
Submitted:
05 August 2025
Posted:
07 August 2025
You are already at the latest version
Abstract
Standard cancer chemotherapy is increasingly being supplemented with novel therapeutics to overcome known chemoresistance pathways. Resistance to treatment is common across various tumor types, driven by multiple mechanisms. One emerging contributor is protein methylation, a post-translational modification mediated by protein methyltransferases (PMTs), which regulate protein function by adding methyl groups, mainly on lysine and arginine residues. Dysregulation of protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) has been linked to cancer progression and drug resistance, making them attractive therapeutic targets. Consequently, several small-molecule PMT inhibitors have been developed, with some progressing to clinical trials. However, many candidates showing promise in preclinical studies fail to demonstrate efficacy or safety in later stages, limiting clinical success. This gap highlights the need to rethink current approaches to PMT inhibitor design. A deeper understanding of PMT mechanisms, catalytic domains, and their roles in chemoresistance is essential for creating more selective, potent, and clinically viable inhibitors. This review will summarize major chemoresistance pathways and PMTs implicated in cancer, then explore current and prospective PMT inhibitor classes. Building on mechanistic insights, we propose strategies to develop next-generation inhibitors with improved therapeutic potential against chemoresistant cancers.
Keywords:
protein methylation
; protein methyltransferases (PMTs)
; PMT inhibitors
; methylproteomics
; chemoresistance
1. Introduction
1.1. Challenge to Treatment
Chemoresistance is recognised as the greatest challenge in the successful treatment of cancer by chemotherapy, with development and proliferation of highly aggressive phenotypes, resulting in an associated poor prognosis.
Given the well-documented impact of methylation on protein expression levels through its action on histones, it is no surprise that protein methyltransferase (PMT) inhibitors have been considered as potential therapeutics to prevent or counter specific chemoresistance mechanisms at least in a sub-set of patients, in the hopes of increasing the possibility of obtaining long-term benefits with the currently administered chemotherapeutic regimens. However, despite this knowledge related to histones, there is at present a lack of understanding as to the biological relevance of non-histone proteins targeted by PMTs as part of chemoresistance mechanisms.
The greatest stumbling block presently is that the functional role played by PMTs in either the development or the maintenance of chemoresistance is close to unknown. Inhibiting the function of a type of methylation without knowing the expected outcome is an issue that magnifies itself when translated to the clinical setting, with patient responses to therapy varying greatly, presenting side effects which might, but are not necessarily, linked to the use of PMT inhibitors in the combinatorial treatment.
For this reason, understanding at least the core mechanism of methylation addition for the different PMT families and their role in the major chemoresistance pathways is essentially needed for designing inhibitors that act with high specificity and sensitivity.
1.2. Chemoresistance Mechanisms
The evasion of cancer therapeutics is generally achieved by one or more of the following strategies employed by malignant cells irrespective of cancer origin. These mechanisms have been reviewed in detail elsewhere (Mansoori et al., 2017; Kannampuzha and Gopalakrishnan, 2023; Khan et al., 2024) but an overview will be provided in order to build the discussion in relation to PMTs.
From a genetic perspective, mutations that result in structural changes to the enzymes that the drugs interact with, such as alterations to catalytic sites that affect either drug binding or enzyme activation. For example mutated topoisomerase II confers resistance to etoposide and doxorubicin (Elton et al., 2020), mutations in the EGFR kinase domain grant resistance against Gefitinib (Kobayashi et al., 2005), mutations of the extracellular receptor smoothened (Zhang et al., 2018) limit cyclopamine effectiveness, mutations in nucleoside transporters (Galmarini et al., 2001; Damaraju et al., 2003) block nucleoside drugs such as cytosine arabinoside (ara-C, cytarabine) or 2-chlorodeoxyadenosine (2-CdA) whilst mutations in folate transporters (Gao et al., 2021) inhibit toxic folate analogues, such as methotrexate.
Another dominant mechanism is increased efflux of chemotherapeutic drugs, often mediated by either overexpression or increased activity of transporter proteins including ATP-binding cassette (ABC) transporters. For example, increased expression of ABCB1 (P-glycoprotein, multidrug resistance 1) provides resistance to various anti-cancer drugs, including paclitaxel, olaparib (Vaidyanathan et al., 2016), and doxorubicin (Hui et al., 2008; Mirzaei et al., 2022). The increased expression of ABCG2 provides resistance to doxorubicin (Roh et al., 2018), oxaliplatin (Hsu et al., 2018), and 5-fluorouracil (Kim et al., 2020). Similarly, ABCC2 (multidrug resistance protein 2; MRP2) confers resistance to cisplatin (Chen et al., 2021) and oxaliplatin (Biswas et al., 2019).
Equally widespread among tumours is enhanced activity of detoxifying enzymes like cytochrome P450, which increases drug inactivation. For example, increased expression of Glutathione transferases (GST) and production of glutathione (GSH) are effectively employed by tumours to detoxify many alkylating agents and platinum-based drugs (Pljesa-Ercegovac et al., 2018).
Epigenetic and epiproteomic alterations are also frequently observed. Epigenetic changes are most commonly DNA methylation changes, including aberrant methylation of the ABCB1, ABCC1 and ABCG2 promoters to increase their expression (Spitzwieser et al., 2016; Li et al., 2016; Sumarpo et al., 2020) but can also involve post-transcriptional modifications through microRNAs (miRNAs) (Mondal and Meeran, 2021). Epiproteomic changes encompass various histone modifications that alter gene expression patterns (Arrigoni et al., 2016), including histone H3 hyperacetylation (Shui et al., 2024) or an enzyme such as Histone deacetylase 2 (HDAC2) upregulating the expression of ABCB1 (Ye et al., 2016).
Other mechanisms include enhanced DNA repair capabilities (Sakthivel and Hariharan, 2017; Abad et al., 2020; Li et al., 2021), activation of other signalling pathways to bypass drug effects (Pan et al., 2016; Alimbetov et al., 2018), and alterations in apoptotic pathways to evade cell death (Alimbetov et al., 2018; Chen et al., 2018).
2. Protein Methylation
2.1. Types and Function
The cell has the ability to broadly modulate protein properties through the addition of post-translational modifications (PTMs), particularly methylation. These epiproteomic changes allow for fast responses at low cellular cost.
Protein methylation involves the transfer of a CH3 group from the methyl donor S-adenosylmethionine (SAM or AdoMet) to an amino acid residue on a protein. The residues altered are mainly lysine (K) and arginine (R) residues and the methyl group is added onto nitrogen making it N-methylation. A number of other amino acids can undergo N-methylation, namely asparagine (N), histidine (H), proline (P) and alanine (A). Two other types of methylation, S- and O- methylation, exist. The amino acids methionine (M) and cysteine (C) can undergo S-methylation, while aspartate (D) and glutamate (E) can undergo O-methylation (Deng et al., 2016; Cloutier et al., 2013; Clarke, 2013).
One or more CH3 groups can be added to both K and R residues with the epsilon (ε) amine of the former being able to undergo mono- (Kme1), di- (Kme2) or tri- (Kme3) methyl addition by protein lysine methyltransferases (PKMTs), while the guanidinium moiety of the latter can undergo mono- (MMA/Rme1), di-symmetrical (SDMA/sRme2) or di-asymmetrical (ADMA/aRme2) methyl addition by protein arginine methyltransferases (PRMTs) (Greer and Yang, 2012).
Multiple CH3 groups can be added to the protein before the protein dissociates from the PMT, which is referred to as a processive addition mechanism. On the other hand, if only a single CH3 group is added before the protein is dissociated from the PMT and subsequent methylations are added by the same or different PMTs, this is referred to as a distributive addition mechanism (Copeland et al., 2009; Al-Hamashi et al., 2020; Fulton et al., 2018). PKMTs and PRMTs implement both these mechanisms.
At a protein level, whilst methylation does not affect the overall charge of the residue, there is an associated increase in both hydrophobicity and bulkiness around the modified residue, which in turn can alter protein properties such as protein-protein interactions and recognitions, having a downstream impact on gene regulation (Kaniskan et al., 2015).
In the context of cancer, the most well-studied substrates for methylation are histones, with exceptional focus on histones 3 and 4. However, studies have in recent years expanded this investigation to non-histone proteins, including proteins such as tumour protein 53 (p53), heat shock proteins 70 or 90 (HSP70 or HSP90), Nuclear factor kappa B (NF-κB) and Retinoblastoma 1 (RB1) (Hamamoto et al., 2015; Wei et al., 2014). Nevertheless, not much is known about the methylation status and functional role of these key cancer proteins following chemoresistance.
2.2. The Methyl Cycle: S-Adenosylmethionine Production
The primary methyl group donor for methylating various substrates, including DNA, RNA, proteins, lipids, and other molecules, is S-adenosylmethionine (SAM). SAM is synthesised from the conjugation of adenosine from adenosine triphosphate (ATP) with methionine. The reaction is catalysed by SAM synthetase (a.k.a. methionine adenosyltransferase; MAT), which dephosphorylates a molecule of ATP in the presence of potassium and magnesium ions (Mato et al., 2002), together with cleavage of the C5'-O5' bond of ATP and changing the conformation of the ribose ring from C4'-exo to C3'-endo such that the S of Met makes a nucleophilic attack on the C5' to form SAM (Komoto et al., 2004).
Methylation reactions are catalysed by methyltransferases and release S-adenosylhomocysteine (SAH or AdoHC). SAH is a competitive inhibitor of MTases and is hydrolysed by adenosylhomocysteinase (AHCY) to homocysteine (Hcy). This reaction is reversible, meaning that increased Hcy levels can lead to the accumulation of SAH, which can bring about a lower methylation capacity. This can be represented as the SAM/SAH ratio (Hao et al., 2016). Homocysteine is re-methylated to regenerate methionine using 5-methyltetrahydrafolate (5-MTHF) or betaine via betaine homocysteine methyltransferase (BHMT) (Pascale at al., 2022). 5-MTHF, produced from dietary folate (dihydrofolate) through the folate cycle, donates a methyl group, catalysed by methionine synthase (MTR) using vitamin B12 (cobalamin, Cbl) as its cofactor (Nazki et al., 2014). These steps all form part of what is called the methyl cycle (Figure 1) (Froese et al., 2019). The essential nutrients for this cycle are thus methionine, folic acid (for CH3-THF), and choline (to generate betaine).
The dysregulation of SAM leads to alterations in over a hundred methyltransferase reactions, which modulate metabolic pathways (Loenen, 2006). SAM has been shown to have anti-cancer properties, limiting proliferative, inducing apoptosis, and preventing metastasis, although the mechanisms were not identified (Mosca et al., 2020; Wang et al., 2017). It has also been shown to sensitise CRC cells to 5-FU treatment via p-glycoprotein inhibition and a reduction in NF-κB acetylation/NF-κB protein ratio, suppressing NF-κB Activation (Mosca et al., 2021). SAM has been tested as both a diagnostic and prognostic biomarker in different cancers in the form of the SAM/SAH ratio and has been studied as a potential chemotherapeutic, both on its own and as a chemosensitising agent in combination with other drugs, in a wide variety of cancer types (Fernández-Ramos et al., 2025).
2.3. Protein Methyltransferases
Methyltransferases across all families, irrespective of their substrates, typically have catalytic domains consisting of a well-conserved SAM-binding pocket, together with highly variable substrate acceptor regions (Richon et al., 2011; Copeland et al., 2009). These two compartments are linked via a hydrophobic channel, allowing the transfer of a CH3 group from SAM to the target residue through an SN2 transition (Kaniskan et al., 2018).
In the most basic arrangement, the Rossmann-like fold consists of regions of alternating β-strands and α-helices. This produces a flat β-sheet at the centre, with a set of helices on either side of the sheet. In such an arrangement, the N-terminal β-strand is located in the middle of the sheet, with the strand topology following the order 3-2-1-4-5-7-6, and having the 7th strand anti-parallel to the other strands. Furthermore, functionally important, conserved residues in such enzymes are often found either at the C-termini of the β-strands or else in the adjoining loops (Kozbial and Mushegian, 2005).
Whilst there is a wide variety of PMTs, most can be clustered based on the overall structure and sequence similarities of their enzymatic domain, forming three superfamilies, namely the Seven-β-strand (7βS) domain family, the SpoU-TrmD (SPOUT) domain family and the Su(var)3-9, enhancer of zeste (E(z)), and trithorax (trx) (SET) domain family (Clarke, 2013; Husmann and Gozani, 2019; Zhang et al., 2015).
The 7βS family features a core structure containing two domains, a seven-stranded Rossmann-fold domain forming the binding site for the methyl donor SAM and a β-barrel for dimerisation (Schapira and de Freitas, 2014). The SET domain family has small β-sheets forming a knot-like structure, surrounded by a pre- and post-SET domain together with an iSET domain forming a helical structure adjacent to the substrate binding cleft (Kozbial and Mushegian, 2005). The SPOUT methyltransferase family contain a distinctive α/β knot-like structure (Petrossian et al., 2010; Tkaczuk et al., 2007; Clarke, 2013; Young et al.,2012).
For the scope of this review the focus will be on the 7βS and SET domain families since the two major groups of PMTs i.e., protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) fall within these families.
2.3.1. Protein Lysine Methyltransferases (PKMTs)
Over 50 PKMTs have been identified in humans (Yang and Bedford, 2013; Hamamoto et al., 2015; Carlson and Gozani, 2016; Husmann and Gozani, 2019). The PKMTs can be grossly subdivided into two groups: those enzymes containing a SET-domain and those that do not (i.e., class 1 canonical Rossmann-fold-like methyltransferases) (Clarke, 2013; Kaniskan et al., 2018). PKMTs are more finely subdivided into eight groups (KMT1-KMT8) based on the sequence similarity flanking the SET domain, activity, and substrates (Allis et al., 2007; Kaniskan et al., 2018; Zhang et al., 2015; Zhang, Wen and Shi, 2012). However, given that numerous KMTs have no known substrates and consequently uncertain activity, categorising such enzymes is speculative and imprecise (Carlson et al., 2016).
Those PKMTs having a SET domain present the typical small β-sheets that produce a knot-like structure, with a pre-SET, post-SET and iSET domains. The helical structure formed by the iSET domain contributes to the formation of a narrow channel connecting the SAM binding surface and the substrate pocket through which the amine of the lysine residue to be methylated on the substrate is oriented and can access the SAM (Kozbial and Mushegian, 2005). All these domains are essential for enzymatic activity. Additionally, the SET proteins also contain four conserved sequence motifs, SET motif I (GXG), SET motif II (YXG), SET motif III (RFINHXCXPN), and SET motif IV (ELXFDY), where each motif is involved in the methylation reaction (Cheng et al., 2005; Kaniskan et al., 2018).
The non-SET domain KMT category is currently composed of disruptor of telomeric silencing-1-like (DOT1L) (which makes up KMT4) and a few members of the Methyltranferase-like (METTL) family (Kaniskan et al., 2018; Richon et al., 2011; Cloutier et al., 2013). DOT1L has a 7βS as part of its N-terminal catalytic domain, which contains seven conserved sequence motifs (I, II, III, IV, VI, VIII and X) as a methyl donor binding site and an active site pocket covered with conserved hydrophobic residues (Falnes et al., 2016; Cheng et al., 2005; McGrath and Trojer, 2015; Wood, Tellier and Murphy, 2018). The lysine methylating METTL enzymes in this group include METTL10, METTL12, METTL13, METTL20, METTL21A, METTL21B, METTL21C, METTL21D, METTL21E and METTL22 (Cloutier et al., 2013; Falnes et al., 2016; Wong et al., 2021; Falnes et al., 2023).
2.3.2. Protein Arginine Methyltransferases (PRMTs)
A total of nine PRMTs have been identified in humans. As part of the 7βS family, the PRMT family members share a common overall structure consisting of a methyltransferase domain, a 7-β-barrel Rossmann fold domain (that interacts with SAM) and a dimerisation arm (Xu and Richard, 2021). In detail, PRMTs harbour a catalytic domain composed of six conserved sequence motifs, where motif I is at the core of the SAM-binding pocket with three highly conserved glycine residues (VLD/VGxGxG), post-motif I hydrogen bonds to SAM either through the use of an glutamic or aspartic acid residue (V/I-X-G/A–X-D/E), motif II forms a β-sheet that stabilises motif I (E/K/VDII), the 'double-E' motif positions the arginine substrate residue via two glutamic acid residues (SExMGxxLxxExM), motif III forms a parallel β-sheet with motif II (LK/xxGxxxP) and finally the THW loop that is essential for both substrate binding and N-terminal stabilisation. (Wei et al., 2014, Tewary et al., 2019).
These 9 enzymes are divided into three sub-types, with all three sub-types giving rise to MMA, but then Type I and Type II PRMTs subsequently proceed further to produce ADMA and SDMA respectively (Obianyo et al., 2011). The Type I group consists of PRMT 1, 2, 3, 4, 6, and 8, the Type II group is made up of PRMT 5 and 9, while Type III only includes PRMT7 (Al-Hamashi et al., 2020; Fulton et al., 2018).
Moreover, PRMTs have preferences for certain motifs flanking the arginine to be methylated. PRMT1, PRMT3, PRMT6, PRMT8 prefer glycine-arginine-rich (GAR/RGG/RG) motifs (Tewary et al., 2019; Al-Hamashi et al., 2020), PRMT4 (also named CARM1) prefers proline-glycine-methionine-rich (PGM) motifs (Tewary et al., 2019), PRMT5 recognises RGG, RXR, and RG motifs (Tewary et al., 2019), PRMT7 can recognise RXRXR or RXR motifs (Tewary et al., 2019; Kaniskan et al., 2018; Al-Hamashi et al., 2020) and PRMT9 uniquely recognises the CFKRKYL motif in SAP145 (Yang et al., 2015).
2.3.3. Cancer Dysregulation
The increased expression of PKMTs, particularly Euchromatic histone-lysine N-methyltransferase 2 (EHMT2), Enhancer of zeste 2 (EZH2), SET domain containing 7 (SETD7), SETD8, SET And MYND Domain Containing 2 (SYMD2) and SYMD3 as well as PRMTs including PRMT1, PRMT3, PRMT5 and PRMT6 in a variety of tumours of different types and origins has been shown to promote chemoresistance through cell cycle progression (Zhang et al., 2019), cancer stem cell (CSC) renewal (Lin et al., 2018) and the alteration of key signalling pathways such as the Wnt or Notch pathways (Shailesh et al., 2021; Chai et al., 2023). The clinical evidence thus supports the targeting of PMTs as a promising cancer treatment (Vougiouklakis et al., 2020; Ning et al., 2023).
2.4. Classes of Methyltransferase Inhibitors
PMTs have a generic structure composed of two key pockets, the SAM binding pocket and the methyl-substrate binding pocket. PMT inhibitors can thus be sub-divided based on the region of the enzyme they target and their mode of action (either covalent and non-covalent binders). More specifically, inhibitors can be designed to compete with the methyl donor, the methylation substrate or an essential cofactor, or alternatively bind at allosteric sites found either within the active or at alternative inactive states (Schapira and de Freitas, 2014).
2.4.1. SAM Competitive Inhibitors
SAM competitors or mimetics compete with the methyl donor for the SAM binding pocket of the PMT thus preventing the enzyme from functioning and adding the methylation to the target protein (Campagna-Slater et al., 2011). A few examples of the more successful SAM inhibitors due to their selective nature include the EHMT2 inhibitor BRD9539 and its methyl ester BRD4770 (Yuan et al., 2012), the SMYD2 inhibitor PFI-5 (Taylor et al., 2019), the EZH2 inhibitors EPZ6438 (tazemetostat) and SKLB1049 (Wei et al., 2024), the DOT1L inhibitors EPZ004777 and its derivative EPZ5676 (Pinometostat) (Waters, 2017), and the PRMT5 inhibitor LLY-283 and its derivative PF-0685580 (Lin and Luengo, 2019).
Despite a large number of SAM competitors having been developed, unfortunately, many such inhibitors present limited specificity given that all methyltransferases use SAM as their methyl donor. Designing SAM competitive inhibitors is a challenge due to the high hydrophilicity of SAM requiring inhibitors to have a high enough polarity to be able to interact with the SAM binding pocket, whilst at the same time presenting sufficient hydrophobicity to be able to traverse cellular membranes in order to reach their target. However, the greatest limitation to such inhibitors is the high intracellular levels of SAM with which they must compete (Ferreira de Freitas et al., 2019).
2.4.2. Substrate Competitive Inhibitors
This class of inhibitors compete with the protein substrate to be methylated for the substrate binding pocket of the PMT. The development of substrate competitive inhibitors is a preferred strategy due to ample structural diversity within the substrate binding pocket of PMTs, allowing for increased specificity (given the selective nature of substrate recruitment) as well as having a lower polarity requirement compared to SAM competitive inhibitors (Ferreira de Freitas et al., 2019).
A few examples of the more potent and promising substrate inhibitors are presented in Table 1.
2.4.3. Bisubstrate Inhibitors
Inhibitors within this class occupy both the SAM and substrate PMT pockets. There are very few confirmed inhibitors within this class, with a few documented examples being the SET7/9 inhibitor set of AzaAdoMet substituted with various alkylamino groups (Mori et al., 2010), the PRMT1 inhibitor decamidine (Zhang et al., 2017), a PRMT4 inhibitor designed as a 5-methylcytosine-adenosine derivative (Halby et al., 2018), the PRMT5 inhibitor CMP5 (Alinari et al., 2017), and the PRMT6 inhibitor 6′-methyleneamine sinefungin (GMS) (Wu et al., 2016).
2.4.4. Allosteric Inhibitors
This class of inhibitors impede enzyme activity by binding to sites on the PMT other than its two active site pockets. The best known of such inhibitors is the PRMT3 inhibitor SGC707. Since PRMT3 forms homodimers to perform its function, SGC707 binds to the pocket at the base of the dimerisation arm of the first monomer and the α-Y segment of the activation helix of the second monomer, inhibiting the formation of the PRMT3 homodimer as a result of its introduction (Kaniskan et al., 2015). One of the few other allosteric inhibitors available is the SMYD3 inhibitor diperodon, which binds to an allosteric site the same allosteric binding site, located within the C-terminus domain, adjacent to the active site containing domain (Talibov et al., 2021).
2.4.5. Complex Disrupting Inhibitors
Many PMTs form multi sub-unit complexes where the integrity of the complex is critical in order for the PMTs to perform their function. This provides alternative targets for inhibition other that the catalytic domain.
EZH1/2 must form part of the PRC2 complex together with at least three other PRC2 sub-units in order to be enzymatically active. One of these sub-units is Embryonic Ectoderm Development (EED). Numerous inhibitors have thus been developed that bind to EED and disrupt the formation of the PRC2 complex (Liu et al., 2022). A few noteworthy inhibitors include SAH-EZH2 (Kim et al., 2013), EED226 (Qi et al., 2017), A-395 (He et al., 2017), BR-001 (Dong et al., 2019) and EEDi-5285 (Rej et al., 2020).
Mixed-Lineage Leukemia protein 1 (MLL1 a.k.a. Histone-lysine N-methyltransferase 2A - KMT2A) or MLL fusion proteins form a complex with the transcriptional regulators Menin or WD repeat-containing protein 5 (WDR5). Although these proteins are not required for MLL1 catalytic activity, they are still critical components of context-specific MLL complexes and as such can be targetted to indirectly block the activity of MLL. Menin interacts with the N-terminal region of MLL proteins (Yokoyama et al., 2005), whilst WDR5 interacts with the WIN motif of MLL proteins (Alicea-Velázquez et al., 2016). Of note are the Menin inhibitors KO-539 (ziftomenib) (Copeland, 2018) and M-808 (Xu et al., 2020), as well as the WDR5 inhibitors OICR-9429 (Grebien et al., 2015) and DDO-2093 (Chen et al., 2021).
PRMT5 forms a complex with methylosome protein 50 (MEP50 a.k.a. WDR77 or p44), which acts as a co-factor, in order to achieve its methyltransferase activity (Ho et al., 2013; Antonysamy, 2017) and this dependence upon a co-factor is unique among the PRMTs. Further proteins are recruited as adaptors to specify substrate selection including Chloride conductance regulatory protein (pICln), RIO kinase 1 (RioK1) and Cooperator of PRMT5 (COPR5) (Mulvaney et al., 2021). So far only one inhibitor has been developed to block the interaction between PRMT5 and MEP50 - by interacting with the hydrophobic pocket within the TIM barrel of PRMT5 (Asberry et al., 2022) whilst another has been developed to block the interaction between PRMT5 and RioK1 or pICln (Krzyzanowski et al., 2022).
2.4.6. Covalent Inhibitors
The vast majority of PMT inhibitors developed so far, irrespective of which region of the enzyme they bind to, tend to be reversible (non-covalent) inhibitors. Covalent inhibitors are designed to consist of a peptide sequence or chemical structure that can covalently bind to a nucleophilic amino acid that is not well conserved within or in the immediate vicinity of the PMT active site via a reactive group, called a warhead, which is generally an alkylating, acylating, phosphonylating, or sulfonylating functional group (Powers et al., 2002). Targeting such a residue improves selective inhibition of the intended PMT over other family members (Li et al., 2019). Most covalent inhibitors are currently designed to target a non-catalytic cysteine residue through an acrylamide or other α,β-unsaturated carbonyl compound (Powers et al., 2002; Gehringer and Laufer, 2018; Gambini et al., 2019).
Targeting PMTs with covalent inhibitors offers several advantages compared to using non-covalent equivalents. Covalent inhibitors often exhibit superior potency and biochemical efficiency due to their irreversible, non-equilibrium-based mechanism of action (Ábrányi-Balogh et al., 2018; Li et al., 2019). Unlike reversible inhibitors, covalent binders do not compete with either the ligands or SAM cofactor, enabling sustained target engagement and reduced dosing amount and frequency, thus reducing off-target effects. Their long residence time on the target translates into extended duration of action, which is especially beneficial when the protein of interest has a slow turnover rate (Ábrányi-Balogh et al., 2018). A diversity of warheads are available for PMT covalent inhibitors targeting cysteine, which can be tailored to exploit the spatial and chemical reactivity of the active site, allowing for the design of smaller, more selective molecules without compromising potency, thereby improving drug-likeness. This is particularly important given that many drug candidates fail due to absorption, distribution, metabolism, and excretion (ADME)-related issues (Gehringer and Laufer, 2018; Li et al., 2019). Moreover, covalent inhibitors can enhance target occupancy even at low plasma concentrations, offering improved therapeutic efficacy and an advantage in overcoming drug resistance (Ábrányi-Balogh et al., 2018).
A few noteworthy examples include the EZH2 inhibitor SKLB-03176 (Zhang et al., 2022), the EHMT2 inhibitor MS8511 (Park et al., 2022), the SETD8 inhibitor MS453 (Butler et al., 2016), the PRMT5 inhibitor hemiaminal 9 (Lin et al., 2019) and the PRMT6 inhibitor MS117 (Shen et al., 2020).
2.4.7. PROTAC Inhibitors
More recently, targeted protein degradation has been applied to PMT inhibition through PROteolysis TArgeting Chimeras (PROTACs). These are bifunctional molecules consisting of a bait moiety that binds to the target protein and a moiety that binds an E3 ubiquitin ligase such as Von Hippel−Lindau (VHL) or Cereblon (CRBN) (Sakamoto et al., 2001; Diehl and Ciulli, 2022). SAM-competitive, substrate-competitive, and allosteric binders can all be utilised as the bait moiety in PROTAC design. Once the PROTAC brings the target protein, it can recruit an E3 ubiquitin ligase, which is then able to polyubiquitinate the target. The polyubiquitin group signals the proteolytic degradation of the target protein by the ubiquitin-proteasome system (UPS).
The inhibition of methylation by PROTACs has so far only been implemented against a small number of PKMTs and PRMTs namely the PKMTs EHMT2, EZH2 and NSD2 and the PRMTs PRMT3, PRMT4 and PRMT5. As for the PKMT PROTACs, MS8709 (Velez et al., 2024), AMB-03-378 (Mukherjee, 2025), G9D-4 (Shi et al., 2024) target EHMT2, MS8847 (Velez et al., 2024), MS8815 (Dale et al., 2022), MS177 (Wang et al., 2022), U3i (Wang et al., 2022), YM181 and YM281 (Tu et al., 2021), MS1943 (Ma et al., 2020), PROTAC E7 (Liu et al., 2012) target EZH2 and MS9715 (Xu et al., 2022), MS159 (Meng et al., 2022), LLC0424 (Liu et al., 2024), together with a handful of unnamed PROTACs (Sun et al., 2022; LegaardAndersson et al., 2023) target NSD2 for degradation. For the PRMTs, there are only degrader ”11” (Zou et al., 2024) for PRMT3, C199 (Ju et al., 2025) for PRMT4 and MS4322 (Shen et al., 2020), MS115 (Zhong et al., 2025) and YZ-836P (Guo et al., 2024) for PRMT5.
Apart from targeting the PMTs directly, interacting sub-units of the PMTs within protein complexes have also been targetted for degradation resulting in the destabilisation of the entire complex. The two proteins targetted in this manner so far are EED as part of the PRC2 complex and WDR5 as part of the MLL (KMT2A) complex. The PROTACs designed to target EED are UNC6852 (Potjewyd et al., 2020), PROTAC 1 and PROTAC 2 (Hsu et al., 2020) and UNC7700 (Bashore et al., 2023), while the ones targeting WDR5 are 8g and 17b (Dölle et al., 2021), MS33 and MS67 (Yu et al., 2021), MS40 and MS169 (Li et al., 2022) and MS132 (Yu et al., 2023).
Compared to conventional inhibitors, these PROTACs have been reported to at times be more potent than their parent inhibitor. However, despite the promise displayed so far, there are still challenges in transitioning these molecules from in vitro to clinical testing, one of which is the development of resistance by tumour cells to the commonly used E3 ubiquitin ligase ligands VHL and CRBN through mutations or down-regulation of the ubiquitin ligase mechanism (Békés et al., 2022). That being said, PROTACs against PMTs are not straightforward to design (Martin et al., 2024), and the general understanding is still in its early stages, with effective development still requiring further optimisation (Wang et al., 2022). Moreover, since the choice of E3 ligase ligand in PROTACs affects selectivity and degradation efficiency, alternative, highly selective ligase ligands need to be sought to further optimise PROTAC design (He et al., 2024).
2.4.8. Inhibitor Limitations
As can be seen from the above exposition, currently selective inhibitors are only available against a very small number of the known PMTs. This is due to the fact that the enzymatic domain is highly conserved. Segregating methyltransferases based on the structural diversity of their SAM-binding site produces two groups, with the first containing SET domain PMTs, whilst a second contains PRMTs, small molecule-, DNA- and RNA-methyltransferases, which all share a cofactor-binding Rossman fold. This clustering underscores the high likelihood of off-target effects when attempting to design an inhibitor that targets the SAM-binding site of a selected PRMT (Schapira and de Freitas, 2014).
Although the relatively high sequence conservation at the SAM-binding site makes inhibitor design challenging, given that the ATP-binding pocket of kinases and the cofactor binding pocket of PMTs have been shown to present a similar level of structural diversity (Campagna-Slater et al., 2011), and selective kinase inhibitors have been successfully developed, isoform-selective inhibitors could be designed for selected PMTs by exploiting the few variable positions available (Schapira and de Freitas, 2014). More selective inhibitors that target specific PMTs as potential therapeutics, especially for application in cancer combinatorial therapy, can be designed by leveraging advances in structure-based design and high-throughput screening (Copeland, 2018).
Furthermore, most of the inhibitors developed thus far are reversible and despite the apparent benefits of covalent inhibitors, it is still difficult to identify adequate warheads to covalently bind to weaker nucleophilic amino acids such as lysine and methionine and since PMT targets do not always contain adequate cysteine, lysine or methionine residues within their active sites, targeting other nucleophilic residues such as tyrosine, serine, threonine, and histidine, is harder since these are less explored (Li et al., 2019), and not so easy to target with the currently available warhead chemistry (Gehringer and Laufer, 2018). For this reason, it is important to optimise the effectiveness of currently available warheads (particularly acrylamide derivatives) as well as explore the development of novel electrophilic warheads in order to facilitate covalent PMT inhibitor discovery and produce potent and selective molecules that can meet the required clinical standards (Li et al., 2019). The investigation of targeting less commonly studied nucleophilic residues (i.e., lysine, tyrosine, serine, threonine, and histidine) may yield alternative options for the development of a new spectrum of covalent inhibitors (Powers et al., 2002; Gambini et al., 2019).
Another issue is the ubiquitous existence and continuous development of pan PMT inhibitors, which offer little clinical benefit, not only because they target PMTs that might be essential for basic cellular function, making it difficult to predict the overall impact on tissues and organs but more so because such inhibitors generally present very different potencies for the respective PMTs they target and finding a balance in the dosing that provides adequate inhibition of the various targets is extremely difficult, which leads to the issue of toxicity.
3. Failure to Clinic
Most of the inhibitors developed so far are only useful for in-vitro experiments since their specificity and sensitivity are not always up to the desired clinical standard. Moreover, even those inhibitors that do make it to clinical trials run the risk of becoming ineffective due to the development of resistance. The basis for designing more effective inhibitors is a clear understanding of why the pre-clinical or clinical trials failed from a biochemical and physiological perspective.
Various reviews have listed the compounds that have entered clinical trials, their use, and failure point (Talukdar et al., 2022; Dempke et al., 2023; Rossi et al., 2024; Tong et al., 2024; Micallef et al., 2024; Zhu et al., 2024). Unfortunately, so far, PMT inhibitors have failed to demonstrate sufficient therapeutic efficacy at non-toxic doses in clinical trials. Having a wide range of protein targets involved in numerous biochemical pathways leads to off-target effects, which bring about significant side effects, outweighing any tangible benefits. In essence, the beneficial therapeutic range, with acceptable side effect and toxicity levels in patients, is very narrow and as a result, it is a significant challenge to achieve the desired therapeutic effect (Micallef et al., 2024).
Computational chemistry, through the implementation of molecular docking, combined with structural biology using PMT crystal structures, provides an invaluable tool set to guide drug design. The data collected from failed trials will provide a stronger basis for in silico prediction using high-throughput screening assays, including virtual screenings (Talukdar et al., 2022; Wang et al., 2022) to confidently shortlist the more promising candidates to be tested further in the wet lab.
4. Considerations for Designing Novel PMT Inhibitors
PMT inhibitors need to have sufficient potency, selectivity, and cell permeability to be successfully implemented in the clinic. Developing selective PMT inhibitors and improving the efficacy of inhibitors, particularly for clinical applications will need to go beyond the production of refined iterations of successful inhibitory molecules in the hope of obtaining something better, ideally by focusing on investigating alternative inhibitory mechanisms. However, this is particularly challenging and the options are somewhat limited considering that all PMTs use SAM as the methyl donor and function through an almost identical mechanism to incorporate the methyl group, given that SAM-binding and residue-binding pockets present a significant similarity across PMTs. In turn, targeting the non-catalytic domains of PMTs is still relatively unexplored, making the assessment of their chemical tractability a compelling area for further research.
Specifically to improve peptide-based inhibitors, one possibility is to replace the peptide backbone with peptide mimics, called peptoids or peptidomimetics. Peptide inhibitors suffer from poor stability and short half-lives due to proteolytic degradation. These peptoids consist of chains of N-substituted glycine residues, such that the side chain is on the nitrogen of the peptide backbone, instead of the α-carbon as in peptides. As a result of this difference, peptoids are less susceptible to degradation by hydrolysis, resulting in improved stability and longer half-lives as well as often having improved cellular permeability and bioavailability compared to their peptide analogues (Powers et al., 2002; Brekker et al., 2022). The success of this approach has been shown in the development of a peptoid inhibitor against SETD3 (Hintzen et al., 2021) and another against PRMT1 (Brekker et al., 2022). Peptoids targeting SETD3 were designed based on a 16-mer β-Actin peptide (residues 66-81) with the histidine at position 73 (H73) being replaced by a panel of methionine analogues, which acted as substrate competitive inhibitors offered varying degrees of inhibition, with selenomethionine being the strongest inhibitor. These molecules showed that a 3-carbon core side chain seemed to be the optimal length and that apart from methionine and selenomethionine, inhibition of SETD3 could also be achieved by β-Actin peptides containing homomethionine, S-methylcysteine, leucine, norleucine, norvaline, and aminobutyric acid (Hintzen et al., 2021). A peptoid that targets PRMT1 was constructed covering the first 16 residues of histone H4 and the guanidine on arginine at position 3 (R3) was replaced with a chloracetamidine group as a warhead, which is well-known to modify cysteine and used to target PRMT1 via cysteine at position 101 (C101) found within the active site, that is not conserved in most other PRMT family members such that it could bring about selective and irreversible modification of PRMT1 (Brekker et al., 2022).
Despite all the available literature, many specifics related to the mode of action of the current inhibitors are still unknown, limiting the design of improved inhibitors. In order to make PMT inhibitor targeting safe for clinical application, the broad use of SAM by a wide variety of enzymes and the similarity between the active sites of enzymes within the same family pose significant challenges to overcome in PMT inhibitor specificity. Considering that each PMT has hundreds of substrate proteins fulfilling very diverse cellular roles, it is expected that perturbing the PMT concentration will have implications on numerous biochemical pathways, some of which may be essential and non-redundant for non-cancerous cells.
Given the growing repertoire of PMT inhibitors, there is a pressing need for comprehensive biochemical and biophysical characterisation, at least for the more successful and promising of the existing PMT inhibitors (Kaniskan et al., 2018). Demonstrating direct interaction between the inhibitor and PMT is imperative and can be achieved through methods such as isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR). Generating structures of the enzyme–inhibitor complexes by using NMR or X-ray crystallography is essential to elucidate the mode of binding and how specificity is being achieved. Functionally important is the demonstration of selectivity of the designed inhibitors for targeting their intended PMT by interrogation against a panel covering a broad range of methyltransferases through robust cellular assays (Kaniskan et al., 2014; Kaniskan et al., 2015).
Inhibitor validation should begin with an in vitro assessment of the level of inhibition of methylation activity. This can be achieved either by quantifying methylated protein products or the by-product SAH, for which both direct or indirect approaches exist (Luo, 2012; Zeng and Wu, 2015). Radiometric assays and top-down mass spectrometry, often in combination with electrophoretic methods such as 2D-PAGE, enable direct quantification of methylated proteins. Alternatively, digested peptides may be analysed using bottom-up or middle-down mass spectrometry, often following antibody enrichment (Luo, 2012).
SAH quantification can be performed directly using anti-SAH antibodies or mass spectrometry. Indirect SAH-quantification methods include radioisotope-labelled assays (using SAM with a tritiated methyl group), a variety of enzyme-coupled spectrophotometric (colorimetric/fluorecence/luminescence) assays (such as reactive thio-mediated chromogenic assays, adenine/xanthine-mediated colorimetric assays, ATP-mediated luminogenic assays, fluorescence polarisation assays and fluorescence lifetime assays), antibody-based assays (namely enzyme-linked immunosorbent assays (ELISA) or dissociation-enhanced lanthanide fluoroimmunoassays (DELFIA)), proximity assays (in the form of proximity homogeneous assays or time-resolved fluorescence resonance energy transfer (TR-FRET) assays), microfluidic capillary electrophoresis, and reporter assays (either using chemical labelling of methyltransferase substrates or cell-based reporter assays) (Luo, 2012; Zeng and Wu, 2015).
Ultimately, the most promising inhibitors will need to be evaluated in disease-relevant or animal models to demonstrate their bioavailability, cell permeability and target engagement within cells as well as their impact on upstream and downstream signalling pathways, thereby bridging the gap from in vitro potency to therapeutic relevance (Luo, 2012; Kaniskan et al., 2015).
Both the mechanistic information on substrate docking and the interaction crystal structure provide key information that not only helps to better understand the mode of action and biological effects that these inhibitors have on their target PMTs but also to identify the strengths and limitations of the different design strategies. Such information should thus become a benchmark for any future inhibitors developed.
A particularly important segment of missing information is related to the impact that the dysregulated PMTs have on the mechanisms of specific chemotherapeutic drug classes. Since each cancer type is treated with a limited number of chemotherapeutics and generally has a handful of commonly dysregulated PMTs, knowing such information would be key towards providing more targeted treatment regimens and avoiding administration of ineffective medication (Micallef et al., 2024).
Another aspect to consider is what the inhibitor is designed to target. In most cases, the inhibitor docking is designed against the canonical sequence and 3D shape; however this does not represent the active form of the PMT. Proteins can be active within cells as functionally different molecules stemming from a combination of genetic polymorphisms, RNA splice variants, and multiple PTMs, and this chemical diversity at a protein level produced from a single gene is collectively described as proteoforms (Aebersold et al., 2018; Smith et al., 2021; Marx et al., 2024). This is particularly relevant to PMT inhibition because proteoform expression generally varies both spatially (across cells and tissues) as well as temporally (with age or over time, as well as after specific events such as disease), affecting both their abundances and interactions, following potentially altered sub-cellular localisation (Smith et al., 2021; Naryzhny, 2024). That being said, it depends very much on the PMT being targetted for inhibition, since some proteins (e.g., actin or histones) present hundreds of proteoforms, while others (and this is probably the category in which most PMTs fall) only present one or two active proteoforms (Naryzhny, 2024). For these reasons, it is important to investigate and identify which proteoform of a specific PMT needs to be targetted in a particular disease context i.e., if it differs from the main proteoform found in healthy cells or other tissues and organs. This would provide a more targetted approach and increase the chances of success in the clinical setting.
As further efforts are made to transition such PMT inhibitors towards clinical application, it is important to also remember that cancer cells will eventually find ways of developing resistance to at least some of these inhibitors, and any prior biochemical and biophysical knowledge will come in handy to overcome such resistance. The best document case so far is for EZH2, where in lymphomas, resistance against inhibitors such as GSK126, EPZ6438 and UNC1999 was acquired through single point mutations at positions I109K, Y111L/N/D, Y661D, C663Y and Y726F, which prevented inhibitor binding (Baker et al., 2015; Gibajaet al., 2016; Bisserier and Wajapeyee, 2018) but could also be due to NSD1 loss (whichmediated H3K36 dimethylation) (Drosos et al., 2022).
Despite the acquisition of resistance against a specific inhibitor, treatment with an inhibitor that targets the same PMT through a different binding mechanism might be able to bypass the acquired resistance in at least some of the tumours (Bisserier and Wajapeyee, 2018). Alternatively, a co-factor within the active complex might need to be targeted (Qi et al., 2017; He et al., 2017) or a different inhibition strategy needs to be adopted (Hsu et al., 2020). Combinatorial therapy blocking epigenetic crosstalk with acetylation (e.g., H3K27ac) and phosphorylation (e.g., Ribosomal S6 kinase 4 (RSK4) or Cyclin-dependent kinase inhibitor (CDKN) 1A/2A/2B) (Pang et al., 2023; Kazansky et al., 2024) and their mediated downstream oncogenic activation (e.g., phosphorylation of Extracellular signal-regulated kinase (ERK) or Aurora kinase B (AURKB)) (Huang et al., 2018; Kazansky et al., 2024) should also be considered for personalised medicine applications.
5. Conclusions and Future Perspectives
In order to gain a more comprehensive understanding of what the roles of at least the major PMTs implicated in cancer chemoresistance are, a collection of patient tumour data from cancer cohorts that are characterised at a protein level, not just genetically, will be essential. So far the literature mostly presents data related to the effects of inhibitors on PMTs dysregulated at the onset of tumour formation. Protein data collected before and after patient treatment with such PMT inhibitors would also provide information on off-target effects as well as key downstream outcomes linked to the combined action of the PMT inhibitor and the chemotherapeutic drug, allowing for improved design or dose administration adjustments. There is, as of yet, very limited literature related to PMT inhibitor efficacy, particularly in relation to countering chemoresistance.
In order to collect reliable data in this regard, there is a need to develop novel experimental techniques for determining PMT inhibitor efficacy, PMT expression quantification, and activity testing. This goes hand in hand with improved or new strategies for higher efficiency protein or peptide enrichment, as well as technological advances in mass spectrometry techniques, which would allow for higher confidence PTM identification and quantification (Deng et al., 2016; Levy, 2019; Micallef and Baron, 2023; Wang et al., 2017). Over and above all this, it is expected that with the current trend of implementing AI-driven design, there will be a push towards improving the currently promising molecules using AI to integrate data from various experimental inputs.
A particularly interesting and impactful future consideration involves the investigation of combinatorial therapies of PMT inhibitors with other small molecule therapeutics (already approved for clinical use), given that tumours often employ multiple chemoresistance mechanisms in parallel. These could include immunomodulatory molecules such as immune checkpoint inhibitors as well as signalling pathway modulators, particularly PI3K, MAPK, or Wnt pathway inhibitors (including via kinase inhibitors).
Author Contributions
Conceptualization, B.B.; writing—original draft preparation, I.M.; writing—review and editing, B.B.; supervision, B.B.; project administration, B.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; E Costello, C.; Cravatt, B.F.; Fenselau, C.; A Garcia, B.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Alicea-Velázquez, N.L.; Shinsky, S.A.; Loh, D.M.; Lee, J.H.; Skalnik, D.G.; Cosgrove, M.S. Targeted disruption of the interaction between WD-40 repeat protein 5 (WDR5) and mixed lineage leukemia (MLL)/SET1 family proteins specifically inhibits MLL1 and SETd1A methyltransferase complexes. Journal of Biological Chemistry 2016, 291, 22357–22372. [Google Scholar] [CrossRef]
- Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1690. [Google Scholar] [CrossRef]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.-H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Antonysamy, S., 2017. The structure and function of the PRMT5: MEP50 complex. Macromolecular Protein Complexes: Structure and Function, pp.185-194.
- Arrigoni, E.; Galimberti, S.; Petrini, M.; Danesi, R.; Di Paolo, A. ATP-binding cassette transmembrane transporters and their epigenetic control in cancer: an overview. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Asberry, A.M.; Cai, X.; Deng, X.; Santiago, U.; Liu, S.; Sims, H.S.; Liang, W.; Xu, X.; Wan, J.; Jiang, W.; et al. Discovery and Biological Characterization of PRMT5:MEP50 Protein–Protein Interaction Inhibitors. J. Med. Chem. 2022, 65, 13793–13812. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.; Nerle, S.; Pritchard, J.; Zhao, B.; Rivera, V.M.; Garner, A.; Gonzalvez, F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 2015, 6, 32646–32655. [Google Scholar] [CrossRef]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.J.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A.; et al. ( R )-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. 2014, 111, 12853–12858. [Google Scholar] [CrossRef]
- Bashore, F.M.; Foley, C.A.; Ong, H.W.; Rectenwald, J.M.; Hanley, R.P.; Norris-Drouin, J.L.; Cholensky, S.H.; Mills, C.A.; Pearce, K.H.; Herring, L.E.; et al. PROTAC Linkerology Leads to an Optimized Bivalent Chemical Degrader of Polycomb Repressive Complex 2 (PRC2) Components. ACS Chem. Biol. 2023, 18, 494–507. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef]
- Biswas, R.; Bugde, P.; He, J.; Merien, F.; Lu, J.; Liu, D.-X.; Myint, K.; Liu, J.; McKeage, M.; Li, Y. Transport-Mediated Oxaliplatin Resistance Associated with Endogenous Overexpression of MRP2 in Caco-2 and PANC-1 Cells. Cancers 2019, 11, 1330. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Brekker, M.A.; Sartawi, T.; Sawatzky, T.M.; Causey, C.P.; Rehman, F.K.; Knuckley, B. A peptoid-based inhibitor of protein arginine methyltransferase 1 (PRMT1) induces apoptosis and autophagy in cancer cells. J. Biol. Chem. 2022, 298, 102205. [Google Scholar] [CrossRef]
- Butler, K.V.; Ma, A.; Yu, W.; Li, F.; Tempel, W.; Babault, N.; Pittella-Silva, F.; Shao, J.; Wang, J.; Luo, M.; et al. Structure-Based Design of a Covalent Inhibitor of the SET Domain-Containing Protein 8 (SETD8) Lysine Methyltransferase. J. Med. Chem. 2016, 59, 9881–9889. [Google Scholar] [CrossRef] [PubMed]
- Campagna-Slater, V.; Mok, M.W.; Nguyen, K.T.; Feher, M.; Najmanovich, R.; Schapira, M. Structural Chemistry of the Histone Methyltransferases Cofactor Binding Site. J. Chem. Inf. Model. 2011, 51, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Pan, C.; Zhang, M.; Huo, H.; Shan, H.; Wu, J. Histone methyltransferase SETD1A interacts with notch and promotes notch transactivation to augment ovarian cancer development. BMC Cancer 2023, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Chen, L., Zeng, Y. and Zhou, S.F., 2018. Role of apoptosis in cancer resistance to chemotherapy. Current understanding of apoptosis-programmed cell death.
- Chen, W.; Chen, X.; Li, D.; Wang, X.; Long, G.; Jiang, Z.; You, Q.; Guo, X. Discovery of a potent MLL1 and WDR5 protein-protein interaction inhibitor with in vivo antitumor activity. Eur. J. Med. Chem. 2021, 223, 113677. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, H.; Yang, S.; Su, D. Increased ABCC2 expression predicts cisplatin resistance in non-small cell lung cancer. Cell Biochem. Funct. 2020, 39, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Protein methyltransferase inhibitors as precision cancer therapeutics: a decade of discovery. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20170080. [Google Scholar] [CrossRef]
- Dale, B.; Anderson, C.; Park, K.-S.; Kaniskan, H.Ü.; Ma, A.; Shen, Y.; Zhang, C.; Xie, L.; Chen, X.; Yu, X.; et al. Targeting Triple-Negative Breast Cancer by a Novel Proteolysis Targeting Chimera Degrader of Enhancer of Zeste Homolog 2. ACS Pharmacol. Transl. Sci. 2022, 5, 491–507. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; A Baldwin, S.; Mackey, J.; Sawyer, M.B.; E Cass, C. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Desole, M.; Chiusolo, P.; Sica, S.; Schmidt-Hieber, M. Targeting the undruggable: menin inhibitors ante portas. J. Cancer Res. Clin. Oncol. 2023, 149, 9451–9459. [Google Scholar] [CrossRef]
- Diehl, C.J.; Ciulli, A. Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem. Soc. Rev. 2022, 51, 8216–8257. [Google Scholar] [CrossRef] [PubMed]
- Dölle, A.; Adhikari, B.; Krämer, A.; Weckesser, J.; Berner, N.; Berger, L.-M.; Diebold, M.; Szewczyk, M.M.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; et al. Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J. Med. Chem. 2021, 64, 10682–10710. [Google Scholar] [CrossRef]
- Dong, H.; Liu, S.; Zhang, X.; Chen, S.; Kang, L.; Chen, Y.; Ma, S.; Fu, X.; Liu, Y.; Zhang, H.; et al. An Allosteric PRC2 Inhibitor Targeting EED Suppresses Tumor Progression by Modulating the Immune Response. Cancer Res. 2019, 79, 5587–5596. [Google Scholar] [CrossRef]
- Drosos, Y.; Myers, J.A.; Xu, B.; Mathias, K.M.; Beane, E.C.; Radko-Juettner, S.; Mobley, R.J.; Larsen, M.E.; Piccioni, F.; Ma, X.; et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol. Cell 2022, 82, 2472–2489.e8. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2015, 7, 162–166. [Google Scholar] [CrossRef]
- Eggert, E.; Hillig, R.C.; Koehr, S.; Stöckigt, D.; Weiske, J.; Barak, N.; Mowat, J.; Brumby, T.; Christ, C.D.; ter Laak, A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. [Google Scholar] [CrossRef]
- Elton, T.S.; Ozer, H.G.; Yalowich, J.C. Effects of DNA topoisomerase IIα splice variants on acquired drug resistance. Cancer Drug Resist. 2020, 3, 161–170. [Google Scholar] [CrossRef]
- Falnes, P.Ø.; Małecki, J.M.; Herrera, M.C.; Bengtsen, M.; Davydova, E. Human seven-β-strand (METTL) methyltransferases - conquering the universe of protein lysine methylation. J. Biol. Chem. 2023, 299, 104661. [Google Scholar] [CrossRef]
- Fernández-Ramos, D.; Lopitz-Otsoa, F.; Lu, S.C.; Mato, J.M. S-Adenosylmethionine: A Multifaceted Regulator in Cancer Pathogenesis and Therapy. Cancers 2025, 17, 535. [Google Scholar] [CrossRef]
- Froese, D. S., Fowler, B. & Baumgartner, M. R. Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 42, 673–685 (2019).
- G. Blum, G. Ibanez, X. Rao, D. Shum, C. Radu, H. Djaballah, J. C. Rice and M. Luo, ACS Chem. Biol., 2014, 9, 2471–2478.
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Gambini, L., Baggio, C., Udompholkul, P., Jossart, J., Salem, A.F., Perry, J.J.P. and Pellecchia, M., 2019. Covalent inhibitors of protein–protein interactions targeting lysine, tyrosine, or histidine residues. Journal of medicinal chemistry, 62(11), pp.5616-5627.
- Gao, J.; Wang, C.; Wei, W. The effects of drug transporters on the efficacy of methotrexate in the treatment of rheumatoid arthritis. Life Sci. 2021, 268, 118907. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2018, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.-P.; de Oca, R.M.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Gibaja, V.; Shen, F.; Harari, J.; Korn, J.; Ruddy, D.; Saenz-Vash, V.; Zhai, H.; Rejtar, T.; Paris, C.G.; Yu, Z.; et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 2015, 35, 558–566. [Google Scholar] [CrossRef]
- Gradl, S.; Steuber, H.; Weiske, J.; Schmees, N.; Siegel, S.; Stoeckigt, D.; Christ, C.D.; Li, F.; Organ, S.; Barsyte-Lovejoy, D.; et al. Abstract 1646: Discovery and characterization of BAY-6035, a novel benzodiazepine-based SMYD3 inhibitor. Cancer Res. 2018, 78, 1646–1646. [Google Scholar] [CrossRef]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D.; et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Y.; Zhou, Z.; Hou, L.; Liu, W.; Ren, W.; Mi, D.; Sun, J.; Dai, X.; Wu, Y.; et al. Targeting PRMT5 through PROTAC for the treatment of triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2024, 43, 1–15. [Google Scholar] [CrossRef]
- H. Nguyen, A. Allali-Hassani, S. Antonysamy, S. Chang, L. H. Chen, C. Curtis, S. Emtage, L. Fan, T. Gheyi, F. Li, S. Liu, J. R. Martin, D. Mendel, J. B. Olsen, L. Pelletier, T. Shatseva, S. Wu, F. F. Zhang, C. H. Arrowsmith, P. J. Brown, R. M. Campbell, B. A. Garcia, D. Barsyte-Lovejoy, M. Mader and M. Vedadi, J. Biol. Chem., 2015, 290, 13641–13653.
- Halby, L.; Marechal, N.; Pechalrieu, D.; Cura, V.; Franchini, D.-M.; Faux, C.; Alby, F.; Troffer-Charlier, N.; Kudithipudi, S.; Jeltsch, A.; et al. Hijacking DNA methyltransferase transition state analogues to produce chemical scaffolds for PRMT inhibitors. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20170072. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Huang, Y.; Qiu, M.; Yin, C.; Ren, H.; Gan, H.; Li, H.; Zhou, Y.; Xia, J.; Li, W.; et al. Immunoassay of S-adenosylmethionine and S-adenosylhomocysteine: the methylation index as a biomarker for disease and health status. BMC Res. Notes 2016, 9, 498. [Google Scholar] [CrossRef]
- He Y et al. 2017 The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 13, 389–395. [CrossRef]
- He, H.; Li, X.; Su, F.; Jin, H.; Zhang, J.; Wang, Y. Current and Emerging Approaches Targeting G9a for the Treatment of Various Diseases. J. Med. Chem. 2024, 68, 1068–1089. [Google Scholar] [CrossRef]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B.; The, J.; Lima-Fernandes, E.; Szewczyk, M.M.; et al. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13, 389–395. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B.; The, J.; Lima-Fernandes, E.; Szewczyk, M.M.; et al. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13, 389–395. [Google Scholar] [CrossRef]
- Hintzen, J.C.J.; Moesgaard, L.; Kwiatkowski, S.; Drozak, J.; Kongsted, J.; Mecinović, J. β-Actin Peptide-Based Inhibitors of Histidine Methyltransferase SETD3. Chem. Med. Chem. 2021, 16, 2695–2702. [Google Scholar] [CrossRef]
- Ho, M.-C.; Wilczek, C.; Bonanno, J.B.; Xing, L.; Seznec, J.; Matsui, T.; Carter, L.G.; Onikubo, T.; Kumar, P.R.; Chan, M.K.; et al. Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS ONE 2013, 8, e57008. [Google Scholar] [CrossRef]
- Hsu, H.; Chen, M.; Baskaran, R.; Lin, Y.; Day, C.H.; Lin, Y.; Tu, C.; Padma, V.V.; Kuo, W.; Huang, C. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell. Physiol. 2017, 233, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.-R.; Rasmusson, T.; Robinson, J.; Pachl, F.; Read, J.; Kawatkar, S.; O'Donovan, D.H.; Bagal, S.; Code, E.; Rawlins, P.; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020, 27, 41–46.e17. [Google Scholar] [CrossRef]
- Huang, X.; Yan, J.; Zhang, M.; Wang, Y.; Chen, Y.; Fu, X.; Wei, R.; Zheng, X.-L.; Liu, Z.; Zhang, X.; et al. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell 2018, 175, 186–199.e19. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.C.-Y.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W.-F. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef]
- Ju, Y.; Song, H.; He, Y.; Lo, Y.; Fan, Z.; Lu, J. Development of a Selective and Potent PRMT4 PROTAC Degrader with Efficacy against Multiple Myeloma in Vitro and in Vivo. J. Med. Chem. 2025, 68, 13973–13989. [Google Scholar] [CrossRef]
- Ju, Y.; Song, H.; He, Y.; Lo, Y.; Fan, Z.; Lu, J. Development of a Selective and Potent PRMT4 PROTAC Degrader with Efficacy against Multiple Myeloma in Vitro and in Vivo. J. Med. Chem. 2025, 68, 13973–13989. [Google Scholar] [CrossRef]
- K. V. Butler, A. Ma, W. Yu, F. Li, W. Tempel, N. Babault, F. Pittella-Silva, J. Shao, J. Wang, M. Luo, M. Vedadi, P. J. Brown, C. H. Arrowsmith and J. Jin, J. Med. Chem., 2016, 59, 9881–9889.
- Kaniskan, H.Ü.; Martini, M.L.; Jin, J. Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev. 2017, 118, 989–1068. [Google Scholar] [CrossRef]
- Kaniskan, H.Ü.; Szewczyk, M.M.; Yu, Z.; Eram, M.S.; Yang, X.; Schmidt, K.; Luo, X.; Dai, M.; He, F.; Zang, I.; et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem. Int. Ed. 2015, 54, 5166–70. [Google Scholar] [CrossRef]
- Kannampuzha, S.; Gopalakrishnan, A.V. Cancer chemoresistance and its mechanisms: Associated molecular factors and its regulatory role. Med Oncol. 2023, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kazansky, Y.; Cameron, D.; Mueller, H.S.; Demarest, P.; Zaffaroni, N.; Arrighetti, N.; Zuco, V.; Kuwahara, Y.; Somwar, R.; Ladanyi, M.; et al. Overcoming Clinical Resistance to EZH2 Inhibition Using Rational Epigenetic Combination Therapy. Cancer Discov. 2024, 14, 965–981. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 2024, 22, 1–26. [Google Scholar] [CrossRef]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; A Kerenyi, M.; Walensky, L.D.; Orkin, S.H. Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, Y.J.; Lee, H.I.; Jeong, S.-H.; Nam, H.J.; Cho, J.H. NRF2 Knockdown Resensitizes 5-Fluorouracil-Resistant Pancreatic Cancer Cells by Suppressing HO-1 and ABCG2 Expression. Int. J. Mol. Sci. 2020, 21, 4646. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; A Kerenyi, M.; Walensky, L.D.; Orkin, S.H. Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Knuhtsen, A.; Legrand, B.; Van der Poorten, O.; Amblard, M.; Martinez, J.; Ballet, S.; Kristensen, J.L.; Pedersen, D.S. Conformationally Constrained Peptidomimetics as Inhibitors of the Protein Arginine Methyl Transferases. Chem. – A Eur. J. 2016, 22, 14022–14028. [Google Scholar] [CrossRef]
- Kobayashi, S., Boggon, T.J., Dayaram, T., Jänne, P.A., Kocher, O., Meyerson, M., Johnson, B.E., Eck, M.J., Tenen, D.G. and Halmos, B., 2005. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. New England Journal of Medicine, 352(8), pp.786-792.
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.-P.; Kuznetsova, E.; et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef]
- Konze, K.D.; Pattenden, S.G.; Liu, F.; Barsyte-Lovejoy, D.; Li, F.; Simon, J.M.; Davis, I.J.; Vedadi, M.; Jin, J. A Chemical Tool for In Vitro and In Vivo Precipitation of Lysine Methyltransferase G9a. ChemMedChem 2014, 9, 549–553. [Google Scholar] [CrossRef]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5, 19–19. [Google Scholar] [CrossRef]
- Kozbial, P.Z.; Mushegian, A.R. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005, 5, 19–19. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Krzyzanowski, A., Esser, L.M., Willaume, A., Prudent, R., Peter, C., ‘t Hart, P. and Waldmann, H., 2022. Development of macrocyclic PRMT5–adaptor protein interaction inhibitors. Journal of Medicinal Chemistry, 65(22), pp.15300-15311.
- Kung, P.P., Bingham, P., Brooun, A., Collins, M., Deng, Y.L., Dinh, D., Fan, C., Gajiwala, K.S., Grantner, R., Gukasyan, H.J. and Hu, W., 2018. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: identification of development candidate (R)-5, 8-Dichloro-7-(methoxy (oxetan-3-yl) methyl)-2-((4-methoxy-6-methyl-2-oxo-1, 2-dihydropyridin-3-yl) methyl)-3, 4-dihydroisoquinolin-1 (2 H)-one (PF-06821497).
- LegaardAndersson, J.; Christensen, J.; Kleine-Kohlbrecher, D.; Comet, I.V.; Støier, J.F.; Antoku, Y.; Poljak, V.; Moretti, L.; Dolberg, J.; Jacso, T.; et al. Discovery of NSD2-Degraders from Novel and Selective DEL Hits. ChemBioChem 2023, 24, e202300515. [Google Scholar] [CrossRef] [PubMed]
- Levy, D. Lysine methylation signaling of non-histone proteins in the nucleus. Cell. Mol. Life Sci. 2019, 76, 2873–2883. [Google Scholar] [CrossRef] [PubMed]
- Li, A., Song, J., Lai, Q., Liu, B., Wang, H., Xu, Y., Feng, X., Sun, X. and Du, Z., 2016. Hypermethylation of ATP-binding cassette B1 (ABCB 1) multidrug resistance 1 (MDR 1) is associated with cisplatin resistance in the A549 lung adenocarcinoma cell line. International journal of experimental pathology, 97(6), pp.412-421.
- Li, B.; Rong, D.; Wang, Y. Targeting Protein-Protein Interaction with Covalent Small-Molecule Inhibitors. Curr. Top. Med. Chem. 2019, 19, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yu, X.; Kottur, J.; Gong, W.; Zhang, Z.; Storey, A.J.; Tsai, Y.-H.; Uryu, H.; Shen, Y.; Byrum, S.D.; et al. Discovery of a dual WDR5 and Ikaros PROTAC degrader as an anti-cancer therapeutic. Oncogene 2022, 41, 3328–3340. [Google Scholar] [CrossRef]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11. [Google Scholar] [CrossRef]
- Lin, H.; Luengo, J.I. Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 1264–1269. [Google Scholar] [CrossRef]
- Lin, H.; Wang, B.; Yu, J.; Wang, J.; Li, Q.; Cao, B. Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. J. Cancer 2018, 9, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, M.; Zhang, Y.W.; Tong, S.; Leal, R.A.; Shetty, R.; Vaddi, K.; Luengo, J.I. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033–1038. [Google Scholar] [CrossRef]
- Liu, F.; Barsyte-Lovejoy, D.; Li, F.; Xiong, Y.; Korboukh, V.; Huang, X.-P.; Allali-Hassani, A.; Janzen, W.P.; Roth, B.L.; Frye, S.V.; et al. Discovery of an in Vivo Chemical Probe of the Lysine Methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. [Google Scholar] [CrossRef]
- Liu, K.-L.; Zhu, K.; Zhang, H. An overview of the development of EED inhibitors to disable the PRC2 function. RSC Med. Chem. 2021, 13, 39–53. [Google Scholar] [CrossRef]
- Liu, L.; Parolia, A.; Liu, Y.; Hou, C.; He, T.; Qiao, Y.; Eyunni, S.; Luo, J.; Li, C.; Wang, Y.; et al. Discovery of LLC0424 as a Potent and Selective in Vivo NSD2 PROTAC Degrader. J. Med. Chem. 2024, 67, 6938–6951. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, X.; Wang, Q.; Wu, X.; Zhang, Q.; Wei, W.; Su, X.; He, H.; Zhou, S.; Hu, R.; et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J. Med. Chem. 2021, 64, 2829–2848. [Google Scholar] [CrossRef]
- Loenen, W. S-Adenosylmethionine: jack of all trades and master of everything? Biochem. Soc. Trans. 2006, 34, 330–333. [Google Scholar] [CrossRef]
- Lu, B.; Shen, X.; Zhang, L.; Liu, D.; Zhang, C.; Cao, J.; Shen, R.; Zhang, J.; Wang, D.; Wan, H.; et al. Discovery of EBI-2511: A Highly Potent and Orally Active EZH2 Inhibitor for the Treatment of Non-Hodgkin’s Lymphoma. ACS Med. Chem. Lett. 2018, 9, 98–102. [Google Scholar] [CrossRef]
- Luo, M. Current Chemical Biology Approaches to Interrogate Protein Methyltransferases. ACS Chem. Biol. 2012, 7, 443–463. [Google Scholar] [CrossRef]
- M. J. Thomenius, J. Totman, D. Harvey, L. H. Mitchell, T. V. Riera, K. Cosmopoulos, A. R. Grassian, C. Klaus, M. Foley, E. A. Admirand, H. Jahic, C. Majer, T. Wigle, S. L. Jacques, J. Gureasko, D. Brach, T. Lingaraj, K. West, S. Smith, N. Rioux, N. J. Waters, C. Tang, A. Raimondi, M. Munchhof, J. E. Mills, S. Ribich, M. Porter Scott, K. W. Kuntz, W. P. Janzen, M. Moyer, J. J. Smith, R. Chesworth, R. A. Copeland and P. A. Boriack-Sjodin, PLoS One, 2018, 13, e0197372.
- Ma, A.; Stratikopoulos, E.; Park, K.-S.; Wei, J.; Martin, T.C.; Yang, X.; Schwarz, M.; Leshchenko, V.; Rialdi, A.; Dale, B.; et al. Discovery of a first-in-class EZH2 selective degrader. Nat. Chem. Biol. 2019, 16, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Yu, W.; Li, F.; Bleich, R.M.; Herold, J.M.; Butler, K.V.; Norris, J.L.; Korboukh, V.; Tripathy, A.; Janzen, W.P.; et al. Discovery of a Selective, Substrate-Competitive Inhibitor of the Lysine Methyltransferase SETD8. J. Med. Chem. 2014, 57, 6822–6833. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Martin, P.L.; Pérez-Areales, F.J.; Rao, S.V.; Walsh, S.J.; Carroll, J.S.; Spring, D.R. Towards the Targeted Protein Degradation of PRMT1. ChemMedChem 2024, 19, e202400269. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Inside the chase after those elusive proteoforms. Nat. Methods 2024, 21, 158–163. [Google Scholar] [CrossRef]
- Mato, J.M.; Corrales, F.J.; Lu, S.C.; Avila, M.A. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002, 16, 15–26. [Google Scholar] [CrossRef]
- Meng, F.; Xu, C.; Park, K.-S.; Kaniskan, H.Ü.; Wang, G.G.; Jin, J. Discovery of a First-in-Class Degrader for Nuclear Receptor Binding SET Domain Protein 2 (NSD2) and Ikaros/Aiolos. J. Med. Chem. 2022, 65, 10611–10625. [Google Scholar] [CrossRef]
- Micallef, I. and Baron, B., 2023. Proteomic strategies for methylation analysis in colorectal cancer chemoresistance. Journal of Proteome Data and Methods, 5, p.16.
- Micallef, I.; Fenech, K.; Baron, B. Therapeutic targeting potential of the protein lysine and arginine methyltransferases to reverse cancer chemoresistance. Front. Mol. Biosci. 2024, 11, 1455415. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2022, 27, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2015, 7, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Meeran, S.M. microRNAs in cancer chemoresistance: The sword and the shield. Non-coding RNA Res. 2021, 6, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Mori, S., Iwase, K., Iwanami, N., Tanaka, Y., Kagechika, H. and Hirano, T., 2010. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9. Bioorganic & medicinal chemistry, 18(23), pp.8158-8166.
- Mosca, L.; Minopoli, M.; Pagano, M.; Vitiello, F.; Carriero, M.V.; Cacciapuoti, G.; Porcelli, M. Effects of S-adenosyl-L-methionine on the invasion and migration of head and neck squamous cancer cells and analysis of the underlying mechanisms. Int. J. Oncol. 2020, 56, 1212–1224. [Google Scholar] [CrossRef]
- Mosca, L.; Pagano, M.; Borzacchiello, L.; Mele, L.; Russo, A.; Russo, G.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine Increases the Sensitivity of Human Colorectal Cancer Cells to 5-Fluorouracil by Inhibiting P-Glycoprotein Expression and NF-κB Activation. Int. J. Mol. Sci. 2021, 22, 9286. [Google Scholar] [CrossRef]
- Mukherjee, A., 2025. Design, Synthesis, and Biological Evaluation of (Doctoral dissertation).
- Mulvaney, K.M.; Blomquist, C.; Acharya, N.; Li, R.; Ranaghan, M.J.; O’kEefe, M.; Rodriguez, D.J.; Young, M.J.; Kesar, D.; Pal, D.; et al. Molecular basis for substrate recruitment to the PRMT5 methylosome. Mol. Cell 2021, 81, 3481–3495.e7. [Google Scholar] [CrossRef]
- Naryzhny, S., 2024. Puzzle of proteoform variety—where is a key?. Proteomes, 12(2), p.15.
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, genes, polymorphisms and the associated diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef]
- Ning, J.; Chen, L.; Xiao, G.; Zeng, Y.; Shi, W.; Tanzhu, G.; Zhou, R. The protein arginine methyltransferase family (PRMTs) regulates metastases in various tumors: From experimental study to clinical application. Biomed. Pharmacother. 2023, 167, 115456. [Google Scholar] [CrossRef]
- Pan, S.; Li, Z.; He, Z.; Qiu, J.; Zhou, S. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Pang, F.; Zhang, L.; Li, M.; Yi, X.; Wang, Y.; Yang, P.; Wen, B.; Jiang, J.; Teng, Y.; Yang, X.; et al. Ribosomal S6 protein kinase 4 promotes resistance to EZH2 inhibitors in glioblastoma. Cancer Gene Ther. 2023, 30, 1636–1648. [Google Scholar] [CrossRef]
- Park, K.-S.; Xiong, Y.; Yim, H.; Velez, J.; Babault, N.; Kumar, P.; Liu, J.; Jin, J. Discovery of the First-in-Class G9a/GLP Covalent Inhibitors. J. Med. Chem. 2022, 65, 10506–10522. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Feo, C.F.; Feo, F. S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells 2022, 11, 409. [Google Scholar] [CrossRef]
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.J.; Di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell. Physiol. 2015, 230, 2447–2460. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [PubMed]
- Potjewyd, F.; Turner, A.-M.W.; Beri, J.; Rectenwald, J.M.; Norris-Drouin, J.L.; Cholensky, S.H.; Margolis, D.M.; Pearce, K.H.; Herring, L.E.; James, L.I. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol. 2020, 27, 47–56.e15. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C., Asgian, J.L., Ekici, Ö.D. and James, K.E., 2002. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chemical reviews, 102(12), pp.4639-4750.
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef]
- Rej, R.K.; Wang, C.; Lu, J.; Wang, M.; Petrunak, E.; Zawacki, K.P.; McEachern, D.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, L.; et al. EEDi-5285: An Exceptionally Potent, Efficacious, and Orally Active Small-Molecule Inhibitor of Embryonic Ectoderm Development. J. Med. Chem. 2020, 63, 7252–7267. [Google Scholar] [CrossRef]
- Roh, Y.-G.; Mun, M.-H.; Jeong, M.-S.; Kim, W.-T.; Lee, S.-R.; Chung, J.-W.; Kim, S.I.; Kim, T.N.; Kil Nam, J.; Leem, S.-H. Drug resistance of bladder cancer cells through activation of ABCG2 by FOXM1. BMB Rep. 2018, 51, 98–103. [Google Scholar] [CrossRef]
- Rossi, A.; Zacchi, F.; Reni, A.; Rota, M.; Palmerio, S.; Menis, J.; Zivi, A.; Milleri, S.; Milella, M. Progresses and Pitfalls of Epigenetics in Solid Tumors Clinical Trials. Int. J. Mol. Sci. 2024, 25, 11740. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.M.; Hariharan, S. Regulatory players of DNA damage repair mechanisms: Role in Cancer Chemoresistance. Biomed. Pharmacother. 2017, 93, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M. and de Freitas, R.F., 2014. Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm, 5(12), pp.1779-1788.
- Shailesh, H.; Siveen, K.S.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) activates WNT/β-catenin signalling in breast cancer cells via epigenetic silencing of DKK1 and DKK3. J. Cell. Mol. Med. 2021, 25, 1583–1600. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.S.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Shen, Y.; Li, F.; Szewczyk, M.M.; Halabelian, L.; Park, K.-S.; Chau, I.; Dong, A.; Zeng, H.; Chen, H.; Meng, F.; et al. Discovery of a First-in-Class Protein Arginine Methyltransferase 6 (PRMT6) Covalent Inhibitor. J. Med. Chem. 2020, 63, 5477–5487. [Google Scholar] [CrossRef]
- Shi, Y.; Shen, Q.; Long, R.; Mao, Y.; Tong, S.; Yang, Y.; Gao, J.; Zhou, H.; Chen, Y.; Zhou, B. Discovery of Potent and Selective G9a Degraders for the Treatment of Pancreatic Cancer. J. Med. Chem. 2024, 67, 13271–13285. [Google Scholar] [CrossRef]
- Shui, X., Tian, L., Zhou, Y. and Zhao, B., 2024. Targeting ACSS2 activity suspends the formation of chemoresistance through suppressed histone H3 acetylation in human breast cancer.
- Smith, L.M.; Kelleher, N.L. Proteoforms as the next proteomics currency. Science 2018, 359, 1106–1107. [Google Scholar] [CrossRef] [PubMed]
- Spitzwieser, M.; Pirker, C.; Koblmüller, B.; Pfeiler, G.; Hacker, S.; Berger, W.; Heffeter, P.; Cichna-Markl, M. Promoter methylation patterns of ABCB1, ABCC1 and ABCG2 in human cancer cell lines, multidrug-resistant cell models and tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. Oncotarget 2016, 7, 73347–73369. [Google Scholar] [CrossRef] [PubMed]
- Sumarpo, A.; Ito, K.; Saiki, Y.; Ishizawa, K.; Wang, R.; Chen, N.; Sunamura, M.; Horii, A. Genetic and epigenetic aberrations of ABCB1 synergistically boost the acquisition of taxane resistance in esophageal squamous cancer cells. Biochem. Biophys. Res. Commun. 2020, 526, 586–591. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Chen, X.; Yu, A.; Du, W.; Huang, Y.; Wu, F.; Yu, L.; Li, J.; Wen, C.; et al. Discovery of a potent and selective proteolysis targeting chimera (PROTAC) degrader of NSD3 histone methyltransferase. Eur. J. Med. Chem. 2022, 239, 114528. [Google Scholar] [CrossRef]
- Sweis, R.F.; Pliushchev, M.; Brown, P.J.; Guo, J.; Li, F.; Maag, D.; Petros, A.M.; Soni, N.B.; Tse, C.; Vedadi, M.; et al. Discovery and Development of Potent and Selective Inhibitors of Histone Methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5, 205–209. [Google Scholar] [CrossRef]
- Sweis, R.F.; Wang, Z.; Algire, M.; Arrowsmith, C.H.; Brown, P.J.; Chiang, G.G.; Guo, J.; Jakob, C.G.; Kennedy, S.; Li, F.; et al. Discovery of A-893, A New Cell-Active Benzoxazinone Inhibitor of Lysine Methyltransferase SMYD2. ACS Med. Chem. Lett. 2015, 6, 695–700. [Google Scholar] [CrossRef]
- Takemoto, Y.; Ito, A.; Niwa, H.; Okamura, M.; Fujiwara, T.; Hirano, T.; Handa, N.; Umehara, T.; Sonoda, T.; Ogawa, K.; et al. Identification of Cyproheptadine as an Inhibitor of SET Domain Containing Lysine Methyltransferase 7/9 (Set7/9) That Regulates Estrogen-Dependent Transcription. J. Med. Chem. 2016, 59, 3650–3660. [Google Scholar] [CrossRef]
- Talibov, V.O.; Fabini, E.; FitzGerald, E.A.; Tedesco, D.; Cederfelt, D.; Talu, M.J.; Rachman, M.M.; Mihalic, F.; Manoni, E.; Naldi, M.; et al. Discovery of an Allosteric Ligand Binding Site in SMYD3 Lysine Methyltransferase. ChemBioChem 2021, 22, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, A.; Mukherjee, A.; Bhattacharya, D. Fascinating Transformation of SAM-Competitive Protein Methyltransferase Inhibitors from Nucleoside Analogues to Non-Nucleoside Analogues. J. Med. Chem. 2022, 65, 1662–1684. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, A.; Mukherjee, A.; Bhattacharya, D. Fascinating Transformation of SAM-Competitive Protein Methyltransferase Inhibitors from Nucleoside Analogues to Non-Nucleoside Analogues. J. Med. Chem. 2022, 65, 1662–1684. [Google Scholar] [CrossRef]
- Taylor, A.P., Swewczyk, M., Kennedy, S., Trush, V.V., Wu, H., Zeng, H., Dong, A., Ferreira de Freitas, R., Tatlock, J., Kumpf, R.A. and Wythes, M., 2019. Selective, small-molecule co-factor binding site inhibition of a Su (var) 3–9, enhancer of zeste, trithorax domain containing lysine methyltransferase. Journal of medicinal chemistry, 62(17), pp.7669-7683.
- Tong, C.; Chang, X.; Qu, F.; Bian, J.; Wang, J.; Li, Z.; Xu, X. Overview of the development of protein arginine methyltransferase modulators: Achievements and future directions. Eur. J. Med. Chem. 2024, 267, 116212. [Google Scholar] [CrossRef]
- Tu, Y.; Sun, Y.; Qiao, S.; Luo, Y.; Liu, P.; Jiang, Z.-X.; Hu, Y.; Wang, Z.; Huang, P.; Wen, S. Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity against Lymphoma. J. Med. Chem. 2021, 64, 10167–10184. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; E Bray, S.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, Rishi G., Victor S. Gehling, Les A. Dakin, Andrew S. Cook, Christopher G. Nasveschuk, Martin Duplessis, Priyadarshini Iyer et al. "Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1, 2-dihydropyridin-3-yl) methyl)-2-methyl-1-(1-(1-(2, 2, 2-trifluoroethyl) piperidin-4-yl) ethyl)-1 H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas." Journal of medicinal chemistry 59, no. 21 (2016): 9928-9941.
- Velez, J.; Dale, B.; Park, K.-S.; Kaniskan, H.Ü.; Yu, X.; Jin, J. Discovery of a novel, highly potent EZH2 PROTAC degrader for targeting non-canonical oncogenic functions of EZH2. Eur. J. Med. Chem. 2024, 267, 116154–116154. [Google Scholar] [CrossRef]
- Velez, J.; Han, Y.; Yim, H.; Yang, P.; Deng, Z.; Park, K.-S.; Kabir, M.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Discovery of the First-in-Class G9a/GLP PROTAC Degrader. J. Med. Chem. 2024, 67, 6397–6409. [Google Scholar] [CrossRef] [PubMed]
- Vougiouklakis, T.; Bernard, B.J.; Nigam, N.; Burkitt, K.; Nakamura, Y.; Saloura, V. Clinicopathologic significance of protein lysine methyltransferases in cancer. Clin. Epigenetics 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Wang, C.; Chen, X.; Liu, X.; Lu, D.; Li, S.; Qu, L.; Yin, F.; Luo, H.; Zhang, Y.; Luo, Z.; et al. Discovery of precision targeting EZH2 degraders for triple-negative breast cancer. Eur. J. Med. Chem. 2022, 238, 114462. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Gong, W.; Liu, X.; Park, K.-S.; Ma, A.; Tsai, Y.-H.; Shen, Y.; Onikubo, T.; Pi, W.-C.; et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 2022, 24, 384–399. [Google Scholar] [CrossRef]
- Wang, M.Y.; Liow, P.; Guzman, M.I.T.; Qi, J. Exploring Methods of Targeting Histone Methyltransferases and Their Applications in Cancer Therapeutics. ACS Chem. Biol. 2022, 17, 744–755. [Google Scholar] [CrossRef]
- Wang, S.E.; Xiong, Y.; Jang, M.-A.; Park, K.-S.; Donahue, M.; Velez, J.; Jin, J.; Jiang, Y.-H. Newly developed oral bioavailable EHMT2 inhibitor as a potential epigenetic therapy for Prader-Willi syndrome. Mol. Ther. 2024, 32, 2662–2675. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef] [PubMed]
- Waters, N.J. Preclinical Pharmacokinetics and Pharmacodynamics of Pinometostat (EPZ-5676), a First-in-Class, Small Molecule S-Adenosyl Methionine Competitive Inhibitor of DOT1L. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 891–901. [Google Scholar] [CrossRef]
- Wei, L.; Mei, D.; Hu, S.; Du, S. Dual-target EZH2 inhibitor: latest advances in medicinal chemistry. Futur. Med. Chem. 2024, 16, 1561–1582. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, W.; Eram, M.S.; Vhuiyan, M.; Dong, A.; Zeng, H.; He, H.; Brown, P.; Frankel, A.; Vedadi, M.; et al. Structural basis of arginine asymmetrical dimethylation by PRMT6. Biochem. J. 2016, 473, 3049–3063. [Google Scholar] [CrossRef]
- Xu, C.; Meng, F.; Park, K.-S.; Storey, A.J.; Gong, W.; Tsai, Y.-H.; Gibson, E.; Byrum, S.D.; Li, D.; Edmondson, R.D.; et al. A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol. 2021, 29, 386–397.e9. [Google Scholar] [CrossRef]
- Xu, S., Aguilar, A., Huang, L., Xu, T., Zheng, K., McEachern, D., Przybranowski, S., Foster, C., Zawacki, K., Liu, Z. and Chinnaswamy, K., 2020. Discovery of M-808 as a highly potent, covalent, small-molecule inhibitor of the Menin–MLL interaction with strong in vivo antitumor activity. Journal of medicinal chemistry, 63(9), pp.4997-5010.
- Yang, Y.; Hadjikyriacou, A.; Xia, Z.; Gayatri, S.; Kim, D.; Zurita-Lopez, C.; Kelly, R.; Guo, A.; Li, W.; Clarke, S.G.; et al. PRMT9 is a Type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Ye, P.; Xing, H.; Lou, F.; Wang, K.; Pan, Q.; Zhou, X.; Gong, L.; Li, D. Histone deacetylase 2 regulates doxorubicin (Dox) sensitivity of colorectal cancer cells by targeting ABCB1 transcription. Cancer Chemother. Pharmacol. 2016, 77, 613–621. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The Menin Tumor Suppressor Protein Is an Essential Oncogenic Cofactor for MLL-Associated Leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Yu, X.; Li, D.; Kottur, J.; Kim, H.S.; Herring, L.E.; Yu, Y.; Xie, L.; Hu, X.; Chen, X.; Cai, L.; et al. Discovery of Potent and Selective WDR5 Proteolysis Targeting Chimeras as Potential Therapeutics for Pancreatic Cancer. J. Med. Chem. 2023, 66, 16168–16186. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, D.; Kottur, J.; Shen, Y.; Kim, H.S.; Park, K.-S.; Tsai, Y.-H.; Gong, W.; Wang, J.; Suzuki, K.; et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 2021, 13, eabj1578–eabj1578. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A Small-Molecule Probe of the Histone Methyltransferase G9a Induces Cellular Senescence in Pancreatic Adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef]
- Zeng, H.; and Xu, W., 2015. Enzymatic assays of histone methyltransferase enzymes. In Epigenetic technological applications (pp. 333-361). Academic Press.
- Zhang, H.; Sun, Z.; Liu, Z.; Song, C. Overcoming the Emerging Drug Resistance of Smoothened: An Overview of Small-Molecule SMO Antagonists with Antiresistance Activity. Futur. Med. Chem. 2018, 10, 2855–2875. [Google Scholar] [CrossRef]
- Zhang, J.; Qian, K.; Yan, C.; He, M.; Jassim, B.A.; Ivanov, I.; Zheng, Y.G. Discovery of decamidine as a new and potent PRMT1 inhibitor. MedChemComm 2017, 8, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, X.; Hu, X.; Duan, X.; Wan, G.; Li, L.; Feng, Q.; Zhang, Y.; Wang, N.; Yu, L. Covalent inhibitors of EZH2: Design, synthesis and evaluation. Biomed. Pharmacother. 2022, 147, 112617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ma, Y.; Hu, X.; Zheng, Y.; Chen, X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J. Cell. Mol. Med. 2018, 23, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Kim, H.; Qian, C.; Xie, L.; Chen, X.; Xiong, Y.; Hu, J.; Chen, M.; Guccione, E.; Shen, Y.; et al. Discovery of a Potent and Selective Protein Arginine Methyltransferase 5 (PRMT5) PROTAC Degrader. J. Med. Chem. 2025. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xia, T.; Chen, D.-Q.; Xiong, X.; Shi, L.; Zuo, Y.; Xiao, H.; Liu, L. Promising role of protein arginine methyltransferases in overcoming anti-cancer drug resistance. Drug Resist. Updat. 2023, 72, 101016. [Google Scholar] [CrossRef]
- Zou, W.; Li, M.; Wan, S.; Ma, J.; Lian, L.; Luo, G.; Zhou, Y.; Li, J.; Zhou, B. Discovery of PRMT3 Degrader for the Treatment of Acute Leukemia. Adv. Sci. 2024, 11, e2405963. [Google Scholar] [CrossRef]
Figure 1.
The methyl cycle.
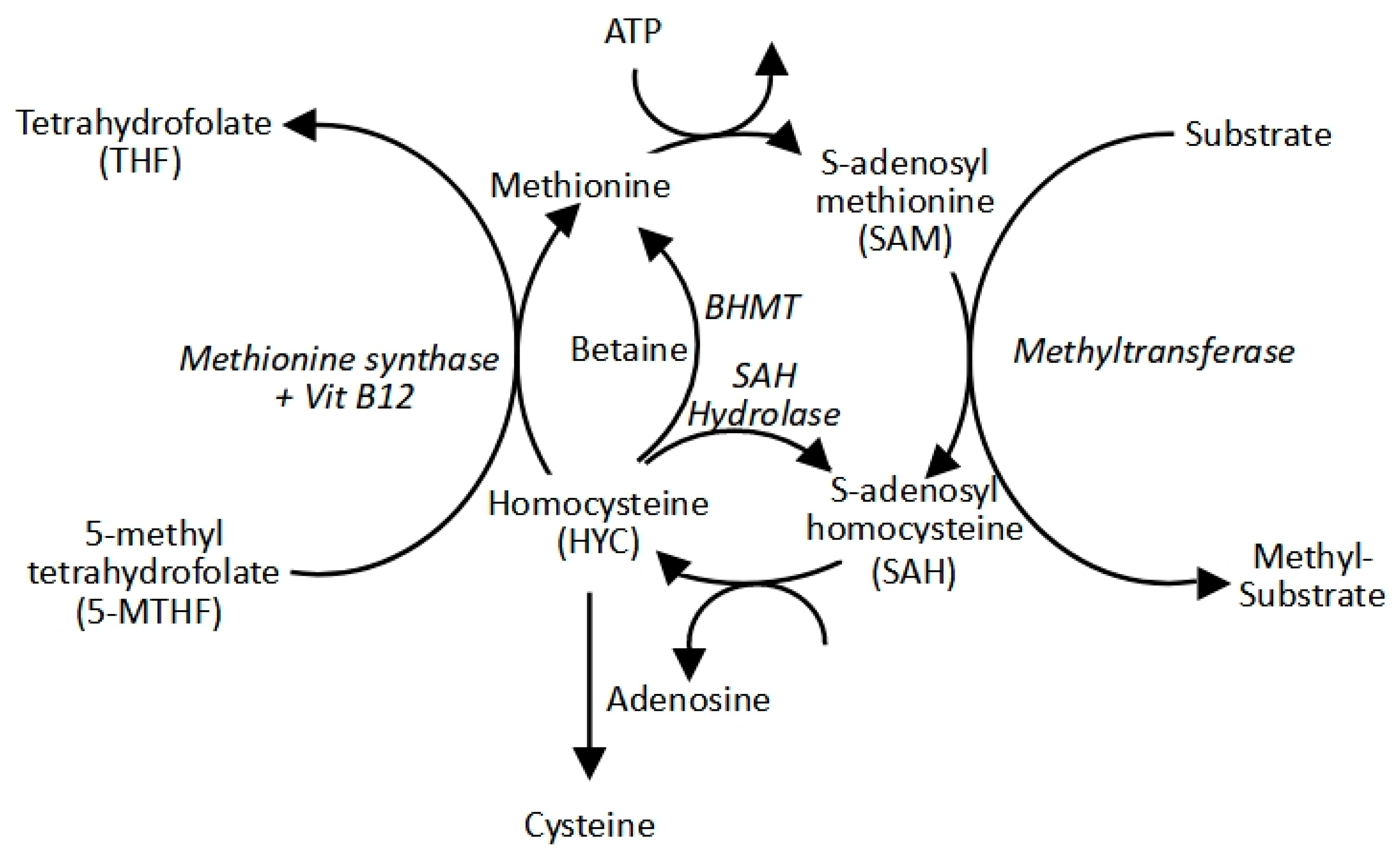

Table 1.
Noteworthy examples of substrate competitive inhibitors for some of the more popular methyltransferases.
Table 1.
Noteworthy examples of substrate competitive inhibitors for some of the more popular methyltransferases.
| Substrate | Inhibitor | Reference |
|---|---|---|
| EHMT2 | UNC0642 | Liu et al., 2013 |
| UNC0925 | Konze et al., 2014 | |
| A-366 | Sweis et al., 2014 | |
| MS152 | Wang et al., 2024 | |
| EZH2 | UNC1999 | Konze et al., 2013 |
| CPI-1205 | Vaswani et al., 2016 | |
| PF-06821497 | Kung et al., 2018 | |
| EBI-2511 | Lu et al., 2018 | |
| SETD7 | (R)-PFI-2 | Barsyte-Lovejoy et al., 2014 |
| Cyproheptadine | Takemoto et al., 2016 | |
| SMYD2 | BAY-598 | Eggery et al., 2016 |
| EPZ030456 | Mitchell et al., 2016 | |
| A-893 | Sweis et al., 2015 | |
| LLY-507 | Nguyen et al., 2015 | |
| EPZ033294 | Thomenius et al., 2018 | |
| SMYD3 | BCI-121 | Peserico et al., 2015 |
| EPZ028862 | Thomenius et al., 2018 | |
| BAY-6035 | Gradl et al., 2018 | |
| SETD8 | UNC0379 | Ma et al., 2014 |
| SPS8I1 | Blum et al., 2014 | |
| MS2177 | Butler et al., 2016 | |
| PRMT5 | GSK3326595/EPZ015938 (Pemrametostat) | Gerhart et al., 2018 |
| GSK3235025/EPZ015666 | Chan-Penebre et al., 2015 | |
| GSK3203591/EPZ015866 | Duncan et al., 2016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



