
Submitted:
17 September 2025
Posted:
18 September 2025
You are already at the latest version
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by loss of dopaminergic neurons and the pathological accumulation of misfolded alpha-synuclein (α-syn) within Lewy bodies and Lewy neurites, Retrospective neuropathologic and biochemical studies have shown that α-syn is not a monolithic structure, but can be assembled as a heterogeneous, structurally distinct fibrillar conformations referred to “strains” with unique biochemical properties, seeding activities, cellular tropisms, and neurotoxic potentials, and strain heterogeneity is gaining increased recognition as contributing to the heterogeneity of PD. Evidence from postmortem brain studies, amplification studies (i.e., PMCA, RT-QuIC) on diseased brain tissue, and biofluid studies also support the notion that α-syn strains lead to distinct clinical presentations, which includes PD, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). Retrospective findings suggest that strain switching may be a phenomenon occurring during the progression of the disease and associated with cognitive decline. Understanding strain diversity not only adds to our understanding of α-syn misfolding and PD pathogenesis, but it also provides an opportunity to develop strain-specific biomarkers and therapeutic targets. The purpose of the current review is to summarize the retrospective evidence for α-syn strain heterogeneity and discuss its potential important implications for diagnosis, prognosis, and precision medicine in PD.
Keywords:
alpha-synuclein
; strains
; Parkinson’s disease
; pathogenesis
; synucleinopathies
; retrospective study
1. Introduction
Alpha-synuclein (α-syn) is a neuronal protein containing 140 amino acids and is encoded by the phosphoprotein gene (SNCA) that is primarily found at presynaptic terminals. SNCAis proposed to regulate synaptic vesicle trafficking, to promote release of neurotransmitters, and to facilitate the formation of SNARE complexes [1,2]. The protein is thought to exist in soluble monomers and memrane-bound multimers in a physiological state to maintain a healthy balance in the synapse [1]. Misfolding of SNCAinto oligomers and fibrils of β-sheet–rich structures is recognized as a pathological event in synucleinopathy, including Parkinson's disease (PD) [3].
PD is the second most common neurodegenerative disorder, where progressive degeneration of dopaminergic neurons occurs in the substantia nigra pars compacta, resulting in classical motor symptoms including bradykinesia, rigidity, and resting tremor, as well as non-motor features which may include cognitive impairment, mood changes, and autonomic disturbance [4,5,6,7,8]. While the etiology of the disease remains multifactorial, there is substantial evidence that aggregation of SNCAis central to the onset of PD and is associated with progression through mitochondrial dysfunction, lysosomal dysfunction, calcium dysregulation, and neuroinflammation [9,10,11,12].
According to the "strain hypothesis" concept, SNCAcan misfold into distinct structural conformations or "strains". Each strain has its own biochemical, biophysical, and pathogenic properties [13].
2. Background
2.1. SNCA Biology and Physiological Role
SNCA is a 140-amino acid protein that is very highly conserved and was first isolated in Torpedo electric organ presynaptic terminals and the nucleus [14]. SNCA is encoded by the SNCA gene found on chromosome 4q22.1 and is primarily expressed in the central nervous system and in presynaptic terminals of neurons [15]. Under physiological conditions, SNCA is an intrinsically disordered protein, which allows for significant conformational flexibility and interactions with a variety of binding partners in various cellular compartments [16]. While this structural plasticity is necessary for its normal functions associated with synaptic vesicle trafficking and neurotransmitter release, it also makes SNCA very likely to misfold and aggregate into distinct pathogenic strains associated with Parkinson's disease and related synucleinopathies.
In its native state of existence, SNCA is important in synaptic vesicle trafficking, regulation of dopamine release, and assembly of the SNARE complex to fine-tune neurotransmission [17]. SNCA also interacts with lipid membranes, and upon binding to membrane structure, can adopt α-helical structure that mediates vesicle docking and recycling [18,19]. SNCA is not only expressed in the CNS, but in peripheral tissue, such as red blood cells, enteric neurons, and the olfactory epithelium, suggesting it has non-localized systemic physiological functions [20,21,22].
2.2. Misfolding and Aggregation in Disease
Pathological SNCA changes its conformation from a mostly unstructured or α-helical monomer to β-sheet-rich oligomers and fibrils [23,24]. This process of misfolding is a primary event in the pathogenesis of synucleinopathies such as PD, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [25]. Genetic mutations in the SNCA gene including A53T, A30P, E46K, H50Q, G51D, or A53E also promote SNCA aggregation and accelerate disease onset when inherited from a parent or the combination of gene duplication or triplication events [26,27,28,29,30].
Exposure to environmental toxins, including the pesticides rotenone and paraquat, oxidative stress, mitochondrial dysfunction, or deficiencies in the proteasomal or autophagic clearance pathways can also induce aggregation [31,32]. Toxicity caused by misfolded SNCA aggregates involves multiple mechanisms, of which comprise the disruption of cellular membranes, mitochondrial depolarization, dysregulation of calcium within the cell, synaptic dysfunction, and the activation of neuroinflammation via microglial-mediated neuroinflammation [33,34,35].
2.3. Concept of Protein Strains
The concept of a protein strain came from prion research [36], when it became apparent that variants exist of the same protein, although they are arranged or structured differently, self-propagated, and had subtle, but important, distinctions with respect to the resulting disease (i.e. infectivity, transmissibility, and tissue preference for a phenotype) [37]. The original idea that SNCA may be prion-like was based upon the observation that misfolded SNCA aggregates can act as templates for misfolding native SNCA [38,39] effectively seeding the conversion of native SNCA into the same 'Seeded' misfolded form, therefore propagating the pathology.
These strains differ in many ways including fibril morphology, stability, seeding ability, and how they prefer to interact with cell types in the brain. It has been shown that when injected in an animal model, various SNCA fibril conformers can cause different phenotypes from a neuropathological perspective, including some strains that prefer dopaminergic neurons and some strains that prefer oligodendrocytes [40,41].
2.4. SNCA Strains in Synucleinopathies
PD, DLB, and MSA all exhibit SNCA pathology; however there are major differences in their clinical characteristics, disease course, and pathology distribution. Notably, the SNCA fibrils from individuals with MSA have a unique and compact fold, when viewed through molecular cryo-electron microscopy (cryo-EM) compared to fibrils isolated from individuals with PD [42,43]. Further biochemistry techniques, such as real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA) show that SNCA seeds from MSA induced significantly more amplification of pathology in oligodendroglial cultures when derived from diseased individuals compared to SNCA seeds derived from individuals with PD [44,45].
Pathological strain differences of SNCA have significant clinical implications, as these strain weightings may explain the different courses of PD and MSA. In MSA, SNCA strains preferentially target oligodendrocytes, causing widespread deposits of glial cytoplasmic inclusions and a rapid clinical course with early and severe autonomic dysfunction. Strains associated with PD primarily affect nigrostriatal neurons, resulting in a slower course, which is initially motor dominant. These mechanistic differences may help explain the more fulminant phenotype of MSA than the slower clinical course of PD [46,48].
2.5. Mechanisms of Strain Diversity
Strain development in SNCA is influenced both endogenously and exogenously. For example, PTMs that impact C-terminal truncation, phosphorylation of Ser129, nitration, and ubiquitination can also influence fibril structure and toxicity [49,51] as well as physiological parameters of fibrillogenesis such as pH, ionic strength or lipid composition [52].
There is an emerging body of evidence indicating that SNCA does not aggregate in vacuum, and is affected by the range of molecular co-factors that provide a dynamic chemical environment for the conformational energetic of SNCA [53]. Even for SNCA strains produced in solitude, the heterogeneity in strains among synucleinopathies and possibly PD (the clinicopathological variations of PD, DLB or MSA) could also be a product of SNCA interactions with one or more host factors modulating aggregation [54].
Cross-seeding studies have shown that tau fibrils can accelerate the aggregation of SNCA by nucleating the formation of some distinct structure altogether with greater cytotoxicity, contributing to its overall complexity [55]. Some amyloid-β variants have been shown to nucleate SNCA fibril formation, that could contribute to the high co-morbidity of both PD and Alzheimer's pathology and lends some explanation regarding the more rapid cognitive decline seen in PD with dementia [56,57,58]. These protein–protein interactions are indicative that the heterogeneity of SNCA strains may be strongly modulated by the molecular context within a host brain.
Aside from proteins, lipid bilayers and membrane composition are also significant in determining the identity of α-synuclein strain. The intrinsic affinity of the protein for negatively charged phospholipids indicates lipid changes (high or low cholesterol, sphingolipid, cardiolipin), can alter fibril morphology and aggregation propensity [59]. Also, experimental studies show specific lipid environments have a role in accelerating fibrillization and they are also solidified with structural traits that dictate tropism to neurons or glia [60].
These findings align with clinical evidence that glial-rich pathology in MSA may also represent lipid-based selection of oligodendrocyte vulnerability, which in turn reflects strain selection based on post-translational modifications (PTMs) of α-synuclein (e.g., phosphorylation, truncation, nitration, and ubiquitination) that will also alter the corresponding aggregation dynamics and cofactor interactions [61]. Additionally, for instance, serine 129-phosphorylation has been shown to alter the β-sheeted regions of α-synuclein and to promote fibril formation while truncation of α-synuclein's C-terminal domain typically produces strains that propagate higher seeding efficiency and increased toxicity [62,63]. It may then be structural remodeling resulting from PTMs in conjunction with tau, Aβ, and lipid cofactors that contributes to variability in strain behavior in a disease-dependent manner.
In conclusion, the relationship between SNCA and its co-factors may provide a plausible model to illustrate how strain heterogeneity might arise at a molecular level. The relationship between SNCA and its co-factors may impact the shape of the amyloid fibril, propensity to "seed" the fibril in host cells, and tropism to specific cellular pathways, and may account for some of the differences in progression between PD (relatively slow and motor dominant) and MSA (relative rapid changes with autonomic dysfunction). If strain heterogeneity arises from co-factor dependent mechanisms as explored in PD and MSA, it may one day provide us with a roadmap for precision diagnostics of clinical phenotypes, and ultimately the development of therapies to inhibit the onset of pathogenic strains. When a strain establishes itself, it continues to maintain a "structural memory", meaning it can continue seeding a particular fold through templated seeding in recipient cells [64]. When considering strain conformations in relation to the cellular context of the host, there may be implications for the distributions of lesions in tissues, trajectories of neurodegeneration, and response to therapies. All of these interactions ultimately create the structural and pathological diversity of α-synuclein that yields different phenotypes between synucleinopathies (Figure 1).
2.5. Strain Diversity Mechanisms
Strain formation is influenced by both internal mechanisms and external mechanisms. The internal mechanisms include post-translational modifications (for example, S129-phosphorylation, truncation, nitration, ubiquitin modifications, etc.),, which can alter physical characteristics of fibrils, seeding activity, and toxicity [65,66]. The external mechanisms comprise environmental influences, such as pH, ionic strength, and lipid environment during fibrillogenesis [67,55] demonstrated that environmental factors could "lock in" certain fibril conformations, resulting in a phenomenon termed structural memory — the tendency of a strain to hold onto a specific 'fold' in successive rounds of propagation. The presence of other amyloidogenic proteins, such as tau and amyloid-β, has also been found to influence strain behavior, potentially contributing to the mixed pathologies in some cases of PD [68] In Table 1 below, we summarize key studies of SNCAstrains in Parkinson's disease in experimental models, methods, strain findings, and clinical conclusions.
3. Materials and Methods
3.1. Study Design
This study was designed as a retrospective, literature-based analysis that synthesized published research on the α-synuclein (α-syn) strains in Parkinson's disease (PD) and related synucleinopathies. Our process followed established methods for systematically gathering data from neuropathological, biochemical, and structural studies (Liberati et al., 2009), using a focus on studies that utilized strain-discriminating assays.
3.2. Literature Search Strategy
We conducted searches in the PubMed, Web of Science, and Scopus databases for studies published from January 1997 to June 2025. Our search terms were "alpha-synuclein" OR "α-synuclein" AND "strain" OR "conformer" OR "polymorph" AND "Parkinson's disease" OR "synucleinopathy" OR "multiple system atrophy" OR "Lewy body" and Boolean operators were used to narrow the search findings and filters were applied for English-language, peer-reviewed papers. The references of the articles and relevant review papers were also screened for additional studies.
3.3. Inclusion and Exclusion Criteria
Inclusion criteria:
-
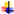
- Original research using human, animal or cell culture models
-
- Studies, demonstrating structural, biochemical, or functional differences between SNCAstrains
-
- Use of advanced detection or amplification methods (cryo-EM, RT-QuIC, PMCA, solid-state NMR)
-
- Data comparing strain characteristics with PD clinical and/or pathological data
Exclusion criteria:
-
- Studies addressing only SNCAexpression with aggregation or strain analysis,
-
- Articles that are non-peer-reviewed or conference abstracts without full text,
-
- Reports without primary data (opinion pieces, narrative reviews)
3.4. Data Extraction and Categorization
Two independent reviewers screened eligible articles and the extracted data were classified under:
-
- Origin and type of SNCA aggregate (humans and brain-derived, recombinant fibrils)
-
- Analytical method (structural, biochemical, seeding assay)
-
- Strain-based characteristics (fold morphology, seeding efficiency, cell tropism, stability)
-
- Clinical correspondence (disease type, disease progression, symptomatology)
Data is displayed in Table 2 for comparison.
3.5. Quality Assessment
The methodological quality of the included studies was evaluated using a modified version of the Newcastle–Ottawa Scale (NOS) for observational studies (Wells et al. 2011), which we adapted for in vitro and structural biology studies. We considered criteria such as reproducibility, sample size, blinding, and relevance to PD pathology.
3.6. Data Synthesis
We utilized a narrative synthesis approach because of the heterogeneity of experimental methods and outcomes (Popay et al., 2006). We then integrated the key findings thematically, to come back and answer the main research question: How do SNCAstrains differ structurally and functionally influencing PD pathogenesis and clinical variability?
An overview of the overall methodological workflow can be seen in Figure 1, which details the sequential framework from the research question to narrative synthesis.
Figure 2.
Flow diagram showing the methodology and decision-making processes for the retrospective literature review of multiple α-synuclein strains in Parkinson's disease. Steps included identifying the research question, systematically searching databases, screening titles/abstracts against inclusion-exclusion criteria, reviewing full texts, extracting structured data to form a Meta dataset, evaluating quality by using modified NOS, and narrative synthesis of findings.
Figure 2.
Flow diagram showing the methodology and decision-making processes for the retrospective literature review of multiple α-synuclein strains in Parkinson's disease. Steps included identifying the research question, systematically searching databases, screening titles/abstracts against inclusion-exclusion criteria, reviewing full texts, extracting structured data to form a Meta dataset, evaluating quality by using modified NOS, and narrative synthesis of findings.
4. Results
4.1. Structural Diversity of α-Synuclein Strains
There have been several high-resolution structural analyses of SNCA which have revealed the existence of strikingly different conformations that SNCA can fold to, with respect to protofilament organization, helical pitch, and β-sheet fabric. Cryo-electron microscopy structure of the SNCA fibrils in post-mortem PD brain tissue showed that the two-protofilament structure is stabilized by a net of inter-protofilament salt bridges [69]. However, Schweighauser et al. [70] referred to MSA-derived fibrils as tighter, characterized by a compact asymmetric fold and fewer solvent-exposed residues, which may explain their increased resistance to proteases.
Bousset et al. (2013) established that these morphologies are not accidental but are conserved in a thoroughly planned manner by being seeded on a template, even when this seeding is followed by several in vitro amplification steps. Further evidence for HCs in solid-state NMR investigations was demonstrated by Tuttle et al. (2016), who indicated that this method detected fear distinguishing the hydrogen bonding population and hydrophobic core aggregation in fibrils of strains differing in pathogenicity and altered significantly by subtle conformational variations. These and other findings underlie the hypothesis that SNCAfibrils, similar to prions, self-existing in vivo also exist as transmissible and structurally stable strains.
4.2. Biochemical and Seeding Properties
Biochemical assays also showed that the SNCAstrains exhibited differences in protease digestion profiles, stability to thermal denaturation, and aggregation kinetics. MSA-derived fibrils were consistently more resistant to proteinase K digestion than PD-derived fibrils. This finding is indicative of a more stable fibril core of the MSA-derived strain (Peng et al., 2018). Thermodynamic stability tests found that fibrils generated from MSA strains retained their fibrillar morphology after heat treatment or exposure to chaotropic agents, while PD strains exhibited partial dissociation after similar treatments (Shahnawaz et al., 2020).
In seeding assays, both PMCA and RT-QuIC provided quantitative evidence for strain-specific propagation efficiencies, as MSA-derived seeds rapidly induced fibril formation and had shorter lag phases and higher plateau fluorescence in Thioflavin T (ThT) kinetics, suggesting a higher intrinsic nucleation rate (Fairfoul et al., 2016; Shahnawaz et al., 2020). These differences were associated with disease aggressiveness as strains diverging in their seeding kinetics were associated with shorter survival times and symptom progression (Koga et al., 2018).
Equally, proteomic analyses of seeded aggregates distinguished differences in co-factors bound to fibrils (e.g., lipids, small heat shock proteins), which may regulate aggregation kinetics and toxicity (Fanning et al., 2019). These biochemical fingerprints may reasonably offer diagnostic features that help differentiate α-synucleinopathies.
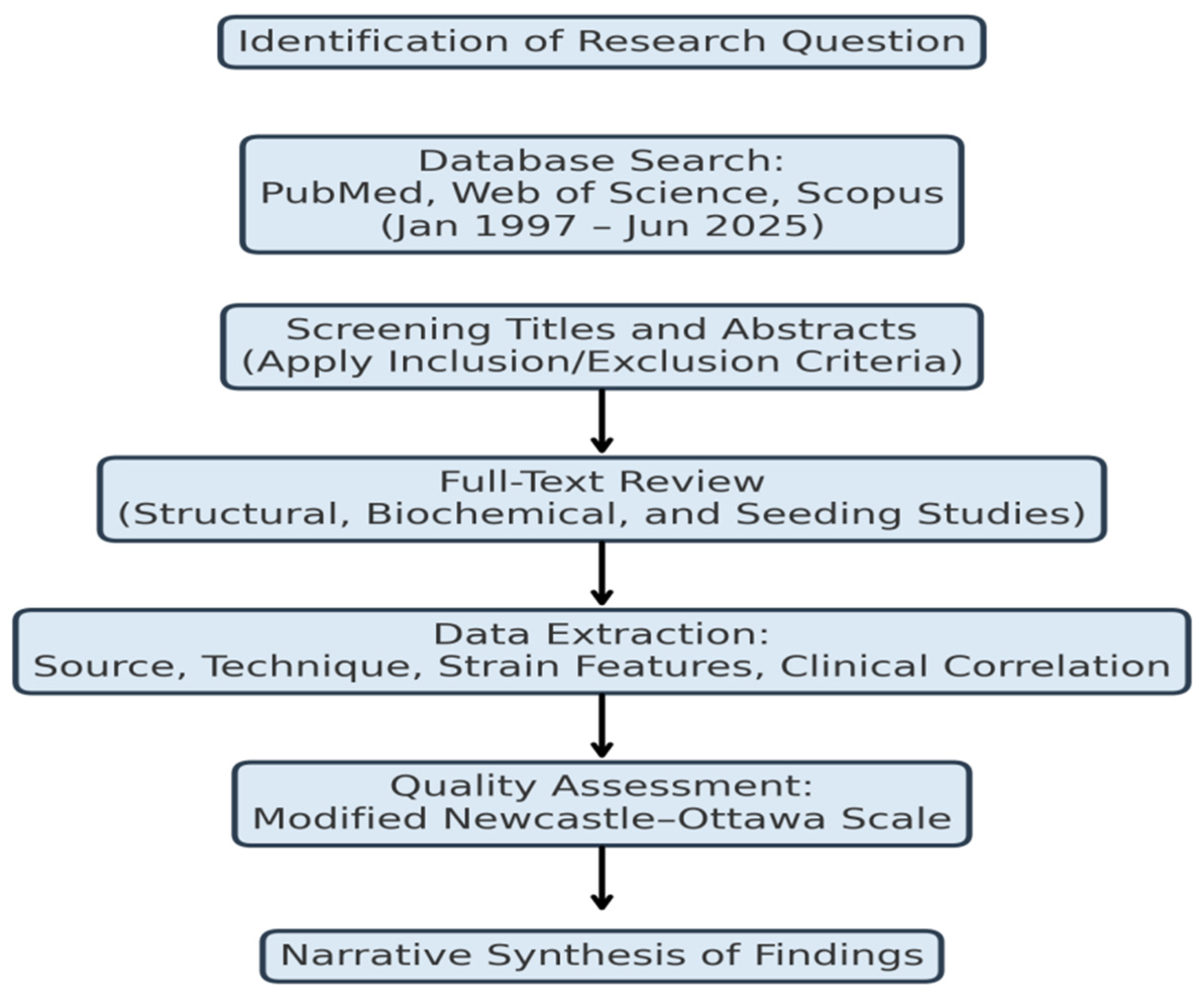
4.3. Cellular Tropism and Pathogenic Spread
Strain diversity extended beyond their biochemical characteristics to cell-type specificity and anatomically diverse spreading of pathology. For instance, in rodent inoculation models, PD-derived fibrils caused intraneuronal inclusions inside the dopaminergic neurons of the substantia nigra pars compacta (in the midbrain) and progressive motor deficits (Luk et al., 2012; Peelaerts et al., 2015). MSA fibrils preferentially targeted oligodendrocytes, causing glial cytoplasmic inclusions (GCIs) and demyelination of white matter tracts (Prusiner et al., 2015).
In vitro studies of human iPSC-derived neurons and glia have demonstrated the differences in tropism may stem from strain specific interactions between strain-specific fibrils and cellular membranes, and uptake pathways (Gribaudo et al., 2019). For example, when PD strains internalized inside the neuron mopde via clathrin-mediated endocytosis, MSA strains appeared to favor macropinocytosis for internalization inside oligodendrocytes.
Further, in vivo tracing experiments have conclusively shown that PD strains spread in nigrostriatal pathways, whereas MSA strains spread by way of cerebellar or corticospinal tracts, which no doubt reflects differences between pathologies in degree of synaptic connectivity and compatibility of axonal transport (Rey et al., 2013; Woerman et al., 2019). These strain-specific routes of propagation may further help explain the clinical syndromes that have converged upon exhibit distinct and non-converging clinical features despite sharing a pathogenic form of α-syn.
Figure 2.
Cell-type specificity and neuroanatomical tropism of α-synuclein strains. Parkinson’s disease (PD) strains selectively target neurons in the nigrostriatal pathway, enter primarily via clathrin-mediated endocytosis, propagate along nigrostriatal and limbic circuits, and form Lewy bodies and neurites. In contrast, multiple system atrophy (MSA) strains preferentially target oligodendrocytes, enter through macropinocytosis, spread via cerebellar and corticospinal tracts, and form glial cytoplasmic inclusions (GCIs). These differences in strain tropism underlie the divergent clinical and pathological features of PD and MSA.
Figure 2.
Cell-type specificity and neuroanatomical tropism of α-synuclein strains. Parkinson’s disease (PD) strains selectively target neurons in the nigrostriatal pathway, enter primarily via clathrin-mediated endocytosis, propagate along nigrostriatal and limbic circuits, and form Lewy bodies and neurites. In contrast, multiple system atrophy (MSA) strains preferentially target oligodendrocytes, enter through macropinocytosis, spread via cerebellar and corticospinal tracts, and form glial cytoplasmic inclusions (GCIs). These differences in strain tropism underlie the divergent clinical and pathological features of PD and MSA.
4.4. Clinical Correlations
Retrospective evaluation of patient data demonstrated that strain type could be associated with clinical phenotypes, clinical course and biomarker profiles. Studies have found PD-associated strains showed a decreased rate of disease progression, asymmetric motor onset, and longer time prior to the onset of non-motor symptoms (Kalia and Lang, 2015; Koga et al., 2018). MSA-associated strains were found to exhibit features such as early autonomic failure, cerebellar ataxia and faster functional decline leading to median survival times often <10 years post-diagnosis (Wenning et al., 2013).
Differences in strain specificity could also be detected in cerebrospinal fluid using RT-QuIC, which had >90% sensitivity and specificity distinguishing between PD and MSA by measuring seeding kinetics (Fairfoul et al., 2016; Shahnawaz et al., 2020). Importantly, these assays were able to detect strain profiles years before being diagnosed clinically in some patients suggesting a future in asymptomatic detection.
Histopathological studies also confirmed the strain–phenotype connection: PD cases generally showed SNCAinclusions directed mainly within brainstem nuclei and limbic structures, while MSA cases had diffuse oligodendroglial inclusions along with myelinated tract degeneration throughout (Ozawa et al., 2004; Koga et al., 2018).
5. Discussion
This retrospective synthesis ultimately provides compelling evidence that α-synuclein (α-syn) does not exist as a singular pathological entity. Rather, SNCAexists as a spectrum of distinct conformational strains, each with its own structural, biochemical, and pathological signatures. This idea is consistent with the formerly established concept of prion strain biology (Prusiner, 1998) and is being increasingly accepted as a means of relating the clinical variability seen among synucleinopathies, including Parkinson's disease (PD), multiple system atrophy (MSA), and dementia with Lewy bodies (DLB).
Our analyses suggest that conformational variation in SNCAstrains is not merely an incidental consequence of SNCAaggregation, but should be thought of as an inherent part of the determinants of disease phenotype, rate of disease progression, and tissue tropism. This also shifts our reasoning from SNCAaggregation being a single-pathway event, to it now being intertwined multi-strain pathogenic process that will have important ramifications for the diagnosis, treatment and modelling of disease.
5.1. Strain-Specific Pathobiology
High-resolution structural studies such as cryo-electron microscopy (cryo-EM) and solid-state nuclear magnetic resonance (ssNMR) have highlighted some significant disparities in protofilament architecture between SNCAvariants (Li et al., 2018; Schweighauser et al., 2020; Guerrero-Ferreira et al., 2019). Typically, PD-derived fibrils have a two-protofilament architecture with solvent-exposed acidic residues that may promote affinity for neuronal membranes, while MSA-derived fibrils are more compact with a tightly folded core that allows for extensive hydrophobic packing, which contributes to resistance to proteolysis (Peng et al., 2018). The structural differences lead to biochemical variation; hence, MSA fibrils are thermodynamically more stable, have much shorter lag phases for seeded aggregation, and cause more aggressive neuropathology in animal models (Peelaerts et al., 2015; Shahnawaz et al., 2020). Conversely, while PD strains are propagating less aggressively, they might deploy their propagation with greater selectivity for oxygenated neurons, resulting in a longer course of the disease. The evidence of templated seeding fidelity in vitro (Bousset et al., 2013) suggests that not only are these structural properties not random, but they are also transmissible through molecular propagation, in a similar manner to the propagation of prion strains.
5.2. Cell-Type Specificity and Neuroanatomical Tropism of α-Synuclein Strains
Among the most notable findings in the study of synucleinopathy is the strong cell-type selectivity of various a-synuclein (a-syn) strains. in PD, pathogenic SNCAstrains infect neurons, primarily those located in the nigrostriatal pathway responsible for the movement features of the disease, creating "classical" pathogenic intraneuronal inclusions such as Lewy bodies and Lewy neurites (Luk et al., 2012). Conversely, in MSA, SNCAstrains will preferentially infect oligodendrocytes resulting in the destruction of the myelin sheath which leads to a rapid and aggressive disease course as widespread glial cytoplasmic inclusions (GCIs) develop and rapid myelin degeneration occurs (Prusiner et al., 2015).
This cell-type specificity is mediated by a combination of biophysical, biochemical and biological factors. The structural differences between strains result in differences in surface charge distributions and corresponding hydrophobicities in comparison to the original protein, and those differences impact their binding affinities to neuronal vs glial lipid membranes (Gribaudo et al., 2019). It is these differential interactions that will dictate the initial recognition and uptake at the cellular interface. Furthermore, the internalization appears to be strain-dependent based on the endocytic pathways; PD-associated strains show a preference for uptake via clathrin-mediated endocytosis and MSA strains prefer to internalize using macropinocytosis, which is a non-specific route of internalization but one that is more aggressive (Reyes et al., 2019).
Once internalized, the strain-dependent distinctions occur in intracellular trafficking and subsequent axonal transport. PD strains move along nigrostriatal and limbic circuits consistently, therefore retaining the motor and non-motor symptom topography. MSA strains tend to disseminate preferentially along cerebellar and corticospinal tracts explaining the associated cerebellar ataxia, autonomic dysregulation, and rapid disability associated with the disease (Rey et al., 2013; Woerman et al., 2019).
These differences in tropism are not just interesting pathological anomalies, they reflect a basic principle: an SNCAstrain's structural identity determines the cells it infects, the neuroanatomy it spreads within, and therefore the clinical phenotype of disease. Consequently, investigation of strain-specific tropism provides mechanistic understanding of the different clinical pathways of these α-synucleinopathies (PD and MSA), and highlights the necessity for utilising strain biology to develop specialised diagnostic and therapeutic approaches.
5.3. Diagnostic Potential and Clinical Translation
On the translation level, the a-synuclein strain profiling technique represents a breakthrough in accurate neurology in that this approach fosters the possibility of differentiating between idiosyncratic synucleinopathies (e.g., PD and MSA) with respect to conformational patterns. This is important as it provides the opportunity to diagnose and treat early and correctly, along with individualized treatments depending on the biology of the strain. Amplification assays, like real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA) leverage the prion-like seeding potential of SNCAstrains to differentiate PD, MSA, and other a-synucleinopathies with sensitivities and specificities greater than 90% (Fairfoul et al., 2016; Shahnawaz et al., 2020). Uniquely, these assays can detect pathological SNCAlong before symptoms are present in navigating motor function, and potentially enable a preclinical diagnosis with early treatment. This is a landmark possibility as it changes how we can develop clinical trials by enabling the potential to screen at risk individuals during the neuroprotective window. Additionally, the strain-specific amplification patterns could guide patient stratification protocols and ensure treatment is adequately designed in relation to the molecular subtype of disease.
5.4. Therapeutic Implications
The strain paradigm is inherently antithetical to "one-size-fits-all" therapeutic strategies for synucleinopathies. Immunotherapies, including monoclonal antibodies, may exhibit strain-directed binding affinities with exacerbated epitope masking conformations that decreased the effectiveness of the monoclonal antibody (Weihofen et al., 2019). Therefore, small-molecule aggregation inhibitors and peptide-based modulators require a panel of strain conformers based on the targeted disorder in order to maintain full-spectrum activity or present a target single-strain milieu directed at clinical circumstances.
In the case of MSA, prevalent oligodendroglial associations exist, and therapies may be subject to myelin incapacitation, promoting oligodendrocyte proteostasis, or displacing strain by filtering glial uptake. On the contrary, PD-directed therapies may focus on neuron-driven delivery systems and the ability of maintaining integrity of the dopaminergic circuit.
5.5. Current Gaps and Future Research Priorities
The emerging field of a-synuclein strain biology presents many unanswered questions, which need targeted investigation. First, we still do not know the nature of strain diversity. The sources of strain diversity could have genetic origins (e.g., SNCA mutations), post-translational modifications (e.g., phosphorylation, truncation), environmental co-factors, or even stochastic processes affecting misfolded protein (Goedert, 2015). Second, regarding the nature of strain evolution, the longitudinal studies currently examining patients will ultimately refine our understanding of whether strain identity is flexible in the course of neurodegeneration, possibly mediating phenotypical change, as has been demonstrated in prion disease (Collinge and Clarke, 2007). Third, we need more relevant disease models. There are no existing in vivo (rodent) or in vitro (disease cell systems, human neurons & brain organoid models) experimental systems in which the behaviours of a-synuclein strains are adequately covered, especially in MSA; it will be important to develop humane and/or 3D organoid models which integrate multiple cell types (e.g., glia and immune cells). Finally, there is an urgent need for developing accessible diagnostic assays. For example, strain-typing mechanisms utilizing minimally invasive instrumentation/nasal mucosa or skin biopsy utilizing RT-QuIC strategies may be simple to deploy within a clinical setting.
6. Conclusions
This retrospective analysis shows that α-synuclein strains represent an important factor contributing to the variation in clinical presentation, pathology, and biochemistry of synucleinopathies. SNCAaggregates are not simply one pathological process, but groups of conformationally-reduced SNCAwith heritable structural and functional properties. The polymorphic strains of α-synuclein vary in proteostability, seeding kinetics, cellular tropism, and neuroanatomical propagation to develop disease-specific phenotypic disease states such as dopaminergic neuron loss in Parkinson's disease (PD) and oligodendroglial inclusions in multiple system atrophy (MSA).
The translational implications of these findings are profound. Strain specific seeding assays, such as RT-QuIC and PMCA, are ultra-sensitive diagnostics and can distinguish between PD, MSA, and the other related disorders, possibly before clinical stages. These modalities will allow us to better identify risk, more efficiently pre-screening primary candidates for possible efficacy/ effectiveness, and target treatments with variable precision. However, if we accept the strain-morphological underpinnings of diversity, this will also complicate drug development; antibody's, aggregation inhibitors, and neuroprotective modalities are not simply conformer-specific, they also have demonstrated strain conformer scarcity. Future organized and directed attempts, using high resolution structural biology and amplification type diagnostics to new rubrics of disease, will ultimately be needed to identify the origin, change, and clinical and prognostic relevance SNCAstrains.
References
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science (New York, NY). 2010;329(5999):1663-7.
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nature reviews Neuroscience. 2013;14(1):38-48.
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839-40.
- Tiwari PC, Pal R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin Neurosci. 2017;19(1):71-80.
- Pal R, Tiwari PC, Nath R, Pant KK. Role of neuroinflammation and latent transcription factors in pathogenesis of Parkinson's disease. Neurol Res. 2016;38(12):1111-22.
- Tiwari PC, Chaudhary MJ, Pal R, Kartik S, Nath R. Pharmacological, Biochemical and Immunological Studies on Protective Effect of Mangiferin in 6-Hydroxydopamine (6-OHDA)-Induced Parkinson's Disease in Rats. Ann Neurosci. 2021;28(3-4):137-49.
- Tiwari PC, Chaudhary MJ. Effects of mangiferin and its combination with nNOS inhibitor 7-nitro-indazole (7-NI) in 6-hydroxydopamine (6-OHDA) lesioned Parkinson's disease rats. 2022;36(6):944-55.
- Tiwari PC, Chaudhary MJ, Pal R. Role of Nitric Oxide Modulators in Neuroprotective Effects of Mangiferin in 6-Hydroxydopamine-induced Parkinson's Disease in Rats. 2024;31(3):186-203.
- Awasthi S, Tiwari PC, Awasthi S, Dwivedi A, Srivastava S. Mechanistic role of proteins and peptides in Management of Neurodegenerative Disorders. Neuropeptides. 2025;110:102505.
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197-211.
- Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nature reviews Neuroscience. 2017;18(2):101-13.
- Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23(2):1-13.
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522(7556):340-4.
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1988;8(8):2804-15.
- George MS, Wassermann EM, Williams WA, Callahan A, Ketter TA, Basser P, et al. Daily repetitive transcranial magnetic stimulation (rTMS) improves mood in depression. Neuroreport. 1995;6(14):1853-6.
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT, Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709-15.
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123(3):383-96.
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443-9.
- Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, et al. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nature Communications. 2014;5(1):3827.
- Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta neuropathologica. 2010;119(6):689-702.
- Doppler K. Detection of Dermal Alpha-Synuclein Deposits as a Biomarker for Parkinson’s Disease. Journal of Parkinson’s Disease. 2021;11(3):937-47.
- Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Santos-García D, Martínez-Valbuena I, Agúndez JAG. Alpha-Synuclein in Peripheral Tissues as a Possible Marker for Neurological Diseases and Other Medical Conditions. Biomolecules. 2023;13(8):1263.
- Conway KA, Harper JD, Lansbury PT, Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39(10):2552-63.
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4(11):1318-20.
- Koszła O, Sołek P. Misfolding and aggregation in neurodegenerative diseases: protein quality control machinery as potential therapeutic clearance pathways. Cell communication and signaling : CCS. 2024;22(1):421.
- Campêlo C, Silva RH. Genetic Variants in SNCA and the Risk of Sporadic Parkinson's Disease and Clinical Outcomes: A Review. 2017;2017:4318416.
- Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier A-L, Ruggeri FS, et al. The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. Journal of Biological Chemistry. 2014;289(32):21856-76.
- Bozi M, Papadimitriou D, Antonellou R, Moraitou M, Maniati M, Vassilatis D, et al. Genetic assessment of familial and early-onset Parkinson's disease in a Greek population. European journal of neurology. 2014;21(7):963-8.
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson's disease. Nature genetics. 1998;18(2):106-8.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science (New York, NY). 1997;276(5321):2045-7.
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson's disease. Environmental health perspectives. 2011;119(6):866-72.
- Huang M, Bargues-Carot A, Riaz Z, Wickham H, Zenitsky G. Impact of Environmental Risk Factors on Mitochondrial Dysfunction, Neuroinflammation, Protein Misfolding, and Oxidative Stress in the Etiopathogenesis of Parkinson's Disease. 2022;23(18).
- Jeon YM, Kwon Y, Jo M, Lee S, Kim S, Kim HJ. The Role of Glial Mitochondria in α-Synuclein Toxicity. Frontiers in cell and developmental biology. 2020;8:548283.
- Brás IC, Xylaki M, Outeiro TF. Chapter 4 - Mechanisms of alpha-synuclein toxicity: An update and outlook. In: Björklund A, Cenci MA, editors. Progress in Brain Research. 252: Elsevier; 2020. p. 91-129.
- Patel JC, Shukla M, Shukla M. Cellular and Molecular Interactions in CNS Injury: The Role of Immune Cells and Inflammatory Responses in Damage and Repair. 2025;14(12).
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science (New York, NY). 1982;216(4542):136-44.
- Vitting-Seerup K. Most protein domains exist as variants with distinct functions across cells, tissues and diseases. NAR genomics and bioinformatics. 2023;5(3):lqad084.
- Tarutani A, Suzuki G, Shimozawa A, Nonaka T, Akiyama H, Hisanaga S, et al. The Effect of Fragmented Pathogenic α-Synuclein Seeds on Prion-like Propagation. J Biol Chem. 2016;291(36):18675-88.
- Kaufman SK, Diamond MI. Prion-like propagation of protein aggregation and related therapeutic strategies. Neurotherapeutics. 2013;10(3):371-82.
- So RWL, Watts JC. α-Synuclein Conformational Strains as Drivers of Phenotypic Heterogeneity in Neurodegenerative Diseases. Journal of Molecular Biology. 2023;435(12):168011.
- Reddy K, Dieriks BV. Multiple system atrophy: α-Synuclein strains at the neuron-oligodendrocyte crossroad. 2022;17(1):77.
- Tarutani A, Kametani F.
- 43.Merz GE.
- Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson's disease and multiple system atrophy. Nature. 2020;578(7794):273-7.
- Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Annals of clinical and translational neurology. 2016;3(10):812-8.
- Fellner L, Jellinger KA, Wenning GK, Haybaeck J. Commentary: Discriminating α-synuclein strains in parkinson's disease and multiple system atrophy. Frontiers in neuroscience. 2020;14:802.
- Malfertheiner K, Stefanova N, Heras-Garvin A. The Concept of α-Synuclein Strains and How Different Conformations May Explain Distinct Neurodegenerative Disorders. Frontiers in neurology. 2021;Volume 12 - 2021.
- Reddy K, Dieriks BV. Multiple system atrophy: α-Synuclein strains at the neuron-oligodendrocyte crossroad. Molecular Neurodegeneration. 2022;17(1):77.
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739-52.
- Shimogawa M. SITE-SPECIFIC INCORPORATION OF POST-TRANSLATIONAL MODIFICATIONS IN ALPHA-SYNUCLEIN: INSIGHTS INTO AGGREGATION, STRUCTURE, AND FUNCTION: University of Pennsylvania; 2025.
- Schaffert L-N, Carter WG. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: a systematic review. Brain sciences. 2020;10(4):232.
- Miraglia F, Ricci A, Rota L, Colla E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regeneration Research. 2018;13(7):1136-44.
- Hoppe SO, Uzunoğlu G. α-Synuclein Strains: Does Amyloid Conformation Explain the Heterogeneity of Synucleinopathies? 2021;11(7).
- Peelaerts W, Baekelandt V. ⍺-Synuclein Structural Diversity and the Cellular Environment in ⍺-Synuclein Transmission Models and Humans. 2023;20(1):67-82.
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575.
- Guerrero-Ferreira R, Taylor NM. Cryo-EM structure of alpha-synuclein fibrils. 2018;7.
- Brás IC, Dominguez-Meijide A, Gerhardt E, Koss D, Lázaro DF, Santos PI, et al. Synucleinopathies: Where we are and where we need to go. Journal of neurochemistry. 2020;153(4):433-54.
- Dominguez-Meijide A, Vasili E, König A, Cima-Omori M-S, Ibáñez de Opakua A, Leonov A, et al. Effects of pharmacological modulators of α-synuclein and tau aggregation and internalization. Scientific reports. 2020;10(1):12827.
- Galvagnion C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson's Disease. Journal of Parkinson's disease. 2017;7(3):433-50.
- Sanderson JM. The association of lipids with amyloid fibrils. J Biol Chem. 2022;298(8):102108.
- Villar-Piqué A, Lopes da Fonseca T, Outeiro TF. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. Journal of neurochemistry. 2016;139 Suppl 1:240-55.
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160-4.
- Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. The American journal of pathology. 2007;170(5):1725-38.
- Zhang S, Lingor P. Strain-dependent alpha-synuclein spreading in Parkinson's disease and multiple system atrophy. 2024;19(12):2581-2.
- Anderson JC, Clarke EJ, Arkin AP, Voigt CA. Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol. 2006;355(4):619-27.
- Srinivasan E, Chandrasekhar G, Chandrasekar P, Anbarasu K, Vickram AS, Karunakaran R, et al. Alpha-Synuclein Aggregation in Parkinson's Disease. Front Med (Lausanne). 2021;8:736978.
- Pieri F, Aldini NN, Fini M, Marchetti C, Corinaldesi G. Immediate fixed implant rehabilitation of the atrophic edentulous maxilla after bilateral sinus floor augmentation: a 12-month pilot study. Clinical implant dentistry and related research. 2012;14 Suppl 1:e67-82.
- Walker L, Attems J. Prevalence of Concomitant Pathologies in Parkinson's Disease: Implications for Prognosis, Diagnosis, and Insights into Common Pathogenic Mechanisms. Journal of Parkinson's disease. 2024;14(1):35-52.
- Yang Y, Garringer HJ, Shi Y, Lövestam S, Peak-Chew S, Zhang X, et al. New SNCA mutation and structures of α-synuclein filaments from juvenile-onset synucleinopathy. Acta neuropathologica. 2023;145(5):561-72.
- Schweighauser M, Shi Y. Structures of α-synuclein filaments from multiple system atrophy. 2020;585(7825):464-9.
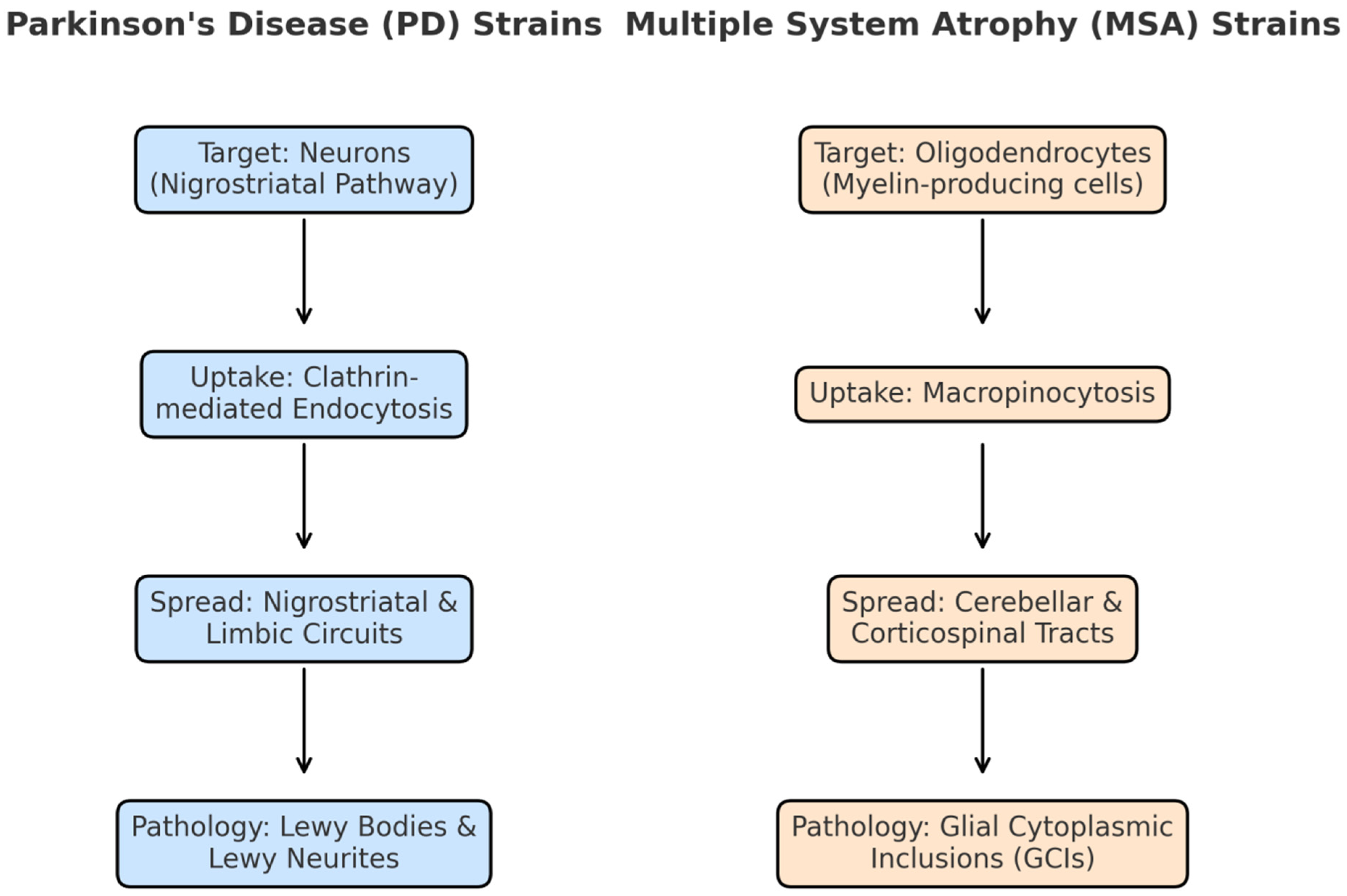
Figure 1.
SNCA strains and co-factor interactions. Tau, amyloid-β, and lipid bilayers/PTMs affect the aggregation of SNCA to generate different strain conformations, which contribute to the motor-dominance of Parkinson's disease (PD) and the rapidly progressive autonomic dysfunction occurring in multiple system atrophy (MSA).
Figure 1.
SNCA strains and co-factor interactions. Tau, amyloid-β, and lipid bilayers/PTMs affect the aggregation of SNCA to generate different strain conformations, which contribute to the motor-dominance of Parkinson's disease (PD) and the rapidly progressive autonomic dysfunction occurring in multiple system atrophy (MSA).

Table 1.
Key studies on α-synuclein strains in PD and related synucleinopathies.
| Year | Author(s) | Model/Method | Key Findings | Clinical Relevance |
| 1997 | Polymeropoulos et al. | Genetic linkage study | Identified A53T mutation in SNCA | Established genetic cause of familial PD |
| 2001 | Conway et al. | In vitro fibrillization | Demonstrated spontaneous SNCAaggregation | Basis for aggregation assays |
| 2013 | Guo et al. | Cell culture, seeding | Demonstrated prion-like propagation of α-syn | Introduced strain concept to α-syn |
| 2015 | Peelaerts et al. | Mouse models | Different SNCAstrains produce distinct pathologies | Explained heterogeneity in synucleinopathies |
| 2016 | Fairfoul et al. | RT-QuIC assay | Discriminated PD vs MSA SNCAin CSF | Potential diagnostic biomarker |
| 2018 | Li et al. | Cryo-EM | Resolved PD vs MSA fibril structures | Structural basis for strain differences |
| 2020 | Shahnawaz et al. | PMCA assay | MSA seeds amplify more efficiently than PD seeds | Supports clinical progression differences |
Table 2.
Data categorization framework for SNCAstrain studies.
| Parameter | Example Categories |
| Source of α-syn | PD brain tissue, MSA brain tissue, recombinant protein |
| Analytical technique | Cryo-EM, solid-state NMR, RT-QuIC, PMCA |
| Strain-specific features | Fibril twist pitch, thermodynamic stability, protease resistance |
| Clinical correlation | PD slow progression, PD rapid progression, MSA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



