Submitted:
12 August 2025
Posted:
13 August 2025
You are already at the latest version
Abstract
Triple-negative breast cancer (TNBC), lacking targetable receptors such as estrogen receptors, progesterone receptors, and human epidermal growth factor receptor 2, is an aggressive subtype with limited treatment options and highlights the need for novel therapeutic strategies. Here, we report that azelastine, a clinically approved H1-antihistamine, suppresses the viability of MDA-MB-231 TNBC cells in a dose- and time-dependent manner with 40% inhibition at 30 µM and 65% at 50 µM after 72 hours. Although histamine receptor 1 (HRH1) is functionally expressed in these cells, neither histamine stimulation nor HRH1 knockdown influenced cell survival, suggesting an HRH1-independent mechanism. Analysis of patient datasets revealed that ADP-ribosylation factor 1 (ARF1) is overexpressed in TNBC and associated with poor survival. Notably, inhibition of ARF1 by golgicide A abolished the cytotoxic effect of azelastine via the cationic amphiphile pathway. These findings suggest that azelastine exerts its anticancer effects through ARF1, not HRH1, and highlight its potential for drug repositioning in ARF1-driven TNBC.
Keywords:
triple-negative breast cancer
; azelastine
; ARF1
; HRH1
; drug repurposing
; golgicide A
; cytotoxicity
1. Introduction
Triple-negative breast cancer (TNBC) is a highly aggressive subtype lacking estrogen, progesterone, and human epidermal growth factor receptor 2 (HER2 receptors), accounting for approximately 15–20% of all breast cancer cases [1]. It is characterized by a high rates of recurrence and distant metastasis and by the most challenging breast cancer subtype to treat due to the lack of effective targeted therapies [2]. Consequently, TNBC patients face a poorer prognosis compared to those with hormone receptor-positive or HER2-amplified tumors [1,2]. The absence of approved targeted therapies for TNBC underscores the urgent need for novel treatment strategies [3,4]. Currently, chemotherapy remains the standard treatment option for TNBC, but its effectiveness is limited, especially in advanced stages. As a result, there is increasing interest in identifying new therapeutic agents and molecular pathways, often through strategies like drug repurposing, that could provide alternative treatment options for TNBC patients [2,9,30].
The histamine system has gained attention in cancer biology, with early studies noting its relevance in clinical oncology [31]. Histamine is a biogenic amine that signals through four well-characterized G-protein–coupled receptors (HRH1–HRH4) [10,32]. The role of histamine receptor H1 (HRH1) in breast cancer is complex and not fully elucidated. Some studies suggest that elevated HRH1 expression is linked to poor clinical outcome and angiogenesis [5,6,7], while other reports and databases suggest its significance across different subtypes is still being explored [33]. Many H1-antihistamines, such as ebastine and terfenadine, have known off-target effects that are independent of their primary histamine-blocking function [9,11,12]. For example, terfenadine, an H1 antagonist, has been shown to induce apoptosis in basal breast cancer cells by activating the mitochondrial pathway [8]. These findings suggest that the anti-tumor activities of H1-antihistamines may involve non–histamine-mediated mechanisms. Although these findings nominate HRH1 as a potential target in TNBC, its precise role remains a point of interest for further investigation [33].
In this context, azelastine, a second-generation HRH1 antagonist widely used as an anti-allergy drug, has also been reported to exert anti-proliferative actions in various cancer cells [13,34]. H1-antihistamines like azelastine are classified as cationic amphiphilic drugs (CADs) due to their specific chemical structure, which include ionizable hydrophobic aromatic rings and hydrophilic side chains containing amine functional groups [11,12,33]. This structure enables them to easily penetrate cell membranes. Beyond its anti-allergic effects, recent studies have shown that azelastine’s anticancer effects are often mediated through its action as a CAD. Notably, previous reports have shown that azelastine strongly inhibited colorectal cancer cell proliferation and mitochondrial fission both in vitro and in vivo by directly targeting ADP-ribosylation factor 1 (ARF1), thereby interfering with downstream oncogenic signaling (IQGAP1–ERK–Drp1 pathway) [25] and it induces anticancer effects through ARF1 activation in colon cancer cells [25,35]. These findings reveal that azelastine can target ARF1, a small GTPase involved in vesicular trafficking, rather than HRH1 blockade to suppress tumorigenesis.
ARF1 plays a central role in intracellular trafficking, Golgi structure maintenance, and mitogenic signaling, and it has recently emerged as a key regulator of tumor progression, with its therapeutic potential being actively explored [20,24,28]. The CAD-associated pathway regulating ARF1 activation has been implicated in cancer cell growth, migration, and survival [27]. High ARF1 expression is observed in aggressive breast cancer subtypes, and ARF1 promotes metastatic behavior. For example, Schlienger et al. showed that ARF1 is highly expressed in advanced breast tumors, and that ARF1 overexpression drives epithelial-to-mesenchymal transition (EMT), invasion, proliferation, and chemoresistance [21]. Its role in regulating cancer cell migration and invasion is a subject of significant research [22]. Conversely, ARF1 knockdown in breast cancer models impairs primary tumor growth and markedly reduces lung metastases, highlighting its role in cellular trafficking and cancer progression [23,29]. In fact, ARF1 is the most frequently amplified ARF family member in breast cancer, and its depletion suppresses metastatic dissemination in mice and zebrafish models [26]. The modulation of ARF1 and its downstream signaling, such as the PI3K-AKT pathway, establishes it as a pro-metastatic signaling hub in breast and other cancers [19,36,37].
Here, we investigated whether azelastine exerts anticancer effects in TNBC and examined whether these effects are mediated through HRH1 antagonism or its activity as a CAD. This study is the first to suggest the possibility that the HRH1 antagonist azelastine may exert anticancer effects as a CAD in TNBC, providing important mechanistic evidence.
2. Results
2.1. Azelastine Decreases TNBC Cell Viability in a Dose- and Time-Dependent Manner
To investigate the anticancer effects of azelastine on MDA-MB-231 cells, we performed cell viability assays using varying concentrations of azelastine (at concentrations of 30 µM and 50 µM for 72 hours). Also, azelastine is primarily metabolized in the liver by the cytochrome P450 (CYP) enzyme system, particularly CYP3A4, to form its major metabolite, N-desmethyl azelastine (Figure 1A). We evaluated the anticancer effects of both azelastine and its metabolite on TNBC cells, finding that the parent compound, azelastine, was the primary driver of cytotoxicity. Treatment with azelastine led to a marked reduction in cell viability in a dose- and time-dependent manner. The inhibitory effect increased over time: viability after 30 and 50 µM azelastine significantly fell progressively at 24, 48, and 72 hours than vehicle-treated controls (n=3 per time point; two-way ANOVA, p<0.0001 for time × dose interaction) (Figure 1B).
To confirm the reproducibility and robustness of the effect at 72 hours, we repeated the assay with an increased sample size, which consistently showed that 50 µM azelastine reduced cell viability by approximately 65% (34.9 ± 3.7% of control, n=6 per dose; one-way ANOVA, p<0.0001), while 30 µM also significantly decreased viability to 61.3 ± 6.1% (n=6, p<0.0001) (Figure 1C). Additionally, 30 µM of N-desmethyl azelastine was also found to have an anticancer effect similar to azelastine, with a cell viability of 52.4 ± 12.5% (n=4, p<0.0001). These data show that azelastine exhibits significant anticancer activity by reducing the viability of MDA-MB-231 cells in a concentration-dependent manner. Notably, this finding is consistent with prior reports that azelastine can inhibit cancer cell growth.
2.2. Downregulated HRH1 Expression in Breast Tumors Does Not Correlate with Patient Survival
We first investigated HRH1 expression across various cancer types using TCGA data. The pan-cancer analysis indicated that HRH1 expression is altered in numerous malignancies (Figure 2A). Specifically focusing on breast cancer (BRCA), RNA-seq data revealed that HRH1 mRNA levels were significantly downregulated in tumor tissues compared to normal breast tissue (p<0.001, Figure 2A).
We further examined the relationship between HRH1 expression and clinical progression. As shown in Figure 2B, HRH1 expression was significantly downregulated in primary tumors and even more so in metastatic samples compared to normal breast tissue (p = 1.64e-25). Furthermore, a clear negative correlation was observed between HRH1 expression and tumor stage, with expression levels decreasing as the disease progressed from Stage I to IV (Kruskal-Wallis p = 2.85e-08).
To determine whether the downregulation of HRH1 is associated with survival outcomes in TNBC patients, the METABRIC dataset was used to analyze Overall Survival (OS) and Disease-Free Survival (DFS). Kaplan-Meier analysis demonstrated that while there was no significant difference in OS between the high and low HRH1 expression groups (log-rank p=0.1346), lower HRH1 expression was significantly associated with poorer DFS (log-rank p<0.05, Figure 2C). These findings suggest that HRH1 expression may serve as a prognostic factor for disease recurrence in TNBC patients.
2.3. HRH1 Is Functionally Expressed in TNBC Cells But Does Not Mediate Azelastine Cytotoxicity
The effects of histamine on the MDA-MB-231 cell line were examined to investigate the observed lack of significant correlation between HRH1 expression and survival outcomes in TNBC patients. First, we confirmed the functional expression of HRH1 in MDA-MB-231 cells by assessing their response to histamine (100 µM) stimulation. Calcium imaging experiments were conducted to monitor calcium flux following histamine treatment, and a transient calcium response by histamine was observed in all treated cells, indicating active HRH1 signaling (representative trace; p<0.01 versus vehicle) (Figure 3A). Next, we evaluated the impact of a high concentration of histamine (100 µM) on MDA-MB-231 cell viability. However, treatment of the cells with histamine (100 µM) for 72 hours had no significant effect on viability compared to vehicle-treated group. Cell viability was 100.6 ± 2.1% at 24 hours, 95.5 ± 6.4% at 48 hours, and 101.7 ± 3.1% at 72 hours (n=4 per time; two-way ANOVA, ns) (Figure 3B).
Finally, to further elucidate the mechanism of action, we used RNA interference (RNAi) to knock down HRH1 expression in MDA-MB-231 cells. First, we observed the effect of HRH1 knockdown on basal cell viability. HRH1 silencing alone significantly reduced cell viability to 82.1 ± 2.5% compared to the siCTRL group (n=6; unpaired t-test, p<0.0001) (Figure 3C), suggesting a potential role for HRH1 in maintaining cell viability. Next, to confirm that its signaling does not mediate azelastine’s action, we assessed azelastine’s effect in these knockdown cells. Despite a reduction in HRH1 expression, azelastine treatment resulted in similar levels of cell viability between the scramble control group (siCTRL, 47.37 ± 3.5% of control, n=6; two-way ANOVA, p<0.0001) and the HRH1 knockdown group (siHRH1, 52.26 ± 4.91% of control, n=6, p<0.0001) (Figure 3D). These findings collectively indicate that while MDA-MB-231 cells express functional HRH1 and its knockdown reduces basal cell viability, this pathway does not mediate azelastine sensitivity.
2.4. ARF1 Is Overexpressed in TNBC and Associated with Poor Patient Survival
Next, we investigated ARF1 expression across various cancer types using TCGA data. The pan-cancer analysis indicated that ARF1 expression is altered in numerous malignancies (Figure 4A). Specifically focusing on breast cancer (BRCA), RNA-seq data revealed that ARF1 mRNA levels were significantly upregulated in tumor tissues compared to normal breast tissue (p<0.001, Figure 4A).
We further examined the relationship between ARF1 expression and clinical progression. As shown in Figure 4B, ARF1 expression was significantly increased in primary tumors and even more so in metastatic samples compared to normal breast tissue (p=5.05e−110). Furthermore, a clear positive correlation was observed between ARF1 expression and tumor stage, with expression levels increasing as the disease progressed from Stage I to IV (Kruskal-Wallis p=4.87e−90).
To determine whether the upregulation of ARF1 is associated with survival outcomes in TNBC patients, the METABRIC dataset was used to analyze Overall Survival (OS) and Disease-Free Survival (DFS). Kaplan-Meier analysis demonstrated that high ARF1 expression was significantly associated with poorer Overall Survival (OS) and Disease-Free Survival (DFS) (log-rank p<0.01 for both, Figure 4C). These findings suggest that ARF1 expression may serve as a powerful predictive factor for poor prognosis in TNBC patients.
2.5. ARF1 Inhibition Rescues Cells from Azelastine-Induced Death
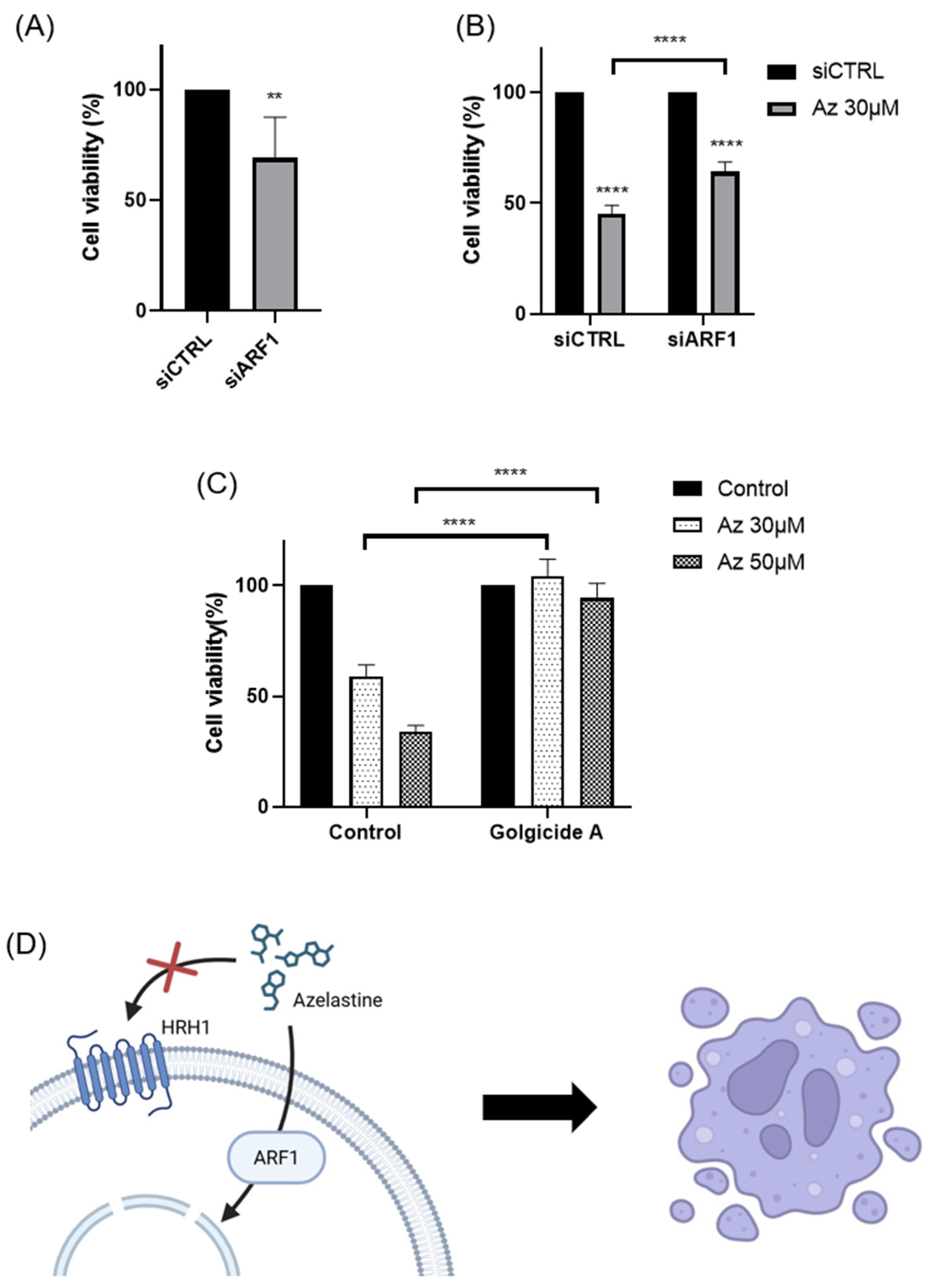
To investigate whether the anticancer effects of azelastine are mediated through ARF1, we first used RNAi to knock down ARF1 expression in MDA-MB-231 cells. ARF1 silencing alone significantly reduced cell viability to ~75 ± 3.5% (n=6; unpaired t test p<0.01) of the control siRNA (siCTRL) group, indicating ARF1’s essential role in TNBC cell proliferation and cell viability (Figure 5A). We then performed ARF1 knockdown to investigate whether this off-target effect was the primary mechanism of azelastine-induced cytotoxicity. After azelastine treatment, ARF1 knockdown significantly rescued cells from cytotoxicity, with a marked increase in cell viability compared to the siCTRL group (siARF1, 63.9 ± 4.9%; siCTRL, 47.4 ± 3.5%, n=6; two-way ANOVA, p<0.0001, Figure 5B).
Finally, to further elucidate the mechanism of action, we conducted additional experiments using golgicide A (GCA), a specific inhibitor of ARF1. Under the same conditions used to evaluate the viability of MDA-MB-231 cells, the effects of azelastine treated at concentrations of 30 µM and 50 µM for 72 hours were compared between cells treated with azelastine alone and those co-treated with GCA. When azelastine was administered alone, cell viability decreased to 58.84 ± 8.5% and 33.83 ± 4.8% at 30 µM and 50 µM, respectively. However, in cells co-treated with GCA, cell viability was restored to 104.27 ± 10.3% and 94.37 ± 8.2% at the same concentrations (n=6; two-way ANOVA, p<0.0001) (Figure 5C). The results demonstrated that GCA completely abrogated the anticancer effects of azelastine, indicating that ARF1 inhibition restored cell viability to control levels despite azelastine treatment. This significant reversal of azelastine’s anticancer effects upon ARF1 inhibition indicates that ARF1 plays a critical role in mediating the cytotoxic activity of azelastine and these results are consistent with previous findings that azelastine fails to inhibit proliferation in ARF1-deficient cells.
These results provide compelling evidence that the anticancer effects of azelastine are specifically mediated through an ARF1-dependent mechanism. This led us to further investigate the expression and clinical relevance of ARF1 in breast cancer.
3. Discussion
Our findings demonstrate that azelastine, a clinically approved antihistamine, and its metabolite N-desmethyl azelastine potently inhibit the viability of the TNBC cell line MDA-MB-231 through an ARF1-dependent mechanism. Treatment with azelastine resulted in a significant, dose- and time-dependent reduction in cell viability. Notably, these cytotoxic effects were unaffected by HRH1 knockdown and were not reproduced by histamine stimulation, indicating that azelastine acts independently of canonical HRH1 signaling. Mechanistically, co-treatment with GCA, a selective ARF1 inhibitor, completely rescued cell viability, and ARF1 knockdown via RNA interference significantly attenuated azelastine-induced cytotoxicity, increasing cell viability from ~45% in control siRNA-transfected cells to nearly 64% in ARF1-silenced cells. These results demonstrate that ARF1 activity is essential for azelastine’s cytotoxic effect. In our additional studies, azelastine also reduced viability in the BT-549 cell line, another TNBC subtype, by 34.37 ± 4.8 % of cell viability after 72 hours at 30 μM (unpublished data), comparable to the effect observed in MDA-MB-231. Although further validation in BT-549 was not performed, these findings suggest potential efficacy across different TNBC subtypes. Collectively, these findings support the conclusion that azelastine induces cytotoxicity through a non-canonical mechanism involving ARF1 rather than histamine receptor blockade, and may represent a promising therapeutic candidate for TNBC, a disease subtype notorious for its aggressive nature and lack of effective molecularly targeted therapies [1,2,3,4].
The role of HRH1 in breast cancer is complex, with our findings contributing a new perspective to a varied body of literature. While some previous studies reported that elevated HRH1 expression correlates with poor prognosis in certain breast cancer contexts [5,6,7] and that its pharmacological inhibition can induce apoptosis [8], our results show that HRH1 is downregulated in the broader breast cancer patient cohort and that its expression has no clear correlation with survival in TNBC. Moreover, our functional assays confirmed that neither HRH1 knockdown nor histamine stimulation affected azelastine’s cytotoxicity. These findings, together with the complete reversal of azelastine’s effect by ARF1 inhibition, strongly suggest that HRH1 is not the primary mediator of azelastine’s anticancer mechanism in this TNBC model. This interpretation aligns with an emerging paradigm where H1-antihistamines exert “off-target” antitumor effects through mechanisms unrelated to histamine receptor signaling [9,10]. For example, the antihistamine ebastine was recently shown to inhibit focal adhesion kinase in TNBC, suppressing metastasis [11,12]. Such receptor-independent mechanisms may reflect interactions with intracellular signaling proteins, highlighting the broader therapeutic potential of this drug class [13]. Our findings further support this concept, demonstrating that azelastine’s efficacy in TNBC does not rely on HRH1.
Azelastine is primarily metabolized in the liver by the CYP450 enzyme system to form its major metabolite, N-desmethyl azelastine. While both compounds function as HRH1 antagonists, N-desmethyl azelastine is known to have a lower binding affinity for the receptor (Ki of 9–14 nM) compared to the parent drug, azelastine (Ki of 1.2–1.9 nM) [14,15]. To determine if the observed anticancer effects were primarily driven by azelastine or its metabolite, we assessed the cytotoxic potential of both compounds. Our study demonstrates that despite azelastine’s higher binding affinity for HRH1, both compounds exhibit similar cytotoxic effects on TNBC cells (Figure 1C). This observation, which is not directly correlated with HRH1 binding affinity, supports our hypothesis that the anticancer effects are mediated through a mechanism independent of HRH1 antagonism.
Beyond its role as an HRH1 antagonist, both azelastine and N-desmethyl azelastine are likely to have similar cell membrane permeability as CADs, defined by two key features: a hydrophobic ring structure and a hydrophilic cationic amine group [16]. When azelastine is metabolized to N-desmethyl azelastine, only a methyl group is removed from the amine [14,15]. The N-demethylation of azelastine to form desmethyl azelastine does not alter the fundamental key structural features that allow both compounds to function as CADs. These properties allow CADs to accumulate in lysosomes and potentially induce phospholipidosis, a common off-target effect [17,18]. N-desmethyl azelastine retains both the cationic amine and the hydrophobic aromatic rings, suggesting that it also fulfills the structural requirements to function as a CAD. Therefore, we hypothesize that the observed anticancer effects of both azelastine and N-desmethyl azelastine are mediated through their shared properties as CADs, rather than exclusively through HRH1 antagonism. Our data suggest that both compounds, due to their structural similarities as CADs, likely contribute to the overall therapeutic response.
Regarding azelastine’s action as a CAD, our data identifies ARF1 as the critical mediator of azelastine’s cytotoxicity. Importantly, RNAi-mediated ARF1 knockdown partially rescued cell viability despite continued azelastine exposure, reinforcing the view that ARF1 is essential for mediating azelastine’s anti-TNBC effects. This observation is consistent with ARF1’s known pro-tumorigenic roles in various cancers. While the functions of ARF family members can be context-dependent [19], ARF1 is frequently implicated in processes that drive malignancy, including cell migration, invasion, and chemo-resistance [20,21,22,23]. Aligning with this predominant view, our analysis of patient datasets did not show a favorable correlation; on the contrary, we found that high ARF1 expression was significantly associated with worse overall and disease-free survival in TNBC patients. This clinical finding provides a strong rationale for our experimental results, where pharmacological inhibition of ARF1 completely abrogated azelastine’s cytotoxic effects. Thus, our study confirms that ARF1 is not only a marker of poor prognosis but also a key functional dependency for TNBC cell survival, making it a compelling therapeutic target [24].
Mechanistically, our results in TNBC are consistent with recent reports in other cancer types. A pivotal study demonstrated that azelastine can directly bind ARF1, disrupting its activity and downstream oncogenic signaling, such as the ERK and PI3K/AKT pathways [22,25,26]. The parallel between our TNBC results and prior studies in other cancers supports the notion that ARF1 is a conserved and druggable vulnerability across cancer types. Future studies should investigate the relevance of azelastine’s binding site in TNBC and further characterize its downstream signaling effects.
A unique aspect of our study is the identification of the CAD–ARF1 signaling axis as a potential therapeutic target. Previous work has implicated CADs in the activation of ARF1 and regulation of tumor cell survival [27]. Our data suggest that azelastine’s cytotoxicity in TNBC cells may be mediated through this CAD–ARF1 axis, offering a new direction for therapeutic intervention and underscoring the therapeutic potential of targeting ARF1 [28,29]. Further studies are warranted to explore how azelastine interacts with CAD-associated effectors and whether this interaction can be optimized.
Given its established safety profile as an FDA-approved antihistamine, azelastine represents a strong candidate for drug repurposing, a strategy with significant logistical and economic advantages [30]. Our findings suggest that TNBC tumors with high ARF1 expression may be especially responsive to this approach. This strategy fits within a broader trend of repositioning histamine receptor antagonists for cancer therapy. These findings, together with our own, support the idea that antihistamines may harbor unexplored anticancer potential via structural features that enable engagement with oncogenic signaling proteins [9]. From a drug development standpoint, our results underscore the promise of repurposing azelastine as an ARF1-targeting therapy for TNBC. This approach may be rapidly translated into clinical settings. Since azelastine has already passed safety evaluation in humans, early-phase trials in TNBC could be expedited. Moreover, the correlation between ARF1 expression and drug responsiveness suggests that patient stratification based on ARF1 status could enhance therapeutic precision. Future work should explore the utility of azelastine in preclinical TNBC models, including in vivo efficacy, pharmacokinetics, and potential synergy with existing chemotherapies or ARF1-pathway inhibitors.
In conclusion, our study provides compelling evidence that azelastine inhibits TNBC cell viability through an ARF1-dependent mechanism, independent of HRH1 signaling. This effect appears to be mediated via the CAD–ARF1 axis, underscoring a novel therapeutic pathway in aggressive breast cancers (Figure 5C). By repurposing azelastine to target ARF1, we propose a promising strategy to expand treatment options for TNBC patients. These findings also advance our understanding of histamine receptor antagonists in oncology and highlight ARF1 as a clinically relevant therapeutic target. Further research is needed to validate this approach in vivo and to elucidate the broader applicability of ARF1-targeted interventions across cancer types.
4. Materials and Methods
4.1. Cell Culture
The TNBC cell line MDA-MB-231 was purchased from the Korean Cell Line Bank (KCLB, Korea). The cells were cultured under standard conditions at 37°C in a humidified atmosphere containing 5% CO2. According to the supplier’s recommendations, the cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin.
4.2. Drug Treatment and Reagents
Azelastine hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in DMSO to prepare 100 mM stock solutions, which were diluted to final concentrations of 30 µM and 50 µM in culture medium. Histamine dihydrochloride (100 µM; Sigma-Aldrich, USA) and golgicide A (10 µM, ARF1 inhibitor; Sellekchem, USA) were used in designated experiments. Final DMSO concentration was kept below 0.1% in all treatment groups, including vehicle controls.
4.3. Cell Viability Assay
Cell viability was measured using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) following the manufacturer’s protocol. Briefly, MDA-MB-231 cells were seeded at 2 × 103 cells/well in 96-well plates and incubated for 18 hours to allow stable attachment. After drug treatment for 24, 48, and 72 hours, 10 µL of CCK-8 reagent was added to each well and incubated for 2 hours at 37°C. Absorbance was measured at 450 nm using a microplate reader (iMark Microplate Reader; Bio-Rad, Hercules, CA, USA). Results were expressed as percentages relative to vehicle-treated controls.
4.4. Calcium Imaging
Calcium responses in the MDA-MB-231 cell line were measured using the fluorescent calcium indicator Fura-2 AM (Invitrogen, USA) dye. Cells were loaded with 5 µM Fura-2 AM in HBSS containing 0.1% Pluronic F-127 for 30 minutes at 37°C. After washing, fluorescence was recorded using an inverted fluorescence microscope (BX-70, Olympus, Japan) following stimulation with 100 µM histamine. Fluorescence intensity changes were analyzed using MetaFluor® Software (Version 8.0, Molecular Devices, France).
4.5. RNA Interference
Small interfering RNAs (siRNAs) targeting human HRH1 (siHRH1) and human ARF1 (siARF1) were purchased as AccuTarget™ Predesigned siRNA from Bioneer (Seoul, Korea). A non-targeting control (siCTRL) was purchased from Dharmacon (Lafayette, CO, USA). The sequences for siHRH1 and siARF1 were 5’-GUAGUUUGGAAAGUUCUUA-3’ and 5’-CUGCAUUCCAUAGCCAUGU-3’, respectively. Cells were transfected with 50 nM siRNA using Lipofectamine 2000 RNAiMAX (Invitrogen, USA) according to the manufacturer’s instructions for siRNA-mediated gene silencing. At 48 hours post-transfection, cells were treated with azelastine (30 µM or 50 µM), and viability was assessed 72 hours later.
4.6. Bioinformatic Analysis of Patient Datasets
HRH1 and ARF1 mRNA expression levels were analyzed in normal breast tissue, tumor tissue, and metastatic tissue using TNMplot (https://tnmplot.com/analysis/), which utilizes a variety of databases including the TCGA and GTEx datasets. We performed a differential gene expression analysis between tumor, metastatic, and normal tissues. In addition, we used the TCGA breast cancer dataset within the TIMER 2.0 platform (http://timer.cistrome.org/) to analyze the expression levels of these markers in normal breast tissue, overall breast cancer patients, and different breast cancer subtypes. Survival analysis for TNBC patients was conducted using the Kaplan–Meier plotter method, and the METABRIC-TNBC dataset was used. Patients were stratified into high- and low-expression groups based on median expression values, and overall survival (OS) and disease-free survival (DFS) were analyzed using the CTGS (Cancer Target Gene Screening) program (http://ctgs.biohackers.net/) with a log-rank test.
4.7. Statistical Analysis
All experiments were performed in at least three independent biological replicates unless otherwise noted. Data are expressed as mean ± standard deviation (SD). Statistical comparisons were conducted using one-way or two-way ANOVA followed by Tukey’s post hoc test, or unpaired Student’s t-test where appropriate. Survival curves were analyzed using the Kaplan–Meier method and log-rank test. P-values less than 0.05 were considered statistically significant. Analyses were conducted using GraphPad Prism 9.0 (GraphPad Software, San Diego, CA, USA).
4.8. Ethics Statement
This study utilized publicly available, de-identified patient data obtained from TCGA, TIMER2.0, and TNMplot databases. As all data were previously collected and anonymized, ethical approval and informed consent were not required under institutional and international guidelines. All experimental procedures using cell lines complied with institutional biosafety and research policies.
Author Contributions
Conceptualization, H.K.J. and S.J.J.; methodology, H.K.J.; validation, S.U.P.; formal analysis, H.J.J. and J.Y.L.; investigation, G.U.J. and E.K.P.; resources, H.K.J. and S.J.J.; data curation, D.C.C and H.J.J.; writing—original draft preparation, S.U.P.; writing—review and editing, S.J.J.; visualization, S.U.P.; supervision, H.K.J., H.J.J and S.J.J.; project administration, S.J.J.; funding acquisition, S.J.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the research fund of Hanyang University (HY-202200000003365) and by the National Research Foundation of Korea(NRF) grant of the Korea government(MSIT) (No. RS-2023-00302489). and The APC was funded by E.K.P.
Conflicts of Interest
Authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ARF1 | ADP-ribosylation Factor 1 |
| HRH1 | Histamine Receptor 1 |
| DFS | Disease-Free Survival |
| OS | Overall Survival |
| TNBC | Triple-Negative Breast Cancer |
| GCA | Golgicide A |
| CTGS | Cancer Target Gene Screening |
| TCGA | The Cancer Genome Atlas |
References
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.M.; Renshaw, L. Triple-negative breast cancer: Challenges and treatment options. Breast 2018, 38, 1–10. [Google Scholar]
- Pandi, A.; Nakka, M.; Maturi, K.; Basha, J. Triple-negative breast cancer: New insights into pathogenesis and therapeutic strategies. Cancer Treat. Rev. 2021, 91, 102108. [Google Scholar]
- Migliorini, D.; Campana, L.; Zazzeroni, F.; Ceccarelli, C.; Soverini, S.; Giannini, A.; et al. Histamine receptors as therapeutic targets in cancer. Cancer Res. 2019, 79, 4596–4604. [Google Scholar]
- Kim, H.; Lee, J.Y.; Park, S.J.; Lee, D.; Youn, H. Histamine receptor expression in breast cancer and its prognostic significance. Oncol. Rep. 2021, 45, 1497–1505. [Google Scholar]
- Choi, S.; Park, M.; Lee, E.; Kim, K. Histamine receptor 1 expression in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 67. [Google Scholar]
- El-Mowafy, A.M.; White, K. Terfenadine, a H1-receptor antagonist, demonstrates a new class of pro-apoptotic activity in human breast cancer cells, which involves protein kinase C-dependent and -independent pathways. Br. J. Pharmacol. 1999, 126, 575–585. [Google Scholar]
- Sarkar, D.; Bouker, K.B.; Sengupta, S.; Sharma, A.; Toth, K.; Koomen, J.M.; et al. Repositioning antihistamines as cancer therapeutics. J. Exp. Med. 2021, 218, e20210453. [Google Scholar]
- Hill, S.J.; Ganellin, C.R.; Timmerman, H.; Schwartz, J.C.; Shankley, N.P.; Young, J.M.; et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 1997, 49, 253–278. [Google Scholar] [CrossRef]
- Li, Q.; Liu, K.Y.; Liu, Q.; Wang, G.; Jiang, W.; Meng, Q.; et al. Antihistamine drug Ebastine inhibits cancer growth by targeting Polycomb group protein EZH2. Mol. Cancer Ther. 2020, 19, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Liu, D.; Wei, L.; Ye, X.; Zhang, L.; Zhang, X.; et al. Ebastine impairs metastatic spread in triple-negative breast cancer by targeting focal adhesion kinase. Cancer Lett. 2023, 561, 216125. [Google Scholar]
- Hill, R.L.; Davis, M.A.; Kim, J.; et al. Investigating the effects of antihistamines on tumor cell proliferation. J. Cancer Res. Clin. Oncol. 2020, 146, 1515–1527. [Google Scholar]
- Simons, F.E. Azelastine: a review of its pharmacodynamics, pharmacokinetics, and clinical efficacy. J. Allergy Clin. Immunol. 1999, 103, 359–367. [Google Scholar]
- Anderson, N.; Borlak, J. Drug-induced phospholipidosis. FEBS Lett. 2006, 580, 5533–5540. [Google Scholar] [CrossRef]
- Hochhaus, G.; Derendorf, H. Pharmacokinetics and metabolism of azelastine. Drugs 1994, 48, 694–703. [Google Scholar]
- Halliwell, W.H. Cationic amphiphilic drug-induced phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef]
- Reasor, M.J.; Hastings, K.L. Drug-induced phospholipidosis: characterization and relevance to the clinical situation. Toxicol. Appl. Pharmacol. 2003, 192, 282–291. [Google Scholar]
- Lee, K.; Bae, Y.; Kim, H.; et al. The involvement of ARF-1 in tumor suppression and its modulation by pharmacological agents. Cancer Lett. 2022, 519, 20–28. [Google Scholar]
- Zhang, S.; Wu, M.; Wang, H.; Bao, Y.; Liu, H. The role of ARF-1 in cancer progression and its therapeutic potential. Front. Cell Dev. Biol. 2022, 10, 835246. [Google Scholar]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates proliferation and the actin cytoskeleton in breast cancer cells. J. Biol. Chem. 2009, 284, 3956–3967. [Google Scholar]
- Donaldson, J.G.; Jackson, C.L. ARF1 and its GEFs in cancer cell migration and invasion. Small GTPases 2018, 9, 17–25. [Google Scholar]
- Roush, W.; Wang, Y.; Wang, H.; et al. ARF-1 activation and its role in tumor cell migration and survival. J. Cell Sci. 2016, 129, 1211–1221. [Google Scholar]
- Tanaka, A.; Satoh, T.; Okada, Y.; et al. Targeting ARF-1 for cancer therapy: Insights from preclinical studies. Mol. Cancer 2019, 18, 99. [Google Scholar]
- Hu, P.; Liu, Q.; Li, J.; Ma, Y.; Wei, W.; Zhang, Y.; Zhang, P. Azelastine directly targets ARF1 to inhibit colorectal cancer cell growth. Cell Death Dis. 2022, 13, 237. [Google Scholar]
- Xie, H.; Sun, L.; Zhao, Y.; Liu, Y.; Lu, Y.; Cui, Y.; Guo, L. ARF1 is amplified in breast cancer and promotes tumorigenesis through regulation of the MEK/ERK pathway. Cancer Lett. 2018, 438, 1–10. [Google Scholar]
- Koch, R.L.; Jensen, H.A.; Engelman, J.A.; et al. The CAD–ARF1 pathway as a therapeutic target in cancer. Mol. Cancer Ther. 2021, 20, 2105–2116. [Google Scholar]
- Zhang, L.; Wang, C.; Liu, Q.; Hu, P. The therapeutic potential of targeting ARF-1 in cancer therapy. Nat. Rev. Cancer 2022, 22, 253–266. [Google Scholar]
- Li, L.; et al. ARF-1’s role in modulating cellular trafficking and cancer progression. Cancer Res. 2020, 80, 987–996. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Lorenz, W.; Doenicke, A.; Weber, D.; Neugebauer, E.; Schmal, A.; Backhaus, B. Histamine and histamine antagonists in experimental and clinical oncology. Pharmacol. Ther. 1985, 27, 319–354. [Google Scholar]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.; et al. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef]
- Lee, M.; Park, S.; Kim, J. Histamine receptor 1 is a modulator of angiogenesis in breast cancer. J. Clin. Oncol. 2020, 38, 1158–1166. [Google Scholar]
- Liu, Y.; Zhang, Y.; Li, J.; Wei, W.; Ma, Y.; Zhang, P.; et al. Azelastine inhibits melanoma cell growth by regulating histamine receptors. Cancer Res. 2019, 79, 3024–3032. [Google Scholar]
- Zhang, J.; Zhao, Z.; Li, W.; et al. Mechanisms of azelastine in cancer treatment: A novel approach. Cancer Chemother. Pharmacol. 2021, 88, 847–857. [Google Scholar]
- Choi, H.; Kim, Y.; Kim, K.; et al. ARF1 promotes tumorigenesis by regulating PI3K-AKT signaling in liver cancer. Exp. Mol. Med. 2020, 52, 821–832. [Google Scholar]
- Jin, S.; et al. Role of ARF-1 in cellular homeostasis and cancer progression. Cell. Mol. Life Sci. 2018, 75, 2761–2773. [Google Scholar]
Figure 1.
(A) Azelastine is metabolized in the liver by the CYP450 enzyme to N-desmethyl azelastine. (B) MDA-MB-231 cells were treated with azelastine (30 µM or 50 µM) or vehicle, and cell viability was assessed at 24, 48, and 72 hours using the CCK-8 assay. Azelastine treatment caused a time- and dose-dependent reduction in cell viability. (C) After 72 hours of treatment, azelastine at 30 µM and 50 µM reduced viability to 61.3 ± 6.1% and 34.9 ± 3.7%, respectively. Treatment with 30 µM N-desmethyl azelastine showed 52.4 ± 12.5% viability. (****p < 0.0001).
Figure 1.
(A) Azelastine is metabolized in the liver by the CYP450 enzyme to N-desmethyl azelastine. (B) MDA-MB-231 cells were treated with azelastine (30 µM or 50 µM) or vehicle, and cell viability was assessed at 24, 48, and 72 hours using the CCK-8 assay. Azelastine treatment caused a time- and dose-dependent reduction in cell viability. (C) After 72 hours of treatment, azelastine at 30 µM and 50 µM reduced viability to 61.3 ± 6.1% and 34.9 ± 3.7%, respectively. Treatment with 30 µM N-desmethyl azelastine showed 52.4 ± 12.5% viability. (****p < 0.0001).
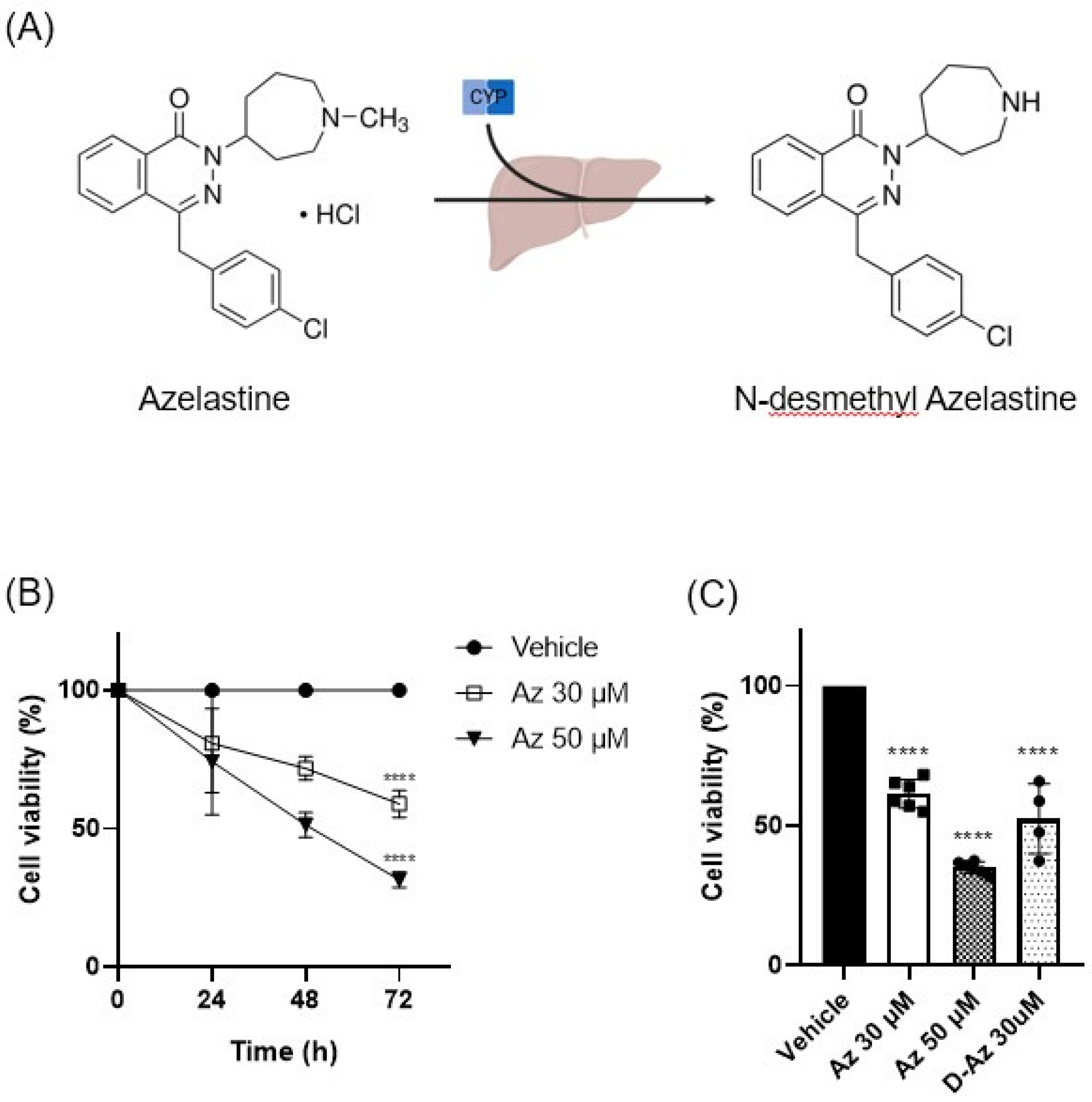
Figure 2.
(A) Pan-cancer analysis of HRH1 gene expression from TCGA data. HRH1 expression was significantly lower in breast cancer (BRCA) tissues than in normal tissues (p<0.001). (B) HRH1 gene expression correlates with clinical progression. Expression is significantly lower in tumor and metastatic tissues compared to normal tissue (top; p=1.64e-25) and is inversely associated with advanced tumor stages (bottom; Kruskal-Wallis p=2.85e-08). (C) Kaplan-Meier analysis of TNBC patients (METABRIC dataset) showed that low HRH1 expression was associated with worse disease-free survival (DFS; log-rank p<0.05), but no significant association was observed for overall survival (OS; p=0.1346).
Figure 2.
(A) Pan-cancer analysis of HRH1 gene expression from TCGA data. HRH1 expression was significantly lower in breast cancer (BRCA) tissues than in normal tissues (p<0.001). (B) HRH1 gene expression correlates with clinical progression. Expression is significantly lower in tumor and metastatic tissues compared to normal tissue (top; p=1.64e-25) and is inversely associated with advanced tumor stages (bottom; Kruskal-Wallis p=2.85e-08). (C) Kaplan-Meier analysis of TNBC patients (METABRIC dataset) showed that low HRH1 expression was associated with worse disease-free survival (DFS; log-rank p<0.05), but no significant association was observed for overall survival (OS; p=0.1346).

Figure 3.
(A) HRH1 functionality was confirmed in MDA-MB-231 cells via calcium imaging after histamine stimulation. Representative calcium flux trace showing transient intracellular calcium rise following histamine (100 µM) stimulation (n=100). (B) Cell viability was assessed every 24 hours after treatment with 100 µM histamine in MDA-MB-231 cells. Histamine treatment for 72 hours does not affect cell viability. (C) The effect of HRH1 knockdown on cell viability in MDA-MB-231 cells. (D) At 30 µM, azelastine showed a cell viability of 47.37 ± 3.5% in the siCTRL group and 52.26 ± 4.91% in the siHRH1 group, respectively. (ns, not significant; ****p <0.0001).
Figure 3.
(A) HRH1 functionality was confirmed in MDA-MB-231 cells via calcium imaging after histamine stimulation. Representative calcium flux trace showing transient intracellular calcium rise following histamine (100 µM) stimulation (n=100). (B) Cell viability was assessed every 24 hours after treatment with 100 µM histamine in MDA-MB-231 cells. Histamine treatment for 72 hours does not affect cell viability. (C) The effect of HRH1 knockdown on cell viability in MDA-MB-231 cells. (D) At 30 µM, azelastine showed a cell viability of 47.37 ± 3.5% in the siCTRL group and 52.26 ± 4.91% in the siHRH1 group, respectively. (ns, not significant; ****p <0.0001).
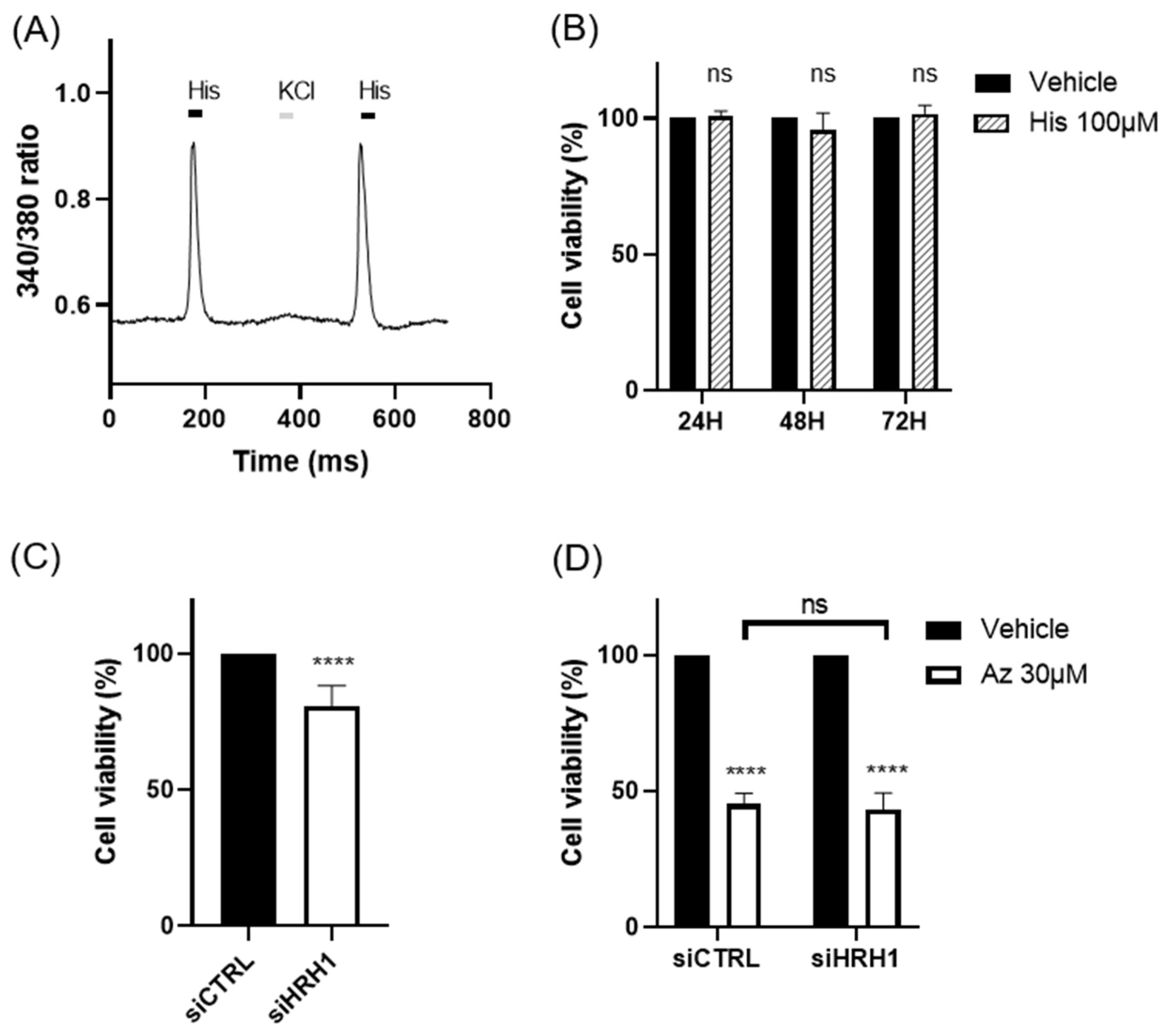
Figure 4.
(A) Pan-cancer analysis of ARF1 gene expression from TCGA data. ARF1 expression was significantly higher in breast cancer (BRCA) tissues than in normal tissues (p<0.001). (B) ARF1 gene expression correlates with clinical progression. Expression is significantly higher in tumor and metastatic tissues compared to normal tissue (top; p=5.05e−110) and shows a positive correlation with advanced tumor stages (bottom; Kruskal-Wallis p=4.87e−90). (C) Kaplan-Meier analysis of TNBC patients (METABRIC dataset) showed that high ARF1 expression was significantly associated with shorter overall survival (OS) and disease-free survival (DFS) (log-rank p<0.01 for both).
Figure 4.
(A) Pan-cancer analysis of ARF1 gene expression from TCGA data. ARF1 expression was significantly higher in breast cancer (BRCA) tissues than in normal tissues (p<0.001). (B) ARF1 gene expression correlates with clinical progression. Expression is significantly higher in tumor and metastatic tissues compared to normal tissue (top; p=5.05e−110) and shows a positive correlation with advanced tumor stages (bottom; Kruskal-Wallis p=4.87e−90). (C) Kaplan-Meier analysis of TNBC patients (METABRIC dataset) showed that high ARF1 expression was significantly associated with shorter overall survival (OS) and disease-free survival (DFS) (log-rank p<0.01 for both).

Figure 5.
(A) ARF1 knockdown alone reduced MDA-MB-231 cell viability. (B) Azelastine treatment (30 µM) further reduced cell viability in both siCTRL (47.4 ± 3.5%) and siARF1 (63.9 ± 4.9%) groups. (C) Pretreatment with Golgicide A (10 µM), an ARF1 inhibitor, significantly restored viability in azelastine-treated cells, suggesting ARF1 involvement in azelastine-induced cytotoxicity. (D) Schematic model illustrating that azelastine penetrates the membrane independently of HRH1 and induces cell death via ARF1 activation. Created using BioRender. (ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Figure 5.
(A) ARF1 knockdown alone reduced MDA-MB-231 cell viability. (B) Azelastine treatment (30 µM) further reduced cell viability in both siCTRL (47.4 ± 3.5%) and siARF1 (63.9 ± 4.9%) groups. (C) Pretreatment with Golgicide A (10 µM), an ARF1 inhibitor, significantly restored viability in azelastine-treated cells, suggesting ARF1 involvement in azelastine-induced cytotoxicity. (D) Schematic model illustrating that azelastine penetrates the membrane independently of HRH1 and induces cell death via ARF1 activation. Created using BioRender. (ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



