Submitted:
06 September 2025
Posted:
08 September 2025
You are already at the latest version
Abstract
Marrubium vulgare L. is a medicinal plant widely used in traditional medicine, with emerging evidence of anticancer potential. This study investigated its bioactive com-pounds as inhibitors of Heat Shock Protein 90 alpha (Hsp90α), a molecular chaperone essential for oncogenic protein stability. Organic and aqueous extracts were profiled using high-performance liquid chromatography–mass spectrometry (HPLC–MS), re-vealing a diverse phytochemical composition. Identified compounds were screened against the full-length crystal structure of Hsp90α using a structure-based computa-tional workflow that included extra-precision and domain-specific molecular docking, molecular dynamics (MD) simulations, and MM/GBSA binding free energy calculations. Pharmacokinetic and toxicity profiles were evaluated through ADMET predictions. This study elucidated the chemical composition of the plant and identified two hit com-pounds: Forsythoside B bound preferentially to the middle domain, potentially inter-fering with client protein interactions, and chlorogenic acid targeted the C-terminal domain, which regulates dimerization and allosteric activity. Both ligands displayed stable protein–ligand interactions during MD and favorable ADMET properties. These findings provide the first integrated chemical and computational analysis of M. vulgare targeting Hsp90α, highlighting its potential as a source of novel anticancer scaffolds and laying the groundwork for experimental validation and drug development.
Keywords:
Marrubium vulgare
; phytochemicals
; HSP90 heat-shock proteins
; computational biology
; liquid chromatography-mass spectrometry
; antineoplastic agents
1. Introduction
Medicinal plants have long served as a foundation for traditional medicine and the development of modern therapeutic agents [1,2]. Among these, Marrubium vulgare L., commonly known as white horehound, has been used extensively for its broad pharmacological properties, including antioxidants, anti-inflammatory, antimicrobial, and antihypertensive effects [3,4]. This perennial member of the Lamiaceae family is native to the Mediterranean basin and has been widely adopted in folk medicine across Europe, North Africa, and the Middle East [5]. In Morocco, where the plant is known as “merriwet” or “M’riwta,” it holds significant ethnopharmacological value and is traditionally prescribed for respiratory disorders, hypertension, diabetes, gastrointestinal issues, and more [5]. Ethnobotanical surveys have demonstrated its continued use in regions like Meknes-Tafilalet and Southeastern Morocco, especially in areas with limited access to modern healthcare [6,7].
The medicinal effects of M. vulgare are attributed to its phytochemical richness, including flavonoids, phenolic acids, diterpenoids, and alkaloids [8]. Notably, marrubiin, a furanoid diterpene, has been studied for its antioxidant, hepatoprotective, and cardioprotective properties [9]. Other components such as apigenin, luteolin, and various phenolics also contribute to its reported antimicrobial and anticancer activities [10,11]. Analytical techniques, such as High-Performance Liquid Chromatography (HPLC), have enabled the detailed chemical profiling of M. vulgare, facilitating the identification of active secondary metabolites [11]. Based on this traditional and biochemical background, our study selected M. vulgare for further investigation. Plant material was collected from its natural Moroccan habitat, and both aqueous and organic extractions were employed to ensure a wide range of compound solubility. Phytochemical composition was analyzed using HPLC linked to Mass Spectrometry (MS).
A particularly promising avenue for such compound screening is the heat shock protein 90 (Hsp90), a molecular chaperone essential for maintaining protein homeostasis and cellular viability [12]. Hsp90 assists in the folding and stabilization of over 200 client proteins, many of which are involved in cell cycle progression, signal transduction, and oncogenesis [13]. Hsp90 is frequently overexpressed in cancer cells, where it protects mutated and unstable oncoproteins from degradation, contributing to tumor survival and progression [14]. This makes it a compelling target in cancer therapy. However, despite the development of numerous Hsp90 inhibitors, especially those targeting the N-terminal ATP-binding domain, none have successfully gained FDA approval, largely due to issues related to toxicity, resistance, and poor selectivity [15]. Consequently, there is growing interest in targeting alternative Hsp90 domains, such as the middle and C-terminal domains, which are involved in client protein binding and dimerization [16].
Given the reported bioactivity of flavonoids, diterpenes, and phenolic acids present in Marrubium vulgare, we hypothesized that some of these compounds may interact with Hsp90 and inhibit its function through novel, non-N-terminal domain (non-NTD) binding mechanisms [17]. This study was designed to identify potential Hsp90-binding compounds from M. vulgare using computational chemistry with particular focus on binding affinity and site specificity beyond the NTD. By integrating ethnopharmacological insights, phytochemical profiling, and bioinformatics analyses, this study aims to contribute to the discovery of innovative, plant-based Hsp90 inhibitors for therapeutic development.
2. Results
2.1. Plant Extraction
The aerial parts (leaves and stems) of Marrubium vulgare L., locally known as “Merriwut,” were collected in March 2022 from the Khenifra region of Morocco. After taxonomic authentication (Voucher ID: RAB114877), the harvested material was air-dried for five days under shaded, ventilated conditions and then ground into a fine powder. A total of 200 grams of powdered material was recovered and subjected to sequential extractions using organic and aqueous solvents to obtain chemically distinct fractions. The organic extractions were carried out using a Soxhlet apparatus with a polarity gradient, starting with hexane (yield ratio r=4.725) followed by dichloromethane (r=3.825), ethyl acetate (r=4.675), ethanol (r=2.125), and methanol (r=4.675). Aqueous extraction was performed separately using cold maceration in distilled water (5% w/v), followed by filtration and freeze-drying.
2.2. Chemical Analysis of Marrubium Vulgare Extracts
High-performance liquid chromatography coupled with mass spectrometry (HPLC-MS) was employed to profile the chemical composition of all six M. vulgare extracts. Across the dataset, a total of 40 distinct secondary metabolites were identified, with molecular weights ranging from 164.16 g/mol to 888.8 g/mol. The ethanol extract was the most chemically diverse, yielding 38 compounds, including several phenylpropanoid glycosides, flavonoids, and diterpenes. The ethyl acetate extract contained 17 compounds, while the methanol and dichloromethane extracts each yielded 10 compounds. The aqueous extract revealed 9 polar metabolites, and the non-polar hexane fraction contained 8 hydrophobic constituents. Each extract showed a unique phytochemical fingerprint reflective of the solvent’s polarity. The ethanol and ethyl acetate fractions were rich in glycosylated flavonoids (e.g., luteolin 7-O-glucuronide, apigenin 7-O-glucuronide), phenolic acids (rosmarinic acid, chlorogenic acid), and iridoid glycosides (forsythoside B, verbascoside). The aqueous extract, while less diverse, included key antioxidant constituents such as p-coumaric acid, apigenin 7-acetate, and eugenol. Non-polar extracts like hexane and dichloromethane were dominated by lipophilic compounds including β-sitosterol, hexadecane, and stigmast-5-en-3-ol.
The structural identities of all compounds were confirmed through MS fragmentation patterns and database matching. A comprehensive list of compounds identified in each extract, along with their molecular weights and MS spectrums, is provided in the Supplementary Table S1. The structures of representative compounds categorized by chemical class is shown in Figure 1.
2.3. Structures Preparation
To investigate potential interactions with Hsp90α, identified Marrubium vulgare compounds and known Hsp90 inhibitors were retrieved from PubChem and prepared using Open Babel and Schrödinger tools. The crystal structure of Hsp90α (PDB ID: 7KRJ) was refined by homology modeling via SWISS-MODEL to reconstruct missing N-terminal loops, and its quality was validated using PROCHECK, through Ramachandran plot analysis (Figure 2), The plot shows that less than 2% of the amino acids (24 residues) are outside the optimal and allowed areas. Since more than 90% of residues of the model fall within the optimal and allowed regions, the generated structure is considered reliable for further computational studies. Ligands were optimized using LigPrep, and the protein was pre-processed with the Protein Preparation Wizard. Molecular docking was then performed to explore their binding profiles.
2.4. Molecular Docking
Molecular docking simulations identified several phytochemicals with favorable binding affinities to non-ATP-binding regions of Hsp90α. The screening was specifically focused on the middle domain of chain A, the inter-chain middle-domain interface, and the C-terminal domain (CTD), deliberately excluding the N-terminal ATP-binding site to prioritize non-ATP-competitive inhibitors.
In the middle domain of chain A, Forsythoside B, Terniflorin, and Rosmarinic Acid exhibited the most favorable GlideScores of –6.84, –6.59, and –6.48, respectively. These compounds formed multiple hydrogen bonds and hydrophobic interactions with residues known to participate in client protein stabilization (Table 1).
In the inter-chain middle domain, Forsythoside B again ranked highest with a docking score of –7.96, significantly outperforming reference compounds such as Enniatin A. Several structurally related analogs from the phenylethanoid glycoside family (Figure 1 and Table 2), also exhibited high docking scores for the middle-domain, suggesting a conserved binding profile within this chemical class. Rosmarinic Acid emerged as the top-ranking compound from a different molecular class, with a score of –6.85 (Table 1).
Docking at the C-terminal domain, a region implicated in chaperone dimerization and allosteric regulation, highlighted Luteolin, Naringenin, and Chlorogenic Acid as the top binders, with GlideScores of –7.82, –7.50, and –7.33 kcal/mol, respectively. These affinities exceeded that of the C-terminal reference inhibitor Novobiocin (Table 1).
Based on their superior docking scores and diverse chemotypes, and to further test their stability and interaction dynamics, Forsythoside B and Rosmarinic Acid were selected for molecular dynamics (MD) simulations at both the inter-chain and chain A of the middle-domain. While Luteolin and Chlorogenic Acid were selected for MD simulations at the CTD (Figure 3).
2.5. Molecular Dynamics (MD) Simulations
The MD simulations were conducted to further investigate the stability and binding behavior of the top-scoring compounds identified from docking studies. The selected ligands included Forsythoside B and Rosmarinic Acid for the middle domain in both intra-chain and interchain sites, as well as Luteolin and Chlorogenic Acid for the CTD. These ligands were simulated in complex with Hsp90α to assess the dynamic nature of their interactions and their impact on protein flexibility over time. The simulation results indicated a stabilizing effect on the protein upon ligand binding, as compared to the unbound Hsp90α homodimer (Table 2).
All the RMSD values have lower averages than Hsp90 (avg = 6.56 Å). More specifically, Forsythoside B bound to the middle domain of the protein’s Chain A showed the lowest RMSD values (avg = 5.25 Å). Still, when bound to the interchain of the middle domain, it showed less prominent stabilization (avg = 6.47 Å), but this value is still lower than the avg of Hsp90 alone. Interestingly, Rosmarinic Acid didn’t replicate the effect of Forsythoside B, as it managed to stabilize Hsp90 when it was bound to its Chain A middle domain, but failed to stabilize it when it was bound to the pocket between the middle domains of chain A and B of the dimer. Its MD simulation results in this domain were eliminated because the ligand continued to interact with multiple residues in various regions throughout the 100 ns simulation (Supplementary Table S3).
As for the C-terminal domain, both Chlorogenic acid and Luteolin exhibited similar patterns, with RMSD values lower than the unbound Hsp90 (avg = 6.16 Å and avg = 6.11 Å) (Figure 4). The RMSF values of all the systems follow the same trend as the RMSD values, as the Hsp90 protein has the highest RMSF values when it’s unbound (avg = 2.16 Å), while the binding of Forsythoside B to the chain A middle domain lowers the average value down to 1.87 Å.
This correlation between RMSD and RMSF values is to be expected, as the presence of a ligand and its engagement in interactions with protein residues, especially when these interactions are stable over time, lowers the fluctuations of these specific residues and thus contributes to the limitation of the overall movement of the protein structure. On the other hand, Forsythoside B bound to the interchain binding pocket induces the second most noticeable effect on the RMSF values (avg = 1.90 Å). The remaining systems keep the same trend going, where the avg RMSF is lower than Hsp90 with no ligand.
A focus on the PLI gives an in-depth insight into the mechanism of action of each ligand and in each domain. Starting with Rosmarinic Acid in the middle domain of chain A, a considerable number of amino acids maintained a strong and consistent interaction with the ligand, including TYR438, GLU439, TYR466, GLU473, MET474, and LYS499. It is worth mentioning that periodically, the ligand would interact with additional proteins, but the afore mentioned ones are those that could maintain stable and continuous interaction throughout the simulation time. The total contact number fluctuated between 1 and 15, with the most prominent values sitting at around 5 and 10 contacts per 0.1 ns. Upon close inspection of RMSF values of these residues, a clear decline in all their individual RMSF was noticed, due to their constant engagement with stabilizing interactions with Rosmarinic Acid. For Forsythoside B in the middle domain of chain A, an interesting case arises, because the ligand interacts very tightly with ASP302, Val326, HIS328, GLU426, GLU429, and ASP430 up until the first 30 ns of the simulation, where it detached from the majority of the amino acids, and kept its interaction with GLU429 and ASP430, while initiating a new set of interactions with different amino acids, such as ASP479, THR482, ARG483, LYS485, GLU486, and GLU537. Coincidentally, the average number of interactions per ps dropped from 12 bonds within the first 30 ns to less than 6 for the remaining simulation time. We thus hypothesize that Forsythoside B in this domain has induced the most stabilizing effect because it targeted essential amino acids that are originally supposed to be flexible, rather than the number of bonds it formed with said residues. Forsythoside B interacts with the protein via a second region, which is the binding pocket between the middle domain of the two chains forming the dimer. The ligand has shown exceptional stability of a multitude of interactions, ranging from 10 to 20 interactions for the majority of the 100 ns simulation, with residues including PHE352, GLU353, ARG355, GLU451, GLN617, ALA618, and LEU619, on both chain A and B of the dimer, a few other residues in chain A such as GLU380, and on chain B such as TYR528, and HIS450.
On the CTD, Chlorogenic acid shows several interactions per 0.1 ns, fluctuating mostly between 10 and 15 bonds, with amino acids from both chains, specifically, GLU497, GLN501, ASN609, ARG612, SER677, and LEU678. On chain A we find LYS615, SER674, and on chain B, GLY675, PHE676. All these amino acids display stable patterns interaction with Chlorogenic Acid. As for Luteolin, the interaction profile appeared less prominent, due to the limited number of amino acids interacting with the ligand through the simulation time. The lasting interactions involve GLU497, ALA506, LEU672, and SER673 in chain A, and only Glu497 on chain B, but with an apparent weakening of the interaction by the end of the simulation time.
Based on the RMSD, RMSF, and PLI interactions, Forsythoside B at the interchain binding pocket and Chlorogenic acid at CTD were selected for MM/GBSA assessment.
The MM/GBSA analysis on the last 200 frames (20 ns) of the trajectory revealed that both molecules display good binding affinities with the protein (Table 3), with Forsythoside B showing a more favorable interaction profile.
2.5. ADMET Assessment
Finally, Chlorogenic acid and forsythoside B were identified as hit compounds based on their favorable binding profiles to Hsp90α observed through molecular docking and MD simulations and validated with MM/GBSA analysis. Following their selection, pharmacokinetic and toxicity predictions were carried out using SwissADME and ProTox 3.0.
According to ProTox 3.0, both compounds exhibited low acute oral toxicity, with a predicted LD50 of 5000 mg/kg and classification under toxicity class 5. Neither compound was predicted to be mutagenic or carcinogenic (Supplementary Table S4 for chlorogenic acid; Supplementary Table S5 for forsythoside B). However, both were flagged for potential immunotoxicity and nephrotoxicity, and forsythoside B was additionally predicted to be cardiotoxic.
SwissADME results indicated low gastrointestinal absorption and no blood–brain barrier (BBB) permeability for both compounds, consistent with their high topological polar surface areas (TPSA = 164.75 Å2 for chlorogenic acid and 304.21 Å2 for forsythoside B). Chlorogenic acid met most drug-likeness rules, with only one Lipinski violation and a bioavailability score of 0.11. In contrast, forsythoside B showed multiple violations of drug-likeness rules due to its large molecular weight (756.7 g/mol), high number of hydrogen bond donors/acceptors, and extensive polarity, with a slightly higher bioavailability score of 0.17 (Table 4). Neither compound was predicted to inhibit any of the major cytochrome P450 enzymes (Supplementary Table S6).
3. Discussion
Marrubium vulgare L. is a widely used medicinal plant across North Africa, the Mediterranean, and Europe due to its reported antioxidant, anti-inflammatory, and anticancer activities [18]. Despite its widespread ethnomedicinal application, this plant remains underexplored from a molecular pharmacology perspective. To valorize both the phytochemical richness of M. vulgare and the ancient ethnopharmacological knowledge surrounding it, this study aimed to identify bioactive compounds from 5 organic and 1 aqueous extract and evaluate their potential against a clinically relevant target: Heat Shock Protein 90 (Hsp90), a key molecular chaperone involved in cancer progression and drug resistance [15,16]. In this study, we combined phytochemical screening, HPLC-MS profiling, and computational analyses to evaluate M. vulgare constituents as potential Hsp90 modulators, given Hsp90’s central role in stabilizing oncogenic client proteins and its relevance as a target in anticancer drug discovery. The idea of targeting Hsp90 with plant-derived compounds is supported by a growing body of literature, including recent works [19,20], which highlights the potential of natural products in modulating Hsp90 pathways in cancer models. This aligns with a broader shift in cancer therapeutics favoring selective modulation of protein networks over broad-spectrum inhibition, particularly when aiming to reduce side effects and improve tolerability.
Phytochemical analyses of M. vulgare revealed several bioactive molecules with potential anticancer activity, including flavonoids (luteolin, apigenin), phenylpropanoids (forsythoside B, rosmarinic acid), and phenolic acids (syringic, para-coumaric, chlorogenic) [21]. HPLC-MS profiling confirmed eight different chemical classes rich in flavonoids, phenolic acids, terpenoids, and phenylpropanoid glycosides. The ethanol and ethyl acetate extracts were abundant in glycosylated flavonoids such as luteolin and apigenin derivatives, phenolic acids like rosmarinic and chlorogenic acids, and iridoid glycosides including forsythoside B and verbascoside. These compound classes have been frequently associated with antioxidants, anti-inflammatory, and anticancer properties [22,23].
In our search for novel Hsp90 modulators, we selected four phytochemicals: Forsythoside A, Rosmarinic Acid, Luteolin, and Chlorogenic Acid based on their docking scores and structural attributes. Forsythoside B and rosmarinic acid showed favorable docking to the middle domain of Hsp90α, a region implicated in client and co-chaperone binding interactions [24]. Their polyhydroxylated aromatic rings and hydrophilic side chains favor electrostatic and hydrogen bonding with polar residues, potentially disrupting client–chaperone interactions. Molecular dynamics simulations revealed that forsythoside B increased rigidity at ASP302, HIS328, and GLU429 and maintained persistent contacts with interdomain residues such as PHE352, GLU353, ARG355, and GLN617, suggesting it may disrupt chaperone-client interactions by reducing protein flexibility and impeding domain communication. In contrast, rosmarinic acid had weaker and more localized interactions, suggesting lower efficacy as a modulator.
Luteolin and chlorogenic acid targeted CTD, a domain essential for dimerization, co-chaperone binding and an emerging target for selective inhibition [24]. Luteolin displayed weak and inconsistent binding during the MD simulations, while chlorogenic acid exhibited a more promising profile, forming consistent hydrogen bonds and hydrophobic interactions with residues such as GLU497, GLN501, and ARG612 across both chains. RMSD and RMSF values for the chlorogenic acid complex averaged 6.16 Å and below 2.0 Å, suggesting moderate stabilization and a selective engagement of the CTD compared to the unbound protein.
Chlorogenic acid has been reported to influence pathways involving Hsp90 client proteins, although it has not been characterized as a direct Hsp90 inhibitor, it has been shown to influence Hsp90-related signaling. A recent study found that it modulates transcription factor interactions with Hsp90: it weakens the SREBP2–Hsp90 complex and stabilizes PXR–Hsp90 binding, thereby altering nuclear translocation of these factors and regulating cholesterol homeostasis [25]. However, the only M. vulgare compound reported with direct evidence of Hsp90 inhibition is luteolin [26,27]. No peer-reviewed reports link forsythoside B or rosmarinic acid to direct Hsp90 modulation, although their predicted binding to the middle domain and known anti-inflammatory properties suggest potential for chaperone regulation.
Collectively, the combination of biochemical activity, drug-like properties, and structural compatibility led us to hypothesize that these compounds may act as Hsp90 modulators. Forsythoside B showed a strong modulatory effect with its ability to bind the middle domain and alter the protein function via rigidification of the amino acids and lowering its internal motion, thereby preventing its enzymatic activity and physically blocking the client protein recruitment pocket. Chlorogenic acid emerged as a moderate modulator; it binds specifically to the C-terminal domain and slightly lowers the protein flexibility, without affecting protein stability as strongly as forsythoside B. Each of these ligands could achieve a different biological response within the cell, depending on the degree to which we want to hinder the functionality of Hsp90. Further in vitro and in vivo studies would help clarify the difference each of these compounds would have on the protein and thus provide a clearer view of their therapeutic potential as Hsp90 modulators.
MM/GBSA free energy calculations confirmed forsythoside B as the most stable binder among the candidates [28], implying that interchain targeting compounds could serve as novel non-ATP-site modulators of Hsp90α [29,30]. ADMET analysis supported low acute oral toxicity for forsythoside B and chlorogenic acid (LD50 = 5000 mg/kg), no predicted mutagenicity or carcinogenicity, and absence of CYP450 inhibition [31]. Forsythoside B, however, showed potential cardiotoxic and immunotoxic risks. Chlorogenic acid demonstrated better gastrointestinal absorption and drug-likeness properties.
In conclusion, this study characterized the phytochemical composition of Marrubium vulgare L. across extracts of increasing polarity and aqueous extract and highlighted three key compounds with potential modulatory effects on Hsp90. Luteolin, a known Hsp90 inhibitor, reinforces the plant’s therapeutic relevance. Chlorogenic acid showed selective interaction with the C-terminal domain, indicating a modulatory but less disruptive role. Forsythoside B demonstrated promising binding to the middle domain, warranting further in vitro validation. These findings provide a phytochemical and computational basis for exploring Marrubium vulgare as a source of Hsp90 modulators, supporting its potential in anticancer drug discovery.
4. Materials and Methods
4.1. Plant Extraction
M. vulgare L. was collected in May 2025 from khenifra, Morocco, the species was confirmed at the RAB-Scientific Institute in Rabat, Morocco (voucher no114877) and dried separately in the shade in a dry and ventilated place at room temperature for 5 days until their weight stabilized then the leaves and stems were grounded using an electric mill and then sieved to obtain a fine powder. The extraction was carried out using Soxhlet apparatus where 40g of the powder was put in a series of solvents from the lowest to the highest polarity successively, starting from hexane, ethyl acetate to ethanol. Another cycle of extraction with a new powder was done with hexane, dichloromethane to methanol. The extraction took about 5 to 12 hours. The filtrate obtained was concentrated in a rotary evaporator to obtain the crude extract. The aqueous extract was prepared by maceration of the powder of M. vulgare L. with a ratio of 5g/100ml (mass/v), for 24 h at room temperature using a magnetic stirrer. The extract was centrifuged and then filtered. All extracts were kept at 4°C until further use.
4.2. Chemical Analysis of Marrubium Vulgare Extracts
The chemical composition of Marrubium Vulgare Extracts was identified using high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS) using an Ultimate 3000-Exactive plus THERMO scientific apparatus, at the National Centre for Scientific and Technical Research (CNRST) in Rabat. The analysis involved six extracts, which were diluted in MeOH (methanol) and analyzed along with the control (MeOH). The separation of compounds was achieved using a BDS HYPERSIL C18, dim. 150X4.6 mm particle size 5microm in polar mobile phase consisting of solvent A (water/0.5% formic acid) and solvent B (acetonitrile/0.5% formic acid). The gradient profile employed a constant flow rate of 0.5 mL/min, with a temperature of 30°C of solvent B as follows: 10% B (3 min), 20% B (10 min), 30% B (30 min), 90% B (12 min), and 10% B (7 min). An injection volume of 20μl was used, and the UV spectrum was monitored in the range of 190 nm to 600 nm. In addition to UV detection, mass spectrometry (MS) analysis was performed with an analysis time of 62 minutes. The scanning range for mass data acquisition was set to 90-1350 m/z in both positive and negative ionization modes.
4.3. Database Preparation
The 3D structures of the identified compounds, as well as the known inhibitors for Hsp90 (17AAG, Geldanamycin, Radicicol, Novobiocin) were obtained from PubChem database and converted to PDB format using Open Babel to ensure compatibility. The 3D structure of Hsp90α (PDB ID: 7KRJ) [32] was obtained from the RCSB Protein Data. As the structure lacked critical loops within the N-terminal domain required for molecular dynamics simulations, homology modeling was carried out using SWISS-MODEL [33] to reconstruct the missing regions. The modeling process was based on the amino acid sequence of Hsp90α (UniProt ID: P07900), with the 7KRJ structure used as a template. Structural quality was evaluated through a Ramachandran plot generated by the PROCHECK tool [34]. The prepared protein structure was then processed using the Protein Preparation Wizard in Maestro (Schrödinger Suite), which included the removal of water molecules, addition of nonpolar hydrogens, creation of salt bridges, and assignment of appropriate charges. Ligands were prepared using the LigPrep module, which included the addition of missing hydrogens, bond order assignment, and energy minimization using the OPLS4 force field within the Schrödinger environment. All chemical structures were constructed and reoriented using ChemDraw Professional (version 23.1.1). All known chiral centers are represented with either dashes or wedges, while chrial centers with unknown absolute stereochemistry are represented with wavy bonds. Atomic labels used Arial 10 with 120° bond angles, 18% bond spacing, 0.2 in bond length, 0.0278 in bold width, and 0.0083 in line width. The figure was exported in 600 dpi resolution as a .cdx file.
4.4. Extra Precision Docking
Extra Precision (XP) docking mode in the Schrodinger Glide module [35] was used to screen the identified compounds against the prepared structure of Hsp90α. The docking grids were generated using a targeted docking approach, where three receptor grid boxes were centered on the centroid of the binding pocket formed between the middle domains of the two chains of the protein dimer, the middle domain of chain A alone, and the C-terminal domain of the protein. The grid boxes were set to exclude the N-terminal domain, since our aim was to avoid screening ATP-competitive ligands and instead focus on compounds that may bind to alternative sites of Hsp90. Docking in XP mode was conducted with default parameters, including a van der Waals radius scaling factor set to 0.8 and a partial charge cutoff set to 0.15 for non-polar atoms. No constraints were applied during docking. The ligand poses were scored and ranked based on their GlideScore. A comparative ranking was used to identify the top two ranking molecules per domain, which were selected for subsequent MD simulations. Ligand-protein interactions were visualized and analyzed using Ligand Interaction module in Maestro to identify key residue interactions contributing to binding affinity.
4.5. Molecular Dynamics Simulation
The top two molecules showing the highest binding affinities with the middle domain interchain or chain A, and C-terminal domain of the protein were selected for molecular dynamics (MD) simulations. The docking-generated complexes were used as the initial conformation for the simulation system preparation using the System Builder wizard in Desmond [37].
The preparation consisted of embedding the Hsp90 dimer complex and ligand-bound complexes, in a TIP3P solvent using a Cubic box system with 10 Å padding, neutralizing the systems with Cl- ions, then adding 0.15 M of NaCl concentration to mimic physiological ionic strength. The generated systems were then subjected to 100 ns MD simulations using OPLS4 force field, which is specifically optimized for proteins and drug-like ligands, including phytochemicals. The multigrator integrator was employed, using the Martyna-Tobias-Klein (MTK) method for pressure coupling and the Nose-Hoover thermostat for temperature regulation. The temperature was set to 300 K, and the pressure to 1.01325 bar, using relaxation times (τ) of 1.0 ps and 2.0 ps, respectively, for thermostat and barostat. A time step of 2 fs was used for bonded and short-range nonbonded interactions, and 6 fs for long-range interactions. Long-range electrostatics were handled using the u-series method, with a cutoff radius of 9.0 Å for non-bonded interactions. The constraints on bond lengths involving hydrogen atoms were applied using the default Desmond algorithm, with a convergence tolerance of 1×10−8 and a maximum of 16 iterations. Initial velocities were randomly assigned from a Maxwell–Boltzmann distribution at 300 K using a fixed seed (2007) to ensure reproducibility. Simulation trajectories were saved every 500 ps, and system energies were recorded every 1.2 ps. Checkpoint files were written every 240.06 ps, and simulation box dimensions were monitored at 1.2 ps intervals.
Following the completion of molecular dynamics (MD) simulations for each system, the resulting trajectories were analyzed using the Simulation Interaction Diagram (SID) in Maestro. This analysis quantified system dynamics through metrics such as root mean square deviation (RMSD), root mean square fluctuation (RMSF). The average (Avg) and standard deviation (SD) of each parameter were calculated. The protein-ligand interactions (PLI) were tracked throughout the simulation for each residue. Additionally, the Prime Molecular mechanics with generalized Born and surface area solvation (MM/GBSA) was calculated to evaluate the free binding affinity between the protein and selected molecules during the last 20 ns of the MD trajectory, to further evaluate the modulatory effect of said molecules on Hsp90α.
4.6. ADMET Assessment
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Table S1_MS spectrums; Table S2: Table S2_PubChem_compound_id_list; Table S3: Table S3_MD_7KRJ_RosmarinicAcid; Table S4: Table S4_ProTox-3.0 - Prediction of TOXicity of chemicals- Chlorogenic acid; Table S5: Table S5_ProTox-3.0 - Prediction of TOXicity of chemicals- Forsytoside B; Table S6: Table S6_SwissADME- Chlorogenic acid and Forsytoside B.
Author Contributions
Conceptualization, Ilham Zarguan, Devan Buchanan, Abdelaziz Benjouad and Lamiae Belayachi; Data curation, Ilham Zarguan, Hanane Abbou, Razana Zegrari, Rihab Festali and Devan Buchanan; Formal analysis, Ilham Zarguan, Hanane Abbou, Razana Zegrari, Rihab Festali and Devan Buchanan; Funding acquisition, Abdelaziz Benjouad and Lamiae Belayachi; Investigation, Ilham Zarguan, Hanane Abbou, Razana Zegrari and Rihab Festali; Methodology, Ilham Zarguan, Hanane Abbou, Razana Zegrari, Rihab Festali, Devan Buchanan and Lamiae Belayachi; Project administration, Ilham Zarguan, Abdelaziz Benjouad and Lamiae Belayachi; Resources, Rihab Festali and Lamiae Belayachi; Software, Ilham Zarguan, Razana Zegrari, Rihab Festali and Devan Buchanan; Supervision, Abdelaziz Benjouad and Lamiae Belayachi; Validation, Ilham Zarguan, Hanane Abbou, Razana Zegrari, Rihab Festali, Devan Buchanan, Abdelaziz Benjouad and Lamiae Belayachi; Visualization, Ilham Zarguan, Hanane Abbou, Razana Zegrari and Devan Buchanan; Writing – original draft, Ilham Zarguan and Hanane Abbou; Writing – review & editing, Ilham Zarguan, Hanane Abbou, Razana Zegrari, Rihab Festali, Devan Buchanan, Abdelaziz Benjouad and Lamiae Belayachi. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this study are included in the supplementary material. Additional details are available from the corresponding author upon request.
Acknowledgments
The authors gratefully acknowledge the Centre National pour la Recherche Scientifique et Technique (CNRST), Rabat for providing access to the HPLC/MS core facilities, and the Agence Nationale des Plantes Médicinales et Aromatiques (ANPMA), Taounate for their support and laboratory resources used for plant extraction. Their valuable contributions in terms of infrastructure and technical assistance were essential for the successful completion of this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CTD | C Terminal Domain |
| HPLC-MS | High-Performance Liquid Chromatography-Mass Spectormetry |
| Hsp90 | Heat-Shock Protein 90 |
| MD | Molecular Dynamics |
| MMGBSA | Molecular mechanics with generalized Born and surface area solvation |
| NTD | N Terminal Domain |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| XP | Extra Precision |
References
- B. B. Petrovska, “Historical review of medicinal plants’ usage,” Pharmacogn. Rev., vol. 6, no. 11, pp. 1–5, 2012. [CrossRef]
- M. Akram and K. Mahmood, “Awareness and current knowledge of medicinal plants,” RPS Pharm. Pharmacol. Rep., vol. 3, no. 4, p. rqae023, Oct. 2024. [CrossRef]
- Z. Ouadghiri et al., “In Vivo and In Vitro Assessment of Marrubium vulgare: Chemical, Antioxidant and Anti-inflammatory Profiles,” Arab. J. Sci. Eng., Mar. 2025. [CrossRef]
- A. Bouyahya et al., “Anti-inflammatory and analgesic properties of Moroccan medicinal plants: Phytochemistry, in vitro and in vivo investigations, mechanism insights, clinical evidences and perspectives,” J. Pharm. Anal., vol. 12, no. 1, pp. 35–57, Feb. 2022. [CrossRef]
- M. Eddouks, M. Ajebli, and M. Hebi, “Ethnopharmacological survey of medicinal plants used in Daraa-Tafilalet region (Province of Errachidia), Morocco,” J. Ethnopharmacol., vol. 198, pp. 516–530, Feb. 2017. [CrossRef]
- L. E. Fakir, V. Bito, A. Zaid, and T. M. Alaoui, “Complimentary Herbal Treatments Used in Meknes-Tafilalet Region (Morocco) to Manage Cancer,” Am. J. Plant Sci., vol. 10, no. 5, Art. no. 5, May 2019. [CrossRef]
- M. Aboufaras, K. Selmaoui, and N. Ouzennou, “Adverse effects of medicinal plants used by cancer patients in Beni Mellal and the communication of this use,” E3S Web Conf., vol. 319, p. 01107, 2021. [CrossRef]
- M. Aćimović et al., “Marrubium vulgare L.: A Phytochemical and Pharmacological Overview,” Molecules, vol. 25, no. 12, p. 2898, Jan. 2020. [CrossRef]
- O. K. Popoola, A. M. Elbagory, F. Ameer, and A. A. Hussein, “Marrubiin,” Molecules, vol. 18, no. 8, pp. 9049–9060, July 2013. [CrossRef]
- J. M. Al-Khayri, G. R. Sahana, P. Nagella, B. V. Joseph, F. M. Alessa, and M. Q. Al-Mssallem, “Flavonoids as Potential Anti-Inflammatory Molecules: A Review,” Molecules, vol. 27, no. 9, p. 2901, May 2022. [CrossRef]
- A. A. Gourich et al., “Insight into biological activities of chemically characterized extract from Marrubium vulgare L. in vitro, in vivo and in silico approaches,” Front. Chem., vol. 11, p. 1238346, Aug. 2023. [CrossRef]
- G. Chiosis, C. S. Digwal, J. B. Trepel, and L. Neckers, “Structural and functional complexity of HSP90 in cellular homeostasis and disease,” Nat. Rev. Mol. Cell Biol., vol. 24, no. 11, pp. 797–815, Nov. 2023. [CrossRef]
- Y. Miyata, H. Nakamoto, and L. Neckers, “The therapeutic target Hsp90 and cancer hallmarks,” Curr. Pharm. Des., vol. 19, no. 3, pp. 347–365, 2013. [CrossRef]
- J. Trepel, M. Mollapour, G. Giaccone, and L. Neckers, “Targeting the dynamic HSP90 complex in cancer,” Nat. Rev. Cancer, vol. 10, no. 8, pp. 537–549, Aug. 2010. [CrossRef]
- J. Trepel, M. Mollapour, G. Giaccone, and L. Neckers, “Targeting the dynamic HSP90 complex in cancer,” Nat. Rev. Cancer, vol. 10, no. 8, pp. 537–549, Aug. 2010. [CrossRef]
- X. Liang, R. Chen, C. Wang, Y. Wang, and J. Zhang, “Targeting HSP90 for Cancer Therapy: Current Progress and Emerging Prospects,” J. Med. Chem., vol. 67, no. 18, pp. 15968–15995, Sept. 2024. [CrossRef]
- S. Riedl et al., “Evolution of the conformational dynamics of the molecular chaperone Hsp90,” Nat. Commun., vol. 15, no. 1, p. 8627, Oct. 2024. [CrossRef]
- B. Amri et al., “Marrubium vulgare L. Leave Extract: Phytochemical Composition, Antioxidant and Wound Healing Properties,” Mol. J. Synth. Chem. Nat. Prod. Chem., vol. 22, no. 11, p. 1851, Oct. 2017. [CrossRef]
- I. Zarguan, S. Ghoul, L. Belayachi, and A. Benjouad, “Plant-Based HSP90 Inhibitors in Breast Cancer Models: A Systematic Review,” Int. J. Mol. Sci., vol. 25, no. 10, p. 5468, May 2024. [CrossRef]
- H. Y. Liew, X. Y. Tan, H. H. Chan, K. Y. Khaw, and Y. S. Ong, “Natural HSP90 inhibitors as a potential therapeutic intervention in treating cancers: A comprehensive review,” Pharmacol. Res., vol. 181, p. 106260, July 2022. [CrossRef]
- M. Aćimović et al., “Marrubium vulgare L.: A Phytochemical and Pharmacological Overview,” Mol. Basel Switz., vol. 25, no. 12, p. 2898, June 2020. [CrossRef]
- S. Puangpraphant, E.-O. Cuevas-Rodríguez, and M. Oseguera-Toledo, “Chapter 9 - Anti-inflammatory and antioxidant phenolic compounds,” in Current Advances for Development of Functional Foods Modulating Inflammation and Oxidative Stress, B. Hernández-Ledesma and C. Martínez-Villaluenga, Eds., Academic Press, 2022, pp. 165–180. [CrossRef]
- M. C. Dias, D. C. G. A. Pinto, and A. M. S. Silva, “Plant Flavonoids: Chemical Characteristics and Biological Activity,” Molecules, vol. 26, no. 17, p. 5377, Sept. 2021. [CrossRef]
- J. Li and J. Buchner, “Structure, function and regulation of the hsp90 machinery,” Biomed. J., vol. 36, no. 3, pp. 106–117, 2013. [CrossRef]
- C. Meng et al., “Chlorogenic acid regulates the expression of NPC1L1 and HMGCR through PXR and SREBP2 signaling pathways and their interactions with HSP90 to maintain cholesterol homeostasis,” Phytomedicine Int. J. Phytother. Phytopharm., vol. 123, p. 155271, Jan. 2024. [CrossRef]
- D. Chen et al., “Luteolin exhibits anti-inflammatory effects by blocking the activity of heat shock protein 90 in macrophages,” Biochem. Biophys. Res. Commun., vol. 443, no. 1, pp. 326–332, Jan. 2014. [CrossRef]
- J. Lv et al., “Effects of luteolin on treatment of psoriasis by repressing HSP90,” Int. Immunopharmacol., vol. 79, p. 106070, Feb. 2020. [CrossRef]
- S. Genheden and U. Ryde, “The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities,” Expert Opin. Drug Discov., vol. 10, no. 5, pp. 449–461, May 2015. [CrossRef]
- J. Gu et al., “Advances in the structures, mechanisms and targeting of molecular chaperones,” Signal Transduct. Target. Ther., vol. 10, no. 1, p. 84, Mar. 2025. [CrossRef]
- Z. Wang, W. Huang, K. Zhou, X. Ren, and K. Ding, “Targeting the Non-Catalytic Functions: a New Paradigm for Kinase Drug Discovery?,” J. Med. Chem., vol. 65, no. 3, pp. 1735–1748, Feb. 2022. [CrossRef]
- A. Daina, O. Michielin, and V. Zoete, “SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules,” Sci. Rep., vol. 7, no. 1, p. 42717, Mar. 2017. [CrossRef]
- C. M. Noddings, R. Y.-R. Wang, J. L. Johnson, and D. A. Agard, “Structure of Hsp90–p23–GR reveals the Hsp90 client-remodelling mechanism,” Nature, vol. 601, no. 7893, pp. 465–469, Jan. 2022. [CrossRef]
- A. Waterhouse et al., “SWISS-MODEL: homology modelling of protein structures and complexes,” Nucleic Acids Res., vol. 46, no. W1, pp. W296–W303, July 2018. [CrossRef]
- R. A. Laskowski, M. W. MacArthur, D. S. Moss, and J. M. Thornton, “PROCHECK: a program to check the stereochemical quality of protein structures,” J. Appl. Crystallogr., vol. 26, no. 2, pp. 283–291, Apr. 1993. [CrossRef]
- R. A. Friesner et al., “Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes,” J. Med. Chem., vol. 49, no. 21, pp. 6177–6196, Oct. 2006. [CrossRef]
- R. A. Laskowski and M. B. Swindells, “LigPlot+: multiple ligand-protein interaction diagrams for drug discovery,” J. Chem. Inf. Model., vol. 51, no. 10, pp. 2778–2786, Oct. 2011. [CrossRef]
- “Scalable algorithms for molecular dynamics simulations on commodity clusters | Proceedings of the 2006 ACM/IEEE conference on Supercomputing,” ACM Conferences. Accessed: Aug. 22, 2025. [Online]. Available: https://dl.acm.org/doi/10.1145/1188455.1188544. [CrossRef]
- A. Daina, O. Michielin, and V. Zoete, “SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules,” Sci. Rep., vol. 7, no. 1, p. 42717, Mar. 2017. [CrossRef]
- P. Banerjee, E. Kemmler, M. Dunkel, and R. Preissner, “ProTox 3.0: a webserver for the prediction of toxicity of chemicals,” Nucleic Acids Res., vol. 52, no. W1, pp. W513–W520, July 2024. [CrossRef]
Figure 1.
Chemical structures of the identified compounds from Marrubium Vulgare L. extracts identified using HPLC-MS arranged based on chemical class. All chemical structures were constructed and reoriented using ChemDraw Professional (version 23.1.1).
Figure 1.
Chemical structures of the identified compounds from Marrubium Vulgare L. extracts identified using HPLC-MS arranged based on chemical class. All chemical structures were constructed and reoriented using ChemDraw Professional (version 23.1.1).
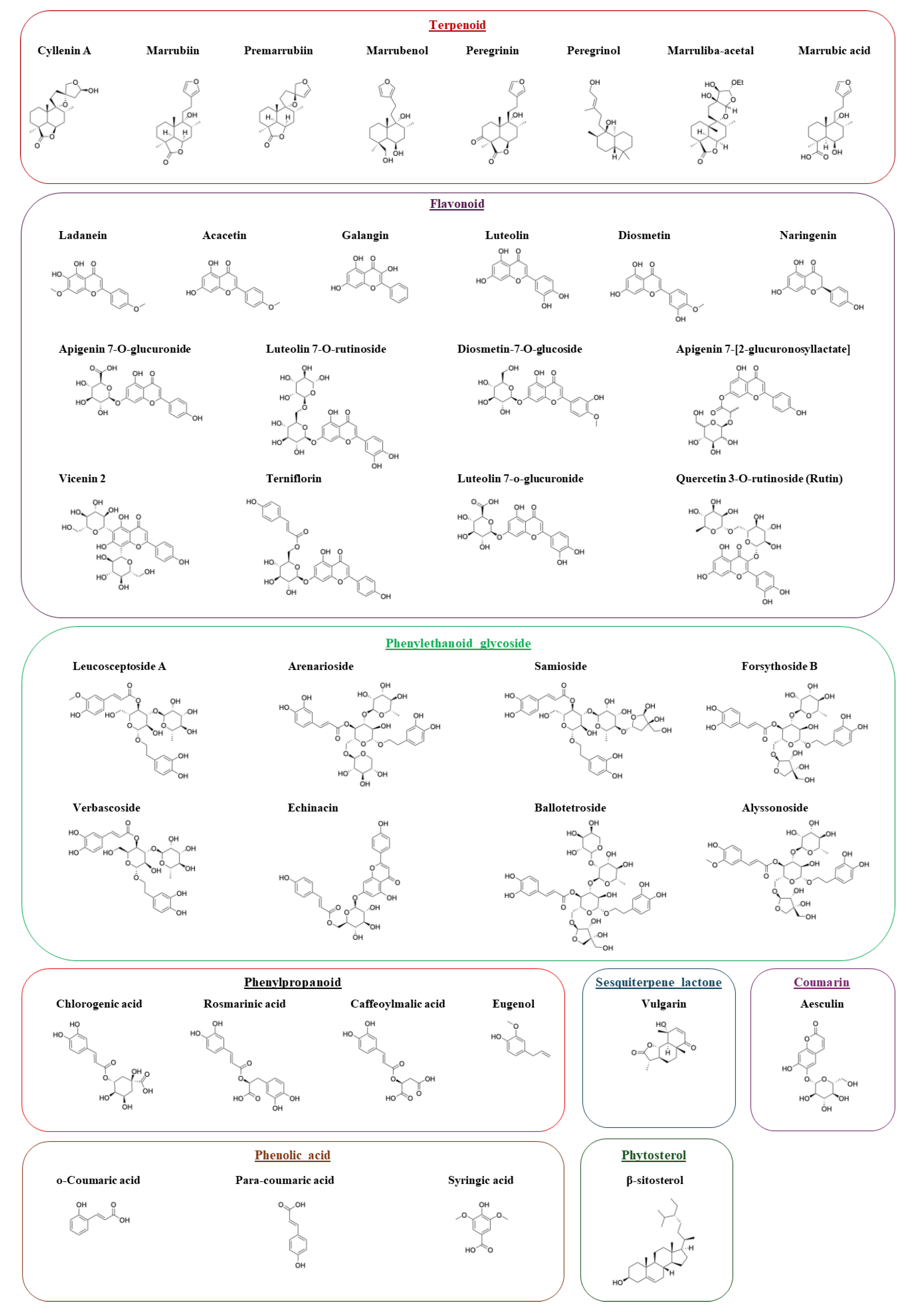
Figure 2.
A) 3D structure of full-length Hsp90α (PDB ID: 7KRJ) refined for molecular docking using SWISS-MODEL and visualized in PyMOL. B) Ramachandran plot validation from PROCHECK.
Figure 2.
A) 3D structure of full-length Hsp90α (PDB ID: 7KRJ) refined for molecular docking using SWISS-MODEL and visualized in PyMOL. B) Ramachandran plot validation from PROCHECK.
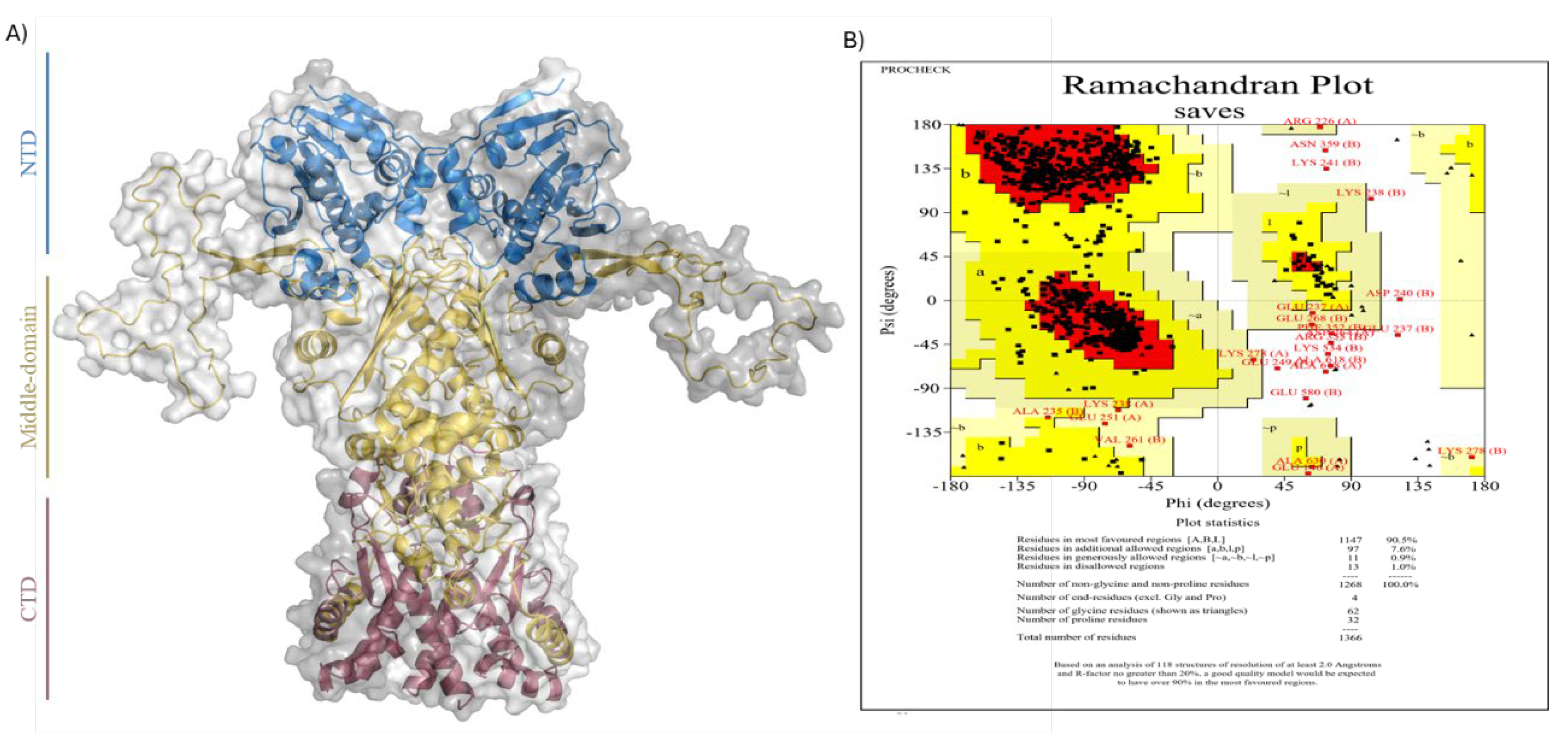
Figure 3.
Binding poses of selected molecules for MD simulation. A.1) Rosmarinic Acid and A.2) Forsythoside B interaction with Hsp90α in the middle-domain chain A. B.1) Rosmarinic Acid and B.2) Forsythoside B bound to the interchain of the middle domain. C.1) Luteolin and C.2) Chlorogenic Acid bound to the CTD. 3D structure of Hsp90α and Ligand–protein interactions were visualized and analyzed using Maestro.
Figure 3.
Binding poses of selected molecules for MD simulation. A.1) Rosmarinic Acid and A.2) Forsythoside B interaction with Hsp90α in the middle-domain chain A. B.1) Rosmarinic Acid and B.2) Forsythoside B bound to the interchain of the middle domain. C.1) Luteolin and C.2) Chlorogenic Acid bound to the CTD. 3D structure of Hsp90α and Ligand–protein interactions were visualized and analyzed using Maestro.

Figure 4.
MD simulation analysis using Desmond. A) RMSD plots of Hsp90α alone vs protein–ligand complexes over 100 ns showing structural stability. B) RMSF plots show residue-level flexibility during the simulation.
Figure 4.
MD simulation analysis using Desmond. A) RMSD plots of Hsp90α alone vs protein–ligand complexes over 100 ns showing structural stability. B) RMSF plots show residue-level flexibility during the simulation.

Table 1.
Docking scores of the identified compounds from Marrubium vulgare L. against the middle-domain chain A, interchain region, and C-terminal domain (CTD) of Hsp90α. Ligand poses were scored and ranked based on GlideScore from Glide XP docking mode.
Table 1.
Docking scores of the identified compounds from Marrubium vulgare L. against the middle-domain chain A, interchain region, and C-terminal domain (CTD) of Hsp90α. Ligand poses were scored and ranked based on GlideScore from Glide XP docking mode.
| Middle domain Chain A | Middle domain Interchain | CTD | |||
|---|---|---|---|---|---|
| Compound | Docking score | Compound | Docking score | Compound | Docking score |
| Forsythoside B | -6.84 | Forsythoside B | -7.96 | Luteolin | -7.82 |
| Terniflorin | -6.59 | Samioside | -7.64 | Naringenin | -7.50 |
| Rosmarinic_acid | -6.48 | Terniflorin | -7.44 | Chlorogenic acid | -7.33 |
| Luteolin7_o_glucuronide | -6.48 | Alyssonoside | -7.33 | Galangin | -7.20 |
| Apigenin | -6.40 | Verbascoside | -6.93 | Forsythoside B | -6.99 |
| Samioside | -6.26 | Acteoside | -6.93 | Diosmetin | -6.91 |
| Acteoside | -5.97 | Rosmarinic_acid | -6.85 | Terniflorin | -6.89 |
| Verbascoside | -5.97 | LeucosceptosideA | -6.71 | Samioside | -6.80 |
| Luteolin7_O_rutinoside | -5.91 | Echinacin | -6.50 | Acacetin | -6.65 |
| Vicenin2 | -5.90 | Vicenin2 | -6.20 | Vulgarin | -6.52 |
| Echinacin | -5.86 | Diosmetin | -6.06 | Ladanein | -6.43 |
| Rutin | -5.73 | Syringic_acid | -6.03 | Rosmarinic_acid | -6.33 |
| Alyssonoside | -5.69 | Luteolin | -5.94 | Alyssonoside | -6.26 |
| Luteolin | -5.62 | Naringenin | -5.77 | Acteoside | -6.26 |
| Aesculin | -5.56 | Diosmetin_7_O_glucoside | -5.70 | Verbascoside | -6.26 |
| Eugenol | -5.38 | Acacetin | -5.57 | cis_Piperitone | -6.13 |
| Diosmetin | -5.18 | Luteolin7_O_rutinoside | -5.53 | Echinacin | -5.71 |
| Chlorogenic acid | -4.95 | Rutin | -5.49 | Syringic_acid | -5.64 |
| Acacetin | -4.90 | Ladanein | -5.41 | Aesculin | -5.63 |
| Galangin | -4.88 | Galangin | -5.34 | Luteolin7_O_rutinoside | -5.60 |
| Vulgarin | -4.86 | Chlorogenic acid | -5.05 | Apigenin | -5.59 |
| Syringic_acid | -4.71 | Aesculin | -4.99 | Rutin | -5.42 |
| Ladanein | -4.62 | Novobiocin | -4.98 | LeucosceptosideA | -5.36 |
| Peregrinin | -4.61 | Apigenin | -4.92 | Diosmetin_7_O_glucoside | -5.27 |
| Diosmetin_7_O_glucoside | -4.54 | Eugenol | -4.87 | Eugenol | -5.24 |
| Marrubiin | -4.48 | Vulgarin | -4.85 | Marrubenol | -4.91 |
| o_Coumaric_acid | -4.48 | Peregrinin | -4.82 | Vicenin2 | -4.78 |
| Oxacyclohexadecan_2_one | -4.44 | o_Coumaric_acid | -4.77 | Luteolin7_o_glucuronide | -4.63 |
| Para_coumaric_acid | -4.44 | Oxacyclohexadecan_2_one | -4.57 | Para_coumaric_acid | -4.46 |
| Cyllenin_A | -4.35 | Luteolin7_o_glucuronide | -4.35 | Radicicol | -4.43 |
| Naringenin | -4.26 | Cyllenin_A | -4.26 | o_Coumaric_acid | -4.16 |
| cis_Piperitone | -4.25 | Para_coumaric_acid | -4.19 | 17AAG | -4.14 |
| Caffeoylmalic acid | -4.08 | Marrubiin | -4.13 | Oxacyclohexadecan_2_one | -3.88 |
| Novobiocin | -3.99 | Marrubenol | -4.10 | Novobiocin | -3.69 |
| Radicicol | -3.86 | cis_Piperitone | -4.10 | Premarrubiin | -3.56 |
| Premarrubiin | -3.79 | Enniatin A | -4.05 | Caffeoylmalic acid | -3.52 |
| 17AAG | -3.47 | Radicicol | -3.99 | Marrubiin | -3.50 |
| Marrubenol | -3.30 | Premarrubiin | -3.92 | Cyllenin_A | -3.42 |
| Geldanamycin | -3.02 | 24_Ethylcholest_5_en_3beta_ol | -3.81 | Peregrinin | -3.39 |
| Leucosceptoside A | -2.74 | Sitosterol | -3.81 | 24_Ethylcholest_5_en_3beta_ol | -3.13 |
| Enniatin A | -2.50 | Stigmast_5_en_3_ol | -3.81 | Sitosterol | -3.13 |
| 24_Ethylcholest_5_en_3beta_ol | -2.23 | Caffeoylmalic acid | -3.61 | Stigmast_5_en_3_ol | -3.13 |
| Sitosterol | -2.23 | Geldanamycin | -3.43 | Geldanamycin | -2.96 |
| Stigmast_5_en_3_ol | -2.23 | Phytol | -1.39 | Phytol | -2.37 |
| Phytol | -0.58 | Methyl_linoleate | 0.14 | Enniatin A | -2.29 |
| Octadecatrienic_acid | 0.86 | Methyl_linoleate | -1.00 | ||
| Octadecatrienic_acid | -0.56 | ||||
Table 2.
The averages (avg) and standard deviations (SD) of the parameters generated from molecular dynamics of the simulated systems.
Table 2.
The averages (avg) and standard deviations (SD) of the parameters generated from molecular dynamics of the simulated systems.
| Unbound structure | Middle domain Chain A |
Middle domain Interchain |
CTD | ||||
|---|---|---|---|---|---|---|---|
| Hsp90α | Forsythoside B | Rosmarinic Acid | Forsythoside B | Chlorogenic acid | Luteolin | ||
| RMSD (Å) | Avg | 6.56 | 5.25 | 6.14 | 6.47 | 6.16 | 6.11 |
| SD | 1.01 | 0.68 | 0.96 | 1.14 | 0.97 | 0.85 | |
| RMSF (Å) | Avg | 2.16 | 1.87 | 1.96 | 1.90 | 2.07 | 2.11 |
| SD | 1.41 | 1.37 | 1.29 | 1.33 | 1.36 | 1.30 | |
Table 3.
MM/GBSA analysis of the interaction between Forsythoside B bound to the middle domain interchain and Chlorogenic acid bound to the CTD of Hsp90α.
Table 3.
MM/GBSA analysis of the interaction between Forsythoside B bound to the middle domain interchain and Chlorogenic acid bound to the CTD of Hsp90α.
| Complex | ΔG Average (kcal mol-1) | ΔG Standard Deviation | ΔG Range |
|---|---|---|---|
|
Forsythoside B (Interchain) |
-71.68 | 14.62 | -107.08 to 0.69 |
| Chlorogenic Acid (CTD) | -51.50 | 10.02 | -64.47 to 0.53 |
Table 4.
ADMET analysis of the hit compounds Chlorogenic acid and Forsythoside B. Data predictions were done using SwissADME.
Table 4.
ADMET analysis of the hit compounds Chlorogenic acid and Forsythoside B. Data predictions were done using SwissADME.
| Chlorogenic acid | 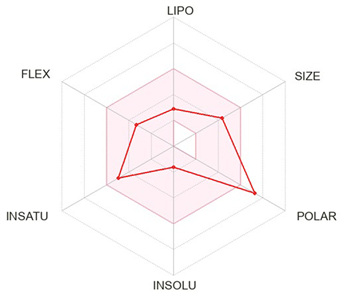 |
Formula | C16H18O9 |
| Molecular weight | 354.31 g/mol | ||
|
Druglikeness (Lipinski) |
Yes; 1 violation: NHorOH>5 | ||
| Log S (ESOL) | -1.62 | ||
| Consensus LogPo/w | -0.38 | ||
| TPSA | 164.75 Å2 | ||
| Bioavailability Score | 0.11 | ||
| Forsythoside B | 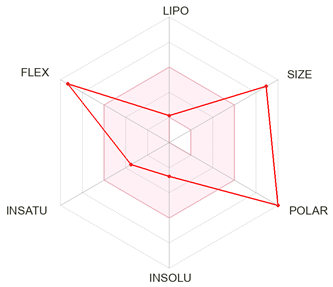 |
Formula | C34H44O19 |
| Molecular weight | 756.70 g/mol | ||
|
Druglikeness (Lipinski) |
No; 3 violations: MW>500, NorO>10, NHorOH>5 | ||
| Log S (ESOL) | -2.69 | ||
| Consensus LogPo/w | -1.49 | ||
| TPSA | 304.21 Å2 | ||
| Bioavailability Score | 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



