Submitted:
17 September 2025
Posted:
18 September 2025
You are already at the latest version
Abstract
Therapeutic strategies targeting “tumor-educated platelets” (TEPs) and platelet-tumor interactions by key signaling pathways (ITAM, P2Y12) may reduce metastasis and cancer.
Using a TEP gene expression dataset originally created to study swarm intelligence-enhanced detection of lung cancer cells (GSE89843), we did do extensive transcriptome analysis to integrate these data with directed protein-protein interactions and build a TEP-specific signaling network. We analyze network topology and controllability and identify critical and indispensable nodes as well as high-weight, usually high-score nodes. We reconstruct (pharmacological) controllable subnetworks of TEP signaling, which we then explore for drugs targets.
We found 111 upregulated and 108 downregulated genes compared to control platelets, enriched in pathways related to extracellular matrix interactions, cytoskeleton organization, immune signaling, and platelet activation. Ribosomal function, apoptosis, and immune signaling were among the downregulated processes, highlighting unique TEP profiles in non-small cell lung cancer (NSCLC). Our integrative analysis of TEPs in NSCLC reveals key transcriptional and network-based alterations harmful for the cancer patient. Using four complementary strategies, we identified five high-confidence genes, ITGA2B, FLNA, GRB2, FCGR2A, and APP, as central to TEP signaling. These can be targeted by FDA-approved drugs. Fostamatinib, a SYK inhibitor, emerged as top candidate drugs to disrupt ITAM-mediated platelet activation selectively, metastasis promoting metalloprotease and cytoskeletal targets influencing adhesion were also identified. A low-dose combination therapy of fostamatinib, Aducanumab, and acetylsalicylic acid (aspirin) may control TEP effects. In conclusion, our pre-clinical in silico approach revealed FDA-approved drugs that allow therapeutic targeting of metastasis-promoting TEPs and target NSCLC at the same time.
Keywords:
tumor-educated platelets (TEP)
; signaling
; ITAM
; P2Y12
; cancer
; non-small cell lung cancer (NSCLC)
1. Introduction
Armand Trousseau was the first (1865) to observe the connection between cancer and platelets [1]. This discovery was followed by many others that advanced the understanding of the relationship between platelets and cancer [2,3,4,5,6]. What’s now referred as “Trousseau’s syndrome” describes increased incidence and severity of thrombotic events with increased cancer progression, sometimes even detected before the cancer diagnosis [7,8,9,10,11,12]. However, this relationship is also observed vice versa, in which higher cancer progression and metastasis is observed with increased platelet numbers/activity [13].
The effect of tumor cells on platelets can be via such as promoting platelet production [4,14], changing platelet transcriptomes and proteomes [15,16,17], or by changing platelet signaling and activity (review in [18]). These are collectively designated as “platelet education”.
One of the main mechanisms of platelet education is the transfer and storage of tumor-associated factors (proteins, DNA, or RNA) into platelets [19,20,187], which are then carried to dormant tumors at distant sites, aiding angiogenesis and preparing the pre-metastatic niche [21,22].
A second mechanism of platelet education involves platelet RNA processing and production [23]. Although anucleate, platelets can still synthesize proteins in response to various signals [24]. This occurs through differential splicing and alternative splicing of pre-RNA to mRNA [25], as well as alterations directly within the gene expression profile of megakaryocytes [17,23]. Alterations in the noncoding RNAs have also been observed in platelets during cancer [26,27,28,29,30]. It is still unclear whether these changes affect platelet gene expression and contribute to their platelet education by tumor cells.
Tumor cells can activate platelets directly by molecules on the tumor cell surface [31,32,33,34] or those released by tumor cells [34,35,36] leading to tumor cell-induced platelet aggregation (TCIPA) [37], which is observed in several cancer types including lung [36], ovarian, pancreatic, and breast cancer [38]. Indirectly, cancer cells can lead to platelet activation by interacting with other cells in the tumor microenvironment, including inducing formation of NETs [39,40], and activating the granulocytes [41].
In return, activated platelets contribute to cancer progression in multiple ways [18], promoting tumor cell proliferation by releasing growth factors, cytokines, and pro- and anti-angiogenic molecules [22,39,42,43,44,45,46], shielding them from immune responses [35,39,40,41,47,48], inducing epithelial-mesenchymal transition (EMT) [39,49], protecting them from shear stress in the bloodstream, supporting tumor cell adhesion to endothelium [39,40,50,51,52], establishing metastatic sites [31,40,41,53], and enabling tumor cell extravasation to the metastasis site [39,50,54,55,56,57,58].
The relationship between platelets and cancer can be explored as various therapeutic opportunities. Increased platelet counts are often observed in cancer patients [59], suggesting that platelets could be used for diagnostics [60] as well as to monitor cancer progression and treatment response. Beyond diagnostics, platelets can also be utilized directly for cancer treatment, either as drug delivery systems or as therapeutic targets [61]. Use of antiplatelet agents for cancer therapy can be beneficial in two ways. First, they can be utilized to target the interactions between cancer cells and platelets. The platelet-tumor crosstalk is mediated through direct cellular contact, activation of cell surface receptors, release of soluble proteins, and shedding of microparticles [39]. Targeting platelet-cancer cell interactions aims to disrupt the protective role that platelets play in shielding tumor cells from immune detection and facilitating metastasis [62,63]. By interfering with the mechanisms that cancer cells use to activate and aggregate platelets, this approach could potentially reduce the spread of circulating tumor cells, enhancing the effectiveness of existing cancer treatments.
Moreover, targeting platelet signaling pathways that contribute to cancer progression, such as those involved in inflammation and thrombosis, offers another therapeutic avenue. Drugs like acetylsalicylic acid (aspirin), which inhibits cyclooxygenase-1 (COX-1), have shown promise in reducing the risk of certain cancers, such as colorectal cancer [62,63,64,65,66]. Other antiplatelet agents such as P2Y12 inhibitor ticagrelor [40,49,67] and antibodies against integrin αIIbβ3 [68,69,297] were shown to have anti-metastatic effects. This approach may provide a broad-spectrum effect by reducing risk of thrombotic complications that arise from sustained activation of platelets [70,71,72], and by slowing down cancer progression and metastasis and thereby improving patient outcomes. However, the long-term use of antiplatelet drugs comes with an increased risk of bleeding, and their efficacy may vary depending on the cancer type and stage. Inhibiting platelet kinases may at the same time target also cancer promoting kinases in cancer cells. Bearing these complex relationships in mind, further research is required to determine the optimal regime of antiplatelet therapies implied by our basic target options from our network analysis: [73].
To identify antiplatelet therapeutic options targeting regulated platelet signaling in cancer, we did transcriptome analysis of experimental data from a unique gene expression dataset on tumor-educated platelets, GEO dataset GSE89843 [177]. This dataset was created to study swarm intelligence-enhanced detection of non-small-cell lung cancer using tumor-educated platelets [177], but the transcriptome data was not yet analyzed. Moreover, we integrated experimental information from OmniPath database [74] regarding the implied protein-protein interactions. This combination of experimental validated data allowed us to construct a TEP-specific signaling network. Network topology and controllability analysis identified key proteins essential to TEP signaling. We reconstructed controllable subnetworks and explored potential drug targets, revealing genes associated with platelet activation, immune signaling, and cytoskeleton organization. Screening these subnetworks identified several FDA-approved drugs. Fostamatinib, a SYK inhibitor controlling platelet hyperactivity [115,126], emerged again as a top candidate drug as it controls and disrupts the wide-spread ITAM-mediated platelet activation. However, metastasis promoting metalloprotease and cytoskeletal targets influencing adhesion were also identified as important targets. In conclusion, our approach revealed FDA-approved drugs that allow therapeutic targeting of metastasis-promoting TEPs and target NSCLC at the same time.
2. Results
2.1. RNA Profiles of Tumor-Educated Platelets in NSCLC
We compared the gene expression profile in platelets from NSCLC patients to platelets from non-cancer donors. 60% of the donors with no known cancer were classified as healthy, and 40% were diagnosed with inflammatory conditions (Figure 1A, Table S1). We found 111 upregulated and 108 downregulated genes in tumor-educated platelets (TEP) compared to platelets from a non-cancer condition (Figure 1A, Datasheet DS1).
Figure 1.
Differential Gene Expression Analysis of TEPs in NSCLC patients reveal key genes in metastasis promotion. (A) Top 30 upregulated (red) and downregulated (blue) genes in TEPs in NSCLC patients (B) Relationship between the expression of DEGs in different conditions. (C) Gene Set Enrichment Analysis of NSCLC-TEPs showing activated and suppressed KEGG pathways. MS: Multiple sclerosis, PulHyp: Pulmonary hypertension, StableAP: Stable angina pectoris, NSCLC: non-small cell lung cancer, ChrPanc: Chronic pancreatitis, nsAth: Non-significant atherosclerosis, UnstableAP: Unstable angina pectoris.
Figure 1.
Differential Gene Expression Analysis of TEPs in NSCLC patients reveal key genes in metastasis promotion. (A) Top 30 upregulated (red) and downregulated (blue) genes in TEPs in NSCLC patients (B) Relationship between the expression of DEGs in different conditions. (C) Gene Set Enrichment Analysis of NSCLC-TEPs showing activated and suppressed KEGG pathways. MS: Multiple sclerosis, PulHyp: Pulmonary hypertension, StableAP: Stable angina pectoris, NSCLC: non-small cell lung cancer, ChrPanc: Chronic pancreatitis, nsAth: Non-significant atherosclerosis, UnstableAP: Unstable angina pectoris.
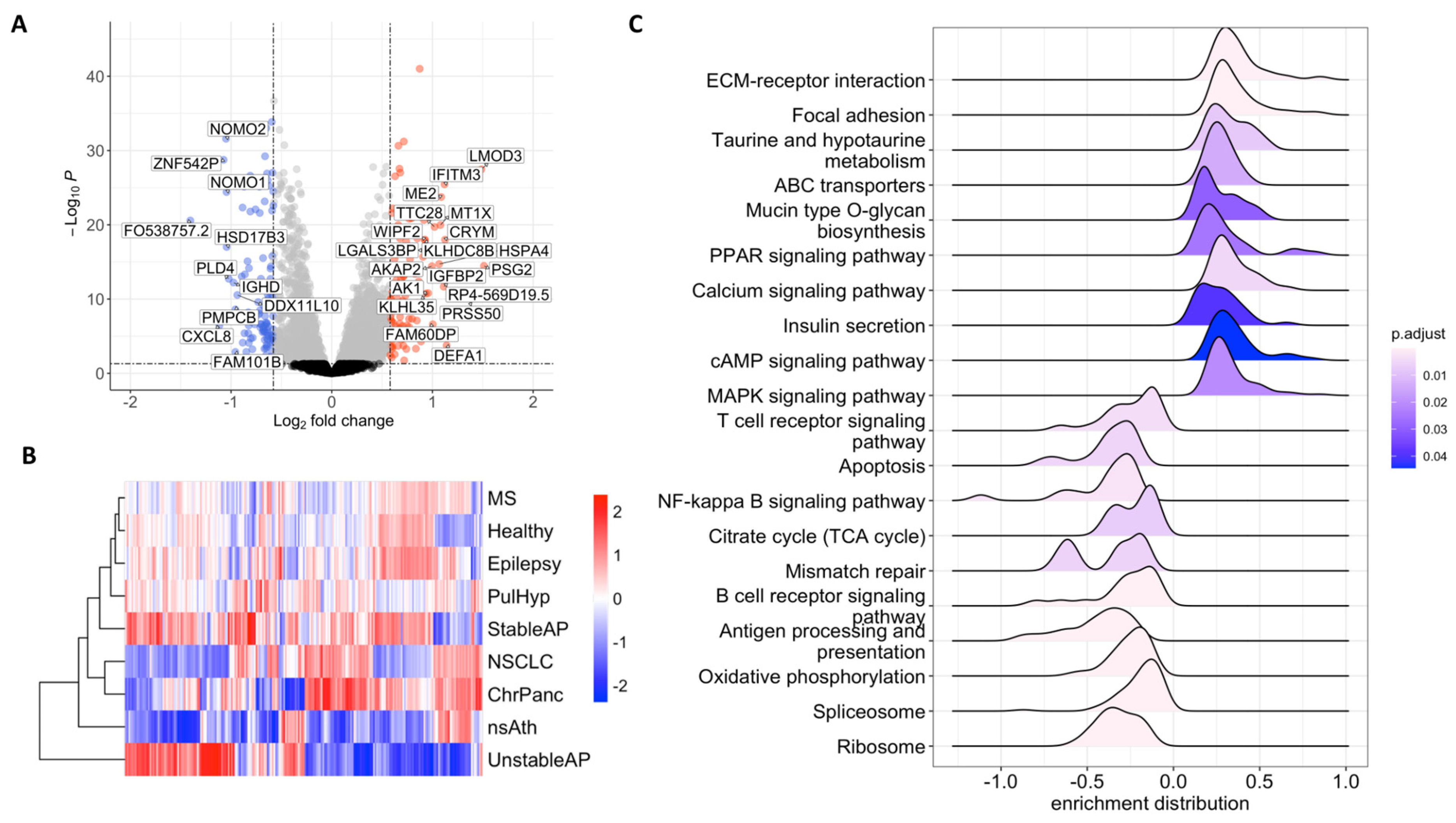
Gene expression of the differentially expressed genes in chronic pancreatitis (ChrPanc) appeared to be the most similar condition to NSCLC in terms of expression profiles of the DEGs. While platelets of multiple sclerosis (MS) patients showed a profile like healthy patients followed by epilepsy patients, patients with pulmonary hypertension (PulHyp) and stable Angina pectoris (stableAP), DEG profiles of platelets from patients with unstable Angina pectoris (unstableAP) and non-significant atherosclerosis (nsAth) appeared to be the most diverse when compared to the rest of the samples (Figure 1B).
Upregulated genes were predominantly involved in KEGG pathways associated with ECM-receptor interaction, Focal adhesion, metabolism of taurine, biosynthesis of mucins, signaling pathways of calcium, PPAR, cAMP and MAPKs, and insulin secretion. Conversely, downregulated genes were enriched in immune-related pathways such as T-cell and B-cell signaling, NF-κB signaling, and antigen processing, as well as apoptosis, TCA cycle and oxidative phosphorylation, ribosome and spliceosome related pathways. These findings highlight a shift in platelet function in NSCLC, favoring pro-adhesive and ECM-remodeling activities while downregulating immune-associated signaling. (Figure 1C).
Gene enrichment analysis using Gene Ontology Biological Process Terms, and Reactome Pathways also highlighted similar pathways associated with apoptosis, B- and T-cell signaling, NF-KB signaling, translation and post-translational processes, and DNA repair as downregulated. Upregulation of pathways and processes associated with cell-cell or cell-ECM interaction, ECM organization, and cytoskeleton continued to be consistently observed (Figure S1, Datasheets DS2-4).
In summary, our findings indicate that TEPs exhibit increased gene expression of genes associated with cell-cell interactions, extracellular matrix (ECM) dynamics, platelet activation, and cytoskeleton organization. Conversely, genes related to ribosomal functions, apoptosis, and immune processes, particularly those involving MHC class II and B and T-cell signaling, are downregulated. These results highlight the altered cellular processes in TEPs and provide insight into their potential role in cancer.
2.2. Therapeutic Target Discovery Based on Transcriptome Data
Using this gene expression dataset, we employed four complementary strategies to identify potential therapeutic targets. The first two strategies are solely based on differential gene expression, while the latter two integrated network-based analyses to enhance target selection.
To ensure that the regulated pathways were NSCLC-specific, we performed k-means clustering of the differentially expressed genes (DEGs), separating patients with inflammatory conditions from healthy donors. This resulted in 9 distinct modules based on different gene expression profiles across different sample groups (Figure 2A). Among these, four modules (1, 3, 7 and 8) were upregulated in TEPs from NSCLC compared to healthy samples and enriched in processes related to protein stability, platelet activation, metabolism, and cytoskeleton. In contrast, modules 2, 5, and 9 were downregulated and associated with processes related to ribosome, apoptosis, cell surface, DNA binding, metabolism, infection, and MHC class II complex (Figure 2B).
Figure 2.
Gene Expression Profile of TEPs in NSCLC compared to the profile of control patients with other disease entities. (A) Hierarchical clustering of the differentially expressed genes according to disease for all different patient groups and data contained in GSE89843. Vertical Black boxes mark cancer (NSCLC) and healthy control. Horizontal boxes mark protein-interaction networks (“modules”) with the most up-regulated genes in NSCLC (modules 1,3,7 and 8) (B) Combined overrepresentation analysis of gene ontology (GO) molecular function. In addition, we show cellular compartment, biological process as well as KEGG and REACTOME pathways in each module (vertical modules, numbered from 1 to 9 as in (A)).
Figure 2.
Gene Expression Profile of TEPs in NSCLC compared to the profile of control patients with other disease entities. (A) Hierarchical clustering of the differentially expressed genes according to disease for all different patient groups and data contained in GSE89843. Vertical Black boxes mark cancer (NSCLC) and healthy control. Horizontal boxes mark protein-interaction networks (“modules”) with the most up-regulated genes in NSCLC (modules 1,3,7 and 8) (B) Combined overrepresentation analysis of gene ontology (GO) molecular function. In addition, we show cellular compartment, biological process as well as KEGG and REACTOME pathways in each module (vertical modules, numbered from 1 to 9 as in (A)).
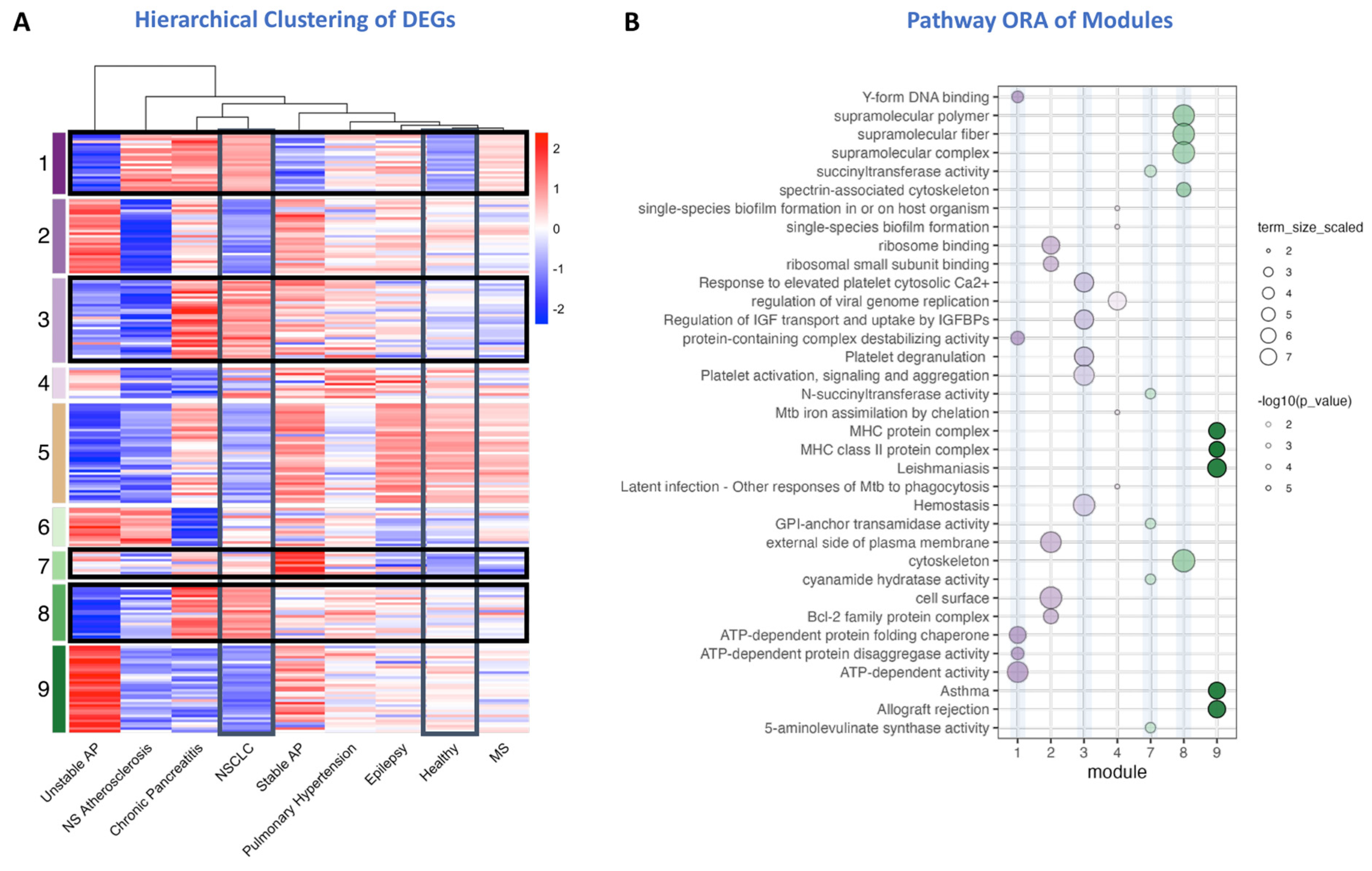
Module 1 was associated with nucleic acid and protein homeostasis as indicated by the overrepresented Gene Ontology Molecular Function terms (GO:MF) such as ATP-dependent chaperone and disaggregating activity and DNA/RNA and G-quadruplex binding terms (Figure 2B). Among the genes encoding proteins involved in these processes we found drug targets like calcium voltage-gated channel subunit alpha1 D (CACNA1D), peroxisome proliferator activated receptor alpha (PPARA), lysophosphatidic acid receptor 4 (LPAR4), malic enzyme 2 (ME2) (Table 1).

Table 1.
Drug targets in the upregulated modules in TEPs.
| Target Name1 | Description | Drugs2 | module |
|---|---|---|---|
| CACNA1D | calcium voltage-gated channel subunit alpha1 D | Ergocalciferol, Enflurane, Ranolazine, Phenytoin, Isradipine, Topiramate, Nimodipine, Nisoldipine, Spironolactone, Nicardipine, Magnesium sulfate, Verapamil, Levomenthol, Ethanol, Felodipine, Miconazole, Nifedipine, Amiodarone, Dronedarone, Clevidipine, Levamlodipine, Fish oil | 1 |
| PPARA | peroxisome proliferator activated receptor alpha | Valproic acid, Indomethacin, Rosiglitazone, Fenoprofen, Fenofibrate, Ibuprofen, Amiodarone, Gemfibrozil, Prasterone, Palmitic Acid, Soybean oil, Fenofibric acid, Fish oil | 1 |
| LPAR4 | lysophosphatidic acid receptor 4 | Promethazine | 1 |
| ME2 | malic enzyme 2 | NADH | 1 |
| MAOB | monoamine oxidase B | Amphetamine, Phentermine, Procaine, Tranylcypromine, Phenelzine, Zonisamide, Selegiline, Pioglitazone, Procarbazine, Isocarboxazid, Rasagiline, Metamfetamine, Flavin adenine dinucleotide, Safinamide, Viloxazine, Flortaucipir F-18 | 3 |
| FCGR2A | Fc gamma receptor IIa | Cetuximab, Etanercept, Human immunoglobulin G, Abciximab, Alemtuzumab, Bevacizumab, Sarilumab | 3 |
| SMPD1 | sphingomyelin phosphodiesterase 1 | Amlodipine, Chlorpromazine, Desipramine | 3 |
| GSTM3 | glutathione S-transferase mu 3 | Glutathione disulfide, Deoxycholic acid | 3 |
| ITGA2B | integrin subunit alpha 2b | Abciximab, Tirofiban | 3 |
| CA1 | carbonic anhydrase 1 | Topiramate, Chlorthalidone, Amlodipine, Methocarbamol, Bendroflumethiazide, Methazolamide, Hydroflumethiazide, Acetazolamide, Dorzolamide, Chlorothiazide, Zonisamide, Diclofenamide, Brinzolamide, Sodium sulfate | 7 |
| HBA1 | hemoglobin subunit alpha 1 | Iron Dextran, Nitrous acid, Copper, Sodium ferric gluconate complex, Ferric pyrophosphate citrate, Zinc acetate, Ferrous fumarate, Zinc chloride, Voxelotor, Ferric derisomaltose | 7 |
| SEC14L2 | SEC14 like lipid binding 2 | Vitamin E | 7 |
| ALAS2 | 5’-aminolevulinate synthase 2 | Glycine | 7 |
| ATOX1 | antioxidant 1 copper chaperone | Cisplatin | 8 |
| MAP1A | microtubule associated protein 1A | Estramustine | 8 |
1 FDA approved drugs from Drugbank v5.1.12.; 2Only upregulated module genes are targeted.
Genes in the high platelet activation-mediating module 3 were mostly upregulated in platelet from NSCLC patients and this module was the only module associated with many platelet-related REACTOME pathways like “Hemostasis”, “Platelet activation, signaling, and aggregation”, “Platelet degranulation”, and “Response to elevated platelet cytosolic Ca2+” (Figure 2B). In module 3, we found genes coding for monoamine oxidase B (MAOB), Fc gamma receptor IIa (FCGR2A), sphingomyelin phosphodiesterase 1 (SMPD1), glutathione S-transferase mu 3 (GSTM3), and integrin subunit alpha 2b (ITGA2B) as potential drug targets (Table 1). Apart from platelet pathways, the only other GO term overrepresented in this module was “Regulation of IGF and transport and uptake by IGFBPs”, and we found Insulin-like growth factor-binding protein 2 (IGFBP2), Laminin subunit beta-2 (LAMB2), Interstitial collagenase (MMP1), and Sulfhydryl oxidase 1 (QSOX1) being involved in this process (Table S2), however no FDA-approved drugs were found targeting these proteins. This pathway also appeared among enriched Reactome pathways in cancer (R-HSA-381426, Datasheet DS4).
Module 7 was predominantly associated with the metabolic adaptations in TEPs with overrepresentation of relevant KEGG and REACTOME pathways as well as GO:MF and GO:CC terms (Figure 2B). We identified several druggable targets in this metabolic module, namely carbonic anhydrase 1 (CA1), hemoglobin subunit alpha 1 (HBA1), SEC14 like lipid binding 2 (SEC14L2), and 5’-aminolevulinate synthase 2 (ALAS2) (Table 1).
Lastly, module 8 included genes involved in cytoskeletal remodeling of platelets, especially the spectrin and actin cytoskeleton (Figure 2B). This cytoskeletal module was enriched in GO terms associated with supramolecular fiber organization, cytoskeletal protein binding, and structural integrity of the platelet cytoskeleton. Among these genes, we identified antioxidant 1 copper chaperone (ATOX1), and microtubule associated protein 1A (MAP1A) as potential drug targets (Table 1).
We identified four upregulated gene modules in NSCLC-derived TEPs: protein homeostasis (Module 1), platelet activation (Module 3), metabolism (Module 7), and cytoskeletal remodeling (Module 8). Each module contained potential drug targets, including CACNA1D, FCGR2A, ITGA2B, CA1, and ATOX1, highlighting key pathways for therapeutic exploration.
Building on the hierarchical clustering approach, we further examined the top upregulated REACTOME pathways (normalized enrichment score (NES) of at least 2, see M&M). identified in gene set enrichment analysis (GSEA) to refine the selection of potential drug targets. Pathways related to extracellular matrix (ECM) remodeling, cell adhesion, and insulin signaling were among the most prominently enriched, indicating platelets’ contribution to cancer beyond their signaling. Given their functional relevance and overlap with KEGG and Gene Ontology categories, we prioritized druggable targets within these pathways. (Table 2). Prevention of thrombosis, inflammation, as well as application of drugs targeting platelet hyperactivity and at the same time directly the cancer cells (e.g. proliferation or metastasis targeting metalloproteases and cytoskeleton) are particularly beneficial for the patient.
Table 2.
Drug targets in the upregulated REACTOME pathways in TEPs.
| ID | Description | Drug Targets |
|---|---|---|
| R-HSA-445095 | Interaction between L1 and Ankyrins |
SCN1A (Sodium channel protein type 1 subunit alpha), SCN2A (Sodium channel protein type 2 subunit alpha), SCN9A (Sodium channel protein type 9 subunit alpha), SCN3A (Sodium channel protein type 3 subunit alpha), SCN11A (Sodium channel protein type 11 subunit alpha), SCN8A (Sodium channel protein type 8 subunit alpha), SCN1B (Sodium channel regulatory subunit beta-1), SCN3B (Sodium channel regulatory subunit beta-3) |
| R-HSA-3000178 | ECM proteoglycans |
APP (Amyloid-beta precursor protein), FN1 (Fibronectin), HAPLN1 (Hyaluronan and proteoglycan link protein 1), HSPG2 (Basement membrane-specific heparan sulfate proteoglycan core protein), ITGA2B (Integrin alpha-IIb) |
| R-HAS-1474228 | Degradation of the extracellular matrix |
ELN (Elastin), FBN2 (Fibrillin-2), FN1 (Fibronectin), HSPG2 (Basement membrane-specific heparan sulfate proteoglycan core protein), NID1 (Nidogen-1), PLG (Plasminogen) |
| R-HSA-446728 | Cell junction organization |
CDH11 (Cadherin-11), FLNA (Filamin-A), TESK1 (Dual specificity testis-specific protein kinase 1) |
| R-HSA-381426 | Regulation o fIGF transport and uptake by IGFBPs |
APP (Amyloid-beta precursor protein), CP (Ceruloplasmin), F5 (Coagulation factor V), FN1 (Fibronectin), ITIH2 (Inter-alpha-trypsin inhibitor heavy chain H2), PLG (Plasminogen), SERPIND1 (Heparin cofactor 2) |
| R-HSA-5173105 | O-linked glycosylation | MUC16 (Mucin-16) |
| R-HSA-1474290 | Collagen formation |
P3H2 (Prolyl 3-hydroxylase 2), P4HB (Protein disulfide-isomerase), PLOD1 (Procollagen-lysine,2-oxoglutarate 5-dioxygenase 1), PLOD2 (Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2) |
| R-HSA-1592389 | Activation of Matrix Metalloproteinases | no FDA-approved drugs found. |
Several proteins involved in ECM remodeling and adhesion were identified as potential therapeutic targets. Fibronectin (FN1) was highly represented in multiple pathways, including ECM proteoglycans and insulin-like growth factor (IGF) regulation, and is targetable by zinc-based compounds. Similarly, amyloid-beta precursor protein (APP) was enriched in ECM-associated processes and IGF transport, with known inhibitors such as deferoxamine and aducanumab. Plasminogen (PLG), which plays a role in ECM degradation and IGF transport, is targeted by fibrinolytic agents including alteplase and urokinase among others. Basement membrane proteoglycan HSPG2 is another key component of ECM pathways and can be inhibited by palifermin and efanesoctocog alfa. Additionally, inter-alpha-trypsin inhibitor heavy chain H2 (ITIH2), present in IGF-related processes, is modulated by zinc acetate and zinc chloride.
Several additional pathway-linked targets were identified. Multiple sodium channel protein subunits (SCN genes) were significantly enriched in L1-ankyrin interactions and are known targets of brivaracetam, ranolazine, and cocaine. Proteins involved in cell junction organization, such as cadherin-11 (CDH11) and filamin A (FLNA), are targetable by celecoxib and artenimol, respectively. Moreover, collagen biosynthesis proteins (P3H2, PLOD1, PLOD2) were identified and can be modulated by ascorbic acid. Finally, coagulation factor V (F5), found in IGF regulatory pathways, is targetable by drotrecogin alfa and thrombin-based therapies. (Datasheet DS5).
These findings highlight key platelet-associated pathways in NSCLC and identify potential drug targets within ECM remodeling, adhesion, and insulin signaling.
In summary, these transcriptome-based findings provide an initial step in identifying potential therapeutic targets in NSCLC. While the clustering approach revealed platelet-intrinsic regulatory modules, GSEA uncovered globally enriched pathways in TEPs, representing broader systemic alterations in platelets in NSCLC. A more integrated perspective considering platelet interactions, pathway networks, and cancer cell targeting could further enhance the understanding and clinical relevance of these targets.
2.3. Therapeutic Target Discovery Based on Network Controllability
To have a broader view into platelet function in NSCLC, we first reconstructed the platelet signaling network using high-quality, directed, and signed interactions from OmniPath [74], which provides a comprehensive representation of key pathways involved in platelet function. This network consists of 962 interactions among 401 platelet-specific proteins, capturing essential signaling processes such as platelet activation, receptor tyrosine kinase signaling, and Rho GTPase signaling. Pathway enrichment analysis further highlights its strong representation of immune-related pathways and cytoskeletal regulation, reinforcing its biological relevance (Figure S2). The detailed construction and analysis of this network, along with its implications for platelet signaling, are presented in the Supplementary Text.
Our next step focused on analyzing the platelet network to determine its controllability, a key factor in understanding how to effectively manipulate the network, in other words, to be able to steer the network from any initial state to any desired final state through external interventions. To achieve this, we first identified critical nodes, nodes that are necessary for network control. These critical nodes are present in every MDS, meaning that they are necessary for modulating this network’s behavior. Our analysis revealed 86 critical nodes within the network. In addition, we identified 196 intermittent nodes that play a supporting role in network modulation and 119 redundant nodes, whose presence is not necessary for the network since alternative pathways exist.
Next, we analyzed the network to classify nodes according to their indispensability. Indispensable nodes are essential for signal transduction through the network; removing these nodes significantly hampers the ability to control the network. We found that the network contains 62 indispensable nodes. In contrast, we identified 127 neutral nodes, which have a moderate impact on control when removed, and 212 dispensable nodes, whose removal has little to no effect on the network’s overall controllability (Figure 3A).
Figure 3.
Network Controllability analysis reveals key proteins to target in TEPs. (A) We show indispensable nodes (yellow) and critical nodes (blue) in the platelet network. (B) Comparison of controllability, degree, betweenness centrality, and logFC between indispensable and critical nodes. The statistical comparisons are made using the Mann-Whitney U test (Wilcoxon rank sum test).
Figure 3.
Network Controllability analysis reveals key proteins to target in TEPs. (A) We show indispensable nodes (yellow) and critical nodes (blue) in the platelet network. (B) Comparison of controllability, degree, betweenness centrality, and logFC between indispensable and critical nodes. The statistical comparisons are made using the Mann-Whitney U test (Wilcoxon rank sum test).
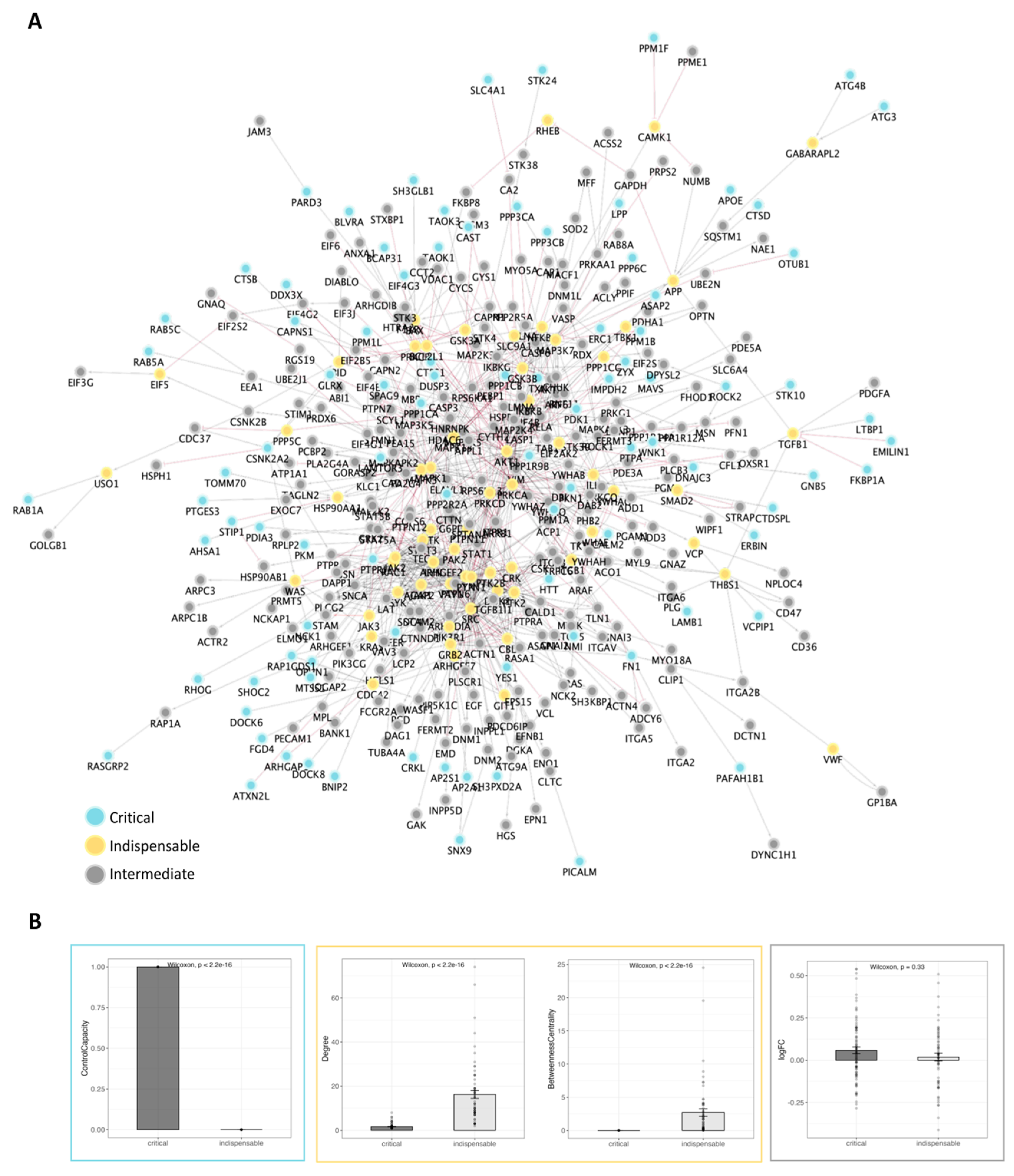
To further understand the roles of critical and indispensable nodes within the platelet network, we compared their controllability and topological measures. Critical nodes are consistently included in the Minimum Steering Node Set (MSS), which is a subset of the Minimal Driver Sets (MDS). Additionally, they show significantly higher control capacity. In contrast, indispensable nodes have a control capacity of zero and are found in none of the MSS. On the other hand, indispensable nodes show a slightly higher average control centrality of 73.5 compared to the average control centrality of 69.3 for critical nodes (Figure 3B).
Topologically, the differences between these two types of nodes are also pronounced. Indispensable nodes have significantly higher values in various measures including degree (number of connections), closeness centrality, betweenness centrality, stress, and clustering coefficients. They also play a key role as partners in multi-edge node pairs. In contrast, critical nodes show higher eccentricity and average shortest path length (Figure S3). Critical nodes also have 0 incoming interactions, slightly lower neighborhood connectivity in comparison to indispensable nodes (Figure S4).
Interestingly, there is no significant difference between the average expression levels and log2FC values of critical and indispensable nodes (Figure 3B). This implies that the observed differences between these node types are not attributed to the inherent expression or differential expression of these nodes but rather stem from their distinct roles and functional positions within the network.
These findings underscore the distinct roles that critical and indispensable nodes play in the platelet network. While critical nodes are essential for network control, indispensable nodes contribute significantly to the network’s structural properties and connectivity.
To identify the most regulated and targetable regions of platelet signaling in NSCLC, we constructed a central TEP subnetwork by calculating edge weights based on the fold changes and mapping the shortest paths from critical to indispensable nodes. This resulted in a focused subnetwork of 188 nodes and 501 interactions (Figure 4), representing the key dysregulated platelet functions in the tumor environment.
Figure 4.
Central TEP subnetwork in the platelet signaling with key nodes and drugs to target them. The central TEP network was constructed by identifying shortest paths from critical to indispensable nodes targeted by FDA-approved drugs. Blue: critical, yellow: indispensable, gray: intermediate, purple: drugs.
Figure 4.
Central TEP subnetwork in the platelet signaling with key nodes and drugs to target them. The central TEP network was constructed by identifying shortest paths from critical to indispensable nodes targeted by FDA-approved drugs. Blue: critical, yellow: indispensable, gray: intermediate, purple: drugs.
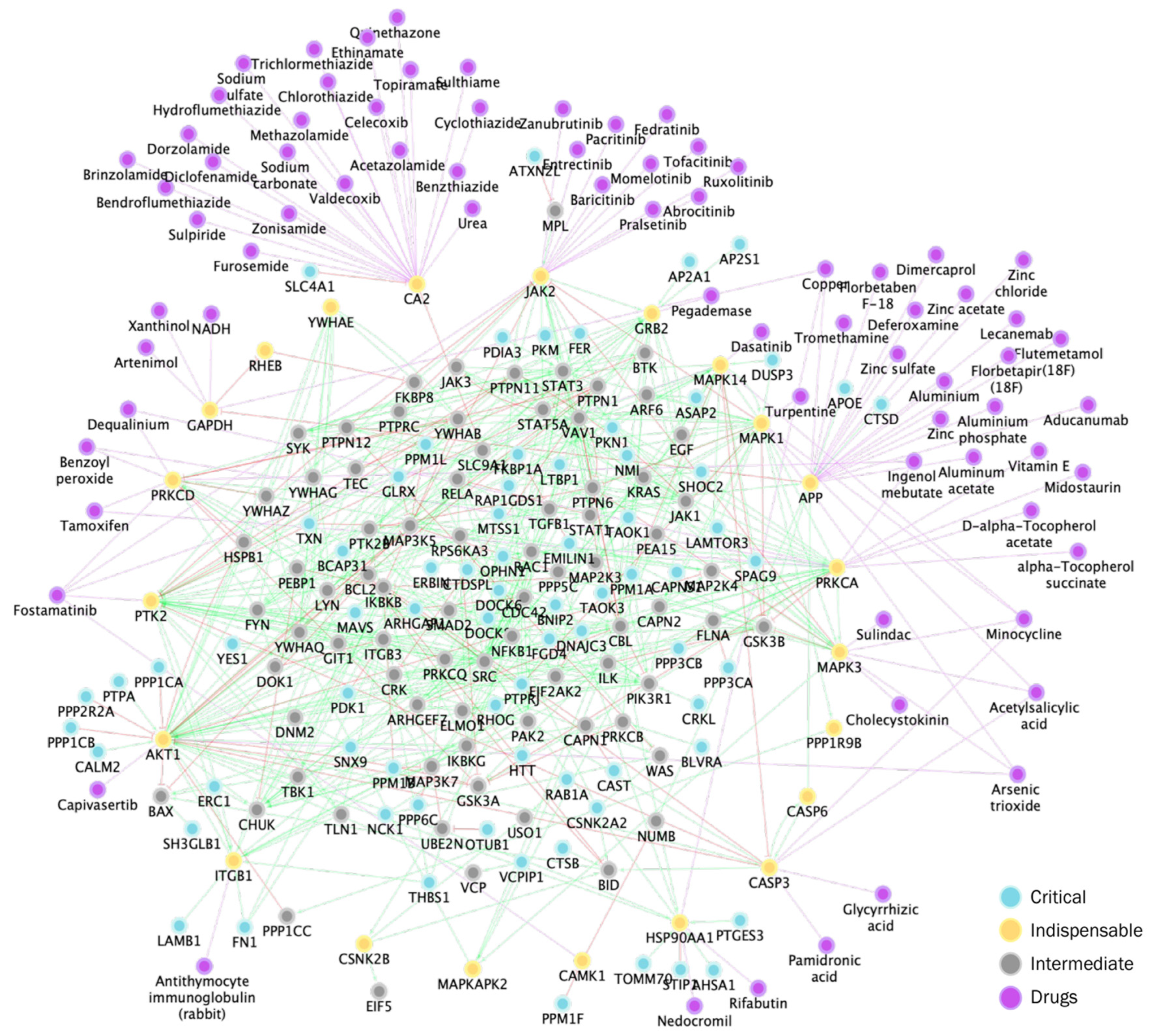
To enhance specificity and safety of therapeutic options, we identified the indispensable nodes in this central TEP. With this approach we aimed to narrow down the targetable space to those nodes that are more specifically involved in TEP functions, potentially offering a more precise and less risky therapeutic strategy.
Among the 22 indispensable nodes we identified in the central TEP subnetwork, 16 are known drug targets, which can be targeted by a total of 75 drugs. Top drugs targeting most indispensable nodes included fostamatinib, minocycline, and acetylsalicylic acid (Table 3). The top targets included were Carbonic anhydrase 2 (CA2), amyloid-beta precursor protein (APP), and Tyrosine-protein kinase JAK2 (JAK2).
Table 3.
FDA approved drugs targeting the reanalyzed Indispensable Nodes in the Controllable Subnetwork.
Table 3.
FDA approved drugs targeting the reanalyzed Indispensable Nodes in the Controllable Subnetwork.
| Drug Name1 | Number of Targets | Targets2 |
| Fostamatinib | 5 | CAMK1, JAK2, MAPK14, PRKCD, PTK2 |
| Minocycline | 4 | CASP3, MAPK1, MAPK14, MAPK3 |
| Acetylsalicylic acid | 3 | CASP3, MAPK1, MAPK3 |
| Arsenic trioxide | 3 | MAPK3, MAPK1, AKT1 |
| Copper | 3 | APP, GAPDH, HSP90AA1 |
| Benzoyl peroxide | 2 | PRKCA, PRKCD |
| Dequalinium* | 2 | PRKCA, PRKCD |
| Ingenol mebutate | 2 | PRKCD, PRKCA |
| Tamoxifen | 2 | PRKCA, PRKCD |
| Abrocitinib | 1 | JAK2 |
1 FDA approved drugs from Drugbank v5.1.12.; 2Only reanalyzed indispensable nodes are targeted.; *Other Approved.
To identify an optimal combination therapy, we filtered for FDA-approved drugs with known mechanisms and minimal interaction profiles. We prioritized drugs based on target coverage, inhibition strength, and network controllability. Fostamatinib emerged as the most promising agent, strongly inhibiting JAK2, PTK2, and CAMK1. Aducanumab was selected for its specificity against APP, and Acetylsalicylic acid for targeting CASP3, both consistently ranked high in control centrality and network influence.
To prioritize clinically actionable therapies, we filtered for FDA-approved drugs and known pharmacological action to ensure clinical applicability. Of the 75 drugs initially identified, only 54 met this criterion. Among the indispensable nodes, there were no FDA-approved drugs with a known mechanism of action and a non-interacting profile for PRKCA, GAPDH, HSP90AA1, AKT1, GRB2, and ITGB1. We provide a list of drugs in the supplement (Datasheet S6) that can target these nodes as their implied function in TEP signaling, these non-FDA approved drugs are an important lead for later drug development, targeting in this way both TEP’s contribution metastasis as well as direct metastatic potential in tumor cells.
To identify the most effective drug combination targeting indispensable nodes, we filtered for drugs with no interaction risk with fostamatinib. We then ranked combinations based on (1) number of indispensable targets covered, (2) target inhibition strength [75], and (3) control metrics of the targets within the network (e.g., control centrality, edge count, and betweenness centrality).
Fostamatinib emerged as the most promising agent, strongly inhibiting JAK2, PTK2, and CAMK1 ( details in Supplemental Text). Aducanumab was selected for its specificity against APP, and Acetylsalicylic acid for targeting CASP3, both targets consistently ranking high in network influence.
While other drugs like minocycline and acetazolamide were considered, they either had weaker evidence or added to regimen complexity ( details in Supplemental Text). By refining based on network metrics and drug interaction profiles, we propose the final, optimized combination of fostamatinib, aducanumab, and acetylsalicylic acid (aspirin) as a clinically actionable, multi-target strategy designed to modulate platelet hyperactivity and inflammation in cancer.
2.4. Expanded Platelet Interactome Reveals Novel Targetable Nodes
To enhance the platelet network analysis, we incorporated all available protein-protein interactions rather than restricting the dataset to signed and directed reactions from the Omnipath database. Filtering specific interactions can increase specificity but also carries the risk of overlooking potentially important correlations. By including all interactions, we reconstructed a more comprehensive platelet network encompassing 1638 interactions among 600 platelet proteins, approximately 1.5 times larger than the initial platelet signaling network.
To prioritize key proteins, we first assigned node weights based on both differential expression and network connectivity. The top 10% highest-weighted nodes (55 in total) included Proto-oncogene tyrosine-protein kinase Src (SRC) as the most connected protein, followed by Protein kinase C alpha (PRKCA), Tyrosine-protein kinase Lyn (LYN), Signal transducer and activator of transcription 1 (STAT1), and Phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) (Table S3, Datasheet DS7).
We then applied a second proximity-based scoring method designed for undirected networks, which ranked nodes by their closeness to high-weight nodes [76] (Table 4). The highest-scoring protein was Phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase 1 (INPP5D), followed by 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-2 (PLCG2) and E3 ubiquitin-protein ligase CBL (CBL), indicating their central roles in the expanded network (Table S4). This approach uncovered new potential drug targets, such as Low affinity immunoglobulin gamma Fc region receptor II-a (FCGR2A), P2Y purinoceptor 12 (P2RY12), Tyrosine-protein kinase Tec (TEC), Peptidyl-prolyl cis-trans isomerase A (PPIA), Tyrosine-protein kinase CSK (CSK), Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit beta (PIK3CB), Tubulin alpha-4A chain (TUBA4A), Integrin alpha-5 (ITGA5), and Tyrosine-protein phosphatase non-receptor type 6 (PTPN6) (Datasheet DS8).
Among the high-score nodes, Tyrosine-protein kinase JAK1 (JAK1) emerged as the most targetable, with ten FDA-approved drugs, including ruxolitinib, tofacitinib, and baricitinib. Low-affinity immunoglobulin gamma Fc region receptor II-a (FCGR2A) was also highly targetable, with drugs such as cetuximab, bevacizumab, and etanercept. Similarly, Tyrosine-protein kinase JAK3 (JAK3) was associated with multiple inhibitors, including ruxolitinib, filgotinib, and upadacitinib, while P2Y purinoceptor 12 (P2RY12) was targeted by ticagrelor, clopidogrel, and prasugrel. Notably, Tyrosine-protein kinase BTK (BTK) was another promising target, with inhibitors such as ibrutinib, acalabrutinib, and zanubrutinib.
In conclusion, combining the results of new transcriptome data on tumor-educated platelets and combining it with latest proteome and protein interaction data as well as applying a complementary prioritization approach revealed novel key nodes active in TEPs that are tumor and metastasis promoting that according to our results, are active in NSCLC lung cancer and we can now influence by FDA-approved drugs.
Table 4.
Targetable high-score nodes in platelet interactome.
| Drug Name1 | Degree | Log2FC | Weight | GeneScore | Drugs2 |
|---|---|---|---|---|---|
| LYN | 48 | -0,24 | 11,34 | 0,48 | Dasatinib, Bosutinib, Ponatinib, Nintedanib, Fostamatinib |
| JAK1 | 21 | -- | 0 | 0,47 | Ruxolitinib, Tofacitinib, Momelotinib, Baricitinib, Fostamatinib, Fedratinib, Filgotinib, Abrocitinib, Upadacitinib, Pralsetinib |
| FCGR2A | 4 | 0,68 | 2,71 | 0,46 | Cetuximab, Etanercept, Human immunoglobulin G, Abciximab, Alemtuzumab, Bevacizumab, Catumaxomab, Sarilumab |
| TEC | 10 | 0,25 | 2,47 | 0,46 | Bosutinib, Fostamatinib, Ritlecitinib, Zanubrutinib |
| TUBA4A | 1 | -0,13 | 0,13 | 0,45 | Vincristine, Podofilox |
| PTPN6 | 29 | -0,09 | 2,73 | 0,45 | Tiludronic acid |
| SYK | 37 | -0,1 | 3,84 | 0,45 | Fostamatinib |
| ITGA5 | 5 | -- | 0 | 0,45 | Tauroursodeoxycholic acid |
| GRB2 | 37 | -- | 0 | 0,45 | Pegademase |
| JAK3 | 16 | 0,45 | 7,3 | 0,44 | Ruxolitinib, Tofacitinib, Momelotinib, Baricitinib, Fostamatinib, Ritlecitinib, Abrocitinib, Zanubrutinib |
| P2RY12 | 1 | -0,49 | 0,49 | 0,44 | Ticlopidine, Treprostinil, Clopidogrel, Promethazine, Epoprostenol, Prasugrel, Cangrelor, Ticagrelor |
| BTK | 19 | -0,271 | 5,08 | 0,44 | Dasatinib, Ibrutinib, Acalabrutinib, Fostamatinib, Ritlecitinib, Zanubrutinib, Pirtobrutinib |
| PPIA | 1 | -- | 0 | 0,44 | Cyclosporine, Copper, Artenimol |
| PIK3CB | 1 | 0,13 | 0,13 | 0,44 | Caffeine, Copanlisib |
| CSK | 18 | 0,11 | 1,95 | 0,44 | Dasatinib, Fostamatinib |
| PTK2B | 37 | 0,18 | 6,76 | 0,44 | Leflunomide, Fostamatinib |
1 FDA approved drugs from Drugbank v5.1.12.; 2Only high-score nodes are targeted.
3. Discussion
Unlike physiological platelet activation triggered by vascular injury, tumor cell-induced platelet aggregation (TCIPA) is mediated by tumor-secreted factors such as coagulation and growth factors, and matrix metalloproteases [35,41]. This tumor-specific signaling presents a therapeutic opportunity to target tumor-educated platelets (TEPs) without disrupting normal platelet function [18].
Integration of the gene expression data with protein-protein interaction information allowed us to build a TEP-specific signaling network. Analyzing the network topology and controllability, we highlighted important proteins to this signaling, e.g., critical and indispensable nodes, high-weight and high-score nodes. These nodes revealed potential drug targets, and fostamatinib came up as the top-ranking with the broadest effect on the central TEP subnetwork.
The original authors (Best et al.) of the GSE dataset had an interesting agenda: “Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets”. Their study primarily analyzed RNA splicing events and identified differentially spliced transcripts in platelets from cancer patients using the thromboSeq method. They further focused on enhancing this approach with a particle swarm optimization (PSO) algorithm. However, their analysis was limited to splicing-related changes and did not include a detailed pathway analysis of platelet-pathophysiology. Instead, we directly investigated how TEP transcriptome differs from non-cancer platelets and activation.
Transcriptome comparisons revealed the highest similarity between TEPs and platelets from chronic pancreatitis patients, suggesting overlapping inflammatory signatures. However, to isolate cancer-specific signals, we pooled all non-cancer samples as controls. Among the most upregulated transcripts were several previously reported TEP biomarkers (e.g., ITGA2B (integrin alpha 2ß) [77], CD79 (B-cell antigen receptor complex) [78,79], PRSS50 (serine protease 50) [80], CRYM (crystallin mu; also known as THBP (NADP-regulated thyroid-hormone-binding protein)) [80], IGFBP2 (insulin-like growth factor binding protein 2) [80,81], LGALS3BP (galectin 3 binding protein) [81], IFITM3 (interferon induced transmembrane protein 3) [81], HPSE (heparanase) [81], LAMB2 (laminin subunit beta 2) [80], IFI27 (interferon alpha inducible protein 27) [81]). A notable novel finding was PSG2 (Pregnancy Specific Beta-1-Glycoprotein 2), a gene typically expressed in pregnancy [82] regulating immune response [83,84]. It was upregulated in certain cancers [85,86] but not previously detected in platelets. It has been noted that cancers exploit pregnancy-induced immunosuppression, which allows embryos and fetuses to express paternal antigens and still evade immune defense [87]. Its strong upregulation in TEPs as well as lack of evidence to its presence in platelets suggests tumor-derived RNA uptake, potentially supporting metastasis.
GSEA analysis using Gene Ontology, Reactome Pathways, and KEGG databases revealed downregulation of RNA splicing and translation, as well as antigen processing and presentation, B- and T-cell receptor signaling, apoptosis, NF-κB signaling, antigen processing, apoptosis, TCA cycle and oxidative phosphorylation, while we consistently observed upregulation of cell-cell or cell-ECM interaction, ECM organization, and cytoskeleton associated processes (Figure 1B). This finding aligns with decreased gene expression observed in lung cancer TEPs [15]. Additionally Best et al. also identified cytoskeletal processes as upregulated, and RNA translation, T cell immunity, and interleukin signaling as downregulated in TEPs from NSCLC as well as from glioblastoma, colorectal, pancreatic, hepatobiliary, and breast cancers [79].
Tumor-educated platelets (TEPs) showed transcriptional changes indicating adhesive interactions, potentially with tumor cells and the vasculature, could promote in this combination metastasis. A key finding was the significant upregulation of ITGA2B, a subunit of the platelet-specific αIIbβ3 integrin complex. This integrin mediates platelet aggregation and binds to ECM proteins such as fibrinogen, von Willebrand factor (VWF), and fibronectin, all mildly upregulated in TEPs. These interactions facilitate tumor–platelet bridging via fibrinogen and αVβ3 on tumor cells [52,88,89], enhancing shear resistance and vascular adhesion representing key steps in extravasation [89,90]. Furthermore, we observed upregulation of other platelet surface proteins like glycoproteins including GP1BB (glycoprotein Ib platelet subunit beta), GPNMB (glycoprotein nonmetastatic melanoma protein B), GPM6A (glycoprotein M6A), GPM6B (glycoprotein M6B) as well as PAPLN (papilin; proteoglycan-like sulfated glycoprotein), RHBG (Rh family B glycoprotein), SV2B (synaptic vesicle glycoprotein 2B), MOG (myelin oligodendrocyte glycoprotein), and PSG (pregnancy-specific glycoprotein) 1, 2, 4, 6, 9, and 11 (Figure S5).
TEPs showed increased expression of genes related to focal adhesion (KEGG hsa04510); proteins DOCK1 (dedicator of cytokinesis protein 1), LAMB2 (laminin subunit beta-2 protein), ITGA2B (integrin alpha 2ß), MYL9 (myosin light chain 9), FLNA (filamin A)) and ECM-receptor interactions (map hsa04512; LAMB2, ITGA2B).
Conversely, key immune-related genes such as CXCL8 (interleukin-8) were downregulated in TEPs. It is involved in neutrophil recruitment and angiogenesis [91,92,93], but its suppression may reflect tumor strategies to evade immune surveillance [94]. Gene set enrichment analysis revealed consistent downregulation of RNA processing, immune signaling (T- and B-cell receptor pathways), and metabolism, while genes related to cytoskeletal remodeling and ECM interactions were upregulated—hallmarks of platelet activation and tumor interaction.
In our analysis of the platelet transcriptome in NSCLC, we observed downregulation of genes encoding several cell surface proteins in TEPs, contradicting increased cell adhesion processes. However, we found here mostly downregulation of cell surface proteins related to immune signaling. CD79B is a component of the B-cell antigen receptor complex, CD79, and plays a critical role in B-cell signaling. It has been identified as an oncogenic driver in lung adenocarcinoma [95] and is included among the eleven genes suggested by Best et al. [79] as biomarkers and then tested by Goswami et al. [78] for the TEPs to diagnose NSCLC. Both CD79A and CD79B are also part of the downregulated gene set of the KEGG pathway “B-cell receptor signaling” (Figure S6). We also found CD8A along with CD8B, CD3D, and CD247 in the downregulated “T-cell receptor signaling” KEGG pathway in our enrichment analysis (Figure S6). Additionally, CXCR1 is a chemokine receptor, and the effect of platelets on T-cell CXCR1 has been shown to be immunosuppressive [96]. We observed CXCR1 in “Viral protein interaction with cytokine and cytokine receptor” KEGG pathway in GSEA (Figure S6).
In metastatic vs. primary cancer comparison (Datasheet DS9), we found platelet derived growth factor subunit A (PDGFA) upregulated (0.67). It was also slightly upregulated in NSCLC TEPs when directly compared to healthy donors with no existing health condition (0.33) (Datasheet DS10). PDGF is both pro-angiogenic and pro-metastatic [97] and increased PDGF secretion from platelets may be distinguishing factor aiding the transition from primary tumor to metastatic one. Increased levels of PDGF, along with TGFβ and MMP1, was shown to result from platelets taking up mRNA and proteins that are secreted by tumors [21,22].
TEPs are enriched in IGF-related pathways, particularly the REACTOME pathway for “IGF transport and uptake via IGF binding proteins (IGFBPs)”, highlighting a potential role in cancer progression. IGF-1 enhances platelet activation through the IGF receptor/IRS/PI3K/PKB pathway [98], contributing to cancer-associated hypercoagulability [99] and metastasis [100]. Autocrine IGF-1 may further drive tumor-mediated platelet “education,” with cancer signals altering RNA splicing to increase IGF-related transcripts [100].
A key mediator is IGFBP2, upregulated in several cancers and involved in processes such as EMT, angiogenesis, and invasion via β-catenin and STAT2 signaling contributing to the malignancy [101]. It promotes platelet-mediated cancer cell communication, particularly in MACC1-driven colorectal cancer metastasis [102,103]. IGFBP2 upregulation is further associated with increased metastasis, larger tumors and poor survival in NSCLC [103,104,105], while enhancing gefitinib resistance through STAT2 signaling [106]. IGFBP2 can be released from platelets upon activation leading to the modulation of IGF signaling in the tumor microenvironment [102] which suggests a role as a functional mediator as well as a biomarker for the disease progression.
3.1. Targeting Tumor-Educated Platelet Signaling
TEPs have emerged as key facilitators of cancer progression, immune evasion, and metastasis. To identify potential therapeutic targets within TEPs, we employed four complementary strategies, integrating transcriptomic profiling with network-based systems biology approaches.
In the following we show that our suggestions of targets and FDA-approved drugs to positively influence TEPs are supported by literature.
The first two strategies focused exclusively on the gene expression data. These analyses identified ITGA2B and FLNA as consistently upregulated and functionally relevant in platelet signaling and cancer-associated thrombosis. Both genes are critical to platelet structure and activation, and represent targets of FDA-approved therapies.
Inhibitors of αIIbβ3 integrin were shown to decrease TCIPA, ECM degradation by tumor cells [88], and colonization of tumor cells in the lungs [69]. Moreover, the strong expression levels of ITGA2B were found to be significantly higher in TEPs from NSCLC patients and its use as a diagnostic marker proved to have high sensitivity and specificity, making it a promising marker to diagnose early-stage NSCLC [77].
To expand beyond expression changes and capture the complexity of protein interactions, we implemented two network-based approaches. In the third strategy, we applied network controllability analysis, identifying optimal intervention points that could efficiently modulate TEP signaling. This approach prioritized a drug combination of fostamatinib, aducanumab, and acetylsalicylic acid, targeting JAK2, PTK2, and CAMK1; APP; and CASP3 respectively.
The fourth strategy involved a network proximity-based scoring method to rank genes by their closeness to high-weight nodes within the platelet–cancer interaction network. Both network-based strategies independently highlighted GRB2, a central adaptor in multiple platelet activation pathways, including ITAM signaling, as a potential therapeutic bottleneck [107,108].
Across all methods, we observed convergence on FCGR2A and APP as key targets, supporting their robust involvement in cancer-platelet signaling. APP, known for its role in amyloid-β processing, also contributes to platelet-mediated thrombus formation and is targetable via monoclonal antibodies such as aducanumab [109].
FCGR2A binds to tumor cell-derived IgG, facilitating platelet activation via the ITAM signaling pathway, which also includes ITGA2B and GRB2, and has been implicated to mediate immuno-thrombosis across multiple cancers via interacting with the platelet integrin αIIbβ3, though not yet in NSCLC [34,110]. Studies have shown that blocking FCGR2A or knocking down IgG in various cancers, including hepatocellular carcinoma (HCC), cervical, and bladder cancers, reduces platelet activation and subsequent metastasis [34]. Similarly, in colon, prostate, and breast cancers, interference with FCGR2A has proven effective in limiting platelet activation [33]. However, the role of FCGR2A in NSCLC remains unexplored. Targeting cancer-derived IgG or blocking FCGR2A could potentially hinder TEP-mediated metastasis and may offer a novel therapeutic strategy for inhibiting cancer-induced platelet activation while minimizing disruption to normal hemostatic processes.
Taken together, our integrative strategy revealed ITGA2B, FLNA, GRB2, FCGR2A, and APP as high-confidence, FDA-targetable candidates that intersect cancer-specific platelet education with pro-metastatic platelet signaling. Importantly, several targets (e.g., GRB2, FCGR2A) are involved in ITAM signaling, which governs immune receptor-driven platelet activation. Selectively disrupting this pathway may suppress TEP-mediated cancer progression while preserving physiological hemostasis.
Along with the targeted therapeutic strategies focusing on specific platelet proteins, most of which proved to be useful targets in antiplatelet and antimetastatic therapy [40,62,63,64,65,66,67,68,69,70,71,72,73,111,112,113,114], we looked at the bigger picture using a more comprehensive strategy by investigating the TEP signaling regulated by cancer employing networks. Across network-based strategies, fostamatinib frequently emerged as a top drug target, as well as in our previous study on platelet hyperactivation in COVID-19 [115]. Although we identified multiple targets of fostamatinib in our networks, it is important to note that it does not inhibit all targets equally, as indicated by varying inhibition constants (Ki) [75]. Through its primary target Syk, it inhibits the ITAM signaling pathways downstream of GPVI, CLEC-2, and FCGR2A [116,117,118]. These three receptors were also shown to be the primary way tumor cells activate platelets, differently from physiological conditions, where platelet activation starts with adhesion receptors [31,32,33,119]. Furthermore, it’s been reported that both liver cancer [34] and SARS-CoV-2 [120,121] activate platelets by IgG binding to the FCGR2A receptor. This suggests that receptors of ITAM signaling are promising drug targets for managing both infection-related coagulopathy and cancer-associated thrombosis. Especially, inhibiting GPVI was shown to inhibit thrombosis with no major impact on hemostasis, making it a potential antithrombotic and anti-metastatic therapeutic option [31].
Furthermore, we suggest a combination therapy of fostamatinib, aducanumab, and aspirin, tailored to selectively target TEP activity instead of only using fostamatinib. As they do not interfere with each other and act independently with different biological targets (see Supplementary Text), this allows a safer low dose regime for administrating the combination.
Fostamatinib has already been shown to be a safe and effective therapeutic agent against primary and secondary immune thrombocytopenia (ITP) [122]. Besides the anti-thrombotic features, it demonstrated anti-cancer activities in multiple in vitro studies inhibiting various molecules and pathways such as receptor tyrosine kinases, PI3K-AKT pathway and immune checkpoints PD-L1 and CD47, decreasing cell proliferation and inducing apoptosis in different cancers including NSCLC and AML [123,124]. These results were also supported by multiple phase I and phase II clinical trials targeting different carcinomas such as NSCLC, advanced colorectal, platinum-resistant ovarian, thyroid and renal cell carcinoma [125,126].
Aspirin has long been known to show anti-platelet activity mainly via the inhibition of COX-1 pathway and reducing thromboxane A2 formation, and thus, has been effective against inflammation-induced platelet aggregation [127,128,129]. It was also suggested to have a protective effect against multiple cancer types including colorectal, prostate, pancreatic cancers as well as lymphomas [130]. A recent study revealed anti-metastatic properties of aspirin via the inhibition of COX-1-TXA2 pathway and releasing T-cells from the TXA2 mediated and intrinsic ARHGEF1 dependent immunosuppression leading to decrease in metastatic rates [131]. When used in combination, the anti-thrombotic and anti-metastatic properties of fostamatinib and aspirin might be boosted as the safety of such a combination was previously assessed positively [117]. Aducanumab has not yet been tested for any effect regarding platelets or cancer, however, it might be a valuable addition against one of the consistently emerging key proteins, APP, that aducanumab was shown to effectively target [132].
Based on these observations, the potential of fostamatinib against platelet hyperactivation in inflammatory contexts is strongly supported. As the next steps, experiments to assess the effects of fostamatinib on the other targets that we identified can help refine our networks and followingly our understanding of their impact. Additionally, measuring fostamatinib’s impact directly on platelet function in platelets derived from NSCLC patients would further reinforce its anti-metastatic use, while in the context of COVID-19 it has already been shown to reverse platelet hyperactivity [118]. It is important to note, that using drugs with broader effects on TEP signaling may have drawbacks, including the possibility of unknown side effects that could lead to bleeding complications. Therefore, Future work should focus on functional validation of these targets, particularly in NSCLC, and assessment of therapeutic combinations in preclinical models.
3.2. Limitations and Outlook
To gain a broader understanding of the cancer side of the platelet-cancer loop, reconstruction of the NSCLC large-scale networks can help identify the significant modules when combined with gene expression or proteomics data. Additionally, as we demonstrated in the analysis of the platelet signaling, network analysis including centrality and controllability measures can assist in identifying key targets that are central to pro-metastatic functions.
Further investigation of platelets’ role in metastasis requires drawing a more specific picture. For this, although more challenging than large-scale analysis, integration of newly found important platelets protein and their simulation may be helpful. Furthermore, a complete annotation of platelet proteins such as receptor, surface or secreted proteins etc. can help to identify important surface proteins. These can be then investigated for their interaction partners on cancer cell surface via analyzing domain-domain interactions, domain-motif interactions, and using machine learning approaches.
Lastly, investigating the indirect role of platelets in cancer through interactions with other cell types may help us understand their functions in a bigger context, including interactions with endothelial cells to promote angiogenesis or natural killer cells to inhibit their cytotoxicity or macrophages to induce their differentiation to M2 phenotype, all of which constitute an important aspect of platelets’ role in metastasis.
Mammalian platelets evolved to be highly efficient in hemostasis and immune defense, offering survival advantages through rapid response to injury; however, this efficiency carries an evolutionary tradeoff, particularly evident under chronic inflammatory conditions that now drive high rates of thrombotic disorders in humans [133]. With advances in medicine significantly increasing life expectancy since the 19th century [134,135], infectious diseases have become less prevalent, shifting major health burdens toward aging-related conditions like cardiovascular disease and cancer [136]. As a result, platelet hyperactivity has emerged as a central factor in these diseases, as well as in acute infections such as COVID-19 [137,138,139]. The very mechanisms that once ensured survival are now co-opted by cancers, which mimic chronic wounds [140] and perpetuate a cycle of inflammation and platelet activation, fostering disease progression through thrombo-inflammation. Thus, our drug screening approach, while directly targeting TEPs, also potentially disrupts pro-metastatic tumor cell behaviors and coagulation-promoting kinase activity, stressing a dual therapeutic benefit.
4. Materials and Methods
4.1. RNAseq Data Analysis
We downloaded 779 samples (826 SRA files/runs) from ENA Bioproject ID: PRJNA353588, GSE89843 [177]. The samples included 402 tumor-educated platelet (TEP) samples from patients with non-small cell lung cancer (NSCLC) and 377 platelet samples from donors without cancer, some with inflammatory conditions. The samples were sequenced on Illumina Hiseq 2500 platform. The quality control of the fastq files were evaluated with FastQC [153] and fastp [152]. Fastp was also used to trim adapters and low-quality reads. After trimming, we indexed human GRCh38 cDNA/transcriptome with kallisto index and performed pseudoalignment and quantification of the reads with kallisto quant function [161].
4.2. Differential Gene Expression Analysis and Data Normalization
Kallisto output files with counts for each sample were imported in Rstudio with tximport [174]. We first collapsed technical replicates (collapseReplicates from DESeq2) and removed one sample from PISA hospital (GSM2391029). Lowly expressed genes were filtered by using filterByExpr from edgeR (min.count = 10, design = ~class, min. group size = 376). We performed variance-stabilizing transformation (vst from DESeq2) to explore the data with principal component analysis (PCA). We removed two outliers we observed in PCA plots (GSM2390915 and GSM2627443). Next, we checked for unwanted variation (UV) in the data using RUVg from RUVSeq. For this, we generated a list of stable (empirical) genes. We fit a Gamma-Poisson generalized linear model to the filtered data and ran a likelihood ratio test (LRT) to find genes that do not have changes in their expression across different levels of the experimental conditions (~class). We then defined the empirical genes as the genes with log2FC between -0.05 and 0.05 and with adjusted p-values larger than 0.05. We ended up with 1491 genes as empirical genes and using this list we performed RUV with k=5. We included the 5 UV factors in the design matrix alongside the condition of interest (~class: non-cancer vs. NSCLC) and performed differential expression (DE) analysis with limma-voom and edgeR. Genes that have log2FC values smaller than -0.58 and larger than 0.58 adjusted p-values smaller than 0.05 were classified as differentially expressed genes.
4.3. Volcano Plots and Heatmaps
We used EnhancedVolcano R package to make the volcano plots of all the genes. Differentially expressed genes were colored blue if downregulated, red if upregulated, and black if significant and gray if not significant.
For the heatmaps we used pheatmap and scaling was done row-wise (to obtain z-scores).
4.4. Gene Set Enrichment Analysis
We used ClusterProfiler for the Gene Set Enrichment Analysis (GSEA) and used enrichGO, enrichKEGG functions for Gene Ontology and KEGG Pathways. For Reactome Pathways the gsePathway function from ReactomePA R package was used. P-values were corrected for multiple testing using Benjamini-Hochberg.
4.5. Clustering of DEGs
We used k-means clustering with k=9 (number of modules) to cluster DEGs. We separated the non-cancer class into subgroups and ended up with 9 groups: NSCLC, Healthy, Chronic Pancreatitis, Epilepsy, Multiple Sclerosis (MS), Non-significant (NS) atherosclerosis, Pulmonary Hypertension, Stable Angina Pectoris (AP), and Unstable Angina Pectoris (AP). We used Pearson correlation to cluster the rows and Spearman correlation for samples. For each module, we then performed functional enrichment analysis using the gost function from gprofiler2 package to find overrepresented Gene Ontology (GO) terms, KEGG, and REACTOME pathways. We visualized top 5 overrepresented pathways for each module using ggplot2.
4.6. Construction of Platelet Signaling Network
We began by importing all post-translational interactions using the OmnipathR [165] package in R. To ensure data quality, we filtered out low-quality interactions and kept only directed and signed interactions that were supported by a curation effort of at least 3 [74]. This quality control step significantly reduced the number of interactions from 134,282 to 12,963.
Next, we constructed the platelet signaling network by filtering the high-quality Omnipath interactions based on gene and protein expression specific to platelets. We selected nodes with an average expression (average log2-expression for the probe over all arrays and channels in limma:topTable) of at least 1 (25th percentile, also defined as first quartile) in the RNA-sequencing dataset or those detected in the two proteomics datasets (unpublished, details in Supplement). We then predicted interactions based on the regulation of the nodes involved by multiplying their respective log2FC values. We used only the log2FCs of significant genes, as the regulation of non-significant genes could be misleading; for these, we assigned NA. If the product of this multiplication was positive, the interaction was classified as “activation”; if negative, it was classified as “inhibition.” We then compared these predicted interactions to the actual interaction types from the Omnipath database. Interactions where the predicted and actual types were contradictory were removed from the network, and the nodes that were not connected with the rest of the network were removed, resulting in the largest connected component of the platelet network containing 962 interactions among 401 platelet proteins.
From this platelet network, we further extracted a subnetwork focused on differentially expressed genes (DEGs). We isolated the interactions involving the 219 DEGs and removed any interactions that contradicted the predicted interaction type, as previously described. This process yielded a DEG network consisting of 23 nodes and 31 interactions.
Pathway Overrepresentation Analysis is done using gProfiler Cytoscape plugin. P-values were adjusted using g:SCS algorithm and whole human genome was used as background.
4.7. Network Controllability and Subnetworks
We identified the nodes that are topologically important for the controllability of the platelet network: indispensable nodes, and critical nodes.
To identify the minimum driver node sets (MDS) in the reconstructed networks, we implemented the concept of controllability and used Maximum Matching (MM), a graph theory-based approach for network analysis [144]. Then using the identified MDSs, we utilized the node classification schemes recommended by Vinayagam et al. [178], i.e., indispensable, neutral, and dispensable and Jia et al. [179], i.e., critical, intermittent, and redundant to identify the key nodes in the network for controllability and targeting.
To get the controllable subnetworks, we first calculated edge weight between nodes i and j as:
A lower edge weight signifies a more important edge, where the interacting nodes exhibit higher relative fold changes between two conditions. Using these edge weights, we identified the shortest paths from critical nodes to indispensable nodes by applying Dijkstra’s algorithm (igraph::shortest_paths) and constructed the controllable subspace.
4.8. Gene Scores and Subnetworks
We first assigned node weights to each node in the platelet network using the multiplication of degree and log2FC. For nodes that are not significantly regulated, we used log2FC value of 1*10-6. High weight genes are the ones that are then highly connected and regulated. Then using these high weight genes, we assigned gene scores to nodes that are connected to them with shortest path.
Where represents the weight of node j and is the average node weight, and is an identity function that equals 1 when node I and node j are within a distance s, and 0 otherwise (SANTA::Knode) [76]. We then obtained the controllable subspaces by combining the shortest paths between the top 10% of the high gene scored nodes and the top 10% high weight nodes. Since the node classification is already based on differential regulation, we did not use edge weights.
4.9. Drug Repurposing
We downloaded all FDA approved drugs and their targets from Drugbank 5.1.12. We searched for all drugs that target the critical nodes (or the high gene score nodes) in the controllable subspace and visualized it with R ggraph. Drug interactions are obtained from Drugbank and drugs.com.
5. Conclusions
Tumor-educated platelets (TEPs) display a cancer-specific transcriptional program distinct from physiological activation. In this study, we investigated TEPs in non-small cell lung cancer (NSCLC) to uncover potential therapeutic vulnerabilities that could curb metastasis without impairing physiological hemostasis. Analyzing an exceptional, recent large-scale transcriptome data set focusing on TEPs, complementing it by latest data on protein-protein interactions we give pathways, molecular markers and target TEPs pharmacologically in a novel, beneficial way which, after appropriate clinical testing, should find its way into the clinic:
Pathways: Transcriptomic analysis of latest TEP data revealed that genes for adhesion, ECM-receptor interactions, and cytoskeletal remodeling are upregulated, whereas RNA processing, antigen presentation, lymphocyte receptor signaling, and core metabolic pathways are suppressed.
Molecular markers: Alongside established TEP biomarkers (ITGA2B, IGFBP2), PSG2 indicates tumor-derived RNA uptake. PDGF/IGF signaling may potentiate platelet activation and pro-metastatic crosstalk. In this platelet state primed for tumor binding, vascular adhesion, and metastatic assistance, central platelet subnetworks involve the ITAM-SYK hyperactivation core (FCGR2A, SYK, GRB2) with JAK2, CAMK1, and PTK2 as potential intervention points; a cytoskeleton/adhesion sub-module (ITGA2B, FLNA, MYL9, and DOCK1), an ECM-remodeling/metalloprotease axis (MMP1, HPSE), and an apoptosis sub-module centered around CASP3. Two approaches based solely on differential gene expression converged on ITGA2B and FLNA, known regulators of platelet structure and function. Network-based analyses, leveraging controllability and proximity scoring, further highlighted GRB2 as a key signaling hub. Importantly, FCGR2A and APP were consistently identified across transcriptomic and network methods, reinforcing their roles in TEP-mediated immune evasion and thromboinflammatory processes.
FDA-approved drugs for mitigation: These five genes—ITGA2B, FLNA, GRB2, FCGR2A, and APP—emerged as high-confidence, FDA-targetable candidates. Many participate in ITAM signaling, which is uniquely co-opted by tumor cells to activate platelets, representing an attractive avenue for targeted disruption of cancer-associated platelet activity. We also identify metalloproteases as key promoters of metastasis and historically challenging yet compelling targets for therapeutic exploration. Notably, fostamatinib, an FDA-approved SYK inhibitor, consistently ranked as a top candidate, suggesting it could suppress ITAM-mediated TEP activation. In light of its previous success in reversing platelet hyperactivation in COVID-19, we propose fostamatinib, alone or in combination with aducanumab and acetyl salicylic acid, as a potential therapeutic strategy to selectively block cancer-platelet crosstalk. Future experimental validation in NSCLC-derived platelets and preclinical models will be critical to assess the efficacy and safety of these targeted interventions and to refine our understanding of TEP signaling in cancer progression.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Ö.O., S.K.G., T.D. and H.S. conceptualized the work and Ö.O., E.Ö., and S.K.G. developed the methodology and implementation. Ö.O. and E.Ö. curated the data and together with S.K.G., visualized the results. T.D., H.S., and K.G.H. provided funding acquisition while T.D. and H.S. additionally supervised the project and administration. Ö.O., E.Ö., S.K.G., and T.D. wrote the initial manuscript, and with the support of H.S. carried out validations. All authors reviewed and edited the manuscript and contributed to the investigation. Ö.O. and E.Ö. contributed equally; T.D. is the corresponding author.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), CRC1583 DECIDE [Project ID 492620490/INF].
Conflicts of Interest
The authors declare no conflict of interest, neither personal nor financial or otherwise.
References
- Metharom, P., M. Falasca, and M.C. Berndt, The History of Armand Trousseau and Cancer-Associated Thrombosis. Cancers (Basel), 2019. 11(2).
- Crissman, J.D., et al., Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab Invest, 1985. 53(4): p. 470-8.
- Gasic, G.J., et al., Aggregation of platelets and cell membrane vesiculation by rat cells transformed in vitro by Rous sarcoma virus. Cancer Res, 1978. 38(9): p. 2950-5.
- Stone, R.L., et al., Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med, 2012. 366(7): p. 610-8.
- Menter, D.G., et al., Platelets and cancer: a casual or causal relationship: revisited. Cancer Metastasis Rev, 2014. 33(1): p. 231-69.
- Haemmerle, M., et al., FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J Clin Invest, 2016. 126(5): p. 1885-96.
- Varki, A., Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood, 2007. 110(6): p. 1723-9.
- Khorana, A.A. and G.C. Connolly, Assessing risk of venous thromboembolism in the patient with cancer. J Clin Oncol, 2009. 27(29): p. 4839-47.
- Lyman, G.H. and A.A. Khorana, Cancer, clots and consensus: new understanding of an old problem. J Clin Oncol, 2009. 27(29): p. 4821-6.
- Bailey, S.E., et al., Clinical relevance of thrombocytosis in primary care: a prospective cohort study of cancer incidence using English electronic medical records and cancer registry data. Br J Gen Pract, 2017. 67(659): p. e405-e413.
- Abdulrahman, G.O., N. Das, and K. Lutchman Singh, The predictive role of thrombocytosis in benign, borderline and malignant ovarian tumors. Platelets, 2020. 31(6): p. 795-800.
- Skorek, P., et al., Preoperative thrombocytosis in surgically treated patients with non-small cell lung cancer. Pol Arch Intern Med, 2018. 128(9): p. 512-517.
- Li, N., Platelets in cancer metastasis: To help the “villain” to do evil. Int J Cancer, 2016. 138(9): p. 2078-87.
- Sasaki, Y., et al., Production of thrombopoietin by human carcinomas and its novel isoforms. Blood, 1999. 94(6): p. 1952-60.
- Calverley, D.C., et al., Significant downregulation of platelet gene expression in metastatic lung cancer. Clin Transl Sci, 2010. 3(5): p. 227-32.
- Sabrkhany, S., et al., Exploration of the platelet proteome in patients with early-stage cancer. J Proteomics, 2018. 177: p. 65-74.
- Denis, M.M., et al., Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell, 2005. 122(3): p. 379-91.
- Li, S., et al., The dynamic role of platelets in cancer progression and their therapeutic implications. Nat Rev Cancer, 2024. 24(1): p. 72-87.
- Banerjee, M., et al., Cellubrevin/vesicle-associated membrane protein-3-mediated endocytosis and trafficking regulate platelet functions. Blood, 2017. 130(26): p. 2872-2883.
- Banerjee, M. and S.W. Whiteheart, The ins and outs of endocytic trafficking in platelet functions. Curr Opin Hematol, 2017. 24(5): p. 467-474.
- Kuznetsov, H.S., et al., Identification of luminal breast cancers that establish a tumor-supportive macroenvironment defined by proangiogenic platelets and bone marrow-derived cells. Cancer Discov, 2012. 2(12): p. 1150-65.
- Kerr, B.A., et al., Platelets govern pre-metastatic tumor communication to bone. Oncogene, 2013. 32(36): p. 4319-24.
- Roweth, H.G. and E.M. Battinelli, Lessons to learn from tumor-educated platelets. Blood, 2021. 137(23): p. 3174-3180.
- Weyrich, A.S., et al., Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci U S A, 1998. 95(10): p. 5556-61.
- Nilsson, R.J., et al., Blood platelets contain tumor-derived RNA biomarkers. Blood, 2011. 118(13): p. 3680-3.
- Ye, B., et al., A panel of platelet-associated circulating long non-coding RNAs as potential biomarkers for colorectal cancer. Genomics, 2022. 114(1): p. 31-37.
- Tabaeian, S.P., et al., Evaluation of tumor-educated platelet long non-coding RNAs (lncRNAs) as potential diagnostic biomarkers for colorectal cancer. J Cancer Res Ther, 2024. 20(5): p. 1453-1458.
- Moonmuang, S., et al., Circulating Long Non-Coding RNAs as Novel Potential Biomarkers for Osteogenic Sarcoma. Cancers (Basel), 2021. 13(16).
- Yuan, M., et al., Screening and validation of platelet activation-related lncRNAs as potential biomarkers for prognosis and immunotherapy in gastric cancer patients. Front Genet, 2022. 13: p. 965033.
- Laffont, B., et al., Activated platelets can deliver mRNA regulatory Ago2*microRNA complexes to endothelial cells via microparticles. Blood, 2013. 122(2): p. 253-61.
- Mammadova-Bach, E., et al., Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood, 2020. 135(14): p. 1146-1160.
- Ichikawa, J., et al., Role of Platelet C-Type Lectin-Like Receptor 2 in Promoting Lung Metastasis in Osteosarcoma. J Bone Miner Res, 2020. 35(9): p. 1738-1750.
- Mitrugno, A., et al., A novel and essential role for FcgammaRIIa in cancer cell-induced platelet activation. Blood, 2014. 123(2): p. 249-60.
- Miao, S., et al., Cancer cell-derived immunoglobulin G activates platelets by binding to platelet FcgammaRIIa. Cell Death Dis, 2019. 10(2): p. 87.
- Strasenburg, W., et al., Tumor Cell-Induced Platelet Aggregation as an Emerging Therapeutic Target for Cancer Therapy. Front Oncol, 2022. 12: p. 909767.
- Heinmoller, E., et al., Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol, 1996. 122(12): p. 735-44.
- Jurasz, P., D. Alonso-Escolano, and M.W. Radomski, Platelet--cancer interactions: mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br J Pharmacol, 2004. 143(7): p. 819-26.
- Cooke, N.M., et al., Increased platelet reactivity in patients with late-stage metastatic cancer. Cancer Med, 2013. 2(4): p. 564-70.
- Tesfamariam, B., Involvement of platelets in tumor cell metastasis. Pharmacol Ther, 2016. 157: p. 112-9.
- Mammadova-Bach, E., et al., Platelets in cancer. From basic research to therapeutic implications. Hamostaseologie, 2015. 35(4): p. 325-36.
- Palacios-Acedo, A.L., et al., Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front Immunol, 2019. 10: p. 1805.
- Filippelli, A., et al., Scoping Review on Platelets and Tumor Angiogenesis: Do We Need More Evidence or Better Analysis? Int J Mol Sci, 2022. 23(21).
- Haemmerle, M., et al., The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell, 2018. 33(6): p. 965-983.
- von Hundelshausen, P., et al., Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood, 2005. 105(3): p. 924-30.
- Ansari, M.J., et al., Cancer combination therapies by angiogenesis inhibitors; a comprehensive review. Cell Commun Signal, 2022. 20(1): p. 49.
- Pinedo, H.M., et al., Involvement of platelets in tumour angiogenesis? Lancet, 1998. 352(9142): p. 1775-7.
- Maurer, S. and L. Ferrari de Andrade, NK Cell Interaction With Platelets and Myeloid Cells in the Tumor Milieu. Front Immunol, 2020. 11: p. 608849.
- Placke, T., et al., Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res, 2012. 72(2): p. 440-8.
- Plantureux, L., et al., Effects of platelets on cancer progression. Thromb Res, 2018. 164 Suppl 1: p. S40-S47.
- Gay, L.J. and B. Felding-Habermann, Contribution of platelets to tumour metastasis. Nat Rev Cancer, 2011. 11(2): p. 123-34.
- Erpenbeck, L. and M.P. Schon, Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood, 2010. 115(17): p. 3427-36.
- Lonsdorf, A.S., et al., Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem, 2012. 287(3): p. 2168-78.
- Labelle, M., S. Begum, and R.O. Hynes, Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A, 2014. 111(30): p. E3053-61.
- Foss, A., et al., The contribution of platelets to intravascular arrest, extravasation, and outgrowth of disseminated tumor cells. Clin Exp Metastasis, 2020. 37(1): p. 47-67.
- Schumacher, D., et al., Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell, 2013. 24(1): p. 130-7.
- Sierko, E. and M.Z. Wojtukiewicz, Inhibition of platelet function: does it offer a chance of better cancer progression control? Semin Thromb Hemost, 2007. 33(7): p. 712-21.
- Seizer, P. and A.E. May, Platelets and matrix metalloproteinases. Thromb Haemost, 2013. 110(5): p. 903-9.
- Vlodavsky, I., et al., Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis, 1992. 12(2): p. 112-27.
- Giannakeas, V., et al., Analysis of Platelet Count and New Cancer Diagnosis Over a 10-Year Period. JAMA Netw Open, 2022. 5(1): p. e2141633.
- Sabrkhany, S., et al., Platelets as messengers of early-stage cancer. Cancer Metastasis Rev, 2021. 40(2): p. 563-573.
- Huong, P.T., et al., The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers (Basel), 2019. 11(2).
- Yu, L., et al., Bidirectional Interaction Between Cancer Cells and Platelets Provides Potential Strategies for Cancer Therapies. Front Oncol, 2021. 11: p. 764119.
- Franco, A.T., A. Corken, and J. Ware, Platelets at the interface of thrombosis, inflammation, and cancer. Blood, 2015. 126(5): p. 582-8.
- Battinelli, E.M., B.A. Markens, and J.E. Italiano, Jr., Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood, 2011. 118(5): p. 1359-69.
- Guillem-Llobat, P., et al., Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget, 2016. 7(22): p. 32462-77.
- Lucotti, S., et al., Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest, 2019. 129(5): p. 1845-1862.
- Gebremeskel, S., et al., The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int J Cancer, 2015. 136(1): p. 234-40.
- Zhang, W., et al., A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood, 2012. 120(14): p. 2889-98.
- Amirkhosravi, A., et al., Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost, 2003. 90(3): p. 549-54.
- Khorana, A.A., et al., Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost, 2007. 5(3): p. 632-4.
- Riedl, J., et al., Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood, 2017. 129(13): p. 1831-1839.
- Sasano, T., et al., Podoplanin promotes tumor growth, platelet aggregation, and venous thrombosis in murine models of ovarian cancer. J Thromb Haemost, 2022. 20(1): p. 104-114.
- Wojtukiewicz, M.Z., et al., Antiplatelet agents for cancer treatment: a real perspective or just an echo from the past? Cancer Metastasis Rev, 2017. 36(2): p. 305-329.
- Turei, D., T. Korcsmaros, and J. Saez-Rodriguez, OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat Methods, 2016. 13(12): p. 966-967.
- Rolf, M.G., et al., In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol Res Perspect, 2015. 3(5): p. e00175.
- Cornish, A.J. and F. Markowetz, SANTA: quantifying the functional content of molecular networks. PLoS Comput Biol, 2014. 10(9): p. e1003808.
- Xing, S., et al., Development and Validation of Tumor-educated Blood Platelets Integrin Alpha 2b (ITGA2B) RNA for Diagnosis and Prognosis of Non-small-cell Lung Cancer through RNA-seq. Int J Biol Sci, 2019. 15(9): p. 1977-1992.
- Goswami, C., et al., Molecular signature comprising 11 platelet-genes enables accurate blood-based diagnosis of NSCLC. BMC Genomics, 2020. 21(1): p. 744.
- Best, M.G., et al., RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell, 2015. 28(5): p. 666-676.
- Ge, X., et al., Identification of seven tumor-educated platelets RNAs for cancer diagnosis. J Clin Lab Anal, 2021. 35(6): p. e23791.
- Zhang, B., et al., Prognostic value of IGFBP2 in various cancers: a systematic review and meta-analysis. Cancer Med, 2022. 11(16): p. 3035-3047.
- Moore, T. and G.S. Dveksler, Pregnancy-specific glycoproteins: complex gene families regulating maternal-fetal interactions. Int J Dev Biol, 2014. 58(2-4): p. 273-80.
- Ingman, W.V. and S.A. Robertson, The essential roles of TGFB1 in reproduction. Cytokine Growth Factor Rev, 2009. 20(3): p. 233-9.
- Zhao, M.R., et al., Dual effect of transforming growth factor beta1 on cell adhesion and invasion in human placenta trophoblast cells. Reproduction, 2006. 132(2): p. 333-41.
- Sorensen, S., J. Andersen, and T. Norgaard, Pregnancy-specific beta 1-glycoprotein (SP1) in serum and tissue from patients with benign and malignant breast tumours. Br J Cancer, 1984. 49(5): p. 663-7.
- Skinner, J.M. and R. Whitehead, Pregnancy-specific beta glycoprotein (SP1) in tumours of the human gastrointestinal tract. Br J Cancer, 1981. 44(3): p. 476-8.
- Zhao, J., R. Rabadan, and A.J. Levine, Pregnancy specific glycoproteins: a possible mediator of immune tolerance of cancers. Journal of Cellular Immunology, 2021. 3(2): p. 109-117.
- Pang, J.H., et al., Activation of tumour cell ECM degradation by thrombin-activated platelet membranes: potentially a P-selectin and GPIIb/IIIa-dependent process. Clin Exp Metastasis, 2015. 32(5): p. 495-505.
- Felding-Habermann, B., et al., Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J Biol Chem, 1996. 271(10): p. 5892-900.
- Burdick, M.M. and K. Konstantopoulos, Platelet-induced enhancement of LS174T colon carcinoma and THP-1 monocytoid cell adhesion to vascular endothelium under flow. Am J Physiol Cell Physiol, 2004. 287(2): p. C539-47.
- Koch, A.E., et al., Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science, 1992. 258(5089): p. 1798-801.
- Strieter, R.M., et al., The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem, 1995. 270(45): p. 27348-57.
- Cambier, S., M. Gouwy, and P. Proost, The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol, 2023. 20(3): p. 217-251.
- Zhu, Y.M., et al., Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br J Cancer, 2004. 91(11): p. 1970-6.
- Zhang, W., et al., Identification of cuproptosis and immune-related gene prognostic signature in lung adenocarcinoma. Front Immunol, 2023. 14: p. 1179742.
- Tyagi, T., et al., Platelet-derived TLT-1 promotes tumor progression by suppressing CD8+ T cells. J Exp Med, 2023. 220(1).
- Pandey, P., et al., New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed Pharmacother, 2023. 161: p. 114491.
- Hers, I., Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood, 2007. 110(13): p. 4243-52.
- Mantini, G., et al., Omics Analysis of Educated Platelets in Cancer and Benign Disease of the Pancreas. Cancers (Basel), 2020. 13(1).
- Eslami, S.Z., et al., In vitro cross-talk between metastasis-competent circulating tumor cells and platelets in colon cancer: a malicious association during the harsh journey in the blood. Front Cell Dev Biol, 2023. 11: p. 1209846.
- Li, T., et al., IGFBP2: integrative hub of developmental and oncogenic signaling network. Oncogene, 2020. 39(11): p. 2243-2257.
- Haschemi, R., et al., Insulin-like Growth Factor Binding Protein-2 (IGFBP2) Is a Key Molecule in the MACC1-Mediated Platelet Communication and Metastasis of Colorectal Cancer Cells. Int J Mol Sci, 2021. 22(22).
- Wei, L.F., et al., IGFBP2 in cancer: Pathological role and clinical significance (Review). Oncol Rep, 2021. 45(2): p. 427-438.
- Guo, C., et al., Insulin-like growth factor binding protein-2 level is increased in blood of lung cancer patients and associated with poor survival. PLoS One, 2013. 8(9): p. e74973.
- Wang, J., et al., Gene expression and prognosis of insulin-like growth factor-binding protein family members in non-small cell lung cancer. Oncol Rep, 2019. 42(5): p. 1981-1995.
- Lu, H., et al., IGFBP2/ITGA5 promotes gefitinib resistance via activating STAT3/CXCL1 axis in non-small cell lung cancer. Cell Death Dis, 2024. 15(6): p. 447.
- Saci, A., et al., Differential effect of the inhibition of Grb2-SH3 interactions in platelet activation induced by thrombin and by Fc receptor engagement. Biochem J, 2002. 363(Pt 3): p. 717-25.
- Vogtle, T., et al., Critical redundant functions of the adapters Grb2 and Gads in platelet (hem)ITAM signaling in mice. Platelets, 2020. 31(6): p. 801-811.
- Visconte, C., et al., Amyloid precursor protein is required for in vitro platelet adhesion to amyloid peptides and potentiation of thrombus formation. Cell Signal, 2018. 52: p. 95-102.
- Patel, P., et al., Platelet FcgammaRIIA in immunity and thrombosis: Adaptive immunothrombosis. J Thromb Haemost, 2021. 19(5): p. 1149-1160.
- Camerer, E., et al., Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood, 2004. 104(2): p. 397-401.
- Palumbo, J.S., et al., Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood, 2005. 105(1): p. 178-85.
- Trikha, M., et al., Multiple roles for platelet GPIIb/IIIa and alphavbeta3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res, 2002. 62(10): p. 2824-33.
- Jain, S., et al., Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc Natl Acad Sci U S A, 2007. 104(21): p. 9024-8.
- Osmanoglu, O., et al., Signaling network analysis reveals fostamatinib as a potential drug to control platelet hyperactivation during SARS-CoV-2 infection. Front Immunol, 2023. 14: p. 1285345.
- Spalton, J.C., et al., The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost, 2009. 7(7): p. 1192-9.
- Harbi, M.H., et al., Antithrombotic Effects of Fostamatinib in Combination with Conventional Antiplatelet Drugs. Int J Mol Sci, 2022. 23(13).
- Apostolidis, S.A., et al., Signaling Through FcgammaRIIA and the C5a-C5aR Pathway Mediate Platelet Hyperactivation in COVID-19. Front Immunol, 2022. 13: p. 834988.
- Saha, B., et al., Human tumor microenvironment chip evaluates the consequences of platelet extravasation and combinatorial antitumor-antiplatelet therapy in ovarian cancer. Sci Adv, 2021. 7(30).
- Iba, T. and J.H. Levy, The roles of platelets in COVID-19-associated coagulopathy and vaccine-induced immune thrombotic thrombocytopenia. Trends Cardiovasc Med, 2022. 32(1): p. 1-9.
- Bye, A.P., et al., Aberrant glycosylation of anti-SARS-CoV-2 spike IgG is a prothrombotic stimulus for platelets. Blood, 2021. 138(16): p. 1481-1489.
- Gonzalez-Lopez, T.J., et al., Fostamatinib effectiveness and safety for immune thrombocytopenia in clinical practice. Blood, 2024. 144(6): p. 646-656.
- Hu, M., et al., Drug repurposing of fostamatinib against cancer via potential cytotoxicity and immune checkpoint regulation. Front Immunol, 2025. 16: p. 1602189.
- Choi, Y., et al., Repurposing of the Syk inhibitor fostamatinib using a machine learning algorithm. Exp Ther Med, 2025. 29(6): p. 110.
- Park, S.R., et al., A multi-histology trial of fostamatinib in patients with advanced colorectal, non-small cell lung, head and neck, thyroid, and renal cell carcinomas, and pheochromocytomas. Cancer Chemother Pharmacol, 2013. 71(4): p. 981-90.
- Gaillard, S., et al., A phase 1 study of the SYK inhibitor fostamatinib and weekly paclitaxel for recurrent platinum-resistant ovarian cancer. 2023, American Society of Clinical Oncology.
- Schror, K., Aspirin and platelets: the antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Semin Thromb Hemost, 1997. 23(4): p. 349-56.
- Pulcinelli, F.M., et al., Inhibition of platelet aggregation by aspirin progressively decreases in long-term treated patients. J Am Coll Cardiol, 2004. 43(6): p. 979-84.
- van Zijverden, L.M., et al., The efficacy of aspirin to inhibit platelet aggregation in patients hospitalised with a severe infection: a multicentre, open-label, randomised controlled trial. Clin Exp Med, 2023. 23(7): p. 3501-3508.
- Florensa, D., et al., Low-dose acetylsalicylic acid for cancer prevention considering risk factors: a retrospective cohort study. Ann Epidemiol, 2023. 84: p. 60-66.
- Yang, J., et al., Aspirin prevents metastasis by limiting platelet TXA(2) suppression of T cell immunity. Nature, 2025. 640(8060): p. 1052-1061.
- Sevigny, J., et al., The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature, 2016. 537(7618): p. 50-6.
- Mussbacher, M., et al., Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front Immunol, 2019. 10: p. 85.
- Crimmins, E.M., Lifespan and Healthspan: Past, Present, and Promise. Gerontologist, 2015. 55(6): p. 901-11.
- Finch, C.E., Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A, 2010. 107 Suppl 1(Suppl 1): p. 1718-24.
- Wendelboe, A.M. and G.E. Raskob, Global Burden of Thrombosis: Epidemiologic Aspects. Circ Res, 2016. 118(9): p. 1340-7.
- Gu, S.X. and S. Dayal, Inflammation mediated platelet hyperactivity in aging. Ann Blood, 2020. 5.
- Price, J., J.M. Lord, and P. Harrison, Inflammaging and platelet hyperreactivity: A new therapeutic target? J Thromb Haemost, 2020. 18(1): p. 3-5.
- Klavina, P.A., et al., Dysregulated haemostasis in thrombo-inflammatory disease. Clin Sci (Lond), 2022. 136(24): p. 1809-1829.
- Byun, J.S. and K. Gardner, Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol, 2013. 182(4): p. 1055-64.
- Martins Castanheira, N., et al., Uptake of platelets by cancer cells and recycling of the platelet protein CD42a. J Thromb Haemost, 2022. 20(1): p. 170-181.
- Inc., A., Anaconda Software Distribution. 2020: Anaconda Documentation.
- Wilke, C., _cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’_. 2024.
- Wu, L., et al., CytoCtrlAnalyser: a Cytoscape app for biomolecular network controllability analysis. Bioinformatics, 2018. 34(8): p. 1428-1430.
- Shannon, P., et al., Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 2003. 13(11): p. 2498-504.
- Ceccarelli, F., et al., Bringing data from curated pathway resources to Cytoscape with OmniPath. Bioinformatics, 2020. 36(8): p. 2632-2633.
- Oleś, A., DEFormats: Differential gene expression data formats converter. 2024.
- Love, M.I., W. Huber, and S. Anders, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014. 15(12): p. 550.
- Chen Y, C.L., Lun ATL, Baldoni P, Smyth GK edgeR 4.0: powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets. bioRxiv, 2024.
- Blighe, K., S. Rana, and M. Lewis, EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. 2023.
- Rainer, J., EnsDb.Hsapiens.v86: Ensembl based annotation package. 2017.
- Chen, S., et al., fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018. 34(17): p. i884-i890.
- Andrews, S. FastQC: a quality control tool for high throughput sequence data. 2010 [cited 2024]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Wickham, H., ggplot2: Elegant Graphics for Data Analysis. 2016: Springer-Verlag New York.
- Kassambara, A., ggpubr: ‘ggplot2’ Based Publication Ready Plots. 2023.
- Pedersen, T.L., ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. 2024.
- Ahlmann-Eltze, C. and W. Huber, glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics, 2021. 36(24): p. 5701-5702.
- Kolberg, L., et al., gprofiler2 -- an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res, 2020. 9.
- Iannone, R., et al., gt: Easily Create Presentation-Ready Display Tables. 2024.
- Csárdi, G., et al., igraph: Network Analysis and Visualization in R. 2024.
- Bray, N.L., et al., Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol, 2016. 34(5): p. 525-7.
- Ritchie, M.E., et al., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res, 2015. 43(7): p. e47.
- Mills, B., MetBrewer: Color Palettes Inspired by Works at the Metropolitan Museum of Art. 2022.
- Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. 32(19): p. 3047-8.
- Valdeolivas, A., D. Turei, and A. Gabor, OmnipathR: client for the OmniPath web service. 2019.
- Carlson, M., org.Hs.eg.db: Genome wide annotation for Human. 2023.
- Kolde, R., pheatmap: Pretty Heatmaps. 2019.
- Sievert, C., Interactive Web-Based Data Visualization with R, plotly, and shiny. 2020: Chapman and Hall/CRC Florida.
- Fischer, B.S., M.; Pau, G., rhdf5: R Interface to HDF5. R package version 2.48.0. 2024.
- Posit team, T., RStudio: Integrated Development Environment for R. 2024, Posit Software, PBC: Boston, MA.
- Risso, D., et al., Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol, 2014. 32(9): p. 896-902.
- Cornish, A.J. and F. Markowetz, SANTA: Quantifying the Functional Content of Molecular Networks. PLOS Computational Biology, 2014. 10(9): p. e1003808.
- Wickham, H.A., M,; Bryan, J.; Chang, W.; McGowan, LD.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; Kuhn, M; Pedersen, TL; Miller, E-; Bache, SM.; Müller, K.; Ooms, J.; Robinson, D.; Seidel, DP.; Spinu, V.; Takahashi, K.; Vaughan, D.; Wilke, C.; Woo, K.; Yutani, H., Welcome to the tidyverse. Journal of Open Source Software, 2019. 4(43).
- Soneson, C., M.I. Love, and M.D. Robinson, Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res, 2015. 4: p. 1521.
- Lauss, M., swamp: Visualization, Analysis and Adjustment of High-Dimensional Data in Respect to Sample Annotations. 2019.
- Wishart, D.S., et al., DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res, 2018. 46(D1): p. D1074-D1082.
- Best, M.G., et al., Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell, 2017. 32(2): p. 238-252 e9.
- Vinayagam, A., et al., Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc Natl Acad Sci U S A, 2016. 113(18): p. 4976-81.
- Jia, T., et al., Emergence of bimodality in controlling complex networks. Nat Commun, 2013. 4: p. 2002.
- Garrido-Mesa, N., A. Zarzuelo, and J. Galvez, Minocycline: far beyond an antibiotic. Br J Pharmacol, 2013. 169(2): p. 337-52.
- Sapadin, A.N. and R. Fleischmajer, Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol, 2006. 54(2): p. 258-65.
- Wei, X., et al., Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience, 2005. 131(2): p. 513-21.
- Chen, M., et al., Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med, 2000. 6(7): p. 797-801.
- Wang, X., et al., Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A, 2003. 100(18): p. 10483-7.
- Festoff, B.W., et al., Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem, 2006. 97(5): p. 1314-26.
- Berens, S.C., C.M. Bird, and N.A. Harrison, Minocycline differentially modulates human spatial memory systems. Neuropsychopharmacology, 2020. 45(13): p. 2162-2169.
- Murphy L, Inchauspé J, Valenzano G, Holland P, Sousos N, Belnoue-Davis HL, Li R, Jooss NJ, Benlabiod C, Murphy E, Etzioni Z, Shepherd E, Denly L, Biswas S, Chen L, O’Sullivan J, Rimmer MP, Khan AO, Karali CS, Nasreddin N, Hitchcock IS, Koupenova M, Kriaucionis S, Hughes JR, O’Neill E, Vatish M, Rees P, Leedham S, Desborough M, Mead AJ, Schuster-Böckler B, Gregory CD, Psaila B. Platelets sequester extracellular DNA, capturing tumor-derived and free fetal DNA. Science. 2025 Aug 14; 389(6761):eadp3971.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



