
Submitted:
05 August 2025
Posted:
06 August 2025
You are already at the latest version
Abstract
Feminizing adrenocortical tumors (FATs) represent an exceedingly rare subset of primary adrenal neoplasms, characterized by elevated estrogen levels and suppressed gonadotrophin concentrations. These tumors predominantly affect adult males and are commonly associated with clinical manifestations such as gynecomastia, features of hy-pogonadism and unintended weight loss. The vast majority of FATs are malignant in nature, exhibiting an unfavorable prognosis and a high likelihood of recurrence.
We present the case of a 24-year-old male diagnosed with adrenocortical carcinoma, characterized by elevated estrogen levels (90.1 pg/mL), who initially presented with symptoms related to abdominal mass effect and bilateral gynecomastia. Notably, the patient reported one year of unexplained infertility, which resolved spontaneously postoperatively at three months following surgical intervention.
Another distinctive feature of this case was the patient's adrenal hormonal profile, which revealed suppressed ACTH (2.6 pg/mL) in the presence of inappropriately normal cortisol levels (12,1 ug/dL). The patient underwent right adrenalectomy, and histo-pathological along with immunohistochemical analyses confirmed a diagnosis of stage II adrenocortical carcinoma (T2, Nx, M0) with autonomous estradiol production. The case was reviewed by a multidisciplinary tumor board for consideration of mitotane therapy; however, the patient was not enrolled due to a completely resected stage T2NxM0 adrenocortical carcinoma with negative margins and a Ki-67 index of 10%.
This case underscores the rare presentation of feminizing adrenocortical carcinoma and emphasizes the importance of early diagnosis and close postoperative surveillance, given the tumor’s high recurrence potential.
In addition, we conducted a systematic review of the literature from 2015 to 2025, identifying 12 male cases of estrogen-secreting adrenocortical carcinoma (ES-ACC) pre-senting initially with gynecomastia. Data on presentation, hormonal profile, treatment, recurrence and survival were analyzed. Our review highlights a potential paradigm shift in the management of selected low-risk ACC cases, where active surveillance may rep-resent a safe and effective alternative to adjuvant mitotane therapy.
Keywords:
feminizing adrenocortical carcinoma
; bilateral gynecomastia
; estrogen secreting tumour
; fertility
; low-risk ACC
1. Introduction
Primary malignancies of the adrenal gland are exceedingly uncommon. In the United States, adrenocortical carcinoma is diagnosed in approximately 200 individuals annually, while pheochromocytomas and paragangliomas are identified in an estimated 500 to 1,600 cases per year [1]. Adrenocortical carcinomas (ACCs) are uncommon malignancies, with a global incidence estimated at approximately 2 cases per million individuals annually [2]. These tumors display a bimodal age distribution, with a first incidence peak occurring in early childhood (under the age of 5) and a second peak typically observed during the fourth to fifth decades of life [3]. Although adrenal adenomas and adrenocortical carcinomas are frequently non-functioning, they can, in certain instances, secrete corticosteroids or sex steroids. Hormone-secreting tumors may give rise to distinct clinical syndromes, with Cushing syndrome being the most prevalent, occurring in approximately 5 to 20% of incidentally discovered adrenal tumors. Another possible presentation includes primary aldosteronism (Conn’s syndrome) [4].
Feminizing adrenocortical tumors (FATs) are a rare subset of functional adrenal cortical neoplasms that produce only estrogens, specifically estradiol and estrone. These tumors can present as either benign estrogen-producing adrenal adenomas or, more frequently, as malignant estrogen-secreting adrenocortical carcinomas. The excess estrogen typically leads to suppression of testosterone production. FATs are extremely uncommon, comprising under 2% of all adrenal tumors [4,5]. Between 1970 and 2015, only 50 cases were reported in the literature [6], with only a few additional cases documented since then.
Presenting signs/symptoms in adult males commonly include gynecomastia, hypogonadism, and weight fluctuation (either loss or gain, with loss being more common); in adult females, there is irregular or postmenopausal bleeding; in pediatric males, there is contrasexual precocious pseudopuberty; and in pediatric females, there is isosexual precocious puberty [7]. In pure FATs, there is usually a lack of signs or symptoms of Cushing syndrome, hypertension, and virilization/hirsutism, differentiating FATs from mixed functional adrenocortical tumors. Diagnosis is made by a combination of clinical factors (including gynecomastia, change in libido, and erectile dysfunction in adult males), serum laboratory values (including elevated estrogen), radiologic imaging findings (i.e., a visualized adrenal mass), and immunohistochemistry (IHC) imaging findings (i.e., α-aromatase positivity). Radiologically and histologically, estrogen-secreting adrenocortical carcinomas (ES-ACCs) appear broadly similar to other primary adrenal masses with the exception of α-aromatase positivity for FATs [4]. Distinguishing malignant from benign FATs is challenging, but can be assisted with radiographic findings (i.e., irregular borders, heterogeneity, invasion, etc.), histology (i.e., a high Weiss score), or recurrence/metastases.
Adrenal adenomas are known to have an excellent prognosis. In contrast, adrenocortical carcinomas are associated with reduced survival rates, and among them, feminizing adrenocortical tumors carry the poorest prognosis. Additionally, advanced disease stage at the time of diagnosis is a strong predictor of unfavorable clinical outcomes. Although no definitive causal mutations have been identified in feminizing adrenocortical tumors , several genetic alterations have been implicated, including dysregulation of IGF2 expression, aberrations in the Wnt/β-catenin signaling pathway, and mutations in the TP53.1 gene [8]. Some authors have discussed a mutation in tumor suppressor genes [9,10].
While some adrenocortical carcinomas have been associated with hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN1), Carney complex, Beckwith–Wiedemann syndrome, and Li-Fraumeni syndrome, many cases appear to occur sporadically [1]. In the context of feminizing adrenocortical tumors, aside from a single reported case linked to MEN1, genetic investigations have not yet clarified a definitive etiology [11]. These tumors are typically marked by elevated aromatase activity, although in some cases the increase is only moderate, despite the presence of significant hyperestrogenism.
2. Materials and Methods
For the case presentation written informed consent has been obtained from the patient to publish this paper.
A comprehensive literature search was conducted to identify relevant studies on FATs, focusing on symptoms, treatment strategies and outcomes. The search strategy encompassed electronic databases and academic repositories from June 1993 to May 2025. The following databases were utilized: PubMed/MEDLINE, Scopus, Web of Science and Embase.
The search strategy combined Medical Subject Headings (MeSH) terms and free-text keywords, including but not limited to: “Feminizing adrenal carcinoma”, “Infertility due to adrenocortical carcinoma with autonomous estradiol secretion”, “Gynecomastia due to feminizing adrenal tumour”, “ Hyperestrogenism”, “Estrogen secreting tumours”, “ Low-risk ACC “, “Treatment outcomes”, “Mitotane”, “Patient outcomes”.
The search paradigm included combinations of Boolean operators (AND, OR) to refine the search results. Example search strings used were: “ Feminizing adrenal carcinoma” OR “Feminizing adrenal tumour “ AND “treatment” OR “mitotane”, “low-risk adrenocortical carcinoma” OR “low-risk ACC”.
To ensure relevance and specificity, the PICO (Population, Intervention, Comparison, Outcome) framework was applied. Inclusion criteria encompassed peer-reviewed articles, reviews, and observational studies published in English. Exclusion criteria included non-English articles, letters to the editor, and studies not directly related to feminizing adrenal carcinoma treatment or outcomes. Case reports were included due to the rarity of the condition and their relevance to clinical presentation and outcomes.
Two independent reviewers screened titles and abstracts for relevance. Full-text articles meeting inclusion criteria underwent detailed review. Data extraction included study design, participant characteristics, interventions, outcomes, and key findings.
3. Results
3.1. Case Presentation
We present the case of a 24-year-old male diagnosed with adrenocortical carcinoma associated with bilateral gynecomastia due to tumor-derived estrogen secretion.
The disease onset was marked by abdominal discomfort in the right upper quadrant, reported approximately one month before surgical intervention.
An initial abdominal ultrasound revealed a nodular mass superior to the right kidney. Further imaging with contrast-enhanced abdominal MRI revealed a well-defined, macronodular right adrenal mass measuring 8.2 × 6 × 5.8 cm, exhibiting internal areas of necrosis and progressive gadolinium enhancement. The lesion was in contact with the superior pole of the right kidney (without evidence of invasion), the inferior vena cava over a 3.4 cm segment, hepatic segments VI and VII, the right diaphragmatic crus, and the duodenum, without a clearly defined plane of separation.
Clinical examination was unremarkable except for bilateral symmetric gynecomastia, noted by the patient approximately five months prior.
Following identification of the retroperitoneal adrenal mass with imaging features suggestive of malignancy, an extensive endocrine evaluation was conducted. The results showed low ACTH (2.6 pg/mL), inappropriately normal cortisol (12,1 ug/dL), and significantly elevated estradiol (331 pmol/L), most likely of adrenal origin. Aldosterone, renin, metanephrines, and normetanephrines were within normal limits.
Given the patient’s age, hormonal profile, and imaging features of the adrenal mass, a clinical and paraclinical diagnosis of right-sided ACC was established. Surgical management involved a right adrenalectomy.
Figure 1.
Gross appearance of the resected adrenal tumour. The mass was large, solitary and circumscribed tumor (9.5×7×3.5 cm).
Figure 1.
Gross appearance of the resected adrenal tumour. The mass was large, solitary and circumscribed tumor (9.5×7×3.5 cm).
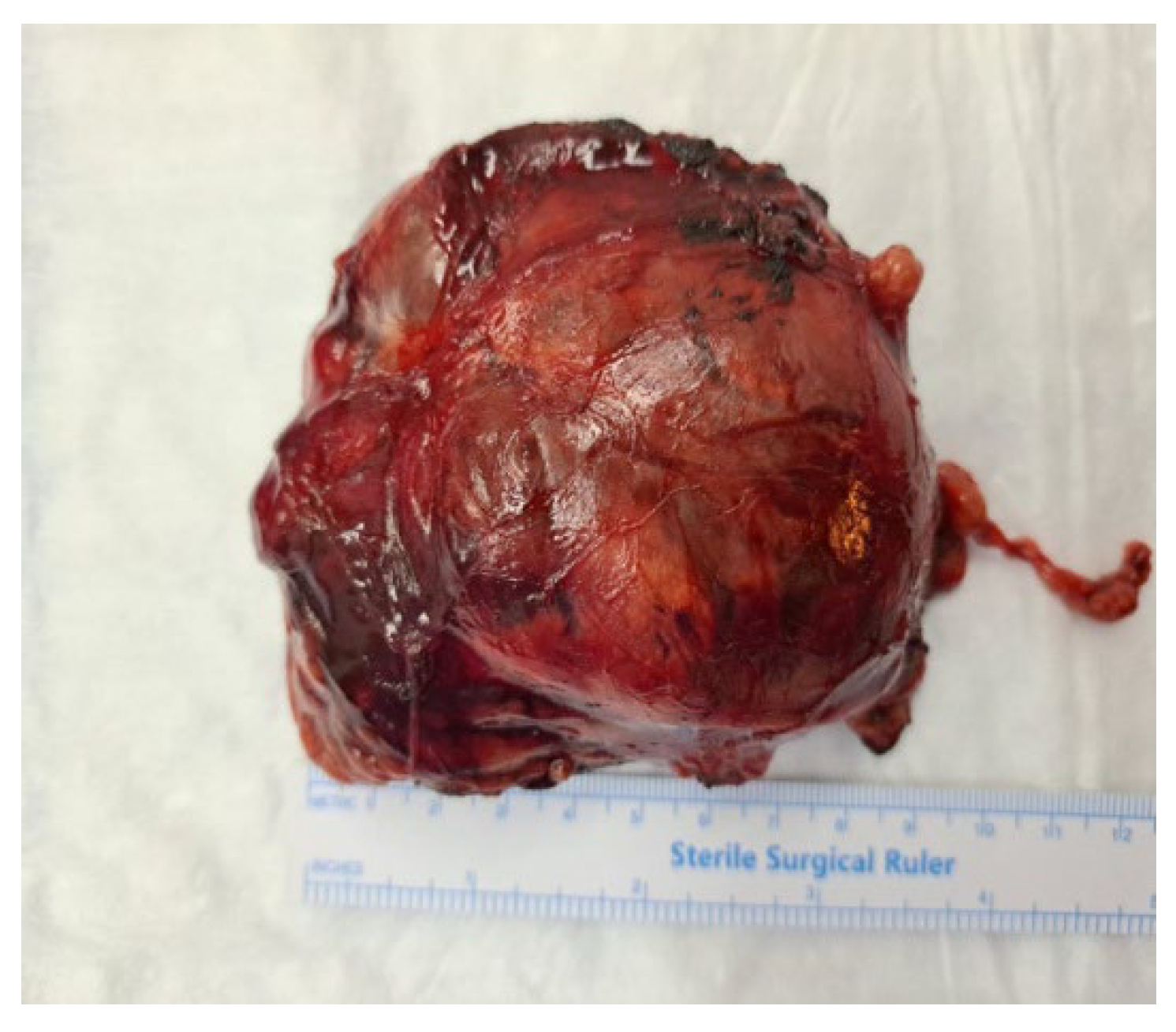
Figure 2.
Gross appearance of the resected adrenal tumour. The mass was large, solitary and circumscribed tumor (9.5×7×3.5 cm).
Figure 2.
Gross appearance of the resected adrenal tumour. The mass was large, solitary and circumscribed tumor (9.5×7×3.5 cm).
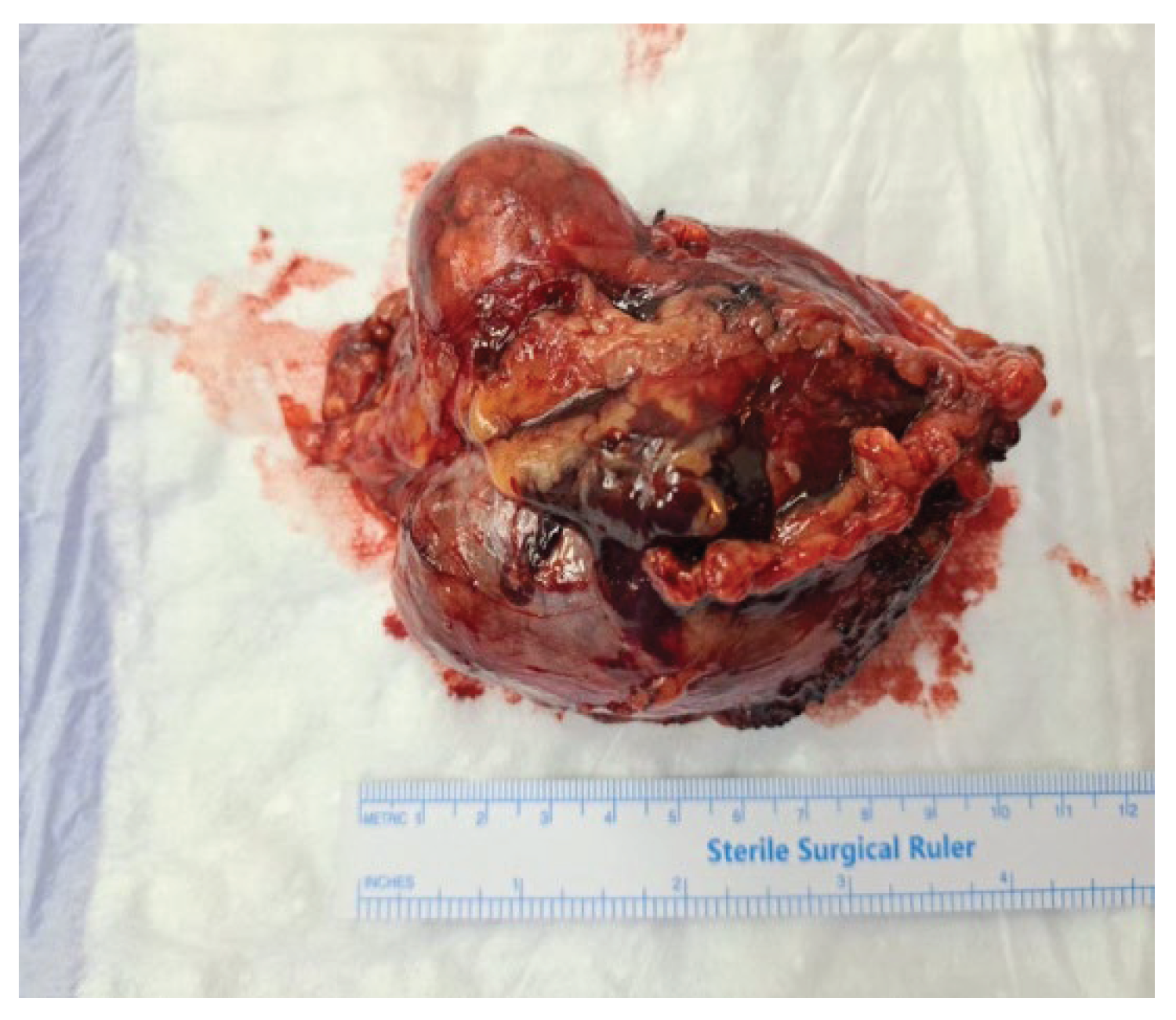
Histopathological evaluation confirmed conventional adrenocortical carcinoma, weighing 162 grams and measuring 9.5 x 7 x 3.5 cm. Weiss criteria assessment revealed five of nine criteria and the patient was diagnosed with a malignant tumor: a Fuhrman nuclear grade of 4, mitotic index of 8 mitoses/50 HPF, presence of atypical mitoses, tumor necrosis, and diffuse architecture with loss of the reticulin framework.
Immunohistochemistry showed a Ki-67 index of 10%, weak cytoplasmic synaptophysin staining in rare tumor cells, Melan-A positivity, granular cytoplasmic inhibin positivity in rare cells, cytoplasmic and focal nuclear calretinin positivity, and p53 positivity in <1% of tumor cells.
Based on histopathological and immunohistochemical findings, the diagnosis was confirmed as stage II adrenocortical carcinoma (T2, Nx, M0) with autonomous estradiol secretion.
Postoperative evaluation at one month revealed no significant clinical changes except for persistent bilateral gynecomastia. Hormonal reassessment demonstrated normalization of the hypothalamic-pituitary-adrenal axis: ACTH 61.8 pg/mL, serum cortisol 10.11 µg/dL, and estradiol decreased to 123.57 pmol/L (over 50% reduction from baseline). DHEA-S, androstenedione, aldosterone, and renin levels were all within normal ranges.
Bilateral breast ultrasound showed retroareolar fibroglandular tissue in a florid phase: right breast with a maximal thickness of 1 cm and length of approximately 2 cm; left breast with a thickness of 1 cm and length of approximately 1.6 cm.
Therapeutic options, including the initiation of adjuvant mitotane therapy, were discussed in detail. Postoperatively, a multidisciplinary team was assembled to evaluate the initiation of mitotane therapy. However, based on the most recent clinical data, the Ki-67 index of 10%, and the patient’s preferences-the patient expressed a preference to defer systemic treatment temporarily in consideration of family planning; it was decided to defer adjuvant treatment for the time being. Follow-up imaging with abdominal-pelvic MRI and thoracic CT was scheduled every 3 months during the first two years, then every 6 months over the next three years in the absence of recurrence.
At the three-month postoperative follow-up, gynecomastia persisted. Hormonal profile showed elevated ACTH (114 pg/mL), elevated cortisol (19,9 ug/dL), normal DHEA-S (128 µUI/mL), and a normalized gonadal axis: LH 1.2 µUI/mL, FSH 2.72 mUI/mL, testosterone 3.1 µg/mL, and estradiol 24.2 pg/mL. (Table 1)
Breast ultrasound indicated a stable glandular appearance: right breast retroareolar gland measuring 15 x 7 mm and left breast 5 x 10 mm, both in a proliferative phase. Follow-up abdominal-pelvic MRI did not reveal any suspicious tumor recurrence.
4. Discussion
This case exhibits several atypical features, including right upper quadrant abdominal pain, most likely related to the large size of the adrenal mass, although the patient was of normal weight. On clinical examination, bilateral gynecomastia was also identified, which the patient reported noticing approximately five months earlier. Regarding paraclinical evaluations, estradiol levels had normalized by three months postoperatively. Although preoperative gonadotropin levels are not available, the most recent assessment showed low-normal FSH and LH levels, suggesting that these were likely suppressed preoperatively due to estrogen hypersecretion by the tumor.
Feminizing adrenocortical carcinomas are typically sizable masses and become palpable in approximately 62% of cases at advanced stages. In most instances, the tumor exceeds 10 cm in diameter by the time it is detected [5,12]. The majority of adrenocortical carcinoma cases described in the literature initially presented with abdominal pain as the primary symptom [13,14].
The underlying mechanisms of abnormal breast development can be grouped into four categories: estrogen excess, androgen deficiency, drug-induced changes, and hepatic dysfunction [5]. In our case, the patient noticed breast enlargement approximately five months before the onset of abdominal pain, without any other associated symptoms. However, this change was not significant enough to prompt him to seek medical attention earlier.
Gynecomastia is sometimes the primary clinical manifestation and represents a ‘true’ form, resembling normal female breast tissue. It is typically characterized by tenderness, palpable glandular lobules and areolar enlargement, though pigmentation is not always present, and there is no nipple discharge [15]. Differential diagnosis should always consider alternative etiologies, including the use of antihypertensive or antidopaminergic medications, digoxin, liver cirrhosis, bronchogenic carcinoma or other ectopic hormone syndromes, as well as testicular disorders such as testicular feminization syndrome [16].
Adrenocortical carcinomas are marked by disordered steroid hormone production, resulting from inconsistent expression patterns of steroidogenic enzymes, which in turn leads to the release of diverse steroid intermediates. Two main pathways have been suggested to account for the elevated estrogen levels seen in feminizing ACCs. The first involves the peripheral conversion of tumor-produced androgens into estrogens by aromatase activity in adipose tissue. The second proposes that this transformation occurs directly within the tumor itself, as supported by the presence of aromatase detected in tumor cells.
The patient and his partner expressed a desire to conceive and reported unprotected sexual intercourse over the past year, without success. Spontaneous conception occurred two months after surgery. It can be hypothesized that the prior infertility was due to suppression of gonadotropin secretion via negative feedback from adrenal-origin hyperestrogenism. This hypothesis is supported by the observation that gonadotropin levels remained at the lower limit of the normal range even three months postoperatively.
Within the endocrinology literature, feminizing adrenal tumors in males are typically associated with gynecomastia and hypogonadotropic hypogonadism; however, to date, there are no documented cases of successful conception by the partner following surgical resection.
Regarding the normalization of the hypothalamic–pituitary–gonadal axis, existing medical literature reports a rapid decline in estrogen levels following surgical resection, with normalization of gonadotropins occurring approximately two months postoperatively. In the article by Joseph M., it is specifically noted that gonadotropin levels returned to normal at two months after surgery [17]—a finding that was also observed in our patient. Therefore, it can be concluded that the hypothalamic–pituitary–gonadal axis tends to recover almost completely within approximately three months following curative and recurrence-free surgical excision.
Regarding treatment, surgical intervention is recommended for all resectable cases of feminizing adrenocortical carcinoma without evidence of metastasis, as histopathological analysis remains the definitive method for confirming malignancy, and surgery represents the only potential curative approach. A laparoscopic approach is not recommended due to the fragile capsule of FACs and the significant risk of rupture. Open laparotomy is the preferred surgical method.
With regard to postoperative adjuvant therapy, a multidisciplinary team was convened to evaluate the available treatment options. In 2017, the research groups led by Massimo Terzolo and Martin Fassnacht published a pivotal study in The New England Journal of Medicine [18], which provided strong evidence supporting the use of mitotane for preventing recurrence in adrenocortical carcinoma. This study established mitotane as the global standard of care for adjuvant therapy, regardless of the presence of risk factors, which were not yet fully defined at that time.
In light of these findings, initiation of mitotane therapy was considered for our patient, but more recent evidence, specifically a clinical trial published in The Lancet Diabetes & Endocrinology in August 2023 by the same group of researchers, challenges the universal need for adjuvant mitotane [19]. Their data suggest that mitotane may be safely omitted in selected low-risk patients, provided three key criteria are met: (1) a complete surgical resection with negative margins (R0), (2) absence of tumor dissemination or advanced staging, and (3) a Ki-67 proliferation index below 10%. In such cases, where the risk of recurrence is minimal, the benefit of adjuvant mitotane is questionable.
Following the establishment of specific criteria to define the low-risk category in adrenocortical carcinoma, the first-ever randomized clinical trial worldwide on adjuvant treatment—ADIUVO—was initiated. This international study enrolled 91 patients across 23 centers in seven countries. Participants with completely resected ACC (R0 resection), stages I–III, and a Ki-67 index of ≤10% were randomized to receive either oral mitotane for two years or active surveillance through regular imaging and laboratory monitoring. The primary endpoint was recurrence-free survival (RFS). At five years, the RFS was 79% in the mitotane group compared to 75% in the surveillance group. Importantly, there was no statistically significant difference in overall survival between the two groups [20].
In our case, these data were presented to the tumor board and after evaluating the patient’s profile—namely, a completely resected stage T2NxM0 adrenocortical carcinoma with negative margins and a Ki-67 index of 10%—the patient was classified as low risk for recurrence. Consequently, the decision was made to delay the initiation of mitotane therapy in favor of active clinical surveillance. This decision also took into account the patient’s preference to postpone adjuvant treatment in order to attempt conception with his partner.
We present in our article a comprehensive review of all reported cases of estrogen-secreting adrenocortical carcinoma in men between 2015 and 2025 and reveals a consistent clinical, biochemical, and histopathologic features.
Gynecomastia was the predominant presenting symptom across all cases, frequently associated with decreased libido, infertility, and, in some instances, testicular atrophy or weight loss. Preoperative hormonal evaluation consistently demonstrated marked hyperestrogenism, with low or suppressed gonadotropin and testosterone levels—supporting a diagnosis of hypogonadotropic hypogonadism secondary to estrogen excess. Several cases also exhibited co-secretion of adrenal androgens or cortisol, underscoring the functional complexity of these tumors.
Tumor sizes ranged from 4.3 cm to over 21 cm, with a majority located in the right adrenal gland. Histopathologic analysis often revealed high Weiss scores and, in some cases, Ki-67 indices exceeding 10%, indicating malignant potential. Immunohistochemistry typically demonstrated positivity for aromatase (CYP19A1), inhibin, synaptophysin, and Melan-A. Open adrenalectomy was the most common surgical approach, although laparoscopic methods were employed in select cases with smaller or well-localized tumors. Adjuvant mitotane therapy was frequently administered, especially in patients with high-risk features, recurrence, or metastatic spread.
While postoperative normalization of estradiol and restoration of gonadotropin levels were commonly observed within weeks to months, gynecomastia persisted in some patients, requiring surgical correction. Notably, fertility recovery was documented in at least one case, suggesting that tumor-induced hypogonadism may be at least partially reversible.
Despite a generally favorable short-term prognosis following complete tumor resection, long-term outcomes varied. Some patients remained recurrence-free under close surveillance, while others developed local recurrence or distant metastases, necessitating systemic chemotherapy. One case was fatal due to hemorrhagic shock shortly after diagnosis, highlighting the potential for acute complications.
Overall, feminizing ACC remains an exceedingly rare but clinically distinct entity, and its timely recognition is essential to improve oncologic and functional outcomes. (Table 2)
5. Conclusions
Feminizing adrenocortical carcinoma is a rare entity with high malignant potential and a generally poor prognosis. The present case highlights the importance of a thorough endocrine evaluation in patients presenting with suggestive clinical signs—such as bilateral gynecomastia and infertility—to enable early detection of hormone-secreting adrenal tumors.
The differential diagnosis of gynecomastia in young males should always include rare tumor-related etiologies, especially in the absence of obvious systemic or drug-related causes. The normalization of the hormonal profile and spontaneous conception after surgery suggest that the hypogonadism induced by tumor-related hyperestrogenism may be reversible—an aspect rarely documented in current medical literature.
Postoperative therapeutic decisions in low-risk cases should be tailored individually, considering emerging evidence supporting active surveillance over universal adjuvant mitotane therapy.
This case adds valuable insight to the current literature by detailing a rare clinical presentation of ACC and documenting the potential restoration of fertility following curative surgical treatment.
The rarity of these tumors underscores the need for increased awareness and an international case registry to improve diagnostic and therapeutic strategies for future cases.
Author Contributions
All authors contributed equally to the conception, drafting, revision, and approval of the final manuscript. All authors have read and agreed to the published version of the manuscript.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Conflicts of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Abbreviations
The following abbreviations are used in this manuscript:
| ES-ACCs | Estrogen-Secreting Adrenocortical Carcinoma |
| 17-OHP | 17-alpha hydroxyprogesterone |
| DHEA-S | Dehydroepiandrosterone Sulfate |
| ACTH | Adrenocorticotropic Hormone |
| FATs | Feminizing adrenocortical tumors |
|
ACCs MEN1 IHC FSH |
Adrenocortical carcinomas Multiple endocrine neoplasia type 1 Immunohistochemistry Follicle stimulating hormone |
| 11-DOC | 11-deoxycorticosterone |
| LH | Luteinizing hormone |
References
- Fancellu, A. Pinna, and A. Porcu, “Feminizing Adrenocortical Carcinoma with Distant Metastases: Can Surgery Be Considered?,” Clin Pract, vol. 4, no. 2, p. 651, Jul. 2014. [CrossRef]
- M. T. Kidd, N. J. Karlin, and C. B. Cook, “Feminizing Adrenal Neoplasms: Case Presentations and Review of the Literature,” Journal of Clinical Oncology, vol. 29, no. 6, pp. e127–e130, Feb. 2011. [CrossRef]
- J. LaFemina and M. F. Brennan, “Adrenocortical carcinoma: Past, present, and future,” J Surg Oncol, vol. 106, no. 5, pp. 586–594, Oct. 2012. [CrossRef]
- J. M. Rich, V. Duddalwar, P. M. Cheng, M. Aron, and S. Daneshmand, “Feminizing Adrenocortical Tumor with Multiple Recurrences: A Case Report,” Case Rep Oncol, vol. 16, no. 1, pp. 1033–1040, Sep. 2023. [CrossRef]
- S. Moreno, M. Guillermo, M. Decoulx, D. Dewailly, R. Bresson, and Ch. Proye, “Feminizing adreno-cortical carcinomas in male adults. A dire prognosis,” Ann Endocrinol (Paris), vol. 67, no. 1, pp. 32–38, Mar. 2006. [CrossRef]
- F. Chentli, I. Bekkaye, and S. Azzoug, “Feminizing adrenocortical tumors: Literature review,” Indian J Endocrinol Metab, vol. 19, no. 3, p. 332, 2015. [CrossRef]
- D. Vurallı et al., “Feminizing Adrenocortical Tumors as a Rare Etiology of Isosexual/Contrasexual Pseudopuberty,” J Clin Res Pediatr Endocrinol, vol. 14, no. 1, pp. 17–28, Mar. 2022. [CrossRef]
- M. Lerario, A. Moraitis, and G. D. Hammer, “Genetics and epigenetics of adrenocortical tumors,” Mol Cell Endocrinol, vol. 386, no. 1–2, pp. 67–84, Apr. 2014. [CrossRef]
- Allolio and M. Fassnacht, “Adrenocortical Carcinoma: Clinical Update,” J Clin Endocrinol Metab, vol. 91, no. 6, pp. 2027–2037, Jun. 2006. [CrossRef]
- F. Chentli, I. Bekkaye, and S. Azzoug, “Feminizing adrenocortical tumors: Literature review,” Indian J Endocrinol Metab, vol. 19, no. 3, p. 332, 2015. [CrossRef]
- Phornphutkul et al., “Aromatase P450 Expression in a Feminizing Adrenal Adenoma Presenting as Isosexual Precocious Puberty,” J Clin Endocrinol Metab, vol. 86, no. 2, pp. 649–652, Feb. 2001. [CrossRef]
- F. Chentli, F. Chabour, D. Bouchibane, and N. Nouar, “Feminizing Adrenocortical Carcinoma Without Gynecomastia,” Oman Med J, vol. 32, no. 4, pp. 349–351, Jul. 2017. [CrossRef]
- N. FUKAI et al., “A Case of Estrogen-secreting Adrenocortical Carcinoma with Subclinical Cushing’s Syndrome,” Endocr J, vol. 53, no. 2, pp. 237–245, 2006. [CrossRef]
- E. B. Visconti, R. W. Peters, A. Cangir, G. L. Zorn, and S. Fisher, “Unusual case of adrenal cortical carcinoma in a female infant.,” Arch Dis Child, vol. 53, no. 4, pp. 342–344, Apr. 1978. [CrossRef]
- P.-H. Savoie et al., “RETRACTED: Recommandations françaises du Comité de Cancérologie de l’AFU — Actualisation 2018—2020 : tumeur de la surrénale,” Progrès en Urologie, vol. 28, no. 12, pp. S175–S193, Nov. 2018. [CrossRef]
- Lanigan, R. G. Choa, and J. Evans, “A feminizing adrenocortical carcinoma presenting with gynaecomastia,” Postgrad Med J, vol. 69, no. 812, pp. 481–483, Jun. 1993. [CrossRef]
- J. M. Rich, V. Duddalwar, P. M. Cheng, M. Aron, and S. Daneshmand, “Feminizing Adrenocortical Tumor with Multiple Recurrences: A Case Report,” Case Rep Oncol, vol. 16, no. 1, pp. 1033–1040, Sep. 2023. [CrossRef]
- M. Terzolo et al., “Adjuvant Mitotane Treatment for Adrenocortical Carcinoma,” New England Journal of Medicine, vol. 356, no. 23, pp. 2372–2380, Jun. 2007. [CrossRef]
- M. Terzolo et al., “Adjuvant mitotane versus surveillance in low-grade, localised adrenocortical carcinoma (ADIUVO): an international, multicentre, open-label, randomised, phase 3 trial and observational study,” Lancet Diabetes Endocrinol, vol. 11, no. 10, pp. 720–730, Oct. 2023. [CrossRef]
- News Medical, “Study shows not all adrenal carcinoma patients need mitotane after surgery,” News-Medical.net.
- J. Sykes, J. L. Ellis, L. Bukavina, C. A. Koch, S. Wei, and A. Kutikov, “Estradiol-secreting adrenal oncocytoma in a 31-year old male,” Urol Case Rep, vol. 44, p. 102138, Sep. 2022. [CrossRef]
- M. Hatano et al., “Feminizing Adrenocortical Carcinoma with Distinct Histopathological Findings,” Internal Medicine, vol. 55, no. 22, pp. 3301–3307, 2016. [CrossRef]
- Fadi Ibrahim, “MALE GYNECOMASTIA: A RARE CASE OF FEMINIZING ADRENAL CORTICAL CARCINOMA,” Journal of Hospital Medicine 2018; April 8-11; Orlando, Fla., Jun. 2018.
- T. Takeuchi et al., “Adrenocortical carcinoma characterized by gynecomastia: A case report.,” Clin Pediatr Endocrinol, vol. 27, no. 1, pp. 9–18, 2018. [CrossRef]
- Y. Jeong et al., “Estrogen-secreting adrenocortical carcinoma,” Yeungnam Univ J Med, vol. 36, no. 1, pp. 54–58, Jan. 2019. [CrossRef]
- H. C. De, E. Philipse, and B. C. De, “An adrenal tumor with gynecomastia,” Endocrine Abstracts, Oct. 2019. [CrossRef]
- S. M. Gibbons, N. Jassam, A. Abbas, K. Stuart, A. Fairhurst, and J. H. Barth, “Gynaecomastia caused by a feminizing adrenal tumour,” Annals of Clinical Biochemistry: International Journal of Laboratory Medicine, vol. 57, no. 1, pp. 99–101, Jan. 2020. [CrossRef]
- C. Vogt et al., “Feminizing adrenal tumor identified by plasma steroid profiling,” Endocrinol Diabetes Metab Case Rep, vol. 2021, Nov. 2021. [CrossRef]
- J. Saini, P. Navin, M. Rivera, and I. Bancos, “Gynecomastia in a Man With Adrenal Mass,” JCEM Case Reports, vol. 2, no. 1, Dec. 2023. [CrossRef]
- M. Abir, “Bilateral gynecomastia revealing adrenocorticaloma,” Endocrine Abstracts, May 2025. [CrossRef]

Table 1.
Evolution of hormonal profile.
| Laboratory findings | Preoperative | One month postoperative | Three month postoperative | Normal ranges |
| ACTH | 2,6 | 61,8 | 114 | 7.2 - 63.3 pg/ml |
| Morning cortisol | 12,1 | 10,1 | 19,9 | 4,2-19,6 ug/dl |
| DHEAS | - | 128,4 | 85 - 690 uUI/ml | |
| Estradiol | 90,1 | 33,7 | 24,2 | 29.8 - 33.1 pg/ml |
| Testosterone, total | - | 3,1 | 2.59 - 8.16 ng/ml | |
| Androstendione | - | 1,68 | 2,05 | 0,50 - 3,50 ng/ml |
| LH | - | 1,2 | 1.24 - 8.62 mIU/ml | |
| FSH | - | 2,72 | 1.27 - 19.26 mIU/ml | |
| Plasmatic Metanephrines | N | 18,8 | 25,7 | < 100 pg/ml |
| Plasmatic Normetanephrines | N | 22,5 | 70,5 | < 216 pg/ml |
| Aldosterone | N | 5,74 | 11,3 | 1.76 - 23.2 ng/dl |
| Renine | N | 13,11 | 36,19 | 2.8-39.9 uUI/ml |
Table 2.
A comprehensive review of all reported cases of estrogen-secreting adrenocortical carcinoma in male cases between 2015 and 2025, including one pediatric case.
Table 2.
A comprehensive review of all reported cases of estrogen-secreting adrenocortical carcinoma in male cases between 2015 and 2025, including one pediatric case.
| Author | Year | Age | Clinical presentation | Tumour size&side | Hormonal profile preoperative | Treatment | Outcome | Follow-up |
| Sykes J et al. [21] | 2015 | 31 | Gynecomastia, infertility | 9 cm, right adrenal | ↑Estradiol (83 pg/mL); ↑ DHEAS (502 μg/dL); ↓FSH (0.8 mlU/mL) | Right modified Makuuchi incision-open adrenalectomy | Hormone normalization after 2 weeks postoperative Normal gonadotrophines at 5 months follow up |
Endocrine labs every 6 months, and CT scans every 6–12 months for at least 5 years |
| Hatano M. et al. [22] | 2016 | 60 | Gynecomastia, right hyponcondriac pain, low libido | 16x11x14 cm, right adrenal | ↑Estradiol (284 pg/mL); ↑ DHEAS (560 μg/dL); ↓FSH (0.2 mlU/mL) and LH (0.1 mIU/mL); ↓ free testosterone <0.6 pg/ml | Right adrenalectomy p0T2N0M0, Weiss score 7, ki 67 18% |
Hormone normalization -locoregional lymph nodes and peritoneum metastases |
No adjuvant therapy until 11 months Initiation of Mitotane after discover of locoregional lymph nodes |
| Ibrahim F et al. [23] | 2018 | 55 | Gynecomastia, testicular hypotrophy, pleuritic left chest pain, hypertension | 6.3 cm, right adrenal | ↑Estradiol 134 pg/mL; ↓testosterone 109 ng/dL; ↓ FSH and LH | Laparoscopic right adrenalectomy | ? | ? |
| Takeuchi et al. [24] | 2018 | 4 | Gynecomastia, acute growth spurt | 8 cm, right adrenal | ↑ Estradiol 28.1 pg/mL;N testosterone (0.82 ng/mL); ↓ LH (<0.1 mIU/mL); ↓ FSH (0.13 mIU/mL); ↑DHEAS 1950 ng/ml, ↑ androstenedione 4.6 ng/ml |
Initial- combined chemoterapy of ARAR0332 with mitotane 120-150 mg/d pre-operative Surgical resection Weiss score 7 |
Gynecomastia disappeared after 1 year post-operative Normal growth velocity after tumor resection |
Followed by administration of GPOH-MET97 therapy and mitotane *mitotane was discontinued after 6 months post-operative Postoperative- replacement therapy with hydrocortisone and mineralocorticoid No relapse after 2 years |
| Jeong Y et al. [25] | 2019 | 53 | Gynecomastia, abdominal discomfort, right-sided flank pain | 21×15.3×12 cm, right retroperitoneum | ↑Estradiol (820 pg/mL); ↑ DHEAS (578 μg/dL); ↓FSH (1.07 mlU/mL); ↓ACTH (4.2pg/mL), inappropriately normal cortisol (16,3 ug/dl), ↑ free cortisol/24 h urine 134 ug/day |
Open adrenalectomy with partial liver resection pT2N0M0, stage 2, Weiss score 6 IHC: positive staining for inhibin α, MART-1, and calretinin and a Ki67 proliferation index of 20% |
Estradiol 70.32 pg/ml; the gynecomastia had almost disappeared at 3 month postoperative Without any local recurrence or metastasis at 21 months after diagnosis and treatment, and the gynecomastia has almost resolved |
Additional adjuvant radiation therapy. Postoperative- replacement therapy with hydrocortisone |
| C De Herdt et al.[26] | 2019 | 42 | Gynecomastia, icterus | 5.1 cm, right adrenal | ↑Estradiol 44 ng/l; hypogonadotropic hypogonadism | Open right adrenalectomy pT3L0V0Pn0R0, stage 3, Weiss score 4 |
Estradiol 9 ng/l; almost complete recovery of the gonadotropic axis after 2 weeks postoperative -disappearance of gynecomastia |
Adjuvant treatment with mitotane |
| Gibbons S et al. [27] | 2020 | 52 | Gynaecomastia, low libido, erectile dysfunction | 8 x 8 cm, right adrenal | ↑ Estradiol 932 pmol/L;↓ testosterone (0.7 nmol/L); N LH (1.2 IU/L); ↓ FSH (<0.1 IU/L) | Right open adrenalectomy pT3 Nx Ki-67 60% |
Hormone normalization -disappearance of gynecomastia |
Adjuvant treatment with mitotane for two years in addition to bi-annual CT scans |
| Vogt E et al. [28] | 2021 | 58 | Gynaecomastia, low libido, | 6.5 cm × 5.2 cm, left adrenal | ↑ Estradiol (208 pmol/L); ↑ 11-deoxyxortisol (23.5 nmol/L), ↑ DHEAS (10.6 µmol/L), ↑ androstenedione (18.1 nmol/L), cortisol overproduction, low normal FSH and LH | Transabdominal laparoscopic Weiss score of 7 IHC: inhibin, synaptophysin, CD31 (PECAM-1) and aromatase (CYP19A1) Ki 67 5% |
Hormone normalization Gynecomastia had not improved significantly 1 year after surgery, and therefore liposuction and perioareolar incision was planned. |
Adjuvant therapy with the adrenolytic drug mitotane was started 8 weeks after resection, |
| Rich J et al.[4] | 2023 | 35 | Gynecomastia, low libido, RUQ pain | 18×8.5×14.5 cm, right adrenal | ↓FSH (<0.1 mIU/mL), ↓ LH(<0.1 mIU/mL), ↓ testosterone (37 ng/dL), ↑ estradiol (181 pg/mL), normal cortisol (8.0–14.5 μg/dL), ↓ ACTH (1 pg/mL) | Open right adrenalectomy |
Hormone normalization after 2 months postoperative - gynecomastia decreased to near baseline, and libido/erections returned to normal |
Local recurence: a new 1 cm nodule in the right adrenalectomy bed -plans includehemotherapy with mitotane or etoposide/doxorubicin/cisplatin |
| Saini J et al. [29] | 2023 | 65 | Gynecomastia | 4.3 cm, right adrenal, with metastatic lessions after 5 years | ↑ Estradiol 72 pg/mL; ↑ Estrone 345 pg/mL; ↑ progesterone 0.59 ng/mL; ↓ total testosterone 157 ng/dL; ↑ 11 DOC 204 ng/dL; ↑ renine plasma activity 204 ng/dL |
Initial- right laparoscopic adrenalectomy locally Reintervention-debulking surgery with the removal of multiple metastatic lesions Weiss score 4 |
Redeveloped gynecomastia after 5 years After reintervention: Hormone normalization after 3 months postoperative - gynecomastia improved |
Adjuvant therapy with mitotne plus replacement therapy with hydrocortisone - progressive disease at 3 the month-follow up - cytotoxic chemotherapy with etoposide, doxorubicin, and cisplatin was initiated, and mitotane was discontinued in an attempt to reduce the overall toxicity |
| Abir M et al. [30] | 2025 | 57 | Gynecomastia, weight loss, decreased libido, abdominal pain | large left adrenal tumor mass | Hyperestrogenism, increase in the precursors of adrenal androgens 17OHP, androstendione, DHEAS | A biopsy of the adrenal mass performed externally came back in favor of a left adrenocorticaloma with positive immunohistochemistry, synaptophysin and Melan A | Patient died following a state of hemorrhagic shock | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



