction
Broadly neutralizing antibodies against HIV-1 have been identified from HIV-infected donors that neutralize over 50% of HIV-1 strains. Such broadly neutralizing antibodies have been proposed as templates for vaccine development [1,2,3,4], and, indeed, vaccine-elicited broadly neutralizing antibodies targeting similar sites of vulnerability have been identified from macaques. These include a broadly neutralizing lineage, LJF-0034, which targets the CD4-binding site and neutralizes almost 70% of a 84-strain panel [5], an interface targeting antibody, 1C2, with 87% breadth [6], and antibodies against the fusion peptide (FP)-site of vulnerability, which have been elicited by vaccination in mice, guinea pigs, and rhesus macaques [7,8]. The best of the FP-directed antibodies, DFPH-a.01, elicited in macaques, has 59% neutralization breadth on a cross-clade 208-strain panel [8]. Recently, macaques vaccinated to induce FP-directed antibodies have been boosted by infection with simian-human immunodeficiency virus (SHIV) to achieve serum-neutralizing titers of 45-77% neutralization breadth with geometric mean potency of ~1:100 ID50 [9]; such titers are of sufficient breadth and potency to suggest that they might prevent HIV-1 infection [10], if they could be achieved by vaccination alone. Questions remain, however, about the breadth limit of FP-directed antibodies, as the template antibody for the FP site, naturally elicited antibody VRC34.01, has only ~50% breadth [11], which is lower than the breadth of template antibodies identified against other HIV-1 sites of vulnerability. For example, antibodies from natural infection show breadth that often exceed 90% against the aforementioned CD4-binding site. Could vaccine-elicited FP-directed antibodies of higher breadth (>60%) be obtained? Or is there an intrinsic breadth limit? And how rare would such breadth-improving mutations be?
B cells sample mutations at varying rates during antibody development in vivo. Many of the possible mutations are rare, and only a small fraction of possible single mutations is effectively sampled in vaccine animal models. In contrast, a yeast antibody library display paired with site-saturation DNA library mutagenesis and next-generation sequencing can efficiently sample all single antibody mutations in large-scale affinity studies [12,13,14]. Yeast display thus provides a valuable platform to explore questions related to HIV antibody engineering and development and provides a mechanism to map potential mutational pathways based on affinity to diverse HIV-1 Env trimeric antigen probes. These library-scale affinity studies can evaluate rare mutations not sampled efficiently in vivo and can also explore combinatorial strategies that include both rare and commonly sampled mutations. Yeast display thus provides a system to address important questions about mechanisms of antibody improvement and identify structural pathways that can enhance performance of vaccine-elicited HIV-1 broadly neutralizing antibodies.
In this study, we identified mutations that improved the breadth of the best FP-directed antibody, DFPH-a.01, which has 59% neutralization breadth with a geometric mean IC50 of 3.12 ug/ml [8]. In addition to being the best vaccine-elicited antibody, this antibody is also a member of a multi-donor antibody class, with antibody lineages with similar recognition and ontogeny being elicited in multiple macaques (including DFPH, 0PV, DJ85, GP6Z, and TRNM) [8,9]. Thus, a breadth or potency limit for DFPH-a.01 represents not only a limit for one particular antibody but also extends to a class of antibodies that makes up more than half of the broad FP-directed antibodies elicited in monkeys thus far. Here we created single- and multi-mutation libraries and screened them for enhanced affinity against HIV Env trimers with diverse FPs. We identified antibodies with improved affinity, which we assessed for neutralization. With an improved multi-mutation variant, we analyzed biochemical stability and affinity to Env trimer for not only the improved variant, but also for its individual component mutants. To provide atomic-level information, we also solved the structure of an improved variant in complex with Env trimer. Overall, we found that we could enhance the neutralization breadth of DFPH-a.01 to 64%, that improving mutations were often rare or distal from the region of antibody that contacted antigen, and that improving mutations were generally plentiful. These findings provide important insights that support the induction of broadly neutralizing immunity at the FP site-of-vulnerability.
2. Materials and Methods
2.1. Site-Saturation Mutagenesis Library Construction for DFPH-a.01
Site-saturation mutagenesis (SSM) libraries were generated using mutagenic primers containing degenerate single codons (NNK or MNN) to express all 20 amino acids at each residue of the heavy (VH) and light (VL) chains [12,13,14,15]. VH-SSM:VLtemplate and VHtemplate:VL-SSM libraries were cloned into a yeast display plasmid vector containing a FLAG expression marker to quantify Fab surface display, as well as a leucine zipper and protein disulfide isomerase expression for enhanced expression of diverse antibody libraries, as reported previously [12,13,14,16]. Plasmid DNA libraries were used to transform AWY101 yeast cells (Eric Shusta Lab, University of Wisconsin), and libraries of at least 2 x 106 clones were maintained in all steps [12,13,14].
To generate multi-mutation variants, high-affinity populations collected after the third round of single-mutation SSM library screens were diversified using another round of SSM and DNA shuffling [12,13,14]. Plasmid DNA libraries were isolated from sorted yeast cells using previously described extraction methods[12,13,14]. VH and VL genes were amplified from library plasmids with Kapa Hifi HotStart ReadyMix (Kapa Biosystems, Roche, Wilmington, MA). Four multi-mutation libraries were designed using template DNA from the enriched single-mutants: Library 1 was generated by pooling FACS-enriched single-mutation VH and single-mutation VL gene libraries, with the VH:VL pairing randomized (“combinatorial,” VH-SSM:VL-SSM); Library 2 was generated by performing DNA shuffling on Library 1, where template DNA was fragmented with DNAseI, followed by homologous reassembly and reamplification of the shuffled genetic material (“shuffled,” VH-shuffled:VL-shuffled); and Libraries 3 and 4 were created by repeating another round of SSM on either VH (“VH-multi,” VH-re-SSM:VL-SSM) or VL (“VL-multi,” VH-SSM:VL-re-SSM), respectively. Yeast cells were transformed with multi-mutation plasmid libraries via electroporation, with library sizes exceeding 3 x 106 maintained in all cloning steps.
2.2. Yeast Display Library Screening
Transformed yeast libraries were cultured in SGCAA (20 g/L galactose, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 5.4 g/L Na2HPO4, and 8.6 g/L NaH2PO4·H2O; TEKnova, Hollister, CA), supplemented with 2 g/L dextrose (SGDCAA) for 36 hours at 20 °C and 225 rpm to induce surface expression of antibody fragment (Fab) libraries. Induced yeast libraries were washed and stained with an anti-FLAG FITC monoclonal to quantify Fab expression (F4049, Clone M2, 1:49 dilution, Sigma-Aldrich, Burlington, MA). The HIV BG505 SOSIP Env trimer was used as antigen probe, expressed with one of three distinct versions of the FP: AVGIGAVF (BG505-FP8v1, the native BG505 FP8 sequence), AIGLGAMF (BG505_FP8-v3), and AVGIGAMI (BG505_FP8-Thai). Trimer antigen probes were generated by fluorescently labeling biotinylated constructs with a streptavidin-PE label (21388, Thermo Scientific, Waltham, MA) and assayed at 10 nM for BG505-FP8v1, and 50 nM for BG505-FP8v3 and BG505_FP8-Thai. Anti-FLAG-FITC-labeled libraries were co-stained with fluorescently conjugated antigen probes to screen Fab display libraries based on antigen affinity. A SONY Multi-Application 900 cell sorter running SONY LE-MA900FP Cell Sorter Software was used to separate VL+ (Fab-expressing cells) into low, medium, and high-affinity fractions across four consecutive rounds of enrichment sorting [12,13,14]. A minimum of 3 × 107 yeast cells were stained in the first round of affinity-based sorting. Libraries of VL+ yeast were also collected to track the prevalence of clonal variants prior to HIV-1 antigen affinity-based selection. Sorted yeast cells were cultured in low-pH SDCAA medium (20 g/L dextrose, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 10.4 g/L trisodium citrate, and 7.4 g/L citric acid monohydrate, pH 4.5) for 24-48 h at 30 °C and shaking at 225 rpm between rounds.
2.3. NGS Analysis of Sorted Yeast Libraries
After each round of FACS enrichment, yeast libraries were expanded via incubation at 30°C for 24-48 hr. An aliquot of this culture was drawn and reserved for high-efficiency yeast plasmid DNA extraction [17]. A high-fidelity polymerase (Kapa Hifi HotStart Mastermix, Kapa Biosystems, Roche, Wilmington, MA) and primers targeting the yeast display vector backbone were used to amplify heavy and light genes from each library [18]. A second round of primer-extension PCR with barcoded primers added a unique identifier to all heavy and light chains from a particular library [19]. Sorted libraries were sequenced on the Illumina 2x300 MiSeq platform and sequencing was performed for each library after each round of FACS enrichment. Raw Illumina FASTQ files were quality-filtered to retain quality scores of ≥30 over 90% of the reads using the Fastx Toolkit (v0.0.14, http://hannonlab.cshl.edu/fastx_toolkit/). Filtered reads were processed as previously reported to extract in-frame antibody amino acid sequences [12,13,14]. Mutant sequences were aligned to the template DFPH-a.01 antibody sequence with Usearch [20]. Mutations were defined by determining the percent identity match to the template gene and denoting the substituted amino acid residue(s) [12,13,14,21]. The enrichment ratio (ER) was calculated to monitor changes in mutation prevalence across successive sorting rounds. Each variant was binned into a high-, medium-, or low-affinity population by comparing prevalence and enrichment ratio values across screening rounds, as we reported previously [12,13,14,21].
2.4. Antibody Expression and Purification
Selected antibody variants were expressed as IgG and purified as described previously10. Briefly, VH and VL sequences were codon-optimized, synthesized, and cloned into a VRC8400 (CMV/R expression vector)-based vector. 50 μg of plasmid encoding VH and 50 μg of plasmid encoding VL were co-transfected into 100 ml of Expi293F cells at 2.5 x 106/ml (ThermoFisher Scientific, Waltham, MA) with Turbo293 transfection reagent (SPEED BioSystems, Gaithersburg, MD) following manufacturer’s protocols. Transfected cells were incubated at 37 °C, 9% CO2, with agitation at 120 rpm. 5 days post-transfection, culture supernatants were harvested and antibodies purified using a Protein A column (GE Healthcare, Chicago, IL), collected with elution buffer (ThermoFisher Scientific, Waltham, MA), and pH adjusted to neutral with 1 M Tris-HCl, pH 8. The molecular weight and purity of expressed antibodies were confirmed by SDS-PAGE.
2.5. Virus Neutralization Assays
2.5.1. 20-Virus Panel
Monoclonal antibodies were assessed one on one against multiple viral strains using entry neutralization assays. Five-fold serial antibody dilutions starting at 500 μg/mL were mixed in 50 μL volumes with stocks of viruses carrying fluorescent luciferase reporter genes. Mixtures were incubated at 37°C for 1 h, followed by the addition of 20 μL TZM-bI cells (0.5x106 cells/mL, NIH AIDS Reagent Program, Bethesda, MD) and incubated overnight at 37°C. After 24 hrs an additional 130 μL of complete Dulbecco’s modified Eagle medium was added to the neutralization test reactions and incubated at 37°C overnight. On day 3, cells were lysed and assessed for luciferase activity indicative of viral infection by measuring in relative light units. The concentration of antibody required to inhibit 50% and 80% of virus infection as determined by comparing relative light units between samples to negative non-neutralized controls were determined using a hill-slope regression analysis as described [12,14].
2.5.2. 208-Virus Panel
To model monoclonal antibody function against globally circulating FP diversity an automated large-batch neutralization panel of 208 HIV-1 Env-pseudotyped viruses was performed using 384-well microneutralization assays as described previously [22].
2.6. Affinity Measurements by Surface Plasmon Resonance
Surface Plasmon Resonance (SPR) binding assays were performed using a T200 biosensor equipped with a Series S CM5 sensor chip at 25° C in a running buffer of HBS-P (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% (v/v) Tween-20). Antibody 2G12 (IgG) was immobilized on all surfaces by using amine-coupling chemistry to ~10,000 RU; this antibody was used to tether BG505 DS-SOSIP to the chip surface at approximately 400 RU. Binding of the wild type DFPH-a.01 and its respective mutants, was tested at five concentrations ranging from 1.11-90 nM, which were prepared in a running buffer using a three-fold dilution series. Binding cycles consisted of a 120 s association phase and 900 s dissociation phase at a flow rate of 50 μL/min followed by a 60 s regeneration step using 3 M MgCl2 at 30 μL/min. A surface immobilized with 2G12 IgG was used as a reference surface to subtract bulk refractive index changes; and buffer blanks, in which running buffer without Fab was flowed over the BG505 DS-SOSIP surfaces, were used to subtract systematic noise drift. Binding responses were globally fit to 1:1 interaction model using Scrubber 2.0 to determine the kinetic parameters ka and kd as well as the equilibrium binding constant KD for each interaction.
2.7. Stability Measurements
The Fab of wildtype DFPH-a.01 and select mutants were assessed for stability using a TychoTM NT.6 to measure the ratio of intrinsic tryptophan fluorescence at 350 and 330 nm. All Fabs were diluted to 1 mg/ml, loaded into capillaries, and melted over a temperature range of 30-95 °C. Each Fab was assessed in experimental triplicate, and data was normalized to the starting 350/330 ratio.
2.8. Cryo-EM Structure Determination
BG505 DS-SOSIP HIV-1 envelope trimer and DFPH-a.01_10R59P-LC Fab were concentrated to the 6.5 mg/ml and 8.0 mg/ml, respectively and mixed at the molar ratio 1:1.3. To prevent preferred orientation, the complex was supplemented with the 0.1 mM of n-Dodecyl β-D-maltoside (DDM; final concentration). Quantifoil R 2/2 gold grids were glow-discharged with PELCO easiGlow glow-discharger (0.39 mBar, 20 mA, and 30 s). 2.7 ul of the mix was applied on the grid and vitrified in liquid ethane using FEI Vitrobot Mark IV plunger (chamber temperature 4C; humidity 95%; blot force -5; blot time 1.5 – 3 s). Cryo-EM datasets were collected at the National Cryo- Electron Microscopy Facility (NICE, Frederick, MD) on an FEI Titan Krios electron microscope equipped with a Gatan K2 summit DED operated in the super-resolution mode (pixel size before binning: 0.415 Å) using SerialEM [23] (Table S5). Cryo-EM reconstruction has been done with CryoSPARC v3.3 [24]. Movies were aligned, dose-weighted and binned to 0.83 Å using patch motion correction, and the micrograph contrast transfer function (CTF) parameters were estimated using patch CTF estimation. Particles were picked using the blob picker, extracted from the micrographs and subjected to 2D classification followed by the selection of the best classes. Ab-initio reconstruction, heterogeneous, homogeneous and non-uniform refinement jobs were run in C1. Local resolution was determined with local resolution module in CryoSPARC.
To obtain initial atomic models, the complex the coordinates of BG505 DS-SOSIP trimer (PDB 8EUV) [12] and the AlphaFold2 [25,26] model of the DFPH-a.01_10R59P-LC Fab were docked into corresponding parts of the cryo-EM map in UCSF Chimera [27]. Atomic models were refined by alternating rounds of model building in Coot [28,29] and real-space refinement in Phenix [30]. Structure validation was performed with Molprobity [31,32] and the PDB validation server. The analysis of HIV-Fab interfaces was done with EMBL PISA server [33]. Summaries of model refinement statistics and quality assessment for cryo-EM reconstructions are given in Supplemental Table S5, and in Supplemental Figure S8. Structure figures were generated with UCSF Chimera [27], ChimeraX [34], and Pymol (Schrodinger, Inc. https://pymol.org/).
2.9. Quantification and Statistical Analysis
IC50 calculations were reported using GraphPad Prism software. Briefly, experimental data was imported and modeled using a least-squares regression method to fit the data to a variable slope (four parameter) inhibitor versus response curve with bottom parameters constrained to zero. Flow cytometry analysis and figures were generated using FlowJo software (version 10.8).
3. Results
3.1. Precision Yeast Display Antibody Engineering of DFPH-a.01
We selected the vaccine-elicited FP-directed antibody with highest neutralization breadth, antibody DFPH-a.01, with an IC50 neutralization breadth of 59% on a 208-strain panel [8], to investigate features of potential mutations that would further improve its breadth (Figure 1A). We performed deep mutational scanning to evaluate the functional impact of every single amino acid (aa) mutation on the heavy (VH) and light (VL) chains of DFPH-a.01 using site-saturation mutagenesis (SSM) [12,13,14,15]. SSM variant libraries were cloned into yeast surface display and stained with BG505 SOSIP trimer antigens for FACS. >250-fold theoretical library coverage was maintained throughout all cloning steps to ensure robust data collection. SSM libraries were screened for their affinity to three BG505 DS-SOSIP.664 HIV-1 Env trimers with a distinct globally circulating FP variant: BG505_FP8-v1, BG505_FP8-v3, and BG505_FP8-Thai (Figure 1A) [12]. Single-mutant libraries for heavy and light chains (VH-SSM and VL-SSM, respectively) were analyzed by flow cytometry gated based on the ratio of surface Fab expression to HIV-1 antigen binding. The established sorting strategy fractionated the antibody library into three different populations based on binding affinity (Figure 1B). Yeast-expressing mutations detrimental to Env binding were enriched in the low-affinity gate, whereas mutations with no significant impact or with binding comparable to the template DFPH-a.01 were sorted into the medium-affinity gate. Any mutations with enhanced affinity were enriched in the high-affinity gates. After four rounds of affinity-based sorting, high-affinity populations showed clear phenotypic binding improvements in both the VH-SSM and VL-SSM libraries, for all three HIV-1 antigens used in the study (Figure 1B, Figure S1).
Sorted libraries were analyzed by next generation sequencing (NGS) to bioinformatically track enriched mutant sequences based on the composition of sorted gates. We evaluated the impact of each mutation based on enrichment ratios across screening rounds. Of the possible single amino acid mutations in the heavy chain, 45% showed a detrimental effect on HIV-BG505_FP8-v1 binding affinity, whereas 38% led to affinity similar to the template antibody. The remaining 0.6% of single mutations had a beneficial effect (Figure 2A; Figure S1). Similarly, 42%, 47%, and 0.8% of the light chain mutations showed deleterious, neutral, or enhancing effects on binding affinity, respectively (Figure 2A; Figure S1). The top 10 single mutants (based on enrichment ratios) were selected for further analysis.
3.2. Multi-Mutation Library Generation and Screening
To identify potentially synergistic multi-mutation combinations, we performed another round of SSM paired with DNA shuffling of enriched single mutants (Figure 1A). We expected that multi-mutation combinations would outperform single mutations in yeast display [12,13,14]. Four multi-mutation libraries were generated by pooling the high-affinity single-mutant libraries isolated after 3 rounds of enrichment. Libraries were enriched for high-affinity binders and after three rounds of sorting, the libraries showed enhanced trimer recognition compared to the template antibody against all three antigens (Figure 2B). NGS data was mined for potentially beneficial mutation combinations, and the top 6 light chain multi-mutants were selected for expression and characterization based on persistence in multi-mutation Rounds 2 and 3 against any of the three antigens, highest enrichment ratios in Round 2 or Round 3, and in some cases containing at least one light chain mutation known to enhance neutralization from single mutation tests (Figure S4).
3.3. HIV-1 Neutralization Analysis of Mutational Variants Identified in Yeast Display
Variants with affinity-improving mutations identified by NGS were expressed as IgG1 to evaluate the potential effects on neutralization potency and breadth. We first assessed single mutation DFPH-a.01 variants for HIV-1 against a panel of 20 viral isolates that were selected based on predictive capacity for broad FP-specific neutralization. The panel included 5 isolates that encoded FP8-v1 (AVGIGAVF), 2 with FP8-v2 (AVGLGAVF), 2 with FP8-v3 (AIGLGAMF), 2 with FP8-v4 (AVGTIGAMF), 4 with FP8-Thai (AVGIGAMI), 1 with FP8-v6 (AVGIGAMF), and 4 with other FP sequences (Table S1).
Ten DFPH-a.01 single-mutation variants were selected based on high-affinity enrichment against at least one FP8 antigen after round 2 and/or 3 (5 mutations in the heavy chain, 5 in the light chain) and were assayed for HIV-1 neutralization in the 20-virus panel. Of these, several showed some enhanced breadth in the 20-virus panel (Figure 2C, Figure S2, Table S1). The most potently improved single amino acid mutant, S10RLC, showed a 1.7-fold better potency against viruses carrying the FP variant FP8-v1 (geomean IC50 = 2.98 μg/mL compared to DFPH-a.01 geomean IC50 = 5.10 μg/mL), 1.4-fold improved against FP8v2 (geomean IC50 = 6.29 μg/mL versus 8.91 μg/mL), and 1.3-fold improved against FP8-Thai variants (geomean IC50 = 7.96 μg/mL versus 10.5 μg/mL). These improvements corresponded to an increase in 20-virus panel IC50 breadth from 75% to 85% (Figure 2C, Figure S2, Table S1).
Next, we analyzed selected variants from the multi-mutation screens to identify synergistic effects (Figure S4). On the 20-strain panel, the top performing variant DFPH-a.01_10R59P-LC (with S10RLC and S59PLC) showed 2.7-fold improved neutralization potency (geomean IC50 = 2.15 μg/mL compared with DFPH-a.01 geomean IC50 = 5.87 μg/mL) (Figure 2D, Figure S3; Table S2). Two other variants (D1G_S10A-LC, and D1G_S10A_T56K-LC) also showed improvements in IC50 and IC80 neutralization data. Based on these results, we sought to better characterize DFPH-a.01_10R59P-LC on a larger panel and learn more about the mechanisms of its improvement.
3.4. Breadth and Potency Analysis of DFPH-a.01_10R59P-LC
Based on the 20-virus panel data, DFPH-a.01_10R59P-LC was selected for evaluation against a broader 208-strain pseudovirus panel that is globally representative of HIV-1 strain diversity [7,12,14] (Table S3). We found that in addition to incremental potency improvements (Figure 3A-B, Table S3), these mutations improved the IC50 neutralization breadth from 59% to 63%, which was comparable to the highest breadth identified to date among non-engineered monoclonal antibodies (PGT151) [7,8,9,11,12,14,35,36,37] (Figure 3C-D, Figure S5, Table S3). DFPH-a.01_10R59P-LC also showed gain-of-function neutralization with the FP8-v3 sequence, and against two strains encoding the FP-Thai sequence (50 μg/mL cutoff) (Figure 3A-C, Figure S5, Table S3).
3.5. Surface Plasmon Resonance and Thermal Stability Analyses
To understand further the impact of the introduced mutations, we used surface plasmon resonance to characterize the affinity of DFPH-a.01 wild-type and variant antibodies to the soluble prefusion-closed Env trimer (BG505 DS-SOSIP). The double mutant DFPH-a.01_10R59P-LC had a KD of 113 pM, roughly twice the binding affinity of wildtype, which had a KD of 209 pM (Figure 4A), suggesting higher affinity as the basis for its increased neutralization breadth. Interestingly, the constituent individual mutations actually had higher affinities than the multi-mutation variant: DFPH-a.01_10R-LC had a KD of 97 pM and DFPH-a.01_59P-LC had a KD of 113 pM, indicating the two underlying mutations were not additive for affinity.
To characterize stability, we monitored intrinsic fluorescence of tryptophan by assessing the ratio of fluorescence at 350 nm versus 330 nm over a temperature range of 30-95º C. We observed both double and constituent single mutations to have increased melting temperatures. Interestingly, single mutations enhanced stability only marginally (0.3º and 1.0º C), whereas the double mutant had a melting temperature that was 2.5º C higher (Figure 4B). Thus, in contrast to affinity, the two single mutations appeared to synergize with respect to increased stability.
3.6. Cryo-EM Structure of Fab DFPH-a.01_10R59P-LC with BG505 DS-SOSIP Env Trimer
To investigate the structural basis for the observed improvements in affinity and stability of DFPH-a.01_10R59P-LC, we purified its antigen-binding fragment, generated complexes with BG505 DS-SOSIP Env trimer, and collected single particle cryo-EM data on a Titian Krios microscope. From 347,691 particles, we obtained a 3.0 Å resolution reconstruction, for which we built an atomic-level model comprising component Fab and Env trimer (Figure 5A; Figure S8; Table S5). We had previously determined the structure of another lineage member (DFPH-a.15, which had 46 amino acid mutations versus DFPH-a.01, and observed highly similar recognition (Figure S6). In terms of the two mutations, both were distal from the paratope, with 10R being 23.4 Å distal and 59P being 9.1 Å distal (Figure 5B).
The S10R mutation was located at the interface between the variable domains and the constant domains, at the elbow of the Fab (Figure 6A). While the constant regions of most other members of the class have not been defined, the structure of the constant regions has been defined for the vaccine-elicited, SHIV-infection boosted TRNM-b.01 antibody, which has a serine at VL position 10. Comparison with TRNM-b.01 revealed residue 10 to influence the elbow angle, with 15 degree difference between DFPH-a.01_10R59P-LC and TRNM-b.01 (Figure 6B). Analysis of the structural context for the S59P mutation revealed its ability to provide isosteric positioning of T56 CDR-L2 contact with Env trimer (Figure 6C).
3.7. A Second Yeast Display-Identified Antibody, DFPH-a.01_1G10A56K-LC, with 64% Neutralization Breadth
To gain insight into the prevalence and increase in neutralization by other DFPH-a.01 variants identified by yeast display, we characterized a second multi-mutation variant, DFPH-a.01_1G10A56K-LC (Figure S4). This antibody showed slightly higher breadth at 64% than DFPH-a.01_10R59P-LC (Figure 7A). This second antibody showed a similar FP neutralization profile, most able to neutralize v1 and v2 variants of FP8 (Figure 7B). SPR analysis revealed a 3-fold increase in KD (to BG505 DS-SOSIP), related mostly to the increased association rate (Figure 7C). A slight increase in stability was also observed (Figure 7D). Overall, several single, double, and multi-mutation variants were found to have increased neutralization breadth on the 20-isolate panel, and several of these showed increased potency (Figure 7E).
3.8. Comparative Analysis of Improving FP Broadly Neutralizing Antibody Mutations Across Macaque, Mouse, and Human Studies
To understand generalizable features for antibody improvement against the FP epitope, we compared the data collected here for DFPH-a.01 (a macaque vaccine-elicited antibody) with prior data from yeast display campaigns with VRC34.01 [12] (a human, naturally elicited antibody) and with vFP16.02 [14] (a mouse, vaccine-elicited antibody) (Figure 8A). Each of these three antibodies are among the very best anti-FP broadly neutralizing antibodies that have been isolated in their respective species. Across all three species, we found that site-saturation mutagenesis and yeast display identified few highly beneficial mutations in the CDR3s. The most beneficial mutations identified were commonly outside of CDRs (5/6 red mutations, Figure 8B) and sometimes occurred in paratope-distal locations, especially for vaccine-elicited antibodies. These paratope-distal substitutions improved DFPH-a.01 by allosterically rigidifying the Fab elbow (DFPH-a.01_S10RLC) and increasing CDR loop flexibility (DFPH-a.01_S59PLC). Similarly, deep mutational scanning of the murine broadly neutralizing antibody vFP16.02 showed that many beneficial mutations were framework changes distant from the paratope [14] (Figure 8B). In particular, the vFP16.02 light-chain S48K mutation enhanced neutralization breadth (from ~28% to 37%) by altering the VH–VL orientation without directly contacting the FP [14]. In contrast, several mutations that improved the naturally elicited human antibody VRC34.01 made direct contact with antigen, including VRC34.01_E2K_VH which generated a new contact site [12].
We also sought to understand the prevalence of identified mutations in natural immune repertoires. For DFPH-a.01 which was isolated from an immunized macaque, we identified both frequent and rare improving mutations in yeast display: S59PLC was common (13% of natural sequences) whereas S10RLC was very rare (<0.1% of sequences). However, the three most important heavy-chain substitutions for the naturally-elicited human broadly neutralizing antibody VRC34.01_mm28 (VH_E2K, VH_A33P, and VH_T58F) had either rare or moderate prevalence, with frequencies in humans of 0.01%, 0.1%, and 0.72%, respectively (Figure 8B).
While improving mutations that made direct contact with antigen were sometimes observed in vaccine-elicited antibodies, we note that many promising single mutations did not make direct contact with antigen; thus, of the 16 promising single mutants identified by yeast display, 14 of these (the majority) were not part of the paratope (Figure 8C). One of these, DFPH-a.01_S59P-LC, allowed for the isosteric positioning of nearby residues that did indeed contact antigen. We note that for VRC34.01, the ratio of single mutations that were or were not part of the paratope is more balanced, with half the identified improving mutations contacting the epitope and half not (Figure 8C), thereby suggesting the ratio of contact versus non-contact improvements can depend on particulars of antibody and recognition. Overall, these data suggest diverse mechanisms by which contact and non-contact residues can contribute to improved FP-directed neutralization breadth and potency across species.
4. Discussion
In this study we used yeast display affinity engineering to enhance the neutralization breadth and potency of the top macaque vaccine-elicited antibody against HIV-1 FP, DFPH-a.01. By further improving DFPH-a.01, we demonstrated here that vaccine-elicited antibodies against HIV-1 FP in macaques are not limited to a neutralization breadth <60%. We identified multiple mutations that enhanced the sorting affinity to Env trimer with diverse FPs. We characterized several of these mutations in depth, including two multi-mutant variants, DFPH-a.01_10R59P-LC and DFPH-a.01_1G10A56K, with breadth of 63% and 64% on a 208-strain panel, respectively. Overall, many individual mutations appeared to improve DFPH-a.01, suggesting improvement mutations to be plentiful for anti-FP antibodies of this class.
The high prevalence of mutations to improve DFPH-a.01 suggests the breadth/potency limit for DFPH-a class antibodies to be substantially higher than the 59% breadth of the best (DFPH-a.01). This is an important finding, as the breadth of the SHIV-infection boosted antibodies was at highest only 56% (for DJ85-a.01). Thus, despite substantial in vivo affinity maturation (SHM rates of 15-20%), the SHIV-infection boosted antibodies still did not appear to achieve maximal breadth. Likely these limits reflect the manner by which these antibodies were selected and matured, and suggests that the immunization process can be further optimized to maximize FP-directed neutralization breadth.
Overall, the ability of yeast display to sample many mutations that improve neutralization breadth of antibody DFPH-a.01, the best of the DFPH-a class of “reproducible’ or ‘multi-donor’ antibodies, suggests the breadth limit for this prevalent class of FP-directed antibodies to be considerably higher than 60%. It will be fascinating to see how high the breadth can go. But whatever the breadth limit, the breadth of FP-directed antibodies already exceeds the target 50% level, which has been suggested as a minimal breadth level that would lead to an impactful vaccine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Bioinformatic analysis of DFPH single mutants, related to Figure 1 and 2; Figure S2: Neutralization breadth of single-mutant DFPH-a.01 variants across diverse HIV-1 strains, related to Figure 2; Figure S3: Neutralization breadth of multi-mutant DFPH-a.01 variants across diverse HIV-1 strains, related to Figure 2; Figure S4: Germline gene alignments for DFPH-a.01, and also TRNM_b.01, an antibody of the same class, related to Figures 2, 3, 7, and 8; Figure S5: Neutralization breadth of multi-mutant DFPH-a.01_10R59P-LC and DFPH-a.01_1G10A56K-LC across 208 HIV-1 strains, related to Figure 3 and Figure 7; Figure S6: DFPHa.01_10R59P-LC has an S59PLC mutation that allows for an isosteric recognition loop. Related to Figure 6; Figure S7: Electrostatic surfaces for DFPH-a.01_10R59P-LC and S10 in TRNM-b.01. Related to Figure 6; Figure S8: Cryo-EM validation of the BG505 DS-SOSIP – DFPH-a.01_10R59P-LC complex. Related to Figure 5 and Table S5; Table S1: IC50 and IC80 neutralization values for single-mutants against a 20-virus panel. Related to Figure 2; Table S2: IC50 and IC80 neutralization values for combination mutants against a 20-virus panel. Related to Figure 2; Table S3: IC50 and IC80 neutralization values for mutant DFPH-a.01_10R59P-LC against a 208-virus panel. Related to Figure 3 and Figure 7; Table S4: Mutations from the study expressed in template numbering and Kabat numbering formats. Related to Figures 2, 3, 7, and 8; Table S5: Cryo-EM data collection, refinement, and validation statistics for BG505 DS-SOSIP – DFPH-a.01_10R59P-LC Fab complex. Related to Figure 5 and Figure S8; Table S6: BG505 DS-SOSIP (HIV4571) - DFPH-a.01_10R59P-LC Fab interface details. Related to Figure 8.
Author Contributions
Designed experiments: S.P., B.M., P.K., N.C.M., K.M., B.Z., T.B., S.O., F.B., S.M., M.K.L., B.C.L., N.A.D-R., P.D.K, L.S., B.J.D. Performed experiments: S.P., B.M., P.K., N.C.M., K.M., B.Z., R.N., S.O., F.B., S.M., M.K.L., B.C.L. Analyzed the data: C.T.F., S.P., B.M., P.K., N.C.M., T.B., N.A. D-R., P.D.K, Z.S., T.Z., B.J.D. Writing: C.T.F., S.P., P.D.K, B.J.D. Reviewing and editing: all authors. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was also provided by NIH grants DP5OD023118, R01AI141452, R21AI143407, R21AI166396, U01AI169587, 1R01AI181684, and R01AI192975, the MIT Research Support Committee, the MIT Department of Chemical Engineering, and by the Ragon Institute of MGH, MIT, and Harvard.
Data Availability Statement
DNA sequences are deposited in NCBI GenBank as PX207628-PX207649. Resolved structure has been added to the PDB and EMDB databases as entries 9Q0W and EMD-72108, respectively. Code for analysis of immune receptor sequences is provided as part of Protocol 1: Clonal Variant Analysis of Antibody Engineering Libraries, https://github.com/dekoskylab/CSHL_protocols. Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This study used the Office of Cyber Infrastructure and Computational Biology High Performance Computing cluster at the National Institute of Allergy and Infectious Diseases, Bethesda, MD.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health (NIH). The contributions of the NIH authors were made as part of their duties as NIH federal employees, are in compliance with agency policy requirements, and are considered Works of the United States Government. However, the findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services. We thank the New York Structural Biology Center for access to the TychoTM NT.6 We thank J. Stuckey for assistance with figures, and members of the Structural Biology Section and Structural Bioinformatics Core, Vaccine Research Center, for discussions and comments on the manuscript. We thank R. Carroll, N. Jean-Baptiste, C. Moore, C. Whittaker, and A.B. McDermott for their assistance with neutralization assessments on the 208-strain panel and J. Baalwa, D. Ellenberger, F. Gao, B. Hahn, K. Hong, J. Kim, F. McCutchan, D. Montefiori, L. Morris, E. Sanders-Buell, G. Shaw, R. Swanstrom, M. Thomson, S. Tovanabutra, C. Williamson, and L. Zhang for contributing the HIV-1 envelope plasmids used in our neutralization panel. We thank the VRC Production Program for providing BG505 and ConC Env trimers. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Å | Ångström (10⁻¹⁰ m, used for structural resolution/length) |
| BSA | Buried Surface Area |
| °C | Degrees Celsius |
| CDR | Complementarity-Determining Region |
| COOT | Model-Building Tools for Molecular Graphics |
| Cryo-EM | Cryogenic Electron Microscopy |
| CTF | Contrast Transfer Function |
| DDM | n-Dodecyl β-D-maltoside |
| DOAJ | Directory of Open Access Journals |
| Env | Envelope Glycoprotein |
| Fab | Fragment Antigen-Binding |
| FACS | Fluorescence-Activated Cell Sorting |
| FP | Fusion Peptide |
| FR | Framework Region |
| HIV | Human Immunodeficiency Virus |
| IC50/ IC80 |
Concentration of Antibody Required to Inhibit 50% / 80% of Infection |
| ID50 | Inhibitory Dilution 50 |
| KD | Equilibrium Dissociation Constant |
| Ka | Association Rate Constant |
| Kd | Dissociation Rate Constant |
| LD | Linear Dichroism |
| mg/ml | Milligrams per milliliter (concentration unit) |
| μg/ml | Micrograms per milliliter (concentration unit) |
| μL | Microliter (10⁻⁶ liter) |
| μM | Micromolar (10⁻⁶ molar concentration) |
| nM | Nanomolar (10⁻⁹ molar concentration) |
| NGS | Next-Generation Sequencing |
| PDB | Protein Data Bank |
| RU | Response Units (Surface Plasmon Resonance Readout) |
| SHIV | Simian-Human Immunodeficiency Virus |
| SHM | Somatic Hypermutation |
| SPR | Surface Plasmon Resonance |
| SSM | Site-Saturation Mutagenesis |
| VH | Variable Heavy Chain |
| VL | Variable Light Chain |
References
- Willis, J.R.; Berndsen, Z.T.; Ma, K.M.; Steichen, J.M.; Schiffner, T.; Landais, E.; Liguori, A.; Kalyuzhniy, O.; Allen, J.D.; Baboo, S.; et al. Human immunoglobulin repertoire analysis guides design of vaccine priming immunogens targeting HIV V2-apex broadly neutralizing antibody precursors. Immunity 2022, 55, 2149-2167 e2149. [CrossRef]
- Haynes, B.F.; Wiehe, K.; Borrow, P.; Saunders, K.O.; Korber, B.; Wagh, K.; McMichael, A.J.; Kelsoe, G.; Hahn, B.H.; Alt, F.; et al. Strategies for HIV-1 vaccines that induce broadly neutralizing antibodies. Nat Rev Immunol 2023, 23, 142-158. [CrossRef]
- Burton, D.R.; Hangartner, L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu Rev Immunol 2016, 34, 635-659. [CrossRef]
- Medina-Ramirez, M.; Garces, F.; Escolano, A.; Skog, P.; de Taeye, S.W.; Del Moral-Sanchez, I.; McGuire, A.T.; Yasmeen, A.; Behrens, A.J.; Ozorowski, G.; et al. Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J Exp Med 2017, 214, 2573-2590. [CrossRef]
- Schleich, F.A.; Bale, S.; Guenaga, J.; Ozorowski, G.; Adori, M.; Lin, X.; Castro Dopico, X.; Wilson, R.; Chernyshev, M.; Cotgreave, A.T.; et al. Vaccination of nonhuman primates elicits a broadly neutralizing antibody lineage targeting a quaternary epitope on the HIV-1 Env trimer. Immunity 2025, 58, 1598-1613 e1598. [CrossRef]
- Dubrovskaya, V.; Tran, K.; Ozorowski, G.; Guenaga, J.; Wilson, R.; Bale, S.; Cottrell, C.A.; Turner, H.L.; Seabright, G.; O'Dell, S.; et al. Vaccination with Glycan-Modified HIV NFL Envelope Trimer-Liposomes Elicits Broadly Neutralizing Antibodies to Multiple Sites of Vulnerability. Immunity 2019, 51, 915-929 e917. [CrossRef]
- Xu, K.; Acharya, P.; Kong, R.; Cheng, C.; Chuang, G.Y.; Liu, K.; Louder, M.K.; O'Dell, S.; Rawi, R.; Sastry, M.; et al. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med 2018, 24, 857-867. [CrossRef]
- Kong, R.; Duan, H.; Sheng, Z.; Xu, K.; Acharya, P.; Chen, X.; Cheng, C.; Dingens, A.S.; Gorman, J.; Sastry, M.; et al. Antibody Lineages with Vaccine-Induced Antigen-Binding Hotspots Develop Broad HIV Neutralization. Cell 2019, 178, 567-584 e519. [CrossRef]
- Wang, H.; Cheng, C.; Dal Santo, J.L.; Shen, C.H.; Bylund, T.; Henry, A.R.; Howe, C.A.; Hwang, J.; Morano, N.C.; Morris, D.J.; et al. Potent and broad HIV-1 neutralization in fusion peptide-primed SHIV-infected macaques. Cell 2024, 187, 7214-7231 e7223. [CrossRef]
- Pegu, A.; Lovelace, S.E.; DeMouth, M.E.; Cully, M.D.; Morris, D.J.; Li, Y.; Wang, K.; Schmidt, S.D.; Choe, M.; Liu, C.; et al. Antibodies targeting the fusion peptide on the HIV envelope provide protection to rhesus macaques against mucosal SHIV challenge. Sci Transl Med 2024, 16, eadh9039. [CrossRef]
- Kong, R.; Xu, K.; Zhou, T.; Acharya, P.; Lemmin, T.; Liu, K.; Ozorowski, G.; Soto, C.; Taft, J.D.; Bailer, R.T.; et al. Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 2016, 352, 828-833. [CrossRef]
- Banach, B.B.; Pletnev, S.; Olia, A.S.; Xu, K.; Zhang, B.; Rawi, R.; Bylund, T.; Doria-Rose, N.A.; Nguyen, T.D.; Fahad, A.S.; et al. Antibody-directed evolution reveals a mechanism for enhanced neutralization at the HIV-1 fusion peptide site. Nat Commun 2023, 14, 7593. [CrossRef]
- Banach, B.B.; Tripathi, P.; Da Silva Pereira, L.; Gorman, J.; Nguyen, T.D.; Dillon, M.; Fahad, A.S.; Kiyuka, P.K.; Madan, B.; Wolfe, J.R.; et al. Highly protective antimalarial antibodies via precision library generation and yeast display screening. J Exp Med 2022, 219. [CrossRef]
- Madan, B.; Zhang, B.; Xu, K.; Chao, C.W.; O'Dell, S.; Wolfe, J.R.; Chuang, G.Y.; Fahad, A.S.; Geng, H.; Kong, R.; et al. Mutational fitness landscapes reveal genetic and structural improvement pathways for a vaccine-elicited HIV-1 broadly neutralizing antibody. Proc Natl Acad Sci U S A 2021, 118. [CrossRef]
- Wrenbeck, E.E.; Klesmith, J.R.; Stapleton, J.A.; Adeniran, A.; Tyo, K.E.; Whitehead, T.A. Plasmid-based one-pot saturation mutagenesis. Nat Methods 2016, 13, 928-930. [CrossRef]
- Meyer, A.J.; Ellefson, J.W.; Ellington, A.D. Library generation by gene shuffling. Curr Protoc Mol Biol 2014, 105, Unit 15 12. [CrossRef]
- Whitehead, T.A.; Chevalier, A.; Song, Y.; Dreyfus, C.; Fleishman, S.J.; De Mattos, C.; Myers, C.A.; Kamisetty, H.; Blair, P.; Wilson, I.A.; et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol 2012, 30, 543-548. [CrossRef]
- Wang, B.; DeKosky, B.J.; Timm, M.R.; Lee, J.; Normandin, E.; Misasi, J.; Kong, R.; McDaniel, J.R.; Delidakis, G.; Leigh, K.E.; et al. Functional interrogation and mining of natively paired human V(H):V(L) antibody repertoires. Nat Biotechnol 2018, 36, 152-155. [CrossRef]
- McDaniel, J.R.; DeKosky, B.J.; Tanno, H.; Ellington, A.D.; Georgiou, G. Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat Protoc 2016, 11, 429-442. [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460-2461. [CrossRef]
- Fahad, A.S.; Gutierrez-Gonzalez, M.F.; Madan, B.; DeKosky, B.J. Clonal Variant Analysis of Antibody Engineering Libraries. Cold Spring Harb Protoc 2025, 2025, pdb prot108626. [CrossRef]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods 2014, 409, 131-146. [CrossRef]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 2005, 152, 36-51. [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 2017, 14, 290-296. [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nat Methods 2022, 19, 679-682. [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583-589. [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25, 1605-1612. [CrossRef]
- Emsley, P.; Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60, 2126-2132. [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486-501. [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 2019, 75, 861-877. [CrossRef]
- Davis, I.W.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 2004, 32, W615-619. [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66, 12-21. [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 2007, 372, 774-797. [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 2021, 30, 70-82. [CrossRef]
- Falkowska, E.; Le, K.M.; Ramos, A.; Doores, K.J.; Lee, J.H.; Blattner, C.; Ramirez, A.; Derking, R.; van Gils, M.J.; Liang, C.H.; et al. Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity 2014, 40, 657-668. [CrossRef]
- van Gils, M.J.; van den Kerkhof, T.L.; Ozorowski, G.; Cottrell, C.A.; Sok, D.; Pauthner, M.; Pallesen, J.; de Val, N.; Yasmeen, A.; de Taeye, S.W.; et al. An HIV-1 antibody from an elite neutralizer implicates the fusion peptide as a site of vulnerability. Nat Microbiol 2016, 2, 16199. [CrossRef]
- Reveiz, M.; Xu, K.; Lee, M.; Wang, S.; Olia, A.S.; Harris, D.R.; Liu, K.; Liu, T.; Schaub, A.J.; Stephens, T.; et al. Vaccine-elicited and naturally elicited antibodies differ in their recognition of the HIV-1 fusion peptide. Front Immunol 2024, 15, 1484029. [CrossRef]
Figure 1.
Precision antibody yeast display to evaluate mutational landscapes for the HIV-1 broadly neutralizing antibody, DFPH-a.01. (a) Site-saturation mutagenesis (SSM) introduced diversity into the DFPH-a.01 variable region genes, which were cloned and displayed as Fab libraries on yeast. Flow cytometry screened variants for binding to BG505 SOSIP Env trimers bearing distinct HIV-1 fusion peptide (FP8) sequences (FP8-v1, FP8-v3, and FP8-Thai). Sorted libraries were sequenced to map each mutation’s impact on affinity. High-affinity variants were expressed and evaluated in neutralization assays. Structural analysis revealed mutational mechanisms underlying improved breadth and potency; (b) Binding for the wild type (WT) antibody DFPH-a.01 compared to the enriched heavy (VH) and light (VL) chain libraries after four rounds of selection against three Env trimers. See also Figure S1.
Figure 1.
Precision antibody yeast display to evaluate mutational landscapes for the HIV-1 broadly neutralizing antibody, DFPH-a.01. (a) Site-saturation mutagenesis (SSM) introduced diversity into the DFPH-a.01 variable region genes, which were cloned and displayed as Fab libraries on yeast. Flow cytometry screened variants for binding to BG505 SOSIP Env trimers bearing distinct HIV-1 fusion peptide (FP8) sequences (FP8-v1, FP8-v3, and FP8-Thai). Sorted libraries were sequenced to map each mutation’s impact on affinity. High-affinity variants were expressed and evaluated in neutralization assays. Structural analysis revealed mutational mechanisms underlying improved breadth and potency; (b) Binding for the wild type (WT) antibody DFPH-a.01 compared to the enriched heavy (VH) and light (VL) chain libraries after four rounds of selection against three Env trimers. See also Figure S1.
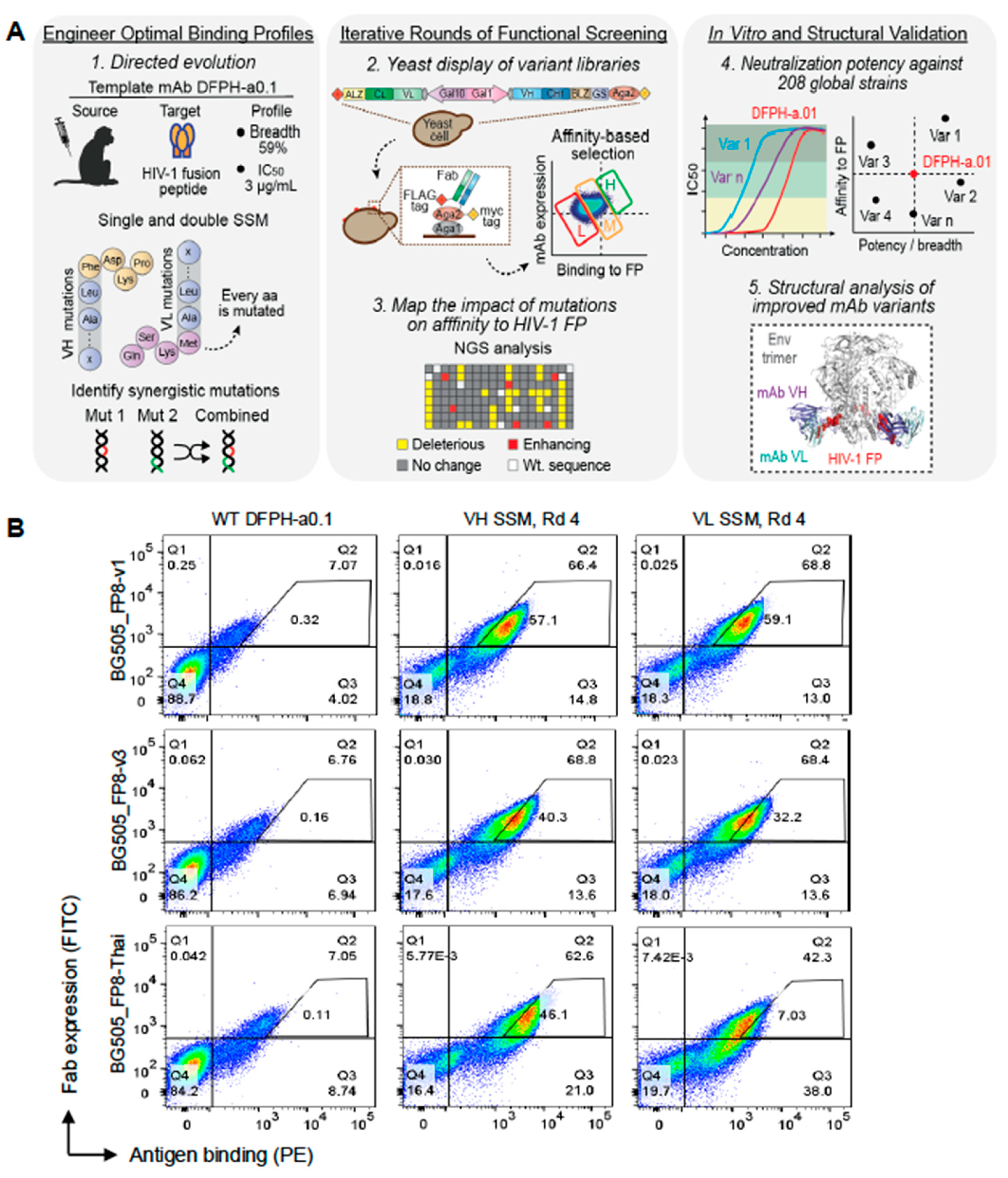
Figure 2.
Bioinformatic analysis of library screening data reveals multiple single mutations that enhance HIV-1 neutralization potency and breadth, with multi-mutation screening revealing efficacious combinations. (a) Heat maps display the functional impact of single mutations on binding to HIV-1 BG505-FP8v1 Env. (b) Flow cytometry profiles show binding of wild type (WT) DFPH-a.01 compared to the enriched heavy (VH) and light (VL) chain libraries after three rounds of selection against three Env trimers. (c) Selected single mutants were expressed as IgG1 and evaluated in pseudovirus IC50 neutralization assays against a 20-virus panel; IC50 ≤ 50 μg/mL was used as the threshold for neutralization sensitivity. (d) Multi-mutation screening identified synergistic combinations that improved neutralization potency and breadth against diverse HIV-1 variants using the same IC50 threshold. See also Figures S1–S4, and Tables S1, S2, and S4.
Figure 2.
Bioinformatic analysis of library screening data reveals multiple single mutations that enhance HIV-1 neutralization potency and breadth, with multi-mutation screening revealing efficacious combinations. (a) Heat maps display the functional impact of single mutations on binding to HIV-1 BG505-FP8v1 Env. (b) Flow cytometry profiles show binding of wild type (WT) DFPH-a.01 compared to the enriched heavy (VH) and light (VL) chain libraries after three rounds of selection against three Env trimers. (c) Selected single mutants were expressed as IgG1 and evaluated in pseudovirus IC50 neutralization assays against a 20-virus panel; IC50 ≤ 50 μg/mL was used as the threshold for neutralization sensitivity. (d) Multi-mutation screening identified synergistic combinations that improved neutralization potency and breadth against diverse HIV-1 variants using the same IC50 threshold. See also Figures S1–S4, and Tables S1, S2, and S4.
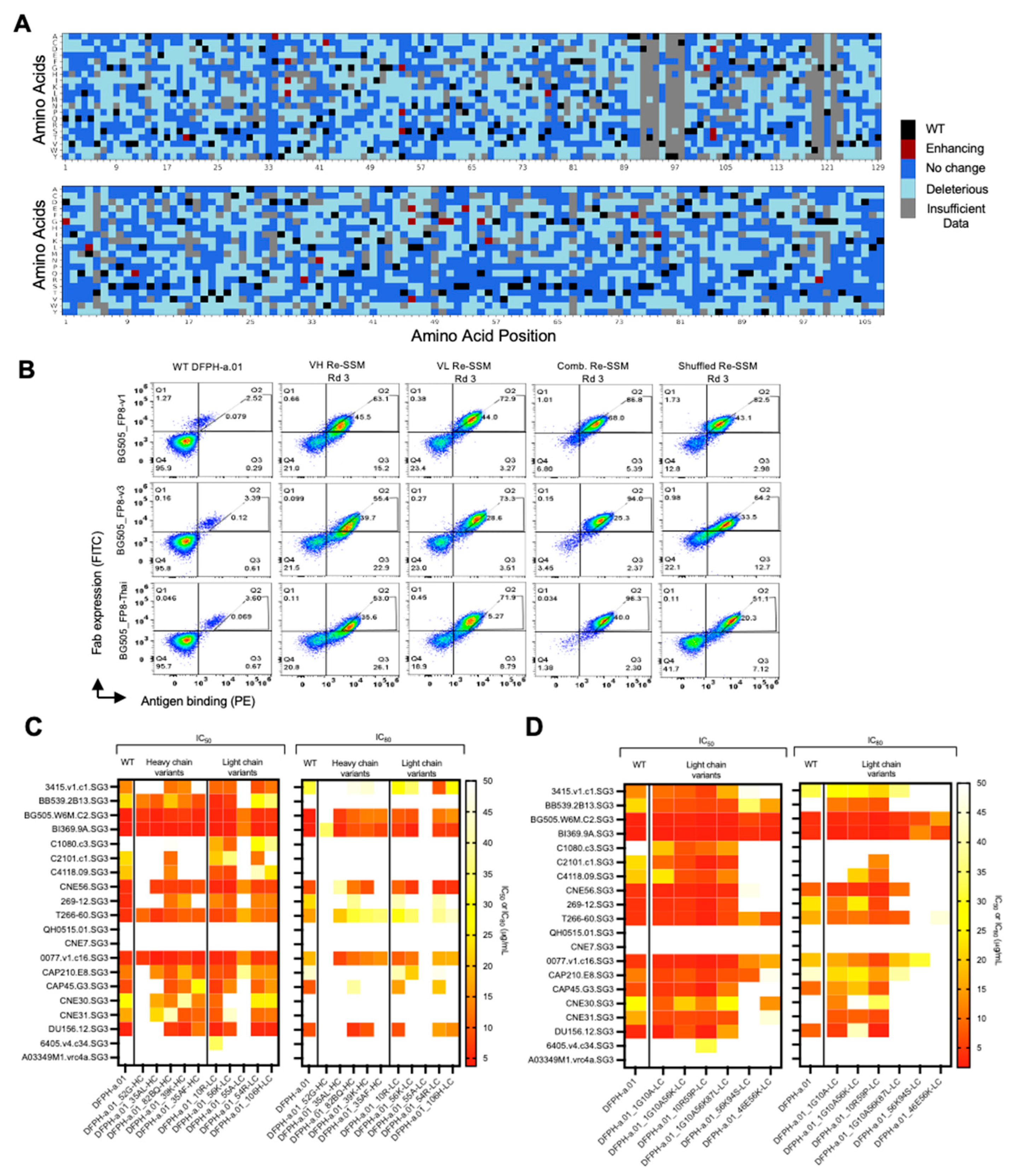
Figure 3.
Assessment of DFPH-a.01_10R59P-LC on a 208-strain panel reveals enhanced neutralization potency and breadth. (a) Dendrogram plot of neutralization reveals that the engineered antibody DFPH-a.01_10R59P-LC increased both cross-clade recognition and neutralization against DFPH-a.01-resistant and sensitive pseudovirus strains. Geometric mean potency is shown. (b) Titration curves show that the engineered antibody gained stepwise improvements in neutralization potency and breadth compared to the template antibody across the panel. (c) 208-virus panel neutralization data showed enhanced breadth for the engineered antibody compared with the template against a globally representative HIV-1 panel. Viral strains are grouped by FP sequence. We used IC50 (left) or IC80 (right) of 50 μg/mL to determine neutralization sensitivity against a viral strain. (d) Comparison of known anti-FP neutralizing antibodies. See also Figures S4 and S5, and Tables S3, and S4.
Figure 3.
Assessment of DFPH-a.01_10R59P-LC on a 208-strain panel reveals enhanced neutralization potency and breadth. (a) Dendrogram plot of neutralization reveals that the engineered antibody DFPH-a.01_10R59P-LC increased both cross-clade recognition and neutralization against DFPH-a.01-resistant and sensitive pseudovirus strains. Geometric mean potency is shown. (b) Titration curves show that the engineered antibody gained stepwise improvements in neutralization potency and breadth compared to the template antibody across the panel. (c) 208-virus panel neutralization data showed enhanced breadth for the engineered antibody compared with the template against a globally representative HIV-1 panel. Viral strains are grouped by FP sequence. We used IC50 (left) or IC80 (right) of 50 μg/mL to determine neutralization sensitivity against a viral strain. (d) Comparison of known anti-FP neutralizing antibodies. See also Figures S4 and S5, and Tables S3, and S4.
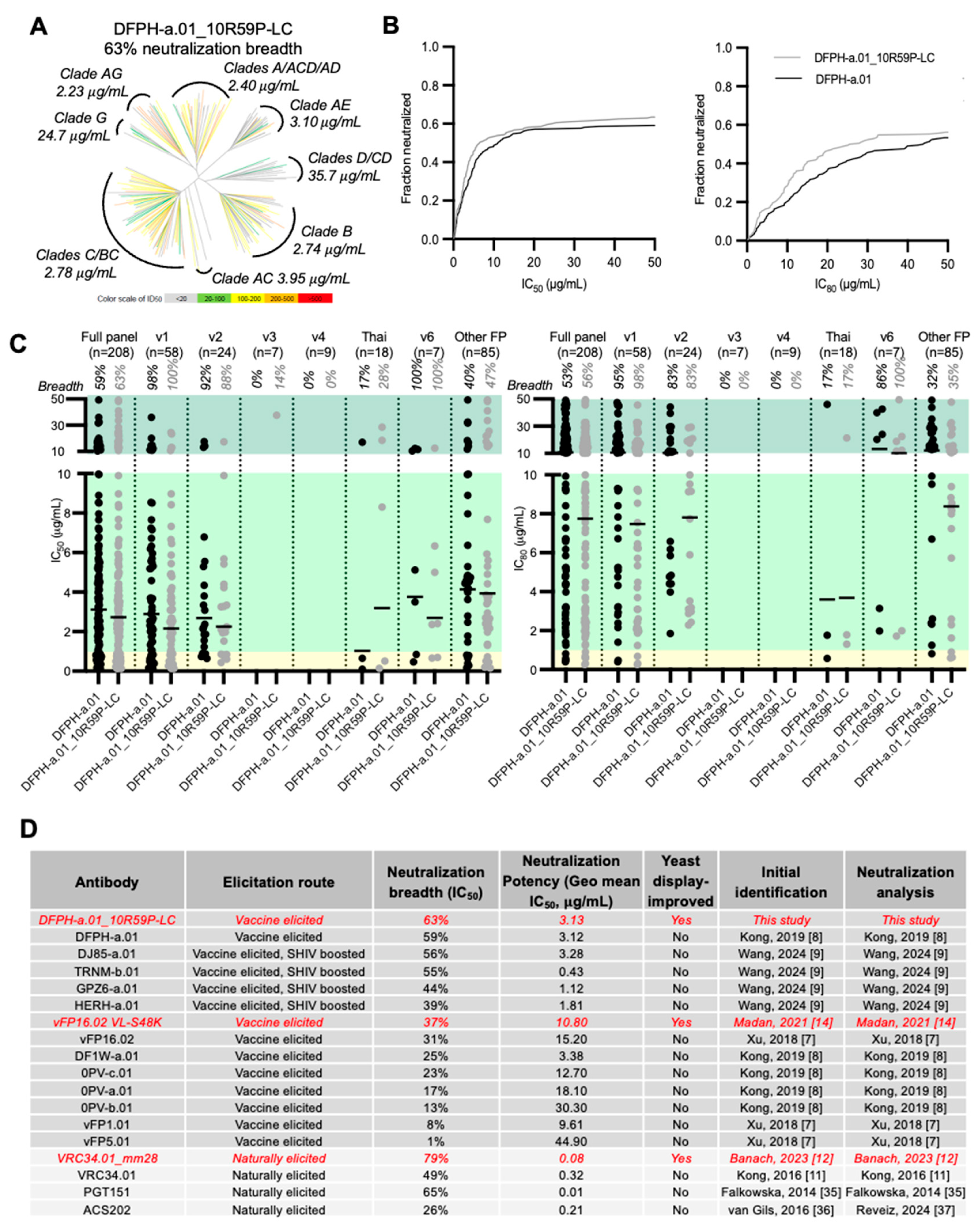
Figure 4.
Surface plasmon resonance and temperature-dependent fluorescent analyses reveal DFPH-a.01_10R-LC to be 4-fold improved in affinity, and DFPH-a.01_10R59P-LC to be improved by 2.5 degrees in stability. (a) Surface plasmon resonance analysis of DFPH-a.01 and the single and double mutant for binding to BG505 Env trimer. Binding analysis was performed using BG505 DS-SOSIP tethered to the sensor chip surface via 2G12, with DFPH-a.01 and its respective mutants tested at five concentrations ranging from 1.11-90 nM. The black traces represent experimental data, and the red lines represent the fit to 1:1 interaction model. The number in brackets show the error of the fit in the last significant digit. (b) DFPH_a.01, DFPH_a.01_10R-LC, DFPH_a.01_59P-LC, and DFPH_a.01_10R59P-LC were assessed for stability by monitoring change to the 350/330 ratio from intrinsic fluorescence of tryptophan during melting. All three variants showed improved stability as compared to wildtype DFPH_a.01. DFPH_a.01_10R-LC increased stability by 0.3 °C while DFPH_a.01_59P-LC increased stability by 1.0 °C. The double mutant displayed increased stability by 2.5 °C, indicating possible structural synergy between the two mutations, as combined they enhance stability greater than either individual mutant. Results shown are averaged from 3 experimental replicates and normalized so that the starting 350/330 ratio is set to 0. See also Figures 5, 6, 8 and S6.
Figure 4.
Surface plasmon resonance and temperature-dependent fluorescent analyses reveal DFPH-a.01_10R-LC to be 4-fold improved in affinity, and DFPH-a.01_10R59P-LC to be improved by 2.5 degrees in stability. (a) Surface plasmon resonance analysis of DFPH-a.01 and the single and double mutant for binding to BG505 Env trimer. Binding analysis was performed using BG505 DS-SOSIP tethered to the sensor chip surface via 2G12, with DFPH-a.01 and its respective mutants tested at five concentrations ranging from 1.11-90 nM. The black traces represent experimental data, and the red lines represent the fit to 1:1 interaction model. The number in brackets show the error of the fit in the last significant digit. (b) DFPH_a.01, DFPH_a.01_10R-LC, DFPH_a.01_59P-LC, and DFPH_a.01_10R59P-LC were assessed for stability by monitoring change to the 350/330 ratio from intrinsic fluorescence of tryptophan during melting. All three variants showed improved stability as compared to wildtype DFPH_a.01. DFPH_a.01_10R-LC increased stability by 0.3 °C while DFPH_a.01_59P-LC increased stability by 1.0 °C. The double mutant displayed increased stability by 2.5 °C, indicating possible structural synergy between the two mutations, as combined they enhance stability greater than either individual mutant. Results shown are averaged from 3 experimental replicates and normalized so that the starting 350/330 ratio is set to 0. See also Figures 5, 6, 8 and S6.
Figure 5.
Cryo-EM structure of Fab DFPH-a.01_10R59P-LC with BG505 DS-SOSIP Env trimer reveals mutations that improve affinity and stability to be located distal from the fusion peptide-epitope. (a) Details of DFPH-a.01_10R59P-LC recognition. BG505 DS-SOSIP trimer is shown in gray, fusion peptide is in red, heavy and light chains of DFPH-a.01_10R59P-LC Fab are in purple and light cyan, respectively. Fusion peptide is threaded between CDR-H1 and CDR-H3 with its N-terminus fixed by CDR-L3. (b) Location of mutations 10R and 59P in DFPH-a.01_10R59P-LC Fab. Both R10 and P59 are not in contact with the antigen. The shortest distances to the paratope for R10 and P59 are 23.4 A and 9.1 A, respectively. See also Figure S6, Figure S8, and Table S5.
Figure 5.
Cryo-EM structure of Fab DFPH-a.01_10R59P-LC with BG505 DS-SOSIP Env trimer reveals mutations that improve affinity and stability to be located distal from the fusion peptide-epitope. (a) Details of DFPH-a.01_10R59P-LC recognition. BG505 DS-SOSIP trimer is shown in gray, fusion peptide is in red, heavy and light chains of DFPH-a.01_10R59P-LC Fab are in purple and light cyan, respectively. Fusion peptide is threaded between CDR-H1 and CDR-H3 with its N-terminus fixed by CDR-L3. (b) Location of mutations 10R and 59P in DFPH-a.01_10R59P-LC Fab. Both R10 and P59 are not in contact with the antigen. The shortest distances to the paratope for R10 and P59 are 23.4 A and 9.1 A, respectively. See also Figure S6, Figure S8, and Table S5.
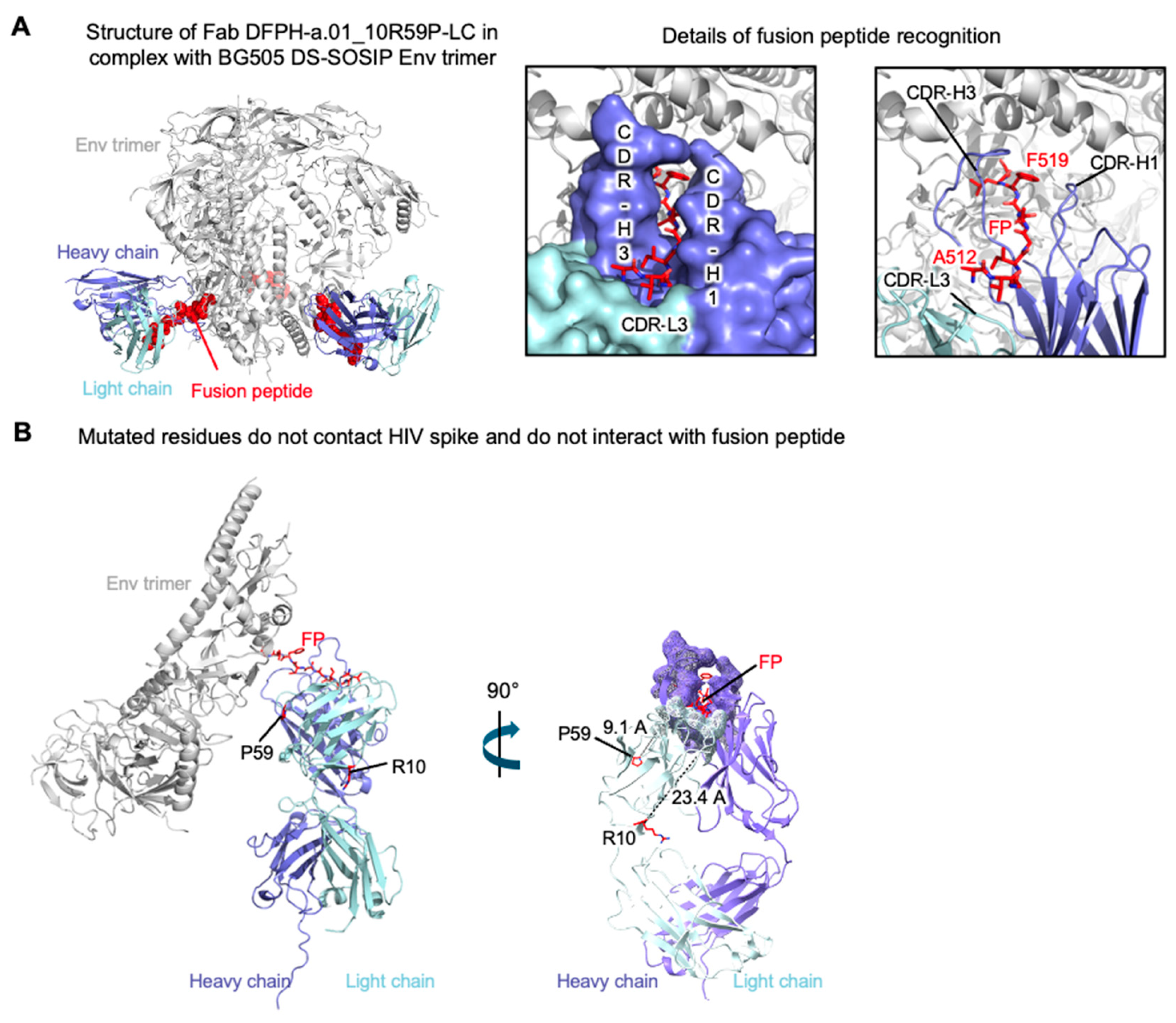
Figure 6.
Cryo-EM structural explanations: S10R influences elbow angle and S59P enables isosteric positioning. (a) Details of residue 10 interactions in DFPH-a.01_10R59P-LC in TRNM-b.01. The distances between 10R and other residues that are shorter than 5 Å are shown in green. (b) Overall view of Fabs, after variable domain superposition, highlighting different elbow angles for DFPH-a.01_10R59P-LC (which has 10R) and TRNM-1.b01 (which has 10S). (c) The conformation of CDR-L2 in DFPH-a.01_10R59P-LC, TRNM-b.01, DJ85-b.01, 0PV-c.01 and DFPH-a.15. The shortest antibody-antigen distances less than and more than 5 Å are shown in green and gray, respectively. The CDR-L2 of DFPH-a.01_10R59P-LC forms a weak contact with H85 and E87 of HIV trimer. The CDR-L2 of TRNM-b.01, DJ85-b.01, 0PV-c.01 and DFPH-a.15 do not interact with HIV trimer. See also Figure S6, Figure S7 and Table S6.
Figure 6.
Cryo-EM structural explanations: S10R influences elbow angle and S59P enables isosteric positioning. (a) Details of residue 10 interactions in DFPH-a.01_10R59P-LC in TRNM-b.01. The distances between 10R and other residues that are shorter than 5 Å are shown in green. (b) Overall view of Fabs, after variable domain superposition, highlighting different elbow angles for DFPH-a.01_10R59P-LC (which has 10R) and TRNM-1.b01 (which has 10S). (c) The conformation of CDR-L2 in DFPH-a.01_10R59P-LC, TRNM-b.01, DJ85-b.01, 0PV-c.01 and DFPH-a.15. The shortest antibody-antigen distances less than and more than 5 Å are shown in green and gray, respectively. The CDR-L2 of DFPH-a.01_10R59P-LC forms a weak contact with H85 and E87 of HIV trimer. The CDR-L2 of TRNM-b.01, DJ85-b.01, 0PV-c.01 and DFPH-a.15 do not interact with HIV trimer. See also Figure S6, Figure S7 and Table S6.
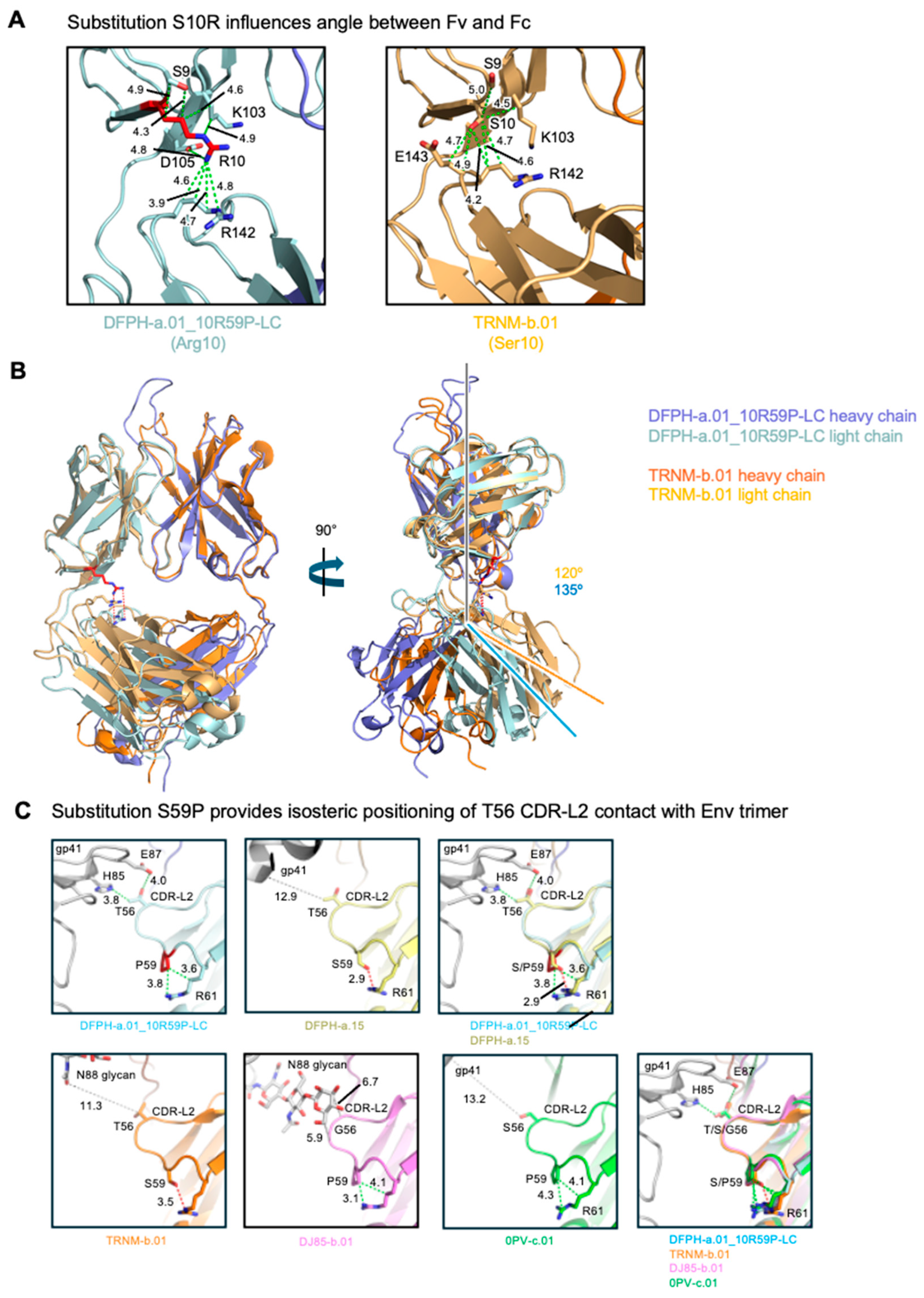
Figure 7.
Antibody DFPH-a.01_1G10A56K-LC: neutralization breadth, Env trimer affinity, stability, and comparison of DFPH-a.01 antibodies. (a) Dendrogram analysis shows that the engineered antibody DFPH-a.01_1G10A56K-LC increased both cross-clade recognition and neutralization against DFPH-a.01-resistant and sensitive strains. (b) 208-virus panel neutralization data showed enhanced breadth engineered antibody compared with the template against a globally representative panel of HIV-1 strains. Viral strains are grouped by FP sequence. We used IC50 (left) or IC80 (right) of 50 μg/mL to determine neutralization sensitivity against a pseudovirus strain. (c) Surface plasmon resonance analysis of DFPH-a.01_1G10A56K-LC binding to BG505 Env trimer. Binding analysis was performed using BG505 DS-SOSIP tethered to the sensor chip surface via 2G12, with DFPH-a.01 and its mutant tested at five concentrations ranging from 1.11-90 nM. The black traces represent experimental data, and the red lines represent the fit to 1:1 interaction model. The number in brackets show the error of the fit in the last significant digit. (d) DFPH_a.01 and DFPH_a.01_1G10A56K-LC were assessed for stability by monitoring change to the 350/330 ratio from intrinsic fluorescence of tryptophan during melting. (e) Summary of single- and multi-mutations with increased neutralization breadth compared to template DFPHa-01. ND-not determined. See also Figures S4 and S5, and Tables S3, and S4.
Figure 7.
Antibody DFPH-a.01_1G10A56K-LC: neutralization breadth, Env trimer affinity, stability, and comparison of DFPH-a.01 antibodies. (a) Dendrogram analysis shows that the engineered antibody DFPH-a.01_1G10A56K-LC increased both cross-clade recognition and neutralization against DFPH-a.01-resistant and sensitive strains. (b) 208-virus panel neutralization data showed enhanced breadth engineered antibody compared with the template against a globally representative panel of HIV-1 strains. Viral strains are grouped by FP sequence. We used IC50 (left) or IC80 (right) of 50 μg/mL to determine neutralization sensitivity against a pseudovirus strain. (c) Surface plasmon resonance analysis of DFPH-a.01_1G10A56K-LC binding to BG505 Env trimer. Binding analysis was performed using BG505 DS-SOSIP tethered to the sensor chip surface via 2G12, with DFPH-a.01 and its mutant tested at five concentrations ranging from 1.11-90 nM. The black traces represent experimental data, and the red lines represent the fit to 1:1 interaction model. The number in brackets show the error of the fit in the last significant digit. (d) DFPH_a.01 and DFPH_a.01_1G10A56K-LC were assessed for stability by monitoring change to the 350/330 ratio from intrinsic fluorescence of tryptophan during melting. (e) Summary of single- and multi-mutations with increased neutralization breadth compared to template DFPHa-01. ND-not determined. See also Figures S4 and S5, and Tables S3, and S4.
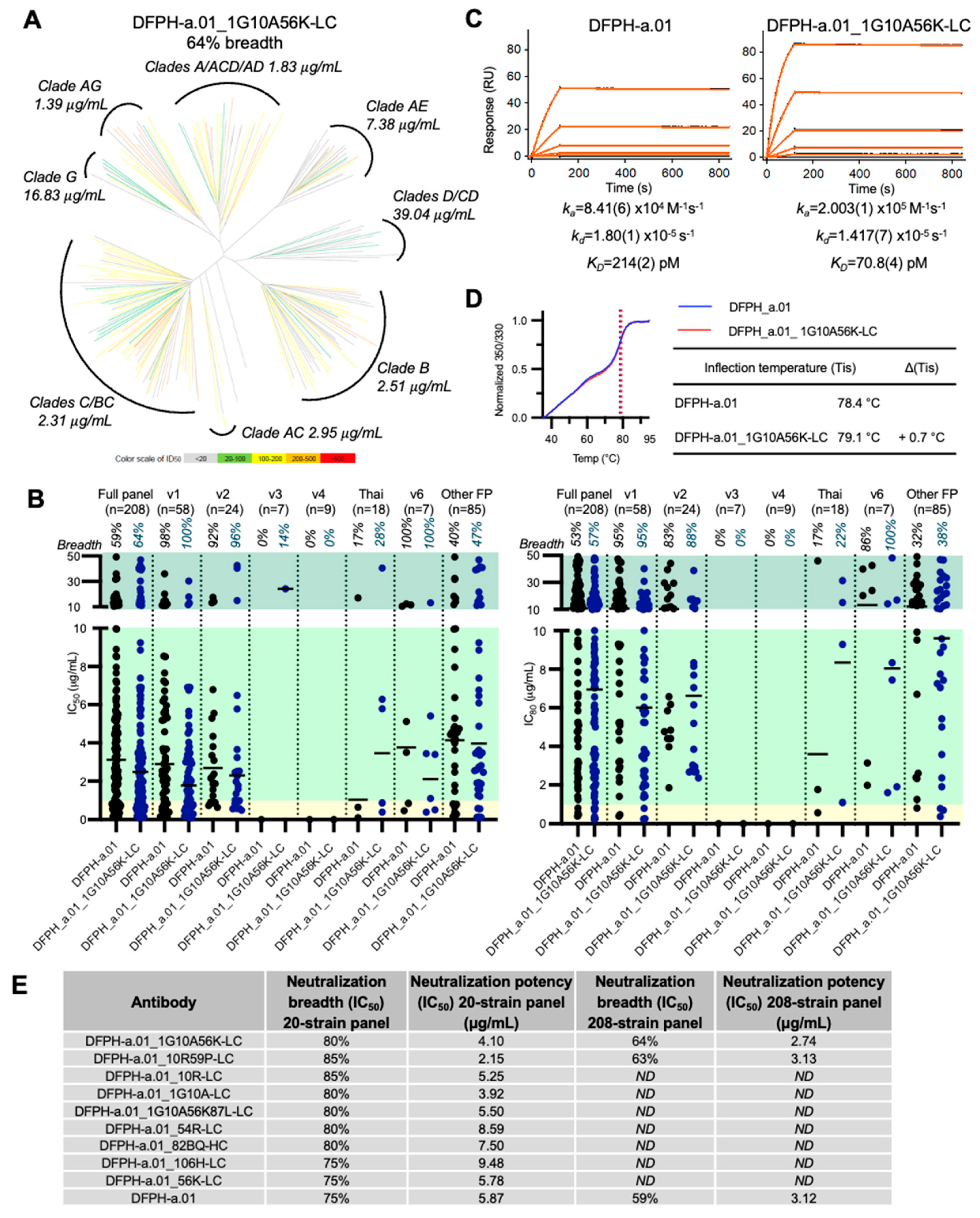
Figure 8.
Paratope distance and mutation frequencies for yeast display-identified mutations that improve broadly neutralizing antibodies for macaque, mouse, and human. (a) Paratope-distal mutations that enhance vaccine-elicited antibodies isolated from macaque, human, and mouse. (b) Overview of mutations observed in anti-FP antibodies. Top (most protective) mutations are shown in red. Tick marks were used for vFP16.02 because mutation prevalence data is not available for mouse germline genes. * Indicates a new contact region established by the heavy chain mutation E2K. Only contact residues with BSA (Buried Surface Area) equal/higher than 15 Å2 were indicated. Mutations are indicated in Kabat numbering. (c) Mutations frequency and distance from paratope for identified mutations. See also Figure S4, and Table S4.
Figure 8.
Paratope distance and mutation frequencies for yeast display-identified mutations that improve broadly neutralizing antibodies for macaque, mouse, and human. (a) Paratope-distal mutations that enhance vaccine-elicited antibodies isolated from macaque, human, and mouse. (b) Overview of mutations observed in anti-FP antibodies. Top (most protective) mutations are shown in red. Tick marks were used for vFP16.02 because mutation prevalence data is not available for mouse germline genes. * Indicates a new contact region established by the heavy chain mutation E2K. Only contact residues with BSA (Buried Surface Area) equal/higher than 15 Å2 were indicated. Mutations are indicated in Kabat numbering. (c) Mutations frequency and distance from paratope for identified mutations. See also Figure S4, and Table S4.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



