
Submitted:
25 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
Lysine succinylation, and its reversal by sirtuin-5 (SIRT5), is known to modulate mitochondrial fatty acid β-oxidation (FAO). We recently showed that feeding mice dodecanedioic acid, a 12-carbon dicarboxylic acid (DC12) that can be chain-shortened four rounds to succinyl-CoA, drives high-level protein hypersuccinylation in the peroxisome, particularly on peroxisomal FAO enzymes. But the ability of SIRT5 to reverse DC12-induced peroxisomal succinylation, or to regulate peroxisomal FAO in this context, remained unexplored. Here, we showed that feeding DC12 strongly recruits SIRT5 into hepatic peroxisomes. Knocking out SIRT5 impaired peroxisomal FAO as evidenced by reduced 14C-DC12 flux in liver homogenates and elevated levels of partially shortened DC12 catabolites in urine. Further, mass spectrometry revealed a trend toward less peroxisomal protein succinylation in SIRT5 knockout liver. This is consistent with reduced flux of DC12 through the peroxisomal FAO pathway, thereby reducing production of the succinyl-CoA that chemically reacts with lysine residues to produce protein succinylation. Mass spectrometry comparisons of site-level succinylation in wildtype and SIRT5 knockout liver did not reveal any clear pattern of SIRT5 target sites in the peroxisome after DC12 feeding. However, SIRT5 co-immunoprecipitated with 15 peroxisomal proteins including the key peroxisomal FAO enzymes acyl-CoA oxidase-1 and enoyl-CoA/3-hydroxyacyl-CoA dehydrogenase (EHHADH). In vitro, recombinant SIRT5 partially desuccinylated chemically modified recombinant ACOX1a, ACOX1b, and EHHADH. Desuccinylation by SIRT5 had no effect on enzyme activity for ACOX1a and EHHADH. For ACOX1b, SIRT5-mediated desuccinylation decreased activity by ~15%. Possible interpretations of this data are discussed.
Keywords:
peroxisome
; sirtuin-5
; fatty acid oxidation
1. Introduction
Lysine succinylation is a post-translational modification arising from a non-enzymatic reaction between succinyl-CoA and solvent-accessible lysine residues on the surface of proteins [1]. Succinylation has been well-characterized in the mitochondria where it has detrimental effects on protein function if left unchecked [2, 3]. Reversing this PTM is handled by sirtuin-5 (SIRT5), an NAD+ dependent lysine deacylase [2]. SIRT5 has an isoform that localizes to the mitochondria, where it desuccinylates, deglutarylates, and demalonylates numerous substrates [3], and an isoform that localizes to the cytoplasm where it demalonylates additional protein substrates [4]. Interestingly, SIRT5 was recently shown to localize to the peroxisome in the hepatic cell lines HepG2 and Huh7 [5]. In the peroxisome, SIRT5 desuccinylated acyl-CoA oxidase-1 (ACOX1), the rate-limiting enzyme of peroxisomal fatty acid β-oxidation (FAO). SIRT5 desuccinylation of ACOX1 destablized the enzyme to reduce its activity. This suggests that SIRT5 may suppress peroxisomal FAO, being opposite of its mitochondrial role in promoting FAO.
The observation that SIRT5 desuccinylates ACOX1 raises the question of how lysine succinylation arises within the peroxisomal compartment. Chen et al [5] postulated that impaired function of mitochondrial succinate dehydrogenase could cause an accumulation of succinate, with subsequent transfer to peroxisomes. In this model, succinate entering peroxisomes would require esterification to coenzyme-A prior to succinylating proteins, as free succinate does not react with lysine residues. The mechanism of peroxisomal succinate esterification was not explored. In contrast, our recent study on dodecanedioic acid suggested a more direct route of succinyl-CoA formation inside the peroxisome [6]. Dodecanedioic acid is a 12-carbon dicarboxylic acid (hereafter, DC12) which is a naturally occurring fatty acid in liver and kidney, formed through a poorly understood ω-oxidation pathway. Synthesis of DC12 by the ω-oxidation pathway begins in the endoplasmic reticulum with the hydroxylation of lauric acid (C12) on the terminal (ω) carbon. Two more steps, catalyzed by an alcohol dehydrogenase and an aldehyde dehydrogenase, complete the conversion of the ω carbon into a carboxyl group, thereby rendering the fatty acid “dicarboxylic”. DC12 is preferentially degraded in the peroxisome rather than the mitochondria [7]. It is activated to DC12-CoA inside the peroxisome and then chain-shortened through four rounds of peroxisomal FAO to succinyl-CoA [8]. We fed mice a diet enriched in DC12 and observed massive increases in peroxisomal protein succinylation, including sites on ACOX1 [6]. We did not, however, interrogate the function of peroxisomal SIRT5 in the context of DC12 feeding. The work of Chen et al suggests that succinylation of ACOX1 during DC12 feeding may enhance peroxisomal FAO via increased ACOX1 activity, while the presence of peroxisomal SIRT5 could hinder it. Here, we set out to investigate the role of SIRT5 during DC12-induced peroxisomal protein hypersuccinylation.
2. Materials and Methods
2.1. Animals and DC12 Administration
All animal protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC), and all experiments were conducted in accordance with the guidelines and regulations set forth in the Animal Welfare Act (AWA) and PHS Policy on Humane Care and Use of Laboratory Animals. SIRT5-/- and wildtype control mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred in-house. The mice were maintained on a 12 hr light/dark cycle in a pathogen-free barrier facility. In initial experiments, the DC12 diet was made by mixing DC12 free acid (Sigma-Aldrich Co., St. Louis, MO) by weight (10% w/w) into powdered standard rodent diet. In experiments that monitored the physiological response to DC12, including EchoMRI and urine collection, the diet was manufactured as pellets by Research Diets, Inc. This diet contained 51% of calories as carbohydrate, 21% as DC12, 12% as soybean oil, and 16% as protein. The isocaloric control diet contained 51% of calories as carbohydrate, 33% as soybean oil, and 16% as protein. For physiological experiments mice were maintained on these diets for five weeks. Afterwards, body composition was determined using the EchoMRI-100 system (Echo Medical Systems, Houston, TX), and overnight urine samples were collected using metabolic caging.
2.2. Density Gradient Centrifugation
Organelles were separated by density gradient centrifugation. Fresh liver tissue (0.3-0.5 g) was washed with cold PBS to remove blood and then minced in Petri dishes on ice. The minced tissue was transferred with 5 mL of cold PBS into ice-cold glass tubes (15x85 mm) and homogenized with 10 slow up and down strokes in 5 mL of ice-cold Extraction Buffer (5 mM MOPS, pH 7.65, with 250 mM sucrose, 1 mM EDTA, 0.5% ethanol and 0.2 mM PMSF). The homogenate was centrifuged at 1000 × g for 10 minutes to remove nuclei and other cell debris. The supernatant was transferred to a 15-ml centrifuge tube and centrifuged at 2000 × g for 10 minutes; the supernatant from this spin was then centrifuged at 25,000 × g for 20 minutes. The pellet was resuspended in Extraction Buffer (0.4 mL per gram of original tissue), diluted with OptiPrep Density Gradient Medium (60% iodixanol, Sigma, D1556) and OptiPrep Dilution Buffer (5 mM MOPS, pH 8.0, with 1 mM EDTA and 0.1% ethanol) to make a diluted sample containing 22.5% OptiPrep. In ultacentrifuge tubes, ¼ volume of 27.5% OptiPrep solution was overlaid with ½ volume of the diluted sample, followed by a ¼ volume of 20% OptiPrep Solution. These layered samples were centrifuged for 3 hours at 50,000 × g or 15 hours at 10,000 × g. A cloudy, floating layer on the top of the gradient was primarily endoplasmic reticulum (ER) and lysosomes, a ring at 22.5%/27.5% interface was mitochondria, and a slightly floating pellet at the bottom of the tube was pure peroxisome fraction.
2.3. SIRT5 Co-Immunoprecipitation
Liver lysate in RIPA buffer with protease inhibitors was centrifuged at 16,100 × g for 15 minutes. The supernatant was transferred to a fresh tube and 20 µL Protein A agarose (Santa Cruz Biotechnologies, sc-2001) slurry was added to pre-clear lysate for 1 hour on a rotating mixer, followed by centrifugation at 1000 × g for 1 minute to pellet the agarose beads. The supernatant was transferred to a fresh tube and 5 µg of recombinant 6His tagged SIRT5 was added and incubated at 37°C for 30 minutes. 50 µL of polyHis-antibody conjugated agarose slurry (Santa Cruz Biotechnologies, sc-803AC) was added and incubated on the rotating mixer overnight at 4°C. The agarose beads were pelleted by centrifugation at 1000 × g for 1 minute. The beads were washed 4 times with the RIPA buffer (1 mL) and pelleted each time at 1000 × g for 1 minute. 40 µL of 2× Laemmli buffer was added to the resulting beads and heated at 80°C for 10 minutes. After centrifugation the supernatant was used for SDS-PAGE. Bands were excised and submitted to the University of Pittsburgh Health Sciences Mass Spectrometry Core Facility for identification of immunoprecipitated proteins.
2.4. Immunoblotting
Tissue lysates were electrophoresed on Criterion SDS polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred to nitrocellulose membranes. Antibodies used were: anti-succinyllysine (PTM-401 and PTM-419, PTM Biolabs); anti-acyl-CoA oxidase-1 (ACOX1; Abcam #ab59964); anti-sirtuin-5 (SIRT5; Cell Signaling #8782); anti-heat shock protein-60 (HSP60; Cell Signaling #12165), anti-catalase (CAT; Sigma #C0979); anti-peroxisomal membrane protein-70 (PMP70; Abcam ab85550); and anti- mitochondrial import inner membrane translocase subunit TIM23 (TIM23; BD Biosciences #611222). After incubation with HRP-conjugated secondary antibodies, blots were visualized with chemiluminescence (Bio-Rad Clarity family ECL reagents), and in some cases, scanned and subjected to densitometric analysis using ImageJ software (NIH).
2.5. Fatty Acid Oxidation Flux Assays
1-14C-labeled lignoceric acid (C24) and 1-14C-dodecanedioc acid (DC12) were sourced from PerkinElmer (Waltham, MA) and Moravek, Inc (Brea, CA), respectively. Just prior to assay, the fatty acids were dried to completion under nitrogen and then solubilized in 10 mg/mL α-cyclodextrin via incubation at 37°C for 30 min. Each was used at 0.5 µCi per mL, with a final substrate concentration of 50 µM. FAO reactions consisted of ~100 µg of liver lysate in a reaction volume of 200 µL containing 100 mM sucrose, 10 mM Tris-HCl, 5 mM KH2PO4, 0.2 mM EDTA, 80 mM KCl, 1 mM MgCl2, 0.1 mM malate, 0.05 mM coenzyme A, 2 mM ATP, and 1 mM DTT. After 2 hr incubation at 37°C, the reactions were stopped by adding perchloric acid to 0.5 M final concentration. Following centrifugation to remove precipitated material, the water-soluble 14C-labeled FAO products were isolated by methanol-chloroform extraction and quantified on a scintillation counter. The results were normalized to protein concentration.
2.6. Urinary Dicarboxylic Acids
Urine was collected during the dark cycle (12 hr) and subjected to gas chromatography and mass spectrometry (GC-MS) at the Children's Hospital of Pittsburgh Clinical Biochemical Genetics Laboratory. Creatinine concentration was determined using a kit (Cayman Chemicals, Inc). For each sample, a volume of urine was used that was equal to 1.0 mmole of creatinine. A 2-phenylbutyrate internal standard was added and organic acids were extracted by sequential ethyl ether and acetoacetate extractions. After trimethylsilane derivatization, the analysis was conducted on an Agilent 7890A gas chromatograph and 5975C mass spectrometer. All peak areas were normalized to that of 2-phenylbutyrate. Fragmentation patterns of urine analytes were compared to an internally compiled fragmentation library and the fragmentation library of the National Institute for Standards and Technology.
2.7. Proteomics and Succinylomics
Pieces of frozen liver (~100 mg) were homogenized, alkylated, trypsinized, clarified, and desalted exactly as described [6]. 100 μg of each peptide elution was set aside for analysis of protein-level changes, and the remaining material used for enrichment of succinylated peptides with the PTMScan Succinyl-Lysine Motif Kit (Cell Signaling Technologies). LC-MS/MS analyses were performed on a Dionex UltiMate 3000 system online coupled to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). All samples were acquired in data-independent acquisition (DIA) mode. DIA data was processed in Spectronaut (version 14.10.201222.47784) using directDIA for both the protein level as well as PTM enriched samples. Differential expression analysis was performed using a paired t-test and p-values were corrected for multiple testing. For whole lysate (protein level) analysis, statistically significant differences were defined as a q-value < 0.01 and an absolute Log2 (fold-change) > 0.58. For the PTM analysis, significance was established as q-value < 0.05 and absolute Log2(fold-change) > 0.58. Peroxisomal proteins were mapped using GO Cellular Component identifiers and verified manually by cross-referencing to Peroxisome-Db [9].
2.8. Recombinant Protein Expression and Purification
Coding sequences for human SIRT5 and the inactive mutant H158Y were incorporated into the pET28a-LIC plasmid with a 6His tag coding sequence at 5’-end. The plasmids were transformed onto E. coli strain BL21(DE3) competent cells. The cells were cultured at 37°C in LB medium and protein expression was induced by adding 0.5 mM IPTG when OD was 0.4. The expression was continued overnight with continuous shaking at 30°C. Cells were lysed on ice by sonication in lysis buffer (50 mM NaPO4, 300 mM NaCl, 10 mM imidazole, pH8.0). Ultracentrifugation was employed to separate lysate supernatant and cell debris. The recombinant proteins were purified on HisTrapTM HP columns (Cytiva Sweden AB) with an AKTA Pure system. The washing buffer was 50 mM NaPO4, 300 mM NaCl, 10% glycerol, 20 mM imidazole, pH8.0. The proteins were eluted with an imidazole gradient.
Human ACOX1a and ACOX1b coding sequences were incorporated into pET21a(+) plasmids with 6His tag coding sequence at 5’- end following a start codon. The plasmids were transformed onto C43(DE3) competent cells and cultured at 37°C in LB medium. Expression was induced with 0.5 mM IPTG at 23°C for 3 hours for ACOX1a and 30°C overnight for ACOX1b. The cell pellets were lysed on ice by sonication in lysis buffer (20 mM NaPO4, 100 mM NaCl, 10% glycerol, 10 mM imidazole, 0.5 mM PMSF, 25 µM FAD, 1 mM benzamidine, 50 ug/ml lysozyme, 0.5% Tween-20, pH 8.0). Ultracentrifugation was employed to separate lysate supernatant and cell debris. The recombinant proteins were purified on HisTrapTM HP columns (Cytiva Sweden AB) by using an AKTA Pure system. The washing buffer is 20 mM NaPO4, 100 mM NaCl, 10% glycerol, 10 mM imidazole, 0.05% Tween 20, pH8.0. The proteins were eluted with an imidazole gradient.
The human EHHADH coding sequence with a 6His tag coding sequence at 5’-end following a start codon was incorporated into pET21a(+) plasmid. The EHHADH expression plasmid and a GroEL/ES (chloramphenicol resistant) plasmid were co-transformed onto C41(DE3) chemically competent cells. The cells were cultured at 37°C in LB medium and protein expression was induced by adding 0.5 mM IPTG when OD was 2. The expression was continued overnight with continuous shaking at 19°C. Harvested cell pellet was lysed in 20 mM NaPO4, 100 mM NaCl, 10% Glycerol, pH 7.7 containing 0.5 mM PMSF, 1mM benzamidine, 50ug/ml lysozyme, 10 mM imidazole and 0.5 % Tween 20. Ultracentrifugation cleared lysate supernatant was loaded on HisTrapTM HP columns (Cytiva Sweden AB) by using an AKTA Pure system. The unbound lysate components were washed away by washing buffer (20 mM NaPO4, 100 mM NaCl, 10% glycerol, 10 mM imidazole, 0.05% Tween 20, pH7.7). The pure EHHADH protein was eluted with an imidazole gradient.
2.9. Enzyme Activity Assays
ACOX1a, ACOX1b or EHHADH were chemically succinylated by 0.1 mM succinyl-CoA in MSH buffer (210 mM mannitol, 70 mM sucrose, 5 mM Hepes, pH 8.0) with 5 mM DTT, 0.05% Tween-20 at 37°C for 30 minutes. Desuccinylation was carried out in 50 mM Tris (pH7.5) with 0.2 µg/ul SIRT5, 1 mM NAD, 2 mM MgCl2, 10% glycerol, 5 mM DTT, 0.05% TW-20. The buffers were changed to 50 mM NaPO4, pH 7.0 (ACOX1) or 50 mM Tris-HCl, pH8.5 (EHHADH), with 10% glycerol and 0.05% Tween-20. Enyzme activity was then measured as follows. ACOX1 activity was assayed with 25 µM of acyl-CoA substrate and following H2O2 production with Amplex Red as described [6, 10]. 1 µg of protein was added to 50 mM phosphate buffer pH 7.0 containing HRP (1 U/ml) and Amplex UltraRed (50 µM) at 30°C. Reactions were started by addition of acyl-CoA substrate and fluorescence was monitored for 30 minutes. The EHHADH activity assay was conducted in 100 ul of 50 mM Tris, pH8.5, with 0.05% Tween-20, 1 mM NAD, 5 µg EHHADH, and 10 µg ACOX1a or ACOX1b. Substrate concentration was 50 µM. The assay was carried out at 37°C on a fluorescence plate reader (FluoStar Omega, BMG LabTech), using excitation wavelength 340 nm and emission wavelength 490 nm.
3. Results
3.1. Dicarboxylic Acid Feeding Recruits SIRT5 to Liver Peroxisomes
SIRT5 localizes in part to peroxisomes in cultured cell lines [5]; however, in vivo, SIRT5 peroxisomal localization has not been directly demonstrated. Therefore, we began by optimizing a density gradient centrifugation protocol to isolate pure mouse liver peroxisomes. Wild-type mouse liver peroxisomes, positive for the peroxisomal marker ACOX1 but negative for the mitochondrial marker HSP60, were immunoblotted for SIRT5. Indeed, trace amounts of SIRT5 were present in wild-type liver peroxisomes under basal conditions (Figure 1a). We estimated the basal amount of peroxisomal SIRT5 to be ~10% of the amount found in either the heavy mitochondrial fraction (HM) or light mitochondria fraction (LM) separated during the gradient centrifugation (Figure 1b). Fasting overnight increased the amount of peroxisomal SIRT5 (Figure 1a), suggesting that localization of SIRT5 to the peroxisomal compartment may be inducible under conditions that drive peroxisomal fatty acid oxidation (FAO). This led us to ask whether feeding mice dodecanedioic acid (DC12), a dicarboxylic peroxisomal FAO substrate, would recruit SIRT5 to the peroxisome. We previously showed that DC12 is chain-shortened in peroxisomes to succinyl-CoA which then reacts with peroxisomal proteins to produce protein hypersuccinylation [6]. Indeed, mice consuming a diet containing 10% w/w DC12 (21% of calories) had an 8-fold increase in the abundance of peroxisomal SIRT5, whereas mitochondrial SIRT5 remained unchanged (Figure 1c).
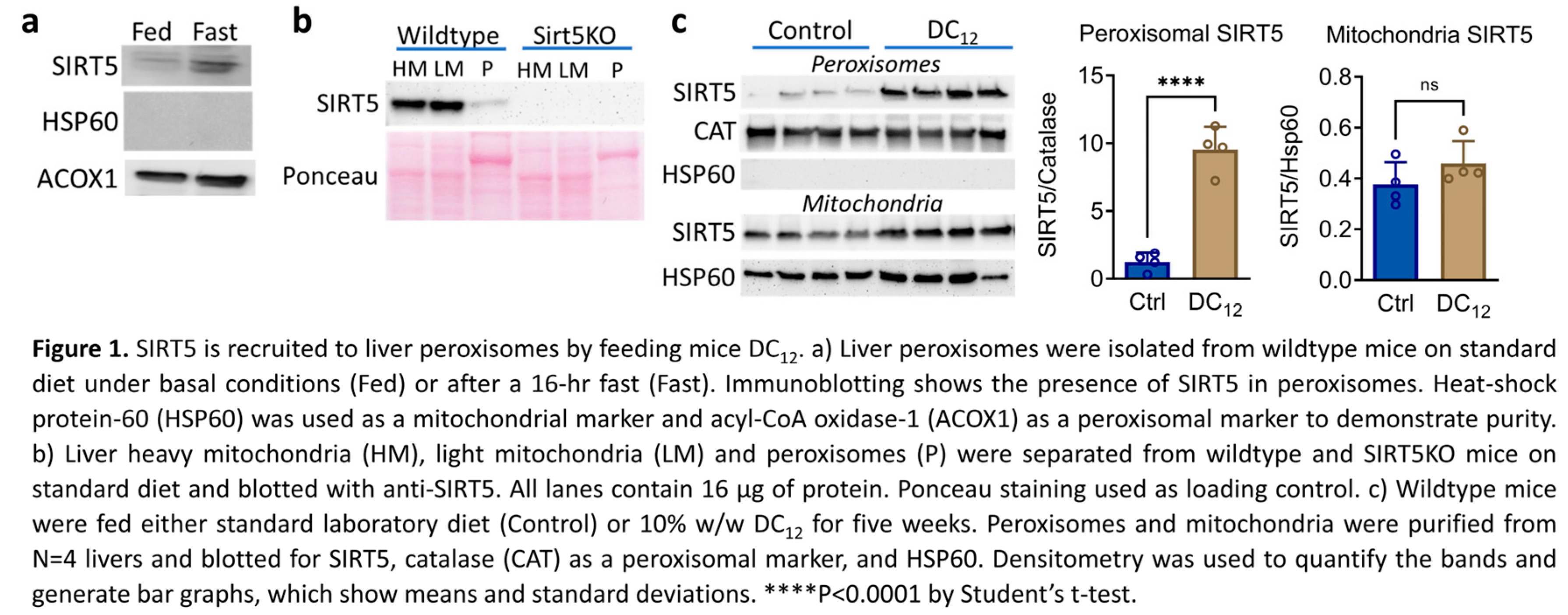
3.2. SIRT5 Knockout Mice Have Impaired DC12 Catabolism
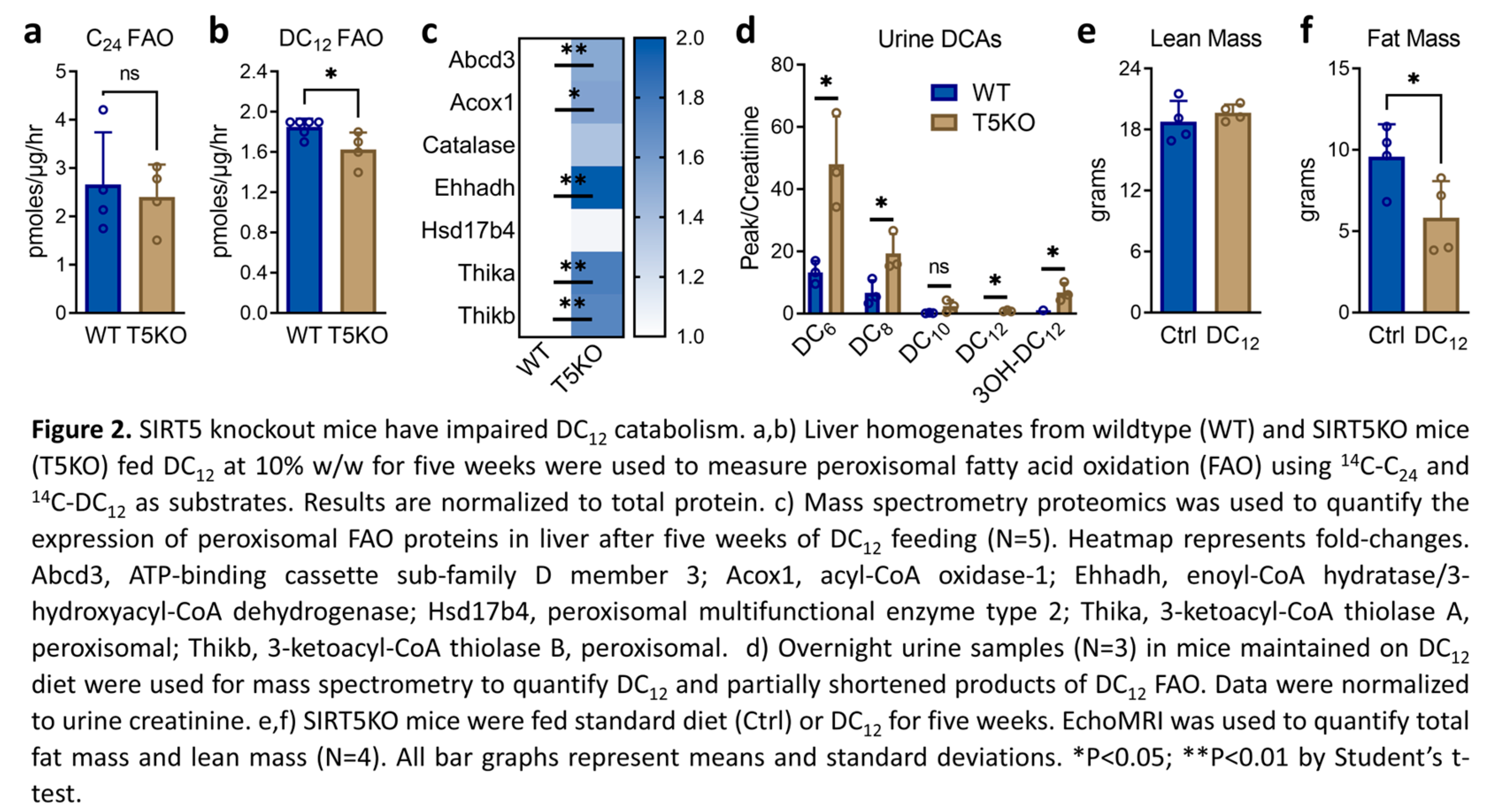
Chen et al [5] showed that succinylation increases the activity of ACOX1, the rate-limiting enzyme for peroxisomal straight-chain FAO, and conversely, desuccinylation by SIRT5 decreases ACOX1 activity. We therefore hypothesized that knocking out SIRT5 would enhance peroxisomal FAO via increased ACOX1 function. To test this, wildtype and SIRT5KO male mice were fed DC12 at 10% w/w for five weeks. Liver homogenates were used to assess the rate of peroxisomal FAO using 14C-DC12 and the very long-chain substrate 14C-C24. Opposite to our hypothesis, the rate of DC12 FAO was significantly reduced, not increased, by ablation of SIRT5 while C24 FAO was not changed (Figure 2a,b). These FAO assay rates were normalized to total liver protein. However, liver proteomics showed that the peroxisomal FAO machinery was upregulated by ~1.5 to 2-fold in DC12-fed SIRT5KO liver compared to DC12-fed wildtype liver (Figure 2c). If the FAO data were normalized to the abundance of the FAO machinery, then C24 FAO would be ~50% lower in SIRT5KO liver and DC12 FAO ~75% lower. In short, these data suggest that peroxisomal FAO is impaired in SIRT5KO liver and that peroxisomal enzymes are upregulated to compensate. In keeping with impaired D12 catabolism, DC12-fed SIRT5KO mice had elevated D12 intermediates in urine, including DC12, 3-hydroxy-DC12, DC8, and DC6 (Figure 2d). Despite this impairment, SIRT5 was not required for the reduced adiposity that we previously showed to be a hallmark of DC12 feeding [6]. SIRT5KO mice were ~40% leaner than wildtype control mice after five weeks on the DC12 diet (Figure 2e,f).
3.3. Knocking Out SIRT5 Has Minimal Effects on the Peroxisomal Succinylome
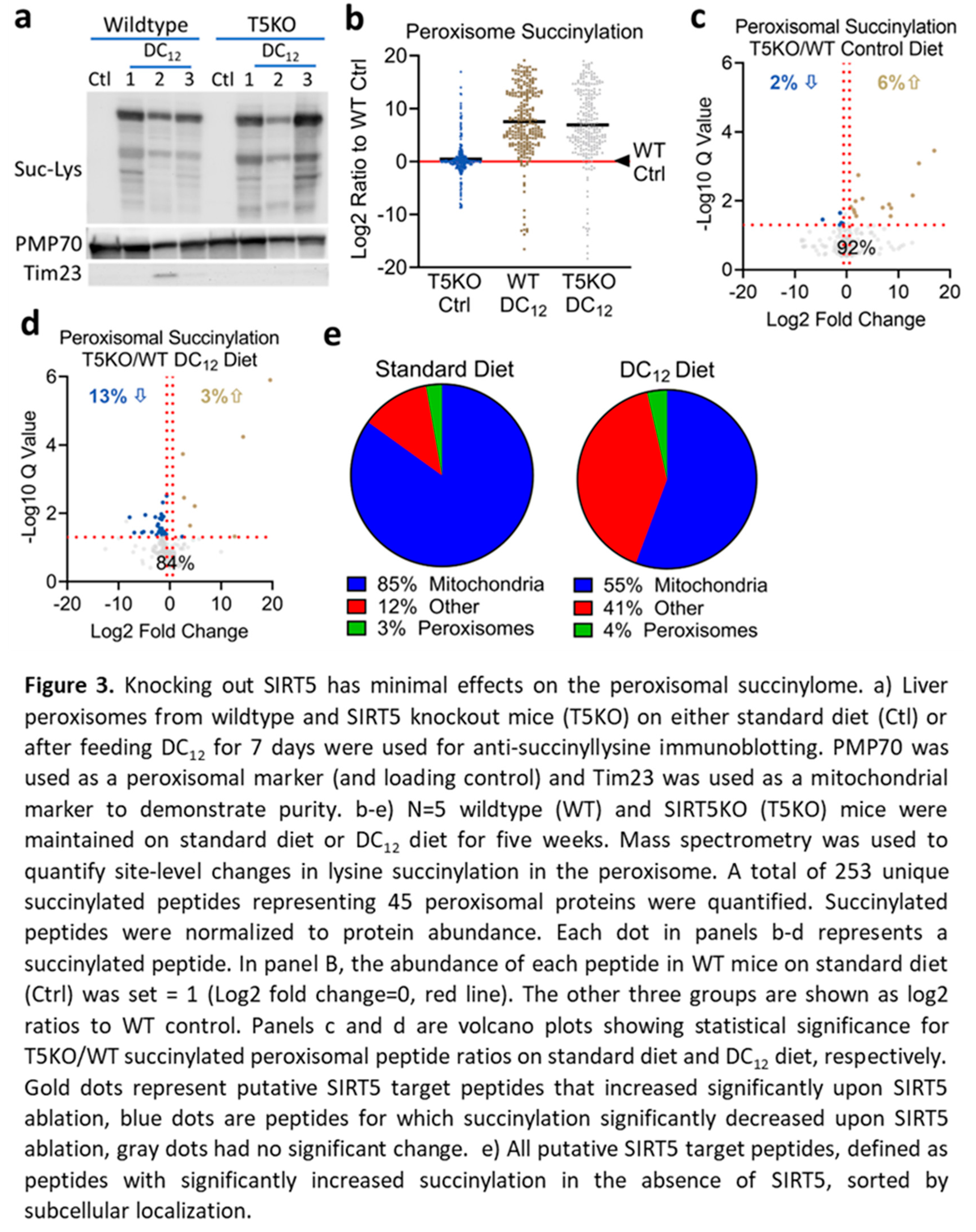
Data presented in Figure 2 suggested that rather than enhancing peroxisomal function, knocking out SIRT5 may impair it. We next sought to probe the mechanisms behind the impairment by profiling SIRT5 actions in the peroxisome. To screen for SIRT5 interactions with peroxisomal proteins, we prepared liver homogenate from a SIRT5KO mouse, incubated the homogenate with His-tagged recombinant SIRT5, and used anti-His antibody to precipitate proteins complexed with SIRT5. Mass spectrometry identified 257 putative SIRT5 interacting proteins with > 10% peptide coverage (Supplemental Table 1). Of the 10 proteins with highest peptide coverage, three were peroxisomal. In total, 15 peroxisomal proteins were identified as SIRT5 interacting partners. Nearly all of these are involved in FAO (Table 1). Of particular interest were ACOX1 and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase (EHHADH) which are required for dicarboxylic acid chain-shortening [7]. Next, we examined the effect of SIRT5 ablation on the in vivo peroxisomal succinylome. First, we fed wildtype and SIRT5KO mice 10% w/w DC12 acutely for 7 days and purified hepatic peroxisomes. Anti-succinyllysine immunoblotting confirmed that lysine succinylation is very low in peroxisomes in mice fed standard laboratory control diet (Figure 3a). DC12 dramatically increased succinylation in both wildtype and SIRT5KO peroxisomes. Densitometry analysis of the total succinylation signal normalized to peroxisomal PMP70 as a loading control showed a non-significant trend toward higher succinyl- ation in SIR5KO peroxisomes. To interrogate this further, we fed another cohort of mice the DC12 diet (or standard diet as control) for five weeks and used mass spectrometry to quantify changes in lysine succinylation at the residue level. A total of 253 succinylated peptides representing 45 peroxisomal proteins were quantified across both genotypes and both diets. To control for potential differences in gene expression, the abundance of the succinylated peptides was normalized to protein abundance. Using wildtype mice on standard chow as the benchmark, SIRT5KO mice had slightly increased peroxisomal lysine succinylation, with an average fold-increase of 1.3-fold (log2 of 0.43 in Figure 3b) across all 253 peptides. Wildtype mice on DC12 diet had an average increase of 185-fold (log2 7.5 in Figure 3b) while SIRT5KO mice had an average increase of 121-fold (log2 of 6.9 in Figure 3b). This suppression of global peroxisomal lysine succinylation in DC12-fed SIRT5KO was unexpected. We further interrogated the mass spectrometry data set by separating the data by diet and statistically analyzing for SIRT5-dependent site-level changes in succinylation. Specifically, we sought to identify lysine residues demonstrating an increase in succinylation upon ablation of SIRT5, which would represent putative SIRT5 target sites in the peroxisome. On standard control diet, 19 out of the total 253 succinylated peroxisomal peptides showed statistically significant differences, defined as q<0.05 and fold-change > 1.5, between genotypes (Figure 3c). Of the 19 peptides, 14 were increased and 5 were decreased in SIRT5KO livers (Table 2). In the DC12-fed groups there were a total of 41 out of 253 succinylated peptides that significantly differed between wildtype and SIRT5KO peroxisomes (Figure 3d). The majority of these (34 peptides) were significantly decreased, not in- creased, in SIRT5KO compared to wildtype (Table 3). Only three lysine residues were hypersuccinylated in peroxisomes across both diets in SIRT5KO mice—HSDL2 K295, NUDT19 K351, and EHHADH K355. Of these three proteins, only EHHADH plays a role in peroxisomal FAO. Of note, ACOX1, which was previously shown to be regulated by SIRT5 [5], had 17 quantified succinylated peptides (not shown). None were significantly different between wildtype and SIRT5KO under either diet. Most trended downward in SIRT5KO. Similarly, while EHHADH was hypersuccinylated on one lysine residue (K355) in the absence of SIRT5, 28 other lysine residues on EHHADH were statistically unchanged, with most trending downward in SIRT5KO. Together, these findings indicate that SIRT5 has minimal impact on the peroxisomal
succinylome. To further illustrate this, we sorted all the significantly increased succinylated peptides in SIRT5KO liver by subcellular compartment. On standard diet, the putative SIRT5-targeted peptides were overwhelmingly mitochondrial (Figure 3e). On DC12 diet, there was a clear shift toward hypersuccinylation in “other” compartments upon SIRT5 ablation. The “other” category represents cytoplasm, endoplasmic reticulum, nucleus, golgi, and plasma membrane combined.
3.4. SIRT5 Desuccinylates Recombinant ACOX1 and EHHADH but the Impact on Enzyme Activity Is Minimal
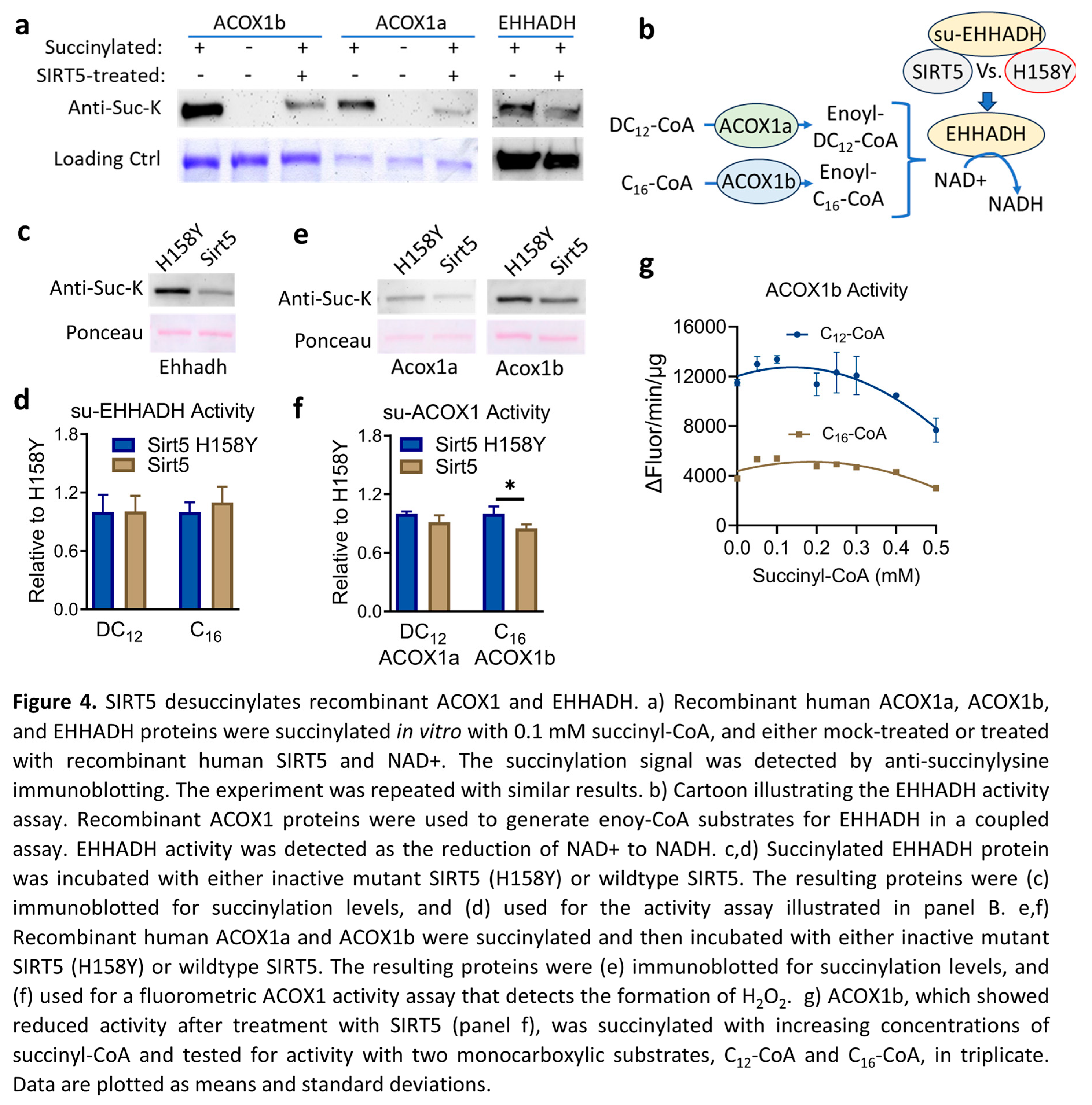
In peroxisomes from DC12-fed mice, EHHADH and ACOX1, which are involved in DC12 chain-shortening [7], were the two most succinylated proteins with 29 and 17 quantified succinylated peptides, respectively. We therefore chose to study these two enzymes in more detail using in vitro methods pioneered by us and others in which recombinant proteins are succinylated chemically via exposure to succinyl-CoA [1, 11, 12]. Regarding ACOX1, we recently showed that of the two major splice variants, only ACOX1a has appreciable activity with dicarboxylic substrates, even though the two isoforms differ by only 27 amino acids [6]. ACOX1b demonstrates a preference for longer monocarboxylic substrates like C16-CoA and C24-CoA [6]. Chen et al studied the interaction between SIRT5 and ACOX1 but did not distinguish between the two splice variants. We therefore expressed and purified both recombinant human ACOX1a and ACOX1b, as well as human EHHADH. The proteins were incubated with 0.1 mM succinyl-CoA for 30 min to induce high-level lysine succinylation, and then used as substrates for recombinant human SIRT5. All three proteins showed evidence of desuccinylation by recombinant SIRT5 in the presence of NAD+ (Figure 4a). Next, we asked whether this desuccinylation by SIRT5 impacts enzymatic activity. The physiological substrates for EHHADH are enoyl-CoAs, the products of the ACOX1 reaction. EHHADH is a bifunctional enzyme that first hydrates and then dehydrogenates enoyl-CoAs, reducing NAD+ to NADH in the process. Enoyl-CoAs are not commercially available. To test the effect of SIRT5 on the activity of EHHADH, we developed an approach in which chemically-succinylated EHHADH was incubated with NAD+ and either inactive mutant SIRT5 (H158Y) or active wildtype SIRT5 for 30 min. Then the succinylated and desuccinylated EHHADH proteins were buffer exchanged and mixed with either 1) recombinant ACOX1a and DC12-CoA or 2) recombinant ACOX1b and C16-CoA, in the presence of NAD+. In this coupled assay the ACOX1 proteins converted their acyl-CoA substrates to enoyl-CoAs, which were then hydrated and dehydrogenated by EHHADH, with concomitant reduction of NAD+ to NADH which was followed fluorometrically (Figure 4b). Desuccinylation of EHHADH by SIRT5 had no effect on EHHADH enzyme activity in this coupled assay, despite substantial desuccinylation by SIRT5 (Figure 4c). We similarly assessed the effect of SIRT5 on ACOX1a and ACOX1b activity, starting with chemically succinylated recombinant proteins and incubating with either SIRT5 or SIRT5 H158Y. SIRT5 had no effect on ACOX1a activity with DC12-CoA (Figure 4d). However, incubation of ACOX1b with SIRT5 did modestly reduce ACOX1b activity with C16-CoA, which agrees with the work of Chen et al [5] who studied ACOX1 in cultured cells. In a final experiment we incubated recombinant ACOX1b with increasing amounts of succinyl-CoA for 30 min prior to measuring enzymatic activity. At 0.1 mM succinyl-CoA, which is the concentration we used to succinylate ACOX1 proteins for the experiments in Figure 4d, there was a clear increase in ACOX1b activity with both C12-CoA, which is the optimum substrate for ACOX1b, and C16-CoA (Figure 4e). With exposure to increasing amounts of succinyl-CoA, activity of ACOX1b declined, particularly with the optimum substrate C12-CoA.
4. Discussion
Our findings confirm and extend upon the previous studies of Chen et al [5] who showed that ACOX1 localizes partly to the peroxisome in multiple cell lines. Under normal, fed-state conditions we saw a modest amount of SIRT5 protein within hepatic peroxisomes. Fasting, which is known to stimulate fatty acid ω-oxidation, dicarboxylic acid formation, and peroxisomal FAO, increased SIRT5 representation in peroxisomes. The fact the peroxisomal localization of SIRT5 was enhanced several-fold by feeding mice an exogenous dicarboxylic acid (DC12) suggests that the mechanism of recruitment is responding to the level of succinyl PTMs in the peroxisome. Future studies are needed to determine the mechanisms of SIRT5 peroxisomal recruitment. There are two known types of peroxisomal targeting signal (PTS), dubbed PTS1 and PTS2. PTS1 signals are variants of the canonical “SKL” motif at the carboxy terminus. PTS2 signals are variable and occur at either a terminus or internally on proteins. For SIRT5, Chen et al found a putative PTS2 signal at the amino terminus, embedded within the mitochondrial targeting signal [5]. Our immunoblotting studies on purified organelles indicated that DC12 feeding induces a small, nonsignificant increase in mitochondrial SIRT5 while peroxisomal SIRT5 increases ~8-fold. This suggests that DC12 is not directing SIRT5 away from mitochondria into peroxisomes, but rather, increasing the total pool of SIRT5 protein. We did not, however, quantify cytosolic SIRT5 in the context of DC12. Future work will interrogate the balance of SIRT5 protein across mitochondria, cytoplasm, and peroxisomes, and seek to understand how this balance might respond to changes in PTMs within each compartment.
One unexplained phenomenon is why DC12-induced peroxisomal recruitment of SIRT5 has such a minimal effect on the peroxisomal succinylome. In mitochondria, protein succinylation is well-known to increase with SIRT5 ablation [2, 13-15]. Indeed, our mass spectrometry survey found significantly increased succinylation in SIRT5KO liver on ~25% of all detected mitochondrial peptides, confirming previous studies (data not shown). But in the peroxisome, succinylation tended to be modestly suppressed in SIRT5KO liver after DC12 feeding, not increased. This is in keeping with reduced flux through the DC12 catabolic pathway. Despite having significantly higher expression of key enzymes required for DC12 catabolism, such as ACOX1 and EHHADH, SIRT5KO liver homogenates showed a slower rate of 14C-DC12 FAO. Further, DC12 catabolic intermediates were higher in urine. Slower flux of DC12 through the pathway would slow the rate of succinyl-CoA production from DC12, thereby lowering the overall level of peroxisomal protein succinylation. The fact that the acylating agent, succinyl-CoA, may have decreased in concentration inside the peroxisome upon SIRT5 ablation may have masked our ability to identify putative SIRT5 target sites on peroxisomal proteins. In other words, we would have expected succinylation to increase on SIRT5-targeted lysine residues in the absence of SIRT5, but an impaired ability to form the succinyl-CoA that is necessary to produce the increased succinylation serves as a confounding variable.
Regarding the mechanism behind reduced DC12 flux through SIRT5KO peroxisomes, we may only speculate. Given that ~95% of putative SIRT5 target sites in DC12-fed mouse liver are in the mitochondria or in “other” compartments such as the cytoplasm, the likely explanation is that an accumulation of succinyl PTMs in one or more of those compartments is responsible. One possibility is that mitochondrial FAO partly contributes to DC12 oxidation and that the reduced flux and accumulation of urinary DC12 metabolites in SIRT5KO mice is attributed to changes in mitochondrial FAO enzymes, not peroxisomal. While mitochondria are capable of oxidizing DC12 in vitro [16], other studies have shown that DC12 is strictly catabolized by peroxisomes [8]. The mitochondrial contribution to DC12 FAO remains controversial. Another possible explanation is that a cytosolic factor is responsible for the reduced flux of DC12 in SIRT5KO liver. For example, increased succinylation of acyl-CoA synthetases, which would be required to activate DC12 to coenzyme A, may suppress the rate of FAO. Alternatively, succinylation of fatty acid binding proteins (FABPs), which convey fatty acids to membrane receptors for organelle import, may be affected in the absence of SIRT5. A final area of speculation relates to the possibility that in wildtype liver, even though DC12 feeding increases the abundance of peroxisomal SIRT5 by ~8-fold, this pool of SIRT5 may be enzymatically handicapped by a lack of available NAD+. All sirtuins are NAD+-dependent [17]. In vitro the Km of SIRT5 for NAD+ is around 200 µM [18]. One biological explanation for this would be to prevent sirtuins from outcompeting metabolic enzymes for the shared NAD+ cofactor. In the peroxisome, each round of FAO chain shortening requires the reduction of NAD+ to NADH by EHHADH and HSD17B4, the two peroxisomal bifunctional enzymes. The Km of EHHADH and HSD17B4 for NAD+ has been estimated at ~5 µM [19], which is 40-fold lower than the Km of SIRT5 for NAD+. It is possible that when we feed animals DC12 the peroxisomal FAO pathway is constitutively activated, and under this context, the NAD+ is preferentially channeled into the chain-shortening of DC12 and SIRT5 is left inactive. Such a mechanism could conceivably explain the apparent lack of difference in the succinylome between wildtype and SIRT5KO mice on DC12 diet—in essence, even the wildtype mice may be deficient in peroxisomal SIRT5 activity due to competition for NAD+. Future work is needed to interrogate these intriguing possibilities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: SIRT5 interacting proteins identified by Co-IP.
Author Contributions
Conceptualization, E.G. and Y.Z.; methodology, E.G., Y.Z., and B.S.; investigation, Y.Z., B.Z., S.B., J.B., J.R., S.S., S.D., and E.G.; formal analysis, Y.Z., E.G., S.D., and B.S.; data curation, Y.Z., E.G., and J.B.; writing—original draft preparation, E.G. and Y.Z.; writing—review and editing, S.D., J.B., and B.S.; project administration, E.G. and B.S.; funding acquisition, E.G. and B.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of University of Pittsburgh (protocol #24014359, approved January 2021).
Data Availability Statement
Raw data and complete MS data sets have been uploaded to the Center for Computational Mass Spectrometry, to the MassIVE repository at UCSD, and can be downloaded using the following FTP link: ftp://MSV000096026@massive.ucsd.edu or via the MassIVE website: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=21320bba927a4e409db5416af1651591 (MassIVE ID number: MSV000096026; ProteomeXchange ID: PXD056506).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wagner, G.R. and R.M. Payne, Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem 2013, 288, 29036–45. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J. , et al., SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 2013, 18, 920–33. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D. and Y. Zhao, Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol Cell Proteomics 2015, 14, 2308–15. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y. , et al., SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol Cell 2015, 59, 321–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F. , et al., SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep 2018, 19. [Google Scholar]
- Goetzman, E.S. , et al., Dietary dicarboxylic acids provide a non-storable alternative fat source that protects mice against obesity. J Clin Invest 2024, 134. [Google Scholar]
- Ranea-Robles, P. and S.M. Houten, The biochemistry and physiology of long-chain dicarboxylic acid metabolism. Biochem J 2023, 480, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z. , et al., Compartmentation of Metabolism of the C12-, C9-, and C5-n-dicarboxylates in Rat Liver, Investigated by Mass Isotopomer Analysis: ANAPLEROSIS FROM DODECANEDIOATE. J Biol Chem 2015, 290, 18671–7. [Google Scholar] [CrossRef] [PubMed]
- Schluter, A. , et al, PeroxisomeDB 2.0: an integrative view of the global peroxisomal metabolome. Nucleic Acids Res 2010, 38, D800–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. , et al., The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol 2019, 26, 101253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. and E. Goetzman, The enzyme activity of mitochondrial trifunctional protein is not altered by lysine acetylation or lysine succinylation. PLoS One 2021, 16, e0256619. [Google Scholar]
- Zhang, Y. , et al., SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain acyl-CoA dehydrogenase. PLoS One 2015, 10, e0122297. [Google Scholar]
- Hershberger, K.A. , et al., Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J Biol Chem 2018, 293, 10630–10645. [Google Scholar] [CrossRef] [PubMed]
- Alleyn, M. , et al., The dawn of succinylation: a posttranslational modification. Am J Physiol Cell Physiol 2018, 314, C228–C232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. , et al., Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem 2017, 292, 10239–10249. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, S.S. , et al., Role of mitochondrial acyl-CoA dehydrogenases in the metabolism of dicarboxylic fatty acids. Biochem Biophys Res Commun 2020, 527, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C. , et al., From sirtuin biology to human diseases: an update. J Biol Chem 2012, 287, 42444–52. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.S. , et al., Investigating the Sensitivity of NAD+-dependent Sirtuin Deacylation Activities to NADH. J Biol Chem 2016, 291, 7128–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y., D. Cuebas, and H. Schulz, Channeling of 3-hydroxy-4-trans-decenoyl coenzyme A on the bifunctional beta-oxidation enzyme from rat liver peroxisomes and on the large subunit of the fatty acid oxidation complex from Escherichia coli. J Biol Chem 1986, 261, 15390–5. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Peroxisomal proteins detected in Sirt5 Co-IP.
| Protein | Function | % Coverage |
|---|---|---|
| Uricase (UOX) | Oxidizes uric acid to 5-hydroxyisourate | 86 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | Steps 2 and 3 of β-oxidation | 74 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | Steps 2 and 3 of β-oxidation | 65 |
| Acyl-CoA dehydrogenase 11 (ACAD11) | Step 1 of very long-chain β-oxidation | 50 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | Reduces double bonds, β-oxidation | 49 |
| Peroxisomal acyl-coenzyme A oxidase 1 (ACOX1) | Step 1 of β-oxidation | 40 |
| Catalase (CAT) | Degrades H2O2 to H2O and O2 | 33 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | Fatty acid transporter | 23 |
| Acyl-coenzyme A thioesterase 4 (ACOT4) | Cleaves CoA from succinyl-CoA | 21 |
| Non-specific lipid-transfer protein (SCP2) | Step 4 of β-oxidation | 21 |
| Peroxisomal sarcosine oxidase (PIPOX) | Metabolizes sarcosine & pipecolic acid | 16 |
| Lon protease homolog 2, peroxisomal (LONP2) | Degrades misfolded proteins | 14 |
| 3-ketoacyl-CoA thiolase B (ACAA1b) | Step 4 of β-oxidation | 13 |
| Very long-chain acyl-CoA synthetase (SLC27A2) | Activates fatty acids into acyl-CoAs | 12 |
| Peroxisomal coenzyme A diphosphatase (NUDT7) | Hydrolyzes CoA moiety of acyl-CoAs | 10 |
Table 2.
Significantly altered succinylated peptides in liver peroxisomes on standard diet.
| Protein | Lysine Residue | Log2 FC KO/WT |
|---|---|---|
| Increased Succinylation (q<0.05) | ||
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K481 | 16.9 |
| Enoyl-CoA isomerase (ECH1) | K320 | 14.0 |
| Enoyl-CoA isomerase (ECH1) | K76 | 12.8 |
| Acyl-coenzyme A diphosphatase (NUDT19) | K351 | 8.6 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K390 | 8.5 |
| Alpha-methylacyl-CoA racemase (AMACR) | K204 | 8.2 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K295 | 7.0 |
| Prostaglandin reductase-3 (ZADH2) | K214 | 3.1 |
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K106 | 2.3 |
| Catalase (CAT) | K522 | 1.9 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | K230 | 1.7 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | K6 | 1.5 |
| Alanine--glyoxylate aminotransferase (AGXT) | K322 | 1.4 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K355 | 0.8 |
| Decreased Succinylation (q<0.05) | ||
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K99 | -0.7 |
| Catalase (CAT) | K98 | -1.0 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K83 | -1.3 |
| Enoyl-CoA isomerase (ECH1) | K230 | -1.3 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K249 | -4.6 |
Table 3.
Significantly altered succinylated peptides in liver peroxisomes on DC12 diet.
| Protein | Lysine Residue | Log2 FC KO/WT |
|---|---|---|
| Increased Succinylation (q<0.05) | ||
| Peroxisomal ATPase (PEX1) | K1205 | 14.3 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K50/K57 | 12.6 |
| Alanine--glyoxylate aminotransferase (AGXT) | K411/K314 | 4.9 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K295 | 4.0 |
| Catalase (CAT) | K477 | 2.8 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K348/K355 | 2.6 |
| Acyl-coenzyme A diphosphatase (NUDT19) | K351 | 2.4 |
| Decreased Succinylation (q<0.05) | ||
| Uricase (UOX) | K27 | -0.6 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K579 | -0.7 |
| Acyl-CoA dehydrogenase-11 (ACAD11) | K422 | -0.9 |
| Uricase (UOX) | K118 | -1.0 |
| Uricase (UOX) | K80 | -1.1 |
| Bile acid-CoA: amino acid N-acyltransferase (BAAT) | K346 | -1.2 |
| Peroxisomal sarcosine oxidase (PIPOX) | K104 | -1.3 |
| ATP-binding cassette sub-family D member 3 (ABCD3) | K260 | -1.4 |
| Bile acid-CoA: amino acid N-acyltransferase (BAAT) | K406 | -1.4 |
| Non-specific lipid-transfer protein (SCP2) | K26 | -1.5 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K527 | -1.6 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K353 | -1.6 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K81 | -1.6 |
| Peroxisomal bifunctional enzyme type 1 (EHHADH) | K459 | -1.6 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K57 | -1.8 |
| Dehydrogenase/reductase SDR family member 4 (DHRS4) | K99 | -1.8 |
| Peroxisomal bifunctional enzyme type 2 (HSD17B4) | K65/K68 | -1.8 |
| Hydroxysteroid dehydrogenase like 2 (HSDL2) | K87 | -1.8 |
| Acyl-CoA dehydrogenase-11 (ACAD11) | K90 | -1.9 |
| Peroxisomal 2,4-dienoyl-CoA reductase (DECR2) | K70/K71 | -1.9 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K32 | -1.9 |
| Peroxisomal sarcosine oxidase (PIPOX) | K153 | -2.1 |
| Peroxisomal sarcosine oxidase (PIPOX) | K98 | -2.1 |
| Alpha-methylacyl-CoA racemase (AMACR) | K57 | -2.3 |
| Bifunctional epoxide hydrolase 2 (EPHX2) | K482 | -2.4 |
| 2-hydroxyacyl-CoA lyase 1 (HACL1) | K21 | -2.5 |
| Enoyl-CoA isomerase (ECH1) | K230 | -2.8 |
| Peroxisomal coenzyme A diphosphatase (NUDT7) | K20 | -3.5 |
| 2-hydroxyacyl-CoA lyase 1 (HACL1) | K238 | -4.8 |
| 2-Hydroxyacid oxidase 1 (HAO1) | K309 | -5.1 |
| Acyl-CoA oxidase-2 (ACOX2) | K678 | -5.4 |
| Peroxisomal trans-2-enoyl-CoA reductase (PECR) | K17 | -6.9 |
| Acyl-CoA oxidase-2 (ACOX2) | K303 | -7.8 |
| NAD-capped RNA hydrolase (NUDT12) | K282 | -8.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



