
Submitted:
01 August 2025
Posted:
04 August 2025
You are already at the latest version
Abstract
This study evaluated the molecular resistance profile to insecticides and the genetic diversity of Anopheles albimanus populations from malaria endemic comarcas in Panama, a country in Mesoamerica aiming to eliminate local malaria transmission. Molecular screening was performed in 891 An. albimanus, distributed in 162 pools, collected between 2011 and 2023. Pools were molecularly examined to detect natural infection with Plasmodium and sequenced to assess mutations in genes (vgsc, ace-1 and rdl) associated with resistance to commonly used insecticides. A high molecular infection rate by Plasmodium vivax was detected in all comarcas throughout the study period, and P. falciparum infections were detected in the last two years (2022-2023) in the east region. Mutations associated with pyrethroids/DDT resistance (H973Y and L1014F/C) and to organophosphorus/carbamate resistance (G119S) were detected at high frequencies (50.8% and 70%, respectively) in eastern comarcas but were absent in comarcas located west of the Panama Canal. Mutations in the rdl gene, associated with resistance to cyclodienes and neonicotinoids, were also frequently present. Anopheles populations from the western side were highly homogenous, suggesting a clonal expansion, contrasting with eastern samples the exhibited a high genetic diversity. Our study provides a valuable baseline for planning future molecular vector surveillance studies in the region. It also provides valuable information to the vector control program in Panama to guide insecticide selection for IRS.
Keywords:
Anopheles albimanus
; insecticide-resistance
; mutations
; malaria
; comarcas
; Panama
1. Introduction
Due to its relatively low number of cases and deaths compared to other regions, the World Health Organization (WHO) has identified Mesoamerica as having the potential for malaria elimination in the short term [1, 2]. Indeed, several countries from this subregion of the Americas, encompassing southeastern Mexico and Central America, have made significant progress in malaria elimination efforts. However, heterogeneity in disease epidemiology, both between and within countries, has resulted in uneven progress towards elimination. While some countries from this subregion, such as El Salvador end Belice, have recently been certified malaria-free by the WHO [1], others like Panama, located at the southernmost point of Mesoamerica, still faces significant transmission challenges.
Although Panama is part of the E-2025 initiative aiming to eliminate local malaria transmission by 2025, the number of indigenous cases has increased dramatically from 375 in 2015 to 15610 in 2024 (Figure 1). This represents an alarming rise of over 4,000% during this period, highlighting major obstacles in achieving national elimination goals. In fact, in recent years, malaria has been considered one of the most important re-emerging vector-borne diseases in the country.
Several factors may have contributed to this uncontrol increase in cases, including but not limited to health threats posed by climate change and the mass human migration crossing the Darien Gap in Panama aiming to reach the United States or Canada [3]. In fact, malaria transmission has risen along migration routes in eastern Panama [1]. This situation is further exacerbated by growing concerns over the reduced effectiveness of interventions, driven by biological threats such as emerging drug and insecticide resistance in local parasite and anopheline vector populations [4].
Considering this epidemiological scenario, completely off-track from elimination targets, the National Malaria Elimination Program (NMEP) in Panama is now reorienting its intervention strategies to regain malaria control, particularly prioritizing high transmission areas. In this context, effective vector control interventions remain a critical strategy for preventing malaria epidemics resulting from local transmission and secondary infections triggered by imported cases [4,5].
Although several species have been documented in Panama [6], Anopheles albimanus is by far the most abundant and widespread malaria vector across endemic regions in the country [6,7]. Indoor residual spraying (IRS) involving the rotation of different insecticides, has been the main vector control intervention in Panama. However, in recent years the NMEP has also implemented the use of insecticide-treated nets (ITN) for beds and hammocks in selected communities with high transmission [8,9] (Figure 1).
Since the beginning of the national malaria control program in Panama (1957), multiple classes of insecticides (organochlorines, organophosphates, carbamates, pyrethroids, and more recently neonicotinoids) have been intensively and repeatedly used, exerting considerably selective pressure on Anopheles populations. However, the selection of insecticides for IRS has often been based on product availability or international recommendations with no previous evaluation, rather than on localized entomological and resistance surveillance evidence. As a result, current knowledge about the status and distribution of insecticide resistance in An. Albimanus population from Panama remains limited.
Given that insecticide resistance poses a significant threat to the efficacy of current malaria vector-control interventions, this study evaluated the molecular resistance profile of An. albimanus populations by detecting the presence, frequency and distribution of resistance alleles in three key genes encoding targets of commonly used insecticides: ace-1 (encoding acetylcholinesterase), vgsc (encoding the voltage-gated sodium channel), and rdl (encoding the GABA receptor). The Plasmodium infection status of collected mosquitoes was evaluated throughout the study period. In addition, we conducted a preliminary analysis of the genetic diversity among An. albimanus populations from various endemic regions to explore potential geographic structuring and population differentiation.
2. Materials and Methods
2.1. Study Area
Panama is located in the southernmost part of the Mesoamerican subregion, which includes southeastern Mexico and all Central American nations (Figure 2). The country is bordered by Costa Rica to the west and Colombia to the east and is flanked by the Caribbean Sea to the north and the Pacific Ocean to the south, forming a biological corridor that joins South America with the rest of Central America. The climate and natural vegetation of most parts of Panamá are typically tropical rainforest, highly suitable for breeding of malaria vectors. The country has a bimodal climate season, with a dry season characterized by an almost total absence of rain from January to March. The rainy season spans from May to November. April and December are considered transition months. Humidity is high throughout the year in most of the country, but during the rainy season, it can reach almost 100% [10].
The Panama Canal pathway has artificially divided the country into Eastern and Western Panama. Administratively, the country is organized in ten provinces and five indigenous semi-autonomous regions called “comarcas” (Figure 2). Four of these comarcas are located East from the Panama Canal (Guna Yala, Madungandí, Wargandí and Emberá Wounáan), and one West of the Panamá Canal (Ngäbe Buglé). Around 17.0% of the estimated population of the country (4,064,780 inhabitants by 2023) lives within these indigenous territories that occupy 22.2% of the country’s area [11]. During the past few decades, malaria transmission in Panama has clustered across these comarcas, representing about 90 percent of all diagnosed cases in the country [12].
2.2. Mosquito Collections and Identification
Populations of An. albimanus collected between 2011 and 2023 were retrospectively analyzed. Mosquitoes were collected by human-landing catches from the peridomicile of historically endemic localities under epidemiological surveillance across the five indigenous comarcas in Panama (Figure 2). Adult mosquitoes were morphologically identified using keys for Anopheles [13]
2.3. Ethical Statement
The retrospective molecular analysis of the mosquito samples for this study was granted exemption from the Comité Institucional para el Buen Uso y Cuidado de los Animales (CIUCAL) del Instituto Conmemorativo Gorgas de Estudios de la Salud (No. 005/CIUCAL/ICGES-2024).
2.4. Genomic DNA Extraction and Plasmodium Infection
Samples were processed in ‘pools’ containing five An. Albimanus per pool. Genomic DNA from these pools was isolated using the commercial DNeasy Blood and Tissue Kits (Qiagen, Hilden, Germany), following the manufacturer’s protocol. Two molecular methods were performed to evaluate the Plasmodium infections in the mosquito samples. Samples collected between 2011 and 2018 were analyzed using a nested PCR targeting the small subunit ribosomal RNA (ssrRNA) genes following a modified methodology from the protocol by Snounou et al. [14]. Specifically, as P. vivax and P. falciparum are the only malaria species circulating in Panama, the second PCR reaction included species-specific primers for these two parasites exclusively. The remaining An. Albimanus pools collected between 2021 and 2023 were also analyzed by a qRT-PCR protocol targeting mitochondrial COX I and COX III genes as previously described [15].
2.5. Amplifications and Sequencing of Insecticide Resistance Genes
Vgsc Gene
A 225 bp fragment of the kdr region in the vgsc gene was amplified using primers designed for An. albimanus, following protocols proposed by Lol et al. (2013) [16]: AAKDRR: (5′-GCAANGCTAAGAANAGRTTNAG-′3), AAKDRF: (5′-AGATGGAAYTTYACNGAYTTC-′3), and AAKDRF2: (5′-CATTCATTTATGATTGTGTTTCGTG-′3). The PCR mixture (25 μL) consisted of 7.7 μL of water, 12.5 μL of PCR Master Mix, 2x (Promega, USA), 1 μL of MgCl2 (25x), 0.4 μL of each primer (AAKDRR and AAKDRF), and 5 μL of DNA template. The primary PCR conditions were set to 95°C for 3 minutes, followed by 35 cycles of 95°C for 45 seconds, 40.5°C for 45 seconds, and 72°C for 1 minute with a final extension at 72°C for 5 minutes in a SimpliAmp Termocycler (Applied Biosystems, USA). A second PCR was performed using the AAKDRR and AAKDRF2 primers with the same reaction specifications as in the primary PCR and the product of the first PCR as template DNA. The second PCR conditions consisted of 95°C for 3 minutes, followed by 40 cycles of 95°C for 45 seconds, 51.5°C for 45 seconds, and 72°C for 1 minute with a final extension at 72°C for 5 minutes in a SimpliAmp Termocycler (Applied Biosystems, USA).
Ace-1 gene
For the ace-1 gene, specific primers for An. albimanus were used to amplify a 193 bp fragment that flanks the target codon position 119 (AAace1F 5’-TGTGGAACCCAAATAC GC’3 and AAace1R 5’-ACGTTCTCTTCCGAGGCG’3). The PCR reaction with a final volume of 25 μL consisted of 5.5 μL of water, 12.5 μL of PCR Master Mix, 2x (Promega, USA), 1 μL of MgCl2 (25x), 0.5 μL of each primer (AAace1F and AAace1R), and 5 μL of DNA template. The cycle conditions were 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 60 seconds, and a final extension at 72 °C for 5 minutes [17].
Rdl gene
Initial attempts to amplify a fragment of the rdl gene of various anopheles’ species from Indonesia following the protocol described by Asih et al. (2012) were unsuccessful [18]. Therefore, we designed a set of specific primers for An. Albimanus based on published sequence of the rdl gene from this species (NCBI RefSeq accessions: NC_050157.1 and XM_035935106.1). The primers specificity of the primers, as well as the identification of secondary structures and potential dimerization were assessed using the OligoAnalyzer tool (OligoAnalyzer Tool - Primer analysis and Tm Calculator | IDT).
The designed primers - Fwd1 (5’AGTATAGCTGTGTAAGAGTC-3’) and Rev2 (5’-AGCAAATACGGAACTGAACC-3’) - were successfully used to amplify a 507 bp fragment encompassing the target codon position at codon 302 from An. albimanus. Amplification reactions were performed in a final volume of 25 μL containing 12.5 μL of PCR Master Mix, 2x (Promega, Madison, WI, USA), 0.4 μL of each primer (Fwd1 and Rev2), and 3μL of DNA purified from An. Albimanus pools. The following PCR cycling conditions were used: 94 ◦C for 3 min; 35 cycles of 95 ◦C for 30 s, 50.5◦C for 30 s, 72 ◦C for 1 min, and a final extension at 72 ◦C for 5 min.
PCR products were confirmed via electrophoresis in a 2% agarose gel with a 0.5X TBE buffer stained with SYBR™ Safe DNA Gel Stain (Invitrogen, USA). The amplicons were then purified using the QIAquick purification kit (Qiagen, Hilden Germany). To ensure suitability for sequencing, the quality and concentration of the DNA were evaluated measuring the absorbance at 260 and 280 nm with a NanoDrop spectrophotometer. Purified PCR products were then directly sequenced in both directions with the Sanger method using the same primers for each gene (vgsc, ace-1, and rdl) described for amplification and an ABI Prism 3500 XL130 sequencer (Applied Biosystems, Foster City, CA, USA). Chromatograms were visually checked, and nucleotide or amino acid consensus sequences were edited, and aligned using Sequencher v4.1.4 [19] and Molecular Evolutionary Genetics Analysis (MEGA) v12.0 software (Pennsylvania State University, Center, PA, USA) [20]. Identity verification of the sequences obtained was performed by comparison with vgsc, ace-1, and rdl gene sequences available in GenBank, using the NCBI Blast tool (https://blast.ncbi.nlm.nih.gov/Blast). Nucleotide sequences from this study were submitted and registered in GenBank (Table S1).
2.6. Phylogenetic and Genetic Diversity Analyses
To explore genetic diversity and conduct a preliminary population structure analysis among An. albimanus populations from various endemic regions, 30 representative sequences of the three insecticide resistance genes (vgsc, ace-1, and rdl) were concatenated into a single sequence for each sample of 629 bp. Multilocus sequences were aligned using MEGA 12.0 software, and a phylogenetic tree was constructed by the maximum likelihood method with the Tamura model and 1000 bootstrap replicates. To create a haplotype network, PopART program version 1.7 were employed [21], utilizing the 30 concatenated samples from the three genes.
To compute genetic diversity indices, samples were grouped by their geographic origin into Western Panama and Eastern Panama. DnaSP Version v6 was used to estimate the number of haplotypes (H), haplotype diversity (Hd), segregating sites (S), nucleotide diversity (π), total number of mutations (Eta), the mean number of pairwise differences (k), and neutrality tests (Fu’s Fs statistics) [22].
2.7. Data Analysis
The data collected was registered in Microsoft Excel. The allelic frequencies from mutations detected in the three resistance genes were calculated using R 2023 (version 4.3.2, Build 513, Posit Software, PBC). To assess the possible association between resistance/susceptible alleles and vector Plasmodium infection, a Fisher´s Exact test was calculated. The significance level for statistical tests was set at 5% [23].
3. Results
Molecular screening was performed in 891 adult female An. albimanus specimens (collected between 2011 and 2023) distributed in 162 pools: 120 pools from Madungandí comarca, 7 from Guna Yala, 2 from Wargandí, 22 from Emberá Wounaan and 11 from Ngäbe Buglé. Pools were molecularly examined to detect natural infection with Plasmodium and were sequenced to assess mutations in genes (vgsc, ace-1 and rdl) associated with resistance to commonly used insecticides for malaria vector control.
3.1. Plasmodium Infection in Collected An. Albimanus
The overall prevalence of Plasmodium infection in female Anopheles mosquitoes was found to be 61.7% (100/162 pools), with a much higher Plasmodium frequency in the Comarca Madungandí (Table S2). The overall estimated prevalence calculated by the method of pooled prevalence was 0.17%. Of the 100 positive pools, 84 were found positive for P. vivax (85.81%), 9 pools for P. falciparum and 7 were mix-infections by both species (5.1%). Plasmodium falciparum infections were detected in pools from Anopheles collected in the Madungandí comarca between 2020 and 2023 (Table S2).
3.2. Frequency and Distribution of Vgsc Genotypes
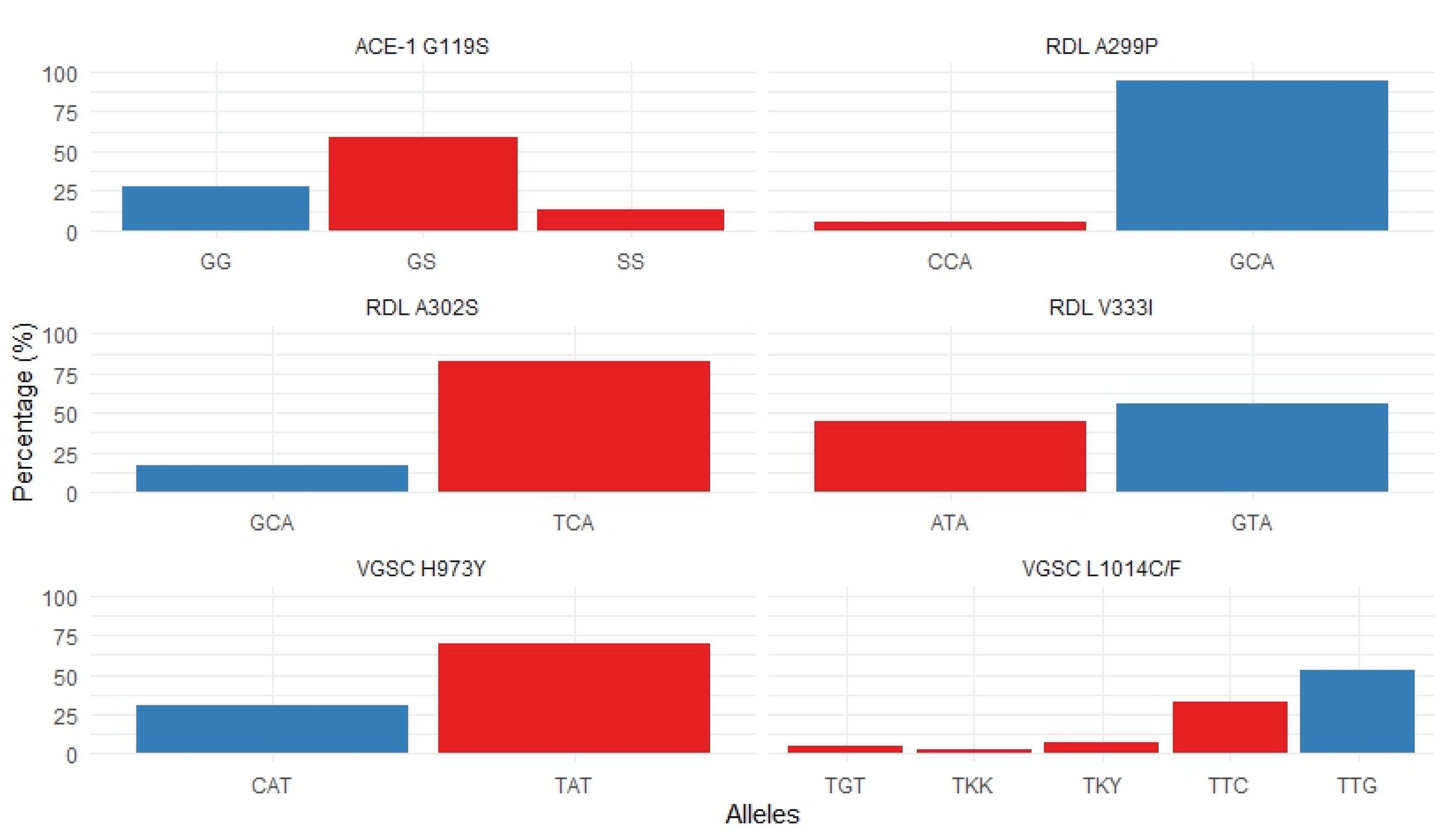
Of the 162 An. albimanus pools, 122 sequences corresponding to segment 6 of domain II of the vgsc gene were successfully genotyped. All sequences were aligned and compared with reference vgsc gene sequences from An. albimanus available in GenBank under accession numbers OL630652.1, KF137581.1, and MN087506.1. The wild-type allele (susceptible to pyrethroid) at codon 1014 (TTG; leucine, L1014) was present in 64 sequences (52.5%), while the wild-type allele at codon 973 (CAT; histidine, H973) was found in 28 sequences (22.9%).
Conversely, pyrethroid resistance-associated mutations resulting in amino acid substitutions, were identified at both codons (1014 and 973). At codon 1014, the well-characterized knockdown resistance (kdr) mutation was detected in almost half of the sequences, including TTC (phenylalanine, L1014F) in 6 sequences (4.9%) and TGT (cysteine, L1014C) in 40 sequences (32.8%). Additionally, heterozygous alleles were identified at this codon: TKY in 9 sequences (7.3%) and TKK in 3 sequences (2.5%); all originating from pools collected in the eastern comarcas of Madungandí and Emberá-Wounaan (Fig. 3 and Fig. S1). The kdr mutation was found to fixed in An. albimanus pools analyzed from eastern regions since 2021 (Table S3).
At codon 973, the mutation TAT (tyrosine, H973Y), also implicated in resistance to pyrethroids, was observed in 64 sequences (52.5%); all of which were homozygotes and originated from eastern comarcas. Overall, 50.8% of the kdr-associated mutant alleles (L1014F/C and H973Y) were detected in pools from communities within the Madungandí Comarca (n = 62), located east of the Panama Canal (Fig. 3 and Fig. S1). In contrast, none of the vgsc sequences analyzed in this study from the Ngäbe-Buglé comarca located west of the Panama Canal, exhibited mutation associated with pyrethroids resistance (Table S3).
Figure 3.
The frequency for each allele within the vgsc, ace-1, and rdl genes is shown. Alleles associated with insecticide resistance are displayed in red, while susceptible alleles are shown in blue. These alleles were detected in pooled samples of Anopheles albimanus from comarcas in Panama.
Figure 3.
The frequency for each allele within the vgsc, ace-1, and rdl genes is shown. Alleles associated with insecticide resistance are displayed in red, while susceptible alleles are shown in blue. These alleles were detected in pooled samples of Anopheles albimanus from comarcas in Panama.
3.3. Frequency and Distribution of Ace-1 Genotypes
All ace-1 sequences from this study were aligned with reference acetylcholinesterase-1 gene sequences from An. albimanus (GenBank accessions AJ566403.1 and AJ566402.1) and An. darlingi (MK477204.1). All three possible genotypes of the ace-1 gene were identified at codon 119 in 132 sequences analyzed. These included the homozygous susceptible genotype (119GG) found in 36 sequences (27%), the heterozygous genotype (119GS) in 77 sequences (58%) and the homozygous resistant genotype (119SS) found in 18 samples (14.7%), which is associated with resistance to organophosphorus (OP) and carbamate (CM) insecticides. The resistance-associated mutation (G119S) was detected in samples from communities in eastern Panama, including Madungandí (n = 15), Emberá-Wounaan (n = 2), and Wargandí (n = 1) (Fig. 3, Fig. S1 and Table S3). Notably, the G119S mutation was absent in samples collected from western Panama.
3.4. Frequency and Distribution of Rdl Genotypes
Three non-synonymous mutations were identified in the rdl gene based on the analysis of 91 An. albimanus sequences from this study. All sequences were aligned with the reference rdl gene sequence from An. funestus, available in GenBank under accession number MN562790.1. Nucleotide variations resulting in corresponding amino acid changes were observed at codons 299, 302, and 333 (Table S3). All identified alleles in the rdl gene from this study were found in homozygous form.
At codon 299, the wild-type allele GCA, which encodes alanine (A299), was found in 86 sequences (94.5%). A mutant variant encoding proline (A299P; CCA) was detected in five sequences (5.5%), including four from eastern comarcas and one from the Ngäbe-Buglé Comarca (n = 1) in western Panama (Fig. 3, Fig. S1 and Table S3).
At codon 302, the well-characterized resistance-associated mutation (A302S) that results in the substitution of the amino acid alanine (GSA) with serine (TCA), was detected in 67 sequences (73.6%) (Fig. 3). This A302S substitution was found predominantly in samples from eastern Panama, including Madungandí (n = 56), Emberá-Wounaan (n = 11) and Wargandí (n = 3), as well as in one sample from the western comarca of Ngäbe-Buglé (n = 1) (Fig S1).
An additional polymorphism was detected at codon 333. The wild-type allele GTA, which encodes valine (V333), was identified in 45 sequences (49.5%), while a variant allele ATA (V333I) encoding isoleucine was found in 36 sequences (39.5%). This V333I variant was observed in An. albimanus pools from communities in Madungandí (n = 29) and Emberá-Wounaan (n = 6) in eastern Panama, and in one sample from the Ngäbe-Buglé Comarca in western Panama (Fig. 3, Fig. S1 and Table S3).
The total number of alleles (resistant and susceptible) detected in the An. albimanus pools from each of the five comarcas studied is summarized in Figure 4. Although there was considerable variability in the number of samples analyzed from each comarca, Madungandí exhibited the highest number of detected alleles, followed by Wargandí and Emberá-Wounaan. In contrast, the eastern comarcas of Guna Yala and Ngäbe-Buglé in the west, displayed lower frequencies. The ratio of resistant to susceptible alleles also varies among comarcas, suggesting regional differences in genetic resistance patterns.
3.5. Plasmodium Infection and Presence of Resistance Alleles
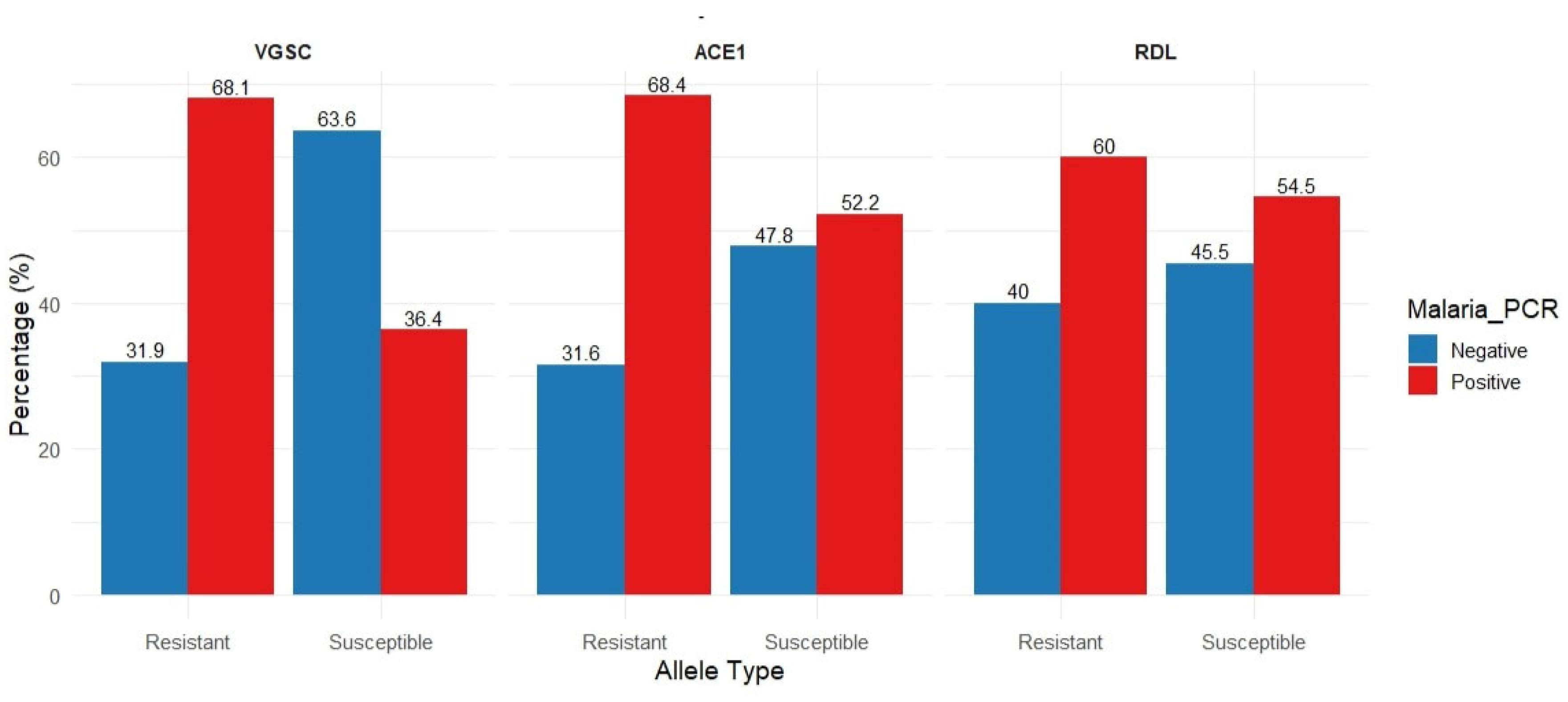
The potential association between Plasmodium infection and the presence of resistance/susceptible alleles in vgsc, ace-1 and rdl genes was assessed in the collected An. Albimanus pools. For all three genes, the frequency of resistant alleles was higher in Plasmodium positive samples (Fig. 5). This difference was statistically significant for vgsc (p= 0.048) and for ace -1 (p= 0.047), but not for rdl gene (p = 0.75) (Table S4).
Figure 5.
The possible assocition between Plasmodium infection and the existence of resistant or susceptible alleles in the vgsc, ace-1, and rdl genes was evaluated in pooled samples of Anopheles albimanus. The bars show the Plasmodium infection rate (%) based on the existence of resistant (red) or susceptible (blue) alleles for each gene examined. The rate of resistant alleles was higher in Plasmodium-positive groups in all three genes. A statistical significance of these differences was observed for vgsc (p = 0.048) and ace-1 (p = 0.047), while it was not significant for the rdl gene (p = 0.75).
Figure 5.
The possible assocition between Plasmodium infection and the existence of resistant or susceptible alleles in the vgsc, ace-1, and rdl genes was evaluated in pooled samples of Anopheles albimanus. The bars show the Plasmodium infection rate (%) based on the existence of resistant (red) or susceptible (blue) alleles for each gene examined. The rate of resistant alleles was higher in Plasmodium-positive groups in all three genes. A statistical significance of these differences was observed for vgsc (p = 0.048) and ace-1 (p = 0.047), while it was not significant for the rdl gene (p = 0.75).
3.6. Phylogenetic and Genetic Diversity Analyses
Phylogenetic tree reconstruction and a haplotype network analysis were conducted using concatenated sequences of the three insecticide resistance markers (vgsc, ace-1 and rdl genes) from An. albimanus collected between 2011 and 2023 across three comarcas (Madungandí and Emberá-Wounaan from the east, and Ngäbe-Buglé from the west). The phylogenetic tree was inferred using the Maximum Likelihood method and Tamura model. The final analysis incorporated 30 nucleotide sequences, covering 629 positions in the final dataset (Fig. 6 and Table S1). The complete sequence alignment of the concatenated genes from this study is available upon request.
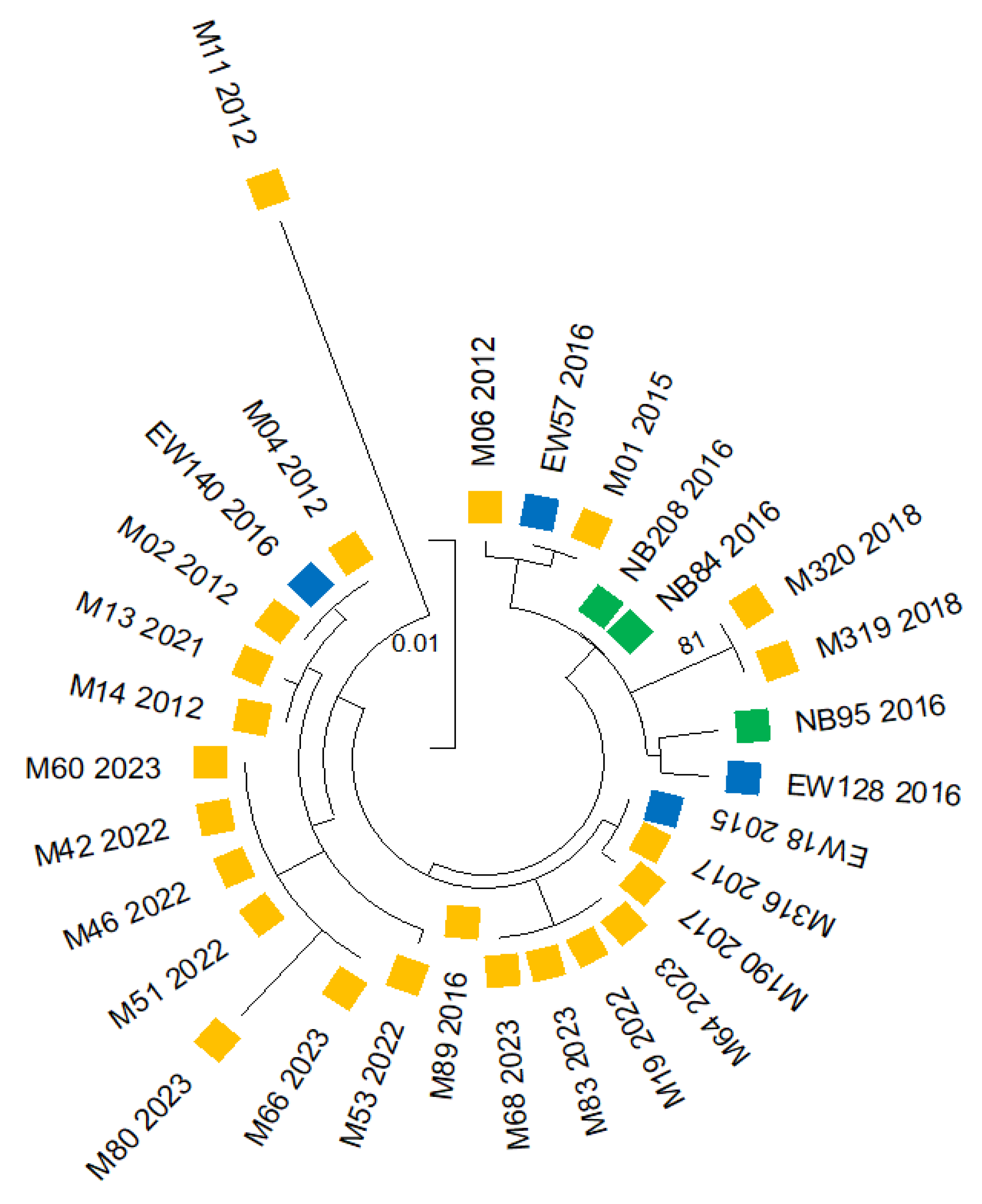
With the exception of a single sample (M11 2012), all samples grouped in a predominant, well-defined clade that encompassed samples from the three comarcas and from the multiple collection years, indicating the absence of a dominant genotype circulating, and instead suggests the persistence of diverse genotypes coexisting over time (Fig 6). Within this predominant clade, several subclades were observed, reflecting a greater genetic diversity among samples generated from eastern comarcas mainly from Madungandí, which clustered into distinct subclades with closer genetic relatedness between them. In contrast, sequences from the Ngäbe-Buglé comarca located in western Panama formed a separate subclade together with some eastern samples from Madungandí and Emberá-Wounaan, indicating some degree of genetic connectivity.
The distinct sample M11 2012 was collected from Puente Bayano, a hiperendemic community located in the eastern comarca of Madugandí. This sample grouped apart in a different clade, highlighting its genetic distance from the rest of the samples analyzed.
Figure 6.
Phylogenetic tree constructed from concatenated sequences of three insecticide resistance markers (vgsc, ace-1 and rdl genes) from 30 An. Albimanus collected between 2011 al 2023 in three comarcas: 23 from Madungandí (yellow color), 4 from Emberá-Wounaan (turquoise color) and 3 from Ngäbe-Buglé (green color). The phylogenetic tree was inferred using the Maximum Likelihood method and Tamura model. The final analysis incorporated 30 nucleotide sequences, covering 629 positions in the final dataset.
Figure 6.
Phylogenetic tree constructed from concatenated sequences of three insecticide resistance markers (vgsc, ace-1 and rdl genes) from 30 An. Albimanus collected between 2011 al 2023 in three comarcas: 23 from Madungandí (yellow color), 4 from Emberá-Wounaan (turquoise color) and 3 from Ngäbe-Buglé (green color). The phylogenetic tree was inferred using the Maximum Likelihood method and Tamura model. The final analysis incorporated 30 nucleotide sequences, covering 629 positions in the final dataset.
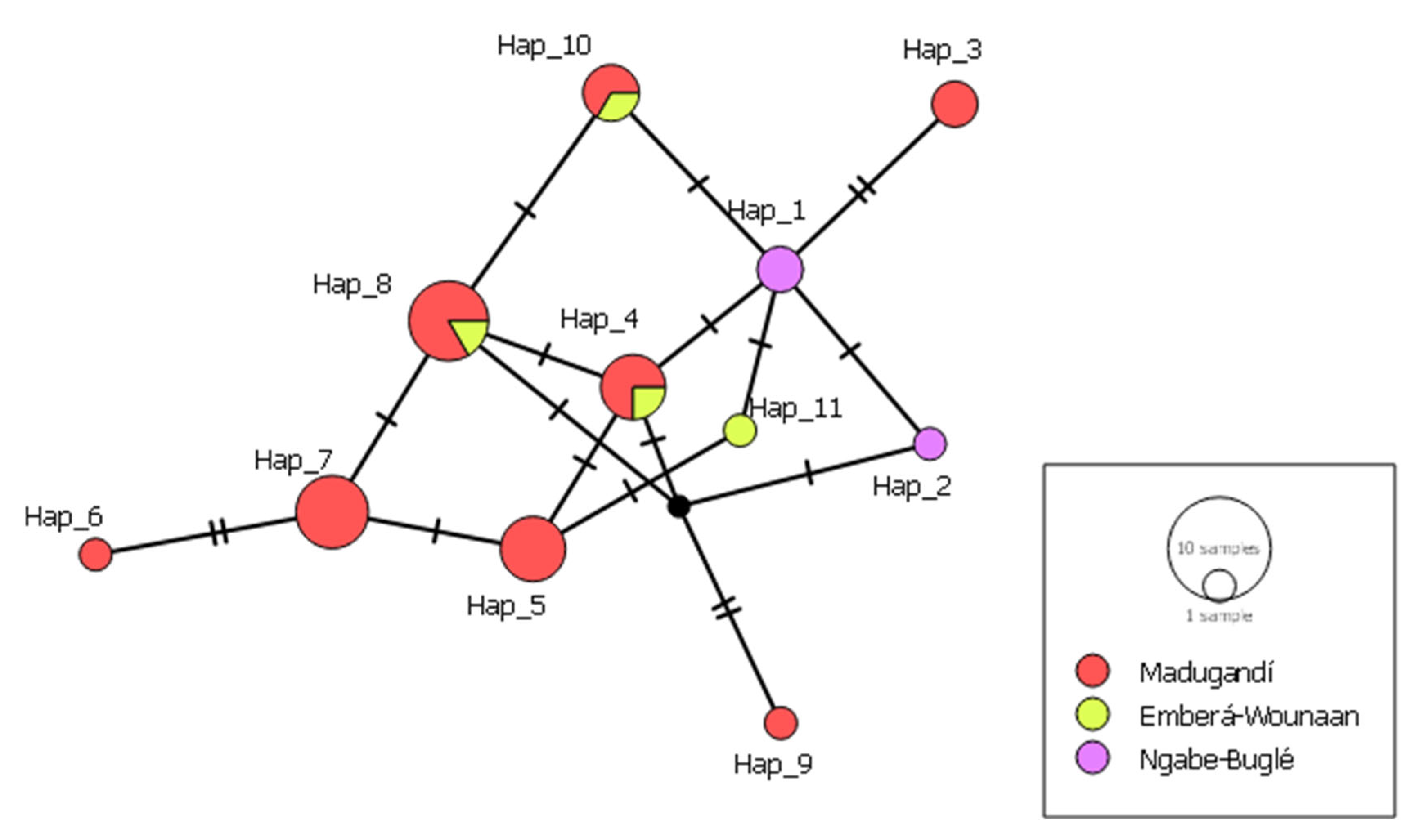
The haplotype network analysis, based on the concatenated gene sequences revealed the presence of 11 distinct haplotypes (Figure 7). Most haplotypes were restricted to a single comarca, indicating localized circulations of specific genetic variants. For example, haplotypes 3, 5, 6, 7 y 9 were only found in Madugandí, while haplotype 11 was restricted to the comarca Emberá-Wounaan, both located in eastern Panama. In contrast, haplotypes 1 y 2 were exclusively identified in the comarca Ngäbe-bugle in western Panama. Haplotypes 4, 8 and 10 contained samples from different eastern comarcas and showed genetic relatedness between them, indicating potential gene flow or shared ancestry among populations in that region.
To explore potential geographic structuring and population differentiation among An. albimanus populations collected in this study, a preliminary analysis of the genetic diversity was performed. For this purpose, samples were grouped according to the collection site into two ecologically and geographically distinct malaria-endemic regions of Panama (Eastern and Western). Genetic diversity indexes were calculated and compared for each of the three resistance-associated genes (vgsc, ace-1 and rdl) (Table S5). Higher diversity indexes were captured with the vgsc gene (kdr) and therefore was chosen for further diversity analysis.
Substantial differences in genetic diversity were observed among both groups. Eastern populations exhibited a high genetic diversity, with 7 segregating sites, 14 haplotypes, and a haplotype diversity of 0.77. The nucleotide diversity (ᴫ = 0.078) and the presence of 8 different mutations, reflect the heterogeneous origins of these specimens. The negative Fu's Fs statistic (-5.89) is consistent with recent population expansion or that Anopheles populations from this region are under selective pressure.
In contrast, western Panama populations displayed low genetic diversity, with only two haplotypes identified. Most specimens were genetically similar l (Hd = 0.48; ᴫ = 0). This homogeneity suggests a clonal population structure or genetic bottleneck, possibly due to limited gene flow, localized vector control pressure or ecological constraints.
4. Discussion
After falling short of meeting the 2025 malaria elimination goals set by the WHO Global Technical Strategy (GTS), the NMEP in Panama is currently adjusting strategies to address issues that might have hindered elimination progress. A key component of this revised strategy needs to include a more effective and evidenced-based vector control program, guided by local epidemiological and entomological data, including a program for monitoring and managing insecticide resistance [24]. In this context, this study provides the first molecular evidence in the country on how allelic variants might be contributing to the resistance status to insecticides in An. albimanus populations, the dominant malaria vector in Panama across malaria endemic comarcas.
Over the past several decades, malaria transmission in Panama has remained disproportionally clustered in indigenous comarcas that together occupy approximately 22.0% of the Panamanian territory [25]. Indeed, malaria cases in these reservations have accumulated more than 90% of all cases diagnosed in the country in the last decades [12]. Indigenous communities living in comarcas are highly underserved, with substantially higher rates of extreme poverty and limited healthcare compared to the rest of the country [12, 25]. These areas are not generally exposed to the high amounts of agrochemicals used in large-scale crop plantations, since their indigenous inhabitants relied mainly on subsistence agriculture and fishing. However, these areas have been subjected to recurrent applications of multiple classes of insecticides used primarily as IRS by vector control programs. This continued exposure might have exerted strong selection pressure over vector populations, therefore favoring the emergence of resistance alleles.
At the initial phases of NMP in Panama (from the early 1960´s to the 1980´s), organochlorides (DDT and dieldrin) and carbamates (Propoxur) were widely used as part of the IRS vector control program [12]. Over the past 25 years, these insecticides have been replaced by chemicals from the pyrethroid, organophosphate, and neonicotinoid classes. Pyrethroids (Deltamethrin) were used between 1996 and 2002, followed by the organophosphate fenitrothion (Sumithion) from 2002 to 2020 (Figure 1). In September 2019, a neonicotinoid insecticide (Sumishield) was introduced as an alternative to fenitrothion in three eastern highly endemic comarcas: Madungandí, Guna Yala and Wargandí. Its use was later expanded to the remaining malaria-endemic regions beginning in 2021, (Figure 1) [9]. However, in most cases, changes in insecticides for IRS have been driven primarily by product availability or international policy recommendations rather than by thorough local evaluations of insecticide effectiveness and resistance status of the local vectors.
To evaluate the molecular resistance status of An. albimanus in Panama, we analyzed mutations in three genes (vgsc, ace-1, and rdl) shown to be strongly associated with resistance to common insecticides. vgsc gene is the primarily target of pyrethroids and DDT (dichlorodiphenyltrichloroethane), and mutations at codons 973 and 1014 have been shown to confer resistance to both insecticides in Anopheles mosquitoes [26, 27,28]. In this study, both mutations (H973Y and L1014F/C) were detected at high frequencies (50.8%) in eastern comarcas but were absent in comarcas located west of the Panama Canal (Figure 3 and Table S3).
Acetylcholinesterase is the primary molecular target of organophosphates and carbamates, and genetic variants in this gene have been clearly recognized to confer resistance to both insecticides in Anopheles populations [28, 29]. The resistance-associated mutation (G119S) was widespread in eastern comarcas (over 70% of the samples evaluated) but was not detected in western comarcas (Figure 3 and Table S3).
Three non-synonymous mutations (A299P, A302S, V333I) were identified in the rdl gene, the primary target for insecticides of several insecticides, including cyclodienes (dieldrin) (Figure 3 and Table S3). This gene has also been identified as a potential secondary target for other insecticide classes, including neonicotinoids and pyrethroids [30]. The resistance-related amino acid substitution A302S was detected at high frequencies (73.6%) across all studied comarcas, particularly in the east. A valine to isoleucine amino acid substitution was detected at codon 333 (V333I) in 39.5% of the samples. A similar amino acid replacement has been described at codon 327 in Anopheles species from Africa and Asia [31,32]. Further studies are needed to assess the impact of the V302I mutation in An. albimanus populations. An additional non-synonymous substitution was detected at low frequencies (5.5%) at codon 299 (alanine to proline) on the rdl gene (A299P). The biological implication of this amino acid substitution in An. albimanus populations warrants further investigation.
Dieldrin or other cyclodienes have not been used in the country by the NMP since the early 1960´s. Therefore, the existence of rdl mutant alleles in An. albimanus populations could be associated with the use of other insecticides by NMP that also target rdl, such as neonicotinoids. It is also possible that these mutant alleles are stable in the absence of insecticide pressure or that they confer a selective advantage compared to the wild types.
Few studies have evaluated the presence and frequency of insecticide resistance-associated mutations in dominant Anopheles populations from Mesoamerica (from Panama to southern Mexico). Resistant kdr alleles have been previously reported in An. albimanus populations from México, Nicaragua and Costa Rica [33, 34]. Conversely, a recent study did not detect kdr or ace-1 mutations in An. albimanus from the State of Quintana Roo in southeastern Mexico [35]. The present study represents the first report of kdr and ace-1 mutations in An. albimanus populations from Panama and is the first description of rdl polymorphisms in Anopheles from Mesoamerica.
A high molecular infection rate by P. vivax was observed in the An. albimanus pools collected from all comarcas (Table S2), a finding consistent with the high P. vivax transmission reported in humans during the collection years. Notably, P. falciparum infections were detected in pools collected in recent years (2022-2023) from communities within the eastern comarca of Madungandí (Table S3). This finding preceded the official confirmation of human P. falciparum cases in 2024 from the same communities where infected vectors were found. Autochthonous P. falciparum transmission was virtually eliminated in the country, but it has now re-emerged in the eastern comarcas of the country. In this context, monitoring infection in mosquito vectors for Plasmodium species can be helpful as an important early warning tool to anticipate changes in human malaria transmission, including the possible resurgence of Plasmodium species in a specific area.
Several studies have suggested that the presence of mutations associated with insecticide resistance in Anopheles can affect their vector competence, influencing the outcome of malaria infection by increasing Plasmodium susceptibility and infection rates [36, 37, 38, 39]. In this study, we also found a positive association between the presence of resistance alleles in vgsc/ ace -1 genes, and an increased infection rates by Plasmodium in An. albimanus populations (Table S4). More studies are needed for a comprehensive understanding of the epidemiological consequences of this relationship, and its importance when designing vector control strategies in different endemic settings.
Although insecticide-resistance genes may not be ideal for conducting genetic diversity studies in Anopheles due to the strong selective pressures to which they are frequently subjected, our results evidence a distinct An. albimanus population structure between eastern and western comarcas (Table S4). Mosquitoes from the western side were highly homogenous, suggesting a clonal expansion, possibly due to limited gene flow or ecological isolation. In contrast, eastern samples exhibited a much higher genetic diversity, possibly influenced by geographical, ecological and epidemiological differences that characterize this region that includes a higher vector species abundance / diversity and intense malaria transmission rate compared to the western side.
Our study has several important limitations. First, the number of pools analyzed was relatively small and with a limited timeframe, spanning from 2011 to 2023. There was also an uneven distribution of the number of pools collected across the comarcas, with a considerable underrepresentation of samples from the western comarca of Ngäbe Buglé. Consequently, our results may not necessarily capture the entire or current insecticide-resistance profile or the diversity of An. albimanus populations in the country. However, it is noteworthy mentioning that collecting mosquitoes in malaria endemic regions in Panama is logistically challenging. Most of these areas are remote and difficult to reach, particularly western comarcas during the rainy season. Transporting mosquito samples from remote areas to specialized laboratories for molecular testing is also time-consuming and costly.
Analyzing Anopheles mosquitoes in pools instead of individually is also major limitation that not only can negatively impact on an accurate estimation of allele frequencies but also can affect the estimation of population genetic parameters, such as determining allele zygosity of the mutations associated with insecticide resistance. Nevertheless, pool analysis can be an efficient and cost-effective approach when working in most malaria endemic settings, characterized by resource constraints. It is also an efficient strategy when the goal is rapid screening of common mutations or a preliminary assessment to identify population structure.
Despite the limited and unbalanced sample size across the comarcas, our study provides a valuable baseline for planning future molecular vector surveillance studies in the region. It also provides valuable information to guide insecticide selection for IRS performed by the NMEP in Panamá. Nevertheless, our molecular results for assessing insecticide resistance in Anopheles should be confirmed with additional susceptibility phenotypic bioassays, such as the WHO tube test and CDC bottle assay.
In conclusion, resistance related mutations to all four main classes of insecticides were present at high frequencies in An. albimanus populations from eastern Panama, while they were absent or at low frequencies in populations from the western region These findings emphasize the need to implement region-specific vector control strategies.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Frequency of resistance -related alleles in vgsc, ace-1 and rdl genes at each study area (comarca) by year of collection; Table 12: Genbank accesion numbers and mutations detected in insecticide- resistance genes from Anopheles albimanus in Panama; Table S2: Prevalence of Plasmodium spp in pools of Anopheles albimanus populations collected in the study; Table S3: Prevalence of Plasmodium and frequency/distribution of resistance -related alleles in vgsc, ace-1 and rdl genes at each study area (comarca) by year of collection; Table S4: Plasmodium infection rate in An. Albimanus pools collected in this study according to the presence of resistant/ susceptible alleles in vgsc, ace-1 and rdl genes; Table S5: Comparison of insecticide-resistance genes diversity among Anopheles albimanus collected from two geographical and ecological separate malaria-endemic regions in Panama.
Author Contributions
Conceptualization, C.A.R., G.G., A.C., J.E.C.; methodology, C.A.R., V.V., A.M.S., J.E.C., G.G., A.C.; software, C.A.R., V.V., L.A.H., G.G.; validation, C.A.R., J.E.C., G.G., A.C. and V.V.; formal analysis, C.A.R., J.E.C., G.G., V.V.; investigation, C.A.R., V.V., A.M.S., L.C.,; resources, C.A.R., A.M.S., L.A.H., L.C.; data curation, C.A.R., J.E.C., G.G., A.C., V.V.; writing—original draft preparation, C.A.R., J.E.C.,V.V.; writing—review and editing, C.A.R., J.E.C., G.G., A.C., V.V.; visualization, C.A.R., J.E.C., G.G., A.C., V.V.; supervision, J.E.C., G.G., A.C.; project administration, C.A.R., J.E.C.; funding acquisition, C.A.R., J.E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received financial support from the Secretaria Nacional de Ciencia, Tecnología e Innovación (Project APY-NI-2023A-65) and the Instituto Conmemorativo Gorgas de Estudios de la Salud in Panama City, Panama (Project 23-173). L.C and J.E.C. are members and received financial support from the Sistema Nacional de Investigacion (SNI No. 173-2021), SENACYT, Panama. AC and GG are members of the Sistema Nacional de Investigadores (SNI-ANII, Uruguay) and the “Programa de Desarrollo de Ciencias Básicas” (PEDECIBA, Uruguay).
Data Availability Statement
All data underlying the results from this study are provided as part of the article in tables and figures. DNA sequences were deposited in GenBank as described in the methodology.
Acknowledgments
We thank the field entomology mosquito collection technicians from the Departamento de Control de Vectores del MINSA y al personal del Plana Estratégico para la Eliminación de la Malaria (PEEM) from the Ministry of Health of Panama. We thank the ICGES for providing administrative and logistical support. We also thank Alberto Cumbrera for map creation and figure edition and Luis Jaén and Randhy Rodríguez for their technical support in the laboratory and Anopheles field collections.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ace-1 | Acetylcholinesterase 1 |
| Hd | haplotype diversity |
| GABA | Gamma aminobutyric acid |
| IRS | Indoor residual spraying |
| ITN | Insecticide-Treated Nets |
| NMPE | National Malaria Elimination Program |
| PEEM | Plan for the Elimination of Malaria |
| rdl | Resistance to dieldrin |
| vgsc | Voltage-gated sodium channel |
| WHO | World Health Organization |
References
- Global Malaria Programme operational strategy 2024-2030. Geneva: World Health Organization; 2024. Licence: CC BY-NC-SA 3.0 IGO.
- World malaria report 2024: addressing inequity in the global malaria response. Geneva: World Health Organization; 2024. Licence: CC BY-NC-SA 3.0 IGO.
- Vásquez, V.; Santamaría, A.M.; Moreno, D.; Ruíz, F.; Rigg, C.A.; Chaves, L.F.; Calzada, J.E. Genetic Diversity of Potential Drug Resistance Markers in Plasmodium vivax Isolates from Panama, Mesoamerica. Pathogens 2025; 27;14(3). [CrossRef] [PubMed]
- WHO guidelines for malaria, 30 November 2024. Geneva: World Health Organization; 2024. [CrossRef]
- Framework for a national plan for monitoring and management of insecticide resistance in malaria vectors. Geneva: World Health Organization; 2017. Licence: CC BY-NC-SA 3.0 IGO.
- Loaiza, J.R.; Bermingham, E.; Scott, M.E.; Rovira, J.R.; Conn, J.E. Species composition and distribution of adult Anopheles (Diptera: Culicidae) in Panama. J Med Entomol. 2008. [CrossRef] [PubMed]
- Rigg, C.A.; Hurtado, L.A.; Calzada, J.E.; Chaves, L.F. Malaria infection rates in Anopheles albimanus (Diptera: Culicidae) at Ipetí-Guna, a village within a region targeted for malaria elimination in Panamá. Infect Genet Evol. 2019; [CrossRef] [PubMed]
- Ministerio de Salud de Panamá (MINSA). Plan Estratégico de Eliminación de la Malaria (PEEM) en Panamá 2018–2022. Panamá, Republica de Panamá. 2018. (accessed on 12 June 2025).
- MINSA / CSS / OPS / MESOAMÉRICA MALARIA. Guía de Abordaje Integral para la Eliminación de la Malaria en la República de Panamá. Available online: https://guia_integrada_malaria_2021.pdf (accessed on 12 March 2025).
- Ministerio de Ambiente. Plan Nacional Contra la Sequía de Panamá; Ministerio de Ambiente: Panamá, Panamá, 2020. [Google Scholar]
- Instituto de Estadísticas y Censo. Resultados Finales Básicos XII Censo Nacional de Población y VIII de Vivienda 2023. https://www.inec.gob.pa/publicaciones/Default3.aspx?ID_PUBLICACION=1199&ID_CATEGORIA=19&ID_SUBCATEGORIA=71.
- Hurtado, L.; Cumbrera, A.; Rigg, C.; Perea, M.; Santamaría, A.M.; Chaves, L.F.; Moreno, D.; Romero, L.; Lasso, J.; Caceres, L.; et al. Long-term transmission patterns and public health policies leading to malaria elimination in Panamá. Malar. J. 2020; 19, 265.
- Wilkerson, R.C.; Strickman, D.; Litwak, T.R. Illustrated key to the female anopheline mosquitoes of Central America and Mexico. J Am Mosq Control Assoc. 1990; 6:7–34. pmid:2324726.
- Snounou, G.; Viriyakosol, S.; Jarra, W.; Thaithong, S.; Brown, K.N. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 1993; 58, 283–292.
- Chaumeau, V.; Andolina, C.; Fustec, B.; Tuikue Ndam, N.; Brengues, C.; et al. Comparison of the Performances of Five Primer Sets for the Detection and Quantification of Plasmodium in Anopheline Vectors by Real-Time PCR. PLOS ONE 2016; 11(7): e015916. [CrossRef]
- Lol, J.C.; Castañeda, D.; Mackenzie-Impoinvil, L.; Romero, C.G.; Lenhart, A.; Padilla, N.R. Development of molecular assays to detect target-site mechanisms associated with insecticide resistance in malaria vectors from Latin America. Malar J. 2019;20;18(1):202. [CrossRef] [PubMed]
- Liebman, K.A.; Pinto, J.; Valle, J.; Palomino, M.; Vizcaino, L.; Brogdon, W.; Lenhart, A. Novel mutations on the ace-1 gene of the malaria vector Anopheles albimanus provide evidence for balancing selection in an area of high insecticide resistance in Peru. Malar J. 2015; 14. [CrossRef] [PubMed]
- Asih, P.B.; Syahrani, L.; Rozi, I.E.; Pratama, N.R.; Marantina, S.S.; Arsyad, D.S.; Mangunwardoyo, W.; Hawley, W. ; Laihad. F.; Shinta- Sukowati, S.; Lobo, N.F.; Syafruddin, D. Existence of the rdl mutant alleles among the anopheles malaria vector in Indonesia. Malar J. 2012; 25. [CrossRef] [PubMed]
- Sequencher® version 4.1.4 DNA sequence analysis software, Gene Codes Corporation, Ann Arbor, MI USA http://www.genecodes.com.
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 12; 2024.
- Leigh, J.W.; Bryant, D. Citing PopART or Integer Neighbour-Joining Networks: PopART: Full-feature software for haplotype network construction. Methods; Ecol Evol. 2015: 6(9):1110–1116.
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico,S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol.2017; 34: 3299-3302.
- R Core Team. R: A language and environment for statistical computing (Version 4.3.2) [Computer software]. 2023. R Foundation for Statistical Computing. https://www.R-project.org/.
- Global technical strategy for malaria 2016–2030, 2021 update. Geneva: World Health Organization 2021. Licence: CC BY-NC-SA 3.0 IGO.
- Cumbrera, A.; Calzada, J.E.; Chaves, L.F.; Hurtado, L.A. Spatiotemporal Analysis of Malaria Transmission in the Autonomous Indigenous Regions of Panama, Central America, 2015–2022. Trop. Med. Infect. Dis. 2024; 9,. [CrossRef]
- Silva, A.P.B.; Santos, J.M.M.; Martins, A.J. Mutations in the voltage-gated sodium channel gene of anophelines and their association with resistance to pyrethroids – a review. Parasites Vectors 2014; 7,. [CrossRef]
- Dong, K.; Du, Y.; Rinkevich, F.; Nomura, Y.; Xu, P.; Wang, L.; Silver, K.; Zhorov, B.S. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol. 2014;50. [CrossRef] [PubMed]
- Hancock, P.A.; Ochomo, E.; Messenger, L.A. Genetic surveillance of insecticide resistance in African Anopheles populations to inform malaria vector control. Trends Parasitol. 2024; 40(7):. [CrossRef] [PubMed]
- Qian, W.; Liu, N.; Yang, Y.; Liu, J.; He, J.; Chen, Z.; Li, M.; Qiu, X. A survey of insecticide resistance-conferring mutations in multiple targets in Anopheles sinensis populations across Sichuan, China. Parasites Vectors. 2021. [CrossRef]
- Taylor-Wells, J.; Brooke, B.D.; Bermudez, I.; Jones, A.K. The neonicotinoid imidacloprid, and the pyrethroid deltamethrin, are antagonists of the insect Rdl GABA receptor. J Neurochem. 2015; 135. [CrossRef]
- Wondji, C.S; Dabire, R. K; Tukur. Z.; Irving. H.; Djouaka, R.; Morgan. J.C. Identification and distribution of a GABA receptor mutation conferring dieldrin resistance in the malaria vector Anopheles funestus in Africa. Insect Biochem Mol Biol. [CrossRef]
- Yang, C.; Huang, Z.; Li, M.; Feng, X.; Qiu, X. RDL mutations predict multiple insecticide resistance in Anopheles sinensis in Guangxi, China. Malar. [CrossRef]
- Lol, J.C.; Castellanos, M.E.; Liebman, K.A.; Lenhart, A.; Pennington, P.M.; Padilla, N.R. Molecular evidence for historical presence of knock-down resistance in Anopheles albimanus, a key malaria vector in Latin America. Parasit Vectors 2013.
- Gutiérrez Pérez, H.M.; Mayorga Marín, F.J. Resistencia a insecticidas mediante ensayos moleculares en Anopheles albimanus en la Costa Caribe Norte de Nicaragua. Revista Científica Estelí 2025; 14. [CrossRef]
- Escobar, D.; González-Olvera, G.; Gómez-Rivera, Á.S. ; Navarrete-Carballo, J;, Mis-Ávila, P.; Baack-Valle, R.; Escalante, G.; Reyes-Cabrera, G.; Correa-Morales, F.; Che-Mendoza, A.; Vazquez-Prokopec, G.; Lenhart, A.; Manrique-Saide, P. Insecticide susceptibility status of Anopheles albimanus populations in historical malaria foci in Quintana Roo, Mexico. Malar J. 2024. [CrossRef] [PubMed]
- Alout, H.; Ndam, N.T.; Sandeu, M.M.; Djégbe, I. .; Chandre, F.; Cohuet, A. Insecticide resistance alleles affect vector competence of Anopheles gambiae s.s. for Plasmodium falciparum. PLoS Pathogens 2014; 10.
- Ndiath, M.O.; Cailleau, A.; Diedhiou, S.M.; Gaye, A.; Boudin, C.; Richard, V.; et al. Effects of the kdr resistance mutation on the susceptibility of wild Anopheles gambiae populations to Plasmodium falciparum: a hindrance for vector control. Malar J. 2014; 3.
- Kabula, B.; Tungu, P.; Rippon, E.J.; Steen, K.; Kisinza, W.; Magesa, S.; et al. A significant association between deltamethrin resistance, Plasmodium falciparum infection and the Vgsc-1014S resistance mutation in Anopheles gambiae highlights the epidemiological importance of resistance markers. Malar J. 2016; 15:289.
- Adams, K.L.; Selland, E.K.; Willett, B.C. ; Carew. J.W.; Vidoudez, C.; Singh, N.; Catteruccia, F. Selection for insecticide resistance can promote Plasmodium falciparum infection in Anopheles. PLoS Pathog. 2023; 20;19(6):e1011448. [CrossRef] [PubMed]
Figure 1.
Annual malaria cases in Panama from 2000 to 2025 (first semester) highlighting major events related to vector control interventions applied in the country since 2000.
Figure 1.
Annual malaria cases in Panama from 2000 to 2025 (first semester) highlighting major events related to vector control interventions applied in the country since 2000.
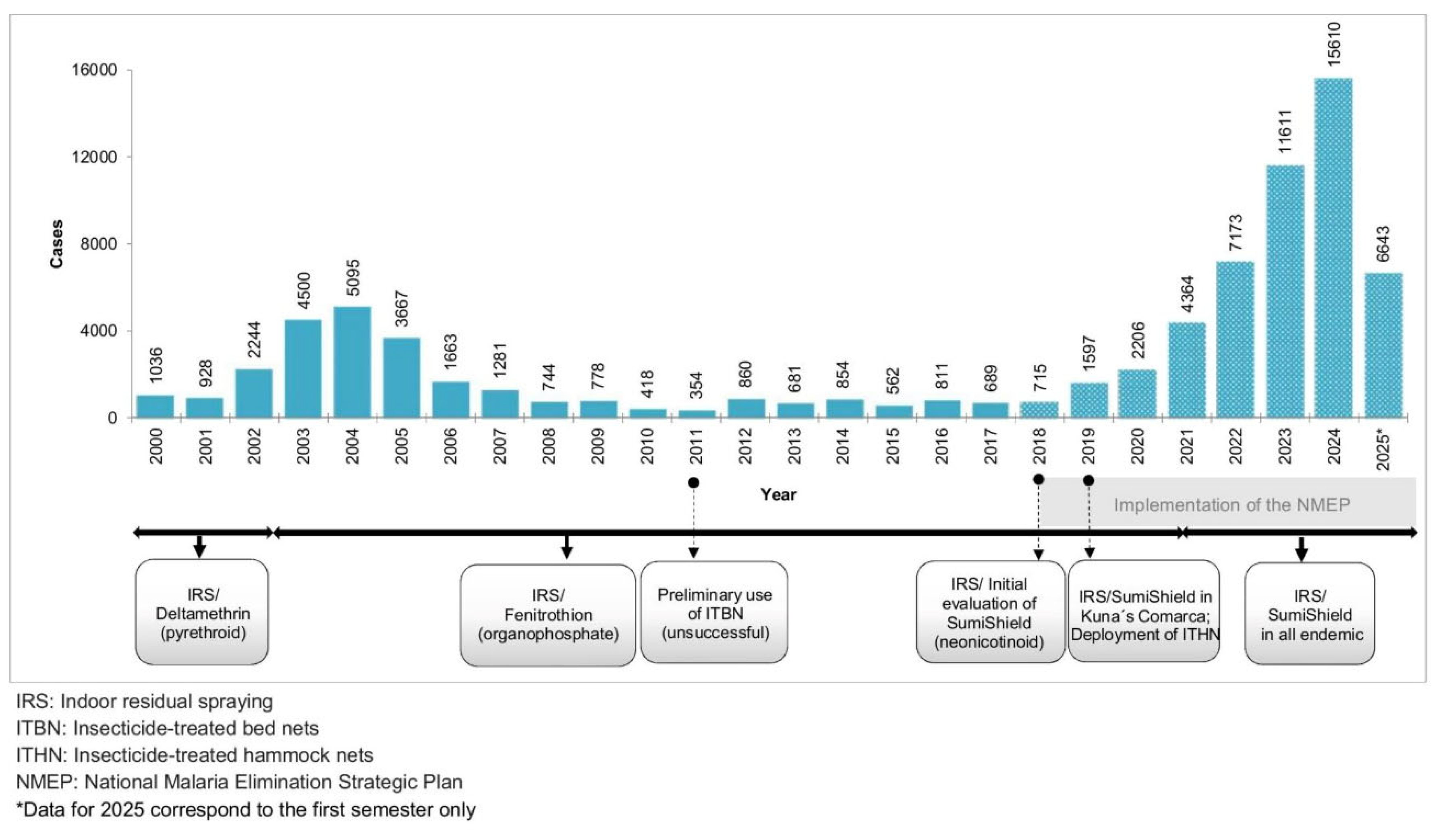
Figure 2.
Map of the Panama showing Anopheles albimanus sampling sites (red dots) within the indigenous regions (“Comarcas”) marked in yellow. The lower inset map shows the location of Panama in the Mesoamerican region, colored in yellow. The red dashed lines indicate the location of the Panama Canal that divides the country into the Eastern and Western regions.
Figure 2.
Map of the Panama showing Anopheles albimanus sampling sites (red dots) within the indigenous regions (“Comarcas”) marked in yellow. The lower inset map shows the location of Panama in the Mesoamerican region, colored in yellow. The red dashed lines indicate the location of the Panama Canal that divides the country into the Eastern and Western regions.
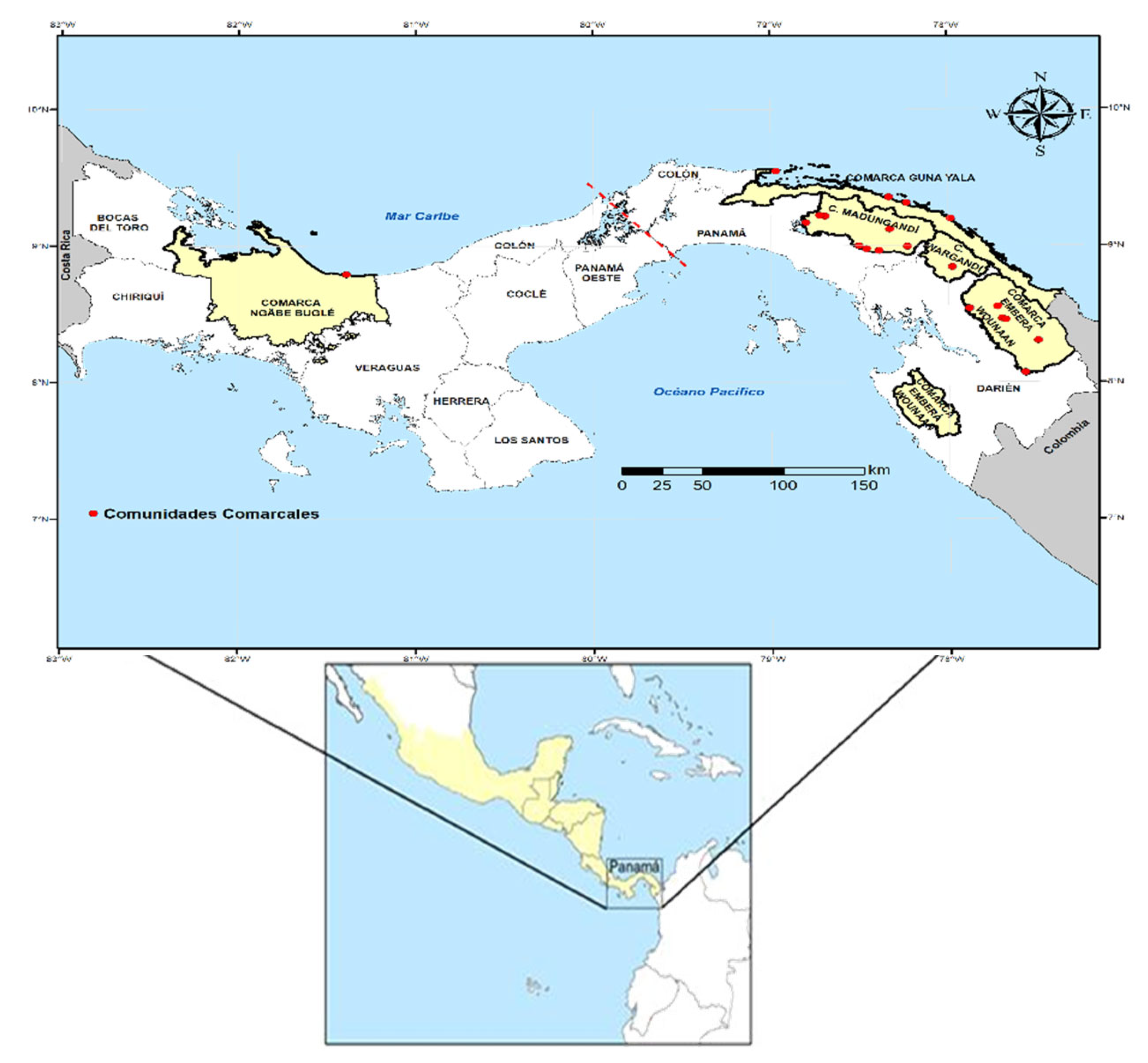
Figure 4.
The relative frequency (%) of resistant and susceptible alleles across the different comarcas analyzed. Resistant alleles (red) predominate in some comarcas, suggesting a higher potential for insecticide resistance, while other comarcas exhibit a greater proportion of susceptible alleles (blue), indicating lower prevalence of resistance-associated variants.
Figure 4.
The relative frequency (%) of resistant and susceptible alleles across the different comarcas analyzed. Resistant alleles (red) predominate in some comarcas, suggesting a higher potential for insecticide resistance, while other comarcas exhibit a greater proportion of susceptible alleles (blue), indicating lower prevalence of resistance-associated variants.
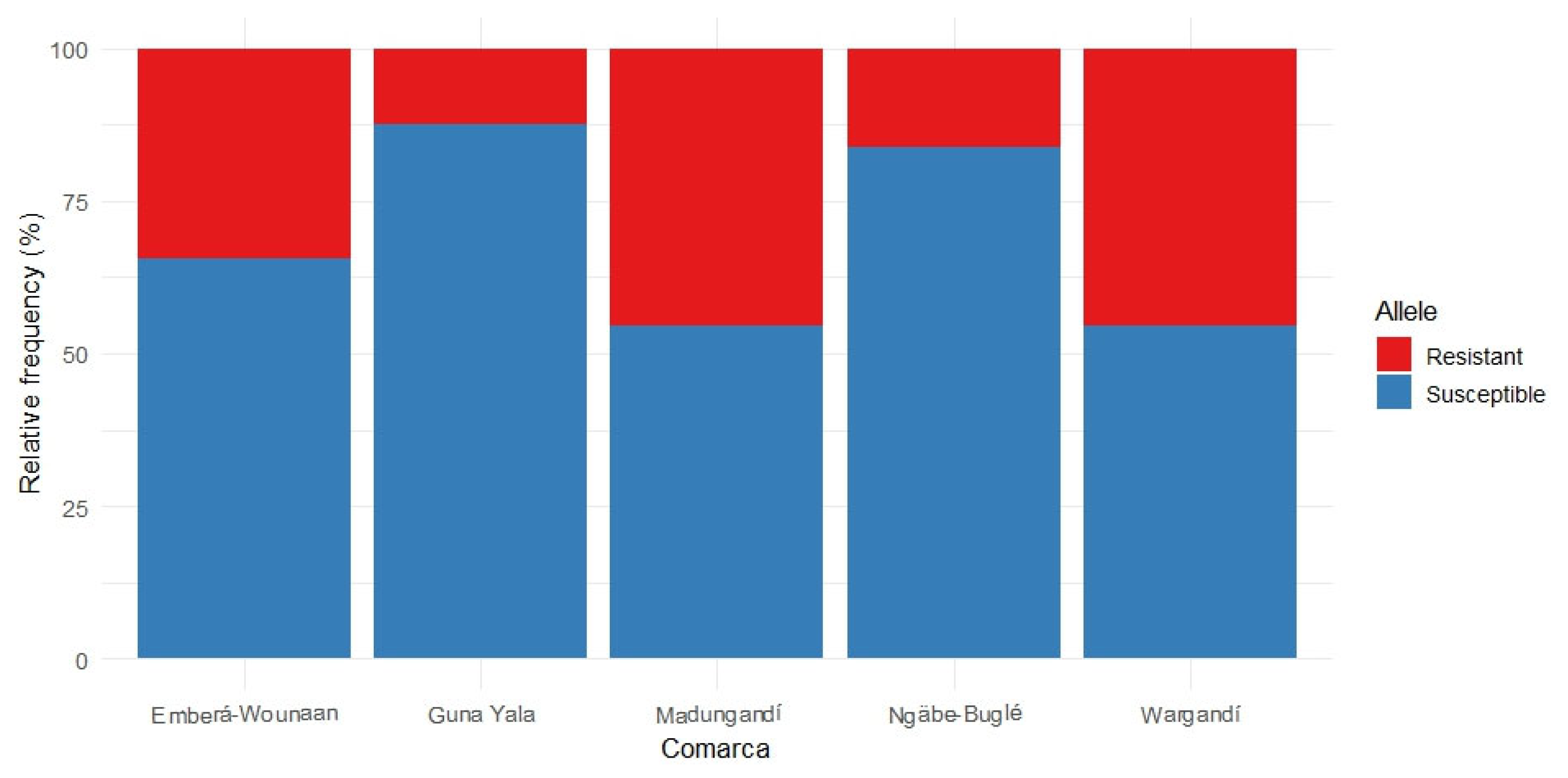
Figure 7.
Haplotype network inferred by a median-joining method using concatenated sequences of three insecticide resistance markers (vgsc, ace-1 and rdl genes) from 30 An. Albimanus collected between 2011 al 2023 across three comarcas. Circles represent an independent sequence haplotype with the color denoting the geographic origin and the size of the circle accounting for its frequency. The lengths of the lines connecting the haplotypes refer to the distance of relatedness.
Figure 7.
Haplotype network inferred by a median-joining method using concatenated sequences of three insecticide resistance markers (vgsc, ace-1 and rdl genes) from 30 An. Albimanus collected between 2011 al 2023 across three comarcas. Circles represent an independent sequence haplotype with the color denoting the geographic origin and the size of the circle accounting for its frequency. The lengths of the lines connecting the haplotypes refer to the distance of relatedness.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



