
Submitted:
28 August 2025
Posted:
01 September 2025
You are already at the latest version
Abstract
Background/Objectives: Colorectal cancer (CRC) remains a major global health challenge and current therapies are not always effective. In addition, certain immune cell populations, such as myeloid-derived suppressor cells (MDSCs) pose a significant barrier to immune-based treatments. Some phytochemicals, particularly compounds derived from Allium spp. like Propyl Propane Thiosulfonate (PTSO), have shown strong immunomodulatory potential in digestive disorders. This study aims to investigate the capacity of PTSO to modulate immune responses and affect tumor progression in CRC models, in vitro e in vivo, with a focus on the immune cell populations that comprise the tumor microenvironment (TME). Methods: Human peripheral blood mononuclear cells (hPBMCs) were incubated with PTSO and characterized by flow cytometry. These cells were then injected into NOD scid gamma (NSG) immunodeficient mice, which were simultaneously induced to develop a subcutaneous tumor by injection of HCT116 enriched cancer stem cells (CSCs) colonospheres. Results: PTSO reduced MDSC populations and increased T cell subpopulations, particularly interferon gamma (IFNG)-producing cytotoxic CD8+ T cells, which are associated with antitumor activity. In the humanized tumor xenograft mouse model, the administration of PTSO-pretreated hPBMCs led to increased infiltration of CD4+ T lymphocytes and Natural Killer (NK) cells into the tumor, accompanied by reduced expression of immunosuppressive genes. These effects resulted in a reduction in tumor size and cancer cell proliferation and invasiveness. Conclusions: The dual effect of PTSO on immune cell populations, reducing immunosuppressive myeloid cells and enhancing effector T lymphocyte and NK cell responses, resulted in an antitumor effect, highlighting this bioactive compound as a promising adjuvant in CRC immunotherapy.
Keywords:
human peripheral blood mononuclear cells
; Propyl Propane Thiosulfonate
; colonospheres
; myeloid-derived suppressor cells
; tumor microenvironment
; immunomodulation
; colorectal cancer
; cancer stem cells
1. Introduction
Colorectal cancer (CRC) remains a major global health concern, ranking as the third most commonly diagnosed malignancy and the second leading cause of cancer-related mortality worldwide [1,2]. Despite therapeutic advancements, the five-year survival rate for patients with advanced-stage CRC remains dismally low, at less than 13% [3]. Immunotherapy, which harnesses and enhances the effect of the immune system against tumor cells, has achieved significant progress through the emergence of immune checkpoint inhibitors (ICIs), CAR-T therapy and the research of new experimental strategies based on therapeutic vaccines, as well as the development of new strategies that modulate the tumor microenvironment (TME) to inhibit those immunosuppressive signals that prevent the immune system from attacking tumor cells [4]. This last strategy involves the modulation of various components of the TME, including immune cells, stromal cells and immunosuppressive signaling molecules. In this scenario, the expansion of immunosuppressive cells is a key mechanism by which tumor cells evade immune destruction. Among these, regulatory T cells, MDSCs and type 2 tumor-associated macrophages (TAM) play a pivotal role. These cell populations exert a strong inhibitory influence on both innate and adaptive anticancer immune responses, thus representing major impediments to actual therapies against cancer, particularly immune-based interventions [5]. Among these cell types, the infiltration of MDSCs into the TME has been linked to chronic inflammation, tumor progression and the suppression of antitumor adaptive immune response. This immunosuppressive activity is mediated through the release of mediators such as arginase-1 (ARG1), reactive oxygen and nitrogen species and inducible nitric oxide synthase (iNOS) [6,7]. Therefore, research into new molecules capable of inhibiting these immunosuppressive components would offer great potential for the treatment of different subtypes of CRC, especially those with high CSCs-immune suppression. Additionally, the development of novel therapeutic strategies that enhance immune cell infiltration into the TME may also benefit CRC subtypes with impaired immune function or low baseline immune infiltration.
Phytochemicals, which are natural bioactive compounds produced by plants, have demonstrated a broad range of biological properties, including antioxidant, anti-inflammatory, and immunomodulatory effects. These compounds can modulate both systemic immunity and gut microbiota composition, thus offering a multifaceted approach for cancer prevention and treatment [8]. Among them, organosulfur compounds derived from Allium species, such as allicin, propyl-propane thiosulfonate (PTSO) and propyl-propane thiosulfinate (PTS), have shown particular promise in preclinical models of gastrointestinal disorders, including CRC [9,10,11,12]. In this scenario, the immunomodulatory effects of these Allium-derived compounds have been reported to exert immunoregulatory effects by reducing inflammatory mediators, modulating immune cell profiles and potentially restoring antitumor immune surveillance [13,14,15]. In this context, the present study aims to evaluate the immunomodulatory properties of PTSO, a bioactive organosulfur compound derived from Allium cepa, and its capacity to modulate immune cell populations and influence tumor progression in CRC. Thus, this study employs an integrated experimental approach combining in vitro immune assays, 3D CSCs-based colonosphere models and an immune-humanized CRC mouse model to investigate the capacity of PTSO to modulate the TME and potentiate antitumor immune responses. Additionally, the study also explores the translational potential of dietary organosulfur compounds as adjuvants in CRC immunotherapy.
2. Materials and Methods
2.1. Reagents and Treatment
PTSO was obtained from Allium cepa as previously reported [16] and provided by DOMCA S.A.U. at a purity of 89.6 %. PTSO was initially dissolved in dimethyl sulfoxide (DMSO) and further diluted in Dulbecco’s Modified Eagle Medium (DMEM) to obtain the final working concentrations. The selection of PTSO concentrations was based on prior in vitro studies [10,17]. Unless otherwise indicated, all reagents were obtained from Sigma-Aldrich (Madrid, Spain).
2.2. Generating CSCs-Enriched Colonospheres
The human colorectal carcinoma cell line HCT116 was used to generate CSCs-enriched colonospheres according to the patented protocol WO2016020572A1 [18]. Cells were obtained from the Center for Scientific Instrumentation (University of Granada, Spain) and cultured in serum-free DMEM/F-12 medium supplemented with 1× B-27 supplement minus vitamin A (Invitrogen, Waltham, MA, USA), 4 ng/mL heparin, 10 µg/mL insulin (Insulin-Transferrin-Selenium, Invitrogen), 1 µg/mL hydrocortisone, 10 ng/mL each of epidermal growth factor, fibroblast growth factor, interleukin-6, and hepatocyte growth factor (Miltenyi Biotec, Bergisch Gladbach, Germany; Auburn, CA, USA). Primary colonospheres were cultured in ultra-low attachment plates (Corning® Costar®) for 72 h. Subsequently, they were washed with PBS, disaggregated using TrypLE™ enzyme (Thermo Fisher Scientific Inc., Waltham, MA, USA) at 37°C for 5 min, and neutralized with FBS-containing medium. After washing, single-cell suspensions were seeded in fresh ultra-low attachment plates with the same conditioned medium to generate secondary CSCs-enriched colonospheres.
2.3. Isolation and Culture of Human PBMCs
Human peripheral blood mononuclear cells (hPBMCs) were isolated from healthy donors following written informed consent. Cells were obtained via density gradient centrifugation using Lymphoprep™ (StemCell Technologies) and cultured in RPMI 1640 (Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamin, 1% penicillin-streptomycin and 1% amphotericin B. hPBMCs were treated with vehicle (DMEM) or 25 μM for 48 h at 37ºC in a 5% CO2 incubator. Following treatment, cells were used for flow cytometry or in vivo experiments.
2.4. Immunophenotyping of PBMCs
PTSO- or vehicle-treated hPBMCs (n = 6 per group), both adherent and non-adherent, were collected and stained with viability dyes (Zombie Aqua™, BioLegend or eFluor™ 780, Invitrogen) and fluorochrome-conjugated antibodies (Table S1). Labelled cells were acquired using a BD FACSymphonyTM A5 SORP (BD Biosciences) flow cytometer of Center for Scientific Instrumentation (University of Granada, Granada, Spain) and analyzed with FlowJo software (v. 10.7.2).
2.5. Immune-Humanized Tumor-Bearing Murine Model
Animal procedures were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and were approved by the Ethics Committee (Ref. No.07/12/2019/127). NOD scid gamma (NSG) mice were obtained from the Center for Scientific Instrumentation (University of Granada, Spain) and maintained under controlled environmental conditions (22 ± 1 °C, 55 ± 10% humidity, 12 h light/dark cycle) with ad libitum access to standard chow (Harlan Laboratories) and water. Animal welfare was continuously monitored by qualified staff to ensure the well-being of the animals throughout all experimental procedures. ARRIVE guidelines were followed throughout the study. Mice (n = 9 per group, based on previous studies) were randomized into control (vehicle-treated hPBMCs) and PTSO (PTSO-treated hPBMCs) groups. Each animal received 1 × 10⁶ hPBMCs via tail vein injection. After three weeks, HCT116 CSCs-derived secondary colonosphere (60,000 cells/mouse) were subcutaneously inoculated in the right flank in a 1:1 mix of Matrigel® (Cat.# 356231) and DMEM High Glucose without phenol red. A second intravenous injection of hPBMCs (1 × 10⁶ cells/mouse) was administered the same day (Figure S1). Tumor growth was assessed twice weekly using a caliper and the tumor volume was calculated by the following formula (1):
V = length2 x width x π/6
The tumor size was controlled to not exceed the maximum permitted by the ethic committee, 3 cm3. At the end of the assay, animal Magnetic Resonance Imaging (MRI) was performed to evaluate tumor size and assess potential metastasis. MRI scan was conducted at the Center for Scientific Instrumentation (University of Granada, Granada, Spain) using a BioSpec 70/20 USR MRI System (Bruker, Billerica, MA, USA). For this purpose, mice (n=3) were anesthetized with isoflurane and scanned. Images were taken and high-intensity regions corresponding to tumor tissue were considered.
After mice sacrifice, tumors were excised and weighed. Blood and tumor samples were taken and stored for further gene expression, flow cytometry and histological analysis.
2.6. Immune Profiling of Blood and Tumor Samples
At the endpoint of the in vivo assay, tumors and blood samples were harvested for immune profiling. Blood was collected via cardiac puncture and subjected to red blood cell lysis. Tumor tissues were cut into small pieces and enzymatically digested in 5 mL of HBSS (40-50 mg tissue/mL) containing 400 U/mL DNase, 500 U/mL collagenase type IV and 0.09 U/mL dispase II (Roche Applied System). Digestion was incubated for 30 minutes at 37ºC with agitation, then washed twice with cold PBS containing 2 mM EDTA and filtered through a 70-μm nylon mesh. Single cell suspensions from blood were stained with anti-C45, anti-CD193, anti-CD14 and anti-CD64 (Table S2); and single cell suspensions from tumors were stained with anti-C45, anti-CD193, anti-CD14 and anti-CD64, anti-CD3, anti-CD4 and anti-CD56 (Table S2). Labelled cells were acquired on a BD FACSymphonyTM A5 SORP (BD Biosciences) flow cytometer of Center for Scientific Instrumentation (University of Granada, Granada, Spain) and analyzed using FlowJo software (v. 10.7.2).
2.7. RNA Extraction and Analysis of Gene Expression
Total RNA from tumor tissues was extracted using the Maxwell® 16 total RNA Purification Kit (Promega, Madison, WI, USA) (Cat.# AS1340) with DNase treatment, according to the manufacturer’s recommendations. Then, RNA was reverse transcribed into cDNA, which was used for qPCR using an EcoTM Real-Time PCR system (Illumina, San Diego, CA, USA). Gene expression was normalized to GAPDH levels using the 2–ΔΔCt method and all samples were measured in triplicate. The specific primer sequences are presented in Supplementary Table S2.
2.8. Histological Studies
Tumor sections were fixed in 4% PFA for 24 h and embedded in paraffin for haematoxylin and eosin (H&E) staining. For immunofluorescence analysis, antigen retrieval was performed prior to incubation with an anti-KI67 primary antibody (Abcam®, Cat.# AB15580 1:500 dilution in blocking buffer) overnight at 4 °C. Subsequently, sections were incubated with a goat anti-rabbit conjugated with AlexaFluor 488 (Abcam®, Cat.# AB170057, 1:1000 dilution in PBS).
Images were acquired from three random fields per slide using a light microscope, for H&E-stained sections, or a fluorescence microscope, for KI67 stained sections. Images were captured and analyzed with ImageJ Software, which was used to count KI67 positive nuclei and quantify the fluorescence intensity of the labeling with anti-KI67 antibody.
2.9. Statistical Analysis
Statistical analysis was conducted using the GraphPad Prism version 8 software (GraphPad Software, Inc, San Diego, CA, USA). All experimental animals were retained for analysis; no exclusion criteria were applied. Data are presented as mean ± SEM from at least three independent experiments or biological replicates unless otherwise specified in the figure legends. For multiple group comparisons, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was applied. A p-value < 0.05 was considered statistically significant.
3. Results
3.1. PTSO Reduces the Levels of Immunosuppressive Myeloid Populations In Vitro
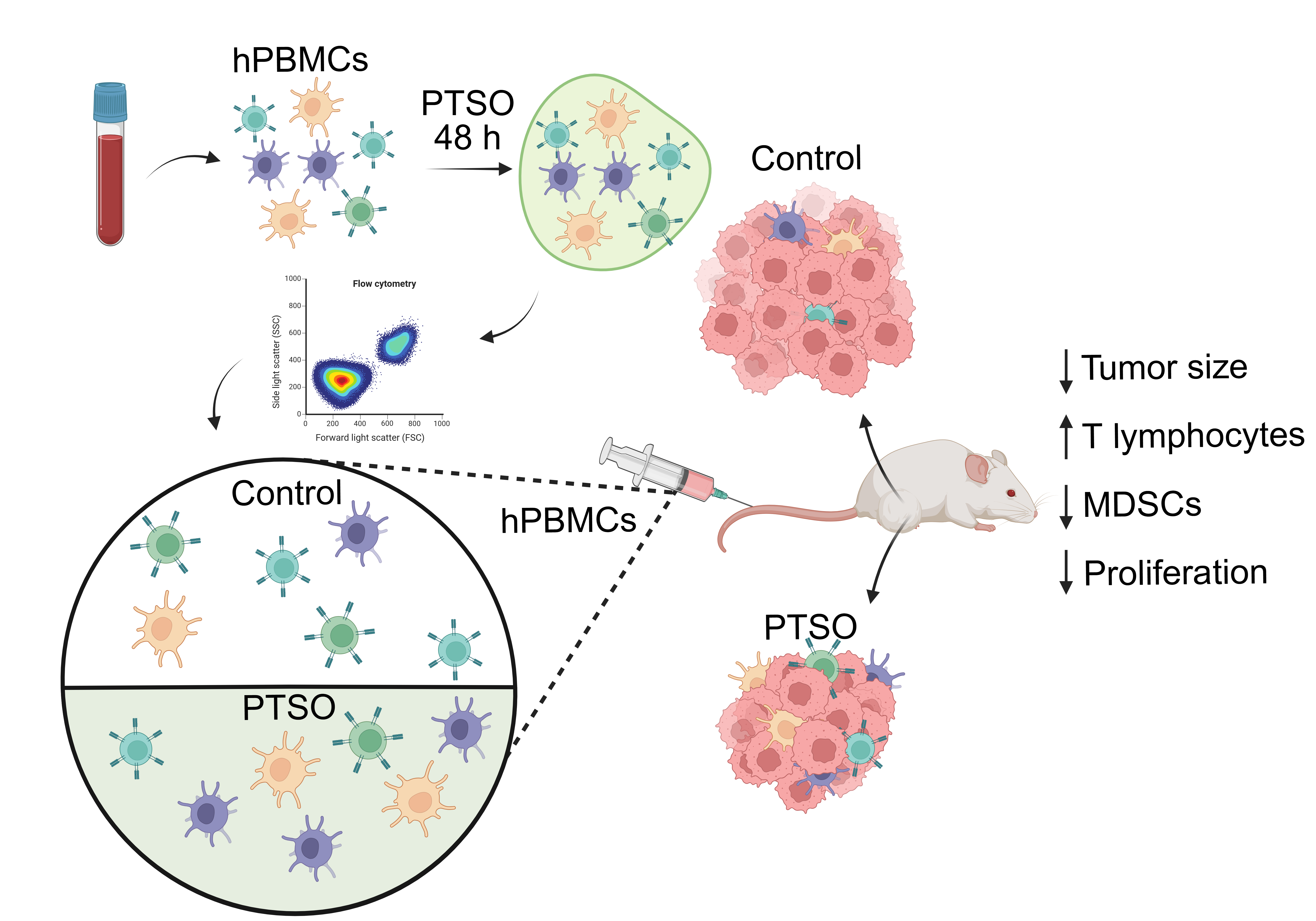
To investigate the immunomodulatory potential of PTSO, we assessed its effects on myeloid subsets within hPBMCs via flow cytometry. The immunomodulatory role of this compound resulted in a significant reduction of the percentage of total CD11b+ myeloid cells compared to control-treated hPBMCs (Figure 1A). Notably, this decrease was predominantly driven by a marked reduction in the CD11b+CD14+ monocyte population.
Next, the effect of PTSO on MDSCs, a heterogeneous group of immunosuppressive cells broadly classified into monocytic (M-MDSC, CD14+HLA-DRlow/–) and granulocytic or polymorphonuclear (PMN-MDSC, SSChighCD14⁻) subtypes, was also evaluated. These populations are known contributors to tumor immune evasion and progression [19]. As shown in Figure 1B, PTSO significantly reduced the proportion of both M-MDSCs and PMN-MDSCs. Within the PMN-MDSC subset, reductions were observed in both CD16+CD11b+ and CD16+CD11b- populations. These findings indicate that PTSO can exert an immunomodulatory effect by selectively depleting immunosuppressive myeloid populations in vitro, which could impact positively on adaptive immune response.
3.2. PTSO Promotes Cytotoxic T Cell Expansion In Vitro
The impact of PTSO on T lymphocyte subsets was also evaluated in vitro in the PBMC populations (Figure 2). As shown in Figure 2A, PTSO treatment did not significantly alter the proportion of total CD3+CD4+ helper T (Th) cells. However, there was a trend toward an increased frequency of Th1 cells (CD4+IFNG+), suggesting a shift toward a more proinflammatory phenotype. More importantly, PTSO significantly increased the frequency of cytotoxic CD3+CD8+ T (Tc) cells, accompanied by a notable rise in IFNG-producing Tc1 cells (CD8+IFNG+), indicating a skewing towards an effector antitumor phenotype (Figure 2A and B). These findings point to a potential role for PTSO in enhancing T cell-mediated immunity by fostering cytotoxic T lymphocyte expansion.
3.3. Immunomodulatory Activity of PTSO Impairs Tumor Growth in a CRC Xenograft Mouse Model
To translate these in vitro findings, we investigated the impact of PTSO-modulated hPBMCs on colorectal tumor development in a xenograft mouse model using HCT116 CSC-enriched colonospheres. Importantly, the tumors of mice that received PTSO-treated hPBMCs showed a smaller volume from day 14 post-injection, being this difference statistically significant from day 20 until the end of the assay (Figure 3A). On the day of sacrifice MRI analysis confirmed these observations, thus revealing a marked reduction in tumor size in mice treated with PTSO-pretreated hPBMCs (Figure 3B). Conversely, the control group exhibited consistently larger volumes with more extensive tumor expansion into adjacent tissue (Figure 3B). Moreover, the greater heterogeneity in MRI signals observed in control mice indicates increased variability in tumor composition, which may be associated with hypoxia and necrotic areas (Figure 3B). Postmortem analysis corroborated these results: mice treated with PTSO-modulated hPBMCs developed significantly lighter and smaller tumors (Figure 3C and D). Moreover, histological analysis of H&E-stained tumor sections reported notable differences between both groups (Figure 3E). In this scenario, tumors from those mice treated with control hPBMCs displayed a higher cellular density and structural integrity. Tumors from control mice exhibited dense cellularity and extensive local invasion, with muscle tissue embedded within the tumor mass (Figure 3E). Conversely, those mice administered with PTSO-pretreated hPBMCs showed prominent necrotic areas associated with loss of tumor integrity. Importantly, no evidence of adjacent tissue invasion was observed in this group (Figure 3E).
3.4. PTSO Modulates Tumor Cell Proliferation and Invasive Potential
To evaluate whether the impact on tumor growth observed in mice receiving PTSO-pretreated hPBMCs was associated with a reduction in tumor cell aggressiveness, tumor sections were analyzed for markers of proliferation and invasion. Immunofluorescence staining of KI67 revealed a significant decrease in the number of KI67-positive nuclei in tumors from the PTSO group, indicating reduced tumor cell proliferation. This was further supported by a decrease in the mean fluorescence intensity (MFI), confirming a diminished proliferative index in these tumors. Additionally, gene expression analysis carried out in tumors from the two experimental groups revealed a significant downregulation of the proliferation and invasion marker WNT5A in mice receiving PTSO-pretreated hPBMCs (Figure 4A). Importantly, this was accompanied by reduced expression of MMP9, a matrix metalloproteinase closely associated with tumor invasion and metastasis (Figure 4B). These findings suggest that the immunomodulatory effect of PTSO on hPBMCs contributes to dampening tumor cell proliferation and invasiveness in vivo.
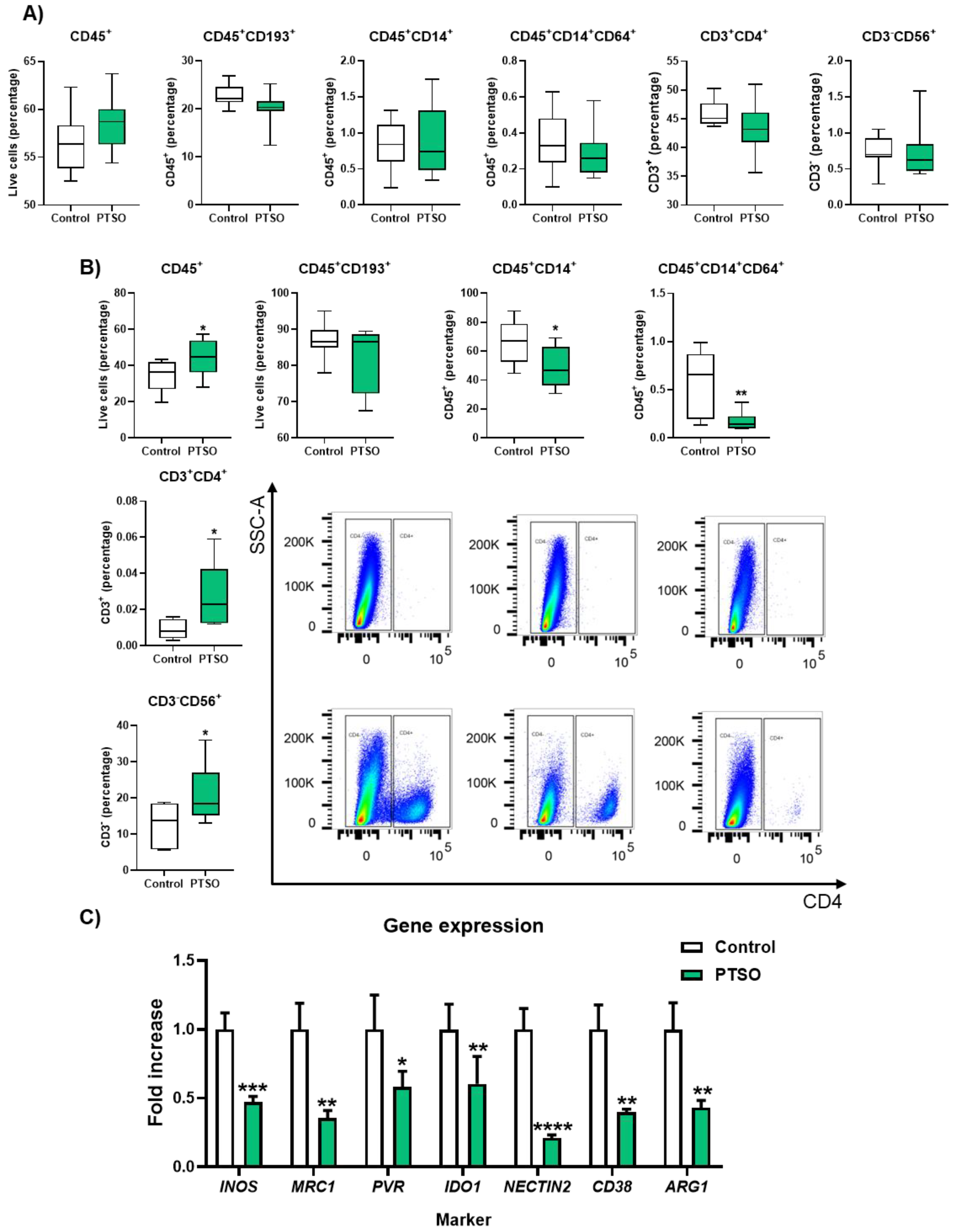
3.5. PTSO-Treated hPBMCs Enhance Tumor Immune Infiltration and Deplete the Intratumoral Myeloid Suppressor Cells
To further dissect the immune mechanisms underlying tumor control by PTSO treatment the presence of different immune cell populations was analyzed both in blood and in tumor tissues of xenografted mice treated with hPBMCs. Flow cytometry analysis of blood samples revealed no significant differences between both groups in the levels of total immune cells (CD45+), CD45+CD193+ granulocytes, neither in the general CD45+CD14+ monocytes nor particularly in those CD45+CD14+CD64+ subset (Figure 5A). Moreover, no significant changes were found in T cells (CD45+CD3+) and NK cells (CD3-CD56+) levels when compared the two experimental groups (Figure 5A). Conversely, the analysis of immune cell infiltration into the tumor of both groups of mice revealed significant differences in the number of CD45+ cells, being this higher in those animals administered with PTSO-treated hPBMCs compared to control (44.5 ± 3.13 vs. 34.2 ± 2.91), indicating an increased immune cell infiltration into the tumor (Figure 5B). Mice that received PTSO-treated hPBMCs exhibited an increased infiltration of total CD45+ immune cells within the tumor that was associated with a significant enrichment of CD3+CD4+ Th cells and CD3-CD56+ NK cells (Figure 5B), thus suggesting enhanced antitumor immune activity within the TME. Moreover, a significant reduction in tumor-infiltrating CD14+ monocytic cells was observed in the group treated with PTSO-modulated hPBMCs compared to the control group (Figure 5B). This depletion of monocytic myeloid cells in tumors was correlated with a marked downregulation in the expression of key immunosuppressive genes including MRC1, NECTIN2 and others typically associated with MDSCs such as ARG1, NOS2, IDO1 and CD38 (Figure 5C). These findings demonstrate that PTSO-pretreated hPBMCs not only enhance immune infiltration into the tumor but also reprogram the immune landscape by reducing immunosuppressive myeloid populations, ultimately fostering a more immunostimulatory TME.
4. Discussion
Immunotherapy has emerged as a transformative approach in cancer treatment, with immune checkpoint inhibitors (ICIs) demonstrating significant clinical benefit in various malignancies [20]. However, their efficacy is notably limited in CRC cases characterized by microsatellite stability (MSS) or proficient mismatch repair (pMMR), where the TME is typically immunologically “cold” due to low immune infiltration [21]. In these contexts, combination strategies employing immunomodulators alongside ICIs are gaining interest as promising therapeutic approaches [22].
This study demonstrates, for the first time, that PTSO, a bioactive organosulfur compound derived from Allium cepa, exerts significant antitumor effects through its immunomodulatory activity. It achieves this by suppressing immunosuppressive signals and promoting the proliferation and infiltration of immune cells with antitumor capabilities, such as T lymphocytes and NK cells. PTSO reduced immunosuppressive myeloid populations, particularly MDSCs, including both M-MDSCs and PMN-MDSCs subsets, which are known to hinder antitumor immunity. This is of particular relevance as depletion or functional reprogramming of MDSCs has been shown to enhance effector T cell infiltration and suppress tumor growth in preclinical and clinical settings [23,24,25]. These findings are consistent with a previous study published by Fujiwara Y. et al. (2016), which demonstrated that another bioactive compound derived from Allium cepa, Onionin A, inhibited the immunosuppressive activity of MDSCs, thereby reducing tumor growth and lung metastasis in a subcutaneous tumor model [26].
High infiltration of MDSCs within tumors has been consistently associated with poor clinical prognosis, due to their potent suppressive effects on effector lymphocytes and NK cells [23,27]. MDSCs exert their immunosuppressive function primarily through the secretion of mediators such as ARG1 and iNOS, which inhibit effector cell activity within the TME. ARG1 depletes arginine, thereby restricting T cell proliferation and activation [28], while nitric oxide produced by iNOS impairs T cell function through nitration of the T-cell receptor and CD8 molecules [29]. Notably, PTSO treatment reduced the in vivo expression of these enzymes, suggesting an attenuation of MDSC-mediated immunosuppression. In CRC, MDSCs not only suppress antitumor immunity but also actively promote CSCs characteristics. Granulocytic MDSCs enhance CRC cell pluripotency by transferring exosomal S100A9 proteins to tumor cells. This promotes CSC formation by upregulating markers such as CD44 and CD133, especially under hypoxic conditions within the TME [30]. Furthermore, MDSCs in CRC are strongly influenced by IL-6 driven STAT3 marker activation. Autocrine IL-6 maintains MDSC survival through the STAT3-DNMT epigenetic axis, helping them evade necroptosis and accumulate in the TME [31]. Moreover, tumor cells can induce MDSC expansion in CRC, by secreting cytokines and other factors such as VEGF, iNOS and ARG1, which reinforces immunosuppressive feedback loops.
CD38 has also been described as a pivotal marker in the MDSCs biology by contributing to their maturation. Moreover, high expression of CD38 in MDSCs has been associated with a higher immunosuppressive capacity that results in a greater ability to suppress activated T cells and promote tumor growth in comparison with CD38low MDSCs [32]. Importantly, PTSO treatment resulted in a reduction of the expression of this marker in the tumors of mice that received the pretreated hPBMCs, thus contributing to the alleviation of the immunosuppressive microenvironment and the promotion of T cell activity against tumor cells. This is noteworthy because there is currently no description in the literature of any bioactive compound derived from Allium that has the ability to reduce the levels of this marker.
Indoleamine 2,3-dioxygenase (IDO) is an enzyme that catalyzes the degradation of tryptophan into N-formyl kynurenine and plays a critical role in mediating immunosuppression within the TME. This immunosuppressive effect is largely attributed to the capacity of IDO to promote an environment conducive to tumor growth by recruiting and activating MDSCs, which in turn contribute to resistance against T cell-targeted immunotherapies [33]. Specifically, IDO suppresses effector T cell activation by depleting tryptophan and increasing kynurenine production, a process that facilitates the induction and expansion of regulatory T cells (Tregs), further reinforcing the immunosuppressive milieu and favoring tumor progression [33]. Notably, in this study, IDO1 expression was significantly reduced in tumors from mice treated with PTSO-pretreated hPBMCs, suggesting that this organosulfur compound may mitigate immune suppression and enhance antitumor immune responses, ultimately promoting tumor immunogenic cell death [22]. These results align with previous in vitro findings reporting that Allium-derived compounds can downregulate IDO expression in vitro [34].
This study also demonstrates the ability of PTSO to attenuate immunosuppressive activity within the TME by downregulating key markers such as MRC1, PVR and NECTIN2. TAMs are known to express the mannose receptor C-type 1 (MRC1, also referred to as CD206) on their surface, a receptor implicated in promoting tumor immunosuppression, angiogenesis, metastasis, and relapse [35]. The PTSO-induced downregulation of MRC1 may therefore enhance tumor immunogenicity, contributing to the observed antitumor effects in treated mice. Additionally, tumor-associated neutrophils (TANs), another abundant TME component involved in tumor progression, have been linked to the upregulation of NECTIN2. NECTIN2 expression has been reported to inhibit CD8+ T-cell infiltration and diminish cytotoxic T cell activity within the TME [36]. Consistently, administration of PTSO-pretreated hPBMCs resulted in reduced NECTIN2 gene expression in tumors, which may have facilitated the increased infiltration of antitumor T cells observed in treated mice relative to controls.
The poliovirus receptor (PVR) or CD155, also plays an important role in CRC progression and immune evasion. In fact, the expression of this receptor has been found to be increased in almost 90 % of CRC patients and has been strongly associated with enhanced tumor cell proliferation, migration, invasion and metastasis. Within the TME, PVR facilitates immune escape through its interaction with the TIGIT receptor on T and NK cells, thereby suppressing their cytotoxic functions. These properties make PVR an attractive immunotherapeutic target in CRC [37]. In this study, mice receiving PTSO-pretreated hPBMCs exhibited significantly reduced PVR gene expression within the tumor, suggesting that PTSO may contribute to tumor immunogenicity by reversing immune suppression mediated by this pathway.
The immunomodulatory capacity of PTSO, demonstrated both in vitro and in vivo, was also linked to a notable increase in antitumor effector immune cells, particularly CD4+ and CD8+ T lymphocytes and NK cells. Importantly, characterization of CD4+ and CD8+ T cell subpopulations in vitro showed that PTSO was able to increase those producing IFNG, thus resulting in an increase in Th1 and Tc1 subsets, which are reported to be linked to a reduction in tumor progression in CRC [38,39]. This shift toward an immune profile rich in antitumor T lymphocytes observed in vitro is important because these cells are the main effectors in mediating tumor cell killing, and their enhanced frequency can directly translate into improved antitumor immunity [40]. This immunomodulatory activity was also demonstrated in vivo, since the administration of PTSO-pretreated hPBMCs led to an increase in the tumor infiltration of CD4+ T lymphocytes and NK cells, which was associated with a reduction in CD14+ monocytic cells and the downregulation of key immunosuppressive genes, as previously discussed. These findings align with the cumulative evidence suggesting that enhanced Th1, CD8+ and NK cells infiltration into tumors is correlated with better prognosis and improved responsiveness to immunotherapies in CRC [41,42]. Extracts derived from Allium sp. have also been reported to exert an immunomodulatory role that results in an antitumor effect derived from an increase in the infiltration of CD4+ and CD8+ lymphocytes in subcutaneous tumors induced in vivo, thus supporting the findings obtained in the present study [26,43].
The observed increase in T and NK cell tumor infiltration further suggests that PTSO may influence the expression of adhesion molecules and chemokines that guide immune cell homing to the tumor site. Immunosuppressive myeloid cells are known to impede T cell trafficking by downregulating molecules such as L-selectin; reversing these effects may help reprogram the TME to support more effective immune responses [44]. Further studies are warranted to elucidate the impact of PTSO on chemokine gradients and adhesion molecule dynamics within the TME.
Importantly, the enhanced immune infiltration induced by PTSO treatment was associated with a significant antitumor effect, as evidenced by reduced tumor volume and compromised tumor architecture in mice receiving PTSO-pretreated PBMCs. This outcome likely reflects increased immune-mediated cytotoxicity, along with impaired angiogenesis. Indeed, MDSCs are known to promote tumor vascularization by secreting pro-angiogenic factors; therefore, their depletion by PTSO may disrupt vascular support, aggravate tumor hypoxia and sensitize cancer cells to immune attack [45].
Moreover, this antitumor action observed in the PTSO groups of mice was corroborated molecularly by a reduction of cancer cell proliferation markers, specifically KI67 and WNT5A, which is a key marker of the Wnt/β-catenin signaling pathway. Importantly, activation of the Wnt signaling pathway is a hallmark of colorectal CSCs, sustaining their self-renewal capacity, pluripotency and tumor-initiating potential [46]. Among its components, WNT5A has been shown to modulate dependent pathways of β-catenin, promoting sphere-forming ability and upregulating key stemness markers, including CD44, LGR5 and ALDH1 [46,47,48,49,50]. Consequently, the downregulation of WNT5A observed in PTSO-treated mice may not only indicate suppression of proliferative signaling, but also a direct impairment of CSC maintenance, potentially lowering the risk of tumor recurrence and therapy resistance. Additionally, these beneficial effects resulted in a reduction of the invasive capacity of tumor cells, which was manifested in the histological analysis of the control group tumor sections, with a greater infiltration of tumor cells into adjacent tissues such as muscle tissue compared to those mice in the group administered hPBMCs pretreated with PTSO. Moreover, this effect was correlated with a significant reduction in molecular markers involved in tumor invasion, especially MMP9. It has been reported that MDSCs can promote the invasive capacity of cancer cells via MMP9, thus enabling these cells to move from the primary tumor to metastatic sites [51]. In addition, single-cell sequencing experiments have reported that an increased MMP9 expression is correlated with decreased infiltration of cytotoxic CD8+ T cells into the tumor, as well as increased anti-PD-1 resistance in MSI-H/dMMR CRC patients [52]. Therefore, the immunomodulatory effect of PTSO may positively impact the expression of these proliferation and invasion markers, thus resulting in the beneficial effects observed against preclinical CRC.
Taken together, our findings provide evidence for considering PTSO as a novel immunomodulatory agent capable of reducing immunosuppressive myeloid populations while boosting T and NK cell-mediated antitumor responses, ultimately resulting in reduced tumor burden in a CRC xenograft model. By reprogramming the TME from a suppressive to an immunologically active state, PTSO holds promise as a complementary agent to improve the efficacy of immunotherapies such as ICIs and adoptive T cell therapies, which are frequently limited by the presence of MDSCs and dysfunctional T cell activity.
Regarding translational implications, these results align with clinical data supporting the targeting of myeloid cell populations in CRC as a strategy to improve patient outcomes [53]. Patients with lower circulating MDSC levels and greater intratumoral T cell infiltration tend to respond more favorably to immunotherapies and exhibit longer survival [54]. In this context, the ability of PTSO to modulate both arms of this immune axis suggests its potential utility as an adjunct to current immunotherapeutic regimens. Nevertheless, certain limitations of this study should be acknowledged. The experimental data are derived from in vitro models and xenograft systems, which, although valuable, do not fully capture the complexity of human tumor-immune interactions. The heterogeneity of the TME in patients receiving immunotherapy involves intricate crosstalk between immune, stromal and tumor cells that cannot be entirely replicated in preclinical models. Furthermore, while PTSO has shown promising immunomodulatory activity and its safety profile has been evaluated in vivo [55], additional studies are needed to validate its efficacy in clinically relevant settings. Future research should incorporate more advanced models, such as patient-derived organoids or ex vivo systems incorporating TME components, to better predict clinical outcomes. Another key consideration is the phenotypic and functional heterogeneity within the myeloid compartment [56]. While this study primarily focused on major myeloid subsets, high-resolution techniques such as single-cell RNA sequencing or mass cytometry could offer deeper insights into the specific immune subpopulations modulated by PTSO in the TME. Despite these limitations, the data presented here support the development of PTSO as a promising immunotherapeutic adjuvant capable of enhancing antitumor immunity and reducing tumor progression in CRC.
5. Conclusions
The present study highlights the immunomodulatory potential of PTSO, evidenced by its ability to reduce immunosuppressive myeloid populations and promote the differentiation of T lymphocytes towards effector phenotypes. These immune changes collectively contribute to a decrease in tumor growth, reduction of cancer cell proliferation and invasiveness. Altogether, these findings position PTSO as a promising nutritional adjuvant to improve the efficacy of immunotherapy in CRC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: In vivo model tumor xenograft treated with modified by PTSO-hPBMCs.; Table S1: Flow cytometry antibodies; Table S2: qPCR primers sequences.
Author Contributions
Conceptualization, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., A.J.R.M., J.G., M.E.R.C. and A.R.N.; methodology, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., J.A.M.T., A.H.P, T.V., L.L.E., C.G.L., M.J.R.S. (María José Rodríguez Sánchez) and A.J.R.M.; software, , M.J.R.S. (María Jesús Rodríguez Sojo) and J.G.G. ; validation, M.J.R.S. (María Jesús Rodríguez Sojo), A.J.R.M., A.B., J.A.M., J.G., M.E.R.C. and A.R.N.; formal analysis, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., A.J.R.M. and A.R.N.; investigation, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., A.J.R.M., J.G. and A.R.N.; resources, A.B., J.A.M., J.G., M.E.R.C. and A.R.N.; data curation, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., A.J.R.M., J.G. and A.R.N.; writing—original draft preparation, M.J.R.S. (María Jesús Rodríguez Sojo), A.J.R.M., J.G. and A.R.N.; writing—review and editing, A.B., J.A.M., J.G., M.E.R.C. and A.R.N.; visualization, M.J.R.S. (María Jesús Rodríguez Sojo), L.G., A.J.R.M., J.G., M.E.R.C. and A.R.N.; supervision, J.G., M.E.R.C. and A.R.N.; project administration, A.B., J.A.M., J.G., M.E.R.C. and A.R.N.; funding acquisition, A.B., J.A.M., J.G., M.E.R.C. and A.R.N. All authors have read and agreed to the published version of the manuscript.
Funding
The manuscript has been funded by the Junta de Andalucia (CTS164) (Spain) and Fondo Europeo de Desarrollo Regional (FEDER), from the European Union, through the CIBER-EHD and the research grants PY20-01157, B-CTS-664-UGR20, P18-RT-4930, predoctoral grant IFI21/00030 to MJRS, postdoctoral grant CD23/00234 to JGG, postdoctoral grant CD23/00089 to AHP, and POSTDOC_21_638 to CGL. This paper was also supported by Instituto de Salud Carlos III (ISCIII) (Spain) through the projects PI19/01058, PI20/01447 and PI22/0163 and cofounded by the European Union and by the Chair “Doctors Galera-Requena in cancer stem cell research” (CMC-CTS963 to JAM). Moreover, the University of Granada Library provided financial support for the publication of this study.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Laboratory Animals of the University of Granada (Spain) (Ref. No. 07/12/2019/127).
Informed Consent Statement
After an explanation of all procedures, risks, and benefits was provided, each participant provided written informed consent before participating.
Data Availability Statement
Data will be available to be shared upon publication by correspondence with either Alba Rodriguez Nogales (albarn@ugr.es) or Julio Galvez (jgalvez@ugr.es), after approval of a proposal, with a signed access agreement, and relevant ethics consent.
Conflicts of Interest
The authors declare that they do not have any competing interests.
Abbreviations
The following abbreviations are used in this manuscript:
| ARG1 | Arginase-1 |
| CRC | Colorectal Cancer |
| CSCs | Cancer stem cells |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| hPBMCs | Human peripheral blood mononuclear cells |
| ICIs | Immune checkpoint inhibitors (ICIs) |
| IFNG | Interferon gamma |
| iNOS | Inducible nitric oxide synthase |
| MDSCs | Myeloid-derived suppressor cells |
| MFI | Mean fluorescence intensity |
| M-MDSCs | Monocytic myeloid-derived suppressor cells |
| MRI | Magnetic Resonance Imaging |
| MSS | Microsatellite stability |
| NK | Natural Killer |
| NSG | NOD scid gamma |
| pMMR | Proficient mismatch repair |
| PMN-MDSCs | Polymorphonuclear Myeloid-derived suppressor cells |
| PTSO | Propyl Propane Thiosulfonate |
| Tc | T cytotoxic |
| Th | T helper |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J Clin 2023, 73, 233-254. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209-249. [CrossRef]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020, 70, 145-164. [CrossRef]
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci 2021, 17, 3837-3849. [CrossRef]
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. Oncoimmunology 2022, 11, 2120676. [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012, 12, 253-268. [CrossRef]
- Allegrezza, M.J.; Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Perales-Puchalt, A.; Nguyen, J.M.; Payne, K.K.; Singhal, S.; Eruslanov, E.B.; Tchou, J.; et al. Trametinib Drives T-cell-Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Res 2016, 76, 6253-6265. [CrossRef]
- De, S.; Paul, S.; Manna, A.; Majumder, C.; Pal, K.; Casarcia, N.; Mondal, A.; Banerjee, S.; Nelson, V.K.; Ghosh, S.; et al. Phenolic Phytochemicals for Prevention and Treatment of Colorectal Cancer: A Critical Evaluation of In Vivo Studies. Cancers (Basel) 2023, 15. [CrossRef]
- Elattar, M.M.; Darwish, R.S.; Hammoda, H.M.; Dawood, H.M. An ethnopharmacological, phytochemical, and pharmacological overview of onion (Allium cepa L.). J Ethnopharmacol 2024, 324, 117779. [CrossRef]
- Guillamon, E.; Mut-Salud, N.; Rodriguez-Sojo, M.J.; Ruiz-Malagon, A.J.; Cuberos-Escobar, A.; Martinez-Ferez, A.; Rodriguez-Nogales, A.; Galvez, J.; Banos, A. In Vitro Antitumor and Anti-Inflammatory Activities of Allium-Derived Compounds Propyl Propane Thiosulfonate (PTSO) and Propyl Propane Thiosulfinate (PTS). Nutrients 2023, 15. [CrossRef]
- Zhu, L.; Myhill, L.J.; Andersen-Civil, A.I.S.; Thamsborg, S.M.; Blanchard, A.; Williams, A.R. Garlic-Derived Organosulfur Compounds Regulate Metabolic and Immune Pathways in Macrophages and Attenuate Intestinal Inflammation in Mice. Mol Nutr Food Res 2022, 66, e2101004. [CrossRef]
- Schafer, G.; Kaschula, C.H. The immunomodulation and anti-inflammatory effects of garlic organosulfur compounds in cancer chemoprevention. Anticancer Agents Med Chem 2014, 14, 233-240. [CrossRef]
- Amani, M.; Shokati, E.; Entezami, K.; Khorrami, S.; Jazayeri, M.H.; Safari, E. The immunomodulatory effects of low molecular weight garlic protein in crosstalk between peripheral blood mononuclear cells and colon cancer cells. Process Biochemistry 2021, 108, 161-168. [CrossRef]
- Xu, J.; Yu, Y.; Zhang, Y.; Dai, H.; Yang, Q.; Wang, B.; Ma, Q.; Chen, Y.; Xu, F.; Shi, X.; et al. Oral administration of garlic-derived nanoparticles improves cancer immunotherapy by inducing intestinal IFNgamma-producing gammadelta T cells. Nat Nanotechnol 2024, 19, 1569-1578. [CrossRef]
- Zhang, R.J.; Rao, Q.R.; Jiang, X.Q.; Ye, N.; Li, N.; Du, H.L.; Zhang, S.J.; Ye, H.Y.; Wu, W.S.; Zhao, M. Exploring the Immunomodulatory Properties of Red Onion (Allium cepa L.) Skin: Isolation, Structural Elucidation, and Bioactivity Study of Novel Onion Chalcones Targeting the A(2A) Adenosine Receptor. J Agric Food Chem 2023. [CrossRef]
- Falcón Piñeiro, A.; Garrido Garrido, D.; Baños Arjona, A. PTS and PTSO, two organosulfur compounds from onion by-products as a novel solution for plant disease and pest management. 2023. [CrossRef]
- Vezza, T.; Algieri, F.; Garrido-Mesa, J.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Banos, A.; Guillamon, E.; Garcia, F.; Rodriguez-Nogales, A.; Galvez, J. The Immunomodulatory Properties of Propyl-Propane Thiosulfonate Contribute to its Intestinal Anti-Inflammatory Effect in Experimental Colitis. Mol Nutr Food Res 2019, 63, e1800653. [CrossRef]
- Jimenez, G.; Hackenberg, M.; Catalina, P.; Boulaiz, H.; Grinan-Lison, C.; Garcia, M.A.; Peran, M.; Lopez-Ruiz, E.; Ramirez, A.; Morata-Tarifa, C.; et al. Mesenchymal stem cell’s secretome promotes selective enrichment of cancer stem-like cells with specific cytogenetic profile. Cancer Lett 2018, 429, 78-88. [CrossRef]
- Xia, X.; Mao, Z.; Wang, W.; Ma, J.; Tian, J.; Wang, S.; Yin, K. Netrin-1 Promotes the Immunosuppressive Activity of MDSCs in Colorectal Cancer. Cancer Immunol Res 2023, 11, 600-613. [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 2020, 20, 651-668. [CrossRef]
- Mao, Y.; Xu, Y.; Chang, J.; Chang, W.; Lv, Y.; Zheng, P.; Zhang, Z.; Li, Z.; Lin, Q.; Tang, W.; et al. The immune phenotypes and different immune escape mechanisms in colorectal cancer. Front Immunol 2022, 13, 968089. [CrossRef]
- Liang, R.; Ding, D.; Li, Y.; Lan, T.; Ryabtseva, S.; Huang, S.; Ren, J.; Huang, H.; Wei, B. HDACi combination therapy with IDO1i remodels the tumor microenvironment and boosts antitumor efficacy in colorectal cancer with microsatellite stability. J Nanobiotechnology 2024, 22, 753. [CrossRef]
- Iglesias-Escudero, M.; Arias-Gonzalez, N.; Martinez-Caceres, E. Regulatory cells and the effect of cancer immunotherapy. Mol Cancer 2023, 22, 26. [CrossRef]
- Zhai, J.; Chen, H.; Wong, C.C.; Peng, Y.; Gou, H.; Zhang, J.; Pan, Y.; Chen, D.; Lin, Y.; Wang, S.; et al. ALKBH5 Drives Immune Suppression Via Targeting AXIN2 to Promote Colorectal Cancer and Is a Target for Boosting Immunotherapy. Gastroenterology 2023, 165, 445-462. [CrossRef]
- Bao, Y.; Zhai, J.; Chen, H.; Wong, C.C.; Liang, C.; Ding, Y.; Huang, D.; Gou, H.; Chen, D.; Pan, Y.; et al. Targeting m(6)A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut 2023, 72, 1497-1509. [CrossRef]
- Fujiwara, Y.; Horlad, H.; Shiraishi, D.; Tsuboki, J.; Kudo, R.; Ikeda, T.; Nohara, T.; Takeya, M.; Komohara, Y. Onionin A, a sulfur-containing compound isolated from onions, impairs tumor development and lung metastasis by inhibiting the protumoral and immunosuppressive functions of myeloid cells. Mol Nutr Food Res 2016, 60, 2467-2480. [CrossRef]
- Limagne, E.; Euvrard, R.; Thibaudin, M.; Rebe, C.; Derangere, V.; Chevriaux, A.; Boidot, R.; Vegran, F.; Bonnefoy, N.; Vincent, J.; et al. Accumulation of MDSC and Th17 Cells in Patients with Metastatic Colorectal Cancer Predicts the Efficacy of a FOLFOX-Bevacizumab Drug Treatment Regimen. Cancer Res 2016, 76, 5241-5252. [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 2010, 70, 68-77. [CrossRef]
- Tsukumo, S.I.; Yasutomo, K. Regulation of CD8(+) T Cells and Antitumor Immunity by Notch Signaling. Front Immunol 2018, 9, 101. [CrossRef]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv Sci (Weinh) 2019, 6, 1901278. [CrossRef]
- Smith, A.D.; Lu, C.; Payne, D.; Paschall, A.V.; Klement, J.D.; Redd, P.S.; Ibrahim, M.L.; Yang, D.; Han, Q.; Liu, Z.; et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFalpha-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res 2020, 80, 3145-3156. [CrossRef]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.B.; Lee, J.S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res 2015, 75, 4074-4085. [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep 2015, 13, 412-424. [CrossRef]
- Nikoo, S.; Bozorgmehr, M.; Namdar Ahmadabad, H.; Hassan, Z.M.; Moazzeni, S.M.; Pourpak, Z.; Ghazanfari, T. The 14kDa protein molecule isolated from garlic suppresses indoleamine 2, 3-dioxygenase metabolites in mononuclear cells in vitro. Iran J Allergy Asthma Immunol 2008, 7, 203-208.
- Scodeller, P.; Simon-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Saalik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Sci Rep 2017, 7, 14655. [CrossRef]
- Luo, H.; Ikenaga, N.; Nakata, K.; Higashijima, N.; Zhong, P.; Kubo, A.; Wu, C.; Tsutsumi, C.; Shimada, Y.; Hayashi, M.; et al. Tumor-associated neutrophils upregulate Nectin2 expression, creating the immunosuppressive microenvironment in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res 2024, 43, 258. [CrossRef]
- Qian, C.J.; He, Y.S.; Guo, T.; Tao, J.; Wei, Z.Y.; Zhang, J.L.; Bao, C.; Chen, J.H. ADAR-mediated RNA editing regulates PVR immune checkpoint in colorectal cancer. Biochem Biophys Res Commun 2024, 695, 149373. [CrossRef]
- Domblides, C.; Crampton, S.; Liu, H.; Bartleson, J.M.; Nguyen, A.; Champagne, C.; Landy, E.E.; Spiker, L.; Proffitt, C.; Bhattarai, S.; et al. Human NLRC4 expression promotes cancer survival and associates with type I interferon signaling and immune infiltration. J Clin Invest 2024, 134. [CrossRef]
- de Oliveira Alves, N.; Dalmasso, G.; Nikitina, D.; Vaysse, A.; Ruez, R.; Ledoux, L.; Pedron, T.; Bergsten, E.; Boulard, O.; Autier, L.; et al. The colibactin-producing Escherichia coli alters the tumor microenvironment to immunosuppressive lipid overload facilitating colorectal cancer progression and chemoresistance. Gut Microbes 2024, 16, 2320291. [CrossRef]
- Chen, Y.; Wang, D.; Li, Y.; Qi, L.; Si, W.; Bo, Y.; Chen, X.; Ye, Z.; Fan, H.; Liu, B.; et al. Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell 2024, 42, 1268-1285 e1267. [CrossRef]
- Nersesian, S.; Schwartz, S.L.; Grantham, S.R.; MacLean, L.K.; Lee, S.N.; Pugh-Toole, M.; Boudreau, J.E. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Transl Oncol 2021, 14, 100930. [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer 2021, 124, 359-367. [CrossRef]
- Ebrahimi, M.; Mohammad Hassan, Z.; Mostafaie, A.; Zare Mehrjardi, N.; Ghazanfari, T. Purif ied Protein Fraction of Garlic Extract Modulates Cellular Immune Response against Breast Transplanted Tumors in BALB/c Mice Model. Cell J 2013, 15, 65-75.
- Park, S.Y.; Pylaeva, E.; Bhuria, V.; Gambardella, A.R.; Schiavoni, G.; Mougiakakos, D.; Kim, S.H.; Jablonska, J. Harnessing myeloid cells in cancer. Mol Cancer 2025, 24, 69. [CrossRef]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J Cell Physiol 2018, 233, 3024-3036. [CrossRef]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J Cell Physiol 2017, 232, 2008-2018. [CrossRef]
- Pothuraju, R.; Rachagani, S.; Krishn, S.R.; Chaudhary, S.; Nimmakayala, R.K.; Siddiqui, J.A.; Ganguly, K.; Lakshmanan, I.; Cox, J.L.; Mallya, K.; et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol Cancer 2020, 19, 37. [CrossRef]
- Chen, Z.; Tang, C.; Zhu, Y.; Xie, M.; He, D.; Pan, Q.; Zhang, P.; Hua, D.; Wang, T.; Jin, L.; et al. TrpC5 regulates differentiation through the Ca2+/Wnt5a signalling pathway in colorectal cancer. Clin Sci (Lond) 2017, 131, 227-237. [CrossRef]
- Fang, Y.; Xiao, X.; Wang, J.; Dasari, S.; Pepin, D.; Nephew, K.P.; Zamarin, D.; Mitra, A.K. Cancer associated fibroblasts serve as an ovarian cancer stem cell niche through noncanonical Wnt5a signaling. NPJ Precis Oncol 2024, 8, 7. [CrossRef]
- Sun, G.; Wu, L.; Sun, G.; Shi, X.; Cao, H.; Tang, W. WNT5a in Colorectal Cancer: Research Progress and Challenges. Cancer Manag Res 2021, 13, 2483-2498. [CrossRef]
- Kusmartsev, S. Metastasis-promoting functions of myeloid cells. Cancer Metastasis Rev 2025, 44, 61. [CrossRef]
- Wu, T.; Zhang, X.; Liu, X.; Cai, X.; Shen, T.; Pan, D.; Liang, R.; Ding, R.; Hu, R.; Dong, J.; et al. Single-cell sequencing reveals the immune microenvironment landscape related to anti-PD-1 resistance in metastatic colorectal cancer with high microsatellite instability. BMC Med 2023, 21, 161. [CrossRef]
- Rogala, J.; Sieminska, I.; Baran, J.; Rubinkiewicz, M.; Zybaczynska, J.; Szczepanik, A.M.; Pach, R. Myeloid-Derived Suppressor Cells May Predict the Occurrence of Postoperative Complications in Colorectal Cancer Patients-a Pilot Study. J Gastrointest Surg 2022, 26, 2354-2357. [CrossRef]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: from molecular mechanisms to targeted therapy. Signal Transduct Target Ther 2024, 9, 274. [CrossRef]
- Garcia-Nicolas, M.; Pastor-Belda, M.; Campillo, N.; Rodriguez-Sojo, M.J.; Ruiz-Malagon, A.J.; Hidalgo-Garcia, L.; Abad, P.; de la Torre, J.M.; Guillamon, E.; Banos, A.; et al. Analytical Platform for the Study of Metabolic Pathway of Propyl Propane Thiosulfonate (PTSO) from Allium spp. Foods 2023, 12. [CrossRef]
- Chen, Y.; Huang, J.; Fan, Y.; Huang, L.; Cai, X. Understanding the cellular and molecular heterogeneity in colorectal cancer through the use of single-cell RNA sequencing. Transl Oncol 2025, 55, 102374. [CrossRef]
Figure 1.
Impact of PTSO on the myeloid fraction of hPBMCs. A) Effect of PTSO on the overall myeloid population (CD11b⁺) and on Monocytes (CD11b⁺CD14⁺). B) Effect of PTSO on myeloid-derived suppressor cell (MDSC) subpopulations. Data (n = 6) are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 1.
Impact of PTSO on the myeloid fraction of hPBMCs. A) Effect of PTSO on the overall myeloid population (CD11b⁺) and on Monocytes (CD11b⁺CD14⁺). B) Effect of PTSO on myeloid-derived suppressor cell (MDSC) subpopulations. Data (n = 6) are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
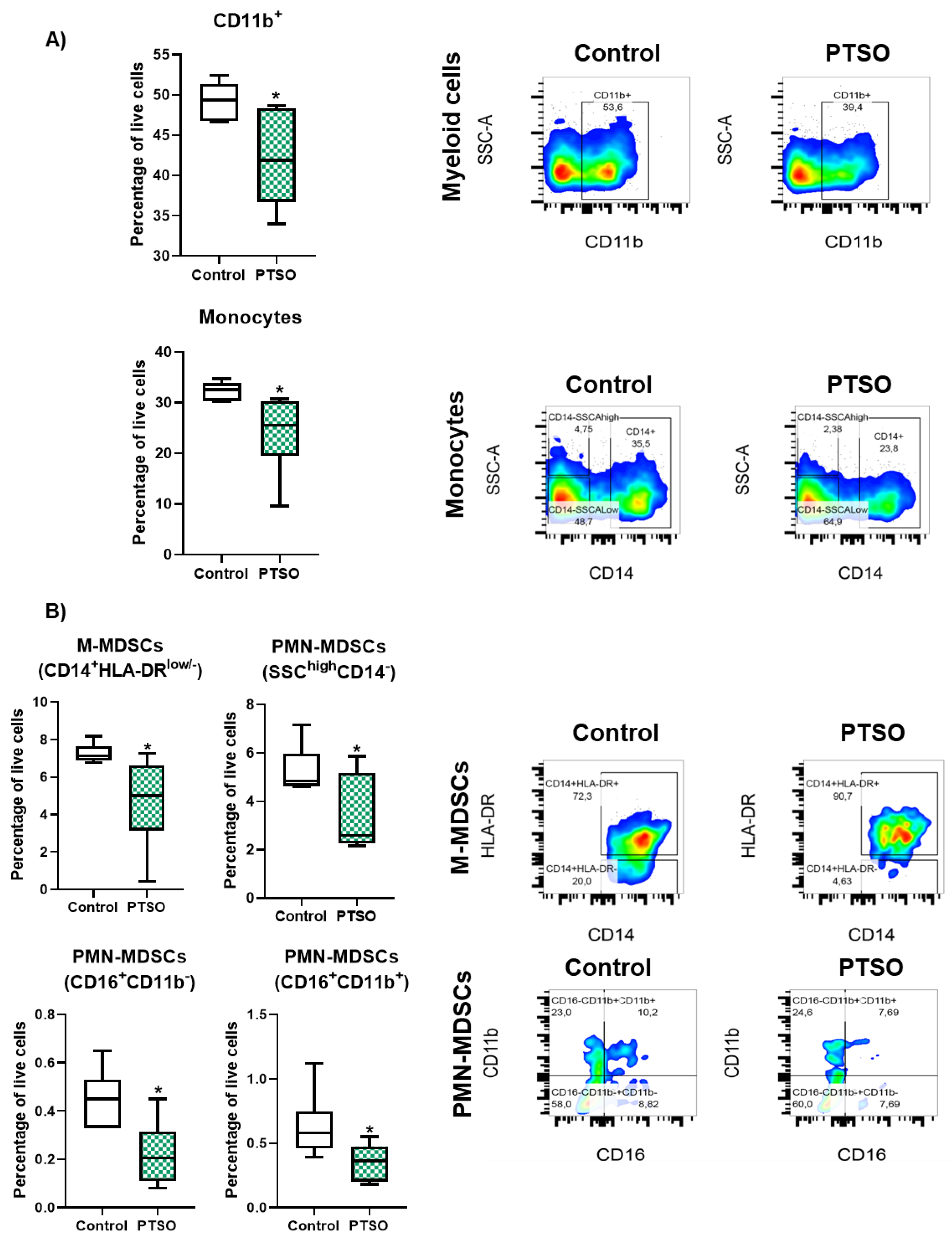
Figure 2.
Effect of PTSO on CD4+ and CD8+ T cell subsets in hPBMCs. A) Quantification of the effect of PTSO on CD4+, Th1, CD8+, and Tc1 populations in hPBMCs. B) Representative gating strategy showing the effect of PTSO on CD4+ T cells and Th1 (CD3+CD4+IFNG+) and Tc1 (CD3+CD8+IFNG+) subpopulations. Data (n = 6) are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 2.
Effect of PTSO on CD4+ and CD8+ T cell subsets in hPBMCs. A) Quantification of the effect of PTSO on CD4+, Th1, CD8+, and Tc1 populations in hPBMCs. B) Representative gating strategy showing the effect of PTSO on CD4+ T cells and Th1 (CD3+CD4+IFNG+) and Tc1 (CD3+CD8+IFNG+) subpopulations. Data (n = 6) are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
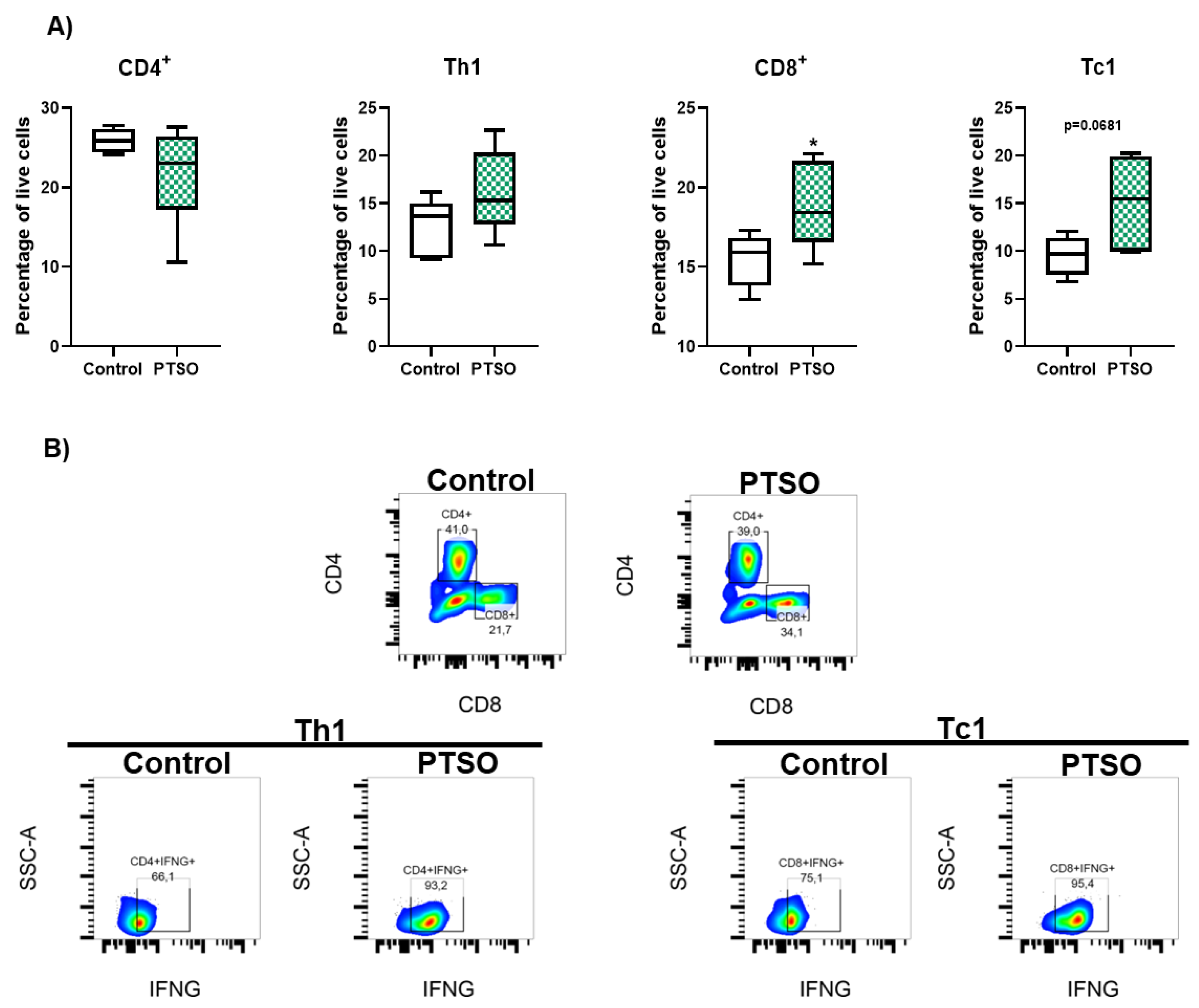
Figure 3.
Characterization of the effect produced by the modulation of human PBMCs by PTSO in xenograft tumors. A) Evolution of tumor volume (n = 9), B) Representative cross-sectional MRI images of both groups. Tumors are outlined by yellow lines. Red arrows indicate a central region of signal enhancement, consistent with necrosis and cell death linked to heightened tumor activity (n = 3). C) Representative images of the tumor sizes of both groups of mice. D) Tumor weight and volume post-sacrifice (n = 9). E) Representative histological sections of xenograft tumors stained with hematoxylin and eosin (H&E) from control and PTSO groups, scale bar: 200 μm for 4× and 50 μm for 10×. White arrows indicate areas of muscle tissue invasion by the tumor (n = 9). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 3.
Characterization of the effect produced by the modulation of human PBMCs by PTSO in xenograft tumors. A) Evolution of tumor volume (n = 9), B) Representative cross-sectional MRI images of both groups. Tumors are outlined by yellow lines. Red arrows indicate a central region of signal enhancement, consistent with necrosis and cell death linked to heightened tumor activity (n = 3). C) Representative images of the tumor sizes of both groups of mice. D) Tumor weight and volume post-sacrifice (n = 9). E) Representative histological sections of xenograft tumors stained with hematoxylin and eosin (H&E) from control and PTSO groups, scale bar: 200 μm for 4× and 50 μm for 10×. White arrows indicate areas of muscle tissue invasion by the tumor (n = 9). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 4.
Proliferation index in xenograft tumors. A) Evaluation of the proliferation process assessed by immunofluorescence using anti-KI67 staining (green); nuclei were counterstained with Hoechst (blue). Images were taken at 40× magnification; scale bar: 20 μm. B) Gene expression analysis of MMP9 and WNT5A. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 4.
Proliferation index in xenograft tumors. A) Evaluation of the proliferation process assessed by immunofluorescence using anti-KI67 staining (green); nuclei were counterstained with Hoechst (blue). Images were taken at 40× magnification; scale bar: 20 μm. B) Gene expression analysis of MMP9 and WNT5A. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
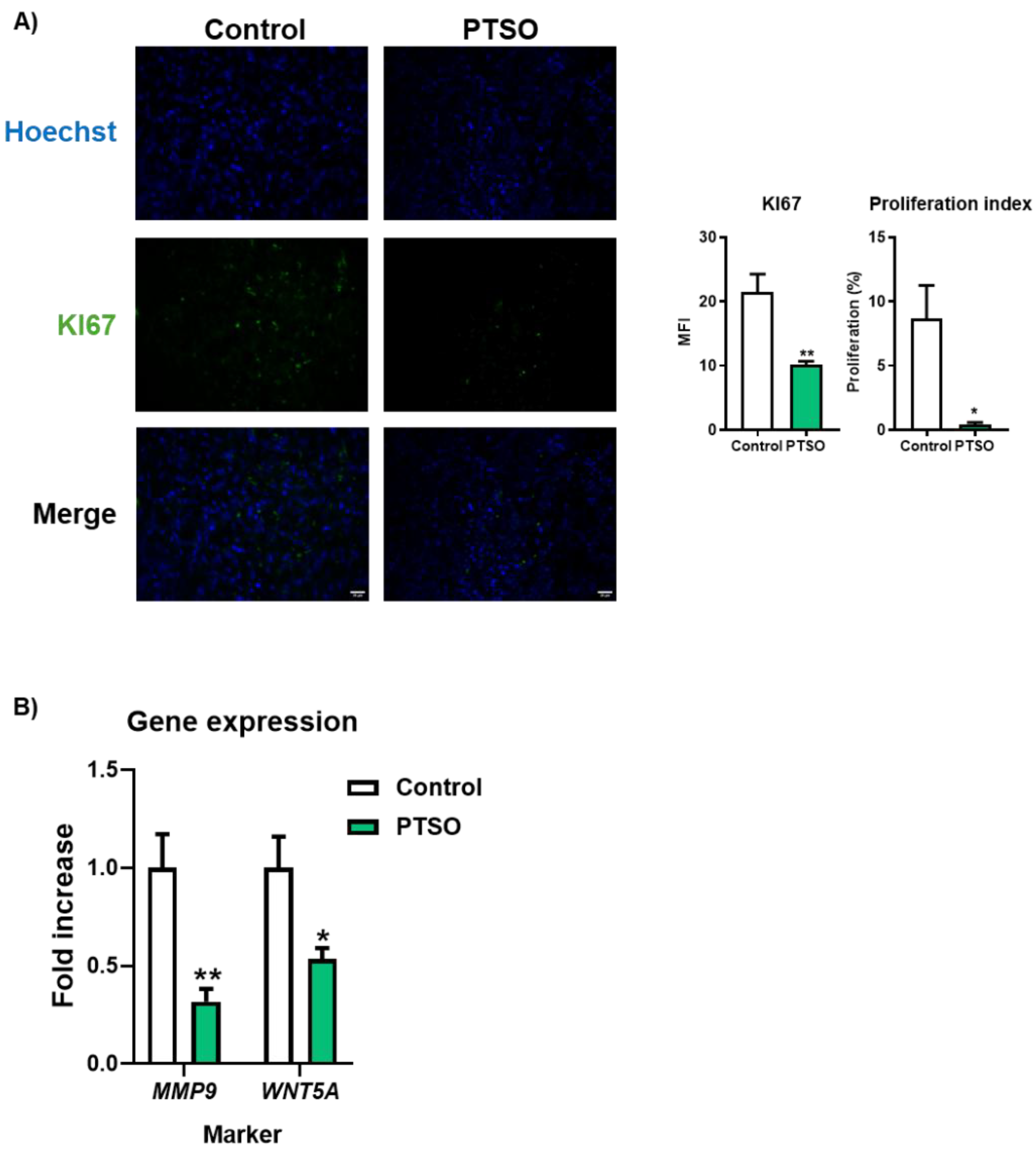
Figure 5.
Characterization of the immunologic profile of tumor tissue of tumors xenograft modulated by human PBMCs model. A) Immunologic profile of peripheral blood analyzed by flow cytometry, B) immune cell composition within tumor tissue determined by flow cytometry, C) Gene expression levels of selected immune-related markers, including INOS, MRC1, PVR, IDO1, NECTIN2, CD38, and ARG1, assessed by qPCR. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Figure 5.
Characterization of the immunologic profile of tumor tissue of tumors xenograft modulated by human PBMCs model. A) Immunologic profile of peripheral blood analyzed by flow cytometry, B) immune cell composition within tumor tissue determined by flow cytometry, C) Gene expression levels of selected immune-related markers, including INOS, MRC1, PVR, IDO1, NECTIN2, CD38, and ARG1, assessed by qPCR. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



