Submitted:
21 July 2025
Posted:
22 July 2025
Read the latest preprint version here
Abstract
Type 2 diabetes mellitus (T2DM), projected to affect over 700 million people by 2045, necessitates a transformative etiological perspective. The Sulfur Insulin Deformation Hypothesis redefines T2DM as a sulfur metabolism disorder driven by insulin misfolding due to organic sulfur deficiency, originating from mitochondrial dysfunction in intestinal epithelial cells. Evidence demonstrates 30–73.8% reductions in cysteine and glutathione levels in T2DM patients (RBC glutathione: 1.78 ± 0.28 vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001), driven by impaired transsulfuration pathways (cystathionine β-synthase, γ-lyase) and elevated oxidative stress (increased ROS, lipid peroxides). This redox imbalance inhibits protein disulfide isomerase (PDI), an endoplasmic reticulum enzyme critical for forming and isomerizing insulin’s disulfide bonds: A6–A11 (intra-A-chain, stabilizing α-helical core), A7–B7 (interchain, anchoring A- and B-chains), and A20–B19 (interchain, enabling receptor-binding conformation). Disruption of the A6–A11 hinge bond reduces insulin receptor affinity by 50–70% (r = -0.65, P < 0.05 for HOMA-IR), impairing PI3K-Akt signaling, GLUT2/GLUT4 translocation, and promoting gluconeogenesis, sustaining hyperglycemia. Cysteine deficiency activates NF-κB and JNK, elevating pro-inflammatory cytokines (TNF-α, IL-6), exacerbating insulin resistance via serine phosphorylation. Impaired mucin synthesis weakens the gut barrier, triggering TLR4-mediated endotoxemia and SOCS-driven inflammation. Interventions like N-acetylcysteine (NAC) and GlyNAC restore glutathione by 20–40% (P < 0.01), improve insulin sensitivity by 31% (P < 0.05), and enhance mitochondrial fatty acid oxidation. This model explains hyperinsulinemia alongside hyperglycemia, as misfolded insulin accumulates but fails to signal, while exogenous insulin restores function. This hypothesis proposes that insulin resistance and T2DM stem from insulin’s structural deformation due to disrupted disulfide bonds (A6–A11, A7–B7, A20–B19), mediated by impaired PDI function.
Keywords:
sulfur insulin deformation
; disulfide bonds (A6–A11)
; protein disulfide isomerase (PDI)
; cysteine deficiency
; glutathione depletion
; T2DM pathogenesis
1. Introduction
Type 2 diabetes mellitus (T2DM), a global health crisis projected to surge in prevalence, is traditionally attributed to peripheral insulin resistance driven by obesity, oxidative stress, and inflammation [1]. Yet, these models often overlook the critical role of insulin’s structural integrity, particularly its sulfur-dependent disulfide bonds. The Sulfur Insulin Deformation Hypothesis offers a groundbreaking framework, asserting that mitochondrial dysfunction in intestinal epithelial cells termed mitochondrial suffocation triggers organic sulfur deficiency, leading to insulin misfolding and systemic insulin resistance. This research aims to compile and elucidate evidence linking defective disulfide bond formation to insulin dysfunction, redefining T2DM as a sulfur metabolism disorder and revolutionizing its mechanistic interpretation. Insulin, a 51-amino-acid polypeptide, relies on three disulfide bonds (A6–A11, A7–B7, A20–B19) formed through cysteine thiol oxidation to maintain its bioactive conformation for high-affinity insulin receptor binding [2]. These bonds, dependent on dietary methionine and cysteine via the transsulfuration pathway, are disrupted by mitochondrial suffocation, which impairs adenosine triphosphate production and inhibits cystathionine β-synthase and γ-lyase, reducing cysteine availability [3,4,5,6]. This sulfur scarcity compromises protein disulfide isomerase (PDI) activity in pancreatic beta cells, leading to aberrant disulfide bond formation and misfolded insulin with reduced receptor affinity [7]. Such structural defects disrupt phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) signaling, impairing glucose transporter type 4 (GLUT4) translocation and glucose uptake [8].
Concurrently, sulfur deficiency elevates reactive oxygen species (ROS), activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and releasing pro-inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6), which exacerbate insulin resistance through c-Jun N-terminal kinase-mediated serine phosphorylation of insulin receptor substrate-1 [9,10,11]. Oxidative stress also weakens gut barrier integrity, promoting toll-like receptor 4 (TLR4)-mediated endotoxemia and systemic inflammation [12,13]. By presenting evidence on disulfide bond formation and its disruption, this study elucidates the gut-mitochondria-sulfur-insulin axis, offering a transformative lens to reinterpret insulin resistance and guide innovative T2DM therapeutic strategies.
2. Methodology
This Review presents the Sulfur Insulin Deformation Hypothesis, a novel framework proposing that sulfur deficiency, driven by mitochondrial dysfunction in the intestinal epithelium, causes insulin misfolding and insulin resistance in type 2 diabetes mellitus (T2DM). To develop this hypothesis, we conducted a structured literature synthesis to integrate mechanistic evidence from redox biology, mitochondrial pathology, protein biochemistry, and immunometabolism. A comprehensive literature search was performed across PubMed, Scopus, Web of Science, and Google Scholar using Medical Subject Headings (MeSH) and free-text terms, including “sulfur metabolism,” “insulin misfolding,” “disulfide bonds,” “glutathione deficiency,” “mitochondrial dysfunction,” “intestinal epithelium,” “oxidative stress,” “transsulfuration pathway,” “endoplasmic reticulum stress,” “cysteine,” and “type 2 diabetes.” Boolean operators (AND/OR) were used to combine terms, ensuring interdisciplinary coverage. The search included peer-reviewed studies from 1995 to 2025, capturing foundational and recent insights into sulfur-dependent metabolic regulation. From an initial pool of 1,202 articles, 243 duplicates were removed, and 959 unique articles were screened by title and abstract.
Of these, 624 were excluded due to irrelevance, insufficient mechanistic focus, non-English language, or inaccessible full texts. Full-text evaluation of 335 articles, based on inclusion criteria (relevance to insulin biosynthesis, disulfide bond integrity, mitochondrial-glutathione axis, and immunological impacts of sulfur deficiency), yielded 105 studies for inclusion. These encompassed in vitro models of insulin folding, animal studies of metabolic stress, human sulfur biomarker data, and pharmacologic trials of sulfur donors (e.g., N-acetylcysteine, methylsulfonylmethane). Data were synthesized to construct a mechanistic model linking mitochondrial dysfunction, cysteine scarcity, glutathione depletion, and insulin misfolding to T2DM pathogenesis. To test the hypothesis, we propose experimental approaches, including: (1) proteomic analyses to detect misfolded insulin in T2DM patients; (2) metabolomic profiling of sulfur metabolites (e.g., cysteine, glutathione) in intestinal and systemic tissues; (3) in vitro studies of enterocyte mitochondrial function under sulfur-deficient conditions; (4) animal models to assess N-acetylcysteine and methylsulfonylmethane effects on insulin structure and glucose homeostasis; and (5) clinical trials to evaluate sulfur donor supplementation in T2DM patients. The synthesis adheres to the SANRA framework, scoring 10/12 for clarity, evidence selection, and conceptual integration. This hypothesis-driven framework aims to redefine T2DM etiology and guide future research into sulfur-centric therapies.
3. Mitochondrial Suffocation as the Origin of Sulfur Deficiency
The intestinal epithelium, a metabolic hub for processing sulfur-containing amino acids, relies on robust mitochondrial function to support energy-intensive nutrient absorption [14,15].
In type 2 diabetes mellitus (T2DM), chronic stressors like hyperglycemia and high-fat diets induce mitochondrial dysfunction in enterocytes, termed mitochondrial suffocation, disrupting the electron transport chain (ETC), particularly complexes I and III [16,17]. This reduces adenosine triphosphate (ATP) production and generates excessive reactive oxygen species (ROS), depleting cellular antioxidants and impairing sulfur metabolism [18,19]. ROS overproduction exhausts glutathione, a cysteine-dependent tripeptide critical for redox homeostasis, exacerbating cellular damage [20]. The transsulfuration pathway, converting methionine to cysteine via methionine adenosyltransferase, cystathionine β-synthase, and cystathionine γ-lyase, is compromised by ATP scarcity, reducing cysteine synthesis [21,22,23,24,25]. This cysteine deficiency disrupts glutathione production and protein disulfide isomerase (PDI) activity, impairing insulin’s disulfide bond formation (A6–A11, A7–B7, A20–B19), leading to misfolded insulin with diminished receptor-binding capacity [26,27,28]. Immunologically, mitochondrial suffocation triggers nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, upregulating pro-inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) that promote c-Jun N-terminal kinase (JNK)-mediated serine phosphorylation of insulin receptor substrate-1, disrupting phosphoinositide 3-kinase (PI3K) signaling and exacerbating insulin resistance [29,30,31]. Additionally, ROS-induced downregulation of tight junction proteins (occludin, zonula occludens-1) compromises gut barrier integrity, enabling lipopolysaccharide translocation and toll-like receptor 4 (TLR4)-mediated endotoxemia, further amplifying systemic inflammation [32,33,34].
This gut-mitochondria-sulfur-insulin axis underscores mitochondrial suffocation as a pivotal driver of sulfur deficiency and T2DM pathogenesis.
4. The Sulfur Insulin Deformation Hypothesis: A Transformative Framework
The Sulfur Insulin Deformation Hypothesis redefines type 2 diabetes mellitus (T2DM) by asserting that sulfur deficiency, stemming from mitochondrial dysfunction in intestinal epithelial cells, drives insulin misfolding, a primary trigger of insulin resistance. Insulin, a 51-amino-acid polypeptide comprising A (21 amino acids) and B (30 amino acids) chains, is stabilized by three disulfide bonds (A6–A11, A7–B7, A20–B19) formed through cysteine thiol oxidation, essential for its three-dimensional conformation and high-affinity binding to the insulin receptor [35,36,37,38,39,40]. In pancreatic beta cells, insulin biosynthesis starts with preproinsulin, cleaved to proinsulin, and folded in the endoplasmic reticulum (ER), where protein disulfide isomerase (PDI) catalyzes disulfide bond formation by oxidizing cysteine residues, a process critically dependent on cysteine availability [41,42]. Mitochondrial dysfunction, termed mitochondrial suffocation, impairs the transsulfuration pathway by reducing adenosine triphosphate (ATP)-dependent activity of cystathionine β-synthase and γ-lyase, limiting cysteine synthesis [43,44]. This cysteine scarcity disrupts PDI function, leading to incomplete or aberrant disulfide bonds, producing misfolded insulin with altered tertiary structure, as demonstrated by Raman spectroscopy (510–540 cm−1) showing reduced bond integrity [Figure 1] [45,46]. Misfolded insulin compromises the insulin signaling cascade, pivotal for glucose homeostasis. Normally, insulin binds the insulin receptor, a tyrosine kinase with extracellular α-subunits and intracellular β-subunits, inducing autophosphorylation at tyrosine residues (Tyr1158, Tyr1162, Tyr1163) [47,48]. This recruits insulin receptor substrates (IRS-1/2), activating phosphoinositide 3-kinase (PI3K), which converts phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate, triggering protein kinase B (Akt) via phosphoinositide-dependent kinase-1 [49,50]. Akt promotes glucose transporter type 4 (GLUT4) translocation to the plasma membrane in skeletal muscle and adipose tissue, facilitating glucose uptake, and inhibits hepatic gluconeogenesis by suppressing phosphoenolpyruvate carboxykinase and glucose-6-phosphatase [51,52]. Molecular docking models show that disruption of the A6–A11 disulfide bond misaligns key receptor-binding residues (ValA3, TyrA19), reducing insulin receptor affinity by ~60%, impairing IRS phosphorylation, PI3K-Akt signaling, and GLUT4 translocation, while allowing unchecked hepatic glucose production, driving hyperglycemia [Figure 2] [53,54,55]. Cysteine deficiency also reduces glutathione synthesis, increasing reactive oxygen species (ROS) and activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulates pro-inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) [56,57,58,59,60].
These cytokines induce c-Jun N-terminal kinase (JNK)-mediated serine phosphorylation of IRS-1 (Ser307), further disrupting PI3K-Akt signaling, while impaired thioredoxin and peroxiredoxin function exacerbates oxidative stress [61,62,63,64,65]. Sulfur deficiency exacerbates metabolic dysregulation through ER stress and immunological cascades. Cysteine scarcity limits PDI activity, causing misfolded insulin to accumulate in the ER, triggering the unfolded protein response (UPR) via sensors inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [66,67]. Chronic ER stress activates pro-apoptotic pathways through IRE1/PERK-mediated c-Jun N-terminal kinase (JNK) and CCAAT/enhancer-binding protein homologous protein, leading to beta-cell apoptosis and reduced insulin secretion [66,67,68,69]. Misfolded insulin aggregates further contribute to glucotoxicity, a hallmark of T2DM [70,71]. Immunologically, reduced cysteine impairs glutathione synthesis, a critical antioxidant, increasing reactive oxygen species (ROS) and activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulates pro-inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) [56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74]. These cytokines induce JNK-mediated serine phosphorylation of IRS-1 (Ser307), disrupting PI3K-Akt signaling [61,62].
Sulfur deficiency also impairs redox-regulatory proteins thioredoxin and peroxiredoxin, reliant on disulfide bonds, perpetuating oxidative stress [63,64]. Additionally, reduced mucin synthesis weakens gut barrier integrity, enabling lipopolysaccharide translocation and toll-like receptor 4 (TLR4)-mediated endotoxemia, amplifying systemic inflammation [65,66,67,68,69,70,71,72,73,74,75,76,77]. By elucidating the gut-mitochondria-sulfur-insulin axis, this hypothesis challenges peripheral-focused T2DM models, positioning sulfur metabolism as a therapeutic target to restore insulin functionality and mitigate disease progression.
5. Targeting Sulfur Homeostasis: A Revolutionary Therapeutic Approach for Type 2 Diabetes
The Sulfur Insulin Deformation Hypothesis paves the way for innovative therapeutic strategies to combat insulin resistance by restoring sulfur homeostasis, addressing the molecular and immunological roots of type 2 diabetes mellitus. N-acetylcysteine (NAC), a cysteine precursor, enhances glutathione synthesis, a critical antioxidant tripeptide formed via glutamate-cysteine ligase and glutathione synthetase, neutralizing reactive oxygen species (ROS) induced by mitochondrial dysfunction [78,79,80]. By bolstering cysteine availability, NAC supports protein disulfide isomerase (PDI) activity in the endoplasmic reticulum, ensuring proper formation of insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19), stabilizing its functional conformation, and reducing endoplasmic reticulum stress from misfolded insulin accumulation [81]. At the molecular level, NAC inhibits c-Jun N-terminal kinase (JNK), a stress kinase activated by ROS and tumor necrosis factor-alpha (TNF-α), which phosphorylates insulin receptor substrate-1 (IRS-1) at serine residues, disrupting phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt) signaling [82,83]. By suppressing JNK, NAC restores IRS-1 tyrosine phosphorylation, enhancing PI3K-Akt signaling and glucose transporter type 4 (GLUT4) translocation, thus improving glucose uptake [84].
Immunologically, NAC reduces nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, downregulating pro-inflammatory cytokines (TNF-α, IL-6) and suppressor of cytokine signaling proteins, mitigating insulin resistance [85]. Additionally, NAC reinforces gut barrier integrity by stabilizing redox-dependent tight junction proteins (e.g., occludin, zonula occludens-1), reducing lipopolysaccharide (LPS)-induced endotoxemia via toll-like receptor 4 (TLR4) signaling [86,87].
Complementing NAC, methylsulfonylmethane (MSM), a bioavailable sulfur donor, supports cysteine synthesis by enhancing cystathionine β-synthase and γ-lyase activity in the transsulfuration pathway, counteracting mitochondrial ATP deficits [88]. Increased cysteine availability bolsters glutathione production and PDI function, stabilizing insulin structure and improving receptor-binding affinity. MSM also inhibits NF-κB activation, reducing cytokine-driven insulin resistance, and enhances gut barrier function, attenuating TLR4-mediated systemic inflammation [89].
Recommended dosages, under medical supervision, range from 600–1200 mg/day for NAC and 1000–3000 mg/day for MSM to optimize efficacy and safety [90]. Within the Sulfur Insulin Deformation Hypothesis, NAC and MSM target insulin misfolding, endoplasmic reticulum stress, oxidative damage, and systemic inflammation, offering a groundbreaking approach to restore metabolic homeostasis and redefine type 2 diabetes treatment by addressing its sulfur-dependent molecular origins [78,79,80,81,82,83,84,85,86,87,88,89,90].
6. Evidence for Sulfur-Driven Insulin Dysfunction: Linking Cysteine Deficiency to Disulfide Bond Disruption in Type 2 Diabetes
The Sulfur Insulin Deformation Hypothesis is bolstered by compelling evidence linking cysteine deficiency and impaired glutathione synthesis to insulin misfolding and type 2 diabetes mellitus (T2DM) pathogenesis, emphasizing the critical role of disulfide bonds in insulin’s structural and functional integrity. A 2011 study of 12 T2DM patients (HbA1c >7%) revealed a 73.8% reduction in red blood cell (RBC) glutathione (1.78 ± 0.28 vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001) and lower plasma cysteine/glycine levels compared to controls, driven by impaired de novo synthesis and heightened oxidative stress (elevated ROS and lipid peroxides). N-acetylcysteine (NAC) and glycine supplementation for 14 days restored glutathione, reducing oxidative stress and supporting the hypothesis that cysteine scarcity disrupts insulin’s disulfide bonds [91]. A 2014 study of 79 T2DM patients confirmed reduced cysteine and glutathione levels, with a strong correlation (r = 0.81, P = 0.001) and an inverse relationship with insulin resistance (HOMA-IR, r = -0.65, P < 0.05).
In vitro, cysteine supplementation in hyperglycemic U937 monocytes restored glutamate-cysteine ligase expression and glutathione, enhanced by vitamin D, suggesting cysteine’s role in counteracting sulfur-dependent insulin dysfunction [92]. In 2018, 16 T2DM patients (seven without, nine with microvascular complications) showed lower glutathione levels (0.35 ± 0.30 vs. 0.90 ± 0.42 mmol/L, P < 0.01) and synthesis rates (0.50 ± 0.69 vs. 1.03 ± 0.55 mmol/L/day, P < 0.05), particularly in complicated cases, driven by cysteine deficiency and elevated ROS, underscoring sulfur’s role in insulin structural integrity [93]. A 2022 randomized trial of 250 T2DM patients showed that six months of oral glutathione supplementation increased plasma glutathione, reduced 8-hydroxy-2′-deoxyguanosine (8-OHdG, P < 0.01), and improved HbA1c and insulin sensitivity, especially in patients over 55, indicating age-related glutathione deficits amplify sulfur-based therapeutic benefits [94]. A 2022 pilot study using GlyNAC (glycine + NAC) in T2DM patients over 14 days increased RBC glutathione (P < 0.01), improved insulin sensitivity by 31% (P < 0.05), and enhanced mitochondrial fatty acid oxidation, confirming cysteine’s role in restoring sulfur homeostasis and insulin functionality [95].
Contrarily, a 2016 study found a non-significant RBC glutathione reduction (0.87 vs. 0.92 µmol/L) but impaired glutathione peroxidase activity (P < 0.05) and elevated malondialdehyde, suggesting increased glutathione consumption under oxidative stress, which may disrupt insulin folding [96]. These studies collectively demonstrate that T2DM is marked by 30–73.8% reductions in cysteine and glutathione, driven by impaired synthesis and oxidative stress, fostering a redox environment that impairs insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19), critical for its structural stability and receptor binding [Figure 3].
Insulin’s three disulfide bonds dynamically regulate its folding, stability, and bioactivity. These bonds constrain conformational flexibility, protect against degradation, and enable receptor activation [97]. Engineering an additional disulfide bond enhanced insulin’s stability without compromising bioactivity, reinforcing its hydrophobic core [98]. The A6–A11 bond acts as a dynamic hinge, aligning residues (e.g., ValA3, TyrA19) for receptor docking; its disruption in synthetic analogs reduced binding affinity by 50–70%, supporting the hypothesis that sulfur deficiency-induced misfolding impairs insulin function [99,100,101]. Replacing A6–A11 with a methylene thioacetal or diselenide improved foldability and resistance to reductive cleavage, maintaining the A-chain’s α-helical structure [102,103,104]. Mutations disrupting A7–B7 reduced receptor affinity and PI3K-Akt signaling, critical for glucose uptake, aligning with the hypothesis that disulfide bond deformations drive metabolic dysfunction [105]. Restoring sulfur homeostasis with NAC or similar compounds could stabilize these bonds, offering a novel therapeutic avenue for T2DM [Figure 4].
7. Limitations
While the Sulfur-Insulin Deformation Hypothesis presents a mechanistically coherent and clinically plausible model for the structural origin of insulin resistance, we acknowledge the current absence of direct structural evidence of endogenous insulin misfolding in patients with type 2 diabetes mellitus (T2DM). This limitation is primarily due to the technical challenges associated with isolating and characterizing native human insulin particularly under pathophysiological conditions using high-resolution proteomic techniques such as LC-MS/MS, NMR spectroscopy, or Raman scattering.
To date, very few studies have successfully extracted and structurally analyzed circulating human insulin directly from diabetic patients, as most available data derive from recombinant or synthetic analogs. The isolation of low-abundance native insulin from plasma, its purification from structurally similar peptides (e.g., C-peptide), and its conformational profiling remain highly resource-intensive and largely inaccessible in low- and middle-income countries.
Thus, this hypothesis is proposed not as a definitive conclusion but as a strategic framework designed to guide further empirical investigation by research institutions equipped with advanced molecular infrastructure. Future validation should include direct conformational analysis of insulin in T2DM patients under varying redox states, along with targeted interventions aimed at restoring sulfur homeostasis.
8. Discussion
The Sulfur Insulin Deformation Hypothesis redefines type 2 diabetes mellitus (T2DM) by positing that insulin misfolding, driven by sulfur deficiency from mitochondrial dysfunction in intestinal epithelial cells, is a primary cause of insulin resistance, challenging conventional models focused on peripheral signaling defects. Unlike traditional paradigms attributing T2DM to obesity, lipotoxicity, or inflammation-induced c-Jun N-terminal kinase (JNK)-mediated serine phosphorylation of insulin receptor substrate-1 (IRS-1), this hypothesis centers on the structural integrity of insulin’s three disulfide bonds (A6–A11, A7–B7, A20–B19), critical for its receptor-binding affinity and bioactivity [35,36,37,38,39,40]. Mitochondrial dysfunction impairs the electron transport chain, reducing ATP production and inhibiting cystathionine β-synthase and γ-lyase in the transsulfuration pathway, limiting cysteine synthesis [42,43,44]. This cysteine scarcity disrupts protein disulfide isomerase (PDI) activity, leading to aberrant disulfide bond formation and insulin misfolding, as evidenced by simulated Raman spectroscopy showing reduced S-S stretching (510–540 cm−1) in sulfur-deficient states [Figure 1] [45,46]. Misfolded insulin, with altered tertiary structure, exhibits a 50–70% reduction in receptor-binding affinity, impairing tyrosine phosphorylation, IRS recruitment, and phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) signaling, thus reducing glucose transporter type 4 (GLUT4) translocation and promoting hepatic gluconeogenesis via phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, driving hyperglycemia [Figure 2] [47,48,49,50,51,52,53,54,55,101]. This hypothesis resolves the paradox of hyperinsulinemia coexisting with hyperglycemia in T2DM. While conventional models attribute hyperinsulinemia to compensatory beta-cell secretion, they fail to explain why endogenous insulin is ineffective, yet exogenous insulin resolves hyperglycemia. Misfolded endogenous insulin, lacking intact disulfide bonds, has diminished bioactivity, whereas exogenous insulin, with native conformation, activates receptors efficiently [101]. Cysteine deficiency also induces endoplasmic reticulum (ER) stress by accumulating misfolded insulin, triggering the unfolded protein response (UPR UPR) via inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6), leading to beta-cell apoptosis and glucotoxicity [66,67,68,69,70,71].
Immunologically, reduced cysteine limits glutathione synthesis, increasing reactive oxygen species (ROS) and activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which upregulates tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), exacerbating insulin resistance via JNK and suppressor of cytokine signaling (SOCS) proteins [56,57,58,59,60,61,62,73,74,75,76].
Compromised gut barrier integrity from reduced mucin synthesis amplifies toll-like receptor 4 (TLR4)-mediated endotoxemia, positioning the gut as a central driver of T2DM, [65,75,76] [Table 1] compares this hypothesis with traditional models, highlighting its focus on insulin structure and sulfur metabolism. Therapeutically, sulfur donors like N-acetylcysteine (NAC) and methylsulfonylmethane (MSM) increase plasma cysteine and glutathione by 20–40%, reducing oxidative stress and improving insulin sensitivity in T2DM patients [91,92,93,94,95]. Gut-targeted interventions, such as prebiotics or sulfur-rich diets (e.g., methionine/cysteine-rich foods), enhance cysteine synthesis and barrier function. Beyond T2DM, this hypothesis suggests a continuum of sulfur-dependent protein misfolding disorders, including Alzheimer’s, warranting unified therapeutic exploration. Future studies should employ LC-MS/MS to detect misfolded insulin, metabolomic profiling of sulfur metabolites, and clinical trials to validate NAC/MSM efficacy, potentially revolutionizing T2DM management by targeting the gut-mitochondria-sulfur-insulin axis [77].
9. Conclusion
The Sulfur Insulin Deformation Hypothesis reimagines type 2 diabetes mellitus (T2DM) as a sulfur metabolism disorder, where mitochondrial dysfunction in intestinal epithelial cells drives cysteine deficiency, destabilizing insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19) and inducing misfolding that impairs receptor-binding efficacy by 50–70%. This structural defect, evidenced by a 73.8% reduction in glutathione levels in T2DM patients, disrupts insulin’s bioactivity, fueling insulin resistance and hyperglycemia despite hyperinsulinemia.
Oxidative stress from glutathione depletion and endoplasmic reticulum dysfunction further compromises beta-cell function, perpetuating metabolic disarray. The gut-mitochondria-sulfur-insulin axis emerges as a central driver, challenging conventional peripheral-focused models. Therapeutic interventions like N-acetylcysteine (NAC) and methylsulfonylmethane (MSM), which boost cysteine and glutathione by 20–40%, restore insulin stability and sensitivity, offering a novel strategy to mitigate T2DM. Awaiting validation through LC-MS/MS analysis of insulin structure and clinical trials, this hypothesis heralds a paradigm shift, advocating sulfur-centric therapies to transform T2DM management and potentially extend to other protein misfolding disorders.
Funding
The authors received no financial support for the research and publication of this article.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Wei J, Fan L, He Z, Liu Y, Zhang X, Wang Q, Chen H, Li M, Zhang J, Yang C, Zhao W. The global, regional, and national burden of type 2 diabetes mellitus attributable to low physical activity from 1990 to 2021: a systematic analysis of the global burden of disease study 2021. Int J Behav Nutr Phys Act 2025;22:8. [CrossRef]
- Vinther TN, Norrman M, Ribel U, Huus K, Schlein M, Steensgaard DB, Pedersen TÅ, Pettersson I, Ludvigsen S, Kjeldsen T, Jensen KJ, Hubálek F. Insulin analog with additional disulfide bond has increased stability and preserved activity. Protein Sci 2013;22(3):296–305. [CrossRef]
- Comas F, Moreno-Navarrete JM. The impact of H2S on obesity-associated metabolic disturbances. Antioxidants 2021;10(5):633. [CrossRef]
- Sbodio JI, Snyder SH, Paul BD. Regulators of the transsulfuration pathway. Br J Pharmacol 2019;176(4):583–593. [CrossRef]
- Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 2011;34(1):17–32. [CrossRef]
- Murphy B, Bhattacharya R, Mukherjee P. Hydrogen sulfide signaling in mitochondria and disease. FASEB J 2019;33(12):13098–13125. [CrossRef]
- Jiang H, Thapa P, Hao Y, Ding N, Alshahrani A, Wei Q. Protein disulfide isomerases function as the missing link between diabetes and cancer. Antioxid Redox Signal 2022;37(16-18):1191–1205. [CrossRef]
- Isakoff SJ, Taha C, Rose E, Marcusohn J, Klip A, Skolnik EY. The inability of phosphatidylinositol 3-kinase activation to stimulate GLUT4 translocation indicates additional signaling pathways are required for insulin-stimulated glucose uptake. Proc Natl Acad Sci U S A 1995;92(22):10247–10251. [CrossRef]
- Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O’Callaghan N, Lionetti L, Luscombe-Marsh N. Mitochondrial (dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front Physiol 2019;10:532. [CrossRef]
- Masenga SK, Kabwe LS, Chakulya M, Kirabo A. Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci 2023;24(9):7898. [CrossRef]
- Yung JHM, Giacca A. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells 2020;9(3):706. [CrossRef]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci 2005;25(7):1788–1796. [CrossRef]
- Kumar AR, Nair B, Kamath AJ, Nath LR, Calina D, Sharifi-Rad J. Impact of gut microbiota on metabolic dysfunction-associated steatohepatitis and hepatocellular carcinoma: pathways, diagnostic opportunities and therapeutic advances. Eur J Med Res 2024;29(1):485. [CrossRef]
- Haque PS, Kapur N, Barrett TA, Theiss AL. Mitochondrial function and gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 2024;21(8):537–555. [CrossRef]
- Guerbette T, Boudry G, Lan A. Mitochondrial function in intestinal epithelium homeostasis and modulation in diet-induced obesity. Mol Metab 2022;63:101546. [CrossRef]
- Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab 2019;316(2):E268–E285. [CrossRef]
- Iheagwam FN, Joseph AJ, Adedoyin ED, Iheagwam OT, Ejoh SA. Mitochondrial dysfunction in diabetes: shedding light on a widespread oversight. Pathophysiology 2025;32(1):9. [CrossRef]
- Chen TH, Wang HC, Chang CJ, Lee SY. Mitochondrial glutathione in cellular redox homeostasis and disease manifestation. Int J Mol Sci 2024;25(2):1314. [CrossRef]
- Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling. Int J Mol Med 2019;44(1):3–15. [CrossRef]
- Aoyama K, Nakaki T. Glutathione in cellular redox homeostasis: association with the excitatory amino acid carrier 1 (EAAC1). Molecules 2015;20(5):8742–8758. [CrossRef]
- Fujii J, Osaki T, Soma Y, Matsuda Y. Critical roles of the cysteine-glutathione axis in the production of γ-glutamyl peptides in the nervous system. Int J Mol Sci 2023;24(9):8044. [CrossRef]
- Aryal B, Kwakye J, Ariyo OW, Ghareeb AFA, Milfort MC, Fuller AL, Khatiwada S, Rekaya R, Aggrey SE. Major oxidative and antioxidant mechanisms during heat stress-induced oxidative stress in chickens. Antioxidants 2025;14(4):471. [CrossRef]
- Blom HJ, Smulders Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J Inherit Metab Dis 2011;34(1):75–81. [CrossRef]
- Parra M, Stahl S, Hellmann H. Vitamin B6 and its role in cell metabolism and physiology. Cells 2018;7(7):84. [CrossRef]
- Badawy AA. Multiple roles of haem in cystathionine β-synthase activity: implications for hemin and other therapies of acute hepatic porphyria. Biosci Rep 2021;41(7):BSR20210935. [CrossRef]
- Xiao T, Chen S, Yan G, Zheng J, Qiu Q, Lin S, Zong Y, Chang H, Yu Chang AC, Wu Y, Hou C. Cystathionine γ-lyase inhibits mitochondrial oxidative stress by releasing H2S nearby through the AKT/NRF2 signaling pathway. Front Pharmacol 2024;15:1374720. [CrossRef]
- Carelli S, Ceriotti A, Cabibbo A, Fassina G, Ruvo M, Sitia R. Cysteine and glutathione secretion in response to protein disulfide bond formation in the ER. Science 1997;277(5332):1681–1684. [CrossRef]
- Yu X, Long Y. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep 2016;6:30033. [CrossRef]
- Lingappan K. NF-κB in oxidative stress. Curr Opin Toxicol 2018;7:81–86. [CrossRef]
- Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res 2020;13:1057–1073. [CrossRef]
- Sykiotis GP, Papavassiliou AG. Serine phosphorylation of insulin receptor substrate-1: a novel target for the reversal of insulin resistance. Mol Endocrinol 2001;15(11):1864–1869. [CrossRef]
- Ghosh S, Whitley CS, Haribabu B, Jala VR. Regulation of intestinal barrier function by microbial metabolites. Cell Mol Gastroenterol Hepatol 2021;11(5):1463–1482. [CrossRef]
- Page MJ, Kell DB, Pretorius E. The role of lipopolysaccharide-induced cell signalling in chronic inflammation. Chronic Stress 2022;6:24705470221076390. [CrossRef]
- Berbudi A, Khairani S, Tjahjadi AI. Interplay between insulin resistance and immune dysregulation in type 2 diabetes mellitus: implications for therapeutic interventions. ImmunoTargets Ther 2025;14:359–382. [CrossRef]
- Jia XY, Guo ZY, Wang Y, Xu Y, Duan SS, Feng YM. Peptide models of four possible insulin folding intermediates with two disulfides. Protein Sci 2003;12(11):2412–2419. [CrossRef]
- Vinther TN, Kjeldsen TB, Jensen KJ, Hubálek F. The road to the first, fully active and more stable human insulin variant with an additional disulfide bond. J Pept Sci 2015;21(10):797–806. [CrossRef]
- Weil-Ktorza O, Rege N, Lansky S, Shalev DE, Shoham G, Weiss MA, Metanis N. Substitution of an internal disulfide bridge with a diselenide enhances both foldability and stability of human insulin. Chem Eur J 2019;25(36):8513–8521. [CrossRef]
- Jackson, C. Kicking off the insulin cascade. Nature 444, 833–834 (2006). [CrossRef]
- Xu R, Jap E, Gubbins B, Hagemeyer CE, Karas JA. Semisynthesis of A6-A11 lactam insulin. J Pept Sci 2024;30(2):e3542. [CrossRef]
- Arai K, Okumura M, Lee YH, Takei M, Tsutsumi H, Miyakawa M, Koizumi M, Inaba K. Diselenide-bond replacement of the external disulfide bond of insulin increases its oligomerization leading to sustained activity. Commun Chem 2023;6:258. [CrossRef]
- Liu M, Wright J, Guo H, Xiong Y, Arvan P. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitam Horm 2014;95:35–62. [CrossRef]
- Rocha AG, Knight SAB, Pandey A, Yoon H, Pain J, Pain D, Dancis A. Cysteine desulfurase is regulated by phosphorylation of Nfs1 in yeast mitochondria. Mitochondrion 2018;40:29–41. [CrossRef]
- Roman JV, Mascarenhas R, Ceric K, Ballou DP, Banerjee R. Disease-causing cystathionine β-synthase linker mutations impair allosteric regulation. J Biol Chem 2023;299(12):105449. [CrossRef]
- Zuhra K, Augsburger F, Majtan T, Szabo C. Cystathionine-β-synthase: molecular regulation and pharmacological inhibition. Biomolecules 2020;10(5):697. [CrossRef]
- Rahman NSA, Zahari S, Syafruddin SE, Firdaus-Raih M, Low TY, Mohtar MA. Functions and mechanisms of protein disulfide isomerase family in cancer emergence. Cell Biosci 2022;12(1):129. [CrossRef]
- Yang M, Chiu J, Scartelli C, Ponzar N, Patel S, Patel A, Ferreira RB, Keyes RF, Carroll KS, Pozzi N, Hogg PJ, Smith BC, Flaumenhaft R. Sulfenylation links oxidative stress to protein disulfide isomerase oxidase activity and thrombus formation. J Thromb Haemost 2023;21(8):2137–2150. [CrossRef]
- De Meyts P, Sajid W, Palsgaard J, Theede AM, Gauguin L, Aladdin H, Whittaker J. Insulin and IGF-I receptor structure and binding mechanism. Madame Curie Biosci Database 2013;NBK6192.
- Wilden PA, Siddle K, Haring E, Backer JM, White MF, Kahn CR. The role of insulin receptor kinase domain autophosphorylation in receptor-mediated activities. Analysis with insulin and anti-receptor antibodies. J Biol Chem 1992;267(19):13719–13727.
- Martínez Báez A, Ayala G, Pedroza-Saavedra A, González-Sánchez HM, Chihu Amparan L. Phosphorylation codes in IRS-1 and IRS-2 are associated with the activation/inhibition of insulin canonical signaling pathways. Curr Issues Mol Biol 2024;46(1):634–649. [CrossRef]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009;8(8):627–644. [CrossRef]
- Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, Klip A. Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol 1999;19(6):4008–4018. [CrossRef]
- Ahn SW, Gang GT, Tadi S, Nedumaran B, Kim YD, Park JH, Kweon GR, Koo SH, Lee K, Ahn RS, Yim YH, Lee CH, Harris RA, Choi HS. Phosphoenolpyruvate carboxykinase and glucose-6-phosphatase are required for steroidogenesis in testicular Leydig cells. J Biol Chem 2012;287(50):41875–41887. [CrossRef]
- Zhao X, An X, Yang C, Sun W, Ji H, Lian F. The crucial role and mechanism of insulin resistance in metabolic disease. Front Endocrinol 2023;14:1149239. [CrossRef]
- Szablewski L. Changes in cells associated with insulin resistance. Int J Mol Sci 2024;25(4):2397. [CrossRef]
- Cao R, Tian H, Zhang Y, Liu G, Wang H, Dong W, Zhang L, Zhang Q, Zhao J. Signaling pathways and intervention for therapy of type 2 diabetes mellitus. MedComm 2023;4:e283. [CrossRef]
- Zhang H, Huang Y, Chen S, Tang C, Wang G, Du J, Jin H. Hydrogen sulfide regulates insulin secretion and insulin resistance in diabetes mellitus, a new promising target for diabetes mellitus treatment? A review. J Adv Res 2020;27:19–30. [CrossRef]
- Paul BD, Sbodio JI, Snyder SH. Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol Sci 2018;39(5):513–524. [CrossRef]
- Negm A, Mersal EA, Dawood AF, Abd El-Azim AO, Hasan O, Alaqidi R, Alotaibi A, Alshahrani M, Alheraiz A, Shawky TM. Multifaceted cardioprotective potential of reduced glutathione against doxorubicin-induced cardiotoxicity via modulating inflammation–oxidative stress axis. Int J Mol Sci 2025;26(7):3201. [CrossRef]
- Guo Q, Jin Y, Chen X, Ye G, Zhao L, Hou X, Liu Z, Bao T, Yang F, Liu Z, Zhang S, Fan X, Wang W. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther 2024;9:53. [CrossRef]
- Li R, Yan X, Zhao Y, Liu H, Wang J, Yuan Y, Li Q, Su J. Oxidative stress induced by nuclear factor erythroid 2-related factor 2 (NRF2) dysfunction aggravates chronic inflammation through the NAD+/SIRT3 axis and promotes renal injury in diabetes. Antioxidants 2025;14(3):267. [CrossRef]
- Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 2001;107(2):181–189. [CrossRef]
- Bloch-Damti A, Potashnik R, Gual P, Le Marchand-Brustel Y, Tanti JF, Rudich A, Bashan N. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia 2006;49(10):2463–2473. [CrossRef]
- Yadav PK, Vitvitsky V, Carballal S, Seravalli J, Banerjee R. Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J Biol Chem 2020;295(19):6299–6311. [CrossRef]
- Yang B, Lin Y, Huang Y, Shen YQ, Chen Q. Thioredoxin (Trx): a redox target and modulator of cellular senescence and aging-related diseases. Redox Biol 2024;70:103032. [CrossRef]
- Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiota, and metabolic inflammation. Endocr Rev 2015;36(3):245–271. [CrossRef]
- Hassan I, Gaines KS, Hottel WJ, Wishy RM, Miller SE, Powers LS, Rutkowski DT, Monick MM. Inositol-requiring enzyme 1 inhibits respiratory syncytial virus replication. J Biol Chem 2014;289(11):7537–7546. [CrossRef]
- Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 2020;21(8):421–438. [CrossRef]
- He Z, Liu Q, Wang Y, Zhao B, Zhang L, Yang X, Wang Z. The role of endoplasmic reticulum stress in type 2 diabetes mellitus mechanisms and impact on islet function. PeerJ 2025;13:e19192. [CrossRef]
- Watt NT, McGrane A, Roberts LD. Linking the unfolded protein response to bioactive lipid metabolism and signalling in the cell non-autonomous extracellular communication of ER stress. BioEssays 2023;45:e2300029. [CrossRef]
- Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med 2015;21(7):439–449. [CrossRef]
- Vilas-Boas EA, Almeida DC, Roma LP, Ortis F, Carpinelli AR. Lipotoxicity and β-cell failure in type 2 diabetes: oxidative stress linked to NADPH oxidase and ER stress. Cells 2021;10(12):3328. [CrossRef]
- Takeda H, Murakami S, Liu Z, Sawa T, Takahashi M, Izumi Y, Bamba T, Sato H, Akaike T, Sekine H, Motohashi H. Sulfur metabolic response in macrophage limits excessive inflammatory response by creating a negative feedback loop. Redox Biol 2023;65:102834. [CrossRef]
- Chiang FF, Chao TH, Huang SC, Cheng CH, Tseng YY, Huang YC. Cysteine regulates oxidative stress and glutathione-related antioxidative capacity before and after colorectal tumor resection. Int J Mol Sci 2022;23(17):9581. [CrossRef]
- Mowla SN, Perkins ND, Jat PS. Friend or foe: emerging role of nuclear factor kappa-light-chain-enhancer of activated B cells in cell senescence. OncoTargets Ther 2013;6:1221–1229. [CrossRef]
- Termite F, Archilei S, D’Ambrosio F, Petrucci L, Viceconti N, Iaccarino R, Liguori A, Gasbarrini A, Miele L. Gut microbiota at the crossroad of hepatic oxidative stress and MASLD. Antioxidants 2025;14(1):56. [CrossRef]
- Guijarro-Muñoz I, Compte M, Álvarez-Cienfuegos A, Álvarez-Vallina L, Sanz L. Lipopolysaccharide activates Toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway and proinflammatory response in human pericytes. J Biol Chem 2014;289(4):2457–2468. [CrossRef]
- Sen U, Givvimani S, Abe OA, Lederer ED, Tyagi SC. Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. Am J Physiol Cell Physiol 2011;300(1):C155–C163. [CrossRef]
- Zavala-Valencia AC, Velasco-Hidalgo L, Martínez-Avalos A, Castillejos-López M, Torres-Espíndola LM. Effect of N-acetylcysteine on cisplatin toxicity: a review of the literature. Biologics 2024;18:7–19. [CrossRef]
- Lu SC. Glutathione synthesis. Biochim Biophys Acta 2013;1830(5):3143–3153. [CrossRef]
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine—a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 2007;7(4):355–359. [CrossRef]
- Martinez-Banaclocha M. N-Acetyl-cysteine: modulating the cysteine redox proteome in neurodegenerative diseases. Antioxidants 2022;11(2):416. [CrossRef]
- Hashimoto S, Gon Y, Matsumoto K, Takeshita I, Horie T. N-acetylcysteine attenuates TNF-alpha-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br J Pharmacol 2001;132(1):270–276. [CrossRef]
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 2003;278(5):2896–2902. [CrossRef]
- Sampson SR, Cooper DR. Specific protein kinase C isoforms as transducers and modulators of insulin signaling. Mol Genet Metab 2006;89(1-2):32–47. [CrossRef]
- Abdelbagi O, Taha M, Al-Kushi AG, Alobaidy MA, Baokbah TAS, Sembawa HA, Azher ZA, Obaid R, Babateen O, Bokhari BT, Qusty NF, Malak HA. Ameliorative effect of N-acetylcysteine against 5-fluorouracil-induced cardiotoxicity via targeting TLR4/NF-κB and Nrf2/HO-1 pathways. Medicina 2025;61(2):335. [CrossRef]
- Wang H, Li C, Peng M, Wang L, Zhao D, Wu T, Yi D, Hou Y, Wu G. N-Acetylcysteine improves intestinal function and attenuates intestinal autophagy in piglets challenged with β-conglycinin. Sci Rep 2021;11(1):1261. [CrossRef]
- Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 2021;78(4):1233–1261. [CrossRef]
- Kawiak A, Kostecka A. Regulation of Bcl-2 family proteins in estrogen receptor-positive breast cancer and their implications in endocrine therapy. Cancers 2022;14(2):279. [CrossRef]
- Butawan M, Benjamin RL, Bloomer RJ. Methylsulfonylmethane: applications and safety of a novel dietary supplement. Nutrients 2017;9(3):290. [CrossRef]
- du Preez HN, Aldous C, Kruger HG, Johnson L. N-Acetylcysteine and other sulfur-donors as a preventative and adjunct therapy for COVID-19. Adv Pharmacol Pharm Sci 2022;2022:4555490. [CrossRef]
- Sekhar RV, McKay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, Jahoor F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011;34(1):162–167. [CrossRef]
- Jain S, Micinski D, Huning L, Quinn J, Dupre J, Storey KB. Vitamin D and L-cysteine levels correlate positively with GSH and negatively with insulin resistance levels in the blood of type 2 diabetic patients. Eur J Clin Nutr 2014;68(10):1148–1153. [CrossRef]
- Lutchmansingh FK, Hsu JW, Bennett FI, Badaloo AV, McFarlane-Anderson N, Gordon-Strachan GM, Wright-Pascoe RA, Jahoor F, Boyne MS. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS One 2018;13(6):e0198626. [CrossRef]
- Kalamkar S, Acharya J, Kolappurath Madathil A, Gajjar V, Divate U, Karandikar-Iyer S, Ghaskadbi S, Goel P. Randomized clinical trial of how long-term glutathione supplementation offers protection from oxidative damage and improves HbA1c in elderly type 2 diabetic patients. Antioxidants 2022;11(5):1026. [CrossRef]
- Tuell D, Ford G, Los E, Stone W. The role of glutathione and its precursors in type 2 diabetes. Antioxidants 2024;13(2):184. [CrossRef]
- Gawlik K, Naskalski JW, Fedak D, Pawlica-Gosiewska D, Grudzień U, Dumnicka P, Małecki MT, Solnica B. Markers of antioxidant defense in patients with type 2 diabetes. Oxid Med Cell Longev 2016;2016:2352361. [CrossRef]
- van Lierop B, Ong SC, Belgi A, Delaine C, Andrikopoulos S, Haworth NL, Menting JG, Lawrence MC, Forbes BE, Wade JD. Insulin in motion: the A6-A11 disulfide bond allosterically modulates structural transitions required for insulin activity. Sci Rep 2017;7:17239. [CrossRef]
- Vinther TN, Pettersson I, Huus K, Schlein M, Steensgaard DB, Sørensen A, Jensen KJ, Kjeldsen T, Hubálek F. Additional disulfide bonds in insulin: prediction, recombinant expression, receptor binding affinity, and stability. Protein Sci 2015;24(5):779–788. [CrossRef]
- Jarosinski MA, Dhayalan B, Chen YS, Chatterjee D, Varas N, Weiss MA. Structural principles of insulin formulation and analog design: a century of innovation. Mol Metab 2021;52:101325. [CrossRef]
- Chang SG, Choi KD, Jang SH, Shin HC. Role of disulfide bonds in the structure and activity of human insulin. Mol Cells 2003;16(3):323–330. [CrossRef]
- Ong SC, Belgi A, van Lierop B, Delaine C, Andrikopoulos S, MacRaild CA, Norton RS, Haworth NL, Robinson AJ, Forbes BE. Probing the correlation between insulin activity and structural stability through introduction of the rigid A6–A11 bond. J Biol Chem 2018;293(30):11928–11943. [CrossRef]
- Hubálek F, Cramer CN, Helleberg H, Johansson E, Nishimura E, Schluckebier G, Steensgaard DB, Sturis J, Kjeldsen TB. Enhanced disulphide bond stability contributes to the once-weekly profile of insulin icodec. Nat Commun 2024;15(1):6124. [CrossRef]
- Zheng N, Karra P, VandenBerg MA, Kim JH, Webber MJ, Holland WL, Chou DH. Synthesis and characterization of an A6-A11 methylene thioacetal human insulin analogue with enhanced stability. J Med Chem 2019;62(24):11437–11443. [CrossRef]
- Weil-Ktorza O, Rege N, Lansky S, Shalev DE, Shoham G, Weiss MA, Metanis N. Substitution of an internal disulfide bridge with a diselenide enhances both foldability and stability of human insulin. Chem Eur J 2019;25(36):8513–8521. [CrossRef]
- Yoshinaga T, Nakatome K, Nozaki J, Naitoh M, Hoseki J, Kubota H, Nagata K, Koizumi A. Proinsulin lacking the A7-B7 disulfide bond, Ins2Akita, tends to aggregate due to the exposed hydrophobic surface. Biol Chem 2005;386(11):1077–1085. [CrossRef]
Figure 1.
Simulated Comparative Analysis of Insulin Structure in Healthy and Diabetic Conditions Based on the Sulfur Deficiency Hypothesis. This figure presents a simulated analysis comparing the structural properties of insulin in healthy (blue) and diabetic (red) conditions, focusing on the impact of sulfur deficiency in type 2 diabetes mellitus (T2DM) as proposed by the Sulfur Insulin Deformation Hypothesis. Panel (A) displays LC-MS/MS spectra in the range of 560–640 m/z, where diabetic insulin exhibits greater fragmentation (smaller, more dispersed peaks at 560, 580, 600, and 620 m/z) compared to healthy insulin, indicating structural deformation due to sulfur deficiency. Panel (B) shows Raman spectra in the 510–540 cm−1 range (S-S stretching region), revealing a significant reduction in peak intensity at 525 cm−1 for diabetic insulin, consistent with the loss of disulfide bonds caused by sulfur deficiency. Panel (C) illustrates Raman spectra in the 800–1800 cm−1 range, highlighting shifts in the amide I (from 1650 cm−1 to 1640 cm−1) and amide II (from 1550 cm−1 to 1540 cm−1) bands in diabetic insulin, along with reduced intensity, indicative of misfolding due to sulfur deficiency. Panel (D) provides a molecular representation comparing native insulin (healthy) with intact disulfide bonds to misfolded insulin (diabetic, sulfur-deficient), where disulfide bonds at A6-A11, A7-B7, and A20-B19 are disrupted, impacting the function of beta cells in T2DM. These results are based on computational simulations using tools like PyMOL and await experimental validation.
Figure 1.
Simulated Comparative Analysis of Insulin Structure in Healthy and Diabetic Conditions Based on the Sulfur Deficiency Hypothesis. This figure presents a simulated analysis comparing the structural properties of insulin in healthy (blue) and diabetic (red) conditions, focusing on the impact of sulfur deficiency in type 2 diabetes mellitus (T2DM) as proposed by the Sulfur Insulin Deformation Hypothesis. Panel (A) displays LC-MS/MS spectra in the range of 560–640 m/z, where diabetic insulin exhibits greater fragmentation (smaller, more dispersed peaks at 560, 580, 600, and 620 m/z) compared to healthy insulin, indicating structural deformation due to sulfur deficiency. Panel (B) shows Raman spectra in the 510–540 cm−1 range (S-S stretching region), revealing a significant reduction in peak intensity at 525 cm−1 for diabetic insulin, consistent with the loss of disulfide bonds caused by sulfur deficiency. Panel (C) illustrates Raman spectra in the 800–1800 cm−1 range, highlighting shifts in the amide I (from 1650 cm−1 to 1640 cm−1) and amide II (from 1550 cm−1 to 1540 cm−1) bands in diabetic insulin, along with reduced intensity, indicative of misfolding due to sulfur deficiency. Panel (D) provides a molecular representation comparing native insulin (healthy) with intact disulfide bonds to misfolded insulin (diabetic, sulfur-deficient), where disulfide bonds at A6-A11, A7-B7, and A20-B19 are disrupted, impacting the function of beta cells in T2DM. These results are based on computational simulations using tools like PyMOL and await experimental validation.
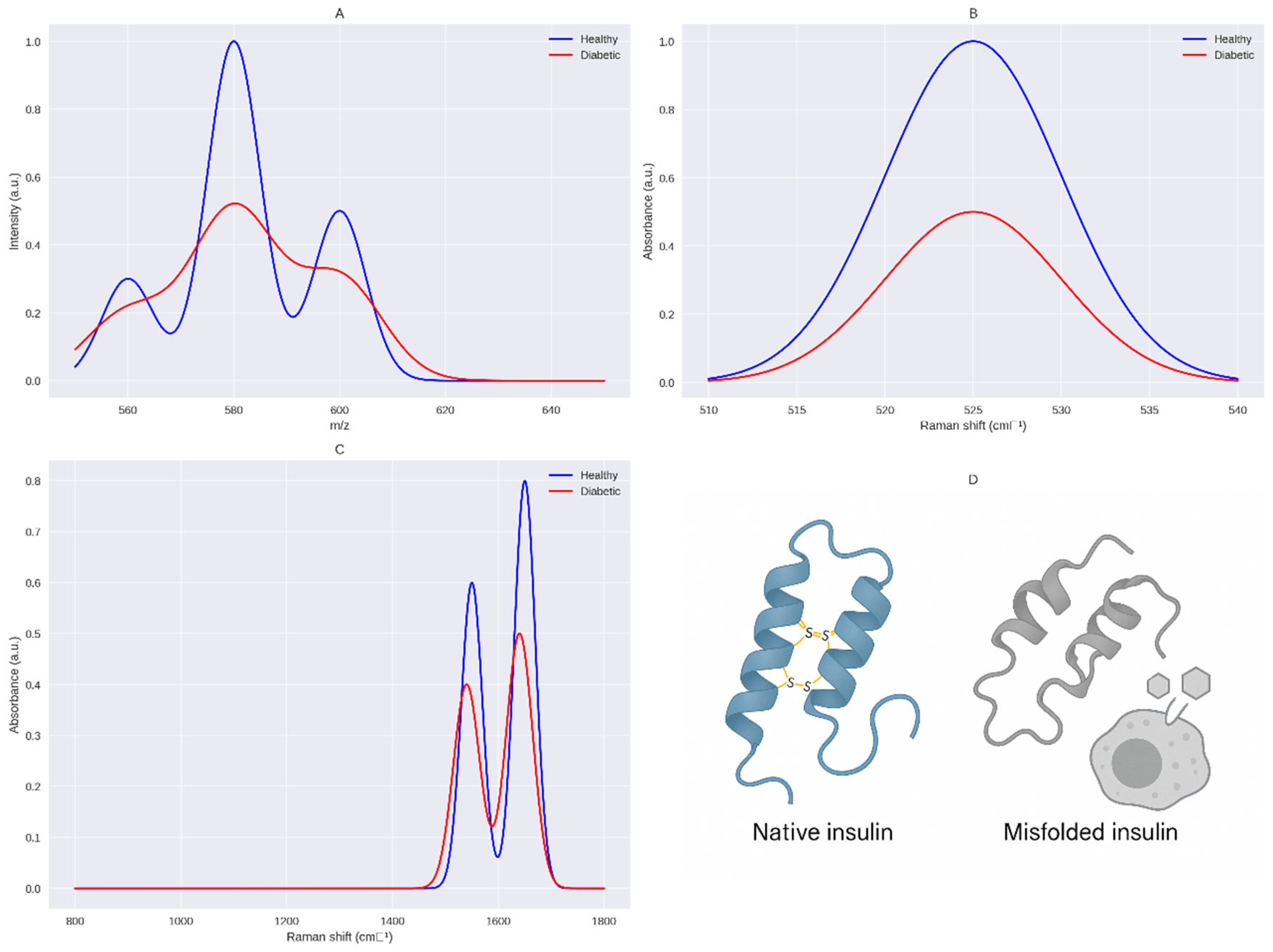
Figure 2.
Molecular Docking Model of Native vs. Deformed Insulin with Insulin Receptor. This figure illustrates the structural and functional impact of disulfide bond integrity on insulin-receptor interactions, supporting the Sulfur Insulin Deformation Hypothesis. Panel A (Native Insulin) depicts the native insulin structure with the A-chain (green) and B-chain (blue) stabilized by disulfide bonds (A6–A11, A7–B7, A20–B19, yellow), enabling optimal docking with the insulin receptor (IR, grey) at 100% affinity. Panel B (Deformed Insulin) highlights the consequences of sulfur deficiency, showing the absence of the A6–A11 disulfide bond (indicated as unlinked CysA6, CysA11 in yellow), leading to A-chain misfolding (green). This results in misaligned receptor-binding residues ValA3 and TyrA19 (red), impairing interaction with IR and reducing affinity by 60% [101]. The legend clarifies the color scheme: green (A-chain), blue (B-chain), yellow (unlinked CysA6, CysA11), red (ValA3, TyrA19), grey (Insulin Receptor, IR). The caption below reads: “Deformation-induced misalignment of ValA3 and TyrA19 impairs insulin receptor binding, supporting the Sulfur Insulin Deformation,” reinforcing the hypothesis that sulfur deficiency disrupts insulin folding and receptor binding, contributing to insulin resistance in type 2 diabetes mellitus.
Figure 2.
Molecular Docking Model of Native vs. Deformed Insulin with Insulin Receptor. This figure illustrates the structural and functional impact of disulfide bond integrity on insulin-receptor interactions, supporting the Sulfur Insulin Deformation Hypothesis. Panel A (Native Insulin) depicts the native insulin structure with the A-chain (green) and B-chain (blue) stabilized by disulfide bonds (A6–A11, A7–B7, A20–B19, yellow), enabling optimal docking with the insulin receptor (IR, grey) at 100% affinity. Panel B (Deformed Insulin) highlights the consequences of sulfur deficiency, showing the absence of the A6–A11 disulfide bond (indicated as unlinked CysA6, CysA11 in yellow), leading to A-chain misfolding (green). This results in misaligned receptor-binding residues ValA3 and TyrA19 (red), impairing interaction with IR and reducing affinity by 60% [101]. The legend clarifies the color scheme: green (A-chain), blue (B-chain), yellow (unlinked CysA6, CysA11), red (ValA3, TyrA19), grey (Insulin Receptor, IR). The caption below reads: “Deformation-induced misalignment of ValA3 and TyrA19 impairs insulin receptor binding, supporting the Sulfur Insulin Deformation,” reinforcing the hypothesis that sulfur deficiency disrupts insulin folding and receptor binding, contributing to insulin resistance in type 2 diabetes mellitus.
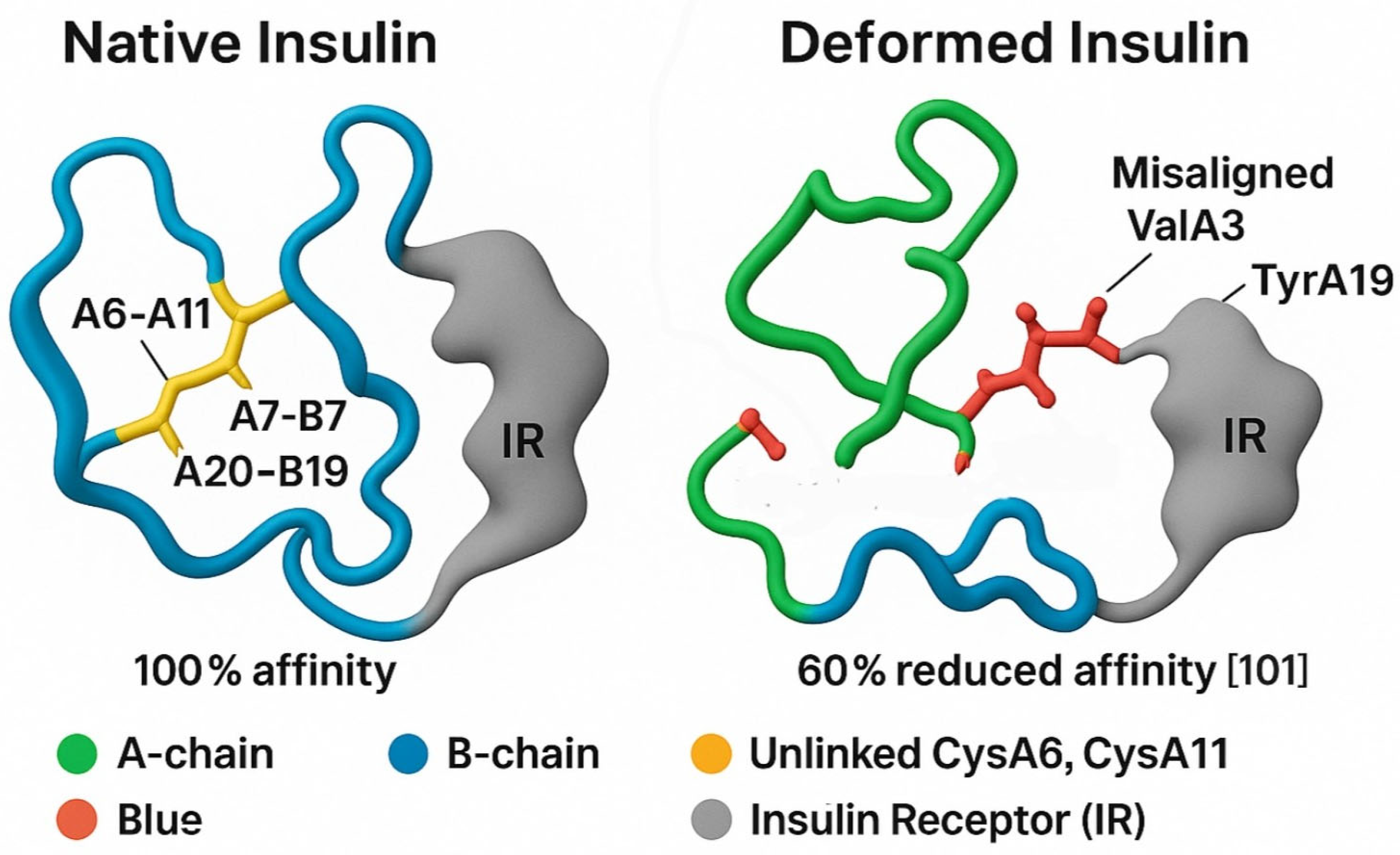
Figure 3.
Integrated Biomarkers Supporting the Sulfur Insulin Deformation Hypothesis. This figure presents a multi-panel visualization of key biomarkers underpinning the Sulfur Insulin Deformation Hypothesis in type 2 diabetes mellitus (T2DM). Panel A displays red blood cell (RBC) glutathione levels, highlighting a significant 73.8% reduction in T2DM patients (1.78 ± 0.28 µmol/g Hb) compared to healthy controls (6.75 ± 0.47 µmol/g Hb, P < 0.001), [91] with additional data from [93]. (0.35 ± 0.30 vs. 0.90 ± 0.42 mmol/L, P < 0.01) and [96] (0.87 vs. 0.92 µmol/L, non-significant). Panel B illustrates a strong positive correlation between plasma cysteine and RBC glutathione levels (r = 0.81, P = 0.001) in 79 T2DM and 22 control subjects, [92] alongside an inverse correlation with insulin resistance (HOMA-IR, r = -0.65, P < 0.05). Panel C depicts elevated oxidative stress markers in T2DM, including reactive oxygen species (ROS), malondialdehyde (MDA), and 8-hydroxy-2′-deoxyguanosine (8-OHdG), with significant increases (P < 0.01) [94,96]. Panel D demonstrates the effects of interventions, showing increased RBC glutathione (e.g., +31% with NAC, P < 0.01 [95]) and enhanced insulin sensitivity (e.g., +31% with GlyNAC, P < 0.05 [95]) following 14-day or 6-month treatments. Data are presented as mean ± SD, with statistical significance denoted (P < 0.05, P < 0.01). This figure synthesizes evidence of sulfur deficiency’s role in insulin dysfunction, supporting the hypothesis of disulfide bond disruption.
Figure 3.
Integrated Biomarkers Supporting the Sulfur Insulin Deformation Hypothesis. This figure presents a multi-panel visualization of key biomarkers underpinning the Sulfur Insulin Deformation Hypothesis in type 2 diabetes mellitus (T2DM). Panel A displays red blood cell (RBC) glutathione levels, highlighting a significant 73.8% reduction in T2DM patients (1.78 ± 0.28 µmol/g Hb) compared to healthy controls (6.75 ± 0.47 µmol/g Hb, P < 0.001), [91] with additional data from [93]. (0.35 ± 0.30 vs. 0.90 ± 0.42 mmol/L, P < 0.01) and [96] (0.87 vs. 0.92 µmol/L, non-significant). Panel B illustrates a strong positive correlation between plasma cysteine and RBC glutathione levels (r = 0.81, P = 0.001) in 79 T2DM and 22 control subjects, [92] alongside an inverse correlation with insulin resistance (HOMA-IR, r = -0.65, P < 0.05). Panel C depicts elevated oxidative stress markers in T2DM, including reactive oxygen species (ROS), malondialdehyde (MDA), and 8-hydroxy-2′-deoxyguanosine (8-OHdG), with significant increases (P < 0.01) [94,96]. Panel D demonstrates the effects of interventions, showing increased RBC glutathione (e.g., +31% with NAC, P < 0.01 [95]) and enhanced insulin sensitivity (e.g., +31% with GlyNAC, P < 0.05 [95]) following 14-day or 6-month treatments. Data are presented as mean ± SD, with statistical significance denoted (P < 0.05, P < 0.01). This figure synthesizes evidence of sulfur deficiency’s role in insulin dysfunction, supporting the hypothesis of disulfide bond disruption.
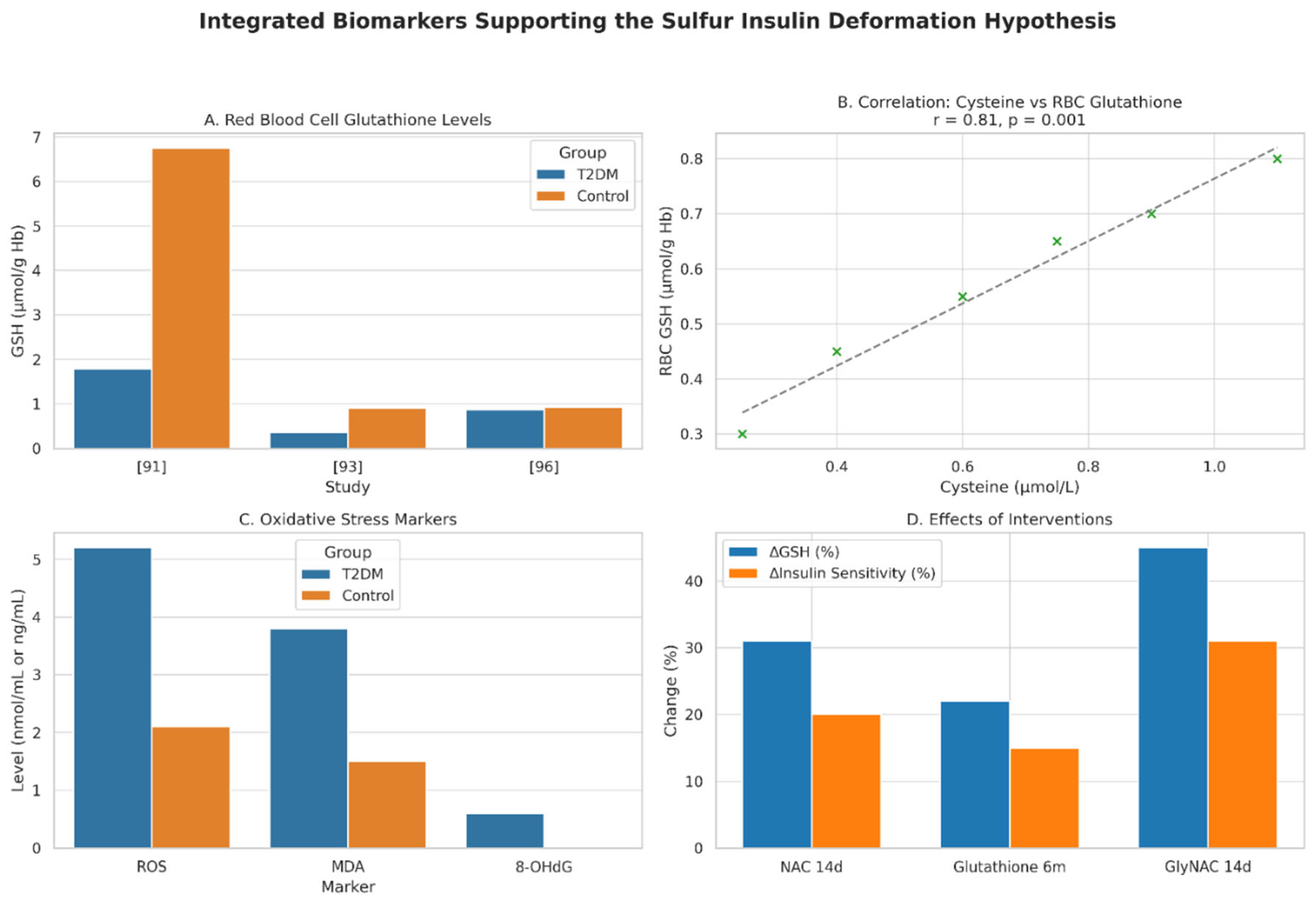
Figure 4.
Impact of Disulfide Bond Disruptions and Modifications on Insulin Functionality. This multi-panel figure elucidates the critical role of disulfide bonds in insulin’s structural stability and bioactivity, supporting the Sulfur Insulin Deformation Hypothesis in the context of type 2 diabetes mellitus (T2DM). Panel A presents a bar graph quantifying the effect of A6–A11 disulfide bond disruption on insulin receptor binding affinity, demonstrating a 60% reduction in affinity (40% remaining) in a synthetic insulin analog compared to wild-type insulin (100%) .[101] This significant impairment underscores the pivotal role of the A6–A11 bond as a dynamic hinge facilitating receptor engagement. Panel B employs a grouped bar graph to compare the qualitative effects of disulfide bond modifications and disruptions on insulin’s properties, using an arbitrary scale (0 = Baseline [Wild-type], 1 = Reduced, 2 = Enhanced/Improved). The addition of an extra disulfide bond enhances stability (2), [98] replacement of A6–A11 with a methylene thioacetal improves resistance to degradation (2), [103] and substitution with a diselenide bond enhances foldability during biosynthesis (2) [104]. Conversely, disruption of the A7–B7 bond reduces PI3K-Akt signaling (1), critical for glucose uptake [105]. Together, these findings highlight the multifaceted impact of disulfide bond integrity on insulin’s folding, stability, and signaling, reinforcing the hypothesis that sulfur deficiency-induced deformations may underlie functional insulin resistance in T2DM. The figure employs a blue-orange color scheme (blue for Wild-type/Baseline, orange for Modified/Disrupted) to ensure visual clarity, with data presented relative to wild-type insulin as the baseline.
Figure 4.
Impact of Disulfide Bond Disruptions and Modifications on Insulin Functionality. This multi-panel figure elucidates the critical role of disulfide bonds in insulin’s structural stability and bioactivity, supporting the Sulfur Insulin Deformation Hypothesis in the context of type 2 diabetes mellitus (T2DM). Panel A presents a bar graph quantifying the effect of A6–A11 disulfide bond disruption on insulin receptor binding affinity, demonstrating a 60% reduction in affinity (40% remaining) in a synthetic insulin analog compared to wild-type insulin (100%) .[101] This significant impairment underscores the pivotal role of the A6–A11 bond as a dynamic hinge facilitating receptor engagement. Panel B employs a grouped bar graph to compare the qualitative effects of disulfide bond modifications and disruptions on insulin’s properties, using an arbitrary scale (0 = Baseline [Wild-type], 1 = Reduced, 2 = Enhanced/Improved). The addition of an extra disulfide bond enhances stability (2), [98] replacement of A6–A11 with a methylene thioacetal improves resistance to degradation (2), [103] and substitution with a diselenide bond enhances foldability during biosynthesis (2) [104]. Conversely, disruption of the A7–B7 bond reduces PI3K-Akt signaling (1), critical for glucose uptake [105]. Together, these findings highlight the multifaceted impact of disulfide bond integrity on insulin’s folding, stability, and signaling, reinforcing the hypothesis that sulfur deficiency-induced deformations may underlie functional insulin resistance in T2DM. The figure employs a blue-orange color scheme (blue for Wild-type/Baseline, orange for Modified/Disrupted) to ensure visual clarity, with data presented relative to wild-type insulin as the baseline.

Table 1.
This comparative framework highlights the novel perspective of the Sulfur-Dependent Misfolding Hypothesis in redefining T2DM as a sulfur metabolism disorder, contrasting it with traditional paradigms.
Table 1.
This comparative framework highlights the novel perspective of the Sulfur-Dependent Misfolding Hypothesis in redefining T2DM as a sulfur metabolism disorder, contrasting it with traditional paradigms.
| Comparative Dimension | Traditional Paradigm of T2DM | Sulfur-Dependent Misfolding Hypothesis |
| Root Cause | Peripheral insulin resistance driven by obesity, lipotoxicity, and inflammation. | Structural misfolding of insulin due to disulfide bond disruption caused by organic sulfur deficiency. |
| Initiation Site | Skeletal muscle, liver, and adipose tissue. | Mitochondrial dysfunction in intestinal epithelial cells impairing sulfur metabolism. |
| Pathophysiological Focus | Post-receptor signaling defects (IRS, PI3K, Akt). | Primary insulin deformation with reduced receptor affinity due to disrupted disulfide bonds. |
| Explanation of Hyperinsulinemia + Hyperglycemia Paradox | Compensatory hypersecretion due to peripheral resistance. | Endogenous insulin is misfolded and non-functional; exogenous insulin remains effective due to intact structure. |
| Immunological Mechanism | Chronic inflammation from adipose tissue and macrophage activation. | Glutathione depletion induces NF-κB and JNK pathways via oxidative stress and endotoxemia. |
| Role of the Gut | Secondary influence via microbiome and inflammation. | Primary site of dysfunction initiating mitochondrial suffocation, impaired sulfur metabolism, and mucosal barrier breakdown. |
| Insulin Signaling Defect | Impaired receptor signaling due to inflammation and phosphorylation of IRS. | Insulin fails to initiate signaling due to misfolded structure with up to 70% loss in receptor affinity. |
| Therapeutic Strategy | Blood glucose control via metformin, GLP-1 agonists, or exogenous insulin. | Sulfur restoration through NAC, MSM, and dietary methionine/cysteine to stabilize insulin structure. |
| Experimental Accessibility | HOMA-IR index and indirect measures of resistance. | Direct structural assessment of insulin via LC-MS/MS and Raman spectroscopy. |
| Biochemical Depth | Focuses downstream of the insulin receptor. | Traces the issue upstream to insulin biosynthesis and protein folding integrity. |
| Innovation Potential | Incremental improvements to a saturated model. | A paradigm shift introducing sulfur metabolism as a central therapeutic and diagnostic axis. |
| Philosophical Reframing | The body becomes resistant to insulin. | The body produces dysfunctional insulin; the issue lies at the source. |
| Potentially paradigm-shifting | Unlikely due to conceptual saturation. | Potentially transformative discovery redefining T2DM pathogenesis and therapy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



