
Submitted:
12 August 2025
Posted:
14 August 2025
You are already at the latest version
Abstract
About twenty previously and newly synthesized amphoteric 8-[(dialkylamino)methyl]-7-hydroxy-4-(2-oxo-2H-chromen-3-yl)-2H-chromen-2-ones were assayed as inhibitors of monoamine oxidases (MAOs A and B) and cholinesterases (AChE and BChE). Five of the tested compounds (2b, 2c, 3c, 5b and 5c), namely those bearing the less bulky alkyls in the Mannich base 8-CH2NR2 (R = Me, Et) and the halogens (Cl, Br) at C6 of the 4-coumarin-3-yl moiety, showed inhibitory potencies towards MAO-A in the single-digit micromolar range (IC50s 1.49÷3.04 µM). Among them, 3c proved to be also the strongest inhibitor of AChE (IC50 = 1.56 µM) in the series. Molecular docking calculations suggested binding modes of the most active compounds to MAO-A and AChE binding sites fairly consistent with the experimental data. Chemoinformatic tools suggest for the most active compounds, including the dual MAO-A/AChE inhibitor 3c, full compliance with Lipinski’s rule-of-five, high probability of gastrointestinal absorption, and no or poor blood-brain barrier (BBB) permeability. While there is room to improve their CNS distribution, herein we identified 4-(2-oxo-2H-chromen-3-yl)-2H-chromen-2-one as a new scaffold of amphoteric MTDLs with potential for neurodegenerative syndromes.
Keywords:
biscoumarin
; monoamine oxidases
; cholinesterases
; inhibitors
; molecular docking
; ADME profile
; Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD) is the leading neurodegenerative disease, with strong impact on health and quality of life. People suffering from AD have learning deficits along with the impairment of memory and language skills due to the disruption of cholinergic transmission in hippocampal areas [1]. As a result of the so-called ‘cholinergic hypothesis’, up to date, the pharmacological therapy of AD has been essentially based on the restoration of adequate levels of acetylcholine (ACh) ensured by acetylcholinesterase (AChE) inhibitors, (i.e., rivastigmine, galantamine and donepezil) [2], but also butyrylcholinesterase (BChE) inhibitors. Memantine, an antagonist of NMDA (N-methyl-d-aspartate) receptor, is also used in the therapy of moderate to severe forms of AD [3]. Nevertheless, all the mentioned drugs do not cure the disease but exert only symptomatic relief. Therefore, the need for new drugs against AD remains a challenging task. Monoclonal antibodies, such as aducanumab, lecanemab and donanemab [4], deserved great hope as disease-resolving therapies, but approved therapies have so far shown contrasting results in terms of efficacy and harmful side effects.
In the last two decades, the multitarget approach to drug design has been considered as promising strategy to discover neurotherapeutics against AD [5], paying attention to other related mechanisms of neurotoxicity. In particular, oxidative stress has been considered an interplaying event with the deposition of amyloid plaques and neurofibrillary tangles [6]. In turn, deregulation of endogenous redox systems and over-production of radical species lead to lipid peroxidation and nucleic acid mutations. In this context, a neuroprotective activity against oxidative stress has been claimed for inhibitors of monoamine oxidases. The flavoenzyme monoamine oxidase (MAO, EC 1.4.3.4), which catalyzes the oxidative deamination of dietary and neurotransmitter amines, including serotonin, tryptamine, dopamine, norepinephrine, epinephrine, β-phenylethylamine, tyramine and octopamine, has been considered as anti-AD drug target. MAO exists in two isoforms, MAO-A and MAO-B, sharing about 70% sequence identity and showing distinct substrate and inhibitor specificities [7]. MAO-A preferentially metabolizes serotonin and norepinephrine, while MAO-B preferentially metabolizes phenylethylamine, benzylamine, and dopamine.
According to their selectivity, MAO subtypes are involved in various neurodegenerative pathologies, including AD, Parkinson’s disease (PD), and depression. Age-related increase in MAO expression levels is a significant causative factor in the etiology of neurodegenerative illnesses and mental disorders. Selective MAO-A inhibitors (MAOAIs) find use in the treatment of depression, whereas MAOBIs are used in alternation to levodopa to treat PD. First-generation MAO inhibitors, such as iproniazid, phenelzine and tranylcypromine, are not isoform-selective and their use was associated to severe side effects [8]. Currently, selective MAOAIs (moclobemide) and MAOBIs (l-deprenyl and safinamide) are preferred because of their better therapeutic index, but with limited prevalence compared with safer and more effective drugs. Recently, a possible repositioning of MAOs as a target in chemoresistant tumors has boosted a renewed attention in MAO inhibitors, particularly for MAOAIs that have demonstrated synergistic efficacy in the antitumor therapy of recurrent prostate cancer, metastatic breast cancer, and glioma [9].
Among the numerous MAOIs developed, coumarin-based derivatives are well-documented MAO inhibitors with good MAO-A/B selectivity ratios, depending upon the nature and pattern of their substitution, and great potential as multitarget agents for neurodegenerative diseases [10]. Coumarins have high electron count and exhibit good charge transport capabilities [11]. The coumarin moiety taken as scaffold is synthetically versatile and widely used in the preparation of biologically active molecules. Indeed, coumarin derivatives have found a wide variety of applications for their anticoagulant, anti-inflammatory, anti-cancer, antidiabetic, anti-neurodegenerative, antioxidant, antidepressive and antimicrobial effects [12]. Coumarin dimers (i.e., biscoumarins), mostly from natural origin, form also a bioactive scaffold. Very often these are symmetrical homodimers, like, for example, dicoumarol and its substituted derivatives [13,14], 3,3’-biisofraxidin (from Sarcandrae Herba) [15] and 4,4’-biisofraxidin (from Impatiens balsamina) [16], 8,8’-biscoumarins like toddalosin (from Toddalia asiatica) [17] and the mycotoxins kotanin and desmethylkotanin (from Aspergillus glaucus) [18] (Figure 1). Biscoumarins attracted as well significant interest due to their wide range of biological activities such as antimicrobial [19], anti-inflammatory [20], anticancer [21], anti-obesity [22], antiviral [23,24], antidiabetic [25], MAO inhibitory [26] and anti-cholinesterase [27,28,29] activities.
Several years ago, Frasinyuk et al. reported the aminomethylation of 7-hydroxy-4-(2-oxo-2H-chromen-3-yl)-2H-chromen-2-one (see Scheme 1) [30], synthesizing diverse amphoteric ortho-hydroxy Mannich bases (i.e., 8-[(dialkylamino)methyl]-7-hydroxy substituents) on the 2H,2’H-3,4’-bichromene-2,2’-dione (hereinafter referred to as 3,4’-biscoumarin) scaffold. The online PASS (prediction of activity spectra for substances) software [31] predicted for forty diverse amphoteric 3,4’-biscoumarin derivatives indicated above MAO inhibitory activity with a Pa value (i.e., the probability of a compound to be active) in the range 0.203–0.895. Note that most of the compounds had Pa values lower than 0.5, indicating on one hand their relative novelty compared to the molecular structures in the PASS training set and on the other hand the low probability to be active as MAOIs.
Due to our interest toward novel coumarin cores as scaffold of novel MAOIs, endowed with further anti-AD, activities, such as the ChE inhibition, herein we screened some twenty previously and newly synthesized nonsymmetrical 3,4’-biscoumarins bearing the amphoteric 8-[(dialkylamino)methyl)-7-hydroxy group as dual MAO/ChE inhibitors, along with docking calculation, aimed at understanding the key factors of MAO and ChE potency, solubility determination and in-silico druglikeness.
2. Results and Discussion
2.1. Chemistry
Nine new 3,4’-biscoumarin-containing amphoteric derivatives (2a-d, 3c, 4d, 5d, 6d and 8d) were synthesized as previously described [30] according to Scheme 1. 7-hydroxy-3,4’-biscoumarins (1a-d) were prepared by condensation of 7-hydroxy-coumarin-4-methylacetate with substituted salicylic aldehydes in dioxane (or DMF for 1d) in the presence of DBU (1,8-diazabicyclo [5.4.0]undec-7-ene) as catalyst. Regarding aminomethylation procedure, the aminals of the secondary amines were refluxed in dioxane with a product yield of 48-82%. In this case the electrophilic substitution took place exclusively at C8 as confirmed by the NMR spectra [30].
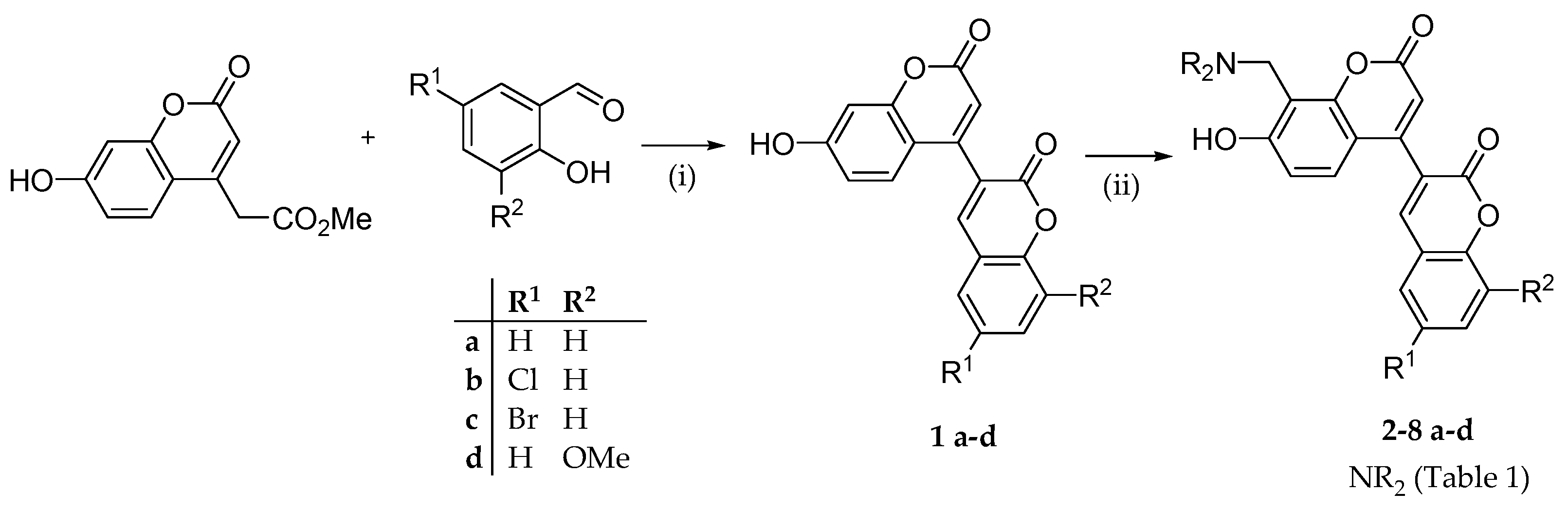
Scheme 1.
Synthesis scheme of 8-[(dialkylamino)methyl]-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one derivatives. Reagents and conditions: (i) DBU, abs. 1,4-dioxane (DMF for 1d), 100-105 °C, 4-10 h; (ii) suitable aminal, 1,4-dioxane, reflux, 3-5h.
Scheme 1.
Synthesis scheme of 8-[(dialkylamino)methyl]-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one derivatives. Reagents and conditions: (i) DBU, abs. 1,4-dioxane (DMF for 1d), 100-105 °C, 4-10 h; (ii) suitable aminal, 1,4-dioxane, reflux, 3-5h.
The structural exploration was primarily focused on the variation in terms of hydrophobicity and steric effects of the alkyls of the aminomethyl group at C8 (NR2) of the coumarin moiety (cmpds 2-8a). The congeners 2-8b-d, bearing the halogens Cl or Br (as R1) at C6 and OMe (as R2) at C8 of the coumarin-3-yl moiety, available in our molecular library, were also investigated.
2.2. Structure-Activity Relationships
2.2.1. In-Vitro Inhibition of Monoamine Oxidases and Cholinesterases
All nineteen compounds were tested against each enzyme, i.e., human MAO-A and MAO-B isoforms, electric eel AChE and horse serum BChE, at 10 μM concentration, and for those exhibiting more than 60% inhibition at that concentration, IC50 values were determined in at least three independent experiments. Clorgiline and donepezil were used as positive controls in the MAO and ChE inhibition assays, respectively. The enzymes’ inhibition data, as means ± SD from three independent experiments, are summarized in Table 1.
For the sake of clarity, the position of the Mannich base (-CH2NR2) on the main coumarin core will be indicated as C8, whereas the positions of Cl or Br and OMe on the 4-coumarin-3-yl moiety will be indicated as C6’ and C8’. From the data in Table 1 it can be deduced that, with different potencies depending on hydrophobicity and steric hindrance of the aminomethyl group at C8, about half of the screened compounds significantly inhibited the two MAO isoforms at 10 µM concentration, achieving a slight selectivity towards MAO-A. The most potent MAO-A inhibitors were found to be 2b (IC50 1.49 µM) and 5b (IC50 1.85 µM) bearing NMe2 and N(Me)nBu, respectively, as NR2 at C8 and Cl as R1 at C6’. Three further compounds, namely the 6’-Br congeners 2c, 3c and 5c proved to act as MAO-A inhibitors with single-digit micromolar IC50s.
Within the limits of the explored property space, the data suggest that the anti-MAO (preferentially anti-MAO-A) activity is primarily affected by the size of the tertiary amine of the Mannich base at C8. As a trend, tertiary amines with double branched alkyl group (subset 7) or larger than Et (subsets 4, 6 and 8) are less tolerated than NMe2 (2), NEt2 (3) and N(Me)nBu (5). The halogens at C6’ appear to enhance MAO-A inhibition potency, with Cl exerting a slightly stronger effect than Br. In contrast, the introduction of a methoxy group at C8’ decreases the inhibitory activity against MAO-A.
Regarding the cholinesterase inhibition, most of the screened derivatives proved to preferentially inhibit AChE over BChE, with only four compounds (3c, 4d, 5c and 5d) achieving a finite IC50 at concentrations less than 10 μM. The most potent AChE inhibitor was found to be 3c (IC50 1.56 μM), which also exhibited MAO-A inhibition at very close IC50 (3.04 μM). The optimal AChE inhibition potency was achieved for -CH2NEt2 and -CH2NnPr2 at C8, with 6’-Br (3c and 5c) or 8’-OMe (4d and 5d) exerting favorable effects.
Overall, in the series examined, 3c appears to be the one that inhibits both MAO-A and AChE with a single digit micromolar IC50 and a slight selectivity over MAO-B and BChE, respectively.
2.2.2. Molecular Docking Calculations
To shed light on the molecular factors mainly responsible for more efficient binding modes and higher inhibition potencies of the examined amphoteric biscoumarins to the target enzymes, molecular docking calculations were carried out using the AutoDock software (release 4.2).The main results obtained from docking simulation of most of the compounds with crystal structure of human MAO-A, in complex with clorgiline (pdb 2BXR), or human MAO-B (pdb 1GOS) are reported in Supp. Mat. (Table S1), whereas in Table 2 the main docking calculation results are summarized, which pertain the highest-scored docking poses to MAO-A of 2a, 2b, 2d, 4a, 5a and 5b, which can help to understand the key factors modulating the inhibition potency of the compounds under investigation.
In Table 2, besides the free energy of binding (FEB, kcal·mol-1), the residues involved in H-bonding, hydrophobic/aromatic and ionic interactions with the ligand are reported. To aid in the interpretation of the potency ranking, the residues that were established as playing a role in substrate and inhibitor selectivity are highlighted in bold [32]. The 2D graphical representations of best scored docking pose of the lead 2a (NR2 = NMe2, Figure 2), compared to that of the less active 4a (NR2 = N(nPr)2, Figure 4), could suggest why and how the different MAO-A inhibitory potency may relate to diverse binding mode.
Unlike MAO-B, whose FAD binding site has two distinct cavities, i.e., the entrance and substrate cavities separated by a gate (Tyr326), MAO-A has one single hydrophobic cavity (about 550 Å3 volume), in which Phe208 and Ile335 play significant roles in substrate and inhibitor selectivity, whereas Tyr407 and Tyr444 may favor ligand binding through aromatic stacking or H-bond formation through their phenolic OH groups [33].
By orienting the coumarin moiety into a flexible cavity primarily lined by Phe208 and Val210 (not shown) through hydrophobic contacts, the biscoumarin 2a may bind MAO-A strongly. The coumarin benzene ring is well accommodated into the catalytic cleft facing the FAD cofactor (Figure 3) by formation of stable hydrogen bonding through Tyr444 (1.59Å). Two residues, namely Ser209 and Ile207, should contribute to stabilizing the complex interacting with the phenolic OH at C7 of the inhibitor as H-bond donor (2.87 Å) and acceptor (3.05 Å), respectively. Moreover, the protonated tertiary amine of 2a may attain a salt bridge strengthened by H-bonds with Glu216. In addition, several hydrophobic residues, such as Phe208, Val303, Ile335, Phe352, Tyr444 and Met445,should form extra hydrophobic interactions with the inhibitor into the active site (Figure 2a). The drug moclobemide, taken as reference MAO-A inhibitor, exhibited similar binding mode (Figure 2b).
Figure 3 highlights an almost parallel aromatic stacking between flavin and distal coumarin moiety.
With increasing size of the alkyls on tertiary amino groups beyond the ethyl groups, like in 4a (Figure 4), it can be reasonably hypothesized that the potency decrease could be due to a change in the binding mode with a decrease in the ligand efficiency. Indeed, the interactions of the n-propyl groups of the tertiary amine in 4a with the hydrophobic residues (Tyr444, Met445, Val 303, Phe352) do not most likely compensate the loss of electrostatic interactions and H-bonding achieved by the NMe2 group with Glu216. Besides the hydrophobic and aromatic interactions around the two coumarin moieties, two key interactions characterize the different (less efficient) binding mode of 4a compared to that of the more active 2a, i.e., Ser209 as H-bond donor to carbonyl O of the second coumarin nucleus (instead of the phenolic OH) and Tyr407 as H-bond donor to O of the first coumarin nucleus.
Compound 2b (6’-Cl congener) achieved higher activity against MAO-A compared to the unsubstituted 2a (Figure 5). Similarly, to moclobemide, it forms two key H-bonds with active site residues Ser209 and Tyr444, which suggests a similar binding mode. Moreover, the presence of the 6’-Cl substituent appears to enhance binding by stabilizing the molecule within the enzyme’s active site, through a halogen bond interaction with Gly66 which might contribute to a more favorable orientation and interaction profile (Figure 5 a). In contrast, compound 2d (8’-OMe congener) shows a different orientation in the active site (Figure 5 b) and a less efficient binding mode. The 8’-OMe group is directed toward the core of the binding pocket in an unfavorable conformation, disrupting optimal positioning and weakening key interactions (Figure 5 c). This might likely explain its reduced activity compared to 2b.
The examined biscoumarin derivatives were also docked against AChE using the crystal structure of the Torpedo californica AChE in complex with donepezil (pdb 1EVE). To validate the docking protocol, donepezil was re-docked using AutoDock, and the resulting RMSD value between the ligand and the re-docked pose was 1.95 Å, which demonstrated the reliability of the docking protocol. Information about all the docking simulation is reported in Supp. Mat. (Table S2), whereas Table 3 summarizes the results (i.e., free binding energy and the contacting residues) of some selected ligands, including the most potent AChE inhibitor 3c identified in the series (Figure 6).
Also in this case, to aid in the interpretation of the docking results, the residues mainly involved in the ligand binding are highlighted in bold, namely Trp84, Phe330 and Phe331 belonging to the anionic site, Phe288 and Phe290 lining the acyl pocket, Gly218, Gly219 and Ala 201 forming the oxyanion hole, and the aromatic residues Trp279, Tyr121, Tyr70 and Tyr334 which together with Asp72 delimit the peripheral anionic site (PAS).
The highest-scored docking pose of donepezil (Figure 6) highlighted the importance of interactions between the dimethoxy phenyl group and Trp279, while the phenyl group demonstrated aromatic interactions with Trp84. The piperidine N attained arene–cation interactions with Trp84 and Phe330. Hydrophobic interactions were found between donepezil and Ile444 and Tyr84.
Compound 3c, the most potent AChE inhibitor in the series, showed the best docking score (FEB = -9.15 kcal mol-1) and the best binding mode (Figure 5). In the best-scored docking pose, 3c may form H-bond between C=O of the first coumarin and Tyr70 (2.54 Å distance) and 7-OH and Phe228 (2.87 Å distance). Moreover, hydrophobic contacts and aromatic stackings with Phe330, Phe331, Tyr334, Leu282, Tyr279 and Ile287 may contribute to the stability of the 3c-AChE complex.
2.3. Physicochemical Properties and Drug-Likeness Assessment
2.3.1. Water Solubility and Lipophilicity Determinations
Aqueous solubility and lipophilicity were experimentally determined for the five compounds for which finite IC50 values could be measured within the threshold concentration close to 10 µM (i.e., 2a-d and 3c). These compounds include the most potent MAO-A inhibitor (2b) and the most potent AChE inhibitor (3c); the biscoumarin derivative 3c achieved, on the other hand, the strongest dual enzymes’ inhibition with single-digit micromolar IC50 values. The kinetic solubility in aqueous buffers was determined in HCl (0.01 M, pH 2.0, KCl 0.15 M) and in PBS (50 mM, pH 7.4, KCl 0.15 M) and at 25 ± 1 °C, using RP-HPLC as the analytical method.
In acidic solution (0.01 M HCl) all the tested compounds achieved a kinetic solubility > 2·10-4 mol·L-1, which means that they can be considered from ‘soluble’ to ‘very soluble’ according to the classification of SwissADME based on the topological approach of the ESOL model [34]. In PBS at pH 7.4 the solubilities were found to be two (2a) to thirteen (2c) to fifty (3c) times higher than that calculated with the ESOL model (Table 4), which means that they fall all within the class of ‘soluble’ compounds (-4 < Log S < -2) and not within the class of ‘moderately soluble’ compounds (-6 < Log S < -4) as predicted by ESOL-based calculations for neutral molecules.
Most likely such a strong solubility difference between calculated and experimental values to the different degree of ionization at pH 7.4 with respect to minimum solubility at the isoelectric pH where these amphoteric molecules should exist as zwitterions. Let’s take the homologs 2c and 3c as examples.
For the amphoteric compound 2c, ACD/Labs software (release 9.00. version 9.04) estimates pKa1 (related to the phenolic OH) and pKa2 (related to CH2NMe2) of about 9.0 and 7.2, respectively, from which an isoelectric pH of 8.1 is calculated. For the NEt2 homolog 3c, the estimated pKa values are about 9.4 and 8.2, and hence an isoelectric pH of 8.8. This could imply that at pH 7.4 the protonated fraction of compound 3c, and then its aqueous solubility, is consistently higher than that of 2c, despite the decrease of hydrophilicity of their neutral forms. Indeed, in contrast with the expected threefold decrease of water solubility, at pH 7.4 the less hydrophilic 3c achieves water solubility 1.3-fold higher than that of 2c.
The lipophilicity of the five compounds in Table 4 was assessed by calculation and experimental approaches. As a calculated lipophilicity parameter (log Pcalc) we took from SwissADME free website (http://www.swissadme.ch) the ‘Consensus Log Po/w’, that is the arithmetic mean of octanol/water log Ps calculated by five methods (iLOGP, XLOGP3, WLOGP, MLOGP, and SILICOS-IT) [34]. To ascertain the consistency of the calculated log P scale, a precise and cost-effective RP-HPLC method [36] was used to determine the polycratic capacity factor (log k’w), that is the log of the capacity factor (k’) extrapolated to 100% water mobile phase, for each compound. The correlation between the RP-HPLC-derived lipophilicity parameter and the predicted log Po/w was proven by the following statistically good linear regression Equation (1):
where R2 is the determination coefficient (i.e., a measure of the explained variance), S the standard deviation of the regression model (i.e., a measure of the distance between the observed datapoints and the regression line), F is the F-statistic indicating the overall significance of the regression model, and in parentheses 95% confidence intervals of the regression coefficients.
log k’w = 0.53 (± 0.10) log Pcalc + 0.67 (± 0.35)
n = 5, R2 = 0.900, S = 0.110, F = 27.08
n = 5, R2 = 0.900, S = 0.110, F = 27.08
In the above linear equation, the lower-than-unity slope value (+0.53) and the higher-than-zero intercept (+0.67) suggest that retention on the C18 column and partitioning between the immiscible 1-octanol and water are differently influenced by the physicochemical properties of the solutes/analytes. Apparently, the retention of the amphoteric biscoumarins in the RP-HPLC system is affected, not only by the hydrophobic interactions between the long alkyl chains (C18) and the biscoumarin analytes, which in turn dominate in the biphasic partitioning, but also by adsorption and silanophilic interactions, mainly governed by van der Waals interactions, H-bonding and electrostatic interactions [37]. However, the good statistics associated with the regression equation supports the consistency of the relative log Po/w scale assessed by the SwissADME website.
Chemoinformatic Assessment of Druglikeness and Bioavailability
For the investigated amphoteric biscoumarins achieving finite IC50 values less than (or very close to) 10 µM against MAO-A and/or AChE (2a-d, 3c, 4d, 5b-d) the physicochemical properties, including lipophilicity and water solubility, pharmacokinetics (GI absorption, BBB permeation and potential as P-gp substrate), druglikeness (as the compliance with the ‘Lipinski’s rule of five’), bioavailability and alerts for pan-assay interference compounds (PAINS) were estimated by querying the free website SwissADME [34] and summarized in Table S3 (Supp. Mat.).
According to predictions, all nine active compounds achieved a bioavailability score of 0.55, which should suggest a high probability of efficient absorption, with no violation of Lipinski’s rule of five and moderate aqueous solubility (at pH 7.4 the experimental water solubility resulted even higher, as in the case of the most dual active 3c). The gastrointestinal absorption is estimated to be high, whereas all the compounds are predicted to be ‘not BBB permeant’ and, with two exceptions (the 8’-methoxy congeners 4d and 5d), to be not P-gp substrates. While solutions for enhancing their BBB crossing can be found at a more advanced stage of lead optimization, exploiting for instance novel prodrug approaches or efficient nanoparticle-based drug delivery systems [38], one issue deals with the PAINS alert related to the presence of the Mannich base at C8 of the first coumarin moiety. This alert requires for sure attention in the next stages of study. Nevertheless, it has to be highlighted that several safe drugs in various therapeutic areas, including anti-inflammation, anti-infective and anti-cancer areas, bear Mannich base groups [39]. An interesting example of multitarget-directed ligand (MTDL) as a lead candidate for AD is provided by ortho-phenol Mannich base of the anti-inflammatory flurbiprofen [40], which showed in vitro good BBB permeability, low neurotoxicity, biometal-chelating and anti-oxidant properties, anti-inflammation, and inhibition of Aβ aggregation and AChE.
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
Starting materials and all chemicals and solvents were purchased from Merck (Darmstadt, Germany). Melting points were determined by using the capillary method on a Stuart Scientific SMP3 electrothermal apparatus and are uncorrected. Mass spectra were obtained by Agilent 1100 Series LC–MSD Trap System VL, equipped with ESI (electrospray ionization) source and the high-resolution molecular masses of test compounds were assessed by Agilent 6530 Accurate Mass Q-TOF spectrometer (Agilent Technologies). IR spectra (KBr disks) were recorded on a Spectrum One FT infrared spectrophotometer (PerkinElmer), and the most significant absorption bands are listed. 1H and 13C NMR spectra were recorded at 400 and 101MHz respectively, on a Varian Mercury instrument in dimethylsulfoxide-d6 (DMSO-d6) solution at 25 °C. Chemical shifts data are expressed in δ and the coupling constants J are in hertz (Hz); the following abbreviations are used for multiplicity: s, singlet; br. s, broad singlet; d, doublet; dd, doublet-doublet; td, triplet of doublets; t, triplet; q, quartet; m, multiplet.
3.1.2. Synthesis of 8-((dialkylamino)methyl)-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-ones(2-8)
To a hot solution of a biscoumarin 1a-d (2.0 mmol) in dioxane (10 mL), the suitable aminal (2.2 mmol) was added and the mixture was refluxed for 3-5 h. After TLC control, the reaction mixture was cooled and the solvent evaporated under reduced pressure. Finally, the residue crystallized from i-PrOH–n-hexane (1:2, v/v) mixture.
Synthesis and characterization of compounds 4a, 5a-c, 6a, 6b, 7a, 7d, 8a and 8b were published previously [30]. For each newly synthesized compound(2a-d, 3c, 4d, 5d, 6d and 8d) uncorrected m.p., main IR bands, 1H- and 13C-NMR spectra, HRMS m/z are reported.
8-((dimethylamino)methyl)-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (2a). M.p: 208-211°C; 1H NMR (400 MHz, DMSO) δ 8.28 (s, 1H), 7.81 (dd, J = 7.7, 1.3 Hz, 1H), 7.76 – 7.68 (m, 1H), 7.51 (d, J = 8.2 Hz, 1H), 7.48 – 7.39 (m, 1H), 7.36 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.8 Hz, 1H), 6.32 (s, 1H), 4.21 (br. s, 1H, OH), 3.96 (s, 2H, CH2), 2.38 (s, 6H, (CH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 163.96, 160.39, 159.17, 154.23, 153.15, 151.05, 144.21, 133.21, 129.52, 127.51, 125.28, 123.84, 119.17, 116.74, 113.79, 111.44, 110.03, 108.76, 54.10, 44.37 (2C) ppm; IR (KBr): ν = 3433, 1710, 1737, 1604cm-1; HRMS (Q-TOF): calcd for C21H17NO5 [M-H]– m/z 362.1034, found 362.1018, [M+Na]+ m/z 386.0999, found 386.1002.
8-((dimethylamino)methyl)-4-(6-chloro-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (2b). M.p: 210-212°C; 1H NMR (400 MHz, DMSO) δ 8.21 (s, 1H), 7.92 (d, J = 2.6 Hz, 1H), 7.76 (dd, J = 8.9, 2.6 Hz, 1H), 7.56 (d, J = 8.9 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.8 Hz, 1H), 3.97 (s, 2H, CH2, br. s, 1H, OH), 2.38 (s, 6H, (CH3)2) ppm;13C NMR (101 MHz, DMSO) δ 164.09, 160.29, 158.80, 153.19, 152.88, 150.67, 142.89, 132.68, 128.91, 128.44, 127.57, 125.02, 120.59, 118.77, 113.81, 111.43, 109.88, 108.69, 54.06, 44.33 (2C) ppm; IR (KBr): ν = 3420, 1714, 1604, 1574cm-1; HRMS (Q-TOF): calcd for C21H16ClNO5[M-H]– m/z 396.0644, found 396.0642, [M+H]+ m/z 398.0790, found 398.0788, [M+Na]+ m/z 420.0609, found 420.0609.
8-((dimethylamino)methyl)-4-(6-bromo-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (2c). M.p: 202-204°C;1H NMR (400 MHz, DMSO) δ 8.20 (s, 1H), 8.05 (d, J = 2.4 Hz, 1H), 7.87 (dd, J = 8.8, 2.4 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 6.66 (d, J = 8.8 Hz, 1H), 6.31 (s, 1H), 3.96 (s, 2H, CH2, br. s, 1H, OH), 2.38 (s, 6H, (CH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 164.12, 160.29, 158.75, 153.30, 153.18, 150.66, 142.81, 135.45, 131.44, 127.55, 124.96, 121.08, 119.03, 116.61, 113.82, 111.42, 109.87, 108.72, 54.10, 44.34 (2C) ppm; IR (KBr): ν = 3435, 1728, 1714, 1601, 1574cm-1; HRMS (Q-TOF): calcd for C21H16BrNO5 [M-H]– m/z 440.0139, found 440.0124, [M+Na]+ m/z 464.0104, found 464.0194.
8-((dimethylamino)methyl)-4-(8-methoxy-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (2d). M.p.: 221-225°C;1H NMR (400 MHz, DMSO) δ 8.26 (s, 1H), 7.52 – 7.24 (m, 4H), 6.67 (d, J = 8.8 Hz, 1H), 6.32 (s, 1H), 3.97 (s, 2H, CH2), 3.96 (s, 3H, OCH3), 3.78 (br. s, 1H, OH), 2.39 (s, 6H, N (CH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 163.84, 160.35, 158.90, 153.17, 151.02, 146.96, 144.43, 143.59, 127.59, 125.24, 123.99, 120.63, 119.71, 115.45, 113.76, 111.49, 110.05, 108.63, 56.74, 53.96, 44.33 (2C) ppm; IR (KBr): ν = 3430, 1708, 1607, 1384 cm-1; HRMS (Q-TOF): calcd for C22H19NO6 [M-H]– m/z 392.1140, found 392.1133, [M+Na]+ m/z 416.1105, found 416.1111.
8-((diethylamino)methyl)-4-(6-bromo-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (3c). M.p.: 198-200°C; 1H NMR (400 MHz, DMSO) δ 8.20 (s, 1H), 8.05 (d, J = 2.4 Hz, 1H), 7.87 (dd, J = 8.8, 2.4 Hz, 1H), 7.49 (d, J = 8.8 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 6.60 (d, J = 8.8 Hz, 1H), 6.28 (s, 1H), 4.09 (s, 2H, CH2), 3.52 (br. s, 1H, OH), 2.71 (q, J = 7.2 Hz, 4H, N-(CH2CH3)2), 1.11 (t, J = 7.2 Hz, 6H, N-(CH2CH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 160.34, 158.75, 153.29, 153.08, 150.69, 142.76, 135.43, 131.43 (2C), 127.32, 125.01, 121.08, 119.03 (2C), 116.69, 114.10, 109.62, 108.50, 49.43, 46.69 (2C), 11.27 (2C) ppm; IR (KBr): ν = 3433, 1731, 1602, 1378 cm-1; HRMS (Q-TOF): calcd for C23H20BrNO5 [M-H]– m/z 468.0452, found 468.0432, [M+Na]+ m/z 492.0417, found 416.0420.
8-((dipropylamino)methyl)-4-(8-methoxy-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (4d). M.p.: 128-130°C; 1H NMR (400 MHz, DMSO) δ 8.25 (s, 1H), 7.47 – 7.27 (m, 4H), 6.62 (d, J = 8.8 Hz, 1H), 6.31 (s, 1H), 4.07 (s, 2H, CH2), 3.96 (s, 3H, OCH3), 3.57 (br., s, 1H, OH), 2.57 (dd, J = 8.5, 6.6 Hz, 4H, N-(CH2CH2CH3)2), 1.65 – 1.46 (m, 4H, N-(CH2CH2CH3)2), 0.87 (t, J = 7.4 Hz, 6H, N-(CH2CH2CH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 164.24, 160.37, 158.90, 153.02, 151.02, 146.96, 144.42, 143.58, 127.30, 125.24, 124.02, 120.63, 119.71, 115.44, 113.86, 111.38, 109.99, 108.89, 56.74, 55.51 (2C), 50.50, 19.34 (2C), 12.05 (2C) ppm; IR (KBr): ν = 3430, 2962, 1712, 1605, 1478,1379 cm-1; HRMS (Q-TOF): calcd for C26H27NO6 [M-H]– m/z 448.1766, found 448.1744, [M+Na]+ m/z 472.1731, found 472.1741.
8-((butyl(methyl)amino)methyl)-4-(8-methoxy-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (5d). M.p.: 163-165°C; 1H NMR (400 MHz, DMSO) δ 8.25 (s, 1H), 7.46 – 7.27 (m, 4H), 6.63 (d, J = 8.8 Hz, 1H), 6.31 (s, 1H), 4.01 (s, 2H, CH2), 3.96 (s, 3H, OCH3), 3.54 (br. s, 1H, OH), 2.64 – 2.54 (m, 2H, N-(CH2CH2CH2CH3)), 2.32 (s, 3H, N-CH3), 1.54 (dt, J = 14.9, 7.4 Hz, 2H, N-(CH2CH2CH2CH3)), 1.32 (dq, J = 14.5, 7.3 Hz, 2H, N-(CH2CH2CH2CH3)), 0.90 (t, J = 7.3 Hz, 3H, N-(CH2CH2CH2CH3)) ppm; 13C NMR (101 MHz, DMSO) δ 160.38, 158.90, 151.02, 146.96, 144.40 (2C), 143.59, 127.40, 125.23 (2C), 124.03, 120.63 (2C), 119.71, 115.44, 113.86, 108.68 (2C), 56.74, 56.37, 53.07, 41.24, 28.66, 20.26, 14.22 ppm; IR (KBr): ν = 3437, 2935, 1720, 1603 cm-1; HRMS (Q-TOF): calcd for C25H25NO6 [M-H]– m/z 434.1609, found 434.1618, [M+Na]+ m/z 454.1574, found 454.1586.
8-((bis(2-methoxyethyl)amino)methyl)-4-(8-methoxy-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (6d). M.p.: 165-167°C; 1H NMR (400 MHz, DMSO) δ 8.26 (s, 1H), 7.54 – 7.13 (m, 4H), 6.68 (d, J = 8.8 Hz, 1H), 6.35 (s, 1H), 4.12 (s, 2H, CH2), 3.96 (s, 3H, OCH3), 3.51 (t, J = 5.5 Hz, 4H, (NCH2-CH2OCH3)2), 3.24 (s, 6H, (NCH2-CH2OCH3)2), 2.81 (t, J = 5.5 Hz, 4H, (NCH2-CH2OCH3)2) ppm; 13C NMR (101 MHz, DMSO) δ 162.97, 160.33, 158.89, 153.05, 151.00, 146.97, 144.46, 143.60, 127.34, 125.24, 123.97, 120.64, 119.71, 115.46, 113.63, 111.95, 110.50, 109.75, 69.63 (2C), 58.50 (2C), 56.74, 53.18 (2C), 49.82 ppm; IR (KBr): ν = 3437, 1725, 1600, 1384 cm-1; HRMS (Q-TOF): calcd for C26H27NO8 [M-H]– m/z 480.1664, found 480.1649, [M+Na]+ m/z504.1629, found 504.1638.
8-(((benzyl(methyl)amino)methyl)-4-(8-methoxy-2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one (8d). M.p.: 204-206°C; 1H NMR (400 MHz, DMSO) δ 8.26 (s, 1H), 7.49 – 7.22 (m, 9H), 6.71 (d, J = 8.8 Hz, 1H), 6.36 (s, 1H), 4.02 (s, 2H), 3.96 (s, 3H, OCH3), 3.74 (s, 2H, Bn), 2.23 (s, 3H, NCH3) ppm; 13C NMR (101 MHz, DMSO) δ 162.73, 160.31, 158.88, 153.20, 150.97, 146.97, 144.48, 143.60, 137.50, 129.73 (2C), 128.94 (2C), 127.99, 127.54, 125.24, 123.95, 120.64, 119.71, 115.46, 113.48, 112.04, 110.59, 109.38, 60.97, 56.74, 51.84 ppm; IR (KBr): ν = 3439, 1723, 1602 cm-1; HRMS (Q-TOF): calcd for C28H23NO6 [M-H]– m/z 468.1453, found 468.1451, [M+Na]+ m/z 492.1418, found 504.1413.
3.2. Determination of Kinetic Aqueous Solubility and Lipophilicity
Water solubility and lipophilicity parameters were measured following previously reported methods [36]. For measuring kinetic solubility in aqueous buffers, a 200 µM sample solution in PBS (50 mM, pH 7.4, KCl 0.15 M), or in HCl (0.01 M, pH 2.00, KCl 0.15 M) prepared from a 10 mM stock solution in DMSO, was incubated at room temperature (25 ± 1 °C) for 2 hours while being continuously shaken at 250 rpm on an orbital shaker. The mixture was then separated by centrifugation at 2500 rpm for 3 minutes. Immediately after filtration, 100 µL of the filtrate was mixed with an equal volume of a 1:1 (v/v) DMSO and PBS (50 mM, pH 7.4, KCl 0.15 M) or HCl (0.01 M, pH 2, KCl 0.15 M) solution to prevent precipitation from the saturated solution and subsequently analyzed by HPLC. The peak area was compared to a calibration curve of the tested compound in MeOH. The analytical parameters were set as follows. Mobile Phase: MeOH/water pH 2.8 (acetic acid 0.5% v/v) at different percentages depending on the retention time of each test compound; stationary phase: Phenomenex, Kinetex 5 µ, C8, (150 × 3 mm); flux: 0.5 µL/min; injection: 2 µL; λ 330 nm. The analyses were performed on the Agilent HPLC 1260 Infinity Series Integrated System (Agilent Technologies, Milan, Italy).
The lipophilicity parameters were determined by an RP-HPLC technique [36]. In short, methanolic solutions of the examined compounds (1 mg·mL-1) were analyzed by the Agilent 1260 infinite HPLC system (Agilent Technologies, Milan, Italy) equipped with a diode array detector (DAD), using a Phenomenex C18-column (Kinetex 5 μ, 100 Å, 150×3 mm) as the stationary phase and MeOH/water buffer mobile phases with different v/v composition (i.e., mobile phases containing 0.05 increments of MeOH volume fractions in 10 mM ammonium formate buffer at pH 4.5; φMeOH ranging between 0.70 and 0.20). The chromatographic measurements were carried out at 25 ± 1 °C, flow rate of 0.5 mL·min−1 and detection at λ 330 nm. The log of capacity factors of each compound at different mobile phase compositions was calculated (log k′= log (tR − t0)/t0, where tR represents the retention time of the solute and t0 is the column dead time). For each compound, the equation of the linear relationship between log k′ and φMeOH (R2 > 0.991) on at least five data points, each from duplicate, was derived by regression analysis and the intercept at the y-axis (i.e., φMeOH = 0) calculated as the lipophilicity parameter log k′w.
3.3. Enzymes Inhibition Assays
3.3.1. Inhibition of Monoamine Oxidases
The inhibition assays of human monoamine oxidases were conducted using a fluorescence-based method with kynuramine as a non-selective substrate for each isoforms MAO-A/B [42,43,44]. Recombinant human MAO-A and MAO-B (microsomes from insect cells infected with baculovirus; Sigma Aldrich) were used. The measurements were performed in triplicate in black round-bottom polystyrene 96-well plates (Greiner Bio-One, Kremsmünster, Austria). Fluorescence readings were performed using an Infinite M1000 Pro plate reader (Tecan, Cernusco s.N., Italy), and inhibition values were calculated using Prism (version 5.01 for Windows; GraphPad Software, San Diego, CA, USA). To evaluate enzymatic activity, clorgiline (a selective MAO-A inhibitor) and selegiline (a selective MAO-B inhibitor) were employed as positive controls. In the MAO inhibition assays all compounds were initially screened at a concentration of 10 µM. Each of them was preincubated at 37°C for 20 minutes with 50 µM kynuramine in a phosphate buffer (PBS, 0.1 M, pH 8.0), adjusted to 0.39 osmolarity with KCl. After adding recombinant human MAO-A/B (250 U/mg and 59 U/mg, respectively) and incubating for another 30 minutes, NaOH was added. The fluorescence was measured at excitation/emission wavelengths of 310/400 nm. Compounds showing at least 60% MAO inhibition at 10 µM were further tested at seven different concentrations to determine the IC50 value, calculated via nonlinear regression. The IC50 is reported as the mean ± SD of three independent experiments, each performed in duplicate.
3.3.2. Inhibition of Cholinesterases
Cholinesterase inhibition assays were carried out using the Ellman spectrophotometric method, modified for a 96-well plate format [43,44]. Acetylcholinesterase from electric eel and butyrylcholinesterase from equine serum (Sigma Aldrich) were employed. The measurements were performed in triplicate using clear, flat-bottom polystyrene plates (Grainer). To assess enzymatic activity, donepezil (AChE-selective) and tacrine (BChE-selective) were used as positive controls. The solutions containing AChE (1.2 U/mL) or BChE (2 U/mL), 5,5’-dithiobis(2-nitrobenzoic acid) (Ellman’s reagent, 0.33 mM), the test compound (10 µM concentration for the initial screening, or seven different concentrations for compounds achieving more 60% enzyme inhibition at 10 µM to determine IC50 value), in PBS (0.1 M, pH 8.0), were incubated for 20 minutes at 25°C. Then, acetylthiocholine or butyrylthiocholine iodide (5 µM) as substrate was added, and the hydrolysis rates were recorded for 5 minutes at 412 nm. The IC50 value, calculated using the nonlinear regression method ‘log[inhibitor] vs. response’, or % inhibition at 10 µM, is reported as the mean ± SD of three independent experiments, each performed in duplicate.
3.4. Molecular Docking Calculations
To comprehend the molecular binding mechanisms of the active compounds toward two distinct biological targets that are associated with human monoamine oxidases enzymes, molecular docking studies were conducted. The human MAO-A (PDB code 2BXR) and human MAO-B (PDB code 1GOS) crystallographic structures from the Protein Data Bank [45] (PDB) were taken into consideration as the enzyme targets for docking simulations. In the docking simulations, the proteins were considered without co-crystallized ligands. The docking protocol was validated by performing the simulation with only the bound ligands and low RMSD between the docked and crystal conformations, prior to screening the active compounds.
All docking calculations were performed using AutoDock 4.2 [46] software. The docking input files were created, and the docking outcomes were analyzed using the AutoDock Tools software. A 90×90×90 grid box was created, covering nearly the whole surface of the protein, with grid points spaced 0.375 Å apart. Before the computations, all non-polar hydrogens and crystallographic water molecules were eliminated. The mass center of the TSA that was bound served as the docking grid’s center. Using genetic algorithm searches, 100 docked structures were produced in each case.
A default protocol was used, with an initial population of 50 randomly arranged conformations. Heavy atom comparison root mean square deviations (RMSD values) were determined and initial ligand binding modes were plotted. Plots of protein-ligand interactions were generated using LigandScout software [47].
3.5. Drug-Likeness and Bioavailability Assessment
Physicochemical properties, such as topological polar surface area (TPSA), consensus logarithm of partition coefficient (Log Po/w) and water solubility (Log S), pharmacokinetics, ADME-related features and drug-likeness of the investigated compounds were estimated using the free web server SwissADME [34].
4. Conclusions
Evaluating the inhibitory activity of MAOs and ChEs of nineteen amphoteric 3,4’-biscoumarins bearing a (dialkylamino)methyl group at C8 and a phenolic hydroxyl at C7 of the coumarin core, some compounds were disclosed which proved to inhibit MAO-A and/or AChE with IC50 values in the low micromolar range. Structure-activity relationships (SARs) suggested that the enzymes’ inhibitory potency depends upon the bulkiness and hydrophobicity of the NR2 group. Indeed, the highest inhibition potency against MAO-A (IC50 = 1.49 µM) was achieved by the 8-(dimethylamino)methyl derivative 2b (6’-Cl congener), whereas the 8-(diethylamino)methyl derivative 3c (6’-Br congener) exhibited the highest inhibition potency against AChE (IC50 = 1.56 µM) as well as a good inhibition of MAO-A (IC50 = 3.04 µM).
Interestingly, molecular docking calculations provided support to understand the key interactions responsible for MAO-A inhibition. The biscoumarin derivative 2b binds into the MAO-A active pocket in a mode like that adopted by the reference MAO-A selective reversible inhibitor moclobemide forming H-bond interactions with Ser209 and Tyr444, as well as hydrophobic/aromatic interactions; the binding affinity of 2b is favored by an almost parallel π-stacking between the coumarin-3-yl moiety and the flavin of the cofactor FAD. The most potent AChE inhibitor 3c in this series achieved the best docking score, but a binding mode quite different from that of the reference drug donepezil. The most potent dual inhibitor 3c, which can be taken as a hit for future molecular optimization studies and candidate for further AD-related targeted biochemical and biophysical assays (e.g., biometal-chelating, antioxidant, β-amyloid aggregation), showed a good water solubility at pH 7.4 (1.8 mol·L-1; fiftyfold more soluble than calculated). Chemoinformatics suggests for 3c full compliance with Lipinski’s rule-of-five, high probability of gastrointestinal absorption, and no or poor SNC distribution due to low BBB permeation. While there is room to work on the last point, herein we identified 4-(2-oxo-2H-chromen-3-yl)-2H-chromen-2-one as a new scaffold of amphoteric MTDLs for treating AD and other neurodegenerative syndromes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.G., V.K. and C.D.A.; methodology, A.P., C.D., R.P. and B:L.; software, A.P. and M.d.C.; validation, A.P., M.C. and M.d.C; formal analysis, R.P., C.D. and A.G.; investigation, M.C., V.K. and A.N.K; resources, M.d.C; data curation, C.D., A.P. and R.P.; writing—original draft preparation, A.G., A.P., C.D. and C.D.A.; writing—review and editing, all the Authors; visualization, all the Authors; supervision, A.G. and C.D.A. All the Authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and supplementary material. Further inquiries can be directed to the corresponding authors.
Acknowledgments
R.P. and M.d.C. acknowledge the financial support of the Italian Ministry of Universities and Research (PRIN, Grant 202297LJZK_003).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, Z.-R.; Huang, J.-B.; Yang, S.-L.; Hong, F.-F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef]
- Llanes, L.C.; Kuehlewein, I.; França, I.V.D.; Da Silva, L.V.; Da Cruz Junior, J.W. Anticholinesterase Agents For Alzheimer’s Disease Treatment: An UpdatedOverview. CMC 2023, 30, 701–724. [Google Scholar] [CrossRef]
- Rafe, M.R.; Saha, P.; Bello, S.T. Targeting NMDA Receptors with an Antagonist Is a Promising Therapeutic Strategy for Treating Neurological Disorders. Behavioural Brain Research 2024, 472, 115173. [Google Scholar] [CrossRef]
- Kim, B.-H.; Kim, S.; Nam, Y.; Park, Y.H.; Shin, S.M.; Moon, M. Second-Generation Anti-Amyloid Monoclonal Antibodies for Alzheimer’s Disease: Current Landscape and Future Perspectives. Transl Neurodegener 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Multi-Target Drug Design Using Chem-Bioinformatic Approaches; Roy, K., Ed.; Methods in Pharmacology and Toxicology; Springer New York: New York, NY, USA, 2019; ISBN 978-1-4939-8732-0. [Google Scholar]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Dostert, P.L.; Benedetti, M.S.; Tipton, K.F. Interactions of Monoamine Oxidase with Substrates and Inhibitors. Medicinal Research Reviews 1989, 9, 45–89. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged Scaffolds as MAO Inhibitors: Retrospect and Prospects. European Journal of Medicinal Chemistry 2018, 145, 445–497. [Google Scholar] [CrossRef]
- Sblano, S.; Boccarelli, A.; Mesiti, F.; Purgatorio, R.; De Candia, M.; Catto, M.; Altomare, C.D. A Second Life for MAO Inhibitors? From CNS Diseases to Anticancer Therapy. European Journal of Medicinal Chemistry 2024, 267, 116180. [Google Scholar] [CrossRef]
- Pisani, L.; Catto, M.; Muncipinto, G.; Nicolotti, O.; Carrieri, A.; Rullo, M.; Stefanachi, A.; Leonetti, F.; Altomare, C. A Twenty-Year Journey Exploring Coumarin-Based Derivatives as Bioactive Molecules. Front. Chem. 2022, 10, 1002547. [Google Scholar] [CrossRef]
- Todorov, L.; Saso, L.; Kostova, I. Antioxidant Activity of Coumarins and Their Metal Complexes. Pharmaceuticals 2023, 16, 651. [Google Scholar] [CrossRef]
- Malamati-Konstantina, K.; Dimitra, H.-L. Coumarin Derivatives as Therapeutic Candidates: A Review of Their Updated Patents (2017–Present). Expert Opinion on Therapeutic Patents 2024, 34, 1231–1254. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva-Reis, R.; Moreira-Pais, A.; Ferreira, T.; Oliveira, P.A.; Ferreira, R.; Cardoso, S.M.; Sharifi-Rad, J.; Butnariu, M.; Costea, M.A.; et al. Dicoumarol: From Chemistry to Antitumor Benefits. Chin Med 2022, 17. [Google Scholar] [CrossRef]
- Ren, Q.-C.; Gao, C.; Xu, Z.; Feng, L.-S.; Liu, M.-L.; Wu, X.; Zhao, F. Bis-Coumarin Derivatives and Their Biological Activities. CTMC 2018, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-T.; Lv, S.-M.; Lu, C.-H.; Gong, J.; An, J.-B. Effect of 3,3′-Biisofraxidin on Apoptosis of Human Gastric Cancer BGC-823 Cells. Trop. J. Pharm Res 2015, 14, 1803. [Google Scholar] [CrossRef]
- Qian, H.; Wang, B.; Ma, J.; Li, C.; Zhang, Q.; Zhao, Y. Impatiens Balsamina: An Updated Review on the Ethnobotanical Uses, Phytochemistry, and Pharmacological Activity. Journal of Ethnopharmacology 2023, 303, 115956. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Tian, R.; Feng, J.; Yang, N.; Yuan, L. A Systematic Review on Traditional Medicine Toddalia Asiatica (L.) Lam.: Chemistry and Medicinal Potential. Saudi Pharmaceutical Journal 2021, 29, 781–798. [Google Scholar] [CrossRef]
- Buechi, G.; Klaubert, D.H.; Shank, R.C.; Weinreb, S.M.; Wogan, G.N. Structure and Synthesis of Kotanin and Desmethylkotanin, Metabolites of Aspergillus Glaucus. J. Org. Chem. 1971, 36, 1143–1147. [Google Scholar] [CrossRef]
- Qu, D.; Li, J.; Yang, X.-H.; Zhang, Z.-D.; Luo, X.-X.; Li, M.-K.; Li, X. New Biscoumarin Derivatives: Synthesis, Crystal Structure, Theoretical Study and Antibacterial Activity against Staphylococcus Aureus. Molecules 2014, 19, 19868–19879. [Google Scholar] [CrossRef]
- Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Naik, N.S.; Shastri, L.A.; Dodamani, S.; Jalalpure, S.; Dixit, S.R.; Joshi, S.D.; Sunagar, V.A. Green, Unexpected Synthesis of Bis-Coumarin Derivatives as Potent Anti-Bacterial and Anti-Inflammatory Agents. European Journal of Medicinal Chemistry 2018, 143, 1744–1756. [Google Scholar] [CrossRef]
- Luo, W.; Chang, G.; Lin, D.; Xie, H.; Sun, H.; Li, Z.; Mo, S.; Wang, R.; Wang, Y.; Zheng, Z. 3,3’-((3,4,5-trifluoropHenyl)Methylene)Bis(4-Hydroxy-2H-Chromen-2-One) Inhibit Lung Cancer Cell Proliferation and Migration. PLoS ONE 2024, 19, e0303186. [Google Scholar] [CrossRef]
- Abdallah, R.M.; Hammoda, H.M.; El-Gazzar, N.S.; Ibrahim, R.S.; Sallam, S.M. Exploring the Anti-Obesity Bioactive Compounds of Thymelaea Hirsuta and Ziziphus Spina-Christi through Integration of Lipase Inhibition Screening and Molecular Docking Analysis. RSC Adv. 2023, 13, 27167–27173. [Google Scholar] [CrossRef]
- Ha, N.X.; Huong, T.T.; Khanh, P.N.; Hung, N.P.; Loc, V.T.; Ha, V.T.; Quynh, D.T.; Nghi, D.H.; Hai, P.T.; Scarlett, C.J.; et al. In Vitro and in Silico Study of New Biscoumarin Glycosides from Paramignya Trimera against Angiotensin-Converting Enzyme 2 (ACE-2) for Preventing SARS-CoV-2 Infection. Chem. Pharm. Bull. 2024, 72, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-X.; Mouscadet, J.-F.; Chiang, C.-C.; Tsai, H.-J.; Hsu, L.-Y. HIV-1 Integrase Inhibition of Biscoumarin Analogues. Chem. Pharm. Bull. 2006, 54, 682–686. [Google Scholar] [CrossRef]
- Asgari, M.S.; Mohammadi-Khanaposhtani, M.; Kiani, M.; Ranjbar, P.R.; Zabihi, E.; Pourbagher, R.; Rahimi, R.; Faramarzi, M.A.; Biglar, M.; Larijani, B.; et al. Biscoumarin-1,2,3-Triazole Hybrids as Novel Anti-Diabetic Agents: Design, Synthesis, in Vitro α-Glucosidase Inhibition, Kinetic, and Docking Studies. Bioorganic Chemistry 2019, 92, 103206. [Google Scholar] [CrossRef]
- Bustamante, D.; Bustamante, L.; Segura-Aguilar, J.; Goiny, M.; Herrera-Marschitz, M. Effects of the DT-Diaphorase Inhibitor Dicumarol on Striatal Monoamine Levels in L-DOPA and L-Deprenyl Pre-Treated Rats. neurotox res 2003, 5, 569–577. [Google Scholar] [CrossRef]
- Hudáčová, M.; Hamuľaková, S.; Konkoľová, E.; Jendželovský, R.; Vargová, J.; Ševc, J.; Fedoročko, P.; Soukup, O.; Janočková, J.; Ihnatova, V.; et al. Synthesis of New Biscoumarin Derivatives, In Vitro Cholinesterase Inhibition, Molecular Modelling and Antiproliferative Effect in A549 Human Lung Carcinoma Cells. IJMS 2021, 22, 3830. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-Y.; Cui, H.; Lu, X.-Y.; Zhang, L.-D.; Ding, X.-Y.; Wu, J.-J.; Duan, L.-X.; Zhang, S.-J.; Liu, Z.; Zhang, R.-R. (+/−)-Dievodialetins A−G: Seven Pairs of Enantiomeric Coumarin Dimers with Anti-Acetylcholinesterase Activity from the Roots of Evodia Lepta Merr. Phytochemistry 2021, 182, 112597. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; De Palma, A.; Farina, R.; Cellamare, S.; Altomare, C.D. Discovery of Potent Dual Binding Site Acetylcholinesterase Inhibitors via Homo- and Heterodimerization of Coumarin-Based Moieties. ChemMedChem 2017, 12, 1349–1358. [Google Scholar] [CrossRef]
- Frasinyuk, M.S.; Bondarenko, S.P.; Khilya, V.P. Chemistry of 3-Hetarylcoumarins 3*. Synthesis and Aminomethylation of 7′-Hydroxy-3,4′- Bicoumarins. Chem Heterocycl Comp 2012, 48, 422–426. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of Conserved Active Site Residues in Monoamine Oxidase A and B and Their Three-Dimensional Molecular Modeling. Journal of Biological Chemistry 2002, 277, 17209–17216. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D. The FAD Binding Sites of Human Monoamine Oxidases A and B. NeuroToxicology 2004, 25, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Nevskaya, A.A.; Purgatorio, R.; Borisova, T.N.; Varlamov, A.V.; Anikina, L.V.; Obydennik, A.Y.; Nevskaya, E.Y.; Niso, M.; Colabufo, N.A.; Carrieri, A.; et al. Nature-Inspired 1-Phenylpyrrolo [2,1-a]Isoquinoline Scaffold for Novel Antiproliferative Agents Circumventing P-Glycoprotein-Dependent Multidrug Resistance. Pharmaceuticals 2024, 17, 539. [Google Scholar] [CrossRef]
- Tan, L.C.; Carr, P.W.; Frechet, J.M.J.; Smigol, V. Liquid Chromatographic Study of Solute Hydrogen Bond Basicity. Anal. Chem. 1994, 66, 450–457. [Google Scholar] [CrossRef]
- Alves, P.A.; Camargo, L.C.; Souza, G.M.D.; Mortari, M.R.; Homem-de-Mello, M. Computational Modeling of Pharmaceuticals with an Emphasis on Crossing the Blood–Brain Barrier. Pharmaceuticals 2025, 18, 217. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, M.; Gul, H.I. Current Pharmaceutical Research on the Significant Pharmacophore Mannich Bases in Drug Design. CTMC 2023, 23, 2590–2608. [Google Scholar] [CrossRef]
- Liu, H.; Qiang, X.; Song, Q.; Li, W.; He, Y.; Ye, C.; Tan, Z.; Deng, Y. Discovery of 4′-OH-Flurbiprofen Mannich Base Derivatives as Potential Alzheimer’s Disease Treatment with Multiple Inhibitory Activities. Bioorganic & Medicinal Chemistry 2019, 27, 991–1001. [Google Scholar] [CrossRef]
- Dubovik, I.P.; Garazd, M.M.; Khilya, V.P. Modified Coumarins. 14. Synthesis of 7-Hydroxy-[4,3?]Dichromenyl-2,2?-Dione Derivatives. Chem Nat Compd 2004, 40, 434–443. [Google Scholar] [CrossRef]
- Kulikova, L.N.; Purgatorio, R.; Beloglazkin, A.A.; Tafeenko, V.A.; Reza, R.G.; Levickaya, D.D.; Sblano, S.; Boccarelli, A.; De Candia, M.; Catto, M.; et al. Chemical and Biological Evaluation of Novel 1H-Chromeno [3,2-c]Pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity. IJMS 2023, 24, 7724. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.A.; Purgatorio, R.; Obydennik, A.Y.; Listratova, A.V.; Borisova, T.N.; de Candia, M.; Catto, M.; Altomare, C.D.; Varlamov, A.V.; Voskressensky, L.G. Synthesis of Isomeric 3-Benzazecines Decorated with Endocyclic Allene Moiety and Exocyclic Conjugated Double Bond and Evaluation of Their Anticholinesterase Activity. Molecules 2022, 27, 6276. [Google Scholar] [CrossRef]
- Purgatorio, R.; Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of Water-Soluble Mannich Base Prodrugs of 2,3,4,5-Tetrahydroazepino [4,3-b]Indol-1(6 H)-one as Multitarget-Directed Agents for Alzheimer’s Disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative structures of biologically active biscoumarin derivatives.
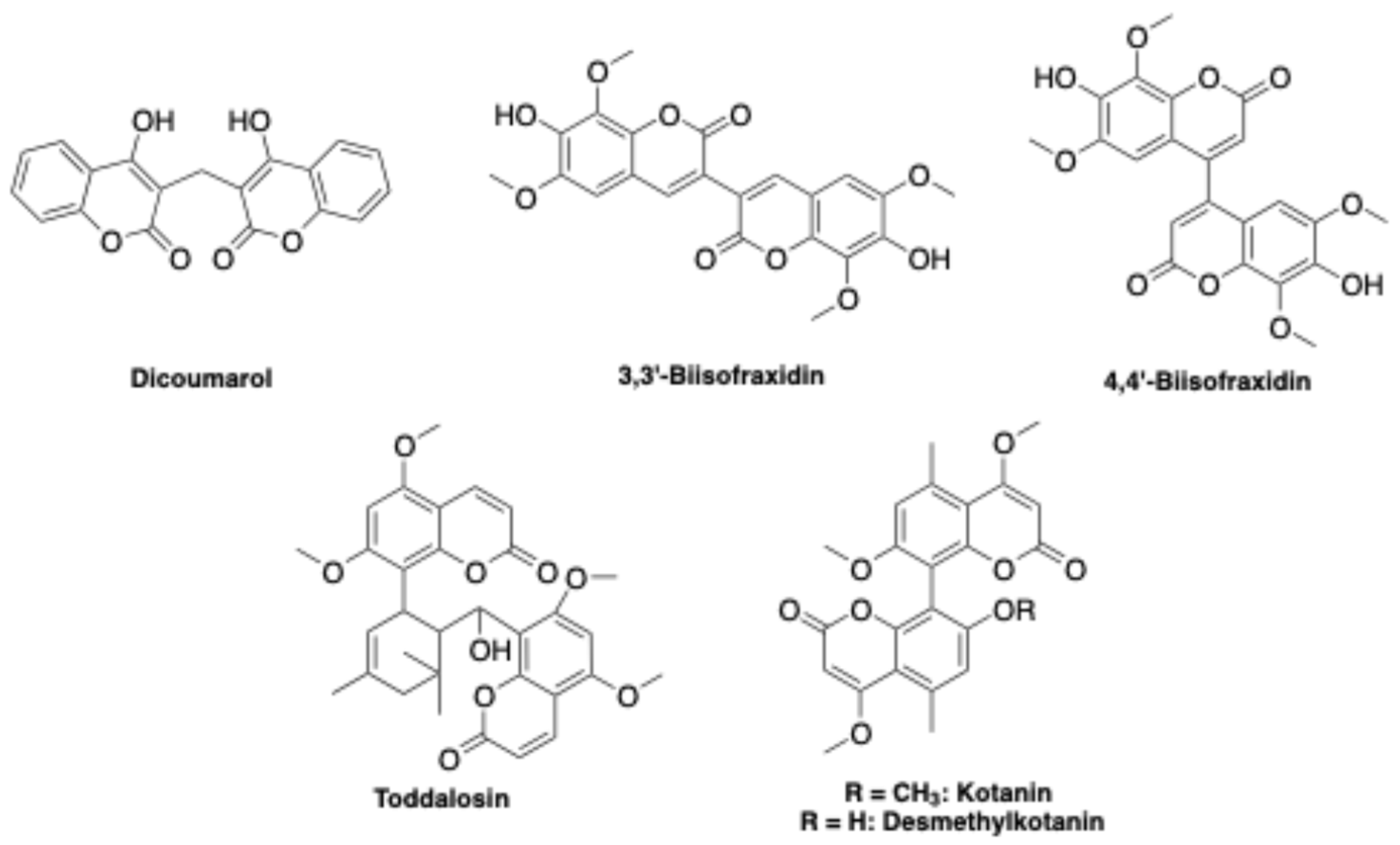
Figure 2.
(a) 2D diagram of the lead compound 2a into the MAO-A binding site. (b) Superposition of the structure of 2a (pink) and moclobemide (blue), a well-known selective MAO-A inhibitor, into the active site of MAO-A enzyme. Red and green dotted arrows indicate H-bond interactions, blue arrows interactions with positive ionizable amino group and yellow curved lines (a) or spheres (b) hydrophobic interactions.
Figure 2.
(a) 2D diagram of the lead compound 2a into the MAO-A binding site. (b) Superposition of the structure of 2a (pink) and moclobemide (blue), a well-known selective MAO-A inhibitor, into the active site of MAO-A enzyme. Red and green dotted arrows indicate H-bond interactions, blue arrows interactions with positive ionizable amino group and yellow curved lines (a) or spheres (b) hydrophobic interactions.
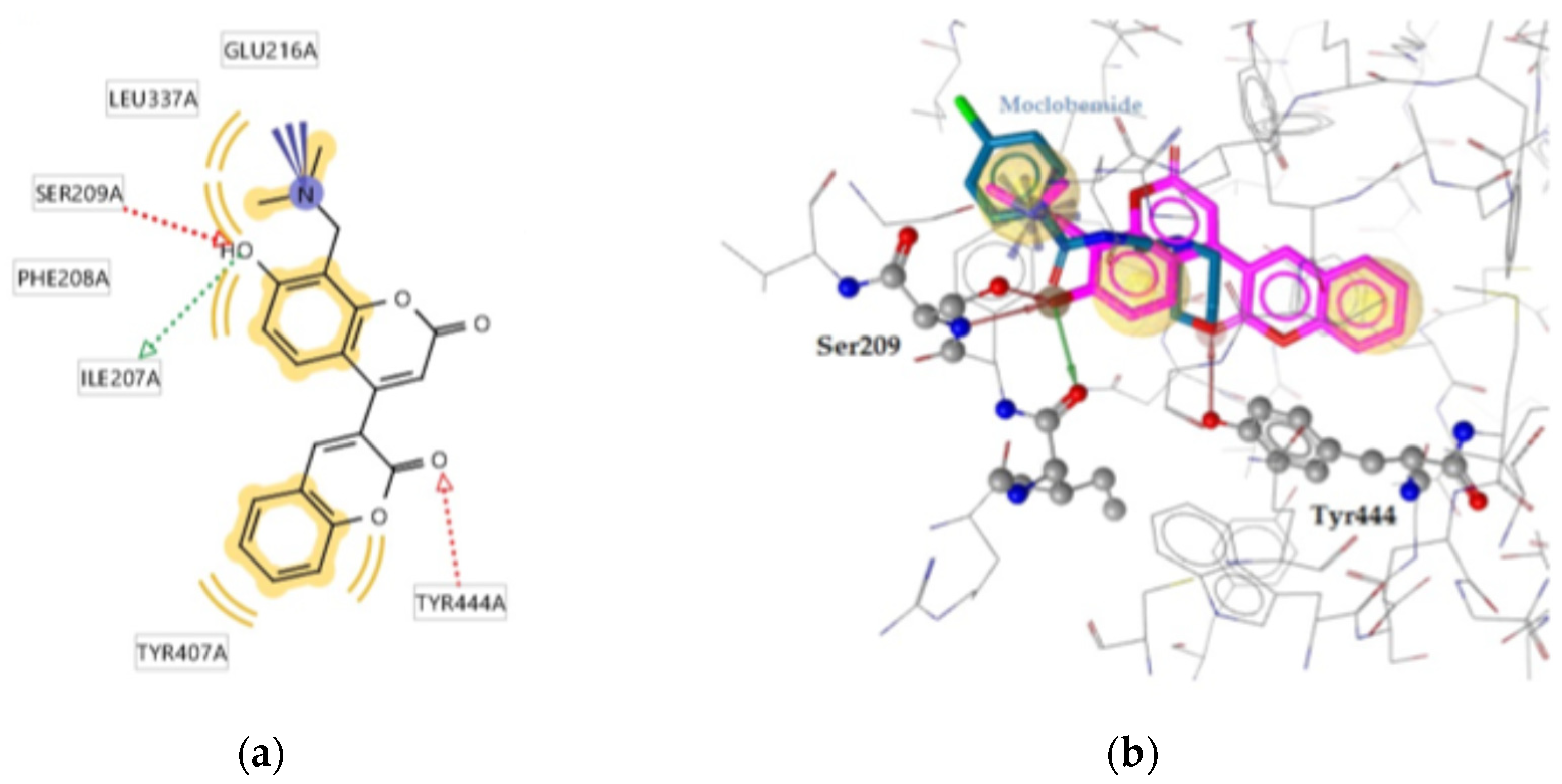
Figure 3.
The highest scored docking pose of the biscoumarin compound 2a (pink) and FAD (blue) into the active site of MAO-A enzyme.
Figure 3.
The highest scored docking pose of the biscoumarin compound 2a (pink) and FAD (blue) into the active site of MAO-A enzyme.
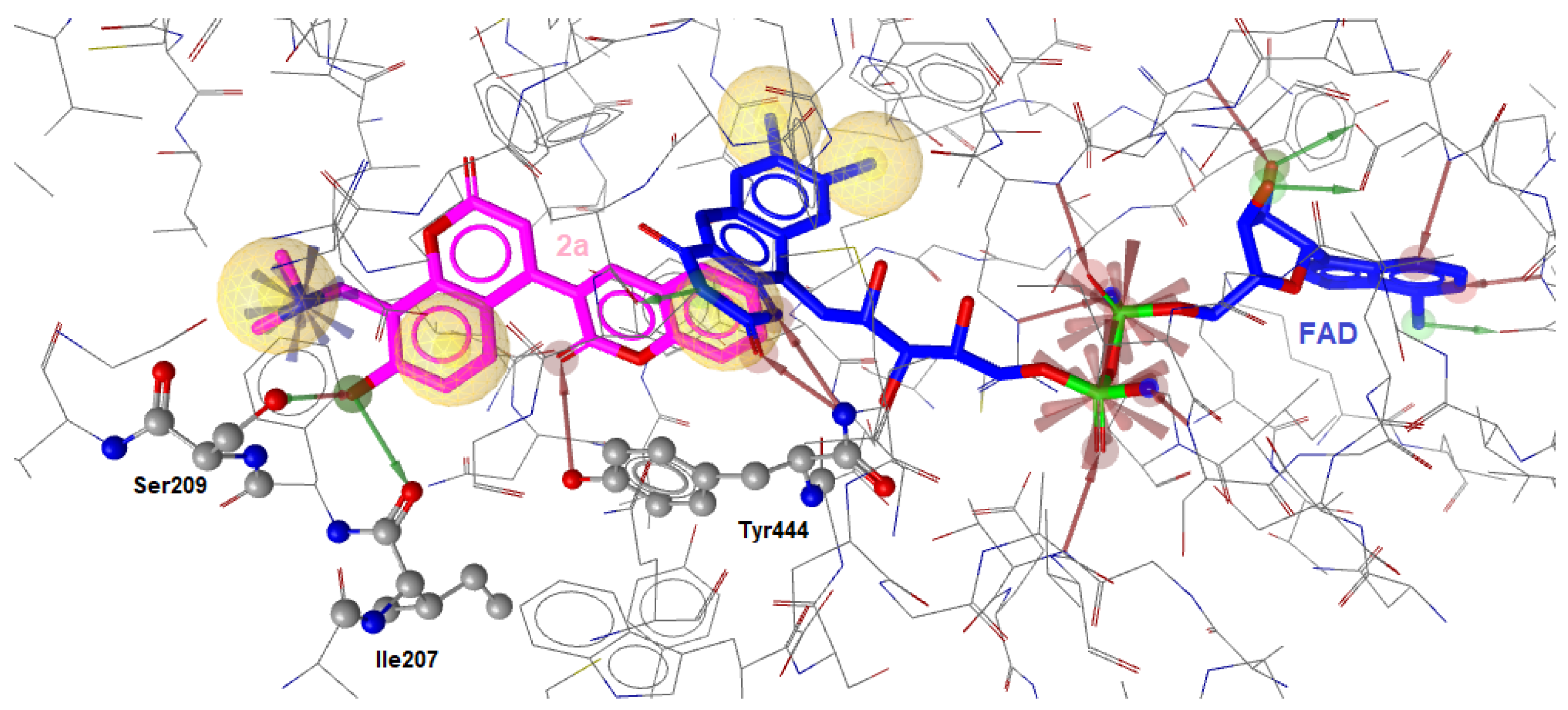
Figure 4.
2D diagram of compound 4a into the binding site of MAO-A. Red and green dotted arrows indicate H-bonds, blue arrows positive ionizable group and yellow curved lines hydrophobic interactions.
Figure 4.
2D diagram of compound 4a into the binding site of MAO-A. Red and green dotted arrows indicate H-bonds, blue arrows positive ionizable group and yellow curved lines hydrophobic interactions.
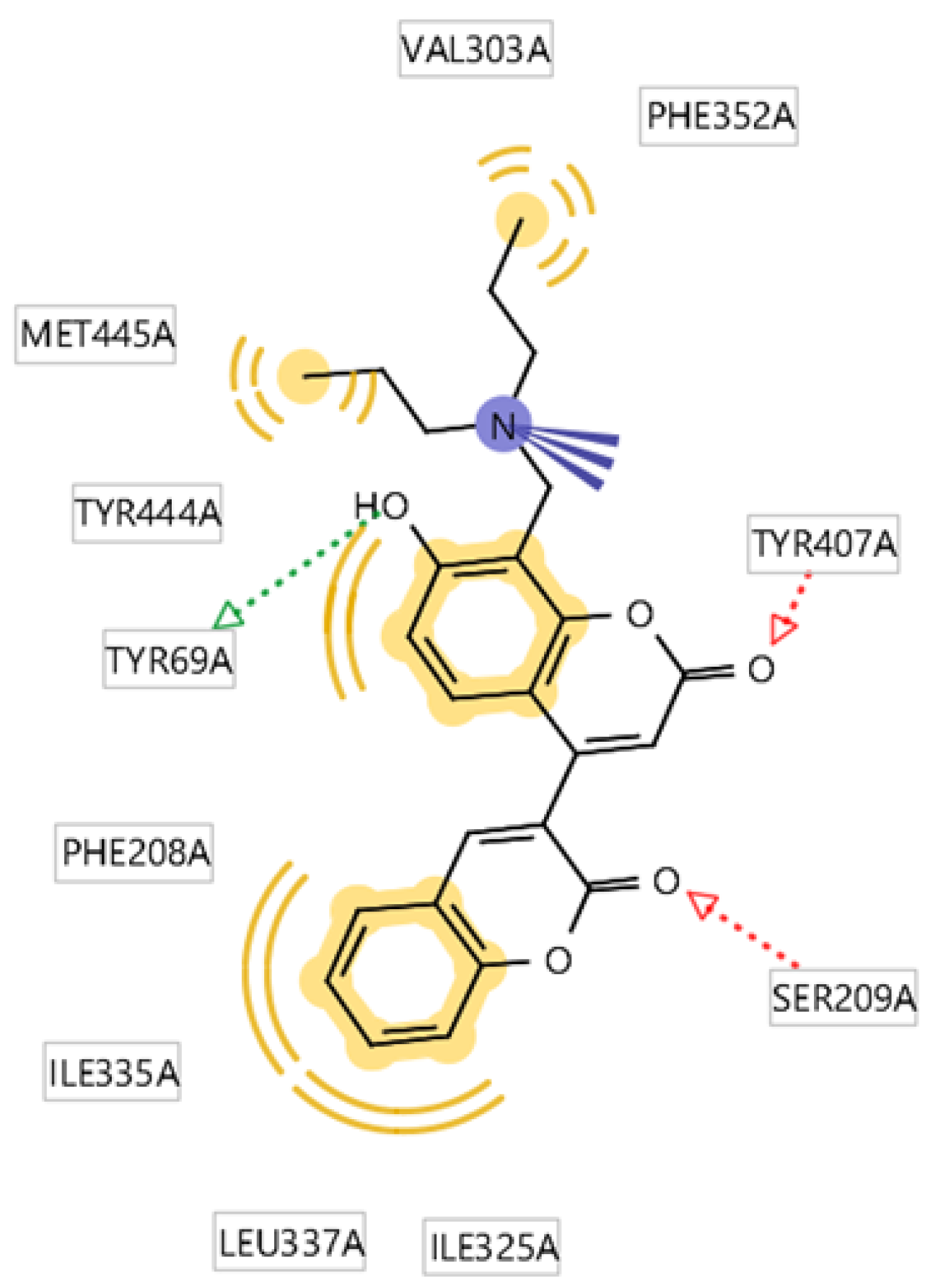
Figure 5.
(a) 2D diagram of compound 2b in the binding site of MAO-A. (b) 2D diagram of compound 2d in MAO-A. (c) Superposition of 2b (green) and 2d (cyan) in the active site of MAO-A enzyme. Red dotted arrows indicate H-bonds, purple halogen bond and yellow spheres hydrophobic interactions.
Figure 5.
(a) 2D diagram of compound 2b in the binding site of MAO-A. (b) 2D diagram of compound 2d in MAO-A. (c) Superposition of 2b (green) and 2d (cyan) in the active site of MAO-A enzyme. Red dotted arrows indicate H-bonds, purple halogen bond and yellow spheres hydrophobic interactions.
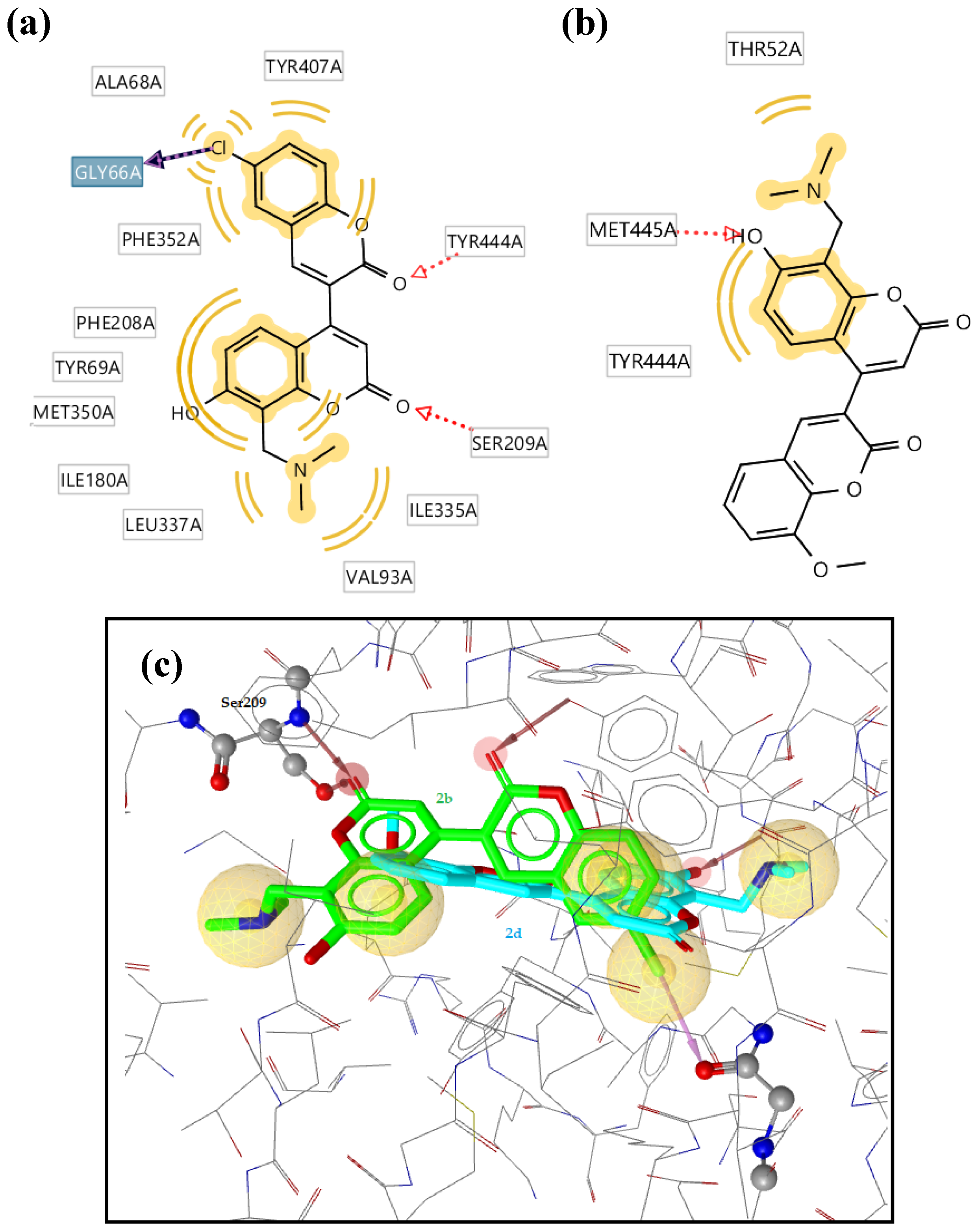
Figure 6.
(a) Superposition of compound 3c (purple) and donepezil (green) into the active site of AChE. B) 2D diagram of compound 3c into the AChE active site. (b) Red and green dotted arrows indicate H-bonds, blue arrows positive ionizable and yellow spheres or curved lines the hydrophobic contacts.
Figure 6.
(a) Superposition of compound 3c (purple) and donepezil (green) into the active site of AChE. B) 2D diagram of compound 3c into the AChE active site. (b) Red and green dotted arrows indicate H-bonds, blue arrows positive ionizable and yellow spheres or curved lines the hydrophobic contacts.

Table 1.
Monoamine oxidases (MAOs A and B) and cholinesterases (AChE and BChE) inhibition data of 8-[(dialkylamino)methyl]-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one derivatives. a.
Table 1.
Monoamine oxidases (MAOs A and B) and cholinesterases (AChE and BChE) inhibition data of 8-[(dialkylamino)methyl]-4-(2-oxo-2H-chromen-3-yl)-7-hydroxy-2H-chromen-2-one derivatives. a.
| Cmpd | NR2 | R1 | R2 | MAO-A | MAO-B | AChE | BChE |
|---|---|---|---|---|---|---|---|
| 2a | NMe2 | H | H | 8.30 ± 0.36 | [36 ± 5] | [22 ± 3] | i.a. |
| 2b | Cl | H | 1.49 ± 0.49 | ~ 10 | ~ 10 | i.a. | |
| 2c | Br | H | 2.23 ± 0.22 | ~ 10 | [20 ± 1] | i.a. | |
| 2d | H | OMe | 11.7 ± 1.3 | [28 ± 3] | [39 ± 9] | [25 ± 3] | |
| 3c | NEt2 | Br | H | 3.04 ± 1.37 | [42 ± 3] | 1.56 ± 0.31 | [37 ± 4] |
| 4a | N(nPr)2 | H | H | [29 ± 6] | [33 ± 1] | [37 ± 3] | [22 ± 3] |
| 4b | H | OMe | [29 ± 5] | [43 ± 5] | 3.05 ± 0.97 | [14 ± 10] | |
| 5a | N(Me)nBu | H | H | [32 ± 3] | [23 ± 3] | [28 ± 3] | [27 ± 2] |
| 5b | Cl | H | 1.85 ± 0.11 | ~ 10 | [25 ± 5] | [17 ± 7] | |
| 5c | Br | H | 2.52 ± 0.70 | ~ 10 | 4.25 ± 0.5 | [25 ± 11] | |
| 5d | H | OMe | [39 ± 4] | ~ 10 | 7.80 ± 1.92 | [40 ± 4] | |
| 6a | N(CH2CH2OMe)2 | H | H | [32 ± 5] | [29 ± 6] | [37 ± 9] | [15 ± 4] |
| 6b | Cl | H | [45 ± 3] | [35 ± 9] | [16 ± 4] | i.a. | |
| 6d | H | OMe | [26 ± 4] | [31 ± 1] | [20 ± 2] | i.a. | |
| 7a | N(iBu)2 | H | H | [27 ± 2] | [28 ± 4] | ~ 10 | ~ 10 |
| 7d | H | OMe | [27 ± 3] | [31 ± 2] | ~ 10 | [34 ± 3] | |
| 8a | N(Me)Bn | H | H | [28 ± 4] | ~ 10 | [40 ± 9] | [31 ± 4] |
| 8b | Cl | H | ~ 10 | [38 ± 3] | [16 ± 6] | i.a. | |
| 8d | H | OMe | [31 ± 4] | [42 ± 3] | ~ 10 | ~ 10 | |
| Clorgiline | 0.0025 ± 0.0002 | 2.51 ± 0.45 | |||||
| Donepezil | 0.051 ± 0.003 | 2.71 ± 0.31 | |||||
a Enzymes are human MAOs A and B, electric eel AChE and horse serum BChE. IC50 values (µM, bold) are listed for compounds achieving more than 60% inhibition at 10 µM concentration or % inhibition at 10 µM concentration in square brackets, expressed as means ± SD of three independent determinations; IC50 reported as ~ 10 for compounds achieving at 10 µM concentration average % inhibition within the range 50÷60%; i.a.: compounds displaying average % inhibition less than 10% at 10 µM concentration.
Table 2.
Molecular docking results with MAO-A (pdb 2BXR).
| Cmpd | Free energy of binding (kcal·mol-1) a |
H-bonding b | Hydrophobic/ aromatic interactions b |
Ionic interactions b |
|---|---|---|---|---|
| 2a | -11.38 | Ile207, Ser209, Tyr447 | Phe208, Leu337, Tyr407, Tyr444 | Glu216 |
| 2b | -11.25 | Ser209, Tyr444 | Ala68, Tyr407, The352, Phe208, Tyr68, Met350, Ile180, Val93, Ile335 | - |
| 2d | -7.62 | Met445 | Thr52, Tyr44 | - |
| 4a | -10.06 | Tyr69, Ser209, Tyr407 | Phe208, Val303, Ile335, Phe352, Tyr444, Met445 | Tyr407 |
| 5a | -7.82 | Ser209, Ile207 | Leu97, Phe208, Leu337 | - |
| 5b | -10.98 | Ser209, Tyr444 | Tyr68, Ala68, Ile180, Phe208, Met350, Ile335 | - |
| Moclobemide | -8.92 | Ser209, Tyr444 | Leu97, Phe208, Ile325, Ile335, Leu337 | - |
a Free energy of binding (kcal·mol-1) estimated by AutoDock 4.2 software for ligands in the highest scored binding pose. b Residues in the binding site of MAO-A mainly involved in the interaction with biscoumarin ligands; residues established as playing a role in substrate and inhibitor selectivity are highlighted in bold.
Table 3.
Molecular docking results with AChE (pdb 1EVE).
| Cmpd | Free binding energy (kcal·mol-1) a |
H-bonding | Hydrophobic/aromatic interactions b | Ionic interactions b |
|---|---|---|---|---|
| 2a | -5.12 | - | Phe331, Tyr334 | - |
| 2c | -2.13 | - | - | - |
| 3c | -9.15 | Tyr70, Phe228 | Phe330, Phe331, Tyr334, Leu282, Trp279, Ile287, Phe290 | - |
| 4d | -7.15 | Tyr70 | Trp279, Phe290 | - |
| 5c | -8.02 | - | Trp84, Trp279, Phe290 | - |
| 5d | -8.51 | Trp84 | Trp84, Tyr279, Ile287, Phe290 | - |
| Donepezil | -9.53 | - | Trp84, Ile144 | Phe330, Trp84 |
a Free energy of binding (kcal·mol-1) estimated by AutoDock 4.2 software for ligands in the highest scored binding pose. b Residues in the binding site of AChE mainly involved in the interaction with biscoumarin ligands. Residues lining the anionic site, acyl pocket, oxyanion hole and peripheral anionic site, which are well-established as playing role in substrate and inhibitor selectivity, are highlighted in bold.
Table 4.
Calculated and experimental solubility and lipophilicity data.
| Cmpd | Solubility (mol/L) | Lipophilicity | ||
|---|---|---|---|---|
| Calc. a | Exp. b | Log Pcalc c | Log k’w d | |
| 2a | 8.79·10-5 | 1.83·10-4 | 2.96 | 2.11 |
| 2b | 2.26·10-5 | 1.47·10-4 | 3.45 | 2.45 |
| 2c | 1.08·10-5 | 1.41·10-4 | 3.58 | 2.65 |
| 2d | 7.49·10-5 | 1.96·10-4 | 2.94 | 2.32 |
| 3c | 3.69·10-6 | 1.81·10-4 | 4.26 | 2.89 |
a Solubility calculated by chemoinformatic tools of the SwissADME free website (http://www.swissadme.ch), applying the topological method (ESOL) implemented by J.S. Delaney [35]. b Average solubility (n = 3, RSD < 5%) determined in PBS (50 mM, pH 7.4, 0.15 M KCl) at 25 ± 1°C. c Consensus Log Po/w calculated by SwissADME as the arithmetic mean of octanol/water log Ps calculated by five methods (iLOGP, XLOGP3, WLOGP, MLOGP, and SILICOS-IT), as reported in ref. [34]. d Log of the polycratic capacity factor (k’w) determined as lipophilicity parameter by a reversed phase (RP) HPLC technique [36].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



