
Submitted:
22 September 2025
Posted:
22 September 2025
You are already at the latest version
Abstract
Oxidative stress (OS), resulting from an imbalance between reactive oxygen species (ROS) generation and antioxidant defenses, plays a pivotal role in vascular diseases such as atherosclerosis and hypertension. ROS derived from NADPH oxidase, mitochondria, and xanthine oxidase promote endothelial dysfunction by inducing lipid and protein oxidation, apoptosis, and pro-inflammatory signaling, thereby enhancing smooth muscle proliferation and atherogenesis.
This review summarizes the molecular mechanisms linking OS to vascular injury and highlights the interplay between ROS and inflammation in the progression of cardiovascular disease. Particular attention is given to biomarkers of oxidative stress, including those assessing antioxidant enzyme activity and oxidative damage products, which hold potential for clinical use. However, challenges remain regarding their standardization and variability influenced by biological and environmental factors.
Therapeutic strategies targeting OS, including dietary and pharmacological antioxidants, show promise in improving vascular health, although clinical outcomes have been inconsistent. Personalized approaches based on accurate redox profiling may enhance efficacy.
Overall, OS is a central mediator in vascular pathology, and progress in biomarker validation and targeted therapies will be essential to translate current knowledge into effective prevention, diagnosis, and treatment of cardiovascular diseases.
Keywords:
reactive oxygen species (ROS)
; cardiovascular disease
; endothelial dysfunction
; vascular inflammation
; biomarkers
1. Introduction
Over the past decades, research on oxidative stress (OS) has provided deeper insights into its impact on health and the development of various pathologies. It has been demonstrated that reactive oxygen species (ROS) are implicated in the pathogenesis of numerous neurodegenerative diseases, cancer, metabolic disorders, and notably, various cardiovascular diseases such as coronary ischemic disease, heart failure, arterial hypertension, and arteriosclerosis [1].
OS is defined as a cellular state where the oxidation-reduction homeostasis is disrupted, indicating an imbalance between ROS production and the body's capacity to detoxify them. These ROS include free radicals like superoxide, characterized by an unpaired electron in their outer atomic orbital, rendering them highly reactive and unstable. This chemical structure enables them to interact extensively with cellular macromolecules such as carbohydrates, lipids, proteins, and nucleic acids [2,3].
The human body has natural antioxidant mechanisms to neutralize ROS. However, excessive ROS production or diminished antioxidant capacity can lead to significant cellular damage. In cardiovascular and metabolic diseases, OS is a key factor in their pathophysiology. Under normal conditions, ROS perform essential functions, such as cellular signaling and pathogen defense. Nevertheless, sustained elevation of ROS can contribute to the development and progression of various pathologies. For example, in arterial hypertension and atherosclerosis, excessive ROS can induce endothelial dysfunction, chronic inflammation, and arterial wall damage. Similarly, in metabolic disorders such as diabetes, OS plays a crucial role in the development of insulin resistance and associated microvascular and macrovascular complications [3,4,5].
OS serves not only as a mechanism of cellular damage but also as a central mediator in the progression of numerous chronic diseases. Understanding its underlying mechanisms and its relationship with specific disorders is essential for developing effective preventive and therapeutic interventions.
Currently, new therapeutic strategies targeting OS modulation include the use of exogenous antioxidants, enhancements in endogenous antioxidant defenses, and ongoing research into new biochemical markers to identify, prevent, and reverse oxidative damage [5,6].
Furthermore, Oxidative stress biomarkers are being developed that may play a promising role in the treatment or diagnosis of diseases where oxidative stress plays a significant role. However, further research is needed to overcome the limitations these markers present, as we will discuss later.
2. Genesis of Reactive Oxigen Species
In aerobic metabolism, between 1% and 5% of the oxygen consumed undergoes partial, incomplete reduction, forming reactive intermediate species, known as ROS. These include the radical anion superoxide (O2-) and hydrogen peroxide (H2O2), which may or may not have unpaired electrons (radical or non-radical species), but are universally characterized by their high instability and reactivity. Once formed, they can further react to produce more complex species such as peroxynitrite (ONOO-) and hypochlorous acid (HOCl), as well as lipid peroxyl radicals.
These reactive species originate from numerous metabolic processes, prominently the mitochondrial electron transport chain. Another reactive species, hydroxyl radical (OH·), considered the most cytotoxic of all ROS, can also form and contribute to oxidative damage. The highly reactive nature of ROS enables them to initiate chain reactions to acquire electrons and stabilize.
ROS are generated in various oxidative metabolism reactions, either as reaction intermediates or products thereof. The activation of neutrophils and macrophages leads to the formation of O2·- and related species, underpinning their biocidal effects [7,8]. O2·- dismutation yields non-radical hydrogen peroxide or oxygenated water (Figure 1).
The role of the O2·- in generating hydroxyl radicals (OH·) has significant biological implications. Hydroxyl radicals are much more reactive than O2·-. The formation of hydroxyl radicals occurs through the Fenton reaction.
All types of vascular cells produce ROS, including endothelial cells, smooth muscle cells, fibroblasts, adventitial cells, adipocytes and phagocytic cells [12,13].
In vascular tissue, the main endogenous sources of ROS generation are (Figure 2):
- Mitochondria, where O2·- and H2O2 are produced from respiratory chain complexes I and II, representing 80% of basal O2·- production. Complexes I and III were traditionally considered most active in ROS generation, but recent studies indicate high activity in complex II as well, with current understanding suggesting similar activities between complexes I and II, and the highest activity in complex III [7,14].
- Nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase or NOX), a multiprotein complex crucial for ROS production in various cells and tissues, especially phagocytic cells (neutrophils and macrophages), involved in pathogen elimination and inflammatory processes [15].
- Xanthine oxidase, an enzyme in purine catabolism catalyzing the oxidation of hypoxanthine to xanthine and then to uric acid, also associated with arteriosclerosis due to its activation of inflammatory pathways, such as NF-κB, which increase vascular inflammation and exacerbate endothelial damage, thereby promoting the progression of atherosclerosis [12].
- Nitric oxide (NO·) production by endothelial nitric oxide synthase (eNOS), which oxidizes L-arginine to citrulline in the presence of calmodulin to form NO·, using BH4 as a cofactor. BH4 oxidation can lead to non-enzymatic O2·- production, limiting eNOS's ability to produce free NO· in the absence of SOD. This enzyme is the major vascular producer of NO·, but OS can cause uncoupling of eNOS, resulting in O2·- production instead of NO·, due to BH4 oxidation and subsequent deficiency [16,17].
- Inducible nitric oxide synthase (iNOS), predominantly producing NO· from arginine, but BH4 deficiency promotes superoxide generation instead of NO·, similar to eNOS.
The mitochondrial electron transport chain is the primary energy source in cardiac cells, driving oxidative-reduction processes that generate ROS. Cells that utilize oxygen for energy also produce ROS, through enzymatic reduction. Enzymes such as NADH/NADPH oxidase, xanthine oxidase, lipoxygenase, cyclooxygenase, and nitric oxide synthase catalyze these reactions. Furthermore, fatty acid oxidation in peroxisomes produces hydrogen peroxide, a significant ROS. [7,15,18,19].
3. Antioxidant Systems and Mechanisms
Just as there are prooxidant molecules, a parallel system of enzymes and molecules known as antioxidants exists. These substances prevent or inhibit the oxidation of other molecules by donating electrons, stabilizing unpaired electrons, or chelating metal ions within their structure. Both endogenous and exogenous antioxidants aim to stabilize reactive species, transforming them into more stable molecules or structures. Under physiological conditions, antioxidant concentrations are significantly higher than those of reactive species, ensuring continuous but controlled ROS formation. However, when ROS production becomes excessive and unregulated, endogenous antioxidant enzymes may not be sufficient to neutralize them. Antioxidant systems are classified into enzymatic and non-enzymatic systems based on their mechanisms of action [1,2,21].
Regarding enzymatic systems, effective antioxidant protection requires the synchronized action of the three main enzymes: Superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx).
- SOD catalyzes the dismutation of superoxide radicals (O2·-) to hydrogen peroxide (H2O2). Despite being more stable, H2O2 remains highly reactive. Different molecular variants of SOD have been identified: Cu/Zn-SOD (cytosolic, SOD1), Mn-SOD (mitochondrial, SOD2), and Cu/Zn-SOD (extracellular, SOD3) [2,9,22].
- Catalases catalyze the dismutation and peroxidation of H2O2 into water and oxygen. This is one of the fastest known catalytic activities, making catalases highly effective antioxidants against H2O2 [23].
- GPx removes hydroperoxides and organic peroxides, simultaneously oxidizing its physiological substrate, glutathione (GSH), to oxidized glutathione (GSSG). There are several molecular variants (GPx1 to GPx5), both selenium-dependent (tetrameric) and selenium-independent (dimeric) [24].
In addition to these enzymes, other low molecular weight molecules exhibit non-enzymatic antioxidant activity. Notably, ascorbic acid (vitamin C) reduces ROS to water, with its oxidized species being non-reactive. Vitamin E (alpha-tocopherol) degrades peroxides to hydroperoxides, which can then be further degraded by reduced glutathione (GSH), one of the most effective endogenous antioxidants. GSH detoxifies xenobiotics via GSH-transferase, maintains the reduced state of many proteins, and neutralizes OH·. Flavonoids also degrade peroxides to non-reactive forms, and carotenoids function as antioxidants due to their system of conjugated double bonds [25,26,27].
Furthermore, proteins capable of metal coordination, such as ferritin, transferrin, albumin, or ceruloplasmin, along with repair mechanisms mediated by enzymes that eliminate or repair biomolecular damage, and molecules like carotenoids, vitamins, flavonoids, and enzymes such as SOD and other metallothioneins, remove excess ROS through enzymatic or antioxidant compound action [27,28]. Heat shock proteins (HSPs) constitute another defense mechanism by safeguarding cellular proteins from OS induced damage, through facilitation of proper folding and repair of damaged proteins [29].
Continuous research in this domain remains essential for the development of novel therapeutic strategies aimed at augmenting endogenous antioxidant defenses and mitigating the deleterious effects of oxidative stress on human health [3].
4. Oxidative Stress and Cardiovascular Disease Pathogenesis
4.1. Endothelial Dysfunction
The endothelium, the inner layer of blood vessels, plays a key role in homeostasis. In response to various stimuli, it releases vasodilatory, anticoagulant, and anti-inflammatory factors like NO, and vasoconstrictive and pro-aggregatory factors such as endothelin-1 (ET-1) and Angiotensin-II [30,31].
OS induces endothelial dysfunction and activates pro-inflammatory pathways, crucial in diseases like arteriosclerosis and hypertension. OS affects endothelial function, diminishing its protective role and promoting vasoconstrictive and atherogenic mediators. Endothelial dysfunction is common in many cardiovascular diseases, including hypertension, arteriosclerosis, heart failure, stroke, and complications of metabolic disorders like diabetes, obesity, and metabolic syndrome [24,32,33].
The effects of ROS on cardiovascular function depend on their concentrations and antioxidant production. Key antioxidants include GSH and thioredoxin (Trx), which detoxify H2O2. Elevated thiol levels protect cells from OS induced death, while depletion leads to disease states [33,34].
NADPH oxidase, found in endothelial cells, vascular smooth muscle cells, and fibroblasts (NOX 1, NOX 2, NOX 4, and NOX 5), is a primary source of O2·- in the vessel wall. Physiologically, it has low activity but is upregulated by angiotensin II, ET-1, and urotensin II, increasing ROS production and contributing to cardiovascular disease [7,8,16,35].
NO· acts as a crucial paracrine regulator of vascular tone, exerting potent vasodilatory effects by increasing cGMP levels through guanylate cyclase activation. Reduced NO· availability in metabolic disorders like hyperglycemia, diabetes, and hypertension leads to endothelial dysfunction and cardiovascular diseases. O2·- generated from these conditions react with NO· to form peroxynitrite, exacerbating endothelial barrier dysfunction and promoting LDL accumulation and leukocyte adhesion in arterial walls, triggering inflammation [36].
NADPH oxidase-driven O2·- production further reduces NO· levels by forming ONOO-, disrupting endothelial function and eNOS activity by oxidizing BH4. This shift favors O2·- production over NO·, contributing to hypertension, atherosclerosis, and other cardiovascular disorders. Xanthine oxidase, activated by NADPH oxidase, amplifies O2·- production, exacerbating OS. Myeloperoxidase catalyzes hypochlorite production, intensifying inflammation and arterial damage, promoting atherosclerotic plaque formation and cardiovascular disease progression [5,37].
The alterations caused by OS disrupt normal endothelial function, leading to an imbalance between vasoconstrictive and vasodilatory substances. This disrupts blood flow regulation, leukocyte adhesion, platelet aggregation, heat shock production, and cell growth control, resulting in changes in vascular diameter and remodeling. These alterations affect the anticoagulant and proinflammatory properties of the endothelium [8,20,38].
Endothelial dysfunction is implicated in the pathophysiology of various cardiovascular diseases, including hypertension. It is characterized by impaired endothelial functions related to vasodilation, increased proinflammatory state, and enhanced prothrombotic activity, collectively contributing to vascular inflammation [12,18].
4.2. Mitochondrial Dysfunction
Mitochondrial ROS production is considered the most significant source of free radicals under physiological conditions due to the electron transport chain located in the inner mitochondrial membrane. These radicals are continuously generated during oxidative phosphorylation and ATP production [39].
Mitochondrial dysfunction in cardiovascular diseases can manifest in several ways:
- Increased ROS Production: Continuous production of O2·- and other ROS in mitochondria increases oxidative damage to biomolecules, particularly nuclear and mitochondrial DNA (mtDNA). mtDNA instability is crucial in mitochondrial dysfunction, leading to mutagenic or cytotoxic effects and subsequent mutations after DNA replication. The accumulation of mtDNA mutations contributes to tissue function loss, including in the heart [40,41,42,43]. Animal models have confirmed this increased mtDNA instability [44].
- Calcium Homeostasis Imbalance: Mitochondria regulate intracellular calcium concentration, essential for cardiac contraction. Mitochondrial dysfunction disrupts calcium homeostasis, affecting cardiac contractility and potentially contributing to arrhythmias [45].
- Inflammation: Dysfunctional mitochondria can activate inflammatory responses in cardiac and other cardiovascular cells, contributing to the progression of diseases such as atherosclerosis and ischemic heart disease. [47]
4.3. Oxidative Modifications Induced by ROS
ROS cause cellular damage to various biomolecules. The primary cytotoxic effects of oxidative/nitrosative stress result from the interaction of oxygen and nitrogen radicals with cell membrane, lipids, proteins, and nucleic acids [48].
Lipid Peroxidation and LDL Oxidation:
Oxidation of low-density lipoproteins (LDL) is a free radical-mediated process causing significant structural changes. The initial event is the peroxidation of polyunsaturated fatty acids (PUFA) in LDL particles. This peroxidation alters membrane properties, potentially inactivating membrane receptors or enzymes, and affecting normal cellular function. Oxidized LDL (oxLDL) stimulate vascular ROS formation, creating a vicious cycle. Myeloperoxidase, expressed in macrophages within atherosclerotic lesions, primarily catalyzes the complete oxidation of LDL, along with glycosylases. The number, composition, and oxidation susceptibility of LDL particles also play crucial roles [49,50].
Stages of lipid peroxidation:
- Initiation: ROS extract a hydrogen ion (H+) from a PUFA double bond, forming conjugated dienes (CD). Antioxidants in LDL particles initially halt this oxidation.
- Propagation: Once antioxidants are depleted, another H+ is abstracted by a peroxyl radical (LOO.) from a PUFA, forming lipid hydroperoxides. This increases LDL's negative charge due to Schiff base formation between positively charged amino groups and aldehyde groups, leading to macrophage recognition via scavenger receptors in arterial intima.
Endothelial Damage and Inflammation:
Endothelial lesions facilitate LDL entry into the vascular intima, where oxidation triggers an inflammatory response, causing cytokine expression (e.g., IL-4, IL-1β, TNF-α) and adhesion proteins (e.g., VCAM-1, ICAM-1, P-selectin). These allow monocyte migration and transformation into macrophages and foam cells, after phagocytosing oxLDL, promoting atheroma plaque formation and its physiological consequences [5,53].
Lipid Peroxidation Propagation:
Lipid peroxidation rapidly propagates through plasma membranes, and the mutagenic potential of its products makes this mechanism significant in OS toxicity [5,53].
High-density lipoproteins (HDL):
Undergo oxidative modifications that impair their anti-atherogenic properties. Oxidation by agents like copper or acrolein reduces HDL's cholesterol transport activity and cellular cholesterol efflux via the ATP Binding Cassette A1 (ABCA1) transporter. Specifically, oxidation by malondialdehyde and myeloperoxidase at the methionine 112 residue of apolipoprotein A1 (Apo A1) diminishes HDL's capacity to inactivate lipid hydroperoxides. Reactive carbonyl-induced covalent modifications further impair HDL-mediated cholesterol efflux [54,55,56,57,58].
5. Consequences of Oxidative Stress: Atherosclerosis, Heart Failure, Ischemic Heart Disease, Diabetic Cardiomyopathy
ROS play a key role in the pathogenesis of hypertension, atherosclerosis, heart failure, stroke, cardiac hypertrophy, and vascular complications of various metabolic diseases such as diabetes, obesity, and metabolic syndrome. However, despite the well-known involvement of OS in certain cardiovascular diseases, the molecular mechanisms remain not fully elucidated.
5.1. Atherosclerosis
Atherosclerosis begins with the infiltration of oxLDL into the arterial wall through the endothelium. This infiltration triggers inflammatory processes involving ROS production by macrophages, which, along with oxLDL, contribute to the formation of atheromatous plaques and foam cells, exacerbating vascular damage [33,59,60].
Furthermore, non-coding RNAs play a crucial role in regulating OS, inflammation, and apoptosis in the development of atherosclerosis. Factors such as high glucose, ANG II, and OS lead to endothelial dysfunction, which is aggravated by the alteration of signaling pathways, including non-canonical Wnt and PI3K/Akt/NOS pathways. This dysfunction triggers the adhesion and infiltration of monocytes, transforming into inflammatory macrophages (M1) under high OS conditions. M1 macrophages, through TLR and NFκB activation, release inflammatory cytokines and oxidize LDL, intensifying inflammation and OS in the arterial wall and promoting the formation of atherosclerotic plaques. Thus, atherosclerosis becomes a self-amplifying cycle of endothelial dysfunction, OS, and inflammation, contributing to the development and progression of cardiovascular diseases (Figure 3) [61].
5.2. Hypertension
Hypertension triggers an inflammatory response due to OS, disrupting the redox balance in endothelial cells. This imbalance induces the expression of vascular adhesion molecules, accelerating atherosclerosis. ROS, such as O2·-, reduce the availability of NO, which is essential for arterial smooth muscle relaxation (62). Additionally, ROS increase the synthesis of ET-1, causing vasoconstriction and elevated blood pressure. ROS also induce apoptosis in endothelial cells, particularly in resistance arteries, and are involved in various pathological mechanisms of stroke. Hypertension stimulates OS, directly correlating with blood pressure levels and target organ damage. Moreover, these species impair endothelium-mediated vascular function, arterial elasticity, and the vascular response to vasodilators, contributing to atherogenic vascular remodeling and chronic hypertension. Activation of angiotensin I receptors triggers NADPH oxidase, generating an oxidative stimulus, crucial for the pathogenesis of hypertension and atherosclerosis. Furthermore, OS is associated with structural changes in resistance arterioles and premature endothelial cell senescence [59,63].
5.3. Heart Failure
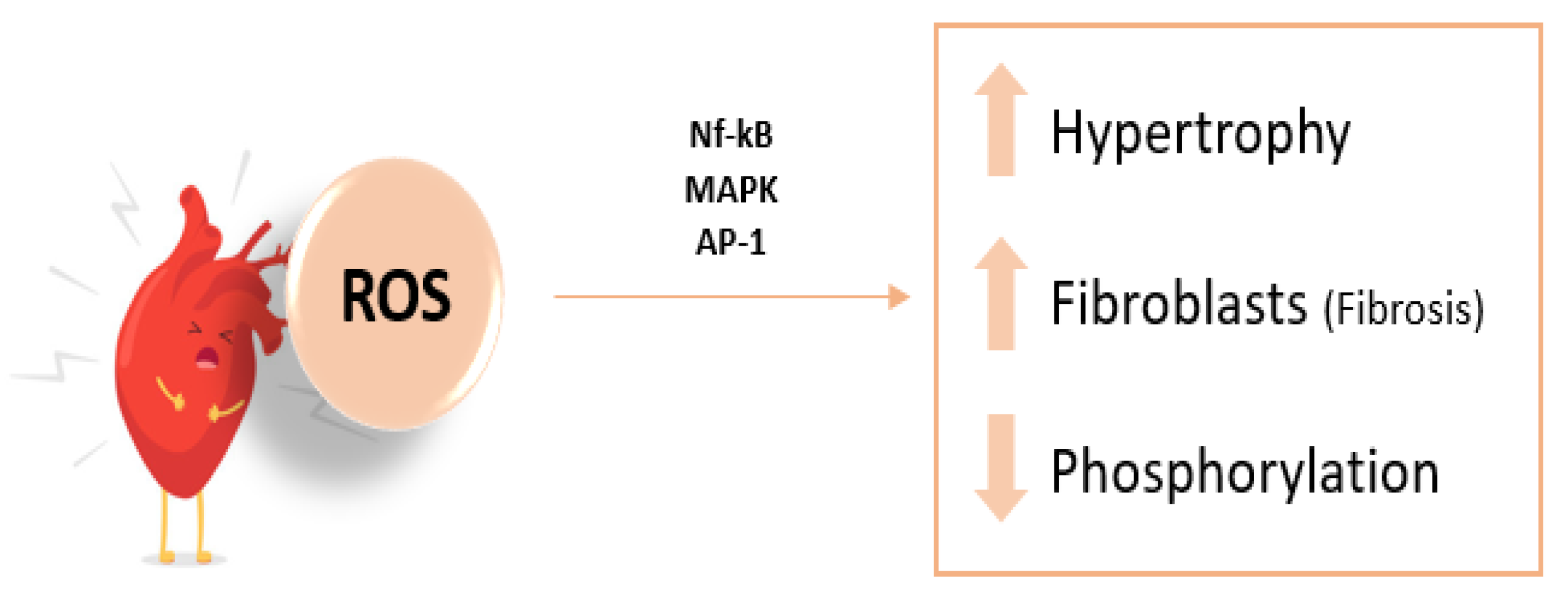
ROS activate various kinases and transcription factors that promote myocardial growth, matrix reorganization, and cellular dysfunction [33,64]. They induce hypertrophy in ventricular myocytes and affect the extracellular matrix by stimulating cardiac fibroblast proliferation and matrix metalloproteinase activation, which are relevant in fibrosis and matrix remodeling. Several studies in animal models have demonstrated that the administration of antioxidants can prevent pathological phenomena associated with heart failure, such as myocyte hypertrophy, apoptosis, ischemia, and reperfusion [65].
Other effects of free radicals in the pathogenesis of HF, through the xanthine oxidase pathway, include endothelial dysfunction and myocardial injury. Increased ROS production can lead to decreased NO bioavailability and cytokine-mediated myocardial contractile dysfunction, which inactivates the sarcoplasmic Ca2+-ATPase, thereby altering calcium homeostasis (Figure 4) [66,67].
5.4. Diabetic Cardiomyopathy
Diabetes mellitus (DM) is a primary risk factor for the development of atherosclerosis, closely associated with increased OS. Hyperglycemia, characteristic of both type 1 and type 2 DM, triggers ROS through various mechanisms, including glucose auto-oxidation and non-enzymatic glycation of proteins. Pathological processes exacerbated in DM, such as the increased production of advanced glycation end-products (AGEs) and their interaction with endothelial and smooth muscle cells, enhance ROS production and activate nuclear factor kappa B (NF-κB), contributing to increased inflammation and thrombosis in the vascular endothelium. Consequently, endothelial dysfunction is observed, characterized by reduced NO bioavailability and calcium homeostasis alterations, leading to increased arterial stiffness and impaired vascular reactivity. These phenomena promote the progression of atherosclerosis and associated cardiovascular complications, underscoring the importance of glycemic control in mitigating these pathological effects [67,68].
Parameters of OS in Cardiovascular Disease
OS biomarkers are valuable tools for the development of new preventive, diagnostic, and therapeutic strategies aimed at preventing or delaying the onset of pathologies such as arteriosclerosis and cardiovascular diseases. The identification of oxidative damage in cells and tissues, and the monitoring of responses to antioxidant treatments and other therapeutic interventions [69,70,71,72,73,74,75,76,77,78,79].
The first studies, emerging in the 1980s, introduced a series of markers that, directly or indirectly, provide information about reactive oxygen and nitrogen species in the body.
The use of biomarkers arose from the discovery of the role of oxidative stress in certain diseases. Their goal is to develop new diagnostic, therapeutic, and preventive strategies for complications such as atherosclerosis and other cardiovascular diseases [70].
The biochemical markers potentially applicable for determining the redox status must meet a series of characteristics [71]
Be a product of oxidative damage that can be related to the onset or progression of a pathology.
- Be accessible in a target tissue or plasma, reflecting oxidative changes in a quantitative manner.
- Be a specific marker for the study of reactive species and independent of external factors.
- Be sensitive, robust, and reproducible.
- Be stable for sample handling, including processing, analysis, and storage.
One of the shortcomings found in clinical trials with antioxidant products is the inadequate selection of oxidative stress markers in biological fluids (plasma, whole blood, urine, or others). Most of the published studies consider measuring the concentration of the antioxidant product itself in a biological fluid, which does not provide information about the intensity of the process related to oxidative stress [70].
Due to the complexity of diseases associated with oxidative stress, it is highly unlikely that a single oxidative stress biomarker could replace the results of a clinical diagnosis.
Although there are direct techniques for determining reactive oxygen and nitrogen species and other free radicals, such as Electron Paramagnetic Resonance (EPR) and the spin trapping method—considered the gold standard—this technique is complicated and not easily applicable, as free radicals are highly reactive and have a very short half-life. Therefore, the most common approach is the determination of biomarkers using indirect techniques, where reactive oxygen species are captured by an appropriate reagent to form a stable chemical entity that is then analyzed by gasometric, spectrophotometric, immunoenzymatic (ELISA), and chromatographic techniques [72].
Lipid peroxidation, which is important in atherosclerosis, inflammation, and mitochondrial functions, is a complex process consisting of three stages: initiation, propagation, and termination. For each stage, methods are available to quantify the progression of the process.
For example, since lipid peroxidation causes the loss of substances such as chains of unsaturated fatty acids, measuring lipid content can indicate the peroxidation of compounds. Additionally, because oxygen is consumed during the propagation stage, measurements of its uptake using oxygen electrodes can serve as a tool to evaluate the progress of oxidation. Another approach involves measuring the formation of peroxide during the process. Among the methods developed for this purpose, some determine the total peroxide concentration, while others determine the concentration of peroxide, which may indicate that the fatty acid is undergoing the peroxidation process. Through the abstraction of a hydrogen atom by a reactive oxygen or nitrogen species, a reorganization of the fatty acid occurs, leading to the formation of a free radical characterized by the formation of a conjugated diene, which can be monitored by spectroscopic methods [73,74].
Numerous publications refer to oxidative stress biomarkers targeted at specific enzymes or damage products that are used in the research of the pathophysiology of toxic or neurodegenerative diseases (such as cancer) or are applied to the development of new drugs. Below are some of these biomarkers potentially useful for studying oxidative stress processes associated with cardiovascular pathology.
Malondialdehyde (MDA): MDA is a ketoaldehyde formed from the breakdown of lipids containing polyunsaturated fatty acids, derived from arachidonic acid metabolism primarily found in cell membranes. Tissue damage can elevate MDA concentrations, which can react with lysine residues, leading to protein alterations that trigger immune mechanisms associated with cardiovascular diseases such as arteriosclerosis or acute myocardial Infarction [81]. The determination of this marker can be performed using colorimetric, immunoenzymatic, or HPLC techniques. [72]
Acrolein and 4-Hydroxy-2-nonenal (HNE): Acrolein, also known as 2-propenal, is a highly reactive unsaturated aldehyde present in the environment from oil combustion, tobacco, gasoline, and petroleum. HNE, derived from acrolein, is another aldehyde with higher toxicity produced by ROS and RNS acting on arachidonic, linoleic, and linolenic acids. Elevated acrolein and HNE levels in pathological lipid peroxidation react with lysine, histidine, and cysteine residues. HNE can interact with phospholipids, proteins, and nucleic acids, acting as a second messenger in cytotoxicity, mutagenesis, genetic toxicity, and apoptosis [82]. The most commonly used techniques for its determination are HPLC or GC-MS [72].
Isoprostanes: Isoprostanes are molecules generated by non-enzymatic lipid peroxidation that release free radicals reacting with arachidonic acid. They are found in various biological fluids, with plasma and urine being the most analyzed for oxidative damage studies. [83]. Its determination can be carried out using colorimetric, fluorometric, or immunoenzymatic techniques [72].
Myeloperoxidase: Its quantification can be done using ELISA assays, but sample collection, handling, and processing significantly influence results, such as using heparinized samples or heparin-containing tubes.
Oxidized LDL Determination: Specific monoclonal antibodies recognizing oxidation-specific epitopes allow for the determination of oxidized LDL, alongside ELISA methods.
Thiols: Early appearance in cardiovascular disease, total thiols represent the overall state of vascular redox control. Their increase correlates with cardiovascular events in middle-aged individuals (45-60 years), although more studies are needed to establish a clear relationship with acute myocardial Infarction [84].
Glutathione and S-glutathionylated proteins: Since blood concentrations of glutathione reflect the status of glutathione in other less accessible tissues, measurements of reduced glutathione (GSH) and glutathione disulfide (GSSG) in blood are considered essential as an index of the GSH status throughout the body and are a useful indicator of oxidative stress status in humans. [75]
Antioxidant Enzymes and Redox Balance: Studies suggest measuring antioxidant enzyme activity as biomarkers of oxidative damage or establishing ratios between oxidative damage markers and antioxidant enzyme activity to assess overall redox status [79,80,81,85].
In the same way, in addition to being able to determine the concentration of products from reactions involving oxidative stress, it is also possible to assess the antioxidant capacity of the body. For example, this can be done by measuring the activity of antioxidant enzymes (Superoxide dismutase, glutathione peroxidase, catalase, or xanthine oxidase) using colorimetric methods or ELISA methods. Non-enzymatic antioxidants can also be analyzed, providing more information on the oxidative process, such as Glutathione, ascorbic acid, or trace elements like Zinc, Selenium, Manganese, among others, using colorimetric techniques or flame photometry, respectively. [72]
One of the most commonly used approaches is the determination of the total antioxidant activity of a biological site. The determination of the ratio between the oxidant and the reductant (for example, ascorbate-dehydroascorbate or reduced glutathione-oxidized glutathione) is an indicator of oxidative damage. The loss of an antioxidant molecule causes changes in the concentration of other antioxidant molecules, which can be evaluated through biochemical, immunohistological, spectroscopic, or electrochemical techniques. The GSH/GSSG ratio is not influenced by external factors or specific damage conditions in the body (as MDA might be, which depends on lipid peroxidation). Glutathione is an essential component in cells, and its relationship with reactive oxygen remains relatively constant across a wide range of physiological conditions. This makes it a more stable and reliable marker for the overall antioxidant status. The GSH/GSSG ratio reflects a dynamic equilibrium, as its proportion changes immediately with fluctuations in oxidative stress or pathophysiological conditions. This type of measurement can provide more functional data on the cellular health status over time. [76]
The biological reference intervals estimated by a laboratory depend on the socio-biological characteristics of the reference population and the availability of a measurement method.
The identification and development of specific oxidative stress biomarkers are important, as this process is crucial for certain human diseases, including cardiovascular diseases. The initial issue that arises in the context of the applicability of oxidative stress biomarkers in clinical practice is that any result lacks validity if it cannot be compared with reference values. These values are established after understanding the distribution of the reference population's figures. Unfortunately, reference values for these types of markers are still unknown. Factors such as age, gender, nutritional status, or health condition can affect these biomarkers, as well as the lack of standardization in measurement techniques, as there is no widely accepted consensus on the most appropriate analytical method for determining these markers. Different analysis techniques may generate results that are not comparable with one another, making clinical interpretation more difficult. Although these biomarkers may indicate cellular damage, it is not always clear to what extent they reflect the progression of specific diseases. Furthermore, in diseases like cardiovascular conditions, oxidative stress is just one of many contributing factors, further complicating the interpretation of these markers as specific indicators of pathology. Additionally, it should be emphasized that many of these oxidative stress biomarkers are highly reactive and can break down quickly in uncontrolled conditions. This means that proper sample handling and storage are essential to obtain reliable results. For all these reasons, currently, the applicability of oxidative stress biomarkers is limited to in-house methods used by a few laboratories, which employ their own reference values based on their study population [72,77,78].
6. Therapeutic Strategies to Reduce Oxidative Stress
Therapeutic strategies targeting OS in cardiovascular disease, particularly arteriosclerosis, aim to influence in its pathogenic mechanisms including inflammation and OS (OS). Various studies have demonstrated that angiotensin-converting enzyme inhibitors (ACEIs) such as Captopril, and calcium channel blockers possess antioxidant properties. Their vascular protective effect is mediated by reducing OS through thiol groups, positively regulating eNOS expression, and inhibiting NADPH oxidase activity, thus restoring vascular defense activity via endogenous antioxidants like extracellular SOD [33,86].
Combination therapies with renin-angiotensin-aldosterone system (RAAS) inhibitors in acute myocardial infarction patients have shown reduced vascular OS and myocardial damage, proving more effective in preventing harmful effects of vascular OS compared to vitamin supplementation.
Furthermore, publications discuss inhibition of ASK1-signalosome, a key protein complex regulating OS that links ROS generation to signaling pathways involved in aging and cardiovascular diseases. Thioredoxin acts by keeping ASK1-signalosome inactive when reduced, thus considered a potential cardioprotective therapy against OS [37,86].
Flavonoids, key bioactive components in the diet, exhibit antioxidant, anti-inflammatory, and cardioprotective properties. They contribute to lipid and blood pressure reduction, myocardial ischemia, and arrhythmias. Epidemiological studies have established an inverse correlation between flavonoid consumption and cardiovascular mortality. These compounds act through multiple mechanisms involving the NO-guanylate cyclase pathway, endothelium-derived hyperpolarizing factors, and ET-1 to protect endothelial cells from apoptosis. They also reduce circulating oxidized LDL concentrations and aortic stiffness, thus improving endothelial function. Clinically, foods such as green tea, dark chocolate, grape anthocyanins, and quercetin from onions have shown improvements in endothelial function in hypertensive and ischemic heart disease Patients [66,88,89,90,91,92].
Various nutritional compounds including vitamin C [93], antioxidant-rich nuts containing selenium, zinc, vitamins A, E, and tocopherols [93], probiotics [95] and minerals like potassium, calcium, and magnesium demonstrate significant positive effects on cardiovascular health [96]. Specifically, Ginkgo Biloba and curcuminoids from turmeric have been studied for their ability to activate the SIRT1 axis, reduce inflammation, preserve cardiomyocytes, increase NO bioavailability, decrease cardiac OS, and myocardial apoptosis [97,98,99,100]. Additionally, research suggests that diets enriched with extra virgin olive oil can reduce the risk of acute myocardial infarction, stroke, and cardiovascular-related mortality by 30% [101]. These dietary strategies contribute to preventing uptake of oxidized LDL by arterial wall cells, promoting vasodilation, improving endothelial function, reducing inflammation, and mitigating OS.
Antioxidants, both natural and synthetic, play a crucial role in early prevention of cardiovascular diseases and are effective when used at optimal concentrations. Compounds with polyphenolic structures and hydroxyl groups (OH) are particularly effective due to their antioxidant activity [88].
7. Conclusions
OS plays a critical role in endothelial damage and the pathogenesis of cardiovascular diseases such as atherosclerosis and hypertension. It arises from an imbalance between ROS production and antioxidant defenses, leading to cellular injury. In atherosclerosis, ROS oxidize low-density lipoproteins, triggering plaque formation through inflammation and immune cell recruitment. This compromises endothelial integrity, induces apoptosis and stimulates smooth muscle cell proliferation, accelerating disease progression. In hypertension, OS reduces nitric oxide availability, crucial for vasodilation, resulting in vasoconstriction, increased vascular resistance, and elevated blood pressure. Despite advances, antioxidant therapies have shown mixed clinical results, necessitating more targeted approaches and deeper molecular understanding. Current research focuses on precise therapeutic targets and optimized antioxidant combinations to enhance clinical efficacy.
Further investigation into OS biomarkers is crucial for personalized treatments and improved outcomes in vascular diseases. Standardizing these biomarkers will aid in assessing disease severity and treatment efficacy. Continued research is essential to refine prevention and treatment strategies, ensuring better management of cardiovascular diseases.
Data Availability Statement
No new data were created or analyzed in this study.
Abbreviations
The following abbreviations are used in this manuscript:
| LDL | Low-density lipoprotein |
| OS | oxidative stress |
| ROS | reactive oxygen species |
| eNOS | endotelial nitric oxide synthase |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| iNOS | inducible nitric oxide synthase |
| BH4 | Tetrahydrobiopterine |
| NADPH | nicotinamide adenine dinucleotide phosphate oxidase |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| SOD | superoxide dismutase |
| GPx | Glutathione peroxidase |
| GSSG | oxidized glutathione |
| GSH | glutathione |
| HSPs | heat shock proteins |
| NO | nitric oxide |
| ET-1 | endothelin-1 |
| Trx | thioredoxin |
| cGMP | guanosie 3’,5’-cyclic monophosphate |
| ONOO-, | Peroxynitrite |
| ATP | adenosine triphosphate |
| mtDNA | mitocondrial DNA |
| oxLDL | oxidized low-density lipoproteins |
| PUFA | peroxidation of polyunsatured fatty acids |
| CD | cojugated dienes |
| LOO. | peroxyl radical |
| MDA | malondialdehyde |
| HNE | 4-hidroxynonenal |
| IL4 | interleukin-4 |
| IL1β | interleukin 1β |
| TNF-α | tumor necrosis factor-α |
| VCAM-1 | vascular cell adhesión molecule 1 |
| ICAM-1 | intercelular adhesión molecule 1 |
| HDL | high-density lipoprotein |
| ABCA1 | ATP binding casette |
| Apo A1 | apolipoprotein A1 |
| M1 | inflammatory macrophages |
| ANGII | angiotensin II |
| AP-1 | activator protein-1 |
| MAPK | mitogen-activated protein kinase |
| DM | diabetes mellitus |
| AGEs | advnaced glycation end-products |
| ACEIs | angiotensin-converting enzyme inhibitors |
| RAAS | renin-angiotensin-aldosterone system |
| ASK1 | apoptosis signal-regulating kinase 1. |
References
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Sies, H. OS: From basic research to clinical application. Am J Med. 1991, 91, S31–S38. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free radicals in biology and medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015; 905p. [Google Scholar]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. ROS in metabolic and inflammatory signaling. Circ Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce ROS. Biochem J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M. ROS and Vascular Biology: Implications in Human Hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial Formation of ROS. J. Physiol. 2003, 552 (Pt 2), 335–344. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free. Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837d. [Google Scholar] [CrossRef]
- Crabtree, M.J.; Tatham, A.L.; Al-Wakeel, Y.; Warrick, N.; Hale, A.B.; Cai, S.; Channon, K.M.; Alp, N.J. Quantitative Regulation of Intracellular Endothelial Nitric-Oxide Synthase (eNOS) Coupling by Both Tetrahydrobiopterin-eNOS Stoichiometry and Biopterin Redox Status: Insights from Cells with Tet-Regulated GTP Cyclohydrolase I Expression. J. Biol. Chem. 2009, 284, 1136–1144. [Google Scholar] [CrossRef]
- Battelli, M.G.; Polito, L.; Bolognesi, A. Xanthine oxidoreductase in atherosclerosis pathogenesis: Not only oxidative stress. Atherosclerosis 2014, 237, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and OS. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in Cigarette Smoke Radicals, Hydrogen Peroxide, Peroxynitrate, and Peroxynitritea. Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Hu, X.; Hao, C.; Cheng, Z.-M.; Zhong, Y. Genome-Wide Identification, Characterization, and Expression Analysis of the Grapevine Superoxide Dismutase (SOD) Family. Int. J. Genom. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef]
- Arthur, J.R. The Glutathione Peroxidases. Cell. Mol. Life Sci. 2000, 57, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Higdon, J.V. Antioxidant Activity of Tea Polyphenols In Vivo: Evidence from Animal Studies. J. Nutr. 2003, 133, 3275S–3284S. [Google Scholar] [CrossRef]
- Wu, G.; Lupton, J.R.; Turner, N.D.; Fang, Y.-Z.; Yang, S. Glutathione Metabolism and Its Implications for Health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Agudo, A.; Cabrera, L.; Amiano, P.; Ardanaz, E.; Barricarte, A.; Berenguer, T.; Chirlaque, M.D.; Dorronsoro, M.; Jakszyn, P.; Larrañaga, N.; et al. Fruit and vegetable intakes, dietary antioxidant nutrients, and total mortality in Spanish adults: findings from the Spanish cohort of the European Prospective Investigation into Cancer and Nutrition (EPIC-Spain). Am. J. Clin. Nutr. 2007, 85, 1634–1642. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Gong, J. Heat Shock Proteins Promote Cancer: It's a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction: Testing and Clinical Relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Griendling, K.K.; FitzGerald, G.A. OS and Cardiovascular Injury: Part I: Basic Mechanisms and In Vivo Monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef]
- Leopold, J.A. Antioxidants and coronary artery disease: from pathophysiology to preventive therapy. Coronary Artery Disease. marzo de 2015, 26, 176–183. [Google Scholar]
- Rotariu, D.; Babes, E.E.; Tit, D.M.; Moisi, M.; Bustea, C.; Stoicescu, M.; et al. OS – Complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomedicine & Pharmacotherapy. agosto de 2022, 152, 113238. [Google Scholar]
- Franco, R.; ACidlowski, J. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Montezano, A.C.; Touyz, R.M. ROS, Vascular Noxs, and Hypertension: Focus on Translational and Clinical Research. Antioxid. Redox Signal. 2012, 20, 164–182. [Google Scholar] [CrossRef]
- Doughan, A.; Harrison, D.; Dikalov, S. Abstract 1229: Molecular Mechanisms of Angiotensin II-Mediated Mitochondrial Dysfunction: Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. Circulation 2007, 116. [Google Scholar] [CrossRef]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef]
- Drexler, H.; Zeiher, A.M. Endothelial function in human coronary arteries in vivo. Focus on hypercholesterolemia. Hypertension 1991, 18, II90–9. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H. Uracil in DNA—General mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Tungekar, M.A.; Jung, M.-Y.; Tyurin, V.A.; Greenberger, J.S.; Stoyanovsky, D.A.; Kagan, V.E. Mitochondria targeting of non-peroxidizable triphenylphosphonium conjugated oleic acid protects mouse embryonic cells against apoptosis: Role of cardiolipin remodeling. FEBS Lett. 2011, 586, 235–241. [Google Scholar] [CrossRef]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Brüning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; Thorén, P.; Larsson, N.G. Dilated Cardiomyopathy and Mitochondrial Dysfunction in Heart Specific Antisense DNA. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Scorrano, L. Two Close, Too Close: Sarcoplasmic Reticulum-Mitochondrial Crosstalk and Cardiomyocyte Fate. Circ. Res. 2010, 107, 689–699. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: gates of life and death. Cardiovasc. Res. 2007, 77, 334–343. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Arbustini, E.; Chandrashekhar, Y. Mechanisms of Disease: apoptosis in heart failure—seeing hope in death. Nat. Clin. Pr. Cardiovasc. Med. 2006, 3, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J. Mitochondria: Sovereign of inflammation? Eur. J. Immunol. 2011, 41, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Sisein, E.A. Biochemistry of free radicals and antioxidants. Sch Acad J Biosci. 2014, 2, 110–118. [Google Scholar]
- Chistiakov, D.A.; Melnichenko, A.A.; Orekhov, A.N. Peroxiredoxin family as redox-sensitive proteins implicated in cardiovascular diseases. Oxid Med Cell Longev. 2017, 2017, 1–20. [Google Scholar]
- Steinberg, D.; Witztum, J.L. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation. 2002, 105, 2107–2111. [Google Scholar] [CrossRef]
- Gęgotek, A.; Skrzydlewska, E. The role of transcription factor Nrf2 in skin cells metabolism. Arch. Dermatol. Res. 2015, 307, 385–396. [Google Scholar] [CrossRef]
- Catalá,, A. ; Díaz, M. Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front. Physiol. 2016, 7, 423–423. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free. Radic. Biol. Med. 2013, 61, 473–501. [Google Scholar] [CrossRef]
- Smith, J.D. Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. J. Clin. Lipidol. 2010, 4, 382–388. [Google Scholar] [CrossRef]
- Shao, B. Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2012, 1821, 490–501. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Holme, R.L.; Chen, Y.; Thomas, M.J.; Sorci-Thomas, M.G.; Silverstein, R.L.; Pritchard KA,, J. r.; Sahoo, D. Acrolein Impairs the Cholesterol Transport Functions of High Density Lipoproteins. PLoS ONE 2015, 10, e0123138. [Google Scholar] [CrossRef]
- Chen, Y.; Arnal-Levron, M.; Hullin-Matsuda, F.; Knibbe, C.; Moulin, P.; Luquain-Costaz, C.; Delton, I. In vitro oxidized HDL and HDL from type 2 diabetes patients have reduced ability to efflux oxysterols from THP-1 macrophages. Biochimie 2018, 153, 232–237. [Google Scholar] [CrossRef]
- Sawada, N.; Obama, T.; Koba, S.; Takaki, T.; Iwamoto, S.; Aiuchi, T.; Kato, R.; Kikuchi, M.; Hamazaki, Y.; Itabe, H. Circulating oxidized LDL, increased in patients with acute myocardial infarction, is accompanied by heavily modified HDL. J. Lipid Res. 2020, 61, 816–829. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Ginckels, P.; Holvoet, P. Oxidative Stress and Inflammation in Cardiovascular Diseases and Cancer: Role of Non-coding RNAs. Yale J Biol Med. 2022, 95, 129–152. [Google Scholar] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular Cell Adhesion Molecule-1 Expression and Signaling During Disease: Regulation by Reactive Oxygen Species and Antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef]
- Shantsila, E.; Wrigley, B.J.; Blann, A.D.; Gill, P.S.; Lip, G.Y. A contemporary view on endothelial function in heart failure. Eur. J. Hear. Fail. 2012, 14, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Colucci, W.S. Regulation of Matrix Metalloproteinases by Cytokines and Reactive Oxygen/Nitrogen Species in the Myocardium. Hear. Fail. Rev. 2004, 9, 43–51. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic metals and oxidative stress Part I: Mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Jamialahmadi, T.; Sahebkar, A. Polyphenols and atherosclerosis: A critical review of clinical effects on LDL oxidation. Pharmacol. Res. 2022, 184, 106414. [Google Scholar] [CrossRef]
- Hold, G.L.; El-Omar, M.E. Genetic aspects of inflammation and cancer. Biochem. J. 2008, 410, 225–235. [Google Scholar] [CrossRef]
- Barja, G. Mitochondrial Oxygen Radical Generation and Leak: Sites of Production in States 4 and 3, Organ Specificity, and Relation to Aging and Longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef]
- Adegoke, O.; Forbes, P.B. Challenges and advances in quantum dot fluorescent probes to detect reactive oxygen and nitrogen species: A review. Anal. Chim. Acta 2015, 862, 1–13. [Google Scholar] [CrossRef]
- Niki, E. Biomarkers of lipid peroxidation in clinical material. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2014, 1840, 809–817. [Google Scholar] [CrossRef]
- Polidori, M.C.; Stahl, W.; Eichler, O.; Niestroj, I.; Sies, H. Profiles of antioxidants in human plasma. Free Radic Biol Med. 2001, 30, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Mahajan, N.; Sah, S.; Nath, S.K.; Paudyal, B. Oxidative stress and its biomarkers in systemic lupus erythematosus. J. Biomed. Sci. 2014, 21, 23–23. [Google Scholar] [CrossRef]
- Lowe, F. Biomarkers of oxidative stress. In Systems Biology of Free Radicals and Antioxidants; Leher, *!!! REPLACE !!!*, Ed.; Springer-Verlag Heidelberg, 2014; pp. 65–87. [Google Scholar]
- Halliwell, B.; Kaur, H. Hydroxylation of salicylate and phenylalanine as assays for hydroxyl radicals: A cautionary note visited for the third time. Free Radic Res. 1997, 27, 239–244. [Google Scholar] [CrossRef]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kB Regulation in Rheumatoid Arthritis and Murine Collagen-Induced Arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Averill-Bates, D.A. The antioxidant glutathione. Vitamins Hormones. 2023, 121, 109–141. [Google Scholar] [PubMed]
- Franco, C.; Sciatti, E.; Favero, G.; Bonomini, F.; Vizzardi, E.; Rezzani, R. Essential Hypertension and Oxidative Stress: Novel Future Perspectives. Int. J. Mol. Sci. 2022, 23, 14489. [Google Scholar] [CrossRef]
- Demirci-Çekiç, S.; Özkan, G.; Avan, A.N.; Uzunboy, S.; Çapanoğlu, E.; Apak, R. Biomarkers of Oxidative Stress and Antioxidant Defense. J. Pharm. Biomed. Anal. 2022, 209, 114477. [Google Scholar] [CrossRef]
- Schöttker, B.; Saum, K.-U.; Jansen, E.H.J.M.; Boffetta, P.; Trichopoulou, A.; Holleczek, B.; Dieffenbach, A.K.; Brenner, H. Oxidative Stress Markers and All-Cause Mortality at Older Age: A Population-Based Cohort Study. Journals Gerontol. Ser. A 2014, 70, 518–524. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.; Verma, H.K.; Lakkakula, S.; Merchant, N.; Kadir, F.; Rahman, S.; Jeffree, M.S.; Lakkakula, B.V.K.S.; Rao, P.V. Biomarkers of Oxidative Stress Tethered to Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2022, 2022, 9154295. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Stevens, P.E. Evaluation and management of chronic kidney disease: synopsis of the Kidney Disease: Improving Global Outcomes 2012 clinical practice guideline. Ann Intern Med. 2013, 158, 825. [Google Scholar] [CrossRef] [PubMed]
- Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ 2nd. Isoprostane generation and function. Chem Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Bobak, M.; Anusruti, A.; Jansen, E.H.J.M.; Pająk, A.; Tamosiunas, A.; Saum, K.-U.; Holleczek, B.; Gao, X.; Brenner, H.; et al. Association of serum markers of oxidative stress with myocardial infarction and stroke: pooled results from four large European cohort studies. Eur. J. Epidemiology 2018, 34, 471–481. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining Oxidative Stress. Antioxidants Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Neha, K.; Haider, R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef]
- Stroev, S.A.; Gluschenko, T.S.; Tjulkova, E.I.; Spyrou, G.; Rybnikova, E.A.; Samoilov, M.O.; Pelto-Huikko, M. Preconditioning enhances the expression of mitochondrial antioxidant thioredoxin-2 in the forebrain of rats exposed to severe hypobaric hypoxia. J. Neurosci. Res. 2004, 78, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.; Pérez-Vizcaíno, F. Protección cardiovascular con flavonoides: enigma farmacocinético. Ars Pharm. 2015, 56, 193–200. [Google Scholar] [CrossRef]
- Perez-Vizcaino, F.; Duarte, J.; Andriantsitohaina, R. Endothelial function and cardiovascular disease: Effects of quercetin and wine polyphenols. Free. Radic. Res. 2006, 40, 1054–1065. [Google Scholar] [CrossRef]
- Tayebati, S.K.; Tomassoni, D.; Mannelli, L.D.C.; Amenta, F. Effect of treatment with the antioxidant alpha-lipoic (thioctic) acid on heart and kidney microvasculature in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2015, 38, 30–38. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, Y.; Yan, F.; Dong, M.; Ren, Y. Research progress of quercetin in cardiovascular disease. Front. Cardiovasc. Med. 2023, 10, 1203713. [Google Scholar] [CrossRef]
- Sánchez, M.; Romero, M.; Gómez-Guzmán, M.; Tamargo, J.; Pérez-Vizcaino, F.; Duarte, J. Cardiovascular effects of flavonoids. CMC. 2019, 26, 6991–7034. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.; Chun, O.K. Vitamin C and Heart Health: A Review Based on Findings from Epidemiologic Studies. Int. J. Mol. Sci. 2016, 17, 1328. [Google Scholar] [CrossRef]
- Lorenzon Dos Santos, J.; Schaan De Quadros, A.; Weschenfelder, C.; Bueno Garofallo, S.; Marcadenti, A. OS biomarkers, nut-related antioxidants, and cardiovascular disease. Nutrients. 2020, 12, 682. [Google Scholar] [CrossRef]
- Romero, M.; Duarte, J. Probiotics and Prebiotics in Cardiovascular Diseases. Nutrients 2023, 15, 3686. [Google Scholar] [CrossRef]
- AAltawili, A.; Altawili, M.; Alwadai, A.M.; Alahmadi, A.S.; AAlshehri, A.M.; Muyini, B.H.; Alshwwaf, A.R.; Almarzooq, A.M.; AAlqarni, A.H.; Alruwili, Z.A.L.; et al. An Exploration of Dietary Strategies for Hypertension Management: A Narrative Review. Cureus 2023, 15, e50130. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Karathanasis, S.K.; Ding, Z.; Arulandu, A.; Varughese, K.I.; Mehta, J.L. LOX-1 in Atherosclerosis and Myocardial Ischemia: Biology, genetics, and modulation. JACC 2017, 69, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Kocaadam, B.; Şanlier, N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit. Rev. Food Sci. Nutr. 2015, 57, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: what’s new to NO? American Journal of Physiology-Cell Physiology. 1 de marzo de 2017, 312, C254–C262. [Google Scholar] [CrossRef]
- Kapakos, G.; Youreva, V.; Srivastava, A.K. Cardiovascular protection by curcumin: molecular aspects. Indian J Biochem. Biophys. 2012, 49, 306–315. [Google Scholar]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet. N Engl J Med. 2013, 368, 1279–1290. [Google Scholar] [CrossRef]
Figure 1.
Fenton and Haber-Weiss reactions.
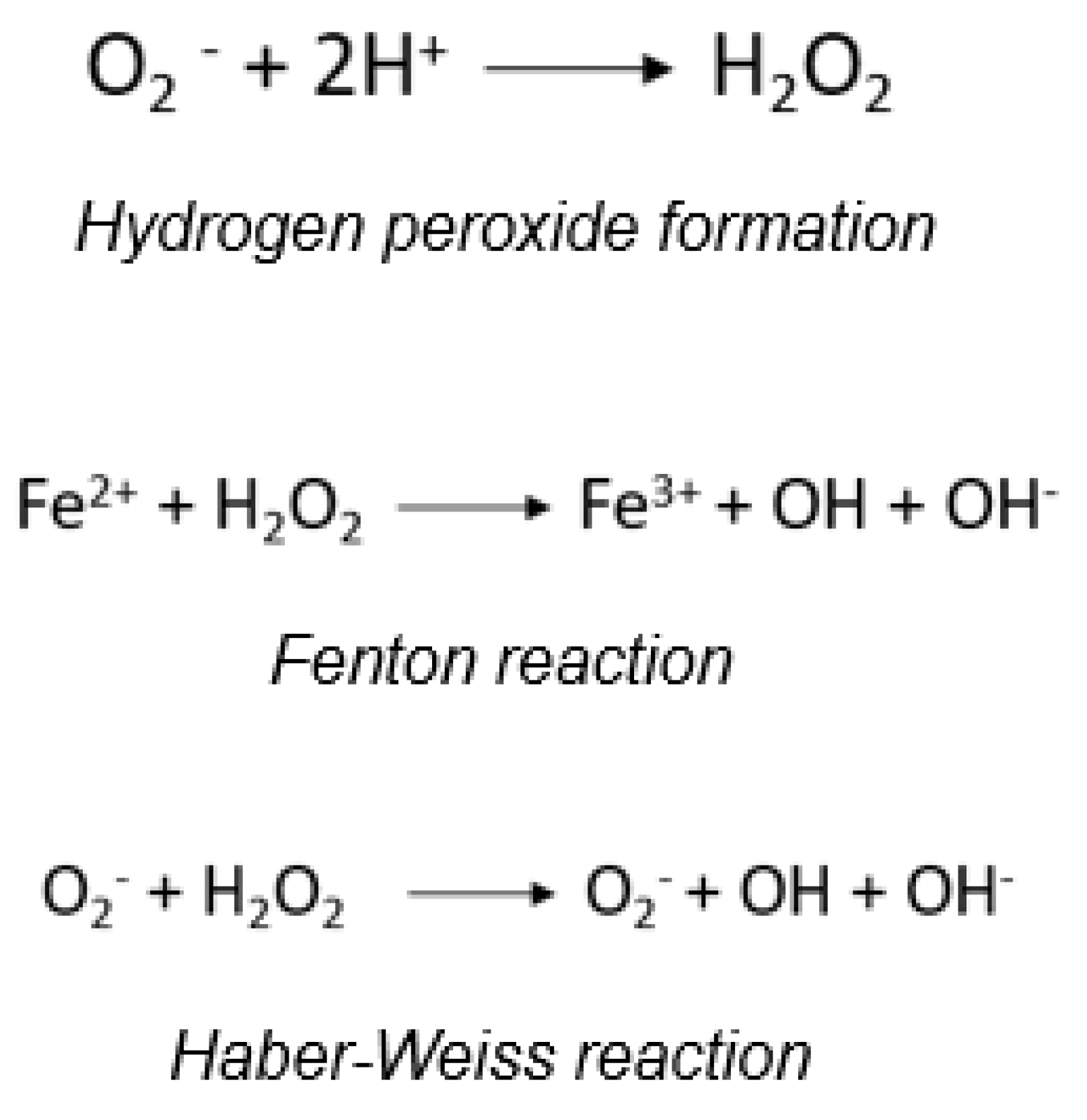
Figure 2.
Primary Endogenous Sources of ROS Production.
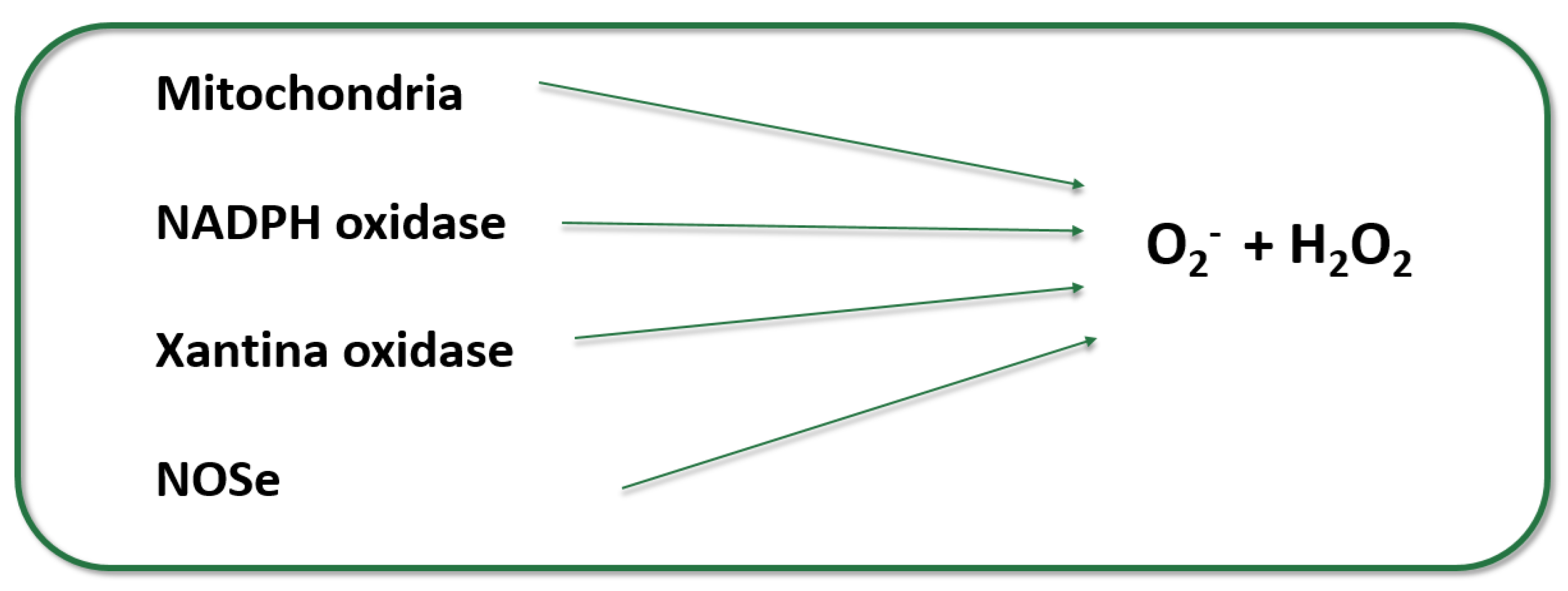
Figure 3.
OS, immune response, inflammation, and apoptosis in atherosclerosis are interconnected processes. High levels of glucose, angiotensin II (ANGII), oxidized LDL (ox-LDL), and stress cause endothelial dysfunction, characterized by mitochondrial OS and ROS release. This stress results from an imbalance favoring oxidants over antioxidants. The damaged endothelium induces monocyte adhesion and infiltration, differentiating into macrophages. ROS provoke macrophage polarization from M2 to M1. Additionally, M1 macrophages release pro-inflammatory cytokines, inducing ROS release and apoptosis in vascular cells. Overexpressed regulators are indicated in yellow circles, underexpressed in blue. Adapted from Ginckels P, Holvoet P. OS and Inflammation in Cardiovascular Diseases and Cancer: Role of Non-coding RNAs. Yale J Biol Med. 2022 Mar 31;95(1):129-152. PMID: 35370493; PMCID: PMC8961704 [61].
Figure 3.
OS, immune response, inflammation, and apoptosis in atherosclerosis are interconnected processes. High levels of glucose, angiotensin II (ANGII), oxidized LDL (ox-LDL), and stress cause endothelial dysfunction, characterized by mitochondrial OS and ROS release. This stress results from an imbalance favoring oxidants over antioxidants. The damaged endothelium induces monocyte adhesion and infiltration, differentiating into macrophages. ROS provoke macrophage polarization from M2 to M1. Additionally, M1 macrophages release pro-inflammatory cytokines, inducing ROS release and apoptosis in vascular cells. Overexpressed regulators are indicated in yellow circles, underexpressed in blue. Adapted from Ginckels P, Holvoet P. OS and Inflammation in Cardiovascular Diseases and Cancer: Role of Non-coding RNAs. Yale J Biol Med. 2022 Mar 31;95(1):129-152. PMID: 35370493; PMCID: PMC8961704 [61].
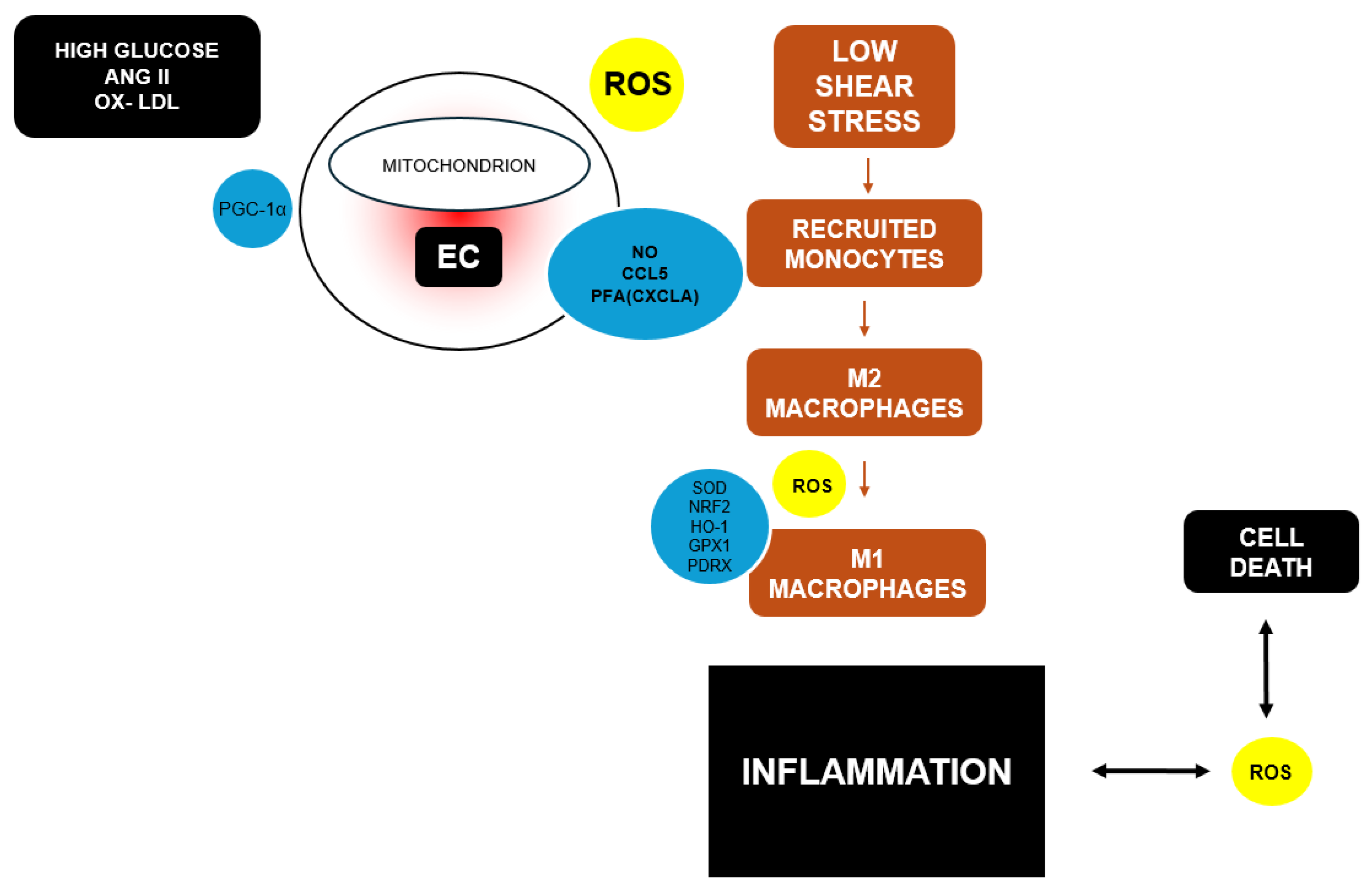
Figure 4.
Pathophysiology of Heart Failure and OS Contribution. NF-κB, nuclear factor kappa beta; AP-1, activator protein-1; MAPK, mitogen-activated protein kinase. Adapted from Rotariu D, et al. Biomed Pharmacother. 2022.
Figure 4.
Pathophysiology of Heart Failure and OS Contribution. NF-κB, nuclear factor kappa beta; AP-1, activator protein-1; MAPK, mitogen-activated protein kinase. Adapted from Rotariu D, et al. Biomed Pharmacother. 2022.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



