
Submitted:
01 August 2025
Posted:
04 August 2025
You are already at the latest version
Abstract
Background: The Asian longhorned tick, Haemaphysalis longicornis, is a vector of significant public health importance, capable of transmitting numerous zoonotic pathogens. In China, comprehensive data on the geographic distribution and genetic diversity of these pathogens, particularly Spotted Fever Group Rickettsiae (SFGR), remain limited. This study aimed to investigate the prevalence and characteristics of key pathogens in questing H. longicornis ticks from two geographically and ecologically distinct regions of China.
Methods: A total of 1,004 questing H. longicornis ticks were collected from Liaoning Province (Northeast China) and Anhui Province (East-Central China) and processed in 670 pools. Tick species was confirmed using mitochondrial cytochrome oxidase I (COI) gene sequencing. Nucleic acids from all tick pools were screened for Dabie bandavirus (formerly SFTSV) and bacteria using universal 16S rRNA gene primers. Bacterial-positive samples were subsequently subjected to multi-gene nested PCR assays for Rickettsia spp. and Coxiella-like endosymbionts (CLE). Phylogenetic analyses, including supertree construction from concatenated gene sequences, were performed to ensure robust species identification and explore genetic relationships.
Results: No Dabie bandavirus was detected. SFGR were identified in 14 of the 670 tick pools, yielding an overall Minimum Infection Rate (MIR) of 1.4% (14/1,004). Notably, the detected SFGR species showed clear geographic separation: Candidatus Rickettsia jingxinensis was found exclusively in Liaoning (MIR 2.0%), while Rickettsia heilongjiangensis was detected only in Anhui (MIR 2.6%). Additionally, CLE were found in 20 tick pools (overall MIR 2.0%), with a significantly higher prevalence in Anhui (17.1%) compared to Liaoning (1.2%).
Conclusions: Our surveillance reveals distinct geographic distributions of SFGR species within H. longicornis populations in China, confirming the circulation of Ca. R. jingxinensis in the northeast and R. heilongjiangensis in the east-central region. These findings provide crucial, multi-gene-supported epidemiological data that enhance our understanding of regional tick-borne disease risks, and underscore the importance of continued, geographically targeted surveillance.
Keywords:
spotted fever group rickettsia (SFGR)
; Rickettsia heilongjiangensis
; Candidatus Rickettsia jingxinensis
; Haemaphysalis longicornis
; Coxiella-like endosymbiont (CLE)
; tick surveillance
; China
1. Introduction
Ticks are obligate hematophagous ectoparasites and rank second only to mosquitoes as vectors of human pathogens globally [1]. Understanding the specific pathogens circulating within local tick populations is fundamental for effective public health surveillance and disease prevention strategies.
Haemaphysalis longicornis, the Asian longhorned tick, is a vector of major medical and veterinary importance [2]. Native to East Asia, including eastern China, Japan, and Korea, this species has demonstrated remarkable ecological adaptability, establishing invasive populations in Australia, New Zealand, and the eastern United States [2,3,4]. Its capacity for parthenogenetic reproduction can facilitate rapid population establishment and massive host infestations [2].
H. longicornis is a competent vector for over 30 human pathogens, including multiple species of Spotted Fever Group Rickettsia (SFGR), agents of anaplasmosis, and viruses such as Dabie bandavirus (the causative agent of Severe Fever with Thrombocytopenia Syndrome, SFTS) [4]. In China, SFGR species like Rickettsia japonica and Rickettsia heilongjiangensis are significant causes of tick-borne rickettsioses [5].
Despite the recognized threat, epidemiological data on the prevalence and genetic diversity of SFGR in H. longicornis across different regions of China are still fragmented. Such information is critical for assessing regional public health risks, and understanding the ecological factors that may influence pathogen distribution. Therefore, this study was designed to investigate and compare the prevalence and genetic characteristics of SFGR and other selected pathogens, in questing H. longicornis ticks collected from two geographically and ecologically distinct provinces: Liaoning in Northeast China and Anhui in East-Central China.
2. Materials and Methods
2.1. Sample Collection
Questing ticks were collected from vegetation by flagging with a 1-m² corduroy cloth in woodland and grassland habitats in Helan Town, Liaoning Province (June 2021) and Hanshan County, Anhui Province (August 2022). Geographic coordinates and habitat characteristics of all collection sites were recorded (Figure 1).
2.2. Host Species Identification
All ticks were first identified to the species level based on morphological keys. To confirm morphological identification, DNA was extracted from a representative subset of ticks, and the mitochondrial cytochrome oxidase I (COI) gene was amplified by nested PCR and sequenced, using primers LCO1490 and HCO2198 [6].
2.3. Sample Processing and Pooling Strategy
A total of 1,004 ticks were processed. To optimize detection sensitivity for low-prevalence pathogens, a pooling strategy was employed. Questing adult ticks (n=503) were processed individually (503 pools). Questing nymphs (n=501) were combined into pools of up to three individuals, resulting in 167 nymphal pools. In total, 670 tick pools were analyzed.
2.4. Tick Surface Decontamination and Homogenization
Prior to homogenization, individual ticks or tick pools were surface-sterilized to minimize contamination from external microbes. Each sample was washed sequentially by vortexing in 70% ethanol for 30 seconds, followed by 1% sodium hypochlorite for 30 seconds, and finally rinsed three times in sterile phosphate-buffered saline (PBS) to remove residual disinfectants [7,8,9]. Each sterilized tick pool was homogenized in 1 mL of sterile PBS using a TissueLyser II (QIAGEN, Germany). An aliquot of the homogenate (300 µl) was reserved for bacterial culture, while the remainder was used for nucleic acid extraction.
2.5. Nucleic Acid Extraction
Total DNA and RNA were co-extracted from 200 µl of each tissue homogenate using the AllPrep DNA/RNA Mini Kit (QIAGEN, Germany) following the manufacturer’s protocol. Nucleic acids were eluted in a final volume of 120 µl (2 x 60 µl elutions) to maximize yield. The concentration and purity of the extracted nucleic acids were measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific, USA), and samples were stored at -40°C.
2.6. Molecular Detection of Pathogens
All nucleic acid samples were first screened for Dabie bandavirus (SFTSV) using a one-step RT-PCR targeting the S segment [10,11]. For bacterial detection, samples were initially screened with universal primers targeting the bacterial 16S ribosomal RNA (rRNA) gene [12]. Samples positive for the 16S rRNA gene were then subjected to a series of nested PCR assays targeting specific genes for pathogens of interest (Table 1). Specifically, tick samples were tested for Rickettsia spp. (targeting rrs, gltA, 17kDa, ompA, ompB, sca4) and Coxiella-like endosymbionts (CLE) (targeting 16S rRNA, groEL, rpoB). All PCR products were visualized by electrophoresis on 1.0% agarose gels. Amplicons of the expected size were purified using the QIAquick PCR Purification Kit (QIAGEN, Germany) and sent for bidirectional Sanger sequencing.
2.7. Bacterial Culture and Identification
An aliquot of the tissue homogenate from each sample was plated onto cysteine heart agar blood (CHAB) medium supplemented with antibiotics (colistin, amphotericin, lincomycin, methicillin, ampicillin) to select for specific bacterial groups. Plates were incubated at 37°C in a 5% CO2 atmosphere and monitored daily. Individual colonies were isolated, sub-cultured for purity, and identified by sequencing the 16S rRNA gene amplified from colony material.
2.8. Phylogenetic and Data Analysis
Obtained nucleotide sequences were compared against the GenBank database using BLASTn. For phylogenetic inference, sequences were aligned with reference sequences using MUSCLE. Initial phylogenetic trees for individual genes were constructed using the Neighbor-Joining (NJ) method with 1,000 bootstrap replicates in MEGA 11 software. To provide more robust phylogenetic placement for key pathogens, “supertrees” were constructed from concatenated sequences of multiple genes using the Maximum Likelihood (ML) method with 1,000 bootstrap replicates. This multi-gene approach increases the confidence of the phylogenetic inference. The pathogen prevalence in pooled tick samples was calculated as the Minimum Infection Rate (MIR), estimated using the formula: MIR = (Number of positive pools / Total number of ticks tested) × 100.
3. Results
3.1. Sample Collection and Host Identification
3.2. Pathogen Prevalence
Molecular screening revealed the presence of SFGR and CLE, while no samples tested positive for Dabie bandavirus (SFTSV). The prevalence of all detected microbes is summarized in Table 2. In H. longicornis ticks, the overall MIR for SFGR was 1.4% (14/1,004). Geographically, the detected species differed: Ca. R. jingxinensis was found exclusively in Liaoning, with an MIR of 2.0% (12/594 positive pools), while R. heilongjiangensis was found only in Anhui, with an MIR of 2.6% (2/76 positive pools). Coxiella-like endosymbionts (CLE) were detected in both locations, with an overall MIR of 2.0% (20/1,004). Notably, the MIR of CLE was substantially higher in Anhui (17.1%) compared to Liaoning (1.2%). Four tick pools were co-infected with both an SFGR species and CLE.
3.3. Supplementary Culture-Based Findings
Bacterial culture from tissue homogenates yielded several isolates, primarily from the genera Pseudomonas, Staphylococcus, and Bacillus. These bacteria were identified via 16S rRNA sequencing of the isolates. As these organisms are common environmental microbes or commensals, and their detection via culture does not distinguish between internal colonization and potential residual surface contaminants surviving sterilization, their pathogenic significance in this context was not further investigated.
3.4. Phylogenetic Analysis of Detected Pathogens
Multi-gene phylogenetic analyses provided robust identification and revealed the genetic relationships of the detected pathogens.
Rickettsia spp.: The six targeted genes (rrs, 17kDa, gltA, ompA, ompB, sca4) from the Ca. R. jingxinensis isolates from Liaoning (represented by isolate tick73) showed high homology to strains previously reported from Shaanxi, Jilin, and Jiangsu provinces, forming a well-supported clade within the R. japonica subgroup of SFGR (Figure 2) [19,20]. The two R. heilongjiangensis isolates from Anhui (represented by tick5) were genetically very close to each other. For five of their genes, they showed highest homology to strain B8, which was isolated from a human patient in Anhui [21]. However, their ompB gene sequences were 100% identical to strain 054 from Heilongjiang, suggesting potential genetic links between geographically distant populations (Figure 2) [22]. The ML supertree analysis, based on concatenated gene sequences, strongly supported these classifications, placing the Anhui isolate within the R. heilongjiangensis cluster and the Liaoning isolate within a broader SFGR clade that includes R. conorii (Figure 3).
Coxiella-like Endosymbionts (CLE): The CLE sequences (16S rRNA, groEL, rpoB) from both Liaoning and Anhui were nearly identical to each other and clustered tightly with a CLE strain previously identified from Jiangxi province, indicating a conserved lineage of this endosymbiont in H. longicornis across eastern China (Figure 4).
4. Discussion
This study provides a focused molecular survey of pathogens in H. longicornis ticks from two geographically distinct provinces in China, yielding valuable data on the regional distribution of SFGR and other bacteria.
Our findings confirm the presence of at least two SFGR species circulating in H. longicornis ticks in China, with a notable geographic separation: Ca. R. jingxinensis in Liaoning and R. heilongjiangensis in Anhui. Ca. R. jingxinensis, first described in China in 2016, has since been reported in multiple provinces, and our multi-gene analysis supports its genetic linkage with strains from other regions like Shaanxi and Jiangsu [19,20]. Similarly, the detection of R. heilongjiangensis in Anhui, a pathogen typically associated with northeastern China, and its genetic similarity to a local human patient isolate (strain B8) [21], reinforces its endemicity and public health relevance in this eastern-central province.
The overall MIR of SFGR in questing H. longicornis was low (1.4%). This is consistent with findings from other studies on free-living, unfed ticks, where pathogen prevalence is often significantly lower than in ticks collected directly from animal hosts [23]. For instance, a recent meta-analysis of SFGR in China reported an average prevalence of 11.5% in questing ticks, but this rate can be highly variable depending on location and tick species [23]. In contrast, prevalence in ticks feeding on livestock can be extremely high, sometimes exceeding 70-90% [20,24]. Our low MIR provides an important baseline for the risk of human exposure from questing ticks in these specific environments, but it also highlights that host-associated ticks likely play a more significant role in amplifying and maintaining these pathogens.
This study has several limitations that must be acknowledged. First, our detection of pathogens was based on nucleic acid amplification (DNA), which confirms the presence of the organism’s genetic material but does not prove viability or infectivity, except in the case of the cultured bacteria. Second, the use of nymph pooling for MIR calculation provides a cost-effective estimate of prevalence but is less precise than individual testing. Finally, while this study provides a valuable snapshot, further longitudinal surveillance is needed to understand the temporal dynamics of these pathogens.
5. Conclusions
In summary, this study provides an updated, multi-gene-supported epidemiological snapshot of pathogen distribution in H. longicornis ticks from Northeast and East-Central China. We confirm the distinct geographic circulation of Ca. R. jingxinensis in Liaoning and R. heilongjiangensis in Anhui. These findings contribute significantly to the understanding of regional SFGR diversity and underscore the necessity for continued surveillance to inform public health risk assessments for tick-borne diseases in China.
Author Contributions
Conceptualization, Yanhua Wang; Methodology, Wen Wang and Rui Zhang; Investigation, Yingwei Sun, Lei Gong, Shuzhen Guo, Lingling Mao, Dandan Song, Yuhui Guan, Yuanyuan Jiang, Kun Yang, Xuesheng Liu and Ming Wu; Resources, Tian Qin and Yanhua Wang; Writing – original draft, Guodong Yang; Writing – review & editing, Yanhua Wang.
Funding
This work was funded by the Natural Science Foundation of Beijing Municipality (Grant No. 7242188) and the National Natural Science Foundation of China (Grant No. 81874275).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting this study are available within the manuscript.
References
- Jongejan, F.; Uilenberg, G. The Global Importance of Ticks. Parasitology 2004, 129, S3–S14. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Cui, X.; Jia, N.; Wei, J.; Xia, L.; Wang, H.; Zhou, Y.; Wang, Q.; Liu, X.; et al. Distribution of Haemaphysalis Longicornis and Associated Pathogens: Analysis of Pooled Data from a China Field Survey and Global Published Data. Lancet Planet. Health 2020, 4, e320–e329. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.B.; Occi, J.; Bonilla, D.L.; Egizi, A.M.; Fonseca, D.M.; Mertins, J.W.; Backenson, B.P.; Bajwa, W.I.; Barbarin, A.M.; Bertone, M.A.; et al. Multistate Infestation with the Exotic Disease–Vector Tick Haemaphysalis Longicornis — United States, August 2017–September 2018. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Rainey, T.; Occi, J.L.; Robbins, R.G.; Egizi, A. Discovery of Haemaphysalis Longicornis (Ixodida: Ixodidae) Parasitizing a Sheep in New Jersey, United States. J. Med. Entomol. 2018, 55, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.; Gong, P.; Wang, W.; Zhao, N.; Jin, X.; Sun, X.; Lu, J.; Lin, X.; Zhou, H.; Wen, B.; et al. The increasing prevalence of Japanese spotted fever in China: A dominant rickettsial threat. J. Infect. 2024, 90, 106387. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Moutailler, S.; Popovici, I.; Devillers, E.; Vayssier-Taussat, M.; Eloit, M.; Ferte, H. Diversity of the tick-associated bacterial communities: impact of the sterilization method. Ticks Tick-Borne Dis. 2016, 7, 140–150. [Google Scholar] [CrossRef]
- Binetruy, F.; Buysse, M.; Lejal, E.; Koadima, O.; Diancourt, L.; Santos, A.; Rose, T.; Durand, P.; Mediannikov, O.; Paddock, C.D.; et al. Ticks (Acari: Ixodida) of the genus Amblyomma, vectors of Rickettsia species in the French Antilles. PLoS Negl. Trop. Dis. 2019, 13, e0007424. [Google Scholar] [CrossRef]
- Sprong, H.; Fonville, M.; van Leeuwen, A.D.; van der-Lelie, D.; van Wieren, S.E.; Gort, G.; Takken, W.; Heyman, P.; van-Vliet, A.J.H. Ticks and tick-borne pathogens in the expanding range of wild boar (Sus scrofa) in the Netherlands. Vector Borne Zoonotic Dis. 2014, 14, 640–647. [Google Scholar] [CrossRef]
- Wen, H.-L.; Zhao, L.; Zhai, S.; Chi, Y.; Cui, F.; Wang, D.; Wang, L.; Wang, Z.; Wang, Q.; Zhang, S.; et al. Severe Fever with Thrombocytopenia Syndrome, Shandong Province, China, 2011. Emerg. Infect. Dis. 2014, 20, 1–5. [Google Scholar] [CrossRef]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Lynch, K. Use of 16S rRNA Gene for Identification of a Broad Range of Clinically Relevant Bacterial Pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef]
- Lv, J.Z.; Wu, S.Q.; Zhang, Y.N.; Chen, Y.; Feng, C.Y.; Yuan, X.F.; Jia, G.L.; Deng, J.H.; Wang, C.X.; Wang, Q.; et al. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasit. Vectors 2014, 7, 93. [Google Scholar] [CrossRef]
- Borsoi, A.B.P.; Bitencourth, K.; De Oliveira, S.V.; Amorim, M.; Gazêta, G.S. Human Parasitism by Amblyomma Parkeri Ticks Infected with Candidatus Rickettsia Paranaensis, Brazil. Emerg. Infect. Dis. 2019, 25, 2339–2341. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, P.H.; Huang, Y.; Du, J.; Cui, N.; Yang, Z.D.; Tang, F.; Fu, F.X.; Li, X.M.; Cui, X.M.; et al. Isolation and Identification of Rickettsia Raoultii in Human Cases: A Surveillance Study in 3 Medical Centers in China. Clin. Infect. Dis. 2018, 66, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.; Lee, Y.S.; Kim, H.C.; Chong, S.T.; Klein, T.A.; Jiang, J.; Richards, A.L.; Lee, H.K.; Kim, S.Y. Molecular Detection of Rickettsia Species in Ticks Collected from the Southwestern Provinces of the Republic of Korea. Parasit. Vectors 2017, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; An, H.; Lee, J.S.; O’Guinn, M.L.; Kim, H.C.; Chong, S.T.; Zhang, Y.; Song, D.; Burrus, R.G.; Bao, Y.; et al. Molecular Characterization of Haemaphysalis Longicornis-Borne Rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 2018, 9, 1606–1613. [Google Scholar] [CrossRef]
- Duron, O.; Noël, V.; McCoy, K.D.; Bonazzi, M.; Sidi-Boumedine, K.; Morel, O.; Vavre, F.; Zenner, L.; Jourdain, E.; Durand, P.; et al. The Recent Evolution of a Maternally-Inherited Endosymbiont of Ticks Led to the Emergence of the Q Fever Pathogen, Coxiella Burnetii. PLoS Pathog. 2015, 11, e1004892. [Google Scholar] [CrossRef]
- He, M.; Zhang, L.; Hu, H.; Liu, X.; Zhang, C.; Xin, Y.; Liu, B.; Chen, Z.; Xu, K.; Liu, Y. Genetic and Phylogenetic Characterization of Spotted Fever Group Rickettsiae in Ticks from Jiangsu Province, Eastern China. Front. Cell. Infect. Microbiol. 2022, 12, 954785. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, W.; Teng, Z.; Zhao, N.; Zhou, Y.; Ma, D.; Ma, L.; Cheng, Y.; Wei, J.; He, J.; et al. Molecular Detection of Rickettsiales and a Potential Novel Ehrlichia Species Closely Related to Ehrlichia Chaffeensis in Ticks (Acari: Ixodidae) from Shaanxi Province, China, in 2022 to 2023. Front. Microbiol. 2023, 14, 1331434. [Google Scholar] [CrossRef]
- He, M.; Zhang, L.; Hu, H.; Liu, X.; Zhang, C.; Xin, Y.; Liu, B.; Chen, Z.; Xu, K.; Liu, Y. Complete Genome Sequencing and Comparative Genomic Analyses of a New Spotted-Fever Rickettsia Heilongjiangensis Strain B8. Emerg. Microbes Infect. 2023, 12, 2153085. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Zhang, J.; Wang, Y.; Wen, B. Complete Genome Sequence of Rickettsia heilongjiangensis. J. Bacteriol. 2011, 193, 4284–4285, https://www.google.com/search?q=https://doi.org/10.1128/JB.05852-11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, W.; Jia, Z.; Wang, X.; Huo, Q.; Wang, Y.; Liu, Q. A meta-analysis of the prevalence of spotted fever group rickettsiae in ticks in China. PLoS Negl. Trop. Dis. 2024, 18, e0012550. [Google Scholar] [CrossRef]
- Lu, M.; Meng, C.; Zhang, B.; Wang, X.; Tian, J.H.; Tang, G.P.; Wang, W.; Li, N.; Li, M.Y.; Xu, X.Y.; et al. High Diversity and Prevalence of Rickettsial Agents in Rhipicephalus microplus Ticks from Livestock in Karst Landscapes of Southwest China. Microorganisms 2024, 12, 1135. [Google Scholar] [CrossRef]
Figure 1.
Map of China showing the locations of Helan Town, and Hanshan County, alongside a phylogenetic tree based on COI gene sequences of the samples.
Figure 1.
Map of China showing the locations of Helan Town, and Hanshan County, alongside a phylogenetic tree based on COI gene sequences of the samples.
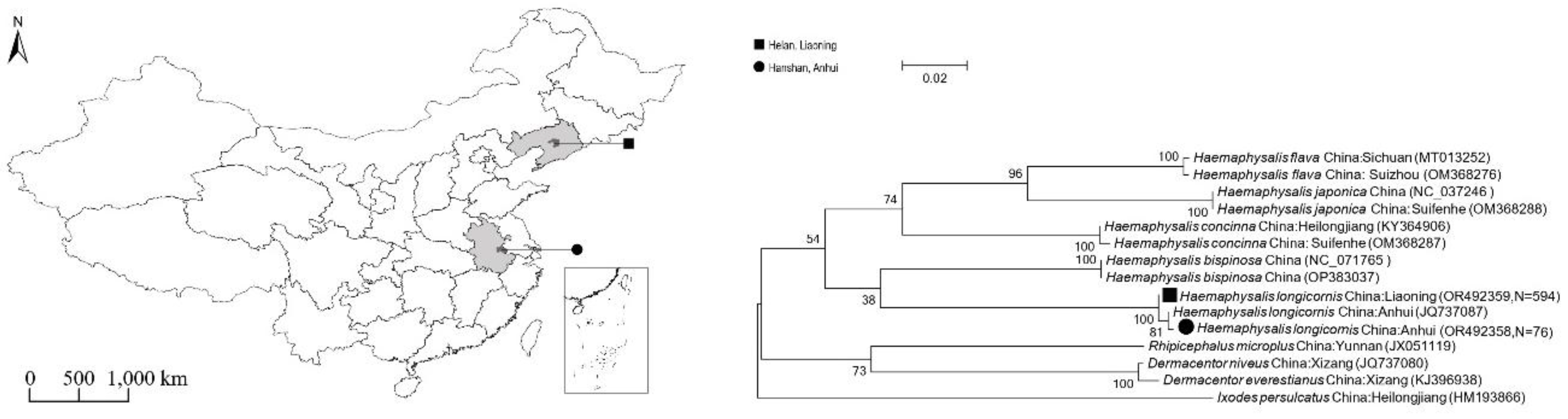
Figure 2.
Phylogenetic trees based on the nucleotide sequences of the 16S rRNA,17kDa, gltA, ompA, ompB and sca4 genes of Rickettsia.Bootstrap values > 60% based on 1000 replicates are shown at the nodes, using neighbor-joiningmethod.▲represents Candidatus Rickettsia jingxinensis detected in this study. ●represents Rickettsia heilongjiangensis detected in this study.
Figure 2.
Phylogenetic trees based on the nucleotide sequences of the 16S rRNA,17kDa, gltA, ompA, ompB and sca4 genes of Rickettsia.Bootstrap values > 60% based on 1000 replicates are shown at the nodes, using neighbor-joiningmethod.▲represents Candidatus Rickettsia jingxinensis detected in this study. ●represents Rickettsia heilongjiangensis detected in this study.
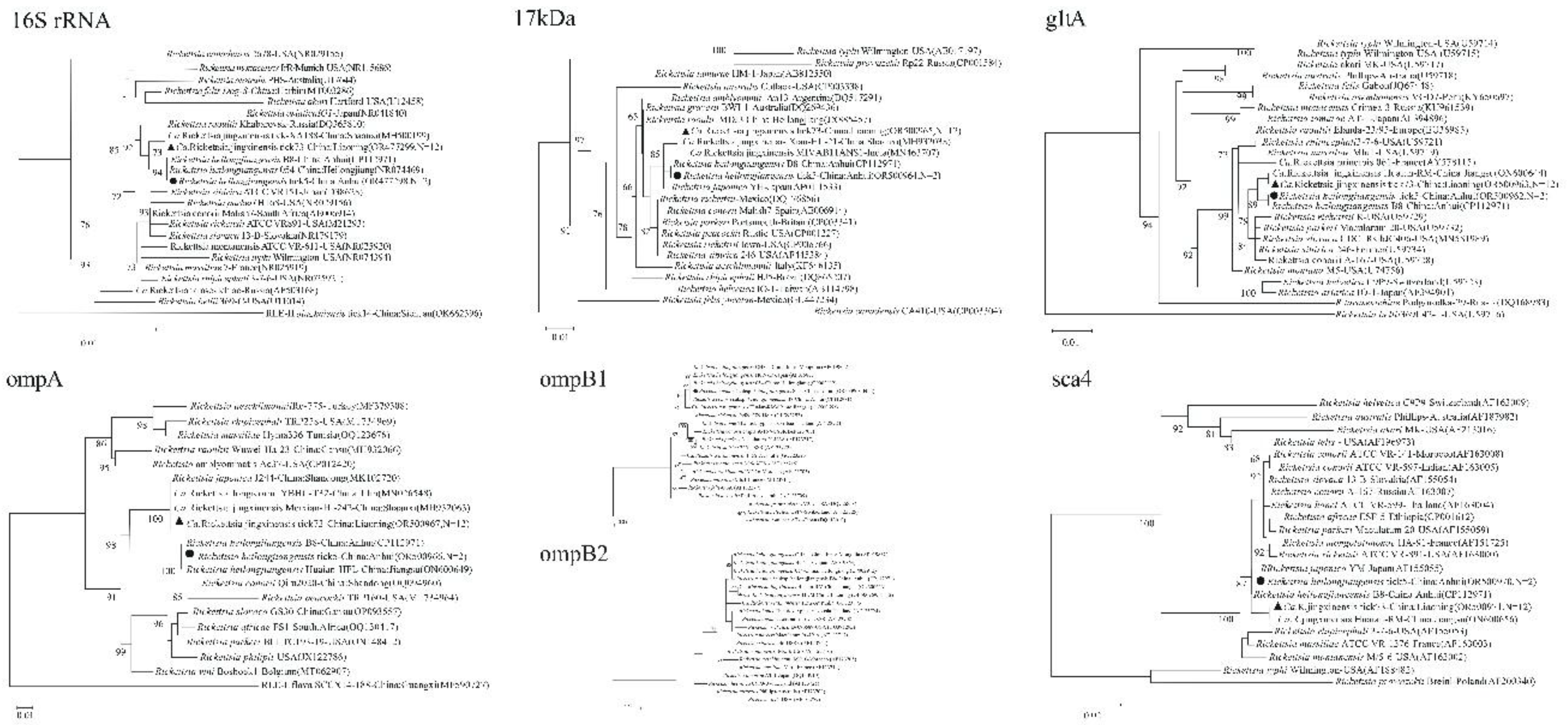
Figure 3.
Phylogenetic supertrees based on the concatenated gene sequences of Rickettsia. A. 16S rRNA+17kDa+gltA+ompA+ompB1+sca4. B. 16S rRNA+17kDa+gltA+ompA+ompB2+sca4.Bootstrap values >60% based on 1000 replicates are shown at the nodes, using maximum likelihood method. ▲represents Candidatus Rickettsia jingxinensis and ● represents Rickettsia heilongjiangensis detected in this study.
Figure 3.
Phylogenetic supertrees based on the concatenated gene sequences of Rickettsia. A. 16S rRNA+17kDa+gltA+ompA+ompB1+sca4. B. 16S rRNA+17kDa+gltA+ompA+ompB2+sca4.Bootstrap values >60% based on 1000 replicates are shown at the nodes, using maximum likelihood method. ▲represents Candidatus Rickettsia jingxinensis and ● represents Rickettsia heilongjiangensis detected in this study.

Figure 4.
Phylogenetic trees based on the nucleotide sequences of the 16S rRNA, groEL and rpoB genes of Coxiella-like endosymbionts (CLE). Bootstrap values >60% based on 1000 replicates are shown at the nodes, using neighbor-joining method. ▲represents CLE detected from Liaoning in this study. ●represents CLE detected from Anhui in this study.
Figure 4.
Phylogenetic trees based on the nucleotide sequences of the 16S rRNA, groEL and rpoB genes of Coxiella-like endosymbionts (CLE). Bootstrap values >60% based on 1000 replicates are shown at the nodes, using neighbor-joining method. ▲represents CLE detected from Liaoning in this study. ●represents CLE detected from Anhui in this study.
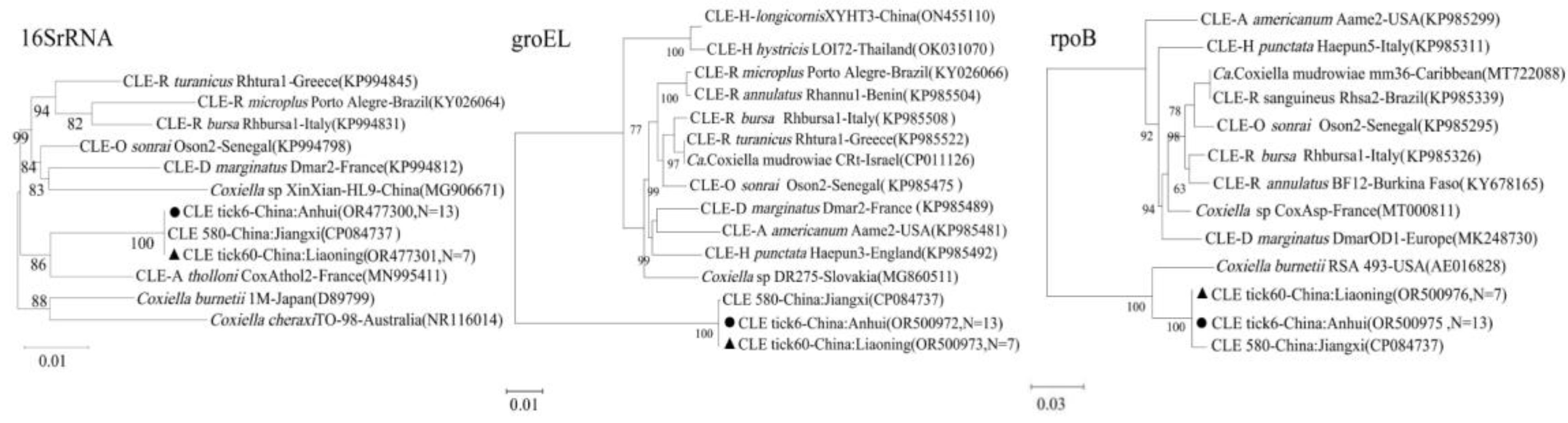

Table 1.
Primers and PCR conditions used for pathogen detection and host identification.
| Organism | Gene Target | Primer Name | Sequence (5’-3’) | PCR Type | Amplicon (bp) | Reference |
| Dabie bandavirus (SFTSV) | S segment | S-F1/S-R1 | CAGCCACTTTACCCGAACAT / GGAAAGACGCAAAGGAGTGA | Conv. | 679 | 10 |
| S-F2/S-R2 | CTGGTCTCTGCCCTCTCAAC / GGATTGCAGTGGAGTTTGGTG | Nested | 560 | |||
| Universal Bacteria | 16S rRNA | 27F/1492R | AGAGTTTGATCMTGGCTCAG / GGTTACCTTGTTACGACTT | Nested | ~1500 | 13 |
| Rickettsia spp. | rrs (16S rRNA) | rrs-F/rrs-R | YTACGGAATAACTTTTAGAAA / CATGATGACTTGACRTCGT | Nested | ~900 | 14 |
| gltA | Ric-glt-F/Ric-glt-R | ACTTAYGAYCCGGGCTTTAT / AGCTGTCTAGGTCTGCTGATT | Nested | ~1100 | 15 | |
| 17kDa | R-17kD-F/R-17kD-R | GCTCTTGCAACTTCTATGTT / CATTGTTCGTCAGGTTGGCG | Nested | ~434 | 15 | |
| ompA | R-ompA-F/R-ompA-R | ATGGCGAATATTTCTCCAAAA / AGTGCAGCATTCGCTCCCCCT | Nested | ~862 | 16 | |
| ompB | R-ompB-F/R-ompB-R | GTAACCGGAAGTAATCGTTTCGTAA / CTTTATAACCAGCTAAACCACC | Nested | ~769 | 14 | |
| sca4 | Ric-sca4-F/Ric-sca4-R | ATGAGTAAAGACGGTAACCT / AAGCTATTGCGTCATCTCCG | Nested | ~928 | 17 | |
| Coxiella-like Endosymbiont (CLE) | 16S rRNA | Cox-F/Cox-R | ACTYYCCAACAGCTAGTTCTCA / GTAGGAATCTACCTTRTAGWGG | Nested | ~600 | 18 |
| groEL | Cox-gro-F/Cox-gro-R | CTCAAGTCCCGAACCATCT / AGCCAACGCAGTCAAAGTA | Nested | ~494 | 18 | |
| rpoB | Cox-rpo-F/Cox-rpo-R | TTTCTCCTTTCGGTGTTAC / GATGCTTCACGGATTGTTA | Nested | ~496 | 18 | |
| Tick Host | COI | LCO1490/HCO2198 | GGTCAACAAATCATAAAGATATTGG / TAAACTTCAGGGTGACCAAAAAATCA | Nested | ~650 | 13 |
Note: Conv. = conventional PCR; Nested = nested PCR.
Table 2.
Prevalence of pathogens detected by molecular methods in ticks.
| Pathogen | Host | Location | No. Positive Pools | Total Ticks (in Pools) | Prevalence (%) |
| SFGR | |||||
| Ca. R. jingxinensis | H. longicornis | Liaoning | 12 | 1004 (in 594 pools) | 2.0 (MIR) |
| R. heilongjiangensis | H. longicornis | Anhui | 2 | 122 (in 76 pools) | 2.6 (MIR) |
| Endosymbiont | |||||
| Coxiella-like Endosymbiont (CLE) | H. longicornis | Liaoning | 7 | 1004 (in 594 pools) | 1.2 (MIR) |
| H. longicornis | Anhui | 13 | 122 (in 76 pools) | 17.1 (MIR) | |
Note: MIR = Minimum Infection Rate, calculated for pooled tick samples.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



