
Submitted:
15 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
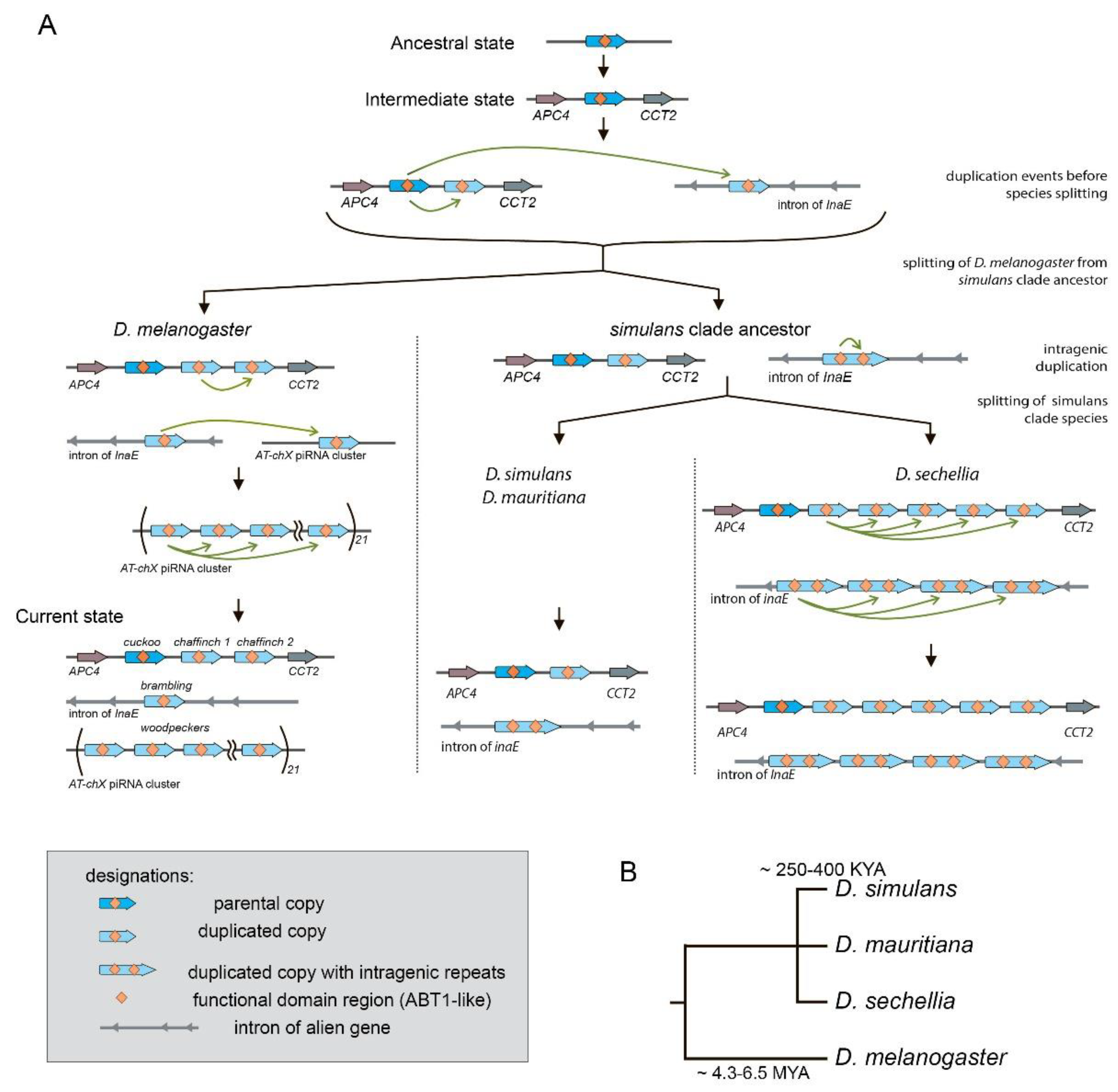
Gene duplications are considered to be the major evolutionary resource of novel functions. The gene family Esf2/ABP1 is conserved in metazoan organisms from yeast to humans. Here we performed a search and characterization of Esf2/ABP1 homologs in the Drosophila genus. Whereas in the majority of Drosophila species this gene family is represented by only a single gene, in the melanogaster and suzukii subgroups recurrent gene duplications arose, providing 47 homologous genes located on the X chromosome. To study the evolutionary history of duplicates, we performed phylogenetic, functional domain, and tissue-specific expression analyses. We revealed a male-specific and testis-biased transcription pattern of duplicated copies in Drosophila melanogaster and Drosophila sechellia compared to ubiquitous expression of the parental gene. The amplification of 21 repeated paralogs within the heterochromatic piRNA cluster resulted in the ovarian-specific transformation of these repeats into piRNAs in D. melanogaster. In three species of the suzukii subgroup, Esf2/ABP1 genes evolved with domain diversification: in addition to RNA-binding ABT1-like domain preservation, all homologous proteins acquired expanded intrinsically disordered regions. By studying the duplicated copies of the Esf2/ABP1 family in Drosophila, we offer insight into how novel gene functions emerge and are maintained, contributing to life's diversity and complexity.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Homolog search and identification
2.2. Phylogenetic analysis
2.3. Z-test of neutrality
2.4. Search for protein domains
2.5. Fly stocks
2.6. RT-qPCR analysis
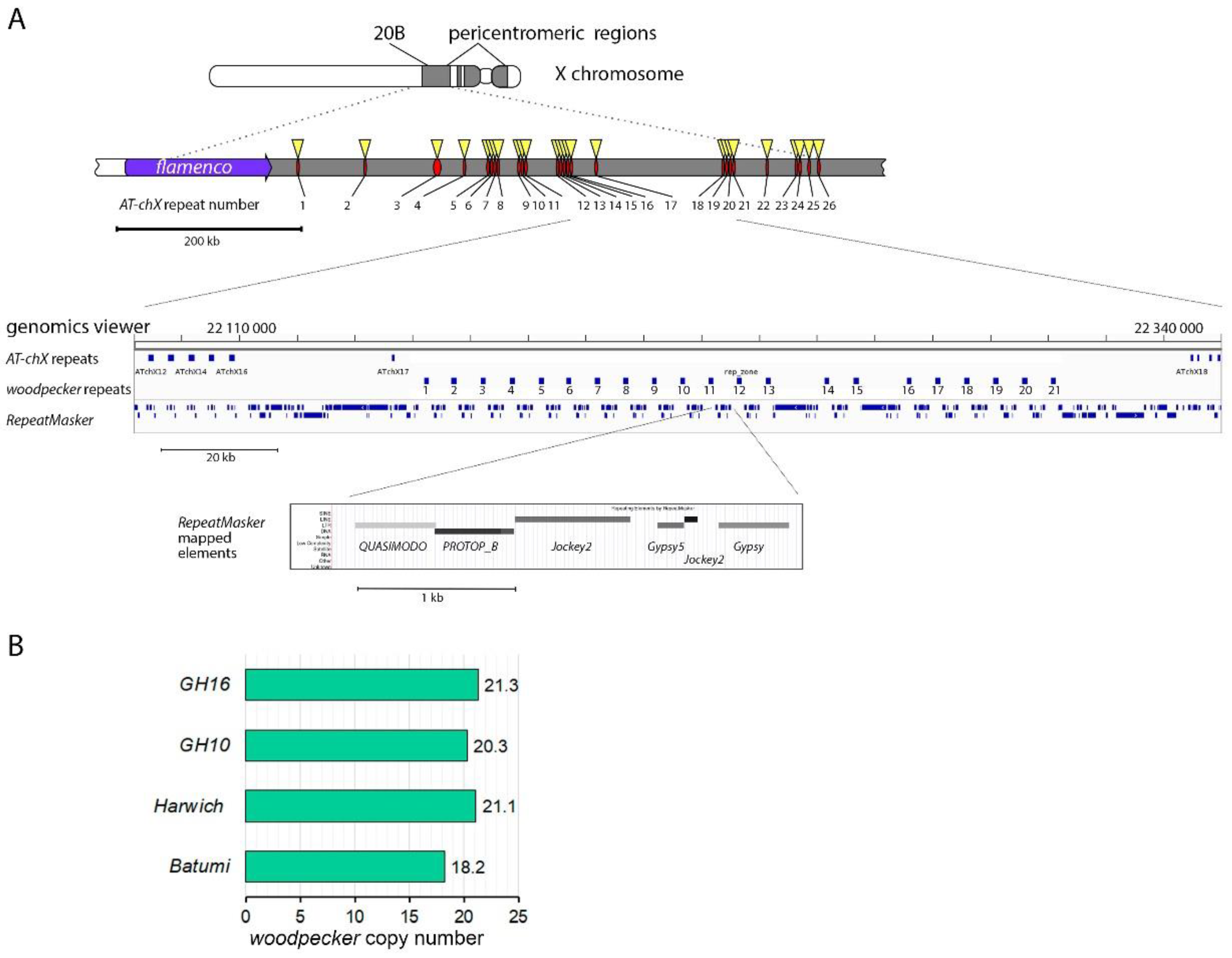
2.7. Copy number estimation of woodpecker genes
2.8. Transcriptomic analyses of Esf2/ABP1 family genes using deep sequencing library data
2.9. Bioinformatics analysis of piRNAs
3. Results
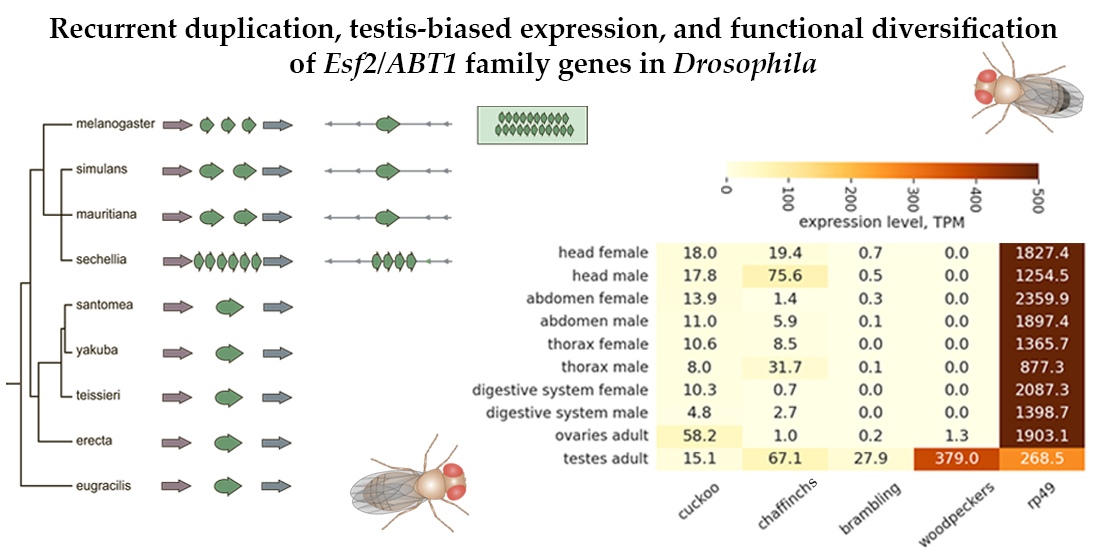
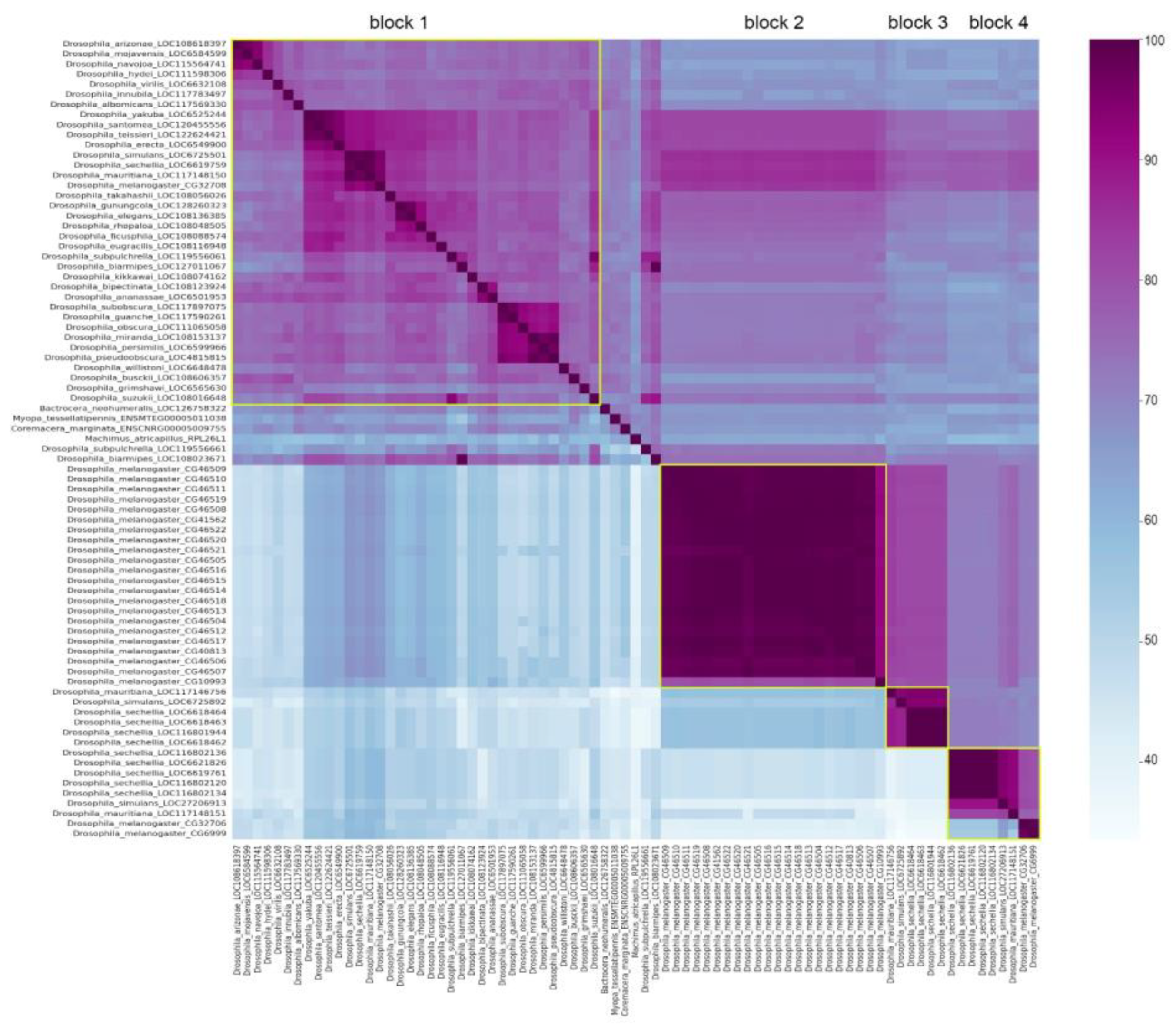
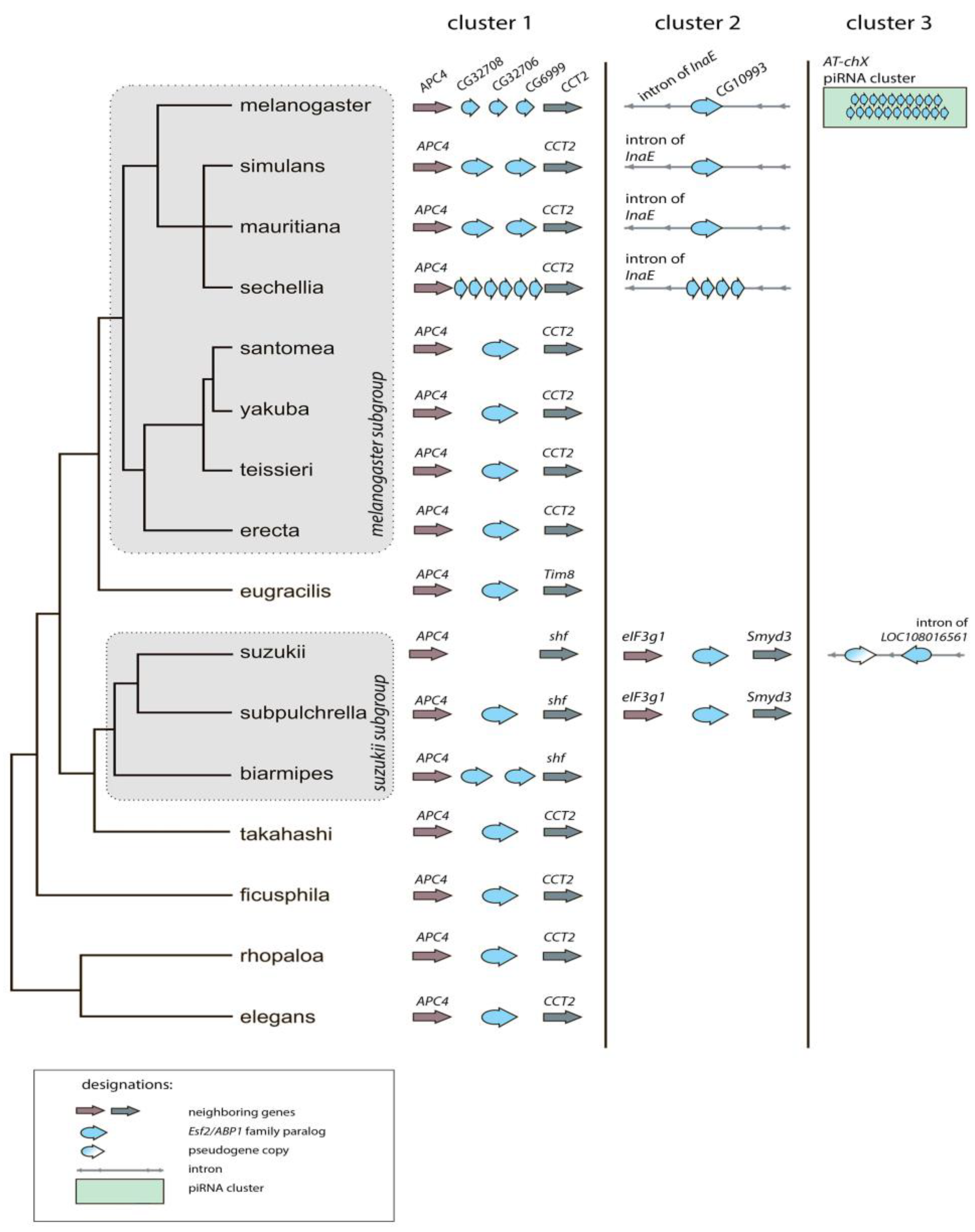
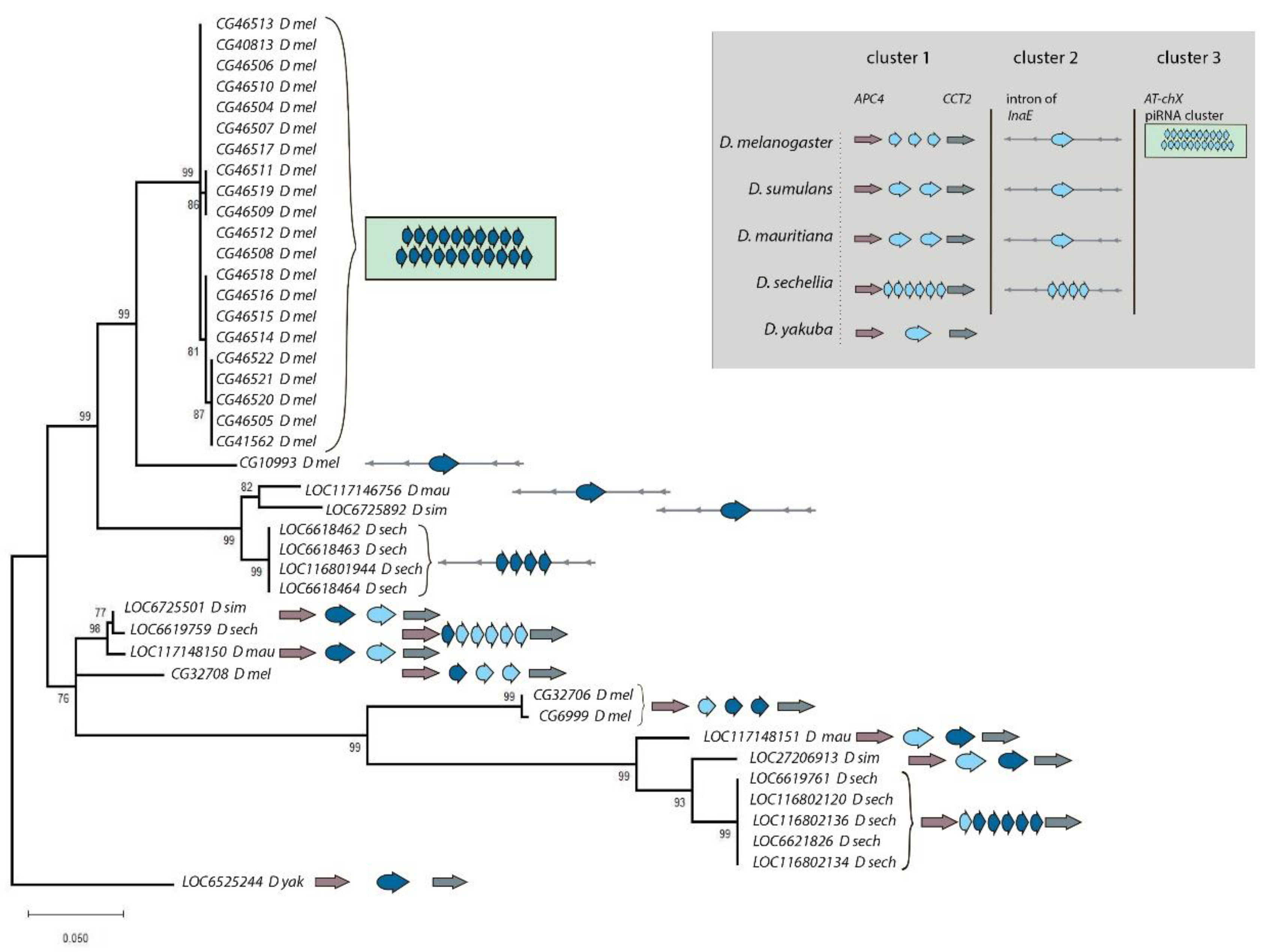
3.1. Survey of recurrent duplications of Esf2/ABP1 family genes in Drosophila
3.2. Phylogenetic analysis for homologous genes of melanogaster subgroup
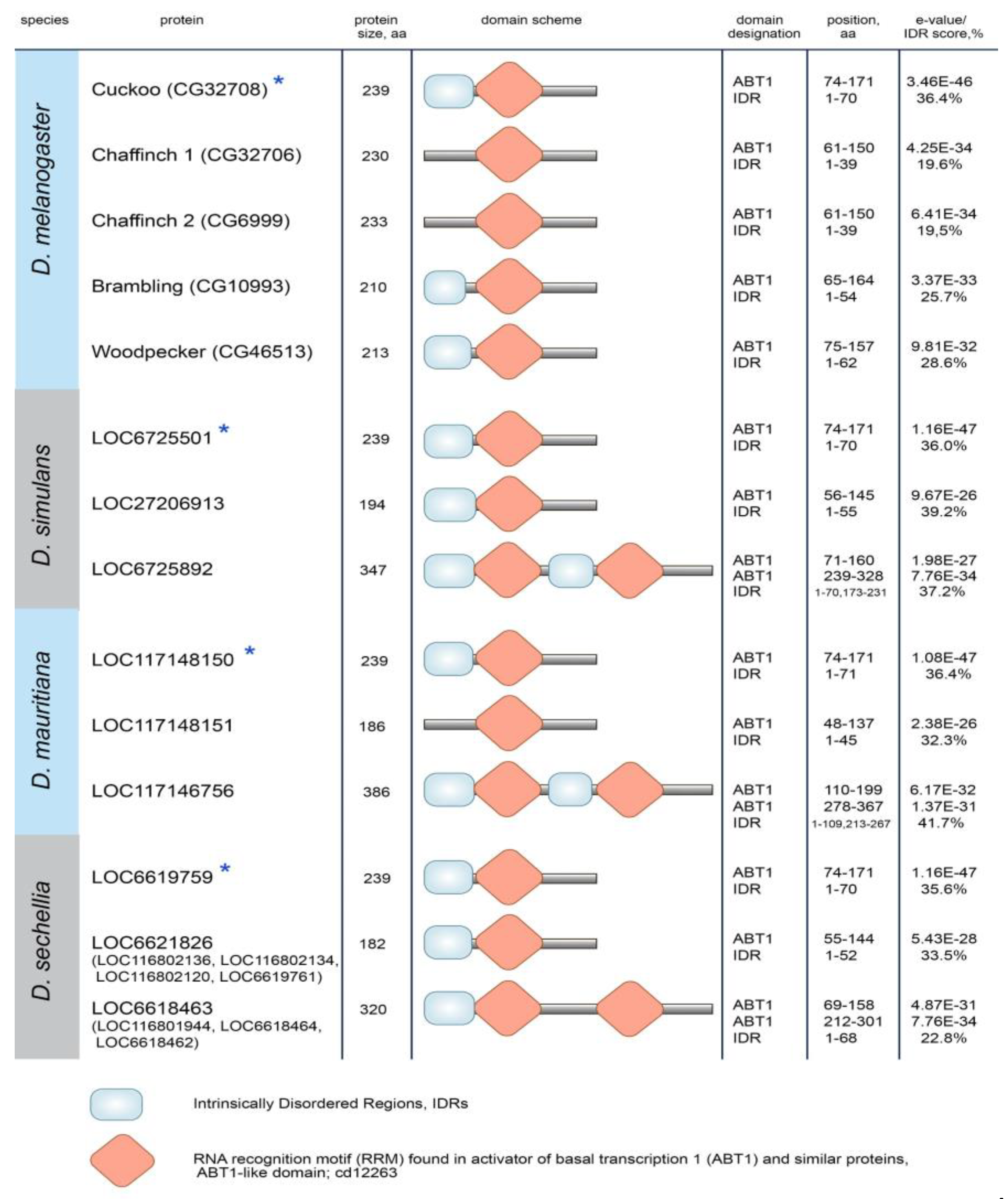
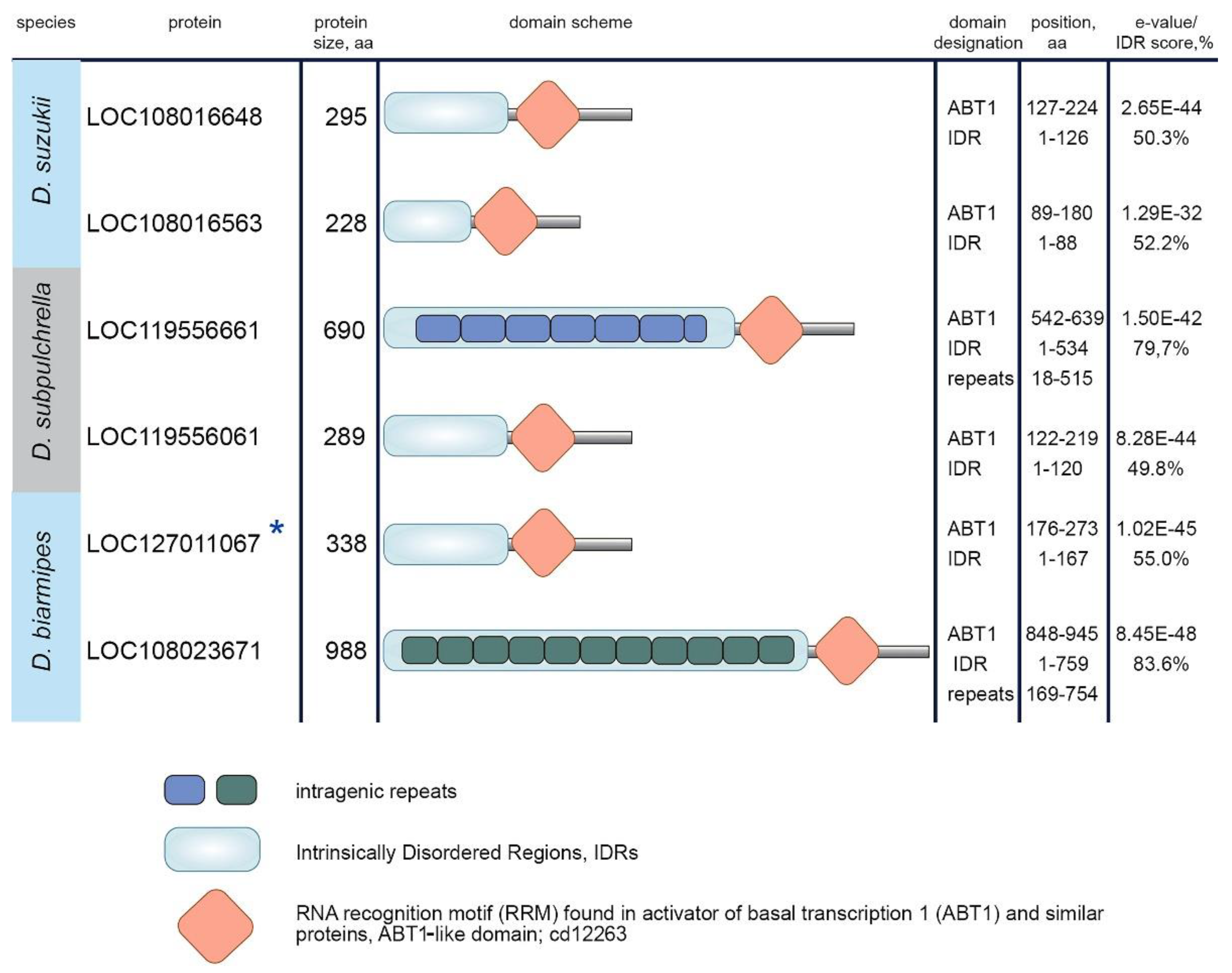
3.3. Protein domain search in Esf2/ABP1 proteins
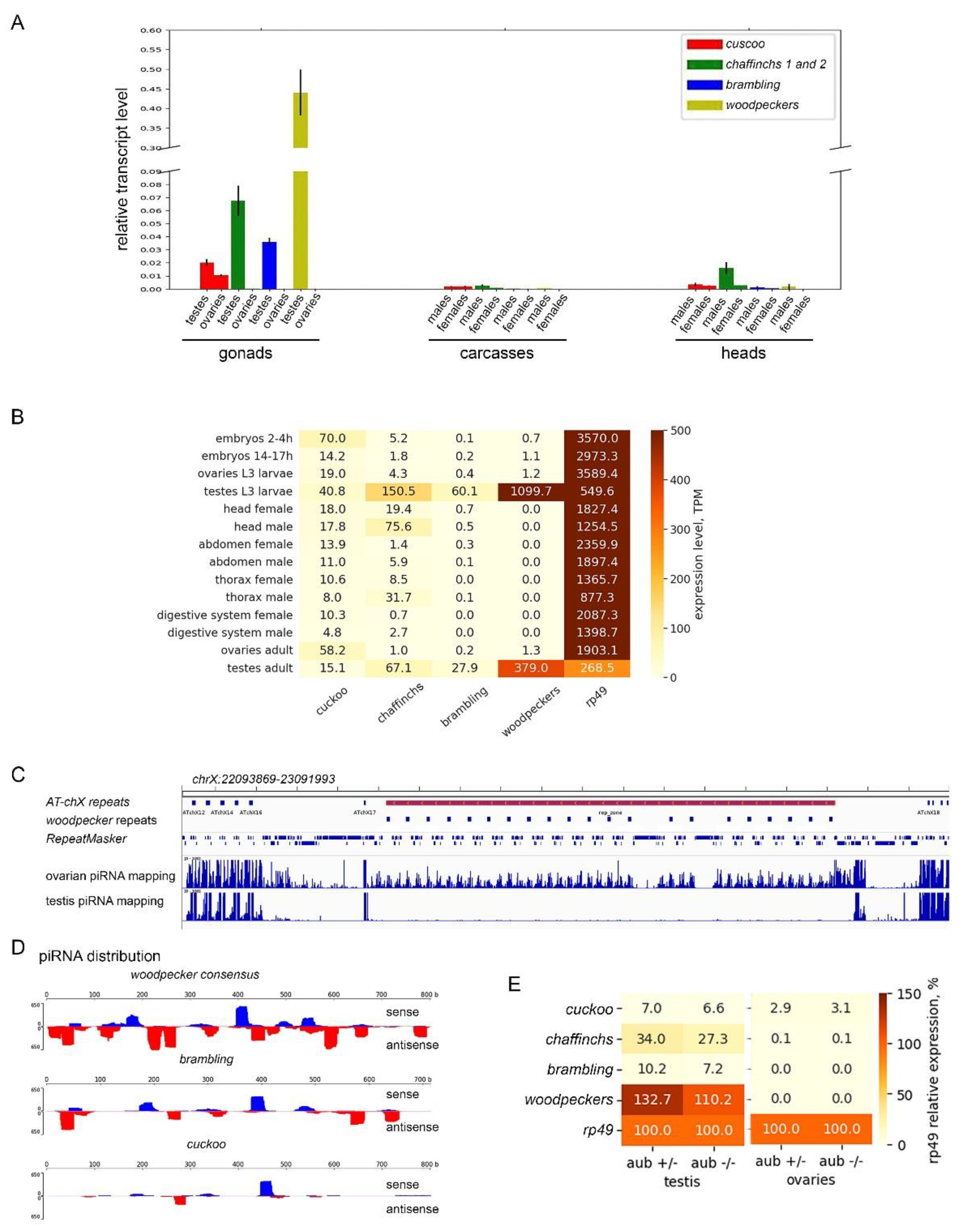
3.4. Diversification of the expression pattern of Esf2/ABP1 paralogs in D. melanogaster and D. sechellia
4. Discussion
Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Evolution by Gene Duplication, Springer: New York, USA, 1970; pp. 1–160.
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Assis, R.; Bachtrog, D. Neofunctionalization of young duplicate genes in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 17409–17414. [Google Scholar] [CrossRef] [PubMed]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef]
- Oda, T.; Kayukawa, K.; Hagiwara, H.; Yudate, H.T.; Masuho, Y.; Murakami, Y.; Tamura, T.A.; Muramatsu, M.A. A novel TATA-binding protein-binding protein, ABT1, activates basal transcription and has a yeast homolog that is essential for growth. Mol. Cell. Biol. 2000, 20, 1407–1418. [Google Scholar] [CrossRef]
- Hoang, T.; Peng, W.T.; Vanrobays, E.; Krogan, N.; Hiley, S.; Beyer, A.L.; Osheim, Y.N.; Greenblatt, J.; Hughes, T.R.; Lafontaine, D.L. Esf2p, a U3-associated factor required for small-subunit processome assembly and compaction. Mol. Cell. Biol. 2005, 25, 5523–5534. [Google Scholar] [CrossRef]
- Mummery-Widmer, J.L.; Yamazaki, M.; Stoeger, T.; Novatchkova, M.; Bhalerao, S.; Chen, D.; Dietzl, G.; Dickson, B.J.; Knoblich, J.A. Genome-wide analysis of Notch signaling in Drosophila by transgenic RNAi. Nature 2009, 458, 987–992. [Google Scholar] [CrossRef]
- Schnorrer, F.; Schönbauer, C.; Langer, C.C.; Dietzl, G.; Novatchkova, M.; Schernhuber, K.; Fellner, M.; Azaryan, A.; Radolf, M.; Stark, A.; Keleman, K.; Dickson, B.J. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 2010, 464, 287–291. [Google Scholar] [CrossRef]
- Neumüller, R.A.; Richter, C.; Fischer, A.; Novatchkova, M.; Neumüller, K.G.; Knoblich, J.A. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 2011, 8, 580–593. [Google Scholar] [CrossRef]
- Viswanatha, R.; Li, Z.; Hu, Y.; Perrimon, N. Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. Elife 2018, 7, e36333. [Google Scholar] [CrossRef]
- Clifton, B.D.; Jimenez, J.; Kimura, A.; Chahine, Z.; Librado, P.; Sánchez-Gracia, A.; Abbassi, M.; Carranza, F.; Chan, C.; Marchetti, M.; Zhang, W.; Shi, M.; Vu, C.; Yeh, S.; Fanti, L.; Xia, X.Q.; Rozas, J.; Ranz, J.M. Understanding the Early Evolutionary Stages of a Tandem Drosophila melanogaster-Specific Gene Family: A Structural and Functional Population Study. Mol. Biol. Evol. 2020, 37, 2584–2600. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Mejia Natividad, I.; Malik, H.S. Expansion and loss of sperm nuclear basic protein genes in Drosophila correspond with genetic conflicts between sex chromosomes. Elife 2023, 12, e85249. [Google Scholar] [CrossRef] [PubMed]
- Kuvaeva, E.E.; Cherezov, R.O.; Kulikova, D.A.; Mertsalov, I.B. The Drosophila toothrin Gene Related to the d4 Family Genes: An Evolutionary View on Origin and Function. Int. J. Mol. Sci. 2024, 25, 13394. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.L.; Oliver, G.T.; Farkas, I.Z.; Buszczak, M.; Levine, M.T. Recurrent Duplication and Diversification of a Vital DNA Repair Gene Family Across Drosophila. Mol. Biol. Evol. 2024, 41, msae113. [Google Scholar] [CrossRef]
- Zakerzade, R.; Chang, C.H.; Chatla, K.; Krishnapura, A.; Appiah, S.P.; Zhang, J.; Unckless, R.L.; Blumenstiel, J.P.; Bachtrog, D.; Wei, K.H. Diversification and recurrent adaptation of the synaptonemal complex in Drosophila. PLoS Genet. 2025, 21, e1011549. [Google Scholar] [CrossRef]
- Clark, A.G.; et al. Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [Google Scholar] [CrossRef]
- Miller, D.E.; Staber, C.; Zeitlinger, J.; Hawley, R.S. Highly Contiguous Genome Assemblies of 15 Drosophila Species Generated Using Nanopore Sequencing. G3 (Bethesda) 2018, 8, 3131–3141. [Google Scholar] [CrossRef]
- Kim, B.Y.; Wang, J.R.; Miller, D.E.; Barmina, O.; Delaney, E.; Thompson, A.; Comeault, A.A.; Peede, D.; D'Agostino, E.R.R.; Pelaez, J.; Aguilar, J.M.; Haji, D.; Matsunaga, T.; Armstrong, E.E.; Zych, M.; Ogawa, Y.; Stamenković-Radak, M.; Jelić, M.; Veselinović, M.S.; Tanasković, M.; Erić, P.; Gao, J.J.; Katoh, T.K.; Toda, M.J.; Watabe, H.; Watada, M.; Davis, J.S.; Moyle, L.C.; Manoli, G.; Bertolini, E.; Košťál, V.; Hawley, R.S.; Takahashi, A.; Jones, C.D.; Price, D.K.; Whiteman, N.; Kopp, A.; Matute, D.R.; Petrov, D.A. Highly contiguous assemblies of 101 drosophilid genomes. Elife 2021, 10, e66405. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; Yang, M.; Zhang, D.; Zheng, C.; Lanczycki, C.J.; Marchler-Bauer, A. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Conte, A.D.; Mehdiabadi, M.; Bouhraoua, A.; Miguel Monzon, A.; Tosatto, S.C.E.; Piovesan, D. Critical assessment of protein intrinsic disorder prediction (CAID) - Results of round 2. Proteins 2023, 91, 1925–1934. [Google Scholar] [CrossRef]
- Bergman, C.M.; Haddrill, P.R. Strain-specific and pooled genome sequences for populations of Drosophila melanogaster from three continents. F1000Res. 2015, 4, 31. [Google Scholar] [CrossRef]
- Teixeira, F.K.; Okuniewska, M.; Malone, C.D.; Coux, R.X.; Rio, D.C.; Lehmann, R. piRNA-mediated regulation of transposon alternative splicing in the soma and germ line. Nature 2017, 552, 268–272. [Google Scholar] [CrossRef]
- Yang, H.; Jaime, M.; Polihronakis, M.; Kanegawa, K.; Markow, T.; Kaneshiro, K.; Oliver, B. Re-annotation of eight Drosophila genomes. Life Sci. Alliance 2018, 1, e201800156. [Google Scholar] [CrossRef]
- Maksimov, D.A.; Laktionov, P.P.; Posukh, O.V.; Belyakin, S.N.; Koryakov, D.E. Genome-wide analysis of SU(VAR)3-9 distribution in chromosomes of Drosophila melanogaster. Chromosoma 2018, 127, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kotov, A.A.; Godneeva, B.K.; Bazylev, S.S.; Olenina, L.V.; Aravin, A.A. piRNA-mediated gene regulation and adaptation to sex-specific transposon expression in D. melanogaster male germline. Genes Dev. 2021, 35, 914–935. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, V.; Natarajan, M.; Johnston, J.; Zeitlinger, J. TATA and paused promoters active in differentiated tissues have distinct expression characteristics. Mol. Syst. Biol. 2021, 17, e9866. [Google Scholar] [CrossRef] [PubMed]
- Mahadevaraju, S.; Pal, S.; Bhaskar, P.; McDonald, B.D.; Benner, L.; Denti, L.; Cozzi, D.; Bonizzoni, P.; Przytycka, T.M.; Oliver, B. Diverse somatic Transformer and sex chromosome karyotype pathways regulate gene expression in Drosophila gonad development. bioRxiv [Preprint]. 2024 2024.08.12.607556.
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Srivastav, S.P.; Feschotte, C.; Clark, A.G. Rapid evolution of piRNA clusters in the Drosophila melanogaster ovary. Genome Res. 2024, 34, 711–724. [Google Scholar] [CrossRef]
- Kotov, A.A.; Adashev, V.E.; Godneeva, B.K.; Ninova, M.; Shatskikh, A.S.; Bazylev, S.S.; Aravin, A.A.; Olenina, L.V. piRNA silencing contributes to interspecies hybrid sterility and reproductive isolation in Drosophila melanogaster. Nucleic Acids Res. 2019, 47, 4255–4271. [Google Scholar] [CrossRef]
- Antoniewski, C. Computing siRNA and piRNA overlap signatures. Methods Mol. Biol. 2014, 1173, 135–146. [Google Scholar] [CrossRef]
- Fan, C.; Chen, Y.; Long, M. Recurrent tandem gene duplication gave rise to functionally divergent genes in Drosophila. Mol. Biol. Evol. 2008, 25, 1451–1458. [Google Scholar] [CrossRef]
- Lachaise, D.; Cariou, M.L.; David, J.R.; Lemeunier, F.; Tsacas, L.; Ashburner, M. Historical biogeography of the Drosophila-Melanogaster species subgroup. Evol. Biol. 1988, 22, 159–225. [Google Scholar] [CrossRef]
- Russo, C.A.; Takezaki, N.; Nei, M. Molecular phylogeny and divergence times of drosophilid species. Mol. Biol. Evol. 1995, 12, 391–404. [Google Scholar] [CrossRef]
- Tamura, K.; Subramanian, S.; Kumar, S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 2004, 21, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Garrigan, D.; Kingan, S.B.; Geneva, A.J.; Andolfatto, P.; Clark, A.G.; Thornton, K.R.; Presgraves, D.C. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012, 22, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Suvorov, A.; Kim, B.Y.; Wang, J.; Armstrong, E.E.; Peede, D.; D'Agostino, E.R.R.; Price, D.K.; Waddell, P.; Lang, M.; Courtier-Orgogozo, V.; David, J.R.; Petrov, D.; Matute, D.R.; Schrider, D.R.; Comeault, A.A. Widespread introgression across a phylogeny of 155 Drosophila genomes. Curr. Biol. 2022, 32, 111–123.e5. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.D.; Brennecke, J.; Dus, M.; Stark, A.; McCombie, W.R.; Sachidanandam, R.; Hannon, G.J. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 2009, 137, 522–535. [Google Scholar] [CrossRef]
- Konstantinidou, P.; Loubalova, Z.; Ahrend, F.; Friman, A.; Almeida, M.V.; Poulet, A.; Horvat, F.; Wang, Y.; Losert, W.; Lorenzi, H.; Svoboda, P.; Miska, E.A.; van Wolfswinkel, J.C.; Haase, A.D. A comparative roadmap of PIWI-interacting RNAs across seven species reveals insights into de novo piRNA-precursor formation in mammals. Cell Rep. 2024, 43, 114777. [Google Scholar] [CrossRef]
- Chang, C.H.; Gregory, L.E.; Gordon, K.E.; Meiklejohn, C.D.; Larracuente, A.M. Unique structure and positive selection promote the rapid divergence of Drosophila Y chromosomes. Elife 2022, 11, e75795. [Google Scholar] [CrossRef]
- Montanari, F.; Shields, D.C.; Khaldi, N. Differences in the Number of Intrinsically Disordered Regions between Yeast Duplicated Proteins, and Their Relationship with Functional Divergence. PLoS ONE 2011, 6, e24989. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; Kim, P.M.; Kriwacki, R.W.; Oldfield, C.J.; Pappu, R.V.; Tompa, P.; Uversky, V.N.; Wright, P.E.; Babu, M.M. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Aravin, A.A.; Naumova, N.M.; Tulin, A.V.; Vagin, V.V.; Rozovsky, Y.M.; Gvozdev, V.A. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 2001, 11, 1017–1027. [Google Scholar] [CrossRef]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef]
- Kotov, A.A.; Bazylev, S.S.; Adashev, V.E.; Shatskikh, A.S.; Olenina, L.V. Drosophila as a Model System for Studying of the Evolution and Functional Specialization of the Y Chromosome. Int. J. Mol. Sci. 2022, 23, 4184. [Google Scholar] [CrossRef]
- Prestel, M.; Feller, C.; Straub, T.; Mitlöhner, H.; Becker, P.B. The activation potential of MOF is constrained for dosage compensation. Mol. Cell 2010, 38, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, J.C.; Kuroda, M.I. Dosage compensation in Drosophila. Cold Spring Harb, Perspect Biol. 2015, 7, a019398. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, L.; Kuroda, M.I. An analysis of maleless and histone H4 acetylation in Drosophila melanogaster spermatogenesis. Mech. Dev. 1998, 71, 107–117. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Landeen, E.L.; Cook, J.M.; Kingan, S.B.; Presgraves, D.C. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 2011, 9, e1001126. [Google Scholar] [CrossRef]
- Mahadevaraju, S.; Fear, J.M.; Akeju, M.; Galletta, B.J.; Pinheiro, M.M.L.S.; Avelino, C.C.; Cabral-de-Mello, D.C.; Conlon, K.; Dell'Orso, S.; Demere, Z.; Mansuria, K.; Mendonça, C.A.; Palacios-Gimenez, O.M.; Ross, E.; Savery, M.; Yu, K.; Smith, H.E.; Sartorelli, V.; Yang, H.; Rusan, N.M.; Vibranovski, M.D.; Matunis, E.; Oliver, B. Dynamic sex chromosome expression in Drosophila male germ cells. Nat. Commun. 2021, 12, 892. [Google Scholar] [CrossRef]
- Chang, C.H.; Larracuente, A.M. Heterochromatin-enriched assemblies reveal the sequence and organization of the Drosophila melanogaster Y chromosome. Genetics 2019, 211, 333–348. [Google Scholar] [CrossRef]
- Adashev, V.E.; Kotov, A.A.; Bazylev, S.S.; Shatskikh, A.S.; Aravin, A.A.; Olenina, L.V. Stellate Genes and the piRNA Pathway in Speciation and Reproductive Isolation of Drosophila melanogaster. Front. Genet. 2021, 11, 610665. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Kragelund, B.B. The molecular basis for cellular function of intrinsically disordered protein regions. Nat. Rev. Mol. Cell Biol. 2024, 25, 187–211. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays 2003, 25, 847–855. [Google Scholar] [CrossRef]

| Gene number | New gene designation |
| CG32708 | cuckoo |
| CG32706 | chaffinch 1 |
| CG6999 | chaffinch 2 |
| CG10993 | brambling |
| CG40813 | woodpecker 1 |
| CG46504 | woodpecker 2 |
| CG46505 | woodpecker 3 |
| CG46506 | woodpecker 4 |
| CG46507 | woodpecker 5 |
| CG46508 | woodpecker 6 |
| CG46509 | woodpecker 7 |
| CG46510 | woodpecker 8 |
| CG46511 | woodpecker 9 |
| CG46512 | woodpecker 10 |
| CG46513 | woodpecker 11 |
| CG46514 | woodpecker 12 |
| CG46515 | woodpecker 13 |
| CG46516 | woodpecker 14 |
| CG46517 | woodpecker 15 |
| CG46518 | woodpecker 16 |
| CG46519 | woodpecker 17 |
| CG46520 | woodpecker 18 |
| CG46521 | woodpecker 19 |
| CG46522 | woodpecker 20 |
| CG41562 | woodpecker 21 |
| gene | woodpeckers | brambling | cuckoo | chaffinch1 | chaffinch2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| piRNA read type |
sense piRNAs | antisense piRNAs | sense piRNAs | antisense piRNAs | sense piRNAs | antisense piRNAs | sense piRNAs | antisense piRNAs | sense piRNAs | antisense piRNAs |
| Mapped reads (0-2 mm), rpm | 201.6 | 568.6 | 115.8 | 203.8 | 68.8 | 47.2 | 0.6 | 0.2 | 0 | 0.2 |
| Mapped reads (0 mm), rpm | 150.3 | 422.7 | 1.1 | 55.2 | 15.9 | 0.9 | 0 | 0 | 0 | 0 |
| Mapped reads (1 mm), rpm | 40.5 | 129.5 | 25.2 | 51.4 | 10.1 | 3.2 | 0 | 0 | 0 | 0 |
| Mapped reads (2 mm), rpm | 10.7 | 16.4 | 89.5 | 97.1 | 45.7 | 43.2 | 0.6 | 0.2 | 0 | 0.2 |
| Overlap_10, pairs | 365 | 155 | 23 | 0 | 0 | |||||
| z10-score | 2.86 | 3.08 | 1.01 | NA | NA | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



