1. Introduction
Sickle cell anemia (SCA) is a clinical form within the spectrum of sickle cell disease (SCD), determined by a point mutation in the beta-globin gene (HBB) in humans, which results in the synthesis of the hemoglobin S (HbS) variant in homozygosis [
1]. The change in the red blood cell (RBC) shape, caused by the polymerization of HbS, leads to endothelial damage and makes cells more prone to intravascular hemolysis due to the increased rigidity of sickle-shaped cells [
2,
3]. The vaso-occlusive processes, primarily in microcirculation, reduce tissue oxygenation, which can result in tissue damage and the ischemia-reperfusion process [
4,
5]. The progression of the disease is influenced by various factors, including the presence of other comorbidities, intrinsic genetic factors, and the response to drug treatment [
6].
Hydroxyurea (HU) is currently recognized as the primary effective drug for the treatment of SCD, acting by increasing plasma levels of fetal hemoglobin (HbF) through gene regulation via pre- and post-transcriptional mechanisms. HU serves as a disease modulator by destabilizing the polymerization of HbS under hypoxic conditions [
7,
8,
9]. Although HU can improve disease progression by directly increasing HbF levels, it is a cytotoxic drug with limited potency and sustainability. Its effectiveness depends on adequate hematopoietic reserves, which are finite [
9,
10]. Furthermore, its efficacy is influenced by genetic factors that exhibit heterogeneous behavior across different populations, which explains why some patient groups do not respond to the drug. This variability highlights the need for alternative treatments that further promote the elevation of HbF levels [
8].
Other treatments for SCD have been tested, but some have yelded controversial results in clinical trials. However, gene therapy offers a promising approach to treating SCD, even though it remains experimental [
10]. In vivo studies involving gene editing with CRISPR-Cas9 and lentiviral vectors have produced promising results, including the correction of the mutation that causes SCD, as well as increased HbF levels through the disruption of the
BCL11A gene [
11,
12].
HbF production ceases after birth and is replaced by HbS in patients with SCA. This switching process is primarily regulated by BCL11A, a zinc finger regulatory protein encoded by the
BCL11A gene, which represses the expression of the
HGB1 and
HBG2 genes, thereby promoting the predominance of β chain synthesis, which forms the adult hemoglobin molecule [
13,
14,
15,
16,
17]. The regulatory protein BCL11A plays a key role in modulating the course of SCD, as it directly regulates HbF synthesis. It is responsible for 20-50% of the common variation in HbF levels in both individuals with SCD and healthy adults, and has been extensively studied in both pharmacological and genetic research [
9,
11,
12,
19,
20].
The presence of SNPs in the
BCL11A gene has been widely associated with its role in HbF induction. An in silico study investigated 11,463 SNPs obtained through the Database of Single Nucleotide Polymorphisms (dbSNP) and elucidated polymorphisms in the zinc finger domain as suitable targets to disrupt the functions of BCL11A in hemoglobin switching. Therefore, understanding the behavior of SNPs on the functionality of the BCL11A repressor is of importance for clinical studies in SCA [
21,
22,
23].
The SNPs rs766432 (C>A) and rs6732518 (C>T), both located in an intron region in the
BCL11A gene, are described in the literature as HbF inductors in several populations, such as Iran, Hong Kong and Thailand. However, their relationship with hematological, biochemical and clinical markers in SCD remains poorly elucidated [
24,
25,
26,
27].
In this scenario, the aim of this study was to associate the occurrence of polymorphisms in the BCL11A gene with clinical phenotypes in SCA patients without the use of HU. Considering the need to expand treatments for SCD, our work may contribute to future studies in personalized medicine and in the search for targets for treatments based on gene therapy through the conclusive data generated for the genetic plasticity of polymorphisms in different populations.
2. Results
The basic characteristics of patients with SCA included in this study are described in
Table 1.
Genetic analyses identify polymorphisms frequencies in the studied group, in which the allele frequency for the variant A allele of the rs766432 polymorphism showed a lower frequency than the wild-type allele. However, the variant T allele of the rs6732518 polymorphism showed a higher occurrence in relation to the wild-type allele. Only rs6732518 frequency is in accord to the Hardy-Weinberg equilibrium (
Table 2).
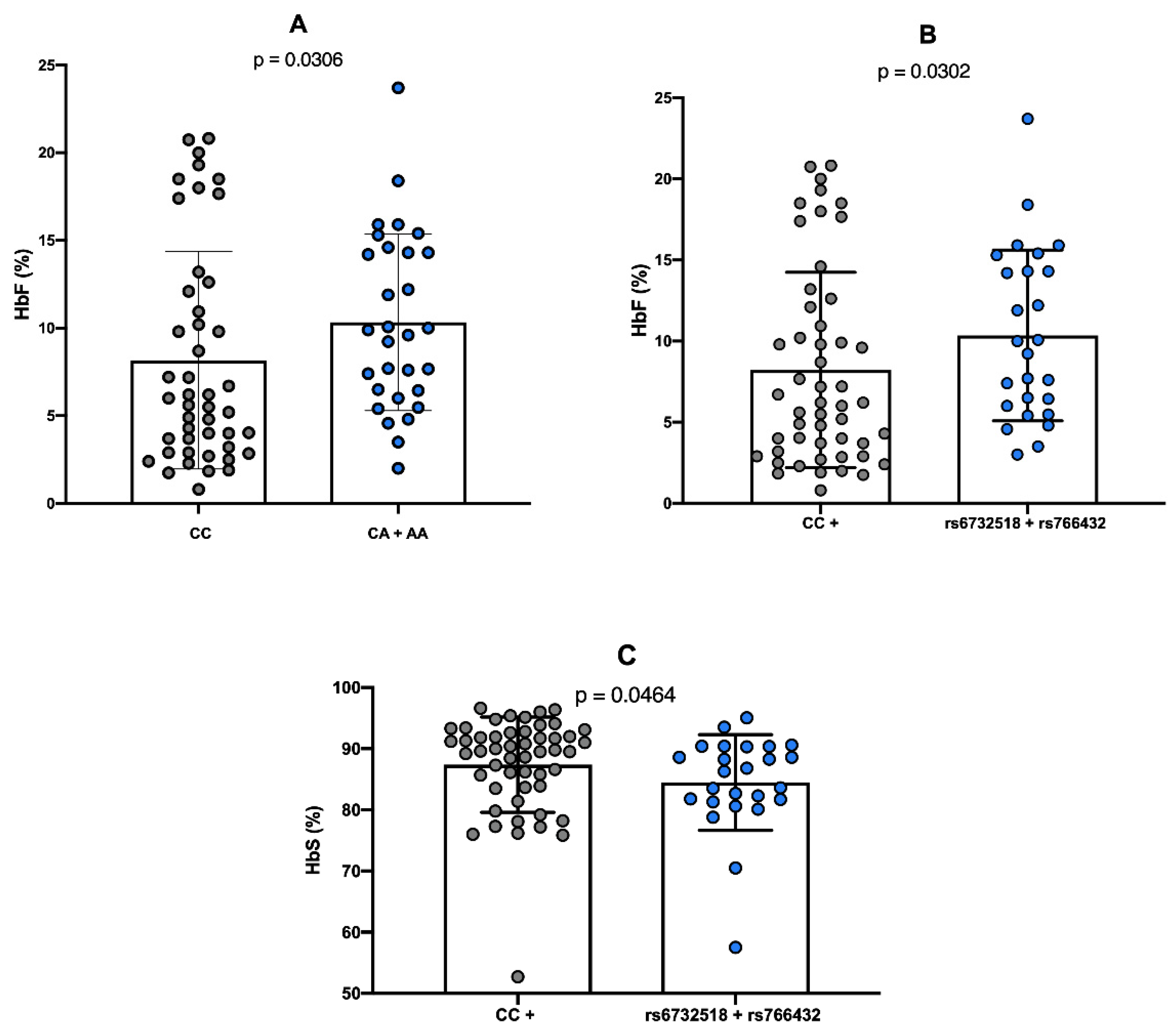
Considering the concentrations of HbF and HbS, the presence of the variant A allele of the rs766432 polymorphism was significantly associated with elevated HbF levels (p = 0.0306), compared to individuals with the wild-type CC genotype (
Figure 1A). In contrast, the rs6732518 polymorphism demonstrated a statistically significant association with HbF levels only when co-inherited with the rs766432 polymorphism (p = 0.0302), relative to individuals carrying one or neither of the polymorphisms (
Figure 1B).
Regarding HbS concentrations, a significant association (p = 0.0464) was observed with the co-inheritance of both rs6732518 and rs766432 polymorphisms, which was linked to reduced HbS concentration in comparison to individuals harboring only one or neither of the variants (
Figure 1C).
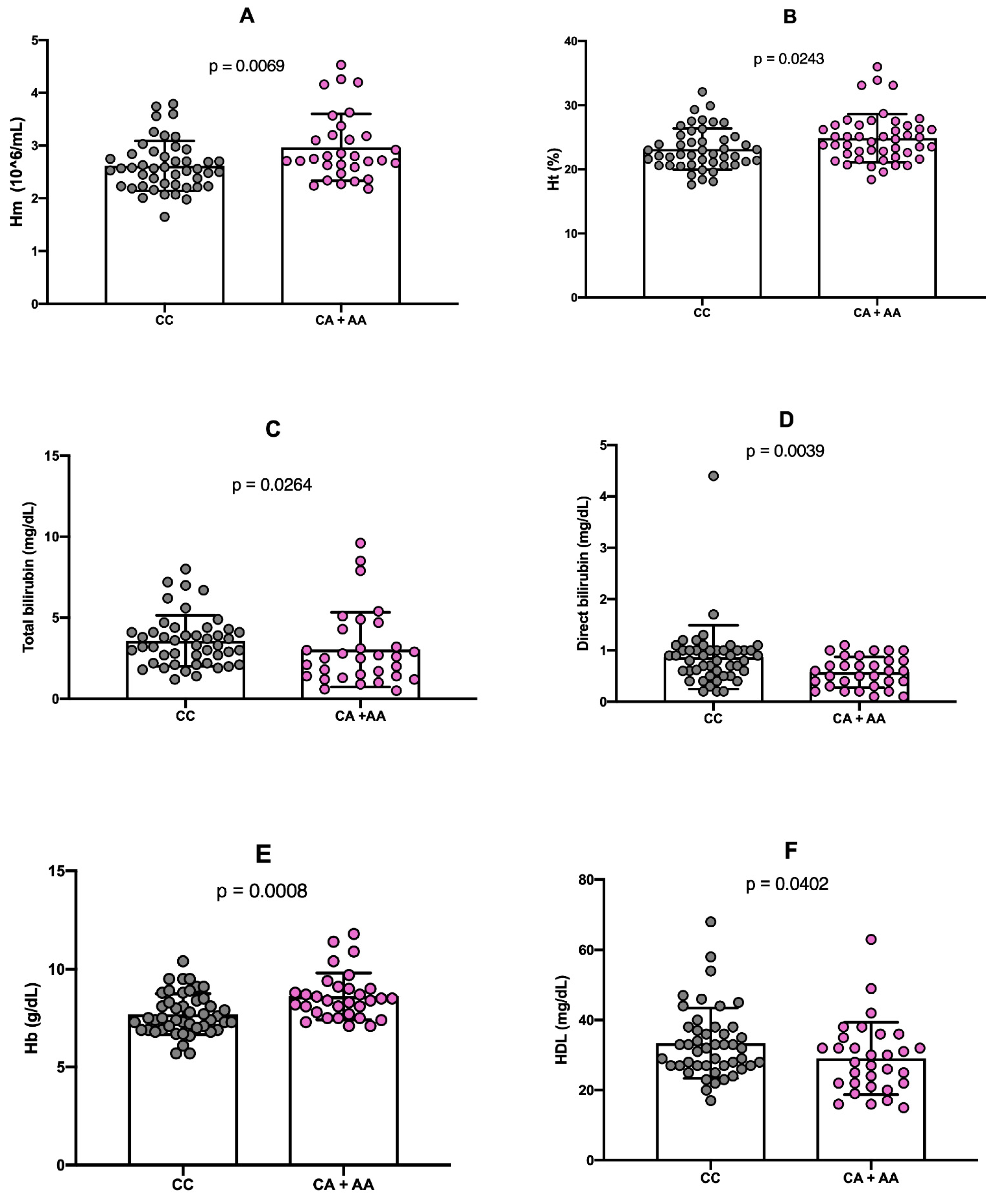
Analysis of hematological parameters in individuals carrying the minor allele (A) of the rs766432 polymorphism revealed statistically significant positive association with the red blood cell count (RBC) (p = 0.0069), hematocrit (Ht) (p = 0.0243), and hemoglobin (Hb) concentration (p = 0.0008). Conversely, significant negative associations were observed with plasma levels of total bilirubin (p = 0.0264), indirect bilirubin (p = 0.0039), and high-density lipoprotein (HDL) cholesterol (p = 0.0402) in the presence of the minor allele (A) of the rs766432 polymorphism (
Figure 2).
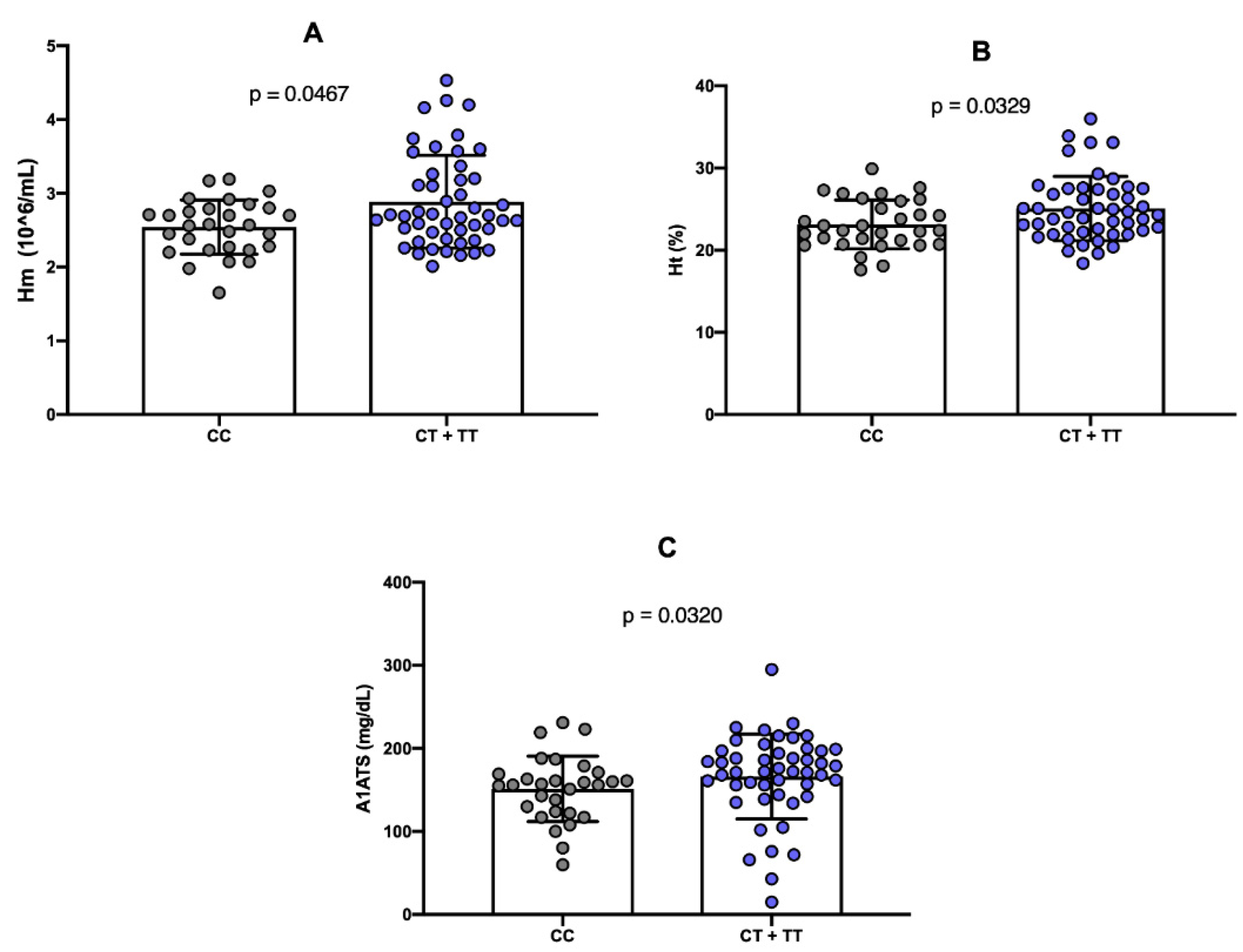
Furthermore, the presence of the minor allele (T) of the rs6732518 polymorphism was significantly associated with increased RBC count (p = 0.0467) and Ht concentration (p = 0.0329), compared to the wild-type genotype (
Figure 3A and 3B). Additionally, the T allele was significantly associated with higher levels of alpha-1 antitrypsin (A1AT) (p = 0.0320) (
Figure 3C).
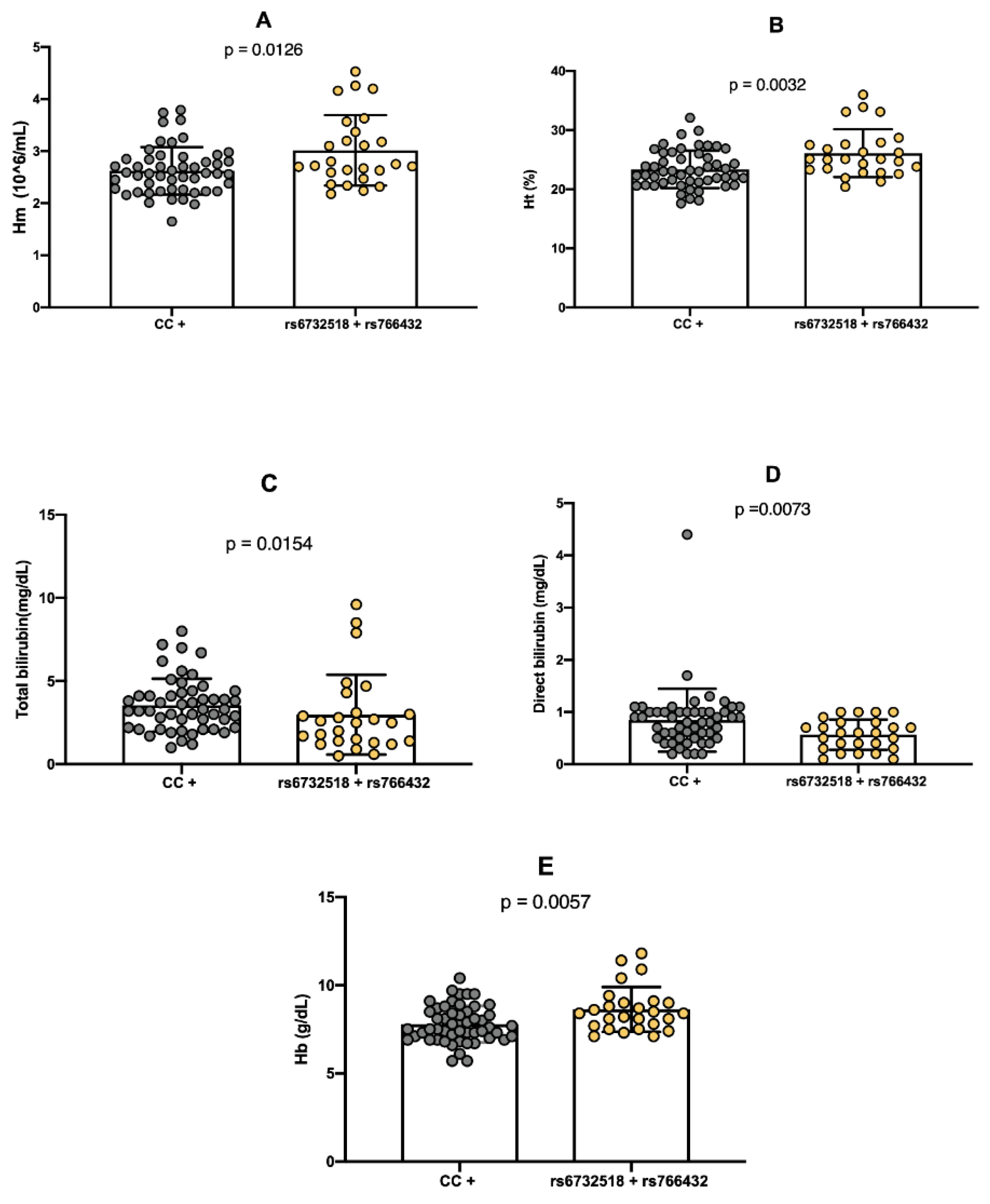
Evaluation of the co-inheritance of the variant alleles A (rs766432) and T (rs6732518) in SCD individuals it was significantly associated with increased RBC count (p = 0.0126), Ht (p = 0.0032), and Hb (p = 0.0057) concentrations (
Figure 4A,B,E). Conversely, significantly lower levels of total bilirubin (p = 0.00154) and direct bilirubin (p = 0.0073) were found in individuals with the dual inheritance of both polymorphism compared to those without this genetic profile characteristic (
Figure 4C,D).
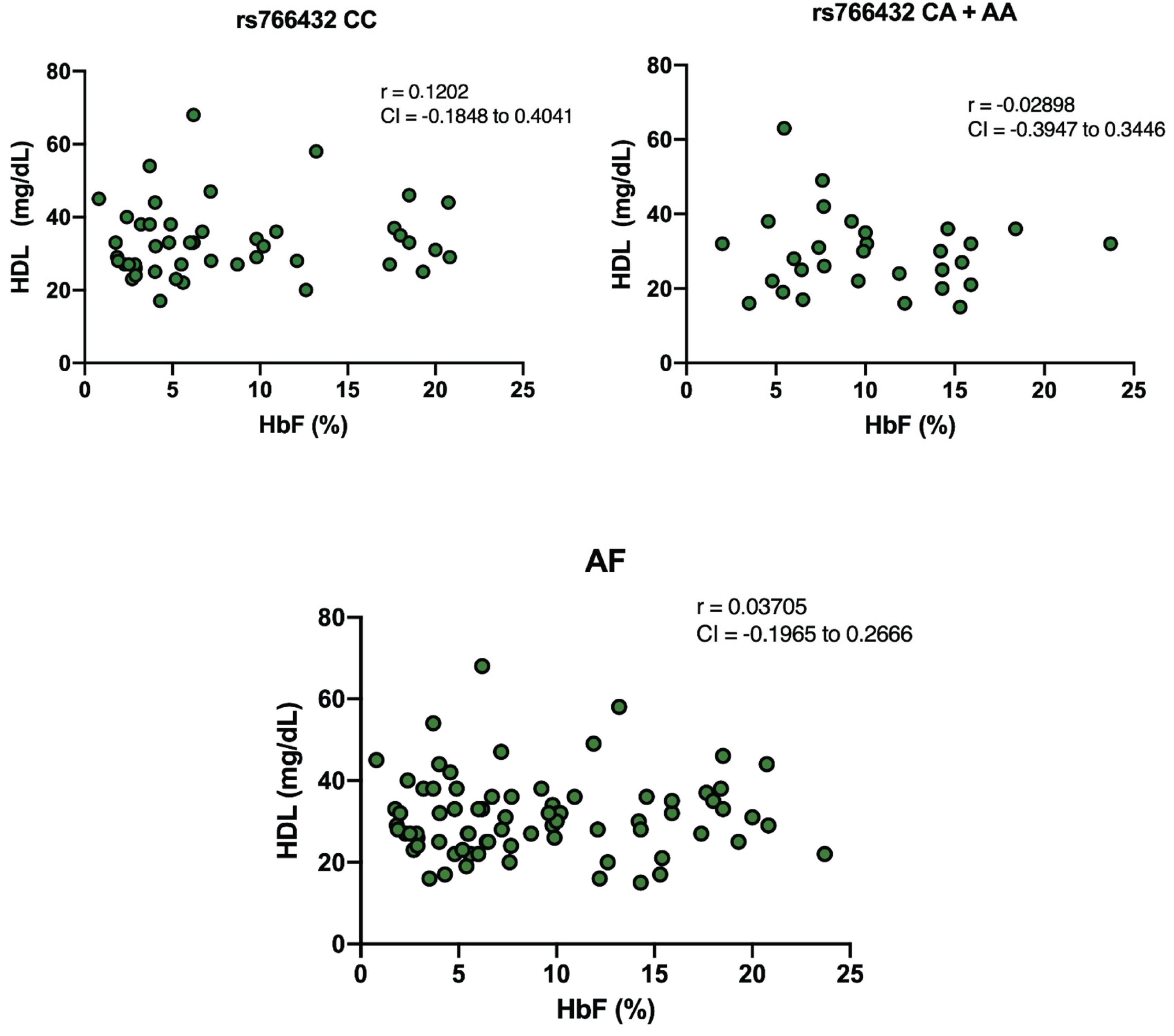
To explore potential associations between the independent variables HbF and HDL cholesterol among SCA individuals, correlation analyses were conducted. No significant correlation was identified with HbF and HDL cholesterol (r = 0.03705), and with the CC wild-type genotype (r = 0.1202), or the variant genotypes CA and AA (r = 0.03705) of the rs766432 polymorphism (
Figure 5).
3. Discussion
Fetal hemoglobin (HbF) is a key modulator of clinical severity of SCD, and its regulation through genetic markers has been increasingly elucidated in the scientific literature [
9,
28,
29]. Among these regulators, the
BCL11A has emerged as a critical gene, identified through by genome-wide association studies (GWAS) as a modulator of HbF levels. It exerts its regulator effect via transcriptional control and epigenetic mechanisms involving chromatin remodeling, positioning it as a major prognostic biomarker for HbF levels [
6]. Consequently, recent therapeutic strategies targeting HbF have focused on understanding the molecular mechanisms involving
BCL11A, including the impact of both synonymous and non-synonymous mutations within its locus. Moreover, recent studies have proposed a pleiotropic role for
BCL11A, accounting for the phenotypic variability observed in individuals with SCD. This is particularly relevant in cases where clinical and laboratory phenotypes exhibit discordant biomarker levels [
21].
The findings of the present study support the previously reported role of the rs6732518 and rs766432 polymorphisms in the positive regulation of HbF concentration across various populations [
24,
25,
26,
27]. However, a novel observation emerged: the rs6732518 polymorphism was significantly associated with elevated HbF levels only when co-inherited with the rs766432 polymorphism. This suggests a potential interaction effect and highlights the need for further in vivo studies to elucidate the independent contribution of rs6732518 to HbF regulation. The observed co-inheritance does not necessarily imply synergistic or additive effects, as the combined presence of both polymorphisms does not translate into a straightforward cumulative impact on HbF levels [
30].
The process of hemoglobin switching, which involves the transition from γ-globin to β-globin chain production, is mediated by
BCL11A through its binding to the promoters of the
HBG1 and
HBG2 genes. This interaction facilitates the repositioning of the locus control region (LCR) to the
HBB gene locus, thereby promoting β-globin expression. Given this mechanism, it is expected that polymorphisms impairing
BCL11A’s repressive function would be associated with reduced levels of sickle hemoglobin (HbS). This study confirmed such an association, but only in individuals with co-inheritance of both rs6732518 and rs766432 polymorphisms [
16].
Furthermore, the presence of rs766432 polymorphism was associated with favorable hematological phenotypes in individuals with SCD. Specifically, the CA and AA genotypes were linked to higher concentrations of hemoglobin, hematocrit, and RBC count. Hematocrit reflects the proportion of blood composed of former elements, while RBC count provides the cellular concentration per microliter of blood. Elevated values of these parameters in the presence of the rs766432 variant suggest an attenuation of the anemic profile typically observed in SCD, potentially indicating a protective effect conferred by this SCD [
31,
32].
These findings are consistent with previous studies that have reported a beneficial association between the rs766432 polymorphism and laboratory markers in SCD, reinforcing its role as a polymorphism presence and improved hematological indices does not necessarily suggest a distinct role for
BCL11A. Rather, it likely reflects the indirect effect of elevated HbF levels on clinical and laboratory parameters. Increased HbF concentrations are known to ameliorate disease severity and improve RBC function, thereby enhancing overall hematological profiles in SCD patients [
33,
34].
In this study, the rs766432 polymorphism was also associated with the altered levels of HDL cholesterol. Specifically, individuals with the CA or AA genotypes exhibited significantly lower HDL concentration. High-density lipoprotein cholesterol is well known for its inverse relationship with cardiovascular risk. Beyond its role in lipid transport, HDL exerts anti-inflammatory and anti-atherogenic effects by facilitating cholesterol efflux, suppressing hematopoietic stem and progenitor cell proliferation, modulating inflammatory responses, and inhibiting inflammasome activation in macrophages [
35]. Therefore, the observed association with favorable hematological parameters initially appears contradictory.
From a border perspective, HDL exhibits pleiotropic functions during the acute phase inflammatory response. These include neutralization of bacterial endotoxins, promoting of corticosteroid release, inhibition of platelet aggregation, protection of endothelial cell from apoptosis, downregulation of monocytes mediated inflammation, and suppression of endothelial adhesion molecule expression [
36]. Prior studies investigating HDL in the context of SCD have reported that hemolytic stress is associated with a significant decline in plasma lipid levels, including HDL cholesterol, which is known to possess both anti-inflammatory and antioxidative properties [
37,
38,
39].
Although the association between the rs766432 and HDL levels has not been extensively described in the literature, the present findings suggest a pleiotropic effect of this polymorphism in the
BCL11A gene. Notably, while
BCL11A is known to induce HbF production thereby improving hematological status and reducing the hemolytic burden, the reduced HDL levels observed in this study conflict with the expected lipid profile in non-hemolytic individuals [
40]. The absence of a significant correlation between HbF and HDL levels in this cohort further supports the notion that these two parameters are independently modulated, despite being linked to the same genetic variant.
Additionally, bilirubin, a key biochemical marker of hemolysis in SCD [
21] was found to be significantly decreased in both its total and direct fractions among individuals carrying the rs766432 variant allele. This finding aligns with the proposed protective role of the polymorphisms, corroborating previous study that associated HbF-inducing polymorphisms in
BCLL1A with improved hematological and biochemical profiles [
21].
In parallel, the T allele of the rs6732518 polymorphism also demonstrated associations indicative of a favorable clinical phenotype. Specifically, individuals with the CT or TT genotypes exhibited higher levels of the anti-inflammatory enzyme alpha-1 antitrypsin (A1AT). A1AT, a well-characterized serine protease inhibitor, is abundant in plasma and serves not only as a protease inhibitor but also as a regulator of inflammation and immune responses [
41,
42,
43,
44]. Interestingly, elevated A1AT levels have previously been associated with more severe inflammatory phenotypes in SCD [
45]. This observation contrasts with our findings, wherein increased A1AT levels were associated with the rs6732518 variant, which is generally considered a favorable prognostic marker due to its link with elevated HbF levels [
24,
25]. These findings suggest that the rs6732518 polymorphism may exert pleitropic effects within the
BCL11A gene.
Although rs6732518 was not independently associated with total or direct bilirubin levels or RBC count, its co-inheritance with rs766432 was associated with lower bilirubin concentrations and higher RBC counts. These results imply that the clinical impact of rs6732518 may be enhanced when inherited alongside other intronic polymorphisms in BCL11A, particularly rs766432. This observation underscored the need for additional studies to clarify the combinatorial effects of these polymorphisms on hematological and biochemical markers in SCD.
Taken together, these findings have significant implications for future clinical research aimed to understanding the genetic modulation of disease severity in SCD. They also contribute to the characterization of BCL11A’s behavior in diverse contexts – a critical objective given that this gene is currently a target in gene therapy strategies for hemoglobinopathies such as SCD. However, it is important to acknowledge the limitations of the present study. As the conclusions are based on in vitro and in silico analyses, further in vivo studies and functional assays are need to validate the biological relevance and mechanistic underpinnings of these associations.
4. Materials and Methods
4.1. Patient Recruitment
A cross-sectional study was conducted to evaluate exposures and outcomes simultaneously. The study was approved by the Human Research Ethics Committee of the Gonçalo Moniz Institute (IGM-FIOCRUZ), under approval number CEP: 29239220.8.3002.8035. Participants with SCA were recruited from the Hematology and Hemotherapy Foundation of the State of Bahia (HEMOBA), where they receive regular follow-up care. This study adhered to the principles of Good Clinical Practice (GCP), the Declaration of Helsinki and its subsequent revisions, as well as Resolution 466/12 and related guidelines of Brazilian National Health Council. All participants provided written informed consent through the Free and Informed Consent Form (FICF) and completed a standardized health questionnaire prior to enrollment.
A total of 77 individuals with SCA were included in this study. Eligibility criteria comprised individuals of any age and sexes with a confirmed HbSS genotype (SCA profile), who had not received hydroxyurea (HU) treatment. Exclusion criteria included prior or current treatment with HU, indeterminate genotyping results for the polymorphisms under investigation, and lack of hemoglobin profile confirmation by high-performance liquid chromatography (HPLC).
4.2. Sample Collection
Venous blood samples (5 mL) were collected in EDTA (sodium ethylenediaminetetraacetic acid) tubes (1.5 mg/ml) for hematological, biochemical, hemoglobin profile and genetic analysis.
4.3. Hematological and Biochemical Analyses
The hemoglobin profile was assessed using high performance liquid chromatography (HPLC) using the ion exchange principle, performed on a VARIANTTM system (Bio-Rad, CA, USA). Hematological parameters and red cell indices were measured using the ABX Pentra 80 automated analyzer (HORIBA Diagnostics, Montpellier, France). Morphological evaluation of red blood cells (RBC) was conducted on Wright-stained peripheral blood smears under optical microscopy. Biochemical analyses, including measurements of total bilirubin and fractions, were performed using an A25 automated analyzer (BioSystems AS, Barcelona, Spain).
4.4. Genetic Analyses
Genomic DNA was extracted from 200µL of peripheral blood using the FlexiGene DNA kit (Qiagen) according to the manufacturer’s instructions. Genotyping of BCL11A polymorphisms and SCD-associated haplotypes were carried out using polymerase chain reaction followed by restriction fragment length polymorphism analysis (PCR-RFLP) techniques. Primers were designed using the NCBI Primer-BLAST tool. DNA was stored at -20ºC until analysis.
4.5. Statistical Analyses
The distribution of quantitative variables was assessed using the Shapiro-Wilk and D’Agostino tests. Depending on the normality of each variable, either parametric tests (Independent t-test) or non-parametric tests (Mann–Whitney U test) were used to compare two groups. All statistical analyses were performed using GraphPad Prism version 9.0. Hardy-Weinberg equilibrium (HWE) was evaluated for each polymorphism using the online tool from Tufts University (USA). A p-value < 0.05 was considered statistically significant for deviations from HWE.
5. Conclusions
This study reinforces the regulatory role of BCL11A in modulating fetal hemoglobin (HbF) levels, a critical factor in the clinical expression of SCD. In addition to this primary role, we identified associations between polymorphisms in the BCL11A gene and several laboratory parameters.
Notably, the rs766432 polymorphism showed characteristics of pleiotropy, as it was associated not only with increased HbF levels but also with reduced HDL concentrations. Similarly, the rs6732518 polymorphism was associated with elevated alpha-1 antitrypsin (A1AT) levels. While low HDL is often linked to inflammation, elevated A1AT may represent a compensatory anti-inflammatory response, highlighting the complex and possibly pleiotropic effects of these polymorphisms.
These findings suggest that the BCL11A gene influences a broader spectrum of clinical and biochemical features in SCD than previously understood. However, additional studies are warranted to replicate these associations in other populations and to elucidate the underlying molecular mechanisms
Author Contributions
Conceptualization, A.M.O. and M.G.; methodology, A.M.O., L.F., C.F., C.G., R.S., S.Y., S.C., I.L. and M.G.; software, A.M.O.; validation, A.M.O., M.G. and A.P.P.; formal analysis, A.M.O. and M.G.; resources, M.G.; data curation, A.M.O., A.P.P. and M.G.; writing—original draft preparation, A.M.O. and M.G.; writing—review and editing, A.M.O., M.G., A.P.P.; supervision, M.G.; project administration, A.M.O. and M.G.; funding acquisition, M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Bahia State Research Support Foundation (FAPESB), grant number 005/2019. The APC was funded by Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Gonçalo Moniz Institute (IGM-FIOCRUZ) (protocol code: 29239220.8.3002.8035, and date of approval: August 14, 2020).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
We would like to thank the HbSS patients and their families for their contribution. We would like to thank also the staffs of the Fundação de Hematologia e Hemoterapia da Bahia (HEMOBA) and the Faculdade de Farmácia da Universidade Federal da Bahia for their support in this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Esoh, K.; Wonkam, A. Evolutionary history of sickle-cell mutation: implications for global genetic medicine. Hum. Mol. Genet. 2021, 30, R119–R128. [Google Scholar] [CrossRef]
- Sikora, J.; et al. Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 2014, 124, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Invest. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Uwaezuoke, S.N.; et al. Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management. J. Pain Res. 2018, 3141–3150. [Google Scholar] [CrossRef]
- Pecker, L.H.; Little, J. Clinical manifestations of sickle cell disease across the lifespan. In Sickle Cell Disease and Hematopoietic Stem Cell Transplantation; Springer: Cham, Switzerland, 2018; pp. 3–39. [Google Scholar]
- Bauer, D.E.; Orkin, S.H. Hemoglobin switching’s surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 2015, 33, 62–70. [Google Scholar] [CrossRef]
- López Rubio, M.; Argüello Marina, M. The current role of hydroxyurea in the treatment of sickle cell anemia. J. Clin. Med. 2024, 13, 6404. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 2007, 7, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Pule, G.D.; et al. A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev. Hematol. 2015, 8, 669–679. [Google Scholar] [CrossRef]
- Lavelle, D.; Engel, J.D.; Saunthararajah, Y. Fetal hemoglobin induction by epigenetic drugs. In Seminars in Hematology; W.B. Saunders, 2018; pp. 60–67. [CrossRef]
- Esrick, E.B.; et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef]
- Frangoul, H.; et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Iarovaia, O.V.; et al. Genetic and epigenetic mechanisms of β-globin gene switching. Biochemistry (Moscow) 2018, 83, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Moi, P. Regulation of the globin genes. Pediatr. Res. 2002, 51, 415–421. [Google Scholar] [CrossRef]
- Ginder, G.D. Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl. Res. 2015, 165, 115–125. [Google Scholar] [CrossRef]
- Liu, N.; et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 2018, 173, 430–442.e17. [Google Scholar] [CrossRef]
- Smith, E.C.; et al. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 2016, 128, 2338–2342. [Google Scholar] [CrossRef]
- Yin, J.; et al. BCL11A: a potential diagnostic biomarker and therapeutic target in human diseases. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L.; et al. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum. Mol. Genet. 2009, 18, R216–R223. [Google Scholar] [CrossRef]
- Métais, J.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef]
- Allard, P.; et al. Genetic modifiers of fetal hemoglobin affect the course of sickle cell disease in patients treated with hydroxyurea. Haematologica 2021, 107, 1577. [Google Scholar] [CrossRef] [PubMed]
- Abdulazeez, S.; et al. The rs61742690 (S783N) single nucleotide polymorphism is a suitable target for disrupting BCL11A-mediated foetal-to-adult globin switching. PLoS One 2019, 14, e0212492. [Google Scholar] [CrossRef]
- Abdulazeez, S. Molecular simulation studies on B-cell lymphoma/leukaemia 11A (BCL11A). Am. J. Transl. Res. 2019, 11, 3689. [Google Scholar]
- Solovieff, N.; et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood 2010, 115, 1815–1822. [Google Scholar] [CrossRef]
- Banan, M.; et al. Utility of the multivariate approach in predicting β-thalassemia intermedia or β-thalassemia major types in Iranian patients. Hemoglobin 2013, 37, 413–422. [Google Scholar] [CrossRef]
- Bhatnagar, P.; et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J. Hum. Genet. 2011, 56, 316–323. [Google Scholar] [CrossRef]
- Aleluia, M.M.; et al. Genetic modulation of fetal hemoglobin in hydroxyurea-treated sickle cell anemia. Am. J. Hematol. 2017, 92, E70. [Google Scholar] [CrossRef]
- Chaouch, L.; et al. New deletion at promoter of HBG1 gene in sickle cell disease patients with high HbF level. J. Pediatr. Hematol. Oncol. 2020, 42, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, L.; et al. rs11886868 and rs4671393 of BCL11A associated with HbF level variation and modulate clinical events among sickle cell anemia patients. Hematology 2016, 21, 425–429. [Google Scholar] [CrossRef]
- Nagel, R.L.; Steinberg, M.H. Role of epistatic (modifier) genes in the modulation of the phenotypic diversity of sickle cell anemia. Pediatr. Pathol. Mol. Med. 2001, 20, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Da Guarda, C.C.; et al. Sickle cell disease: a distinction of two most frequent genotypes (HbSS and HbSC). PLoS One 2020, 15, e0228399. [Google Scholar] [CrossRef] [PubMed]
- Olatunya, O.S.; et al. Red blood cell microparticles are associated with hemolysis markers and may contribute to clinical events among sickle cell disease patients. Ann. Hematol. 2019, 98, 2507–2521. [Google Scholar] [CrossRef]
- Shireen, Z.N.; Elshazali, W.A.; Hiba, K. Association of BCL11A genetic polymorphisms with fetal haemoglobin level in Sudanese patients with sickle cell anaemia. J. Genom. Gene Study 2019, 2, 2. [Google Scholar]
- Sales, R.R.; Nogueira, B.L.; Luizon, M.R. Pharmacogenomics of hydroxyurea therapy and fetal hemoglobin (HbF) levels in sickle cell anemia. Pharmacogenomics 2022. [CrossRef]
- Groenen, A.G.; et al. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.; et al. HDL and persistent inflammation immunosuppression and catabolism syndrome. Curr. Opin. Lipidol. 2021, 32, 315–322. [Google Scholar] [CrossRef]
- Seixas, M.O.; et al. Levels of high-density lipoprotein cholesterol (HDL-C) among children with steady-state sickle cell disease. Lipids Health Dis. 2010, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; et al. Association of pro-inflammatory high-density lipoprotein cholesterol with clinical and laboratory variables in sickle cell disease. Hematology 2015, 20, 289–296. [Google Scholar] [CrossRef]
- Rahimi, Z.; et al. Plasma lipids in Iranians with sickle cell disease: hypocholesterolemia in sickle cell anemia and increase of HDL-cholesterol in sickle cell trait. Clin. Chim. Acta 2006, 365, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, A.; Unal, S.; Oztas, Y. Altered HDL particle in sickle cell disease: decreased cholesterol content is associated with hemolysis, whereas decreased apolipoprotein A1 is linked to inflammation. Lipids Health Dis. 2019, 18, 1–7. [Google Scholar] [CrossRef]
- Bergin, D.A.; et al. Alpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch. Immunol. Ther. Exp. 2012, 60, 81–97. [Google Scholar] [CrossRef]
- Brantly, M. α1-Antitrypsin: not just an antiprotease: extending the half-life of a natural anti-inflammatory molecule by conjugation with polyethylene glycol. Am. J. Respir. Cell Mol. Biol. 2002, 27, 652–654. [Google Scholar] [CrossRef]
- Churg, A.; et al. Alpha-1-antitrypsin and a broad-spectrum metalloprotease inhibitor, RS113456, have similar acute anti-inflammatory effects. Lab. Invest. 2001, 81, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.M.; et al. Rosuvastatin alters the proteome of high-density lipoproteins: generation of alpha-1-antitrypsin enriched particles with anti-inflammatory properties. Mol. Cell. Proteomics 2015, 14, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.O.S.; et al. Evaluation of alpha-1 antitrypsin levels and SERPINA1 gene polymorphisms in sickle cell disease. Front. Immunol. 2017, 8, 1491. [Google Scholar] [CrossRef] [PubMed]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).