
Submitted:
04 August 2025
Posted:
04 August 2025
You are already at the latest version
Abstract
Alzheimer’s disease (AD) is a complex neurodegenerative condition characterized by progressive cognitive decline and neuronal dysfunction. In addition to the known, genetic factors, such as the APOE ε4 allele, growing evidence points to the influence of environmental elements, particularly polluting metals and air pollution, in the development and progression of AD. This review explores how metals like iron (Fe), copper (Cu), and manganese (Mn), when present in excess or dysregulated, contribute to oxidative stress, neuroinflammation, mitochondrial damage, and pathological protein aggregation in the brain. Moreover, air pollution, especially exposure to particulate matter, has been associated with higher AD risk. The role of gut dysbiosis and systemic inflammation is also discussed as a potential link between environmental exposure and neurodegeneration. This synthesis of current literature highlights the importance of recognizing environmental metals as modifiable risk factors for AD and the urgent need to incorporate environmental health considerations into dementia prevention strategies.
Keywords:
Alzheimer's disease
; air pollution
; pathways
; metals
; public health
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cortical atrophy, enlargement of the ventricles, and profound cognitive decline. At the cellular level, AD pathology involves the extracellular aggregation of β-amyloid (Aβ) into senile plaques, intracellular accumulation of hyperphosphorylated tau forming neurofibrillary tangles, and the dysregulation of glial cells such as microglia and astrocytes. These alterations disrupt synaptic plasticity and neural networks, ultimately leading to neuronal death and functional impairment [1,2,3].
Motor dysfunction is increasingly recognized in AD, often preceding cognitive decline. Gait disturbances, balance issues, and reduced strength are early indicators linked to amyloid and tau pathology. Neuroimaging and digital tools enable early detection. While drug options are limited, physical therapy and dual-task training improve mobility and may delay progression, enhancing patient autonomy and quality of life [4].
Recent advancements in immunoassay technologies have enabled more precise quantification of biofluid biomarkers. Brain-derived tau (BD-tau), a tau isoform specific to the central nervous system (CNS), has emerged as a promising biomarker with high specificity for Alzheimer's disease (AD). In addition, elevated levels of neurofilament light chain (NFL), indicative of axonal damage, are observed in AD and are even more pronounced in other neurodegenerative conditions [5].
Using the Nucleic Acid Linked Immuno-Sandwich Assay, plasma p-tau217 and NFL have been identified as key blood-based biomarkers in both AD and GRN mutation carriers [6].
The global burden of AD and related dementias (ADRD) is projected to surpass 140 million cases by 2050, driven largely by increasing life expectancy [7]. Despite extensive research, the precise etiology of AD remains elusive, though numerous contributing factors have been identified.
The APOE ε4 allele remains the most significant genetic risk factor for AD. Carriers demonstrate greater hippocampal atrophy and accelerated cognitive decline, not only in individuals with AD and mild cognitive impairment (MCI) but also in cognitively normal populations [8].
Notably, the effects of APOE ε4 exhibit sex-based dimorphism, with female carriers showing more pronounced hippocampal volume loss than their male counterparts [9]. However, the impact of APOE ε4 varies across populations; among older adults in sub-Saharan Africa, its influence appears attenuated [10].
Environmental and inflammatory factors are increasingly recognized as contributors to AD pathogenesis. Exposure to air pollutants such as nitrogen dioxide and particulate matter (PM) has been linked to a heightened risk of dementia. In sub-Saharan Africa, chronic inflammation and co-infections, including HIV and helminthiases, are associated with elevated serum iron and hepcidin levels, which may contribute to neuronal injury [11,12]. In individuals with HIV, increased inflammatory markers also correlate with poorer cognitive performance [13].
Iron (Fe) dysregulation represents a key mechanistic link between systemic inflammation and neurodegeneration. Hepcidin, the central regulator of Fe metabolism, orchestrates iron absorption, storage, and recycling. In the absence of a physiological excretory pathway, excess iron accumulates in tissues such as the brain, where it catalyzes the formation of reactive oxygen species and exacerbates neuronal damage [14,15].
Notably, elevated dietary iron intake, particularly common among adult men and older individuals through red meat consumption, supplements, and fortified foods, has been associated with multiple chronic conditions, including AD [16,17,18].
Expanding on this, the gut–liver–brain axis offers additional insight into how iron excess contributes to neurodegeneration. Elevated dietary iron levels disrupt gut microbial composition, promoting dysbiosis, impairing intestinal barrier function, and triggering hepatic and systemic inflammation [19]. This dysbiotic state has been linked to increased LDL-C levels and plays a role in amplifying neuroinflammatory processes associated with AD pathogenesis [20,21,22].
The absorption and systemic distribution of essential trace metals such as Fe, zinc (Zn), copper (Cu), and manganese (Mn) are tightly regulated to maintain cellular homeostasis and support enzymatic functions [23]. These divalent cations frequently utilize shared transport mechanisms in the gastrointestinal tract, including DMT1 (divalent metal transporter 1), ZIP family proteins, and ferroportin [24,25].
Notably, these metals also compete for binding to gastrointestinal mucins, which modulate their bioavailability for intestinal absorption. Zinc exhibits the highest mucin-binding affinity, followed by iron, copper, and manganese. Their mutual interactions can influence each other’s accessibility to enterocytes, with iron showing a relatively stable binding unless displaced by manganese. Thus, metal competition begins at the mucus barrier, acting as a selective filter that regulates nutrient uptake and may contribute to trace metal imbalances or deficiencies [26].
Consequently, dietary competition among these metals can significantly influence their respective bioavailability and systemic levels [27].
Excessive intake of a single divalent metal can impair the absorption of others due to competitive inhibition at shared transporters. For instance, high oral zinc intake has been shown to reduce copper absorption, potentially resulting in hypocupremia [28]. Similarly, elevated dietary iron may interfere with the uptake of Mn [29] and zinc [30], thereby compromising antioxidant defenses and metabolic processes reliant on these cofactors.
This interaction becomes particularly significant in populations with unbalanced diets, widespread use of supplements, or altered gastrointestinal physiology. In addition, competition among essential metals can influence the absorption and distribution of toxic heavy metals such as lead (Pb) and cadmium (Cd), which exploit the same transport pathways to enter cells [31].
Although competition between divalent metals such as Fe, zinc, Mn, and Cd is well documented in the duodenum, similar mechanisms also occur in the brain. Transporters including DMT1, ZIP8, and ZIP14 are expressed in neurons and glial cells and facilitate the uptake of these metals across the blood–brain barrier (BBB). Due to their overlapping substrate specificity, these transporters mediate competitive interactions that influence cerebral metal homeostasis and may promote neurotoxicity, particularly in the presence of non-essential or toxic metals like cadmium and lead. This phenomenon is especially relevant in neurodegenerative diseases, where altered metal levels and disrupted transport have been implicated in oxidative stress and neuronal injury [32,33,34].
Inadequate levels of essential metals can increase the brain’s vulnerability to toxic metal accumulation and oxidative damage [35]. Specifically, the disruption of metal balance due to Mn dysregulation through competition with essential elements such as Fe, magnesium (Mg), zinc, Cu, and calcium (Ca), as well as with neurotoxic metals like lead (Pb) or vanadium (V) excess can contribute to neurodegenerative processes. This disturbance may impair metal-dependent enzymatic functions and neuronal signaling. Therefore, neurotoxicity may not result solely from excess or deficiency of a single metal, but from systemic imbalances driven by competitive displacement [36,37,38].
Therefore, maintaining balanced intake and homeostasis of divalent metals is crucial not only for optimizing essential nutrient function but also for mitigating the toxicological risks of environmental metal exposure.
A recent bibliometric review highlights growing global interest in the roles of inflammation, immune dysregulation, and gut-brain interactions in AD, emphasizing the importance of biomarker development and neuroprotective interventions [39].
Accordingly, this narrative review aims to synthesize current evidence on the contribution of polluting metals to the development and progression of AD. It explores their role in triggering inflammation, oxidative stress, gut dysbiosis, and neurodegeneration, and considers implications for biomarker discovery and environmental health policy.
2. Air Pollution as an Environmental Risk Factor in AD
AD and related dementias result from gene-environmental interactions. Air pollution is a key modifiable risk factor. Proposed mechanisms include peripheral inflammation, neuroinflammation, and epigenetic changes, though significant data gaps remain [40].
Fewer than 5% of AD cases are demonstrably directly inherited, indicating that environmental factors may be important in initiating and/or promoting the disease [41].
Global data (1990–2019) show air pollution, especially PM2.5 and ozone (O₃), is significantly associated with increased AD and dementia burden. Stronger effects were seen in the Global South. Findings highlight the need for stricter pollution control measures [42]. South Asia hosts 37 of the 40 most polluted cities, with PM2.5 levels far exceeding WHO limits, reducing life expectancy by five years, unlike high-income countries with stricter regulations and improved air quality [43].
Latin America, part of the Global South, generally experiences higher levels of air pollution compared to North America. According to the 2023 World Air Quality Report by IQAir, many Latin American cities exceed the World Health Organization's recommended limits for pollutants such as PM2.5, ozone, nitrogen dioxide, and sulfur dioxide, posing greater public health risks than in North American cities [44,45]
Neighborhood factors like housing, green space, and pollution influence AD prevalence. Geographically weighted random forest (GWRF) modeling reveals strong spatial variability, highlighting the need for targeted, county-specific interventions to address key local risk factors [46]. AD is more frequent in the global south, particularly in women [47].
Global South accounts for around 80% of deaths due to air pollution. These findings highlight the importance of considering environmental inequality between the Global North and the Global South, as well as the co-benefits of air pollution-climate change mitigation during policymaking processes [48].
From what has been described, the southern part of the world faces a high risk of increasing its incidence of AD in the future due to the high presence of polluted air and the inequity that still exists.
2.1. Sources of Air Pollution: Fine Particulate Matter, PM2.5 (≤2.5 µm) and PM10 (≤10 µm), Organic Compounds, Heavy Metals
Airborne magnetite nanoparticles (<200 nm) from traffic can enter the brain via olfactory pathways, bypassing the blood-brain barrier. Their neurotoxicity and prevalence may pose a global environmental risk factor for AD development [41]. Figure 1 shows how easily the entry of metals in PM into the human brain is through the olfactory pathway.
Magnetite nanoparticles, capable of catalyzing the formation of reactive oxygen species, occur in AD plaques and tangles; they are thought to form in situ, from pathological iron dysfunction [41].
Excess brain iron and air pollution exposure increase neurodegenerative disease risk. Iron, abundant in air pollution, especially in subways, may enter the brain via olfactory pathways, contributing to lifelong accumulation linked to neurological disorders [49].
Higher hippocampal iron uniformity in mild cognitive impairment suggests a pathological continuum from aging to AD, supporting non-heme iron accumulation as an early biomarker of cognitive decline and AD risk [50].
Iron in PM₁₀ was linked to higher AD and dementia rates in Lima, Peru, a city with high air contamination [51]. Inhaled iron nanoparticles accumulate in the brain, showing sex-dependent neurodegenerative effects, Alzheimer-like in females and Parkinson-like in males. Prolonged exposure may raise brain Fe and disease risk, highlighting the need for regulation [49].
Although iron has been linked with different neurodegenerative diseases, this may be because it is differentially distributed across brain regions. For example, a study demonstrates a region-specific distribution of brain iron in amyotrophic lateral sclerosis (ALS). Significant susceptibility differences across hippocampal, amygdala, and thalamic regions were observed based on disease progression rate and motor phenotype. These findings suggest localized iron dyshomeostasis and spatial iron changes in neurodegeneration [52].
Moreover, increased thalamic iron levels have also been observed in other neurodegenerative diseases, particularly in younger individuals and in studies using high-resolution imaging, further supporting a regionally differentiated pattern of iron accumulation in the brain [53].
The reasons why iron is differentially distributed in the brain are not yet fully elucidated; however, it may determine the generation of different neurodegenerative diseases in men and women.
The primary neurotransmitter system affected in Parkinson’s disease (PD) is the nigrostriatal dopaminergic pathway, whose dysfunction gives rise to the hallmark motor symptoms. Substantial evidence indicates sex-based differences in PD prevalence and progression, with men more commonly affected and exhibiting faster symptom progression than women [54,55]. In contrast, AD is more prevalent in women (35% vs. 17%), whereas vascular dementia is slightly more frequent in men (21% vs. 17%) [56].
Environmental air pollution remains a critical issue in large cities due to anthropogenic activities like fossil fuel combustion, which emit suspended particulate matter (PM) [57].
Trace metals, particularly Fe, Zn, and Cu, are key drivers of PM2.5 toxicity, contributing up to 52% of ROS production in Hong Kong [58]. A meta-analysis confirmed that PM2.5 constituents such as iron are consistently linked to increased mortality and cardiovascular and respiratory diseases, underscoring the need to regulate specific components to protect health [59].
Long-term exposure to PM2.5 notably more so than PM₁₀ is associated not only with increased respiratory disease risk, particularly in low- and middle-income countries [60], but also with adverse neurological outcomes. In Shijiazhuang, seasonal spikes in PM2.5 and Mn during heating periods further elevating health risks, despite overall air quality improvements, underscoring the need for stricter seasonal pollution controls [61]. Importantly, PM2.5 can cross the blood–brain barrier via inhalation or olfactory pathways, triggering neuroinflammation, oxidative damage, and microglial activation. These effects are strongly linked to neurodegenerative disease risk, cognitive decline, and structural brain changes in older adults [62,63].
Aerosol particles impact health due to their oxidative potential. On Fukue Island, Japan, anthropogenic sources contributed 17% of Fe and 44% of Mn in PM2.5, mainly transported from industrial regions in Asia [64].
In Seoul (urban, traffic), the main sources of Mn, Fe, V, and Zn in PM2.5 were road dust and tire wear. In Wonju (rural, industrial), the high levels were attributed to industrial emissions [65]. This shows how different anthropogenic sources (traffic, industry) determine the concentration of Fe and Mn in the air, with negative implications for health.
A ferromanganese (FeMn) smelter located in Meyerton, South Africa, has been in operation since 1959 and produces approximately 540 kilotons of FeMn annually. PM2.5 levels in nearby residential areas have been found to contain elevated manganese (Mn) concentrations in both indoor and outdoor environments. Fine particles (<2.5 µm) predominated, which can readily enter the respiratory tract, reach the bloodstream, and translocate to the brain via the olfactory pathway. These findings highlight the impact of metallurgical industries on the bioavailability of environmentally relevant manganese [66].
Air pollution involving trace metals such as Fe and Mn has become a growing public health concern due to their toxic potential, especially in soluble or ultrafine particulate forms that can bypass respiratory defenses. Although both elements are essential micronutrients, chronic inhalation exposure can lead to adverse pulmonary, cardiovascular, and neurological outcomes. In Peru, several regions face heightened risk from combined mining, industrial, and vehicular emissions including La Oroya, Cerro de Pasco, El Callao, and the mining zones of Junín [67].
Complementing environmental data, experimental evidence further supports the biological impact of Mn and Fe co-exposure on the brain.
A study conducted in 2025 showed that Mn exposure alone induces oxidative stress in astrocytes by downregulating the STAT3/SOD2 pathway, whereas co-exposure with iron (Fe) activates the NRF2/NQO1 antioxidant pathway, leading to a reduction in reactive oxygen species (ROS) levels. These findings suggest a complex interaction between Mn and Fe in neural tissue that may involve protective mechanisms [68].
2.2. General Mechanisms by Which Air Pollution Impacts Neurological Health
Air pollution is a critical global concern with far-reaching effects beyond respiratory health, notably impacting neurological and psychiatric disorders. Growing evidence links exposure to PM2.5, sulfur dioxide (SO₂), and nitrogen dioxide (NO₂) with increased risks of dementia, AD, schizophrenia, ADHD, stroke, PD, and multiple sclerosis. In Figure 2, air pollution is observed as a risk factor for the development of AD.
PM2.5 can impair cognitive function through direct entry into the brain via the nose-to-brain route. Short-term exposure in mice induced learning and memory deficits, along with neuronal damage and glial activation, without marked systemic inflammation. Transcriptomic analysis revealed that PM2.5 primarily disrupts synaptic function rather than triggering classic neuroinflammatory pathways. Moreover, microglial depletion did not reverse cognitive impairment, reinforcing the role of synaptic dysregulation as a central mechanism. These findings highlight a direct neurotoxic effect of PM2.5 on brain function, independent of peripheral inflammation [69].
Air pollution, particularly ultrafine particles (UFPs), disrupts the PI3K/AKT signaling pathway, which regulates neuronal survival, especially in AD. UFPs increase oxidative stress and DNA damage without causing immediate cell death. In AD olfactory mucosa (OM) cells, altered microRNA and DNA methylation profiles impair gene regulation, making them more susceptible to air pollutant-induced toxicity [70].
Air pollution accelerates cognitive decline and may contribute to AD by targeting glutamatergic synapses. Chronic exposure to diesel exhaust particles reduces postsynaptic markers and the glutamate receptor, N-methyl-D-aspartate (NMDA) receptor subunits in the cortex, while upregulating antioxidant enzymes involved in ferroptosis mitigation. These synaptic changes suggest a molecular link between air pollution and AD pathogenesis [71].
Gamma-secretase modulator (GSM-15606) attenuates the amyloidogenic amyloid beta (Aβ)42 peptide during exposure to air pollution, which may be a mechanism by which air pollution increases AD risk [72].
Proposed mechanisms underlying neurodegenerative disorders include neuroinflammation, oxidative stress, microglial activation, cerebrovascular dysfunction, and disruption of the blood–brain barrier [73]. Although the precise pathways by which air pollutants contribute to neurological disorders are not fully elucidated, current evidence highlights the roles of BBB compromise, glial activation, and sustained oxidative and inflammatory responses [74].
Microglia, the brain’s resident immune cells, act as key environmental sensors and play a central role in air pollution-induced neuroinflammation. In response to various stimuli, microglia can polarize into either a pro-inflammatory (M1) or an anti-inflammatory (M2) phenotype. A shift toward the M1 state promotes the release of inflammatory cytokines, oxidative stress, and synaptic dysfunction, all of which contribute to neuronal damage and cognitive decline in AD [75].
Chronic PM2.5 exposure promotes Alzheimer’s-like pathology by shifting microglia to a pro-inflammatory state that releases extracellular vesicles (EVs) rich in miR-34a-5p. This suppresses neuronal DUSP10, activates p38 MAPK, and triggers tau phosphorylation, β-amyloid buildup, and neuronal death. Silencing miR-34a-5p mitigates these effects, linking PM2.5 to neurodegeneration [1].
Ultrasound stimulation has been shown to modulate microglial polarization in AD models by reducing M1 and enhancing M2 populations, thereby lowering amyloid plaque burden and increasing hippocampal IL-10 levels. Proteomic analyses link these changes to enhanced mitochondrial oxidative phosphorylation, suggesting a novel mechanism for therapeutic intervention [76].
Circular RNA circAPP, encoded by the amyloid precursor protein gene, is upregulated in AD and promotes M1 polarization by acting as a molecular sponge for miR-1906, which in turn increases Chloride Intracellular Channel 1 (CLIC1) expression and activity. CLIC1 is a redox-sensitive chloride channel activated in microglia during inflammation. Its upregulation contributes to oxidative stress and neuroinflammation in AD, promoting a pro-inflammatory microglial phenotype. The circAPP/miR-1906/CLIC1 axis exacerbates neuroinflammation and cognitive impairment, identifying circAPP as a promising therapeutic target [77].
Similarly, the microglia-enriched circRNA circDlg1 contributes to neuroinflammation in APP/PS1 mice by regulating the degradation of PDE4B. Loss of circDlg1 enhances microglial responses to Aβ deposition and reduces neuroinflammation, underscoring its potential as a microglia-specific therapeutic target in AD [78].
Finally, the Methyltransferase-like 3/ Insulin-like Growth Factor 2 mRNA-binding Protein 2/ Inhibitor of kappa B alpha (METTL3/IGF2BP2/IκBα) signaling pathway also modulates microglial polarization, offering further insight into molecular targets for regulating neuroinflammation in AD [79]. This epigenetic signaling pathway regulates microglial polarization by enhancing inflammatory gene expression through m6A RNA methylation. It contributes to sustained neuroinflammation in AD by activating the NF-κB pathway.
Disruption of microglial metabolism, particularly glycolysis impairs their neuroprotective functions and promotes pro-inflammatory phenotypes, contributing to synaptic damage and cognitive decline characteristic of AD. Understanding how environmental pollutants influence microglial phenotypes is essential for developing targeted prevention and treatment strategies. Modulating microglial glycolytic pathways may represent a promising therapeutic approach for pollution-related neurodegenerative disorders [80,81].
Particulate matter (PM) represents a major environmental health risk due to its complex mixture of organic and inorganic compounds of varying sizes, compositions, and sources. Its ability to penetrate deep into tissues and cross the BBB contributes to neurotoxicity and the pathogenesis of neurodegenerative diseases. Beyond initiating oxidative stress, inflammation, and excitotoxicity, air pollution also induces epigenetic alterations that can affect the expression of genes involved in stress responses, neuroprotection, and synaptic plasticity [82].
PM2.5 exposure has been linked to epigenetic modifications, such as changes in DNA methylation, histone structure, and non-coding RNAs which are associated with cognitive impairment through mechanisms involving inflammation, synaptic dysfunction, cardiovascular factors, and neuronal structural changes [83].
AD is characterized by the progressive misfolding of proteins and the formation of plaques, which initially develop in specific brain regions before spreading to the entire cortex [84]. The inhalation of diesel engine exhaust (DEE), a major source of particulate air pollution, has been shown to accelerate Aβ plaque formation in the cortex and hippocampus [85].
Noise exposure may also contribute to ADRD. A systematic review highlighted the need for standardized methodologies and adjustment for confounders [86]. In the UK Biobank study, night-time road traffic noise above 45 dB was linked to higher AD risk, while railway noise was associated with PD-related dementia. Cardiovascular health may mediate these effects [87]. Simultaneous exposure to noise and carbon monoxide increases the risk of AD [88].
Soluble PM, such as metals and nano-sized particles, may translocate across the olfactory, trigeminal, or vagal nerves through retrograde axonal transport, or systemic circulation, which may disrupt the BBB and deposit in neural tissue. Systemic hormonal and immune changes may further affect the brain through adrenal hormone-dependent pathways [89].
In mice, postnatal exposure to ultrafine particulate matter (UFPs) leads to neurotoxicity marked by brain iron accumulation. Given iron’s redox activity and neurotoxicity, the study suggests that inhaled iron contributes significantly to UFP-induced brain damage. The neurotoxic effects may vary depending on the chemical composition of the particulate mixture, underscoring iron’s role in air pollution toxicity. [90].
Exposure to ultrafine particles (UFPs) during adolescence disrupts neurotransmitter systems and reduces myelination and astrocytes in the corpus callosum, particularly in male mice, indicating high vulnerability. Altered glutamate and corticosterone levels correlated with structural brain damage. These findings support the biological plausibility of links between UFPs and neurodegenerative or psychiatric disorders, highlighting the need for stricter environmental regulation of UFPs [91].
The health risk associated with magnetic iron oxide nanoparticles (FexOy NPs, mainly Fe3O4 and γ-Fe2O3) have garnered escalating attention due to their presence in human blood and brain tissues, especially for their potential association with neurodegenerative diseases like AD [92]. An ecological relationship between Fe in PM10 and AD has been suggested in human beings [51].
Inhaled iron oxide nanoparticles accumulate in the olfactory bulb and induce sex-specific neurodegenerative features, Alzheimer-like in females and Parkinson-like in males. These findings suggest that chronic airborne iron exposure may elevate brain iron by aging and increase the risk of neurodegenerative diseases, warranting further investigation into its long-term effects [49]. These data suggest a need for regulation of Fe in the air for public health protection.
3. Epidemiological Studies for Alzheimer´s Diseases (AD) and Alzheimer´s Diseases-Related Dementia (ADRD)
AD is the most common form of dementia worldwide. AD and ADRD disproportionately affect Black Americans compared to non-Hispanic White individuals. Economic hardships, especially low income and unemployment, reduce access to physical and mental healthcare, drive dementia risk. Those from more deprived areas highlighted the need for improved health education, public transportation, healthcare access, and job opportunities [93].
Women suffer more frequently from AD, whereas men suffer more frequently from PD and ALS. Research suggests neuroprotective effects of estrogens and confirms that factors reducing their level may contribute to a higher morbidity rate of neurodegenerative diseases (NDs). Adverse effects of androgens on NDs have been noticed; however, some data suggest their beneficial actions [94].
The cumulative evidence hitherto collected suggests that sex and gender are factors to be considered in explaining the heterogeneity of these NDs. Clarifying the role of sex and gender in AD, PD, and ALS is a key topic in precision medicine, which will facilitate sex-specific prevention and treatment strategies to be implemented soon [95].
A Mendelian randomization study demonstrates a causal protective role of bioavailable testosterone (BT) against neurodegeneration. Genetic analysis revealed that higher BT levels significantly reduce the risk of developing ALS (OR = 0.794, p = 0.006), while no associations were found for estradiol or sex hormone-binding globulin (SHBG) with ALS, PD, or AD. These findings support a sex hormone–dependent mechanism in ALS susceptibility and suggest that testosterone may be a modifiable factor for neuroprotection [96].
A Mendelian randomization study identifies a potential causal role of sex hormone-binding globulin (SHBG) in multiple sclerosis (MS). Genetically predicted higher SHBG levels were significantly associated with increased MS risk (OR = 1.634, p = 0.038), and bidirectional analysis confirmed that MS may, in turn, influence SHBG levels (OR = 1.005, p = 0.003). No causal links were found for estradiol, total testosterone, or bioavailable testosterone. These findings suggest SHBG as a potential biomarker or modifiable factor in MS pathophysiology [97].
AD is increasingly recognized as a systemic disorder with significant contributions from both hepatic and cardiovascular dysfunction [98,99]. Through the liver-brain axis, the liver influences AD progression by clearing peripheral β-amyloid (Aβ), modulating systemic metabolism, and secreting hepatokines such as SHBG, FGF-21, SELENOP, and ApoJ. These proteins regulate insulin sensitivity, lipid metabolism, oxidative stress, and immune responses, which in turn contribute to Aβ accumulation, tau hyperphosphorylation, and neuronal damage [98].
Cardiovascular disease contributes to AD through shared genetic risks (e.g., APOE, presenilin), impaired cerebral perfusion, and oxidative stress, which promote Aβ and tau pathology. Systemic protein aggregation, microvascular damage, and blood-brain barrier disruption link neurodegeneration and cardiac dysfunction, highlighting the liver-brain and brain-heart axes as therapeutic targets in AD [99].
Body iron content increases progressively in women after menopause due to the cessation of menstrual blood loss, leading to iron accumulation with advancing age. Iron stores in women remain low during the reproductive years but increase progressively from the fifth decade, reaching levels like those observed in men by the seventh decade. This age-related accumulation of iron has been associated with oxidative stress and neurodegenerative processes. The higher lifetime risk of AD observed in women may, in part, be influenced by postmenopausal iron buildup, which could contribute to greater vulnerability through mechanisms such as amyloid-beta aggregation, tau hyperphosphorylation, and neuroinflammation [100].
3.1. Epidemiological Studies That Link Exposure to Polluted Air with Cognitive Impairment and AD
Alzheimer’s disease (AD), a neurodegenerative disorder, accounts for 50–75% of all dementia cases. Numerous studies have identified a significant association between air pollution and increased AD and dementia risk [42,101]. The 2018 Lancet Commission on Pollution described the evidence linking particulate matter (PM) exposure to dementia as compelling [102].
In a cohort of 30,247 adults, air pollution exposure was associated with elevated AD risk [103]. Epidemiological studies further confirm that exposure to fine particulate matter (PM2.5) not only increases the likelihood of developing AD but also accelerates its progression, particularly in highly polluted regions [104,105]. In Figure 3, epidemiological evidence linking air pollution to cognitive impairment and AD.
As life expectancy continues to rise, the global elderly population is expected to increase, leading to a higher prevalence of age-related diseases, including AD. Ambient air pollution has substantially contributed to the global burden of disease over the past 25 years, largely due to population aging, increased rates of non-communicable diseases, and rising pollution levels in low- and middle-income countries [106]. The burden of dementia is expected to increase globally, given the continuous expansion of the ageing population.
A life-course study found that in-utero and early-life exposure to PM2.5 and NO₂ was associated with increased risk of AD and all-cause dementia. Cumulative PM2.5 exposure across the lifespan also elevated dementia risk, highlighting sensitive periods of vulnerability [107]. Differences in brain volume may also mediate the relationships between air pollution and AD [108].
Iron-rich UFPM and industrial nanoparticles from air pollution present in the brains of children and young adults, were early signs of AD, PD, and TDP-43 proteinopathy observed. These particles cross biological barriers, accumulate in sleep–arousal centers, and localize in neurovascular units. Their magnetic reactivity and oxidative potential may trigger early, irreversible neurodegeneration, sleep disruption, and cognitive decline. Neurodegenerative changes begin in pediatric ages, underscoring the need for preventive strategies [109,110].
Sporadic Alzheimer's disease (AD) occurs in 99% of all cases and can be influenced by air pollution such as diesel emissions and more recently, an iron oxide particle, magnetite, detected in the brains of AD patients. There are mechanistic links between inflammation and oxidative stress to pollutant particle-induced AD pathologies, with magnetite apparently inducing the most pathological effects [111].
Long-term exposure to air pollutants, especially PM2.5 and O3, has been associated with increased risk of AD and PD in several countries [112,113]. More than 50 studies over the past two decades have examined the link between air pollution and AD and related dementias (ADRD) [114].
A recent systematic review and meta-analysis of 28 longitudinal cohort studies assessed the association between long-term PM2.5 exposure and dementia risk using the Burden of Proof framework. A nonlinear relationship was found, with at least a 14% increased risk of dementia across PM2.5 levels ranging from 4.5 to 26.9 µg/m³. The association was stronger for AD than for vascular dementia, emphasizing the role of air pollution in brain aging and dementia development [115].
Global ecological data from 162 countries revealed that each 10-unit increase in PM2.5 or ozone levels was associated with increased incidence, mortality, and disability-adjusted life years (DALYs) from AD and other dementias, with greater impact observed in countries with lower gross national income and socio-demographic indices [116]. Recent findings show that among individuals previously hospitalized for AD/ADRD, post-discharge exposure to air pollution is significantly associated with higher risk of readmission and mortality [117].
In the U.S., a geographically weighted model showed AD prevalence is influenced by mobile home residency (19.9%), low greenness (17.4%), physical inactivity (12.9%), lack of vehicle access (11.3%), and PM2.5 exposure (10.4%), highlighting the need for region-specific environmental and lifestyle interventions [46].
Vegetation may have a protective role. Higher levels of greenness attenuate the harmful effects of PM2.5 and O3 on dementia outcomes, underscoring the importance of natural environments in mitigating cognitive decline [116].
Moreover, interactions between ambient air pollution and social vulnerability may also influence ADRD mortality [118].
In Latin America and the Caribbean (LAC), more than 172 million people are exposed to unhealthy air. Air pollution contributes to five established dementia risk factors (obesity, hypertension, stroke, diabetes, and cardiovascular disease) and is linked to three additional factors (physical inactivity, cognitive inactivity, and depression), some of which may mediate the pollution-dementia relationship [119].
While PM2.5 is a known contributor to cognitive decline, ultrafine particles (UFPs <100 nm) are even more toxic due to their small size and high reactivity [120]. In addition, O3 exposure has been implicated in neurodegeneration. In China, during 2023, elevated O3 levels were estimated to have caused approximately 110,000 new AD cases. Reducing O3 concentrations to 70 μg/m³ could prevent over 210,000 cases, highlighting the need for urgent interventions [121].
Nitrogen oxides (NOx) are also emerging as key contributors. In dual-pollutant models, only NOx remained significantly associated with AD risk, particularly with exposures during the previous five years. No association was observed for vascular dementia, indicating the need to differentiate dementia subtypes and use multi-pollutant models [103].
A recent U.S.-based cohort study (2000–2018) identified sulfate (SO₄²⁻), organic carbon (OC), copper (Cu), and iron (Fe), mainly from fossil fuel combustion and traffic, as key PM2.5 components linked to higher AD risk [122]. In Lima, Peru an ecological study showed link between SO₄²⁻, iron, manganese and Cu with AD and ADRD risks [51], public health strategies targeting these sources may reduce the neurodegenerative burden.
Furthermore, combined exposure to air pollution and artificial light at night has been associated with memory disorders, suggesting potential synergistic effects mediated by biomarkers [123].
A nationwide U.S. cohort study found that extreme heat increases the risk of ADRD-related hospitalizations. Risk was significantly higher among Asian, Black, and Hispanic populations, indicating disproportionate climate vulnerability [124].
Prevalence of AD is higher in women than in men. Menopause before age 45 years was associated with a 33% greater hazard of AD dementia compared with menopause after age 50 years (hazard ratio [HR] = 1.33, 95% confidence interval [CI]: 1.10-1.59). Hysterectomy was not associated with hazard of AD dementia (HR = 1.08, CI: 0.94-1.26). The association between age at menopause and hysterectomy status and AD dementia was not different for White and Black women [125]. Therefore, the high longevity of women does not explain the mechanisms underlying the biological differences between the sexes causing a female predominance in the development of AD.
In experimental studies, female APP/PS1 mice had greater amyloid deposition, hyperactivity, lower body weight, and reduced cerebral blood flow, as well as less neuroinflammation, which the authors suggest may have potential neuroprotection [126].
Interleukin (IL)-6 was identified as a key pro-inflammatory marker, consistently linked to AD, PD, and dementia, underscoring its significant role in neurodegenerative disease progression. Brain-derived neurotrophic factor levels were associated with improved cognitive performance, particularly in African American participants. Observational studies identified sex-based differences in IL-10 levels, particularly among older African American women [127].
Systemic comorbidities are common in AD and may influence disease progression, severity, and management. Men had a higher prevalence of heart disease, diabetes, chronic obstructive pulmonary disease and smoking, whereas thyroid disease, hypertension and depression were more common in women (all p<0.05) [128].
Astrocytic TNFR2 signaling is essential for maintaining hippocampal synaptic function and plasticity. Selective deletion of TNFR2 in astrocytes disrupted synaptic protein expression and glial-neuronal communication, leading to cognitive deficits. These effects were more pronounced in male mice, who showed increased astrocyte and microglial reactivity, impaired memory, and reduced LTP, highlighting a sex-dependent regulation of astrocyte-mediated synaptic function [129].
In Long-Evans rats, O₃ altered glucocorticoid signaling, microglial activity, and oxidative stress markers differently in males and females. Notably, female rats exhibited greater oxidative damage, with higher levels of protein carbonyls and increased expression of microglia-related genes such as Aif1, compared to males. Additionally, antioxidant and glucocorticoid-related genes (Cat, Gpx1, Nfe2l2, Sod1) were more strongly modulated in females. These findings suggest that females are more vulnerable to the neurotoxic effects of ozone exposure [130]. Main findings of research on airborne metals and neurodegenerative diseases are shown in Table 1.
4. Metal Homeostasis in the Brain
Metal ions are widely distributed throughout the brain and are essential for numerous central nervous system (CNS) functions, including neuronal activity and synaptic transmission [131]. Trace elements such as Fe, Cu, Zn, Mn, and selenium (Se) play critical roles in neurotransmitter synthesis, myelination, and antioxidant defense mechanisms. Under normal physiological conditions, the homeostasis of these metals is tightly regulated, as both deficiency and excess can impair neuronal function [132].
The brain regions most consistently affected by metal exposure include the globus pallidus, caudate nucleus, frontal cortex, and cerebellum. Some studies exhibit structural or functional reductions in these areas that correlate with increased levels of metal exposure, suggesting a dose-dependent neurotoxic effect [133].
In AD, pathological alterations are closely linked to disruptions in trace element balance. These elements are also fundamental to key physiological processes such as DNA and protein synthesis, energy metabolism, and enzymatic activity, processes essential for cognition and brain development. Dysregulation in the homeostasis of elements including copper, iron, zinc, selenium, rubidium, silicon, chromium, and vanadium has been implicated in the pathogenesis of neurodegenerative diseases such as AD [134].
Human exposure to heavy metals occurs through inhalation, ingestion, or the food chain. These metals impair antioxidant defenses by interacting with intracellular glutathione (GSH) or sulfhydryl groups of key enzymes like SOD, catalase, glutathione peroxidase (GPx), glutathione reductase (GR), and related systems [135].
Environmental toxins promote oxidative stress, neuroinflammation, mitochondrial dysfunction, and altered APP/tau processing. Exposure increases GSK-3β, BACE-1, TNF-α, and caspases, while reducing BDNF and GAP-43 expression. Dysregulated PARP-1, PGC-1α, and MAPK/ERK signaling contribute to AD [136].
An in vitro study showed that PM2.5 promotes neuronal necroptosis via LncRNA Gm16410 downregulation through p38 MAPK, contributing to AD. The SVHRSP peptide mitigated this effect, revealing a novel LncRNA-mediated mechanism and potential therapeutic target for AD [137].
Copper, iron and zinc are accumulated in post-mortem amyloid plaques of Alzheimer's patients (5.7, 2.8 and 3.1 times more than in normal brains, respectively). The use of chelators is proposed to restore metal homeostasis and reduce oxidative damage [138].
The pathophysiology of neurodegenerative diseases is significantly influenced by metal-induced oxidative stress and toxic buildup. For such reasons, therapeutic approaches point to reduce oxidative damage and chelating excess metals. The regulation of these pathways reduces neuronal damage and improves neurons' survival and functionality [139].
Iron participates in a wide array of cellular functions and is essential for normal neural development and physiology. However, if inappropriately managed, the transition metal can generate neurotoxic reactive oxygen species [140].
Mutations in genes such as C19orf12, which regulate iron and lipid metabolism, are implicated in neurodegeneration with brain iron accumulation (NBIA), a group of rare, inherited disorders characterized by excessive iron deposition in the brain. Although NBIA is genetically driven, it provides mechanistic insights into how iron dyshomeostasis leads to mitochondrial dysfunction and neurodegeneration. Similar pathways may be environmentally triggered by chronic exposure to air pollutants, suggesting a shared vulnerability in iron-regulating systems between genetic and environmental etiologies [141,142].
Exposure to heavy metals is linked to increased AD risk, triggering oxidative stress, mitochondrial dysfunction, protein aggregation, neuroinflammation, and tau pathology. Metals like Pb, Cd, As, and Mn affect specific AD pathways [143].
Dysregulation of redox-active metals, especially Fe, Cu, and Mn contribute significantly to AD and PD pathogenesis by promoting oxidative stress, mitochondrial dysfunction, and apoptosis [144]. In the hippocampus, Fe, Zn, and Ca are most abundant, with Cu and Mn present in lower concentrations but exhibiting distinct subcellular localization, particularly in CA1 neurons [145].
In Alzheimer’s disease, Fe, Cu, and Zn accumulate in amyloid plaques and neurofibrillary tangles. Mn impairs dopaminergic neurogenesis, Fe induces ferroptosis, and Cu disrupts antioxidant defenses. Combined exposure enhances β-amyloid aggregation and neuroinflammation [146].
Fe homeostasis in the brain is essential for maintaining neuronal viability. However, excessive iron accumulation induces oxidative stress and ferroptosis, contributing to poor outcomes not only in AD but also in stroke and other neurological disorders [147]. As a divalent metal, Fe may compete with other divalent metals, such as Zn and Mn, for shared transporters, highlighting the importance of maintaining a balanced trace element profile.
After Fe(II) exposure, labile Fe (II) is stored in lysosomes and transported through neurites. This may aid detoxification and supply iron to neuronal compartments. Iron dyshomeostasis, particularly labile iron contributes to neurodegeneration [148].
Type 2 diabetes patients show increased iron accumulation in the caudate and putamen, correlating with poorer cognitive performance, including slower processing speed, memory decline, and executive dysfunction. HbA1c levels were the strongest predictors of iron deposition, especially in the anterior putamen and posterior caudate, highlighting brain iron’s role in T2DM-related cognitive impairment [149].
While Cu is essential for CNS function, its dysregulation contributes to neurodegeneration. Emerging evidence identifies cuproptosis, a Cu-induced form of cell death, as a potential mechanism in neurological disorders [150]. Trace element imbalances, particularly elevated Fe and Cu have been linked to cognitive decline in older adults. Increased Fe and Cu levels, along with reduced glutamic acid decarboxylase activity, correlate with moderate to severe cognitive impairment [151].
Disruptions in intracellular calcium (Ca²⁺) balance are another hallmark of AD pathology. Accumulation of amyloid-β₁₋₄₂ (Aβ₁₋₄₂) disrupts Ca²⁺ homeostasis, promoting neuronal death. In vitro studies have shown that dantrolene, a Ca²⁺ antagonist, significantly enhances neuronal survival and reduces apoptosis and necrosis when co-applied with Aβ₁₋₄₂ [152].
Fe overload in the AD brain extends beyond plaque formation, contributing to glial activation, oxidative stress, and ferroptosis. Targeting iron homeostasis is a promising therapeutic approach, and novel iron chelators are showing encouraging cognitive outcomes in preclinical studies [153].
At the molecular level, transporters such as ZIP14, part of the SLC39A family, play a critical role in the uptake of divalent metals like Mn, Zn, and Fe. ZIP14 knockout models exhibit elevated Mn accumulation in blood and brain, altered Zn/Fe ratios in cerebrospinal fluid (CSF), and decreased CSF Ca²⁺, indicating compensatory transport mechanisms that disrupt CNS metal and calcium homeostasis [154].
The SLC39A family (also known as ZIP transporters, for Zrt-, Irt-like Protein) is a group of membrane proteins that facilitate the influx of metal ions, primarily Zn, but also iron, Mn, and cadmium (Cd²⁺), from the extracellular space or intracellular organelles into the cytoplasm. These transporters play a critical role in maintaining cellular metal homeostasis, especially in tissues with high metal demand like the brain [154,155].
Lastly, potassium (K⁺) homeostasis is also compromised in AD. In 5xFAD mouse models, impaired K⁺ clearance and reduced expression of astrocytic Kir4.1 channels have been linked to neuronal dysfunction and disease progression [156], reinforcing the broader role of glial cells in maintaining ionic balance and neuroprotection.
Manganese (Mn), the third most abundant transition metal in Earth’s crust, is essential for antioxidant defense, energy metabolism, and immune function. While deficiency impairs growth, excessive Mn common in acidic soils and occupational exposure can cause neurotoxicity, including manganism [157,158].
Brain Mn homeostasis relies on a network of transporters (e.g., DMT1, ZIP8/14, SLC30A10, SPCA1, GPP130) [159]. Disruption leads to Mn accumulation, oxidative stress, mitochondrial dysfunction, Endoplasmic reticulum (ER) stress, and neuroinflammation. Overexposure is linked to neurodegenerative diseases such as AD, PD, Huntington’s, and ALS. Therapeutics like metformin, curcumin, and resveratrol show protective potential [36,160].
Environmental Mn exposure is a recognized risk factor for PD, with neurotoxic effects linked to dopaminergic neuron injury and neuroinflammation [161]. Recent studies highlight the HIF-1α/p53/SLC7A11 axis as a mediator of Mn-induced ferroptosis, contributing to Parkinsonian pathology. HIF-1α’s role varies depending on hypoxic context, and its pharmacological modulation is being explored as a therapeutic strategy [162,163].
Mn also interacts with insulin and IGF receptors, activating neuroprotective signaling pathways such as Akt, MAPK, and mTOR, which counteract apoptosis and promote neuronal survival. Conversely, Mn triggers neuroinflammatory pathways including cGAS-STING, NLRP3-CASP1, NF-κB, SIRT, and JAK-STAT, with downstream effects on autophagy, either protective or harmful depending on context [164].
A U-shaped relationship has been observed between Mn levels and health outcomes, where both deficiency and excess are detrimental [36]. Similar patterns are reported for iron (Fe) and cognitive decline (Figure 4). Inorganic Fe above 45% of total iron is linked to increased dementia risk, while levels between 25–40% show the lowest risk [165,166,167].
4.1. Oxidative Stress and Mitochondrial Damage
Mitochondria fundamental in eukaryotic have cell-type specific phenotypes in response to their environment to perform dozens of interconnected functions and undergo dynamic and often reversible physiological recalibrations [168]. Changes in mitochondrial function and morphology contribute to the development of many neurological diseases. PD is one of the neurodegenerative diseases suspected to be associated with defects in mitochondrial function and quality control [169].
Environmental stressors, particularly air pollutants, have been shown to increase mitochondrial DNA (mtDNA) heteroplasmy, promote reactive oxygen species (ROS) accumulation, and disrupt mitochondrial structure and contact sites [170].
Air pollution, particularly ultrafine particles (UFPs ≤0.1 μm) from traffic emissions, has been increasingly linked to AD. Mitochondria are identified as primary targets of environmental toxicants. Exposure of primary human olfactory mucosa (OM) cells to traffic-related UFPs has been shown to impair mitochondrial function by disrupting oxidative phosphorylation, reducing ATP production, and altering redox balance, resulting in increased oxidative stress. These effects differ between cognitively healthy and AD-derived cells. The findings suggest that mitochondrial dysfunction induced by air pollution may contribute to AD pathogenesis via the olfactory-brain axis [171].
ROS are double-edged swords in biological systems, whereas they are essential for normal cellular functions but can cause damage when accumulated due to oxidative stress. Manganese superoxide dismutase (MnSOD), located in the mitochondrial matrix, is a key enzyme that neutralizes superoxide radicals (O2•-), maintaining cellular redox balance and integrity [172]. Overproduction of ROS disrupts the body's antioxidant defense, compromising redox homeostasis and increasing oxidative stress, leading to several diseases [173].
Current evidence indicates that epigenetic mechanisms are key mediators of Mn-induced neurotoxicity in both in vivo and in vitro models. The specific target genes and downstream signaling pathways involved in Mn-associated epigenetic regulation have not fully characterized [174]. However, presenilin-1 (PSEN1) mutations and elevated F2-isoprostanes contribute to brain inflammation, mitochondrial dysfunction, and cognitive decline in AD progression [3].
Environmental exposure to UFPM and industrial nanoparticles (NPs) has been linked to early hallmarks of Alzheimer’s, Parkinson’s, frontotemporal lobar degeneration (FTLD), and ALS, even in youth. These particles reach the brain via nasal, pulmonary, and placental routes, accumulating in regions like the frontal cortex, olfactory bulb, and substantia nigra. Mitochondria are primary targets. NPs also affect the ER, Golgi, and lysosomes, damaging neurons, glia, and endothelium. These findings suggest mitochondrial disruption from pollution contributes to early neurodegeneration [175].
Short, repeated exposure to PM2.5 and heat stress, two key climate change-related stressors, disrupt brain energy metabolism, mitochondrial function, and hippocampal synaptic activity in a mouse model of early-onset AD. It also alters gut permeability and immune signaling, suggesting that climate stress may impair the gut-brain axis and promote neuroinflammation, without immediate cognitive decline [176].
4.2. Activated Microglia and Alzheimer Disease
Glial cells, once considered mere support for neurons, have emerged as key players in brain function across vertebrates. Glial cells include ependymal cells, astrocytes, oligodendrocytes, and microglia [177].
Astrocytes
Astrocytes, the most abundant glial cells in the brain, are essential for synapse formation, neurotransmitter regulation, energy metabolism, and trophic support. They facilitate amyloid-β (Aβ) degradation and clearance through protease production and the expression of receptors, transporters, and gliotransmitters that mediate intercellular signaling. In AD, the Aβ accumulation induces astrocyte polarization toward the neurotoxic A1 phenotype, leading to elevated cytokine release and mitochondrial ROS production. These changes impair gliotransmission, glutamate homeostasis, AMPA receptor trafficking, and synaptic plasticity, processes critical for learning and memory [178].
Astrocytes also preserve neuronal homeostasis and BBB integrity. However, exposure to neurotoxicants such as pesticides and heavy metals promotes astrocyte senescence via oxidative stress, DNA damage, and NF-κB activation, driving inflammation, synaptic dysfunction, and neurodegeneration [179].
Moreover, dopamine significantly modulates astrocyte physiology beyond its classical neuronal functions. Disrupted dopaminergic signaling contributes to neurodegenerative conditions like PD by promoting reactive astrogliosis, glutamate imbalance, and neuroinflammation following dopamine depletion [180].
The brain’s high metabolic demand requires tight iron regulation. Both iron deficiency and excess impair function and are linked to neurological disorders. Astrocytes sense iron levels and signal brain microvascular endothelial cells to regulate iron import. Ceruloplasmin (Cp) upregulation and ferritin light chain 1 (Ftl1) downregulation suggest astrocytes promote iron uptake during deficiency, possibly via paracrine signaling involving CD9 and RACK1 [181].
Elevated iron levels, as seen in neuroferritinopathy, drive astrocyte reactivity, inflammation (↑IL-6, IL-1β, glutamate), and ferroptosis. iPSC-derived astrocytes showed iron dysregulation, altered morphology, and gene expression. While inflammation decreased over time, senescence and ferroptosis increased, underscoring iron’s role in astrocytic dysfunction and neurodegenerative disease progression [182].
Astrocytes, which regulate brain Cu homeostasis, are central to this process. Their dysfunction impairs synaptic signaling and promotes AD progression [156,183,184]. Notably, decreased Cu in postmortem AD brains, but increased Cu and non-ceruloplasmin-bound Cu (non-Cp Cu) is associated with a 3–4-fold greater risk of AD despite unchanged total ceruloplasmin levels [185].
Microglia
Microglia, the immune cells of the CNS, play a dual role in AD, contributing to both neuroprotection and pathology. Their activation states (M1/M2), Aβ and tau interactions, and genetic factors like Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) influence disease progression [186]. Recent studies have indicated that microglial efferocytosis is an important mechanism for clearing apoptotic cells and cellular debris, facilitating the resolution of neuroinflammation [187].
Activated microglia exhibits a spectrum of phenotypes broadly categorized as pro-inflammatory (M1) or anti-inflammatory (M2), though significant overlap exists between these states [188].
In AD, microglia play a central role by responding to amyloid-beta and hyperphosphorylated tau accumulation through inflammatory activation. M1 microglia secrete cytokines such as IL-1β, IL-6, and TNF-α, promoting neurodegeneration. Dysregulated pathways in AD such as MyD88, CSF1R, and DOCK2 further impair microglial function. Although their precise causal role remains unresolved, microglia-driven neuroinflammation is strongly associated with AD pathology [189].
Aβ oligomers trigger microglial activation, leading to the release of pro-inflammatory cytokines, which further exacerbate neuroinflammation and neuronal damage. Importantly, the presence of activated microglia surrounding amyloid plaques is correlated with heightened production of cytokines such as interleukin (IL)-1β and tumor necrosis factor-alpha (TNF-α), creating a vicious cycle of inflammation. While microglia play a protective role by clearing Aβ plaques during the early stages of AD, their chronic activation can lead to detrimental outcomes, including enhanced tau pathology and neuronal apoptosis [190].
LR4-Lyn interaction drives neuroinflammation in AD; inhibiting Lyn enhances Aβ clearance, reduces neuronal damage, and boosts protective microglial responses [191].
Exposure to Mn and α-synuclein fibrils upregulates the m6A methyltransferase METTL3 in microglia and astrocytes, enhancing m6A RNA methylation and a proinflammatory secretome. This mechanism may link environmental neurotoxins to chronic neuroinflammation and epitranscriptomic dysregulation in PD and AD. [192].
The pathogenesis of PD is thought to involve microglia-mediated neuroinflammatory injury, with mitochondrial dysfunction playing a role in aberrant microglial activation [157]. In vivo and in vitro experiments showed that excessive Mn exposure resulted in microglial mitochondrial dysfunction, manifested by increased mitochondrial ROS, decreased mitochondrial mass, and membrane potential. Additionally, with the escalating Mn dose, PINK1/Parkin-mediated mitophagy changed from activation to suppression [157].
Chronic Mn exposure causes Parkinson-like manganism. Estradiol (E2) protects microglia by downregulating specific protein 1 (Sp1), reducing leucine-rich repeat kinase 2 (LRRK2) expression. Sp1 mediates Mn-induced toxicity, while E2 promotes Sp1 degradation via ubiquitin and SUMOylation pathways [193].
Oligodendrocytes
Oligodendrocytes and their precursors are involved in Aβ generation and myelin homeostasis, and their disturbance is responsible for white matter lesions and cognitive impairment [194].
Air pollution affects human health and may disrupt brain maturation, including axon myelination, critical for efficient neural signaling.
The impact of prenatal and long-term exposure to air pollutants (PM and NO₂) on cortical myelination was evaluated in children from the ABCD (USA) and NeuroSmog (Poland) studies. No consistent associations were identified between pollution and T1w/T2w ratios. Subtle regional effects and methodological limitations indicate the need for further investigation across developmental stages [195].
Ambient exposure to PM2.5 and NO₂ at ages 9–10 was significantly associated with alterations in white matter microstructure, including key tracts related to the prefrontal cortex, suggesting disrupted neural conduction. Although no consistent links with emotional problems were found, these findings underscore the potential neurotoxic effects of low-level air pollution on brain connectivity during early adolescence [196].
Nanoscale particulate matter (nPM ≤200 nm), a key air pollutant, induces white matter toxicity. In mice, nPM reduced mature oligodendrocytes and increased precursors, impairing myelination. Combined with cerebral hypoperfusion, nPM triggered a 700% rise in apoptosis, highlighting oligodendrocytes' selective vulnerability to airborne particles [197].
Myelin basic protein (MBP) was decreased (corpus callosum: 28%, external capsule: 29%), and degraded MPB increased (corpus callosum: 32%, external capsule: 53%) in the diesel exhaust particulate (DEP) group. White matter is highly susceptible to chronic DEP exposure [198]. In Figure 5 are shown three kind of glial Cells and their association with AD.
Synapsis and AD
Lipids are essential for synapse formation, neurotransmitter release, and signal transmission in the central nervous system. They regulate membrane structure, vesicle trafficking, and glial-neuronal interactions. Dysregulated lipid metabolism contributes to synaptic dysfunction in neurodegenerative diseases like Alzheimer's and Parkinson’s. Targeting lipid pathways offer promising therapeutic strategies, but mechanisms remain under investigation [199,200].
Synaptic dysfunction is a common feature across a broad spectrum of brain diseases, spanning from psychopathologies such as posttraumatic stress disorder and substance use disorders to neurodegenerative diseases such as AD and PD. Changes in the structural interaction between astrocytes and synapses not only play a pivotal role in modulating synaptic function and behavioral states but also are implicated in the initiation and progression of various brain diseases [201].
Emerging evidence highlights synaptic dysfunction as a central feature of AD. The pathological effects of amyloid β oligomers (Aβo) may be mediated through the metabotropic glutamate receptor subtype 5 (mGluR5), leading to synaptic loss in AD. mGluR5 reduction in AD is closely linked to synaptic loss [202].
This study identifies the Wnt pathway antagonist Dickkopf-3 (DKK3) as a key contributor to synapse loss in AD. Elevated DKK3 levels were found in human AD brains and mouse models, where it disrupted excitatory synapses via Wnt/GSK3β inhibition and enhanced inhibitory synapses through Wnt/JNK activation. Notably, reducing DKK3 restored synapse number and memory, underscoring its role in synaptic degeneration and cognitive decline in AD [203].
AD is pathologically featured by the aberrant accumulation of amyloid-β plaques, neurofibrillary tangles formed by hyperphosphorylated tau, synaptic loss, and dysfunction of neurotransmitter systems. Evidence from in vivo and autopsy studies has consistently shown that synaptic dysfunction and loss are strongly correlated with cognitive decline in AD, particularly in brain regions such as the hippocampus and cortex, which are critical for memory formation and processing [204].
This study revealed a 49% reduction in the levels of SV2A in the middle frontal gyrus in early-onset AD, a decrease that is more pronounced than that in late-onset AD [205]. Cholinergic and glutamatergic systems are critical for learning and memory and are impaired early in AD and are targets for current symptomatic treatments. The cholinergic system is characterized by a reduction in acetylcholinesterase activity, nicotinic receptors, and degeneration of basal forebrain cholinergic neurons. Glutamate excitotoxicity, particularly through the activation of N-methyl-D-aspartate receptors, contributes to synaptic damage and neuronal loss [206].
Positron emission tomography (PET) imaging with ligands for synaptic vesicle glycoprotein 2A (SV2A) has emerged as a promising methodology for measuring synaptic density in AD. Synaptic vesicle glycoprotein 2A (SV2A) measured by a novel cerebrospinal fluid (CSF) enzyme-linked immunosorbent assay (ELISA) was lower in participants with symptomatic Alzheimer's disease (AD). CSF SV2A was highly correlated with SV2A measured by positron emission tomography (PET) in participants with AD [207].
5. Public Health Implications
Growing evidence shows that PM2.5 exposure plays a key role in neurological disorders, especially dementia and AD. It activates glial cells, induces neuroinflammation, increases ROS production, and leads to neuronal apoptosis, synaptic changes, and elevated AD biomarkers such as amyloid-beta and phosphorylated tau. PM2.5 also raises enzymes linked to the amyloidogenic pathway. Clinical and animal studies confirm their association with dementia and AD, with increased biomarkers in cerebrospinal fluid, vascular injury indicators, and BBB disruption in at-risk individuals [208].
Air pollution has serious consequences for human health and the environment. However, it can be controlled and reduced through public action and policies. Industry, transportation, and the burning of solid waste are major sources of air pollution that can be managed and reduced. Solutions are required at both the individual and collective levels.
The regulation of metals in the air and epidemiological surveillance are crucial to protecting public health, as exposure to these pollutants can cause serious health problems. Epidemiological surveillance identifies exposure patterns and their health effects, while regulation sets pollution limits and promotes preventive measures.
Achieving stronger air quality targets has the potential to reduce population-level dementia risk. Neighborhood (i.e., greenness and chronic noise) and occupational (i.e., shift work) characteristics are associated with dementia and are viable public health intervention points [209].
The accumulating evidence linking air pollution, particularly fine and ultrafine metal-rich particles to AD, underscores an urgent need for public health action. First, air pollution must be formally recognized as a modifiable environmental risk factor for dementia, warranting inclusion in national and global dementia prevention frameworks such as the WHO’s public health approach to dementia.
Primary prevention strategies should focus on enforcing stricter air quality standards, particularly targeting emissions of neurotoxic metals like iron, copper, and manganese. Regulatory interventions are especially critical in low- and middle-income countries (LMICs), where weak environmental protections and high urban pollution levels contribute disproportionately to AD risk and health inequities. Urban planning policies that promote green infrastructure, reduce traffic emissions, and mitigate heat and noise exposure are also essential to reduce cumulative environmental insults to brain health.
From a clinical and surveillance perspective, there is an emerging need to integrate environmental exposure assessments into AD risk screening. This includes the development and deployment of non-invasive biomarkers of exposure and early neurodegenerative change, especially in high-risk populations such as older adults, women, and socioeconomically disadvantaged groups. Public health systems should also support longitudinal monitoring of pollution exposure across the life course, including prenatal and developmental windows, to inform precision prevention efforts.
Public health programs must further address sex-specific vulnerabilities by promoting research into hormonal and immune differences that affect neuroinflammatory responses and metal homeostasis. Tailored prevention and treatment strategies, considering sex, age, and cumulative exposure profiles, are key to effective intervention.
Lastly, health equity must be at the center of AD prevention. Environmental justice frameworks should guide resource allocation, policy enforcement, and community-level interventions in underserved areas most burdened by pollution. This includes investments in public education, pollution monitoring infrastructure, and accessible healthcare services for early detection and management of dementia-related conditions.
In summary, the intersection between environmental exposure and neurodegenerative disease demands a transdisciplinary public health response integrating environmental science, neuroscience, urban policy, and equity-focused health planning to mitigate the growing burden of AD worldwide.
6. Conclusions and Future Perspectives
Conclusions
This article reviews the progress of research into the association between air pollution and AD. AD, the most common form of dementia, arises from a multifactorial interplay of genetic, hormonal, environmental, and socioeconomic factors. Among these, air pollution particularly fine (PM2.5) and ultrafine particles enriched with metals like iron, copper, and manganese have emerged as a critical and modifiable risk factor. These airborne pollutants contribute to neurodegeneration through converging mechanisms, including oxidative stress, neuroinflammation, mitochondrial dysfunction, BBB disruption, protein aggregation, and epigenetic alterations.
Disruption of brain metal homeostasis, especially involving Fe, Cu, Mn, and Zn, plays a pivotal role in AD pathogenesis by promoting ferroptosis, cuproptosis, and synaptic dysfunction. These metals often accumulate in key brain regions (hippocampus, corpus callosum, frontal cortex), where they impair glial function, particularly in astrocytes, microglia, and oligodendrocytes, affecting synaptic plasticity and myelination.
Furthermore, sex-specific biological differences, including hormonal changes and differential glial responses, contribute to the higher susceptibility observed in women. Disparities in disease burden are also evident among socioeconomically disadvantaged and racially marginalized populations, particularly in regions with high environmental pollution and weak regulatory controls.
Overall, current evidence underscores the urgent need for precision public health strategies that integrate environmental protection, metal homeostasis monitoring, and targeted interventions to mitigate the growing global impact of AD.
Future Perspectives
Advancing our understanding of how air pollution contributes to AD requires deeper exploration of the cellular and molecular mechanisms underlying neurotoxicity. Particular attention should be given to the roles of metal-rich particulate matter, especially iron and manganese, which contribute to oxidative stress, ferroptosis, neuroinflammation, and glial dysfunction. Key areas of mechanistic research include iron homeostasis, microglial polarization, astrocytic TNFR2 signaling, and transporters such as the SLC39A/ZIP family, which regulate metal uptake and detoxification in neural tissue.
Emerging evidence suggests that biological sex and developmental stage strongly modulate brain vulnerability to air pollution. Future studies should investigate sex-specific immune responses, hormonal regulation, and their impact on metal metabolism and synaptic plasticity, particularly during critical windows of brain development and aging. Understanding these interactions will enable the design of personalized prevention and treatment strategies based on sex and life course exposure.
To detect early pollution-related neurodegenerative changes, there is a pressing need for the development of sensitive and accessible biomarkers, including epigenetic signatures, extracellular vesicle content, iron-sensitive imaging modalities, and inflammatory markers such as IL-6 and IL-10. Coupling these tools with advanced neuroimaging and CSF biomarker platforms may improve early diagnosis and disease monitoring, especially in high-risk and underserved populations.
Epidemiological research should move toward multi-exposure and longitudinal models that account for combined effects of airborne pollutants, noise, heat, artificial light, and socioeconomic stressors. Integrating environmental, genetic, and epigenetic data will improve risk stratification and guide targeted interventions. A life-course approach, beginning from prenatal stages and continuing through old age, is critical to identify windows of heightened vulnerability.
At the population level, environmental policy and urban planning must play a central role. Stricter regulation of airborne metal emissions and enforcement of air quality standards, especially in low- and middle-income countries, are necessary to curb rising pollution-related AD cases. Expanding green spaces and promoting climate-resilient infrastructure can also reduce cumulative environmental exposure.
Finally, therapeutic innovation should focus on chelation strategies that restore metal balance without disrupting physiological functions, as well as lifestyle, dietary, and pharmacological interventions that modulate inflammation and oxidative stress. Integrating these with broader environmental justice frameworks will ensure equitable prevention and care for marginalized populations disproportionately affected by neurotoxic exposures.
Author Contributions
Conceptualization, investigation, methodology and writing original draft preparation, review and editing: GFG. and COA. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Our data does not include human data.
Data Availability Statement
The authors permit them to share all the data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, L.; Lin, Y.; Yang, Z.; Zhang, K.; Gong, H.; Zheng, Y.; Wang, B.; Zhang, X.; Sun, M. Microglia-derived extracellular vesicles mediate fine particulate matter-induced Alzheimer's disease-like behaviors through the mir-34a-5p/dusp10/p-p38 mapk pathway. J Hazard Mater 2025, 495, 138853. [Google Scholar] [CrossRef]
- Renesteen, E.; Boyajian, J.; Islam, P.; Kassab, A.; Abosalha, A.; Makhlouf, S.; Santos, M.; Chen, H.; Shum-Tim, C.; Prakash, S. Microbiome engineering for biotherapeutic in Alzheimer's disease through the gut-brain axis: potentials and limitations. Int J Mol Sci 2025, 26, 5351. [Google Scholar] [CrossRef]
- Timalsina, D.; Abichandani, L.; Ambad, R. A review article on oxidative stress markers f2-isoprostanes and presenilin-1 in Alzheimer's disease. J Pharm Bioallied Sci 2025, 17(Suppl 1), S109–S112. [Google Scholar] [CrossRef]
- Marogianni, C.; Siokas, V.; Dardiotis, E. Recent Advances in the Detection and Management of Motor Dysfunction in Alzheimer's Disease. Int. J. Mol. Sci. 2025, 36, 97–100. [Google Scholar] [CrossRef]
- Blennow, K.; Hansson, O. . Blodtest – fönster till hjärnan vid Alzheimers sjukdom [blood biomarkers open a window to brain pathophysiology in Alzheimer's disease]. Lakartidningen 2024, 121, 23150. [Google Scholar]
- Ashton, N.J.; Benedet, A.L.; Molfetta, G.D.; Pola, I.; Anastasi, F.; Fernández-Lebrero, A.; Puig-Pijoan, A.; Keshavan, A.; Schott, J.; Tan, K.; et al. Biomarker discovery in Alzheimer's and neurodegenerative diseases using Nucleic Acid Linked Immuno-Sandwich Assay. Alzheimers Dement. 2025, 21, e14621. [Google Scholar] [CrossRef]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.; Wu, Y.; Prina, M. World Alzheimer report 2015 - the global impact of dementia: an analysis of prevalence, incidence, cost and trends. London: Alzheimer's Disease International; 2015, 84. A: London.
- Manning, E.; Barnes, J.; Cash, D.; Bartlett, J.; Leung, K.; Ourselin, S.; Fox, N. Apoe ε4 is associated with disproportionate progressive hippocampal atrophy in ad. PLoS One 2014, 9, e97608. [Google Scholar] [CrossRef]
- Shen, S.; Zhou, W.; Chen, X.; Zhang, J. Sex differences in the association of APOE ε4 genotype with longitudinal hippocampal atrophy in cognitively normal older people. Eur J Neurol 2019, 26, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Donkor, D.; Marfo, E.; Bockarie, A.; Tettevi, E.; Antwi, M.; Dogah, J.; Osei, G.; Simpong, D. Genetic and environmental risk factors for dementia in African adults: a systematic review. Alzheimers Dement 2025, 21, e70220. [Google Scholar] [CrossRef] [PubMed]
- Essone, P.; Adegbite, B.; Mbadinga, M.; Mbouna, A.; Lotola-Mougeni, F.; Alabi, A.; Edoa, J.; Lell, B.; Alabi, A.; Adegnika, A.; et al. Creatine kinase-(mb) and hepcidin as candidate biomarkers for early diagnosis of pulmonary tuberculosis: a proof-of-concept study in Lambaréné, Gabon. Infection 2022, 50, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Mpaka-Mbatha, M.N.; Naidoo, P.; Islam, M.M.; Singh, R.; Mkhize-Kwitshana, Z.L. Anaemia and nutritional status during HIV and helminth coinfection among adults in South Africa. Nutrients 2022, 14, 4970. [Google Scholar] [CrossRef]
- Milicic, A.; Wilson, S.; Javandel, S.; Allen, I.; Tsoy, E.; Ndhlovu, L.; Kibuuka, H.; Semwogerere, M.; Langat, R.; Daud, I.; et al. Plasma inflammatory biomarkers link to worse cognition among Africans with HIV. J Acquir Immune Defic Syndr 2025. [CrossRef]
- Obeagu, E.I. Iron homeostasis and health: understanding its role beyond blood health—a narrative review. Ann. Med. Surg. (Lond.) 2025, 87, 3362–3371. [Google Scholar] [CrossRef]
- Zou, Z.; Shen, Q.; Pang, Y.; Li, X.; Chen, Y.; Wang, X.; et al. The synthesized transporter K16APoE enabled the therapeutic HAYED peptide to cross the blood–brain barrier and remove excess iron and radicals in the brain, thus easing Alzheimer's disease. Drug Deliv. Transl. Res. 2019, 9, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Milman, N. Dietary iron intakes in men in Europe are distinctly above the recommendations: a review of 39 national studies from 20 countries in the period 1995 - 2016. Gastroenterology Res 2020, 13, 233–245. [Google Scholar] [CrossRef]
- Turck, D.; Bohn, T.; Castenmiller, J.; de Henauw, S.; Hirsch-Ernst, K.I.; Knutsen, H.K.; et al. Scientific opinion on the tolerable upper intake level for iron. EFSA J. 2024, 22, e8819. [Google Scholar] [CrossRef]
- Habudele, Z.; Chen, G.; Qian, S.; Vaughn, M.; Zhang, J.; Lin, H. High dietary intake of iron might be harmful to atrial fibrillation and modified by genetic diversity: a prospective cohort study. Nutrients 2024, 16, 593. [Google Scholar] [CrossRef]
- Liu, Y.; Li, G.; Lu, F.; Guo, Z.; Cai, S.; Huo, T. Excess iron intake induced liver injury: the role of gut-liver axis and therapeutic potential. Biomed Pharmacother 2023, 168, 115728. [Google Scholar] [CrossRef]
- Wei, M.; Feng, D.; Zhang, Y.; Zuo, Y.; Li, J.; Wang, L.; Hu, P. Effect and correlation of rosa roxburghii tratt juice fermented by lactobacillus paracasei sr10-1 on oxidative stress and gut microflora dysbiosis in streptozotocin (stz)-induced type 2 diabetes mellitus mice. Foods 2023, 12, 3233. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, B.; Biswas, P.; Gulati, M.; Rani, P.; Gupta, R. Gut microbiome and Alzheimer's disease: what we know and what remains to be explored. Ageing Res Rev 2024, 102, 102570. [Google Scholar] [CrossRef] [PubMed]
- Wingo, A.; Vattathil, S.; Liu, J.; Fan, W.; Cutler, D.; Levey, A.; Schneider, J.; Bennett, D.; Wingo, T. LDL cholesterol is associated with higher ad neuropathology burden independent of APOE. J Neurol Neurosurg Psychiatry 2022, 93, 930. [Google Scholar] [CrossRef]
- Maret, W. The Metals in the Biological Periodic System of the Elements: Concepts and Conjectures. Int. J. Mol. Sci. 2016, 17, 66. [Google Scholar] [CrossRef]
- Iyengar, V.; Pullakhandam, R.; Nair, K.M. Coordinate Expression and Localization of Iron and Zinc Transporters Explain Iron-Zinc Interactions during Uptake in Caco-2 Cells: Implications for Iron Uptake at the Enterocyte. J. Nutr. Biochem. 2012, 23, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Kondaiah, P.; Yaduvanshi, P.S.; Sharp, P.A.; Pullakhandam, R. Iron and Zinc Homeostasis and Interactions: Does Enteric Zinc Excretion Cross-Talk with Intestinal Iron Absorption? Nutrients 2019, 11, 1885. [Google Scholar] [CrossRef] [PubMed]
- Maares, M.; Einhorn, V.; Behrendt, J.; Marczynski, M.; Schüßler, C.; Lieleg, O.; Haase, H. Investigation of competitive binding of the essential trace elements zinc, iron, copper, and manganese by gastrointestinal mucins and the effect on their absorption in vitro. J Nutr Biochem 2025, 144, 109983. [Google Scholar] [CrossRef]
- Noordine, M.L.; Seyoum, Y.; Bruneau, A.; Baye, K.; Lefebvre, T.; Cherbuy, C.; Canonne-Hergaux, F.; Nicolas, G.; Humblot, C.; Thomas, M. The Microbiota and the Host Organism Switch between Cooperation and Competition Based on Dietary Iron Levels. Gut Microbes 2024, 16, 2361660. [Google Scholar] [CrossRef]
- Fischer, P.W.; Giroux, A.; L’Abbé, M.R. Effect of zinc supplementation on copper status in adult man. Am. J. Clin. Nutr. 1984, 40, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A Role for Divalent Metal Transporter (DMT1) in Mitochondrial Uptake of Iron and Manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef]
- Stiles, L.I.; Ferrao, K.; Mehta, K.J. Role of Zinc in Health and Disease. Clin. Exp. Med. 2024, 24, 38. [Google Scholar] [CrossRef]
- Somsuan, K.; Phuapittayalert, L.; Srithongchai, Y.; Sonthi, P.; Supanpaiboon, W.; Hipkaeo, W.; Sakulsak, N. Increased DMT-1 Expression in Placentas of Women Living in High-Cd-Contaminated Areas of Thailand. Environ. Sci. Pollut. Res. Int. 2019, 26, 141–151. [Google Scholar] [CrossRef]
- McCabe, S.M.; Zhao, N. Expression of Manganese Transporters ZIP8, ZIP14, and ZnT10 in Brain Barrier Tissues. Int. J. Mol. Sci. 2024, 25, 10342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steimle, B.L.; Smith, F.M.; Kosman, D.J. The Solute Carriers ZIP8 and ZIP14 Regulate Manganese Accumulation in Brain Microvascular Endothelial Cells and Control Brain Manganese Levels. J. Biol. Chem. 2019, 294, 19197–19208. [Google Scholar] [CrossRef]
- Kosman, D.J. A Holistic View of Mammalian (Vertebrate) Cellular Iron Uptake. Metallomics 2020, 12, 1323–1334. [Google Scholar] [CrossRef]
- Mazzocco, J.C.; Jagadapillai, R.; Gozal, E.; Kong, M.; Xu, Q.; Barnes, G.N.; Freedman, J.H. Disruption of essential metal homeostasis in the brain by cadmium and high-fat diet. Toxicol. Rep. 2020, 7, 1164–1169. [Google Scholar] [CrossRef]
- Baj, J.; Flieger, W.; Barbachowska, A.; Kowalska, B.; Flieger, M.; Forma, A.; Teresiński, G.; Portincasa, P.; Buszewicz, G.; Radzikowska-Büchner, E.; et al. Consequences of disturbing manganese homeostasis. Int J Mol Sci 2023, 24, 14959. [Google Scholar] [CrossRef]
- Ścibior, A.; Llopis, J.; Dobrakowski, P.P.; Męcik-Kronenberg, T. CNS-Related Effects Caused by Vanadium at Realistic Exposure Levels in Humans: A Comprehensive Overview Supplemented with Selected Animal Studies. Int. J. Mol. Sci. 2023, 24, 9004. [Google Scholar] [CrossRef]
- Ścibior, A.; Llopis, J.; Dobrakowski, P.P.; Męcik-Kronenberg, T. Magnesium (Mg) and Neurodegeneration: A Comprehensive Overview of Studies on Mg Levels in Biological Specimens in Humans Affected Some Neurodegenerative Disorders with an Update on Therapy and Clinical Trials Supplemented with Selected Animal Studies. Int. J. Mol. Sci. 2024, 25, 12595. [Google Scholar] [CrossRef] [PubMed]
- Azman, K.; Ismail, C.; Shafin, N.; Zakaria, R. Alzheimer's disease and inflammation research: a systematic bibliometric review and network visualization of the published literature between 2000 and 2023. Recent Adv Inflamm Allergy Drug Discov.
- Park, H.; Armstrong, M.; Gorin, F.; Lein, P. Air pollution as an environmental risk factor for Alzheimer's disease and related dementias. Med Res Arch 2024, 12, 5825. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. Airborne magnetite- and iron-rich pollution nanoparticles: potential neurotoxicants and environmental risk factors for neurodegenerative disease, including Alzheimer's disease. J Alzheimers Dis. 2019;71, 361-375. [CrossRef] [PubMed]
- Guo, C.; Wu, D.; Yang, J.; Lu, X.; Chen, X.Y.; Ma, J.; Lin, C.; Lau, A.K.H.; Jin, Y.; Li, R.; et al. Ambient air pollution and Alzheimer's disease and other dementias: a global study between 1990 and 2019. BMC Public Health 2025, 25, 371. [Google Scholar] [CrossRef] [PubMed]
- World Bank. Urgent action needed in south Asia to curb deadly air pollution. 2022. (Accessed on July 21, 2025).
- IQAir. 2023 world air quality report. 2023. Available on: (Accessed on July 21, 2025).
- U.S. Environmental protection agency. Air Quality Trends. 2023. Available online: (Accessed on July 21, 2025).
- Mollalo, A.; Grekousis, G.; Florez, H.; Neelon, B.; Lenert, L.A.; Alekseyenko, A.V. Alzheimer's disease dementia prevalence in the United States: a county-level spatial machine learning analysis. Am. J. Alzheimers Dis. Other Demen. 2025, 40, 15333175251335570. [Google Scholar] [CrossRef]
- Baez, S.; Castro-Aldrete, L.; Britton, G.; Ibañez, A.; Santuccione-Chadha, A. Enhancing brain health in the global south through sex and gender lens. Nat Ment Health 2024, 2, 1308–1317. [Google Scholar] [CrossRef]
- Huang, T.; Li, Y.; Li, J.; Sung, J.; Yim, S. Pm2.5-associated premature mortality attributable to hot-and-polluted episodes and the inequality between the global north and the global south. Geohealth 2025, 9, e2024GH001290. [Google Scholar] [CrossRef]
- George, J.; Hornburg, K.; Merrill, A.; Marvin, E.; Conrad, K.; Welle, K.; Gelein, R.; Chalupa, D.; Graham, U.; Oberdörster, G.; et al. Brain iron accumulation in neurodegenerative disorders: does air pollution play a role? part fibre toxicol. 2025, 22, 9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kagerer, S.; Vionnet, L.; van, B.J.; Meyer, R.; Gietl, A.; Pruessmann, K.; Hock, C.; Unschuld, P. Hippocampal iron patterns in aging and mild cognitive impairment. Front Aging Neurosci 2025, 17, 1598859. [Google Scholar] [CrossRef] [PubMed]
- Fano-Sizgorich, D.; Vásquez-Velásquez, C.; Ordoñez-Aquino, C.; Sánchez-Ccoyllo, O.; Tapia, V.; Gonzales, G.F. Iron trace elements concentration in PM10 and Alzheimer's disease in Lima, Peru: ecological study. Biomedicines 2024, 12, 2043. [Google Scholar] [CrossRef]
- Pirozzi, M.A.; Canale, F.; Di Nardo, F.; Sharbafshaaer, M.; Passaniti, C.; Siciliano, M.; Cirillo, M.; Tessitore, A.; Tedeschi, G.; Esposito, F.; et al. Quantitative susceptibility mapping for investigating brain iron deposits in amyotrophic lateral sclerosis: correlations with clinical phenotype and disease progression. Amyotroph. Lateral Scler. Frontotemporal Degener. 2025, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, S.; Mohammadi, S.; Ahmadzadeh, A.; Darmiani, K.; Arab, B.M.; Jashirenezhad, N.; Helfi, M.; Alibabaei, S.; Azadi, S.; Heidary, S.; et al. Thalamic magnetic susceptibility (χ) alterations in neurodegenerative diseases: a systematic review and meta-analysis of quantitative susceptibility mapping studies. J Magn Reson Imaging 2025, 62, 271–294. [Google Scholar] [CrossRef]
- De Souza Lima, R.; Fornaguera, J. Sex differences in 6-ohda lesioned rats in a preclinical model for parkinson's disease. Behav Brain Res 2025, 490, 115642. [Google Scholar] [CrossRef]
- Hanff, A.; McCrum, C.; Rauschenberger, A.; Aguayo, G.; Pauly, C.; Jónsdóttir, S.; Tsurkalenko, O.; Zeegers, M.; Leist, A.; Krüger, R. Sex-specific progression of Parkinson's disease: a longitudinal mixed-models analysis. J Parkinsons Dis 1877718X251339201, 2025. [CrossRef]
- Abzhandadze, T.; Hoang, M.; Raaschou, P.; Norgren, J.; Mo, M.; Molnar, C.; Xu, H.; Kananen, L.; Åkerman, M.; Religa, D.; et al. Temporal trends in sex differences in dementia care- results from the Swedish registry for cognitive/dementia disorders, SveDem. Sci Rep 2025, 15, 18890. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Valencia, M.; Rojas-Lemus, M.; López-Valdez, N.; González-Villalva, A.; Fortoul, T. Genotoxic and cytotoxic effect of vanadium inhalation and oral administration of hintonia latiflora methanolic extract in mice. J Trace Elem Med Biol 2025, 89, 127677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xie, J.; Zhang, L.; Zhen, Y.J.; Wang, Y.; Li, X. Traffic-related pm2.5 pollution in hong kong: component-specific and source-resolved health risks and cytotoxicity. Environ Pollut 2025, 380, 126525. [Google Scholar] [CrossRef]
- Yan, J.; Li, Z.; Wang, K.; Xie, C.; Zhu, J.; Wu, S. Association between ambient fine particulate matter constituents and mortality and morbidity of cardiovascular and respiratory diseases: a systematic review and meta-analysis. Environ Pollut 2025, 379, 126476. [Google Scholar] [CrossRef]
- Guo, J.; Chai, G.; Song, X.; Hui, X.; Li, Z.; Feng, X.; Yang, K. Long-term exposure to particulate matter on cardiovascular and respiratory diseases in low- and middle-income countries: a systematic review and meta-analysis. Front Public Health 2023, 11, 1134341. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zou, S.; Hou, D.; An, Q.; Wang, P.; Wu, Y.; Zhang, R.; Huang, J.; Xue, J.; Gu, L. Characteristics and health risks of trace metals in pm2.5 before and during the heating period over three years in shijiazhuang, china. Toxics 2025, 13, 291. [Google Scholar] [CrossRef]
- Cristaldi, A.; Fiore, M.; Oliveri Conti, G.; Pulvirenti, E.; Favara, C.; Grasso, A.; Copat, C.; Ferrante, M. Possible association between PM2.5 and neurodegenerative diseases: A systematic review. Environ. Res. 2022, 208, 112581. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Tanaka, M.; Nezu, N.; Ishihara, N.; Oguro, A.; Vogel, C.F.A. Pathways to the Brain: Impact of Fine Particulate Matter Components on the Central Nervous System. Antioxidants 2025, 14, 730. [Google Scholar] [CrossRef]
- Miyakawa, T.; Ito, A.; Zhu, C.; Shimizu, A.; Matsumoto, E.; Kanaya, Y. Trace elements in PM2.5 aerosols in East Asian outflow in the spring of 2018: emission, transport, and source apportionment, Atmos. Chem. Phys.., 23, 14609–14626. 2023. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeon, J.I.; Jung, J.Y.; Yoon, S.W.; Lee, C.M. PM2.5 and heavy metals in urban and agro-industrial areas: health risk assessment considerations. Asian J. Atmos. Environ. 2024, 18, 16. [Google Scholar] [CrossRef]
- Mbazima, S.J.; Masekameni, M.D.; Nelson, G. Physicochemical Properties of Indoor and Outdoor Particulate Matter 2.5 in Selected Residential Areas near a Ferromanganese Smelter. Int. J. Environ. Res. Public Health 2021, 18, 8900. [Google Scholar] [CrossRef]
- Ordoñez-Aquino, C.; Gonzales-, A.C.; Gonzales, G.F. Manganeso, otro contaminante en el aire que afecta el rendimiento escolar en el Perú. Rev Soc Peru Med Interna. 2023, 36, 115–118. [Google Scholar] [CrossRef]
- Wong, M.; Ahmed, A.; Luo, W.; Bowman, A.; Tizabi, Y.; Aschner, M.; Ferrer, B. Combined manganese-iron exposure reduced oxidative stress is associated with the nrf2/nqo1 pathway in astrocytic c8-d1a cells. Biol Trace Elem Res 2025. [CrossRef]
- Liang, C.; Jiang, Y.; Zhang, T.; Ji, Y.; Zhang, Y.; Sun, Y.; Li, S.; Qi, Y.; Wang, Y.; Cai, Y.; Lai, T.; Cui, L. Atmospheric Particulate Matter Impairs Cognition by Modulating Synaptic Function via the Nose-to-Brain Route. Sci. Total Environ. 2023, 857, 159600. [Google Scholar] [CrossRef]
- Mussalo, L.; Afonin, A.M.; Zavodna, T.; Krejcik, Z.; Honkova, K.; Fayad, C.; Shahbaz, M.A.; Kalapudas, J.; Penttilä, E.; Löppönen, H.; Koivisto, A.M.; Malm, T.; Topinka, J.; Jalava, P.; Lampinen, R.; Kanninen, K.M. Traffic-Related Ultrafine Particles Influence Gene Regulation in Olfactory Mucosa Cells Altering PI3K/AKT Signaling. Environ. Int. 2025, 199, 109484. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Lugo, J.A.; Hicks, D.; Durra, S.; Massey, E.R.; Shkirkova, K.; Sauri, A.I.; Kerstiens, E.; Chen, S.; Zhao, L.; Sioutas, C.; Mack, W.J.; Finch, C.E.; Thorwald, M.A. Air Pollution Decreases Postsynaptic PSD-95 and NMDA Receptor Subunits in Synaptosomes from Mouse Cerebral Cortex. Environ. Pollut. 2025, 126845. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Lugo, J.A.; Thorwald, M.A.; Cacciottolo, M.; D'Agostino, C.; Chakhoyan, A.; Sioutas, C.; Tanzi, R.E.; Rynearson, K.D.; Finch, C.E. Air pollution amyloidogenesis is attenuated by the gamma-secretase modulator GSM-15606. Alzheimers Dement. 2024, 20, 6107–6114. [Google Scholar] [CrossRef]
- Sethi, Y.; Agarwal, P.; Vora, V.; Gosavi, S. Impact of air pollution on neurological and psychiatric health. Arch Med Res 2024, 55, 103063. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.; Oyster, E.; Luo, Y.; Wang, H. The effects of air pollution on neurological diseases: a narrative review on causes and mechanisms. Toxics 2025, 13, 207. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J.; Jia, X.; Yang, Y.; Fang, Y.; Ying, X.; Li, H.; Zhang, M.; Wei, J.; Pan, Y. Microglial Polarization in Alzheimer's Disease: Mechanisms, Implications, and Therapeutic Opportunities. J. Alzheimers Dis. 2025, 104, 3–13. [Google Scholar] [CrossRef]
- Lu, X.; Sun, W.; Leng, L.; Yang, Y.; Gong, S.; Zou, Q.; Niu, H.; Wei, C. Ultrasound Stimulation Modulates Microglia M1/M2 Polarization and Affects Hippocampal Proteomic Changes in a Mouse Model of Alzheimer's Disease. Immun. Inflamm. Dis. 2024, 12, e70061. [Google Scholar] [CrossRef]
- Wu, D.P.; Wei, Y.S.; Hou, L.X.; Du, Y.X.; Yan, Q.Q.; Liu, L.L.; Zhao, Y.D.; Yan, R.Y.; Yu, C.; Zhong, Z.G.; Huang, J.L. Circular RNA APP Contributes to Alzheimer's Disease Pathogenesis by Modulating Microglial Polarization via miR-1906/CLIC1 Axis. Alzheimers Res. Ther. 2025, 17, 44. [Google Scholar] [CrossRef]
- Shi, J.; Song, C.; Zhang, P.; Wang, J.; Huang, W.; Yu, T.; Wei, Z.; Wang, L.; Zhao, L.; Zhang, R.; Hou, L.; Zhang, Y.; Chen, H.; Wang, H. Microglial circDlg1 Modulates Neuroinflammation by Blocking PDE4B Ubiquitination-Dependent Degradation Associated with Alzheimer's Disease. Theranostics 2025, 15, 3401–3423. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, C.; Wang, Y.; Zhu, X.; Wu, L.; Chen, L.; Zhou, J.; Wang, F. METTL3/IGF2BP2/IκBα axis participates in neuroinflammation in Alzheimer's disease by regulating M1/M2 polarization of microglia. Neurochem. Int. 2025, 186, 105964. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, C.; Chen, X.; Cai, Y.; Cui, L. Interplay Between Microglia and Environmental Risk Factors in Alzheimer's Disease. Neural Regen. Res. 2024, 19, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, L.; Jiang, W.; Zhang, X.; Yao, Z. Glycolytic Dysregulation in Alzheimer's Disease: Unveiling New Avenues for Understanding Pathogenesis and Improving Therapy. Neural Regen. Res. 2025, 20, 2264–2278. [Google Scholar] [CrossRef]
- Kalenik, S.; Zaczek, A.; Rodacka, A. Air pollution-induced neurotoxicity: the relationship between air pollution, epigenetic changes, and neurological disorders. Int J Mol Sci 2025, 26, 3402. [Google Scholar] [CrossRef]
- Jiang, L.; Shao, M.; Song, C.; Zhou, L.; Nie, W.; Yu, H.; Wang, S.; Liu, Y.; Yu, L. The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment. Toxics 2025, 13, 119. [Google Scholar] [CrossRef]
- Peters, A. Ambient air pollution and Alzheimer’s disease: the role of the composition of fine particles. Proc. Natl. Acad. Sci. USA 2023, 120, e2220028120. [Google Scholar] [CrossRef]
- Hullmann, M.; Albrecht, C.; van, B.D.; Gerlofs-Nijland, M.; Wahle, T.; Boots, A.; Krutmann, J.; Cassee, F.; Bayer, T.; Schins, R. Diesel engine exhaust accelerates plaque formation in a mouse model of Alzheimer's disease. Part Fibre Toxicol 2017, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.A.; Singhvi, A.; Patil, L.; Gohari, K.; Yitshak Sade, M.; Colicino, E.; Aldridge, M.D.; Baccarelli, A.A.; Kloog, I.; Schwartz, J.; et al. A Comprehensive Systematic Review and Meta-Analysis to Unravel the Noise-Dementia Nexus. Public Health Rev. 2025, 46, 1607355. [Google Scholar] [CrossRef] [PubMed]
- Havyarimana, E.; Gong, X.; Jephcote, C.; Johnson, S.; Suri, S.; Xie, W.; Clark, C.; Hansell, A.L.; Cai, Y.S. Residential Exposure to Road and Railway Traffic Noise and Incidence of Dementia: The UK Biobank Cohort Study. Environ. Res. 2025, 279, 121787. [Google Scholar] [CrossRef]
- Bagheri, F.; Rashedi, V. Simultaneous exposure to noise and carbon monoxide increases the risk of Alzheimer's disease: a literature review. Med Gas Res 2020, 10, 85–90. [Google Scholar]
- Rentschler, K.; Kodavanti, U. Mechanistic insights regarding neuropsychiatric and neuropathologic impacts of air pollution. Crit Rev Toxicol 2024, 54, 953–980. [Google Scholar] [CrossRef]
- Sobolewski, M.; Conrad, K.; Marvin, E.; Eckard, M.; Goeke, C.; Merrill, A.; Welle, K.; Jackson, B.; Gelein, R.; Chalupa, D.; et al. . The potential involvement of inhaled iron (fe) in the neurotoxic effects of ultrafine particulate matter air pollution exposure on brain development in mice. Part Fibre Toxicol 2022, 19, 56. [Google Scholar] [CrossRef]
- Cory-Slechta, D.A.; Marvin, E.; Welle, K.; Goeke, C.; Chalupa, D.; Oberdörster, G.; Sobolewski, M. Male-Biased Vulnerability of Mouse Brain Tryptophan/Kynurenine and Glutamate Systems to Adolescent Exposures to Concentrated Ambient Ultrafine Particle Air Pollution. Neurotoxicology 2024, 104, 20–35. [Google Scholar] [CrossRef]
- Yang, H.; Liu, L.; Shu, Z.; Zhang, W.; Huang, C.; Zhu, Y.; Li, S.; Wang, W.; Li, G.; Zhang, Q.; Liu, Q.; Jiang, G. Magnetic Iron Oxide Nanoparticles: An Emerging Threat for the Environment and Human Health. J. Environ. Sci. (China) 2025, 152, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Trani, J.; Singh, R.; Walker, A.; Bekena, S.; Zhu, Y.; Williams, J.; Buckner, T.; Millsap, M.; Chen, C.; Taylor, K.; et al. . Structural and social determinants of dementia risk among adults racialized as black: results from a community-based system dynamics approach. Alzheimers Dement 2025, 21, e70494. [Google Scholar] [CrossRef]
- Sadowska, A.; Grzesiak, M. Rola steroidów płciowych w chorobach neurodegeneracyjnych [the role of steroid hormones in the neurodegenerative diseases]. Postepy Biochem 2022, 68, 387–398. [Google Scholar]
- Nicoletti, A.; Baschi, R.; Cicero, C.E.; Iacono, S.; Re, V.L.; Luca, A.; Schirò, G.; Monastero, R.; Gender Neurology Study Group of the Italian Society of Neurology. Sex and gender differences in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis: a narrative review. Mech. Ageing Dev. 2023, 212, 111821. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, Q.; Guo, J. Causal relationship between sex hormones and risk of developing common neurodegenerative diseases: a mendelian randomization study. J Integr Neurosci 2024, 23, 78. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Tang, Y. Association between levels of sex hormones and risk of multiple sclerosis: a mendelian randomization study. Acta Neurol Belg 2024, 124, 1913–1922. [Google Scholar] [CrossRef]
- Song, D.; Li, Y.; Yang, L.; Luo, Y.; Yao, X. Bridging systemic metabolic dysfunction and Alzheimer's disease: the liver interface. Mol Neurodegener 2025, 20, 61. [Google Scholar] [CrossRef]
- Huang, S.; Nunez, J.; Toresco, D.; Wen, C.; Slotabec, L.; Wang, H.; Zhang, H.; Rouhi, N.; Adenawoola, M.; Li, J. Alterations in the inflammatory homeostasis of aging-related cardiac dysfunction and alzheimer's diseases. FASEB J 2025, 39, e70303. [Google Scholar] [CrossRef]
- Cook, J.D.; Flowers, C.H.; Skikne, B.S. The quantitative assessment of body iron. Blood 2003, 101, 3359–3364. [Google Scholar] [CrossRef]
- Qin, S.J.; Zeng, Q.G.; Zeng, H.X.; Meng, W.J.; Wu, Q.Z.; Lv, Y.; Dai, J.; Dong, G.H.; Zeng, X.W. Novel Perspective on Particulate Matter and Alzheimer's Disease: Insights from Adverse Outcome Pathway Framework. Environ. Pollut. 2025, 367, 125601. [Google Scholar] [CrossRef]
- Serafin, P.; Zaremba, M.; Sulejczak, D.; Kleczkowska, P. Air pollution: a silent key driver of dementia. Biomedicines 2023, 11, 1477. [Google Scholar] [CrossRef] [PubMed]
- Alzhrani, A.; Stockfelt, L.; Xu, Y.; Harari, F.; Gustafsson, S.; Engström, G.; Hansson, O.; Oudin, A. Long-term exposure to air pollution and validated cases of Alzheimer's disease and vascular dementia in the Malmö diet and cancer cohort. J Alzheimers Dis 13872877251360225, 2025. [CrossRef]
- Shi, L.; Wu, X.; Danesh, Y.M.; Braun, D.; Abu, A.Y.; Wei, Y.; Liu, P.; Di, Q.; Wang, Y.; Schwartz, J.; et al. . Long-term effects of pm2·5 on neurological disorders in the american medicare population: a longitudinal cohort study. Lancet Planet Health 2020, 4, e557–e565. [Google Scholar] [CrossRef]
- Fu, P.; Yung, K. Air pollution and alzheimer's disease: a systematic review and meta-analysis. J Alzheimers Dis. 2020, 77, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-Year Trends of the Global Burden of Disease Attributable to Ambient Air Pollution: An Analysis of Data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Jutila, O.I.; Mullin, D.; Vieno, M.; Tomlinson, S.; Taylor, A.; Corley, J.; Deary, I.J.; Cox, S.R.; Baranyi, G.; Pearce, J.; et al. Life-course exposure to air pollution and the risk of dementia in the Lothian Birth Cohort 1936. Environ Epidemiol. 2024, 9, e355. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Tong, X.; Shen, X.; Ran, J.; Sun, S.; Yao, X.I.; Shen, C. Longitudinal Associations between Air Pollution and Incident Dementia as Mediated by MRI-Measured Brain Volumes in the UK Biobank. Environ. Int. 2025, 195, 109219. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Cejudo-Ruiz, F.R.; Stommel, E.W.; González-Maciel, A.; Reynoso-Robles, R.; Silva-Pereyra, H.G.; Pérez-Guille, B.E.; Soriano-Rosales, R.E.; Torres-Jardón, R. Sleep and Arousal Hubs and Ferromagnetic Ultrafine Particulate Matter and Nanoparticle Motion Under Electromagnetic Fields: Neurodegeneration, Sleep Disorders, Orexinergic Neurons, and Air Pollution in Young Urbanites. Toxics 2025, 13, 284. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; González-Maciel, A.; Reynoso-Robles, R.; Cejudo-Ruiz, F.R.; Silva-Pereyra, H.G.; Gorzalski, A.; Torres-Jardón, R. Alzheimer's, Parkinson's, Frontotemporal Lobar Degeneration, and Amyotrophic Lateral Sclerosis Start in Pediatric Ages: Ultrafine Particulate Matter and Industrial Nanoparticles Are Key in the Early-Onset Neurodegeneration: Time to Invest in Preventive Medicine. Toxics 2025, 13, 178. [Google Scholar] [CrossRef]
- Gunawan, C.; Fleming, C.; Irga, P.J.; Wong, R.J.; Amal, R.; Torpy, F.R.; Golzan, M.; McGrath, K.C. Neurodegenerative effects of air pollutant particles: Biological mechanisms implicated for early-onset Alzheimer's disease. Environ. Int. 2024, 185, 108512. [Google Scholar] [CrossRef]
- Trentalange, A.; Badaloni, C.; Porta, D.; Michelozzi, P.; Renzi, M. Association between Air Quality and Neurodegenerative Diseases in River Sacco Valley: A Retrospective Cohort Study in Latium, Central Italy. Int. J. Hyg. Environ. Health 2025, 267, 114578. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, H.C.; Shekha, T.A.M.; Annajigowda, H.H.; Charly, D.; Jessy, A.; Suresh, S.; Bhardwaj, S.; Anirudhan, A.; Mensegere, A.; Issac, T.G. Current Status of Research on the Modifiable Risk Factors of Dementia in India: A Scoping Review. Asian J. Psychiatr. 2025, 105, 104390. [Google Scholar] [CrossRef]
- Kwon, D.; Paul, K.C.; O'Sharkey, K.; Paik, S.A.; Yu, Y.; Bronstein, J.M.; Ritz, B. Challenges in Studying Air Pollution to Neurodegenerative Diseases. Environ. Res. 2025, 278, 121597. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Steinmetz, J.; Marsh, E.K.; Aravkin, A.Y.; Ashbaugh, C.; Murray, C.J.L.; Yang, F.; Ji, J.S.; Zheng, P.; Sorensen, R.J.D.; et al. A Systematic Review with a Burden of Proof Meta-Analysis of Health Effects of Long-Term Ambient Fine Particulate Matter (PM2.5) Exposure on Dementia. Nat. Aging 2025, 5, 897–908. [Google Scholar] [CrossRef]
- Peng, W.; Liu, T. Greenness Modified the Association of PM2.5 and Ozone with Global Disease Burden of Alzheimer's Disease and Other Dementias. Sci. Rep. 2025, 15, 26823. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Braun, D.; Wu, X.; Yitshak-Sade, M.; Blacker, D.; Kioumourtzoglou, M.A.; Schwartz, J.; Mork, D.; Dominici, F.; Zanobetti, A. The Impacts of Air Pollution on Mortality and Hospital Readmission among Medicare Beneficiaries with Alzheimer's Disease and Alzheimer's Disease-Related Dementias: A National Retrospective Cohort Study in the USA. Lancet Planet. Health 2025, 9, e114–e123. [Google Scholar] [CrossRef]
- Moorthy, S.; Dazard, J.E.; Chen, Z.; Charak, R.; Palanivel, S.; Deo, S.; Al-Kindi, S.G.; Rajagopalan, S. Non-Traditional Socio-Environmental and Geospatial Determinants of Alzheimer's Disease-Related Dementia Mortality. Sci. Total Environ. 2025, 984, 179745. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.; Yariwake, V.; Marques, K.; Veras, M.; Fajersztajn, L. Air pollution: a neglected risk factor for dementia in Latin America and the Caribbean. Front Neurol 2021, 12, 684524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Deng, Y.L.; Liu, Y.; Steenland, K. Associations between Ultrafine Particles and Incident Dementia in Older Adults. Environ. Sci. Technol. 2025, 59, 5443–5451. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ye, H.; Dong, Z.; Amujilite; Zhao, M.; Xu, Q.; Xu, J. The health and economic burden of ozone pollution on Alzheimer's disease and mild cognitive impairment in China. Environ Res 2024, 259, 119506. [CrossRef]
- Zhang, H.; Wang, Y.; Li, H.; Zhu, Q.; Ma, T.; Liu, Y.; Steenland, K. The Role of the Components of PM2.5 in the Incidence of Alzheimer's Disease and Related Disorders. Environ. Int. 2025, 200, 109539. [Google Scholar] [CrossRef]
- Tao, H.; Chen, G.; Wu, L.; Lou, H. Synergistic Impact of Air Pollution and Artificial Light at Night on Memory Disorders: A Nationwide Cohort Analysis. BMC Public Health 2025, 25, 1591. [Google Scholar] [CrossRef]
- Delaney, S.W.; Stegmuller, A.; Mork, D.; Mock, L.; Bell, M.L.; Gill, T.M.; Braun, D.; Zanobetti, A. Extreme Heat and Hospitalization among Older Persons with Alzheimer Disease and Related Dementias. JAMA Intern. Med. 2025, 185, 412–421. [Google Scholar] [CrossRef]
- Peterson, A.; Miller, E.C.; De Jager, P.L.; Avila-Rieger, J.; Arce Rentería, M.; Reed, A.; Hohman, T.J.; Jefferson, A.L.; Barnes, L.L.; Arvanitakis, Z.; et al. The Association Between Age of Menopause and Hysterectomy Status and Alzheimer's Disease Risk in a Cohort of Older White and Black Women. J Womens Health (Larchmt). 2025. [Google Scholar] [CrossRef]
- Pluta, R. Gender-Dependent Modulation of Alzheimer's Disease by Brain Ischemia. Comment on Lohkamp et al. Sex-Specific Adaptations in Alzheimer's Disease and Ischemic Stroke: A Longitudinal Study in Male and Female APPswe/PS1dE9 Mice. Life (Basel). 2025, 15, 1146. [Google Scholar] [CrossRef] [PubMed]
- Antwi, M.H.; Bockarie, A.; Osei, G.N.; Donkor, D.M.; Simpong, D.L. Cytokines and immune biomarkers in neurodegeneration and cognitive function: A systematic review among individuals of African ancestry. Alzheimers Dement. 2025, 21, e70514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Luca, A.; Luca, M.; Ferri, R.; Barbanti, M.; Malaguarnera, R.; Pecorino, B.; Scollo, P.; Serretti, A. Medical Comorbidities in Alzheimer's Disease: An Autopsy Confirmed Study with a Focus on Sex-Differences? Clin Neuropsychiatry. 2025, 22, 207–214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carney, B.N.; Illiano, P.; Pohl, T.M.; Desu, H.L.; Mini, A.; Mudalegundi, S.; Asencor, A.I.; Jwala, S.; Ascona, M.C.; Singh, P.K; et al. Astroglial TNFR2 signaling regulates hippocampal synaptic function and plasticity in a sex dependent manner. Brain Behav Immun. 2025, 129, 757–777. [Google Scholar] [CrossRef]
- Valdez, M.C.; Freeborn, D.L.; Vulimiri, P.; Valdez, J.M.; Kodavanti, U.P.; Kodavanti, P.R.S. Acute Ozone-Induced Transcriptional Changes in Markers of Oxidative Stress and Glucocorticoid Signaling in the Rat Hippocampus and Hypothalamus Are Sex-Specific. Int. J. Mol. Sci. 2023, 24, 6404. [Google Scholar] [CrossRef]
- Bush, A. The metallobiology of Alzheimer's disease. Trends Neurosci 2003, 26, 207–14. [Google Scholar] [CrossRef]
- Ramos, P.; Pinto, E.; Santos, A.; Almeida, A. Reference values for trace element levels in the human brain: a systematic review of the literature. J Trace Elem Med Biol 2021, 66, 126745. [Google Scholar] [CrossRef]
- Corbo, D.; Gasparotti, R.; Renzetti, S. Exploring the Neural Correlates of Metal Exposure in Motor Areas. Brain Sci. 2025, 15, 679. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, L.; Zhang, C.; Qin, L.; Liao, X.; Zhao, L. The Close Relationship between Trace Elements (Cu, Fe, Zn, Se, Rb, Si, Cr, and V) and Alzheimer's Disease: Research Progress and Insights. Int. J. Mol. Sci. 2025, 90, 127692. [Google Scholar] [CrossRef]
- Jomova, K.; Alomar, S.; Nepovimova, E.; Kuca, K.; Valko, M. Heavy metals: toxicity and human health effects. Arch Toxicol 2025, 99, 153–209. [Google Scholar] [CrossRef]
- Dhapola, R.; Sharma, P.; Kumari, S.; Bhatti, J.S.; HariKrishnaReddy, D. Environmental toxins and Alzheimer’s disease: a comprehensive analysis of pathogenic mechanisms and therapeutic modulation. Mol. Neurobiol. 2024, 61, 3657–3677. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Li, D.; Zhang, J.; Yin, Z.; Li, F. Scorpion venom heat-resistant synthetic peptide alleviates neuronal necroptosis in Alzheimer's disease model by regulating Lnc Gm6410 under PM2.5 exposure. Int. J. Mol. Sci. 2025, 26, 4372. [Google Scholar] [CrossRef]
- Singh, S.K.; Balendra, V.; Obaid, A.A.; Esposto, J.; Tikhonova, M.A.; Gautam, N.K.; Poeggeler, B. Copper-mediated β-amyloid toxicity and its chelation therapy in Alzheimer's disease. Metallomics 2022, 14, mfac018. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Rauf, A.; Akter, S.; Akter, H.; Al-Imran, M.I.K.; Fakir, M.N.H.; Thufa, G.K.; Islam, M.T.; Hemeg, H.A.; Abdulmonem, W.A.; et al. Neuroprotective Potential of Curcumin in Neurodegenerative Diseases: Clinical Insights into Cellular and Molecular Signaling Pathways . J. Biochem. Mol. Toxicol. 2025, 39, e70369. [Google Scholar] [CrossRef]
- Schipper, D.A.; Schipper, H.M. Neurodegeneration with Brain Iron Accumulation. Int. J. Mol. Sci. 2025, 1480, 291–309. [Google Scholar] [CrossRef]
- Wydrych, A.; Pakuła, B.; Jakubek-Olszewska, P.; Janikiewicz, J.; Dobosz, A.M.; Cudna, A.; Rydzewski, M.; Pierzynowska, K.; Gaffke, L.; Cyske, Z.; et al. Metabolic Alterations in Fibroblasts of Patients Presenting with the MPAN Subtype of Neurodegeneration with Brain Iron Accumulation (NBIA). Int. J. Mol. Sci. 2025, 1871, 167541. [Google Scholar] [CrossRef]
- Wydrych, A.; Pakuła, B.; Janikiewicz, J.; Dobosz, A.M.; Jakubek-Olszewska, P.; Skowrońska, M.; Kurkowska-Jastrzębska, I.; Cwyl, M.; Popielarz, M.; Pinton, P.; et al. Metabolic Impairments in Neurodegeneration with Brain Iron Accumulation. Int. J. Mol. Sci. 2025, 1866, 149517. [Google Scholar] [CrossRef]
- Ahmed, G.; Rahaman, M.; Perez, E.; Khan, K. Associations of environmental exposure to arsenic, manganese, lead, and cadmium with alzheimer's disease: a review of recent evidence from mechanistic studies. J Xenobiot 2025, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Zucca, F.; Duyn, J.; Crichton, R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 2014, 13, 1045–60. [Google Scholar] [CrossRef]
- Colvin, R.A.; Jin, Q.; Lai, B.; Kiedrowski, L. Visualizing metal content and intracellular distribution in primary hippocampal neurons with synchrotron X-ray fluorescence. PLoS One 2016, 11, e0159582. [Google Scholar] [CrossRef]
- Mo, Y.; Zhong, J.; Teng, M.; Peng, J.; Huang, H.; Aschner, M.; Jiang, Y. Advances in research on neurotoxicity and mechanisms of manganese, iron, and copper exposure, alone or in combination. J Appl Toxicol 2025. [Google Scholar] [CrossRef]
- Long, H.; Zhu, W.; Wei, L.; Zhao, J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm 2023, 4, e298. [Google Scholar] [CrossRef]
- Kittilukkana, A.; Carmona, A.; Normand, L.; Gibout, C.; Somogyi, A.; Pilapong, C.; Ortega, R. Storage and transport of labile iron is mediated by lysosomes in axons and dendrites of hippocampal neurons. Metallomics 2025, 17, mfaf021. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yang, L.; Li, M.; Zhang, Q.; Li, J.; Zhang, N.; Che, Y.; Chen, Y.; Liang, P.; Wang, Y.; et al. The Role of Iron Homeostasis Imbalance in T2DM-Associated Cognitive Dysfunction: A Prospective Cohort Study Utilizing Quantitative Susceptibility Mapping. Hum. Brain Mapp. 2025, 46, e70263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, Y.; Wu, J.; Wang, L.; Ji, G.; Dang, Y. Copper homeostasis and cuproptosis in health and disease. MedComm (2020). 2024, 5, e724. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alghadir, A.; Gabr, S.; Al-Eisa, E. . Assessment of the effects of glutamic acid decarboxylase antibodies and trace elements on cognitive performance in older adults. Clin Interv Aging 2015, 10, 1901–7. [Google Scholar]
- Rozumna, N.; Hanzha, V.; Shkryl, V.; Lukyanetz, E. Dantrolene protects hippocampal neurons against amyloid-β₁₋₄₂-induced calcium dysregulation and cell death. Neurochem Res 2025, 50, 198. [Google Scholar] [CrossRef]
- Xu, J.; Shen, R.; Qian, M.; Zhou, Z.; Xie, B.; Jiang, Y.; Yu, Y.; Dong, W. Ferroptosis in Alzheimer's disease: the regulatory role of glial cells. J. Integr. Neurosci. 2025, 24, 25845. [Google Scholar] [CrossRef]
- McCabe, S.M.; Zhao, N. ZIP14 deletion disrupts divalent metal homeostasis in mouse cerebrospinal fluid. Cell Biochem. Funct. 2025, 43, e70086. [Google Scholar] [CrossRef]
- Liu, H.; Chen, M.; Duan, J.; Lu, R.; Li, L. ZnTs: Key Regulators of Zn²⁺ Homeostasis in Diseases. Cell Biol. Int. 2025, 49, e70056. [Google Scholar] [CrossRef] [PubMed]
- Samokhina, E.; Mangat, A.; Malladi, C.; Gyengesi, E.; Morley, J.; Buskila, Y. Potassium homeostasis during disease progression of alzheimer's disease. J Physiol 2025, 603, 3405–3424. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gao, L.; Yang, Y.; Shi, D.; Zhang, Z.; Wang, X.; Huang, Y.; Wu, J.; Meng, J.; Li, H.; et al. Protective role of mitophagy on microglia-mediated neuroinflammatory injury through mtDNA-sting signaling in manganese-induced parkinsonism. J Neuroinflammation 2025, 22, 55. [Google Scholar] [CrossRef] [PubMed]
- Obeng, S.; Kulhánek, M.; Balík, J.; Černý, J.; Sedlář, O. Manganese: from soil to human health-a comprehensive overview of its biological and environmental significance. Nutrients 2024, 16, 3455. [Google Scholar] [CrossRef]
- Chen, P.; Chakraborty, S.; Mukhopadhyay, S.; Lee, E.; Paoliello, M.; Bowman, A.; Aschner, M. Manganese homeostasis in the nervous system. J Neurochem 2015, 134, 601–10. [Google Scholar] [CrossRef]
- Martins, A.; Oliveira-Paula, G.; Tinkov, A.; Skalny, A.; Tizabi, Y.; Bowman, A.; Aschner, M. Role of manganese in brain health and disease: focus on oxidative stress. Free Radic Biol Med 2025, 232, 306–318. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Xia, J.; Jia, J.; Zhang, S.; Wang, W.; He, W.; Song, X.; Chen, L.; Niu, P.; et al. Small extracellular vesicles-derived from 3d cultured human nasal mucosal mesenchymal stem cells during differentiation to dopaminergic progenitors promote neural damage repair via mir-494-3p after manganese exposed mice. Ecotoxicol Environ Saf 2024, 280, 116569. [Google Scholar] [CrossRef]
- Wang, C.; Lv, S.; Zhao, H.; He, G.; Liang, H.; Chen, K.; Qu, M.; He, Y.; Ou, C. Hypoxia-inducible factor-1 as targets for neuroprotection : from ferroptosis to Parkinson's disease. Neurol Sci 2025, 46, 1111–1120. [Google Scholar] [CrossRef]
- Chen, J.; Tao, Z.; Zhang, X.; Hu, J.; Wang, S.; Xing, G.; Ngeng, N.A.; Malik, A.; Appiah-Kubi, K.; Farina, M.; et al. Manganese overexposure results in ferroptosis through the HIF-1α/p53/SLC7A11 pathway in ICR mouse brain and PC12 cells. Ecotoxicol. Environ. Saf. 2024, 279, 116481. [Google Scholar] [CrossRef]
- Cheng, H.; Villahoz, B.; Ponzio, R.; Aschner, M.; Chen, P. Signaling pathways involved in manganese-induced neurotoxicity. Cells 2023, 12, 2842. [Google Scholar] [CrossRef]
- Dewey, K.; Oaks, B. U-shaped curve for risk associated with maternal hemoglobin, iron status, or iron supplementation. Am J Clin Nutr 2017, 106(Suppl 6), 1694S–1702S. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.F.; Valverde-Aparicio, V.M. Impacto de la nueva definición de anemia por parte de la Organización Mundial de la Salud: el rol en investigación de la Universidad Peruana Cayetano Heredia. Acta Herediana 2024, 67, 73–80. [Google Scholar] [CrossRef]
- Urbano, T.; Michalke, B.; Chiari, A.; Malagoli, C.; Bedin, R.; Tondelli, M.; Vinceti, M.; Filippini, T. Iron species in cerebrospinal fluid and dementia risk in subjects with mild cognitive impairment: a cohort study. Neurotoxicology 2025, 110, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted Mitochondria: Moving Mitochondrial Science Beyond Function and Dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Yokota, M. Analysis of Dopaminergic Neuron-Specific Mitochondrial Morphology and Function Using Tyrosine Hydroxylase Reporter iPSC Lines. Anat. Sci. Int. 2025, 100, 155–162. [Google Scholar] [CrossRef]
- Neikirk, K.; Harris, C.; Le, H.; Oliver, A.; Shao, B.; Liu, K.; Beasley, H.K.; Jamison, S.; Ishimwe, J.A.; Kirabo, A.; et al. Air Pollutants as Modulators of Mitochondrial Quality Control in Cardiovascular Disease. Physiol. Rep. 2024, 12, e70118. [Google Scholar] [CrossRef] [PubMed]
- Mussalo, L.; Lampinen, R.; Avesani, S.; Závodná, T.; Krejčík, Z.; Kalapudas, J.; Penttilä, E.; Löppönen, H.; Koivisto, A.M.; Malm, T.; et al. Traffic-Related Ultrafine Particles Impair Mitochondrial Functions in Human Olfactory Mucosa Cells—Implications for Alzheimer's Disease. Redox Biol. 2024, 75, 103272. [Google Scholar] [CrossRef]
- Grujicic, J.; Allen, A.R. MnSOD mimetics in therapy: exploring their role in combating oxidative stress-related diseases. Antioxidants 2024, 13, 1444. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, X.; Chen, B.; Dai, R.; Xi, Z.; Xu, H. Insights into manganese superoxide dismutase and human diseases. Int J Mol Sci 2022, 23, 15893. [Google Scholar] [CrossRef]
- Aschner, M.; Skalny, A.; Rongzhu, L.; Santamaria, A.; Lee, E.; Bowman, A.; Tizabi, Y.; Zhou, J.; Tinkov, A. The role of epigenetics in manganese neurotoxicity: an update with a focus on non-coding RNAs and histone modifications. Neurochem Res 2025, 50, 195. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Stommel, E.W.; Torres-Jardón, R.; Hernández-Luna, J.; Aiello-Mora, M.; González-Maciel, A.; Reynoso-Robles, R.; Pérez-Guillé, B.; Silva-Pereyra, H.G.; Tehuacanero-Cuapa, S.; et al. Alzheimer and Parkinson Diseases, Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Overlapping Neuropathology Start in the First Two Decades of Life in Pollution Exposed Urbanites and Brain Ultrafine Particulate Matter and Industrial Nanoparticles, Including Fe, Ti, Al, V, Ni, Hg, Co, Cu, Zn, Ag, Pt, Ce, La, Pr and W Are Key Players. Metropolitan Mexico City Health Crisis Is in Progress. Front. Hum. Neurosci. 2024, 17, 1297467. [Google Scholar] [CrossRef]
- Iban-Arias, R.; Portela, A.S.D.; Masieri, S.; Radu, A.; Yang, E.J.; Chen, L.C.; Gordon, T.; Pasinetti, G.M. Role of Acute Exposure to Environmental Stressors in the Gut-Brain-Periphery Axis in the Presence of Cognitive Resilience. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167760. [Google Scholar] [CrossRef]
- Lucini, C.; Gatta, C. Glial diversity and evolution: insights from teleost fish. Brain Sci. 2025, 15, 743. [Google Scholar] [CrossRef]
- Muñoz de León-López, C.A.; Navarro-Lobato, I.; Khan, Z.U. The role of astrocytes in synaptic dysfunction and memory deficits in Alzheimer’s disease. Biomolecules 2025, 15, 910. [Google Scholar] [CrossRef]
- Gonzalez, P.R.; Ay, M.; Langley, M.; Plunk, E.; Strazdins, R.; Abu-Salah, A.; Anchan, A.; Shah, A.; Sarkar, S. Neurotoxicants Driving Glial Aging: Role of Astrocytic Aging in Non-Cell Autonomous Neurodegeneration. Toxicol. Sci. 2025, kfaf088. [Google Scholar] [CrossRef]
- Favetta, G.; Bubacco, L. Beyond Neurons: How Does Dopamine Signaling Impact Astrocytic Functions and Pathophysiology? Prog. Neurobiol. 2025, 251, 102798. [Google Scholar] [CrossRef]
- Duhaini, M.; Shamroukh, H.S.; Zhang, Z.; Kondapalli, K.C. Astrocyte Secretome Remodeling under Iron Deficiency: Potential Implications for Brain Iron Homeostasis. Int. J. Mol. Sci. 2025, 14, bio062057. [Google Scholar] [CrossRef]
- Moro, A.S.; Balestrucci, C.; Cozzi, A.; Santambrogio, P.; Levi, S. Neuroferritinopathy Human-Induced Pluripotent Stem Cell-Derived Astrocytes Reveal an Active Role of Free Intracellular Iron in Astrocyte Reactivity. Int. J. Mol. Sci. 2025, 26, 6197. [Google Scholar] [CrossRef]
- Pal, A.; Prasad, R. Recent discoveries on the functions of astrocytes in the copper homeostasis of the brain: a brief update. Neurotox. Res. 2014, 26, 78–84. [Google Scholar] [CrossRef]
- Tan, X.; Su, X.; Wang, Y.; Liang, W.; Wang, D.; Huo, D.; Wang, H.; Qi, Y.; Zhang, W.; Han, L.; et al. Comm domain containing 4 inhibits hephaestin and ferroportin to enhance neuronal ferroptosis by disturbing the cu-fe balance in amyotrophic lateral sclerosis. Brain Res 2025, 1861, 149707. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper imbalance in Alzheimer's disease: meta-analysis of serum, plasma, and brain specimens, and replication study evaluating atp7b gene variants. Biomolecules 2021, 11, 960. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Chen, B.; Yang, H.; Zhou, X. The Dual Role of Microglia in Alzheimer’s Disease: From Immune Regulation to Pathological Progression. Front. Aging Neurosci. 2025, 17, 1554398. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kou, Y.; Ni, Y.; Yang, H.; Xu, C.; Fan, H.; Liu, H. Microglia efferocytosis: an emerging mechanism for the resolution of neuroinflammation in Alzheimer's disease. J. Neuroinflammation 2025, 22, 96. [Google Scholar] [CrossRef]
- Valiukas, Z.; Tangalakis, K.; Apostolopoulos, V.; Feehan, J. Microglial activation states and their implications for Alzheimer's disease. J. Prev. Alzheimers Dis. 2025, 12, 100013. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Kumari, S.; Dhapola, R.; Sharma, P.; Beura, S.K.; Singh, S.K.; Vellingiri, B.; HariKrishnaReddy, D. Shedding Light on Microglial Dysregulation in Alzheimer’s Disease: Exploring Molecular Mechanisms and Therapeutic Avenues. Inflammopharmacology 2025, 33, 679–702. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, R.; Shahrokhi, N.S.; Falah, T.M.; Karimi, Z.; Sadr, S.; Ramadhan, H.D.; Talebian, N.; Esmaeilpour, K. Microglial activation as a hallmark of neuroinflammation in Alzheimer's disease. Metab Brain Dis 2025, 40, 207. [Google Scholar] [CrossRef]
- Islam, R.; Choudhary, H.H.; Zhang, F.; Mehta, H.; Yoshida, J.; Thomas, A.J.; Hanafy, K. Microglial TLR4-Lyn kinase is a critical regulator of neuroinflammation, Aβ phagocytosis, neuronal damage, and cell survival in Alzheimer's disease. Sci. Rep. 2025, 15, 11368. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Ealy, A.; Gregory, A.; Janarthanam, C.; Albers, W.; Richardson, G.; Jin, H.; Zenitsky, G.; Anantharam, V.; Kanthasamy, A.; et al. Pathological α-synuclein dysregulates epitranscriptomic writer mettl3 to drive neuroinflammation in microglia. Cell Rep 2025, 44, 115618. [Google Scholar] [CrossRef] [PubMed]
- Nyarko-Danquah, I.; Pajarillo, E.; Kim, S.; Digman, A.; Multani, H.; Ajayi, I.; Son, D.; Aschner, M.; Lee, E. Microglial sp1 induced lrrk2 upregulation in response to manganese exposure, and 17β-estradiol afforded protection against this manganese toxicity. Neurotoxicology 2024, 103, 105–114. [Google Scholar] [CrossRef]
- Sil, M.; Mukherjee, N.; Chatterjee, I.; Ghosh, A.; Goswami, A. Glial cells in Alzheimer's disease: pathogenic mechanisms and therapeutic frontiers. J. Mol. Neurosci. 2025, 75, 87. [Google Scholar] [CrossRef]
- Szwed, M.; de Jesus, A.V.; Kossowski, B.; Ahmadi, H.; Rutkowska, E.; Mysak, Y.; Baumbach, C.; Kaczmarek-Majer, K.; Degórska, A.; Skotak, K.; et al. Air pollution and cortical myelin T1w/T2w ratio estimates in school-age children from the ABCD and NeuroSmog studies. Dev. Cogn. Neurosci. 2025, 73, 101538. [Google Scholar] [CrossRef]
- Herting, M.M.; Burnor, E.; Ahmadi, H.; Eckel, S.P.; Gauderman, W.; Schwartz, J.; Berhane, K.; McConnell, R.; Chen, J.C. Air pollution exposure, prefrontal connectivity, and emotional behavior in early adolescence. Res. Rep. Health Eff. Inst. 2025, 2025, 1–56. [Google Scholar] [PubMed] [PubMed Central]
- Lamorie-Foote, K.; Liu, Q.; Shkirkova, K.; Ge, B.; He, S.; Morgan, T.E.; Mack, W.J.; Sioutas, C.; Finch, C.E.; Mack, W.J. Particulate matter exposure and chronic cerebral hypoperfusion promote oxidative stress and induce neuronal and oligodendrocyte apoptosis in male mice. J. Neurosci. Res. 2023, 101, 384–402. [Google Scholar] [CrossRef]
- Chakhoyan, A.; Shkrkova, K.; Sizdahkhani, S.; Huuskonen, M.T.; Lamorie-Foote, K.; Diaz, A.; Chen, S.; Liu, Q.; D'Agostino, C.; Zhang, H.; et al. Magnetic resonance imaging of white matter response to diesel exhaust particles. Res. Sq. Preprint, rs.3.rs-3087503. 3087. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, Y.; Xia, X.; Que, X.; Liu, X.; Zhao, G.; Chen, J.; Chen, Q.; Xu, Z.; Tang, Y.; Qin, Q. Regulation of synaptic function and lipid metabolism. Neural Regen. Res. 2026, 21, 1037–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, L.; Qin, C. Alzheimer's disease pathogenesis: standing at the crossroad of lipid metabolism and immune response. Mol Neurodegener 2025, 20, 67. [Google Scholar] [CrossRef]
- Badia-Soteras, A.; Mak, A.; Blok, T.M.; Boers-Escuder, C.; van den Oever, M.C.; Min, R.; Smit, A.B.; Verheijen, M.H.G. Astrocyte-Synapse Structural Plasticity in Neurodegenerative and Neuropsychiatric Diseases. , Biol. Psychiatry 2025, S0006-3223(25)01125-4. [Google Scholar] [CrossRef]
- Salardini, E.; O'Dell, R.S.; Tchorz, E.; Nabulsi, N.B.; Huang, Y.; Carson, R.E.; van Dyck, C.H.; Mecca, A.P. Assessment of the Relationship between Synaptic Density and Metabotropic Glutamate Receptors in Early Alzheimer’s Disease: A Multi-Tracer PET Study. Alzheimers Res. Ther. 2025, 17, 98. [Google Scholar] [CrossRef]
- Martin Flores, N.; Podpolny, M.; McLeod, F.; Workman, I.; Crawford, K.; Ivanov, D.; Leonenko, G.; Escott-Price, V.; Salinas, P.C. Downregulation of Dickkopf-3, a Wnt Antagonist Elevated in Alzheimer's Disease, Restores Synapse Integrity and Memory in a Disease Mouse Model. eLife 2024, 12, RP89453. [Google Scholar] [CrossRef] [PubMed]
- Ni, R. Biomarkers for Synaptic Dysfunction in Alzheimer’s Disease. Neural Regen. Res. 2026, 21, 683–684. [Google Scholar] [CrossRef]
- Shanaki Bavarsad, M.; Spina, S.; Oehler, A.; Allen, I.E.; Suemoto, C.K.; Leite, R.E.P.; Seeley, W.S.; Green, A.; Jagust, W.; Rabinovici, G.D.; Grinberg, L.T. Comprehensive Mapping of Synaptic Vesicle Protein 2A (SV2A) in Health and Neurodegenerative Diseases: A Comparative Analysis with Synaptophysin and Ground Truth for PET-Imaging Interpretation. Acta Neuropathol. 2024, 148, 58. [Google Scholar] [CrossRef]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic Degeneration in Alzheimer Disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef]
- Mecca, A.P.; Ashton, N.J.; Chen, M.K.; O'Dell, R.S.; Toyonaga, T.; Zhao, W.; Young, J.J.; Salardini, E.; Bates, K.A.; Ra, J.; et al. Cerebrospinal Fluid and Brain Positron Emission Tomography Measures of Synaptic Vesicle Glycoprotein 2A: Biomarkers of Synaptic Density in Alzheimer's Disease. Alzheimers Dement. 2025, 21, e70344. [Google Scholar] [CrossRef]
- Thiankhaw, K.; Chattipakorn, N.; Chattipakorn, S. . Pm2.5 exposure in association with ad-related neuropathology and cognitive outcomes. Environ Pollut 2022, 292(Pt A), 118320. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Ali, M.U.; Mayhew, A.; Aryal, K.; Correia, R.H.; Dash, D.; Manis, D.R.; Rehman, A.; O'Connell, M.E.; Taler, V.; et al. Environmental risk factors for all-cause dementia, Alzheimer's disease dementia, vascular dementia, and mild cognitive impairment: An umbrella review and meta-analysis. Environ. Res. 2025, 270, 121007. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Entry of metals in particulate matter into the human brain directly from olfactory pathway.
Figure 1.
Entry of metals in particulate matter into the human brain directly from olfactory pathway.
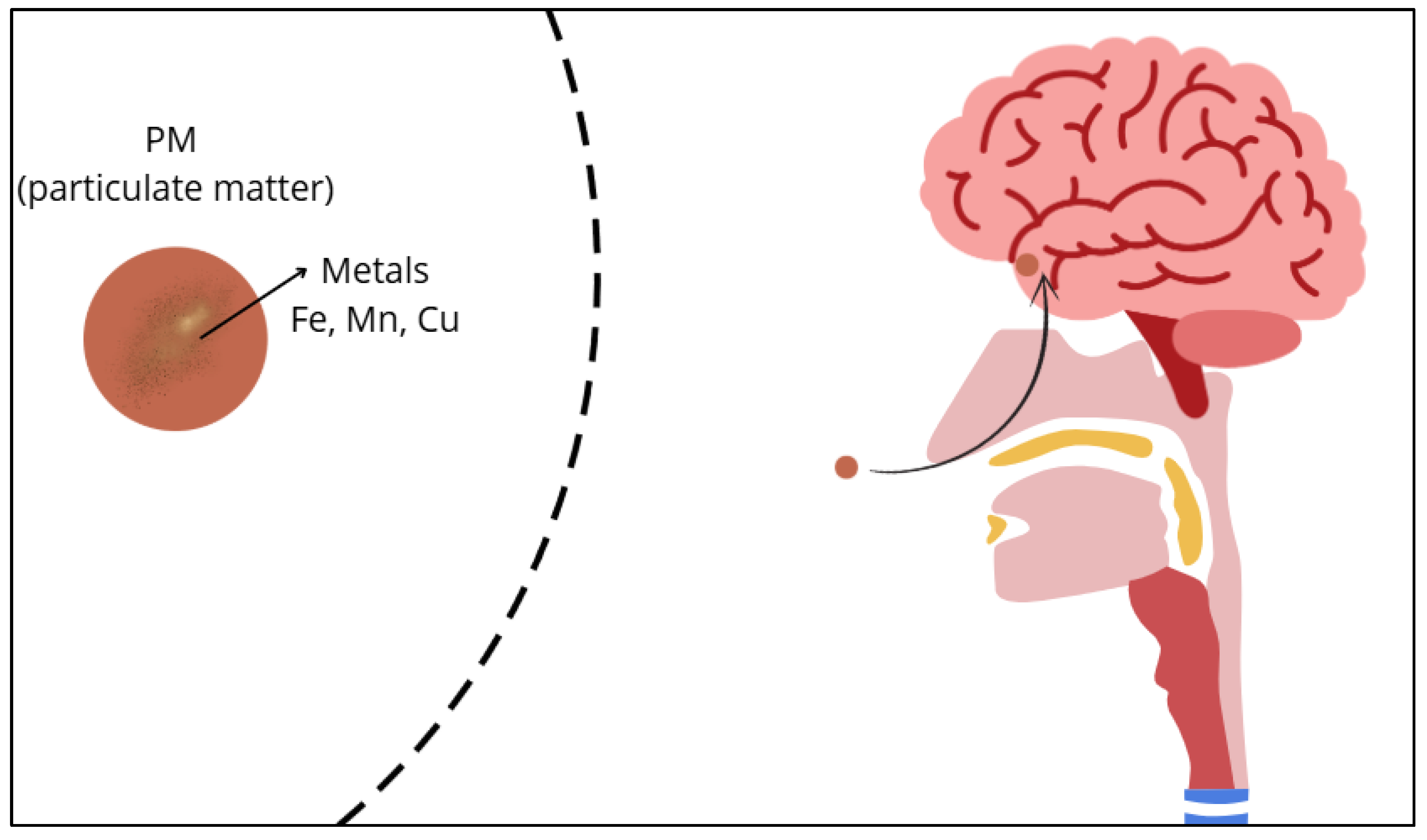
Figure 2.
Air pollution as a risk factor for the development of Alzheimer's disease.
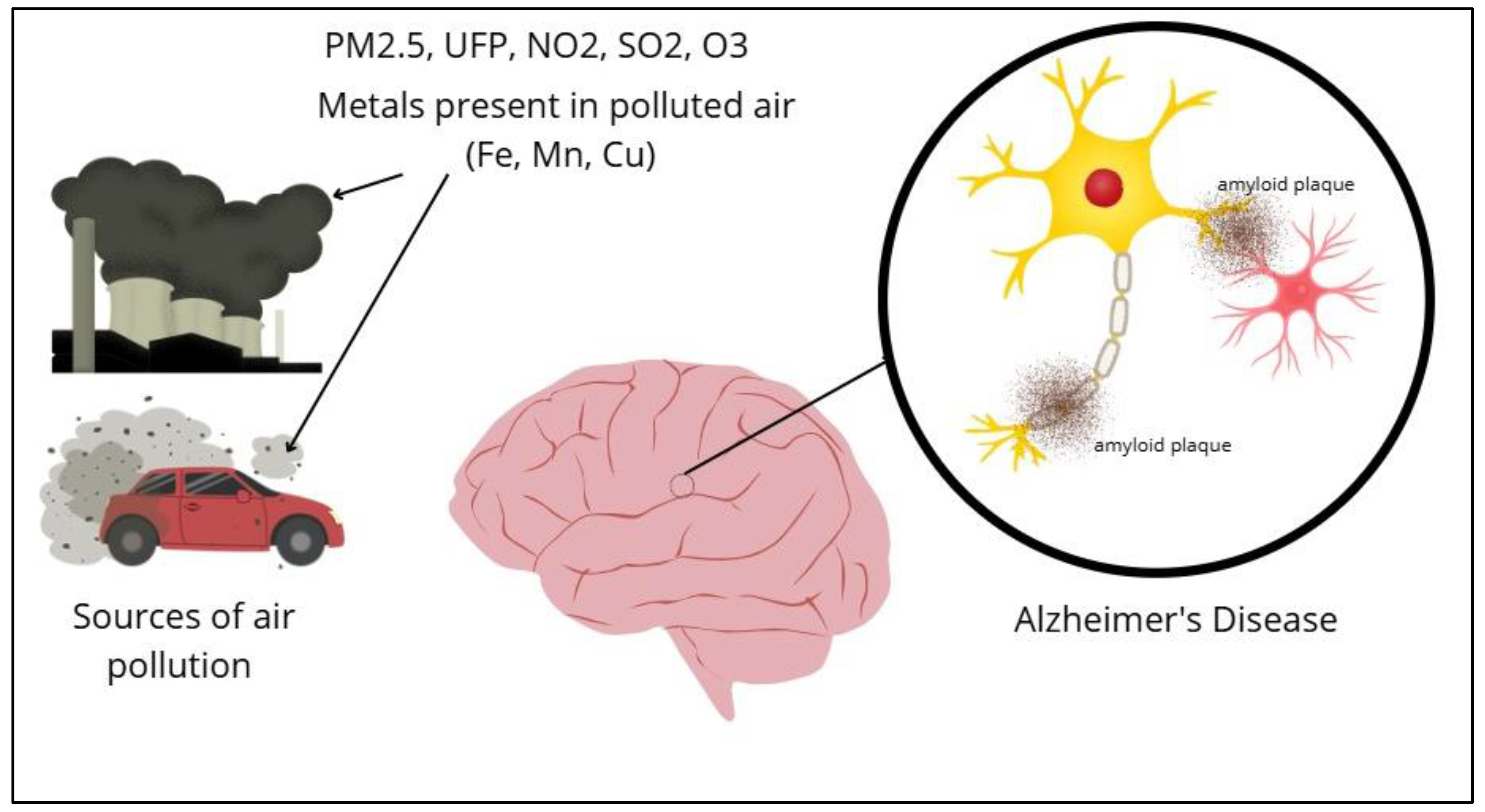
Figure 3.
Epidemiological evidence linking air pollution to cognitive impairment and Alzheimer’s Disease.
Figure 3.
Epidemiological evidence linking air pollution to cognitive impairment and Alzheimer’s Disease.
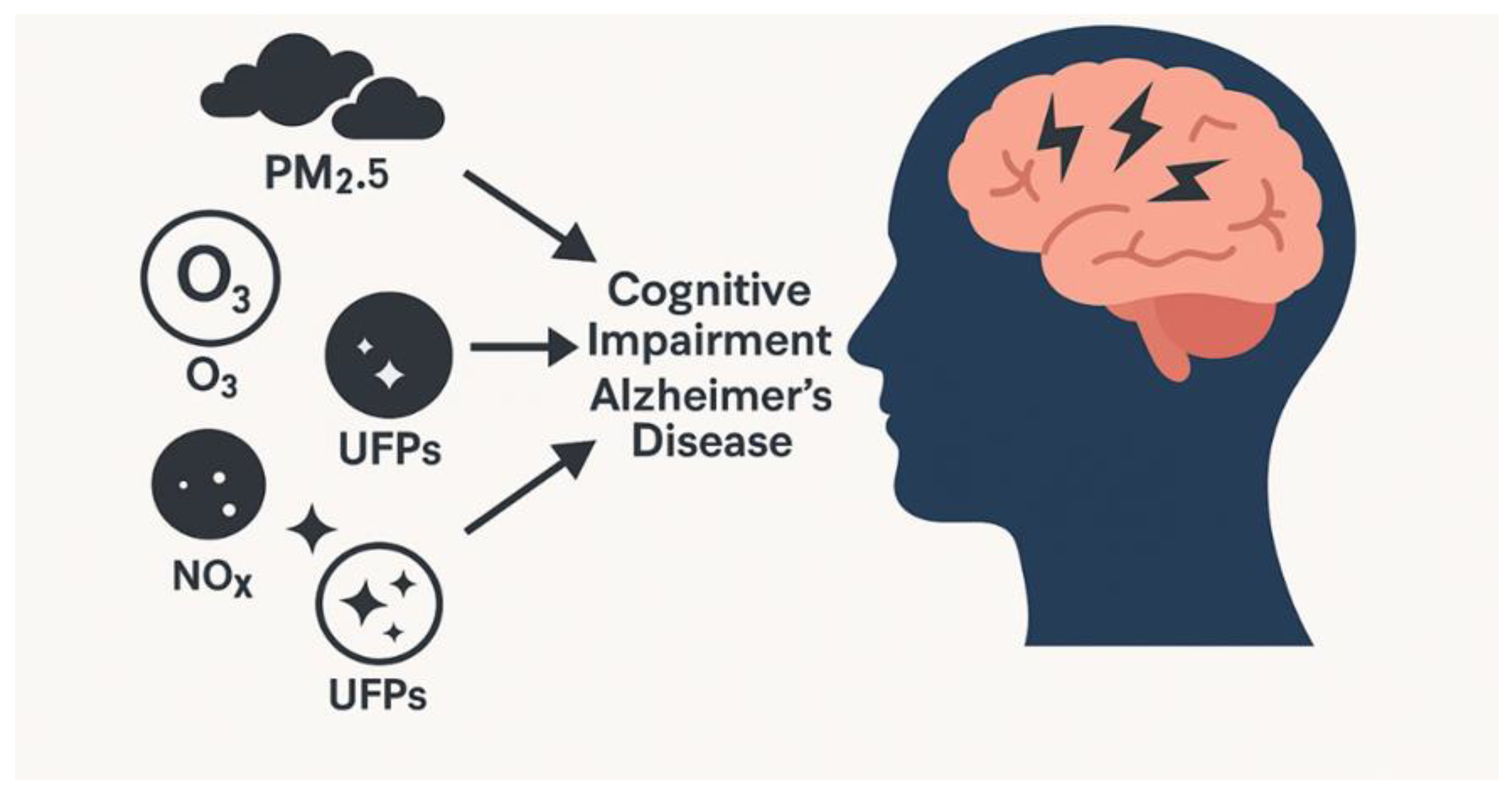
Figure 4.
U-shaped relationship between iron levels and risk of neurodegeneration. Iron deficiency is associated with impaired synaptic plasticity, neurotransmitter dysfunction, and vulnerability to toxic metals. Excess iron contributes to oxidative stress, mitochondrial dysfunction, glial activation, and ferroptosis. An adequate iron balance is essential for maintaining cognitive function and minimizing neurodegeneration risk.
Figure 4.
U-shaped relationship between iron levels and risk of neurodegeneration. Iron deficiency is associated with impaired synaptic plasticity, neurotransmitter dysfunction, and vulnerability to toxic metals. Excess iron contributes to oxidative stress, mitochondrial dysfunction, glial activation, and ferroptosis. An adequate iron balance is essential for maintaining cognitive function and minimizing neurodegeneration risk.
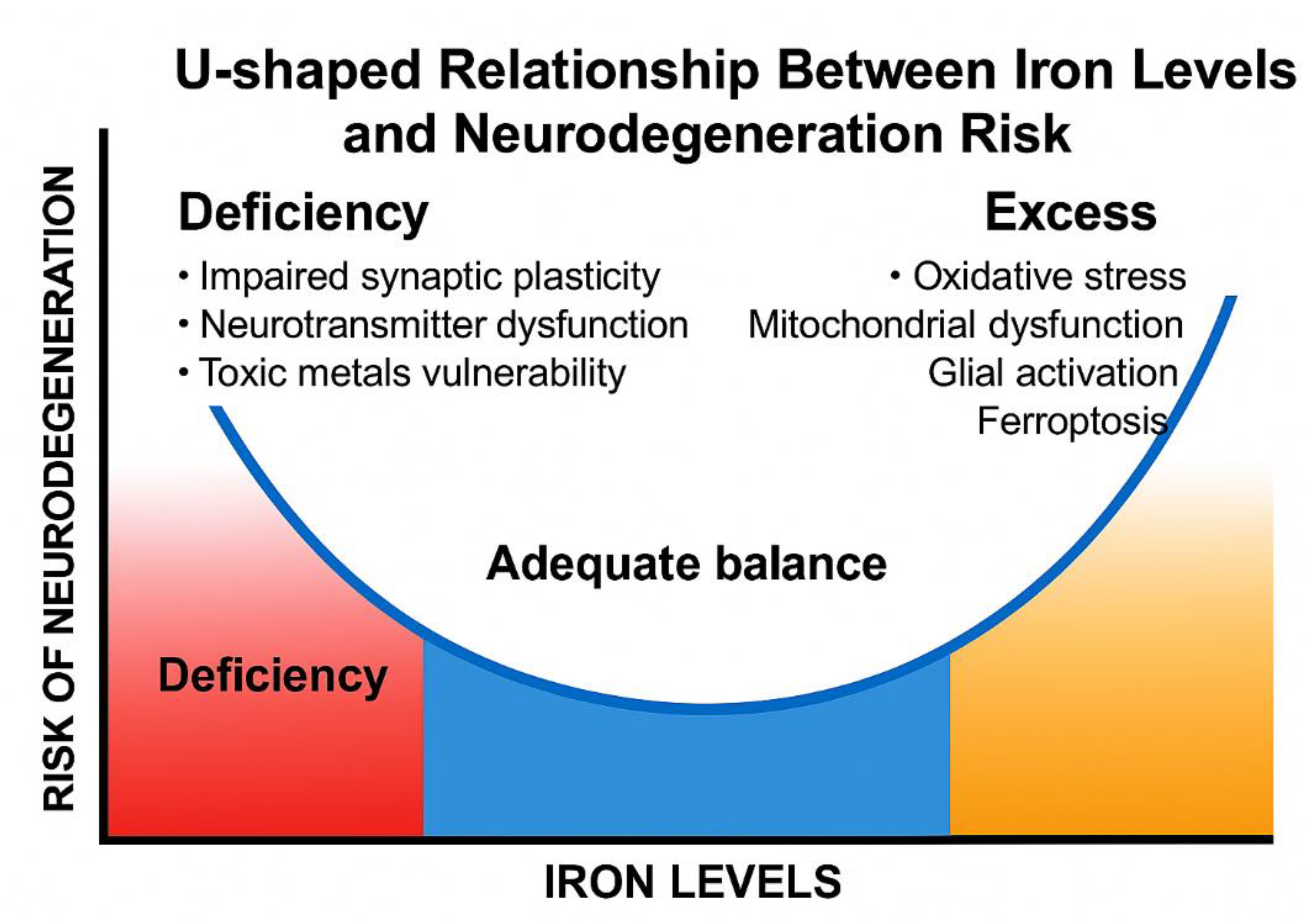
Figure 5.
Glial Cells and Their Role in Alzheimer’s Disease.
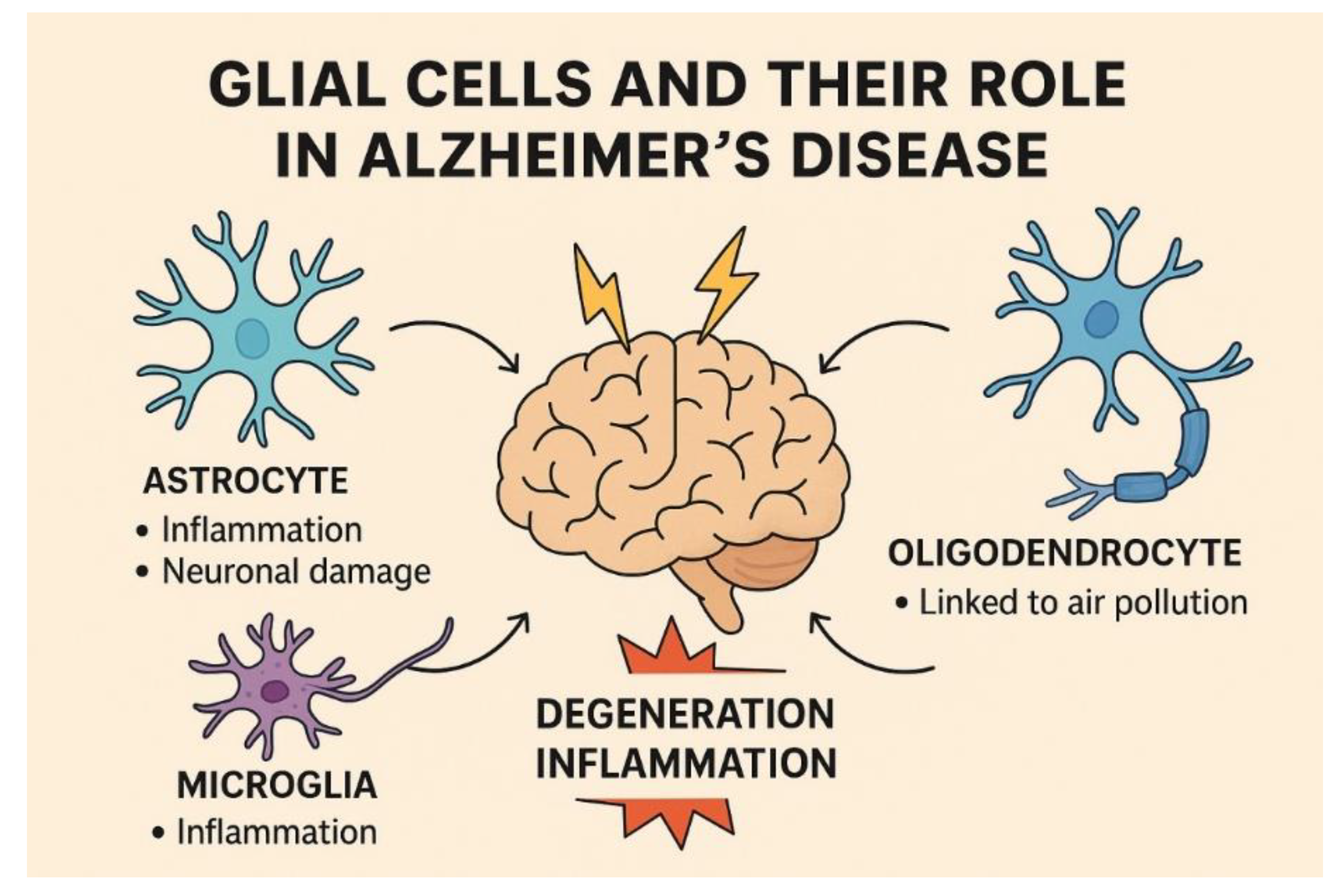

Table 1.
Research on airborne metals and neurodegenerative diseases.
| MATRIX | METALS FOUND / RESULTS | AGE | LOCATION | METHOD | REFERENCE |
| PM2.5 (EXPERIMENTAL EXPOSURE) | Fe, Mn | Mice (juvenile and adult) | Controlled experimental settings | Experimental | [68] |
| AIRBORNE PM10 | Fe, Cu, Mn, Zn, SO2 | ≥ 60 years | Lima, Peru | Ecology | [51] |
| AIRBORNE ULTRAFINE PM | Fe, Ti, Hg, Ni, Co, Cu, Zn, Cd, Al, Mg, Ag, Ce, La, Pr, W, Ca, Cl, K, Si, S, Na y C | Pediatric to older adults | Mexico City | Pathology, Imaging | [109] |
| UFPS | Adolescent mice | Laboratory setting | Experimental Animal Study | [91] | |
| PM2.5 | SO42- , OC, Cu | ≥ 60 years | USA | Cohort study, modeling | [122] |
| PM10 | Fe, Mn | All ages | La Oroya, Cerro de Pasco, Callao, Junín, Lima (Peru) | Narrative synthesis | [67] |
| FE EXPOSURE | Magnetite (Fe₃O₄) | Mice: juvenile | University of Rochester Medical Center, USA | Pathology & Imaging | [49] |
| DIESEL EXHAUST PARTICLES | altered myelin composition in multiple regions | Mice: adult | Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, USA | Pathology & Imaging | [198] |
| IN BLOOD (FER-1 , MNCL2 , FER-1 + MNCL2 ) | Mn-exposed mice exhibited movement impairment and encephalic pathological changes, with decreased HIF-1α, SLC7A11, and GPX4 proteins and increased p53 protein levels. Fer-1 exhibited protective effects against Mn-induced both behavioral and biochemical changes | Mice: juvenile | Department of Preventive Medicine and Public Health Laboratory Sciences, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu 212013, China | Experimental Animal Study | [163] |
| POLLUTANT IRON-BASED PARTICLE, HYDROCARBON-BASED DIESEL COMBUSTION PARTICLE AND MAGNETITE PARTICLES | Neuronal cell loss was shown in the hippocampal and somatosensory cortex, with increased detection of Aβ plaque, | Mice: juvenile | Australian Institute for Microbiology and Infection, University of Technology Sydney, Sydney, Australia | Experimental Animal Study | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



