Submitted:
17 August 2025
Posted:
18 August 2025
You are already at the latest version
Abstract
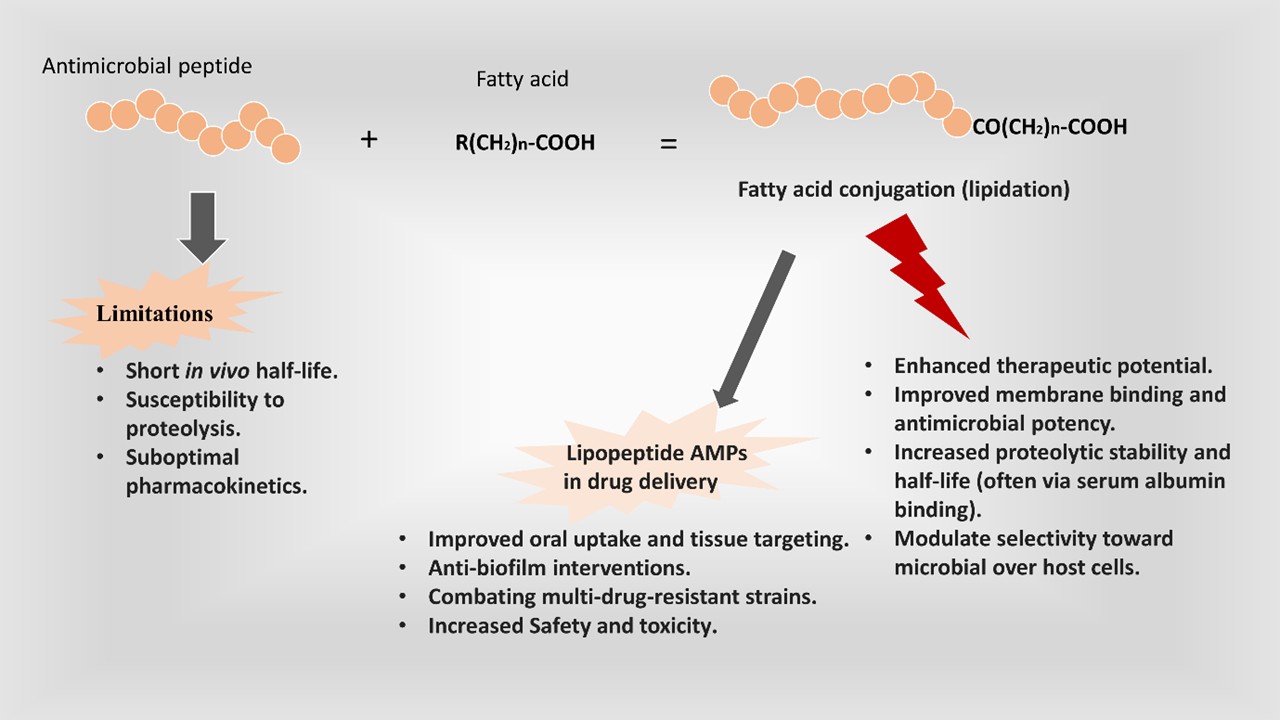
Antimicrobial resistance has emerged as a critical threat to global health, with drug-resistant infections. Antimicrobial peptides (AMPs) are a promising class of therapeutics to address this crisis due to their broad-spectrum activity, ability to target multiple sites, and reduced susceptibility to resistance development. However, native AMPs often face limitations, including short in vivo half-life, susceptibility to proteolysis, and suboptimal pharmacokinetics. Fatty acid conjugation (lipidation) of AMPs has emerged as an effective strategy to enhance their therapeutic potential. Covalently attaching hydrophobic fatty acid chains to peptide sequences is used to improve membrane binding and antimicrobial potency, increase proteolytic stability and half-life (often via serum albumin binding), and modulate selectivity toward microbial over host cells. This review provides an overview of fatty acid-conjugated AMPs (FAMPs). We first summarize the fundamental mechanisms of AMP action and then discuss the rationale for fatty acid modification. We describe common conjugation strategies (e.g., N-terminal acylation, side-chain attachment chemistry) and examine how lipidation affects antimicrobial activity, spectrum, stability against degradation, and selectivity. We highlight applications of lipopeptide AMPs in drug delivery (including improved oral uptake and tissue targeting), anti-biofilm interventions, and combating multi-drug-resistant strains. Safety and toxicity considerations are addressed, emphasizing the balance between increased hydrophobicity and host cell compatibility. Finally, we discuss future directions and perspectives in this field, such as optimizing fatty acid structure to maximize efficacy and safety, using artificial intelligence in design, and integrating FAMPs into next-generation therapeutic strategies.
Keywords:
antibacterial
; antimicrobial
; antifungal
; fatty acids
; peptides
; resistance
; toxicity
1. Introduction
The rise of antimicrobial resistance has rendered many conventional antibiotics ineffective, creating an urgent need for alternative therapeutic strategies [1]. Antimicrobial resistance is associated with about 1.27 million deaths in 2019 [2]. This toll could climb to 10 million annually by 2050 without new interventions [3].
For example, AMR in Enterococcus faecium infections is spreading due to inadequate treatment options for resistant strains, with the high prevalence in Asia [4]. In the USA, about 19% of Extended-Spectrum Beta-Lactamases (ESBL)-Klebsiella spp. in dairy cattle, feed, and water were multidrug resistant [5]. Varied resistance genes of azithromycin have been identified in Escherichia coli and Salmonella from food-producing animals and meat investigated in Europe [6]. A significant incidence of bacterial infections with antimicrobial resistance, mostly E. coli, was detected among pregnant women in East Africa [7].
The link between antimicrobial resistance and cancer is determined from the most frequently reported outcome, the mortality rate, indicated in 47% of studies, according to a systematic review, which highlighted the important influence of AMR on cancer patients’ survival [8]. The high risk of attaining antibiotic-resistant infections in cancer patients is due to numerous hospital visits and admissions for either therapy/or surgeries. In addition, the treatment causes cancer patients to have compromised immune systems; consequently, they are more susceptible to fatal infections such as Methicillin-resistant Staphylococcus aureus and Carbapenem resistance in Acinetobacter baumannii, and Klebsiella pneumoniae [9].
In this context, risks to global health require improving the effectiveness of antimicrobial agents and discovering novel approaches to fight infectious diseases. Amongst these approaches is the antimicrobial peptides (AMPs) that have developed as capable candidates against resistant pathogenic strains [10]. AMPs are naturally occurring or synthetic short peptides that can kill a broad range of bacteria, fungi, and even viruses. The main advantages of using AMPs over current antibiotics include their ability to target multiple sites, reduction in susceptibility to develop resistance, and their high effectiveness at low doses. Indeed, AMPs have demonstrated efficacy against notorious multidrug-resistant pathogens such as the ESKAPE group (Enterococcus faecium, S. aureus, K. pneumoniae, A. baumannii, Pseudomonas aeruginosa, and Enterobacter spp.). Moreover, AMPs can be effective at relatively low concentrations, and some modulate host immune responses, aiding in infection control [11].
AMPs are recognized for their mechanism of action, including membrane permeabilization, inhibiting multiple targets, and innate immunity modulation, which causes their activity against common resistance mechanisms of pathogenic bacteria. Consequently, AMPs, such as polymyxin B, cathelicidins, temporins, defensins, and magainins, which are in clinical use, exhibited effectiveness against a broad range of multidrug-resistant pathogens, comprising the ESKAPE organisms [12]. Both phenotypic and genetic resistance to AMPs may be overcome by identifying specific metabolic pathways and their associated genes, and then targeting them for antimicrobial therapy [13].
Despite their promise, the therapeutic development of AMPs faces challenges. Unmodified peptides are often rapidly degraded by proteases and cleared from the bloodstream, resulting in short half-lives. Many AMPs have suboptimal pharmacokinetic properties and can exhibit toxicity or lack stability under physiological conditions [14]. To overcome these limitations, various peptide engineering approaches have been explored, including cyclization, D-amino acid substitution, and N-terminus capping [14,15,16,17,18,19,20,21,22].
One particularly effective modification is fatty acid conjugation, also known as lipidation, wherein a fatty acyl chain is covalently attached to the peptide. Nature provides precedent for this approach. Several clinically used lipopeptide antibiotics, like daptomycin (a cyclic lipopeptide with an N-terminal decanoyl) [23] and polymyxins (containing hydrophobic fatty acyl tails), including polymyxin B [24] and colistin (polymyxin E) [25] (Figure 1) owe their potent membrane-disruptive activity in part to their lipid moieties.
In general, fatty monoacid derivatives are associated with a higher affinity to bind bacterial membranes and facilitate entry [26]. Drug bioactivity can also be enhanced through fatty acid-modified nanocarriers by fatty acid transporters, facilitating their passage across the intestinal barrier [27]. Fatty acids and monoglycerides, which are assigned to target the bacterial cell membranes, can use three modes of action through increasing the permeability of the membrane and inducing cell lysis, electron transport chain disruption, and oxidative phosphorylation uncoupling, and inhibiting the enzymatic activities of membranes and uptake of nutrients [28].
Figure 2 illustrates the effects of the peptide’s lipidation. Fatty acid conjugation can provide AMPs with greater hydrophobic character, promoting their association with bacterial membranes and improving uptake into target cells. Additionally, attaching a fatty acid can shield peptides from proteolytic enzymes and facilitate binding to plasma proteins like albumin, thereby extending the peptide’s plasma half-life [29]. Early studies showed that acylation of cationic AMPs often broadened their spectrum of activity [30,31].
Over the past decade, numerous modified lipopeptide AMPs have been designed, revealing how the length and nature of the fatty acid, as well as the site of attachment, critically influence antimicrobial efficacy and selectivity. In this review, we detail the mechanisms by which AMPs act on microbes and the rationale behind fatty acid modification. We then examine the effects of lipidation on antimicrobial activity, stability, and cell selectivity, drawing on recent examples from the literature. Key applications of FAMPs, including improved drug delivery, anti-biofilm properties, and activity against resistant pathogens, are highlighted. We also discuss safety and toxicity considerations, since increasing hydrophobicity can impact biocompatibility. Finally, we outline future directions and perspectives for FAMPs, such as optimizing fatty acyl chain properties and combination therapies. By integrating findings from the past decade with foundational studies, this review aims to provide a cohesive understanding of how fatty acid conjugation can enhance AMP therapeutic potential.
2. Mechanisms of Antimicrobial Peptide Action
AMPs exhibit diverse mechanisms of action (Figure 3) that collectively make them effective against a broad array of pathogens and less prone to resistance. The primary and best-studied mechanism is the disruption of microbial membranes [32]. Due to their cationic and amphipathic nature, AMPs are attracted to the negatively charged bacterial membrane surface and can insert into the lipid bilayer, leading to membrane perturbation and cell lysis.
This membrane-targeting action can occur via several models (carpet-like membrane coverage, toroidal pore formation, or barrel-stave pore formation), ultimately compromising the integrity of the pathogen’s cell envelope [33,34,35]. Cationic AMPs can modify membrane permeabilization. AMPs bind to bacterial membranes (rich in acidic phospholipids) and integrate into the lipid bilayer, leading to pore formation or wholesale membrane disruption. This causes leakage of cellular contents and cell death. Membrane depolarization and loss of barrier function are typically rapid, explaining the fast bactericidal action of many AMPs [36].
Membrane-disruptive mechanisms are fundamentally distinct from those of most conventional antibiotics, and thus, bacteria have difficulty developing resistance to them. The electrostatic interaction between positively charged AMPs and anionic bacterial phospholipids facilitates rapid binding, while the hydrophobic portions of the peptide insert into the lipid layer to induce permeability changes. Because this mode of action does not rely on a single protein target, even multidrug-resistant bacteria (which often have altered enzymes or ribosomes) remain susceptible to membrane-active AMPs [37,38].
In addition to membrane disruption, many AMPs can translocate into the cell and interact with internal targets. For example, some AMPs bind to nucleic acids or ribosomes, inhibiting DNA, RNA, or protein synthesis in the microbe [32]. Others interfere with the key enzymes or induce the production of reactive oxygen species inside the cell [39]. A number of AMPs also have immunomodulatory effects in the host, such as recruiting immune cells or neutralizing endotoxins [40]. This multi-faceted mode of action, attacking the pathogen from outside and inside, as well as enlisting host immunity, underlies why AMPs can kill even pathogens that are resistant to multiple drugs [11].
Certain AMPs can avoid or survive membrane translocation and then bind intracellular components. For instance, some proline-rich AMPs inhibit chaperone or protease functions inside bacteria, while others bind DNA or rRNA, halting replication and translation [32].
Beyond direct microbicidal effects, AMPs (especially those in host innate immunity, like defensins or cathelicidins) can modulate immune responses. They may attract immune cells to the infection site, promote wound healing, or neutralize microbial toxins (such as lipopolysaccharide from Gram-negative bacteria) [41,42].
Due to these mechanisms, AMPs retain activity against many drug-resistant strains. Even pathogens like MRSA or carbapenem-resistant A. baumannii that withstand conventional antibiotics can be susceptible to AMPs that disrupt membranes or target conserved cellular components [11]. Additionally, because AMPs attack fundamental structural features (e.g., the membrane), the emergence of resistance is less common; a bacterium would need to extensively remodel its membrane composition to evade AMP action, which comes at a high fitness cost [43]. Indeed, clinical lipopeptide antibiotics like daptomycin and polymyxin B remain effective against many otherwise resistant bacteria decades after their introduction, although cases of evolved resistance (via changes in cell envelope charge or composition) have been documented [44,45]. Overall, the multi-pronged mechanism of AMPs, especially membrane disruption, provides a strong foundation to build upon with chemical modifications such as fatty acid conjugation, aimed at further enhancing their antimicrobial performance.
3. Role and Rationale for Fatty Acid Conjugation to AMPs
3.1. Improving Proteolytic Stability and Half-Life
The concept of using fatty acids to extend peptide half-life was pioneered in drug development for diabetes (insulin detemir uses a C14 fatty acid to hitch onto albumin) [46] and has been applied to AMPs to achieve longer-lasting antimicrobial effects in vivo.
Fatty-acylation generally increases the proteolytic stability of AMPs. By blocking the N-terminus (a common site for exopeptidases) and sometimes by inducing self-association or membrane binding, lipidation protects peptides from enzymatic degradation. The result is often a significant increase in stability in serum or other biological fluids.
Zhong et al. (2020)’s lipidated peptides (C8-C12 on an anoplin analogue) were shown to be highly stable against challenges like trypsin digestion, serum proteases, high salt, and extreme pH. The lipopeptides retained their activity in the presence of physiological salt and serum, a crucial attribute for systemic use [47].
Liao et al. (2025) directly demonstrated enhanced serum stability of their fatty-acylated peptide B1 analogues. In practical terms, this means a lipidated AMP can persist longer in the bloodstream or at infection sites, maintaining its antimicrobial action over extended periods relative to the unmodified peptide [48].
A lipidated peptide may bind to serum proteins such as albumin. this not only prolongs circulation time but also reduces renal clearance. In 1990, the idea of peptide derivatization with a fatty acid was presented in order to extend the duration of peptide drugs’ action through albumin binding. This multifunctional reversible binding to albumin plays a main role in the transportation of endogenous fatty acids, in addition to other substances such as small-molecule drug compounds [26]. A similar concept has been used in designing Semaglutide is a modified glucagon-like peptide-1 (GLP-1) analogue engineered [49,50] for prolonged activity through strategic chemical modifications. It includes an acylation with a C18 fatty diacid attached via a spacer, which enhances reversible binding to serum albumin. This modification shields semaglutide from rapid enzymatic degradation (particularly by dipeptidyl peptidase-4) and significantly extends its half-life, enabling once-weekly dosing for the management of type 2 diabetes.
Another stability aspect is thermal and storage stability. The hydrophobic interactions introduced by fatty acids can sometimes stabilize the peptide’s tertiary structure or self-assembled state, making them less prone to unfolding [51]. Some lipopeptides have shown longer shelf-life or sustained activity upon storage, although this can vary.
3.2. Enhanced Cellular Uptake and Tissue Penetration
Fatty acid moieties can facilitate the uptake of peptides into cells or across biological barriers. The lipid tail may enable AMPs to traverse bacterial envelopes more easily or even penetrate host cell membranes if targeting intracellular infections [47]. There is also evidence that certain fatty acid transport mechanisms can ferry fatty-acylated molecules across barriers; for instance, fatty acid-modified nanocarriers can exploit intestinal fatty acid transporters to improve oral absorption. In essence, a fatty acid can act as a Trojan horse lipidic handle, taking advantage of the body’s normal lipid transport pathways [52]. Incorporating a medium-chain fatty acid into an AMP was shown to facilitate its translocation across the gut epithelium in this context [53]. Figure 4 illustrates the effect of fatty acid conjugation on crossing cell membranes.
Linear peptides composed of more than seven arginine residues were reported to enhance drug delivery in addition to maintaining the ability of cell penetration. The combination of acylation by long-chain fatty acids and cyclization on short arginine-containing peptides showed improvement in their ability of cell-penetrating property, probably through effective interaction between rigid, positively charged R and hydrophobic dodecanoyl moiety with the corresponding residues located in the cell membrane phospholipids [54].
3.3. Increasing Membrane Affinity and Potency
In most cases, adding a fatty acyl chain to an AMP significantly boosts its antimicrobial potency. Many lipidated peptides show lower MIC values and broader spectra compared to their parent (unmodified) peptides. Attachment of hydrophobic fatty acids to the N/C terminal or lysine residues of the AMPs through lipidation can improve their antimicrobial activity, such as daptomycin, colistin, and polymyxin B (Figure 1). The improvement in antimicrobial activity could be via enhanced interaction with cell membranes and increased proteolytic stability of the AMP [55]. The fatty acid acts as a lipid anchor, promoting insertion into the lipid bilayer of pathogens and thereby enhancing membrane disruption. Even a relatively short acyl chain can dramatically improve a peptide’s ability to partition into the membrane interface. This often translates to lower minimum inhibitory concentrations (MICs) and broader spectra of activity [56]. Polymyxin’s N-terminal fatty acid is essential for its potent membrane activity. Likewise, synthetic lipidated analogues of AMPs frequently show improved bactericidal potency compared to their non-lipidated counterparts.
The electrostatic interaction between AMPs and the anionic bacterial membrane surface allows the insertion of the hydrophobic portion of the fatty acid conjugates into the phospholipid layer, which explains the increased production of membrane disruption through conjugating with various lengths of fatty acid chains, because of the enhanced hydrophobicity of the peptides [57,58].
Liao et al. (2025) modified a 16-residue cationic peptide “B1” by attaching a hexanoic (C6) or octanoic (C8) acid to its N-terminus. The introduction of fatty acid chains into the naturally derived antimicrobial peptide B1 VKRFKKFFRKLKKSV-NH2 to B1-C6 (C5H11CO-VKRFKKFFRKLKKSV-NH2) and B1-C8 (C7H15CO-VKRFKKFFRKLKKSV-NH2) demonstrated their significant enhancement in antimicrobial activities against S. aureus and K. pneumoniae, with representative improved stability and biocompatibility. There were lower MIC values of B1-C6 and B1-C8 to four-fold against S. aureus and reached 4 μM from >32 μM against K. pneumoniae [48].
Avrahami and Shai (2002) demonstrated that linking lipophilic (fatty) acids to the N-terminal of magainin-2 analogue GIGKFLHSAKKWGKAFVGEIMNS-NH2 endows it with antifungal activity, an effect achieved without altering the peptide’s amino-acid sequence [59]. The conjugations brought about well-defined shifts in hydrophobicity, secondary structure, and self-association behavior. At physiological concentrations, the nature of the attached fatty chain determined the peptide’s structural outcome and proteolytic stability: Heptanoic acid produced a monomeric unordered structure. Undecanoic acid led to concentration-dependent oligomerization into α-helices. Palmitic acid yielded α-helical monomers, whose structure remained independent of peptide concentration and exhibited resistance to proteolysis. This modulation of hydrophobicity and assembly dynamics translated directly into antifungal potency. The undecanoic acid (C11) conjugate of the magainin 2 analogue lowered the MIC against C. neoformans from 25 µM in maganinin-2 from 25 to 3.1 µM. Palmitic acid (C16) conjugation also markedly enhanced antifungal activity against C. neoformans (MIC = 1.6 µM) but was less effective than C11 against C. albicans and A. fumigatus, while the shorter heptanoic acid (C7) chain produced only a modest improvement. These activity differences correlated with distinct structural effects where the C7 derivative adopted an unordered monomeric conformation, C11 induced concentration-dependent oligomeric α-helices, and C16 yielded concentration-independent monomeric α-helices.
The conjugation of undecanoic acid (UA) or palmitic acid (PA) to otherwise inactive diastereomers of magainin, specifically [D]-4-magainin, a magainin-2 analogue containing four D-amino acid substitutions ([D]-L6, [D]-K10, [D]-F16, [D]-E19, sequence: GIGKFlHSAkKWGKAfVGeIMNS-NH2) (lowercase letters = D-amino acids), also led to significant antimicrobial activation. In addition, they modified a short, weakly active diastereomeric lytic peptide composed of lysine and leucine, [D]-K₅L₇ (sequence: KKllKLLlKlLK-NH2, where lowercase L = D-leucine at positions 3, 4, 8, and 10. All resulting lipopeptides displayed potent activity against C. neoformans and various bacteria. The most potent derivative, [D]-K₅L₇-UA, exhibited broad-spectrum activity against E. coli, S. aureus, C albicans, and C. neoformans, with MIC values in the low micromolar range (1.5-6.2 μM for bacteria and yeast. The peptides also disrupted model membranes mimicking bacterial lipids (phosphatidylethanol (PE)/phosphatidylglycerol (PG)) and fungal lipids (phosphatidylcholine (PC)/PE/phosphatidylinositol (PI)/ergosterol), confirming that the fatty acid moiety enhanced peptide-membrane interactions, oligomerization, and insertion [60].
Aliphatic acids with different lengths (10, 12, 14, or 16 carbon atoms) attached to the N-terminus of a biologically inactive cationic peptide [D]-L6K6 resulted in active lipopeptides with lytic activity against bacterial and fungal cells. [D]-L6K6-decanoic acid showed activity against S. aureus (ATCC6538P), P. aeruginosa (ATCC 27853), and E. coli (ATCC 35218) with MIC values 6.25, 12.5, and 12.5 µM, respectively. [D]-L6K6-myristic acid and [D]-L6K6-palmitic acid were active against C. albicans (ATCC 10231), C. neoformans (ATCC 26430), and A. fumigatus (ATCC MY A-422) with MIC values 1.26, 0.78, and 1.56 µM [61] while [D]-L6K6 exhibited MIC values of >50 µg/mL in all bacterial and fungal strains.
The enhancement in activity is often dramatic for Gram-positive bacteria. A classic example is the peptide SC4 (KLFKRHLKWKII), a 12-mer helix-forming AMP. Lockwood et al. (2004) conjugated dodecanoic (C12) and octadecanoic (C18) fatty acids to the N-terminus of SC4, creating peptide-amphiphiles. The conjugation of dodecyl and octadecyl fatty acids to the N-terminus of the potent bactericidal, helix-forming peptide 12-mer SC4 resulted in a 30-fold increase in the bactericidal activity against Gram-positive strains (S. aureus, Streptococcus pyogenes, and Bacillus anthracis), including resistant strains of S. aureus when the resultant SC4 peptide-amphiphile molecules were tested. On the other hand, SC4 peptide-amphiphiles exhibited slight or no rise in bactericidal activity against the Gram-negative bacteria (E. coli and P. aeruginosa). These lipo-SC4 analogues also neutralized endotoxic lipopolysaccharide (LPS) from Gram-negative bacteria 3–6 fold more effectively. These effects reflected the advantages of the conjugated fatty acid chain to the SC4 peptide in the enhancement of membrane interactions, stabilization of helical structure in the membrane-bound state, and an increase in the bactericidal potency. Their activity against Gram-negative E. coli and P. aeruginosa was not significantly improved [62].
This highlights a pattern noted in several other studies. While lipidation boosts Gram-positive activity (due to easier penetration of the single membrane), Gram-negative efficacy can be limited by the outer membrane acting as a permeability barrier to overly hydrophobic compounds [63]. Some lipopeptides may aggregate or get sequestered by LPS in the outer membrane, reducing their ability to reach the inner membrane target in Gram-negative bacteria. To address this, either moderate chain lengths or cyclic/branched architectures (as in polymyxins) can be considered for Gram-negative activity. Nonetheless, by careful design, lipopeptides can achieve broad-spectrum action.
For instance, Zhong et al. (2020) created a series of anoplin analogues (GLLKRIKTLL-NH₂) with fatty acids of 8–12 carbons and observed potent activity against a range of multidrug-resistant bacteria. Synthetic peptides with conjugation of various lengths of fatty acid chains onto the side chain of the position 4 or 7 D-amino acid of Ano-D4,7 (analogue of anoplin with D-amino acid substitutions at positions 4 and 7 with D-lysine (k) instead of L-lysine (K)) showed excellent antimicrobial activity against a range of bacteria, especially multidrug-resistant bacteria. Peptides conjugated with fatty acid chains ranging from 8 to 12 carbons in length offered desirable selective antimicrobial activity in addition to anti-biofilm activity. Furthermore, the high stability to trypsin, serum, salts, and different pH environments, in addition to low tendency for bacterial resistance development, are the main advantages of those peptides. Ano-D4,7–4C12 (GLLk(C12)RIkTLL-NH2) showed activity with an MIC value of 4 μM against E. coli and P aeruginosa, and antimicrobial potency against other multidrug-resistant strains [47].
Several studies on previously weak or narrow-spectrum peptides demonstrate that lipidation can activate them. Avrahami and Shai showed that conjugating a C11 (undecanoic) or C16 (palmitic) acid to otherwise inactive diastereomeric peptides (including a magainin analogue with D-amino acids and a short Lys-Leu peptide) bestowed potent antibacterial and antifungal activities. Lipopeptides were derivatives of positively charged peptides comprising D- and L-amino acids (diastereomers), palmitoylated (PA) at their N terminus. The sequence K(4)-X(7)-W, where X designates Gly (G), Ala (A), Val (V), or Leu (L)(designated D-X peptides). The data revealed that palmitoyl–KKKKGGGGGGGW (D-form) (PA-D-G) and palmitoyl–KKKKAAAAAAAAW (D-form) (PA-D-A) exhibited strong antibacterial and antifungal activity despite their unmodified parent peptides (KKKKGGGGGGGW (D-form stereochemistry (D-G) and KKKKAAAAAAAAW (D-form stereochemistry) (D-A)) being essentially inactive (MIC > 100 µM) against S. aureus, B. Subtilis, P. aeruginosa, A. baumannii, and E. coli. Specifically, PA-D-G displayed MIC values of 12.5, 3.12, 6.25, 25, and 25 µM, respectively, against these bacterial strains, while PA-D-A showed MIC values of 12.5, 3.12, 6.25, 6.25, and 6.25 µM, respectively. The enhanced potency was attributed to increased cell membrane permeability and differences in oligomeric state and membrane insertion dynamics between variants [64].
Similarly, a fragment of human lactoferrin corresponding to residues 21–31 (LF12: FQWQRNIRKVR-homoserine lactone), which on its own exhibited only weak antibacterial activity (MICs of 40–200 µM against E. coli and 200–>600 µM against S. aureus), became dramatically more potent upon N-terminal acylation. Conjugation with a 12-carbon chain (LF12-C12) yielded the highest antibacterial potency, reducing MICs to 0.3–4 µM against E. coli and 8 µM against S. aureus in solution assays, representing 50- to 78-fold enhancements over the parent peptide. This derivative also showed a 14-fold increase in lipid A-binding affinity (Kd = 1.5 µM) and the strongest LPS-neutralizing capacity (ENC₅₀ = 2.43 µM), approaching the activity of polymyxin B. Activity declined for both shorter and longer acyl chains, establishing C12 as the optimal length for balancing hydrophobicity, antibacterial potency, and LPS-binding strength [65].
The multifunctional bovine lactoferricin (LfcinB) derivatives Lfcin4 (FKAWRWAWRWKKLAAPS) and Lfcin5 (FKAFRWAWRWKKLAAPS) were modified by conjugating the unsaturated fatty acid linoleic acid (18:2) to their N-terminus and further acylating their C-terminus. The resulting peptides of Lin-Lf4NH2 and Lin-Lf5NH2 displayed improved antibacterial activity, with MICs of 3.27 to 6.64 μM when compared to their parent peptides, with MICs of 1.83 to 59.57 μM against Staphylococcus hyicus. In a mouse skin infection model with S. hyicus, topical application of Lin-Lf4NH₂ and Lin-Lf5NH₂ significantly alleviated abscess symptoms. The abscess recovery rates for Lin-Lf4NH₂ (73.25%) and Lin-Lf5NH₂ (71.71%) were 38.8-fold and 37.9-fold higher, respectively, than in the untreated control group (1.89%), and were superior to those of Lf4NH₂ (46.87%) and Lf5NH₂ (58.75%) [66].
Mechanistically, the enhanced killing activity of lipidated peptides is linked to their ability to adopt an active α-helical conformation upon membrane interaction, thereby disrupting microbial membranes. For example, Mak et al. (2003) modified a synthetic peptide corresponding to residues 117–136 of human lysosomal cathepsin G (CG 117–136) by covalently attaching saturated, linear fatty acids to either its N- or C-terminus. Fatty acids with chain lengths of C8, C10, and C12 significantly increased bactericidal activity. The improved potency correlated with the peptide’s ability to form an α-helical structure in a membrane-like environment and to disrupt liposomal membranes. The fatty acyl moiety facilitated proper folding upon membrane contact, promoted membrane partitioning, and enhanced disruption of the bacterial cytoplasmic membrane. The MIC values of CG 117-136 and C-12-CG117-136 against two strains of S. aureus were decreased from ˃500 to 31.25 and 15.6 µg/ml, respectively, and from 500 to 7.8 µg/ml against Neisseria gonorrhoeae strain and 31.25 to 7.8 mg/l against P. aeruginosa [67].
In another study, Chu-Kung et al. (2004) demonstrated that lipidating antimicrobial peptides with lauric acid (C₁₂) can markedly improve their activity. Two peptides were tested: AKK (YGAA[KKAAKAA]₂) and SC4 (KLFKRHLKWKII). Conjugation of lauric acid to the N-terminus of these peptides resulted in substantial increases in antimicrobial potency compared to the unconjugated forms. For AKK, lipidation not only lowered the MIC from > 100 μM to approximately 6.25 μM but also significantly increased α-helicity in membrane-mimicking environments, a structural change that likely facilitated stronger interactions with lipid bilayers and enhanced membrane disruption. For SC4, lauric acid conjugation reduced the MIC from > 100 μM to about 3.1 μM, yielding potent activity without major structural alteration in α-helicity [68].
A dermaseptin derivative ALWKTLLKKVLKACONH₂ was systematically modified by increasing the length of its N-terminal acyl chain while stabilizing its α-helical structure. As the acyl chain lengthened, Gram-positive activity against S. aureus showed a gradual decline, intermediate chain lengths caused partial loss of activity, while longer chains resulted in a more pronounced reduction. Against E. coli, however, activity was reduced across all acylated derivatives. The parent peptide ALWKTLLKKVLKACONH₂ exhibited MIC values of 9 ± 3 μM against S. aureus and 4.5 ± 1.5 μM against E. coli. Acylation with different chain lengths altered these activities in distinct ways: C8-P (octanoyl) increased potency against S. aureus to 1.5 μM but reduced activity against E. coli to 6.25 μM. C10-P (decanoyl) yielded MIC values of 3.12 μM for S. aureus and 12.5 μM for E. coli [69].
Wakabayashi, Matsumoto et al. (1999) designed N-acylated and D-enantiomer derivatives based on a short antimicrobial motif from bovine lactoferricin B. From this, they derived the core active fragment RWQWRMKK. In CH3(CH2)8CO-D-(RRWQWRMKK)-NH2 (Acyl-10-D-(4–12)-NH₂), this D-peptide was N-terminally modified with a 10-carbon fatty acid chain (decanoyl) and C-terminally amidated to improve stability and activity. he lipidated D-form showed markedly improved antimicrobial potency, with MIC values (μg/ml) of 3 (E. coli), 6 (P. aeruginosa), 3 (S. aureus), 12 (C. albicans), and 6 (Trichophyton mentagrophytes), compared to the full-length lactoferricin B, which had MICs of 6, 12, 12, 25, and 25 μg/ml, respectively. These results demonstrate that N-acylation with a C10 fatty acid, combined with D-amino acid substitution, significantly enhanced activity against Gram-negative bacteria, Gram-positive bacteria, and fungi [70].
Chu-Kung, Nguyen et al. (2010) demonstrated that conjugating fatty acids to the amphipathic peptide YGAAKKAAKAAKKAAKAA markedly enhanced its antimicrobial potency. Attachment of lauric acid (C12) significantly improved activity against both Gram-negative (E. coli DH5α and ML-35) and Gram-positive (Staphylococcus epidermidis) bacteria. Conjugation with myristic acid (C14) further enhanced efficacy, reducing the minimum bactericidal concentration (MBC) from >65 μM (unmodified peptide) to 4 μM for E. coli DH5α, 5 μM for E. coli ML-35, and 26 ± 2.5 μM for S. epidermidis. This enhanced activity was attributed to the increased affinity of the lipidated peptides for lipid membranes, achieved through strengthened hydrophobic interactions imparted by the fatty acid moiety. These stronger membrane interactions likely promoted peptide insertion, destabilization of the bilayer, and subsequent bacterial killing [71].
In another study, Li et al. (2023) investigated fatty acid modifications of the antifungal peptide CGA-N9 (RILSILRH) and found that conjugation with n-octanoic acid (C8) produced the most potent analogue (Figure 5). This derivative, CGA-N9-C8, exhibited a minimum inhibitory concentration (MIC) of 7.8 ± 0.03 µg/mL against C. albicans, compared to 246.4 ± 2.3 µg/mL for the unmodified peptide. In addition to its greatly improved planktonic antifungal activity, CGA-N9-C8 showed the strongest capacity among all tested chain lengths to inhibit biofilm formation and eradicate mature biofilms. It also demonstrated high stability against protease hydrolysis in serum [72].
These examples illustrate that lipidation can turn marginal peptides into effective antimicrobials, often expanding their target range to include fungi and yeast in addition to bacteria.
3.4. Increasing Selectivity and Reducing Toxicity
A crucial goal in AMP design is to maximize bacterial cell toxicity while minimizing harm to host (human) cells. Modulating selectivity is a multifaceted aspect of fatty acid conjugation. On one hand, increasing a peptide’s hydrophobicity tends to increase its activity against bacteria, but it can also raise its propensity to interact with and disrupt mammalian cell membranes (causing cytotoxicity or hemolysis). Thus, there is an optimal window of hydrophobicity for selective antimicrobial action [73].
Many studies report that medium-length fatty acids confer the best selectivity. For example, the anoplin analogues with C8–C12 chains [47] were described above as having desirable selective antimicrobial activity, meaning they were strongly antibacterial yet had relatively low toxicity toward mammalian cells. In contrast, peptides with longer acyl chains sometimes showed a loss of activity or selectivity.
Radzishevsky et al. (2005) systematically varied acyl chain length on a dermaseptin peptide and found that while intermediate chain lengths (around C10–C12) were optimal for killing S. aureus, longer chains (C16 and above) caused the peptide to aggregate and resulted in a total loss of activity against that bacterium [69]. Moreover, all acyl derivatives in that study lost activity against E. coli, likely due to aggregation and possibly increased self-association that prevented effective membrane interaction. Such aggregation could also translate to a higher tendency to stick to serum components or to cell membranes nonspecifically, which can increase cytotoxic side effects.
The lesson is that excess hydrophobicity can be counterproductive, promoting self-assembly of peptides in solution (forming inert aggregates) and increasing nonspecific binding to eukaryotic membranes [73]. Indeed, one report noted that overly hydrophobic lipopeptides showed reduced potency precisely because they aggregated; the investigators were able to restore potency by using unsaturated fatty acids that made the aggregates less stable and allowed the lipopeptides to disaggregate in the presence of bacteria [73]. This clever tweak (using, e.g., oleoyl vs stearoyl chains) improved the therapeutic index of those lipopeptides.
Several lipidated AMPs have shown excellent safety profiles in both in vitro and in vivo studies. Posa et al. (2023) designed truncated analogues of battacin, an antimicrobial lipopeptide produced by Paenibacillus species, by introducing novel N-terminal fatty acid moieties. The parent lipopeptide battacin (Figure 6), structurally related to polymyxins (Figure 1), contains 2,4-diaminobutyric acid (Dab) residues and a long-chain fatty acid at its N-terminus. In acute toxicity tests in mice, battacin displayed an LD₅₀ value 2.3-fold higher than that of polymyxin B, indicating lower acute toxicity. These truncated, non-cyclic battacin analogues retained potent antibacterial activity comparable to polymyxin B while exhibiting significantly reduced toxicity. This demonstrates that simplified, linear lipopeptide structures can preserve the antimicrobial potency of more complex cyclic peptides while improving their safety profile [74].
Likewise, Li et al. (2023)’s octanoylated CGA-N9 peptide achieved the optimal balance of potency and biosafety, outperforming other chain lengths in killing fungi while being the least hemolytic to human cells. Moderate chain lengths like C8 in that case were key to high selectivity [72].
We should note that some FAMPs can still be quite harsh on mammalian cells if the hydrophobicity is too great or if the peptide inherently has some toxicity. Hemolysis (lysis of red blood cells) is a common measure of AMP cytotoxicity. many lipopeptides show a threshold concentration above which hemolysis sharply increases. For instance, lauroyl and myristoyl analogues of certain peptides were found to have acceptable hemolysis at their active concentrations, whereas palmitoyl analogues caused unacceptable hemolysis, aligning with the idea that C12–C14 might be a “sweet spot” for selectivity [69].
The inclusion of D-amino acids in the peptide (making them less recognizable to proteases) in combination with fatty acids has also been used to improve stability without raising toxicity; D-amino acids sometimes reduce recognition by host receptors, possibly mitigating unwanted immunogenic responses [47].
In summary, fatty acid conjugation generally enhances antimicrobial efficacy and stability of AMPs. However, achieving optimal selectivity requires tuning the fatty acid properties. With careful design (appropriate chain length, saturation, and attachment position), one can greatly improve the therapeutic index of AMPs: killing microbes more effectively while keeping host cell toxicity low. Table 1 provides a summary of selected studies that exemplify these effects, highlighting the peptide in question, the fatty acid used, method of conjugation, target organisms, and outcomes on activity and safety.
4. Applications in Drug Delivery, Biofilms, and Resistant Strains
Beyond general improvements in activity and stability, FAMPs have enabled specific applications and advantages in challenging contexts such as drug delivery across barriers, eradication of biofilms, and tackling infections by highly resistant organisms.
4.1. Enhanced Drug Delivery and Pharmacokinetics
Lipidation can modify the pharmacological profile of AMPs, allowing novel routes of administration or better tissue targeting. One application is oral delivery. Normally, peptide drugs are poorly absorbed in the gastrointestinal tract due to their polar nature and enzymatic degradation. However, FAMPs may be absorbed via mechanisms akin to dietary lipids [75].
Huang et al. (2024) showed that fatty acid-modified liposomes could encapsulate bioactive peptides and improve their stability and absorption via intestinal fatty acid transport pathways. This concept suggests that an orally administered lipopeptide might hitch a ride through enterocytes, potentially reaching systemic circulation. While clinical translation is yet to be realized, the prospect of oral AMP formulations is on the horizon [27].
Additionally, the strong binding of many lipopeptides to serum albumin can be leveraged for extended release. Once bound to albumin, a FAMPcan circulate longer and act as a reservoir that slowly releases the active peptide to tissues [26,29]. This is advantageous for maintaining therapeutic levels over time from a single dose (reducing dosing frequency). Moreover, albumin-bound lipopeptides might preferentially accumulate in infection sites where albumin tends to extravasate (e.g., inflamed tissues), thereby delivering the peptide where it's most needed.
Lipidation also enables self-assembly [73] into nanoparticles or micelles, which can be useful in drug delivery. Some FAMPs spontaneously form micellar or vesicular structures in aqueous environments at sufficient concentrations. These nano-aggregates can potentially carry hydrophobic drugs or imaging agents, functioning as dual-action antimicrobial nanocarriers. Although this is currently a subject of interest, one could envision a lipopeptide that not only attacks bacteria but also delivers a co-encapsulated antibiotic or adjuvant directly to a biofilm or infection site.
4.2. Anti-Biofilm Activity
Bacterial biofilms, communities of bacteria embedded in a protective extracellular matrix, are notoriously resistant to antibiotics and immune clearance [76]. AMPs are known for their ability to penetrate and disrupt biofilms [77], and fatty-acylation can further enhance this property. The hydrophobic tail may help lipopeptides to insert into the dense biofilm matrix and reach deeply embedded bacteria. For example, the octanoylated CGA-N9 peptide [72] discussed earlier was not only potent against C. albicans planktonic cells but also showed the highest efficacy in preventing biofilm formation and in eradicating pre-formed biofilms among the tested analogues. Its fatty acid likely aided in breaching the polysaccharide-rich biofilm layers of Candida. Likewise, Zhong et al. (2020) [47] reported that their C8–C12 lipopeptides had significant antibiofilm activity against P. aeruginosa, a pathogen known for robust biofilm-based resistance.
Natural lipopeptides, like daptomycin, are already used against biofilm-associated infections (e.g., endocarditis caused by biofilm-forming staphylococci). New FAMPs could be employed as surface-coating agents on medical devices to prevent biofilm formation [78], or as therapeutics to disperse established biofilms [79] on implants or tissues. Because lipopeptides can both kill bacteria and perturb biofilm matrix (due to their surfactant-like properties), they are attractive candidates for such roles. Additionally, some lipopeptides can interfere with quorum sensing in biofilms, indirectly inhibiting biofilm development (this has been observed with certain cyclic lipopeptides from Bacillus species) [80].
4.3. Combating Resistant Strains
The impetus for AMP development is strongest in the context of drug-resistant bacteria. FAMPs have shown efficacy against a spectrum of resistant pathogens: MRSA, vancomycin-resistant enterococci (VRE), carbapenem-resistant Gram-negatives, and others. For instance, the lauric acid-conjugated peptide AKK from Chu-Kung’s study [68] was active against E. coli strains and S. epidermidis that are often resistant to multiple drugs. The SC4 lipopeptide was active against MRSA and even B. anthracis, which is a potential biothreat organism. Some designed lipopeptides, like those discussed by Avrahami & Shai [60,64] were able to kill not only bacteria but also opportunistic fungi, highlighting their utility against organisms (like Candida or Aspergillus) that cause infections in immunocompromised or hospitalized patients.
Another interesting angle is resistance modulation. Bacteria exposed long-term to sublethal levels of lipopeptides might adapt by altering their membrane fatty acid composition. A recent study by Rasković et al. (2025) [81] found that growing S. aureus in the presence of oleic acid (C18:1) led to changes in the bacterial membrane glycolipid fatty acid profile and increased resistance to AMPs. This suggests that bacteria can sometimes respond to external fatty acids by stiffening or changing their membranes to become less susceptible. While this is a potential concern, it also provides insight that the interplay between bacterial membrane adaptation and lipopeptide structure is complex. Using unsaturated fatty acids in FAMPs [81] might make it harder for bacteria to develop resistance, or combination therapies could be used to prevent such adaptation.
In practical terms, FAMPs are being explored for treating infections where conventional antibiotics fail. For example, polymyxin-resistant P. aeruginosa (which has modifications in LPS that reduce polymyxin binding) might still be targeted by a different FAMP that circumvents that mechanism [82]. Some lipopeptides also synergize with conventional antibiotics. By disrupting the membrane, they can increase antibiotic uptake. There is ongoing research into conjugates that link an AMP with an antibiotic (sometimes through a fatty spacer) to get a dual-function molecule [83,84].
4.4. Topical and Targeted Applications
Many FAMPs are particularly well-suited for topical use (skin infections, wound coatings, etc.) because the added hydrophobicity helps them persist at the site of application. Liu et al. (2021) [66] demonstrated that a linoleoyl-lactoferricin analogue could effectively treat a Staph. hyicus skin infection in mice. Its lipid tail likely helped it partition into skin layers and resist being washed away, providing sustained local antimicrobial activity.
In a hospital context, there is a potential that lipopeptide formulations could be used for decolonization of MRSA [85] (e.g., nasal swabs), as they might be more potent and longer-lasting than non-lipidated counterparts.
Another emerging application is using lipopeptides in nanoparticle drug delivery systems [86]. A FAMP can be anchored on the surface of a nanoparticle (liposome, niosome, polymeric nanoparticle) to serve as a targeting ligand that directs the nanoparticle to bacteria. The fatty acid part can insert into the lipid bilayer of a liposomal nanoparticle, effectively decorating it with AMPs that then target and disrupt bacterial cells upon contact. This approach can combine the advantages of both nanomedicine (protection and co-delivery of drugs) and the active targeting/killing ability of AMPs [87].
Fatty acid conjugation broadens the applicability of AMPs in several domains. It enables improved delivery (longer circulation, potential oral uptake, self-assembly into delivery vehicles), empowers AMPs against biofilms (through enhanced penetration and disruption of matrices), and reinforces the fight against resistant pathogens (by boosting potency and sometimes evading known resistance mechanisms). These applications are particularly relevant as we face more chronic infections (like device-associated biofilms) and aggressive, resistant outbreaks where traditional antibiotics are impotent.
5. Safety and Toxicity Considerations
While fatty-acylation can enhance AMP efficacy, it is imperative to consider safety and toxicity, as modifications may affect how these molecules interact with host tissues. The balance between hydrophobicity-driven potency and potential host cell damage is a central theme in FAMP development.
5.1. Hemolysis and Cytotoxicity
A common side effect of cationic amphipathic peptides (especially when made more hydrophobic) is lysis of red blood cells or toxicity to mammalian cell membranes [88]. Fatty acid conjugation can increase this risk if the peptide becomes too membrane-active indiscriminately. Thus, extensive testing is required to determine the concentration at which a lipopeptide causes unacceptable hemolysis or kills human cell lines. A lipopeptide might have an MIC of 5 μM against bacteria but start causing significant hemolysis at, say, 20 μM. The goal is to design lipopeptides with a high therapeutic index (ratio of host toxic dose to bacterial inhibitory dose).
Dawgul et al reported that moderate chain lengths (C10-C12) often yield lipopeptides that maintain a good safety margin [89]. These molecules are hydrophobic enough to target bacterial membranes (which are rich in negatively charged lipids and have a high surface area-to-volume ratio) but not so hydrophobic that they indiscriminately disrupt the more cholesterol-rich, zwitterionic membranes of human cells. Additionally, the initial electrostatic attraction of cationic AMPs to bacteria (absent in neutral mammalian cell surfaces) provides some selectivity, a feature retained in FAMPs. As long as the peptide remains sufficiently cationic and does not aggregate in serum, it tends to preferentially bind to bacteria over erythrocytes.
5.2. Immune Response and Allergenicity
Fatty acids are naturally occurring moieties, so adding them to peptides typically does not introduce obvious immunogenic functional groups. However, any altered peptide could potentially be seen as foreign by the immune system. To the best of our knowledge, there have not been reports of strong antigenicity specifically due to fatty-acylation of AMPs, but it’s an area to monitor, especially for repeated therapeutic use.
Some lipopeptides (like tripalmitoyl-S-glycerylcysteine used in vaccines) are known to be potent immune stimulators via Toll-like receptors [90]. Shorter lipopeptides like those in this review are less likely to trigger such responses, but one should be aware that lipidated peptides might interact with immune cells differently. On the beneficial side, certain lipopeptides can serve as self-adjuvanting antimicrobial agents, enhancing immune clearance of infections [91].
5.3. In Vivo Toxicity and Pharmacology
The in vivo behavior of lipopeptides can differ from in vitro. A peptide that is non-toxic to cultured cells might still cause issues in an organism due to distribution or accumulation in certain organs. Polymyxin B and colistin, for example, can cause kidney toxicity at higher doses due to accumulation in renal tissue [92]. Structural analogues that are too hydrophobic risk similar outcomes. Some new FAMPs have shown improved safety in animal models. The battacin analogues mentioned earlier retained potent antibacterial action but with reduced kidney and overall toxicity compared to polymyxins [74]. By removing some positively charged residues and introducing a different fatty acid, the designers likely reduced non-specific binding to kidney cells and avoided the nephrotoxic mechanism of polymyxin (which is linked to its specific fatty acid and cyclic structure). This indicates that judicious modification can yield lipopeptides that are both effective and safer.
Another safety consideration is the effect on the host’s normal flora. A very broad-spectrum, potent FAMP might wipe out beneficial microbiota if used systemically, similar to broad antibiotics [93]. While not a toxicity in the classical sense, this dysbiosis could have downstream health consequences. Therefore, targeted delivery (like inhalation for lung infections, topical for skin, etc.) might be preferred for FAMPs to limit systemic exposure. If used systemically, it may be advantageous if the FAMP is selectively active (for instance, some are more active on Gram-negatives vs Gram-positives or vice versa, which might spare certain flora).
5.4. Bacterial Resistance to FAMPs
From a safety perspective for long-term use, one should consider if bacteria can develop resistance to these FAMPs, and if so, what the implications are. While AMPs are generally considered less prone to resistance, prolonged sub-lethal exposure can select for resistant subpopulations. Some mechanisms include changes in membrane charge (e.g., S. aureus adding alanine to teichoic acids or E. coli modifying LPS to be less anionic) to repel cationic peptides, or upregulation of efflux pumps [94,95,96]. The presence of exogenous fatty acids might enable bacteria to modify their own membrane composition. The earlier noted study by Rasković et al. (2025) [81] showed S. aureus could incorporate oleic acid into its membrane lipids and become more rigid, thus more resistant to certain AMPs. This suggests that high concentrations of fatty acids in the environment (for instance, on skin or in certain diets) might induce a temporary state of AMP tolerance. However, such adaptations often come at a fitness cost and may be reversible.
In developing lipopeptides as drugs, researchers often perform serial passage experiments to see if resistance emerges. Many FAMPs require multiple, multi-step mutations for bacteria to grow in their presence, and often those mutants have impaired virulence or growth. This is promising, but it highlights the importance of using these agents at adequate doses to fully eradicate infections and minimize the chance of resistance development.
5.5. Mitigating Toxicity
A number of strategies can be used to mitigate the potential toxicity of FAMPs. As noted, unsaturated chains (like oleic, linoleic) can reduce peptide aggregation and possibly make the interaction with membranes more “fluid” and less damaging to host cells [73]. Branched lipids might similarly disturb bacterial membranes effectively while being less able to insert into tightly packed cholesterol-rich domains of animal cell membranes.
A promising strategy to increase selectivity is to design environmentally responsive lipopeptides. For example, LL-37 conjugated with oleic acid formed inactive structures at physiological pH but transformed into active antimicrobial aggregates at acidic pH, mimicking infected tissue conditions [97].
Using FAMPs in a localized manner (inhalation for lung infections, topical gels for wound or skin infections, bladder instillation for urinary tract infections, etc.) can maximize the drug concentration at the target site while minimizing systemic exposure. This takes advantage of the potent action of lipopeptides and circumvents some toxicity concerns.
If a FAMP is used in combination with a traditional antibiotic or another AMP, sometimes a synergistic effect is observed (like membrane disruption improving antibiotic uptake) [98]. This could allow using a lower dose of the FAMP, reducing toxicity risk. Combining AMPs with conventional antibiotics or biofilm matrix-degrading enzymes is a potential strategy to enhance efficacy [11].
In conclusion, safety considerations are integral when advancing FAMPs toward clinical use. While these molecules can be extremely potent antimicrobials, their design must strike a careful balance to avoid harm to the host. The encouraging news from recent studies is that through molecular design, altering fatty acid structure, peptide sequence, and dosing strategies, it is feasible to create lipopeptides with a high degree of selectivity. Ongoing research continues to refine these properties, bringing us closer to FAMP therapeutics that are both effective and safe.
6. Future Perspectives
The field of FAMPs is rapidly evolving, and several future directions hold promise for enhancing their therapeutic potential. Future research will delve deeper into finding the “perfect” lipid tail for each peptide. This includes exploring unsaturated, branched, or cyclic fatty acids as alternatives to linear saturated chains. As discussed, unsaturated tails can improve potency by preventing overly tight self-assembly [73]. Branched chains might reduce toxicity by altering how the lipid inserts into membranes. Even aromatic fatty acid analogues or steroidal lipids could be tried to impart unique properties. By systematically varying lipid chemistry, one can map structure-activity relationships and identify motifs that maximize bacterial killing while minimizing aggregation and toxicity.
Different pathogens might be best targeted by different FAMP designs. For example, Gram-negative bacteria with robust outer membranes might require either shorter or more polar fatty acids. On the other hand, persistent Gram-positive pathogens or mycobacteria might benefit from longer, more hydrophobic tails to penetrate thick cell walls or mycolic acid layers. In the future, we may see pathogen-specific lipopeptides, where the fatty acid and peptide are co-optimized for, say, Pseudomonas lung infections versus Staphylococcal skin infections.
Another perspective is using FAMPs in combination or as part of hybrid molecules. One concept is an AMP-antibiotic conjugate, for instance, linking a FAMP with a conventional antibiotic in a single molecule. The AMP part could target the membrane and increase uptake of the attached antibiotic into bacteria. There have been initial studies along these lines (e.g., a peptide tethered to a β-lactam antibiotic) showing potent activity [99]. Additionally, combining FAMPs with other antimicrobial strategies, like peptidomimetic polymers or quorum quenchers, could yield synergistic effects. Girdhar et al. (2024) also suggested that structural modifications (like lipidation) combined with using AMPs alongside traditional drugs and biofilm matrix inhibitors is a compelling future strategy [11].
Building on the self-assembly of FAMPs, we may see nanostructured delivery systems where lipopeptides form the backbone. For instance, peptide amphiphiles can form nanofibers or micelles that could be used to coat wounds or surgical sites, releasing AMPs over time to prevent infection. Hydrogels containing FAMPs might serve as wound dressings that both seal a wound and continuously elute antimicrobials. Inhalable aerosols of lipopeptide nanoparticles might be developed for respiratory infections. The fatty acid component could help nanoparticles fuse with bacterial membranes on contact, acting as a sort of seek-and-destroy targeting mechanism.
Future research will also focus on understanding and circumventing any resistance mechanisms that bacteria employ against FAMPs. This could involve studying transcriptomic responses of bacteria exposed to sublethal FAMP levels to see what defenses are activated (e.g., do they alter fatty acid synthesis, efflux pumps, etc.). With that knowledge, next-gen FAMPs can be designed to avoid triggering those defenses or to counteract them. Additionally, monitoring for cross-resistance (e.g., if use of a FAMP induces resistance to a natural host defense peptide, or vice versa) will be important if these agents are to be widely used.
On the practical side, translating FAMPs into approved therapeutics will require extensive toxicological evaluation and likely modifications to improve pharmaceutical properties. Many peptides have issues with solubility or stability in formulation, especially when hydrophobic. Efforts will be directed at finding formulation excipients or prodrug forms that make FAMPs more drug-like (perhaps using salt forms, or encapsulating in liposomes to mitigate any local irritation). As some candidates advance to clinical trials, their performance will inform the field on what designs work in human medicine. The success of daptomycin and polymyxin (despite their limitations) provides a precedent, and newer FAMPs aim to be even safer and effective.
A forward-looking idea is to engineer lipopeptides that have additional functionalities, such as immune modulation or wound-healing properties. For example, a FAMP that not only kills bacteria but also promotes tissue regeneration or reduces excessive inflammation could be valuable for infected wounds or burns. This could be achieved by conjugating sequences that have immunomodulatory activity together with fatty acids. The fatty acid might also help target the peptide to certain cell types (for instance, some immune cells uptake fatty acid-tagged molecules).
Advances in Artificial Intelligence (AI), especially machine learning (ML) and deep learning (DL), are revolutionizing the discovery and optimization of AMPs, yet AI-driven FAMPs design remains underexplored. Although much of fatty acid optimization remains experimental, AI can be leveraged to predict optimal lipid characteristics (e.g., chain length, hydrophobicity, branching) that maximize antimicrobial potency without excessive toxicity.
QSAR and descriptor-based models already offer insights into how fatty-acid properties (chain length, hydrophobicity) influence activity and toxicity [100]. Experimental datasets, such as those characterizing how tail length affects membrane disruption, hemolysis, or aggregation, can be used to train ML models that predict a “sweet spot” in lipid tail properties. Recent molecular dynamics studies (e.g., SNAPP variants with C6, C12, C18 chains) demonstrated a C12 sweet-spot for membrane disruption and stability [101]. While documented cases of AI directly choosing fatty acid moieties are rare, this conceptual framework is rapidly emerging as a powerful computational strategy.
Generative AI platforms like AMP-Designer (foundation-model-based) [102] and AMPGen (diffusion-driven) [103] have recently produced novel AMPs with high validation rates, stability, and low toxicity in rapid design-to-validation cycles. Structure-aware ML also enhances prediction accuracy by addressing α-helical versus coiled conformations [104]. Collectively, these tools demonstrate how AI can propose peptide sequences optimized for multiple properties, potent antimicrobial activity, low toxicity, and phenotypic stability, effectively accelerating design cycles before wet-lab synthesis.
We propose the next frontier: integrating AI-driven sequence generation and multi-objective optimization with fatty-acid design. A hybrid framework could predict optimal lipid tail features (e.g., chain length, branching, conjugation chemistry) and AMP sequence simultaneously, balancing antimicrobial potency, stability, selectivity, and self-assembly behavior. Leveraging lipidation’s known PK and albumin-binding advantages, combined with data-driven design tools from liposomal ML models [105], this approach promises to streamline FAMP development from in silico design to preclinical validation.
AI is also starting to influence formulation and self-assembly prediction. ML models, for example, have been successfully utilized in liposome manufacturing to forecast critical quality attributes like size and encapsulation efficiency, suggesting promising potential for future adaptation for FAMP delivery applications[106]. Advances in formulation and quantum ML–based structure prediction offer a promising pathway. Consequently, integrating ML predictions for micelle formation, aggregation behavior, or excipient interactions may soon become feasible, further bridging molecular design with practical delivery platforms.
In summary, the future of FAMPs lies in refinement and integration. Refinement of molecular design will yield lipopeptides optimized for specific uses with minimal downsides. Integration refers to combining FAMPs into broader therapeutic regimens or multi-functional constructs. With the continued rise of AMR, the impetus to innovate in this area is strong. Interdisciplinary efforts drawing on peptide chemistry, lipid biology, pharmacology, and clinical medicine will drive the next wave of FAMP development.
Conclusion
Fatty acid conjugation has emerged as a powerful strategy to enhance the therapeutic potential of antimicrobial peptides. By attaching hydrophobic lipid chains to AMPs, researchers have been able to substantially improve antimicrobial potency, broaden the spectrum of activity, and increase the stability of peptides in biological environments. The acylated lipopeptide analogues leverage improved membrane interactions and extended half-life (often via albumin binding) to overcome many limitations of native peptides. As documented in numerous studies, fatty-acylation can convert modest peptides into potent antimicrobials, endow peptides with anti-biofilm capabilities, and retain efficacy against drug-resistant pathogens.
However, successful implementation of this strategy requires careful optimization to maintain selectivity and safety. The design of FAMPs involves balancing hydrophobic and hydrophilic characteristics to achieve strong bacterial killing without significant host toxicity. Lessons from past research confirm the importance of choosing an appropriate fatty acid (in terms of length and saturation) and attachment site to reach this balance. Indeed, small changes in the lipid moiety can tilt the scale between an efficacious drug and a hemolytic compound. The optimization of all parameters is required for the production of AMP based on structure activity equation. The significant point in the modulation of AMPs is studying the differences in bacterial and eukaryotic membranes' structure, in addition to the characterization of the specific mode of action of the target peptide design.
Overall, fatty acid conjugation expands the chemical space and functionality of antimicrobial peptides, offering a versatile platform for next-generation anti-infectives. With continued innovation, from fine-tuning molecular designs to developing advanced delivery methods, FAMPs are poised to play an important role in the antibiotic arsenal. As we face an era of increasing antimicrobial resistance, such engineered peptides provide a timely and adaptable solution. They are inspired by nature’s own lipopeptide antibiotics yet are readily customizable in the laboratory to meet modern clinical needs. Continued research and collaborative efforts will pave the way for translating these scientific advancements into safe and effective therapies, helping to secure our ability to combat infections in the years to come.
Author Contributions
Conceptualization, N.M.H. and K.P.; writing—original draft preparation, N.M.H. and K.P.; review and editing, N.M.H. and K.P. All authors have read and agreed to the published version of the manuscript.
Funding
Conceptualization, N.M.H. and K.P.; writing—original draft preparation, N.M.H. and K.P.; writing—review and editing, N.M.H., D.D.D., and K.P; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare (Basel) 2023, 11. [CrossRef]
- Ho, C.S.; Wong, C.T.H.; Aung, T.T.; Lakshminarayanan, R.; Mehta, J.S.; Rauz, S.; McNally, A.; Kintses, B.; Peacock, S.J.; de la Fuente-Nunez, C.; et al. Antimicrobial resistance: a concise update. Lancet Microbe 2025, 6, 100947. [CrossRef]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. The Review on Antimicrobial Resistance; 2014.
- Guan, L.; Beig, M.; Wang, L.; Navidifar, T.; Moradi, S.; Motallebi Tabaei, F.; Teymouri, Z.; Abedi Moghadam, M.; Sedighi, M. Global status of antimicrobial resistance in clinical Enterococcus faecalis isolates: systematic review and meta-analysis. Ann Clin Microbiol Antimicrob 2024, 23, 80. [CrossRef] [PubMed]
- Gelalcha, B.D.; Gelgie, A.E.; Kerro Dego, O. Antimicrobial resistance and prevalence of extended-spectrum beta-lactamase-producing Klebsiella species in East Tennessee dairy farms. Microbiol Spectr 2024, 12, e0353723. [CrossRef] [PubMed]
- Ivanova, M.; Ovsepian, A.; Leekitcharoenphon, P.; Seyfarth, A.M.; Mordhorst, H.; Otani, S.; Koeberl-Jelovcan, S.; Milanov, M.; Kompes, G.; Liapi, M.; et al. Azithromycin resistance in Escherichia coli and Salmonella from food-producing animals and meat in Europe. J Antimicrob Chemother 2024, 79, 1657-1667. [CrossRef] [PubMed]
- Mehari, M.G.; Yeshiwas, A.G.; Esubalew, D.; Azmeraw, Y.; Delie, A.M.; Limenh, L.W.; Worku, N.K.; Hailu, M.; Melese, M.; Abie, A.; et al. Dominance of antimicrobial resistance bacteria and risk factors of bacteriuria infection among pregnant women in East Africa: implications for public health. J Health Popul Nutr 2025, 44, 98. [CrossRef]
- Danielsen, A.S.; Franconeri, L.; Page, S.; Myhre, A.E.; Tornes, R.A.; Kacelnik, O.; Bjørnholt, J.V. Clinical outcomes of antimicrobial resistance in cancer patients: a systematic review of multivariable models. BMC Infect Dis 2023, 23, 247. [CrossRef]
- Ntim, O.K.; Awere-Duodu, A.; Osman, A.H.; Donkor, E.S. Antimicrobial resistance of bacterial pathogens isolated from cancer patients: a systematic review and meta-analysis. BMC Infect Dis 2025, 25, 296. [CrossRef]
- Bucataru, C.; Ciobanasu, C. Antimicrobial peptides: Opportunities and challenges in overcoming resistance. Microbiol Res 2024, 286, 127822. [CrossRef]
- Girdhar, M.; Sen, A.; Nigam, A.; Oswalia, J.; Kumar, S.; Gupta, R. Antimicrobial peptide-based strategies to overcome antimicrobial resistance. Arch Microbiol 2024, 206, 411. [CrossRef]
- Gani, Z.; Kumar, A.; Raje, M.; Raje, C.I. Antimicrobial peptides: An alternative strategy to combat antimicrobial resistance. Drug Discovery Today 2025, 30, 104305. [CrossRef] [PubMed]
- Elbediwi, M.; Rolff, J. Metabolic pathways and antimicrobial peptide resistance in bacteria. J Antimicrob Chemother 2024, 79, 1473-1483. [CrossRef] [PubMed]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front Microbiol 2020, 11, 563030. [CrossRef]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim Biophys Sin (Shanghai) 2017, 49, 916-925. [CrossRef] [PubMed]
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: influences on stability and bioactivity. Chembiochem 2013, 14, 617-624. [CrossRef]
- Li, D.; Yang, Y.; Li, R.; Huang, L.; Wang, Z.; Deng, Q.; Dong, S. N-terminal acetylation of antimicrobial peptide L163 improves its stability against protease degradation. J Pept Sci 2021, 27, e3337. [CrossRef]
- Lohan, S.; Konshina, A.G.; Mohammed, E.H.M.; Helmy, N.M.; Jha, S.K.; Tiwari, R.K.; Maslennikov, I.; Efremov, R.G.; Parang, K. Impact of stereochemical replacement on activity and selectivity of membrane-active antibacterial and antifungal cyclic peptides. NPJ Antimicrob Resist 2025, 3, 56. [CrossRef]
- Mohammed, E.H.M.; Lohan, S.; Ghaffari, T.; Gupta, S.; Tiwari, R.K.; Parang, K. Membrane-Active Cyclic Amphiphilic Peptides: Broad-Spectrum Antibacterial Activity Alone and in Combination with Antibiotics. J Med Chem 2022, 65, 15819-15839. [CrossRef]
- Lohan, S.; Konshina, A.G.; Efremov, R.G.; Maslennikov, I.; Parang, K. Structure-Based Rational Design of Small α-Helical Peptides with Broad-Spectrum Activity against Multidrug-Resistant Pathogens. J Med Chem 2023, 66, 855-874. [CrossRef]
- Lohan, S.; Konshina, A.G.; Tiwari, R.K.; Efremov, R.G.; Maslennikov, I.; Parang, K. Broad-spectrum activity of membranolytic cationic macrocyclic peptides against multi-drug resistant bacteria and fungi. Eur J Pharm Sci 2024, 197, 106776. [CrossRef]
- Mohammed, E.H.M.; Lohan, S.; Tiwari, R.K.; Parang, K. Amphiphilic cyclic peptide [W(4)KR(5)]-Antibiotics combinations as broad-spectrum antimicrobial agents. Eur J Med Chem 2022, 235, 114278. [CrossRef]
- Reid, D.J.; Dash, T.; Wang, Z.; Aspinwall, C.A.; Marty, M.T. Investigating Daptomycin-Membrane Interactions Using Native MS and Fast Photochemical Oxidation of Peptides in Nanodiscs. Anal Chem 2023, 95, 4984-4991. [CrossRef] [PubMed]
- Ledger, E.V.K.; Sabnis, A.; Edwards, A.M. Polymyxin and lipopeptide antibiotics: membrane-targeting drugs of last resort. Microbiology (Reading) 2022, 168. [CrossRef]
- El-Sayed Ahmed, M.A.E.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the Era of antibiotic resistance: an extended review (2000-2019). Emerg Microbes Infect 2020, 9, 868-885. [CrossRef] [PubMed]
- Kurtzhals, P.; Østergaard, S.; Nishimura, E.; Kjeldsen, T. Derivatization with fatty acids in peptide and protein drug discovery. Nat Rev Drug Discov 2023, 22, 59-80. [CrossRef]
- Huang, R.; Song, H.; Wang, X.; Shen, H.; Li, S.; Guan, X. Fatty acids-modified liposomes for encapsulation of bioactive peptides: Fabrication, characterization, storage stability and in vitro release. Food Chemistry 2024, 440, 138139. [CrossRef]
- Yoon, B.K.; Jackman, J.A.; Valle-González, E.R.; Cho, N.-J. Antibacterial Free Fatty Acids and Monoglycerides: Biological Activities, Experimental Testing, and Therapeutic Applications. International Journal of Molecular Sciences 2018, 19, 1114. [CrossRef] [PubMed]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: similarities and differences in cell specificities and modes of action. Cell Mol Life Sci 2011, 68, 2267-2280. [CrossRef]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12-Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int J Mol Sci 2020, 21. [CrossRef]
- Krishnakumari, V.; Nagaraj, R. N-Terminal fatty acylation of peptides spanning the cationic C-terminal segment of bovine β-defensin-2 results in salt-resistant antibacterial activity. Biophys Chem 2015, 199, 25-33. [CrossRef]
- K, R.G.; Balenahalli Narasingappa, R.; Vishnu Vyas, G. Unveiling mechanisms of antimicrobial peptide: Actions beyond the membranes disruption. Heliyon 2024, 10, e38079. [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1999, 1462, 55-70. [CrossRef]
- Lee, J.; Lee, D.G. Antimicrobial Peptides (AMPs) with Dual Mechanisms: Membrane Disruption and Apoptosis. J Microbiol Biotechnol 2015, 25, 759-764. [CrossRef]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451-463. [CrossRef]
- Kumari, S.; Booth, V. Antimicrobial Peptide Mechanisms Studied by Whole-Cell Deuterium NMR. International Journal of Molecular Sciences 2022, 23, 2740. [CrossRef]
- Vejzovic, D.; Piller, P.; Cordfunke, R.A.; Drijfhout, J.W.; Eisenberg, T.; Lohner, K.; Malanovic, N. Where Electrostatics Matter: Bacterial Surface Neutralization and Membrane Disruption by Antimicrobial Peptides SAAP-148 and OP-145. Biomolecules 2022, 12. [CrossRef]
- Khavani, M.; Mehranfar, A.; Mofrad, M.R.K. Antimicrobial peptide interactions with bacterial cell membranes. J Biomol Struct Dyn 2025, 43, 4615-4628. [CrossRef] [PubMed]
- Vatansever, F.; de Melo, W.C.M.A.; Avci, P.; Vecchio, D.; Sadasivam, M.; Gupta, A.; Chandran, R.; Karimi, M.; Parizotto, N.A.; Yin, R.; et al. Antimicrobial strategies centered around reactive oxygen species – bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiology Reviews 2013, 37, 955-989. [CrossRef] [PubMed]
- Zughaier, S.M.; Shafer, W.M.; Stephens, D.S. Antimicrobial peptides and endotoxin inhibit cytokine and nitric oxide release but amplify respiratory burst response in human and murine macrophages. Cell Microbiol 2005, 7, 1251-1262. [CrossRef]
- Niyonsaba, F.; Nagaoka, I.; Ogawa, H. Human defensins and cathelicidins in the skin: beyond direct antimicrobial properties. Crit Rev Immunol 2006, 26, 545-576. [CrossRef]
- Guaní-Guerra, E.; Santos-Mendoza, T.; Lugo-Reyes, S.O.; Terán, L.M. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin Immunol 2010, 135, 1-11. [CrossRef] [PubMed]
- Lofton, H.; Pränting, M.; Thulin, E.; Andersson, D.I. Mechanisms and Fitness Costs of Resistance to Antimicrobial Peptides LL-37, CNY100HL and Wheat Germ Histones. PLOS ONE 2013, 8, e68875. [CrossRef] [PubMed]
- Assoni, L.; Milani, B.; Carvalho, M.R.; Nepomuceno, L.N.; Waz, N.T.; Guerra, M.E.S.; Converso, T.R.; Darrieux, M. Resistance Mechanisms to Antimicrobial Peptides in Gram-Positive Bacteria. Front Microbiol 2020, 11, 593215. [CrossRef]
- Nawrocki, K.L.; Crispell, E.K.; McBride, S.M. Antimicrobial Peptide Resistance Mechanisms of Gram-Positive Bacteria. Antibiotics (Basel) 2014, 3, 461-492. [CrossRef]
- Ryberg, L.A.; Sønderby, P.; Barrientos, F.; Bukrinski, J.T.; Peters, G.H.J.; Harris, P. Solution structures of long-acting insulin analogues and their complexes with albumin. Acta Crystallogr D Struct Biol 2019, 75, 272-282. [CrossRef]
- Zhong, C.; Zhu, N.; Zhu, Y.; Liu, T.; Gou, S.; Xie, J.; Yao, J.; Ni, J. Antimicrobial peptides conjugated with fatty acids on the side chain of D-amino acid promises antimicrobial potency against multidrug-resistant bacteria. Eur J Pharm Sci 2020, 141, 105123. [CrossRef]
- Liao, Z.; Wu, Y.; Liu, M.; Zhang, J.; Cui, Y.; Zhangsun, D.; Luo, S. Fatty acid chain modification enhances the serum stability of antimicrobial peptide B1 and activities against Staphylococcus aureus and Klebsiella pneumoniae. Bioorg Chem 2025, 154, 108015. [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J Med Chem 2015, 58, 7370-7380. [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front Endocrinol (Lausanne) 2019, 10, 155. [CrossRef]
- Kiran, G.S.; Priyadharsini, S.; Sajayan, A.; Priyadharsini, G.B.; Poulose, N.; Selvin, J. Production of Lipopeptide Biosurfactant by a Marine Nesterenkonia sp. and Its Application in Food Industry. Frontiers in Microbiology 2017, Volume 8 - 2017. [CrossRef]
- Plaza-Oliver, M.; Santander-Ortega, M.J.; Lozano, M.V. Current approaches in lipid-based nanocarriers for oral drug delivery. Drug Deliv Transl Res 2021, 11, 471-497. [CrossRef] [PubMed]
- Trier, S.; Linderoth, L.; Bjerregaard, S.; Strauss, H.M.; Rahbek, U.L.; Andresen, T.L. Acylation of salmon calcitonin modulates in vitro intestinal peptide flux through membrane permeability enhancement. Eur J Pharm Biopharm 2015, 96, 329-337. [CrossRef] [PubMed]
- Oh, D.; Nasrolahi Shirazi, A.; Northup, K.; Sullivan, B.; Tiwari, R.K.; Bisoffi, M.; Parang, K. Enhanced cellular uptake of short polyarginine peptides through fatty acylation and cyclization. Mol Pharm 2014, 11, 2845-2854. [CrossRef]
- Selvaraj, S.P.; Chen, J.Y. Conjugation of antimicrobial peptides to enhance therapeutic efficacy. Eur J Med Chem 2023, 259, 115680. [CrossRef] [PubMed]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Brzozowski, K.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim Biophys Acta Biomembr 2018, 1860, 2242-2251. [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS infectious diseases 2016, 2, 442-450. [CrossRef]
- Dou, X.; Zhu, X.; Wang, J.; Dong, N.; Shan, A. Novel design of heptad amphiphiles to enhance cell selectivity, salt resistance, antibiofilm properties and their membrane-disruptive mechanism. Journal of Medicinal Chemistry 2017, 60, 2257-2270. [CrossRef]
- Avrahami, D.; Shai, Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry 2002, 41, 2254-2263. [CrossRef]
- Avrahami, D.; Shai, Y. Bestowing antifungal and antibacterial activities by lipophilic acid conjugation to D,L-amino acid-containing antimicrobial peptides: a plausible mode of action. Biochemistry 2003, 42, 14946-14956. [CrossRef]
- Malina, A.; Shai, Y. Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem J 2005, 390, 695-702. [CrossRef]
- Lockwood, N.A.; Haseman, J.R.; Tirrell, M.V.; Mayo, K.H. Acylation of SC4 dodecapeptide increases bactericidal potency against Gram-positive bacteria, including drug-resistant strains. Biochem J 2004, 378, 93-103. [CrossRef] [PubMed]
- Stephani, J.C.; Gerhards, L.; Khairalla, B.; Solov'yov, I.A.; Brand, I. How do Antimicrobial Peptides Interact with the Outer Membrane of Gram-Negative Bacteria? Role of Lipopolysaccharides in Peptide Binding, Anchoring, and Penetration. ACS Infect Dis 2024, 10, 763-778. [CrossRef]
- Avrahami, D.; Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J Biol Chem 2004, 279, 12277-12285. [CrossRef]
- Majerle, A.; Kidric, J.; Jerala, R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J Antimicrob Chemother 2003, 51, 1159-1165. [CrossRef]
- Liu, H.; Yang, N.; Teng, D.; Mao, R.; Hao, Y.; Ma, X.; Wang, X.; Wang, J. Fatty acid modified-antimicrobial peptide analogues with potent antimicrobial activity and topical therapeutic efficacy against Staphylococcus hyicus. Appl Microbiol Biotechnol 2021, 105, 5845-5859. [CrossRef]
- Mak, P.; Pohl, J.; Dubin, A.; Reed, M.S.; Bowers, S.E.; Fallon, M.T.; Shafer, W.M. The increased bactericidal activity of a fatty acid-modified synthetic antimicrobial peptide of human cathepsin G correlates with its enhanced capacity to interact with model membranes. Int J Antimicrob Agents 2003, 21, 13-19. [CrossRef] [PubMed]
- Chu-Kung, A.F.; Bozzelli, K.N.; Lockwood, N.A.; Haseman, J.R.; Mayo, K.H.; Tirrell, M.V. Promotion of Peptide Antimicrobial Activity by Fatty Acid Conjugation. Bioconjugate Chemistry 2004, 15, 530-535. [CrossRef] [PubMed]
- Radzishevsky, I.S.; Rotem, S.; Zaknoon, F.; Gaidukov, L.; Dagan, A.; Mor, A. Effects of acyl versus aminoacyl conjugation on the properties of antimicrobial peptides. Antimicrob Agents Chemother 2005, 49, 2412-2420. [CrossRef] [PubMed]
- Wakabayashi, H.; Matsumoto, H.; Hashimoto, K.; Teraguchi, S.; Takase, M.; Hayasawa, H. N-Acylated and D enantiomer derivatives of a nonamer core peptide of lactoferricin B showing improved antimicrobial activity. Antimicrob Agents Chemother 1999, 43, 1267-1269. [CrossRef]
- Chu-Kung, A.F.; Nguyen, R.; Bozzelli, K.N.; Tirrell, M. Chain length dependence of antimicrobial peptide-fatty acid conjugate activity. J Colloid Interface Sci 2010, 345, 160-167. [CrossRef]
- Li, R.; Wang, X.; Yin, K.; Xu, Q.; Ren, S.; Wang, X.; Wang, Z.; Yi, Y. Fatty acid modification of antimicrobial peptide CGA-N9 and the combats against Candida albicans infection. Biochem Pharmacol 2023, 211, 115535. [CrossRef]
- Meir, O.; Zaknoon, F.; Cogan, U.; Mor, A. A broad-spectrum bactericidal lipopeptide with anti-biofilm properties. Scientific Reports 2017, 7, 2198. [CrossRef] [PubMed]
- Posa, L.; Tomek, P.; Lamba, S.; Sarojini, V.; Barker, D. Development of truncated Battacin antimicrobials featuring novel N-terminal fatty acids with an excellent safety profile. Bioorganic & Medicinal Chemistry Letters 2023, 96, 129535. [CrossRef]
- Hamman, J.H.; Enslin, G.M.; Kotzé, A.F. Oral delivery of peptide drugs: barriers and developments. BioDrugs 2005, 19, 165-177. [CrossRef] [PubMed]
- Azeem, K.; Fatima, S.; Ali, A.; Ubaid, A.; Husain, F.M.; Abid, M. Biochemistry of Bacterial Biofilm: Insights into Antibiotic Resistance Mechanisms and Therapeutic Intervention. Life (Basel) 2025, 15. [CrossRef]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of Antimicrobial Peptides against Bacterial Biofilms. Materials (Basel) 2018, 11. [CrossRef]
- Lamba, S.; Wang, K.; Lu, J.; Phillips, A.; Swift, S.; Sarojini, V. Polydopamine-Mediated Antimicrobial Lipopeptide Surface Coating for Medical Devices. ACS applied bio materials 2024, 7. [CrossRef]
- Casadidio, C.; Butini, M.E.; Trampuz, A.; Di Luca, M.; Censi, R.; Di Martino, P. Daptomycin-loaded biodegradable thermosensitive hydrogels enhance drug stability and foster bactericidal activity against Staphylococcus aureus. Eur J Pharm Biopharm 2018, 130, 260-271. [CrossRef]
- K P, S.; Rajeswari, E.; Devadason, A.; Thiruvengadam, R. Exploring Quorum Sensing Loci and Biofilm Formation in Bacillus Isolates from Pigeonpea Rhizosphere. International Journal of Current Microbiology and Applied Sciences 2017, 6, 3252-3262. [CrossRef]
- Raskovic, D.; Alvarado, G.; Hines, K.M.; Xu, L.; Gatto, C.; Wilkinson, B.J.; Pokorny, A. Growth of Staphylococcus aureus in the presence of oleic acid shifts the glycolipid fatty acid profile and increases resistance to antimicrobial peptides. Biochim Biophys Acta Biomembr 2025, 1867, 184395. [CrossRef] [PubMed]
- de Gier, M.G.; Bauke Albada, H.; Josten, M.; Willems, R.; Leavis, H.; van Mansveld, R.; Paganelli, F.L.; Dekker, B.; Lammers, J.-W.J.; Sahl, H.-G.; et al. Synergistic activity of a short lipidated antimicrobial peptide (lipoAMP) and colistin or tobramycin against Pseudomonas aeruginosa from cystic fibrosis patients. MedChemComm 2016, 7, 148-156. [CrossRef]
- Han, W.; Wei, Z.; Camesano, T.A. New antimicrobial peptide-antibiotic combination strategy for Pseudomonas aeruginosa inactivation. Biointerphases 2022, 17, 041002. [CrossRef]
- Ridyard, K.E.; Elsawy, M.; Mattrasingh, D.; Klein, D.; Strehmel, J.; Beaulieu, C.; Wong, A.; Overhage, J. Synergy between Human Peptide LL-37 and Polymyxin B against Planktonic and Biofilm Cells of Escherichia coli and Pseudomonas aeruginosa. Antibiotics (Basel) 2023, 12. [CrossRef]
- Reddersen, K.; Greber, K.E.; Korona-Glowniak, I.; Wiegand, C. The Short Lipopeptides (C(10))(2)-KKKK-NH(2) and (C(12))(2)-KKKK-NH(2) Protect HaCaT Keratinocytes from Bacterial Damage Caused by Staphylococcus aureus Infection in a Co-Culture Model. Antibiotics (Basel) 2020, 9. [CrossRef]
- Imperlini, E.; Massaro, F.; Buonocore, F. Antimicrobial Peptides against Bacterial Pathogens: Innovative Delivery Nanosystems for Pharmaceutical Applications. Antibiotics 2023, 12, 184.
- Makowski, M.; Silva Í, C.; Pais do Amaral, C.; Gonçalves, S.; Santos, N.C. Advances in Lipid and Metal Nanoparticles for Antimicrobial Peptide Delivery. Pharmaceutics 2019, 11. [CrossRef] [PubMed]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide-membrane interactions of three related antimicrobial peptides. Colloids Surf B Biointerfaces 2016, 141, 528-536. [CrossRef]
- Dawgul, M.A.; Greber, K.E.; Bartoszewska, S.; Baranska-Rybak, W.; Sawicki, W.; Kamysz, W. In Vitro Evaluation of Cytotoxicity and Permeation Study on Lysine- and Arginine-Based Lipopeptides with Proven Antimicrobial Activity. Molecules 2017, 22, 2173. [CrossRef] [PubMed]
- Metzger, J.; Wiesmüller, K.H.; Schaude, R.; Bessler, W.G.; Jung, G. Synthesis of novel immunologically active tripalmitoyl-S-glycerylcysteinyl lipopeptides as useful intermediates for immunogen preparations. Int J Pept Protein Res 1991, 37, 46-57. [CrossRef]
- Spohn, R.; Buwitt-Beckmann, U.; Brock, R.; Jung, G.; Ulmer, A.J.; Wiesmüller, K.H. Synthetic lipopeptide adjuvants and Toll-like receptor 2--structure-activity relationships. Vaccine 2004, 22, 2494-2499. [CrossRef] [PubMed]
- Zavascki, A.P.; Nation, R.L. Nephrotoxicity of Polymyxins: Is There Any Difference between Colistimethate and Polymyxin B? Antimicrob Agents Chemother 2017, 61. [CrossRef]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med 2013, 5, 1465-1483. [CrossRef]
- Costa, S.S.; Junqueira, E.; Palma, C.; Viveiros, M.; Melo-Cristino, J.; Amaral, L.; Couto, I. Resistance to Antimicrobials Mediated by Efflux Pumps in Staphylococcus aureus. Antibiotics (Basel) 2013, 2, 83-99. [CrossRef]
- Pantosti, A.; Sanchini, A.; Monaco, M. Mechanisms of antibiotic resistance in Staphylococcus aureus. Future Microbiol 2007, 2, 323-334. [CrossRef]
- Sonbol, F.I.; El-Banna, T.E.; Abd El-Aziz, A.A.; El-Ekhnawy, E. Impact of triclosan adaptation on membrane properties, efflux and antimicrobial resistance of Escherichia coli clinical isolates. J Appl Microbiol 2019, 126, 730-739. [CrossRef]
- Gontsarik, M.; Yaghmur, A.; Ren, Q.; Maniura-Weber, K.; Salentinig, S. From Structure to Function: pH-Switchable Antimicrobial Nano-Self-Assemblies. ACS Appl Mater Interfaces 2019, 11, 2821-2829. [CrossRef] [PubMed]
- Wang, H.; Niu, M.; Xue, T.; Ma, L.; Gu, X.; Wei, G.; Li, F.; Wang, C. Development of antibacterial peptides with efficient antibacterial activity, low toxicity, high membrane disruptive activity and a synergistic antibacterial effect. Journal of Materials Chemistry B 2022, 10, 1858-1874. [CrossRef]
- Selvaraj, S.P.; Lin, K.H.; Lin, W.C.; You, M.F.; Li, T.L.; Chen, J.Y. Rejuvenation of Meropenem by Conjugation with Tilapia Piscidin-4 Peptide Targeting NDM-1 Escherichia coli. ACS Omega 2024, 9, 29756-29764. [CrossRef] [PubMed]
- Mwangi, J.; Kamau, P.M.; Thuku, R.C.; Lai, R. Design methods for antimicrobial peptides with improved performance. Zool Res 2023, 44, 1095-1114. [CrossRef]
- Jayawardena, A.; Hung, A.; Qiao, G.; Hajizadeh, E. Lipidation-induced bacterial cell membrane translocation of star-peptides; 2025.
- Wang, J.; Feng, J.; Kang, Y.; Pan, P.; Ge, J.; Wang, Y.; Wang, M.; Wu, Z.; Zhang, X.; Yu, J.; et al. Discovery of antimicrobial peptides with notable antibacterial potency by an LLM-based foundation model. Sci Adv 2025, 11, eads8932. [CrossRef] [PubMed]
- Jin, S.; Zeng, Z.; Xiong, X.; Huang, B.; Tang, L.; Wang, H.; Ma, X.; Tang, X.; Shao, G.; Huang, X.; et al. AMPGen: an evolutionary information-reserved and diffusion-driven generative model for de novo design of antimicrobial peptides. Communications Biology 2025, 8, 839. [CrossRef]
- Aguilera-Puga, M.D.C.; Plisson, F. Structure-aware machine learning strategies for antimicrobial peptide discovery. Sci Rep 2024, 14, 11995. [CrossRef] [PubMed]
- Verma, D.P.; Tripathi, A.K.; Thakur, A.K. Innovative Strategies and Methodologies in Antimicrobial Peptide Design. J Funct Biomater 2024, 15. [CrossRef] [PubMed]
- Eugster, R.; Orsi, M.; Buttitta, G.; Serafini, N.; Tiboni, M.; Casettari, L.; Reymond, J.-L.; Aleandri, S.; Luciani, P. Leveraging machine learning to streamline the development of liposomal drug delivery systems. Journal of Controlled Release 2024, 376, 1025-1038. [CrossRef] [PubMed]
Figure 1.
Lipopeptide antibiotics containing a fatty acyl chain (e.g., daptomycin, colistin, and polymyxin B).
Figure 1.
Lipopeptide antibiotics containing a fatty acyl chain (e.g., daptomycin, colistin, and polymyxin B).
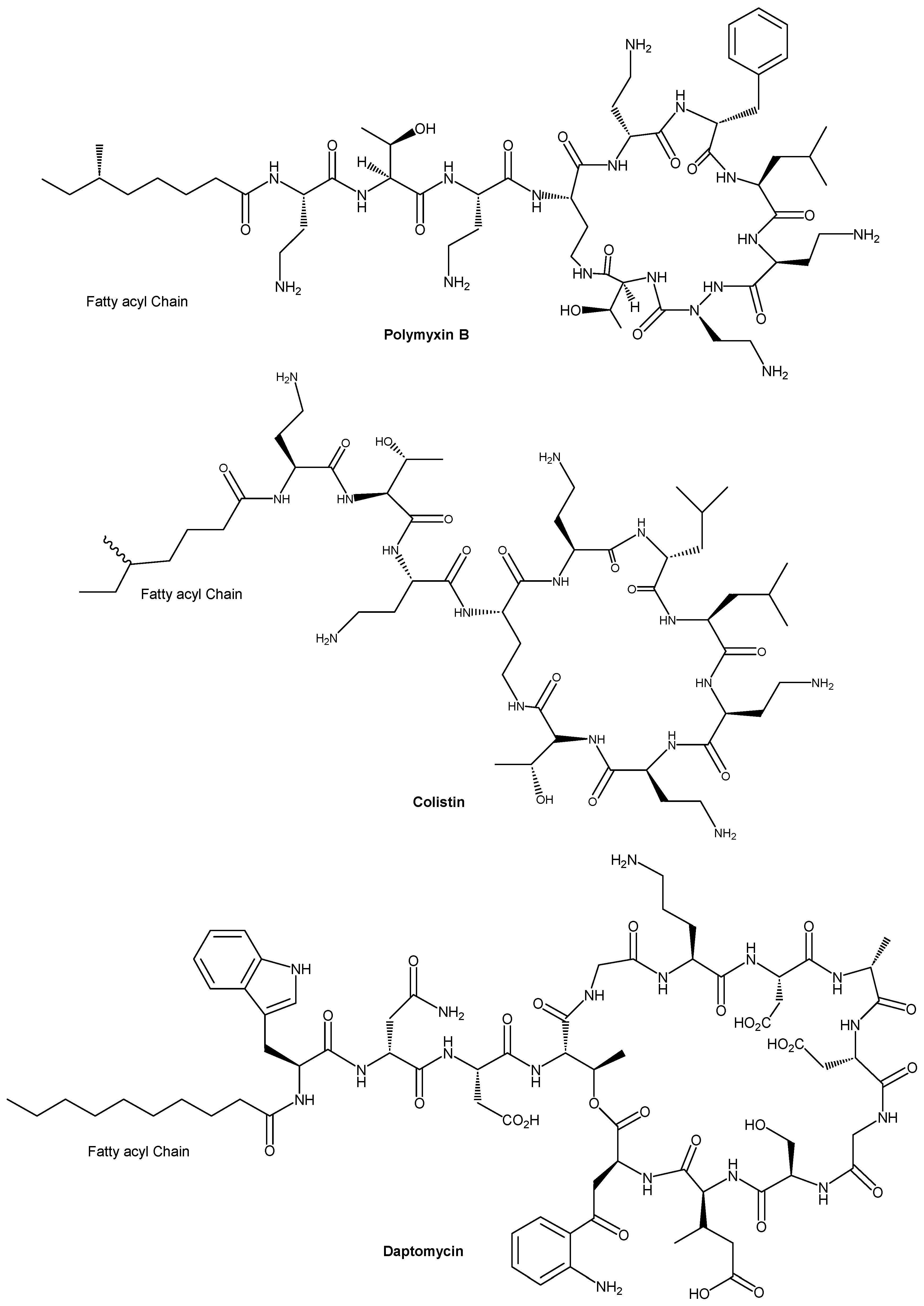
Figure 2.
Effect of peptide lipidation.
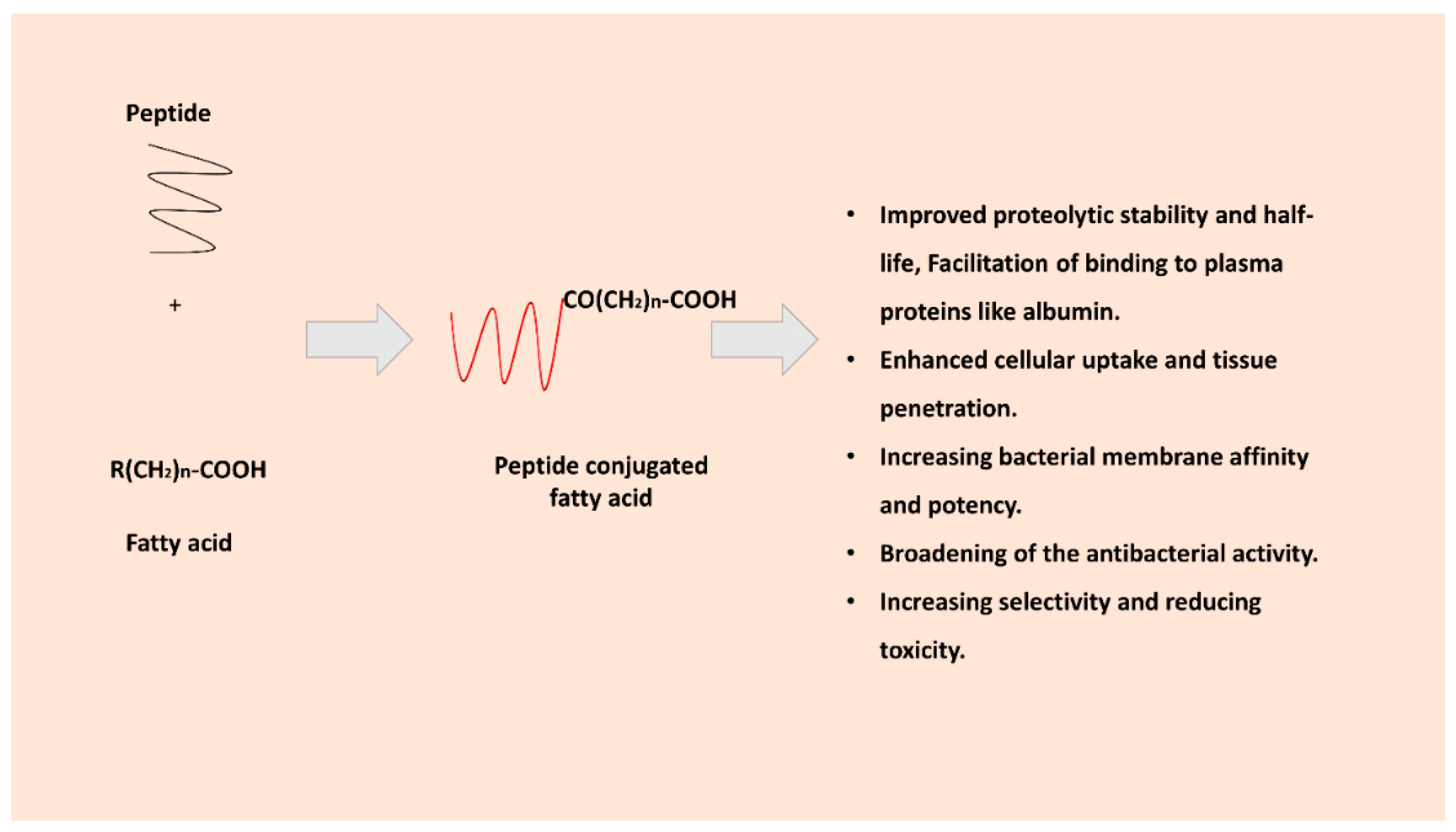
Figure 3.
The main mechanisms of antimicrobial peptides' activity.
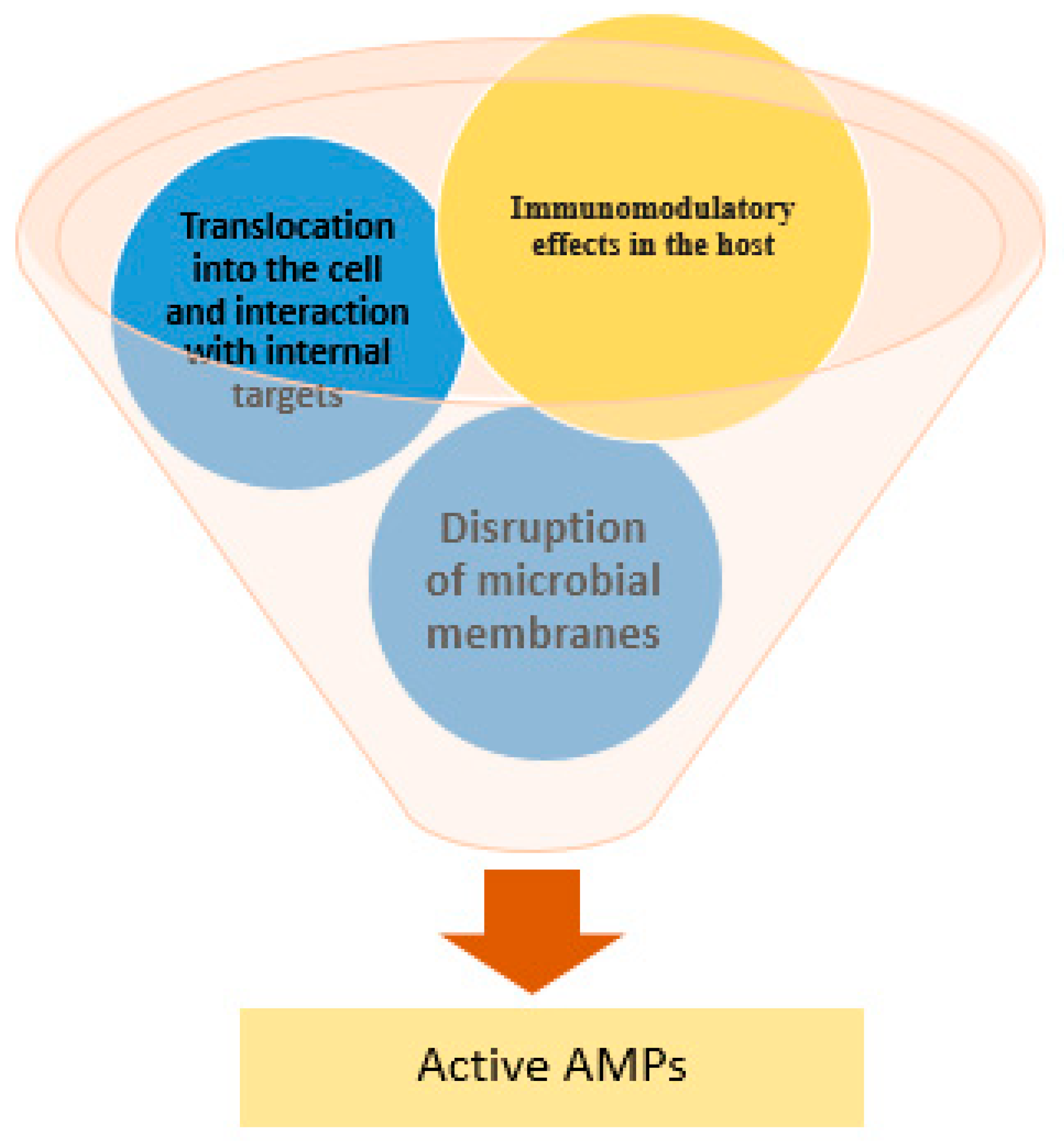
Figure 4.
Effect of fatty acid conjugation on bacterial cell membrane passage. A: Conjugated fatty acids increase binding and facilitate entry to cells. B: FAMP remains on the phospholipid bilayer, causing disintegration.
Figure 4.
Effect of fatty acid conjugation on bacterial cell membrane passage. A: Conjugated fatty acids increase binding and facilitate entry to cells. B: FAMP remains on the phospholipid bilayer, causing disintegration.
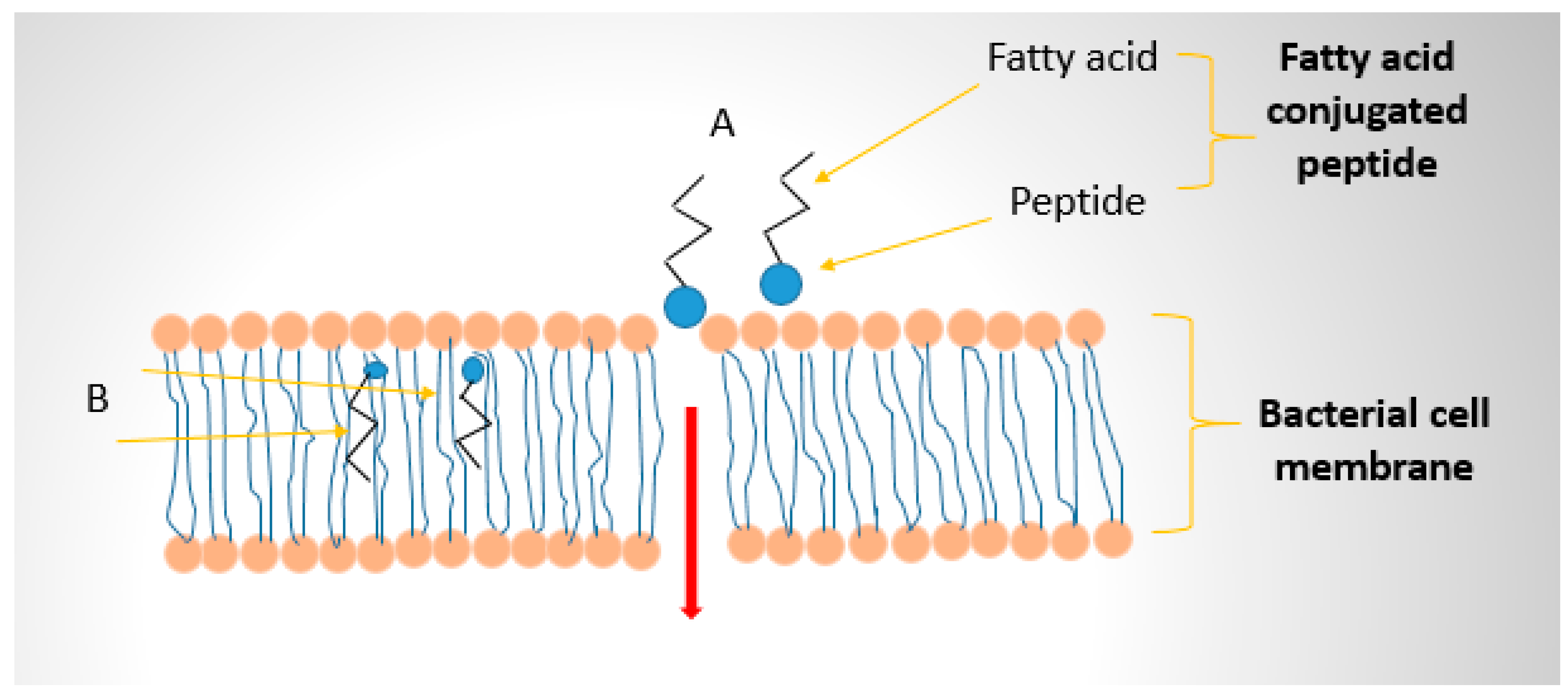
Figure 5.
Chemical structures of CGA-N9 and CGA-N9-C8.
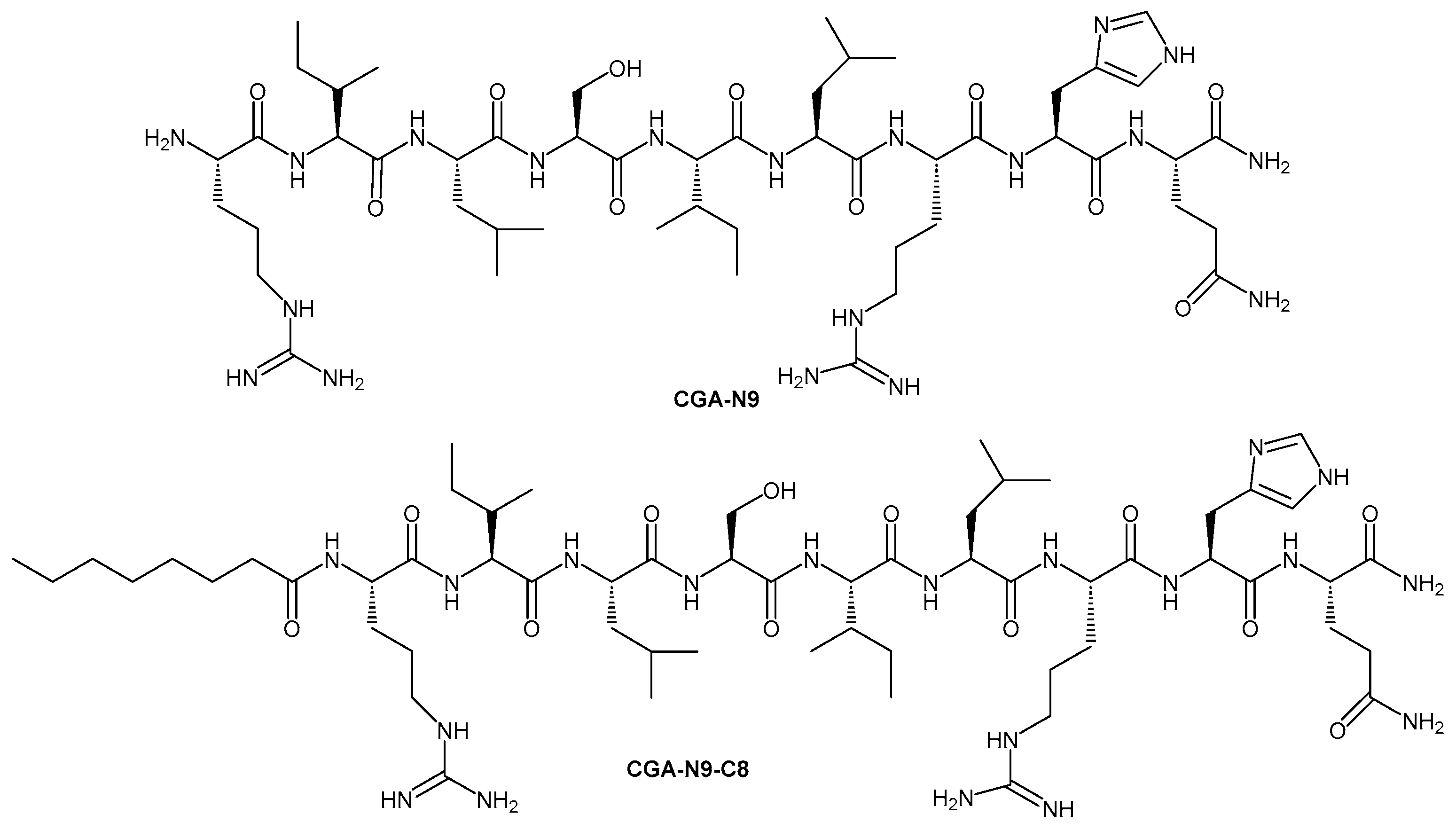
Figure 6.
Chemical structure of Battacin and truncated Battacin analogue.
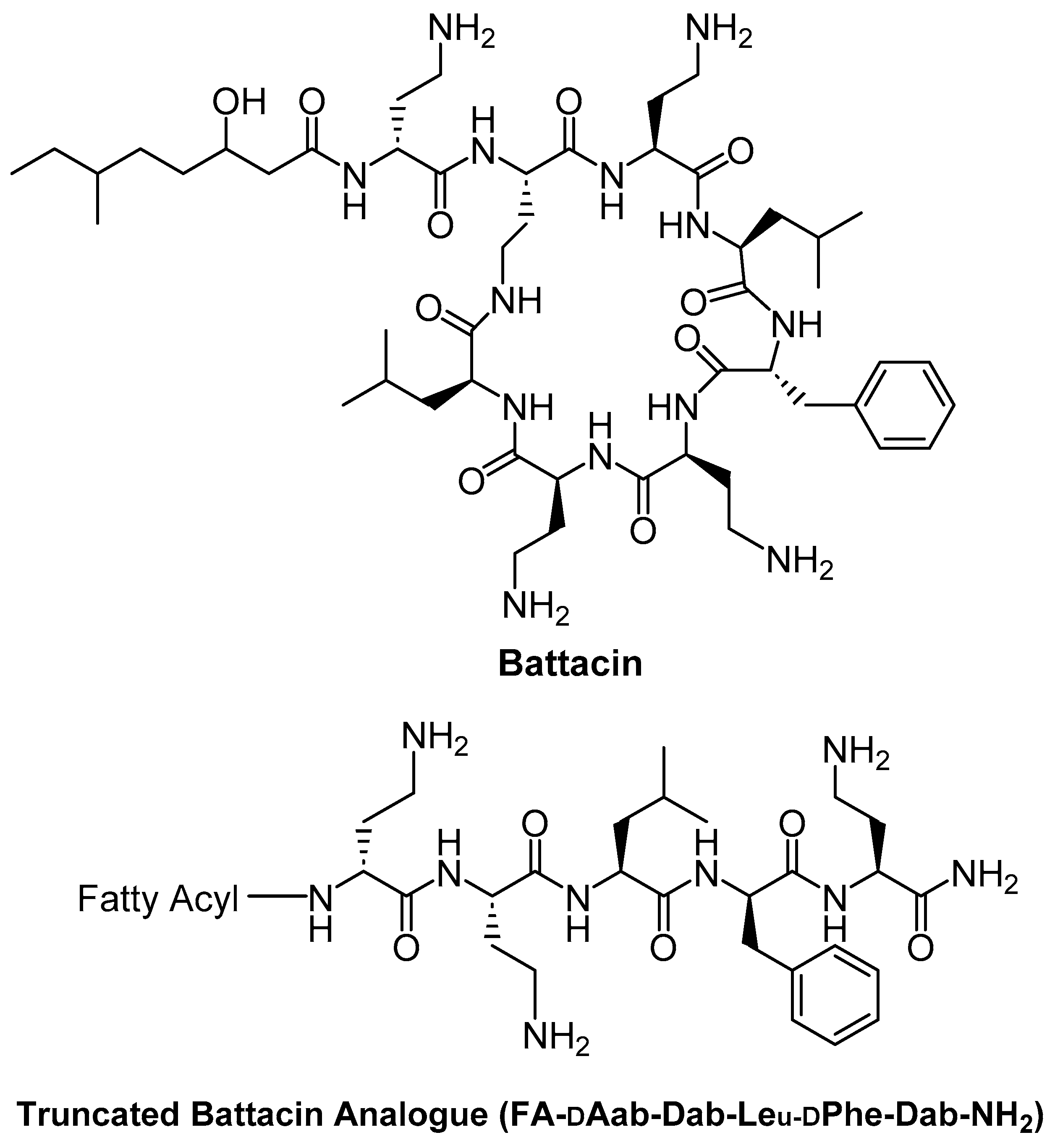

Table 1.
Selected examples of FAMPs from recent studies (last ~10 years, with some landmark older studies). Each entry details the peptide, the fatty acid attached, conjugation strategy, target pathogens, observed effects on activity, and stability.
Table 1.
Selected examples of FAMPs from recent studies (last ~10 years, with some landmark older studies). Each entry details the peptide, the fatty acid attached, conjugation strategy, target pathogens, observed effects on activity, and stability.
| Antimicrobial Peptide (origin/description) | Fatty Acid Conjugated | Conjugation Method | Target Organisms | Effects on Activity & Stability | Reference |
| B1 peptide (16-residue cationic AMP) | Hexanoic (C6) and Octanoic (C8) acids | N-terminal acylation (amide bond) | S. aureus, K. pneumoniae (Gram+ and Gram–) | Significantly enhanced antimicrobial activity; improved serum stability and biocompatibility compared to parent peptide. | [48] |
| CGA-N9 (9-aa fragment of Chromogranin A) | Octanoic acid (C8) | N-terminal acylation (solid-phase synthesis) | C. albicans (yeast; planktonic and biofilm) | Optimal anti-Candida efficacy; highest biofilm inhibition and eradication; strong stability against proteolysis; excellent biosafety profile. | [72] |
| Lfcin4 & Lfcin5 (16-aa bovine lactoferricin derivatives) | Linoleic acid (C18:2, unsaturated) at N-terminus (C-terminal amidation) | N-terminal acylation + C-terminal NH2 | S. hyicus (Gram+; pig pathogen) in an infected mouse model | Improved antibacterial activity (MIC 3–6 µM vs up to 60 µM in parent peptides); in vivo efficacy with membrane disruption and leakage in bacteria; extended duration of action. | [66] |
| Anoplin analogue (Ano-D4,7) (10-aa peptide with D-lys at positions 4,7) | C8–C12 saturated fatty acids (e.g., octanoic, decanoic, lauric) on the side chain | Side-chain acylation on D-amino acid (via amide bond) | Multiple MDR bacteria (e.g., drug-resistant E. coli, S. aureus); also tested against biofilms | Excellent broad-spectrum antimicrobial activity, especially against MDR strains; fatty chain length 8-12 gave the best results: potent antibacterial and anti-biofilm effects, high salt/serum stability, and low propensity for resistance development. | [47] |
| Truncated Battacin analogue (cyclic lipopeptide antibiotic derivative, linear form) | Novel fatty acids (modified long-chain lipids) at the N-terminus | N-terminal acylation (on linear peptide, replacing cyclic lipidated core) | S. aureus (including MRSA); Gram-negative bacteria (E. coli, P. aeruginosa, etc.) | Maintained or improved broad antibacterial activity (comparable to polymyxin/battacin); significantly improved safety, e.g., ~2.3-fold higher LD50 in mice vs polymyxin B. Effective against MRSA and showed an excellent safety profile (low host cell toxicity). | [74] |
| Magainin analogue & [D]-K5L7 (inactive short peptides) | Undecanoic (C11) and Palmitic (C16) acids | N-terminal acylation (amide) | Gram+ bacteria, Gram– bacteria, yeast/fungi (C. neoformans, Candida) | Inactive peptides became highly potent: broad-spectrum antibacterial and antifungal activity was achieved. Lipopeptides caused increased membrane permeability in bacterial and fungal model membranes. Optimal chain (~C11–C12) maximized activity. | [60] |
| Lactoferricin B core (9-mer) RRWQWRMKK | N-acyl groups (various lengths) and D-amino acid substitution | N-terminal acylation; D-enantiomer analogues | E. coli, P. aeruginosa, S. aureus; C. albicans, Trichophyton (fungi) | N-acylated and/or D-form peptides showed improved antimicrobial activity across bacteria and fungi compared to the native peptide. Demonstrated one of the earliest proofs that fatty-acylation enhances AMP efficacy against diverse microbes. | [70] |
MDR, multidrug-resistant; MIC, minimum inhibitory concentration; LD50 – median lethal dose; NH2 – C-terminal amide.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



