
Submitted:
24 July 2025
Posted:
25 July 2025
You are already at the latest version
Abstract
Ohdo syndrome is a rare congenital disorder caused by pathogenic KAT6B variants, with fewer than 30 cases reported globally. We present the first genetically confirmed case in the Philippines: a 16-year-old female with intellectual disability, blepharophimosis, and other craniofacial features. Skeletal survey revealed micrognathia, scoliosis, negative ulnar variance, and digital abnormalities. This case highlights the diagnostic utility of skeletal surveys in documenting phenotypic features, guiding referrals, and supporting clinical recognition of KAT6B-related syndromes like the SBBYS variant.
Keywords:
intellectual disability
; craniofacial abnormalities
; Ohdo syndrome
; case report
; radiography
Case Presentation
A 16-year-old Filipino female presented with intellectual disability and syndromic features. She was born full-term via normal spontaneous delivery to a healthy 35-year-old G3P3 mother. Her birthweight was approximately 2500 grams. She had a poor cry and hypotonia at birth. The parents are non-consanguineous, and her two siblings are unaffected. No known family history of similar conditions was reported.
Postnatally, the patient exhibited marked blepharophimosis and ptosis. Developmental delays became apparent: she began walking at age 2 but never developed speech. Though responsive to commands, she was unable to attend special education due to frequent tantrums. Psychological evaluation revealed an IQ below 70, confirming intellectual disability. Menarche and secondary sexual development began at 14 but remain immature.
Her mother, an overseas worker in Cyprus, sought information from rare disease networks and was advised genetic testing. Whole exome sequencing revealed a pathogenic KAT6B mutation, confirming Say-Barber-Biesecker-Young-Simpson (SBBYS) type Ohdo syndrome. Initial labs including CBC, urinalysis, and thyroid function tests were normal. A skeletal survey was requested to evaluate associated skeletal anomalies.
On physical examination (Figure 1), the patient had a stooped posture, joint laxity, blepharophimosis, ptosis, intermittent nystagmus, broad nasal bridge, rounded nose tip, micrognathia, long thumbs, single palmar crease, atrophic thenar/hypothenar muscles, stiff finger joints, and hallux valgus. A grade 3/6 holosystolic murmur and widely spaced teeth were also noted.
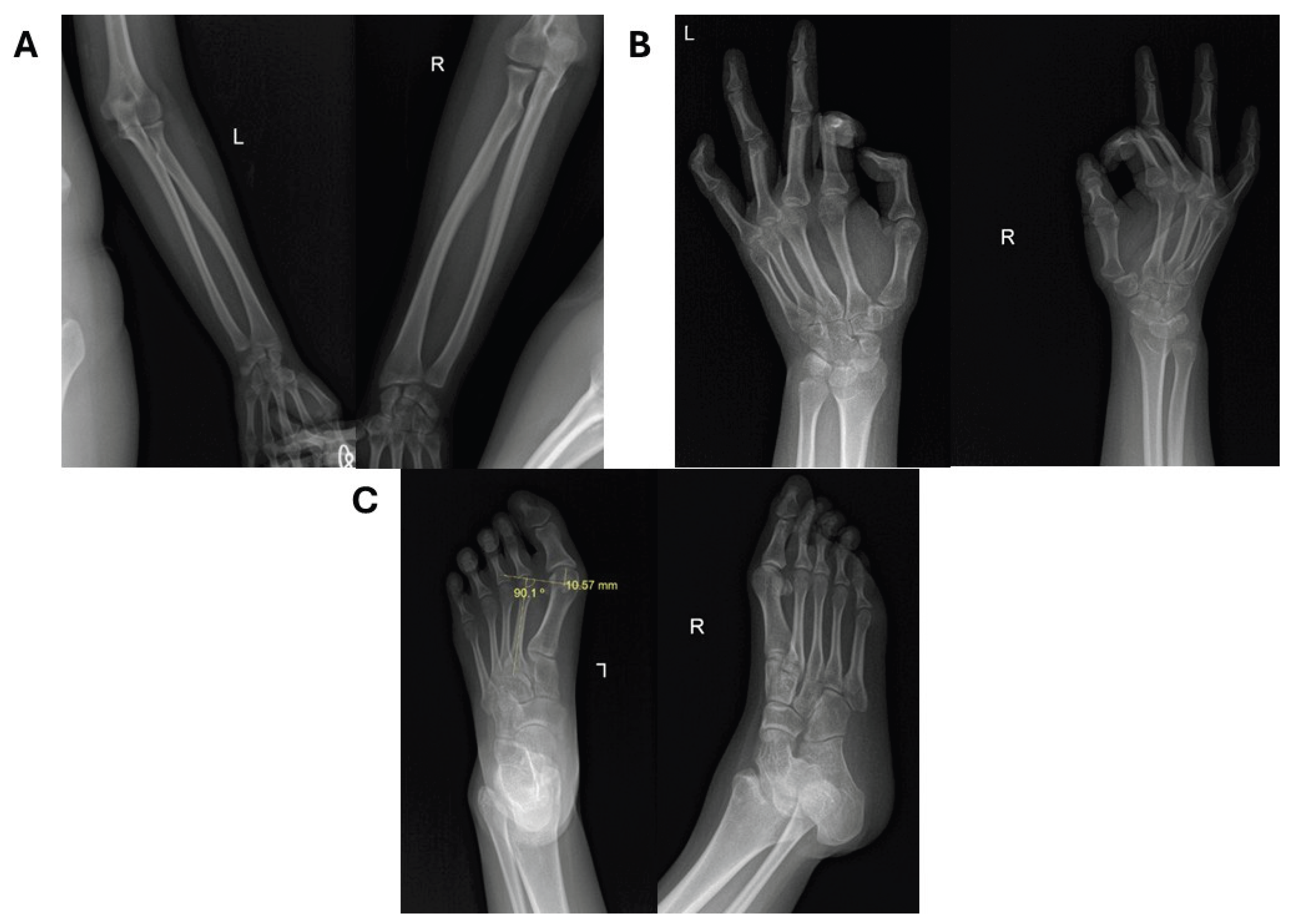
Skeletal survey revealed micrognathia, scoliosis, and intact clavicles and ribs (Figure 2). A proximally placed ulna indicated negative ulnar variance. The 2nd digit distal phalanges were short with flexed DIP joints; PIP joints of 2nd and 3rd fingers were hyperextended. Bone age was consistent with a 17-year-old female. The knees were unremarkable. Foot radiographs showed bilateral long 1st metatarsals and hallux valgus (Figure 3).
These radiologic findings are consistent with SBBYS-type Ohdo syndrome. The presence of normal patellae helped rule out genitopatellar syndrome (Table 1), which shares the same genetic etiology but includes absent patellae and genital anomalies. MRI and panoramic dental imaging were recommended but not completed due to uncooperativeness. The patient was eventually lost to follow-up.
Discussion
Ohdo syndrome was first reported in 1986 by Shozo Ohdo, who described two sisters with intellectual disability, congenital heart disease, ptosis, and dental anomalies [1]. Several variants have since been described. The Say-Barber-Biesecker-Young-Simpson (SBBYS) variant, now recognized as a distinct clinical entity, was described in the late 1980s and early 1990s [2,3]. It is characterized by blepharophimosis, midface hypoplasia, hypotonia, hyperextensible joints, dental anomalies, and intellectual disability [4].
In 2006, Verloes et al. proposed grouping blepharophimosis–mental retardation syndromes (BMRS) into five categories, with SBBYS being the most distinct and recognizable [5]. It is caused by heterozygous mutations in KAT6B, a gene located on chromosome 10q22.2 that encodes a histone acetyltransferase involved in embryonic development [6]. The MYST-type acetyltransferase domain of KAT6B regulates gene transcription via histone H3 acetylation at lysines 9 and 14, impacting skeletal, craniofacial, and neurologic development.
KAT6B plays a vital role in mesenchymal cell differentiation and osteoblast maturation, partly through its control of RUNX2, a master regulator of osteogenesis [7]. Pathogenic variants, typically truncating mutations in the gene's C-terminal region, impair normal protein interactions with co-regulators such as BRPF1, ING5, and MEAF6 [8]. This dysregulation leads to phenotypic features such as scoliosis, hallux valgus, joint contractures, and distinctive craniofacial anomalies.
KAT6B mutations also disrupt neural crest cell migration, resulting in midface hypoplasia, micrognathia, and blepharophimosis. The condition may impair FOXL2-dependent eyelid separation in early fetal life [9]. In the oral-maxillofacial region, panoramic imaging may show hypoplastic maxilla, delayed tooth eruption, and dental malocclusion. Cephalometric analysis may reveal Class III skeletal relationships.
KAT6B is expressed in fetal brain tissue, including the ventricular zone, explaining the intellectual disability. Patients may have global developmental delay, hypotonia, and behavioral disturbances. Diagnostic workup typically includes genetic testing (e.g., whole exome sequencing) and supporting radiologic studies.
Clinical diagnostic criteria for SBBYS include mandatory findings: (1) blepharophimosis, (2) ptosis, and (3) intellectual disability [10]. Supporting features include hypotonia, hypoplastic teeth, undescended testes, midface hypoplasia, and skeletal anomalies. The presence of two major features or one major and two minor features should prompt testing for KAT6B mutations.
Differential Diagnosis

Table 1.
Differential diagnosis of the case.
| Syndrome | Gene(s) | Key Features | Differentiating Radiologic Findings |
| SBBYS-type Ohdo syndrome | KAT6B | Intellectual disability, blepharophimosis, ptosis, long thumbs, dental anomalies | Hallux valgus, scoliosis, normal patellae, shortened phalanges, ulnar variance |
| Genitopatellar syndrome | KAT6B | ID, absent patellae, genital anomalies, contractures | Absent patellae, flexion contractures, pelvic anomalies |
| Kabuki syndrome | KMT2D, KDM6A | Long palpebral fissures, arched eyebrows, cardiac defects | Vertebral anomalies, hip dislocation, short stature |
| Smith-Lemli-Opitz syndrome | DHCR7 | Microcephaly, ambiguous genitalia, syndactyly | Postaxial polydactyly, short long bones, abnormal vertebrae |
| Cornelia de Lange syndrome | NIPBL, others | Synophrys, limb abnormalities, growth retardation | Micromelia, rib anomalies, vertebral segmentation defects |
Conclusions
In this case, skeletal survey confirmed multiple typical findings and ruled out genitopatellar syndrome. Although KAT6B mutations underlie both conditions, genitopatellar syndrome presents with additional features such as agenesis of the corpus callosum, genital abnormalities, and absent patellae. Our patient had normal patellae and no noted genital anomalies. MRI, echocardiography, and thyroid function testing are helpful adjuncts. In this case, a holosystolic murmur suggested a possible septal defect, which is commonly associated with the syndrome. Unfortunately, echocardiography was not performed due to noncooperation. No curative treatment exists for KAT6B-related disorders. Management is multidisciplinary and supportive.
Teaching Points
- KAT6B-related disorders such as Say-Barber-Biesecker-Young-Simpson (SBBYS) syndrome can be differentiated from genitopatellar syndrome by key skeletal findings, such as the presence or absence of patellae and characteristic craniofacial and digital anomalies.
- The presence of subtle but specific hand and foot anomalies, such as long thumbs, single palmar crease, and bilateral hallux valgus, can offer early diagnostic clues in syndromic intellectual disability, even before genetic confirmation.
Multiple Choice Questions
1. What is the genetic basis of both Say-Barber-Biesecker-Young-Simpson and Genitopatellar syndromes?
A. Trisomy 18
B. Mutations in RUNX2
C. Mutations in KAT6B
D. Methylation defects on 15q11
Answer: C. Mutations in KAT6B
2. Which of the following features best distinguishes Say-Barber-Biesecker-Young-Simpson (SBBYS) syndrome from Genitopatellar syndrome, despite both being caused by KAT6B mutations?
A. Intellectual disability
B. Hypoplastic teeth
C. Normal patellae
D. Blepharophimosis
Answer: C. Normal patellae
3. Which of the following best explains the craniofacial and skeletal anomalies seen in KAT6B-related disorders?
A. Failure of neural tube closure during neurulation
B. Impaired collagen cross-linking in bone matrix
C. Disruption of histone acetylation during organogenesis
D. Abnormal migration of neural crest cells due to cilia defects
Answer: C. Disruption of histone acetylation during organogenesis
Funding
No funding was received.
Institutional Review Board Statement
This report was conducted in accordance with institutional ethical standards of Manila Doctors Hospital. Its institutional review board did not require ethical committee approval.
Informed Consent Statement
Written informed consent was obtained from the mother of the patient for publication of this case report and accompanying images.
Data Availability Statement
All photographs and radiographs pertaining to this study are available in the Harvard Dataverse repository at https://doi.org/10.7910/DVN/ISCGOQ.
Conflicts of Interest
The authors declare no competing interests.
References
- Ohdo, S.; Madokoro, H.; Sonoda, T.; Hayakawa, K. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J Med Genet 1986, 23, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Say, B.; Barber, N. Mental retardation with blepharophimosis. J Med Genet 1987, 24, 511. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G. The Ohdo blepharophimosis syndrome: a third case. J Med Genet 1991, 28, 131. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Simpson, K. Unknown syndrome: abnormal facies, congenital heart defects, hypothyroidism, and severe retardation. J Med Genet 1987, 24, 715. [Google Scholar] [CrossRef] [PubMed]
- Verloes, A.; Bremond-Gignac, D.; Isidor, B.; David, A.; Baumann, C.; Leroy, M.A.; et al. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A 2006, 140, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Champagne, N.; Bertos, N.R.; Pelletier, N.; Wang, A.H.; Vezmar, M.; Yang, Y.; et al. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. Journal of Biological Chemistry 1999, 274, 28528–28536. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, N.; Champagne, N.; Stifani, S.; Yang, X.J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 2002, 21, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Cayrou, C.; Ullah, M.; Landry, A.J.; Côté, V.; Selleck, W.; et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 2006, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Geiger, E.A.; Medne, L.; Zackai, E.H.; Shaikh, T.H. An Individual with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES) and Additional Features Expands the Phenotype Associated with Mutations in KAT6B. Am J Med Genet A 2014, 0, 950. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Adès, L.C.; Amor, D.; Liebelt, J.; Bankier, A.; Baker, E.; et al. Two further cases of Ohdo syndrome delineate the phenotypic variability of the condition. Clin Dysmorphol 2003, 12, 109–113. [Google Scholar] [CrossRef]
Figure 1.
Physical examination findings reveal craniofacial and skeletal abnormalities, including (A) blepharophimosis and ptosis, intermittent nystagmus, broad nasal bridge, (B) rounded nose tip, micrognathia, (C) relatively long thumbs, single palmar crease, atrophic hypothenar and thenar muscles, stiff phalangeal joints, and (D) large first toes.
Figure 1.
Physical examination findings reveal craniofacial and skeletal abnormalities, including (A) blepharophimosis and ptosis, intermittent nystagmus, broad nasal bridge, (B) rounded nose tip, micrognathia, (C) relatively long thumbs, single palmar crease, atrophic hypothenar and thenar muscles, stiff phalangeal joints, and (D) large first toes.
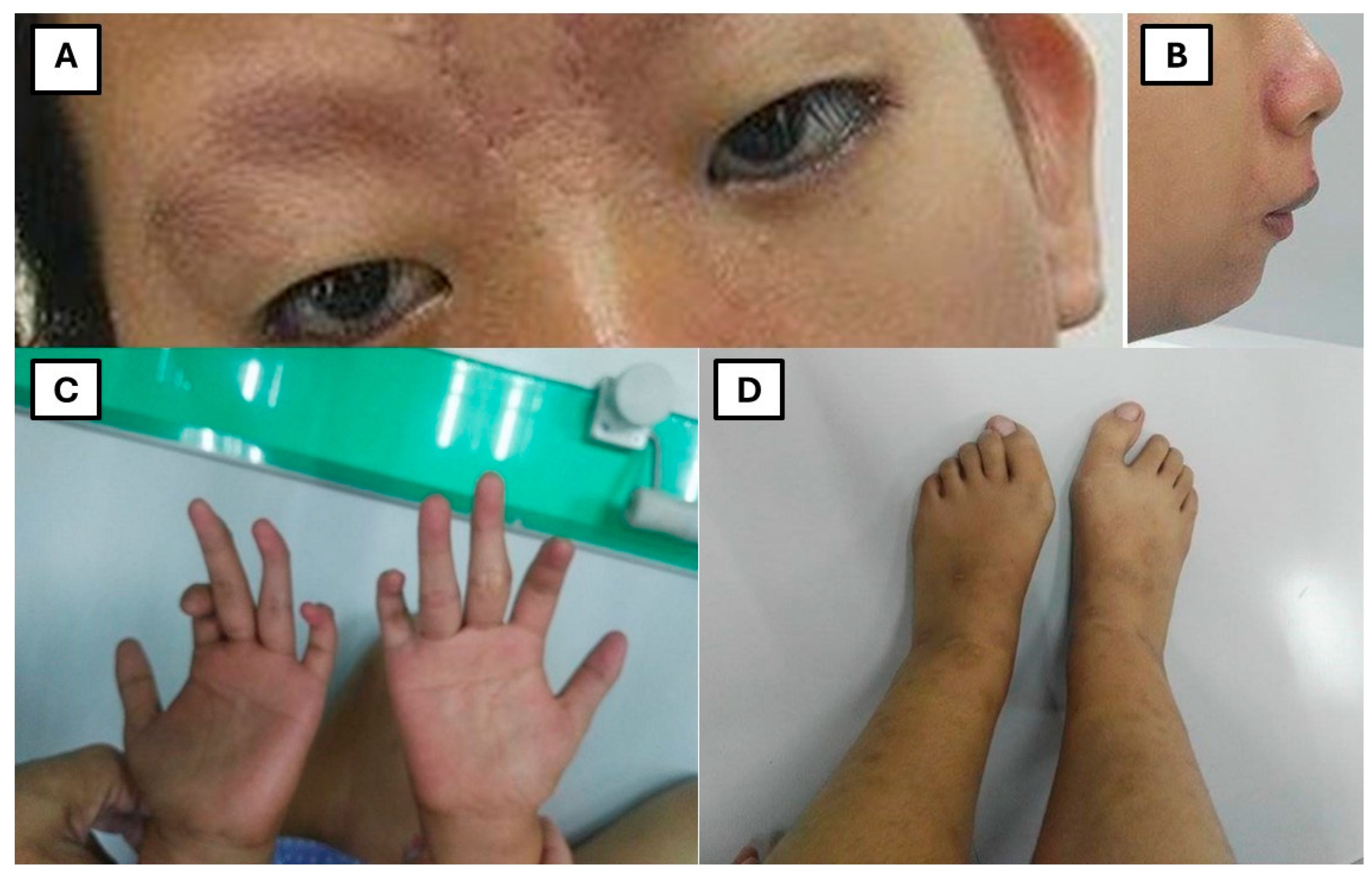
Figure 2.
Composite survey results of the axial skeleton. Radiographs of the skull AP (A), and thoracolumbar spine AP (B) reveal abnormalities. Notable findings include micrognathia (A) and scoliosis (B).
Figure 2.
Composite survey results of the axial skeleton. Radiographs of the skull AP (A), and thoracolumbar spine AP (B) reveal abnormalities. Notable findings include micrognathia (A) and scoliosis (B).
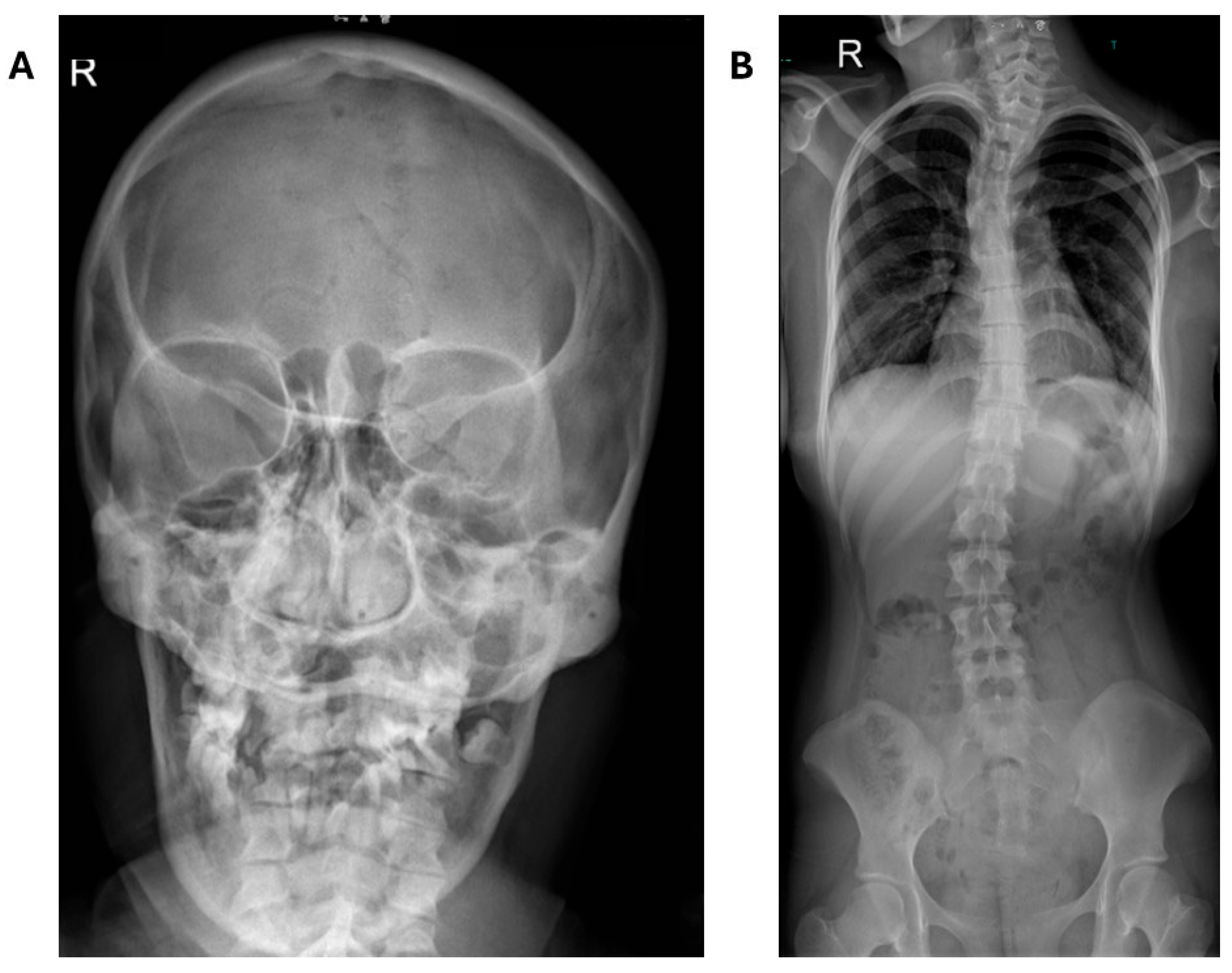
Figure 3.
Composite skeletal survey results of the appendicular skeleton. Radiographs of the radius-ulna AP (A), hands AP (B), and feet APL (C) reveal further generalized abnormalities. Notable findings include negative ulnar variance (A) and bilateral hallux valgi deformity (C). The distal phalanges of both 2nd digits of the hands are shortened (B).
Figure 3.
Composite skeletal survey results of the appendicular skeleton. Radiographs of the radius-ulna AP (A), hands AP (B), and feet APL (C) reveal further generalized abnormalities. Notable findings include negative ulnar variance (A) and bilateral hallux valgi deformity (C). The distal phalanges of both 2nd digits of the hands are shortened (B).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



