Submitted:
28 July 2025
Posted:
30 July 2025
You are already at the latest version
Abstract
Breast cancer (BC) remains a leading cause of cancer related mortality among women worldwide. Despite significant advances in early detection and targeted therapies, treatment of resistant cancers continues to be a major hindrance in long term patient outcomes. Therefore, a comprehensive understanding of the underlying mechanisms and strategies to overcome resistance in BC is urgently needed. Resistance is likely driven by diverse processes such as drug efflux transporter upregulation; genetic mutations and signaling rewiring (e.g., ESR1, PIK3CA/PTEN, PI3K/Akt/mTOR); cancer stem cells and exosome RNA transfer; modulation of tumor microenvironment; epigenetic and metabolic reprogramming; and immune evasion—all of which compromise treatment efficacy. As tumors evolve, monotherapies rapidly lose effectiveness, and metastatic progression further worsens outcomes. In this review, we systematically examine the key metabolic, genetic, epigenetic, immunological, and microenvironmental drivers of BC resistance and emerging strategies to treatments. Further, we also explore the role that artificial intelligence (AI) can play in enhancing early detection, clinical-decision making, developing targeted therapies, and designing vaccines, which can aid in overcoming resistance. By integrating mechanistic insights with cutting-edge technologies, our review highlights the potential of next-generation interventions to overcome resistance and improve patient prognosis and outcome.
Keywords:
breast cancer
; tumor resistance
; AI
; immunotherapy
; experimental models
1. Introduction
Breast cancer (BC) remains a paramount global health challenge and a leading cause of cancer-related morbidity and mortality worldwide. Globally, approximately 2.3 million new female cases were diagnosed and 670,000 deaths were reported for the year 2022 [1,2,3], underscoring the urgent need for continued research and improved clinical management. BC incidence increases with age; the median age of early diagnosis among women in the US is 62 years. It is projected that global incidence of BC is likely to increase 40% by the year 2040 [4], primarily driven by demographic shifts including population growth and increasing longevity. Significant disparities in patient outcomes persist; for example, in the U.S., black women exhibit a 41% higher mortality rate compared to white women despite comparable incidence rates [5]. In 2025, the American Cancer Society projects approximately 316,950 new cases of invasive BC and 59,080 cases of ‘ductal carcinoma in situ’ (DCIS) with 42,170 women dying from BC in the US alone [5,6].
Despite significant advances in early detection and adjuvant therapies that have improved outcomes for localized disease, the prognosis for metastatic breast cancer (MBC) remains a critical area of unmet clinical need. Notably, metastasis is the primary driver for BC-related mortality, accounting for approximately 90% of deaths, highlighting failure of treatment [3,5]. Within BC subtypes, triple-negative breast cancer (TNBC) is particularly aggressive, with a median survival of only 13.3 months following metastasis [7]. The current challenge underpinning this poor prognosis is the frequent development of ‘therapeutic resistance’, a complex biological process wherein cancer cells adapt and evolve to evade the cytotoxic or cytostatic effects of anti-cancer treatments. While initial favorable responses to endocrine therapy, chemotherapy, and targeted agents may be observed, tumors often acquire resistance, leading to disease progression, recurrence, and ultimately, diminished patient survival [8,9,10].
Given the profound impact of therapeutic resistance on clinical outcomes in BC, a comprehensive understanding of the mechanisms underlying resistance is paramount to develop effective therapeutic strategies and intervention[11,12]. This review aims to provide a systematic and in-depth analysis of the landscape of BC resistance mechanisms. While Xiong et al. (2025) [13] and Dhiman et al. (2024) [14] offered broad overviews of BC pathophysiology and nanocarrier applications, this review delivers a more granular analysis—especially of the BC–associated microbiome, exosome dynamics, and resistance mechanisms. It also delves deeply into cutting-edge innovations—AI-assisted diagnostics, functionalized nanocarriers, and novel targets—to showcase how modern technologies are breaking through therapeutic barriers and improve patient outcomes [12,15,16].
1.1. Biological Characteristics of Breast Cancer
BC originates from the epithelial lining of mammary ducts or lobules. Histologically, it is predominantly classified as ductal or lobular carcinoma [17,18]. Invasive ductal carcinoma is the most common subtype, accounting for 70–80% of all BC cases. Cancers, including BC, originate from uncontrolled cell growth and spread, typically driven by oncogenic mutations or epigenetic dysregulation. Increasingly, it is recognized that BC cells, similar to other cancers, can reactivate developmental programs inherent to their tissue of origin. Consequently, BC often exhibits a gland-like growth pattern and is histologically classified as adenocarcinoma [17].
The molecular classification of BC is primarily based on the expression of hormone receptors (estrogen receptor [ER], progesterone receptor [PR]) and human epidermal growth factor receptor 2 (HER2) [19,20]. Approximately 70% of BCs are hormone receptor-positive (HR+), encompassing molecular subtypes such as luminal A and luminal B (both HR+), HER2-enriched, and TNBC. Each of these subtypes exhibits distinct molecular characteristics and therapeutic vulnerabilities [21]. Tumors exhibiting HER2 overexpression (15–20%) are effectively managed with targeted anti-HER2 therapies, including trastuzumab. Notably, the ’HER2-low’ expressing subtype has recently emerged as a clinically relevant entity [22], warranting further investigation into optimal management strategies. Conversely, TNBC, constituting 10–15% of cases, is defined by the absence of HER2, ER, and PR expression. Due to its aggressive biological behavior, treatment for TNBC primarily relies on systemic chemotherapy regimens (e.g., anthracyclines, taxanes), although recent advancements in immunotherapy and combination therapies have demonstrated promising outcomes [23]. Hormone receptor-positive tumors (70%) are characterized by their dependence on estrogen and/or progesterone signaling pathways and are effectively treated with endocrine therapies, such as tamoxifen and aromatase inhibitors [24].
The literature included in this review were primarily selected from electronic database. Peer reviewed research papers were sourced from PubMed, Medline and Scopus, spanning from 2000 to July 2025.
1.2. Genetic Risk Factors
Before discussing resistance-related genetic changes, it is important to outline inherited mutations that increase BC risk. Germline mutations in BRCA1 and BRCA2 genes significantly elevate lifetime BC risk—up to 70% by age 80—and are often linked to earlier onset and bilateral disease [25]. Mutations in PALB2 confer a 14% risk by age 50 and up to 35% by age 70 [26]. Carriers of inactivating mutations in the ATM gene face a twofold increase in risk. Other moderate-risk genes include BARD1, RAD51C, and RAD51D, which are associated with ER-negative BC, while CDH1 and CHEK2 gene mutations are more often linked to ER-positive disease [20]. Recognizing these genetic predispositions, the National Comprehensive Cancer Network (NCCN) recommends genetic counseling and testing for high-risk individuals to guide early detection and preventive strategies. The high, moderate and low-risk penetrating genes involved in BC are shown in Table 1.
1.3. Diagnosis and Progression
BC diagnosis typically follows a three-step process: (a) evaluation of medical history and clinical breast examination, (b) imaging via diagnostic mammography or ultrasound, and (c) confirmation through histological analysis of biopsy specimens (National Cancer Institute, 2025). Furthermore, Magnetic Resonance Imaging (MRI) is a valuable tool in diagnosing and managing BC. Breast MRI boasts high sensitivity in detecting BC, often surpassing mammography and ultrasound in finding smaller lesions and those in dense breast tissue. This is a major advantage for women with dense breasts where mammography might be less effective [source: www.hopkinsmedicine.org]. To minimize artifacts and misdiagnosis associated with traditional MRI, Li et al [27] have developed TabPFN algorithm that has increased sensitivity.
Localized disease is classified as in situ when malignant cells remain confined within the ductal or lobular basement membrane. Invasion occurs when these cells breach the membrane and infiltrate surrounding tissue [17,24]. Metastatic BC involves distant spread—commonly to the lungs, liver, bones, and brain—and requires comprehensive treatment strategies. These may include systemic chemotherapy, symptom-directed radiotherapy, and targeted immunotherapies, such as immune checkpoint inhibitors, depending on the tumor’s molecular subtype.
Current trend in Artificial intelligence (AI) is increasingly advancing the accuracy of histopathological classification, diagnosis and enhancing treatment response prediction through the integrated analysis of genomic profiles and medical imaging [28]. Machine learning algorithms now achieve sensitivity and specificity rates exceeding 90% in mammographic image interpretation—often outperforming human radiologists [29].
1.4. Metastasis and Disease Complexity
Metastatic BC accounts for nearly 90% of all BC-related deaths, underscoring its clinical urgency. The complexity arises from an interplay of chronic inflammation, immune suppression, and organ-specific factors that collectively create a supportive niche for tumor cell colonization and growth [16]. Recent discoveries also point to a surprising dimension: the involvement of the nervous system. Emerging studies in cancer neuroscience suggest that tumor cells may exploit neuro-immune signaling and neural remodeling to facilitate metastasis and immune evasion [30]. These findings open exciting and urgent avenues of investigation, highlighting the need for integrative strategies that consider both systemic and neurobiological contributors to metastatic progression.
Several established risk factors increase the likelihood of developing BC. Advancing age is linked to cumulative DNA damage and declining repair mechanisms [31]. A family history, especially BRCA1 or BRCA2 gene mutations, confers a high hereditary risk [32]. Women previously diagnosed with BC are at greater risk of developing contralateral tumors [31]. Early menarche (before age 12) and late menopause (after age 55) prolong hormonal exposure, increasing risk [31]. Nulliparity or first childbirth after age 30 is associated with hormonal imbalance and delayed breast tissue maturation [33]. High breast density, marked by excess glandular tissue, is another independent risk and complicates mammographic detection [31].
Lifestyle factors—including alcohol, obesity (postmenopausal), and smoking contribute through increased estrogen, inflammation, and exposure to carcinogens [34]. Hormone replacement therapy, especially estrogen-progestin combinations, also raises risk by elevating circulating hormones [31]. Radiation exposure during youth, such as treatment for Hodgkin’s lymphoma, can damage breast DNA and elevate long-term risk [31]. Lastly, utero exposure to diethylstilbestrol (DES) increases the lifetime risk for both exposed women and their daughters [31,35]. While these factors influence cancer development, resistance to therapy poses a greater clinical challenge. This resistance is often driven by genetic instability, tumor heterogeneity, drug efflux pumps, and changes in cancer cell metabolism [36]. Figure 1 illustrates common resistance mechanisms contributing to BC progression, metastasis and recurrence. Table 2 summarizes risk factors involved in developing BC.
2. Early Detection and Diagnostic Technologies
Early detection of BC is critical for improving outcomes. Cutting-edge technologies, including high-resolution imaging, molecular profiling, and liquid biopsies, are transforming diagnostic precision and resistance monitoring [44]. Non-invasive tools like circulating tumor DNA (ctDNA) assays, circulating tumor cells (CTCs), and exosomal biomarkers show promise for early detection, real-time disease tracking, and predicting resistance, especially in aggressive subtypes like TNBC [45,46,47]. However, challenges such as resistance mechanisms in advanced or heterogeneous tumors continue to hinder therapeutic success.
Emerging solutions—AI-driven diagnostics, multi-omics integration, and personalized treatment planning—offer hope but face barriers to widespread adoption, including standardization, sensitivity, and equitable access [48,49,50]. These advancements herald a shift toward precision oncology, though rigorous clinical validation and thoughtful integration into practice are vital to unlock their full potential.
3. Therapeutic Strategies and Ongoing Challenges
BC treatment involves a multimodal approach, including surgery, radiotherapy, chemotherapy, endocrine therapy, and targeted agents [24]. Surgical management of BC has evolved toward precision and de-escalation: breast-conserving procedures (lumpectomy, wide excision, quadrantectomy) are now preferred in early-stage tumors, typically followed by radiotherapy to reduce recurrence [51]. Oncoplastic techniques enhance cosmetic outcomes without compromising control. Sentinel lymph node biopsy (SLNB) remains standard for staging in clinically node-negative women, while axillary lymph node dissection (ALND) is limited to those with extensive nodal disease or residual disease after neoadjuvant therapy. Recent trials—including SOUND (Sentinel Node vs Observation After Axillary Ultra-Sound) [52] and INSEMA (Comparison of Axillary Sentinel Lymph Node Biopsy Versus no Axillary Surgery) [53] — support further omission of axillary interventions in selected low-risk patients. Multidisciplinary evaluation now guides surgical planning, balancing tumor biology, systemic therapy timing, and quality-of-life outcomes.
For hormone receptor-positive (HR+) cancers, endocrine therapies such as tamoxifen and aromatase inhibitors remain the standard [54]. Targeted therapies like ‘trastuzumab’ are essential in HER2-positive BC. In postmenopausal HR+ patients, extended aromatase inhibitor therapy has shown superior outcomes to tamoxifen [55]. Immunotherapy, particularly ‘pembrolizumab’, has shown benefit in high-risk TNBC [56].
In MBC, systemic therapies dominate. Options include chemotherapy (e.g., capecitabine), endocrine therapy combined with CDK4/6 inhibitors (e.g., palbociclib) for HR+ disease, ‘trastuzumab’ for HER2+ tumors, and PARP inhibitors (e.g., olaparib) for BRCA-mutant cancers [57]. Immunotherapy benefits PD-L1+ TNBC, while ‘denosumab’ helps manage bone metastases, often alongside palliative care [58]. Treatment selection is guided by molecular subtype (HR+, HER2+, TNBC), patient comorbidities, and genomic markers. Clinical trials continue to explore antibody-drug conjugates (ADCs) and personalized cancer vaccines. HER2-directed regimens (e.g., trastuzumab + pertuzumab) have improved outcomes in HER2+ BC [59]. CDK4/6 inhibitors (palbociclib, ribociclib, abemaciclib) have significantly prolonged progression-free survival in HR+/HER2− MBC [60]. PARP inhibitors show efficacy in BRCA-mutated BC. In TNBC, combining ‘pembrolizumab’ with chemotherapy has improved pathological complete response in neoadjuvant settings [61].
Advances in early detection and targeted treatments have raised the five-year survival rate for localized BC to nearly 90% [62]. Molecular profiling and AI-enhanced diagnostics enable personalized treatment strategies, reducing toxicity and improving outcomes [44,63] Endocrine and HER2-targeted therapies have decreased recurrence in HR+ and HER2+ subtypes. However, therapeutic resistance remains a major hurdle, often driven by somatic mutations and tumor heterogeneity [16,24,64]. TNBC remains particularly challenging due to its aggressive nature and limited targeted options, though immunotherapy offers new hope [32]. Long-term toxicities—such as cardiotoxicity from anthracyclines and bone loss from aromatase inhibitors—continue to affect survivorship [65,66]. Additionally, disparities in access to advanced treatments hinder equitable care. Addressing resistance, improving tolerability, and ensuring global access remain critical goals in advancing BC therapy.
4. Resistance Mechanisms in Breast Cancer
BC resistance to conventional therapies remains a significant obstacle in achieving durable remissions and cures across all metastatic subtypes including hormone receptor-positive (HR+), HER2-positive, and triple-negative (TNBC) [8,10,32,67]. While intricate molecular and cellular resistance mechanisms have been delineated, innovative strategies targeting metabolic vulnerabilities, epigenetic alterations, immune dynamics, and computational advancements are emerging to counteract these barriers. This section focuses on the underlying mechanisms of BC resistance and novel therapeutics and technologies to overcome them. The mechanisms emphasized include cancer metabolism, mitochondrial function, cancer stem cells (BCSCs), the TME, immunometabolism, immunotherapies, and single-cell RNA sequencing (scRNA-seq). While these resistance barriers present considerable obstacles, emerging strategies offer hope for overcoming them [8,10,32,67] Recent advances in AI-medicine aid in early detection, whereas nanotechnology enables targeted drug delivery [63]. Collectively, these approaches aim to disrupt resistance pathways and enhance treatment efficacy across BC subtypes that are ultimately likely to improve clinical outcomes for patients.
4.1. Experimental Models of Resistance Mechanisms
The experimental models play an indispensable role. They provide controlled systems to dissect the underlying biology of drug resistance, explore tumor-stromal interactions, and evaluate genetic and epigenetic drivers of resistance. Unlike clinical practice—where access to patient tumor samples at resistance onset is limited—preclinical models allow for longitudinal, mechanistic studies, enabling earlier detection of resistance pathways and the assessment of potential interventions [68]. However, there exists species differences between mouse models and human situation. The extrapolation of data becomes a challenge in such a situation.
As technologies evolve, CRISPR/Cas9 gene editing, humanized mouse models, 3D bioprinting, and patient-derived organoids, the gap between preclinical findings and clinical applications is becoming narrower, if not totally absent [68,69,70]. Thus, many experimental models are applicable to human situation and enable drug discovery [70].
Over the years, a spectrum of experimental models has been developed and refined to capture the complexity of BC resistance. These include conventional 2D cell line models, more physiologically relevant 3D spheroids and organoids, patient-derived xenografts (PDXs), genetically engineered mouse models (GEMMs), circulating tumor cell (CTC)-derived models, and in silico computational models driven by artificial intelligence [68,69,70,71]. Each of these models offers distinct insights: while 2D models have illuminated key signaling pathways like estrogen receptor (ER) signaling, HER2 amplification, and ABC transporter-mediated drug efflux, organoid and spheroid systems better replicate the spatial architecture and TME-induced drug gradients observed in vivo.
Importantly, these models have not only elucidated mechanisms of resistance but also directly contributed to the discovery and validation of novel therapeutic strategies:
- i)
- ii)
- iii)
- iv)
- AI-based in silico models have facilitated target prediction and drug repurposing strategies, although biological validation remains a bottleneck [71].
Thus, experimental models (Figure 2) serve a dual purpose: they are essential not only for uncovering mechanisms driving drug resistance but also for screening, optimizing, and validating next-generation therapeutics aimed at overcoming these resistance barriers. Despite their utility, no single model fully recapitulates human BC complexity. Each model has inherent limitations in terms of physiological relevance, scalability, cost, and translational predictability (as summarized in Table-3). Therefore, an integrative, multi-model approach combining in vitro, in vivo, and in silico systems represents the most promising strategy to accelerate the discovery of resistance mechanisms and the development of effective therapies. Taken together, the advancement and optimization of experimental models remain foundational to combating BC drug resistance and reducing the staggering mortality associated with it.
4.2. Resistance Due to Drug Efflux
Chemotherapy, a cornerstone of BC treatment, frequently encounters resistance often mediated by the activation of survival pathways, and the upregulation of drug efflux pumps [79,80,81]. Overexpression of ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp/ABCB1) and multidrug resistance-associated protein 1 (MRP1/ABCC1), is a major mechanism of multidrug resistance (MDR) in BC [82,83]. These transporters actively pump drugs, such as doxorubicin and paclitaxel, out of the cell, resulting in intracellular drug concentrations falling below therapeutic levels [82]. In TNBC, the overexpression of ABCB1 is a significant driver of multidrug resistance (MDR). This adaptive response is often triggered by initial exposure to chemotherapy [30,83]. The ATP-dependent efflux process can be further amplified by inflammation, with cytokines like IL-6 and TNF-α upregulating the expression of these transporters [84].
Additionally, alterations in drug metabolism, mediated by changes in the activity of cytochrome P450 enzymes, can reduce chemotherapeutic efficacy by affecting drug activity or clearance [85]. Recent literature demonstrates that nanoparticle-based drug delivery systems can bypass these efflux pumps through various mechanisms [86], thus enhancing treatment efficacy.
4.3. Resistance Due to Genetic Mutations
Mutations in the genes encoding drug targets can directly impede drug binding and reduce efficacy. For instance, mutations in ERBB2 (HER2) gene can lead to constitutive activation of the HER2 protein, conferring resistance to the targeted therapy by trastuzumab [87,88]. Similarly, specific mutations in the estrogen receptor gene (ESR1), such as Y537S, are well-known drivers of endocrine therapy resistance [89]. Beyond direct target alterations, the overexpression of a target like HER2 can sometimes overwhelm the inhibitory capacity of a drug. Conversely, the loss of a target, such as the downregulation of ER expression, can render the corresponding targeted therapy ineffective [90]. Enhanced DNA damage response (DDR) pathways also play a critical role in such resistance. These pathways, including homologous recombination repair (HRR), enable BC cells to repair DNA damage induced by chemotherapies (e.g., cisplatin) and PARP inhibitors. In TNBC with BRCA mutations, PARP inhibitors target single-strand break repair. However, epigenetic modifications, such as BRCA1 promoter hypermethylation, can initially sensitize cells to PARP inhibition but may subsequently contribute to resistance upon prolonged exposure [91,92]. These findings highlight the significant role of epigenetic regulation in driving resistance.
Circulating tumor DNA (ctDNA) offers a non-invasive, real-time window into tumor evolution, enabling dynamic monitoring of resistance mechanisms. In hormone receptor–positive (HR⁺) BC, ctDNA frequently detects ESR1 mutations (e.g., Y537S) [84], which confer endocrine resistance, as well as PIK3CA mutations [85,86] that predict resistance to CDK4/6 inhibitors and guide the use of alpelisib. In TNBC, ctDNA reveals TP53 and RB1 alterations [87] associated with chemotherapy resistance and may detect acquired HER2 amplification [45], indicating therapeutic escape. Emerging AI-driven ctDNA analytics [88,89] can predict immunotherapy resistance with an estimated accuracy of ~78%. Collectively, ctDNA profiling facilitates personalized treatment by capturing the molecular dynamics of resistance in a minimally invasive and clinically actionable format [90].
4.4. Molecular Signaling in Drug Resistance
Activation of the PI3K/AKT/mTOR cell survival pathway within the TME can promote proliferation, leading to resistance to various therapies, including endocrine therapy and chemotherapy [93]. Similarly, activation of the MAPK pathway can contribute to resistance through multiple mechanisms, including increased cell proliferation and survival [94]. The CXCL12/CXCR4 signaling pathway promotes immune suppression, increases fibrosis, and limits infiltration of cytotoxic immune cells in breast tumors [95]. PI3K/AKT and MAPK pathways mediate resistance by promoting tumor cell proliferation and survival, particularly in response to targeted therapies [93,94]. The TGF-β signaling pathway is involved in immune evasion and enhances the epithelial-to-mesenchymal transition (EMT), leading to increased metastatic potential [96]. These molecular signaling events are depicted in Figure 3.
4.5. Role of Microbiota in Therapeutic Resistance
BC tissues harbor a distinct and dysbiotic microbiome compared to normal breast tissue, significantly influencing tumor biology, immune modulation, and therapeutic outcomes [141,142]. While normal breast tissue contains a conserved microbial community (Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes), BC tissues exhibit reduced α-diversity comprising Escherichia-Shigella, Staphylococcus, and Fusobacterium [143,144]. Specifically, Methylobacterium radiotolerans and Sphingomonas yanoikuyae are elevated in BC tissues [145], the former potentially supporting tumor survival by modulating oxidative stress and lipid metabolism, while the latter’s depletion is associated with dysbiosis and cancer progression [144,148]. These microbial shifts appear subtype-specific, with TNBC and estrogen receptor-positive (ER+) tumors displaying distinct microbial signatures that may influence immune evasion and therapy resistance [146]. In this regard, Fusobacterium nucleatum promotes BC cell migration, metastasis, and immune suppression via the miR-21-3p/FOXO3 axis [147]. Stenotrophomonas maltophilia has also been linked to reduced CD8⁺ T-cell infiltration, further contributing to immune suppression within the TME [97,98].
Importantly, intratumoral bacteria are not limited to extracellular niches but are also localized intracellularly within epithelial cells, fibroblasts, and immune cells (tumor-associated macrophages (TAMs), dendritic cells (DCs), and neutrophils). The intracellular microbes interact with innate immune sensors such as Toll-like receptors (TLRs) and NOD-like receptors, inducing low-grade inflammation, facilitating immune evasion, and promoting metastatic dissemination [145]. These interactions further impair cytotoxic CD8⁺ T-cell infiltration and drive resistance to chemotherapy, endocrine therapy, and immune checkpoint blockade [145,146] (See Figure 4). Angiogenesis, a hallmark of BC progression, is also shaped by tumor-resident and gut microbiota through inflammatory and metabolic signaling pathways [149]. These findings establish that the breast tumor microbiota serve as key modulators of the TME, influencing immune tone, vascular dynamics, metastasis, and therapeutic resistance [142,150,151,152].
4.6. Tumor Microenvironment in Cancer Resistance
The BC-TME is a dynamic and complex ecosystem comprising cancer cells, stromal components, vasculature, and infiltrating immune cells. Increasing evidence underscores the TME’s pivotal role in tumor progression, immune evasion, and resistance to both conventional and targeted therapies [99,100,101]. Across subtypes—HR⁺, HER2⁺, and TNBC—the TME contributes to resistance through mechanisms such as immunosuppression, fibrosis, and dysregulated stromal-tumor signaling [102].
Key cellular contributors include cancer-associated fibroblasts (CAFs), which deposit dense extracellular matrix (ECM) and secrete pro-survival cytokines (e.g., TGF-β, HGF, IL-6), thereby enhancing drug resistance via PI3K/AKT and MAPK pathways [103]. Tumor-associated macrophages (TAMs), predominantly M2-polarized, promote immune suppression and metastasis through cytokines like IL-10 and TGF-β, while regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) inhibit cytotoxic T-cell responses. Cancer stem cells (CSCs) contribute further by maintaining tumorigenic capacity and resisting therapies through self-renewal and plasticity.
In HER2⁺ BC, TAMs suppress antibody-dependent cellular cytotoxicity (ADCC), thereby compromising trastuzumab efficacy [104]. Concurrently, PIK3CA mutations—found in 30–40% of HER2⁺ tumors—sustain PI3K/AKT/mTOR signaling despite HER2 blockade, promoting resistance [105,106]. In TNBC, CCL2-mediated recruitment of immunosuppressive myeloid cells limits immunotherapeutic responses [107,108]. Hypoxic TME conditions—driven by HIF-1α—exacerbate resistance by inducing ECM remodeling, angiogenesis, and direct modulation of drug metabolism [109,110].
The TME supports BC stem cell (BCSC) maintenance, a key driver of therapeutic resistance and recurrence. IL-6 signaling reinforces BCSC stemness; targeting this axis with IL-6 inhibitors such as tocilizumab is under exploration, though direct clinical evidence in BC is still limited [111,112]. Hypoxia, another key contributor, promotes BCSC plasticity. Agents like evofosfamide and salinomycin have shown preclinical efficacy in targeting hypoxia-adapted and stem-like cells, respectively [113,114]. Additional strategies targeting Notch signaling (e.g., ATRA, γ-secretase inhibitors) reduce the stem-like subpopulation [115]. Despite promising preclinical data, clinical translation remains challenging. Rational combinations of BCSC-targeted agents with standard therapies, supported by robust biomarkers to track BCSC dynamics, are needed to ensure durable responses [116,117,118].
4.7. Tumor Vasculature in Resistance
Aberrant angiogenesis, driven by vascular endothelial growth factor (VEGF) overexpression, is a hallmark of BC progression and therapeutic resistance [119]. Tumor vasculature exhibits structural and functional abnormalities—leakiness, tortuosity, and poor perfusion—that impair drug delivery, exacerbate hypoxia, and foster an immunosuppressive TME [119,120]. This section examines the role of dysfunctional vasculature in resistance, its interplay with immunosuppression, and emerging strategies to enhance treatment efficacy [120,121].
VEGF-driven angiogenesis in BC creates disorderly, permeable vessels that elevate interstitial fluid pressure and induce severe hypoxia within the TME [119,122,123]. Hypoxia activates hypoxia-inducible factor-1α (HIF-1α), promoting cancer cell survival and resistance to chemotherapy and targeted therapies, such as trastuzumab, particularly in triple-negative BC (TNBC) [119]. The TME subsequently recruits immunosuppressive cells, including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2-polarized tumor-associated macrophages (TAMs), which inhibit cytotoxic T-cell activity and reduce immune checkpoint inhibitor efficacy [121]. VEGF further suppresses immunity by inhibiting dendritic cell maturation and expanding immunosuppressive populations, amplifying resistance to immunotherapies, including exploratory CAR-T-cell therapies [124,125]. These concepts were analyzed and explored in detail by Fukumura et al [121,124].
Dysfunctional vasculature creates both physical and biochemical barriers to treatment. Poor perfusion limits drug penetration, reducing the efficacy of chemotherapy and targeted agents [119,123]. Hypoxia-driven HIF-1α pathways upregulate survival genes (e.g., BCL-2, MDR1), enhancing resistance in TNBC and HER2-positive BC [122,126]. Taken together, tumor angiogenesis often results in a low-pH and hypoxic TME, which eventually limits drug efficacy. Stromal components, such as cancer-associated fibroblasts (CAFs) and extracellular matrix (ECM), compress vessels, further restricting drug and immune cell access, including CAR-T cells, which face infiltration barriers due to the immunosuppressive TME [127,128]. These vascular and stromal barriers exacerbate resistance across BC subtypes, necessitating targeted interventions.
The seminal work on vascular normalization demonstrates that anti-VEGF therapies, such as bevacizumab, transiently restore vessel function, improving perfusion and drug delivery while reprogramming the TME to enhance immunotherapy outcome [120,121,129]. By reducing hypoxia and immunosuppressive cell recruitment, normalization sensitizes tumors to immune checkpoint inhibitors and may improve CAR-T-cell infiltration in TNBC, though toxicities remain a challenge [127,128]. Emerging strategies combine vascular normalization with stroma-targeting agents (e.g., FAP or TGF-β inhibitors) and immune checkpoint blockade to dismantle TME barriers [130]. Dr. Jain hypothesized that not only blood vessels, but also other components of the TME are abnormal and all these abnormalities in concert fuel tumor progression and treatment resistance [120]. Integrated approaches combining vascular normalization with stroma-targeting agents (e.g., FAP or TGF-β inhibitors) and immune checkpoint blockade, as evidenced by clinical trials (e.g., NCT03394287 for VEGFR2 inhibitor and anti-PD-1 combinations), aim to overcome resistance and enhance outcomes in BC [131].
4.8. Role of Tumor Associated Macrophages in Immunosuppression
Persistent antigen exposure and immunosuppressive cues from TAMs drive T-cell exhaustion in the TME. This includes upregulation of inhibitory receptors (PD-1, CTLA-4, TIGIT) and suppression of cytokine production (IFN-γ, IL-2, TNF) in CD8⁺ T cells—even in early-stage BC [125,132,133]. These exhausted T cells also exhibit metabolic dysfunction, shifting toward oxidative phosphorylation in nutrient-deprived niches and showing impaired effector function, further limiting the efficacy of immune checkpoint blockade (ICB) therapies [134]. TAM reprogramming is a therapeutic focus. In HR⁺ BC, CSF1R inhibition (e.g., pexidartinib) reduces M2-like TAMs and enhances CD8⁺ T-cell infiltration [135]. In TNBC, CCL2/CCR2 axis blockade (e.g., bindarit) disrupts TAM and MDSC recruitment, improving chemotherapy response [136]. Engineering exhaustion-resistant CD8⁺ T cells (e.g., via TOX knockdown) and combining ICB with TME modulators represent synergistic strategies [137]. These mechanisms are illustrated in Figure 5. Cold TMEs are immunologically inert, featuring stromal exclusion, hypoxia, and limited immune infiltration. In contrast, hot TMEs exhibit T-cell infiltration, normalized vasculature, and enhanced immune responsiveness. Emerging omics-driven technologies and rationally designed therapies aim to convert cold tumors into hot, immuno-responsive states, offering new avenues to enhance clinical efficacy in BC.
4.9. Role of Exosomes in TME Modulation
The TME also facilitates resistance through exosome-mediated signaling and metabolic reprogramming. Exosomes play a pivotal role in propagating drug resistance by transferring key molecular cargo—such as miRNAs, lncRNAs, and proteins—that reprogram recipient cells toward epithelial–mesenchymal transition (EMT), stemness, and drug efflux phenotypes [46,138]. In BC, tumor-derived exosomes contribute to doxorubicin resistance by stabilizing HIF-1α and delivering lncRNA H19 to adjacent cells, thereby reinforcing a resistant microenvironment [139,140]. Notably, silencing HIF-1α diminishes exosomal H19 levels and restores sensitivity to chemotherapy. Similarly, exosomes from CAFs enhance tumor stemness and therapy resistance by transferring regulatory miRNAs and signaling proteins. Metabolically, competition for nutrients and mitochondrial dysfunction in immune cells—such as T-cell exhaustion via defective mitochondrial transfer through tunneling nanotubes [141]—further weaken anti-tumor immunity. NK cells are also impaired by Treg-derived IL-10 [142]. These complex intercellular interactions underscore the role of the TME as a central driver of resistance, as shown in Figure 5.
5. Metabolic Reprogramming in BC Resistance
Metabolic homeostasis is a cornerstone of normal physiological function at both organismal and cellular levels. In BC, this homeostatic balance is profoundly disrupted within the tumor microenvironment, driving malignant progression, therapeutic resistance, and immune evasion[142,143,144] . Tumor cells, stromal components, and immune effectors undergo subtype-specific metabolic reprogramming—a hallmark of cancer—to sustain proliferation, adapt to therapeutic stress, and subvert anti-tumor immunity [142]. In BC, tumor cells rewire glycolysis, glutaminolysis, fatty acid oxidation, and mitochondrial dynamics, while stromal cells enhance lactate production and immune cells face nutrient deprivation, collectively fostering a metabolically hostile TME [12,143,144,145]. Hypoxia, nutrient scarcity, and TME acidosis impose selective pressures that promote metabolic plasticity, enabling resistance to chemotherapy, targeted therapies, and immunotherapies across hormone receptor-positive (HR+), HER2-positive (HER2+), and TNBC subtypes. This section elucidates how these metabolic shifts, intricately linked to immune suppression and therapeutic failure, pose formidable barriers to effective BC treatment.
Otto Warburg proposed that cancer cells prefer glycolysis over oxidative phosphorylation (OXPHOS) due to dysfunctional mitochondria [146]. While groundbreaking, this hypothesis is now recognized as incomplete. Later studies, including Warburg’s own, did not consistently demonstrate defective mitochondrial respiration as a hallmark of malignancy [147]. In fact, mitochondrial respiration and related functions are now understood to be critical for tumor progression and immune evasion [144]. In BC, the preference for aerobic glycolysis—commonly referred to as the ‘Warburg effect’—is largely driven by oncogenic signaling pathways such as PI3K/AKT, MYC, and HIF-1α. These pathways upregulate key glycolytic enzymes, including hexokinase 2, pyruvate kinase M2, and lactate dehydrogenase A (LDHA), supporting anabolic processes such as nucleotide and lipid synthesis rather than merely producing ATP [144,148]. Importantly, most BC cells retain intact mitochondria and the capacity for OXPHOS. This metabolic flexibility—often termed metabolic plasticity—allows cancer cells to switch between energy sources depending on environmental pressures. Imaging studies using fluorodeoxyglucose positron emission tomography (FDG-PET) have linked high glucose uptake not only to rapid proliferation but also to dynamic remodeling of the tumor microenvironment [144].
Lactate, a byproduct of cancer cell metabolism, is exported from tumor cells via monocarboxylate transporters (MCT1 and MCT4), leading to acidification of the TME to a pH of 6.5–6.8. This acidic environment impairs the function of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, suppresses interferon-γ production, and promotes an ‘immune-cold’ TME. These effects are especially pronounced in hormone receptor-positive (HR+) and HER2-positive BCs, which often show poor responses to immune checkpoint inhibitors [149,150]. In TNBC, cancer cells rely heavily on glycolysis, which is linked to a pathway that helps them neutralize harmful reactive oxygen species (ROS) produced by chemotherapy drugs like anthracyclines [144]. Concurrently, IL-6/HIF-1α signaling amplifies glycolytic flux and lactate export, compounding extracellular acidification and further hindering drug diffusion [151].
This dual function of aerobic glycolysis—fueling biosynthetic pathways while shaping an immunosuppressive TME—underscores its central role in breast cancer’s therapeutic resistance. Nutrient depletion within the TME impairs the function of cytotoxic T lymphocytes and natural killer cells by limiting the energy required for their proliferation and tumor-killing activity [152]. Accumulated lactate further disrupts T-cell receptor signaling and glycolytic metabolism, while IL-6–mediated stabilization of HIF-1α drives excessive glycolysis. Together, these changes create a self-reinforcing loop that promotes immunosuppression and therapeutic resistance. In TNBC, this ‘metabolic-immune axis’ not only shields tumors from immune surveillance but also reduces drug penetration by altering TME pH and upregulating angiogenic factors like VEGF [153]. This dual role—immune evasion and chemoresistance—positions metabolic rewiring as a linchpin of BC’s adaptive resilience, necessitating integrated therapeutic strategies to disrupt this vicious cycle [149,151].
As glycolytic capacity saturates, BC cells, particularly HR+ subtypes, exhibit glutamine addiction to sustain mitochondrial bioenergetics under therapeutic stress [154]. Glutaminase 1 (GLS1) converts glutamine to glutamate, replenishing the tricarboxylic acid (TCA) cycle for anaplerosis and glutathione synthesis to counter oxidative stress [155,156]. In HR+ BC treated with aromatase inhibitors, GLS1 and ASCT2 upregulation maintain redox homeostasis and biomass production. In TNBC, glutaminolysis supports rapid proliferation and ROS detoxification, enabling survival during chemotherapy [157]. On the other hand, glutamine starvation of T-cells significantly hinders T-cell proliferation and cytokine production. Collectively, metabolic plasticity—toggling between glycolysis, glutaminolysis, and fatty acid oxidation—allows BC cells to adapt to nutrient scarcity and therapeutic pressures, rendering single-pathway targeting ineffective [158].
5.1. Therapeutic Targeting of BC Metabolism
Targeting the metabolic vulnerabilities of BC offer a promising strategy to overcome therapeutic resistance. However, translating preclinical successes to the clinic remains challenging. Inhibitors like 2-deoxyglucose (2-DG) disrupt glycolysis by blocking hexokinase, thus enhancing the efficacy of chemotherapy in BC models by starving tumors of energy and biosynthetic precursors [148]. Similarly, CB-839 (telaglenastat), a glutaminase 1 (GLS1) inhibitor, synergizes with mTOR inhibitors and DNA-damaging agents in TNBC, reducing tumor growth by approximately 40% in xenograft models [157]. Dichloroacetic acid (DCA) shifts metabolism from glycolysis to oxidative phosphorylation (OXPHOS) by inhibiting pyruvate dehydrogenase kinase, promoting apoptosis in BC cells when combined with photodynamic therapy [143]. Despite these advances, clinical trials, such as a phase II study of CB-839 in TNBC (NCT02771626), have shown modest efficacy, hampered by systemic toxicity and the metabolic heterogeneity of BC subtypes [159]. Furthermore, metabolic- epigenetic interactions complicate targeting efforts, as metabolites like acetyl-CoA fuel histone acetylation, driving adaptive gene expression that sustains resistance [160]. Combinatorial approaches, integrating metabolic inhibitors with targeted therapies or epigenetic modulators, are more effective to overcome these barriers and restore therapeutic sensitivity.
Thus, metabolic reprogramming is a primary driver of BC resistance and immune evasion, demanding integration into precision oncology. Combining metabolic inhibitors (e.g., CB-839, mdivi-1) with targeted therapies or ICIs could disrupt the immunosuppressive TME. Biomarkers like GLS1 expression, 18F-fluoroglutamine PET, or lactate levels are critical for patient stratification [159]. Targeting metabolic-epigenetic interactions and TNT-mediated immune sabotage offers novel avenues to overcome resistance, particularly in immunologically refractory TNBC.
Targeting TME heterogeneity remains a major challenge. Lifestyle interventions—such as diet, exercise, and stress reduction—may positively modulate TME composition [161]. Clinically, PD-1/PD-L1 inhibitors (e.g., pembrolizumab) have shown efficacy in TNBC, with the KEYNOTE-522 trial demonstrating improved event-free survival in early-stage disease [75,162]. Other promising avenues include: (a) IL-15 agonists to activate NK cells [163]; (b) CXCL12/CXCR4 axis blockade to enhance immune infiltration [164]; (c) TGF-β inhibition, which restores T-cell function and blocks EMT [165,166,167]; (d) TAM repolarization from M2 to M1 phenotypes [168,169]. Combination regimens involving immunotherapy, chemotherapy, and epigenetic modulators are under active investigation. Liquid biopsy tools offer non-invasive means to monitor resistance mechanisms and tailor therapy in real time. Together, these strategies provide a comprehensive framework to overcome immune evasion and resistance driven by the TME.
6. Breast Cancer Stem Cells (BCSCs) in Resistance
BC stem cells (BCSCs) are a rare tumor subpopulation with stem-like properties, notably self-renewal and multilineage differentiation. These traits allow them to initiate, sustain, and regenerate tumors, driving heterogeneity, metastasis, and relapse [170,171]. Unlike bulk tumor cells, BC-stem cells (BCSCs) are highly resilient, persisting after therapy and replenishing the tumor mass, which drives treatment failure. Their resistance arises from mechanisms such as enhanced DNA repair, drug efflux through transporters like ABCB1 and ABCG2, quiescence, and activation of survival signaling pathways, including PI3K/AKT and Bcl-2. These traits are regulated by developmental pathways—Notch, Wnt/β-catenin, Hedgehog, and Hippo—that sustain BCSC stemness and survival [172,173,174].
Each dormancy state is associated with unique metabolic adaptations—including OXPHOS, ROS scavenging, High ALDH activity, particularly ALDH1, further aids in drug detoxification [175]. This includes cytokines (e.g., IL-6, CXCL8), growth factors, hypoxia, and stromal cells like CAFs, which maintain BCSC stemness and promote immune evasion [174,176]. EMT, often induced by TME signals, triggers stem-like traits and enhances BCSC migratory capacity, directly contributing to metastasis [177]. These properties are reinforced by cues from the TME. The TME autophagy, and unfolded protein response (UPR)—that enable tumor cells to evade therapy and immune surveillance. Reactivation of these dormant cells may lead to tumor relapse or secondary metastases. BCSCs can invade tissues, intravasate, survive circulation, and colonize distant sites, underscoring their role in the metastatic cascade. These cells also evade immune attack by expressing PD-L1, secreting immunosuppressive cytokines (e.g., TGF-β, IL-10), and recruiting Tregs and MDSCs, shaping an immunosuppressive niche [178,179].
Dormancy is a critical BCSC feature that allows them to escape therapy and persist as minimal residual disease [178]. Dormant BCSCs exist in two main forms: (1) cellular dormancy (quiescent G0-phase cells) and (2) angiogenic dormancy (small clusters without sufficient vasculature). These cells may lie latent for years and reactivate under favorable TME cues, triggering relapse [180]. Their survival across time and treatment makes them key mediators of late recurrence and metastasis (Figure 6).
Targeting BCSCs is essential for durable BC control [181] Dittmer, 2018). Approaches include: (a) Pathway Inhibition: Small molecules or antibodies targeting Notch (e.g., MK-0752), Wnt (e.g., LGK974), and Hedgehog signaling are in early-phase trials for reducing BCSC populations [179,182]; (b) Immunotherapy: Preclinical efforts using CAR-T cells directed at BCSC-specific antigens show promise [117]; (c) Differentiation Therapy: Agents like all-trans retinoic acid (ATRA) may induce BCSC differentiation, sensitizing them to chemotherapy [182].
7. Role of Mitochondria in BC Resistance
Mitochondria have emerged as key modulators of therapeutic resistance in BC, influencing not only cellular metabolism but also apoptosis, redox balance, and immune responses [183,184]. Beyond their canonical role as ATP generators, mitochondria act as integrative hubs for signaling pathways that support tumor progression and adaptation to treatment. Mechanistically, mitochondrial dysfunction and plasticity contribute to BC resistance through a variety of pathways—including somatic mitochondrial DNA (mtDNA) mutations, metabolic heterogeneity, dysregulated mitochondrial dynamics, and intercellular mitochondrial transfer [141,183]. This section critically examines how mitochondrial genetics, functional variability, and organelle exchange contribute to therapeutic failure across BC subtypes. These mechanisms not only fuel metabolic adaptability but also compromise anti-tumor immunity, particularly through mitochondrial hijacking of T cells and other immune effectors [141,185].
The mitochondrial genome encodes 13 essential proteins of the electron transport chain (ETC), yet it remains highly susceptible to damage due to its proximity to ROS, lack of histone protection, and limited DNA repair capacity [186]. In BC, recurrent somatic mutations in genes such as MT-ND1, MT-ND4, and MT-ND5 (Complex I subunits) have been documented [187]. Rather than impairing respiration, these mutations often promote metabolic plasticity, enabling cancer cells to tolerate oxidative stress or shift towards an OXPHOS-favorable state under treatment pressure. Some mtDNA variants are associated with aggressive phenotypes, altered redox signaling, and resistance to genotoxic therapies [148,188,189]. Additionally, mtDNA mutations may influence immune recognition by altering mitochondrial antigen presentation. Altogether, mtDNA instability contributes to BC progression, and its signatures may serve as both biomarkers and therapeutic targets.
‘Mitochondrial heterogeneity’ represents variations in organelle mass, membrane potential, ROS output, and metabolic behavior, is a defining feature of treatment-resistant breast tumors. HR+ subtypes often rely on OXPHOS and fatty acid oxidation, whereas TNBCs display heightened glycolytic and glutamine metabolism [190]. Even within a single tumor, diverse mitochondrial phenotypes coexist, allowing subpopulations of cells to escape metabolic or drug-induced stress [188,191]. A well-characterized resistance mechanism involves dysregulated mitochondrial dynamics. Dynamin-related protein 1 (Drp1)-mediated mitochondrial fission leads to fragmented mitochondria with reduced ROS output, attenuated pro-apoptotic signaling, and increased resistance to stress. In HER2-positive tumors, elevated Drp1 activity has been linked to trastuzumab resistance [192].
Additionally, overexpression of anti-apoptotic Bcl-2 family proteins—including Bcl-2, Mcl-1, and Bcl-xL—prevents cytochrome c release by inhibiting mitochondrial outer membrane permeabilization (MOMP), thereby blunting intrinsic apoptosis pathways [193]. In TNBC, PGC-1α-driven mitochondrial biogenesis augments OXPHOS capacity, facilitating survival under chemotherapy-induced metabolic stress [194]. Somatic mutations in MT-ND4 and other ETC genes further enhance ETC efficiency, promoting metastasis and potentially altering immunogenicity [195]. These mitochondrial adaptations form a multifaceted resistance system—supporting energy production, limiting apoptosis, and facilitating immune escape.
7.1. Intercellular Mitochondrial Transfer
One of the most remarkable features of tumor mitochondrial biology is their ability to traffic between cells. Intercellular mitochondrial transfer occurs via tunneling nanotubes (TNTs), microvesicles, gap junctions, and cell fusion, forming a dynamic exchange network between tumor cells and stromal or immune cells [196,197]. TNTs, acquire functional mitochondria from stromal cells—enhancing survival—or export damaged mitochondria to immune cells, impairing their effector function. This process is mediated by cytoskeletal regulators and trafficking proteins such as Miro1/2 and Mitofusins. While the implications of this mitochondrial "networking" are still being defined, it clearly contributes to bioenergetic resilience and cellular reprogramming under therapy-induced pressure. As shown in Figure 7, mitochondria can play a multifaceted role of in BC resistance.
Key mechanisms include:(1) mtDNA mutations in Complex I (e.g., MT-ND4) that promote ETC efficiency and metastasis; (2) Mitochondrial heterogeneity and dynamic remodeling that enable metabolic plasticity and apoptotic resistance; (3) Intercellular mitochondrial transfer through tunneling nanotubes (TNTs), vesicles, and fusion events. An emerging resistance axis involves mitochondrial hijacking, where BC cells transfer depolarized or dysfunctional mitochondria into CD4⁺ and CD8⁺ T lymphocytes, particularly within the TME [141]. These mitochondrial adaptations contribute to treatment resistance and immune evasion, highlighting potential therapeutic targets in mitochondrial trafficking and metabolism.
Importantly, immune suppression in BC may occur independently of classical immune checkpoint pathways, potentially explaining the poor response to checkpoint blockade in some patients with apparent T-cell infiltration [198]. Recent studies suggest that tunneling nanotubes (TNTs) enable the transfer of functional mitochondria from stromal or immune cells to cancer cells, contributing to immune evasion and metabolic reprogramming [199,200]. Disruption of TNTs using cytoskeletal inhibitors such as cytochalasin B or latrunculin A, or inhibition of mitochondrial trafficking proteins like Miro1, has shown potential in preclinical models to restore immune competence [197,201] . However, the physiological roles of TNTs in normal tissues raise safety concerns for systemic targeting. Mitochondrial transfer thus plays a dual role in the TME: (1) conferring metabolic flexibility to cancer cells, and (2) attenuating immune effector function through intercellular energy redistribution—highlighting the immunometabolic complexity of the TME.
Taken together, mitochondria in BC serve roles beyond ATP generation: they actively contribute to therapy resistance, immune escape, and cellular plasticity. Their heteroplasmic variation, dynamic morphological behavior, and capacity for intercellular trafficking complicate efforts to develop durable therapies. Future therapeutic strategies should focus on disrupting key mitochondrial resistance mechanisms. These include inhibiting mitochondrial fission and fusion dynamics (e.g., Drp1 inhibitors such as Mdivi-1), blocking tunneling nanotube (TNT)-mediated organelle trafficking, targeting anti-apoptotic mitochondrial pathways (e.g., BCL-2 family proteins), and designing biomarker-guided combinations of metabolic and immunotherapeutic agents. Emerging platforms such as live-cell mitochondrial imaging, spatial metabolomics, and single-cell transcriptomic profiling will be instrumental in identifying mitochondrial phenotypes in clinical samples. These tools may enable a precision oncology framework that exploits mitochondrial vulnerabilities as next-generation therapeutic targets.
8. Immunotherapy in Breast Cancer
The immune system plays a critical role in surveilling and eliminating BC cells through cancer immunoediting, a process involving antigen release, T-cell priming, and tumor elimination [202,203]. However, the TME in BC disrupts this cycle, promoting immune evasion and therapeutic resistance. Furthermore, BC TME establishes a supportive niche where cancer cells can interact with immune cells and neighboring endothelial cells and is thus a feasible target for cancer therapy [204]. TNBC shows partial responsiveness to immunotherapy, while hormone receptor-positive (HR+) and HER2-positive (HER2+) tumors often remain immune-cold due to TME-mediated suppression [205]. Driven by immune checkpoints (PD-1/PD-L1, CTLA-4), pro-tumoral cells (TAMs, CAFs), cytokines (TGF-β, IL-6), and immunometabolic alterations, the TME creates an immunosuppressive niche. This section explores immune evasion mechanisms, current and emerging immunotherapies, and strategies to overcome TME barriers, emphasizing integrative approaches to restore immune surveillance and address resistance across BC subtypes.
The PD-1/PD-L1 axis suppresses T-cell activity in the BC TME, promoting immune evasion [206,207]. Immune checkpoint inhibitors (ICIs), such as pembrolizumab and atezolizumab, enhance T-cell responses, with the KEYNOTE-522 trial showing a 13.6% increase in pathological complete response (pCR) rates in early-stage TNBC when pembrolizumab is added to chemotherapy [23,56]. However, resistance persists due to MHC class I downregulation, alternative checkpoints (e.g., LAG-3, TIM-3), and TME immunosuppression [49,208]. CTLA-4 inhibitors, less effective in HR+ tumors, show potential in combination with PD-1 inhibitors to enhance T-cell infiltration [205]. Combining ICIs with chemotherapy induces immunogenic cell death, improving antigen presentation, though TGF-β-mediated immunosuppression limits broader efficacy [209,210].
8.1. Immunometabolism and TME Modulation
Metabolic reprogramming in the BC TME impairs anti-tumor immunity. Tumor glycolysis (Warburg effect) produces excess lactate, acidifying the TME (pH ~6.5–6.8) and suppressing CD8⁺ T-cell function via mTOR inhibition while promoting regulatory T-cell (Treg) expansion [204,211]. Indoleamine 2,3-dioxygenase 1 (IDO1), a tryptophan-catabolizing enzyme, enhances immunosuppression in metastatic BC, with its inhibition reducing TNBC invasiveness [204,212]. Targeting TME cellular components is critical. CSF1R blockade reprograms tumor-associated macrophages (TAMs) from pro-tumor (M2-like) to anti-tumor (M1) phenotypes, enhancing T-cell infiltration in TNBC [169,213]. Inhibiting fibroblast activation protein (FAP) or TGF-β signaling reduces immune exclusion by cancer-associated fibroblasts (CAFs) [213,214]. Blocking IL-6, TGF-β, or the CXCL12/CXCR4 axis further enhances T-cell trafficking and synergizes with ICIs [215].
8.2. Emerging Immunotherapies
Personalized neoantigen vaccines targeting tumor-specific mutations show immunogenicity in TNBC, using antigens like HER2 and MUC1, though low tumor immunogenicity and TME tolerance limit efficacy [216]. Novel vaccine formulations and delivery strategies are under investigation to address scalability and timing challenges [216]. Chimeric antigen receptor T-cell (CAR-T) therapy, transformative in hematologic malignancies, is exploratory in BC due to the immunosuppressive TME, antigen heterogeneity, and significant toxicities, including cytokine release syndrome (CRS), neurotoxicity (ICANS), and off-target effects causing inflammation from cellular debris [217,218].
In TNBC, CAR-T cells targeting MUC1 (NCT04025216) and mesothelin (MSLN; NCT02792114) show preclinical promise, with MUC1 overexpressed in ~90% of BCs and MSLN effective in chemoresistant models [219,220,221]. HER2-targeted CAR-T cells, including bispecific HER2/MUC1 CARs, reduce antigen escape in HER2+ BC (NCT04660929) [222]. Multi-armored CAR-T cells with PD-1 or TGFBR2 knockouts enhance T-cell activity in TNBC models [223]. Emerging CAR-based therapies, such as CAR-macrophages (CAR-M) and CAR-NK cells, show potential in navigating the TME [223]. [223]. Combination therapies (e.g., CAR-T with anti-PD-L1 or CDK7 inhibitors) improve efficacy but face challenges from toxicity and high costs [224,225]. These therapies, while promising, require overcoming TME barriers, antigen escape, and manufacturing hurdles to become viable BC treatments.
9. Exosomes in Breast Cancer Resistance and Therapy
Exosomes are nanosized (30–150 nm) lipid-bilayer vesicles originating from multivesicular bodies (MVBs), which fuse with the plasma membrane to release their cargo into extracellular fluids. Both normal and cancer cells secrete exosomes for intercellular communication; however, BC cells secrete significantly more exosomes, positioning them as key mediators of therapy resistance. These vesicles deliver diverse bioactive cargo—including mRNAs, miRNAs, long non-coding RNAs (lncRNAs), proteins, and lipids—that modulate the TME to support tumor survival and therapy evasion [46]. Exosome-mediated signaling enhances angiogenesis, immune suppression, and extracellular matrix (ECM) remodeling, contributing to systemic therapy failure [138,139]. In TNBC, exosomes from chemoresistant cells transfer resistance-conferring molecules. For example, ABCB1 mRNA-enriched exosomes enhance P-glycoprotein expression in recipient cells, reducing cisplatin and doxorubicin efficacy [226]. Similarly, exosomal miR-21 promotes resistance by suppressing PTEN and activating PI3K/AKT signaling [226,227].
Beyond chemoresistance, exosomal lncRNA SNHG16 upregulates PD-L1, attenuating T-cell responses to anti-PD-1 therapies [228]. In hormone receptor-positive (HR+) BC, miR-221 and miR-222 delivered via exosomes downregulate estrogen receptor (ER), promoting endocrine resistance [226,227]. Pro-inflammatory cytokines such as IL-6 and TNF-α further amplify exosome release and oncogenic signaling [229]. Additionally, exosomal TGF-β promotes epithelial-to-mesenchymal transition (EMT), enhancing metastatic potential [230,231].
Cancer-associated fibroblasts (CAFs), abundant in BC stroma, facilitate tumor progression and resistance through secretion of growth factors, tumor-promoting exosomes, ECM remodeling, and immunosuppression [232]. CAF-derived exosomes enriched in IL-8 sustain stem-like phenotypes in TNBC cells, reinforcing resistance [233]. CAF-derived exosomes also reprogram tumor metabolism by transferring detoxification enzymes and metabolic intermediates, enhancing survival under therapeutic stress [234]. These insights position CAFs and their exosomal signaling as therapeutic targets, with emerging strategies aimed at disrupting exosome release or blocking downstream signaling pathways [234,235].
Targeting exosome biogenesis, release, or cargo is an emerging approach to counter exosome-mediated resistance. Rab27a inhibition, for instance, limits exosome secretion and sensitizes BC cells to chemotherapy [236]. Engineered exosomes carrying miR-134 suppress chemoresistance in TNBC by downregulating STAT5B and Hsp90, enhancing cisplatin sensitivity[237]. Similarly, exosomes from drug-sensitive BC cells delivering miR-765 show potential in re-sensitizing resistant cells [238]. Combining exosome-targeted strategies with TME-modulating agents, such as TGF-β inhibitors or CSF1R antagonists, may offer synergistic benefits [239]. However, clinical translation remains challenged by exosome production scalability and tumor heterogeneity.
9.1. Role of MinPP1 in Carcinogenesis
While exosomes traditionally originate from MVBs, emerging evidence implicates the endoplasmic reticulum (ER) in exosome biogenesis, particularly under ER stress [240,241]. ER-derived proteins detected within exosomes suggest additional roles in cancer progression and therapy resistance. In this context, our laboratory has identified secretion of multiple inositol polyphosphate phosphatase (MinPP1), typically an ER-resident enzyme, within exosomes (Figure 8) under ER stress conditions [242]. MinPP1, located near the tumor suppressor PTEN on chromosome 10q23, regulates inositol phosphate metabolism and is linked to PI3K/AKT signaling. Loss of this locus, encompassing both MinPP1 and PTEN, is common in cancers. Inositol hexakisphosphate (InsP6), a key substrate of MinPP1, is known to inhibit proliferation and induce apoptosis [243,244,245]. Our work demonstrates that MinPP1 hydrolyzes InsPs and modulates apoptosis in BC cells [246]. Thus, exosomal secretion of an isoform of MinPP1 represents a novel mechanism of TME modulation supporting tumor progression. Targeting exosomal MinPP1 could restore InsP6-mediated tumor suppression. Ongoing research in our laboratory focuses on developing MinPP1-specific inhibitors as potential therapeutic agents against BC.
10. Nanotechnology in Breast Cancer
Nanotechnology offers innovative solutions to overcome therapeutic resistance in BC by addressing limitations of conventional chemotherapy, such as non-specific biodistribution, off-target toxicity, and poor tumor penetration [247,248]. Leveraging the TME leaky vasculature via the ‘enhanced permeability and retention (EPR)’ effect, nanotechnology enhances drug delivery to resistant tumors [249,250,251]. This section explores nanoparticle mechanisms, clinical applications, and their role in overcoming resistance, integrating insights from vascular normalization strategies [116].
Nanoparticles (NPs) employ passive and active targeting to improve drug specificity. ‘Passive targeting’ exploits the EPR effect, where abnormal tumor vasculature, characterized by leakiness and poor perfusion, allows NP accumulation in the TME [252]. ‘Active targeting’ enhances selectivity by functionalizing NPs with ligands (e.g., antibodies, peptides, aptamers) that bind tumor-specific receptors, such as HER2 or folate receptors in BC [252]. Nanoparticle platforms include liposomes, polymeric NPs, silica NPs, and gold NPs (AuNPs), with AuNPs offering photothermal ablation and tunable surface chemistry but facing translational challenges like dose-dependent toxicity and uncertain long-term clearance [253].
FDA-approved nanocarriers, such as Doxil® (PEGylated liposomal doxorubicin) and Abraxane® (albumin-bound paclitaxel), improve pharmacokinetics, reduce systemic toxicity, and enhance tumor accumulation in BC [254,255,256,257]. These formulations leverage the EPR effect and benefit from vascular normalization strategies that improve perfusion, enhancing drug delivery to resistant tumors [256]. By increasing intratumoral drug concentrations, nanocarriers address resistance driven by poor penetration, supporting their use in frontline and refractory BC settings [254,256].
10.1. Nanocarriers: Overcoming Cellular Resistance
BC resistance, driven by efflux pump overexpression (e.g., P-glycoprotein), altered drug metabolism, and defective apoptosis, limits therapeutic efficacy [250]. Nanocarriers bypass efflux transporters and enhance intracellular drug delivery through modified release profiles [252,253]. Co-delivery nanocarriers, combining chemotherapeutics with resistance-modulating agents (e.g., siRNAs targeting MDR1 or PI3K inhibitors), target multiple resistance pathways, improving outcomes in resistant BC [258]. Stimuli-responsive NPs, triggered by tumor-specific cues (e.g., acidic pH, enzymatic activity), offer precise drug release [255]. Emerging stimuli-responsive “smart” nanocarriers offer further precision, with drug release triggered by tumor-specific cues such as acidic pH, enzymatic activity, or oxidative conditions within the TME [259].
Theranostic NPs (multifunctional nanosystems) integrate diagnostic and therapeutic functions, enabling real-time monitoring of drug delivery and response, facilitating personalized treatment adjustments [260]. RNA-based therapeutics, such as siRNAs targeting TME immunosuppressive genes (e.g., TGF-β), enhance immunotherapy efficacy, including for exploratory CAR-T-cell therapies limited by TME barriers [258]. Machine learning algorithms can analyze vast datasets of nanocarrier properties and biological interactions to predict optimal designs. As a result, AI algorithms can predict how nanocarriers will behave in the body, including their biodistribution and release kinetics. Integration of AI into nanoparticle design is an evolving field, promising to optimize nanocarrier size, shape, surface charge, and drug release kinetics for individualized patient applications [261]. Despite these innovations, challenges include scalability, reproducibility, immune clearance (e.g., macrophage uptake), and long-term safety [252,262]. Robust regulatory frameworks and clinical trials are essential for translation. In conclusion, nanotechnology redefines BC treatment by enhancing specificity, overcoming resistance, and minimizing toxicity, with AI-driven and TME-targeted innovations paving the way for personalized care.
11. Epigenetic Mechanisms Driving Resistance
Epigenetic modifications—such as DNA methylation, histone modifications, and non-coding RNAs—play pivotal roles in breast BC progression and therapy resistance by regulating gene expression without altering the DNA sequence. These mechanisms influence critical pathways governing drug metabolism, apoptosis, immune evasion, and stemness. One well-documented mechanism is hypermethylation of the ESR1 promoter, which suppresses estrogen receptor-α (ERα) expression and leads to poor response to endocrine therapies like tamoxifen [263]. Likewise, aberrant histone modifications—such as increased H3K27 trimethylation mediated by EZH2 and reduced H3K27 acetylation—silence tumor suppressor genes and have been implicated in therapy resistance. EZH2 overexpression further promotes epithelial–mesenchymal transition, invasion, and drug resistance in BC [264].
Histone demethylase KDM2A, targeting H3K36me2, is upregulated in aggressive BC subtypes like TNBC. KDM2A enhances cancer stem cell traits and angiogenesis through JAG1 activation and represses tumor suppressors such as E-cadherin by inhibiting TET2. MicroRNAs, notably miR-155, also contribute to resistance; miR-155 can modulate apoptosis and proliferation, although its precise mechanisms in therapy resistance require further elucidation [265]. Epigenetic dysregulation also affects the TME leading to CD8+ T-cell exhaustion and immune evasion [266]. For example, chromatin changes in tumor-infiltrating T cells drive exhaustion by upregulating immune checkpoint genes, reducing immunotherapy efficacy. Moreover, KDM2A contributes to metabolic rewiring of CSCs via regulation of PGC-1α and promotes stromal remodeling through activation of cancer-associated fibroblasts, further impairing immune and stromal surveillance in BC [266].
The reversible nature of epigenetic alterations presents an attractive avenue for overcoming BC resistance [267,268,269]. DNA methyltransferase inhibitors (DNMTis)—including azacitidine and decitabine—have demonstrated efficacy in reversing promoter hypermethylation of tumor suppressor genes, thereby restoring expression and reversing drug resistance in preclinical BC models (e.g., sequential decitabine followed by doxorubicin sensitizes MCF-7/ADR cells via p21 reactivation) [270]. Histone deacetylase inhibitors (HDACis) such as vorinostat and panobinostat similarly remodel chromatin to enhance tumor immunogenicity and immune-mediated clearance [271].
Emerging evidence suggests that epigenetic agents can re-sensitize endocrine-resistant BC. Recent reports show that decitabine can reconfigure 3D chromatin looping in estrogen receptor–positive cells, restoring sensitivity to hormonal therapies [272]. Additionally, cancer-derived interleukin-6 (IL-6) activates KDM2A in cancer-associated fibroblasts via the STAT3/NF-κB p50 pathway, promoting stromal-mediated resistance [273]. Refined therapeutic strategies that incorporate epigenetic profiling of tumor and immune compartments can enable biomarker-driven, personalized epigenetic therapy—potentially overcoming BC resistance with enhanced precision (see Figure 9).
12. Artificial Intelligence in Breast Cancer Diagnosis and Therapy
Artificial intelligence (AI) is reshaping the landscape of BC research and care, offering transformative capabilities across early detection, diagnostics, treatment planning, and therapeutic innovation. Through the integration of high-dimensional data—ranging from medical imaging and pathology to genomics, spatial transcriptomics, and electronic health records—AI empowers a ‘precision oncology framework’ that is adaptive, scalable, and deeply personalized.
In diagnostics, AI excels at pattern recognition, uncovering subtle imaging features often missed by conventional approaches. Deep learning (DL) algorithms outperform traditional radiology in detecting microcalcifications, architectural distortions, and asymmetries in mammography and MRI, especially in dense breast tissue and early-stage tumors [15,274,275,276]. Radiomics-based models can predict BC risk from screening images and identify aggressive subtypes such as TNBC and HER2-positive disease [275,276]. In digital pathology, DL platforms analyze whole-slide images to quantify tumor-infiltrating lymphocytes (TILs), assess mitotic activity, and classify BC subtypes with high reproducibility. These systems also standardize immunohistochemistry (IHC) scoring for ER, PR, and HER2, enhancing diagnostic consistency [276,277]. Integration with liquid biopsy data including ctDNA and methylation signatures, further supports non-invasive diagnosis and monitoring [278,279].
The clinical utility of AI is underscored by several recent FDA approvals across oncology. Notably, Ibex Prostate Detect, an AI-powered digital pathology system for prostate biopsy interpretation, and OnQ™ Prostate, an MRI post-processing tool using Restriction Spectrum Imaging (RSI), were granted FDA 510(k) clearance in 2024–2025. In BC, the FDA has also cleared two AI-based solutions aimed at improving detection and risk prediction:
(1) ProFound AI Detection Version 4.0 (iCAD), a mammography-based AI tool integrating prior exams to boost sensitivity by up to 22%, with reports of a 23% increase in overall cancer detection, a 4% rise in invasive cancers, and a doubling of lobular cancer detection. In dense breasts, detection was improved by 32%, with a 40% reduction in T2-stage tumors—all achieved without increasing DCIS detection or recall rates.
(2) Clairity Breast, the first AI platform to receive FDA De Novo clearance (June 2025), uses routine screening mammograms to predict a patient’s 5-year risk of developing BC, offering high-precision prognostic modeling directly from imaging data (Table 4). These examples illustrate how AI is enhancing diagnostic accuracy, revealing imaging biomarkers imperceptible to the human eye, increasing reproducibility across clinicians, and supporting improved patient stratification—hallmarks of radiology-informed precision oncology.
Beyond detection, AI also plays a growing role in identifying resistance mechanisms and optimizing therapy. Machine learning (ML) models can predict resistance to endocrine therapies or HER2-targeted agents based on genomic alterations (e.g., BRCA1/2, PIK3CA) and tumor microenvironmental factors [280]. Immune phenotyping algorithms help identify immune evasion signatures and stratify patients for checkpoint inhibitors, particularly in immunologically “cold” TNBC subtypes [281]. AI is also revolutionizing nanomedicine. By refining nanoparticle architecture for improved targeting, AI reduces off-target toxicity and enhances drug delivery to challenging microenvironments such as hypoxic or fibrotic tumors [282,283]. These advances support the development of customized therapies, including antibody-drug conjugates (ADCs) and endocrine regimens guided by real-time molecular profiling [284]. In drug development, AI accelerates discovery by mapping synthetic lethality (e.g., BRCA/PARP), simulating protein folding (e.g., AlphaFold), and optimizing synergistic combinations such as PI3K inhibitors in hormone receptor-positive BC [280,285].
Despite the momentum, significant barriers remain. Data heterogeneity, limited generalizability, privacy concerns, and lack of standardized multi-institutional datasets challenge model deployment [286,287]. Large language models (LLMs), including ChatGPT, offer promise in oncology education and decision support but currently lack rigorous domain-specific calibration and often produce inconsistent or hallucinated outputs [286,288].
Future innovation will depend on: (1) Federated learning, enabling decentralized AI training across institutions while preserving patient privacy [289]; (2) Explainable AI (XAI), promoting interpretability and clinical trust [290]; (3) Digital Twins, simulating tumor evolution and treatment response in silico [291]; and (4) EHR-integrated AI, for real-time prediction and clinical decision-making [291]. Together, these developments suggest that AI is not simply an adjunct, but a core driver of precision oncology, capable of evolving with tumor biology and personalizing cancer care in unprecedented ways.
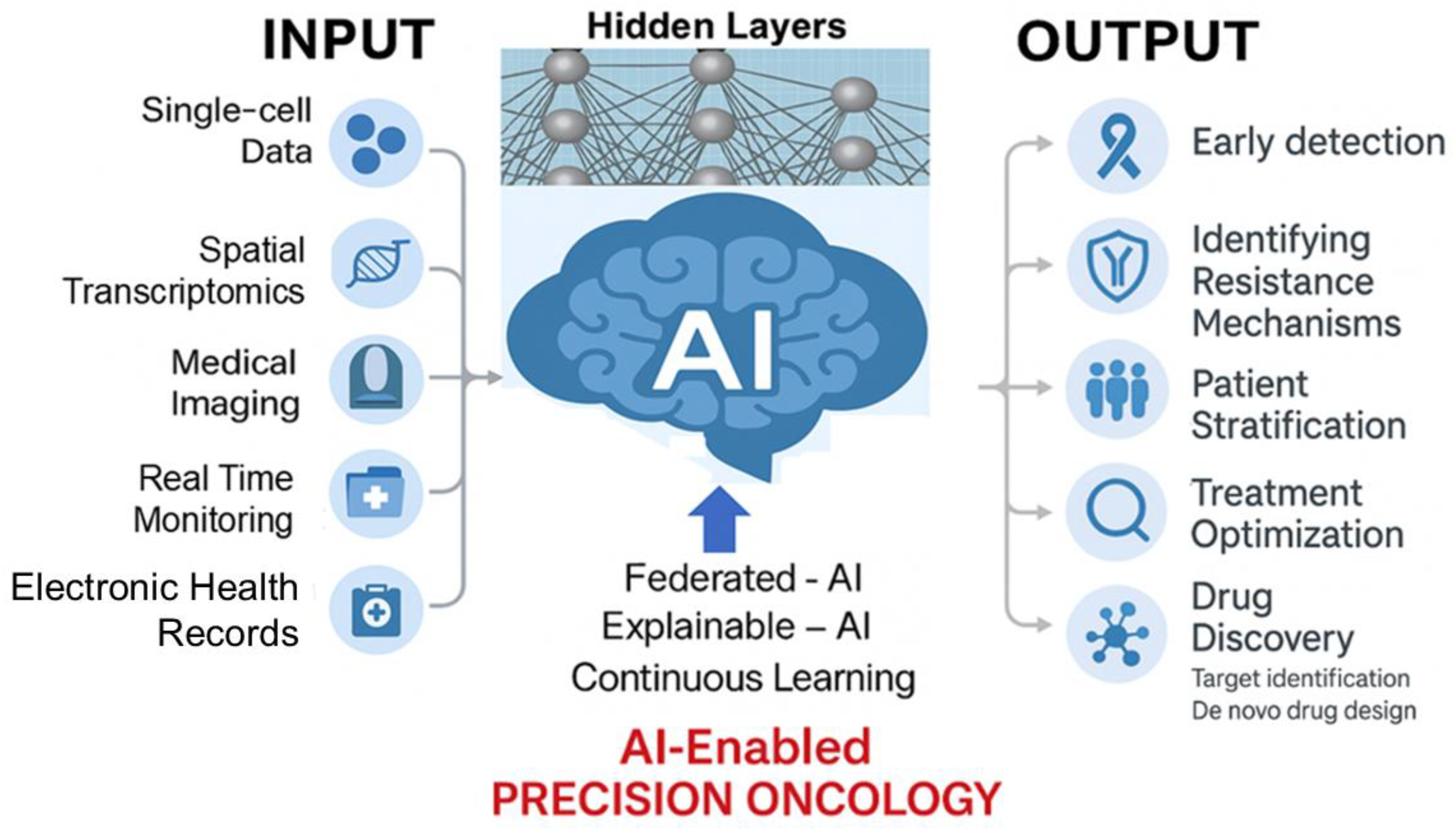
Figure 10.
AI-enabled precision oncology in breast cancer. Multi-modal inputs such as single-cell data, spatial transcriptomics, imaging, EHRs, and real-time patient monitoring are processed through federated and explainable AI models to generate outputs across early detection, resistance mapping, stratification, treatment optimization, and de novo drug discovery.
Figure 10.
AI-enabled precision oncology in breast cancer. Multi-modal inputs such as single-cell data, spatial transcriptomics, imaging, EHRs, and real-time patient monitoring are processed through federated and explainable AI models to generate outputs across early detection, resistance mapping, stratification, treatment optimization, and de novo drug discovery.
13. Personalized Medicine in Overcoming Resistance
Personalized medicine is redefining BC therapy by aligning treatment strategies with the unique molecular profile of each tumor. By integrating genomic, proteomic, and metabolomic data, clinicians can identify resistance-driving mutations—such as ESR1 in HR+ disease or BRCA in TNBC—and tailor interventions like elacestrant or PARP inhibitors accordingly [69,73,74]. Beyond single-gene targeting, multi-omic approaches support rational drug combinations and pathway-specific inhibitors, including PI3K-targeted agents in PIK3CA-mutant tumors [72]. Emerging modalities—from nanocarrier-enhanced delivery systems and exosome-based interventions to CAR-T-cell therapies and macrophage reprogramming—aim to overcome barriers posed by tumor heterogeneity and an immunosuppressive microenvironment [202,273]. When layered with AI-driven analytics, this evolving landscape offers a multidimensional framework to counter therapeutic resistance and advance precision oncology in BC care [288,290,292].
14. Concluding Remarks and Future Perspectives
Despite substantial progress in early detection and targeted therapies, resistance to treatment remains a central challenge in BC management, particularly in aggressive subtypes and metastatic disease. The multifaceted nature of resistance, due to tumor heterogeneity, metabolic adaptation, immune evasion, and dynamic changes within the TME, demands integrated and adaptive therapeutic strategies. Emerging technologies are reshaping the therapeutic landscape. AI is accelerating advances in multi-omics integration, radiomic profiling, and rational drug design, enabling early identification of resistance and personalized treatment selection. In parallel, innovations in nanomedicine, exosome-based delivery, and TME targeted immunotherapies are expanding the treatment options against resistant tumors.
Importantly, this review underscores four areas of translational significance: (1) the use of sophisticated experimental models that have deepened our understanding of resistance biology and enabled high-fidelity drug screening; (2) tumor vasculature as a physical and metabolic modulator of the TME; (3) metabolic rewiring in cancer and immune cells that shapes both disease trajectory and treatment response; and (4) the breast tissue microbiome as an emerging player in carcinogenesis and therapeutic failure. Together, these domains are not only revealing the complexity of resistance but also actively defining the next generation of druggable targets.
Looking ahead, bridging the gap between mechanistic insights and clinical translation will require interdisciplinary collaboration, systems-level thinking, and rigorous clinical validation. With a patient-centered framework and a commitment to innovation, the convergence of molecular oncology, immunotherapy, AI, and bioengineering would offer a promising path to overcome resistance and transform the future of breast cancer care.
Acknowledgments
All figures were developed through active collaboration among K.K., N.A., and co-authors with the aid of graphic tools in Microsoft PowerPoint. Several figures were further refined and modified for publication with the assistance of A.K. and A.S.
Author Contributions
Conceptualization: K.K., N.A., and A.S.; Writing—original draft preparation: K.K., N.A., and A.S.; Writing—critical review and editing: A.S., N.A., and A.K.; Figure refinement: N.A. and A.K. All authors reviewed and approved the final version of the manuscript for submission and agreed to its publication.
Funding
This work was partly supported by the Department of Biology, University of Arkansas at Little Rock, Little Rock. N.A. thanks NASA-EPSCoR-Arkansas Space Grant Consortium for grant funding (AWD-102354; GR-021631) and Arkansas Research Alliance for support during summer 2025 (ARA-GR023610).
Conflict of Interests
The authors declare no conflicts of interest.
References
- Harbeck, N., et al., Breast cancer. Nat Rev Dis Primers, 2019. 5(1): p. 66.
- WHO, Breast Cancer. 2024.
- Sung, H., et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021. 71(3): p. 209-249. [CrossRef]
- Arnold, M., Morgan, E., Rumgay, H., Mafra, A., Singh, D., Laversanne, M., Vignat, J., Gralow, J. R., Cardoso, F., Siesling, S., & Soerjomataram, I. , Current and future burden of breast cancer: Global statistics for 2020 and 2040. The Breast, 2022. 66: p. 15-23. [CrossRef]
- Siegel, R.L., Kratzer, T. B., Giaquinto, A. N., Sung, H., & Jemal, A., Cancer statistics, 2025. CA: A Cancer Journal for Clinicians, 2025. 75(1): p. 4-35.
- Society, A.C., Cancer facts & figures 2025. 2025, Atlanta, GA: American Cancer Society.
- Garrido-Castro, A.C., N.U. Lin, and K. Polyak, Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov, 2019. 9(2): p. 176-198. [CrossRef]
- Dagogo-Jack, I. and A.T. Shaw, Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol, 2018. 15(2): p. 81-94. [CrossRef]
- Hanahan D, W.R.A., Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646-674.
- Zheng, H.C., The molecular mechanisms of chemoresistance in cancers. Oncotarget, 2017. 8(35): p. 59950-59964.
- Hanahan, D., Hallmarks of Cancer: New Dimensions. Cancer Discov, 2022. 12(1): p. 31-46. [CrossRef]
- Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646-74.
- Xiong, X., et al., Breast cancer: pathogenesis and treatments. Signal Transduct Target Ther, 2025. 10(1): p. 49.
- Dhiman, R., et al., Enhanced drug delivery with nanocarriers: a comprehensive review of recent advances in breast cancer detection and treatment. Discov Nano, 2024. 19(1): p. 143. [CrossRef]
- Ahn, J.S., et al., Artificial Intelligence in Breast Cancer Diagnosis and Personalized Medicine. J Breast Cancer, 2023. 26(5): p. 405-435. [CrossRef]
- Boire, A., et al., Why do patients with cancer die? Nat Rev Cancer, 2024. 24(8): p. 578-589.
- Makki, J., Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin Med Insights Pathol, 2015. 8: p. 23-31. [CrossRef]
- Sinn, H.P. and H. Kreipe, A Brief Overview of the WHO Classification of Breast Tumors, 4th Edition, Focusing on Issues and Updates from the 3rd Edition. Breast Care (Basel), 2013. 8(2): p. 149-54. [CrossRef]
- Lukasiewicz, S., et al., Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers (Basel), 2021. 13(17).
- Zubair, M., S. Wang, and N. Ali, Advanced Approaches to Breast Cancer Classification and Diagnosis. Front Pharmacol, 2020. 11: p. 632079. [CrossRef]
- Perou, C.M., et al., Molecular portraits of human breast tumours. Nature, 2000. 406(6797): p. 747-52. [CrossRef]
- Modi, S., et al., Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med, 2022. 387(1): p. 9-20. [CrossRef]
- Schmid, P., et al., Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer. N Engl J Med, 2024. 391(21): p. 1981-1991. [CrossRef]
- Waks, A.G. and E.P. Winer, Breast Cancer Treatment: A Review. JAMA, 2019. 321(3): p. 288-300.
- Kuchenbaecker, K.B., et al., Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA, 2017. 317(23): p. 2402-2416.
- Antoniou, A.C., et al., Breast-cancer risk in families with mutations in PALB2. N Engl J Med, 2014. 371(6): p. 497-506. [CrossRef]
- Li, Y., et al., MRI Delta-Radiomics and Morphological Feature-Driven TabPFN Model for Preoperative Prediction of Lymphovascular Invasion in Invasive Breast Cancer. Technol Cancer Res Treat, 2025. 24: p. 15330338251362050. [CrossRef]
- Ehteshami Bejnordi, B., et al., Diagnostic Assessment of Deep Learning Algorithms for Detection of Lymph Node Metastases in Women With Breast Cancer. JAMA, 2017. 318(22): p. 2199-2210. [CrossRef]
- Le, E.P.V., et al., Artificial intelligence in breast imaging. Clin Radiol, 2019. 74(5): p. 357-366.
- Monje, M. and F. Winkler, Cancer research needs neuroscience and neuroscientists. Nat Neurosci, 2025. 28(5): p. 915-917. [CrossRef]
- Steiner, E., D. Klubert, and D. Knutson, Assessing breast cancer risk in women. Am Fam Physician, 2008. 78(12): p. 1361-6.
- Bianchini, G., et al., Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol, 2016. 13(11): p. 674-690. [CrossRef]
- Nkondjock, A. and P. Ghadirian, [Risk factors and risk reduction of breast cancer]. Med Sci (Paris), 2005. 21(2): p. 175-80.
- Hoxha, I., et al., Breast Cancer and Lifestyle Factors: Umbrella Review. Hematol Oncol Clin North Am, 2024. 38(1): p. 137-170.
- Collaborative Group on Hormonal Factors in Breast, C., Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol, 2012. 13(11): p. 1141-51.
- Kim, C., et al., Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell, 2018. 173(4): p. 879-893 e13. [CrossRef]
- Kim, G. and M. Bahl, Assessing Risk of Breast Cancer: A Review of Risk Prediction Models. J Breast Imaging, 2021. 3(2): p. 144-155. [CrossRef]
- Almasi-Hashiani, A., et al., The causal effect and impact of reproductive factors on breast cancer using super learner and targeted maximum likelihood estimation: a case-control study in Fars Province, Iran. BMC Public Health, 2021. 21(1): p. 1219. [CrossRef]
- Sherman, M.E., et al., Benign Breast Disease and Breast Cancer Risk in the Percutaneous Biopsy Era. JAMA Surg, 2024. 159(2): p. 193-201. [CrossRef]
- Sun, Y.S., et al., Risk Factors and Preventions of Breast Cancer. Int J Biol Sci, 2017. 13(11): p. 1387-1397.
- Chlebowski, R.T., et al., Breast Cancer After Use of Estrogen Plus Progestin and Estrogen Alone: Analyses of Data From 2 Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol, 2015. 1(3): p. 296-305.
- Smith-Bindman, R., et al., Projected Lifetime Cancer Risks From Current Computed Tomography Imaging. JAMA Intern Med, 2025. 185(6): p. 710-719. [CrossRef]
- Hilakivi-Clarke, L., Maternal exposure to diethylstilbestrol during pregnancy and increased breast cancer risk in daughters. Breast Cancer Res, 2014. 16(2): p. 208. [CrossRef]
- Freitas, A.J.A., et al., Liquid Biopsy as a Tool for the Diagnosis, Treatment, and Monitoring of Breast Cancer. Int J Mol Sci, 2022. 23(17).
- Amato, O., N. Giannopoulou, and M. Ignatiadis, Circulating tumor DNA validity and potential uses in metastatic breast cancer. NPJ Breast Cancer, 2024. 10(1): p. 21. [CrossRef]
- Mashouri, L., et al., Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer, 2019. 18(1): p. 75. [CrossRef]
- Malla, R., et al., Exosome-Mediated Cellular Communication in the Tumor Microenvironment Imparts Drug Resistance in Breast Cancer. Cancers (Basel), 2025. 17(7). [CrossRef]
- Chia, J.L.L., et al., Harnessing Artificial Intelligence to Enhance Global Breast Cancer Care: A Scoping Review of Applications, Outcomes, and Challenges. Cancers (Basel), 2025. 17(2). [CrossRef]
- Vatsavai, N., et al., Advances and challenges in cancer immunotherapy: mechanisms, clinical applications, and future directions. Front Pharmacol, 2025. 16: p. 1602529. [CrossRef]
- Xie, H.Y., Z.M. Shao, and D.Q. Li, Tumor microenvironment: driving forces and potential therapeutic targets for breast cancer metastasis. Chin J Cancer, 2017. 36(1): p. 36. [CrossRef]
- Clough, K.B., et al., Improving breast cancer surgery: a classification and quadrant per quadrant atlas for oncoplastic surgery. Ann Surg Oncol, 2010. 17(5): p. 1375-91.
- Gentilini, O.D., et al., Sentinel Lymph Node Biopsy vs No Axillary Surgery in Patients With Small Breast Cancer and Negative Results on Ultrasonography of Axillary Lymph Nodes: The SOUND Randomized Clinical Trial. JAMA Oncol, 2023. 9(11): p. 1557-1564.
- Hersh, E.H. and T.A. King, De-escalating axillary surgery in early-stage breast cancer. Breast, 2022. 62 Suppl 1(Suppl 1): p. S43-S49. [CrossRef]
- Burstein, H.J., et al., Adjuvant Endocrine Therapy for Women With Hormone Receptor-Positive Breast Cancer: ASCO Clinical Practice Guideline Focused Update. J Clin Oncol, 2019. 37(5): p. 423-438. [CrossRef]
- Goss, P.E., et al., Extending Aromatase-Inhibitor Adjuvant Therapy to 10 Years. N Engl J Med, 2016. 375(3): p. 209-19. [CrossRef]
- Schmid, P., et al., Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med, 2020. 382(9): p. 810-821. [CrossRef]
- Cardoso, F., et al., 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol, 2020. 31(12): p. 1623-1649. [CrossRef]
- Cortes, J., et al., Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N Engl J Med, 2022. 387(3): p. 217-226. [CrossRef]
- Swain, S.M., et al., Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol, 2020. 21(4): p. 519-530. [CrossRef]
- Hortobagyi, G.N., et al., Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med, 2016. 375(18): p. 1738-1748. [CrossRef]
- Robson, M., et al., Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med, 2017. 377(6): p. 523-533. [CrossRef]
- Siegel, R.L., et al., Cancer statistics, 2025. CA Cancer J Clin, 2025. 75(1): p. 10-45.
- Ibrahim, A., et al., Artificial intelligence in digital breast pathology: Techniques and applications. Breast, 2020. 49: p. 267-273. [CrossRef]
- Patel, A., N. Unni, and Y. Peng, The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers (Basel), 2020. 12(8). [CrossRef]
- Cardinale, D., et al., Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation, 2015. 131(22): p. 1981-8. [CrossRef]
- Hadji, P., Aromatase inhibitor-associated bone loss in breast cancer patients is distinct from postmenopausal osteoporosis. Crit Rev Oncol Hematol, 2009. 69(1): p. 73-82. [CrossRef]
- Zhao, J., Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther, 2016. 160: p. 145-58. [CrossRef]
- Das, S., et al., Advancing breast cancer research: a comprehensive review of in vitro and in vivo experimental models. Med Oncol, 2025. 42(8): p. 316. [CrossRef]
- Roarty, K. and G.V. Echeverria, Laboratory Models for Investigating Breast Cancer Therapy Resistance and Metastasis. Front Oncol, 2021. 11: p. 645698. [CrossRef]
- Hidalgo, M., et al., Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov, 2014. 4(9): p. 998-1013. [CrossRef]
- Chamorey, E., et al., Critical Appraisal and Future Challenges of Artificial Intelligence and Anticancer Drug Development. Pharmaceuticals (Basel), 2024. 17(7). [CrossRef]
- McKenna, M., S. McGarrigle, and G.P. Pidgeon, The next generation of PI3K-Akt-mTOR pathway inhibitors in breast cancer cohorts. Biochim Biophys Acta Rev Cancer, 2018. 1870(2): p. 185-197. [CrossRef]
- Lee, S.Y., et al., In Vitro three-dimensional (3D) cell culture tools for spheroid and organoid models. SLAS Discov, 2023. 28(4): p. 119-137. [CrossRef]
- Nur Husna, S.M., et al., Inhibitors targeting CDK4/6, PARP and PI3K in breast cancer: a review. Ther Adv Med Oncol, 2018. 10: p. 1758835918808509. [CrossRef]
- Jin, M., et al., PD-1/PD-L1 immune checkpoint blockade in breast cancer: research insights and sensitization strategies. Mol Cancer, 2024. 23(1): p. 266. [CrossRef]
- Rosenbluth, J.M., et al., Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nat Commun, 2020. 11(1): p. 1711. [CrossRef]
- Pedroza, D.A., et al., Leveraging preclinical models of metastatic breast cancer. Biochim Biophys Acta Rev Cancer, 2024. 1879(5): p. 189163. [CrossRef]
- Fina, E., et al., Gene signatures of circulating breast cancer cell models are a source of novel molecular determinants of metastasis and improve circulating tumor cell detection in patients. J Exp Clin Cancer Res, 2022. 41(1): p. 78. [CrossRef]
- Cao, J., et al., Chemoresistance and Metastasis in Breast Cancer Molecular Mechanisms and Novel Clinical Strategies. Front Oncol, 2021. 11: p. 658552. [CrossRef]
- Moitra, K., H. Lou, and M. Dean, Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther, 2011. 89(4): p. 491-502. [CrossRef]
- Holohan, C., et al., Cancer drug resistance: an evolving paradigm. Nat Rev Cancer, 2013. 13(10): p. 714-26. [CrossRef]
- Gottesman, M.M., T. Fojo, and S.E. Bates, Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer, 2002. 2(1): p. 48-58. [CrossRef]
- Robey, R.W., et al., Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer, 2018. 18(7): p. 452-464. [CrossRef]
- Lu, S., et al., Managing Cancer Drug Resistance from the Perspective of Inflammation. J Oncol, 2022. 2022: p. 3426407. [CrossRef]
- Luo, B., et al., Cytochrome P450: Implications for human breast cancer. Oncol Lett, 2021. 22(1): p. 548. [CrossRef]
- Rizwanullah, M., et al., Receptor-Mediated Targeted Delivery of Surface-ModifiedNanomedicine in Breast Cancer: Recent Update and Challenges. Pharmaceutics, 2021. 13(12). [CrossRef]
- Bose, R., et al., Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov, 2013. 3(2): p. 224-37.
- Wang, Z.H., et al., Trastuzumab resistance in HER2-positive breast cancer: Mechanisms, emerging biomarkers and targeting agents. Front Oncol, 2022. 12: p. 1006429. [CrossRef]
- Harrod, A., et al., Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene, 2017. 36(16): p. 2286-2296. [CrossRef]
- Nahta, R., et al., Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol, 2006. 3(5): p. 269-80. [CrossRef]
- Menghi, F., et al., Genomic and epigenomic BRCA alterations predict adaptive resistance and response to platinum-based therapy in patients with triple-negative breast and ovarian carcinomas. Sci Transl Med, 2022. 14(652): p. eabn1926. [CrossRef]
- Tung, N. and J.E. Garber, PARP inhibition in breast cancer: progress made and future hopes. NPJ Breast Cancer, 2022. 8(1): p. 47. [CrossRef]
- Presti, D. and E. Quaquarini, The PI3K/AKT/mTOR and CDK4/6 Pathways in Endocrine Resistant HR+/HER2- Metastatic Breast Cancer: Biological Mechanisms and New Treatments. Cancers (Basel), 2019. 11(9). [CrossRef]
- Lee, S., J. Rauch, and W. Kolch, Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int J Mol Sci, 2020. 21(3). [CrossRef]
- Zielinska, K.A. and V.L. Katanaev, The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers (Basel), 2020. 12(10). [CrossRef]
- Kim, B.G., et al., Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol, 2021. 14(1): p. 55. [CrossRef]
- Banerjee, S., et al., Prognostic correlations with the microbiome of breast cancer subtypes. Cell Death Dis, 2021. 12(9): p. 831. [CrossRef]
- Parhi, L., et al., Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun, 2020. 11(1): p. 3259.
- Akinpelu, A., et al., The impact of tumor microenvironment: unraveling the role of physical cues in breast cancer progression. Cancer Metastasis Rev, 2024. 43(2): p. 823-844. [CrossRef]
- Deepak, K.G.K., et al., Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol Res, 2020. 153: p. 104683. [CrossRef]
- Hanahan, D. and L.M. Coussens, Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell, 2012. 21(3): p. 309-22. [CrossRef]
- Salemme, V., et al., The role of tumor microenvironment in drug resistance: emerging technologies to unravel breast cancer heterogeneity. Front Oncol, 2023. 13: p. 1170264. [CrossRef]
- Zheng, J. and H. Hao, The importance of cancer-associated fibroblasts in targeted therapies and drug resistance in breast cancer. Front Oncol, 2023. 13: p. 1333839. [CrossRef]
- Qian, K. and Q. Liu, Narrative review on the role of immunotherapy in early triple negative breast cancer: unveiling opportunities and overcoming challenges. Translational Breast Cancer Research, 2023. 4. [CrossRef]
- Esteva, F.J., et al., PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol, 2010. 177(4): p. 1647-56.
- Junttila, T.T., et al., Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell, 2009. 15(5): p. 429-40. [CrossRef]
- Kinnel, B., et al., Targeted Therapy and Mechanisms of Drug Resistance in Breast Cancer. Cancers (Basel), 2023. 15(4). [CrossRef]
- Zhang, L., et al., eNAMPT/Ac-STAT3/DIRAS2 Axis Promotes Development and Cancer Stemness in Triple-Negative Breast Cancer by Enhancing Cytokine Crosstalk Between Tumor-Associated Macrophages and Cancer Cells. Int J Biol Sci, 2025. 21(5): p. 2027-2047.
- Zhi, S., et al., Hypoxia-inducible factor in breast cancer: role and target for breast cancer treatment. Front Immunol, 2024. 15: p. 1370800. [CrossRef]
- Kim, I., et al., Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers (Basel), 2022. 14(14).
- Chung, A.W., et al., Tocilizumab overcomes chemotherapy resistance in mesenchymal stem-like breast cancer by negating autocrine IL-1A induction of IL-6. NPJ Breast Cancer, 2022. 8(1): p. 30. [CrossRef]
- Liu, H., et al., Targeting DCLK1 attenuates tumor stemness and evokes antitumor immunity in triple-negative breast cancer by inhibiting IL-6/STAT3 signaling. Breast Cancer Res, 2023. 25(1): p. 43.
- Novohradsky, V., et al., An anticancer Os(II) bathophenanthroline complex as a human breast cancer stem cell-selective, mammosphere potent agent that kills cells by necroptosis. Sci Rep, 2019. 9(1): p. 13327. [CrossRef]
- Avancini, G., et al., Keratin nanoparticles and photodynamic therapy enhance the anticancer stem cells activity of salinomycin. Mater Sci Eng C Mater Biol Appl, 2021. 122: p. 111899. [CrossRef]
- Gupta, P.B., et al., Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell, 2009. 138(4): p. 645-659. [CrossRef]
- Owens, T.W. and M.J. Naylor, Breast cancer stem cells. Front Physiol, 2013. 4: p. 225.
- Batlle, E. and H. Clevers, Cancer stem cells revisited. Nat Med, 2017. 23(10): p. 1124-1134. [CrossRef]
- Liu, Y., et al., Drug resistance and tumor immune microenvironment: An overview of current understandings (Review). Int J Oncol, 2024. 65(4). [CrossRef]
- Jain, R.K., Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science, 2005. 307(5706): p. 58-62. [CrossRef]
- Jain, R.K., Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell, 2014. 26(5): p. 605-22. [CrossRef]
- Fukumura, D., et al., Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol, 2018. 15(5): p. 325-340. [CrossRef]
- Mou, J., et al., Research progress in tumor angiogenesis and drug resistance in breast cancer. Cancer Biol Med, 2024. 21(7): p. 571-85. [CrossRef]
- Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet, 1997. 350(9084): p. 1047-59.
- Gabrilovich, D.I., et al., Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med, 1996. 2(10): p. 1096-103. [CrossRef]
- Wherry, E.J. and M. Kurachi, Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol, 2015. 15(8): p. 486-99.
- Li, Y., et al., HIF-1alpha inhibitor YC-1 suppresses triple-negative breast cancer growth and angiogenesis by targeting PlGF/VEGFR1-induced macrophage polarization. Biomed Pharmacother, 2023. 161: p. 114423. [CrossRef]
- Juillerat, A., et al., An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep, 2017. 7: p. 39833. [CrossRef]
- Supper, V.M., et al., Secretion of a VEGF-blocking scFv enhances CAR T-cell potency. Cancer Immunol Res, 2025. [CrossRef]
- Gu, S., et al., Lack of acquired resistance in HER2-positive breast cancer cells after long-term HER2 siRNA nanoparticle treatment. PLoS One, 2018. 13(6): p. e0198141. [CrossRef]
- Brennen, W.N., et al., Overcoming stromal barriers to immuno-oncological responses via fibroblast activation protein-targeted therapy. Immunotherapy, 2021. 13(2): p. 155-175. [CrossRef]
- Liu, J., et al., Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label phase II trial. J Immunother Cancer, 2020. 8(1). [CrossRef]
- Wherry, E.J., T cell exhaustion. Nat Immunol, 2011. 12(6): p. 492-9.
- Xie, H., et al., CD8(+) T cell exhaustion in the tumor microenvironment of breast cancer. Front Immunol, 2024. 15: p. 1507283. [CrossRef]
- Blank, C.U., et al., Defining ’T cell exhaustion’. Nat Rev Immunol, 2019. 19(11): p. 665-674.
- Zhu, M., et al., Silence of a dependence receptor CSF1R in colorectal cancer cells activates tumor-associated macrophages. J Immunother Cancer, 2022. 10(12). [CrossRef]
- Zollo, M., et al., Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin Exp Metastasis, 2012. 29(6): p. 585-601. [CrossRef]
- Khan, O., et al., TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature, 2019. 571(7764): p. 211-218. [CrossRef]
- Kalluri, R. and V.S. LeBleu, The biology, function, and biomedical applications of exosomes. Science, 2020. 367(6478).
- Dong, X., et al., Exosomes and breast cancer drug resistance. Cell Death Dis, 2020. 11(11): p. 987. [CrossRef]
- Wang, X., et al., Exosome-mediated transfer of long noncoding RNA H19 induces doxorubicin resistance in breast cancer. J Cell Physiol, 2020. 235(10): p. 6896-6904. [CrossRef]
- Saha, T., et al., Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol, 2022. 17(1): p. 98-106. [CrossRef]
- Liu, S., et al., Metabolic reprogramming and therapeutic resistance in primary and metastatic breast cancer. Mol Cancer, 2024. 23(1): p. 261. [CrossRef]
- Alkarakooly, Z., et al., Metabolic reprogramming by Dichloroacetic acid potentiates photodynamic therapy of human breast adenocarcinoma MCF-7 cells. PLoS One, 2018. 13(10): p. e0206182. [CrossRef]
- DeBerardinis, R.J. and N.S. Chandel, Fundamentals of cancer metabolism. Sci Adv, 2016. 2(5): p. e1600200.
- Hanahan, D., O. Michielin, and M.J. Pittet, Convergent inducers and effectors of T cell paralysis in the tumour microenvironment. Nat Rev Cancer, 2025. 25(1): p. 41-58. [CrossRef]
- Warburg, O., On the origin of cancer cells. Science, 1956. 123(3191): p. 309-14. [CrossRef]
- Koppenol, W.H., P.L. Bounds, and C.V. Dang, Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer, 2011. 11(5): p. 325-37.
- Martinez-Outschoorn, U.E., et al., Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol, 2017. 14(1): p. 11-31. [CrossRef]
- Fischer, G.M., et al., Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma Res, 2018. 31(1): p. 11-30. [CrossRef]
- Cao, Z., et al., Lactate oxidase nanocapsules boost T cell immunity and efficacy of cancer immunotherapy. Sci Transl Med, 2023. 15(717): p. eadd2712. [CrossRef]
- Jin, J., et al., Cardamonin inhibits breast cancer growth by repressing HIF-1alpha-dependent metabolic reprogramming. J Exp Clin Cancer Res, 2019. 38(1): p. 377. [CrossRef]
- Wu, S., et al., Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front Oncol, 2020. 10: p. 591342. [CrossRef]
- Jiang, M., et al., From metabolic byproduct to immune modulator: the role of lactate in tumor immune escape. Front Immunol, 2024. 15: p. 1492050. [CrossRef]
- Edwards, D.N., et al., Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest, 2021. 131(4). [CrossRef]
- Spinelli, J.B. and M.C. Haigis, The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol, 2018. 20(7): p. 745-754. [CrossRef]
- Ni, R., et al., Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front Oncol, 2023. 13: p. 1143798. [CrossRef]
- Gross, M.I., et al., Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther, 2014. 13(4): p. 890-901. [CrossRef]
- Lehuede, C., et al., Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res, 2016. 76(18): p. 5201-8. [CrossRef]
- Vander Heiden, M.G. and R.J. DeBerardinis, Understanding the Intersections between Metabolism and Cancer Biology. Cell, 2017. 168(4): p. 657-669. [CrossRef]
- Kinnaird, A., et al., Metabolic control of epigenetics in cancer. Nat Rev Cancer, 2016. 16(11): p. 694-707. [CrossRef]
- Yuan, S., J. Almagro, and E. Fuchs, Beyond genetics: driving cancer with the tumour microenvironment behind the wheel. Nat Rev Cancer, 2024. 24(4): p. 274-286. [CrossRef]
- Schmid, P., et al., Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N Engl J Med, 2022. 386(6): p. 556-567. [CrossRef]
- Ma, S., M.A. Caligiuri, and J. Yu, Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol, 2022. 43(10): p. 833-847. [CrossRef]
- Chen, X., et al., Mechanisms and Strategies to Overcome PD-1/PD-L1 Blockade Resistance in Triple-Negative Breast Cancer. Cancers (Basel), 2022. 15(1).
- Bhola, N.E., et al., TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest, 2013. 123(3): p. 1348-58. [CrossRef]
- Liu, M., et al., TGF-beta suppresses type 2 immunity to cancer. Nature, 2020. 587(7832): p. 115-120.
- Li, S., et al., Cancer immunotherapy via targeted TGF-beta signalling blockade in T(H) cells. Nature, 2020. 587(7832): p. 121-125.
- Georgoudaki, A.M., et al., Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep, 2016. 15(9): p. 2000-11. [CrossRef]
- Cassetta, L. and J.W. Pollard, Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov, 2018. 17(12): p. 887-904. [CrossRef]
- Al-Hajj, M., et al., Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A, 2003. 100(7): p. 3983-8. [CrossRef]
- Liu, S., et al., Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports, 2014. 2(1): p. 78-91. [CrossRef]
- Crabtree, J.S. and L. Miele, Breast Cancer Stem Cells. Biomedicines, 2018. 6(3).
- Phi, L.T.H., et al., Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int, 2018. 2018: p. 5416923. [CrossRef]
- Plaks, V., N. Kong, and Z. Werb, The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 2015. 16(3): p. 225-38.
- Khoury, T., et al., Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod Pathol, 2012. 25(3): p. 388-97.
- Korkaya, H., S. Liu, and M.S. Wicha, Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest, 2011. 121(10): p. 3804-9.
- Ansieau, S., EMT in breast cancer stem cell generation. Cancer Lett, 2013. 338(1): p. 63-8. [CrossRef]
- Son, B., et al., Targeted therapy of cancer stem cells: inhibition of mTOR in pre-clinical and clinical research. Cell Death Dis, 2024. 15(9): p. 696. [CrossRef]
- Song, K. and M. Farzaneh, Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res Ther, 2021. 12(1): p. 245. [CrossRef]
- Francescangeli, F., et al., Dormancy, stemness, and therapy resistance: interconnected players in cancer evolution. Cancer Metastasis Rev, 2023. 42(1): p. 197-215. [CrossRef]
- Butti, R., et al., Breast cancer stem cells: Biology and therapeutic implications. Int J Biochem Cell Biol, 2019. 107: p. 38-52. [CrossRef]
- Palomeras, S., S. Ruiz-Martinez, and T. Puig, Targeting Breast Cancer Stem Cells to Overcome Treatment Resistance. Molecules, 2018. 23(9). [CrossRef]
- Gaude, E. and C. Frezza, Defects in mitochondrial metabolism and cancer. Cancer Metab, 2014. 2: p. 10. [CrossRef]
- Hekmatshoar, Y., et al., The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem J, 2018. 475(14): p. 2305-2328. [CrossRef]
- Singh, K.K. and M. Kulawiec, Mitochondrial DNA polymorphism and risk of cancer. Methods Mol Biol, 2009. 471: p. 291-303.
- Jimenez-Morales, S., et al., Overview of mitochondrial germline variants and mutations in human disease: Focus on breast cancer (Review). Int J Oncol, 2018. 53(3): p. 923-936. [CrossRef]
- Vikramdeo, K.S., et al., Profiling mitochondrial DNA mutations in tumors and circulating extracellular vesicles of triple-negative breast cancer patients for potential biomarker development. FASEB Bioadv, 2023. 5(10): p. 412-426. [CrossRef]
- Martinez-Reyes, I. and N.S. Chandel, Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun, 2020. 11(1): p. 102. [CrossRef]
- Nahacka, Z., et al., Miro proteins and their role in mitochondrial transfer in cancer and beyond. Front Cell Dev Biol, 2022. 10: p. 937753. [CrossRef]
- Sotgia, F., M. Fiorillo, and M.P. Lisanti, Mitochondrial markers predict recurrence, metastasis and tamoxifen-resistance in breast cancer patients: Early detection of treatment failure with companion diagnostics. Oncotarget, 2017. 8(40): p. 68730-68745. [CrossRef]
- Stewart, J.B., Chinnery, P.F., , Extreme heterogeneity of human mitochondrial dNA from organelles to populations. Nat Rev Genet, 2021. 22: p. 106-118. [CrossRef]
- Sharma, A., et al., Mitochondrial signaling pathways and their role in cancer drug resistance. Cell Signal, 2024. 122: p. 111329.
- Delbridge, A.R., et al., Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer, 2016. 16(2): p. 99-109. [CrossRef]
- LeBleu, V.S., et al., PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol, 2014. 16(10): p. 992-1003, 1-15. [CrossRef]
- Ma, Y., et al., Mitochondrial DNA methylation is a predictor of immunotherapy response and prognosis in breast cancer: scRNA-seq and bulk-seq data insights. Front Immunol, 2023. 14: p. 1219652. [CrossRef]
- Pasquier, J., et al., Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med, 2013. 11: p. 94. [CrossRef]
- Patheja, P. and K. Sahu, Macrophage conditioned medium induced cellular network formation in MCF-7 cells through enhanced tunneling nanotube formation and tunneling nanotube mediated release of viable cytoplasmic fragments. Exp Cell Res, 2017. 355(2): p. 182-193. [CrossRef]
- Togashi, Y., K. Shitara, and H. Nishikawa, Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol, 2019. 16(6): p. 356-371. [CrossRef]
- Valdebenito, S., et al., Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci Rep, 2021. 11(1): p. 14556. [CrossRef]
- Zampieri, L.X., et al., Mitochondrial Transfer in Cancer: A Comprehensive Review. Int J Mol Sci, 2021. 22(6). [CrossRef]
- Chen, R. and J. Chen, Mitochondrial transfer - a novel promising approach for the treatment of metabolic diseases. Front Endocrinol (Lausanne), 2023. 14: p. 1346441. [CrossRef]
- Chen, D.S. and I. Mellman, Oncology meets immunology: the cancer-immunity cycle. Immunity, 2013. 39(1): p. 1-10. [CrossRef]
- Lambert, A.W., Y. Zhang, and R.A. Weinberg, Cell-intrinsic and microenvironmental determinants of metastatic colonization. Nat Cell Biol, 2024. 26(5): p. 687-697. [CrossRef]
- Sarangi, P., Role of indoleamine 2, 3-dioxygenase 1 in immunosuppression of breast cancer. Cancer Pathog Ther, 2024. 2(4): p. 246-255. [CrossRef]
- Emens, L.A., Breast Cancer Immunotherapy: Facts and Hopes. Clin Cancer Res, 2018. 24(3): p. 511-520. [CrossRef]
- Setordzi, P., et al., The recent advances of PD-1 and PD-L1 checkpoint signaling inhibition for breast cancer immunotherapy. Eur J Pharmacol, 2021. 895: p. 173867. [CrossRef]
- Nihira, N.T., et al., Nuclear PD-L1 triggers tumour-associated inflammation upon DNA damage. EMBO Rep, 2025. 26(3): p. 635-655. [CrossRef]
- Anderson, A.C., N. Joller, and V.K. Kuchroo, Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity, 2016. 44(5): p. 989-1004. [CrossRef]
- van Schaik, T.A., K.S. Chen, and K. Shah, Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front Oncol, 2021. 11: p. 678562.
- Kalfeist, L., et al., Co-targeting TGF-beta and PD-L1 sensitizes triple-negative breast cancer to experimental immunogenic cisplatin-eribulin chemotherapy doublet. J Clin Invest, 2025. 135(13). [CrossRef]
- Zhang, H., et al., Novel insight into the Warburg effect: Sweet temptation. Crit Rev Oncol Hematol, 2025. 214: p. 104844. [CrossRef]
- Kuo, L.W., et al., Blocking Tryptophan Catabolism Reduces Triple-Negative Breast Cancer Invasive Capacity. Cancer Res Commun, 2024. 4(10): p. 2699-2713. [CrossRef]
- Kundu, M., et al., Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol Cancer, 2024. 23(1): p. 92. [CrossRef]
- Chakravarthy, A., et al., TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun, 2018. 9(1): p. 4692. [CrossRef]
- Akinsipe, T., et al., Cellular interactions in tumor microenvironment during breast cancer progression: new frontiers and implications for novel therapeutics. Front Immunol, 2024. 15: p. 1302587. [CrossRef]
- Blass, E. and P.A. Ott, Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol, 2021. 18(4): p. 215-229. [CrossRef]
- Jan, A., et al., An update on cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol, 2025. 21(6): p. 715-735.
- Stuber, T., et al., Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer, 2020. 8(1). [CrossRef]
- Lee, D.H., Update of early phase clinical trials in cancer immunotherapy. BMB Rep, 2021. 54(1): p. 70-88. [CrossRef]
- Wilkie, S., et al., Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol, 2012. 32(5): p. 1059-70. [CrossRef]
- Heater, N.K., S. Warrior, and J. Lu, Current and future immunotherapy for breast cancer. J Hematol Oncol, 2024. 17(1): p. 131. [CrossRef]
- Priceman, S.J., et al., Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res, 2018. 24(1): p. 95-105. [CrossRef]
- Suarez, E.R., et al., Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget, 2016. 7(23): p. 34341-55. [CrossRef]
- Cherkassky, L., et al., Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest, 2016. 126(8): p. 3130-44. [CrossRef]
- June, C.H., et al., CAR T cell immunotherapy for human cancer. Science, 2018. 359(6382): p. 1361-1365.
- Kwon, Y., et al., Exosomal MicroRNAs as Mediators of Cellular Interactions Between Cancer Cells and Macrophages. Front Immunol, 2020. 11: p. 1167. [CrossRef]
- Bahrami, A., et al., The prognostic and therapeutic application of microRNAs in breast cancer: Tissue and circulating microRNAs. J Cell Physiol, 2018. 233(2): p. 774-786. [CrossRef]
- Ni, C., et al., Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+gammadelta1 Treg cells. Signal Transduct Target Ther, 2020. 5(1): p. 41.
- Wang, Y., et al., The involvement and application potential of exosomes in breast cancer immunotherapy. Front Immunol, 2024. 15: p. 1384946. [CrossRef]
- Hao, Y., D. Baker, and P. Ten Dijke, TGF-beta-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int J Mol Sci, 2019. 20(11). [CrossRef]
- Ruksha, T. and N. Palkina, Role of exosomes in transforming growth factor-beta-mediated cancer cell plasticity and drug resistance. Explor Target Antitumor Ther, 2025. 6: p. 1002322. [CrossRef]
- Hu, D., et al., Cancer-associated fibroblasts in breast cancer: Challenges and opportunities. Cancer Commun (Lond), 2022. 42(5): p. 401-434. [CrossRef]
- Gu, J., et al., The role of PKM2 nuclear translocation in the constant activation of the NF-kappaB signaling pathway in cancer-associated fibroblasts. Cell Death Dis, 2021. 12(4): p. 291. [CrossRef]
- Tao, S., et al., CAF-derived exosomal LINC01711 promotes breast cancer progression by activating the miR-4510/NELFE axis and enhancing glycolysis. FASEB J, 2025. 39(7): p. e70471.
- Mortezaee, K., Exosomes in bridging macrophage-fibroblast polarity and cancer stemness. Med Oncol, 2025. 42(6): p. 216. [CrossRef]
- Wang, L., Z. Yan, and Y. Xia, [Silencing RAB27a inhibits proliferation, invasion and adhesion of triple-negative breast cancer cells]. Nan Fang Yi Ke Da Xue Xue Bao, 2023. 43(4): p. 560-567.
- O’Brien, K., et al., miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget, 2015. 6(32): p. 32774-89. [CrossRef]
- Halvaei, S., et al., Exosomes in Cancer Liquid Biopsy: A Focus on Breast Cancer. Mol Ther Nucleic Acids, 2018. 10: p. 131-141. [CrossRef]
- Abdul-Rahman, T., et al., Extracellular vesicle-mediated drug delivery in breast cancer theranostics. Discov Oncol, 2024. 15(1): p. 181. [CrossRef]
- Verweij, F.J., et al., ER membrane contact sites support endosomal small GTPase conversion for exosome secretion. J Cell Biol, 2022. 221(12). [CrossRef]
- He, C., et al., Exosomes derived from endoplasmic reticulum-stressed liver cancer cells enhance the expression of cytokines in macrophages via the STAT3 signaling pathway. Oncol Lett, 2020. 20(1): p. 589-600. [CrossRef]
- Zubair, M., et al., Identification and functional characterization of multiple inositol polyphosphate phosphatase1 (Minpp1) isoform-2 in exosomes with potential to modulate tumor microenvironment. PLoS One, 2022. 17(3): p. e0264451. [CrossRef]
- Agarwal, R., S. Hassen, and N. Ali, Changes in cellular levels of inositol polyphosphates during apoptosis. Mol Cell Biochem, 2010. 345(1-2): p. 61-8. [CrossRef]
- Agarwal, R., H. Mumtaz, and N. Ali, Role of inositol polyphosphates in programmed cell death. Mol Cell Biochem, 2009. 328(1-2): p. 155-65. [CrossRef]
- Vucenik, I., A. Druzijanic, and N. Druzijanic, Inositol Hexaphosphate (IP6) and Colon Cancer: From Concepts and First Experiments to Clinical Application. Molecules, 2020. 25(24). [CrossRef]
- Kilaparty, S.P., et al., Endoplasmic reticulum stress-induced apoptosis accompanies enhanced expression of multiple inositol polyphosphate phosphatase 1 (Minpp1): a possible role for Minpp1 in cellular stress response. Cell Stress Chaperones, 2016. 21(4): p. 593-608. [CrossRef]
- Al-Thani, A.N., et al., Nanoparticles in cancer theragnostic and drug delivery: A comprehensive review. Life Sci, 2024. 352: p. 122899. [CrossRef]
- Puttasiddaiah, R., et al., Emerging Nanoparticle-Based Diagnostics and Therapeutics for Cancer: Innovations and Challenges. Pharmaceutics, 2025. 17(1). [CrossRef]
- Chen, H., et al., Rethinking cancer nanotheranostics. Nat Rev Mater, 2017. 2. [CrossRef]
- Elumalai, K., Srinivasan, S., Shanmugam S,, Review of the efficacy of nanoparticle-based drug delivery systems for cancer treatment,. Biomedical Technology, 2023. 5: p. 109-122. [CrossRef]
- Reda, A., S. Hosseiny, and I.M. El-Sherbiny, Next-generation nanotheranostics targeting cancer stem cells. Nanomedicine (Lond), 2019. 14(18): p. 2487-2514. [CrossRef]
- Fan, D., et al., Nanomedicine in cancer therapy. Signal Transduct Target Ther, 2023. 8(1): p. 293.
- Singh, S.K., et al., Drug delivery approaches for breast cancer. Int J Nanomedicine, 2017. 12: p. 6205-6218.
- Aldhubiab, B., R.M. Almuqbil, and A.B. Nair, Harnessing the Power of Nanocarriers to Exploit the Tumor Microenvironment for Enhanced Cancer Therapy. Pharmaceuticals (Basel), 2025. 18(5). [CrossRef]
- O’Brien, M.E., et al., Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol, 2004. 15(3): p. 440-9.
- Sharifi-Rad, J., et al., Paclitaxel: Application in Modern Oncology and Nanomedicine-Based Cancer Therapy. Oxid Med Cell Longev, 2021. 2021: p. 3687700. [CrossRef]
- Montero, A.J., et al., Nab-paclitaxel in the treatment of metastatic breast cancer: a comprehensive review. Expert Rev Clin Pharmacol, 2011. 4(3): p. 329-34. [CrossRef]
- Gu, S., et al., Therapeutic siRNA for drug-resistant HER2-positive breast cancer. Oncotarget, 2016. 7(12): p. 14727-41. [CrossRef]
- Rahim, M.A., et al., Recent Advancements in Stimuli Responsive Drug Delivery Platforms for Active and Passive Cancer Targeting. Cancers (Basel), 2021. 13(4). [CrossRef]
- Mitchell, M.J., et al., Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov, 2021. 20(2): p. 101-124. [CrossRef]
- Noury, H., et al., AI-driven innovations in smart multifunctional nanocarriers for drug and gene delivery: A mini-review. Crit Rev Oncol Hematol, 2025. 210: p. 104701. [CrossRef]
- Kemp, J.A., et al., "Combo" nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Deliv Rev, 2016. 98: p. 3-18.
- Shen, J., et al., Crosstalk of methylation and tamoxifen in breast cancer (Review). Mol Med Rep, 2024. 30(4). [CrossRef]
- Gan, L., et al., Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res, 2018. 6: p. 10. [CrossRef]
- Feng, Q., et al., An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res, 2014. 24(7): p. 809-19. [CrossRef]
- Blake, M.K., P. O’Connell, and Y.A. Aldhamen, Fundamentals to therapeutics: Epigenetic modulation of CD8(+) T Cell exhaustion in the tumor microenvironment. Front Cell Dev Biol, 2022. 10: p. 1082195. [CrossRef]
- Achinger-Kawecka, J., et al., The potential of epigenetic therapy to target the 3D epigenome in endocrine-resistant breast cancer. Nat Struct Mol Biol, 2024. 31(3): p. 498-512. [CrossRef]
- Hattori, N., et al., Novel prodrugs of decitabine with greater metabolic stability and less toxicity. Clin Epigenetics, 2019. 11(1): p. 111. [CrossRef]
- Dai, W., et al., Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct Target Ther, 2024. 9(1): p. 332. [CrossRef]
- Li, S.Y., et al., Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release, 2015. 205: p. 7-14. [CrossRef]
- Ranganna, K., et al., Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach. Int J Mol Sci, 2020. 21(21). [CrossRef]
- Guefack, M.F. and S. Bhatnagar, Advances in Epigenetic Therapeutics for Breast Cancer. Adv Exp Med Biol, 2024. 1465: p. 89-97.
- Chen, J.S., et al., A novel STAT3/ NFkappaB p50 axis regulates stromal-KDM2A to promote M2 macrophage-mediated chemoresistance in breast cancer. Cancer Cell Int, 2023. 23(1): p. 237.
- Soliman, A., Z. Li, and A.V. Parwani, Artificial intelligence’s impact on breast cancer pathology: a literature review. Diagn Pathol, 2024. 19(1): p. 38. [CrossRef]
- LeCun, Y., Y. Bengio, and G. Hinton, Deep learning. Nature, 2015. 521(7553): p. 436-44.
- Madani, M., M.M. Behzadi, and S. Nabavi, The Role of Deep Learning in Advancing Breast Cancer Detection Using Different Imaging Modalities: A Systematic Review. Cancers (Basel), 2022. 14(21). [CrossRef]
- Walsh, E., et al., A Deep-Learning Solution Identifies HER2 Negative Cases and Provides ER and PR Results From H&E-Stained Breast Cancer Specimens: A Blind Validation Study. Clin Breast Cancer, 2025. [CrossRef]
- Esteva, A., et al., Deep learning-enabled medical computer vision. NPJ Digit Med, 2021. 4(1): p. 5. [CrossRef]
- Nicolis, O., D. De Los Angeles, and C. Taramasco, A contemporary review of breast cancer risk factors and the role of artificial intelligence. Front Oncol, 2024. 14: p. 1356014. [CrossRef]
- Yala, A., et al., A Deep Learning Mammography-based Model for Improved Breast Cancer Risk Prediction. Radiology, 2019. 292(1): p. 60-66. [CrossRef]
- Ling, J., et al., Artificial intelligence-driven discovery of YH395A: A novel TGFbetaR1 inhibitor with potent anti-tumor activity against triple-negative breast cancer. Cell Commun Signal, 2025. 23(1): p. 326. [CrossRef]
- Bendani, H., et al., Revolutionizing breast cancer immunotherapy by integrating AI and nanotechnology approaches: review of current applications and future directions. Bioelectron Med, 2025. 11(1): p. 13. [CrossRef]
- Ren, S., et al., A carrier-free ultrasound-responsive polyphenol nanonetworks with enhanced sonodynamic-immunotherapy for synergistic therapy of breast cancer. Biomaterials, 2025. 317: p. 123109. [CrossRef]
- Irfan, M., U. Habiba, and A. Maryam, Next-generation cancer therapeutics: unveiling the potential of liposome-based nanoparticles through bioinformatics. Mikrochim Acta, 2025. 192(7): p. 428. [CrossRef]
- Ghandikota, S.K. and A.G. Jegga, Application of artificial intelligence and machine learning in drug repurposing. Prog Mol Biol Transl Sci, 2024. 205: p. 171-211.
- Witten, I., et al., Future views on neuroscience and AI. Cell, 2024. 187(21): p. 5809-5813. [CrossRef]
- Sun, Q., et al., HER2-Targeted Nanoliposome Therapy Activates Immune Response by Converting Cold to Hot Breast Tumors. Technol Cancer Res Treat, 2025. 24: p. 15330338251356387. [CrossRef]
- Topol, E.J., High-performance medicine: the convergence of human and artificial intelligence. Nat Med, 2019. 25(1): p. 44-56. [CrossRef]
- Guan, H., et al., Federated learning for medical image analysis: A survey. Pattern Recognit, 2024. 151.
- Arravalli, T., et al., Detection of breast cancer using machine learning and explainable artificial intelligence. Sci Rep, 2025. 15(1): p. 26931. [CrossRef]
- Shen, S., et al., From virtual to reality: innovative practices of digital twins in tumor therapy. J Transl Med, 2025. 23(1): p. 348. [CrossRef]
- Gupta, M.K., G. Gouda, and R. Vadde, Role of the Tumor Microenvironment in Mediating Resistance to Anti-HER2 Antibodies. Crit Rev Oncog, 2024. 29(4): p. 43-54. [CrossRef]
Figure 1.
Biological Characteristics of Breast cancer development: The common mechanisms involved in BC progression, therapy resistance and tumor recurrence are shown in the schematic representation. A healthy female breast is made up of 12–20 sections called lobes. Each of these lobes is made up of many smaller lobules. Both the lobes and lobules are connected by milk ducts. BC originates either at the lobules or in the ducts. .
Figure 1.
Biological Characteristics of Breast cancer development: The common mechanisms involved in BC progression, therapy resistance and tumor recurrence are shown in the schematic representation. A healthy female breast is made up of 12–20 sections called lobes. Each of these lobes is made up of many smaller lobules. Both the lobes and lobules are connected by milk ducts. BC originates either at the lobules or in the ducts. .
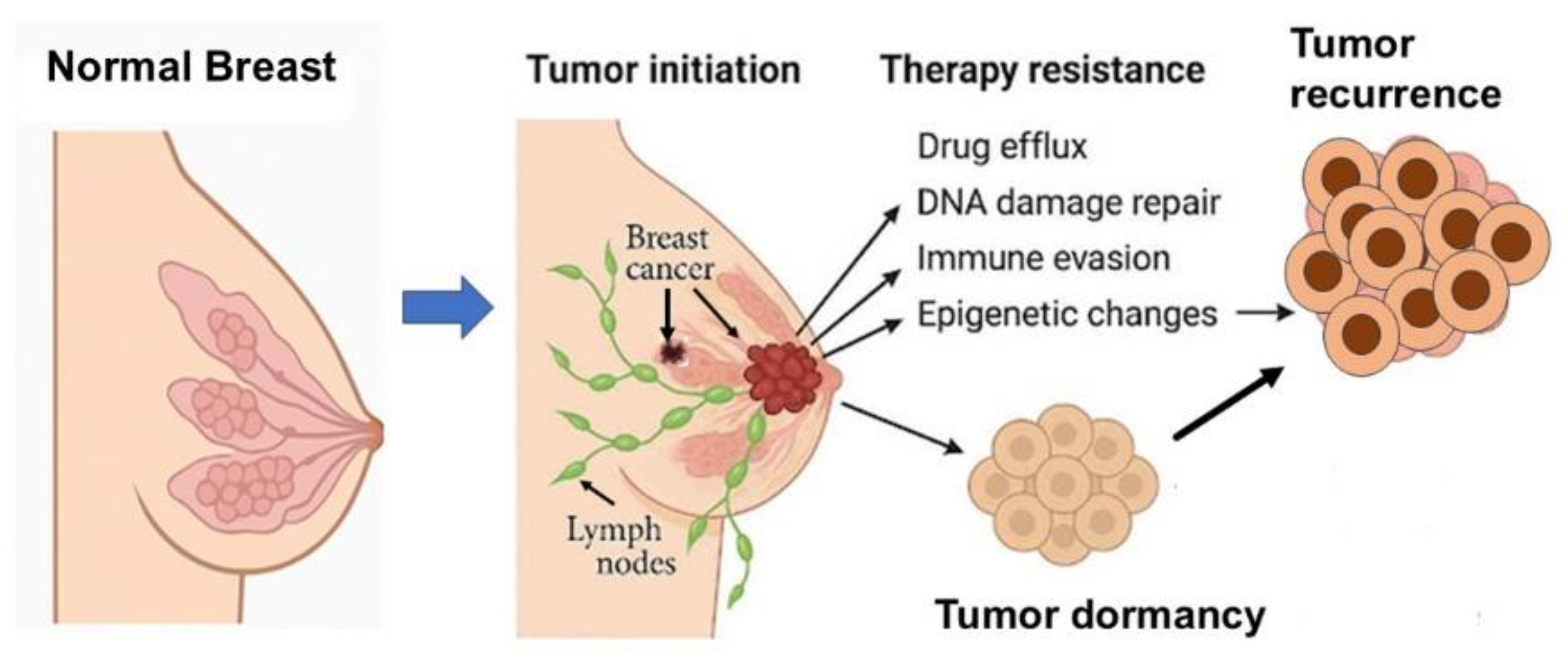
Figure 2.
Experimental models for investigating BC, genetically engineered mouse models (GEMMs), circulating tumor cell (CTC)-derived models, and AI-based in silico platforms.
Figure 2.
Experimental models for investigating BC, genetically engineered mouse models (GEMMs), circulating tumor cell (CTC)-derived models, and AI-based in silico platforms.
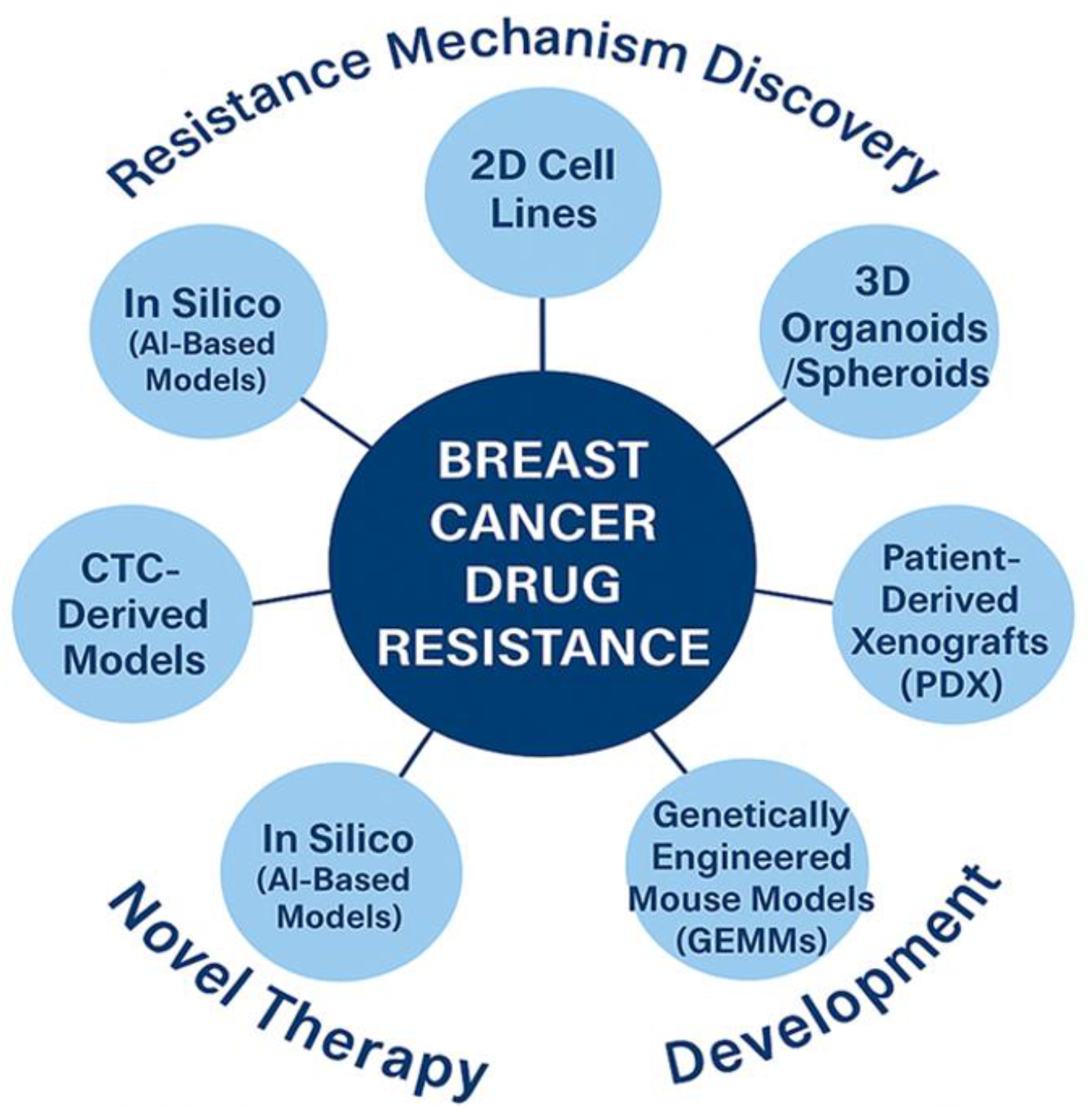
Figure 3.
Key molecular signaling pathways involved in BC that contribute to therapeutic resistance mechanisms. Schematic illustration of six major oncogenic signaling pathways contributing to BC resistance, metastasis, and immune evasion. .
Figure 3.
Key molecular signaling pathways involved in BC that contribute to therapeutic resistance mechanisms. Schematic illustration of six major oncogenic signaling pathways contributing to BC resistance, metastasis, and immune evasion. .

Figure 4.
Schematic representation of the effect of Microbiota on breast cancer.
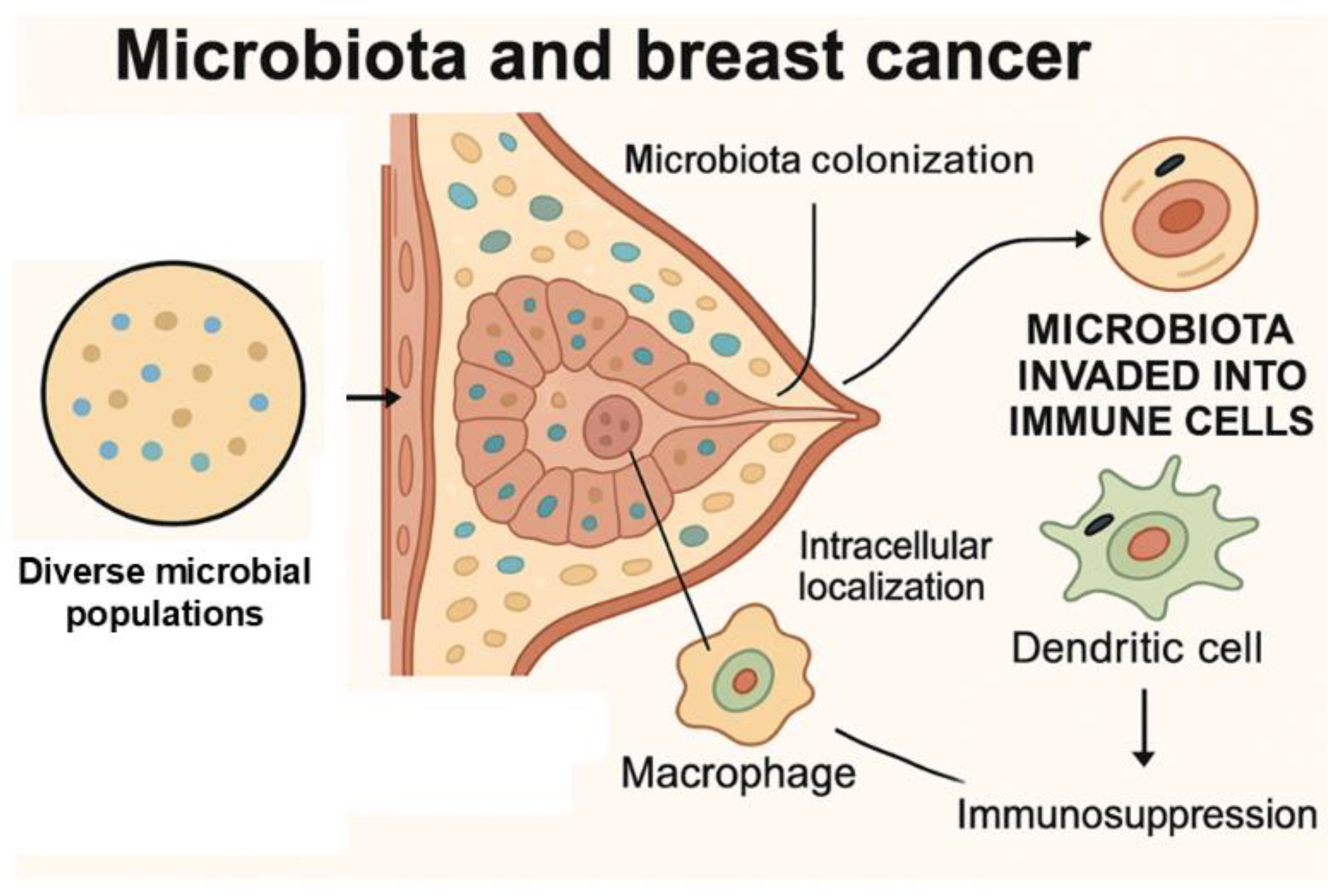
Figure 5.
Tumor Microenvironment (TME) Modulation: Converting ‘Cold’ to ‘Hot’. Simplified depiction of the transition from an immunologically inert (‘cold’) to an immune-responsive (‘hot’) tumor microenvironment, highlighting therapeutic strategies aimed at overcoming immune exclusion and enhancing anti-tumor responses.
Figure 5.
Tumor Microenvironment (TME) Modulation: Converting ‘Cold’ to ‘Hot’. Simplified depiction of the transition from an immunologically inert (‘cold’) to an immune-responsive (‘hot’) tumor microenvironment, highlighting therapeutic strategies aimed at overcoming immune exclusion and enhancing anti-tumor responses.
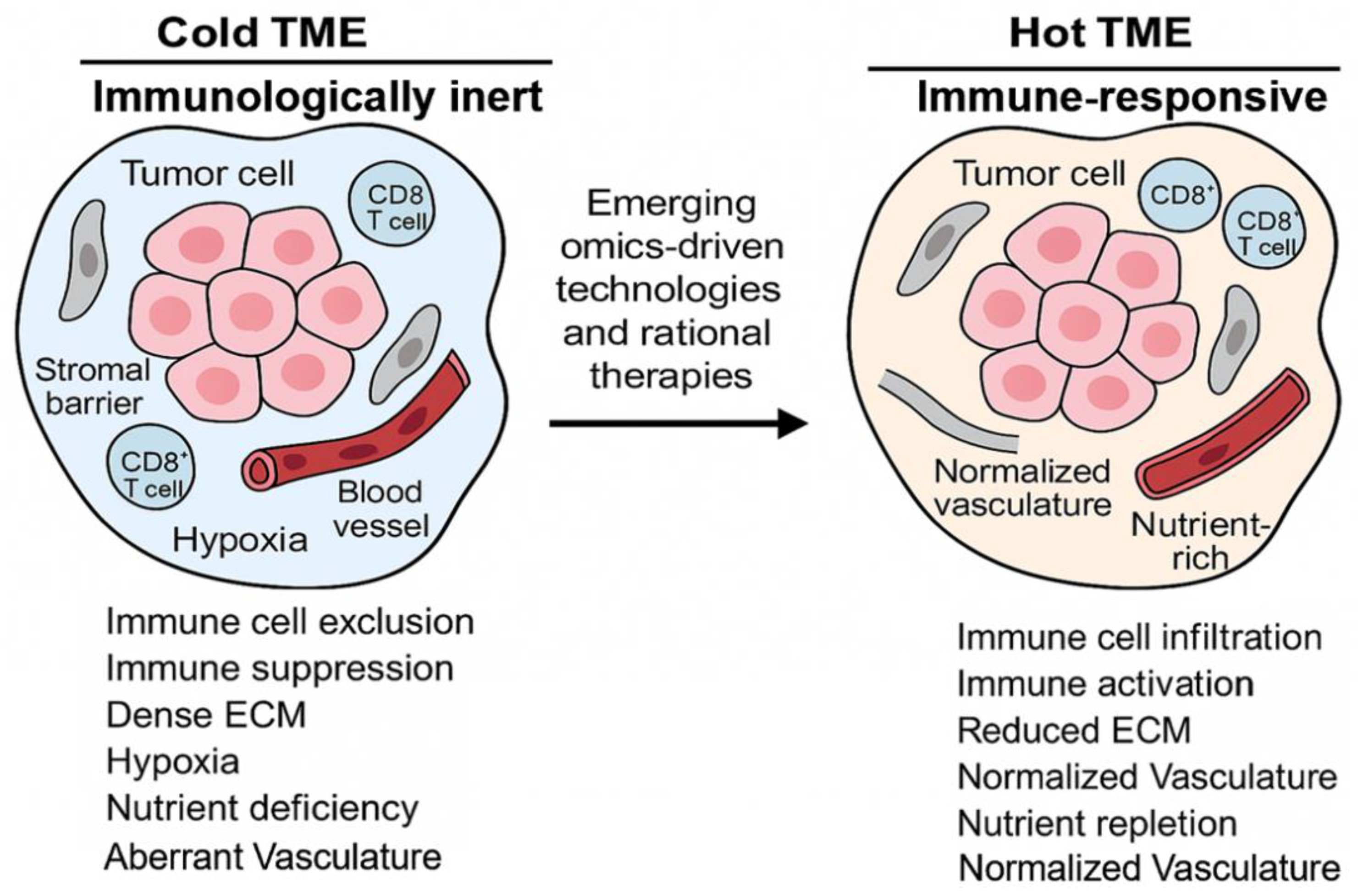
Figure 6.
Mechanisms of Tumor Dormancy and Relapse in Breast Cancer. Schematic illustration of three key dormancy states in BC: primary dormancy driven by microenvironmental cues, therapy-induced dormancy following anticancer treatment, and metastatic dormancy involving disseminated tumor cells (DTCs) that persist in distant sites.
Figure 6.
Mechanisms of Tumor Dormancy and Relapse in Breast Cancer. Schematic illustration of three key dormancy states in BC: primary dormancy driven by microenvironmental cues, therapy-induced dormancy following anticancer treatment, and metastatic dormancy involving disseminated tumor cells (DTCs) that persist in distant sites.
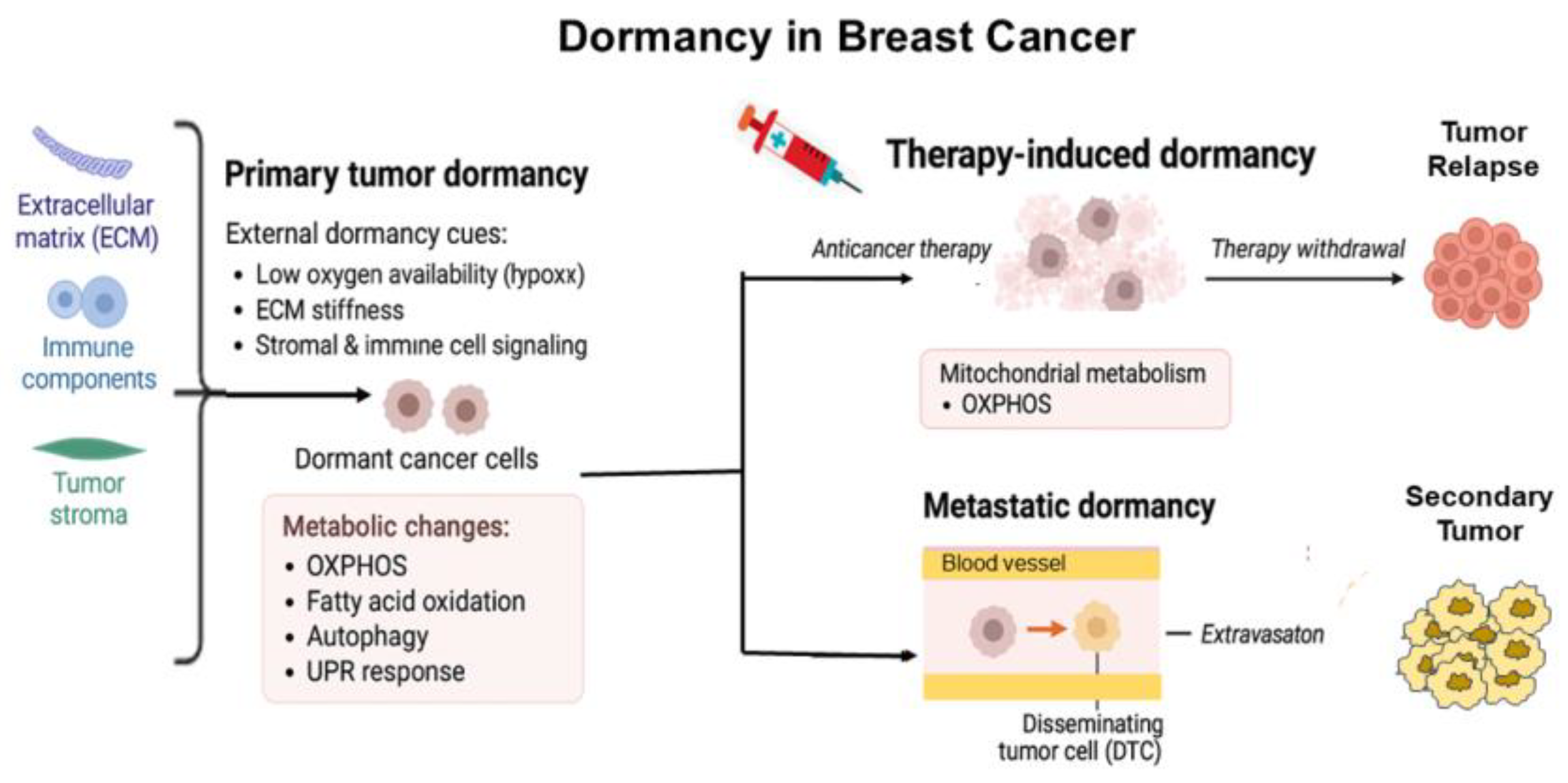
Figure 7.
Mitochondrial Dynamics and Intercellular Transfer Driving Immune Evasion and Tumor Resistance in Breast Cancer. BC cells exhibit mitochondrial DNA mutations, heterogeneity, and abnormal mitochondrial dynamics. Through tunneling nanotubes (TNTs), these cells transfer dysfunctional mitochondria to CD4⁺ and CD8⁺ T cells, leading to T-cell dysfunction and immune evasion.
Figure 7.
Mitochondrial Dynamics and Intercellular Transfer Driving Immune Evasion and Tumor Resistance in Breast Cancer. BC cells exhibit mitochondrial DNA mutations, heterogeneity, and abnormal mitochondrial dynamics. Through tunneling nanotubes (TNTs), these cells transfer dysfunctional mitochondria to CD4⁺ and CD8⁺ T cells, leading to T-cell dysfunction and immune evasion.

Figure 8.
Exosome Biogenesis and MinPP1 Secretion in Cancer Cells. Schematic illustration showing conventional multivesicular body (MVB)-derived exosomes and the emerging role of endoplasmic reticulum (ER) in exosome biogenesis. Note the secretion of MinPP1 isoform-2 in extracellular vesicles (EVs). This ER-to-exosome pathway might potentially contribute to the TME modulation and cancer progression. (Adapted with permission from Zubair, PhD Dissertation, 2023).
Figure 8.
Exosome Biogenesis and MinPP1 Secretion in Cancer Cells. Schematic illustration showing conventional multivesicular body (MVB)-derived exosomes and the emerging role of endoplasmic reticulum (ER) in exosome biogenesis. Note the secretion of MinPP1 isoform-2 in extracellular vesicles (EVs). This ER-to-exosome pathway might potentially contribute to the TME modulation and cancer progression. (Adapted with permission from Zubair, PhD Dissertation, 2023).
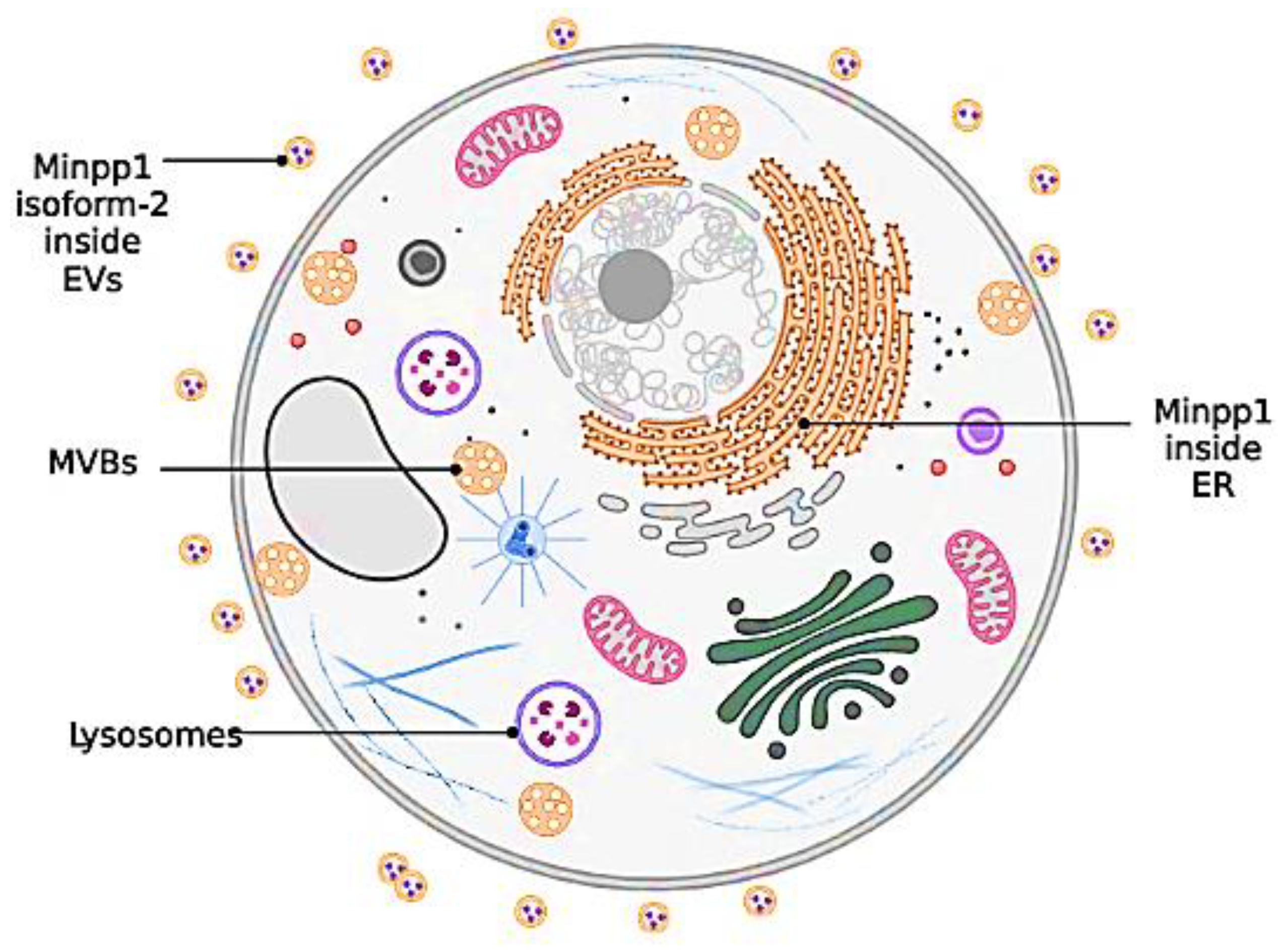
Figure 9.
Epigenetic mechanisms contributing to BC resistance and targeted therapies. DNA hypermethylation, histone modifications (EZH2, KDM2A), and dysregulated non-coding RNAs (e.g., miR-155) promote ERα silencing, chromatin remodeling, and immune escape. Targeted therapies include DNMT inhibitors, HDAC inhibitors, and EZH2 inhibitors aimed at reversing these resistance mechanisms.”.
Figure 9.
Epigenetic mechanisms contributing to BC resistance and targeted therapies. DNA hypermethylation, histone modifications (EZH2, KDM2A), and dysregulated non-coding RNAs (e.g., miR-155) promote ERα silencing, chromatin remodeling, and immune escape. Targeted therapies include DNMT inhibitors, HDAC inhibitors, and EZH2 inhibitors aimed at reversing these resistance mechanisms.”.
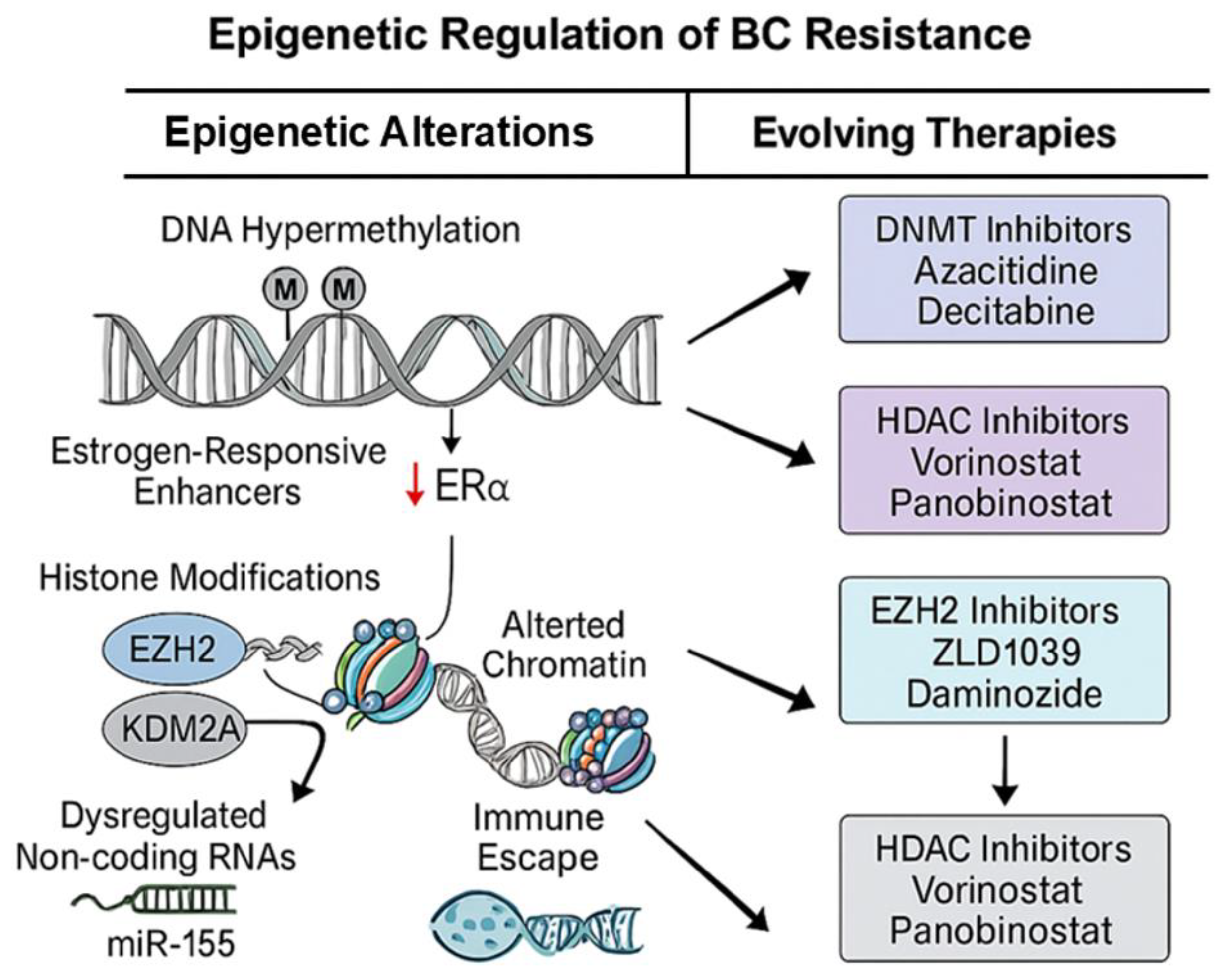

Table 1.
List of genes with driver-mutations in Breast Cancer.
| Gene Penetrance | Genes Involved |
|---|---|
| High Penetrance Genes | BRCA1, BRCA2, PTEN, CDH1, STK11, TP53 |
| Moderate Penetrance Genes | CHEK2, BRIP1, ATM, PALB2 |
| Low Penetrance Genes | FGFR2, LSP1, MAP3K1, TGF-β1, TOX3, RECQL, MUTYH, MSH6, NF1, NBN |
Table 2.
Risk Factors for Developing Breast Cancer are shown.
| Risk Factor(s) | Mechanism | Reference |
|---|---|---|
| Age | Increased incidence with age likely due to cumulative DNA damage and reduced cellular repair mechanisms. | [4,31] |
| Family History | Inherited germline mutations in genes such as BRCA1 and BRCA2 predispose individuals to hereditary C. | [32] |
| Personal History | A prior diagnosis of BC in one breast significantly increases the risk of developing cancer in the contralateral breast. | [37] |
| Early Menarche/Late Menopause | Prolonged lifetime exposure to endogenous estrogen and progesterone, increasing the number of menstrual cycles (Menarche <12 years or menopause >55 years). | [35] |
| Reproductive History | Nulliparity (never having given birth) or first childbirth after age 30 are associated with increased risk, possibly due to altered hormonal profiles and breast tissue development. | [38] |
| Dense Breast Tissue | Higher mammographic density indicates a greater proportion of glandular and fibrous tissue compared to fatty tissue, which is associated with an increased risk and can complicate early detection by mammography. | [39] |
| Lifestyle Factors | Alcohol consumption, obesity (particularly postmenopausal), and smoking are established modifiable risk factors. Alcohol can increase estrogen levels; obesity is linked to chronic inflammation and altered hormone metabolism; smoking introduces carcinogens. | [34,40] |
| Hormonal Factors | Hormone replacement therapy (HRT), particularly combined estrogen and progestin formulations, can increase BC risk by increasing circulating hormone levels. | [41] |
| Radiation Exposure | Exposure to ionizing radiation, especially during youth (e.g., treatment for Hodgkin’s lymphoma), can damage breast tissue DNA and increase the long-term risk. | [42] |
| Diethylstilbestrol (DES) | In utero exposure to DES (prescribed between 1940–1971 in the U.S. to prevent miscarriage) is a known risk factor for BC in both the exposed women and their daughters. | [43] |
Table 3.
Experimental Models in Breast Cancer Resistance Research.
| Model | Key contributions | Pros | Cons | Ref |
|---|---|---|---|---|
| 2D Cell Lines | Mechanistic discoveries in ER, HER2, and efflux resistance | Cheap, reproducible, high-throughput | Poor physiological relevance | [68,69,70] |
| 3D Spheroids/ Organoids | Hypoxia, CSC-driven resistance, TME influence | Mimics architecture, patient-derived | Complex culture, batch variability | [68,69,73,76], |
| Patient-Derived Xenografts (PDX) | Resistance in heterogeneous tumors, therapy validation | High translational relevance | Expensive, lacks human immunity | [69,76,77], |
| Genetically Engineered Mice (GEMMs) | DNA repair defects, immune-competent resistance models | Immune-competent, spontaneous tumors | Genetically rigid, costly | [69,77], |
| CTC Models | Insights into metastasis, mesenchymal resistance | Real-time, metastatic focus | Difficult to culture and expand | [77,78] |
| In Silico Models (AI-based) | Predictive modeling of resistance, target discovery | Fast, scalable, cost-effective | Needs biological validation | [69] |
Table 4.
Next-generation FDA-cleared AI Solutions in Cancer Diagnostics.
| Feature | Ibex Prostate Pathology | OnQ™ Prostate Imaging | AI in Breast Cancer (ProFound 4.0) | AI in Breast Cancer (Clairity Breast) |
|---|---|---|---|---|
| FDA Status | 510(k) cleared (May 2024) | 510(k) cleared (Feb 2025) | 510(k) cleared (Nov 2024) | De Novo clearance (June 2025) |
| Modality | AI-based analysis of H&E-stained biopsy slides | RSI-enhanced diffusion-weighted MRI | Mammography (with or without prior imaging) | AI-based analysis of screening mammograms |
| Purpose | Digital pathology interpretation, cancer detection | Improved lesion characterization, biopsy targeting | Enhanced sensitivity and risk prediction | Detection of subtle imaging features predictive of future cancer |
| Clinical Utility | Gleason scoring, decision support for pathologists | Improves PI-RADS accuracy, reduces inter-reader variability | Improves detection in dense breasts, risk stratification | Predicts 5-year BC risk from routine mammography |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



