Submitted:
11 August 2025
Posted:
12 August 2025
You are already at the latest version
Abstract
This review covers article and patent data obtained mostly within the period 2013−2024 on the synthesis and biological activity of quinazolines [a]-annelated by five-membered heterocycles. Pyrrolo-, (iso)indolo-, pyrazolo-, indazolo-, (benz)imidazo-, (benz)thiazolo- and triazolo- [a]quinazoline systems have shown multiple potential activities against numerous targets. We highlight that most research efforts are directed to design of anticancer, antibacterial, anti-inflammatory and other agents of azolo[a]quinazoline nature. This review emphases both on the medicinal chemistry aspects of pyrrolo[a]-, (iso)indolo[a]- and azolo[a]quinazolines and comprehensive synthetic strategies of quinazolines annelated at N(1)–C(2) bond in the perspective of drug development and discovery.
Keywords:
pyrroloquinazolines
; indoloquinazolines
; azoloquinazolines
; biological activity
; advanced synthesis
1. Introduction
Quinazoline and quinazolinone are medicinally important nitrogen heterocycles, exhibiting diverse biological activities [1,2]. The varied and complex effects that quinazoline compounds have on different biological systems have made them interesting for drug researchers who want to uncover all possible medical uses they might offer [3]. More than 150 naturally occurring alkaloid molecules contain the quinazolinone structure either by itself or within conjugated systems, most of natural products of fused quinazoline origin are [b]-annelated quinazolines [4].
Quinazolinone natural products display a wide range of medicinal properties, including antibacterial. One of them, Luotonin A (Figure 1) proved to exhibit significant cytotoxicity toward the murine leukemia P-388 cell line (IC50 = 1.8 μg/mL); this agent acts as topoisomerase-I inhibitor [5]. Considering the directions of quinazolines modification for potential use in medicinal chemistry, in some cases, chemists designed the angular counterparts of [b]-annelated quinazoline biologically active compounds.
Synthetically created annelated quinazolines have become very popular among scientists worldwide because they show many useful health-related properties [6,7]. The careful utilization of the quinazoline core for designing and synthesizing novel antibacterial agents has recently received considerable attention. Several newly developed fused quinazolinone derivatives displaying antimicrobial properties, particularly those incorporating a triazolo[a]-annelation, have been documented [5]. Scientists combine parts from different active molecules, like quinazolines, using special techniques to create new hybrid compounds that work better and stronger [8].
Quinazolines [c]-annelated by five- and six-membered heterocycles were analyzed recently [9]. Different triazoloquinazoline derivatives were overviewed [10], it was stated that the condensation of 2-hydrazino-3H-quinazolin-4-ones with carbonic acids represents the most common synthetic approach to 1,2,4-triazolo[4,3-a]quinazolines, whereas their isomers, [1,2,4]triazolo[1,5-a]quinazolines are obtained by cyclocondensation of N-cyanoimido(dithio)carbonates with 2-hydrazinobenzoic acid. A variety of biological applications of triazoloquinazolines and imidazoquinazolines are highlighted in the book chapters [11,12]. Synthesis and bioactivity evaluation of some pyrazolo[c]quinazolines were described in reviews [13,14].
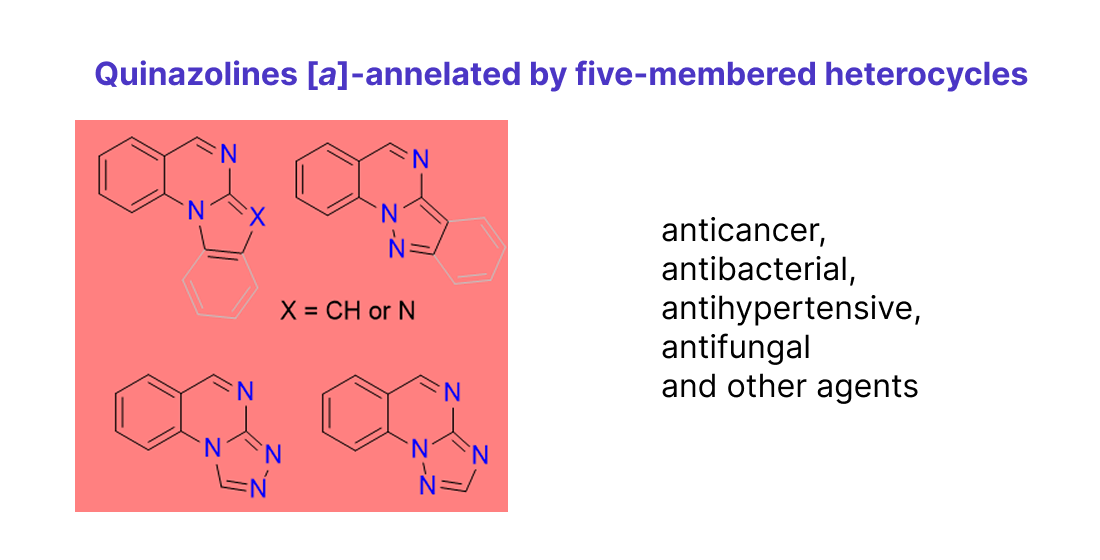
The annelation of an additional ring at the N(1)–C(2) bond represents a crucial modification of quinazolines that enables fine-tuning of their biological activity. Consequently, [a]-annelated quinazolines attract substantial interest owing to their notable pharmacological properties. To date, compounds of this type have not been introduced into medical practice. Vaskevych et al [15] provided the comprehensive analysis of quinazolin-4-one heteroanalogues as biologically relevant substances and drugs, they paid special attention to cyclizations of alkenyl(alkynyl)-functionalized quinazolinones into pyrrolo-, thiazolo- and imidazo[a]- and [b]-annelated quinazolin-4-ones; structural and electronic effects of reagents on the regio- and stereoselectivity of the cyclizations are elucidated, and relevant reaction mechanisms are clarified.
In this review, we focus on the medicinal chemistry aspects of various quinazolines [a]-annelated by five-membered heterocycles (Figure 2), encompassing both synthetic pathways and biological activities. We compile relevant literature reports within the context of drug development and discovery efforts.
A wide variety of methods are essential for constructing such annelated systems. Traditional methods for cyclocondensation may work well but also come with some drawbacks and limitations. Innovative syntheses utilizing appropriate reagents, multicomponent reactions, intramolecular C–H amidations, N-arylation/2-amidation cascades, along with metal-free, catalyst-free eco-friendly strategies, are becoming increasingly prevalent.
2. Pyrrolo[1,2-a]quinazolinones
In the review article [16], one section specifically highlights pyrrolo[1,2-a]quinazolines. This chapter explains synthetic methodologies and gives some clear examples. After its release, other scientific groups looked at both creating similar compounds and testing their biological activities, especially structural analogs of Luotonin and Vasicinone were considered.
Rasapalli et al [17] have investigated the cyclizations of quinazolinonyl enone 1, using super acids, to access the C-aryl luotonins via the intermediacy of tricyclic ketones. It was found that depending on substoichiometric amounts of TfOH in TFA as a solvent the cyclization of quinazolinonyl enone 1 proceeds with the formation of [a]- or [b]-annelated quinazolinones 2 or 3 as triflate salts of the enol tautomers (Scheme 1). It was found that in a more acidic medium, compound 2 is capable of effective conversion to regioisomer 3. Based on data from quantum chemical calculations, the probable mechanism for the formation of regioisomeric tricyclic enols was discussed, and it was suggested that the cyclization process follows an aza-Nazarov pathway.
In subsequent work [18], the same team successfully developed a facile one-pot synthesis of novel angular luotonins 5a-l via a methanesulfonic acid mediated aza-Nazarov-Friedlander cyclization sequence of quinazolinonyl enones 1a-l (Scheme 2). Optimal conditions for the formation of tricyclic enols 4 with a yield of up to 59% were selected, and two-step synthesis of polycyclic compounds 5 was carried out without isolation of intermediates 4. The structure of polycyclic compounds 5f and 5k was confirmed by X-ray data.
A series of angular luotonins 5a-l were assayed as topo-I inhibitors at four different concentrations (0.1, 1.0, 10, and 100 μM), camptothecin (CPT, at 1 μM) was used as the standard. However, tests showed that compound 5 doesn't inhibit human topoisomerase I significantly, this means polycyclic derivatives 5 could be used safely in other areas, such as insecticides, nematicides, and fungicides.
Vaskevych et al [19] developed an efficient approach to 1-(hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones 7, the method is based on oxidative cyclization of 2-(3-butenyl)quinazolin-4(3Н)-ones 6 initiated by iodosobenzene bis(trifluoroacetate) (PIFA) (Scheme 3). The reaction proceeds with high regioselectivity when 2.5 eq of PIFA are used in 2,2,2-trifluoroethanol solution at 0 °С over 24 h. Quinazolinone derivatives 6 were obtained in good yields (63−91%) in two stages: acylation of anthranilamides with α-allylacetyl chloride and subsequent thermal cyclocondensation. The structure of compounds 7 was confirmed by spectral data, including X-ray for derivative 7f. Compounds 7 represent a new class of vasicinone analogs.
Arylsulfenylation (selenylation) of 2-(3-butenyl)quinazolin-4(3Н)-ones 6 in nitromethane in the presence of an equimolar amount of lithium perchlorate proceeds as an electrophilic cyclization involving N(1) or N(3) atoms of quinazolone core to yield 1-arylthio(selenyl)methyl substituted angular-annulated 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones 8 as major products and isomeric linear-annulated 2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-one derivatives 9 as minor products (Scheme 3) [20].
Vaskevych et al developed a selective synthetic approach to pyrroloquinazolinones based on the proton- and halogen-induced electrophilic cyclization of 2-(3-butenyl)quinazolin-4(3H)-ones 6 [21]. Several protic acids (HCl, CF3CO2H, H2SO4, polyphosphoric acid, CF3SO3H) were tested, and trifluoromethanesulfonic acid was chosen as the most efficient reagent. Compounds 6a-h dissolved in CF3SO3H at room temperature were converted into 1-methyl-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-ones 10a-h with high chemoselectivity and in excellent yields (Scheme 4). The linear structure was confirmed by XRD data using compound 10g as an example.
Iodine, bromine, NIS and NBS were used as halogenating agents in the synthesis of pyrrolo[1,2-a]quinazolinones 12 and 13. The reaction of compounds 6a-h with 3 equiv. of iodine in acetic acid followed by the treatment with sodium iodide led to pyrrolo[1,2-a]quinazolinonium salts 11a-h, which when treated with sodium acetate gave 1-iodomethyl-substituted pyrrolo[1,2-a]quinazolinones 12a-h in an overall yield of up to 80% (Scheme 4). The selective cyclization of compounds 6 using bromine failed due to the formation of bromination products at the double bond of quinazolinones 6. The iodination of substrates 6a-e with 1 eq of NIS in acetonitrile solution at 0 oС allows one-step preparation of target products 12a-e in high yields. The treatment of 6a-e with NBS under the same conditions led to the formation of 1-bromomethyl-substituted pyrrolo[1,2-a]quinazolinones 13a-e.
Singh et al developed a highly regioselective synthesis of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-one derivatives 16, using such building blocks as N′-aryl benzohydrazides 14 and cyclopropane aldehydes 15 [22]. Optimal conditions for the annulation of N′-phenyl-2-aminobenzohydrazide 14a with aldehydes 15 were found. The process was realized in DCM in the presence of PTSA (30 mol%) as catalyst at room temperature. Interaction of differentially substituted cyclopropane aldehydes 15a-j with N-phenylbenzohydrazide 14a yielded a series of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones 16aa-ja as two diastereoisomers (trans and cis) in an overall yield of 78−85% (Scheme 5).
The universality of the reaction involving N'-aryl-substituted anthranyl hydrazides 14a−f was studied by their interaction with 4-methoxyphenyl-cyclopropane aldehyde 15a [22]. The formation of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones 16ab-ag was also shown as trans and cis isomers with the same ratio, except 16ag, and an overall yield of 78−81% (Scheme 6). It should be noted that N-benzyl anthranilamide and N-phenyl anthranilamide participate in the reaction with 15a, which also led to the formation of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones in good yields.
The relative configuration of both the diastereomers (cis and trans) was established based on the nuclear Overhauser effect (NOE) experiment data of 16aa and 16aa′ and was supported by the X-ray analysis of both diastereomers 16ag. The authors discussed the proposed mechanism of easily scaled reaction and concluded that the process follows a domino sequence of imination/intramolecular cycling/nucleophilic ring opening.
3. Indolo[1,2-a]quinazolinones and isoindolo[2,1-a]quinazolinediones
Indoles are widely found in pharmaceuticals and nature products, the indole-containing quinazoline derivatives were reported as protein kinase CK2 inhibitors and poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors [24]. Isoindoloquinazolines have shown Tumor Necrosis Factor-alpha (TNF-α) inhibitory [25].
Synthetic approaches to bioactive indolo[1,2-a]quinazolinones are limited. Kotipalli et al developed an easy and convenient two-step method for the synthesis of indolo[1,2-a]quinazolinone derivatives, starting from 2-iodobenzamides 18 and indole derivatives [26]. The first step involves the N-arylation of indole by 2-iodobenzamide derivatives via Ullman coupling. Optimal conditions for this stage were found, which allowed obtaining 2-(1H-indol-1-yl)-N-substituted benzamide derivatives 19 in up to 85% yield. The second step involves intramolecular C–H amidation using a palladium catalyst and AgOAc as oxidant and proceeds in toluene at 100 °C in the presence of TFA (Scheme 8). The series of indolo[1,2-a]quinazolinone derivatives 20 with different substituents R, R1, R2 was obtained. It was shown that long alkyl chain in R1 or the p-methoxy group in the aryl fragment leads to a decrease in yield to 30−34%, in the case R = NO2 the cyclization does not occur.
Abe et al reported a self-relay copper (I)-catalyzed Ullmann N-arylation/2-amidation cascade to form functionalized indolo[1,2-a]quinazolinones 23 in one-pot from easily available 2-bromobenzamides 21 with indoles 22 in high yields [27] (Scheme 9). It was found that the optimal conditions for the cascade are the following: the presence of CO2Me group at the position 3 of indole, the use of CuBr as a catalyst, Cs2CO3 as a base and DMSO as a solvent, boiling for 16 hours. It was also shown that in the case R = NO2 or R1 = t-Bu indolo[1,2-a]quinazolinones could not be obtained even under boiling for 72 hours. The authors discuss the proposed process pathways and note that methyl carboxylate could act as activating group in this Ullmann N-arylation/2-amidation cascade.
Badigenchala et al developed a transition-metal- and base-free approach to indolo[1,2-a]quinazolinones [28]. It was demonstrated that N-iodosuccinimide (NIS) can be used for intramolecular cross-coupling of C(sp2)−H and N−H bonds, leading to the formation of indolo[1,2-a]quinazolinones 25 (Scheme 9). Optimal cyclization conditions were found for 2-(1H-indol-1-yl)-N-methylbenzamide, the effect of substituents R, R1, R2 on reaction ability was studied. It was shown that the presence of strong electron withdrawing (R2 = 5-NO2, 7-azaindole derived) or bulky (R = t-Bu) group suppressed the reaction completely. Gram scale synthesis has been performed to examine the synthetic utility of the protocol.
Jiang et al described Pd-catalyzed domino synthesis of 5-amine-indolo[1,2-a]quinazolines 27 from readily available 2-aminobenzonitriles 26 and 2-(2-bromophenyl)acetonitriles (Scheme 10) [29]. The approach involves a Buchwald-Hartwig type coupling and a base-promoted intramolecular nucleophilic reaction. It was found that in the case of EWG = COOEt, the yield of products 27 is reduced to 55−62%, and in the case of R = CF3, to 37%.
One approach to the synthesis of isoindoloquinazolinediones is based on a multi-component reaction (MCR) of isatoic anhydride 28, amines and 2-formylbenzoic acid under different conditions (Scheme 11). The reaction was carried out by heating the reagents in acetic acid at 110 °C, aliphatic and aromatic amines were used, the highest yields of 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-diones 29 were obtained for the more nucleophilic aliphatic amines [30]. Reddy et al [31] described the faster and greener synthesis of the analogues of compounds 29 via a β-сyclodextrin mediated MCR of the same reagents in water under microwave irradiation.
Esmaeili-Marandi et al [32] described novel 1,2,3-triazole-containing isoindolo[2,1-a]-quinazolines via a convenient three-step reaction starting from isatoic anhydride 28. Interaction of 28 with 1-aminoprop-2-yne in water at room temperature gives 2-amino-N-(prop-2-yn-1-yl)benzamide, which further undergoes cyclization into isoindolo[2,1-a]-quinazoline 30 in the presence of TsOH (20 mol%) in EtOH under reflux (Scheme 11). Product 30 was readily converted to the target compound 31 via click reaction with organic azides, obtained from the corresponding benzyl halogenides and sodium azide.
Mahdaviet al developed a four-step effective and simple synthetic approach for the synthesis of novel isoinodolo[2,1-a]quinazolino[1,2-c]quinazolinones 33 (Scheme 12) [33], the method can be extended for the preperation of a library of potentially bioactive compounds. Reaction of isatoic anhydride 28 and amines led to various 2-aminobenzamide derivatives, their interaction with o-nitrobenzaldehyde and subsequent reduction of nitro group resulted in the formation of 2-(2-aminophenyl)-3-R-2,3-dihydroquinazolin-4(1H)-ones 32. Optimal conditions for the reaction of compounds 32 with 2-formylbenzoic acid were found: boiling in dry ethanol in the presence of p-toluenesulfonic acid (p-TSA) and tetrabutylammonium bromide (TBAB) at a ratio of p-TSA/TBAB = 0.5 [33].
Recently, performing chemical experiments in water with help from plant-based materials has become more popular because it's good for the environment and sustainable. Madhubabu et al [34] developed a short and efficient metal free, catalyst-free greener approach for the synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 34 in water (Scheme 13). Isatoic anhydride, an amine, and 3-bromoisobenzofuran-1(3H)-one served as starting materials. A mixture of water and polyethylene glycol 400 (PEG-400) in a ratio of 4:1 provided the most efficient solvent system. After completing the reaction, the product underwent filtration, and the recovered solvent could be effectively reused multiple times without noticeable loss of activity compared to fresh solvent. This reliable methodology tolerates both aliphatic and aromatic amines and exhibits exceptional scalability.
Abbasian et al [35] reported the synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-diones 35 using the catalyst on silica-based ordered mesoporous material (SBA-15) functionalized by imidazolium ionic liquid sulfonic acid (ImIL-Sul-SBA-15) (Scheme 13). The reaction between isatoic anhydride 28, amines and substituted 2-formylbenzoic acids was conducted in the ethanol-water mixture (1:1) in the presence of ImIL-Sul-SBA-15 (20 mol%) at 50 oС for 1 hour. Once the reaction finished, a mixture containing desired product 35 along with the catalyst (ImIL-Sul-SBA-15) was filtered out. Then this solid was cleaned with ethanol to remove the product, leaving behind just the catalyst. Testing showed that the catalyst could be reused effectively six times before losing any noticeable activity. Benefits include easy recovery of the catalyst, quick reaction time, and high yields of the final product.
Rayatzadeh et al described the synthesis of a series of 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 36 using sulfonic acid functionalized nanoporous silica (SBA-Pr-SO3H) as a reusable catalyst (Scheme 13) [36]. Derivatives 36 were obtained via a two-step process, once the reaction was completed, the catalyst was separated, and the final products were isolated by recrystallization from ethanol. Key advantages are high yields and easy purification process.
The synthesis of isoindoloquinazolinediones 38 was carried out in a single step starting from 2-aminobenzamides 37 and 2-formylbenzoic acid, conditions for cyclization were selected (Scheme 14) [37,38,39]. Devi et al [37] used a mixture of choline chloride (ChCl) and TsOH as promising green deep eutectic solvent. The reaction proceeds rapidly (15 min) at room temperature leading to dihydroisoindolo[2,1-a]quinazolin-5,11-diones 38a-g in high yields.
Lohar et al developed an efficient and green mechanochemical method for the synthesis of isoindolo[2,1-a]quinazolines 38a,b,d,e,g-o via 2,2,2-trifluoroethanol (TFE)-catalyzed liquid-assisted grinding (LAG) (Scheme 14) [39]. A mixture of equimolar amounts of reagents and TFE was ground together for 9–11 min at room temperature; the progress of the reaction was monitored by TLC, compounds 38a,b,d,e,g-o were isolated in high yields.
Dihydroisoindolo[2,1-a]quinazolin-5,11-diones 39 were prepared by cyclization in the xylene/acetic acid mixture at reflux for 3–5 h and isolated as racemates (Scheme 14). The biological activity of compounds 39 was studied by high-throughput screening (HTS) [38]. It was found that the 6aS-enantiomer of derivative 39a inhibits TDP1 and ELG1 protein. Moreover, compound 39a can decrease the intensity of tumor cell proliferation by suppressing the ATG4B protease function. The racemic mixture of the tetracyclic compound 39a was found to be toxic for cardiomyocytes. Compound 39a leads to decomposition of TDP-43 protein. Thus, quinazolinone 39a is promising anticancer agent through inhibition of DNA repair systems and proliferative activity, it also can be useful in neurodegenerative diseases therapy.
A series of 6-(arylamino)-6,6a-dihydroisoindolo[2,1-a]quinazolin-5,11-diones 41a-g was synthesized through iodine-catalyzed reaction of N’-aryl-2-aminobenzohydrazides 40 and 2-formylbenzoic acid in ionic liquid (Scheme 15) [40]. The structure of the compounds was confirmed by X-ray data, for compound 41b as an example. The reaction was shown to be chemoselective, the interaction of unsubstituted 2-aminobenzohydrazide with 2-formylbenzoic acid under the same conditions gave 5H-phthalazino[1,2-b]quinazoline.
A fast and eco-friendly technique was created to prepare a series of important, versatile isoindolo[2,1-a]quinazolines 43 with multiple functions for biology research (Scheme 15) [41]. The reaction of 2-aminobenzhydrazide 42 (R = H) with 2-formylbenzoic acid was studied under solvent free conditions using graphene oxide (GO) as mild heterogeneous carbocatalyst. Optimal conditions have been identified, expanding the scope of 2-aminobenzohydrazides. It has been demonstrated that 2-aminobenzamides give high yield of products 43 under the same conditions. The structures of compounds 43 have been validated through X-ray analysis for two compounds 43 (R = NHPh, R1 = H, and R = CH2C6H4-4-Me, R1 = 3-Cl). The GO nanosheets were prepared from natural graphite powder. A possible reaction mechanism with GO as catalyst was discussed, it was noted that the acidity of GO plays an important role in the studied transformation.
Guo et al [42] reported the synthesis of dihydroisoindolo[2,1-a]quinazolin-5,11-diones 45 without using 2-formylbenzoic acid. The preparation process includes palladium-catalyzed three-component carbonylative cyclization of 2-aminobenzamides 42 with 2-bromobenzaldehydes 44 under an atmospheric pressure of carbon monoxide (Scheme 16). Optimal synthetic conditions, including selection of catalyst, ligand, base, and solvent, were found, and broad substrate scope was demonstrated. A plausible mechanism involves a palladium-catalyzed cyclocondensation/cyclocarbonylation sequence. According to Guo et al [42], this method works well with different types of chemical groups, uses easily available ingredients, demonstrates high regioselectivity, and is straightforward to perform.
Kolotaev et al [43] described synthetic approaches to dihydroisoindolo[2,1-a]quinazolin-5,11-diones, containing hydroxamic acids, as potential HDAC/VEGFR inhibitors. The first method involved the alkylation of the tetracyclic derivatives 46, 47 obtained by the interaction of substituted anthranilamides and 2-formylbenzoic acids, with ethyl 6-bromohexanoic acid (Scheme 17). An attempt to obtain tetracyclic quinazolines 48 from NH-derivatives 46a,b was unsuccessful. In the case of hydroxy derivatives 47a,b, the alkylation in the presence of cesium carbonate in DMF resulted in the selective formation of products 49a,b in good yields.
Another approach was selected for the synthesis of dihydroisoindolo[2,1-a]quinazolin-5,11-diones 48a,b (Scheme 18). The reaction between isatoic anhydride 28 and ethyl 6-aminohexanoic acid led to substituted anthranilamide, which further converted into N-substituted tetracycles 48a,b under the action of 2-formylbenzoic acids. The reaction of compounds 48a,b with hydroxylamine allowed someone to obtain the expected derivatives 50a,b, in which hydroxamic acids attached through a linker to tetracyclic quinazolinone-containing fragments.
Mondal et al described one-pot synthesis of isoindole fused quinazolin-4-ones in the presence of an acid catalyst [44]. Interaction of substituted anthranilamides 51a-d with o-phthalaldehyde in methanol and 2N HCl in 3:1 ratio at room temperature led to the formation of range of isoindolo[2,1-a]quinazolin-5(11H)-ones 52a-d (Scheme 19). It should be noted that compound 52a was isolated from the reaction mixture as the salt 52a‧HCl, and 52a was obtained only after the treatment with sodium bicarbonate solution. The structure of 52a‧HCl was confirmed by X-ray data, the tetracyclic skeleton is planar, the hydrogen bonding of the amide hydrogen through a water molecule forms a network in the solid. Derivatives 52b-d were isolated from the reaction as major products.
Moreover, Mondal et al [44] performed the mechanistic study under deuterated solvent and revealed that the reaction proceeds through intramolecular 1,3-hydride shift. In addition to benzamides 51a-d, various other amides such as 2-aminobenzenesulfonamide and 2-aminotetrahydrobenzothiophenamide were involved in this reaction, leading to corresponding analogs of compounds 52 with similarly high yields. Notably, the nature of substituents within the amide moiety exerts minimal influence on product yield, thus offering substantial flexibility for structural variation of the bicyclic core. Additionally, this method could potentially be applied to many different variations of o-phthaldialdehyde compounds.
4. Pyrazolo[1,5-a]quinazolines and indazolo[2,3-a]quinazolines
In the review article [13], some data on the synthetic approaches and bioactivity of pyrazoloquinazolines in which the pyrazole ring is attached to different edges of pyrimidine core are discussed. Regarding the synthesis of pyrazolo[1,5-a]quinazolines, two strategies were reported: the interaction of 2-hydrazinylbenzoic acid with 3-oxoalkanenitrile in the presence of CH3COOH [45] and the three-component reaction of 3(5)-amino-4-phenylpyrazole with aromatic aldehydes and cyclohexanone in acetic acid [46]. Some pyrazolo[1,5-a]quinazolines were described as negative allosteric modulators of metabotropic glutamate receptors [47], other tricyclic derivatives exhibit the topoisomerase-1 inhibitory activity [48]. Example synthesis of indazoloquinazoline system with a, b or c patterns of annelation of indazole fragment to quinazoline core are provided in the microreview [49].
A series of new manuscripts on pyrazolo[1,5-a]- and indazolo[2,3-a]quinazolines has been published later. Zhang et al [50] presented a convenient and simple synthetic procedure for obtaining several pyrazolo[1,5-a]quinazolin-5(4H)-ones 54 via copper-catalyzed cascade reactions of 2-bromobenzoates 53 with 1H-pyrazol-5-amines under ligand-free conditions in water (Scheme 20). This method is based on commercially available starting materials; water is used as solvent, so the procedure responds to the urgent need for "greener" and "cleaner" chemistry.
Gnanasekaran et al [51] incorporated 2-fluoroaroyl chlorides 55 into reaction with 5-aminopyrazoles (Scheme 20). The formation of pyrazolo[1,5-a]quinazolin-5(4H)-ones 56 is described as two-step sequence: initial acylation of the C5 amino group of the pyrazole was performed in DMF at -10 oC and further heating to 140 oC, then SNAr ring closure between N1 of the pyrazole and the 2-fluoroarylamide was performed. Products 56 were obtained in high yields regardless of the nature of substituents.
An efficient method for the synthesis of pyrazolo[1,5-a]quinazolines 58, indazolo[2,3-a]quinazoline 59, and 10-azaindazolo[2,3-a]quinazoline 60 was described using various 2-fluorobenzaldehydes 57 interacting with 1H-pyrazol-3-amines or 1H-indazol-3-amine (and its 7-aza derivative) under metal-free conditions (Scheme 21) [52]. The process involves an intermolecular condensation step followed by a metal-free base-promoted intramolecular C–N coupling reaction. This methodology opens wide opportunities for preparation of various polycyclic quinazoline derivatives. Importantly, the bromine atom stays unchanged during the reaction, so it can later be used for further chemical changes or transformations.
Gao et al [53] developed regioselective synthesis of indazolo[2,3-a]quinazolines 62 via a sequential annulation and dehydrogenative aromatization of cyclohexanones 61 (Scheme 22). The reaction is carried out in 1-methyl-pyrrolidin-2-one (NMP) as a solvent in the presence of iodine and sulfur at 120 °C for 20 hours. A control experiment verified the sequential nature of the process steps. This reaction works well with many kinds of aminoindazole compounds, including ones with aromatic and heterocyclic aldehydes, as well as differently modified cyclohexanones. Additionally, this method was also used to prepare pyrazolo[1,5-a]quinazolines 63.
A series of pyrazolo[1,5-a]quinazolines and their aza-analogues were synthesized and studied as inhibitors of histone lysine demethylase 4D (KDM4D) [54]. Initially, Fang et al performed molecular docking for 30 compounds, two tricyclic derivatives showed an inhibition rate greater than 50% against KDM4D at a concentration of 10 µM; one of leading compounds was pyrazolo[1,5-a]quinazoline derivative (A), its activity was measured towards three additional KDM enzymes (KDM2B, KDM3B, and KDM5A), excellent selectivity toward KDM4D was demonstrated. Structural modifications focusing on three fragments of compound A (highlighted in green color) led to the synthesis of a series of derivatives 64a–t. These derivatives were prepared by reacting 2-chloroacetyl chlorides, derived from either benzoic acid or picolinic acid, with corresponding 3-amino-1H-pyrazole-4-carbonitriles (Figure 3).
All target compounds were initially tested for their inhibitory activity against KDM4D at a concentration of 10 μM. Only three compounds (64p, 64r, and 64s) exhibited higher potencies compared to A. For these three compounds, IC50 values were determined, derivative 64r was chosen as lead compound (Table 1). Moreover, quinazoline 64r exhibits good selectivity for KDM4D; molecular docking was used to predict the binding model of compound 64r in the active pocket of KDM4D (Figure 4).
Kovács et al [55] reported the synthesis of pyrazolo[1,5-a]quinazoline derivatives through the reaction of 2-hydrazinobenzenecarboxylic acids or their esters (compounds 65) with α-oxo-cyanides (Scheme 23). Refluxing a mixture of compound 65 and 4-oxotetrahydrothiophene-3-carbonitrile in ethanol led to the formation of 6,7-dihydro-5H,9H-thieno[3’,4’:3,4]pyrazolo[1,5-a]quinazolin-5-ones 66, which were subsequently converted into amines 67.
Interaction of 2-oxocyclopentanecarbonitrile (n = 1) or 2-oxocyclohexanecarbonitrile (n = 2) with arylhydrazines 65 in ethanol afforded a series of novel tetracyclic pyrazolo[1,5-a]quinazoline derivatives 68, the reduction of the latter leads to formation of amines 69. Notably, when R2 = Me, Br or R5 = Me, the yield of pyrazolo[1,5-a]quinazoline derivatives 66 and 68 significantly decreased, and their corresponding amines 69 were not detected.
5-Substituted pyrazolo[1,5-a]quinazolines were synthesized and evaluated as ligands of GABAA receptor [56], only one compound showed receptor recognition in the nanomolar range. To find derivatives with higher activity, novel 3- and/or 8-substituted pyrazolo[1,5-a]quinazolines were synthesized [57]. Using a copper-catalyzed tandem reaction, appropriate 2-bromo-4-R-benzaldehyde and 5-amino-1H-pyrazole-4-carbonitrile, 3-cyanoderivatives 70a-c were isolated in low yields (Scheme 24). Later [58], derivative 70b was obtained in 55% yield by fusion of 5-amino-1H-pyrazole-4-carbonitrile with 2-chlorobenzaldehyde at 160 °C for 6 hours. Interaction of ethyl 1-(3-methoxyphenyl)-5-aminopyrazole-3-carboxylate 71 with formaldehyde or paraformaldehyde in an acidic medium afforded 8-methoxy-pyrazolo[1,5-a]quinazoline 72a, however, separating it from its 6-methoxy isomer was difficult. The key intermediate 72a was synthesized in higher yield from 2-carboxy-5-methoxyphenylhydrazine via intermediate 8-methoxy-pyrazolo[1,5-a]quinazolines 73, 74, and 75 (Scheme 24). Derivative 72a and ethyl pyrazolo[1,5-a]quinazoline-3-carboxylate 72b were hydrolyzed to give the respective 3-carboxylic acids 76a,b, which were further transformed into 3-ester derivatives 77a-e and 78a-c. Finally, compounds 77a,b,d were converted into the corresponding 4,5-dihydroderivatives 79a,b,d.
The in vitro study of the BZ site/GABAA-R binding affinity of synthesized pyrazolo[1,5-a]quinazolines derivatives showed that the new compounds have binding recognition in the nanomolar range (3.16 < Ki (nM) < 529.3) at fixed concentrations of 10 μM, raising the subnanomolar affinity value of 0.27 nM for compound 77b. The pharmacological tests evidenced for compound 77b a profile of positive allosteric modulator with anxiolytic and antihyperalgesic activity, lacking toxicity when tested in human neuronal-like cells and in vivo models.
In the next work of the same team [59], modification of pyrazolo[1,5-a]quinazolines 77 was performed by shortening or removing the linker between aryl(hetaryl) ring and the pyrazolo[1,5-a]quinazoline core, 3-aryl(heteryl)-pyrazolo[1,5-a]quinazolines 82 and 3-(hetero)aroyl-pyrazolo[1,5-a]quinazolines 83 were developed. Cyclization of hydrazinobenzoic acid with appropriate propanenitrile resulted in the formation of pyrazolo[1,5-a]quinazolines bearing at position 3 (hetero)aryl group (compounds 80a-e) or (hetero)aroyl group (compounds 80f-h) (Scheme 25). Treatment of compounds 80a-e with LiAlH4 in anhydrous THF followed by oxidation led to 3-(hetero)aryl derivatives 82a-e. Compounds 80f-h were transformed into 3-(hetero)aroylpyrazolo[1,5-a]quinazolines 83a-c via intermediate chloroquinazolines 81.
The synthesis of compounds 82f-i was accomplished using the Suzuki cross-coupling based on halogenated derivatives 86a,b, which were obtained from 8-methoxypyrazolo[1,5-a]quinazoline 85 (Scheme 25). The starting quinazolinone 84 was synthesized by decarboxylation of derivative 73. Derivative 76a used for the preparation of quinazolines 82f and 83d-g (Scheme 25). Treating 3-(hetero)aroyl-pyrazolo[1,5-a]quinazolines 83a,b,d-g with sodium borohydride complex (NaBH3CN) in AcOH allowed obtaining their dihydro analogs 87a,b,d-g.
All target compounds were previously evaluated for their ability to displace [3H]flumazenil (Ro-151788) from its specific binding to Bz receptors in bovine membrane samples. Electrophysiological studies on recombinant α1ꞵ2ɤ2L GABAA receptors were carried out for more promising compounds 82d–f, 83a–d, 6f,g, 87a,b,d. Quinazolines 82 bearing Ar/Het group could not modulate the GABAA function, but they were found to act as null modulators or antagonists. Among aroyl/Het derivatives 83a and 83b can modulate the GABAA receptor in an opposite manner: 83b enhances and 83a reduces the variation of the chlorine current. The most potent derivative 87d reached a maximal activity at 1 μM (+54%) and enhanced the chlorine current at ≥ 0.01 μM. Moreover, compound 83g demonstrated the ability to antagonize the full agonist diazepam.
Guerrini et al [60] evaluated the effect of the shift of the methoxy group from position 8 to position 6, with the same (hetero)aryl ester groups at position 3. 6-Acetyl-7-(2-dimethylaminovinyl)pyrazolo[1,5-a]pyrimidine 3-cyano or 3-ethoxycarbonyl 88a,b, obtained according to the described method [61], were used for the synthesis of 3,6-disubstituted pyrazolo[1,5-a]quinazolines. Boiling of these compounds in an acetic buffer solution led to the formation of 6-hydroxypyrazolo[1,5-a]quinazolines 89a,b, whose alkylation yielded derivatives 90a,b (Scheme 26). Compound 90b was hydrolyzed to produce the corresponding 3-carboxylic acid, which was esterified to ethers 91a-c, and then reduced to 4,5-dihydroderivatives 92a-d. Compound 90a under the treating with hydroxylamine hydrochloride in ethanol and potassium carbonate, followed by cyclization was transformed into 3-(5-methyl-1,2,4-oxadiazol-3-yl)-derivative 93. To prepare derivatives 96a,b, compounds 89a and 90a were converted into 3-carboxyamide derivatives 94a,b, which upon reaction with dimethylformamide-di-tert-butylacetate (DMF-DtBA) in toluene formed acylamidines 95a,b. Boiling these acylamidines with hydrazine hydrate yielded compounds 96a,b. 3-Iodo-6-benzyloxypyrazolo[1,5-a]quinazoline 97 was obtained from compound 89b after decarboxylation, alkylation, and iodination (Scheme 26).
Novel 3,6-disubstituted pyrazolo[1,5-a]quinazolines 89–94, 96, 97were studied as ligands to GABAA receptor. The GABAA-binding affinity of compounds was evaluated for their ability to displace [3H]flumazenil (Ro-151788) from its specific binding in a bovine membrane. From the obtained results, it follows that the compounds demonstrated the percent of inhibition of specific [3H]Ro15-1788 binding at 10 μM concentration from 1% to 43.7%, compound 97 showed the highest inhibition. Based on experimental data and molecular modeling study on compound 91b authors concluded that the movement of substituents from position 8 to position 6 is essential for binding.
New 8-methoxypyrazolo[1,5-a]quinazolines 98a-i and their dihydro derivatives 99a-i bearing the amide fragment at the position 3 were synthesized [62] as analogues of 8-methoxypyrazolo[1,5-a]quinazoline 3-ester [56,57], 3-(hetero)aroyl and 3-(hetero)aryl derivatives [59] identified as modulators of GABAA receptors. 8-Methoxypyrazolo[1,5-a]quinazoline-3-carboxylic acid 78a after the treatment with thionyl chloride or trichloroacetonitrile/PPh3 in CCl4 led to the formation of Het-C(O)Cl intermediate, which without isolation interacted with the appropriate amine, giving compounds 98a-i, which were transformed into the corresponding 4,5-dihydroderivatives 99a-i by the treatment with sodium cyanoborohydride (NaBH3CN) in acetic acid (Figure 5).
All the new compounds 98a-i and 99a-i have been evaluated in vitro for their ability to modulate the chlorine current on recombinant GABAA receptors of the α1β2γ2L type (expressed in frog oocytes of the Xenopus laevis species). Two groups of compounds were identified from electrophysiological test: positive modulators agonists (98e,h,i and 99e,h) and null modulators antagonist (98a,b,d,f,g and 99a-d,f,g).
In the next work [63], new 8-chloropyrazolo[1,5-a]quinazoline derivatives 100–107 (Figure 5) were presented as GABAA receptor modulators. Their synthesis are like ones demonstrated in Scheme 24 and Scheme 25. Compounds 100–107 underwent molecular dynamics simulations performed on an isolated segment of the GABAA receptor protein located between α and γ chains, where the benzodiazepine-binding site is identified. Using the 'Proximity Frequencies' model (PF), Crocetti et al [63] predicted that compounds 100a, 103a, and 106b belong to the agonist class with 93.1% probability. On the contrary, derivatives 101c, 103c, and 107 fall into the antagonist class, with 62–73% prediction. Thus, two types of compounds occupy different areas in the binding site. The virtual prediction for 106b and 107 as agonist and antagonist, respectively, was confirmed through electrophysiological assays.
3,8-Disubstituted pyrazolo[1,5-a]quinazoline 108–113 (Figure 6), bearing oxygen or nitrogen function at position 8, were synthesized and studied as GABAA receptor modulators [64]. The synthesis is based on approaches presented in Scheme 24 and Scheme 25.
Compounds 108–113 were screened through electrophysiological techniques on recombinant α1β2γ2L-GABAA receptors expressed in Xenopus laevis oocytes, some compounds exhibited certain ability to bind the receptor. The most promising electrophysiological results were obtained for compounds 108d, 109a, 109b, and 112. Among the 3-iododerivatives, compound 108d, which does not modulate the chlorine current, was evaluated for its ability to antagonize the full agonist lorazepam (1 μM). Compounds 109a and 112 were found to exhibit agonist profile while quinazolines 109b and 108d act as antagonists. Molecular modelling studies and Hierarchical Cluster Analysis (HCA) data have collocated these ligands in the class corresponding to their pharmacological profile (Figure 7).
A large series (102 compounds) of pyrazolo[1,5-a]quinazoline derivatives were designed and synthesized to investigate their activity as SIRT6 activators [65]. SIRT6 is a particularly important member of Sirtuins and has emerged as a novel therapeutic target for various diseases. Previously [66], a 2-methyl-5-(4-methylpiperazin-1-yl)pyrazolo[1,5-a]quinazoline (Hit41) had been found as a new SIRT6 de-fatty-acylation activator. Zhang at al [65] discovered highly active SIRT6 de-fatty-acylation activators by structural modifications of Hit41, focusing on expanding substituents at positions 2 (R1) and 5 (R2) of the pyrazolo[1,5-a]quinazoline scaffold. Three series of derivatives were synthesized (Scheme 27): 115a–117a (R1 = H); 115b–117b (R1 = Me); 115c–117c (R1 = tBu). In each series, R2 represented a broad set of Ar or Het substituents (number indicated in parentheses). Chlorinated precursors 114a-c, obtained using one of the approaches shown in Scheme 24, were used as starting materials.
Fluor de Lys (FDL) assays were performed for compounds 115–117, for some compounds additional calculations were made to determine the concentration of the compound (μM) at which the compound can increase the enzymatic activity by 50% values (EC1.5), structure-activity relationship (SAR) was studied. A set of novel SIRT6 activators was obtained (13 compounds with EC1.5<50.79 μM); among them, 2-methyl-N-(4-phenoxyphenyl)pyrazolo[1,5-a]quinazolin-5-amine 116b(q) is the most potent one, which exhibited excellent defatty-acylation activation activity against SIRT6 (EC1.5 1.85±0.41 μM and EC50 11.15±0.33 μM). Notably, that the calculated EC1.5 value for Hit41 is only 49.30±0.74 μM. The bioactivity of 116b(q) was further verified by differential scanning fluorimetry (DSF) and surface plasmon resonance (SPR) assays. Molecular docking showed that the pyrazolo[1,5-a]quinazoline 116b(q) formed a hydrogen bond with Val115 and four π–π interactions with Phe64, Phe82 and Phe86; 116b(q) can significantly improve the thermal stability of SIRT6 protein and inhibit the PI3K/Akt signaling pathway in mouse embryonic fibroblasts (MEFs), thereby inhibiting the proliferation of MEFs. As a result, Zhang at al [65] discovered a new potent SIRT6 activator, which can be taken as a lead compound for later studies.
A library of new pyrazolo[1,5-a]quinazoline compounds substituted at positions 3, 7 or 8 was synthesized and their anti-inflammatory activity was studied [67]. The key intermediates 118a-d, 120a-e, and 122 were obtained by the condensation of disubstituted 2-hydrazinobenzoic acid with ethoxymethylene-malononitrile, ethyl-2-cyano-3-ethoxyacrylate, and 3-oxo-2-(3-thienyl)proponitrile, respectively (Scheme 28). Acid hydrolysis of nitriles 118 yielded amides 119, while carboethoxy derivatives 120 were converted into unsubstituted counterparts 121. Pyrazolo[1,5-a]quinazolines 118–122 were subjected to further transformations, and 45 compounds were synthesized for screening. For example, from derivative 119a, a series of 5-alkoxy-substituted pyrazolo[1,5-a]quinazolines 123a-i was prepared, the compound 123c was oxidized to yield sulfoxide 124 and then sulfone 125, whereas 123b was transformed into 3-(1,2,4-triazole) derivative 126 (Scheme 28).
Compounds 123–126 were tested for their ability to inhibit lipopolysaccharide (LPS)-induced nuclear factor κB (NF-κB) transcriptional activity in human THP-1Blue monocytic cells. Only 13 compounds were able to inhibit NF-κB activity with IC50<50 μM, two of them showed the highest activity: 123i (5-[(4-sulfamoylbenzyl)oxy]pyrazolo[1,5-a]quinazoline-3-carboxamide) and 124 (5-[(4-(methylsulfinyl)benzyloxy]pyrazolo[1,5-a]quinazoline-3-carboxamide). Pharmacophore mapping of potential targets and molecular modeling allowed to conclude that these compounds could effectively bind to extracellular signal-regulated kinase 2 (ERK2), p38α, and c-Jun N-terminal kinase 3 (JNK3) with the highest complementarity to JNK3. Moreover, compounds 123i and 124 exhibited micromolar binding affinities for JNK1, JNK2, and JNK3. Obtained results [67] demonstrate the potential for developing lead anti-inflammatory drugs of pyrazolo[1,5-a]quinazoline nature.
Kumar et al presented the synthesis of tetracyclic hybrid structures 127–132, combining pyrazolo[3,4-b]pyridine and pyrazolo[1,5-a]quinazoline fragments, and data of their biological activity [68]. Reaction of 3-amino-pyrazolo[3,4-b]pyridine-5-carboxylate 127 with 2-fluorobenzaldehyde resulted in the formation of ethyl pyrido[2’,3’:3,4]pyrazolo[1,5-a]quinazoline-8-carboxylate 128 in 89% yield. Hydrolysis of compound 128 under alkaline conditions produced carboxylic acid derivative 129, which upon reaction with various substituted anilines, cyclic secondary amines, and primary aliphatic amines led to the corresponding amide-substituted pyridopyrazolo-quinazolines 130a-f, 131a-d, and 132a-h (Scheme 29). In all cases, the reaction between 129 and amines was carried out in the presence of benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP).
Pyridopyrazolo-quinazolines 128–132 were evaluated for antibacterial activity against Gram-positive and Gram-negative bacterial strains, compounds 129, 130a, 130c, 130f, 131a, 132c, 132f and 132h exhibited promising antibacterial activity against various bacterial strains. Compound 129 showed high antibacterial (MIC 3.9 μg/mL) and broad-spectrum anti-biofilm activity. Compounds 129 and 132b also showed good antifungal activity against various Candida strains. Compound 129 was found to be promising as a broad-spectrum biofilm inhibitor both against bacterial pathogens and against C. albicans MTCC 3017. Further, compound 129 reduced the ergosterol content in three Candida strains (C. albicans MTCC 227, C. albicans MTCC 1637 and C. albicans MTCC 3017). Both compound 129 and Miconazole showed the same amount of inhibition of ergosterol content in C. albicans MTCC 227, modelling studies were also performed for validation. Compounds 131d, 132a, 132b and 132d exhibited inhibition >90% against MCF7 (breast) cancer cell line; compounds 130d, 131a, 132a, 132b, 132f and 132g exhibited inhibition >90% against SKOV3 (ovarian) cancer cell line, Doxorubicin was used as a standard.
Synthesis and insecticidal activities of polysubstituted 4,5-dihydropyrazolo[1,5-a]quinazolines 133–136 (Scheme 30) were described [69,70,71]. 5,5-Disubstituted 4,5-dihydropyrazolo[1,5-a]quinazoline derivative 133a, obtained by interaction of 5-amino-1H-phenylpyrazole with dialkyl bromomalonate, exhibited promising insecticidal activity against P. xylostella [69]. Later the same team [70] performed some modifications of compound 133a: substituents in benzene ring (direction A), position 3 of the pyrazole ring (direction B), position 5 of the quinazoline ring (direction C), synthesized three series of novel 4,5-dihydropyrazolo[1,5-a]quinazoline derivatives (133–135). Afterwords, an expanded series of analogues 136 was obtained [71].
Insecticidal activities of compounds 133–135 against insect pests P. xylostella, S. frugiperda and S. invicta were evaluated. The most compounds exhibited good (>50% mortality) to excellent (100% mortality) insecticidal effect against P. xylostella; 15 compounds from the series showed significant mortality rate >50% at 100 mg/L, and 9 compounds were able to show mortality rate >50% at 25 mg/L, the LC50 values were found to be 3.87−24.35 mg/L. Compound 133 (R1 = CF3, Cl, R2 = Me) exhibited the best insecticidal activity, its LC50 value against P. xylostella (3.87 mg/L) was comparable to that of indoxacarb (4.82 mg/L), which is one of the major commercial pesticides to control P. xylostella. Insecticidal activities of compounds 133a and 135 (X = NH, 2-Cl-5-Me-thiazole) against S. frugiperda (mortality rate 79.63% and 72.12%) were comparable to that of fipronil (mortality rate 68.44%). The compounds 133 (R1 = CF3, Cl, R2 = Me) and 135 (X = O, R = Me) showed high insecticidal activities against S. invicta (mortality rate 96.67% and 95.56%) comparable to that of fipronil (mortality rate 100%) after 5 days of treatment at 1.0 mg/L. Moreover, electrophysiological studies indicated that compound 136 (R1 = CO2Me, R2 = H, R3 = CN) could act as a potent GABA receptor antagonist (2 μΜ, inhibition 68.25%).
5. Imidazo[1,2-a]quinazolines, benzimidazo[1,2-a]quinazolines
Synthetic approaches to imidazoquinazolines, in which imidazole cycle is attached to с, b or g edges of quinazoline core, as well as to benzo[4,5]imidazo[1,2-c]- and benzo[4,5]imidazo[1,2-a]quinazolines are described in book chapter [12]. Two main approaches to [a]-annelated derivatives are used: condensation of 2-aminobenzimidazole with various aromatic aldehydes bearing Hal or NO2; and intramolecular C ̶ N bond formation. This section of the current manuscript contains data on the synthesis and biological activity of imidazo- and benzimidazo[a]quinazolines, which were not included in the book chapter [12] or appeared later.
Annareddygari et al [52] developed an efficient method for the synthesis of benzo[4,5]imidazo[1,2-a]quinazolines 137 from different 2-fluoro-benzaldehydes with 2-aminobenzimidazole (its 7-aza derivative) under metal-free conditions in high yields (Scheme 31). The process includes an intermolecular condensation followed by metal-free base-promoted intramolecular C–N coupling reaction.
Using this approach, a series of trisubstituted benzo[4,5]imidazo[1,2-a]quinazolines 138 was obtained to study their anti-fungal activities against six plant pathogenic fungi and analyze SAR [72]. It was found that compound 138 (R = R1 = H) demonstrated broad-spectrum antifungal activities with EC50 = 4.43 μg/mL.
Microwave-assisted synthesis of 1,2,3,4-tetrahydrobenzo[4,5]imidazo[1,2-a]quinazolines 140 was presented [73]. The reaction of 2-bromocyclohex-1-ene carbaldehydes (139a-c) and 2-aminobenzimidazoles in the presence of both base and magnesium sulfate under microwave irradiation led to the formation of derivatives 140 in 58–70% yields (Scheme 32).
In the next work [74] 2-bromocyclohex-1-enecarbaldehydes 139a-c were incorporated into the reaction with 4,7-dimethoxy-1H-benzimidazole-2-amine under optimized conditions (microwave irradiation) for the obtaining of tetrahydrobenzo[4,5]imidazo[1,2-a]quinazolines 141 and subsequent oxidation (Scheme 32). It was demonstrated that 4,7-dimethoxy-1H-benzimidazole-2-amine also interacts effectively with 2-bromobenzaldehydes 143 with the formation of benzimidazo[1,2-a]quinazolines 144. Oxidation of quinazoline-fused dimethoxybenzimidazoles 141 and 144 was conducted using ceric ammonium nitrate (СAN) in the MeCN/H2O medium. The corresponding benzo[4,5]imidazo[1,2-a]quinazoline-8,11-diones 142 and 145 were obtained in good yields.
Dao et al [75] reported the synthesis of 1,2,3,4-tetrahydrobenzo[4,5]imidazo[1,2-a]quinazolin-5-ones 147 from ꞵ-bromo-α,ꞵ-unsaturated amides and benzimidazole via copper powder catalyzed C–N coupling and subsequent C–N bond formative cyclization by C–H activation under microwave irradiation conditions (Scheme 33). Interaction of amide 146 with imidazole under the same conditions led to the formation of 1,2,3,4-tetrahydroimidazo[1,2-a]quinazolin-5-one 148. It was also demonstrated that benzamides 149 under similar conditions allowed to obtain benzimidazo[1,2-a]quinazolin-5-ones 150a-c (Scheme 33). This reaction provides the first known way to make these unique hybrid nitrogen-containing structures using common starting materials.
Heterogeneous amorphous Cu–MOF-74 catalyst was developed for C–N сoupling reaction and applied for the synthesis of benzimidazo/imidazo[1,2-a]quinazolin-5-ones 151a,b [76] (Scheme 33). Ma et al demonstrated that crystalline Cu–MOF-74 can be used as a catalyst precursor to synthesize aCu-MOF-74 under the action of alkali and high temperature in the reaction solution, and then it acts as a true heterogeneous catalyst to catalyze С–N coupling between 2-iodobenzamides 149 and benzimidazole/imidazole, which gives higher isolated yields. The catalyst can be simply removed after the reaction and reused up to six times without losing much effectiveness.
An approach based on the Ullman coupling of 2-chlorobenzamide 149d with imidazole followed by oxidative ring formation was used for the synthesis of imidazo[1,2-a]quinazolin-5-one 152 [77] (Scheme 34). When 4-phenylimidazole was applied, the yield of compound 153 was only 23%, while functionalization of derivative 152 by palladium-catalyzed direct C–H arylation with bromobenzene led to the formation of 2-phenyl-4-ethyl-imidazo[1,2-a]quinazolin-5-one 153 in 55% yield.
Rapid and efficient microwave-assisted metal-free base-mediated synthetic approach to the series of benzimidazo[1,2-a]quinazolines 156 (15 compounds) (Scheme 34) from readily available building blocks was developed [12,78]. In the presence of groups X = NO2 or R = 5-Br in the aldehyde 155, the yields of compounds 156 decreased to 38% and 44%, respectively. This method uses easy-to-get starting materials, works with many different substrates, is simple to carry out, and allows making various benzimidazo[1,2-a]quinazoline compounds without needing metals.
One more strategy for the synthesis of imidazole[1,2-a]quinazolin-5(4H)-ones was described [79]. Interaction of isatoic anhydride 28 with o-chlorobenzyl amine gave 2-amino-N-(2-chlorobenzyl)benzamide, which under the reaction with CS2 in the alkali medium was converted into 3-(2-chlorobenzyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one 157 (Scheme 35). Heating this compound with a mixture of POCl3 and PCl5 yielded the corresponding 2-chloro derivative 158, which upon reaction with 2,2-dimethoxyethylamine in the presence of NEt3 afforded 4-(2-chlorobenzyl)imidazo[1,2-a]quinazolin-5(4H)-one 159. The structure of compound 159 was confirmed by various spectroscopic methods including X-ray crystallography.
In the next work [80] 4-(2-chlorobenzyl)imidazolo[1,2-a]quinazolin-5(4H)-one 159 was functionalized at imidazole cycle by the Vilsmeier-Hacck reaction with DMF and POCl3 and then reductive amination with a secondary amine reagent to obtain compounds 161a-e (Scheme 35). Second series of 4-(2-chlorobenzyl)imidazole[1,2-a]quinazolin-5(4H)-ones (164, 27 compounds) was synthesized based on 2-amino-3-(2-chlorobenzyl)quinazolin-4(3H)-one 162, obtained from chloro derivative 158 in the reaction with p-methoxy-benzylamine. Removing the benzyl group, cyclizing 162 with ethyl bromopyruvate and condensing with amine compounds led to the formation of compounds 164 in good yields (Scheme 35).
Imidazoquinazolinone derivatives 161 and 164 were studied as allosteric inhibitors of SHP2 phosphatase, in vitro enzymatic assays were conducted. The inhibitory activities of 161 and 164 against SHP2 protease at 100 μM and 200 μM were evaluated; results demonstrated that most of the compounds have certain inhibitory activity against SHP2 protease. Imidazoquinazolinones 161a (22.14% inhibition) and 164 (R = 4-HO-piperidinyl) (29.81% inhibition) showed better inhibition of SHP2 protein activity than SHP244 as a positive control, (14.95% inhibition) at 100 mM. The in vitro cytotoxicity of the compounds 161, 164 100 μM on the melanoma cell line A375 was evaluated, according to results, compared with SHP244 and sorafenib (inhibition 13.81% and 14.79%), most of the compounds showed significant cytotoxicity to A375. Among them 161a (76.15% inhibition) and 164 (R = m-CF3-aniline) (27.93% inhibition) showed effective activity. The IC50 values were also measured for the series of compounds and generally it was shown that compared with SHP244, the tumor cell activity of 161 and 164 compounds is significantly better than enzyme activity. The docking studies revealed that C=O groups of imidazoquinazolinones form hydrogen bond interactions with Lys274 and His84, respectively (Figure 8).
Ghashghaei et al [81] presented selective multiple multicomponent Groebke–Blackburn–Bienaymé reaction (GBBR), which allows to obtain imidazoazines by acid-catalyzed interaction of α-aminoazines, aldehydes and isocyanides. Thus, diaminoquinazoline 165 underwent a regioselective GBBR providing mono-adduct 166 at components ratio 1:1:1 (Scheme 36). In a different reaction, substrate 165 afforded the symmetrical bis-adduct 167 (components ratio 1:2:2). Moreover, a second GBBR, performed upon 166, gave the non-symmetrical compound 168 (components ratio 1:1:1). This method can be widely applied and create complicated molecules selectively, adjustably, and directly.
Annareddygari et al [52] developed the general approach to azoloquinazolines. Thus, the reaction of 2-aminobenzothiazole with 2-fluorobenzaldehyde led to the formation of benzo[4,5]thiazolo[3,2-a]quinazoline (169) in 73% yield under the optimized conditions (Scheme 37).
The series of thiazolo[3,2-a]quinazolin-5-one derivatives 172a-f containing a sulfonamide group on C7 was described [82]. Interaction of chlorosulfonyl-substituted benzoyl halogenides 170a-d with aminothiazoles led to the formation of amides 171 (Scheme 37). The treatment them with amine under mild conditions and subsequent cyclization under reflux in DMF or diphenyl ether allowed to obtain thiazolo[3,2-a]quinazolin-5-ones 172a-f. The influence of substituent R1 and the nature of halogen on the yield of target products has been analyzed.
170, 171: X, R1 = F, H (a); Cl, H (b); Cl, Cl (c); Br, H (d); 172: R1 = H (a-c), Cl (d-f); R2R3N = morpholin-4-yl (a, d); R2 = H, R3 = Ph (b, e); R2R3N = piperidin-4-yl (c, f).
6. Triazolo[a]quinazolines
6.1. [1,2,4]-. Triazolo[4,3-a]quinazolines
Book chapter [11] discloses approaches to triazolo[a]- and triazolo[c]-annelated quinazolines based on traditional transformations of chloroquinazolines, condensations between quinazoline and azide or nitrile, copper-catalyzed alkyne-azide cycloaddition reaction, multicomponent reactions, and microwave-assisted procedures. In the book chapter [11], some data on biological activity was also presented. The current section of the manuscript contains data on the synthesis and biological activity of triazolo[a]quinazolines, which were not included in the book chapter [11] or appeared later.
A series of 5-substituted-[1,2,4]triazolo[4,3-a]quinazolines 173 was synthesized, and anticonvulsant activity was tested [83]. Among compounds 173 5-heptyloxy-derivative was found to be especially potent (ED50 = 39.4 mg/kg, PI = 8.3). Continuing research in this direction, the same team has presented a new series of 4-aryl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 175 and 1-substituted derivatives 176–180 (Scheme 38) and evaluated their anticonvulsant [84] and antidepressant activities [85]. In these compounds, an aryl substituent is introduced into the NH group of quinazolin-4(3H)-one, unlike derivatives 173, while the triazole ring is modified. 2-Hydrazino-3-phenyl-2,3-dihydroquinazolin-4(1H)-one derivatives 174, obtained by slightly modifying a previously reported method [86], were used as starting materials for the synthesis of [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 175–180. The treatment of derivatives 174 with formic acid under reflux overnight led to 4-aryl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 175 (24 compounds). To obtain the target compounds 176, 178–180, derivative 174 was reacted with diethyl oxalate, urea, carbon disulfide, and acetic acid, respectively (Scheme 38). The amido derivative 177 was obtained by treating ethoxycarbonyl derivative 176 with aqueous ammonia.
Compounds 175–180 were evaluated for their anticonvulsant activity and neurotoxicity by maximal electroshock (MES) and rotarod neurotoxicity tests [84]. The compounds were dissolved in DMSO and Kunming mice in the 18–22 g weight range were used. According to preliminary data, 11 compounds from the series of 175 possessed anticonvulsant activity against MES-induced seizures at a dose of 100 mg/kg, and five of these (R = o(m,p)-Cl, p-Br, 3,4-Me2) were found to remain active at a dose of 30 mg/kg. Further studies demonstrated that two derivatives 175 (R = p-Cl and p-Br) displayed wide spectrum activity in several models? demonstrated wide margin of safety with a protective index (PI) exceeding one for existing drugs (>25.5 and >26.0), they also showed significant oral activity against MES-induced seizures in mice, with ED50 88.02 and 94.60 mg/kg, respectively.
Compounds 175–180 were evaluated for antidepressant activities in mice [85]. Most of them showed antidepressant activity in the forced swimming test (FST). It was found that three compounds from the series of 175 (R = o-F, m-Me and p-Me) at a dose of 50 mg/kg significantly reduced the immobility time in the FST. Compound 175 (R = p-Me) proved to be the most active, the immobility time decreased by 82.69 % at 50 mg/kg dose. Also, this substance didn't change normal behavior in tests, and its positive impact was like the antidepressant fluoxetine.
1-Methyl[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 182, 183 were obtained by the reaction of 2-hydrazinoquinazolin-4(3H)-ones 181a,b with acetylacetone (Scheme 39) [87]. Notably, that the 7-hydrazinocarbonyl group in quinazolin-4(3H)-one 181a is transformed into a pyrazole derivative, which can be replaced by amine with the amide formation.
1-Substituted-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones, bearing at position 4 pyridine-4-yl (185a-e) or p-nitrophenyl (186a-e) fragment (Scheme 39) were described [88,89]. Compounds 185a-d and 186a-d were obtained by the reaction of 2-hydrazino-3-R-quinazolin-4(3H)-one 184 with the corresponding acid as one carbon donor, whereas interaction of 184 with chloroacetyl chloride in glacial acetic acid was used for the preparation of compounds 185e and 186e. Both series of derivatives have been evaluated for their in vivo antihistaminic and sedative–hypnotic activities. The protection against histamine-induced bronchospasm on conscious guinea pigs method was adopted to determine the antihistaminic potential of the test compounds. All compounds exhibit significant antihistaminic activity, the percentage protection for compounds 185a-e is 69–73%, and for derivatives 186a-e 68–71.6%. The highest activity was shown by derivatives 185a and 186a. Sedative–hypnotic activity was determined by measuring the reduction in motor activity. The results showed that almost all the test compounds were found to exhibit mild activity (less than 10%).
The synthesis, crystal structure, DFT study, and molecular docking of two new 4-(2-chlorobenzyl)-containing [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones (189, 190) were presented [90,91]. Both derivatives were synthesized according to Scheme 40 starting from the corresponding 4(3H)-quinazolinone-3-[(2-chlorophenyl)methyl]-2-hydrazonyl 187, obtained via hydrazinolysis of quinazoline thione 157 (see Scheme 35). The structures of derivatives 189 and 190 have been confirmed by X-ray diffraction data. The crystal structures of 189, 190 were compared with the conformers optimized by DFT calculation, and they were found to be consistent. Studies on the molecular electrostatic potential and frontier molecular orbital (FMO) of 189 show that this compound has a certain nucleophilic reactivity and large hardness.
Molecular docking study for compound 189 demonstrated the formation of seven hydrogen bonds with SHP2 protein, indicating that this compound has a better binding effect with SHP2 [90]. Molecular docking of 190 suggests favorable interactions with SHP2 protein [91]. The SHP2 enzyme inhibitory activity of 190 was evaluated at 10 μM, (SHP244 was used as a reference compound [92]) and it was shown that activity (inhibition rate 12.40%) was lower than that of SHP244 (19.67%). The antitumor activity of compound 190 was evaluated in human hepatoma cells SMMC7721, human melanoma cells A375 and human breast cancer cells MCF-7 using an MTT assay (SHP244 was used as a reference compound). It was found that the IC50 values were 757, 70.19 and >1000 μM, respectively. Compound 190 has better selectivity for melanoma cells. The antitumor activity of 190 was better than that of the reference compound in SMMC7721 and A375, which may be due to 190 has better physical and chemical properties.
A series of [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one derivatives were synthesized and studied as SHP2 protein inhibitors [92]; among them, compound SHP244 showed good results. Continuing these studies, new groups of [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones (compounds 192(1-28), 193a,b, 195a-c) were obtained through modification of the SHP244 structure at positions 1 and 4 (Scheme 41) [80]. The benzene ring in the triazole cycle was replaced with a nitrogen-containing side chain to enhance the binding effect with the receptor and the 2-chlorobenzene ring was replaced with an aromatic ring containing different substituents to investigate the effect of this part of the structure on the activity.
The inhibitory activities of triazoloquinolinone derivatives 192, 193, 195 against SHP2 protease at 100 mM and 200 mM were evaluated in vitro. Self-isolated and purified SHP2 was used as the target protein, and SHP244 was used as a positive control. It was shown that most compounds have certain inhibitory activity against SHP2 protease. Compared with SHP244, some compounds show similar or higher sensitivity to SHP2, which indicates that the modification of the three parts of SHP244 has a profound influence on the activity. For example, compound 193b (28.20% inhibition) and compound 195b (26.38% inhibition) showed better SHP2 protein inhibitory activity than comparable structures at 100mM, and better than SHP244 (23.52% inhibition). To study the antitumor activity of the compounds 192, 193, 195, SHP244 and sorafenib were used as positive controls, and the MTT method was used to evaluate the in vitro cytotoxicity of the compounds 100 mM on the melanoma cell line A375. Compared with SHP244 and sorafenib, most of the compounds showed significant cytotoxicity to A375, such as 192(5) (27.02% inhibition), 192(21) (29.60% inhibition). Metabolic stability of compound 192(22) in human and rat liver microsome in vitro was studied, SHP244 was used as a positive control. It was found that this compound showed considerable stability in human and rat liver microsomes, and the clearance rate for both human and rat liver microsomes was less than 9.6 (mL/min/mg) with similar remaining (82.0% and 78.9%, t1/4 60 min). At the same time, t1/2 are both greater than 145 min, which is much greater than t1/2 of SHP244 for human and rat liver microsomes (14.2 and 12.2 min). Docking studies of the compound 195c was performed, and it was shown that C(O) group of 2-(pyrrolidin-1-yl)acetyl fragment and nitrogen of triazole cycle form hydrogen with the amino acid residues Asn281 and Tyr80, respectively (Figure 9).
Some [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one derivatives exhibit other types of biological activity. Thus, compounds 196a-c (Figure 10) were screened for their in vitro antimicrobial and antitubercular activities against pathogenic strain [93]. Compounds were obtained by the reaction of 3-aryl-2-hydrazinoquinazolines with carbon disulfide in good yields. Compound 196b was found to be nearly equipotent with Ciprofloxacin against both Gram-positive and Gram-negative bacteria (P. aeruginosa and S. aureus) with MIC 2.6 μg/mL and demonstrated very promising antifungal activity against A. niger with MIC 2.6 μg/mL. The anti-tubercular activity of compounds 196 reveal that compound 196b showed better activity than the other compounds with a MIC of 5.2 μg/mL against M. tuberculosis H37Rv.
A 3D similarity-based virtual screen to search for ligands of Toll-like receptor (TLR7), an important target for drug discovery, was performed [94]. Six new compounds were identified as interesting initial hit compounds that can act as TLR7 antagonists with micromolar potencies, as determined using a reporter assay. Among them, [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one 197 (Figure 10), possessing good solubility, was progressed into biological evaluation and showed antagonistic activity (IC50 =185 μM).
A series of 1-aryl-[1,2,4]triazolo[4,3-a]quinazolin-5-ones 198 (11 derivatives) designed as conformationally restricted of CA-4 (well-known antimitotic agent) analogues was synthesized to study their biological efficiency toward tubulin polymerization, their cytotoxicity toward various cancer cell lines as well as their antivascular effects [95]. Derivatives 198 were obtained by the reaction of 3-benzyl(methyl)-2-hydrazino-quinazolinones with corresponding aryl aldehydes followed by oxidative cyclization. Only two of them (198a,b) were potent in vitro tubulin polymerization inhibitors with IC50 values of 4.26 and 0.15 mM respectively, but only the N-methyl substituted counterpart 198b displayed strong growth inhibitions toward the NCI-60 human cancer cell lines panel. This compound has shown remarkable activity in the HUVEC shape change assays as cell invasion and endothelial tube formation experiments which are good indicators for potential in vivo antivascular activity. These results suggest that compound 198b might be lead compound for the development of novel vascular disrupting agents and is promising candidates for in vivo evaluation.
Triazolo[4,3-a]quinazolin-5-one derivative 199 demonstrated potent NF-κB inducing kinase (NIK) inhibitory effect with IC50 values of 103.4±5.1 μM and 57% [96]. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking was performed, 70 compounds from different classes were selected and tested by the NIK adenosine triphosphate (ATP) consumption essay for NIK inhibitory activities. Three of them, including derivative 199, have an inhibiting rate over 50%.
Identification of a new heterocyclic scaffold for inhibitors of the Polo-Box Domain of Polo-like Kinase 1 (PBD of Plk1) was performed [97]. A small chemical library of ∼400 drug-like molecules was screened for the ability to bind to the PBD of Plk1. Among four hits identified was triazoloquinazolinone 200 (Figure 11), which inhibited PBD binding in the ELISA assay with an IC50 of 4.38 μM. When compared to the previously characterized phosphopeptide, PLHSpT (IC50 of 14.74 μM), the affinity of the compound 200 is anticipated to be at least threefold higher than that of peptide. To find compounds with improved characteristics within this series, a large range of triazolo[4,3-a]quinazolin-5-ones 201 (147 derivatives) was synthesized by modifying different fragments of structure 200. All derivatives were obtained through the reaction of 3-R-2-hydrazino-quinazolinones with carbon disulfide. Various substituents were introduced as R1 groups (7(8,9)-F, 7-Br, 7-I, 7-Me, 7-NHAc, 7-NMe2) and aza-analogues of the phenyl ring were also introduced. Many alkyl, heteroalkyl, and arylalkyl modifications of the R2 group (phenylethyl in compound 200) were included.
All compounds were evaluated for their efficacy against the full-length human Plk1 in an ELISA assay and for their in vitro physiochemical properties. Variation of substituent R2 (in the case R1 and R3 = H) demonstrated that compound containing R2 = Pr exhibited the best inhibitory activity (IC50 of 1.03 μM). Replacing R3 in this compound with 2-oxo-2-phenylethyl led to a sharp decrease in inhibitory activity (IC50 >50 μM). Variation of substituent R1 (with R2 = Ph and R3 = H) resulted in changes in inhibitory activity ranging from IC50 1.54–30.71 μM. As a result, it was found that derivative 201a demonstrated the best efficacy against the full-length human Plk1 in an ELISA assay. Moreover, MeS-containing prodrugs 202 effectively inhibited mitotic progression and cell proliferation and their metabolic stability was determined.
6.2. [1,2,4]-. Triazolo[1,5-a]quinazolines
Book chapter [11] discloses approaches to triazolo[a]-annelated quinazolines based on traditional transformation of chloroquinazolines, condensations between quinazoline and azide, copper-catalyzed alkyne-azide cycloaddition reaction, through the coupling of dialkyl/phenyl N-cyanoimidocarbonates with the substituted 2-hydrazinobenzoic acids. Some data on biological activity was also provided in the book chapter [11]. The current section of the manuscript contains data on the synthesis and biological activity of triazolo[a]quinazolines, which were not included in the book chapter [11] or appeared later.
An approach based on the Ullman coupling of 2-chloroarylamides 203 with triazoles followed by oxidative ring formation was applied for the synthesis of [1,2,4]-triazolo[1,5-a]quinazolin-5-ones 204 and their aza-analogs 205 (Scheme 42) [77].
2-(4-Chlorophenyl)[1,2,4]triazolo[1,5-a]quinazoline 207, obtained by the reaction of 2-chlorobenzaldehyde with substituted 1H-1,2,4-triazol-5-amine (Scheme 42), was evaluated for its in vitro anticancer activity against liver cancer HepG2 and breast cancer MCF7 cell lines, unfortunately, the compound demonstrated low activity [98].
The synthesis of [1,2,4]triazolo[1,5-a]quinazolinone derivatives 209, 211 by reaction of hydrazono-nitriles with hydroxylamine hydrochloride in presence of sodium acetate was described [99] (Scheme 43). The influence of the nature of the heterocyclic fragment in hydrazononitriles 208 has been demonstrated. Benzimidazolyl-containing hydrazono-nitrile 208a reacts with hydroxylamine hydrochloride upon boiling in DMF for 3 h leading to triazolo[1,5-a]quinazolinone 209, whereas 208b containing a benzothiazole fragment under the same conditions forms an amidoxime derivative 210, that did not cyclize even over a longer period (48 h). However, heating compound 210 in glacial acetic acid at reflux yielded the cyclized product 211 in good yield.
2-Phenoxybenzo[g][1,2,4]triazolo[1,5-a]quinazolin-5(4H)-one 212 was synthesized in good yield by the reaction of diphenyl-N-cyanoimidocarbonate with hydrazinonaphthoic acid (Scheme 44) [100]. Alkylation, chlorination and thionation of the lactam group in this compound led to the range of derivatives 213–215. Compounds 212–215 were characterized by NMR and HREI-MS analyses.
A series of 2-methylthio-benzo[g][1,2,4]triazolo[1,5-a]quinazolines 216, obtained according to Scheme 44, was tested against a variety of Gram-positive bacterial species, Gram-negative bacteria and against ten types of fungi [101]. Most derivatives showed significant antimicrobial activity against six bacterial and six fungal strains. Al-Salahi et al [101] concluded that the promising compounds could be employed as useful scaffolds for building of new derivatives with more potent antimicrobial effects.
Various substituted 1,2,4-triazolo[1,5-a]quinazolines 217–220 (Figure 12) were synthesized [102], and evaluation of their biological effects on heart rate and blood pressure was performed. Compounds 217 were obtained by interaction of the corresponding 2-hydrazino-benzoic acid with diphenyl-N-cyanoimidocarabonate, the modification of structure 217 led to triazoloquinazolines 218–220. The structures of derivatives 217b, 217d and 2-benzyloxy-1,2,4-triazolo[1,5-a]quinazolin-5-one was confirmed later by X-ray data [103,104].
In vivo antihypertensive activity study of the compounds 217–220 was performed for rats and mice. The results demonstrated that derivatives 217a,c and 218b were found to increase the heart rate (33, 14 and 40%, respectively), whereas derivatives 217d and 218a decrease the heart rate (23 and 15%, respectively). Moreover, compound 217c decreases arterial pressure in the rats (6.5 mm H). Derivative 219 has shown to decrease blood pressure (7.8 mm H) and tachycardia effects. Compound 220a decreases the heart rate (5.9%), at the same time derivative 220b increases the heart rate (28.5%), both these compounds demonstrate decreasing arterial pressure (6.5 mm H). Al-Salahi et al [102] suggested that certain compounds could be tested as drugs blocking adrenaline receptors, while others appear to stimulate heart function or could be altered to improve blood pressure reduction.
A series of some previously obtained methylsulfanyl-triazoloquinazoline derivatives 221–223 (21 compounds, Figure 13) [105] have been tested against Coxsackievirus B4 and Adenovirus type 7 [106]. The antiviral activity of these compounds against Coxsackie B4 and Adeno type 7 was evaluated in BGM and Hep-2 cell lines. Most tested derivatives were found to possess weak effects on Coxsackie B4 and Adeno type 7, compounds 223 proved to be the most promising. High reduction percentages were observed to viral titers 63.3–83.3% by compounds 223с, 223b, and 223d, which indicates promising effect against Coxsackie B4. The reduction of Adeno type 7 titters were 63.3, 50, and 66.6% demonstrated by 223b, 223с, and 223d, respectively.
Screening and evaluation of antioxidant activity was conducted for the expanded series of 1,2,4-triazolo[1,5-a]quinazoline derivatives 221–226 (40 compounds) [107]. The antioxidant activity of 1,2,4-triazolo[1,5-a]quinazolines was evaluated using the 1,1-diphenyl-2-picryl-hydrazyl (DPPH) reagent. The obtained findings were compared with a standard synthetic antioxidant, 2,6-bis(1,1-dimethylethyl)-4-methylphenol (BHT). All tested compounds exhibited antioxidant activity ranged from weak to moderate and high. It was found that triazoloquinazolines 224, 225a,b, 226a,b demonstrated the highest capacity to deplete DPPH, from them derivatives 224, 226a,b showed percent inhibitions of 85.9, 91.0 and 91.2, respectively, when compared with BHT (93%).
Triazoloquinazolines 227 (14 compounds, Figure 14) were reported as a new class of potent α-glucosidase inhibitors [108]. All compounds were synthesized and characterized previously [102,105]. The structure of derivative 229b was later confirmed by X-ray data [109]. Abuelizz et al [108] assayed the enzymatic inhibitory activity of compounds 227 against α-glucosidase.
Triazoloquinazolines 227 demonstrated significant activity with the IC50 values ranging between 12.70±1.87 and 180.34±1.28 μM. Among them derivatives 231, 230, 229a, 229b and 228 showed the highest inhibitory activity (IC50 = 12.70±1.87, 28.54±1.22, 45.65±4.28, 72.28±4.67, and 83.87±5.12 μM, respectively) in relation to that of acarbose (IC50 = 143.54±2.08 μM) as a reference drug. Molecular modeling study confirmed the importance of binding energy in the stability of complex formed between the docked triazoloquinazolines and the amino acid residues in the active site of the enzyme.
7. Conclusions
Looking at current research articles about the discovery of pyrrolo-, indolo-, and azolo[a]-quinazoline compounds shows that different types of these molecules have caught researchers' interest. Although these compounds exhibit markedly distinct structures, most featured molecules remain either unsubstituted at position 4 of the quinazoline nucleus or incorporate oxo-, amino, aryl, or alkoxy substituents.
Considerable prospects are associated with such synthons for [a]-annelated quinazolines as 2-(3-butenyl)quinazolin-4(3Н)-ones, N′-phenyl-2-aminobenzohydrazides, 2-halogeno-benzamides, 2-halogenobenzaldehydes, 2-aminobenzonitriles, anthranilamide, isatoic anhydride, 2-hydrazinobenzoic acid, 2-aminoquinazolines and 2-hydrazinoquinazolines.
A promising line of research is the discovery of new approaches such as cyclocondensations, three-component carbonylative cyclization, Pd-catalyzed domino synthesis or direct C–H arylation, metal-free base-promoted intramolecular C–N coupling reaction. Some green methods were applied, for example, deep eutectic solvent, mechanochemical activation, graphene oxide as mild heterogeneous carbocatalyst, acid functionalized nanoporous silica (SBA-Pr-SO3H) as a reusable catalyst.
Noteworthy, many [a]-annetaled quinazoline derivatives have specific activities in addition to antitumor activity, for example, antibacterial, hypotensive, antidepressant. Pyrazolo[1,5-a]quinazolines exhibit anxiolytic, antihyperalgesic and anti-inflammatory activity, whereas pyrido[2’,3’:3,4]pyrazolo[1,5-a]quinazoline demonstrate anticancer, antibacterial and antifungal activity. Isoindolo[a]- and imidazo[a]-annelated quinazolines show cytotoxic effect, while benzimidazo[1,2-a]quinazoline derivatives have been reported as anticancer, antimicrobial and antitubercular agents. Different [1,2,4]-triazolo[4,3-a]quinazolines proved to exhibit anticancer, antimicrobial, antitubercular, antidepressant, anticonvulsant, antihistaminic, sedative–hypnotic, vascular disrupting activities. Some [1,2,4]triazolo[1,5-a]quinazoline derivatives demonstrated themselves as antimicrobial, antifungal, hypotensive agents or potent α-glucosidase inhibitors.
We can conclude that quinazolines [4,3-a]-annelated by 1,2,4-triazole cycle exhibit more diverse types of biological activity than other quinazolines [a]-annelated by azoles.
Author Contributions
Conceptualization, V.N.C.; methodology, G.N.L.; writing—original draft preparation, G.N.L.; writing—review and editing, E.V.N.; visualization, E.V.N. All authors have read and agreed to the published version of the manuscript.
Funding
Funding from Ministry of Science and Higher Education of the Russian Federation, projects FEUZ-2023-0021 (Chapters 1, 2, 5, 6) and FUWM-2024-0009 (Chapters 3, 4).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TFA | Trifluoroacetic acid |
| CPT | Camptothecin |
| NIS | N-iodosuccinimide |
| NBS | N-bromosuccinimide |
| NOE | Nuclear Overhauser effect |
| DMSO | Dimethyl sulfoxide |
| TBAB | Tetrabutylammonium bromide |
| PEG | Polyethylene glycol |
| TFE | 2,2,2-Trifluoroethanol |
| LAG | Liquid-assisted grinding |
| HTS | High-throughput screening |
| DMF | N,N-Dimethylformamide |
| GABA | Gamma-aminobutyric acid |
| HCA | Hierarchical Cluster Analysis |
| ERK | Extracellular signal-regulated kinase |
| GBBR | Groebke–Blackburn–Bienaymé reaction |
| MES | Maximal electroshock |
| ATP | Adenosine triphosphate |
| SAR | Structure-Activity Relationship |
| DFT | Density Functional Theory |
| FMO | Frontier molecular orbital |
References
- Shagufta, I.A. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Med. Chem. Commun. 2017, 8, 871–885. [Google Scholar] [CrossRef]
- Halappanavar, V.; Teli, S.; Sannakki, H.B.; Teli, D. Quinazoline scaffold as a target for combating microbial resistance: Synthesis and antimicrobial profiling of quinazoline derivatives. Results in Chemistry 2025, 13, 101955. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.T.; Narendhar, B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.F.; Morris-Natschke, S.L.; Liu, Y.Q.; Guo, X.; Xu, X.S.; Goto, M.; Li, J.C.; Yang, G.Z.; Lee, K.H. Biologically active quinoline and quinazoline alkaloids Part II. Med. Res. Rev. 2018, 38, 1614. [Google Scholar] [CrossRef] [PubMed]
- Gatadi, S.; Lakshmi, T.V.; Nandur, S. 4(3H)-Quinazolinone derivatives: Promising antibacterial drug leads. Eur. J. Med. Chem. 2019, 170, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. Crystallographic analysis, Hirshfeld surface investigation, and DFT calculations of 2-phenoxy-triazoloquinazoline molecule: Implications for drug design. J. Mol. Struct. 2025, 1319, 139436. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. A DFT study and Hirshfeld surface analysis of the molecular structures, radical scavenging abilities and ADMET properties of 2-methylthio(methylsulfonyl)-[1,2,4]triazolo[1,5-a]quinazolines: guidance for antioxidant drug design. Crystals 2023, 13, 1086. [Google Scholar] [CrossRef]
- Auti, P.S.; George, G.; Paul, A.T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Advances 2020, 10, 41353–41392. [Google Scholar] [CrossRef]
- Nosova, E.V.; Lipunova, G.N.; Permyakova, Yu.V.; Charushin, V.N. Quinazolines annelated at the N(3)–C(4) bond: synthesis and biological activity. Eur. J. Med. Chem. 2024, 271, 116411. [Google Scholar] [CrossRef]
- Abuelizz, H.A.; Al-Salahi, R. An overview of triazoloquinazolines: pharmacological significance and recent developments. Bioorg. Chem. 2021, 115, 105263. [Google Scholar] [CrossRef]
- Jabeen, T.; Aslam, S.; Ahmad, M.; ul Haq, A.; Al-Hussain, S.A.; Zaki, M.E.A. Triazoloquinazoline: synthetic strategies and medicinal importance. In: Recent Advances on Quinazoline, IntechOpen, 2023, pp. 1–27.
- Rafiq, A.; Aslam, S.; Ahmad, M.; Saif, M.J.; Al-Hussain, S.A.; Zaki, M.E.A. Recent approaches for the synthesis of imidazoquinazolines and benzimidazoquinazolines. In: Recent Advances on Quinazoline, IntechOpen, 2023, pp. 1–19.
- Garg, M.; Chauhan, M.; Singh, P.K.; Alex, J.M.; Kumar, R. Pyrazoloquinazolines: synthetic strategies and bioactivities. Eur. J. Med. Chem. 2015, 97, 444–461. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, X.; Zhang, Y.; Tang, C.; Feng, B. Progress in synthesis and bioactivity evaluation of pyrazoloquinazolines. Lett. Drug Des. Discov. 2020, 17, 104–113. [Google Scholar] [CrossRef]
- Vaskevych, A.I.; Dekhtyar, M.; Vovk, M. Cyclizations of alkenyl(alkynyl)-functionalized quinazolinones and their heteroanalogues: a powerful strategy for the construction of polyheterocyclic structures. The Chemical Record 2024, 24, e202300255. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascu, F.; Georgescu, F.; Georgescu, E.; Cairad, M.R. Chapter Three — Pyrroloquinolines, Imidazoquinolines, and Pyrroloquinazolines with a Bridgehead Nitrogen. Adv. Heterocycl. Chem. 2019, 129, 155–244. [Google Scholar]
- Rasapalli, S.; Sammeta, V.R.; Murphy, Z.F.; Huang, Y.; Boerth, J.A.; Golen, J.A.; Savinov, S.N. Synthesis of C-ring-substituted vasicinones and luotonins via regioselective aza-Nazarov cyclization of quinazolinonyl enones. Org. Lett. 2019, 21, 9824–9828. [Google Scholar] [CrossRef]
- Rasapalli, S.; Sammeta, V.R.; Murphy, Z.F.; Golen, J.A.; Agama, K.; Pommier, Y.; Savinov, S.N. Design and synthesis of C-aryl angular luotonins via a one-pot aza-Nazarov–Friedlander sequence and their topo-I inhibition studies along with C-aryl vasicinones and luotonins. Bioorg. Med. Chem. Lett. 2021, 41, 127998. [Google Scholar] [CrossRef]
- Vaskevych, A.I.; Savinchuk, N.O.; Vaskevych, R.I.; Rusanov, E.B.; Grygorenko, O.O.; Vovk, M.V. PIFA-Initiated oxidative cyclization of 2-(3-butenyl)quinazolin-4(3H)-ones—an efficient approach to 1-(Hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones. Beilstein J. Org. Chem. 2021, 17, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Vaskevych, A.I.; Savinchuk, N.O.; Vaskevych, R.I.; Rusanov, E.B.; Vovk, M.V. Chalcogenation/Pyrrolo(Pyrido)Annulation of 2-(3-butenyl)quinazolin-4(3H)-ones by Arylsulfenyl(Selenyl) Chlorides. Tetrahedron 2022, 111, 132722. [Google Scholar] [CrossRef]
- Vaskevych, R.I.; Savinchuk, N.O.; Vaskevych, A.I.; Rusanov, E.B.; Bylina, D.V.; Kyrylchuk, A.A.; Vovk, M.V. Proton- and halogen-induced cyclizations of 2-(3-butenyl) quinazolin-4(3H)-ones in the synthesis of pyrrolo[2,1-b]- and pyrrolo[1,2-a]quinazolinone derivatives. J. Heterocyclic Chem. 2023, 60, 431–448. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, N.; Banerjee, P. Regioselective Bronsted Acid-Catalyzed Annulation of Cyclopropane Aldehydes with N'-Aryl Anthranil Hydrazides: Domino Construction of Tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones. J. Org. Chem. 2020, 85, 3393–3406. [Google Scholar] [CrossRef] [PubMed]
- Grinev, V.S.; Amalchieva, О.А.; Egorova, А.Yu. Features of Reaction of Substituted 4-Oxobutanoic Acids and 3Н-Furan-2-ones with 1,3-binucleophiles. Russ. J. Org. Chem. 2017, 53, 1669–1674. [Google Scholar] [CrossRef]
- Ferraris, D.V. Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J. Med. Chem. 2010, 53, 4561. [Google Scholar] [CrossRef]
- Kumar, K.S.; Kumar, P.M.; Kumar, K.A.; Sreenivasulu, M.; Jafar, A.A.; Rambabu, D.; Krishna, G.R.; Reddy, C.M.; Kapavarapu, R.; Shivakumar, K.; Parsa, K.V.L.; Pal, M. A new three-component reaction: green synthesis of novel isoindolo[2,1-a]quinazoline derivatives as potent inhibitors of TNF-α. Chem. Commun. 2011, 57, 5010–5012. [Google Scholar] [CrossRef]
- Kotipalli, T.; Kavala, V.; Janreddy, D.; Kuo, C.-W.; Kuo, T.-S.; Huang, H.-N.; He, C.-H.; Yao, C.-F. Syntheses of indolo[1,2-a]quinazolinone derivatives via palladium-catalyzed intramolecular C–H amidation. RSC Adv. 2014, 4, 2274–2283. [Google Scholar] [CrossRef]
- Abe, T.; Takahashi, Y.; Matsubara, Y.; Yamada, K. Ullmann N-Arylation/2-Amidation Cascade by Self-Relay Copper Catalysis: One-pot Synthesis of Indolo[1,2-a]quinazolinones. Org. Chem. Front. 2017, 4, 2124–2127. [Google Scholar] [CrossRef]
- Badigenchala, S.; Sekar, G. NIS-mediated cross-coupling of C(sp²)–H and N–H bonds: a transition metal-free approach towards indolo[1,2-a]quinazolinones. J. Org. Chem. 2017, 82, 7657–7665. [Google Scholar] [CrossRef]
- Jiang, M.; Xiang, H.; Zhu, F.; Xu, X.; Deng, L.; Yang, C. Efficient Pd-catalyzed domino synthesis of 1-phenyl-1H-indol-2-amine and 5-amino-indolo[1,2-a]quinazoline derivatives. Org. Biomol. Chem. 2015, 13, 10122. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Palnati, G.R.; Dodda, R.P.; Avula, S.R.; Swami, P. Studies on novel synthetic methodologies, part XII: an efficient one-pot access to 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-diones and 5-phenylisoindolo[2,1-a]quinazolin-11(6aH)-ones. Synlett 2013, 24, 105–113. [Google Scholar] [CrossRef]
- Reddy, G.R.; Reddy, T.R.; Chary, R.G.; Joseph, S.C.; Mukherjee, S.; Pal, M. β-Cyclodextrin-mediated MCR in water: synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives under microwave irradiation. Tetrahedron Lett. 2013, 54, 6744–6746. [Google Scholar] [CrossRef]
- Esmaeili-Marandi, F.; Saeedi, M.; Yavari, I.; Mahdavi, M.; Shafiee, A. Synthesis of novel isoinodolo[2,1-a]quinazolinedione derivatives containing a 1,2,3-triazole ring system. Helv. Chim. Acta 2016, 99, 37–40. [Google Scholar] [CrossRef]
- Mahdavi, M.; Lotfi, V.; Saeedi, M.; Kianmehr, E.; Shafiee, A. Synthesis of novel fused quinazolinone derivatives. Mol. Divers. 2016, 20, 677–685. [Google Scholar] [CrossRef]
- Madhubabu, M.V.; Shankar, R.; Reddy, G.R.; Rao, T.S.; Rao, M.V.B.; Akula, R. Metal-catalyst-free green and efficient synthesis of five and six-membered fused N-heterocyclic quinazoline derivatives. Tetrahedron Lett. 2016, 57, 5033–5037. [Google Scholar] [CrossRef]
- Abbasian, S.; Kabirifard, H.; Mahdavi, M. The synthesis of 2,3-dihydroquinazoline-4(1H)-one and dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives in the presence of imidazolium ionic liquid sulfonic acid functionalized SBA-15: a novel feature of SBA-15. Arkivoc 2018, III, 302–314. [Google Scholar] [CrossRef]
- Rayatzadeh, A.; Haghipour, S. Efficient synthesis of 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives catalyzed by functionalized nanoporous silica. Monatshefte für Chemie 2021, 152, 103–107. [Google Scholar] [CrossRef]
- Devi, R.V.; Garande, A.M.; Bhate, P.M.; Sirisha, K.; Ganapathi, T. Synthesis of isoindolo[2,1-a]quinazoline, isoindolo[2,1-a]pyrrolo[2,1-c]quinoxalinone, and indolo[1,2-a]isoindolo[1,2-c]quinoxalinone derivatives in a deep eutectic solvent. Synlett 2016, 27, 2807–2810. [Google Scholar] [CrossRef]
- Khachatryan, D.S.; Belus, S.K.; Misyurin, V.A.; Baryshnikova, M.A.; Kolotaev, A.V.; Matevosyan, K.R. Synthesis and properties of 1,2-dihydro-4(3H)-quinazolinones. Russ. Chem. Bull. 2017, 66, 1044–1058. [Google Scholar] [CrossRef]
- Lohar, T.; Mane, A.; Kamat, S.; Kumbhar, A.; Salunkhe, R. Trifluoroethanol and liquid-assisted grinding method: a green catalytic access for multicomponent synthesis. Res. Chem. Intermed. 2017, 44, 1919–1933. [Google Scholar] [CrossRef]
- Jin, R.-Z.; Zhang, W.-T. ; Zhou, Yu-J. ; Wang, X.-S. Iodine-catalyzed synthesis of H-phthalazino[1,2-b]quinazoline and isoindolo[2,1-a]quinazoline derivatives via chemoselective reaction of 2-aminobenzohydrazide and 2-formylbenzoic acid in ionic liquids. Tetrahedron Lett. 2016, 57, 2515–2519. [Google Scholar]
- Bodhak, C.; Hazra, S.; Pramanik, A. Graphene oxide: an efficient carbocatalyst for the facile synthesis of isoindolo[2,1-a]quinazoline-5,11-diones via domino condensation under solvent-free conditions. ChemistrySelect 2018, 3, 7707–7712. [Google Scholar] [CrossRef]
- Guo, S.; Zhai, J.; Wang, F.; Fan, X. One-pot three-component selective synthesis of isoindolo[2,1-a]quinazoline derivatives via a palladium-catalyzed cascade cyclocondensation/cyclocarbonylation sequence. Org. Biomol. Chem. 2017, 15, 3674–3680. [Google Scholar] [CrossRef] [PubMed]
- Kolotaev, A.V.; Matevosyan, K.R.; Osipov, V.N.; Khachatryan, D.S. Synthesis of new quinazoline-containing hydroxamic acids as potential HDAC/VEGFR inhibitors. Unusual rearrangements with pyrrolidone ring opening and dehydration of 3-N-hydroxyquinazoline fragment containing tetracycles. Tetrahedron Lett. 2019, 60, 151315. [Google Scholar] [CrossRef]
- Mondal, M.A.; Mondal, S.; Khan, A.A. Mechanistic insight into the acid-catalyzed, one-pot synthesis of isoindole-fused quinazolin-4-ones. J. Chem. Sci. 2020, 132, 63–68. [Google Scholar] [CrossRef]
- Orvieto, F.; Branca, D.; Giomini, C. Identification of substituted pyrazolo[1,5-a]quinazolin-5(4H)-one as potent poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Bioorg. Med. Chem. 2009, 19, 4196–4200. [Google Scholar] [CrossRef]
- Petrov, A.; Kasatochkin, A. Synthesis of 6,7,8,9-tetrahydropyrazolo[1,5-a]quinazolines containing a 5-phenylamine fragment. Russ. J. Org. Chem. 2013, 49, 1250–1252. [Google Scholar] [CrossRef]
- Ferraguti, F.; Shigemoto, R. Metabotropic Glutamate Receptors. Cell. Tissue Res. 2006, 326, 483–504. [Google Scholar] [CrossRef]
- Taliani, S.; Pugliesi, I.; Barresi, E. Phenylpyrazolo[1,5-a]quinazolin-5(4H)-one: a suitable scaffold for the development of noncamptothecin topoisomerase I (Top1) inhibitors. J. Med. Chem. 2013, 56, 7458–7462. [Google Scholar] [CrossRef] [PubMed]
- Roopan, S.M.; Palaniraja, J. Synthesis of indazoloquinazoline system (microreview). Chem. Heterocyclic Comp. 2016, 52, 93–95. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, L.; Wang, Z.; Fan, X. Water-mediated selective synthesis of pyrazolo[1,5-a]quinazolin-5(4H)-ones and [1,2,4]triazolo[1,5-a]quinazolin-5(4H)-one via copper-catalyzed cascade reactions. Synth. Commun. 2015, 45, 2426–2435. [Google Scholar] [CrossRef]
- Gnanasekaran, K.K.; Muddala, N.P.; Bunce, R.A. Pyrazoloquinazolinones and pyrazolopyridopyrimidinones by a sequential N-acylation–SNAr reaction. Tetrahedron Lett. 2015, 56, 1367–1369. [Google Scholar] [CrossRef]
- Annareddygari, S.; Kasireddy, V.R.; Reddy, J. Transition-metal-free N-arylation: a general approach to aza-fused poly-heteroaromatics. J. Heterocyclic Chem. 2019, 56, 3267–3276. [Google Scholar] [CrossRef]
- Gao, Q.; Guo, Y.; Cao, P.; Fan, G.; Xu, Y. Regioselective synthesis of indazolo[2,3-a]quinazolines enabled by I₂/S-facilitated annulation relay dehydrogenative aromatization of cyclohexanones. Chem. Commun. 2023, 59, 13835–13838. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, T.-Q.; Li, H.; Zhang, G.; Wu, X.-A.; Yang, L.; Peng, Y.-L.; Zou, J.; Li, L.-L.; Xiang, R.; Yang, S.-Y. Discovery of pyrazolo[1,5-a]pyrimidine-3-carbonitrile derivatives as a new class of histone lysine demethylase 4D (KDM4D) inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3201–3204. [Google Scholar] [CrossRef]
- Kovács, D.; Molnár-Tóth, J.; Blaskó, G.; Fejes, I.; Nyerges, M. Synthesis of new pyrazolo[1,5-a]quinazoline derivatives. Synth. Commun. 2015, 45, 1675–1680. [Google Scholar] [CrossRef]
- Guerrini, G.; Ciciani, G.; Ciattini, S.; Crocetti, L.; Daniele, S.; Martini, C.; Melani, F.; Vergelli, C.; Giovannoni, M.P. Pyrazolo[1,5-a]quinazoline scaffold as 5-deaza analogue of pyrazolo[5,1-c][1,2,4]benzotriazine system: synthesis of new derivatives, biological activity on GABAA receptor subtype and molecular dynamic study. J. Enzym. Inhib. Med. Chem. 2016, 31, 195. [Google Scholar] [CrossRef]
- Guerrini, G.; Ciciani, G.; Crocetti, L.; Daniele, S.; Ghelardini, C.; Giovannoni, M.P.; Iacovone, A.; Mannelli, L.D.C.; Martini, C.; Vergelli, C. Identification of a new pyrazolo[1,5-a]quinazoline ligand highly affine to γ-aminobutyric type A (GABAA) receptor subtype with anxiolytic-like and antihyperalgesic activity. J. Med. Chem. 2017, 60, 9691–9702. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Saleh, N.M.; Kadh, M.S.; Abou-Amra, E.S. New fused pyrazolopyrimidine derivatives; heterocyclic styling, synthesis, molecular docking and anticancer evaluation. J. Heterocyclic Chem. 2020, 57, 2704–2721. [Google Scholar] [CrossRef]
- Guerrini, G.; Vergelli, C.; Cantini, N.; Giovannoni, M.P.; Daniele, S.; Mascia, M.P.; Martini, C. Synthesis of new GABAA receptor modulator with pyrazolo[1,5-a]quinazoline (PQ) scaffold. Int. J. Mol. Sci. 2019, 20, 1438–1458. [Google Scholar] [CrossRef]
- Guerrini, G.; Crocetti, L.; Daniele, S.; Lacovone, A.; Cantini, N.; Martini, C.; Melani, F.; Vergelli, C.; Giovannoni, M.P. New 3,6-disubstituted pyrazolo[1,5-a]quinazolines as ligands to GABAA receptor subtype. J. Heterocyclic Chem. 2019, 56, 1571–1589. [Google Scholar] [CrossRef]
- Bruni, F.; Chimichi, S.; Cosimelli, B.; Costanzo, A.; Guerrini, G.; Selleri, S. A new entry to pyrazolo[1,5-a]pyrido[3,4-e]pyrimidine derivatives. Heterocycles 1990, 31, 1141. [Google Scholar] [CrossRef]
- Crocetti, L.; Guerrini, G.; Cantini, N.; Vergelli, C.; Melani, F.; Mascia, M.P.; Giovannoni, M.P. 'Proximity frequencies': a new parameter to evaluate the profile of GABAAR modulators. Bioorg. Med. Chem. Lett. 2021, 34, 127755. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, L.; Guerrini, G.; Melani, F.; Vergelli, C.; Mascia, M.P.; Giovannoni, M.P. GABAA receptor modulators with a pyrazolo[1,5-a]quinazoline core: synthesis, molecular modelling studies and electrophysiological assays. Int. J. Mol. Sci. 2022, 23, 13032. [Google Scholar] [CrossRef]
- Crocetti, L.; Guerrini, G.; Melani, F.; Mascia, M.P.; Giovannoni, M.P. 3,8-Disubstituted pyrazolo[1,5-a]quinazoline as GABAA receptor modulators: synthesis, electrophysiological assays, and molecular modelling studies. Int. J. Mol. Sci. 2024, 25, 10840. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, W.; Zhang, G.; Fang, Z.; Chen, X.; Li, L. Design, synthesis, and biological screening of a series of pyrazolo[1,5-a]quinazoline derivatives as SIRT6 activators. Eur. J. Pharm. Sci. 2023, 185, 106424. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, W.; Huang, S.; Zhang, H.; Lin, G.; Li, H.; Qiao, J.; Li, L.; Yang, S. Discovery of potent small-molecule sirt6 activators: structure-activity relationship and anti-pancreatic ductal adenocarcinoma activity. J. Med. Chem. 2020, 63, 10474–10495. [Google Scholar] [CrossRef] [PubMed]
- Crocetti, L.; Khlebnikov, A.I.; Guerrini, G.; Schepetkin, I.A.; Melani, F.; Giovannoni, M.P.; Quinn, M.T. Anti-inflammatory activity of pyrazolo[1,5-a]quinazolines. Molecules 2024, 29, 2421. [Google Scholar] [CrossRef]
- Shlenev, R.M.; Filimonov, S.I.; Tarasov, A.V.; Danilova, A.S.; Agat’ev, P.A. Synthesis of new sulfonamide derivatives of thiazolo[3,2-a]quinazolin-5-one. Russ. J. Org. Chem. 2016, 52, 69–75. [Google Scholar] [CrossRef]
- Deng, X.Q.; Xiao, C.R.; Wei, C.X.; Quan, Z.S. Synthesis and anticonvulsant activity of substituted [1,2,4]triazolo[4,3-a]quinazolines. Chin. J. Org. Chem. 2011, 31, 2082–2087. [Google Scholar]
- Zhang, H.-J.; Jin, P.; Wang, S.-B.; Li, F.-N.; Guan, L.-P.; Quan, Z.-S. Synthesis and anticonvulsant activity evaluation of 4-phenyl[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one and its derivatives. Arch. Pharm. Chem. Life Sci. 2015, 348, 564–574. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Wang, S.-B.; Quan, Z.-S. Synthesis and antidepressant activities of 4-(substituted-phenyl)triazolo[1,5-a]quinazolin-5(4H)-ones and their derivatives. Mol. Divers. 2015, 19, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, V.; Raja, S.V.; Dhanabal, K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg. Med. Chem. 2007, 15, 235–241. [Google Scholar] [CrossRef]
- Yu, S.; Danylchenko, O.G.; Drushlyak, S.S.; Kovalenko, S.M. Formation of 1-methyl[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones by reaction of 2-hydrazinoquinazolin-4(3H)-ones with acetylacetone. Heterocycl. Commun. 2015, 21, 195–197. [Google Scholar]
- Gobinath, M.; Subramanian, N.; Alagarsamy, V.; Nivedhitha, S.; Solomon, V.R. Synthesis of 1-substituted-4-(pyridin-4-yl)[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones as a new class of H₁-antihistaminic agents. Trop. J. Pharm. Res. 2015, 14, 271–277. [Google Scholar] [CrossRef]
- Gobinath, M.; Subramanian, N.; Alagarsamy, V.; Nivedhitha, S.; Solomon, V.R. Design and synthesis of 1-substituted-4-(4-nitrophenyl)[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones as a new class of antihistaminic agents. Russ. J. Bioorg. Chem. 2020, 46, 403–408. [Google Scholar] [CrossRef]
- Hu, W.; Liao, T.; Deng, L.; Sun, H.; Zhou, Z.; Chai, H.; Zhao, C. Synthesis, crystal structure, and density functional theory study of a new compound 4-(2-chlorobenzyl)-1-(5-fluoro-2-hydroxy-3-(thiomorpholinomethyl)phenyl)[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one. J. Heterocyclic Chem. 2021, 58, 2102–2108. [Google Scholar] [CrossRef]
- Zhou, Z.-X.; Wu, Q.-M.; Huang, Z.-Y.; Yu, D.-H.; Lu, H.-G. Synthesis, crystal structure, DFT, molecular docking and antitumor activity of 4-(2-chlorobenzyl)-1-(5-fluoro-2-hydroxy-3-((4-methylpiperidin-1-yl)methyl)phenyl)[1,2,4]-triazolo[4,3-a]quinazolin-5(4H)-one. Res. Chem. Intermed. 2021, 47, 3609–3627. [Google Scholar] [CrossRef]
- Luo, R.; Wang, Z.; Luo, D.; Qin, Y.; Zhao, C.; Yang, D.; Lu, T.; Zhou, Z.; Huang, Z. Design, synthesis, and biological evaluation of novel triazoloquinazolinone derivatives as SHP2 protein inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Yadava, U.; Upadhyay, A.; Sharma, M.L. Synthesis, biological evaluation and molecular docking studies of novel quinazolinones as antitubercular and antimicrobial agents. Bioorg. Chem. 2021, 108, 104611. [Google Scholar] [CrossRef]
- Švajger, U.; Horvat, Ž.; Knez, D.; Rožman, P.; Turk, S.; Gobec, S. New antagonists of toll-like receptor 7 discovered through 3D ligand-based virtual screening. Med. Chem. Res. 2015, 24, 362–371. [Google Scholar] [CrossRef]
- Driowya, M.; Leclercq, J.; Verones, V.; Barczyk, A.; Lecoeur, M.; Renault, N.; Flouquet, N.; Ghinet, A.; Berthelot, P.; Lebegue, N. Synthesis of triazoloquinazolinone based compounds as tubulin polymerization inhibitors and vascular disrupting agents. Eur. J. Med. Chem. 2016, 115, 393–405. [Google Scholar] [CrossRef]
- Cheng, G.; Mei, X.-B.; Yan, Y.-Y.; Chen, J.; Zhang, B.; Li, J.; Dong, X.-W.; Lin, N.-M.; Zhou, Y.-B. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch. Pharm. Chem. Life Sci. 2019, 352, e1800374. [Google Scholar] [CrossRef]
- Álvarez, C.N.; Park, J.-E.; Toti, K.S.; Xia, Y.; Krausz, K.W.; Rai, G.; Bang, J.K.; Gonzalez, F.J.; Jacobson, K.A.; Lee, K.S. Identification of a new heterocyclic scaffold for inhibitors of the polo-box domain of polo-like kinase 1. J. Med. Chem. 2020, 63, 14087–14117. [Google Scholar] [CrossRef]
- Hassan, A.Y.; Sarg, M.T.; Bayoumi, A.H.; El-Deebb, M.A. Synthesis and anticancer evaluation of some novel 5-amino[1,2,4]triazole derivatives. J. Heterocyclic Chem. 2018, 55, 1450–1478. [Google Scholar] [CrossRef]
- Abdelhamid, I.A.; Abdelmoniem, A.M.; Ghozlan, S.A.S.; Butenschön, H. Hydrazononitriles as precursors for 4-aminotriazoles and 3-aminoisoxazoles: one pot synthesis of triazolo[1,5-a]quinazoline derivatives. J. Heterocyclic Chem. 2016, 53, 1251–1258. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Raja, S.V.; Dhanabal, K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg. Med. Chem. 2007, 15, 235–241. [Google Scholar] [CrossRef]
- Yu, S.; Danylchenko, O.G.; Drushlyak, S.S.; Kovalenko, S.M. Formation of 1-methyl[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones by reaction of 2-hydrazinoquinazolin-4(3H)-ones with acetylacetone. Heterocycl. Commun. 2015, 21, 195–197. [Google Scholar] [CrossRef]
- Gobinath, M.; Subramanian, N.; Alagarsamy, V.; Nivedhitha, S.; Solomon, V.R. Synthesis of 1-Substituted-4-(Pyridin-4-yl) [1,2,4]Triazolo[4,3-a]Quinazolin-5(4H)-ones as a New Class of H₁-Antihistaminic Agents. Trop. J. Pharm. Res. 2015, 14, 271–277. [Google Scholar] [CrossRef]
- Gobinath, M.; Subramanian, N.; Alagarsamy, V.; Nivedhitha, S.; Solomon, V.R. Design and Synthesis of 1-Substituted-4-(4-Nitrophenyl)-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones as a New Class of Antihistaminic Agents. Russ. J. Bioorg. Chem. 2020, 46, 403–408. [Google Scholar] [CrossRef]
- Hu, W.; Liao, T.; Deng, L.; Sun, H.; Zhou, Z.; Chai, H.; Zhao, C. Synthesis, crystal structure, and density functional theory study of a new compound 4-(2-chlorobenzyl)-1-(5-fluoro-2-hydroxy-3-(thiomorpholinomethyl)phenyl)[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one. J. Heterocyclic Chem. 2021, 58, 2102–2108. [Google Scholar] [CrossRef]
- Zhou, Z.-X.; Wu, Q.-M.; Huang, Z.-Y.; Yu, D.-H.; Lu, H.-G. Synthesis, crystal structure, DFT, molecular docking and antitumor activity of 4-(2-chlorobenzyl)-1-(5-fluoro-2-hydroxy-3-((4-methylpiperidin-1-yl)methyl)phenyl)[1,2,4]-triazolo[4,3-a]quinazolin-5(4H)-one. Res. Chem. Intermed. 2021, 47, 3609–3627. [Google Scholar] [CrossRef]
- Luo, R.; Wang, Z.; Luo, D.; Qin, Y.; Zhao, C.; Yang, D.; Lu, T.; Zhou, Z.; Huang, Z. Design, synthesis, and biological evaluation of novel triazoloquinazolinone derivatives as SHP2 protein inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 2170–2182. [Google Scholar] [CrossRef]
- Pandey, S.K.; Yadava, U.; Upadhyay, A.; Sharma, M.L. Synthesis, biological evaluation and molecular docking studies of novel quinazolinones as antitubercular and antimicrobial agents. Bioorg. Chem. 2021, 108, 104611. [Google Scholar] [CrossRef]
- Švajger, U.; Horvat, Ž.; Knez, D.; Rožman, P.; Turk, S.; Gobec, S. New antagonists of toll-like receptor 7 discovered through 3D ligand-based virtual screening. Med. Chem. Res. 2015, 24, 362–371. [Google Scholar] [CrossRef]
- Driowya, M.; Leclercq, J.; Verones, V.; Barczyk, A.; Lecoeur, M.; Renault, N.; Flouquet, N.; Ghinet, A.; Berthelot, P.; Lebegue, N. Synthesis of triazoloquinazolinone based compounds as tubulin polymerization inhibitors and vascular disrupting agents. Eur. J. Med. Chem. 2016, 115, 393–405. [Google Scholar] [CrossRef]
- Cheng, G.; Mei, X.-B.; Yan, Y.-Y.; Chen, J.; Zhang, B.; Li, J.; Dong, X.-W.; Lin, N.-M.; Zhou, Y.-B. Identification of new NIK inhibitors by discriminatory analysis-based molecular docking and biological evaluation. Arch. Pharm. Chem. Life Sci. 2019, 352, e1800374. [Google Scholar] [CrossRef]
- Álvarez, C.N.; Park, J.-E.; Toti, K.S.; Xia, Y.; Krausz, K.W.; Rai, G.; Bang, J.K.; Gonzalez, F.J.; Jacobson, K.A.; Lee, K.S. Identification of a new heterocyclic scaffold for inhibitors of the polo-box domain of polo-like kinase 1. J. Med. Chem. 2020, 63, 14087–14117. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Y.; Sarg, M.T.; Bayoumi, A.H.; El-Deebb, M.A. Synthesis and anticancer evaluation of some novel 5-amino[1,2,4]triazole derivatives. J. Heterocyclic Chem. 2018, 55, 1450–1478. [Google Scholar] [CrossRef]
- Abdelhamid, I.A.; Abdelmoniem, A.M.; Ghozlan, S.A.S.; Butenschön, H. Hydrazononitriles as precursors for 4-aminotriazoles and 3-aminoisoxazoles: one pot synthesis of triazolo[1,5-a]quinazoline derivatives. J. Heterocyclic Chem. 2016, 53, 1251–1258. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Marzouk, M.S. Synthesis of novel 2-phenoxy-benzo[g][1,2,4]triazolo[1,5-a]quinazoline and its derivatives starting with diphenyl-N-cyanoimidocarbonate. Russ. J. Gen. Chem. 2016, 86, 1741–1746. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Abuelizz, H.A.; Wadi, M.; El Dib, R.A.; Alotaibi, M.A.; Marzouk, M. Antimicrobial activity of synthesized 2-methylthiobenzo[g][1,2,4]-triazolo[1,5-a]quinazoline derivatives. Medicinal Chem. (Sharjah) 2016, 12, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Al-Salahi, R.; El-Tahir, K.-E.; Alswaidan, I.; Lolak, N.; Hamidaddin, M.; Marzouk, M. Biological effects of a new set 1,2,4-triazolo[1,5-a]quinazolines on heart rate and blood pressure. Chem. Cent. J. 2014, 8, 1–8. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. A DFT study and Hirshfeld surface analysis of the molecular structures, radical scavenging abilities and ADMET properties of 2-methylthio(methylsulfonyl)-[1,2,4]triazolo[1,5-a]quinazolines: guidance for antioxidant drug design. Crystals 2023, 13, 1086–1109. [Google Scholar] [CrossRef]
- Bakheit, A.H.; Abuelizz, H.A.; Al-Salahi, R. Hirshfeld surface analysis and density functional theory calculations of 2-benzyloxy-1,2,4-triazolo[1,5-a]quinazolin-5(4H)-one: a comprehensive study on crystal structure, intermolecular interactions, and electronic properties. Crystals 2023, 13, 1410–1431. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Geffken, D. Synthesis of novel 2-methylsulfanyl-4H-[1,2,4]triazolo[1,5-a]quinazolin-5-one and derivatives. Synth. Commun. 2011, 41, 3512–3523. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Al-Omar, M.A.; Alswaidan, I.; Marzouk, M.; El-Senousy, W.M.; Amr, A.E.-G.E. Antiviral activities of some synthesized methylsulfanyltriazoloquinazoline derivatives. Res. Chem. Intermed. 2015, 41, 151–161. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Anouar, E.-H.; Marzouk, M.; Taie, H.A.A.; Abuelizz, H.A. Screening and evaluation of antioxidant activity of some 1,2,4-triazolo[1,5-a]quinazoline derivatives. Future Med. Chem. 2017, 10, 379–390. [Google Scholar] [CrossRef]
- Abuelizz, H.A.; Anouar, E.-H.; Ahmad, R.; Azman, N.I.I.N.; Marzouk, M.; Al-Salahi, R. Triazoloquinazolines as a new class of potent α-glucosidase inhibitors: in vitro evaluation and docking study. PLoS ONE 2019, 14, e0220379. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Soliman, S.M.; Ghabbour, H.A.; Marzouk, M.; Abdellatif, M.M.; Al-Salahi, R. DFT calculation, Hirshfeld analysis and X-ray crystal structure of some synthesized N-alkylated (S-alkylated) [1,2,4]triazolo[1,5-a]quinazolines. Crystals 2021, 11, 1195–1208. [Google Scholar] [CrossRef]
Figure 1.
Polycyclic quinazolines of nature origin, which can be regarded as prototypes of biologically important [a]-annelated quinazolines.
Figure 1.
Polycyclic quinazolines of nature origin, which can be regarded as prototypes of biologically important [a]-annelated quinazolines.
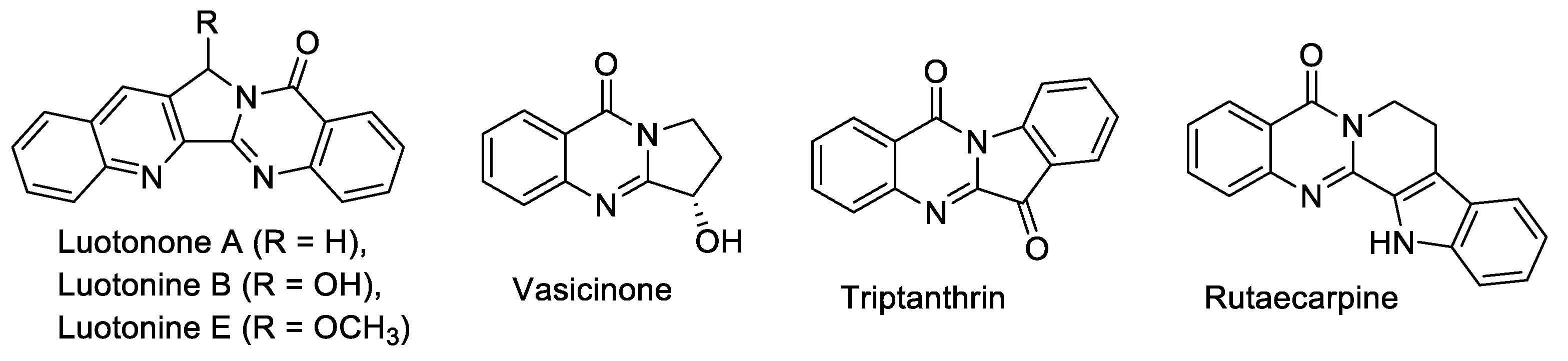
Figure 2.
Types of polycyclic quinazolines considered in the current review.
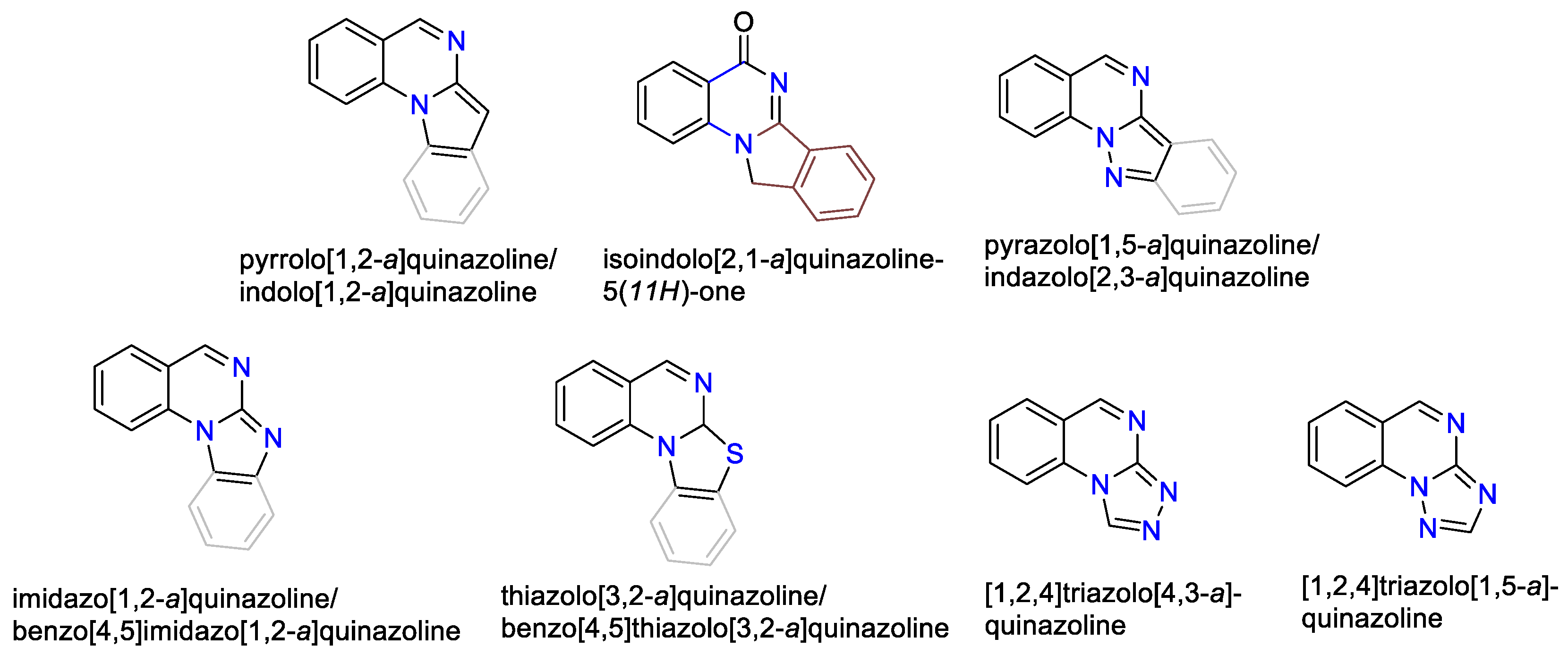
Scheme 1.
Synthetic approach to [a]- or [b]-annelated quinazolinones 2 and 3.
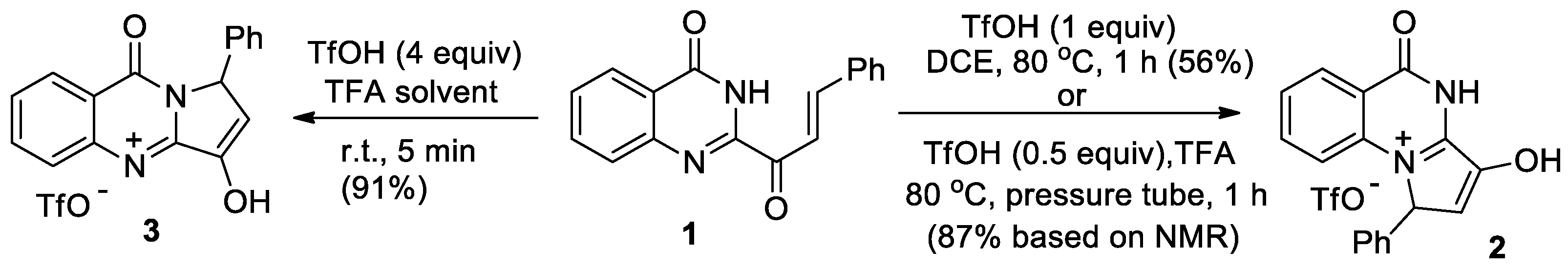
Scheme 2.
Synthesis of angular luotonins 5a-l. 1, 4, 5: R = H (a), 4-OMe (b), 4-Cl (c), 3-OMe (d), 3,4,5-(OMe)3 (e), 4-Br (f), 4-F (g), 4-CF3 (h), 3-Br (i), 4-SMe (j), 2-Me (k), 4-Me (l).
Scheme 2.
Synthesis of angular luotonins 5a-l. 1, 4, 5: R = H (a), 4-OMe (b), 4-Cl (c), 3-OMe (d), 3,4,5-(OMe)3 (e), 4-Br (f), 4-F (g), 4-CF3 (h), 3-Br (i), 4-SMe (j), 2-Me (k), 4-Me (l).
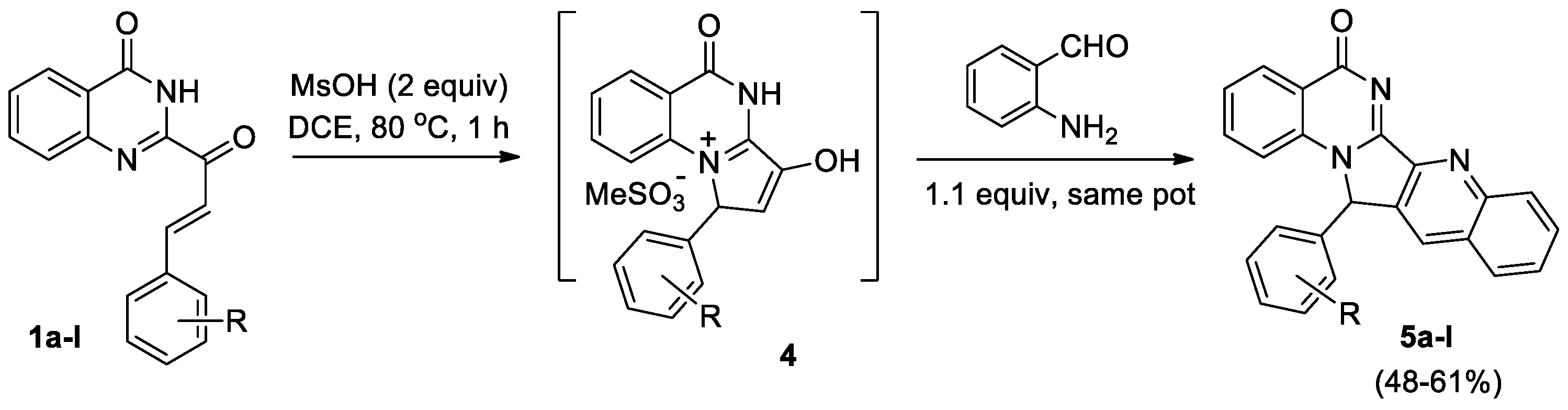
Scheme 3.
Synthesis of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones 7, 8, 9. 7: R = H (a), 5-NO2 (b), 5-F (c), 6-Me (d), 6-OMe (e), 6-Cl (f), 6-NO2 (g), 6,7-(OMe)2 (h), 7-Cl (i), 8-Me (j), 8-Br (k), 8-F (l).
Scheme 3.
Synthesis of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones 7, 8, 9. 7: R = H (a), 5-NO2 (b), 5-F (c), 6-Me (d), 6-OMe (e), 6-Cl (f), 6-NO2 (g), 6,7-(OMe)2 (h), 7-Cl (i), 8-Me (j), 8-Br (k), 8-F (l).
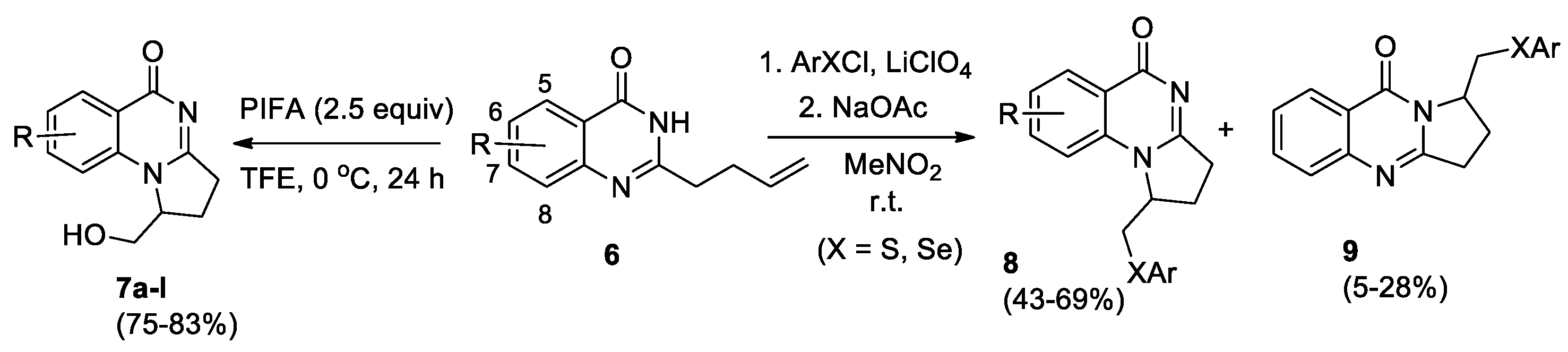
Scheme 4.
Synthetic approach to pyrroloquinazolinones 10, 12, 13. R = H (a), 5-F (b), 6-Cl (c), 7-Cl (d), 8-Me (e), 6-OMe (f), 6-NO2 (g), 6,7-(OMe)2 (h).
Scheme 4.
Synthetic approach to pyrroloquinazolinones 10, 12, 13. R = H (a), 5-F (b), 6-Cl (c), 7-Cl (d), 8-Me (e), 6-OMe (f), 6-NO2 (g), 6,7-(OMe)2 (h).
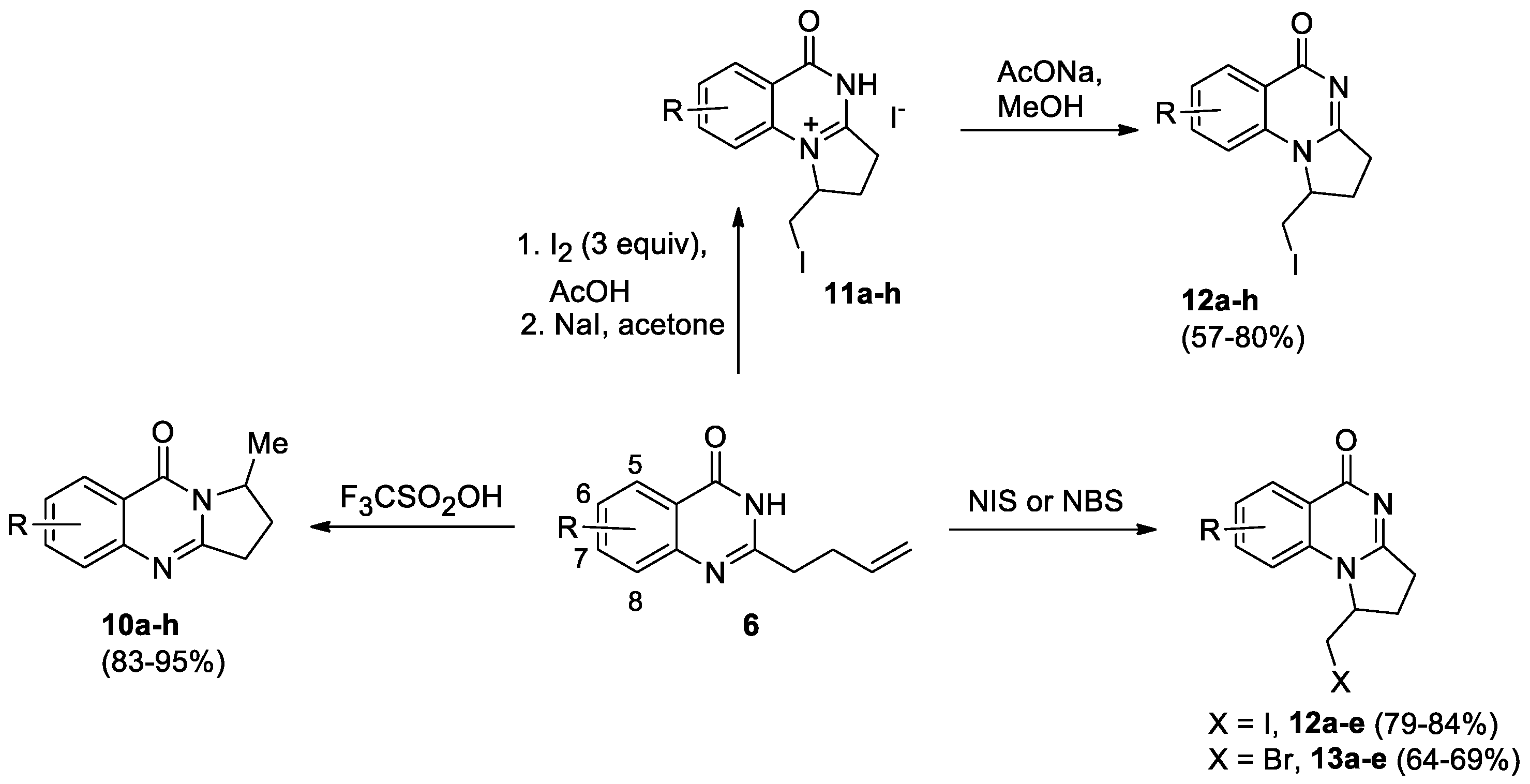
Scheme 5.
Synthesis of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones 16aa-ja.
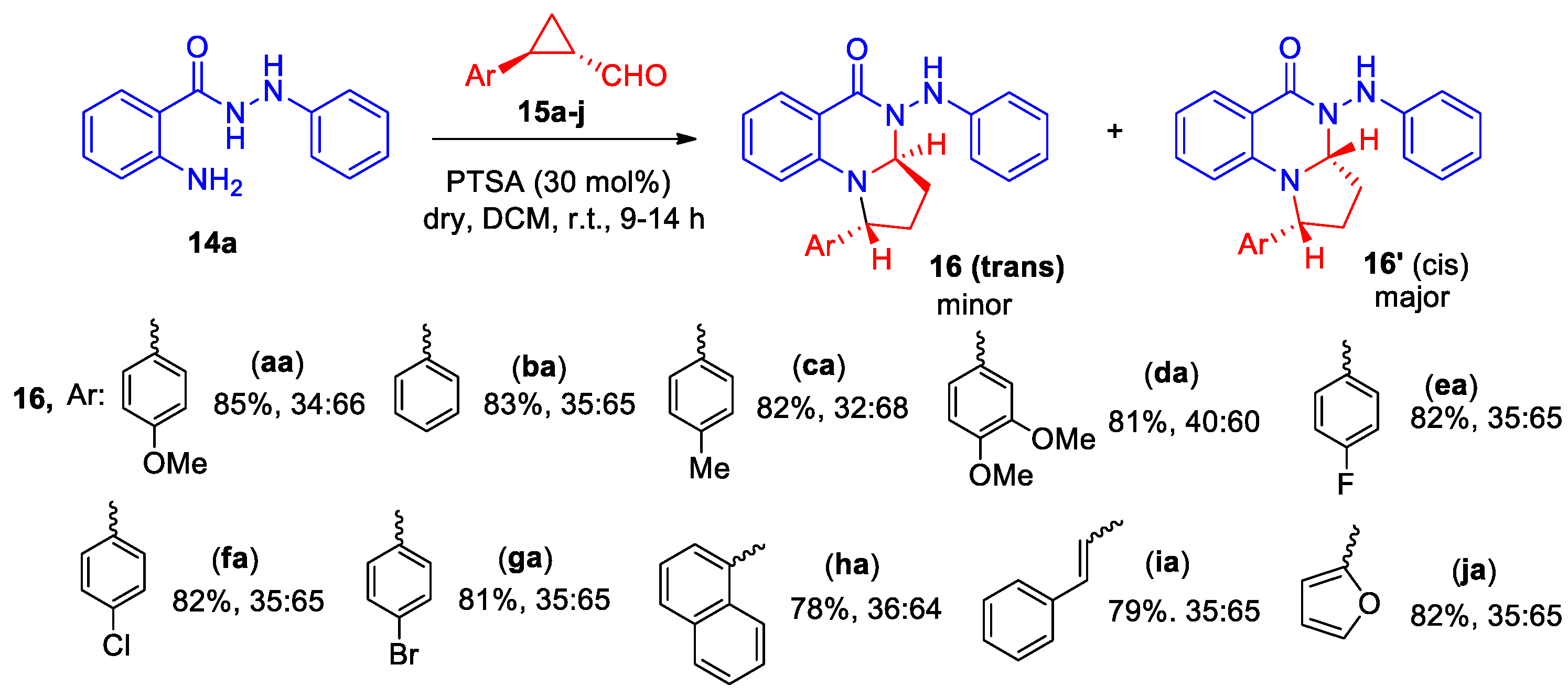
Scheme 6.
Synthesis of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)-ones 16ab-ag.
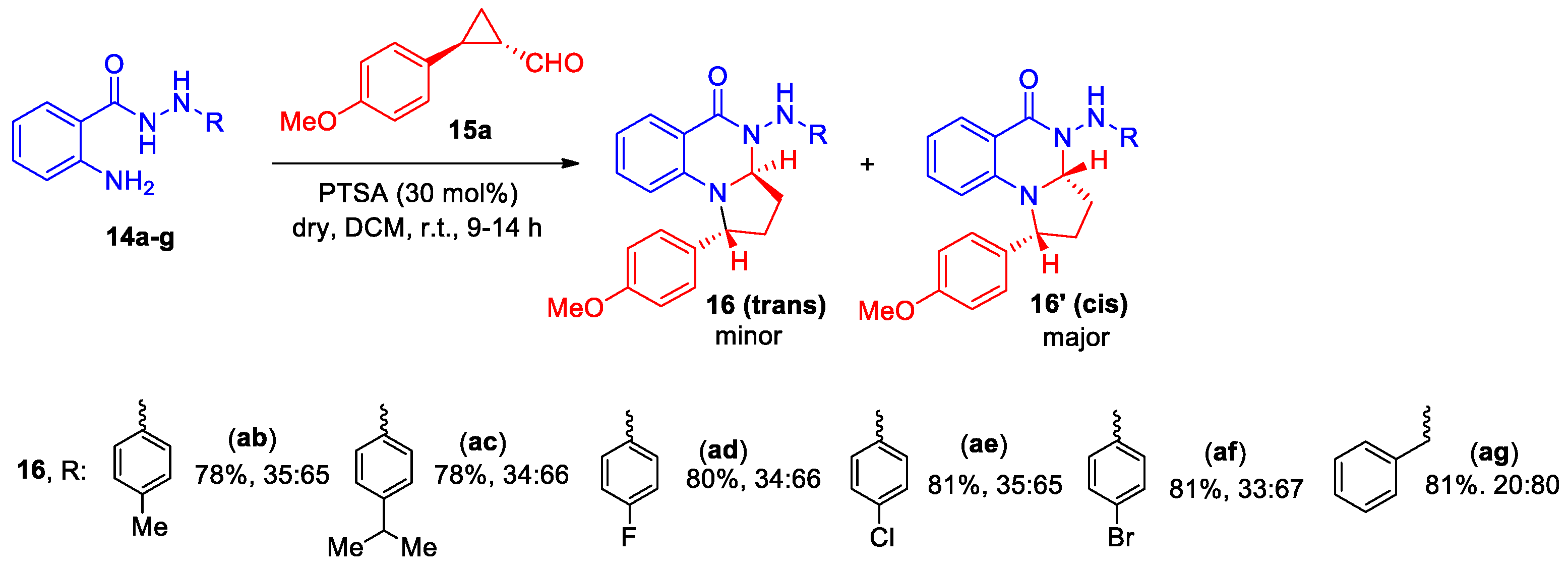
Scheme 7.
This is a figure. Schemes follow the same formatting. Synthesis of 1-R-4,5-dihydropyrrolo[1,2-a]quinazolines 19. R = Ph (a), 4-MeC6H4 (b).
Scheme 7.
This is a figure. Schemes follow the same formatting. Synthesis of 1-R-4,5-dihydropyrrolo[1,2-a]quinazolines 19. R = Ph (a), 4-MeC6H4 (b).
Scheme 8.
Synthetic approach to indolo[1,2-a]quinazolinone derivatives 20. R = H, Cl, (MeO)2; R1 = Me, Et, iso-Pr, Bn, Ph, Ar; R2 = H, OMe, OBn, Cl, Br.
Scheme 8.
Synthetic approach to indolo[1,2-a]quinazolinone derivatives 20. R = H, Cl, (MeO)2; R1 = Me, Et, iso-Pr, Bn, Ph, Ar; R2 = H, OMe, OBn, Cl, Br.
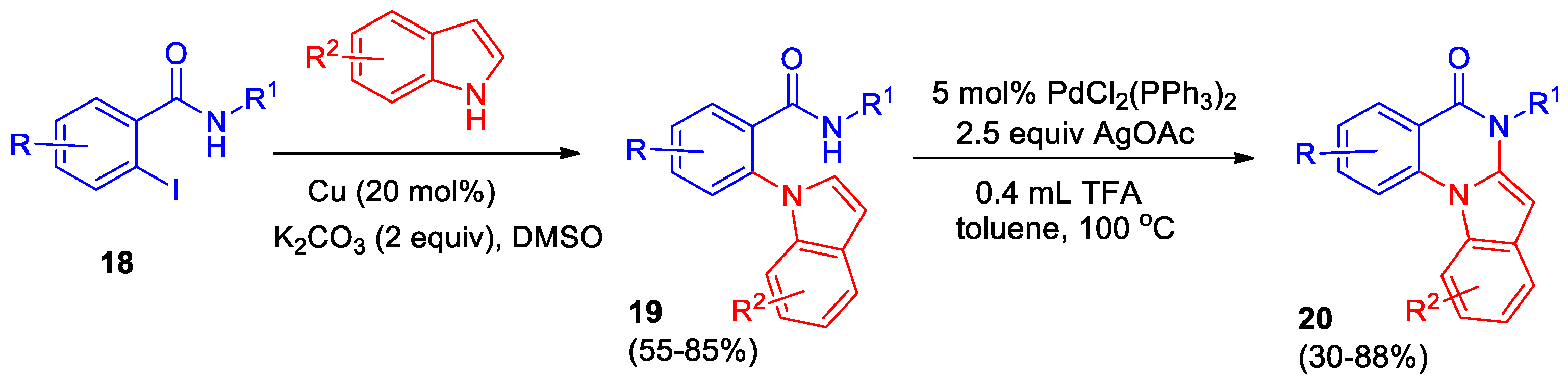
Scheme 9.
Synthesis of indolo[1,2-a]quinazolinones 23 and 25. 23: R = H, MeO, Cl, Br; R1 = H, Me, Bn, Ph; R2 = H, Me, Cl; 25: R = H, Me, Et, Bn, Ar; R1 = H, Me; R2 = H, Me, Et, MeO, Cl, Br.
Scheme 9.
Synthesis of indolo[1,2-a]quinazolinones 23 and 25. 23: R = H, MeO, Cl, Br; R1 = H, Me, Bn, Ph; R2 = H, Me, Cl; 25: R = H, Me, Et, Bn, Ar; R1 = H, Me; R2 = H, Me, Et, MeO, Cl, Br.
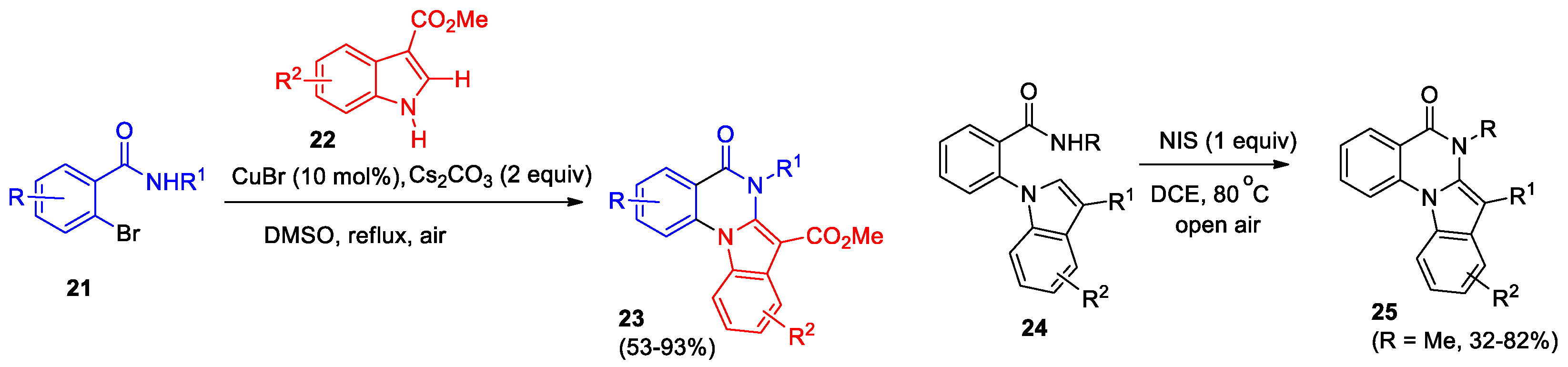
Scheme 10.
Synthesis of 5-amine-indolo[1,2-a]quinazolines 27. R = H, Me, F, CF3; R1 = H, F, MeO; EWG = CN, COOEt.
Scheme 10.
Synthesis of 5-amine-indolo[1,2-a]quinazolines 27. R = H, Me, F, CF3; R1 = H, F, MeO; EWG = CN, COOEt.
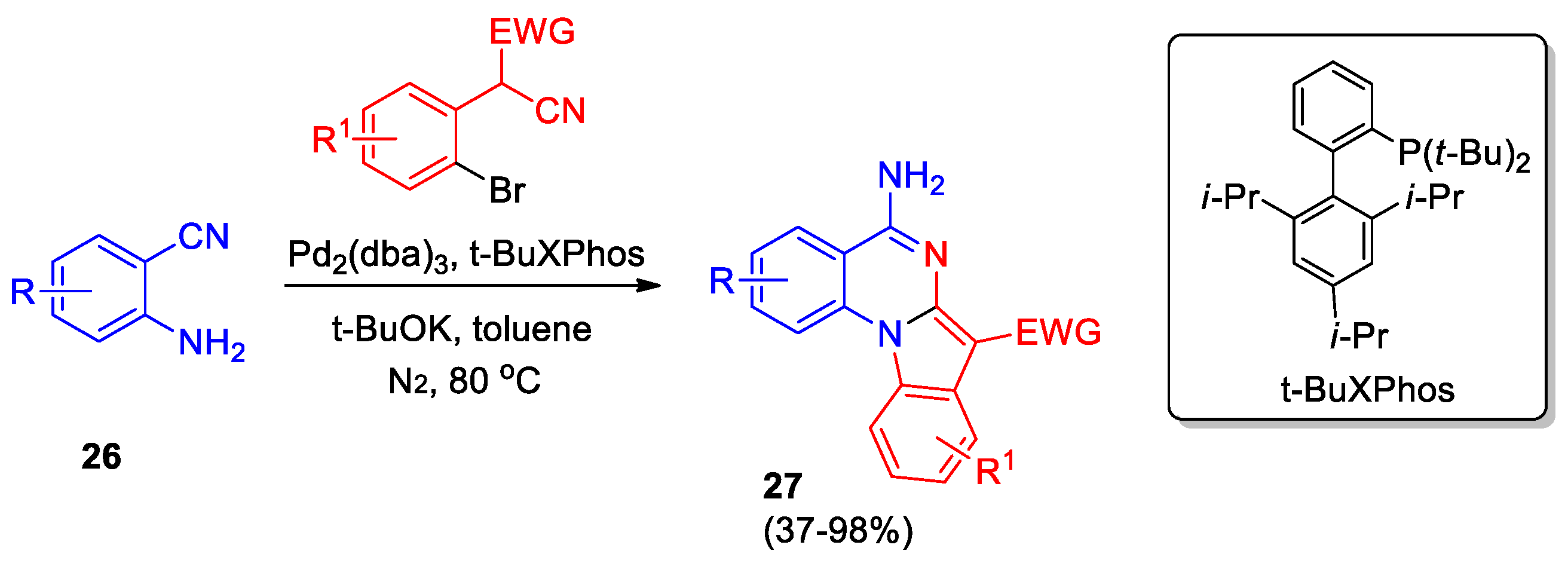
Scheme 11.
Synthetic approach to 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 29, 31. 29: R = H, Me, Et, n-Pr, (CH2)2-OH, (CH2)3-OH, (CH2)3-COOH; 31: Ar = Ph, 2-Me-C6H4, 4-F-C6H4, 2-Cl-C6H4, 2-Br-C6H4, 2-NO2-C6H4, 2,3-Cl2-C6H3, 3,4-Cl2-C6H3.
Scheme 11.
Synthetic approach to 6,6a-dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 29, 31. 29: R = H, Me, Et, n-Pr, (CH2)2-OH, (CH2)3-OH, (CH2)3-COOH; 31: Ar = Ph, 2-Me-C6H4, 4-F-C6H4, 2-Cl-C6H4, 2-Br-C6H4, 2-NO2-C6H4, 2,3-Cl2-C6H3, 3,4-Cl2-C6H3.
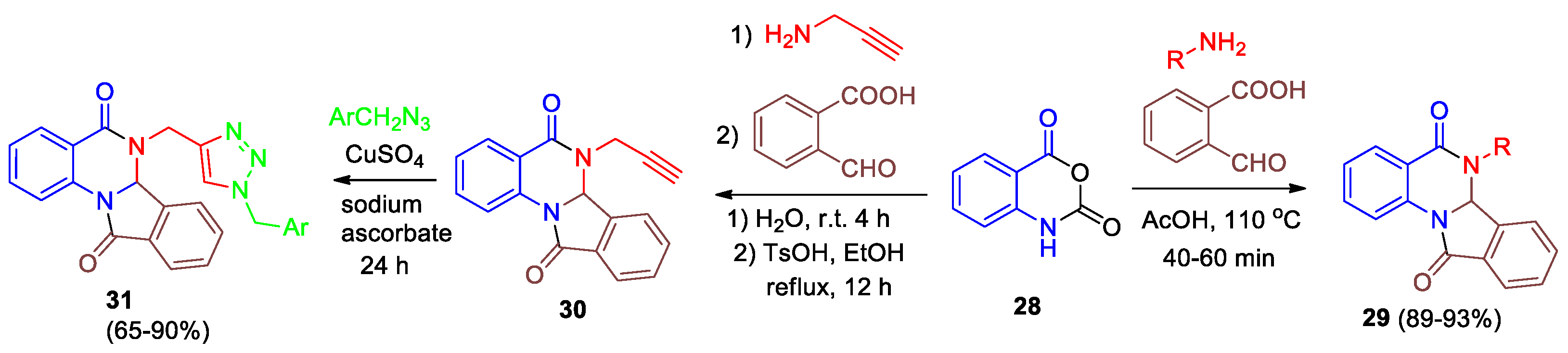
Scheme 12.
Synthetic approach to isoinodolo[2,1-a]quinazolino[1,2-c]quinazolineones 33. R = (CH2)2Ph, cyclopentyl, cyclopropyl, allyl, hexyl, butyl, propyl, ethyl.
Scheme 12.
Synthetic approach to isoinodolo[2,1-a]quinazolino[1,2-c]quinazolineones 33. R = (CH2)2Ph, cyclopentyl, cyclopropyl, allyl, hexyl, butyl, propyl, ethyl.
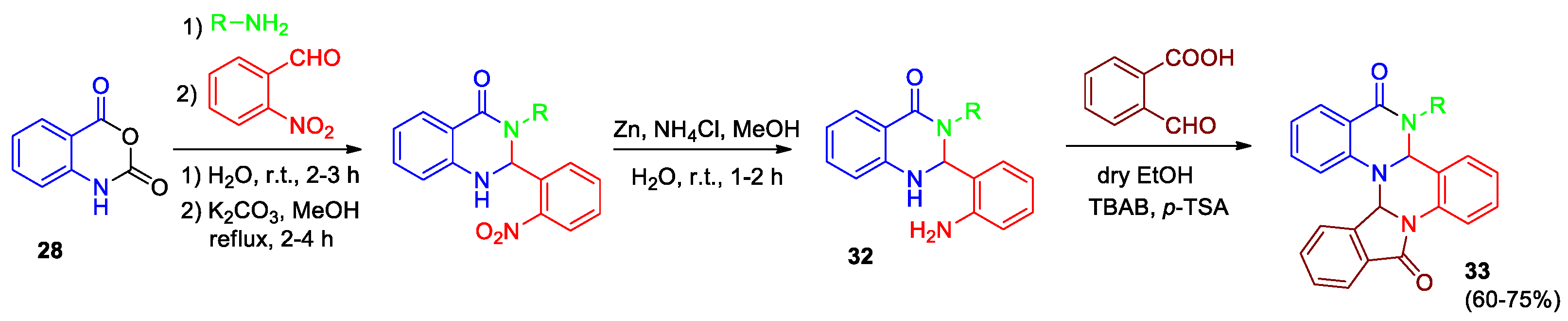
Scheme 13.
Synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 34, 35, 36. 34: R = Me, Bu, prop-2-yne, cyclopropyl, (CH2)2Ph, 1-Nf, Bn-4-Cl, Bn-4-OMe, Bn-2,4-Cl2, 4-Me-C6H4, 4-CN-C6H4, 2-MeO-C6H4, 2,3-(Me)2-C6H3, 2-Cl,3-Me-C6H3, 4-COOMe-C6H4, 4-t-Bu-C6H4; 35: R = propen-2-yl-methyl, Bn, Bn-2-Cl, Bn-4-Cl, furan-2-yl-methyl, Py-2-yl-methyl; X = H, 2,3-(MeO)2; 36: R = propen-2-yl, n-Pr, n-Bu, cyclopropyl, Bn, Bn-4-Me, Bn-2-OMe, Ph, 4-Me-C6H4, 4-MeO-C6H4.
Scheme 13.
Synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 34, 35, 36. 34: R = Me, Bu, prop-2-yne, cyclopropyl, (CH2)2Ph, 1-Nf, Bn-4-Cl, Bn-4-OMe, Bn-2,4-Cl2, 4-Me-C6H4, 4-CN-C6H4, 2-MeO-C6H4, 2,3-(Me)2-C6H3, 2-Cl,3-Me-C6H3, 4-COOMe-C6H4, 4-t-Bu-C6H4; 35: R = propen-2-yl-methyl, Bn, Bn-2-Cl, Bn-4-Cl, furan-2-yl-methyl, Py-2-yl-methyl; X = H, 2,3-(MeO)2; 36: R = propen-2-yl, n-Pr, n-Bu, cyclopropyl, Bn, Bn-4-Me, Bn-2-OMe, Ph, 4-Me-C6H4, 4-MeO-C6H4.
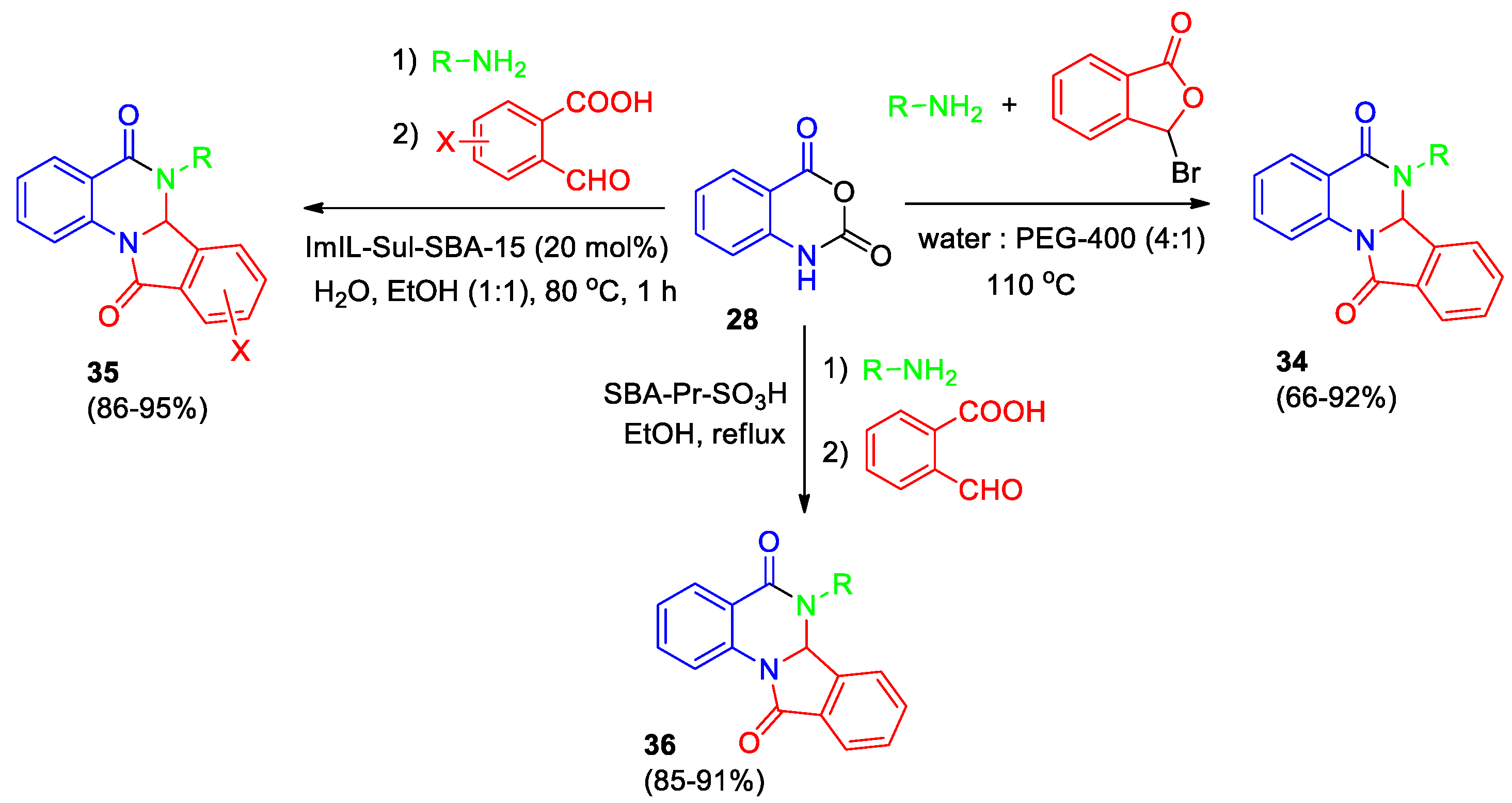
Scheme 14.
Synthetic approach to dihydroisoindolo[2,1-a]quinazolin-5,11-diones 38, 39. 37: R = H, Alk, Bn, Ar; 38: R = H (a), Bn (b), Me (c), Ph (d), 4-Me-C6H4 (e), 3-Me-C6H4 (f), 4-MeO-C6H4 (g), 4-ClC6H4 (h), 4-BrC6H4 (i), 4-NO2C6H4 (k), 3-NO2C6H4 (l), 2-Nf (m), cyclohexyl (n), Bu (o); 39: R2 = R1 = H (a), R2 = R1 = OMe (b).
Scheme 14.
Synthetic approach to dihydroisoindolo[2,1-a]quinazolin-5,11-diones 38, 39. 37: R = H, Alk, Bn, Ar; 38: R = H (a), Bn (b), Me (c), Ph (d), 4-Me-C6H4 (e), 3-Me-C6H4 (f), 4-MeO-C6H4 (g), 4-ClC6H4 (h), 4-BrC6H4 (i), 4-NO2C6H4 (k), 3-NO2C6H4 (l), 2-Nf (m), cyclohexyl (n), Bu (o); 39: R2 = R1 = H (a), R2 = R1 = OMe (b).
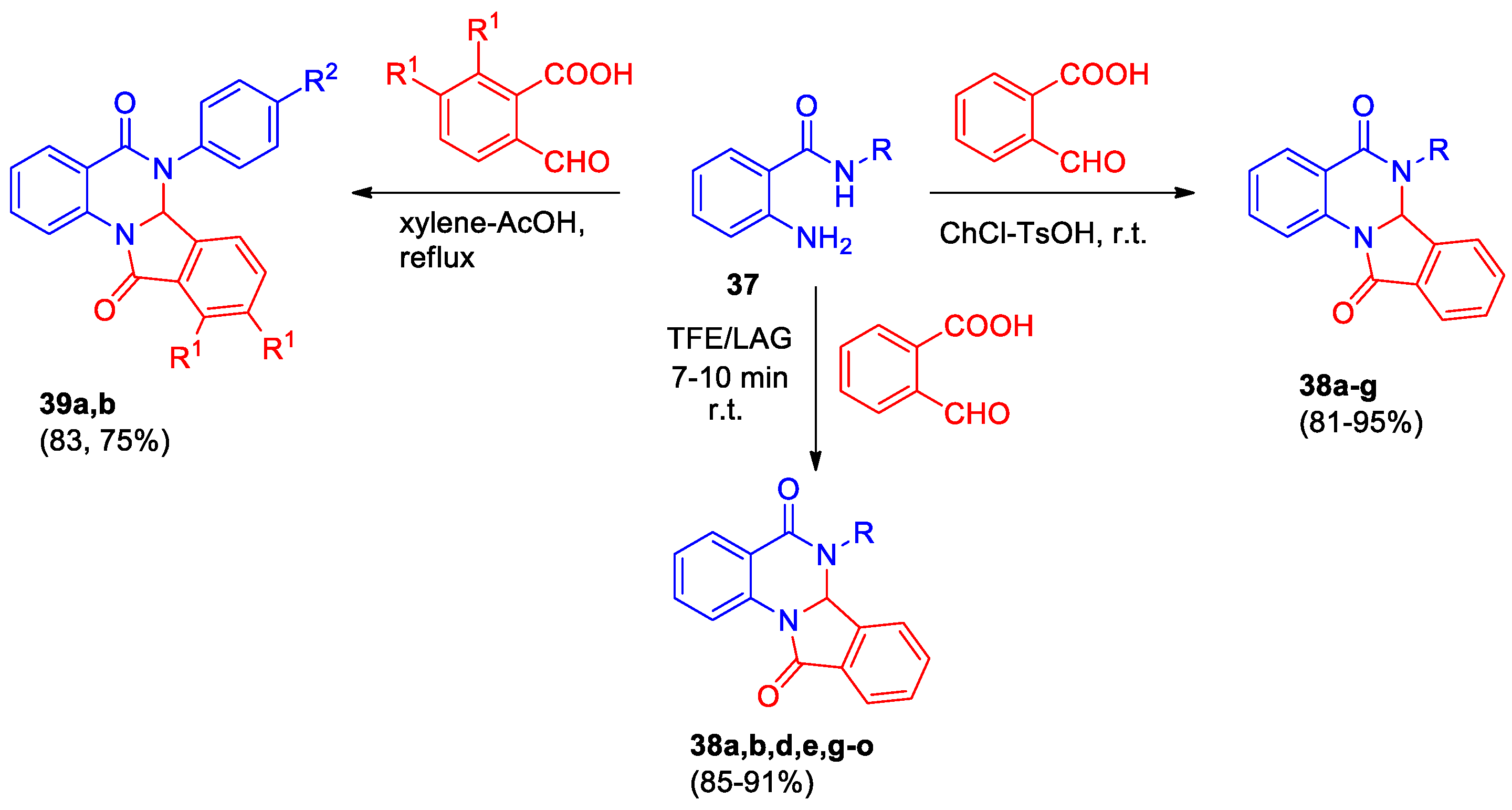
Scheme 15.
Synthetic approach to dihydroisoindolo[2,1-a]quinazoline-5,11-diones 41, 43. 41: R, R1 = Me, 5-Br (a); H, 5-Me (b); Me, H (c); Cl, 5-Br (d); H, H (e); H, 5-Cl (f); Me, 4-Cl (g); 43: R = NHAr, CH2Ar, allyl, cyclopropyl, etc.; R1 = H, Br, Cl.
Scheme 15.
Synthetic approach to dihydroisoindolo[2,1-a]quinazoline-5,11-diones 41, 43. 41: R, R1 = Me, 5-Br (a); H, 5-Me (b); Me, H (c); Cl, 5-Br (d); H, H (e); H, 5-Cl (f); Me, 4-Cl (g); 43: R = NHAr, CH2Ar, allyl, cyclopropyl, etc.; R1 = H, Br, Cl.
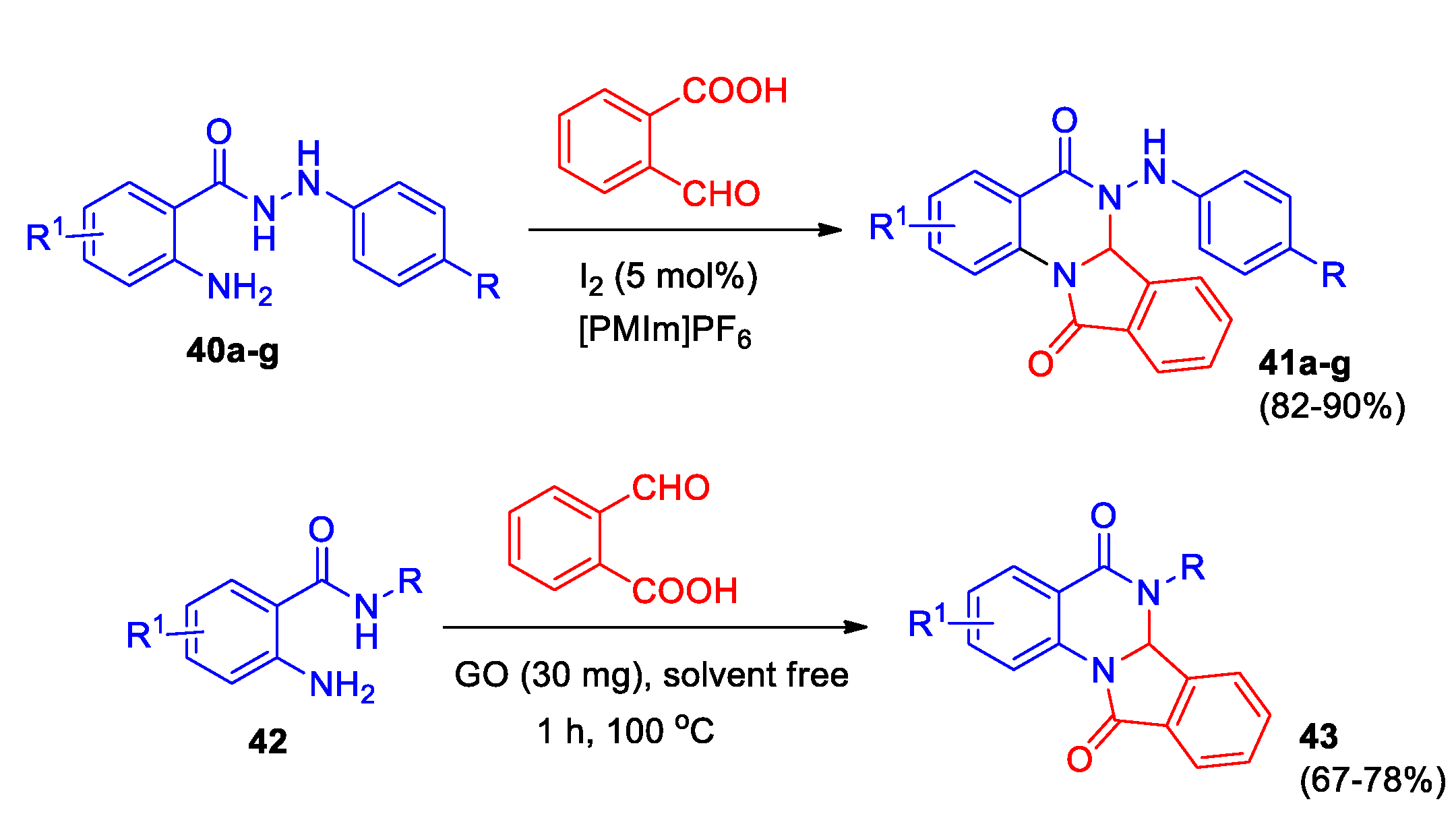
Scheme 16.
Synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 45. 42, 45: R = H, Bn, NHPh; R1 = Me, MeO, Cl; 44, 45: R2 = Me, OMe, F, Cl, CF3.
Scheme 16.
Synthesis of dihydroisoindolo[2,1-a]quinazoline-5,11-dione derivatives 45. 42, 45: R = H, Bn, NHPh; R1 = Me, MeO, Cl; 44, 45: R2 = Me, OMe, F, Cl, CF3.
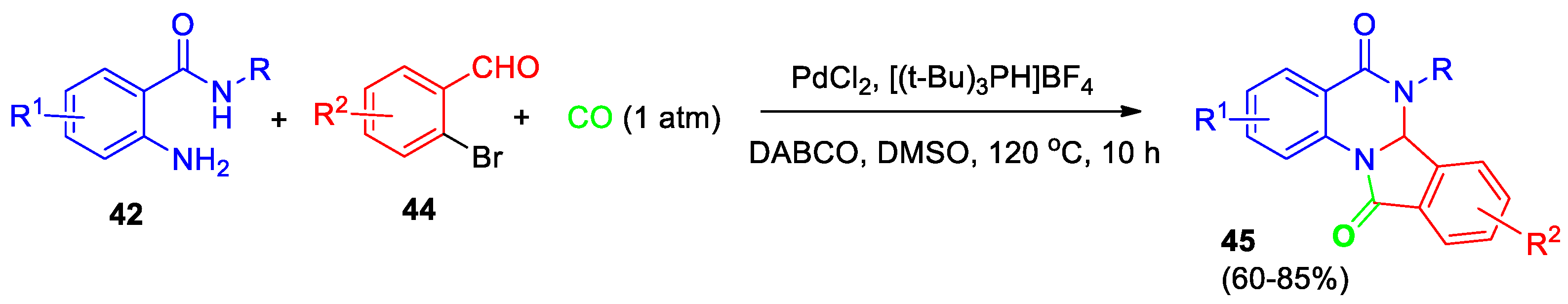
Scheme 17.
Synthetic approach to dihydroisoindolo[2,1-a]quinazoline-5,11-diones 49a,b. 46: X = H, R = H (a), OMe (b); 47: X = OH, R = H (a), OMe (b); 49: R = H (a), OMe (b).
Scheme 17.
Synthetic approach to dihydroisoindolo[2,1-a]quinazoline-5,11-diones 49a,b. 46: X = H, R = H (a), OMe (b); 47: X = OH, R = H (a), OMe (b); 49: R = H (a), OMe (b).
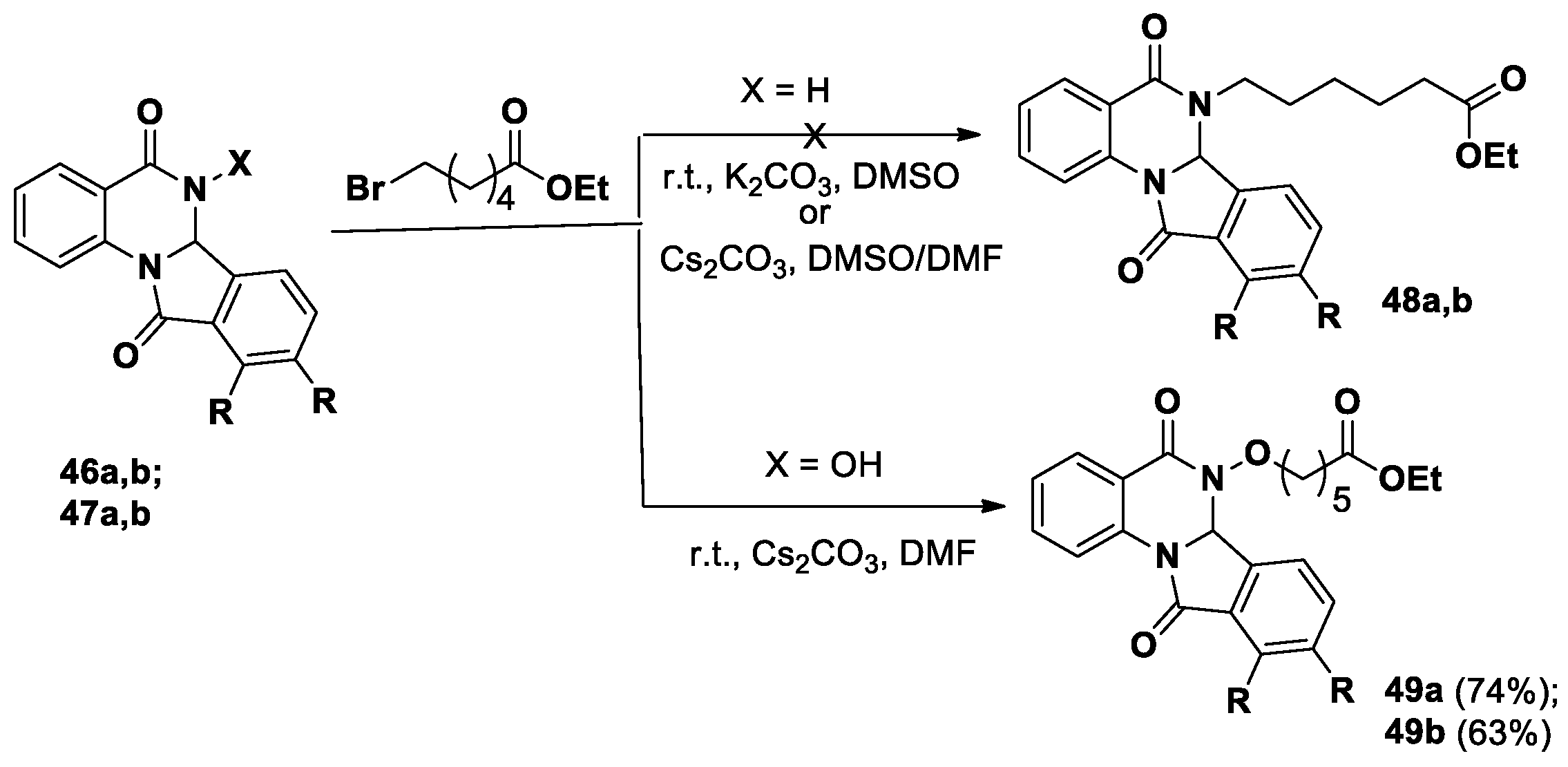
Scheme 18.
Synthetic approach to dihydroisoindolo[2,1-a]quinazolin-5,11-diones 50a,b. 48, 50: R = H (a), OMe (b).
Scheme 18.
Synthetic approach to dihydroisoindolo[2,1-a]quinazolin-5,11-diones 50a,b. 48, 50: R = H (a), OMe (b).
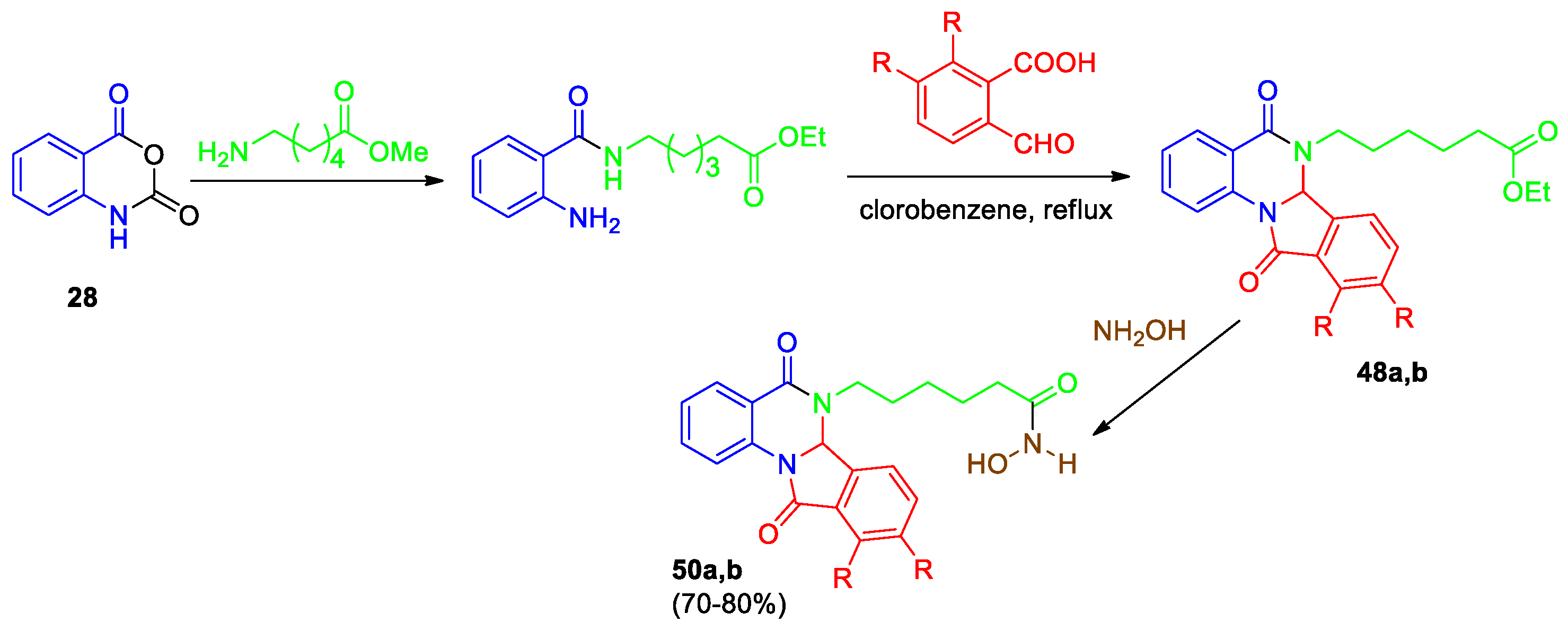
Scheme 19.
Synthesis of isoindolo[2,1-a]quinazolin-5(11H)-ones 52a-d. 51, 52: R1, R2 = H, H (a); 3-NO2, H (b); 3-Br, H (c); Br, Br (d).
Scheme 19.
Synthesis of isoindolo[2,1-a]quinazolin-5(11H)-ones 52a-d. 51, 52: R1, R2 = H, H (a); 3-NO2, H (b); 3-Br, H (c); Br, Br (d).
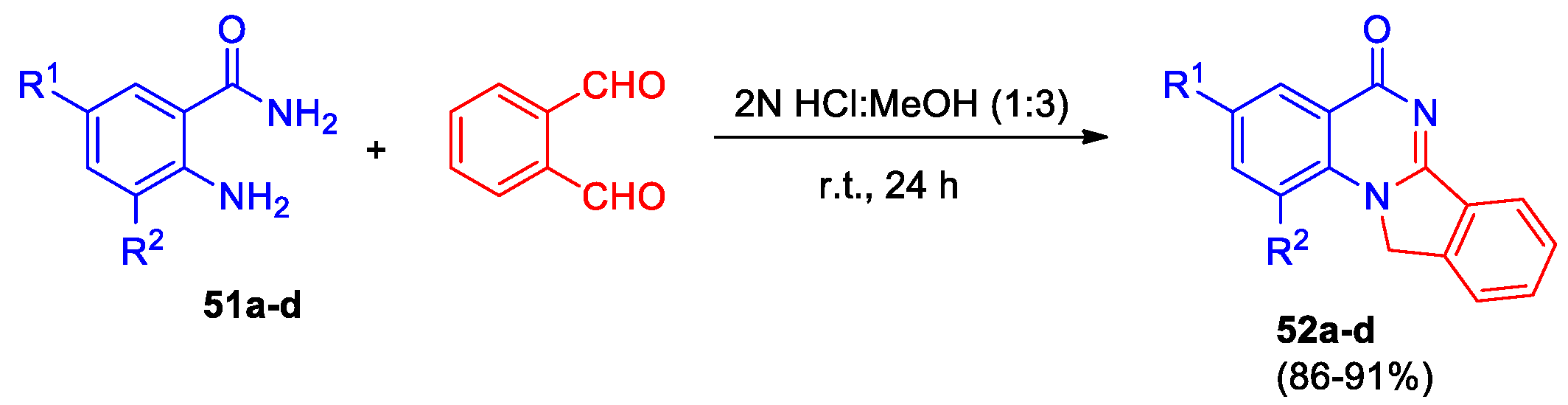
Scheme 20.
Synthetic approach to pyrazolo[1,5-a]quinazolines 54 and 56. 54: R1 = H, Cl, OMe, NO2; R2 = H, Me, CN, Ph, cyclopropyl, 2-thienyl; 56: R = H, NO2; R1 = H, Me, COOEt, cyclopropyl, 4-MeC6H4, 2-thienyl; R2 = H, CN.
Scheme 20.
Synthetic approach to pyrazolo[1,5-a]quinazolines 54 and 56. 54: R1 = H, Cl, OMe, NO2; R2 = H, Me, CN, Ph, cyclopropyl, 2-thienyl; 56: R = H, NO2; R1 = H, Me, COOEt, cyclopropyl, 4-MeC6H4, 2-thienyl; R2 = H, CN.
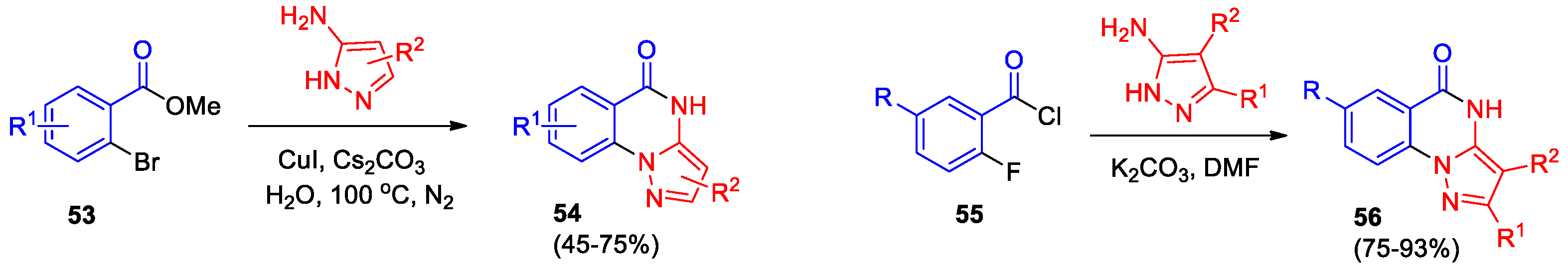
Scheme 21.
Synthesis of pyrazolo[1,5-a]quinazolines 58, indazolo[2,3-a]quinazoline 59, 10-azaindazolo[2,3-a]quinazoline 60. 57, 58: R = H, Br, CF3.
Scheme 21.
Synthesis of pyrazolo[1,5-a]quinazolines 58, indazolo[2,3-a]quinazoline 59, 10-azaindazolo[2,3-a]quinazoline 60. 57, 58: R = H, Br, CF3.
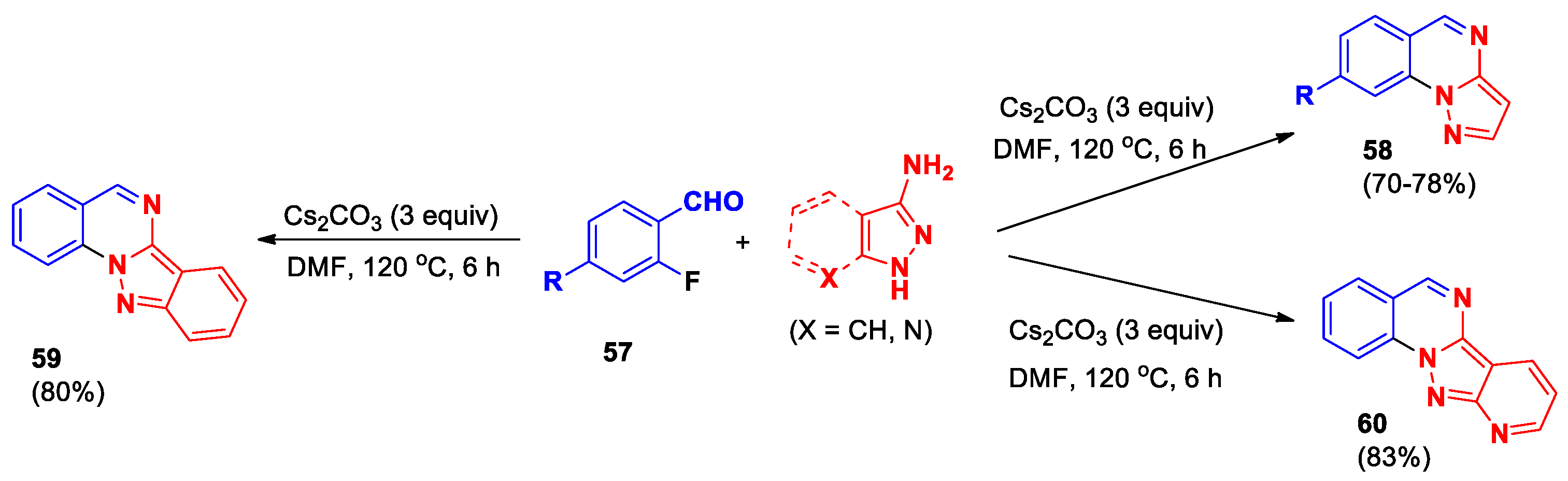
Scheme 22.
Synthesis of indazolo[2,3-a]- and pyrazolo[1,5-a]quinazolines 62, 63. 62: R = H, F, OH, Cl, Br, Me, CN; R1 = H, Me, Et, n-Pr, CF3, OMe, COOEt, Ph, cyclohexyl-4-Pr; Ar = Ph, substituted Ar, naphthalene-2-yl, antracen-9-yl, hetaryl; 63: R2 = H, CONH2, CN, COOEt.
Scheme 22.
Synthesis of indazolo[2,3-a]- and pyrazolo[1,5-a]quinazolines 62, 63. 62: R = H, F, OH, Cl, Br, Me, CN; R1 = H, Me, Et, n-Pr, CF3, OMe, COOEt, Ph, cyclohexyl-4-Pr; Ar = Ph, substituted Ar, naphthalene-2-yl, antracen-9-yl, hetaryl; 63: R2 = H, CONH2, CN, COOEt.
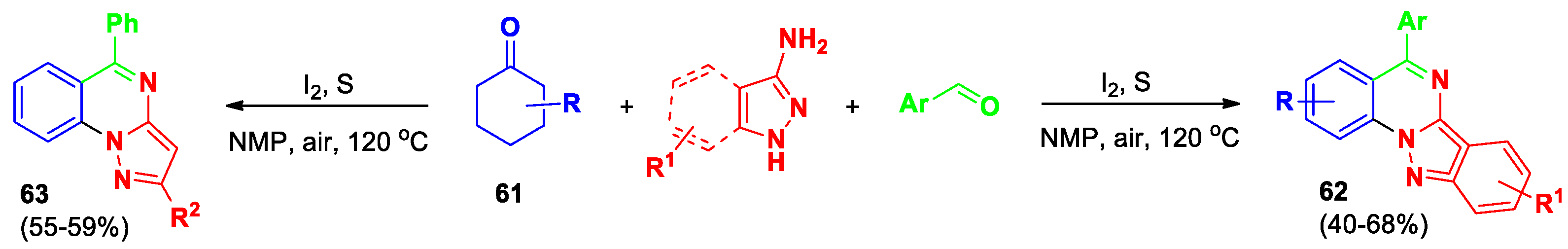
Figure 3.
Chemical structures of the compounds A and 64a-t. 64a-l: X = N, R = 8-Cl (a), 7-NO2 (b), 8-CF3 (c), H (d), 8-Me (e); X = CH, R = H (f), 8-MeSO2 (g), 8-CF3 (h), 9-CF3 (i), 8-NO2 (j), 9-NH2 (k), 8-NH2 (l).
Figure 3.
Chemical structures of the compounds A and 64a-t. 64a-l: X = N, R = 8-Cl (a), 7-NO2 (b), 8-CF3 (c), H (d), 8-Me (e); X = CH, R = H (f), 8-MeSO2 (g), 8-CF3 (h), 9-CF3 (i), 8-NO2 (j), 9-NH2 (k), 8-NH2 (l).
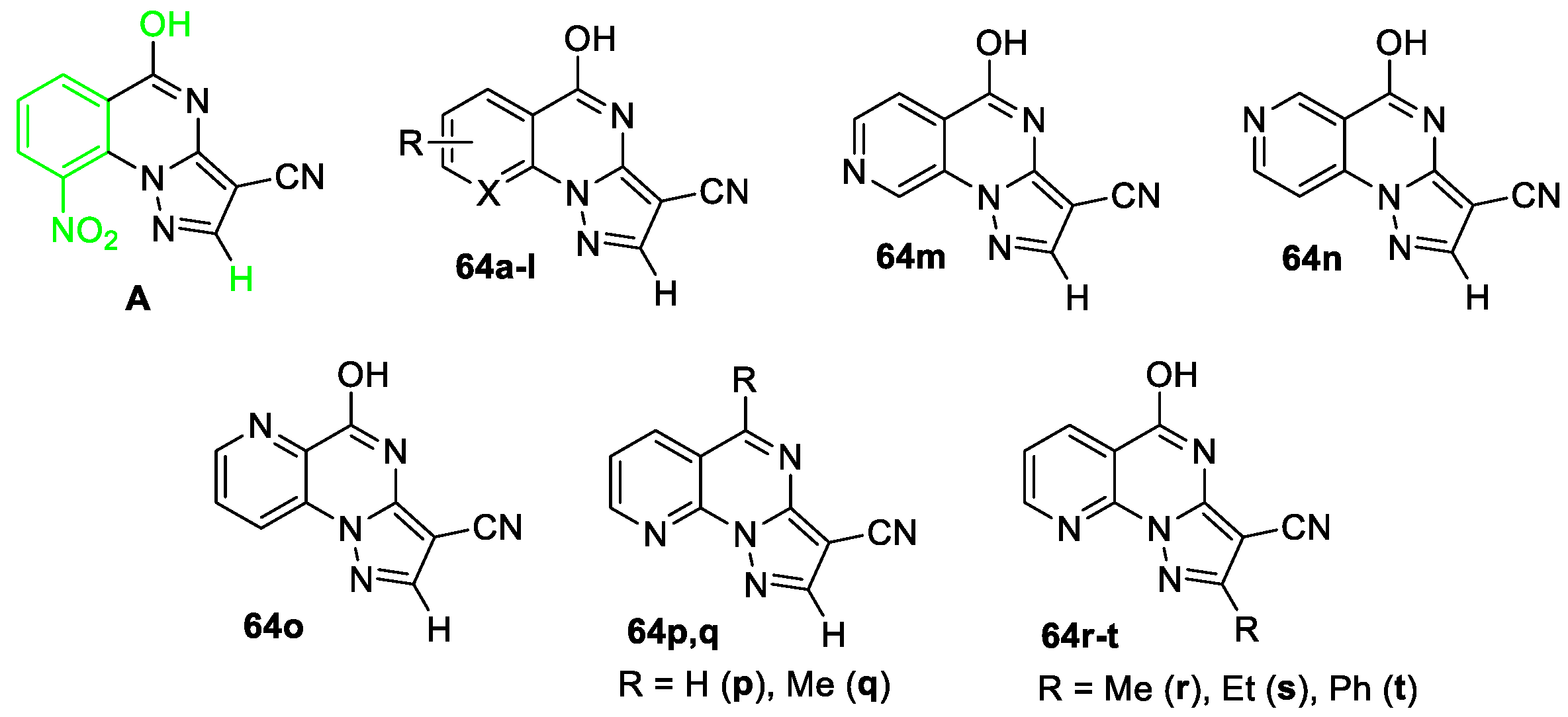
Figure 4.
3D docking model of compound 64r in the active pocket of KDM4D. Reproduced with permission of Elsevier [54].
Figure 4.
3D docking model of compound 64r in the active pocket of KDM4D. Reproduced with permission of Elsevier [54].
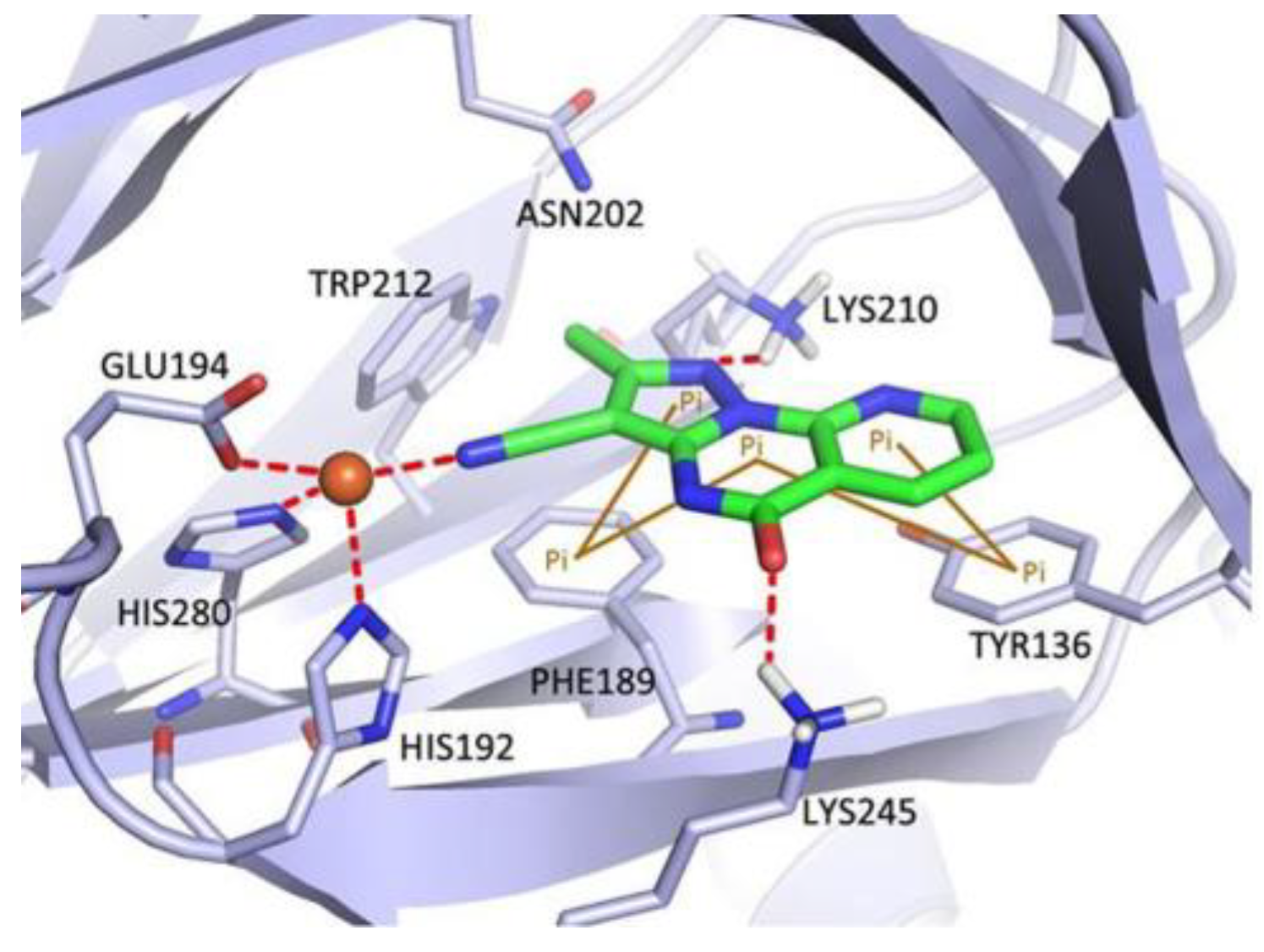
Scheme 23.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 66–69. 65–69: R1 = H, Me; R2 = H, Me, Br; R3 = H, Me, OMe; R4 = H, Me; R5 = H, Me; 68, 69: n = 1, 2.
Scheme 23.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 66–69. 65–69: R1 = H, Me; R2 = H, Me, Br; R3 = H, Me, OMe; R4 = H, Me; R5 = H, Me; 68, 69: n = 1, 2.
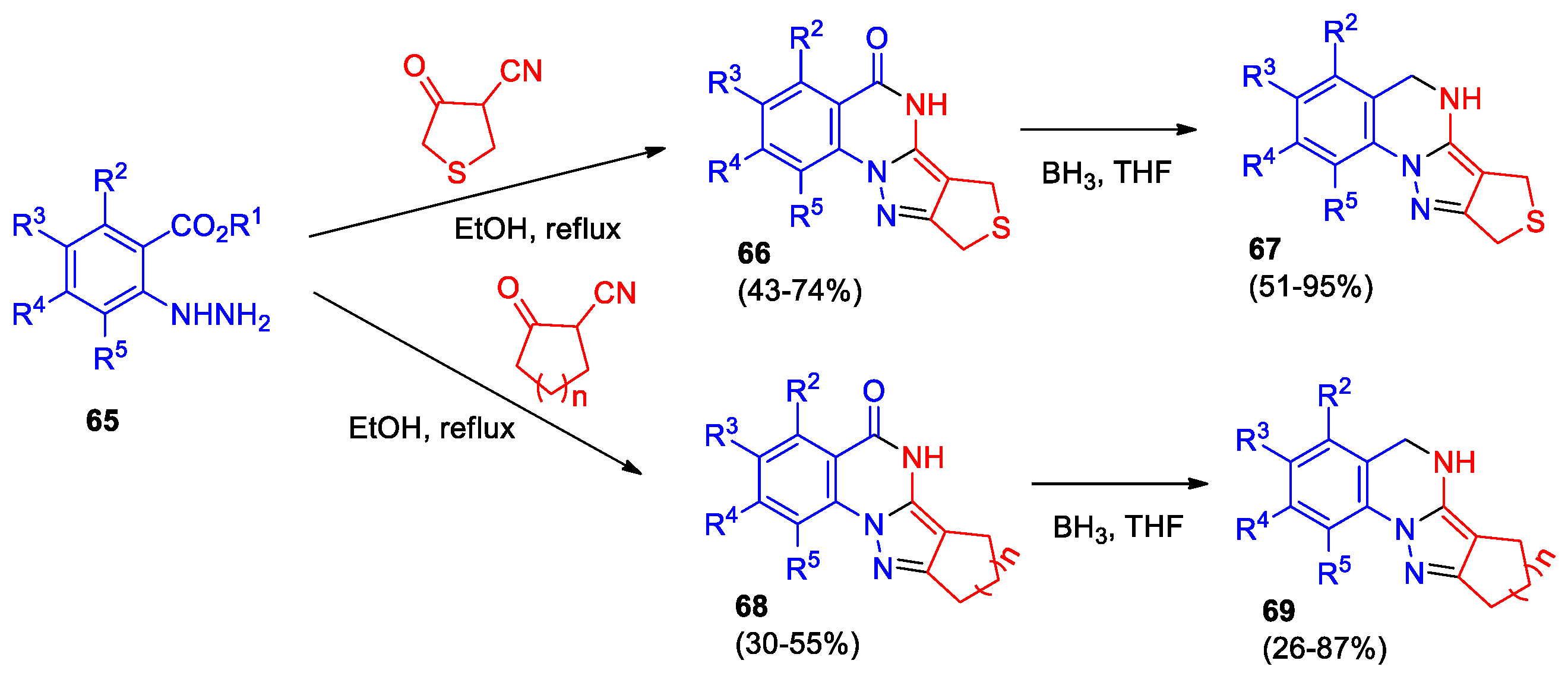
Scheme 24.
Synthetic approach to pyrazolo[1,5-a]quinazolines 70, 72, 73-75, 76, 77, 78, 79. 70: R = OMe (a), H (b), O(CH2)2OMe (c); 72: R = OMe (a), H (b); 76: R = OMe (a), H (b); 77: R = OMe, R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-thienyl (c), CH2-2-furyl (d), CHMe2 (e); 78: R = H, R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-thienyl (c); 79: R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-2-furyl (d).
Scheme 24.
Synthetic approach to pyrazolo[1,5-a]quinazolines 70, 72, 73-75, 76, 77, 78, 79. 70: R = OMe (a), H (b), O(CH2)2OMe (c); 72: R = OMe (a), H (b); 76: R = OMe (a), H (b); 77: R = OMe, R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-thienyl (c), CH2-2-furyl (d), CHMe2 (e); 78: R = H, R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-thienyl (c); 79: R1 = CH2Ph (a), CH2-(2-OMe)Ph (b), CH2-2-furyl (d).
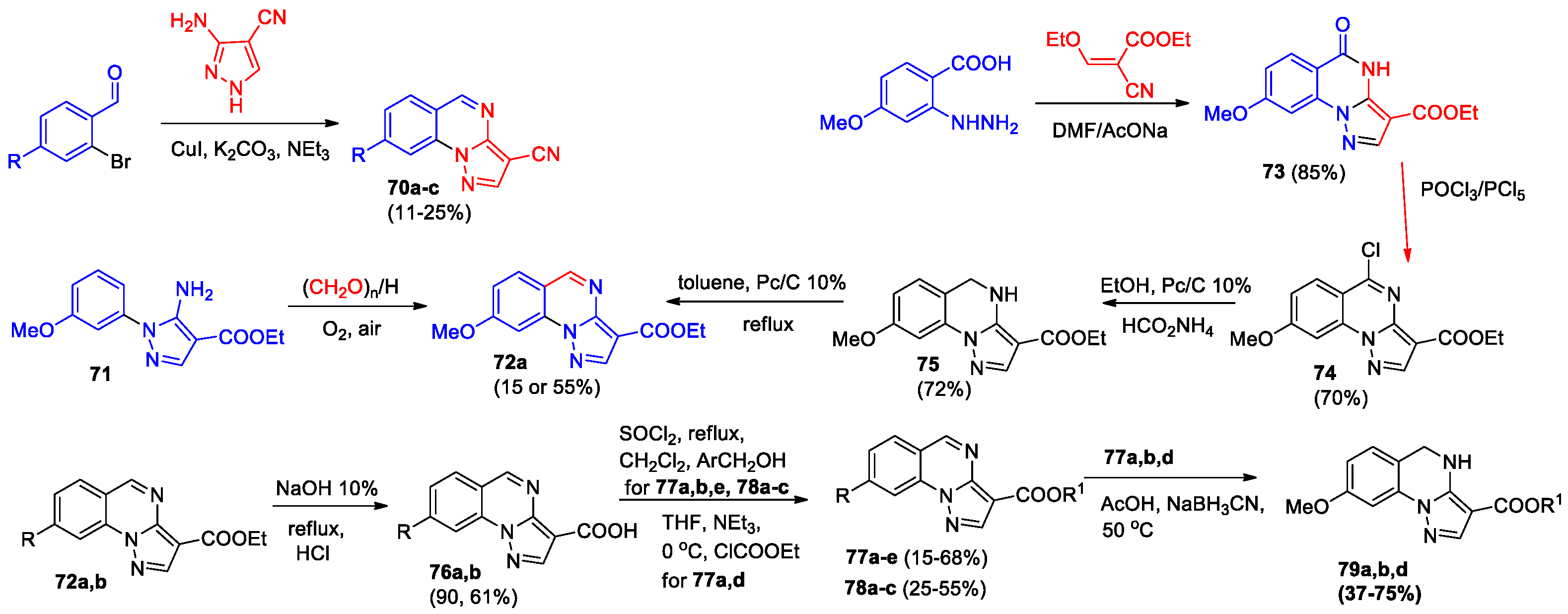
Scheme 25.
Synthetic approach to pyrazolo[1,5-a]quinazolines 80, 82, 83, 87. 80: R = H, R1 = Ph (a), 2-thienyl (b), 3-thienyl (c); R = OMe, R1 = 2-thienyl (d), 3-thienyl (e), CO-(2-OMe)-Ph (f), 2-CO-2-thienyl (g), CO-3-thienyl (h); 82: R = H, Ar/Het = Ph (a), 2-thienyl (b), 3-thienyl (c); R = OMe, Ar/Het = 2-thienyl (d), 3-thienyl (e), 2-MeOPh (f), 3-furyl (g), 1-Boc-2-pyrrolyl (h), 1H-2-pyrrolyl (i); 83: R = OMe, Ar/Het = (2-MeO)Ph (a), 2-thienyl (b), 3-thienyl (c), (4-MeO)Ph (d), 2-furyl (e), 2(1H)-pyrrolyl (f), 2-(1-methyl)pyrrolyl (g); 86: X = Br (a), I (b); 87: Ar/Het = 2-MeOPh (a), 2-thienyl (b), 4-MeOPh (d), 2-furyl (e), 2(1H)-pyrrolyl (f), 2-(1-methyl)pyrrolyl (g).
Scheme 25.
Synthetic approach to pyrazolo[1,5-a]quinazolines 80, 82, 83, 87. 80: R = H, R1 = Ph (a), 2-thienyl (b), 3-thienyl (c); R = OMe, R1 = 2-thienyl (d), 3-thienyl (e), CO-(2-OMe)-Ph (f), 2-CO-2-thienyl (g), CO-3-thienyl (h); 82: R = H, Ar/Het = Ph (a), 2-thienyl (b), 3-thienyl (c); R = OMe, Ar/Het = 2-thienyl (d), 3-thienyl (e), 2-MeOPh (f), 3-furyl (g), 1-Boc-2-pyrrolyl (h), 1H-2-pyrrolyl (i); 83: R = OMe, Ar/Het = (2-MeO)Ph (a), 2-thienyl (b), 3-thienyl (c), (4-MeO)Ph (d), 2-furyl (e), 2(1H)-pyrrolyl (f), 2-(1-methyl)pyrrolyl (g); 86: X = Br (a), I (b); 87: Ar/Het = 2-MeOPh (a), 2-thienyl (b), 4-MeOPh (d), 2-furyl (e), 2(1H)-pyrrolyl (f), 2-(1-methyl)pyrrolyl (g).
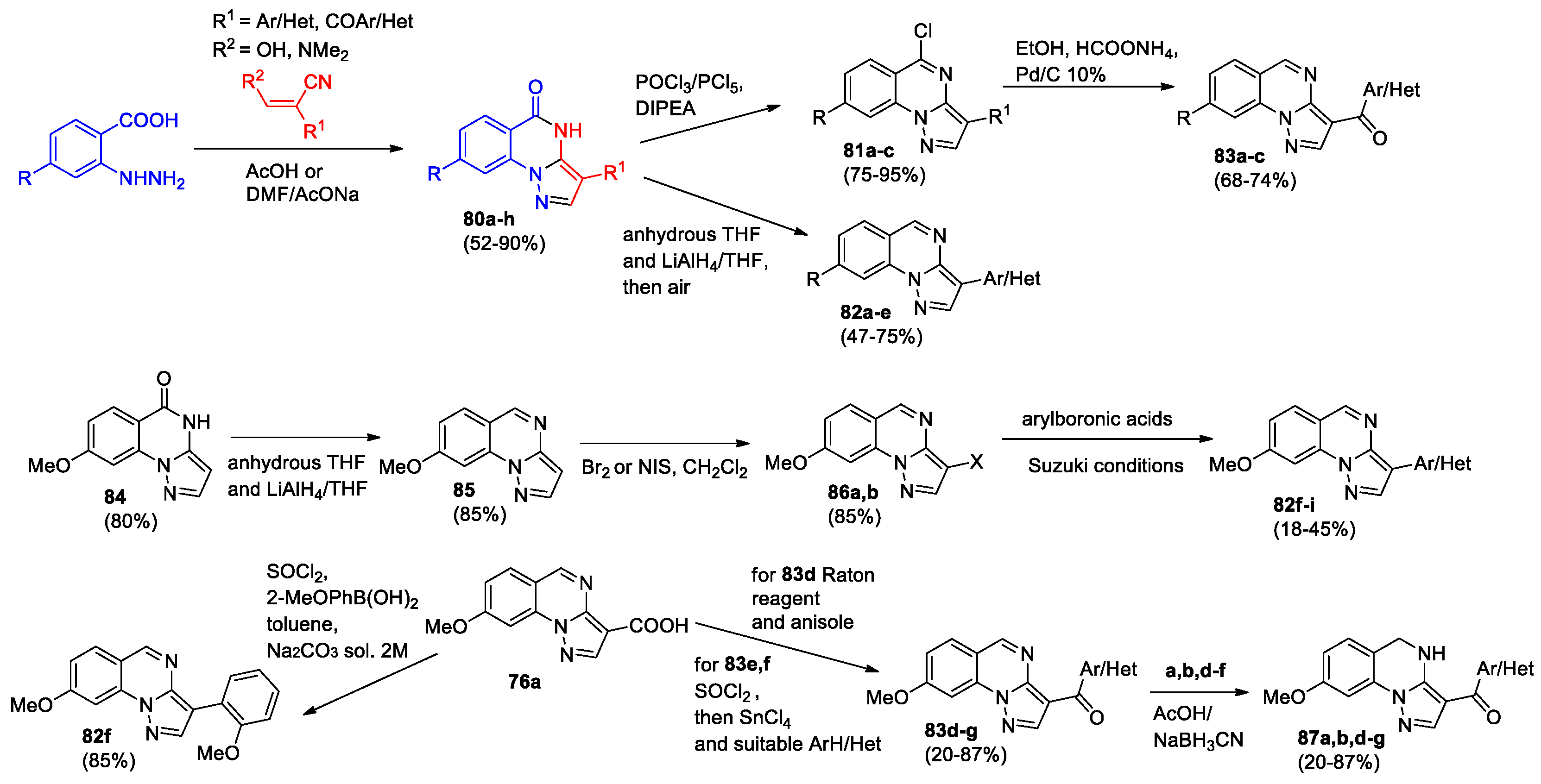
Scheme 26.
Synthetic approach to pyrazolo[1,5-a]quinazolines 89–94, 96, 97. 89: R = CN (a), COOEt (b); 90: R, R1 = CN, CH2Ph (a), COOEt, Me (b); 91: R = CH2Ph (a), CH2(2-OMe-Ph) (b), CH2-2-furyl (c); 92: R = CH2Ph (a), CH2(2-OMe-Ph) (b), CH2-2-furyl (c), Et (d); 94: R = H (a), CH2Ph (b); 96: R = H (a), CH2Ph (b).
Scheme 26.
Synthetic approach to pyrazolo[1,5-a]quinazolines 89–94, 96, 97. 89: R = CN (a), COOEt (b); 90: R, R1 = CN, CH2Ph (a), COOEt, Me (b); 91: R = CH2Ph (a), CH2(2-OMe-Ph) (b), CH2-2-furyl (c); 92: R = CH2Ph (a), CH2(2-OMe-Ph) (b), CH2-2-furyl (c), Et (d); 94: R = H (a), CH2Ph (b); 96: R = H (a), CH2Ph (b).
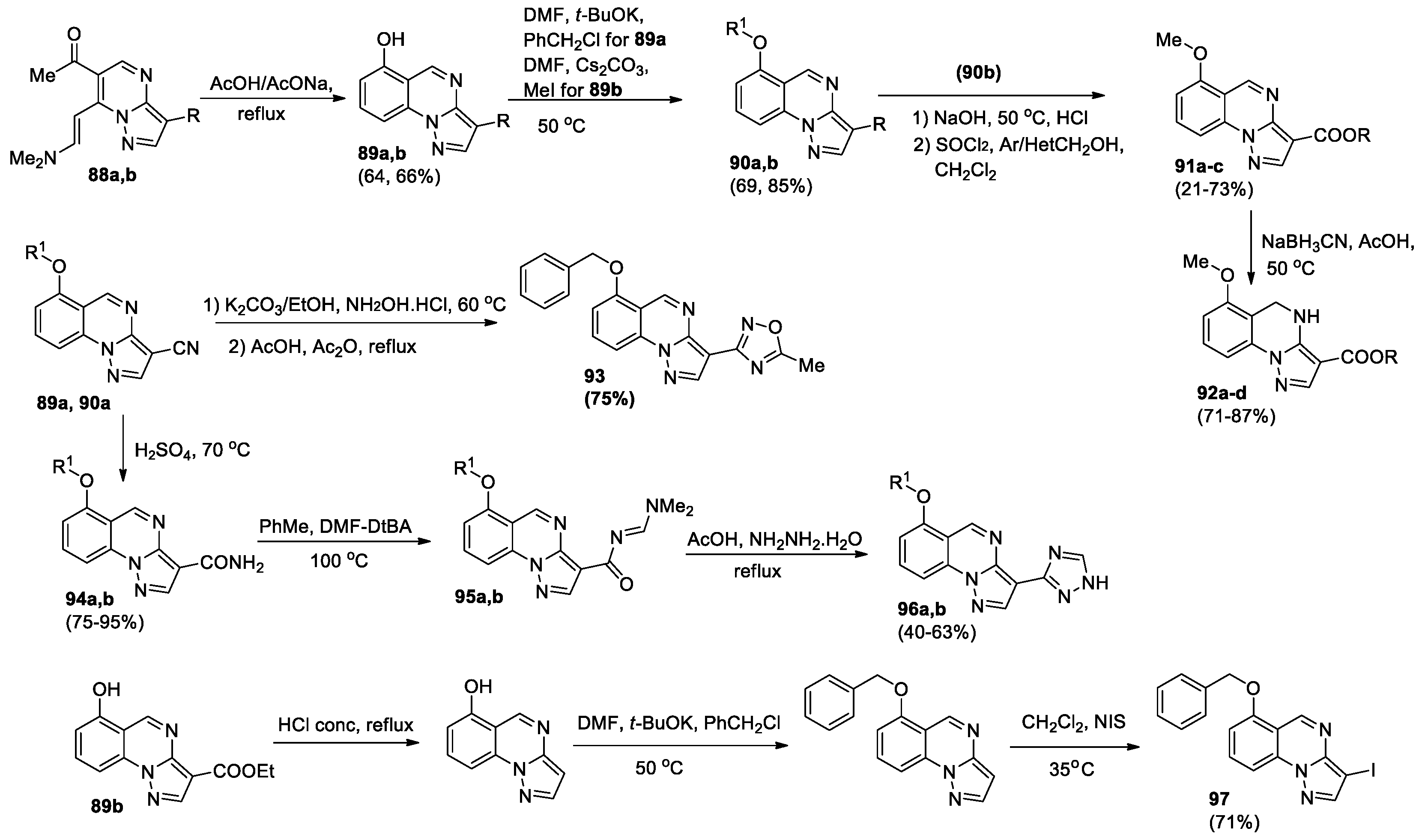
Figure 5.
Chemical structures of the compounds 98, 99, 100-107. 98, 99: R = H (a), CHMe2 (b), cyclohexyl (c), CH2cyclohexyl (d), Ph (e), CH2Ph (f), CH2(2-OMe)Ph (g), CH2(2-furyl) (h), CH2(2-thienyl) (i); 100: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), t-Bu (c); 101: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), Et (c); 102: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), t-Bu (c); 103: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), Et (c); 104: R1 = H (a), Me (b); 106: R1 = H (a), Me (b).
Figure 5.
Chemical structures of the compounds 98, 99, 100-107. 98, 99: R = H (a), CHMe2 (b), cyclohexyl (c), CH2cyclohexyl (d), Ph (e), CH2Ph (f), CH2(2-OMe)Ph (g), CH2(2-furyl) (h), CH2(2-thienyl) (i); 100: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), t-Bu (c); 101: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), Et (c); 102: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), t-Bu (c); 103: R = CH2(2-OMe)Ph (a), CH2(2-thienyl) (b), Et (c); 104: R1 = H (a), Me (b); 106: R1 = H (a), Me (b).
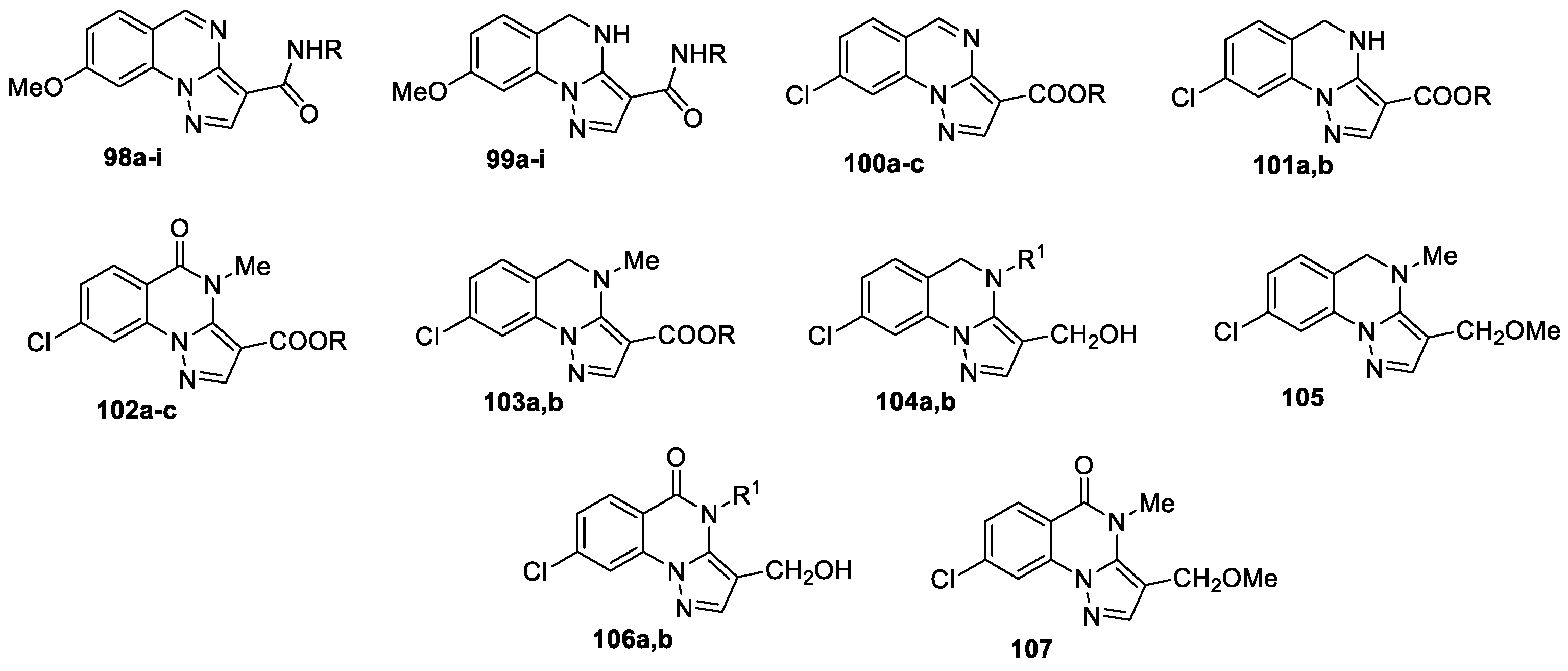
Figure 6.
Chemical structures of the compounds 108–113. 108: R = Me (a), CH2cPr (b), propyne (c), CH2Ph (d), CH2(2-MePh) (e), CH2(2-MeOPh) (f), CH2(pyridin-4-yl) (g); 109: R7 = H, R8 = NO2 (a), R7 = NO2, R8 = H (b); 110: R7 = H, R8 = NO2 (a), R7=NO2, R8=H (b); 111: R7 = H, R8 = NO2, R = CH2(2-MeOPh) (a), R7 = H, R8 = NO2, R = CH2-thien-2-yl (b), R7 = NO2, R8 = H, R = CH2(2-MeOPh) (c), R7 = NO2, R8 = H, R = CH2-thien-2-yl (d); 113: R = CH2(2-MeOPh) (a), CH2-thien-2-yl (b).
Figure 6.
Chemical structures of the compounds 108–113. 108: R = Me (a), CH2cPr (b), propyne (c), CH2Ph (d), CH2(2-MePh) (e), CH2(2-MeOPh) (f), CH2(pyridin-4-yl) (g); 109: R7 = H, R8 = NO2 (a), R7 = NO2, R8 = H (b); 110: R7 = H, R8 = NO2 (a), R7=NO2, R8=H (b); 111: R7 = H, R8 = NO2, R = CH2(2-MeOPh) (a), R7 = H, R8 = NO2, R = CH2-thien-2-yl (b), R7 = NO2, R8 = H, R = CH2(2-MeOPh) (c), R7 = NO2, R8 = H, R = CH2-thien-2-yl (d); 113: R = CH2(2-MeOPh) (a), CH2-thien-2-yl (b).
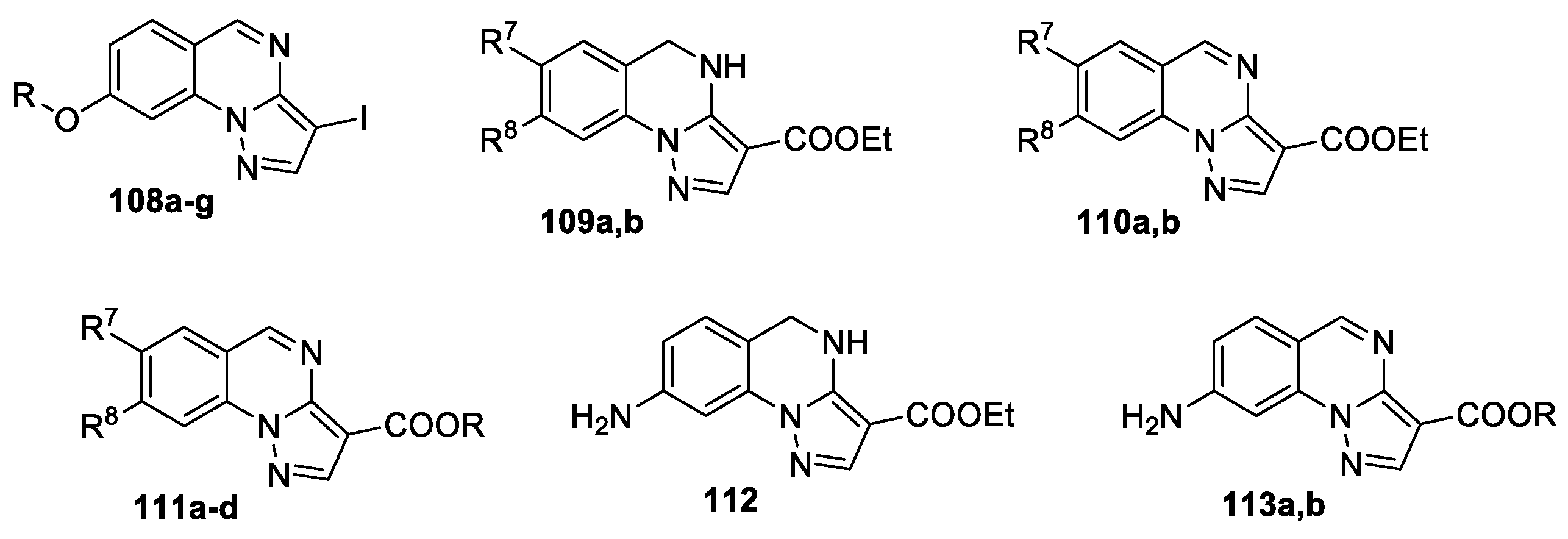
Figure 7.
Hydrogen bonds among the agonist 112 (blue), the antagonist 108d (red), and γThr142, αHis102, and αSer205 in the binding site. Reproduced with permission of MDPI [64].
Figure 7.
Hydrogen bonds among the agonist 112 (blue), the antagonist 108d (red), and γThr142, αHis102, and αSer205 in the binding site. Reproduced with permission of MDPI [64].
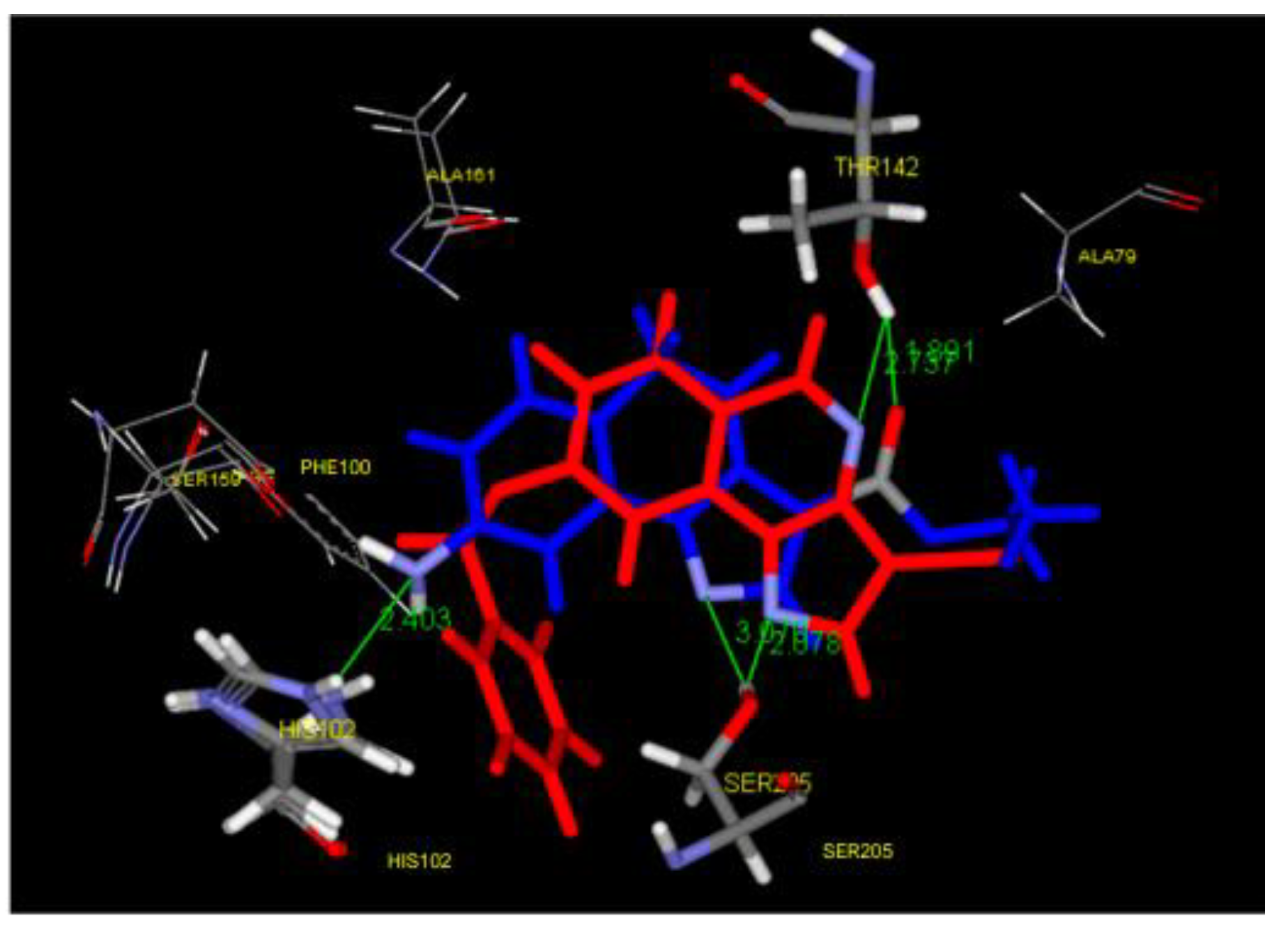
Scheme 27.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 115a (a-f), 115b (a-o),. 115c (a-g); 116a (a-o), 116b (a-y), 116c (a-j); 117a (a-h), 117b (a-h), 117ca (a-h). R1 = H (a), Me (b), tBu (c); R2 = Ar, Ar-Ar, Ar-O-Ar, thiazolyl, benzothiazolyl, benzooxazolyl, pyrazinyl etc.
Scheme 27.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 115a (a-f), 115b (a-o),. 115c (a-g); 116a (a-o), 116b (a-y), 116c (a-j); 117a (a-h), 117b (a-h), 117ca (a-h). R1 = H (a), Me (b), tBu (c); R2 = Ar, Ar-Ar, Ar-O-Ar, thiazolyl, benzothiazolyl, benzooxazolyl, pyrazinyl etc.
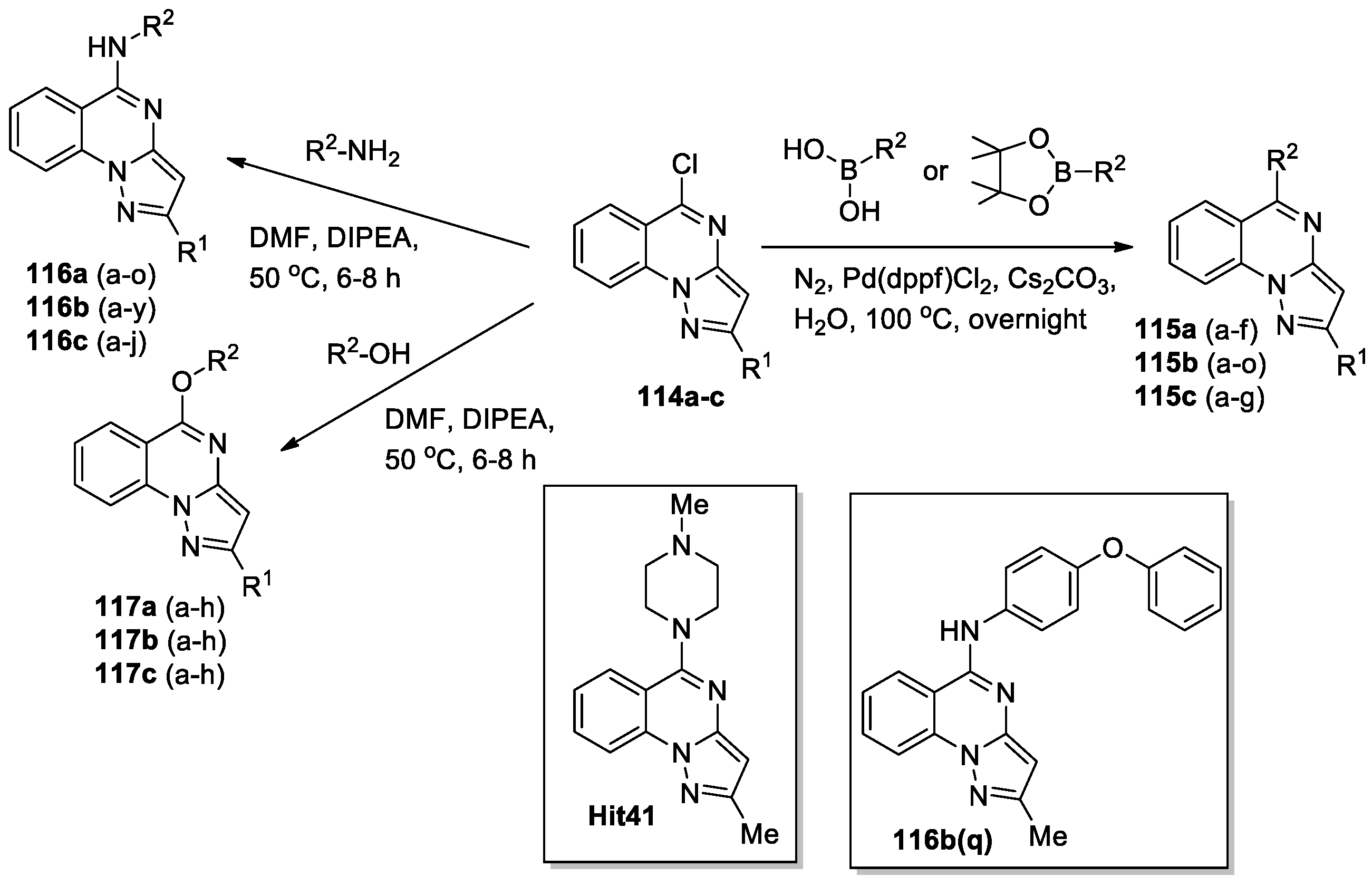
Scheme 28.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 118a-d, 119a-d, 120a-e, 121a-c, 122, 123a-e, 124, 125, 126. 118–121: R7, R8 = H, H (a); H, Cl (b), H, NO2 (c); NO2, H (d); H, OMe (e); 123: R = Me (a), CH2-Ph (b), CH2-4-SMe-Ph (c), CH2-2-Cl-Ph (d), CH2-3-Cl-Ph (e), CH2-4-Br-Ph (f), CH2-2-Me-Ph (g), CH2-3-OMe-Ph (h), CH2-4-SO2NH2-Ph (i).
Scheme 28.
Synthesis of pyrazolo[1,5-a]quinazoline derivatives 118a-d, 119a-d, 120a-e, 121a-c, 122, 123a-e, 124, 125, 126. 118–121: R7, R8 = H, H (a); H, Cl (b), H, NO2 (c); NO2, H (d); H, OMe (e); 123: R = Me (a), CH2-Ph (b), CH2-4-SMe-Ph (c), CH2-2-Cl-Ph (d), CH2-3-Cl-Ph (e), CH2-4-Br-Ph (f), CH2-2-Me-Ph (g), CH2-3-OMe-Ph (h), CH2-4-SO2NH2-Ph (i).
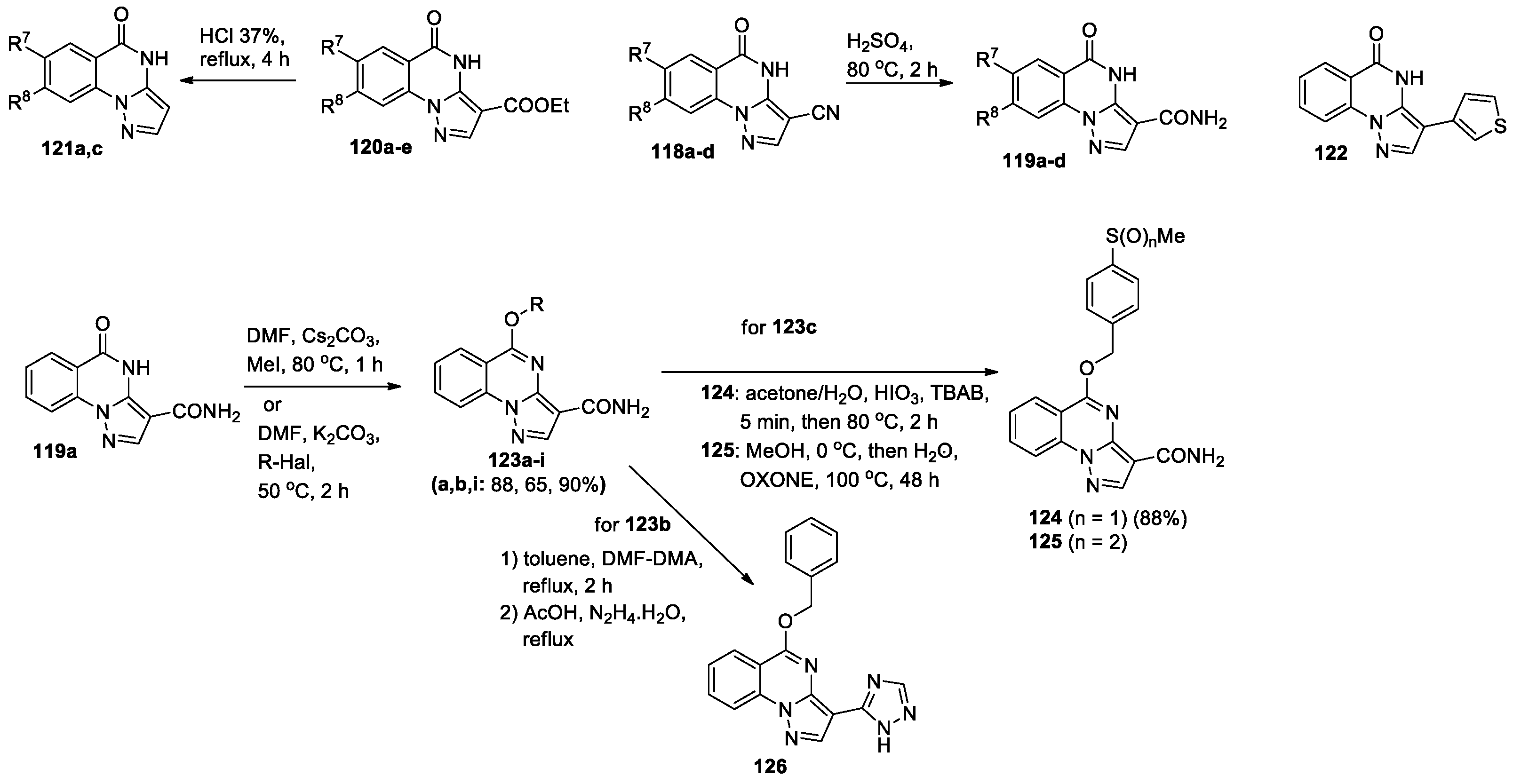
Scheme 29.
Synthetic approach to 9-(trifluoromethyl)-pyrido[2’,3’:3,4]pyrazolo[1,5-a]quinazoline derivatives 128–132.
Scheme 29.
Synthetic approach to 9-(trifluoromethyl)-pyrido[2’,3’:3,4]pyrazolo[1,5-a]quinazoline derivatives 128–132.
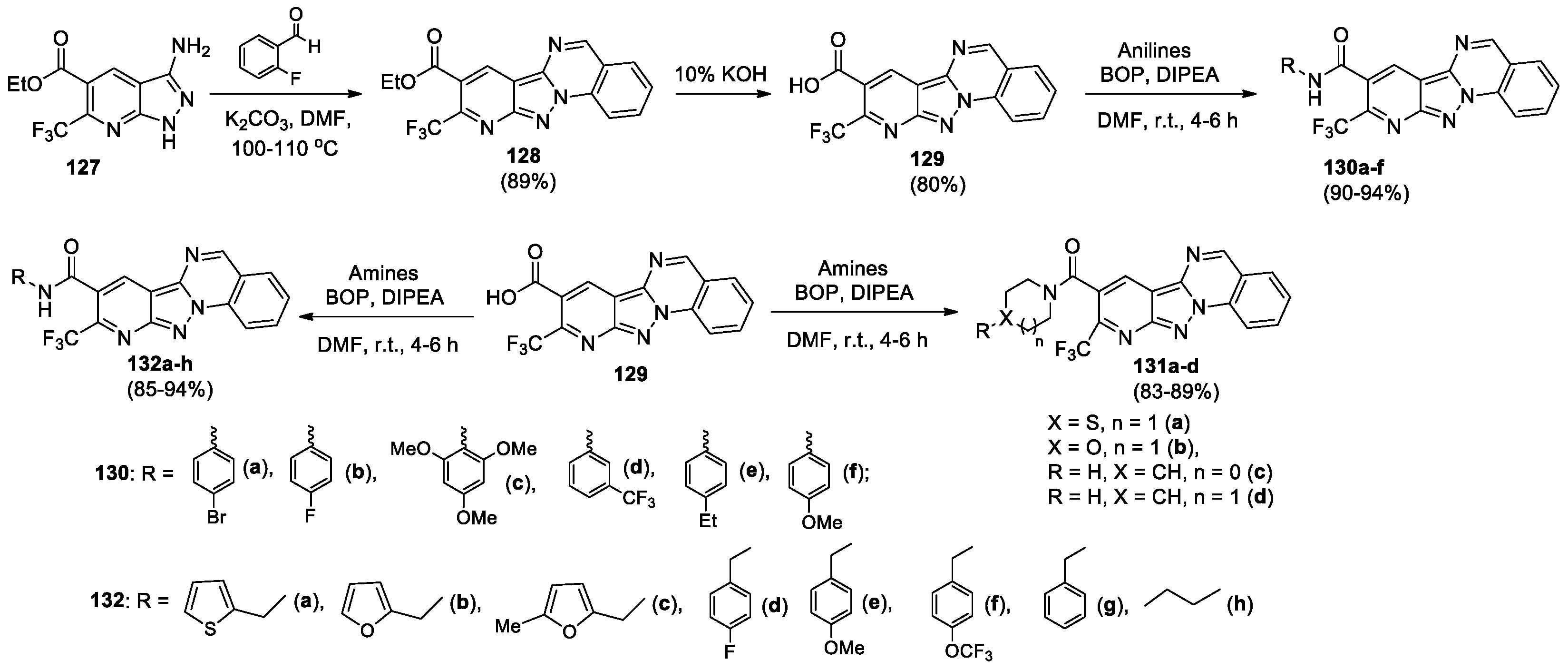
Scheme 30.
Synthetic approach to 4,5-dihydropyrazolo[1,5-a]quinazolines 133–136. 133: R1 = CF3, NO2, CN, Cl, Br, F, OCF3; R2=Et, Me; 134: R2 = Me, Et, n-Pr, cyclobutylmethyl, 4-chlorobenzyl; 135: X = O, NH; R = Me, Et, CH2CF3, 2,4-Me2Ph, 2-Cl-5-Me-thiazole, 2-Cl-5-Me-Py; 136: R1 = esters, amides, acylsulfur; R2 = H, esters; R3 = SOCF3 or SO2CF3.
Scheme 30.
Synthetic approach to 4,5-dihydropyrazolo[1,5-a]quinazolines 133–136. 133: R1 = CF3, NO2, CN, Cl, Br, F, OCF3; R2=Et, Me; 134: R2 = Me, Et, n-Pr, cyclobutylmethyl, 4-chlorobenzyl; 135: X = O, NH; R = Me, Et, CH2CF3, 2,4-Me2Ph, 2-Cl-5-Me-thiazole, 2-Cl-5-Me-Py; 136: R1 = esters, amides, acylsulfur; R2 = H, esters; R3 = SOCF3 or SO2CF3.
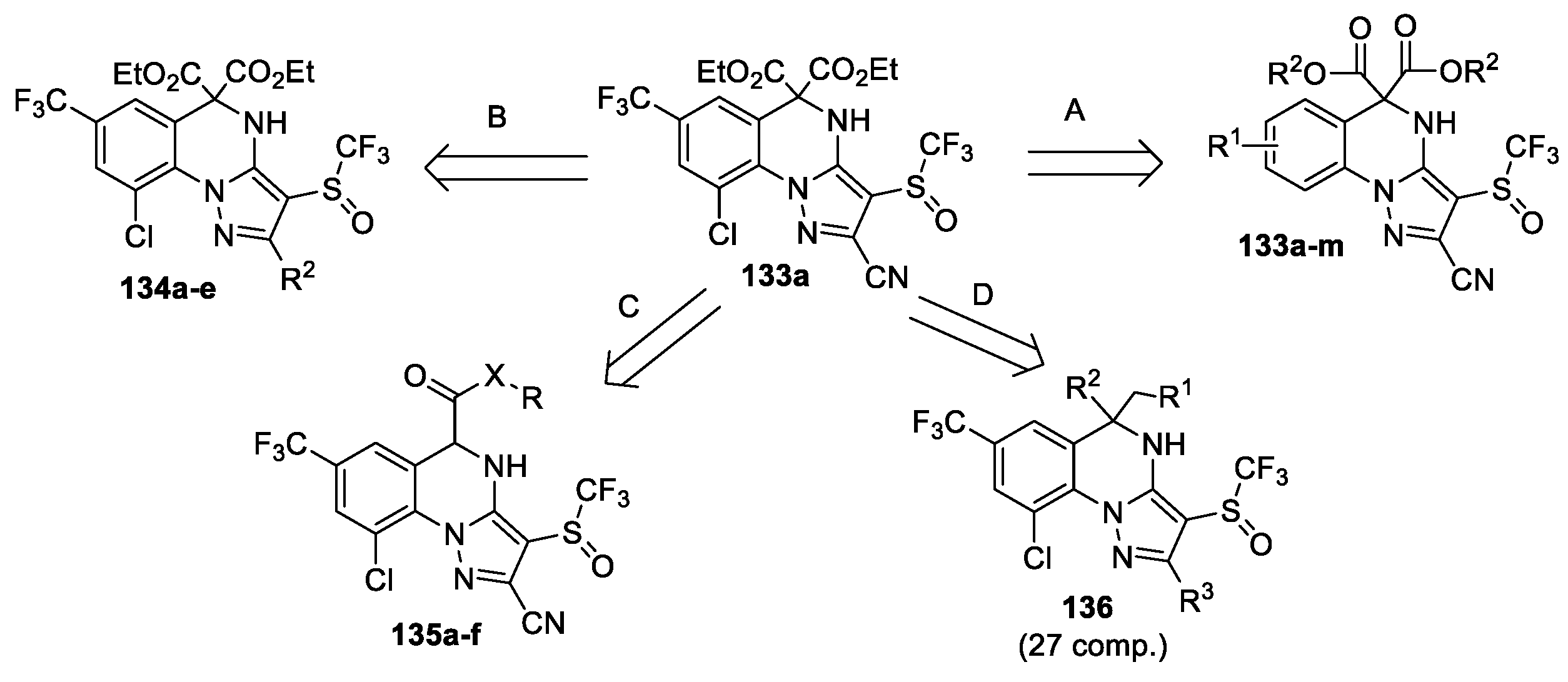
Scheme 31.
Synthetic approach to benzimidazo[1,2-a]quinazolines 137, 138. 137: X = CH, R = H (a), Br (b), OMe (c), CF3 (d), NO2 (e); X = N, R = H (f), Br (g). 138: R = F, Cl, Br, CF3, Me, OMe; R1 = H, Cl, Me.
Scheme 31.
Synthetic approach to benzimidazo[1,2-a]quinazolines 137, 138. 137: X = CH, R = H (a), Br (b), OMe (c), CF3 (d), NO2 (e); X = N, R = H (f), Br (g). 138: R = F, Cl, Br, CF3, Me, OMe; R1 = H, Cl, Me.
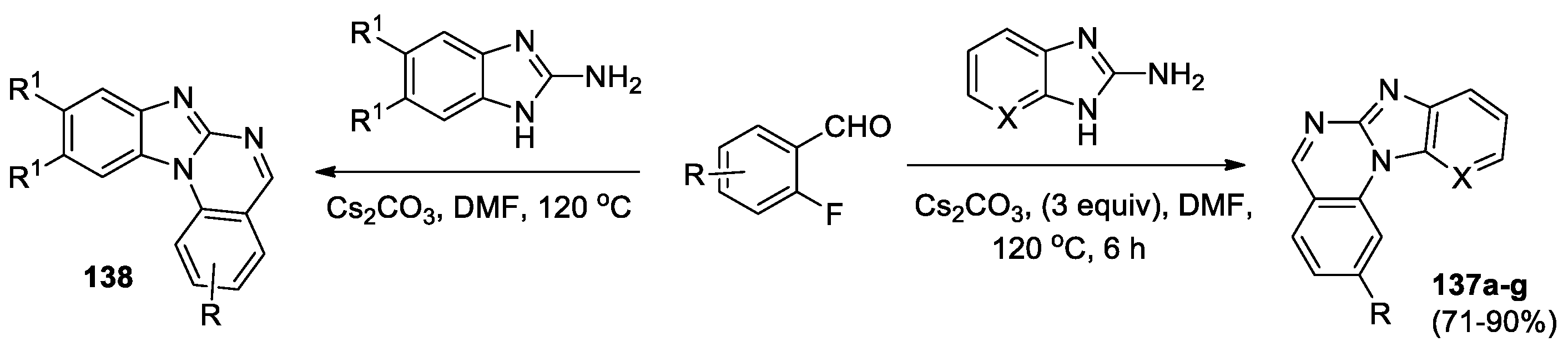
Scheme 32.
Synthetic approach to tetrahydrobenzo [4,5]imidazo[1,2-a]quinazolines 140, 141, benzimidazo[1,2-a]quinazolines 144 and benzimidazole-4,7-dione derivatives 142a-c, 145. 140: R, R1 = H, H (a), H, Me (b), Me, H (c), Ph, H (d); 141, 142: R = H (a), Me (b), Ph (c); 143-145: R = H (a), F (b).
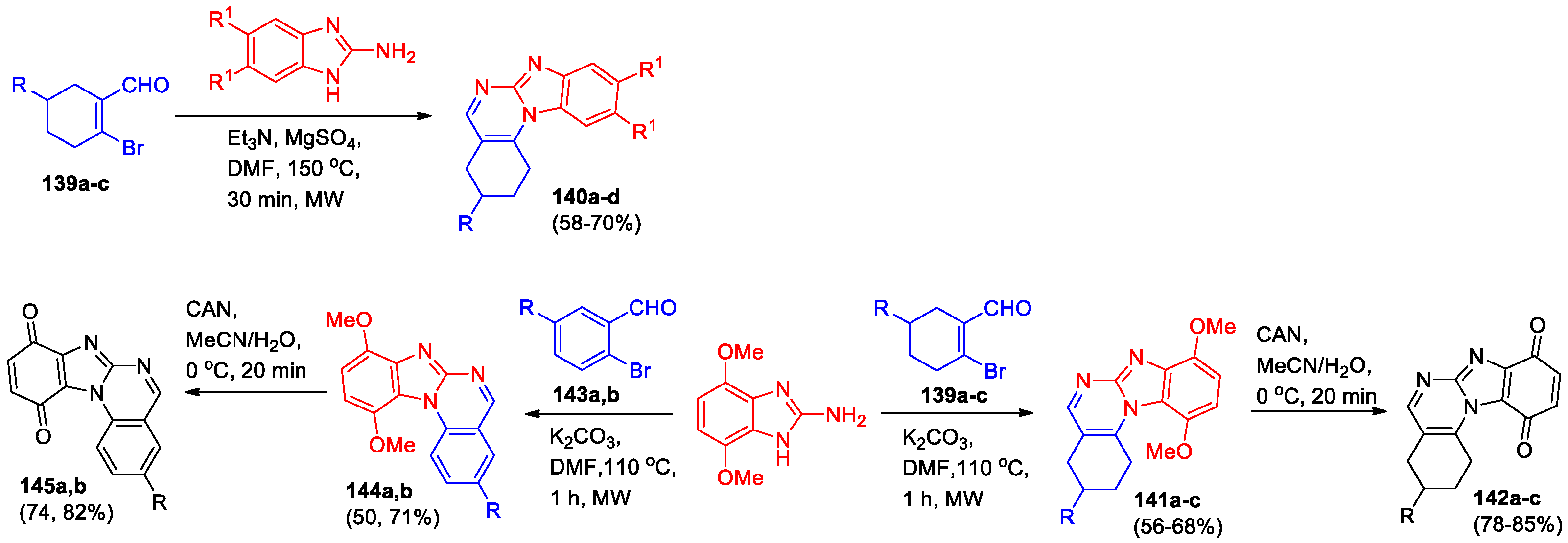
Scheme 33.
Synthetic approach to tetrahydrobenzo[4,5]imidazo[1,2-a]quinazolines 147, tetrahydroimidazo[1,2-a]quinazolin-5-one 148, benzimidazo[1,2-a]quinazolin-5-ones 150a-c, 151a, imidazo[1,2-a]quinazolin-5-one 151b. 147: R, R1 = Bz, H (a); Bu, H (b); Bz, Me (c); Bz, Ph (d); 150: R = Bz (a), Bu (b), Ph (c); 151a: R = n-Pr, benzimidazole; 151b: R = n-Bu, imidazole.
Scheme 33.
Synthetic approach to tetrahydrobenzo[4,5]imidazo[1,2-a]quinazolines 147, tetrahydroimidazo[1,2-a]quinazolin-5-one 148, benzimidazo[1,2-a]quinazolin-5-ones 150a-c, 151a, imidazo[1,2-a]quinazolin-5-one 151b. 147: R, R1 = Bz, H (a); Bu, H (b); Bz, Me (c); Bz, Ph (d); 150: R = Bz (a), Bu (b), Ph (c); 151a: R = n-Pr, benzimidazole; 151b: R = n-Bu, imidazole.
Scheme 34.
Synthesis of imidazo[1,2-a]quinazolin-5-one153 and benzimidazo[1,2-a]quinazolines 156. 154: R1 = H, 5,6-Cl; 155: X = F, Cl, Br, NO2; R = H, 4-F, 5-F, 4-Cl, 3-OMe, 5-Br, 4-Br, 3,6-Br.
Scheme 34.
Synthesis of imidazo[1,2-a]quinazolin-5-one153 and benzimidazo[1,2-a]quinazolines 156. 154: R1 = H, 5,6-Cl; 155: X = F, Cl, Br, NO2; R = H, 4-F, 5-F, 4-Cl, 3-OMe, 5-Br, 4-Br, 3,6-Br.
Scheme 35.
Synthetic approach to 4-(2-chlorobenzyl)imidazole[1,2-a]quinazolin-5(4H)-ones 159, 161a-e, 164. 161: R = 4-Me-piperazino (a), pyrrolidinyl (b), piperidinyl (c), morpholino (d), thiomorpholino (e); 164: R = cycloimines, cycloamines, arylamines.
Scheme 35.
Synthetic approach to 4-(2-chlorobenzyl)imidazole[1,2-a]quinazolin-5(4H)-ones 159, 161a-e, 164. 161: R = 4-Me-piperazino (a), pyrrolidinyl (b), piperidinyl (c), morpholino (d), thiomorpholino (e); 164: R = cycloimines, cycloamines, arylamines.
Figure 8.
Interaction of compound 164 (R = 3-(OCF3)C6H4NH) with SHP2 protein. Reproduced with permission of Taylor & Francis [80].
Figure 8.
Interaction of compound 164 (R = 3-(OCF3)C6H4NH) with SHP2 protein. Reproduced with permission of Taylor & Francis [80].
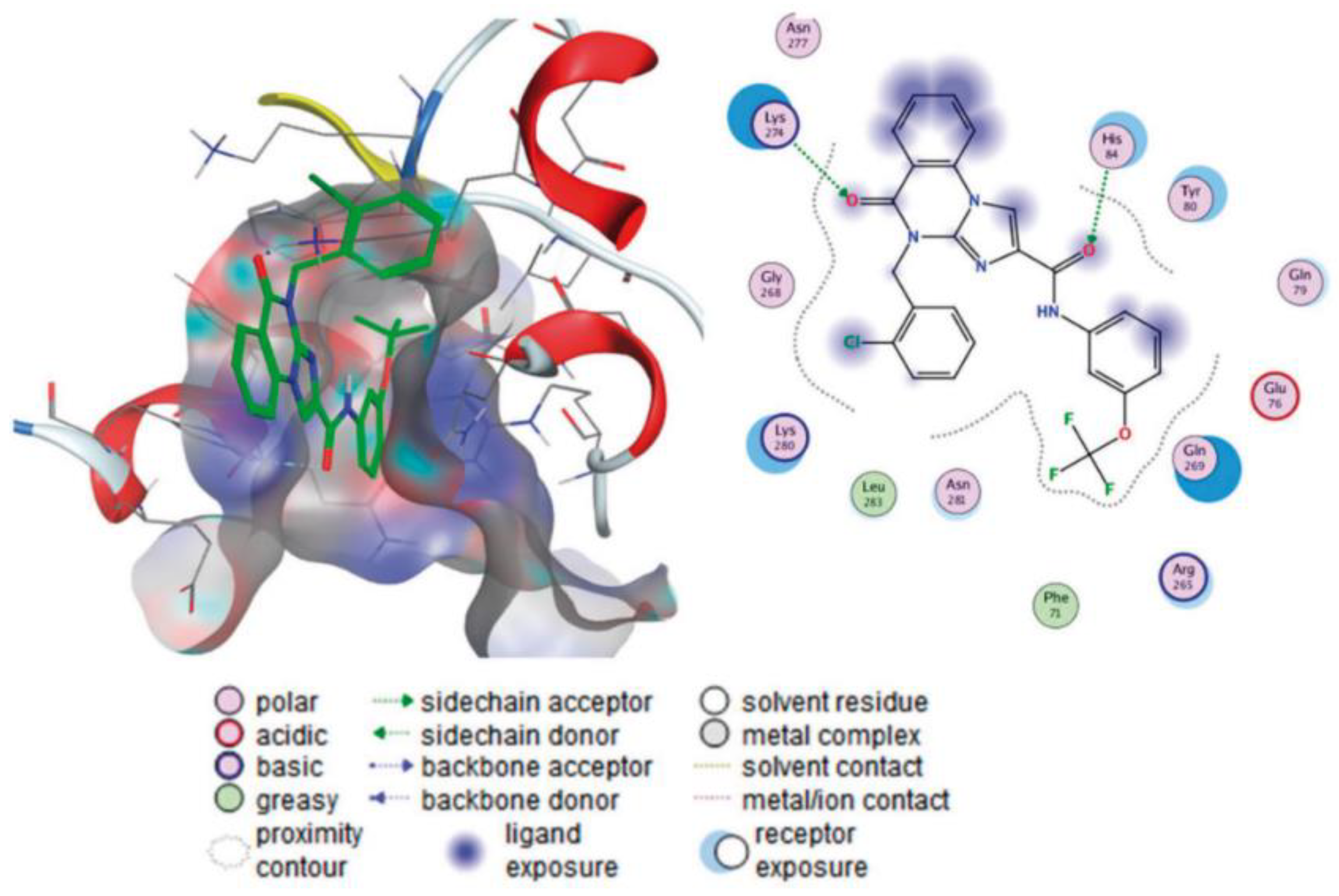
Scheme 36.
Synthesis of imidazo[1,2-a]quinazoline derivative 166, diimidazo[1,2-a:1',2'-c]quinazoline derivatives 167, 168.
Scheme 36.
Synthesis of imidazo[1,2-a]quinazoline derivative 166, diimidazo[1,2-a:1',2'-c]quinazoline derivatives 167, 168.
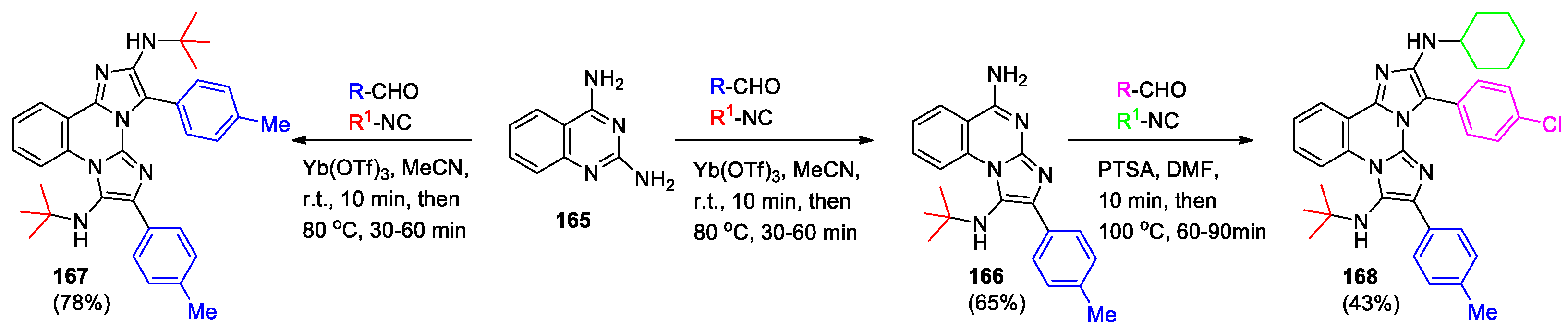
Scheme 37.
Synthesis of benzo[4,5]thiazolo[3,2-a]quinazoline 169, thiazolo[3,2-a]quinazolin-5-ones 172.
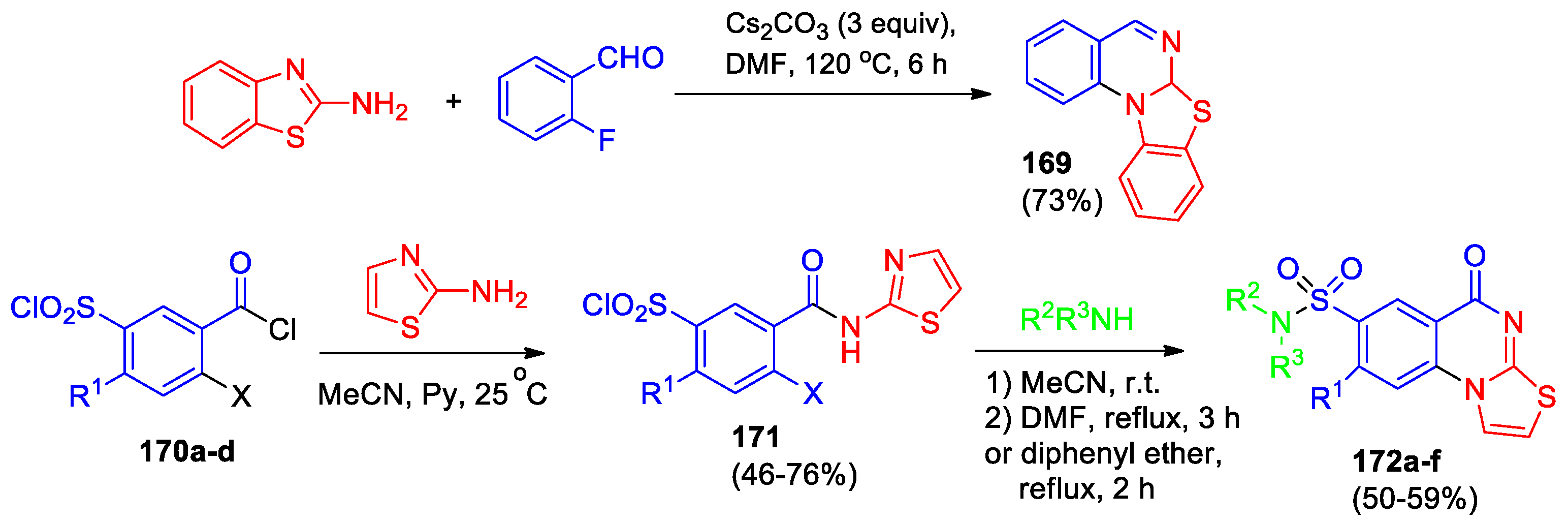
Scheme 38.
Synthesis of 4-aryl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 175 (24 compounds) and 176–180. 174, 175: R = H, o(p)-OMe, p-OC3H7, p-OC4H9, p-OC5H11, p-OC6H13, p-OC8H17, p-OC10H21, p-OC12H25, p-OC14H29, o(m,p)-F, o(m,p)-Cl, m(p)-Br, o(m,p)-Me, p-CF3, o,p-Cl2.
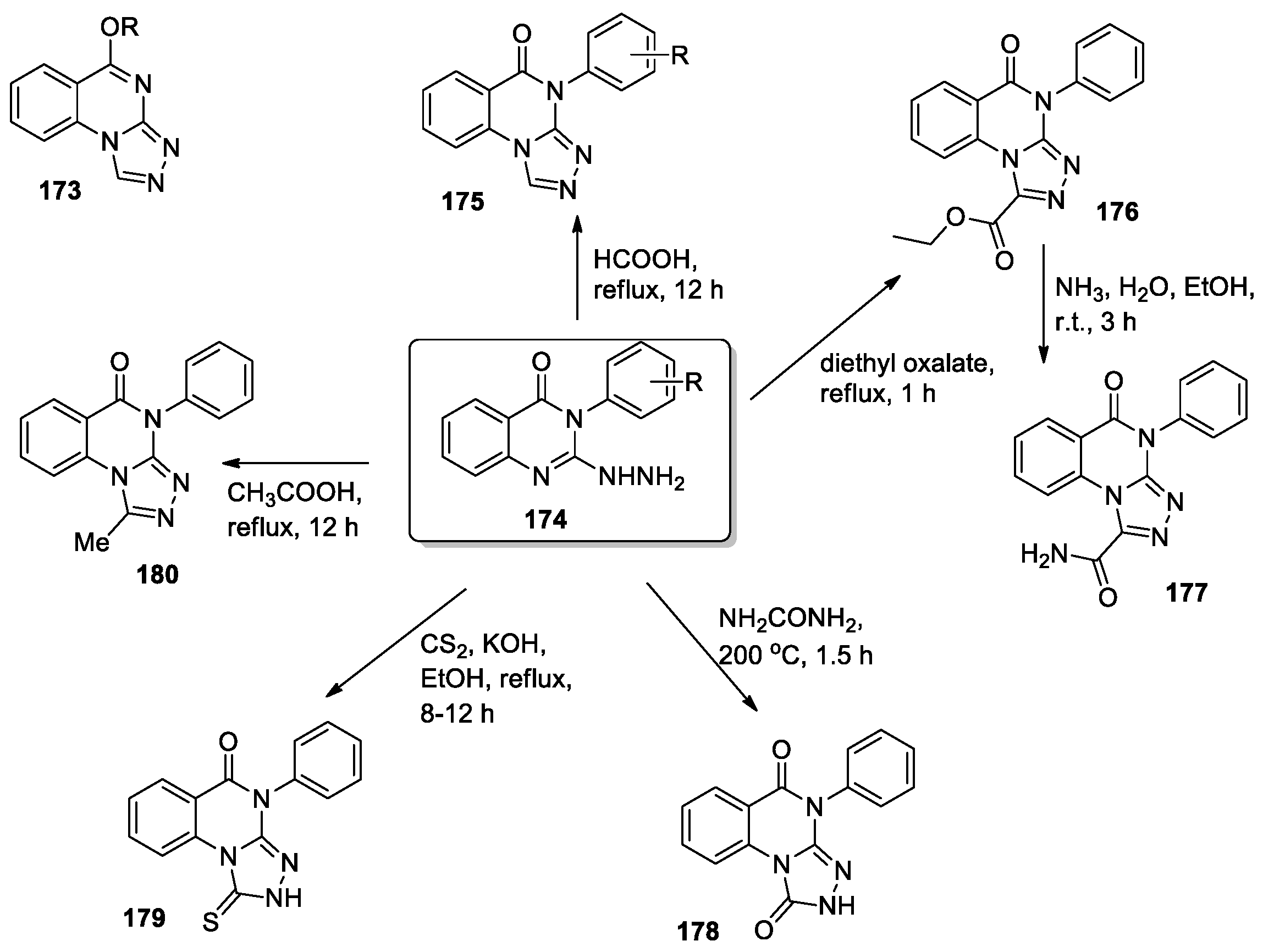
Scheme 39.
Synthesis of 1-substituted-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 182, 183, 185a-e, 186a-e. 181: R = NHNH2 (a), NH-iC5H11 (b); 184: R = pyridine-4-yl (a), 4-nitrophenyl (b); 185, 186: R1 = H (a), Me (b), Et (c), n-Pr (d), CH2Cl (e).
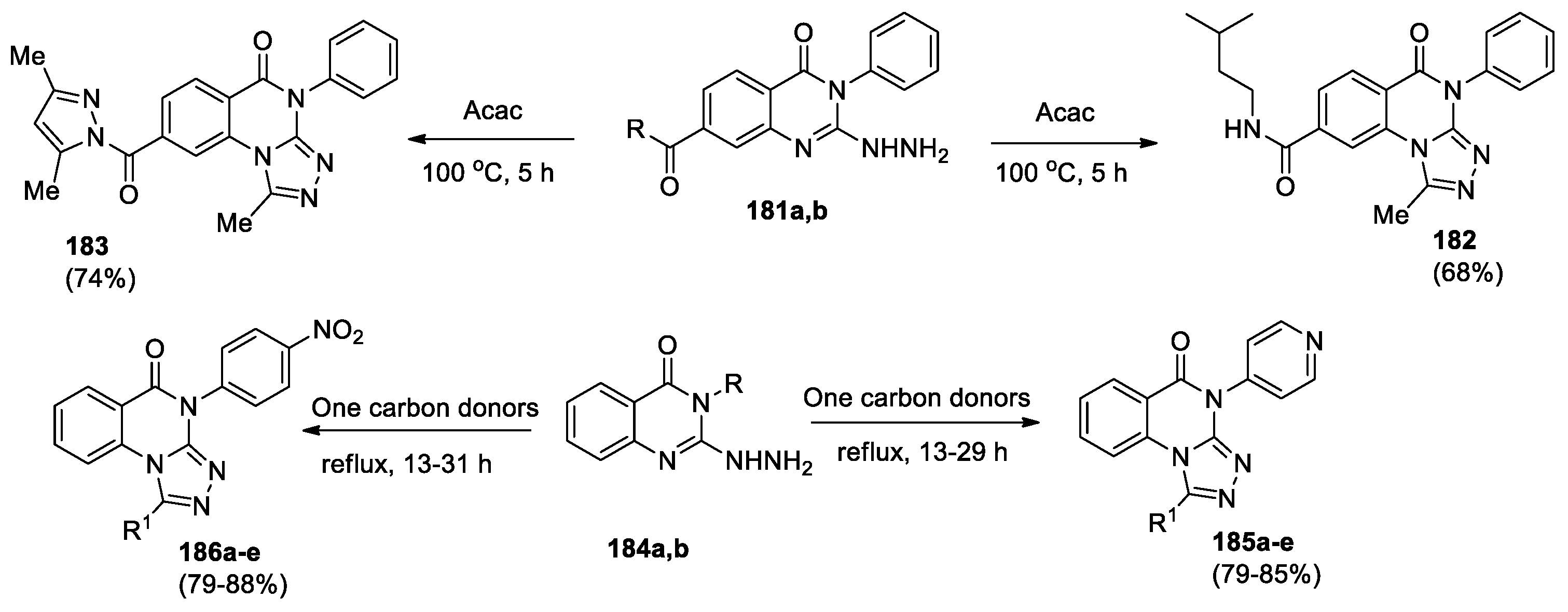
Scheme 40.
Synthesis of 4-(2-chlorobenzyl)containing [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-ones 189 and 190.
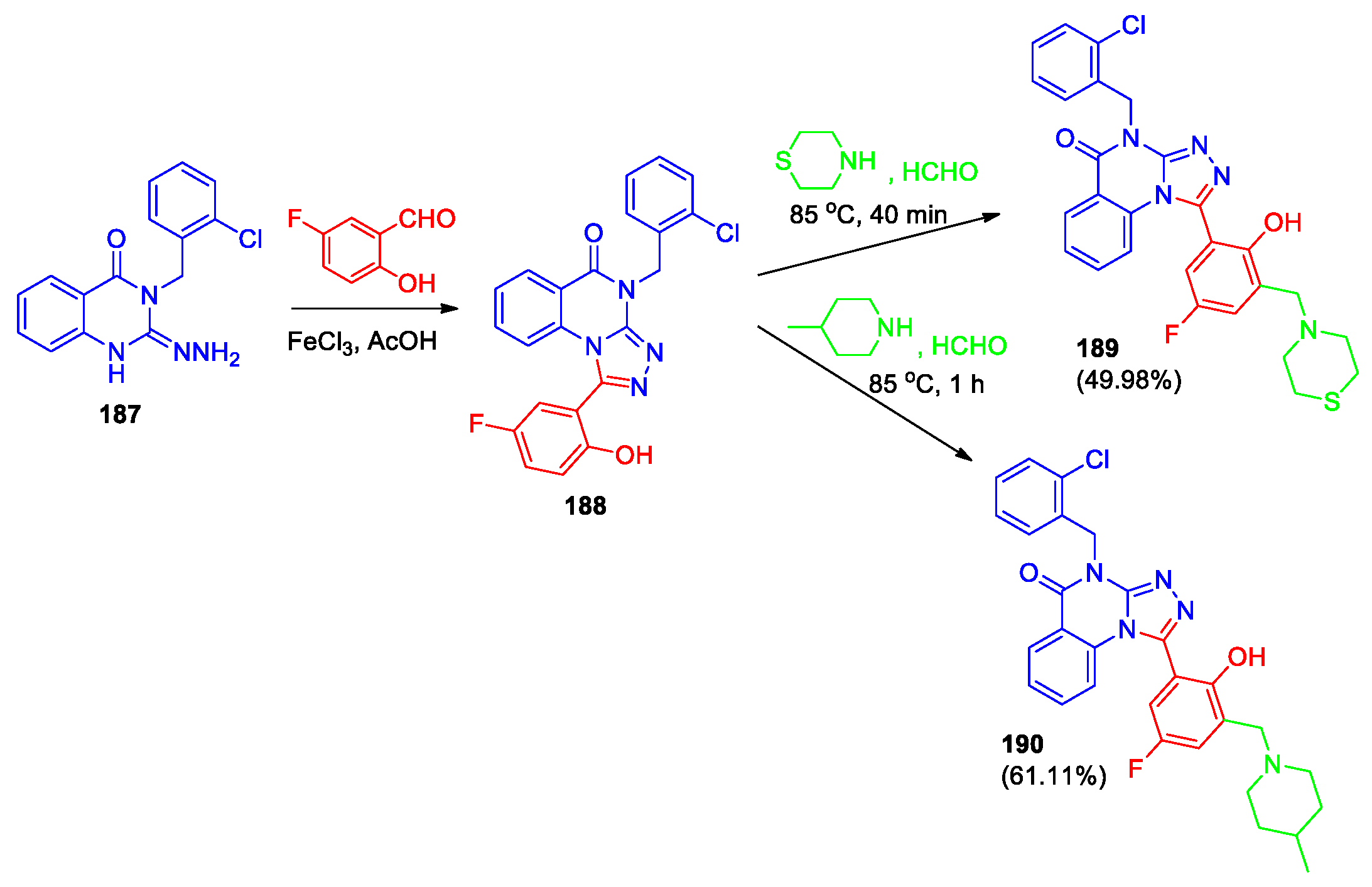
Scheme 41.
Synthesis of [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one derivatives 192, 193, 195. 192: Ar = furan-2-ylmethyl (1-6), thiophen-2-ylmethyl (7-10), 2-chlorobenzyl (11), 4-fluorobenzyl (12-16, 27), 4-methoxybenzyl (17-26, 28); 193, 194: Ar = 4-fluorobenzyl (a), 4-methoxybenzyl (b); 195: Ar, R1 = 4-fluorobenzyl, 2-morpholinoacetyl (a), 4-methoxybenzyl, 2-morpholinoacetyl (b), 4-methoxybenzyl, 2-(pyrrolidin-1-yl)acetyl (c).
Scheme 41.
Synthesis of [1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one derivatives 192, 193, 195. 192: Ar = furan-2-ylmethyl (1-6), thiophen-2-ylmethyl (7-10), 2-chlorobenzyl (11), 4-fluorobenzyl (12-16, 27), 4-methoxybenzyl (17-26, 28); 193, 194: Ar = 4-fluorobenzyl (a), 4-methoxybenzyl (b); 195: Ar, R1 = 4-fluorobenzyl, 2-morpholinoacetyl (a), 4-methoxybenzyl, 2-morpholinoacetyl (b), 4-methoxybenzyl, 2-(pyrrolidin-1-yl)acetyl (c).
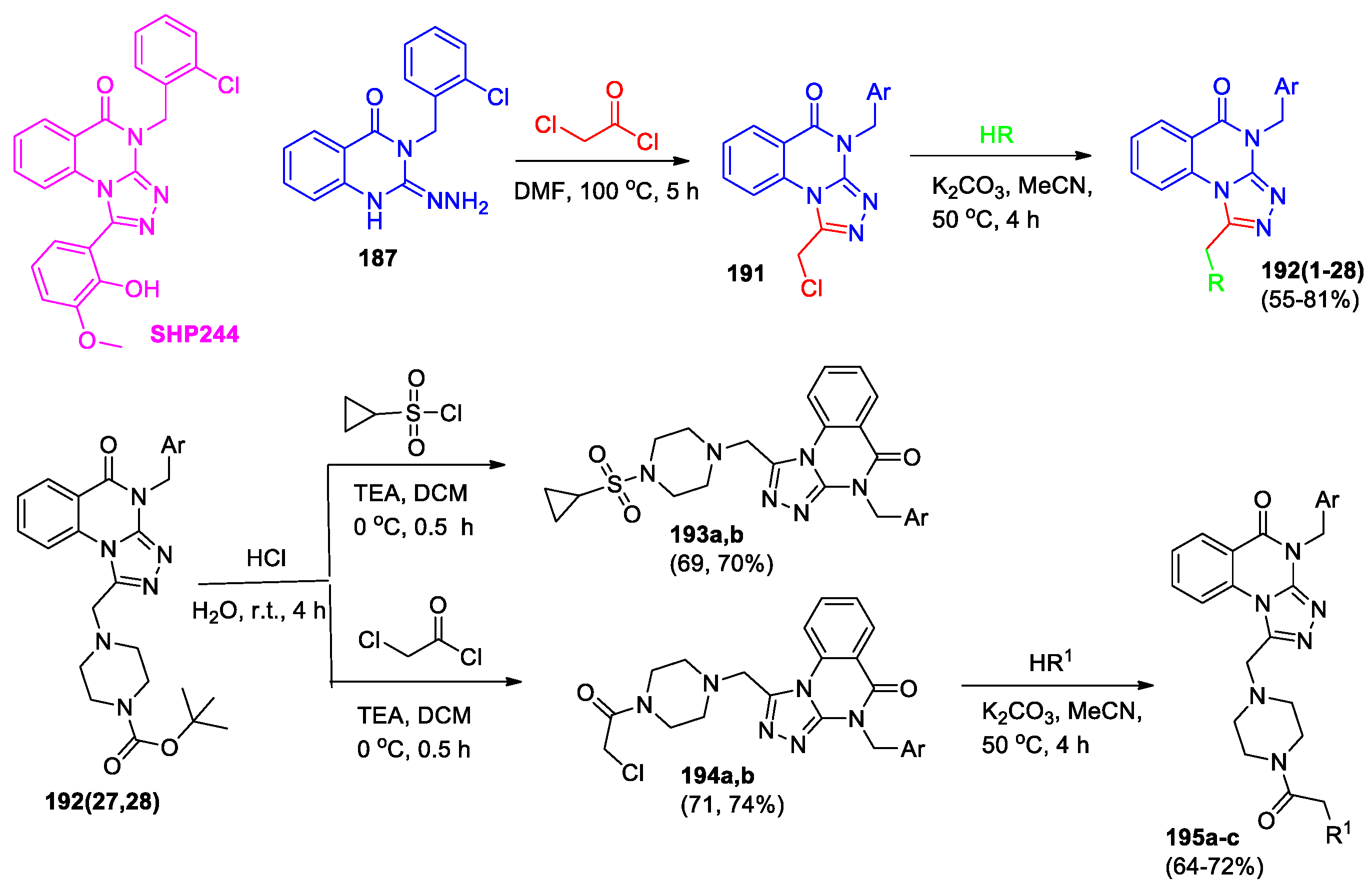
Figure 9.
Interaction of compound 195c with SHP2 protein. Reproduced with permission of Taylor & Francis [80].
Figure 9.
Interaction of compound 195c with SHP2 protein. Reproduced with permission of Taylor & Francis [80].
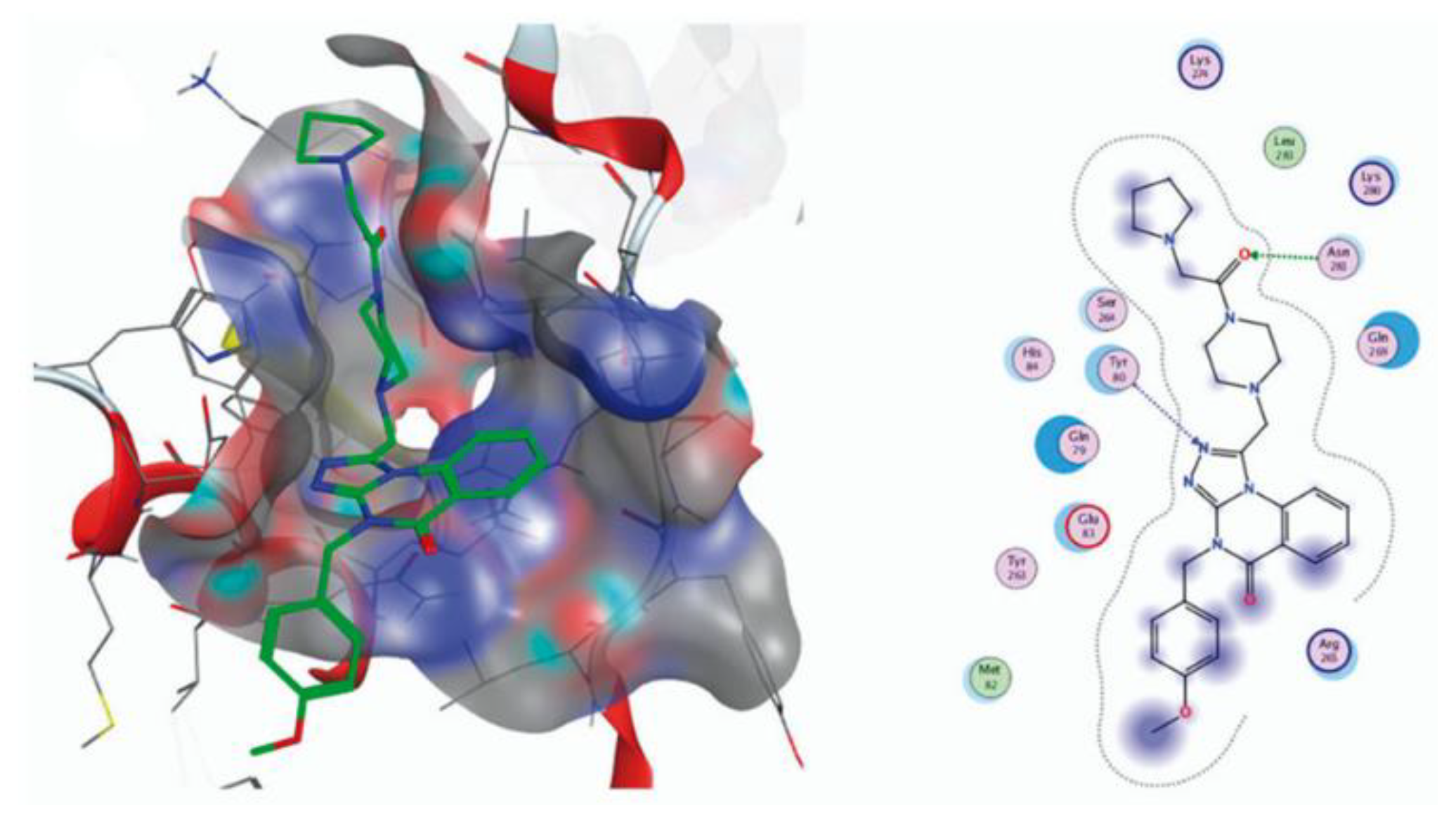
Figure 10.
Chemical structures of the compounds 196, 197, 198a,b, 199. 196: R = H (a), 4-OEt (b), 4-Me (c).
Figure 10.
Chemical structures of the compounds 196, 197, 198a,b, 199. 196: R = H (a), 4-OEt (b), 4-Me (c).
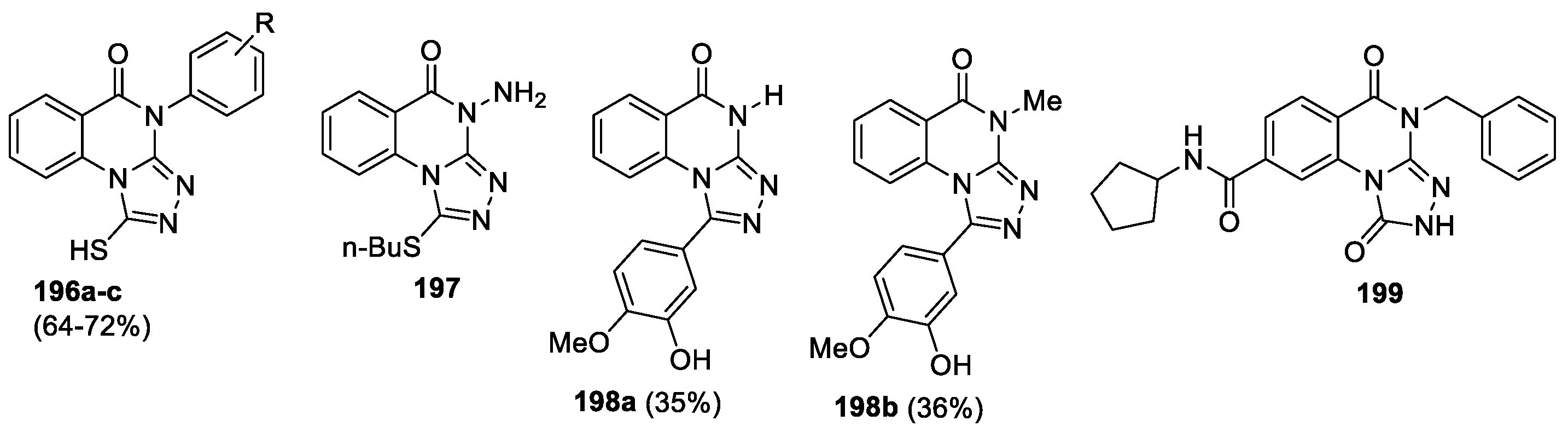
Figure 11.
Chemical structures of the compounds 200, 201, 202.
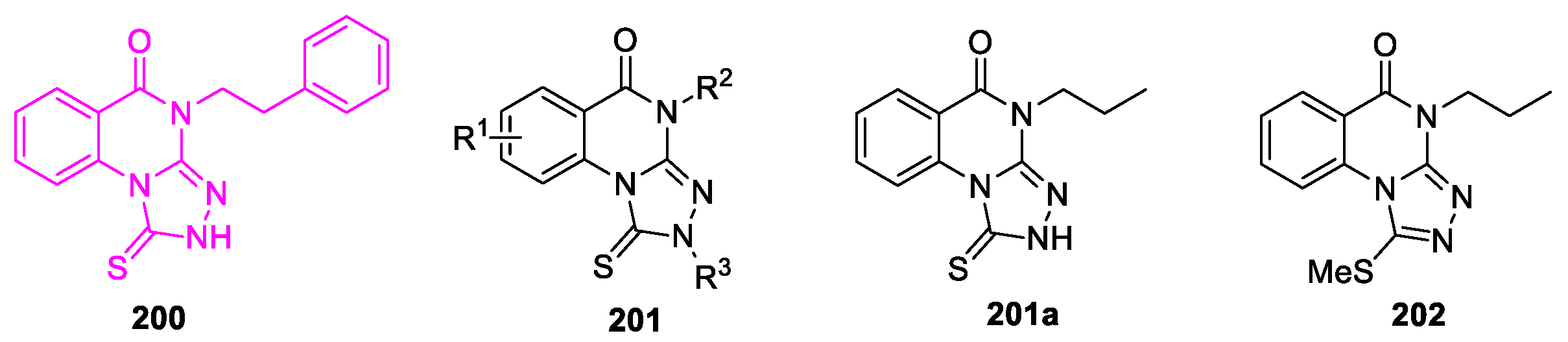
Scheme 42.
Synthesis of [1,2,4]triazolo[1,5-a]quinazolin-5-ones204 and their aza-analogs 205, [1,2,4]triazolo[1,5-a]quinazolines 206, 207. 204, 205: R1 = H (a), Ph (b); 206: R = H (a), Br (b), Me (c). Efficient method was developed for the synthesis of [1,2,4]triazolo[1,5-a]quinazolines 206 from 2-fluoro benzaldehydes 57 with 1H-1,2,4-triazol-3-amine under metal-free conditions in high yields (Scheme 42) [52]. The process includes an intermolecular condensation followed by metal-free base-promoted intramolecular C–N coupling reaction. It's worth mentioning that the bromine atom stays intact during the reaction and can later be modified or used in additional steps.
Scheme 42.
Synthesis of [1,2,4]triazolo[1,5-a]quinazolin-5-ones204 and their aza-analogs 205, [1,2,4]triazolo[1,5-a]quinazolines 206, 207. 204, 205: R1 = H (a), Ph (b); 206: R = H (a), Br (b), Me (c). Efficient method was developed for the synthesis of [1,2,4]triazolo[1,5-a]quinazolines 206 from 2-fluoro benzaldehydes 57 with 1H-1,2,4-triazol-3-amine under metal-free conditions in high yields (Scheme 42) [52]. The process includes an intermolecular condensation followed by metal-free base-promoted intramolecular C–N coupling reaction. It's worth mentioning that the bromine atom stays intact during the reaction and can later be modified or used in additional steps.
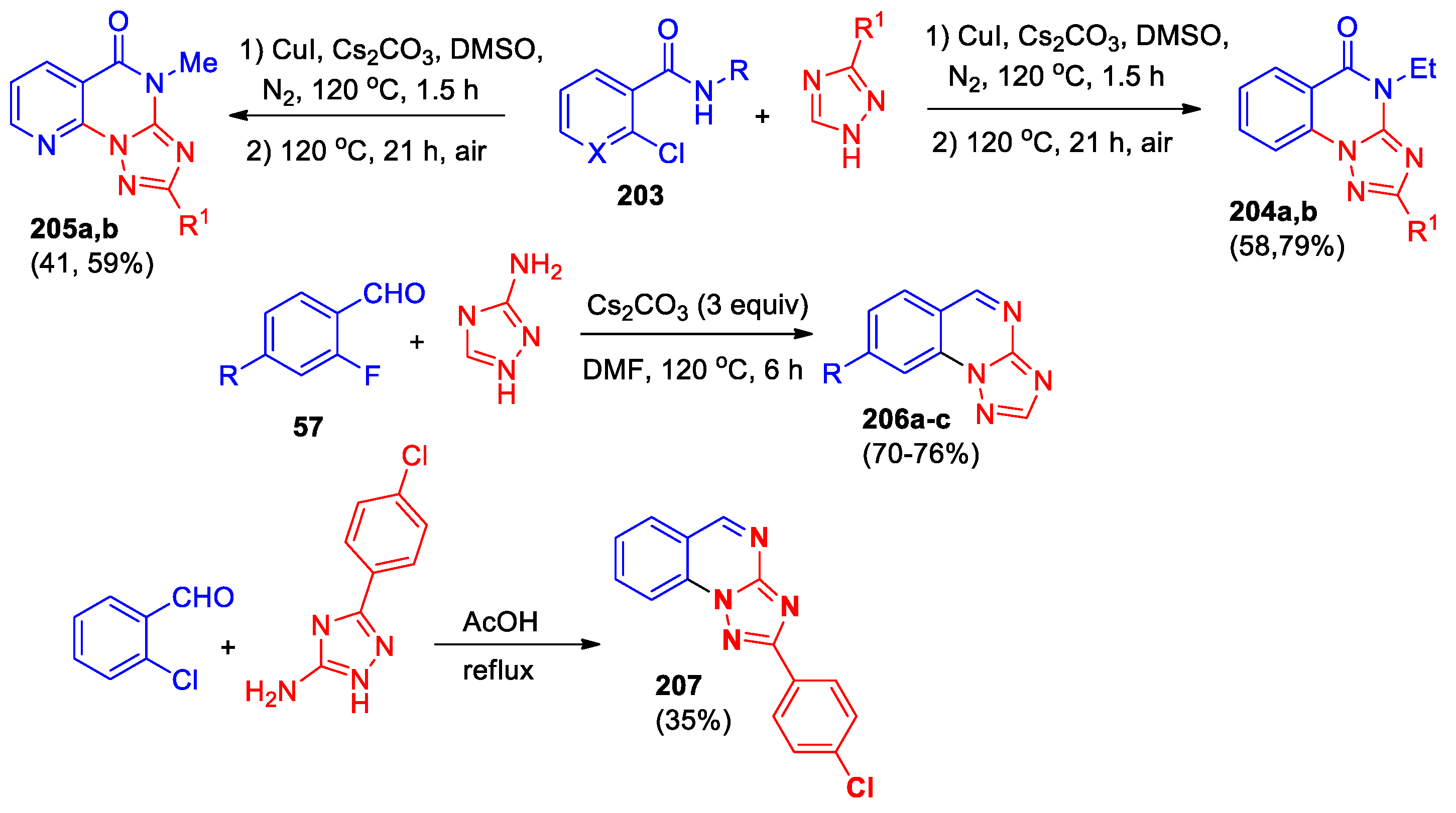
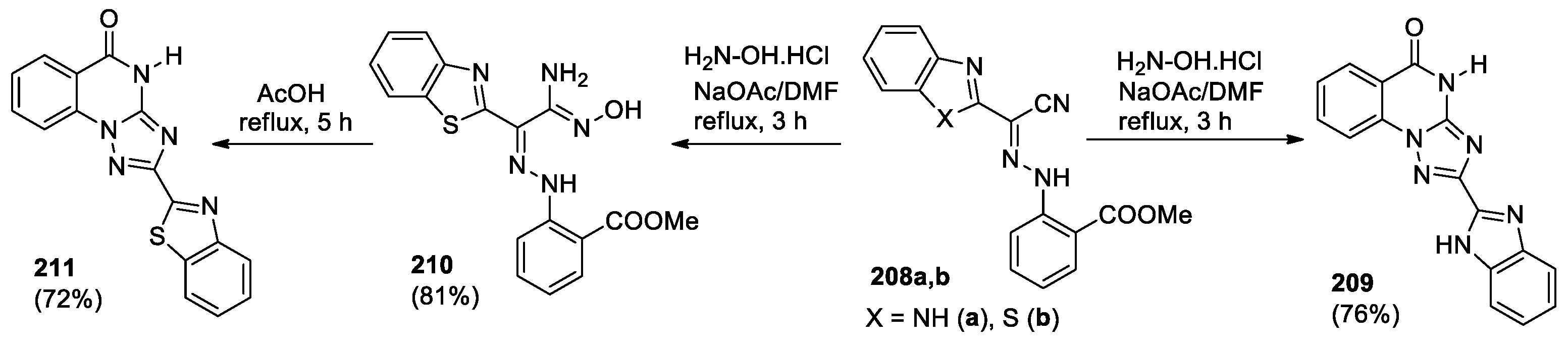
Scheme 44.
Synthesis of 2-phenoxybenzo[g][1,2,4]triazolo[1,5-a]quinazolin-5(4H)-ones 212–215. 213: R = Et, allyl, 2(3)-Me-benzyl, 3-OMe-benzyl, 3(4)-CN-benzyl, 4-Cl-benzyl, 4-NO2-benzyl, 2-(piperidin-1-yl)ethyl, 2-morpholinoethyl, 2-(phtalimido-2-yl)propyl.
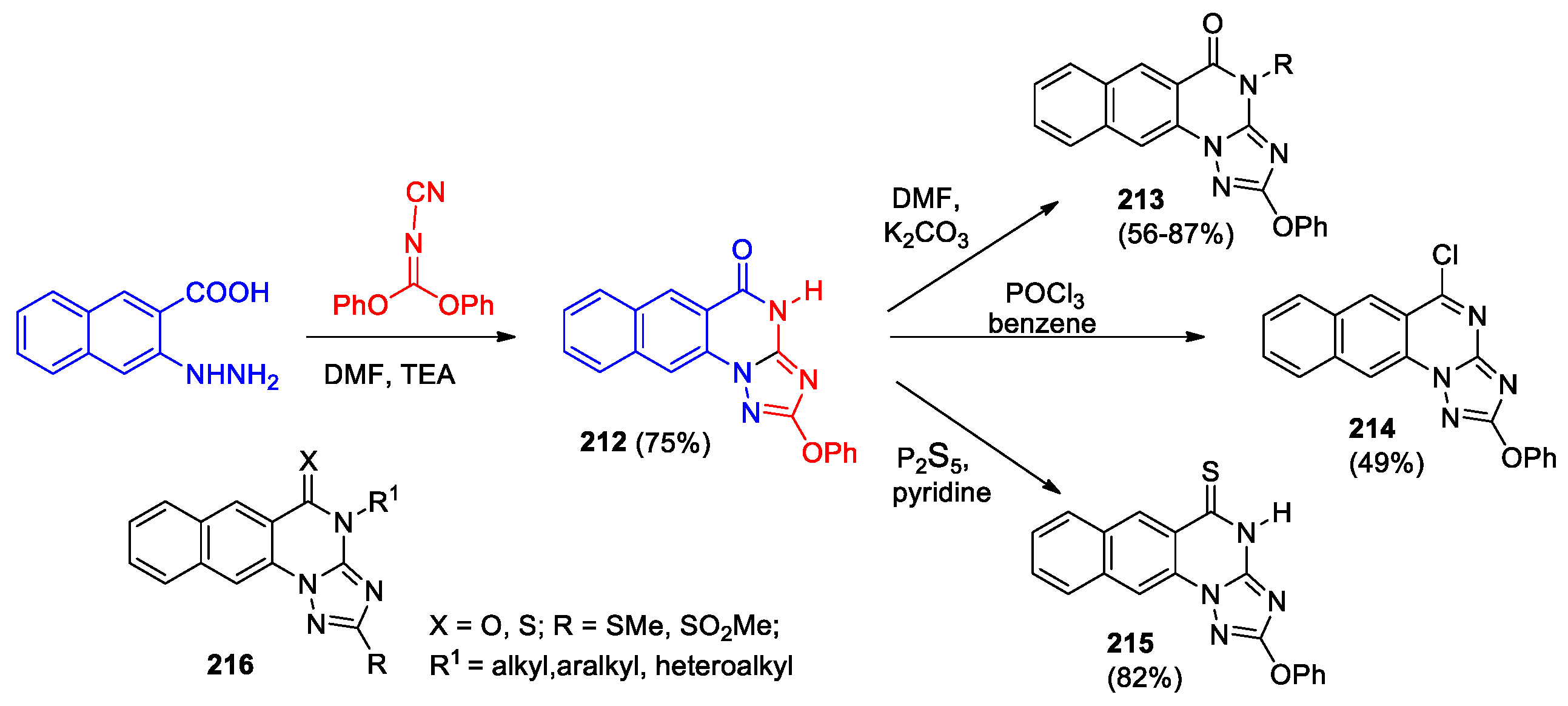
Figure 12.
Chemical structures of the compounds 217–220. 217: R, R1 = H, OPh (a); H, SMe (b); Me, OPh (c); H, SO2Me (d); 218: R, R1 = OPh, 4-NO2-Bn (a); OPh, Et (b); SO2Me, allyl (c); SO2Me, 4-NO2-Bn (d); 220: R = Et (a), n-Pr (b).
Figure 12.
Chemical structures of the compounds 217–220. 217: R, R1 = H, OPh (a); H, SMe (b); Me, OPh (c); H, SO2Me (d); 218: R, R1 = OPh, 4-NO2-Bn (a); OPh, Et (b); SO2Me, allyl (c); SO2Me, 4-NO2-Bn (d); 220: R = Et (a), n-Pr (b).
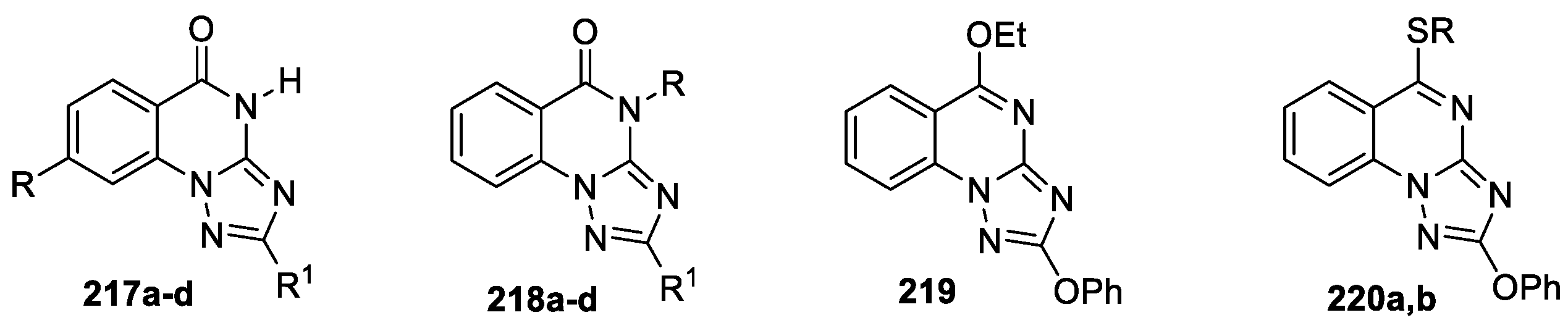
Figure 13.
Chemical structures of the compounds 221, 222, 223a-d, 224, 225a,b, 226a,b. 223: R, R1 = H, allyl (a); Me, Bn (b); H, Bn (c); H, 4-NO2-Bn (d); 225: R1, R2 = H, 3-Py (a); Me, 4-NO2-Ph (b); 226: R1 = allyl (a), cyclohexyl (b).
Figure 13.
Chemical structures of the compounds 221, 222, 223a-d, 224, 225a,b, 226a,b. 223: R, R1 = H, allyl (a); Me, Bn (b); H, Bn (c); H, 4-NO2-Bn (d); 225: R1, R2 = H, 3-Py (a); Me, 4-NO2-Ph (b); 226: R1 = allyl (a), cyclohexyl (b).
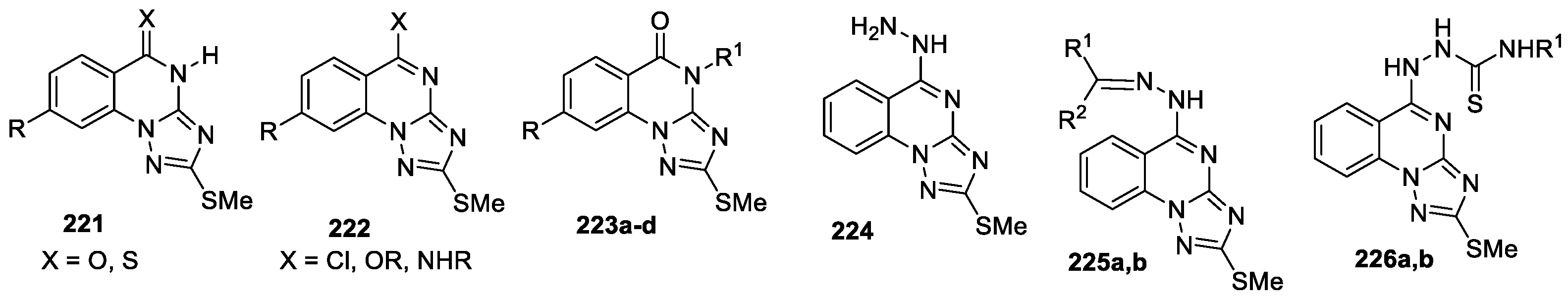
Figure 14.
Chemical structures of the compounds 227, 228, 229a,b, 230, 231. 229: R1 = Et (a), allyl (b).
Figure 14.
Chemical structures of the compounds 227, 228, 229a,b, 230, 231. 229: R1 = Et (a), allyl (b).
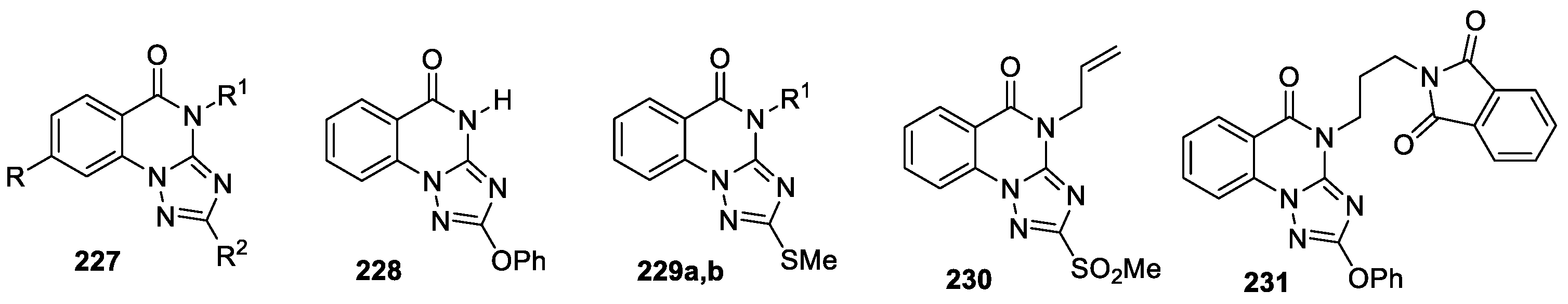

Table 1.
Inhibitory activities of compounds A, 64p, 64r, 64s against KDM4D.
| Compound | Inhibition rate at 10 μM | IC50 (μM) |
|---|---|---|
| A | 73.33 ± 19.14 | 3.14 ± 0.18 |
| 64p | 85.06 ± 3.54 | 4.03 ± 1.02 |
| 64r | 88.82 ± 9.58 | 0.41 ± 0.03 |
| 64s | 78.06 ± 6.27 | 1.85 ± 0.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



