Submitted:
06 September 2025
Posted:
08 September 2025
You are already at the latest version
Abstract
Access to genome sequences is crucial to investigate and engineer traits in plants, explore biodiversity, and support phylogenetic studies. During the last decade, affordable sequencing devices have substantially increased the size of the genomics community by enabling individual research groups to sequence the genomes of their favourite plants. There has also been a quick development of novel tools for sequencing data analysis. Here, we share experiences with plant long-read genomics methods for Oxford Nanopore Technologies (ONT) and provide hands-on guidelines to support newcomers’ dive into plant genomics. The full workflow from planning a plant genome sequencing project to public release of associated data sets is covered.
Keywords:
plant genomics
; long read sequencing
; ONT sequencing
; pore C
Introduction
The first ‘complete’ plant genome sequence, that of Arabidopsis thaliana, was released 25 years ago by a large international consortium as the result of an expensive process [1]. Thanks to the emergence and rapid improvement of long read sequencing technologies, individual research groups can now generate A. thaliana genome sequences of superior quality [2,3]. Currently, Pacific Biosciences and Oxford Nanopore Technologies (ONT) offer technologies for the generation of long and highly accurate sequences by analysing individual DNA molecules [4]. ONT sequencing works by measuring changes in an electrical signal as a DNA strand passes through the nanopore. These changes in the electrical signal are caused by different nucleotide compositions that partially block the pore [4]. Hundreds of plant genomes, including those of numerous crops, have been sequenced with long reads [4,5,6]. The ability to generate genome sequences for plant species of interest is crucial to harness the potential of orphan crops and crop wild relatives [5]. Engineering attempts, for example utilizing genome editing, can also profit from a high-quality genome sequence for experimental design [7]. As a single reference genome sequence cannot capture the full genetic diversity of a species, pangenome projects have been conducted to explore the intraspecific diversity [8,9,10]. Despite all these efforts in plant genomics, we are still only seeing the tip of the iceberg. Based on an estimated number of 522,945 plant species [11] and 6,676 sequenced genomes accessible through NCBI, less than two percent of all plant genomes have been sequenced (Figure 1). Given that multiple sequenced genomes may represent the same species, the actual proportion of covered plant species is likely considerably lower, highlighting significant opportunities for future genome sequencing initiatives. Due to legal restrictions (e.g. the Nagoya protocol), not all scientists are allowed to study plant species native to biodiversity hotspots in the tropics. Therefore, it is important to enable local scientists to conduct genome sequencing projects before species go extinct, and their genomic resources are irreversibly lost.
Understanding the terminology associated with genome sequencing is crucial, as it highlights the differences between genomes and their in silico representations. This distinction is particularly important when interpreting scientific results and drawing conclusions about biological processes in nature based on these reconstructed sequences. A ‘plant gene’ is a segment of DNA present in a plant's genome that contains the instructions for producing a specific protein or RNA molecule. Unlike bacterial genes in polycistronic operons, plant genes also include regulatory regions, such as promoters, and often contain introns, non-coding regions, making their structure more complex, these regulatory elements control when and where genes are expressed. Many genes also contain untranslated regions (UTRs) that help regulate translation efficiency and mRNA stability, these non-coding elements play essential roles in gene regulation, allowing plants to respond to environmental changes. The gain, modification, or loss of these regulatory elements over time contributes to the diversity and adaptability of plant species. The term 'genome' refers to the complete set of DNA molecules present within a cell. Consequently, 'genome sequencing' is the methodology employed to determine the order of nucleotides of this genetic material. Since a genome found in nature cannot be accessed with 100% accuracy, the in silico reconstructed sequence is designated as a 'genome sequence' or 'assembly'. The term 'assembly' also refers to the process of combining smaller reads into larger sequences. Genomes typically consist of multiple distinct 'chromosomes'. A high-quality assembly can accurately represent these chromosomes, which are then referred to as 'pseudochromosomes'. A ‘Phred’ or ‘Q’ quality score is the probability that a nucleotide base is basecalled incorrectly in the sequencing, when this score is greater than 20 (Q20+), it means that the probability of an incorrect basecall is less than 1%. The ‘coverage depth’ (or short ‘coverage’) refers to how many times a genomic region was read during sequencing, i.e. how many reads are later including a specific position in the genome sequence. For example, if a genome is sequenced with 40x coverage, it means that, on average, each base is represented in 40 individual reads. Typically, for ONT sequencing, coverage less than 20x is described as low coverage whereas around 30-50x is defined as moderate coverage. Higher coverage (more than 60x) is usually beneficial and sometimes necessary for large repeat-rich plant genomes. In plants, the genome is usually diploid or polyploid. The various copies of one chromosome are termed 'haplotypes', while their in silico reconstructions are referred to as 'haplophases' [4]. The process of resolving haplophases is known as 'phasing'. When haplophases cannot be resolved, for instance, due to the underlying organisms being highly homozygous, this phenomenon is termed ‘merged haplophases' and results in a single haplophase which represents both haplotypes.
This review covers the major workflow steps including preparing a genome sequencing project, DNA extraction, sequencing, genome sequence assembly, structural annotation, functional annotation, quality control for every step, and the submission of analysis results (Figure 2). The guidelines and example commands shared here are intended to facilitate genomic sequencing projects conducted by individual research groups.
Step 1: Preparations for a Plant Genomics Project
Importance of Botanical Gardens as Source of Material for Sequencing Projects
In the age of plant genomics, botanical gardens are essential as repositories of a vast array of plant genetic diversity, being much more than simple displays of flora. These living collections contain a significant amount of the genetic diversity of plants worldwide and give scientists unparalleled access to a wide variety of documented plant material for genomic research [17]. When maintaining plant collections over long periods, botanical gardens inevitably maintain inbred or double haploid plant lines, and these populations have high homozygosity, which can be beneficial for genome assembly and further analyses by reducing allelic variation and assembly ambiguities caused by heterozygous regions [18]. Examples of high-quality reference genome sequences utilising haploid organisms or inbred lines are numerous [19,20,21,22], homozygous genomes often lead to more contiguous and accurate reference assemblies. This homogeneity is especially advantageous because lower heterozygosity makes it possible to resolve structural variants and repetitive regions more clearly, which improves chromosome-level assemblies and facilitates functional genomics research [23,24,25].
The value of botanical garden collections in furthering genomic research is revealed as large-scale sequencing projects continue to progress. Advances in genome sequencing technologies are laying the foundation for large-scale projects like the Earth BioGenome Project. Specimens in botanical gardens are helpful resources for comparative genomics, as demonstrated in a study by Liu et al., 2019, which generated sequences of nearly 700 vascular plant species from the Ruili Botanical Garden [26]. Botanical Gardens can be considered living bridges between taxonomy, conservation, and genomics, and will continue to be essential for plant science and for safeguarding the genetic repertoire of the world’s flora.
Evaluating Existing Genomic Data
Before initiating a sequencing project, it is essential to assess whether genome sequencing is necessary. In certain applications, RNA-seq data may be sufficient to assemble sequences of expressed protein-coding genes, which can provide adequate information for specific projects [27,28]. The required RNA-seq data sets may already be available in a public database. If genomic reads or a complete genome sequence are required, the subsequent step involves determining whether a genome sequence of sufficient quality is already available. This question can be answered by consulting databases maintained by members of the International Nucleotide Sequence Database Collaboration (INSDC), including GenBank and the Sequence Read Archive (SRA), the European Nucleotide Archive (ENA), and the DNA Data Bank of Japan (DDBJ) [29]. Since some of these databases offer reads in addition to genome sequences, it is essential to search for both assembled genome sequences and raw reads. Repositories hosting reads include the SRA and ENA. Accessing reads can facilitate projects by bypassing the sequencing step and allow for the immediate start of dry lab work. Other big databases are Phytozome and the National Genomics Data Center (NGDC) [30,31]. Additionally, a simple internet search may be helpful, as other specialized databases or data repositories like FigShare [32], Dryad [33], e!DAL [34] and PGP [35] exist, and some authors may make their data available outside major databases.
Resource Estimation Utilising the Plant DNA C-Values Database
To prepare for the sequencing of a plant genome, it is essential to estimate the required resources, which are mostly dependent on the genome size. A preliminary assessment of the genome size can be achieved using the C-value, which represents the amount of DNA in picograms per haploid nucleus and can be obtained from the Kew Garden Plant DNA C-value database which in its current release (Release 7.1, April 2019) contains data for 12,273 species [36].
Resource needs are determined by the project’s specific aims and the characteristics of the plant material, both of which affect the volume of sequencing data required. The availability of homozygous or double haploid organisms is often limited, unless the plant of interest is subject to extensive cultivation or breeding efforts. For instance, the sugar beet reference genome sequence (RefBeet) was established using a double haploid line to reduce assembly complexity [37]. The study by Shi et al., 2023, provided a complete reference genome sequence for grapevine, using the PN40024 line (from the cultivar Helfensteiner) originated from self-fertilization carried out for nine generations, resulting in a 99.8% homozygous genome [38]. When such resources are inaccessible, a haplotype-resolved genome assembly becomes necessary, effectively doubling the data requirements based on the C-value. This demand may further escalate if the genome of the plant in question is polyploid, which is a very common property of plant genomes [4,39]. In our experience, a coverage depth, i.e. the number of reads spanning each genomic position, of 30x per haplophase was sufficient to obtain high continuity assemblies. This is also recommended by others for haplotype-resolved assemblies [40,41]. While the initial calculation based on biochemical data like a C-value is only a rough estimate, first sequencing data can be used for a genome sequence assembly attempt that allows a more accurate assessment of the actual obtained coverage. This analysis can be repeated as more data become available to ensure that a sufficient amount of sequencing data is generated. Generating more data than needed based on the coverage recommendation above should generally improve the assembly quality. By using the full capacity of committed flow cells, it might be possible to generate this additional data by just letting the run continue until all nanopores are broken/blocked.
For example, Victoria cruziana has a 1C value of 4.10 [42], indicating the need to obtain a total of 123 gigabasepairs (Gbp) of long read sequencing data. Following a general guideline suggesting an average yield of about 10 Gbp per MinION flow cell, at least 12 flow cells are estimated to be necessary. Additionally, further read correction processes may lead to a decrease in the overall yield of final sequencing data that can be subjected to the assembly process. The data output heavily depends on the DNA quality, which can be influenced by the quality of the individual plant material. Therefore, it is imperative to optimize the DNA extraction process to ensure optimal sequencing results.
Step 2: DNA Extraction Protocols
The fundamental step in plant genomics is the extraction of high molecular weight DNA to obtain long sequencing reads that enable the resolution of complex and/or repetitive genomic regions [43,44]. Since the sequencing technology imposes no limit on the read length, the read length distribution depends directly on the DNA fragment size subjected to the sequencing. Extracting DNA from plants is challenging due to particularities such as the cell wall, starch and other polysaccharides, and specialized metabolites like polyphenols [45]. With the introduction of the Short Read Eliminator (SRE) kit (originally by Circulomics), it became very important to ensure the presence of long DNA fragments in the sample, while the depletion of short fragments, can be reliably performed in a final step using the SRE kit.
Over the years, a wide range of optimized protocols have been developed, including cetyltrimethylammonium bromide (CTAB)-based methods, which have been widely adopted by the plant science community due to their effectiveness with diverse plant species [46]. This method begins with the disruption of the tissue in liquid nitrogen, followed by grinding into a fine powder with a pestle and mortar. The tissue is then treated with a CTAB buffer, which facilitates cell lysis and prevents specialized metabolites from interfering with the DNA [47]. Customized CTAB protocols can have several modifications to address different challenges faced with material from different plant species, since plants differ substantially in their content of sugar, acids, or specialized metabolites. An example of effective modifications that provide high molecular weight DNA for long read sequencing was applied in the study of Siadjeu et al., 2020, in conjunction with an SRE kit to enhance long DNA fragments [48,49]. The use of fresh plant material is essential and young leaves are recommended due to a high ratio between the DNA containing nuclei and metabolites accumulated in the central vacuole. Keeping plants in the dark prior to extraction is recommended to prevent starch accumulation and reduce plastid content [50].
For some challenging plants, it may be necessary to perform nuclei isolation prior to DNA extraction to reduce the complexity of the biological system, such as the presence of specialized metabolites. Additionally, nuclei isolation can help reduce the amount of organelle DNA in the final sample [51,52,53]. This type of protocol involves disrupting cell walls and membranes while preserving nuclear integrity, followed by the isolation of nuclei using density gradient centrifugation or filtration methods [52,54,55,56]. Nuclei isolation has been shown to be especially beneficial for long-read sequencing technologies that require high-molecular-weight DNA, with applications demonstrating that nuclei-based DNA extraction protocols can produce larger DNA fragments with fewer contaminants [53,56,57,58,59]. Another notable method is magnetic disk DNA extraction, which uses a silica-coated magnetic disk to isolate DNA through standard lysis, binding, washing, and elution steps. This protocol is recommended for applications that require high molecular weight DNA, as the disk-binding mechanism allows DNA to bind and release with minimal fragmentation [60,61,62].
Step 3: Library Preparation and Sequencing
Library preparation is a set of steps that converts purified nucleic acids into a collection of fragments ready for sequencing. In our hands, a ligation-based approach was most successful in generating data for plant genome assemblies. The process involves repairing the DNA prior to adapter ligation. An alternative approach aims at preserving longer fragments and prioritizing speed by omitting the DNA repair and adding the adapters through a transposase [63]. In our experience, the ligation-based approach results in substantially larger amounts of data and filtering short reads allows to improve the read length distribution. Although the successive sequencing of both strands of a DNA molecule is possible with dedicated kits (duplex sequencing), this is usually not beneficial for plant genomics. For ONT sequencing, the ideal is to retain the longest possible DNA throughout sample preparation and library protocol, maximizing read length and fully leveraging the platform’s capacity for very long reads [64]. Post library preparation, the library is loaded onto flow cells, and sequencing is initiated through the MinKNOW control software. Generated data is monitored in real-time and can be analysed during the sequencing run. The ideal run time depends on genome size, desired coverage, and flow cell health. For small genomes, a few hours are sufficient to achieve a high coverage. It is important to monitor the flow cell's health (available and actively sequencing pores) using MinKNOW to determine the best time to stop a run, because a flow cell can be washed and reused as long as the pores are available. Since sequencing is monitored in real time by MinKNOW, the run can be terminated when the required coverage is reached to save nanopores for the next sequencing project.
Step 4: Pore-C
Pore-C is a technique combining chromatin conformation capture (3C) and nanopore long-read sequencing. The three-dimensional chromatin structures [65] can be characterized by Pore-C. Compared to previously established 3C techniques [66], Pore-C does not require DNA amplification steps prior to sequencing and displays a simpler and scalable method for chromatin analysis [67]. The provided protocol [56] represents an adapted end-to-end workflow demonstrated to work well on the water lily Victoria cruziana. Completing all steps of the protocol takes three days (Figure 3). The first step is a chemical crosslinking of DNA and proteins, such as histones, which conserves the spatial arrangement within the nucleus. Compared to animal cell line cultures, plant samples require vacuum pressure for adequate infiltration of formaldehyde during the crosslinking process. After a few infiltration periods, glycine is used to terminate the crosslinking reaction [68]. For plant samples, subsequent cryogrinding in liquid nitrogen is mandatory to disrupt the robust cell wall. For efficient chromatin denaturation, nuclei are isolated and permeabilized using various buffers. These buffers include components such as PVP-40 to remove phenolic compounds [69] for nuclei permeabilization. Subsequently, a low dosed SDS dilution is utilized combined with low heat incubation to ensure that crosslinked interactions are maintained as well as the accessibility for restriction enzymes [70]. By default, ONT suggests NlaIII as it is suitable for many species and generates high density contact outputs with optimal fragment lengths. In theory, an in silico analysis of restriction sites in the genome sequences could reveal areas of reduced cleavage within the genome or repeat rich genomic regions to determine other restriction enzyme candidates. Obviously, this does not have practical relevance in genome sequencing projects aiming to generate a reference sequence for a new species. During overnight restriction enzyme incubation, clusters of DNA fragments, maintained in proximity through crosslinking, are formed, thereby preserving the native interactions present at the time of crosslinking. Based on the utilized restriction enzyme, either heat or chemical inactivation is performed before starting the proximity ligation. Following DNA ligase administration, the cohesive ends of the in proximity digested DNA are ligated into chimeric polymers [66,70]. After completed ligation, remaining enzymes and proteins are degraded with proteinase K and SDS overnight, resulting in de-crosslinking and the release of chimeric Pore-C polymers as dsDNA [66,71]. Finally, DNA obtained with the Pore-C protocol can be isolated using a phenol-chloroform extraction with subsequent ethanol precipitation. The phenol-chloroform mixture removes the remaining peptides, whereas ethanol is utilized to purify DNA from retained buffers. Additional supplementation of 5 M NaCl and 3 M sodium acetate, pH 5.5 can enhance DNA precipitation [72]. After extraction, a mixture of chimeric dsDNA molecules that were originally in proximity is obtained. The processed DNA can now be used for size selection followed by library preparation and sequencing. Protocols for these steps slightly deviate from the standard protocols aiming at the longest possible reads, because chimeric DNA fragments are likely to be substantially shorter [56].
Prior to library preparation (day 4+), DNA obtained using the Pore-C protocol undergoes an additional size selection step to enrich for fragments greater than 2 kb, which is necessary for optimal Pore-C data generation. This method has been originally published by Schalamun & Schwessinger, 2017, and was adapted in the context of Pore-C fragment size selection [73,74]. The method is based on solid-phase reversible immobilization (SPRI) in which coated paramagnetic beads are utilized to reversibly bind nucleic acids [75], for example AMPure XP beads (Beckman Coulter). For the size selection process, a custom prepared buffer with e.g. AMPure XP beads containing PEG 8000 is utilized. PEG 8000 is responsible for the selective DNA precipitation, whereas smaller fragments are recovered in higher PEG 8000 concentration and larger fragments within lower concentration [76]. Therefore, an accurate adjustment of PEG 8000 in the custom SPRI solution is crucial to ensure optimal Pore-C fragment selection [77]. However, to maximise the output of chimeric Pore-C fragments, it is recommended to use 0.85X volume of the custom prepared SPRI ratio after appropriate DNA dilution. Prior to library preparation, it is recommended to re-quantify the DNA, as an approximate loss of 50% should be expected [78]. For library preparation, the ligation sequencing kit V14 (SQK-LSK114) can be used with minor variations regarding the DNA repair and end-prep steps [79].
Step 5: Basecalling
Nucleic acids are measured as they pass through a protein nanopore, generating an electrical signal that reflects the chemical composition of the nucleotides within the pore [80]. This signal fluctuates over time as the movement of the nucleic acid alters the nucleotides positioned within the nanopore. Basecalling is the computational step, which converts the measured electrical signals into nucleotide sequences that can be used in further analysis. Dorado is the latest basecalling software package from ONT, built on highly optimized deep neural networks that feature both simplex and duplex basecalling modes [81]. Specific models need to be used depending on the flow cell type and sequencing chemistry (example commands in Additional file 1, Step 5). The input format is POD5 and the output format is FASTQ. Dorado provides modification detection capabilities that are valuable for plant epigenomics research since it can detect nucleotide modifications like 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), and N6-methyladenosine (6mA) directly from raw nanopore signals [82]. Since the technology is capable of measuring any modification, it is also possible to develop customized models to detect other DNA modifications. Plant genome studies have demonstrated that sequence reads produced by Dorado basecalling support high-quality chromosome-scale assemblies and precise DNA methylation analysis [83,84,85].
However, it is important to note that Dorado’s models require a powerful GPU to run efficiently, which can be a significant challenge for many users who may not have access to high performance computational resources locally. To address this limitation, groups without the necessary hardware for bioinformatic analyses can access cloud computing resources for the computationally challenging steps of genomic analyses. Resources dedicated to academics are usually more affordable options and sometimes free of charge. For example, at the national level, the German Network for Bioinformatics Infrastructure (de.NBI) offers free access to cloud-based computing resources and training for researchers, facilitating data analysis without the need for local hardware or advanced computer knowledge. ELIXIR [86] provides support across Europe, while Cyverse [87] can be a valuable resource for researchers in the USA. If no such ‘academic resources’ are available, commercial computing services can be used. Users often pay based on their usage and do not book specific hardware. This makes hardware automatically scalable to the requirements.
Step 6: Read Correction
Although the accuracy of ONT raw reads has substantially improved to >99% (Phred score >20, Q20+) over the last years, further correction can be helpful. Therefore, sequences obtained through basecalling can be corrected prior to assembly. While assembly tools such as Canu [88], NextDenovo [89] or HiCanu [90] typically utilize an all-vs-all alignment-based read correction step before the actual assembly process, a recent advancement is HERRO [91]. Notably, HERRO was developed to be haplotype-aware, enabling chromosome-scale assemblies using HERRO-corrected reads without the need for additional data (example commands in Additional file 1, Step 6). A critical factor in this process is the availability of longer reads, exceeding 100 kbp [92]. While HERRO supports both R9.4.1 and R10.4.1 ONT simplex reads, it is recommended to use it exclusively for R10 reads, particularly given that the developers classify their R9 model as experimental.
Step 7: Genome Sequence Assembly
The reads generated during sequencing typically do not represent an entire native chromosome and are subject to varying degrees of error. To accurately reconstruct the genome sequence, assembler tools require information on how to connect the reads, which is primarily derived from overlaps between them. Consequently, each segment of the genome must be represented multiple times within the reads. Errors at specific positions in the reads may occur in only a small fraction of them and can be corrected during the assembly process. However, haplotype-resolving assembly programs must be carefully calibrated to avoid misinterpreting native differences between haplotypes as sequencing errors.
Typically, these assembly tools integrate sequence alignment and graph theory-based algorithms to assemble the reads into larger units known as contigs (contiguous sequences). Current state-of-the-art long read assemblers include Shasta [93], NextDenovo [89], Verkko2 [94], and Hifiasm [95]. Shasta is recognized for its rapid processing speed, making it suitable for preliminary assessments during sequencing projects to determine whether the sequencing reads are adequate or if additional data generation is necessary. Hifiasm was initially developed for PacBio HiFi reads, but has since been updated to also support ONT R10 reads starting from versions later than 0.21.0-r686 [96]. Verkko2 offers the capability to incorporate Pore-C reads, which are utilized exclusively for non-haplotype-aware scaffolding following the primary assembly process. Different assemblers are optimized for different types of data and genome characteristics. Based on our experience with multiple plant genome sequencing projects, Shasta performs well on high-coverage, high-accuracy R10 HERRO-corrected reads, exhibiting extremely fast runtimes. However, it may produce more fragmented assemblies (lower N50 values) for repeat-rich plant genomes. NextDenovo is not yet optimized for high-accuracy R10 reads but excels on R9 data, generating highly contiguous assemblies even with moderate coverage [97,98]. Verkko2 can generate T2T assemblies when combining ONT reads with PacBio HiFi reads in a hybrid approach, but performs suboptimally on ONT-only moderate coverage datasets. Hifiasm works well on high-coverage, high-accuracy ONT R10 reads, especially for a heterozygous genome, where haplotype resolution is beneficial. Given these differences, it is advisable to deploy multiple assemblers (example commands in Additional file 1, Step 7) and select the best assembly based on comprehensive quality metrics (discussed in the next section), tailoring the choice to the species and dataset at hand.
Step 8: Assembly Evaluation
Once an assembly is obtained, it is essential to evaluate its quality to determine if it meets the project requirements. Assembly quality can be assessed across three major dimensions: continuity, completeness, and correctness.
The contig or scaffold N50 is the most commonly used metric for assessing assembly continuity, as it reflects the length distribution of assembled sequences. It is calculated by sorting all contigs or scaffolds in descending order by length and identifying the shortest sequence among those that collectively comprise at least 50% of the total assembly size. A higher N50 length, approaching the expected length of a chromosome, signifies greater assembly continuity. Related metrics such as N90 (using a 90% threshold) can provide further insights into the continuity of the assembly.
For assembly completeness, Benchmarking Universal Single-Copy Orthologs (BUSCO) [99] and LTR Assembly Index (LAI) [100] can be used. BUSCO measures the presence of highly conserved reference orthologous genes in the target genome sequence. It is essential to select a lineage-specific dataset, as more specific datasets encompass a greater number of reference genes, allowing for a more granular benchmark. Given that these datasets typically include genes that are present in 90% of the species within the specified lineage, a BUSCO score of 90% indicates that the assembly is likely to be of high completeness. Additionally, the BUSCO report provides information on the number of genes found to be duplicated. Analyzing these duplicated genes can offer insights into the ploidy status of the sequenced material, particularly when the percentage of duplicates approaches the overall percentage of genes identified. However, since BUSCO relies on gene prediction tools, it can overestimate completeness by incorrectly identifying fragmented and duplicated BUSCOs as complete [99] and there is a risk that future assembly and annotation tools might be optimized towards high BUSCO scores. LAI, on the other hand, estimates the assembly completeness by calculating the percentage of intact LTR-retrotransposons (LTR-RT) in the repetitive regions, which are known to be challenging to resolve in an assembly. This is particularly useful in plant genomes, which often contain a high proportion of LTR-RTs, and has gained popularity recently. Additionally, comparing the assembly size to the expected genome size is an important complementary check. If the assembly size is significantly smaller than expected, it may be incomplete, and additional sequencing data might be needed. Conversely, if the assembly size is substantially larger (e.g., ~2x or 4x), this may indicate that the genetic material is polyploid relative to the anticipated genome size. It is also important to consider that the assembly may inadvertently resolve haplotypes (fully or partially) even when such resolution is not desired, resulting in an assembly size that is approximately twice the expected size. In contrast, the assembly may be roughly half the expected size if haplotypes are merged. MGSE (Mapping-based genome size estimation) has been observed to work well for genome size estimation based on long reads, but requires an initial assembly [101].
Measuring the assembly quality and correctness is also important and involves evaluating both base-level accuracy and structural accuracy. Tools such as Merqury [102] and QUAST [103] can provide a comprehensive understanding of the assembly quality. Mercury provides reference-free estimates of base-level accuracy using k-mer-based methods, comparing assembly and high-accuracy reads. The QUAST toolkit combines evidence from both k-mers and long reads to estimate both base-level accuracy and structural accuracy. However, it works best if a closely related reference genome sequence is provided.
In summary, evaluating all three aspects, continuity, completeness, and correctness, is essential for interpretation and improving a genome assembly (example commands to run all mentioned tools in Additional file 1, Step 8). We recommend using multiple tools to capture the full picture of assembly quality and make an informed decision.
Step 9: Scaffolding with Pore-C Data
Although modern sequencing technologies increasingly enable chromosome-scale assemblies composed of long contigs, achieving this level of continuity is not always feasible. Following assembly, an additional step called scaffolding links contigs into larger sequences known as scaffolds. Contigs in scaffolds are typically interconnected by multiple "N" bases, which denote an unknown quantity and type of nucleotides at those positions. Scaffolding can be reference-assisted, utilizing a reference genome sequence from the same or closely related species, or it can be enhanced by supplementary data, such as chromosome conformation capture data.
In the context of long-read genome sequencing projects, Pore-C data plays a crucial role in scaffolding assemblies to a chromosome-scale resolution if individual contigs do not represent entire chromosomes. Due to their chimeric nature, Pore-C reads consist of multiple (potentially) distant monomeric sequences [66]. However, the likelihood of these disparate sequences being in spatial proximity is increased when they originate from the same chromosome and, consequently, from the same haplotype [104]. This probability is leveraged during scaffolding. The relationships of contigs can be visualized using a contact map, which highlights the co-occurrences of sequences. Furthermore, Pore-C data facilitates the resolution of haplophases, a process known as phasing, which can produce assemblies that more accurately represent the distinct haplotypes. Tools that leverage Pore-C data, such as the CPhasing tool [105], typically generate a contact map, offering a comprehensive overview for assessing the status of scaffolding.
Step 10: Structural Annotation
Genome sequence assembly, once the bottleneck of genomics, has now become a routine task. The next and more crucial step is to make sense of these massive strings of A’s, T’s, C’s, and G’s by identifying genes. A gene is defined as a nucleotide sequence that encodes a functional product, which may be either a protein or RNA. This gene prediction process is also referred to as structural annotation. Three major approaches are commonly employed for gene prediction: (a) homology-based predictions using known gene sequences from closely related species, (b) ab initio gene prediction using intrinsic sequence features, and (c) evidence-based predictions incorporating transcriptomic and/or proteomic hints from the same species (Figure 4). For protein-coding genes, the primary challenge is accurate identification of the exon-intron boundaries, i.e. the splice sites, and the extent of the untranslated regions at the 5’ and 3’ ends. RNA-seq data provide critical evidence for gene prediction, ideally using cDNA-derived sequences from the target species, available from public databases such as NCBI SRA [106] or generated de novo if necessary. For homology-based annotation, gene models from closely related species serve as valuable references. GeMoMa [107] utilizes a homology-based approach, integrating MMseqs2 [108] for fast alignments, and optionally incorporating RNA-seq data to improve splice site detection. We recommend selecting high-quality annotations from the closest species available, preferably within the same family, and expanding taxonomic distance only if no closer high-quality references are available. BRAKER (107) is a widely used tool offering flexible combinations of ab initio, homology and transcriptomics-based evidence. It uses GENEMARK-ETP [110] and AUGUSTUS [111] for gene model training. Similarly, Funannotate [112] integrates multiple tools such as Trimmomatic [113], Trinity [114], PASA [115], HISAT2 [116], and Kallisto [117] for transcriptome processing, followed by AUGUSTUS [111], GlimmerHMM [118], SNAP [119] for prediction, and EvidenceModeler [120] for consensus gene model generation. In our experience, across multiple plant species, BRAKER provides faster runtimes and better performance compared to Funannotate when evaluated using BUSCO scores as a proxy for annotation quality [121].
In exceptional cases where transcriptomic data is unavailable and generating new data is not feasible, multi-species RNA-seq mapping (preferably within the same genus or at least the same family) has been seen to improve annotation quality alongside homology-based predictions [121]. The most accurate and complete annotations are typically achieved by combining multiple approaches, especially homology and transcriptome-based approaches [84,121]. Ab initio approach should mainly be reserved for non-model species lacking transcriptomic data and high quality annotations from closely related taxa [122,123,124]. For ease and accuracy, we recommend combining GeMoMa and BRAKER predictions, followed by filtering with GeMoMa Annotation Filter (GAF) to obtain a high-confidence gene set (see Additional file 1, Step 10.1.5 for example commands).
Since there is no truly “perfect” genome annotation available and some genes can be species-specific, assessing the validity or near-completeness of a genome annotation is tricky. A simple yet effective strategy is to compare the BUSCO score of the genome assembly with that of the produced annotation, using the same lineage dataset and parameters. A well-annotated genome sequence typically leads to a BUSCO completeness score within ±2% of the assembly score, indicating that the annotation successfully recovered nearly all genes that BUSCO detected in the assembly. Additionally, visual inspection of predicted gene models using a genome browser like Integrative Genomics Viewer (IGV) [125], ideally along with RNA-seq alignments (in BAM format), provides qualitative validation.
In plants, protein-coding genes often account for only ~1-10% of total genome size. The majority of the plant genome consists of non-coding DNA, including transposable elements (TEs), regulatory elements, and various classes of non-coding RNAs (ncRNAs). Accurate identification of these genomic components is equally important. For transposable element annotation, Extensive de novo TE Annotator (EDTA) [126] pipeline provides a comprehensive, automated solution by integrating several high-performing TE annotation tools with appropriate filtering steps. For non-coding RNAs, Infernal [127] offers a sensitive and efficient method for homology-based detection of ncRNAs. Additional specialized tools include tRNAscan-SE [128] for tRNAs, SSU-ALIGN [129] and RNAmmer [130] for rRNA annotation, among others.
Step 11: Functional Annotation
With the rapid progress in long read sequencing and the frequent release of new plant genome sequences, understanding the functions of genes as the genetic units becomes the new challenge [131]. The classical approach of knocking out genes and investigating the mutant phenotype is not suitable for all plant species and is not scalable. Therefore, alternative approaches like the inference of gene functions based on orthology are required. The underlying assumption is that orthologs, the same gene in different species, have still maintained the same function since their split in the last common ancestor. A number of tools utilize sequence similarity as a proxy for orthology, because it can be easily measured with tools like BLAST [132] or DIAMOND [133]. Reciprocal best BLAST hits (RBHs) are a popular strategy to improve the accuracy of ortholog identification via local sequence similarity analysis without generating prohibitive computational costs [134]. The identification of conserved domains allows inferring functional annotation beyond orthologs. An established tool that can integrate protein domain information from different databases is InterProScan5 [135]. When it comes to well characterized enzymes, detailed knowledge about functionally important amino acid residues, e.g. in the active centre, can be utilized [136]. For a general annotation, the annotate function of the Funannotate pipeline [112] offers a comprehensive solution by integrating diverse resources. It can pull and combine functional information from PFAM [137], InterPro [138], EggNog [139], UniProtKB [140], MEROPS [141], CAzyme [142], GO terms [143], and user-provided custom annotations. Additionally, Funannotate can produce NCBI submission-ready files, making it useful for genome projects aiming at public deposition.
Step 12: Data Submission
After sequencing and data analysis, the resulting data should be made publicly available to comply with the FAIR data principles [144]. Open data is important to maintain trust in the analysis results by enabling others to reproduce findings and also to enable future data reuse studies [145,146]. Releasing the electrical signal data would allow future studies to benefit from improvements in basecalling algorithms, which might lead to new discoveries within the same dataset. Additionally, the reads generated through basecalling should be made available to facilitate re-use without computationally expensive basecalling. Typically, this can be accomplished by submitting data to one of the databases of the INSDC, namely ENA [147], SRA [106], or DRA [148]. The basic structure of an INSDC data submission is organized into a BioProject that contains BioSample(s) and sequencing runs. For ONT reads, data of a sequencing run include POD5 (electrical signal data) and FASTQ files (reads). Furthermore, a project may encompass analysis objects or elements that represent genome assemblies and annotation data. Genome assemblies can be submitted as FASTA files. Submission of annotated genome sequences to the INSDC databases necessitates that a locus tag must be registered for the study, and the data is formatted according to the database specifications. However, before formatting, the annotation must be in a valid GFF3 format, free of structural errors. We recommend using AGAT toolkit’s [149] agat_convert_sp_gxf2gxf.pl script to standardize the GFF3 file, followed by agat_sp_fix_features_locations_duplicated.pl to remove features with duplicated locations, which are commonly flagged by ENA and NCBI. Some issues, such as incorrect exons, often require manual curation, and it is advisable to inspect and manually update erroneous gene model(s) before submission. The structural (and functional, if available) annotation results formatted in GFF3 must be integrated with the assembly FASTA into the appropriate submission format, i.e .tbl file for NCBI submission or an EMBL flat file format for ENA. This can be accomplished using the Genome Annotation generator (GAG) [150] for NCBI or the EMBLmyGFF3 tool [151] for ENA. As INSDC members synchronize their data daily, submissions to any one database are accessible through all three. For NCBI submission, the resulting .tbl file needs to be converted to .ASN format using table2asn, which also outputs validation and discrepancy reports, which should be thoroughly reviewed and corrected. Similarly, for EMBL submission, the validation can be done using Webin-CLI. Final submissions are made via the NCBI submission portal [152] (web interface) or through Webin-CLI (command-line interface) [153] for ENA. Once submitted, the assemblies undergo automated validation checks, and if no significant issues are found, the submission is assigned an accession number and becomes publicly accessible.
Summary
Here we provide a practical and comprehensive guide to plant genomics, covering the entire workflow from project planning to data submission. We begin by outlining strategies for evaluating existing genomic data and estimating required resources, ensuring project efficiency. We then detail optimized protocols for high-quality DNA extraction, adapted to long-read sequencing technologies, followed by best practices for library preparation. The subsequent sections address genome assembly, and structural and functional annotations, to transform sequence data into relevant biological information. Finally, we address data submission procedures and recommend best practices to ensure robust and reproducible plant genomics research. To facilitate practical application, we provide a comprehensive ‘cheat sheet’ with specific command-line interface (CLI) commands in the Additional file 1 [16] covering all key steps of the workflow discussed in this review. Given that the majority of land species remain unsequenced, this field is expected to remain dynamic and expansive. This review offers a timely and practical framework for researchers to initiate and execute long-read plant genome sequencing projects effectively, laying a strong foundation for future functional and evolutionary studies, as well as practical applications in agriculture, biotechnology, and beyond.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Additional file 1: PDF “Cookbook for Plant Genome Sequences: Command-Line Protocols for Plant Genome Sequencing Analysis Using Oxford Nanopore Technologies (ONT) data” with example commands for the steps in a typical plant genomics workflow, https://github.com/bpucker/PlantGenomicsGuide.
Author Contributions
JAVSdO organized the manuscript writing and contributed the general sections. NC and SNM contributed experiences with genome sequence assembly, assembly evaluation, structural annotation, and data submission. SNM also contributed experiences with Pore-C data analysis. MSN contributed experiences with nanopore sequencing and Pore-C. MH designed the figures. BP contributed experience with nanopore sequencing and plant genomics and supervised the work. All authors revised the manuscript and agreed to its submission.
Data Availability Statement
The hands-on commands accompanying this article are openly available in the GitHub repository https://github.com/bpucker/PlantGenomicsGuide and in the Codeberg repository https://codeberg.org/PuckerLab/PlantGenomicsGuide. The hands-on guide is also provided as Additional file 1.
Acknowledgements
We thank all members of the research group Plant Biotechnology and Bioinformatics for discussion and support. Open Access funding enabled and organized by Project DEAL and the University of Bonn.
References
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815.
- Michael TP, Jupe F, Bemm F, Motley ST, Sandoval JP, Lanz C, et al. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun. 2018;9:541.
- Pucker B, Kleinbölting N, Weisshaar B. Large scale genomic rearrangements in selected Arabidopsis thaliana T-DNA lines are caused by T-DNA insertion mutagenesis. BMC Genomics. 2021;22:599.
- Pucker B, Irisarri I, Vries J de, Xu B. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quant Plant Biol. 2022;3:e5.
- Marks RA, Hotaling S, Frandsen PB, VanBuren R. Representation and participation across 20 years of plant genome sequencing. Nat Plants. 2021;7:1571–8.
- Schwacke R, Bolger ME, Usadel B. PubPlant -a continuously updated online resource for sequenced and published plant genomes. Front Plant Sci. 2025;16.
- Saha D, Panda AK, Datta S. Critical considerations and computational tools in plant genome editing. Heliyon. 2025;11:e41135.
- Hyun JC, Monk JM, Palsson BO. Comparative pangenomics: analysis of 12 microbial pathogen pangenomes reveals conserved global structures of genetic and functional diversity. BMC Genomics. 2022;23:7.
- Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, et al. A draft human pangenome reference. Nature. 2023;617:312–24.
- Meng Q, Xie P, Xu Z, Tang J, Hui L, Gu J, et al. Pangenome analysis reveals yield- and fiber-related diversity and interspecific gene flow in Gossypium barbadense L. Nat Commun. 2025;16:4995.
- GBIF Secretariat. GBIF Backbone Taxonomy. 2023.
- Bachman SP, Brown MJM, Leão TCC, Nic Lughadha E, Walker BE. Extinction risk predictions for the world’s flowering plants to support their conservation. New Phytol. 2024;242:797–808.
- The Angiosperm Phylogeny Group, Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181:1–20.
- Janssens SB, Couvreur TLP, Mertens A, Dauby G, Dagallier L-PMJ, Abeele SV, et al. A large-scale species level dated angiosperm phylogeny for evolutionary and ecological analyses. Biodivers Data J. 2020;8:e39677.
- McTavish EJ, Hinchliff CE, Allman JF, Brown JW, Cranston KA, Holder MT, et al. Phylesystem: a git-based data store for community-curated phylogenetic estimates. Bioinformatics. 2015;31:2794–800.
- de Oliveira JAVS, Choudhary N, Meckoni SN, Pucker B. Plant Genomics Guide [Internet]. 2025 [cited 2025 Aug 14]. Available from: https://github.com/bpucker/PlantGenomicsGuide.
- Chen G, Sun W. The role of botanical gardens in scientific research, conservation, and citizen science. Plant Divers. 2018;40:181–8.
- Aleza P, Juárez J, Hernández M, Pina JA, Ollitrault P, Navarro L. Recovery and characterization of a Citrus clementinaHort. ex Tan. “Clemenules” haploid plant selected to establish the reference whole Citrus genome sequence. BMC Plant Biol. 2009;9:110.
- Hirsch CN, Hirsch CD, Brohammer AB, Bowman MJ, Soifer I, Barad O, et al. Draft Assembly of Elite Inbred Line PH207 Provides Insights into Genomic and Transcriptome Diversity in Maize. Plant Cell. 2016;28:2700–14.
- Schwartz JC, Farrell CP, Freimanis G, Sewell AK, Phillips JD, Hammond JA. A genome assembly and transcriptome atlas of the inbred Babraham pig to illuminate porcine immunogenetic variation. Immunogenetics. 2024;76:361–80.
- Wang B, Jiao Y, Chougule K, Olson A, Huang J, Llaca V, et al. Pan-genome Analysis in Sorghum Highlights the Extent of Genomic Variation and Sugarcane Aphid Resistance Genes. bioRxiv; 2021. p. 2021.01.03.424980.
- Yang N, Liu J, Gao Q, Gui S, Chen L, Yang L, et al. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat Genet. 2019;51:1052–9.
- Ma H, Liu Y, Liu D, Sun W, Liu X, Wan Y, et al. Chromosome-level genome assembly and population genetic analysis of a critically endangered rhododendron provide insights into its conservation. Plant J. 2021;107:1533–45.
- Pavese V, Cavalet-Giorsa E, Barchi L, Acquadro A, Torello Marinoni D, Portis E, et al. Whole-genome assembly of Corylus avellana cv “Tonda Gentile delle Langhe” using linked-reads (10X Genomics). G3 GenesGenomesGenetics. 2021;11:jkab152.
- Ben Romdhane W, Ben Saad R, Guiderdoni E, Ali AA mohamed, Tarroum M, Al-Doss A, et al. De novo, high-quality assembly and annotation of the halophyte grass Aeluropus littoralis draft genome and identification of A20/AN1 zinc finger protein family. BMC Plant Biol. 2025;25:556.
- Liu H, Wei J, Yang T, Mu W, Song B, Yang T, et al. Molecular digitization of a botanical garden: high-depth whole-genome sequencing of 689 vascular plant species from the Ruili Botanical Garden. GigaScience. 2019;8:giz007.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
- Li Z, Zhang Z, Yan P, Huang S, Fei Z, Lin K. RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genomics. 2011;12:540.
- Arita M, Karsch-Mizrachi I, Cochrane G, on behalf of the International Nucleotide Sequence Database Collaboration. The international nucleotide sequence database collaboration. Nucleic Acids Res. 2021;49:D121–4.
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–86.
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2025. Nucleic Acids Res. 2025;53:D30.
- Wani ZA, Bhat A. Figshare: A One-Stop Shop for Research Data Management with Diverse Features and Services. J Inf Knowl. 2022;59:391–7.
- Vision, T. The Dryad Digital Repository: Published evolutionary data as part of the greater data ecosystem. Nat Preced. 2010;1.
- Arend D, Lange M, Chen J, Colmsee C, Flemming S, Hecht D, et al. e!DAL - a framework to store, share and publish research data. BMC Bioinformatics. 2014;15:214.
- Arend D, Junker A, Scholz U, Schüler D, Wylie J, Lange M. PGP repository: a plant phenomics and genomics data publication infrastructure. Database. 2016;2016:baw033.
- Pellicer J, Leitch IJ. The Plant DNA C-values database (release 7.1): an updated online repository of plant genome size data for comparative studies. New Phytol. 2020;226:301–5.
- Dohm JC, Minoche AE, Holtgräwe D, Capella-Gutiérrez S, Zakrzewski F, Tafer H, et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature. 2014;505:546–9.
- Shi X, Cao S, Wang X, Huang S, Wang Y, Liu Z, et al. The complete reference genome for grapevine (Vitis vinifera L.) genetics and breeding. Hortic Res. 2023;10:uhad061.
- Heslop-Harrison JS (Pat), Schwarzacher T, Liu Q. Polyploidy: its consequences and enabling role in plant diversification and evolution. Ann Bot. 2023;131:1–10.
- Duan H, Jones AW, Hewitt T, Mackenzie A, Hu Y, Sharp A, et al. Physical separation of haplotypes in dikaryons allows benchmarking of phasing accuracy in Nanopore and HiFi assemblies with Hi-C data. Genome Biol. 2022;23:84.
- Sarashetti P, Lipovac J, Tomas F, Šikić M, Liu J. Evaluating data requirements for high-quality haplotype-resolved genomes for creating robust pangenome references. Genome Biol. 2024;25:312.
- Pellicer J, Kelly LJ, Magdalena C, Leitch IJ. Insights into the dynamics of genome size and chromosome evolution in the early diverging angiosperm lineage Nymphaeales (water lilies). Genome. 2013;56:437–49.
- Espinosa E, Bautista R, Larrosa R, Plata O. Advancements in long-read genome sequencing technologies and algorithms. Genomics. 2024;116:110842.
- Russo A, Mayjonade B, Frei D, Potente G, Kellenberger RT, Frachon L, et al. Low-Input High-Molecular-Weight DNA Extraction for Long-Read Sequencing From Plants of Diverse Families. Front Plant Sci. 2022;13.
- Friar, EA. Isolation of DNA from Plants with Large Amounts of Secondary Metabolites. Methods Enzymol. Academic Press; 2005. p. 1.
- Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshaar B. An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol. 2003;53:247–59.
- Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–5.
- Siadjeu C, Pucker B, Viehöver P, Albach DC, Weisshaar B. High Contiguity de novo Genome Sequence Assembly of Trifoliate Yam (Dioscorea dumetorum) Using Long Read Sequencing. Genes. 2020;11:274.
- Recinos MFM, Winnier S, Lagerhausen K, Ajayi B, Wolff K, Friedhoff R, et al. Cacao genome sequence reveals insights into the flavonoid biosynthesis. bioRxiv; 2024. p. 2024.11.23.624982.
- Pucker B. Plant DNA extraction and preparation for ONT sequencing. protocols.io. 2020.
- Zhang Y, Zhang Y, Burke JM, Gleitsman K, Friedrich SM, Liu KJ, et al. A Simple Thermoplastic Substrate Containing Hierarchical Silica Lamellae for High Molecular Weight DNA Extraction. Adv Mater Deerfield Beach Fla. 2016;28:10630–6.
- Butto T, Mungikar K, Baumann P, Winter J, Lutz B, Gerber S. Nuclei on the Rise: When Nuclei-Based Methods Meet Next-Generation Sequencing. Cells. 2023;12:1051.
- Kang M, Chanderbali A, Lee S, Soltis DE, Soltis PS, Kim S. High-molecular-weight DNA extraction for long-read sequencing of plant genomes: An optimization of standard methods. Appl Plant Sci. 2023;11:e11528.
- Ling G, Waxman DJ. Isolation of Nuclei for use in Genome-wide DNase Hypersensitivity Assays to Probe Chromatin Structure. Methods Mol Biol Clifton NJ. 2013;977:13–9.
- Zerpa-Catanho D, Zhang X, Song J, Hernandez AG, Ming R. Ultra-long DNA molecule isolation from plant nuclei for ultra-long read genome sequencing. STAR Protoc. 2021;2:100343.
- Nowak MS, Pucker B. Pore-C Protocol for Plant Samples. protocols.io. 2025.
- Zhang M, Zhang Y, Scheuring CF, Wu C-C, Dong JJ, Zhang H-B. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat Protoc. 2012;7:467–78.
- Workman R, Fedak R, Kilburn D, Hao S, Liu K, Timp W. High Molecular Weight DNA Extraction from Recalcitrant Plant Species for Third Generation Sequencing. protocols.io. 2019. 2019.
- Li Z, Parris S, Saski CA. A simple plant high-molecular-weight DNA extraction method suitable for single-molecule technologies. Plant Methods. 2020;16:38.
- Mayjonade B, Gouzy,Jérôme, Donnadieu,Cécile, Pouilly,Nicolas, Marande,William, Callot,Caroline, et al. Extraction of High-Molecular-Weight Genomic DNA for Long-Read Sequencing of Single Molecules. BioTechniques. 2016;61:203–5.
- Jones A, Torkel C, Stanley D, Nasim J, Borevitz J, Schwessinger B. Scalable high-molecular weight DNA extraction for long-read sequencing. protocols.io. 2020.
- Jones A, Torkel C, Stanley D, Nasim J, Borevitz J, Schwessinger B. High-molecular weight DNA extraction, clean-up and size selection for long-read sequencing. PLOS ONE. 2021;16:e0253830.
- Sauvage T, Cormier A, Delphine P. A comparison of Oxford nanopore library strategies for bacterial genomics. BMC Genomics. 2023;24:627.
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–45.
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing Chromosome Conformation. Science. 2002;295:1306–11.
- Deshpande AS, Ulahannan N, Pendleton M, Dai X, Ly L, Behr JM, et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat Biotechnol. 2022;40:1488–99.
- Sati S, Cavalli G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma. 2017;126:33–44.
- Hoffman EA, Frey BL, Smith LM, Auble DT. Formaldehyde Crosslinking: A Tool for the Study of Chromatin Complexes. J Biol Chem. 2015;290:26404–11.
- Sikorskaite S, Rajamäki M-L, Baniulis D, Stanys V, Valkonen JP. Protocol: Optimised methodology for isolation of nuclei from leaves of species in the Solanaceae and Rosaceae families. Plant Methods. 2013;9:31.
- Belton J-M, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–76.
- Weber K, Kuter DJ. Reversible Denaturation of Enzymes by Sodium Dodecyl Sulfate. J Biol Chem. 1971;246:4504–9.
- Zhong J-Y, Niu L, Lin Z-B, Bai X, Chen Y, Luo F, et al. High-throughput Pore-C reveals the single-allele topology and cell type-specificity of 3D genome folding. Nat Commun. 2023;14:1250.
- Schalamun M, Schwessinger B. DNA size selection (>1kb) and clean up using an optimized SPRI beads mixture. protocols.io. 2017.
- Schalamun M, Nagar R, Kainer D, Beavan E, Eccles D, Rathjen JP, et al. Harnessing the MinION: An example of how to establish long-read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Mol Ecol Resour. 2018;19:77–89.
- DeAngelis MM, Wang DG, Hawkins TL. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995;23:4742–3.
- Lis JT, Schleif R. Size fractionation of double-stranded DNA by precipitation with polyethylene glycol. Nucleic Acids Res. 1975;2:383–90.
- He Z, Zhu Y, Gu H. A new method for the determination of critical polyethylene glycol concentration for selective precipitation of DNA fragments. Appl Microbiol Biotechnol. 2013;97:9175–83.
- Restriction enzyme Pore-C info sheet [Internet]. Oxf. Nanopore Technol. 2019 [cited 2025 July 23]. Available online: https://nanoporetech.com/document/restriction-enzyme-pore-c.
- Ligation sequencing DNA V14 (SQK-LSK114) [Internet]. Oxf. Nanopore Technol. 2022 [cited 2025 July 23]. Available online: https://nanoporetech.com/document/genomic-dna-by-ligation-sqk-lsk114.
- Pagès-Gallego M, de Ridder J. Comprehensive benchmark and architectural analysis of deep learning models for nanopore sequencing basecalling. Genome Biol. 2023;24:71.
- Dorado Documentation [Internet]. [cited 2025 July 23]. Available online: https://dorado-docs.readthedocs.io/en/latest/.
- Dorado [Internet]. Oxford Nanopore Technologies; [cited 2025 July 23]. Available online: https://github.com/nanoporetech/dorado.
- Koren S, Bao Z, Guarracino A, Ou S, Goodwin S, Jenike KM, et al. Gapless assembly of complete human and plant chromosomes using only nanopore sequencing. Genome Res. 2024;34:1919–30.
- Nowak MS, Harder B, Meckoni SN, Friedhoff R, Wolff K, Pucker B. Genome sequence and RNA-seq analysis reveal genetic basis of flower coloration in the giant water lily Victoria cruziana. bioRxiv; 2024. p. 2024.06.15.599162.
- Krawczyk K, Szablińska-Piernik J, Paukszto Ł, Maździarz M, Sulima P, Przyborowski JA, et al. Chromosome-scale telomere to telomere genome assembly of common crystalwort (Riccia sorocarpa Bisch.). Sci Data. 2025;12:77.
- ELIXIR [Internet]. [cited 2025 July 23]. Available online: https://elixir-europe.org/.
- CyVerse [Internet]. [cited 2025 July 23]. Available online: https://cyverse.org/.
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36.
- Hu J, Wang Z, Sun Z, Hu B, Ayoola AO, Liang F, et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 2024;25:107.
- Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020;30:1291–305.
- HERRO [Internet]. Šikić lab; [cited 2025 July 23]. Available online: https://github.com/lbcb-sci/herro.
- Stanojevic D, Lin D, Nurk S, Florez De Sessions P, Sikic M. Telomere-to-Telomere Phased Genome Assembly Using HERRO-Corrected Simplex Nanopore Reads. bioRxiv; 2024.
- Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 2020;38:1044–53.
- Antipov D, Rautiainen M, Nurk S, Walenz BP, Solar SJ, Phillippy AM, et al. Verkko2: Integrating proximity ligation data with long-read De Bruijn graphs for efficient telomere-to-telomere genome assembly, phasing, and scaffolding. bioRxiv; 2024.
- Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18:170–5.
- hifiasm [Internet]. 2025 [cited 2025 July 23]. Available online: https://github.com/chhylp123/hifiasm.
- Hakim SE, Choudhary N, Malhotra K, Peng J, Bültemeier A, Arafa A, et al. Phylogenomics and metabolic engineering reveal a conserved gene cluster in Solanaceae plants for withanolide biosynthesis. Nat Commun. 2025;16:6367.
- Horz JM, Wolff K, Friedhoff R, Pucker B. Genome sequence of the ornamental plant Digitalis purpurea reveals the molecular basis of flower color and morphology variation. bioRxiv; 2024. p. 2024.02.14.580303.
- Manni M, Berkeley MR, Seppey M, Zdobnov EM. BUSCO: Assessing Genomic Data Quality and Beyond. Curr Protoc. 2021;1:e323.
- Ou S, Chen J, Jiang N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018;46:e126.
- Natarajan S, Gehrke J, Pucker B. Mapping-based genome size estimation. BMC Genomics. 2025;26:482.
- Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:245.
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5.
- McCord RP, Kaplan N, Giorgetti L. Chromosome Conformation Capture and Beyond: Toward an Integrative View of Chromosome Structure and Function. Mol Cell. 2020;77:688–708.
- CPhasing [Internet]. 2025 [cited 2025 ]. Available from: https://github.com/wangyibin/CPhasing. 23 July.
- SRA - NCBI [Internet]. [cited 2025 ]. Available from: https://www.ncbi.nlm.nih.gov/sra/. 23 July.
- Keilwagen J, Hartung F, Grau J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. In: Kollmar M, editor. Gene Predict Methods Protoc [Internet]. New York, NY: Springer; 2019 [cited 2025 ]. p. 161–77 Available from: https://doi.org/10.1007/978-1-4939-9173-0_9. Available from, 23 July. [CrossRef]
- Kallenborn F, Chacon A, Hundt C, Sirelkhatim H, Didi K, Dallago C, et al. GPU-accelerated homology search with MMseqs2. bioRxiv; 2024. p. 2024.11.13.623350.
- Gabriel L, Brůna T, Hoff KJ, Ebel M, Lomsadze A, Borodovsky M, et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 2024;34:769–77.
- Brůna T, Lomsadze A, Borodovsky M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 2024;34:757–68.
- Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinforma Oxf Engl. 2008;24:637–44.
- Palmer JM, Stajich J. Funannotate v1.8.1: Eukaryotic genome annotation. Zenodo; 2020.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
- Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–66.
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinforma Oxf Engl. 2004;20:2878–9.
- Korf, I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59.
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7.
- Karbstein K, Choudhary N, Xie T, Tomasello S, Wagner ND, Barke BH, et al. Efficient assembly of plant genomes: A case study with evolutionary implications in Ranunculus (Ranunculaceae). bioRxiv; 2024. p. 2023.08.08.552429.
- Testa AC, Hane JK, Ellwood SR, Oliver RP. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics. 2015;16:170.
- Vuruputoor VS, Monyak D, Fetter KC, Webster C, Bhattarai A, Shrestha B, et al. Welcome to the big leaves: Best practices for improving genome annotation in non-model plant genomes. Appl Plant Sci. 2023;11:e11533.
- Woldesemayat AA, Ntushelo K, Modise DM. Identification and characterization of protein coding genes in monsonia (Monsonia burkeana Planch. ex harv) using a combination of approaches. Genes Genomics. 2017;39:245–59.
- Robinson JT, Thorvaldsdottir H, Turner D, Mesirov JP. igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics. 2023;39:btac830.
- Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20:275.
- Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–5.
- Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–96.
- Nawrocki, E. Structural RNA Homology Search and Alignment Using Covariance Models. Theses Diss ETDs. 2009;256.
- Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
- Pucker, B. Functional Annotation – How to Tackle the Bottleneck in Plant Genomics. Preprints; 2024.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
- Pucker B, Holtgräwe D, Sörensen TR, Stracke R, Viehöver P, Weisshaar B. A De Novo Genome Sequence Assembly of the Arabidopsis thaliana Accession Niederzenz-1 Displays Presence/Absence Variation and Strong Synteny. PLOS ONE. 2016;11:e0164321.
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40.
- Rempel A, Choudhary N, Pucker B. KIPEs3: Automatic annotation of biosynthesis pathways. PLOS ONE. 2023;18:e0294342.
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–9.
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5.
- Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, et al. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008;36:D250–4.
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023;51:D523–31.
- Rawlings ND, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2000;28:323–5.
- Siva Shanmugam NR, Yin Y. CAZyme3D: A Database of 3D Structures for Carbohydrate-active Enzymes. J Mol Biol. 2025;437:169001.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
- Wilkinson MD, Dumontier M, Aalbersberg IjJ, Appleton G, Axton M, Baak A, et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data. 2016;3:160018.
- Hafner A, DeLeo V, Deng CH, Elsik CG, S Fleming D, Harrison PW, et al. Data reuse in agricultural genomics research: challenges and recommendations. GigaScience. 2025;14:giae106.
- Sielemann K, Hafner A, Pucker B. The reuse of public datasets in the life sciences: potential risks and rewards. PeerJ. 2020;8:e9954.
- ENA - European Nucleotide Archive [Internet]. [cited 2025 July 23]. Available online: https://www.ebi.ac.uk/ena/browser/home.
- Sequence Read Archive [Internet]. 2025 [cited 2025 July 23]. Available online: https://www.ddbj.nig.ac.jp/dra/index-e.html.
- Dainat J, Hereñú D, Murray DKD, Davis E, Ugrin I, Crouch K, et al. NBISweden/AGAT: AGAT-v1.4.1. Zenodo; 2024.
- Geib SM, Hall B, Derego T, Bremer FT, Cannoles K, Sim SB. Genome Annotation Generator: a simple tool for generating and correcting WGS annotation tables for NCBI submission. GigaScience. 2018;7:giy018.
- Norling M, Jareborg N, Dainat J. EMBLmyGFF3: a converter facilitating genome annotation submission to European Nucleotide Archive. BMC Res Notes. 2018;11:584.
- Submission Portal | NCBI | NLM | NIH [Internet]. [cited 2025 July 23]. Available online: https://submit.ncbi.nlm.nih.gov/.
- Webin command line submission interface (Webin-CLI) [Internet]. European Nucleotide Archive; 2025 [cited 2025 July 23]. Available online: https://github.com/enasequence/webin-cli.
Figure 1.
The percentage of threatened (left - in light red and dark red) and non-threatened (right - in light and dark blue) angiosperm species from Bachman et al., 2024 [12]. The species are grouped per order, representing the 64 orders of Angiosperms according to Angiosperm Phylogeny Group IV [13]. The darker red bars represent the threatened and sequenced species, while the darker blue bars represent the non-threatened and sequenced species. The numbers at the right represent the total number of known species in each order according to Bachman et al., 2024 [12]. Overall, based on angiosperm extinction prediction, around 45% of species are estimated to be threatened. In terms of genome sequencing, for the majority of angiosperm orders, less than 2% of species are sequenced, highlighting the dire need for conservation genomics prioritizing threatened species. The phylogenetic tree on the left is estimated from Janssens et al., 2020 [14], accessed using Open Tree of Life [15].
Figure 1.
The percentage of threatened (left - in light red and dark red) and non-threatened (right - in light and dark blue) angiosperm species from Bachman et al., 2024 [12]. The species are grouped per order, representing the 64 orders of Angiosperms according to Angiosperm Phylogeny Group IV [13]. The darker red bars represent the threatened and sequenced species, while the darker blue bars represent the non-threatened and sequenced species. The numbers at the right represent the total number of known species in each order according to Bachman et al., 2024 [12]. Overall, based on angiosperm extinction prediction, around 45% of species are estimated to be threatened. In terms of genome sequencing, for the majority of angiosperm orders, less than 2% of species are sequenced, highlighting the dire need for conservation genomics prioritizing threatened species. The phylogenetic tree on the left is estimated from Janssens et al., 2020 [14], accessed using Open Tree of Life [15].

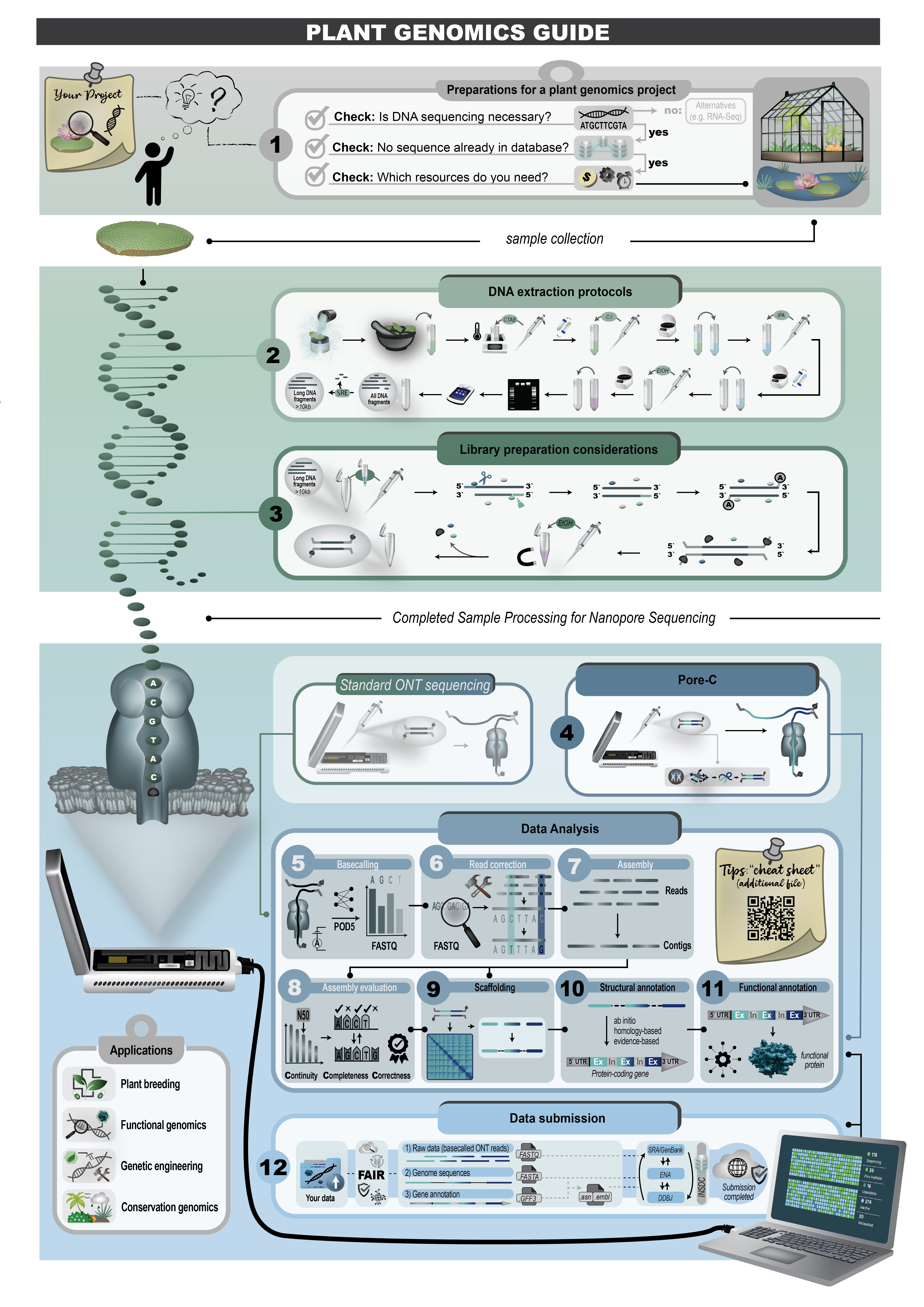
Figure 2.
Figure 2. Major steps in a plant genome sequencing project using Oxford Nanopore Technologies (ONT). This workflow illustrates the twelve essential stages from project conception to data release. The process begins with assessing whether a new sequencing project is necessary, followed by sample collection and extraction of high-molecular-weight (HMW) DNA. Next, key library preparation considerations must be addressed before sequencing. An optional step involves chromosome conformation capture sequencing (e.g., Pore-C) to enable scaffolded assemblies. Sequencing output then undergoes a series of computational analyses: basecalling, read correction, genome assembly, and subsequent evaluation of assembly quality. Further steps include scaffolding, structural annotation, and functional annotation to generate a complete and biologically meaningful genome assembly. Finally, the resulting assemblies and associated data should be submitted to public repositories such as the INSDC databases. Genome sequencing has broad applications, including plant breeding, functional genomics, genetic engineering, and conservation genomics. To support newcomers, a hands-on guide for the data analysis steps is available as a supplementary file [16].
Figure 2.
Figure 2. Major steps in a plant genome sequencing project using Oxford Nanopore Technologies (ONT). This workflow illustrates the twelve essential stages from project conception to data release. The process begins with assessing whether a new sequencing project is necessary, followed by sample collection and extraction of high-molecular-weight (HMW) DNA. Next, key library preparation considerations must be addressed before sequencing. An optional step involves chromosome conformation capture sequencing (e.g., Pore-C) to enable scaffolded assemblies. Sequencing output then undergoes a series of computational analyses: basecalling, read correction, genome assembly, and subsequent evaluation of assembly quality. Further steps include scaffolding, structural annotation, and functional annotation to generate a complete and biologically meaningful genome assembly. Finally, the resulting assemblies and associated data should be submitted to public repositories such as the INSDC databases. Genome sequencing has broad applications, including plant breeding, functional genomics, genetic engineering, and conservation genomics. To support newcomers, a hands-on guide for the data analysis steps is available as a supplementary file [16].
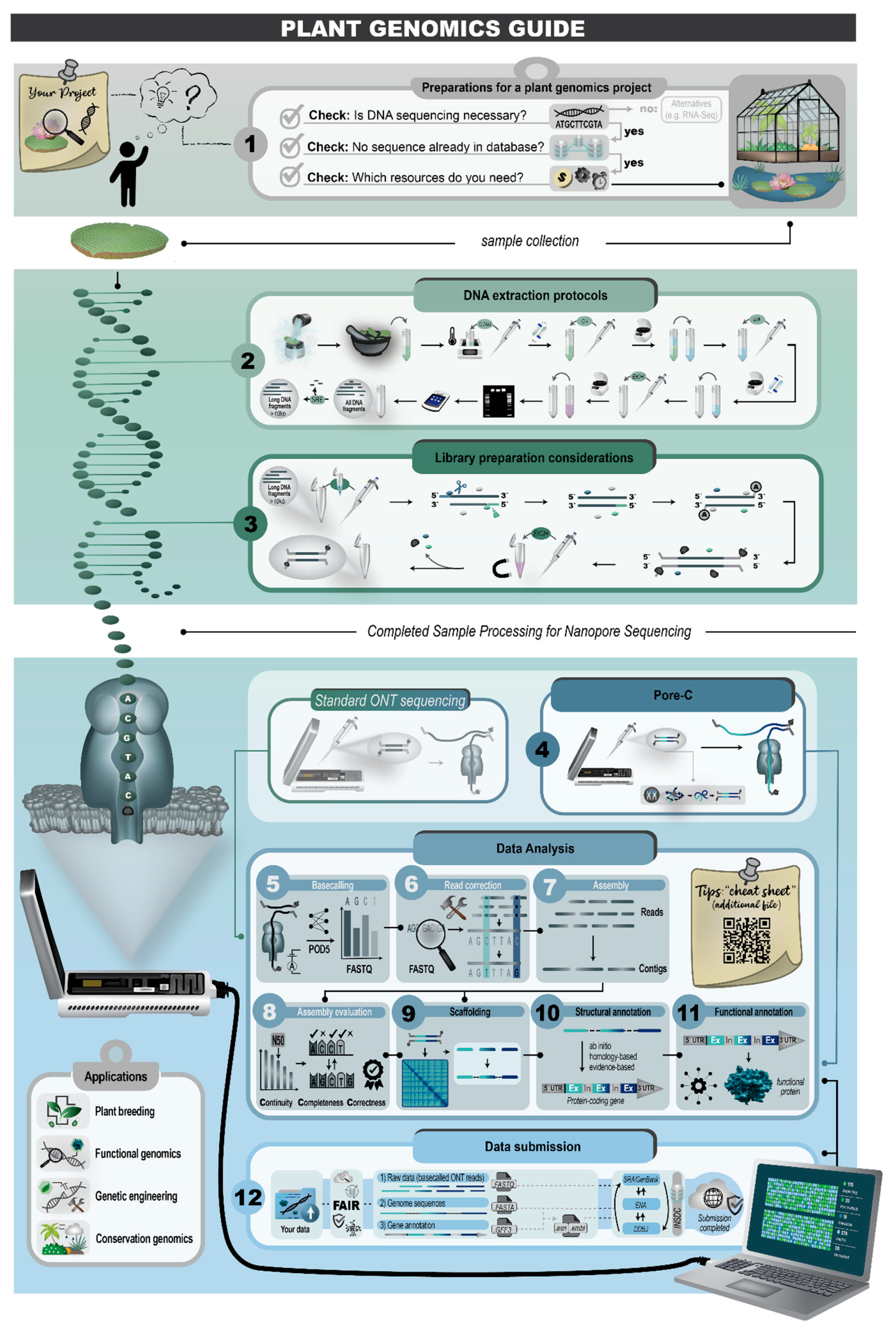
Figure 3.
Illustration depicting the typical Pore-C workflow for plant samples. Steps performed on the first day are highlighted in green boxes and include: sample collection, crosslinking, cryogrinding, nuclei isolation and permeabilization followed by chromatin denaturation and digestion. Second-day procedures, indicated in blue boxes, involve proximity ligation as well as protein degradation and de-crosslinking. The final wet-lab step performed on the third day is DNA extraction, highlighted in yellow. Subsequent sequencing and data analysis steps are shown in grey. The chromatin is illustrated as three grey circles (proteins) surrounded by coloured strands (DNA). The black short lines crossing the illustrated DNA strands mark the restriction enzyme cutting sites.
Figure 3.
Illustration depicting the typical Pore-C workflow for plant samples. Steps performed on the first day are highlighted in green boxes and include: sample collection, crosslinking, cryogrinding, nuclei isolation and permeabilization followed by chromatin denaturation and digestion. Second-day procedures, indicated in blue boxes, involve proximity ligation as well as protein degradation and de-crosslinking. The final wet-lab step performed on the third day is DNA extraction, highlighted in yellow. Subsequent sequencing and data analysis steps are shown in grey. The chromatin is illustrated as three grey circles (proteins) surrounded by coloured strands (DNA). The black short lines crossing the illustrated DNA strands mark the restriction enzyme cutting sites.
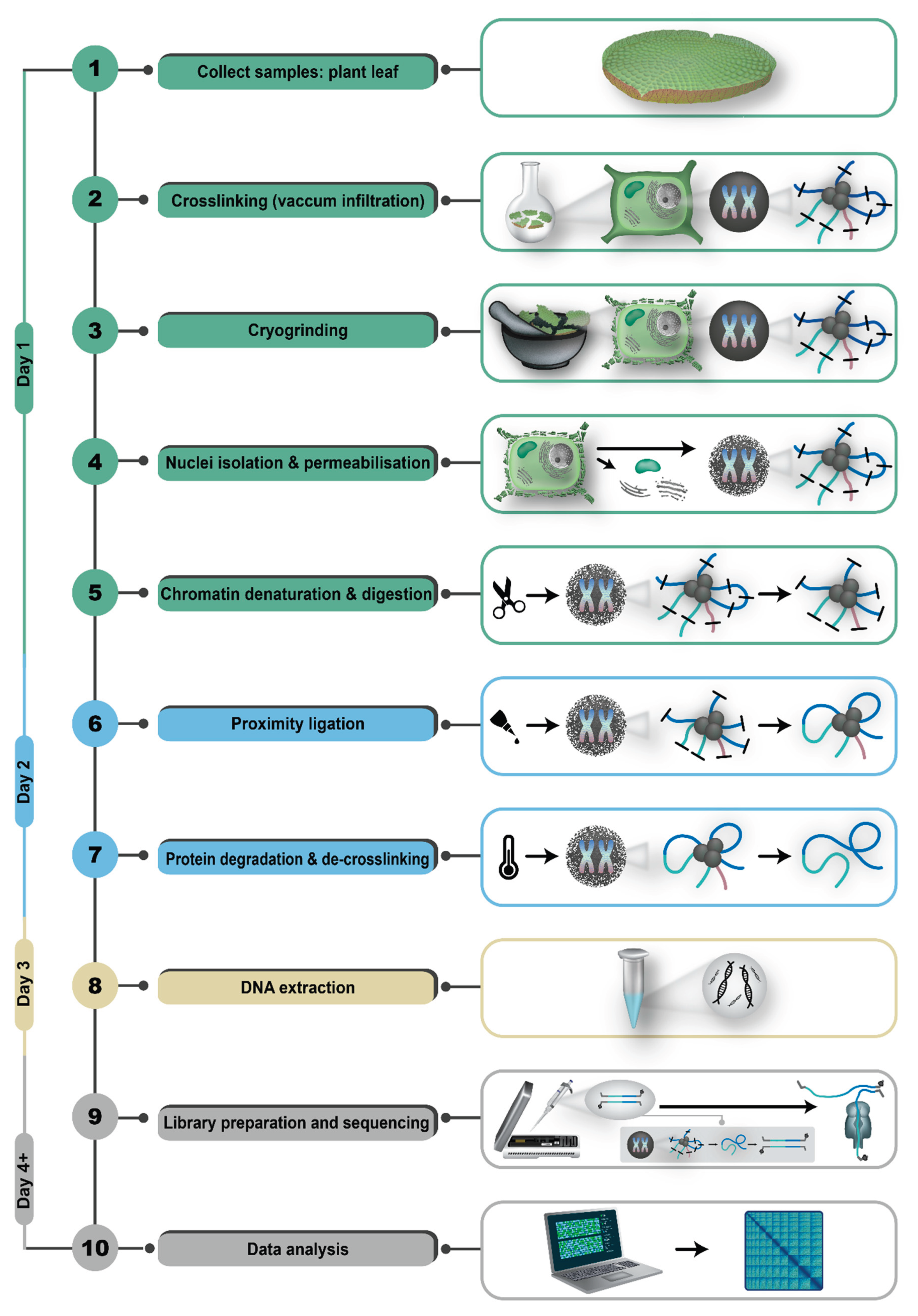
Figure 4.
The data analysis steps in a genome sequencing project can be broadly categorized into assembly, structural annotation, and functional annotation. After sequencing and genome assembly, the next step is to efficiently annotate the genome sequence. A genome comprises not only protein-coding genes but also non-coding genes, such as ncRNAs and transposable elements (TEs). Non-coding genes can be annotated either through sequence homology, using known sequences from literature and databases, or more reliably via de novo structural annotation, which leverages known sequence patterns and, in the case of ncRNAs, secondary structure and pairing relationships. For protein-coding genes, multiple approaches like ab initio predictions, homology-based predictions, and evidence-based predictions can be combined to generate a high-confidence gene set. Functional annotation can be achieved by transferring annotations from homologous sequences and integrating them with knowledge of conserved domains and residues, resulting in more reliable functional assignments. Abbreviations: ncRNA, non-coding RNA; TE, transposable element; ORF, open reading frame; TSS, transcription start site; UTR, untranslated region; LTR, long terminal repeat.
Figure 4.
The data analysis steps in a genome sequencing project can be broadly categorized into assembly, structural annotation, and functional annotation. After sequencing and genome assembly, the next step is to efficiently annotate the genome sequence. A genome comprises not only protein-coding genes but also non-coding genes, such as ncRNAs and transposable elements (TEs). Non-coding genes can be annotated either through sequence homology, using known sequences from literature and databases, or more reliably via de novo structural annotation, which leverages known sequence patterns and, in the case of ncRNAs, secondary structure and pairing relationships. For protein-coding genes, multiple approaches like ab initio predictions, homology-based predictions, and evidence-based predictions can be combined to generate a high-confidence gene set. Functional annotation can be achieved by transferring annotations from homologous sequences and integrating them with knowledge of conserved domains and residues, resulting in more reliable functional assignments. Abbreviations: ncRNA, non-coding RNA; TE, transposable element; ORF, open reading frame; TSS, transcription start site; UTR, untranslated region; LTR, long terminal repeat.
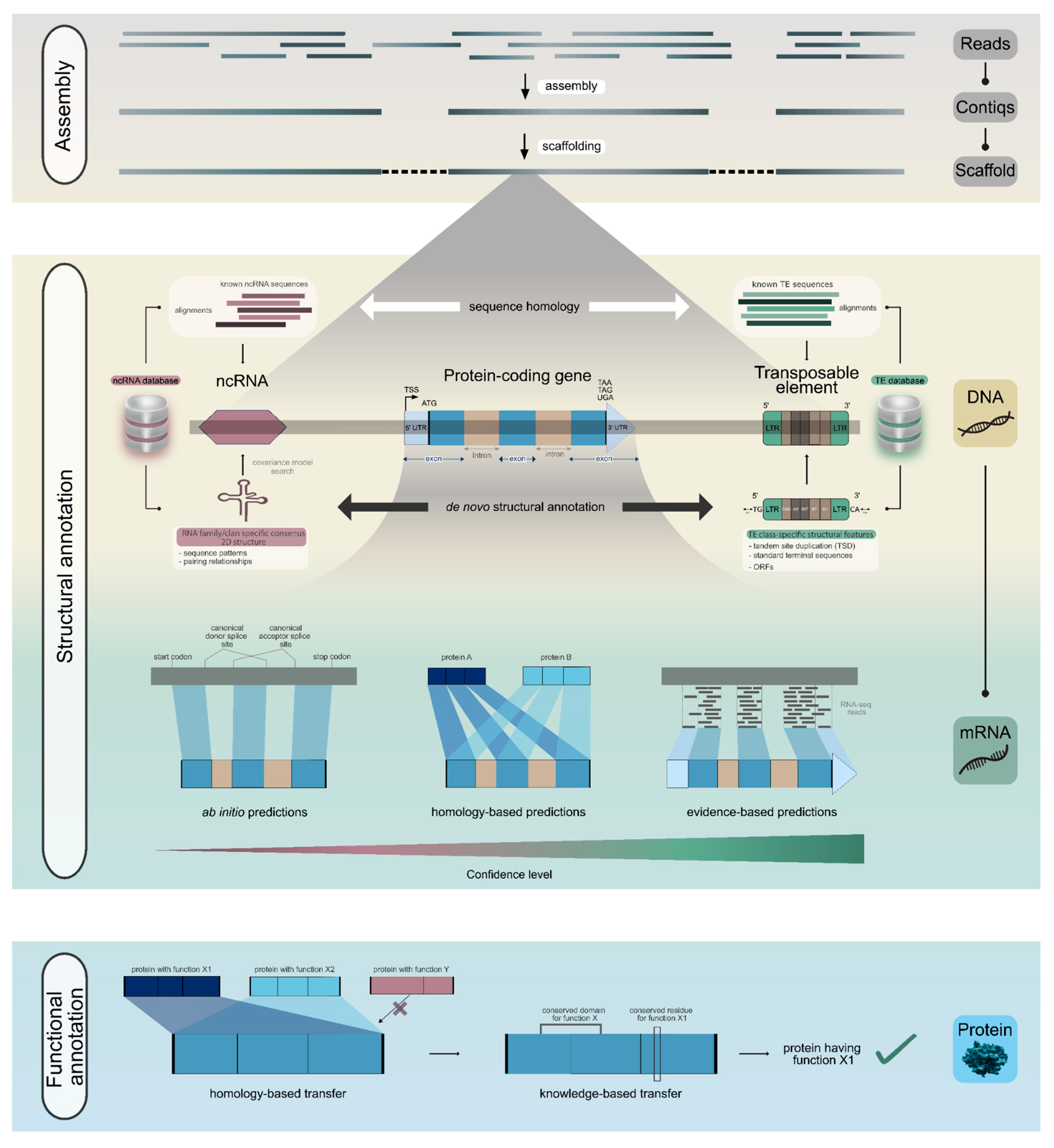
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



