Submitted:
21 July 2025
Posted:
22 July 2025
You are already at the latest version
Abstract
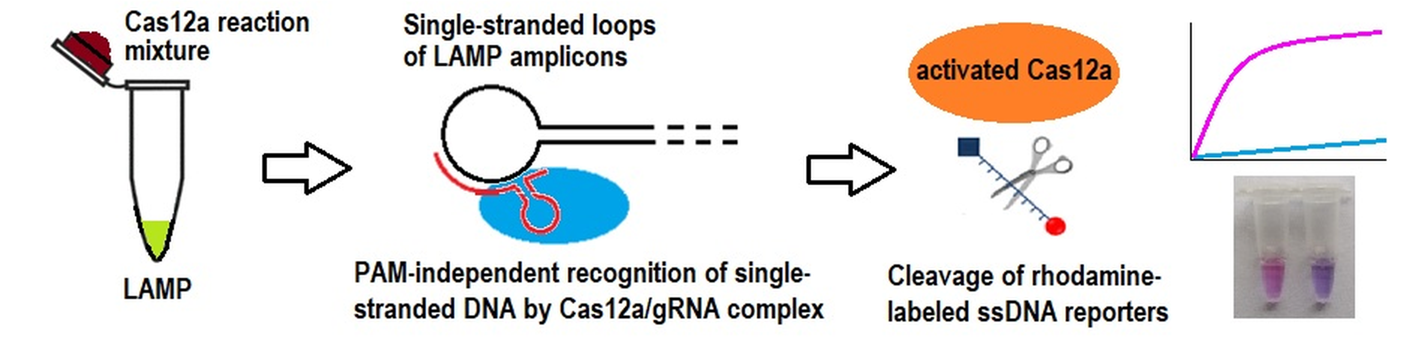
A straightforward approach is suggested to selectively recognize specific products of loop-mediated isothermal amplification (LAMP) by Cas12a nuclease without a need for a protospacer adjacent motif (PAM) in the sequence of LAMP amplicons (LAMPlicons). The strategy is based on the presence of single-stranded DNA loops in LAMPlicons and the ability of Cas12a to be trans-activated via binding of guide RNA (gRNA) to single-stranded DNA in the absence of PAM. The approach feasibility is demonstrated on Clavibacter species – a bunch of bacterial plant pathogens causing harmful diseases of agriculturally important plants. In regard to Clavibacter species, the detection sensitivity of the developed PAM-independent LAMP/Cas12a system was determined by that of LAMP, while the overall detection selectivity was enhanced by the Cas12a analysis of LAMPlicons. It was shown that the LAMP/Cas12a detection system can be fine-tuned by carefully designing gRNA so to selectively distinguish C. sepedonicus among other Clavibacter species, based on single nucleotide substitutions in the targeted LAMPlicon loop. The suggested loop-based Cas12a analysis of LAMPlicons was compatible with a format of single test tube assay with the option of naked-eye detection. The findings broaden the palette of approaches to designing PAM-independent LAMP/Cas12a detection systems with a potential for on-site testing.
Keywords:
loop-mediated isothermal amplification
; Cas12a
; amplicon loops
; Clavibacter species
1. Introduction
Today, selective amplification of unique nucleic acid sequences in genomes/transcriptomes of pathogenic microorganisms or viruses has become an indispensable tool in DNA diagnostics of infectious diseases and beyond [1]. Although polymerase chain reaction (PCR) still dominates DNA diagnostics and stays as the “gold standard” for the field, there is a growing trend towards on-site (out-of-laboratory) testing with numerous applications in medicine, ecology, and agriculture [2,3]. For the on-site testing, the isothermal amplification of nucleic acids with simplified instrumentation requirements but reaction times and sensitivity comparable to PCR appears as an attractive approach [4]. Among isothermal amplification methods, loop-mediated isothermal amplification (LAMP) is mostly used, with numerous examples of practical applications [5]. LAMP introduced in 2000 by Notomi et al. [6] employes a single DNA polymerase with a strand-displacing activity and a set of primers (from 4 to 6) to provide a remarkably high yield of peculiar cauliflower-like DNA amplicons (often referred to as LAMPlicons) which are composed of varying number of stem-loop inverted DNA repeats [5].
Recently, the further development in pathogen-specific detection assays for on-site diagnostics has been made by coupling LAMP with RNA-guided CRISPR/Cas12 analysis of LAMPlicons (CRISPR, clustered regularly interspaced short palindromic repeats; Cas, CRISPR associated protein) [7]. The interest to CRISPR/Cas12 biosensing systems based on both functional activity of Cas12 nucleases alone or via coupling them to isothermal amplification steadily grows [8,9]. By complexing Cas12 nuclease with a particular RNA molecule (guide RNA; gRNA), specific LAMP products can be selectively targeted by binding the gRNA segment known as a “spacer” to a complementary sequence (“protospacer”) in double-stranded sections of LAMPlicons. The binding results in acquiring of trans-activity by the Cas12 nuclease that can be detected via an increase in fluorescence upon nonspecific cleavage of short DNA oligonucleotides labeled with a fluorophore and a fluorescence quencher (molecular reporters, MRs) [7,8,9].
The binding of gRNA spacer to a target’s protospacer requires the presence of the protospacer adjacent motif (PAM) – a short specific nucleotide motif, recognized by a Cas12 nuclease and positioned at the 5′-end next to the sequence complementary to the protospacer in the double-stranded DNA (dsDNA) target sequence [10]. The PAM requirement imposes a restriction on the selection of LAMP primers, making it often difficult to integrate the most effective primer set with CRISPR/Cas analysis of LAMPlicons. To circumvent this limitation, the use of Cas12a mutants and orthologs was suggested to increase diversity of PAM sequences [11,12]. This approach alleviates the PAM requirement but not eliminates it. Another way to solve the problem has been recently demonstrated in [13] by introducing a PAM sequence into a LAMP primer. However, LAMP primers are selected by a LAMP primer designing software as a “self-consistent” set based on thermodynamic parameters and a change in a sequence of one of LAMP primer can potentially affect the performance of the whole set.
In the previous attempts to solve the problem of PAM requirement in LAMP/Cas12 detection systems, the focus was on dsDNA segments of LAMPlicons, which were targeted by a gRNA spacer [11,12,13]. However, LAMPlicons have a loop-stem structure, with loops presented by single-stranded DNA (ssDNA) [5,6]. Advantageously, Cas12a nuclease which is mostly combined with LAMP [7,9] can be trans-activated by a protospacer sequence in a single-stranded DNA (ssDNA) in the absence of PAM [10]. Thus, it appears straightforward to direct the gRNA spacer to a sequence in a single-stranded loop rather than to that in a double-stranded stem to make the CRISPR/Cas analysis of LAMPlicons truly PAM-independent. In the frame of such strategy, sequences of the so-called “loop primers” could potentially serve as a basis to design gRNA spacers. Indeed, the routinely used software for selection of LAMP primers can generate loop primers [14] which anneal to ssDNA sequences in the loops and are employed to speed up the LAMP reaction [15]. To date, the practical feasibility of developing PAM-independent LAMP/Cas detection systems by targeting single-stranded loops of LAMPlicons with a Cas12a/gRNA complex has never been experimentally explored.
The aim of the present work was to test whether a truly PAM-independent LAMP/Cas12a detection system can be developed by targeting single-stranded loops of LAMPlicons with gRNA. The species of Clavibacter genus [16] and a set of primers previously designed and verified by Dobhal et al. [17] to detect all known Clavibacter species by LAMP have been selected as a convenient model for the study. The advantage of the chosen LAMP primer set is that it produces LAMPlicons with a loop sequence differing by single nucleotide substitutions among some Clavibacter species. That additionally allowed us to evaluate whether and how different Clavibacter species can be differentiated by fine-turning the length of gRNA spacer directed to a LAMPlicon loop containing nucleotide substitutions. Aside from being a convenient experimental model for our study, Clavibacter species are well-known bacterial plant pathogens causing extremely harmful diseases of agriculturally important plants, such as bacterial wilt and canker of tomatoes (C. michiganensis; Cm), potato ring rot (C. sepedonicus; Cs), wilting and stunting of alfalfa (C. insidiosus; Ci), wilt and blight of corn (C. nebraskensis; Cn), leaf spots and leaf freckles in wheat (C. tessellarius; Ct), leaf yellowing in beans (C. phaseoli; Cp) [16]. Cm, Cs, and Ci belong to a list of quarantine pathogens in the European Union and some other countries [16]. The potential results of the study could serve as a basis for development of assays aimed at their on-site diagnostics to control these agricultural plant diseases and prevent the pathogens spread.
2. Results
2.1. Sensitivity and Selectivity of LAMP Detection
The overall analytical performance of the LAMP/Cas12a system is determined by that of its constituents, viz. LAMP and the subsequent Cas12a-based detection of specific LAMPlicons. As a first step, the analytical performance of LAMP with the primer set developed by Dobhal et al. for detection of all Clavibacter species [17] was assessed under our experimental conditions in terms of limit of detection (LOD). For that, serial 10-fold dilutions of Cs genomic DNA (strain As-1405, Table S1) were used, allowing for loads of 37 pg down to 3.7 fg of genomic DNA per a 10 µL LAMP reaction. Since the size of Cs genome is 3.4·106 base pairs (bp) [18], that would correspond to the loads of 104 copies to 1 copy of bacterial genome per reaction. The number of genomes was calculated based on the mass of a single genome m equal to about 3.7 fg that was estimated as follows: m (g) = 650·3.4·106 / NA, where 650 is the average molecular weight of bp (g/mol) and NA = 6.02·1023 mol–1 is Avogadro’s number. The representative amplification curves are demonstrated in Figure 1a. The Cs genomic DNA was consistently detected down to the load of 37 fg (10 genome copies) per reaction within the time interval of 30 min when the full set of LAMP primers (including loop primers) was used (Table S2). For other Clavibacter species under study (Cm, Ci, Cn, Ct, and Cp; Table S1), the load of 37 fg of genomic DNA per LAMP reaction also consistently resulted in an occurrence of sigmoidal amplification curves with the approximately the same time of fluorescence rise as for Cs (exemplified in Figure 1b). The similar results for Cs were obtained when 200 ng of potato DNA were additionally present in LAMP reaction (Figure S1). When amplification kinetics was quantified for purposes of comparison by using “characteristic amplification time” (tc), taken as time when fluorescence reaches a half of the maximal level at the plateau, no statistically significant differences (p ≥ 0.119) were found between mean tc values obtained for the same loads of bacterial DNA in the absence and presence of potato DNA (Table S3). Though both in the absence and presence of potato DNA the mean tc values gradually decreased with the DNA load, the statistically significant difference (p = 0.0496) was found only between the minimal (10 copies) and maximal (104 copies) DNA loads in the presence of potato DNA. In the absence of potato DNA, all mean tc values were found statistically indistinguishable (p ≥ 0.07).
By extending the reaction time up to 60 min, a typical sigmoidal amplification curve was observed for a load of 3.7 fg (1 copy of Cs genome) per reaction in 2 out of 5 repetitions, both in the presence and absence of 200 ng of potato DNA (data not shown). The similar result was also obtained at that DNA load for other Clavibacter species under study. When the reaction time was increased up to 90 min for the load of 3.7 fg of Cs DNA per reaction, the result was the same: in 3 repetitions out of 5 no sigmoidal amplification curves were still observed. The observed irreproducibility in the occurrence of sigmoidal fluorescence rise even within the extended 90 min reaction time interval makes the detection of bacterial DNA at the load of 3.7 fg unreliable. Consequently, 10 genome copies have to be taken as LOD for detection of Clavibacter species by LAMP under our experimental conditions (for reaction times of up to 90 min). No rise of fluorescence was observed for no template control of LAMP (NTC LAMP, an aliquot of 1×LAMP buffer instead of DNA sample) in all cases.
To evaluate the LAMP selectivity under our experimental conditions, species of other genera were tested. No increase of fluorescence within the amplification time of 30 min was observed for strains representing species of Pectobacterium, Dickeya, and Escherichia (Table S1), even at a relatively high load of 37 pg per reaction, except for P. odoriferum, for which a beginning of fluorescence rise was observed. When amplification time has been extended to 60 min, the sigmoidal amplification curves were observed for a number of Pectobacterium and Dickeya species at that load (Figure S2).
As known, LAMP reaction is able to proceed without loop primers but can be speeded up by their presence [15]. Indeed, in the absence of loop primers LF and LB (Table S2), the amplification was about two to three times slower (exemplified in Figure S3). Interesting, while the sequence of LAMPlicon loop targeted by primer LB (loop B) was identical in the studied Clavibacter species, the sequence of loop F (the loop targeted by primer LF) for Cs differed from that in other Clavibacter species by 2 to 3 single nucleotide substitutions (Table 1). For Ct, the sequence of the “loop F annealing site” (the section of loop F to which LF anneals) differs by a single nucleotide from that in other Clavibacter species, except for Cs (Table 1). Yet, these differences did not result in an appreciable discrepancy of amplification times (Figure 1b), thus not allowing to reliably differentiate Clavibacter species among themselves with LAMP alone.
2.2. The Loop-Targeted Cas12a Analysis of LAMP Products
Figure 2a shows representative kinetic curves for a cleavage of FAM-MRs (FAM-labeled molecular reporters, Table S2) by Cas12a nuclease from the bacterium Francisella novicida (FnCas12a), activated by LAMP products (1 μL of completed LAMP reaction). The products were recognized by gRNA-B which targets the loop B and designed based on the sequence of primer LB (Table 1, Table 2, and Table S2). As seen, the increase of fluorescence was observed for LAMP products of all species of Clavibacter genus under study. Also, LAMPlicons generated by both using and not using loop primers were recognized by FnCas12a/gRNA-B complex (Figure 2b). Although the kinetic curves are provided for LAMP products generated at a load of 37 fg of Clavibacter genomic DNA per reaction (Figure 2a), the results can be extended to higher loads as well. Indeed, the amplification curves (exemplified by Figure 1) demonstrated approximately equal level of fluorescence for the DNA intercalating fluorescent dye EvaGreen at the reaction saturation plateau, thus suggesting that nearly the same amount of LAMPlicons is generated by the end of LAMP reaction, regardless of DNA load.
In contrast to Clavibacter species, LAMP products generated with genomic DNA from the tested species of other genera (at the extended amplification time as exemplified by Figure S2) were not recognized by the FnCas12a/gRNA-B complex as specific LAMPlicons (Figure 2a). The similar results were obtained for non-specific LAMP products generated in the absence of loop primers (data not shown). In all cases, no appreciable rise of fluorescence was observed for controls (aliquots of LAMP buffer instead of those of completed LAMP reactions) and NTC LAMP.
To demonstrate the utility of the suggested approach for detection of bacteria in potato tuber tissue, DNA extracted from the potato samples artificially contaminated with Cs was examined with the commercial real-time PCR test for Cs detection and the developed PAM-independent LAMP/Cas12a system. For all examined samples, there was a complete concordance between PCR results and results obtained with the PAM-independent LAMP/Cas12a detection system (Table S4), taking into account that sensitivity of the developed system was limited by that of LAMP and equal to 10 Cs genome copies per reaction.
The sequence of loop F in LAMPlicons generated with Cs genomic DNA is expected (based on sequences retrieved from NCBI databases) to differ from that of other Clavibacter species under study (Table 1). To examine whether this difference can be utilized to differentiate Cs from Ct, Cm, Ci, Cn, and Cp by means of Cas12a analysis of LAMPlicons, a series of gRNAs has been designed, with spacers of varying length and sequences identical to that of loop F segments of Cs LAMPlicons (gRNAs-F, Table 1 and Table 2). As seen from Figure 3, the complex of FnCas12a/gRNA-F-20 (gRNA with a 20 nucleotide (nt) long spacer designed in part from the sequence of primer LF; Table 1, Table 2 and Table S2) can specifically recognize LAMPlicons produced from Cs genomic DNA that results in a notably different kinetics of MR cleavage. No rise of fluorescence was observed for LAMP products generated with genomic DNA from species of other genera, similar to that as in the case of the FnCas12a/gRNA-B complex (Figure 2a).
For convenience of presentation, the differences in MR cleavage kinetics for Clavibacter strains are further analyzed using the initial cleavage rate V0 (defined as a slope of an approximately linear segment of kinetic curves in the beginning of a kinetic curve; the slope was obtained by using a linear regression fit). When the spacer length has been extended to 22 and 24 nt, the selectivity of Cas12a analysis in regard to Cs detection seems to worsen (Figure 4). In contrast, by decreasing the length of the spacer to 16 nt, the selectivity can be improved so that all Clavibacter strains others than those of Cs are more evidently distinguished by the Cas12a analysis with gRNA-F-16 (16 nt long spacer, Table 2). FnCas12a nuclease in a complex with gRNA-F-14 (14 nt long spacer, Table 2) was unable to recognize the protospacer and be trans-activated. It is worth noting that a shortening of the spacer length from 20 to 16 nt steadily decreased the degree of trans-activation of FnCas12a nuclease as manifested by the slower MR cleavage. Still, even at that low cleavage rate, the reliable instrumental detection of the differences was possible (Figure 3, Figure 4, and Figure S4, Table S5). Also, from a statistical point of view, the mean V0 values for Cs strains Ac-1405 and Ac-2753 differ from those for the tested strains of other Clavibacter species for all gRNA-F variants used (p-values were less than 0.05; Table S5). The single exception was observed for the Cas12a/gRNA-F-22 complex which was unable to statistically differentiate the strain Ac-2753 of Cs from Cn by the V0 value (p = 0.164; Table S5). Another interesting (though expected) observation is that 3 mismatches in the spacer/protospacer duplex in the case of Ct (Table 1) had a higher impact on the cleavage kinetics that 2 mismatches as it is for Cm, Cn, Ci, and Cp when gRNA-F20 used. The mean V0 value for Ct was the lowest among Clavibacter species tested, Figure 2C and Figure 3C. The same was kept for the 18 nt long spacer but not for the 16 nt long one (Figure 3 and Figure 4), thus indicating a complex interplay between the spacer length and the number and positions of mismatches.
2.3. Visual Detection of C. sepedonicus in a Single Reaction Tube
To demonstrate that the developed PAM-independent LAMP/Cas12a detection system is compatible with a single reaction tube format, the following procedure has been adopted. First, LAMP was conducted for 45 min in 0.2 mL PCR tube, using a 10 µL reaction volume (under a layer of mineral oil, in a dry block thermostat that simplifies instrumentation requirements). Then, upon LAMP completion, the tube was carefully open (so that not to disturb the LAMP reaction volume preserved under the oil layer) and 50 µL of FnCas12a reaction mixture was placed inside the tube lid. The tube was closed and then hand-shaken or shortly centrifuged to combine the volumes.
However, in such a setup, no trans-activation of FnCas12a nuclease was initially observed. The reason is likely to be the accumulation of relatively large amount of pyrophosphate by the end of LAMP reaction (as a result of extensive DNA synthesis), accompanied by a formation of magnesium pyrophosphate [19]. Since the LAMP reaction volume combined with the Cas12a reaction mixture has increased 10-fold in the single reaction tube format, pyrophosphate could effectively sequester Mg2+ ions away from solution, making them unavailable for complexing with Cas12a nuclease. The formation of Cas12a/Mg2+ complexes is known to be essential for Cas12a trans-activity [20]. Indeed, in our case, the FnCas12a trans-cleavage activity was found to be restored by elevating concentration of Mg2+ ions in the Cas12a reaction mixture from the commonly used 6 mM to 12 mM, apparently reaching a plateau at higher magnesium concentrations (Figure S5). The magnesium concentration of 18 mM has been chosen as the working one for further experiments to ensure that Mg2+ ions are present in excess.
Clearly, the on-site testing could benefit from a visual (naked-eye) detection of MR cleavage by trans-activated Cas12a nuclease. The FAM-labeled MRs are most often employed for both instrumental and naked-eye detection [7,9]. However, in the case of naked-eye detection with FAM-labeled MRs, a source of either ultraviolet or blue light is usually needed to illuminate the test tubes. To further simplify the instrumentation requirements, the naked-eye detection was carried out by employing a colorimetric assay with ROX-labeled MRs (Table S2). The cleavage of such reporters by Cas12a nuclease results in a change of color from blue to purple that can be observed simply under daylight [21] and captured with a smartphone camera. As seen from Figure 5, the concentration of ROX-MR-5 (the 5 nt long ssDNA reporter identical to that of FAM-MR except for the fluorophore and quencher, Table S2) in the Cas12a reaction mixture is crucial for the effective visual detection: only the elevated concentration such as 18 µM, compared to 1 µM which was sufficient for instrumental detection (as exemplified in Figure S5), resulted in a clearly visible change of color.
However, the time required for the color development under such conditions was unsatisfactorily long (more than an hour). To further optimize the naked-eye detection conditions, the concentration of Cas12a/gRNA-B complexes was varied and ROX-MRs of different length (ROX-MR-5 and ROX-MR-8 of 5 and 8 nt long, respectively; Table S2) were tested. The elevation of Cas12a/gRNA-B concentration in the reaction mixture by 3-fold led to about 3-fold increase of V0 (although on the expense of a rise in the background fluorescence level, Figure S6). Figure 6 shows the color change of the reaction mixture with the elevated FnCas12a concentration (180 nM) and 18 µM of either ROX-MR-5 or ROX-MR-8 at various incubation times. For ROX-MR-8, the color change is already obvious in 5 min of incubation, while for ROX-MR-5 – only in 45 min. Interesting, by that time the color of control tube (with NTC LAMP) with ROX-MR-8 became practically indistinguishable from that of tube where LAMP was conducted with Cs genomic DNA (Figure 6).
The LAMP/Cas12a analysis in a single test tube format under optimized conditions of the naked-eye detection was applied to discriminating Cs from other Clavibacter species under study, using ROX-MR-8 cleavage with the Cas12a/gRNA-F-20 complex. As seen from Figure 7, the samples of LAMP conducted with Cs genomic DNA do develop purple color in 5 min after combining with the Cas12a reaction mixture. At the same time, no obvious transition from blue to purple was observed for samples of LAMP conducted with DNA extracted from Clavibacter species other than Cs or for NTC LAMP and the control (Figure 7).
3. Discussion
The requirement for PAM in an amplicon sequence can constitute a significant obstacle to development of assays relying on coupling isothermal amplification with Cas12a-based analysis of amplicons [7,8,9]. The consensus PAM sequence for Cas12a nucleases is “TTTN” [22] and the probability to find such sequence at any given position in genome is about 1%, assuming that the distribution of nucleotides is random and uniform. The PAM requirement imposes additional restrictions on selected LAMP primer sets and may not allow to employ the most effective primers when developing a LAMP/Cas12a-based assay. As mentioned in the Introduction section, the problem can be alleviated by using engineered Cas12a mutants [11,12], but presently they are not easily available to the research community interested in developing of innovative LAMP/Cas12a-based diagnostic tests. The approach relying on introduction of a PAM sequence into LAMPlicons by modifying LAMP primers [13] appears to be more universal. However, LAMP is known to be prone to nonspecific amplification, mostly due to the larger number of LAMP primers (4 to 6) that increases a probability of undesirable primer-primer interactions [23]. The modification of primers by inserting additional sequences can further heighten the chance of such primer-primer interactions. In contrast to strategies realized in [11,12,13], the loop-targeted Cas12a analysis of specific LAMPlicons seems to be also universal, but without a need for a potentially harmful modification of the otherwise effective LAMP primer set. Furthermore, the design of gRNA is simplified since sequences of loop primers can be utilized as a starting point for a selection of gRNA spacer sequence.
One might expect that loop primers, after annealing to complementary sections of LAMPlicon loops and extension, can convert ssDNA into dsDNA. As result, the recognition by the FnCas12a/gRNA complex could become impossible since it would require the presence of PAM [10]. However, in the most LAMP reaction mixtures (including that used in our study), the concentration of loop primers is twice lower than that of inner primers FIP and BIP [23]. The LAMP reaction seems to proceed further after the loop primers are exhausted. As a result, the significant number of LAMPlicons with single-stranded loops are always present among LAMP products, allowing for the PAM-independent activation of Cas12a nuclease (Figure 2b). Besides, LAMP can be conducted without loop primers, although at the expense of longer amplification time [23].
In general, one purpose of integrating LAMP with Cas12a-based analysis of LAMPlicons is to enhance detection selectivity by additionally targeting a DNA sequence different from sequences of primer annealing sites [7,8,9]. In our experimental setup, annealing sites for loop primers were chosen as protospacers when designing gRNA. It should be noted that LAMP with loop primers per se was unable to reliably differentiate Clavibacter species among themselves (Figure 1b) despite the presence of nucleotide substitutions in the annealing site for the loop primer F (Table 1). Apparently, selectivity of LAMP is governed by primers F3, B3, FIP, and BIP, while the loop primers serve to simply speed up the amplification. The loop-targeted Cas12a-based analysis of LAMPlicons obviously enhanced the detection selectivity, allowing (1) to discriminate Clavibacter species from some species of other genera, which produced non-specific amplification with the used LAMP primer set and (2) to reliably distinguish Cs among other Clavibacter species, based on nucleotide substitutions in one of the LAMPlicon loops. As to the former, it is thought that annealing of LAMP primers to off-target sequences, although imperfect, can still generate non-specific LAMP products at large loads of non-target genomic DNA, which differ in loop sequences from specific LAMP products and are unrecognizable by the Cas12a/gRNA complex. It appears that the previously reported selectivity of the used LAMP primer set [17] was achieved via properly adjusted amplification time. Yet, such approach can potentially pose a risk of false positives in practical applications if non-target microorganisms are present in a tested sample in large amounts.
Also, our findings demonstrate that the selectivity of the loop-targeted LAMP/Cas12a detection system can be further enhanced by appropriately adjusting the length of gRNA spacer (Figure 3 and Figure 4). For Cas12a nucleases, the gRNA spacer length is typically 20-24 nt [24]. However, only with spacers of 20 nt to 16 nt in length, the selective detection of Cs has been achieved by targeting the loop with nucleotide substitutions, with the best result for gRNA-F-16 (16 nt long spacer). It is known that mismatches in the spacer/protospacer duplex can alter functional activities of Cas12a nucleases [25,26]. For FnCas12a, a mismatch in the seed region (the first 8-10 nt at the 3′-end of gRNA) was reported to reduce its on-target (cis-) cleavage activity but to increase the trans-cleavage activity [27]. The effect of more than one mismatch in the spacer/protospacer duplex depends in a complex and presently unpredictable manner on the number of mismatches and their positions, as well as the spacer length and even the spacer sequence, and can manifest itself by both increase or decrease in the level of Cas12a trans-cleavage activity [28,29,30]. As a result, the fine-turning of gRNA has to be done on a case-by-case basis. In our case, the shortening of gRNA spacer allowed to specifically distinguish Cs from other Clavibacter species. Clearly, the loop-targeted Cas12a analysis of LAMPlicons has also a potential for fine-turning the detection selectivity by also varying the position of priming site of gRNA spacer in the loop (and, thus, the number of mismatches and the sequence of spacer/protospacer duplex). Although not experimentally tested in the present study, such approach appears possible and straightforward, since the priming of gRNA spacer on ssDNA is not tied to a PAM location. In contrast, the use of Cas12a mutants [11,12] or the introduction of a PAM sequence into a LAMP primer [13] ties the priming of gRNA spacer to a location of either natural or artificial PAMs.
Aside from selectivity, another important characteristic of analytical performance is LOD. Since LAMP produced approximately the same amount of LAMP products at various loads of genomic DNA for the subsequent Cas12a-based analysis of LAMPlicons, namely LAMP determines LOD of the LAMP/Cas12a detection system. Rigorously, LOD is defined as the minimal level of analyte, which can be reliably detected [31]. It is commonly to calculate LOD as a ratio of 3.3× standard deviation for a blank to a slope of the initial linear section of calibration curve [31]. However, we observed a week dependence of the tc-reciprocal (the tc-reciprocal, 1/tc, appears as a more convenient function than tc to build a calibration curve in the case of LAMP) on the target amount over the tested range of DNA loads and their significant scatter at the lower end of the range (Table S3). Such behavior was not surprising and is common for LAMP which is known to be rather a qualitative method than a quantitative one, suitable for applications where a simple qualitative result is sufficient [23]. So, it was hardly possible to reliably determine a linear range of calibration curve. Moreover, the tc value for the blank (no template control) is undefinable. Though for the blank sample the tc-reciprocal can be set to zero (assuming tc = ∞), one cannot exclude that the spurious amplification due to, e.g., low probability primer-primer interactions could be observed for the blank sample at the finite time but beyond the reaction time set. Thus, the standard deviation for the blank cannot be determined in practical terms. Nonetheless, based on the LOD definition as a reliably detectable minimal amount of analyte, we took LOD for LAMP under our experimental conditions as 37 pg of genomic DNA (10 copies of Cs genomes) per reaction. Indeed, that load of genomic DNA consistently provided a sigmoidal amplification curve within 30 min reaction time (overall, in 31 LAMP reactions, using DNA extracted from various Clavibacter species) whereas no amplification was observed within this time period for NTC LAMP in all cases. The LOD of 10 genome copies per reaction is comparable with LODs reported for detection of Clavibacter species in other studies, using nucleic acid amplification methods such as PCR, LAMP, or NASBA (nucleic acid sequence based amplification). The LOD values varied from a few to tens bacterial genomes or from less than one to a few viable bacteria per reaction (when ribosomal RNA was used as a target in NASBA) [32,33,34,35,36,37,38].
It deserves discussing that the LOD value was reported as 1 fg of bacterial DNA per reaction in the original study where the employed set of LAMP primers was designed [17]. According to our calculations, 1 fg of Cs genomic DNA would correspond to about 0.3 copy of bacterial genome per reaction. It is though that a consistent detection at so low target concentrations is hardly possible. Indeed, the probability to have a targeted gene in a sample volume added to a test tube should follow the Poisson distribution and would be significantly below unity at so low DNA concentrations, unless the target gene is present in genome in multiple copies. However, that is not a case for the targeted gene which codes ABC transporter ATP-binding protein/ABC transporter permease [17] and is present in Cs genome as a single copy gene (https://www.ncbi.nlm.nih.gov/nuccore/MZMM00000000). Actually, for 0.3 target copies per reaction at average, the probability not to have the target in a particular tube can be calculated as 0.74, based on Poisson distribution. That means merely 1 positive response in 4 repetitions should be expected. Even if the commercial mixture OptiGene Master Mix from OptiGene (UK), used in [17] to perform LAMP is more effective than our home-made LAMP reaction mixture with recombinant Bst polymerase from Biolabmix (OptiGene Master Mix contains a different strand-displacing polymerase, viz. the OptiGene’s original GspSSD LF DNA polymerase with improved characteristics; https://www.optigene.co.uk/gspssd-lf-dna-polymerase), the so low LOD value appears to be unreachable due to purely stochastic reasons.
Another important point for discussion is a design of the single test tube format, implemented in this work. Rigorously, a single test tube assay assumes that all reactions are carried out in a test tube without its opening, including the assessment of reaction outcome. The reason is to prevent contamination of work area by amplification products. However, FnCas12a nuclease cannot tolerate temperature used to conduct LAMP and has to be added post-reaction. Also, the LAMP reaction volume is commonly small (10 µL in our case) and can evaporate at 65 °C when a dry block thermostat with no lid heating is used. As a way to solve both these problems, we employed a technical trick previously widely practiced in PCR. Namely, we covered a LAMP reaction mixture with a layer of mineral oil prior to starting the amplification reaction. After the reaction was completed, the tube was opened, the Cas12a reaction mixture was placed inside the tube lid, and the tube was again closed. Such procedure allowed us to avoid any pipetting of the reaction volume after LAMP is completed, including a transfer of LAMP products to another test tube, that is a major cause of aerosol contamination. Moreover, the top layer of mineral oil is immiscible with the aqueous LAMP reaction mixture and served as a “liquid cap”. Thus, both reactions were subsequently conducted inside a single test tube while the presence of “liquid cap” ensured that no potential aerosol contamination of work area by LAMPlicons could occur upon the tube opening.
Though Clavibacter species were used, first of all, as a convenient experimental model, the obtained results provide a good ground for development of practical assays for on-site detection of Clavibacter species as a genus or as a single species. For the later, the potential of the fine-turning of gRNA spacer by varying its priming site in the loop F may be explored. In particular, we demonstrated that Cs can be successfully detected in potato tuber samples by combining LAMP and the loop-targeted Cas12a analysis of LAMPlicons with LOD, comparable to that of a commercial PCR kit (Table S4). That was achieved by simply varying the length of gRNA-F spacer. Furthermore, the compatibility of the developed loop-targeted LAMP/Cas12a system with testing in a format of a single test tube assay and the naked-eye detection under daylight was clearly demonstrated.
4. Materials and Methods
4.1. Reagents
Bst polymerase and 10× LAMP buffer (300 mM Tris-HCl, pH 8.9, 50 mM (NH4)2SO4, 0.5 mg/mL bovine serum albumin, 2% Twin-20) were from Biolabmix (Novosibirsk, Russia; https://biolabmix.ru/en/). The DNA oligonucleotides used, including LAMP primers developed in [17], were synthesized and HPLC purified by Lumiprobe (Moscow, Russia, https://ru.lumiprobe.com) and are listed in Table S2. 100 mM dNTPs in water were supplied by Evrogen (Moscow, Russia; https://evrogen.com/). The EvaGreen fluorescent dye (20× stock solution in water) was purchased from Lumiprobe. Agar, yeast extract, and casein-peptone were from Becton Dickinson ( Franklin Lakes, NJ, USA), glucose – from Fluka (Buchs, Switzerland), PCR-grade mineral oil, RNase A, and lysozyme were from Merck (Rahway, NJ, USA). Other chemicals used were of ACS grade or higher and also received from Merck. Deionized (18 MΩ) Milli-Q water was used to prepare solutions. The recombinant Francisella tularensis CRISPR-nuclease Cas12a (FnCas12a) was expressed and purified as described in [39]. gRNAs (Table 1) were synthesized enzymatically with “TranscriptAid T7 High Yield Transcription Kit” (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instruction, using an equimolar mixture of T7P DNA oligonucleotide and one of the template DNA oligonucleotides (Table S2), and purified as in [39].
4.2. Bacteria Culturing and DNA Isolation
19 different strains representing species of genera Clavibacter (8 strains; 6 species), Pectobacterium (9 strains; 8 species), and Dickeya (2 strains; 2 species), as well as a strain of Escherichia coli were used in the study. The species and host names, strain numbers and strain source, as well as the countries of origin are listed in Table S1. Clavibacter species were cultivated on agar medium at 28 °C. The agar composition was: 20 g/L of agar, 5 g/L of yeast extract, 10 g/L of casein–peptone, 5 g/L of glucose, 5 g/L of NaCl, pH 7.0–7.2. The suspensions of Clavibacter species were prepared by harvesting bacteria from the agar plates and resuspending them in sterile distilled water. Other bacteria were grown at 28 °С as suspensions in LB medium (Dia-M, Moscow, Russia). Bacteria were pelleted by centrifugation and DNA was extracted with the “innuPREP Bacteria DNA Kit” (IST Innuscreen GmbH, Berlin, Germany) in accordance with the manufacturer manual (including lysozyme and RNase A treatments). To extract potato DNA, 100 mg of potato tuber tissue and a spin column-based “SKYSuper Plant Genomic DNA” isolation kit (SkyGen, Moscow, Russia; https://www.skygen.com/) were used. The DNA isolation was carried out following the manufacturer’s protocol. The potato samples artificially contaminated with Cs were prepared by spiking 100 mg potato tuber tissue samples with 10 µL of 10-fold dilutions of Cs suspension in sterile distilled water, followed by DNA extraction with the “SKYSuper Plant Genomic DNA” isolation kit. Since Cs is a Gram-positive bacterium, the extraction procedure was supplemented with the lysozyme treatment, similar to that as recommended for the “innuPREP Bacteria DNA Kit”. DNA concentrations were measured on a Qubit fluorimeter (Thermo Fisher Scientific) with a Qubit dsDNA BR Assay kit (Thermo Fisher Scientific) and DNA preparations were aliquoted and stored at –20 °С until further use. The number of bacterial genomes in DNA preparations from Cs contaminated potato samples were determined by real-time PCR on a DTprime5 thermal cycler (DNA-Technology, Moscow, Russia) with a commercial kit for Cs detection in plant tissue (“Clavibacter michiganensis subsp. sepedonicus-РВ”; Syntol, Moscow, Russia), according to the kit’s instruction and using the standard curve kindly provided by the manufacturer.
4.3. LAMP Procedure
LAMP was conducted in 1× LAMP buffer, supplemented with dNTPs (1.4 mM each) and 6 mM MgSO4. Concentrations of outer (F3 and B3), inner (FIP and BIP), and loop (LF and LB) primers (Table S2) developed in [17] were 0.2 µM, 0.8 µM, and 0.4 µM each, respectively. Amplification was performed for various time periods in a 10 µL reaction volume at a constant temperature of 65 °C, using Bst polymerase (0.32 unit/µL). The reaction was carried out either in a DTprime5 thermal cycler equipped with an optical system for the real-time detection of fluorescence or in a dry block thermostat Bio TDB-100 (BioSan, Riga, Latvia). Where necessary, either EvaGreen fluorescent dye (40-fold dilution of the stock solution) in the reaction mixture or a mineral oil layer (5 µL in volume) on the top of the reaction mixture were present.
4.4. Real-Time Cas12a Cleavage Assay
To carry out the real-time monitoring of FnCas12a cleavage activity induced by LAMP products, FnCas12a nuclease and one of gRNAs (Table 2) were mixed at equimolar concentrations of 60 nM, if not indicated differently, in the trans-cleavage reaction buffer (40 mM Tris-HCl, pH 8.6, 6 mM MgCl2, 1 mM dithiothreitol, 0.4% polyethylene glycol, 0.001% Triton X-100, 40 mM glycine) and incubated for 10 min at room temperature. Afterwards, either fluorescein or rhodamine labeled MRs (FAM-MR and ROX-MR, respectively; Table S2) were added and the volume was adjusted to 50 µL with the reaction buffer so that the final concentration of MRs was 1 µM, unless specified otherwise. 1 µL (or 10 µL where indicated) of the completed LAMP reaction was combined with 50 µL of FnCas12a reaction mixture and fluorescence intensity was measured over time in DTprime5 thermal cycler at a constant temperature of 37 °C. Where necessary, the concentrations of FnCas12a/gRNA complex and Mg2+ ions in the trans-cleavage reaction buffer varied in the range of 60 to 180 nM and 6 to 18 mM, respectively.
4.5. A Single Test Tube Assay
To conduct assay in a single test tube format, LAMP was carried out for 40 min in the dry block thermostat Bio TDB-100 in 10 µL reaction volume covered with a layer of mineral oil (5 µL), using a 0.2 mL PCR tube. Next, the PCR tube was open and 50 µL of FnCas12a reaction mixture was placed inside the tube lid. The tube was closed and hand-shaken or shortly centrifuged to mix the volumes. The MR cleavage reaction was carried in the dry block thermostat Bio TDB-100. In the latter case, the result of ROX-MRs cleavage was monitored by a visual observation of test tubes under daylight. The results were documented with a smartphone camera (Samsung Galaxy S8; Samsung Electronics, Seoul, Republic of Korea).
4.6. Statistical Treatment
The performance of LAMP and CRISPR/Cas12a detection system was evaluated based on results of 3 to 5 independent experiments. Arithmetic means, standard deviations, and confidence level p-values were calculated using Microsoft Excel’s statistical functions.
5. Conclusions
The present study demonstrates the feasibility of Cas12a-based analysis of LAMP products in a truly PAM-independent fashion by targeting sections of single-stranded LAMPlicon loops. The specific LAMP products generated both with and without loop primers were shown to be effectively recognized by the FnCas12a/gRNA complex in the absence of PAM. While sensitivity of the LAMP/FnCas12a detection system in regard to Clavibacter species was determined in practical terms by that of LAMP per se and can be taken as 10 copies of bacterial genome per reaction, the detection selectivity is enhanced by the loop-targeted Cas12a analysis of LAMPlicons. Furthermore, as shown on the example of Clavibacter species, the loop-targeted LAMP/FnCas12a detection system can be fine-tuned by carefully designing the gRNA spacer so to selectively discriminate one Clavibacter species from others, based on the presence of single nucleotide substitutions in a targeted LAMPlicon loop. The developed PAM-independent LAMP/FnCas12a detection system can be adapted for use as a single test tube assay with options for both the instrumental and visual (under daylight) detection. The overall time of the assay in a single test tube format with visual detection did not exceed 1 hour. Our findings relax the selection of LAMP primers compatible with Cas12a analysis of LAMPlicons, allowing to use the most effective primer sets, and widen the palette of the present strategies for designing PAM-independent LAMP/Cas12a detection systems with a potential for on-site testing.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, S.P.R.; methodology, S.P.R., K.G.P. and L.K.K.; validation, L.K.K. and O.S.T.; formal analysis, E.V.S. and O.S.T.; investigation, K.G.P., S.A.K., L.K.K. and D.D.M.; resources, A.V.L.; writing—original draft preparation, S.P.R. and E.V.S.; writing—review and editing, A.V.L.; visualization, S.A.K.; supervision, S.P.R. and A.V.L.; project administration, A.V.L.; funding acquisition, A.V.L. All authors have read and agreed to the published version of the manuscript.
Funding
The study was performed within the framework of the Program for Basic Research in the Russian Federation for a long-term period (2021–2030) (No. 122030100170-5).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Acknowledgments
We are thankful to Mikhail G. Divashuk (All-Russia Research Institute of Agricultural Biotechnology) for kindly providing us with a strain of D. chrysonthemi from the German Collection of Microorganisms.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Liu, Q.; Jin, X.; Cheng, J.; Zhou, H.; Zhang, Y.; Dai, Y. Advances in the application of molecular diagnostic techniques for the detection of infectious disease pathogens (Review). Mol. Med. Rep. 2023, 27, 104. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Abd El Wahed, A. Point-Of-Care or Point-Of-Need Diagnostic Tests: Time to Change Outbreak Investigation and Pathogen Detection. Trop. Med. Infect. Dis. 2020, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.R.; Carson, M.S.; Lao, A.; Maran, K.; Yang, E.J.; Kamei, D.T. Point-of-Need Diagnostics for Foodborne Pathogen Screening. SLAS Technol. 2021, 26, 55–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, S.; Kim, B.N.; Kwon, O.S.; Rho, W.-Y.; Jun, B.-H. Emerging ultrafast nucleic acid amplification technologies for next-generation molecular diagnostics. Biosens. Bioelectron. 2019, 141, 111448. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Zhang, H.; Han, X.; Liu, Z.; Lu, Y. Advancements and applications of loop-mediated isothermal amplification technology: A comprehensive overview. Front. Microbiol. 2024, 15, 1406632. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, 63e. [Google Scholar] [CrossRef] [PubMed]
- Atceken, N.; Yigci, D.; Ozdalgic, B.; Tasoglu, S. CRISPR-Cas-Integrated LAMP. Biosensors (Basel) 2022, 12, 1035. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhou, C.; Gao, B.; Liu, F.; Ma, L.; Guo, F. Engineering CRISPR/Cas12-based biosensors: Recent advances and future perspectives. TrAC Trends Anal. Chem. 2025, 191, 118296. [Google Scholar] [CrossRef]
- Khmeleva, S.A.; Ptitsyn, K.G.; Kurbatov, L.K.; Timoshenko, O.S.; Suprun, E.V.; Radko, S.P.; Lisitsa, A.V. Biosensing platforms for DNA diagnostics based on CRISPR/Cas nucleases: Towards the detection of nucleic acids at the level of single molecules in non-laboratory settings. Biomed. Khim. 2024, 70, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Goswami, H.N.; Whyms, C.T.; Sridhara, S.; Li, H. Structural principles of CRISPR-Cas enzymes used in nucleic acid detection. J. Struct. Biol. 2022, 214, 107838. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Liu, Y.; Yang, M.; Zheng, J.; Liu, C.; Ye, W.; Song, S.; Bai, T.; Song, C.; Wang, M.; Shi, J.; Wan, R.; Zhang, K.; Hao, P.; Feng, J.; Zheng, X. The engineered CRISPR-Mb2Cas12a variant enables sensitive and fast nucleic acid-based pathogens diagnostics in the field. Plant Biotechnol. J. 2023, 21, 1465–1478. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Guo, J.; Guo, X.; Li, Q.; Li, X.; Sun, Z.; Zhao, Z.; Weng, J.; Wu, J.; Zhang, R.; Li, B. Fast and visual detection of nucleic acids using a one-step RPA-CRISPR detection (ORCD) system unrestricted by the PAM. Anal. Chim. Acta 2023, 1248, 340938. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, R.; Zhang, H.; Wang, J.; Lu, Y.; Zhang, D.; Yang, L. PAM-free loop-mediated isothermal amplification coupled with CRISPR/Cas12a cleavage (Cas-PfLAMP) for rapid detection of rice pathogens. Biosens. Bioelectron. 2022, 204, 114076. [Google Scholar] [CrossRef] [PubMed]
- Ptitsyn, K.G.; Khmeleva, S.A.; Kurbatov, L.K.; Timoshenko, O.S.; Suprun, E.V.; Radko, S.P.; Lisitsa, A.V. Lamp Primer Designing Software: The Overview. Biomed. Chem. Res. Meth. 2024, 7, e00226. [Google Scholar] [CrossRef]
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 2002, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Eichenlaub, R.; Gartemann, K.-H.; Burger, A. Clavibacter michiganensis, a group of gram-positive phytopathogenic bacteria. In Plant-Associated Bacteria; GnanamanickamS.S.Springer International Publishing: Dordrecht, The Netherlands, 2006; pp. 385–421. ISBN 978-1-4020-4536-3. [Google Scholar] [CrossRef]
- Dobhal, S.; Larrea-Sarmiento, A.; Alvarez, A.M.; Arif, M. Development of a loop-mediated isothermal amplification assay for specific detection of all known subspecies of Clavibacter michiganensis. J. Appl. Microbiol. 2019, 126, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Tambong, J.T. Comparative genomics of Clavibacter michiganensis subspecies, pathogens of important agricultural crops. PLoS ONE 2017, 12, e0172295. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C. Making the cut(s): How Cas12a cleaves target and non-target DNA. Biochem. Soc. Trans. 2019, 47, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Xie, L.; Yang, T.; Huo, Z.; Liu, G.; Liu, Y.; Xiong, W.; Zeng, Z. Naked-eye on-site detection platform for Pasteurella multocida based on the CRISPR-Cas12a system coupled with recombinase polymerase amplification. Talanta 2023, 255, 124220. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Kancharla, N.; Javalkote, V.S.; Dasgupta, S.; Brutnell, T.P. CRISPR-Cas12a (Cpf1): A Versatile Tool in the Plant Genome Editing Tool Box for Agricultural Advancement. Front. Plant Sci. 2020, 11, 584151. [Google Scholar] [CrossRef] [PubMed]
- Moehling, T.J.; Choi, G.; Dugan, L.C.; Salit, M.; Meagher, R.J. LAMP Diagnostics at the Point-of-Care: Emerging Trends and Perspectives for the Developer Community. Expert Rev. Mol. Diagn. 2021, 21, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Montoya, G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 2020, 43, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; van der Oost, J.; Jinek, M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell. 2017, 66, 221–233.e4. [Google Scholar] [CrossRef] [PubMed]
- Strohkendl, I.; Saifuddin, F.A.; Rybarski, J.R.; Finkelstein, I.J.; Russell, R. Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell. 2018, 71, 816–824.e3. [Google Scholar] [CrossRef] [PubMed]
- Baranova, S.V.; Zhdanova, P.V.; Pestryakov, P.E.; Chernonosov, A.A.; Koval, V.V. Key thermodynamic characteristics of Cas9 and Cas12a endonucleases’ cleavage of a DNA substrate containing a nucleotide mismatch in the region complementary to RNA. Biochem. Biophys. Res. Commun. 2025, 768, 151892. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Jiang, J.; Li, Q.; Yao, P.; Jiang, J.; Gong, L.; Dong, Y.; Tu, B.; Wang, R.; Tang, H.; Yao, F.; Wang, F. Iterative crRNA design and a PAM-free strategy enabled an ultra-specific RPA-CRISPR/Cas12a detection platform. Commun. Biol. 2024, 7, 1454. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Smith, B.M.; Jain, P.K. Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection. Nat. Commun. 2020, 11, 4906. [Google Scholar] [CrossRef] [PubMed]
- Rananaware, S.R.; Vesco, E.K.; Shoemaker, G.M.; Anekar, S.S.; Sandoval, L.S.W.; Meister, K.S.; Macaluso, N.C.; Nguyen, L.T.; Jain, P.K. Programmable RNA detection with CRISPR-Cas12a. Nat. Commun. 2023, 14, 5409. [Google Scholar] [CrossRef] [PubMed]
- Taleuzzaman, M. Limit of Blank (LOB), Limit of Detection (LOD), and Limit of Quantification (LOQ). Org. Med. Chem. Int. J. 2018, 7, 127–131. [Google Scholar] [CrossRef]
- Pastrik, K.-H. Detection of Clavibacter michiganensis subsp. sepedonicus in Potato Tubers by Multiplex PCR with Coamplification of Host DNA. Eur. J. Plant Pathology 2000, 106, 155–165. [Google Scholar] [CrossRef]
- Gudmestad, N.C.; Mallik, I.; Pasche, J.S.; Anderson, N.R.; Kinzer, K. A Real-Time PCR Assay for the Detection of Clavibacter michiganensis subsp. sepedonicus Based on the Cellulase A Gene Sequence. Plant Dis. 2009, 93, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Park, D.H.; Namgung, M.; Ahn, T.-Y.; Park, D.S. Validation and Application of a Real-time PCR Protocol for the Specific Detection and Quantification of Clavibacter michiganensis subsp. sepedonicus in Potato. Plant Pathol. J. 2015, 31, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Sagcan, H.; Turgut Kara, N. Detection of Potato ring rot Pathogen Clavibacter michiganensis subsp. sepedonicus by Loop-mediated isothermal amplification (LAMP) assay. Sci. Rep. 2019, 9, 20393. [Google Scholar] [CrossRef] [PubMed]
- van Beckhoven, J.R.C.M.; Stead, D.E.; van der Wolf, J.M. Detection of Clavibacter michiganensis subsp. sepedonicus by AmpliDet RNA, a new technology based on real time monitoring of NASBA amplicons with a molecular beacon. J. Appl. Microbiol. 2002, 93, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Khmeleva, S.A.; Kurbatov, L.K.; Ptitsyn, K.G.; Timoshenko, O.S.; Morozova, D.D.; Suprun, E.V.; Radko, S.P.; Lisitsa, A.V. Detection of Potato Pathogen Clavibacter sepedonicus by CRISPR/Cas13a Analysis of NASBA Amplicons. Int. J. Mol. Sci. 2024, 25, 12218. [Google Scholar] [CrossRef] [PubMed]
- Suprun, E.V.; Ptitsyn, K.G.; Khmeleva, S.A.; Kurbatov, L.K.; Shershov, V.E.; Kuznetsova, V.E.; Chudinov, A.V.; Radko, S.P.; Lisitsa, A.V. Loop-mediated isothermal amplification with tyrosine modified 2′-deoxyuridine-5′-triphosphate: Detection of potato pathogen Clavibacter sepedonicus by direct voltammetry of LAMPlicons. Microchem. J. 2025, 212, 113458. [Google Scholar] [CrossRef]
- Kurbatov, L.K.; Radko, S.P.; Khmeleva, S.A.; Ptitsyn, K.G.; Timoshenko, O.S.; Lisitsa, A.V. Application of DETECTR for Selective Detection of Bacterial Phytopathogen Dickeya solani Using Recombinant CRISPR-Nuclease Cas12a Obtained by Single-Stage Chromatographic Purification. Appl. Biochem. Microbiol. 2024, 60, 17–25. [Google Scholar] [CrossRef]
Figure 1.
Detection of Clavibacter species by LAMP. Representative amplification curves (fluorescence in arbitrary units, a.u.). (a) LAMP conducted with Cs genomic DNA (stain 1405). Curve 1 – NTC (no template control), curves 2, 3, 4, 5, and 6 – Cs DNA loads of 3.7 fg, 37 fg, 370 fg, 3.7 pg, and 37 pg per reaction, respectively. (b) LAMP conducted with 37 fg of genomic DNA of Clavibacter species per reaction. Curve 1 – NTC; curves 2 and 3 – strains Ac-1403 and Ac-1144 of Cm, respectively; curves 4 to 6 – Cn, Cp, and Ct, respectively; curves 7 and 8 – strains Ac-2753 and Ac-1405 of Cs, respectively; curve 9 – Ci.
Figure 1.
Detection of Clavibacter species by LAMP. Representative amplification curves (fluorescence in arbitrary units, a.u.). (a) LAMP conducted with Cs genomic DNA (stain 1405). Curve 1 – NTC (no template control), curves 2, 3, 4, 5, and 6 – Cs DNA loads of 3.7 fg, 37 fg, 370 fg, 3.7 pg, and 37 pg per reaction, respectively. (b) LAMP conducted with 37 fg of genomic DNA of Clavibacter species per reaction. Curve 1 – NTC; curves 2 and 3 – strains Ac-1403 and Ac-1144 of Cm, respectively; curves 4 to 6 – Cn, Cp, and Ct, respectively; curves 7 and 8 – strains Ac-2753 and Ac-1405 of Cs, respectively; curve 9 – Ci.
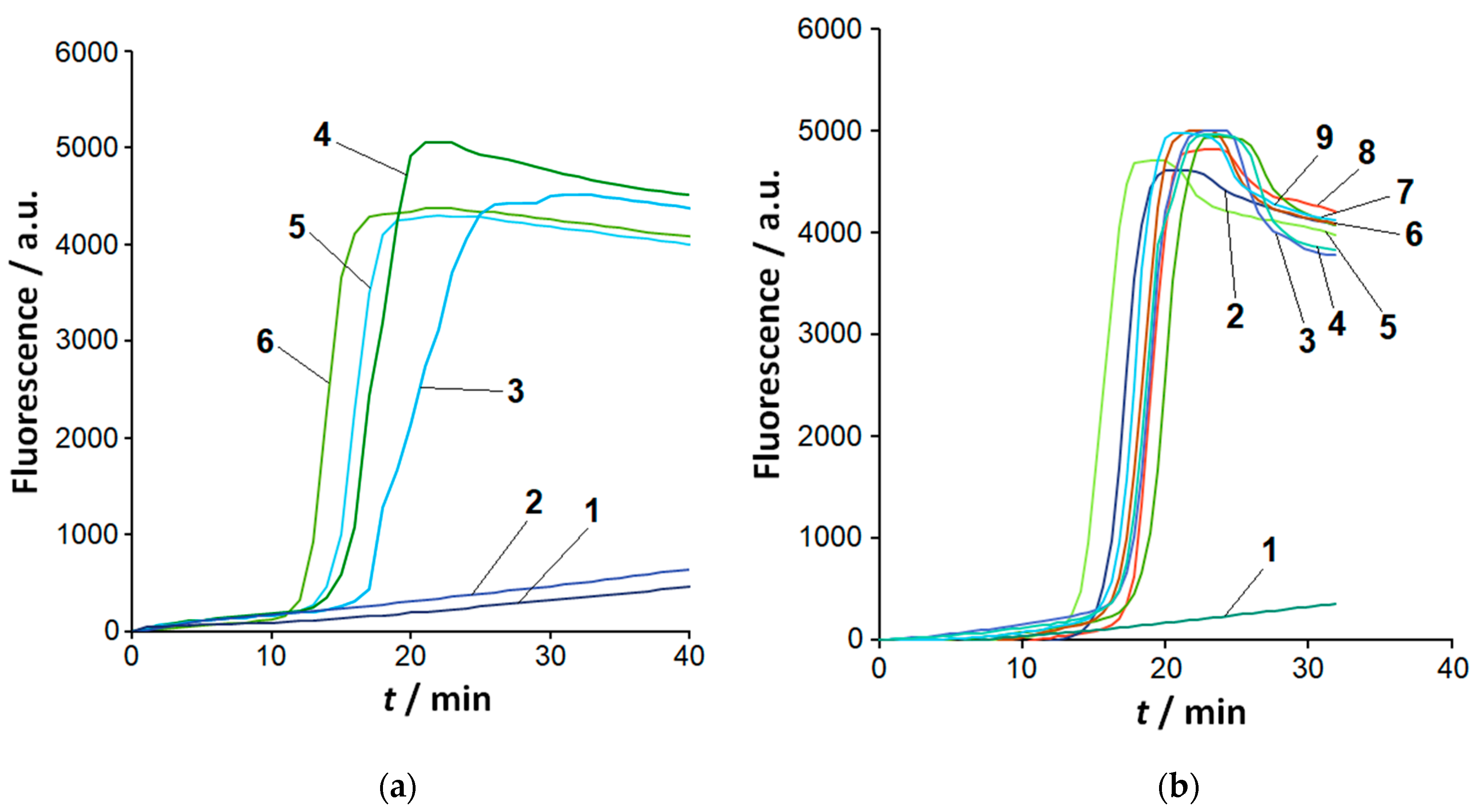
Figure 2.
Analysis of LAMP products with the FnCas12a/gRNA-B complex. Representative kinetic curves of FAM-MR cleavage by FnCas12a nuclease. 1 µL of the completed LAMP reaction, 1 µM of FAM-MR, 60 nM of FnCas12a/gRNA-B. (a) Different Clavibacter species. Curves 1 to 8 – LAMP products for Cn, Cp, Ct, Ci, Cs (strains Ac-2753 and Ac-1405), and Cm (strains Ac-1403 and Ac-1144) genomic DNA, respectively. Curves marked by 9 correspond to species other than Clavibacter (LAMP conducted for 60 min with 37 pg of genomic DNA), NTC LAMP, and control (LAMP buffer instead of completed LAMP reaction). (b) The effect of loop primers on the recognition of LAMPlicons by Cas12a nuclease. Curves 1 and 2 – LAMP products generated with and without loop primers, respectively; curves 3 to 5 – corresponding NTC LAMP and control. LAMP as in Figure S3.
Figure 2.
Analysis of LAMP products with the FnCas12a/gRNA-B complex. Representative kinetic curves of FAM-MR cleavage by FnCas12a nuclease. 1 µL of the completed LAMP reaction, 1 µM of FAM-MR, 60 nM of FnCas12a/gRNA-B. (a) Different Clavibacter species. Curves 1 to 8 – LAMP products for Cn, Cp, Ct, Ci, Cs (strains Ac-2753 and Ac-1405), and Cm (strains Ac-1403 and Ac-1144) genomic DNA, respectively. Curves marked by 9 correspond to species other than Clavibacter (LAMP conducted for 60 min with 37 pg of genomic DNA), NTC LAMP, and control (LAMP buffer instead of completed LAMP reaction). (b) The effect of loop primers on the recognition of LAMPlicons by Cas12a nuclease. Curves 1 and 2 – LAMP products generated with and without loop primers, respectively; curves 3 to 5 – corresponding NTC LAMP and control. LAMP as in Figure S3.
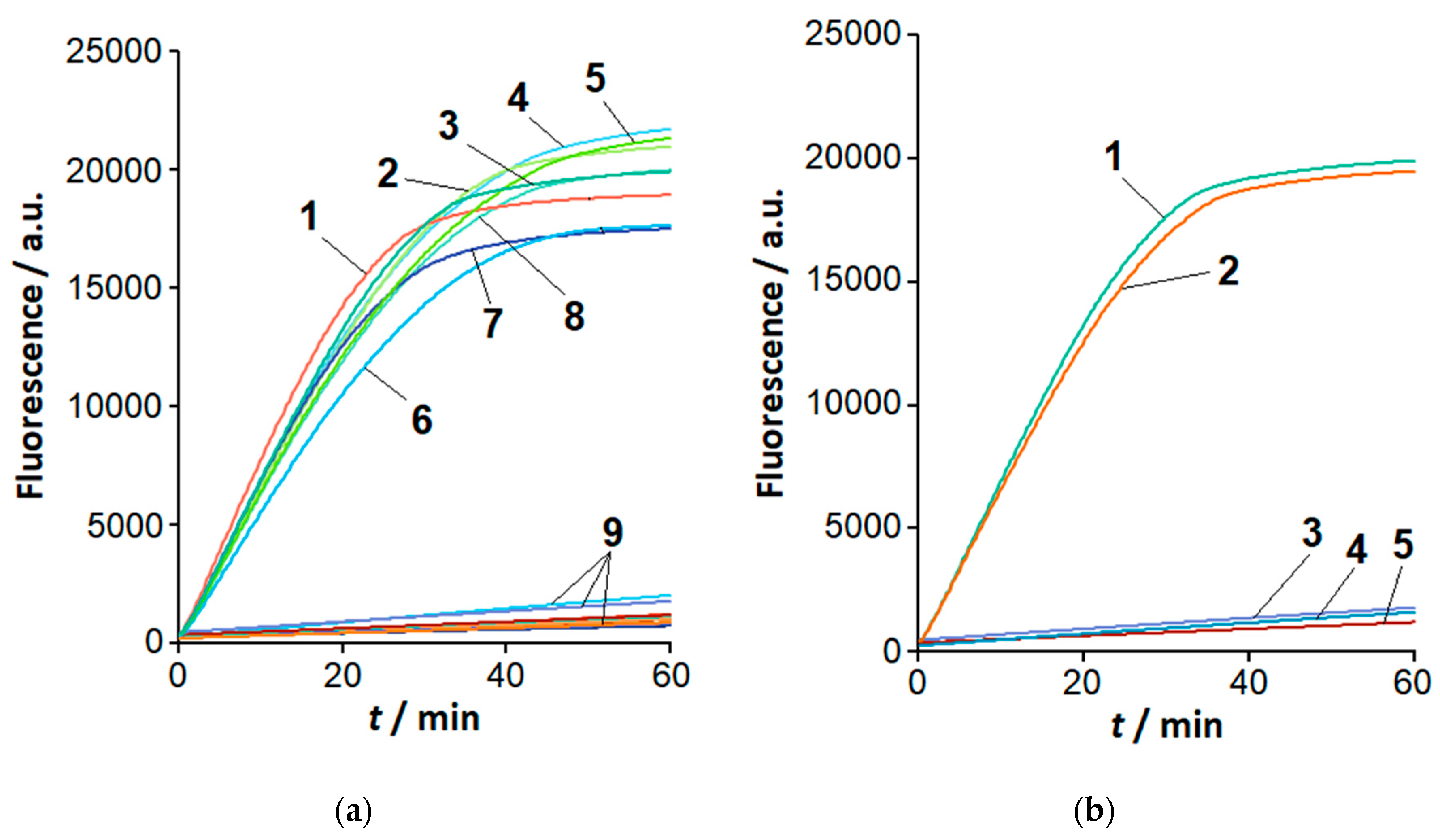
Figure 3.
Analysis of LAMP products with the FnCas12a/gRNA-F-20 complex. Representative kinetic curves of FAM-MR cleavage by FnCas12a nuclease. 1 µL of the completed LAMP reaction, 1 µM of FAM-MR, 60 nM of FnCas12a/gRNA-F-20. Curves 1 to 10 – Cs (Ac-2753 and Ac-1405), Cn, Cm (Ac-1403), Ci, Cp, Cm (Ac-1144), Ct, NTC LAMP, and control, respectively.
Figure 3.
Analysis of LAMP products with the FnCas12a/gRNA-F-20 complex. Representative kinetic curves of FAM-MR cleavage by FnCas12a nuclease. 1 µL of the completed LAMP reaction, 1 µM of FAM-MR, 60 nM of FnCas12a/gRNA-F-20. Curves 1 to 10 – Cs (Ac-2753 and Ac-1405), Cn, Cm (Ac-1403), Ci, Cp, Cm (Ac-1144), Ct, NTC LAMP, and control, respectively.
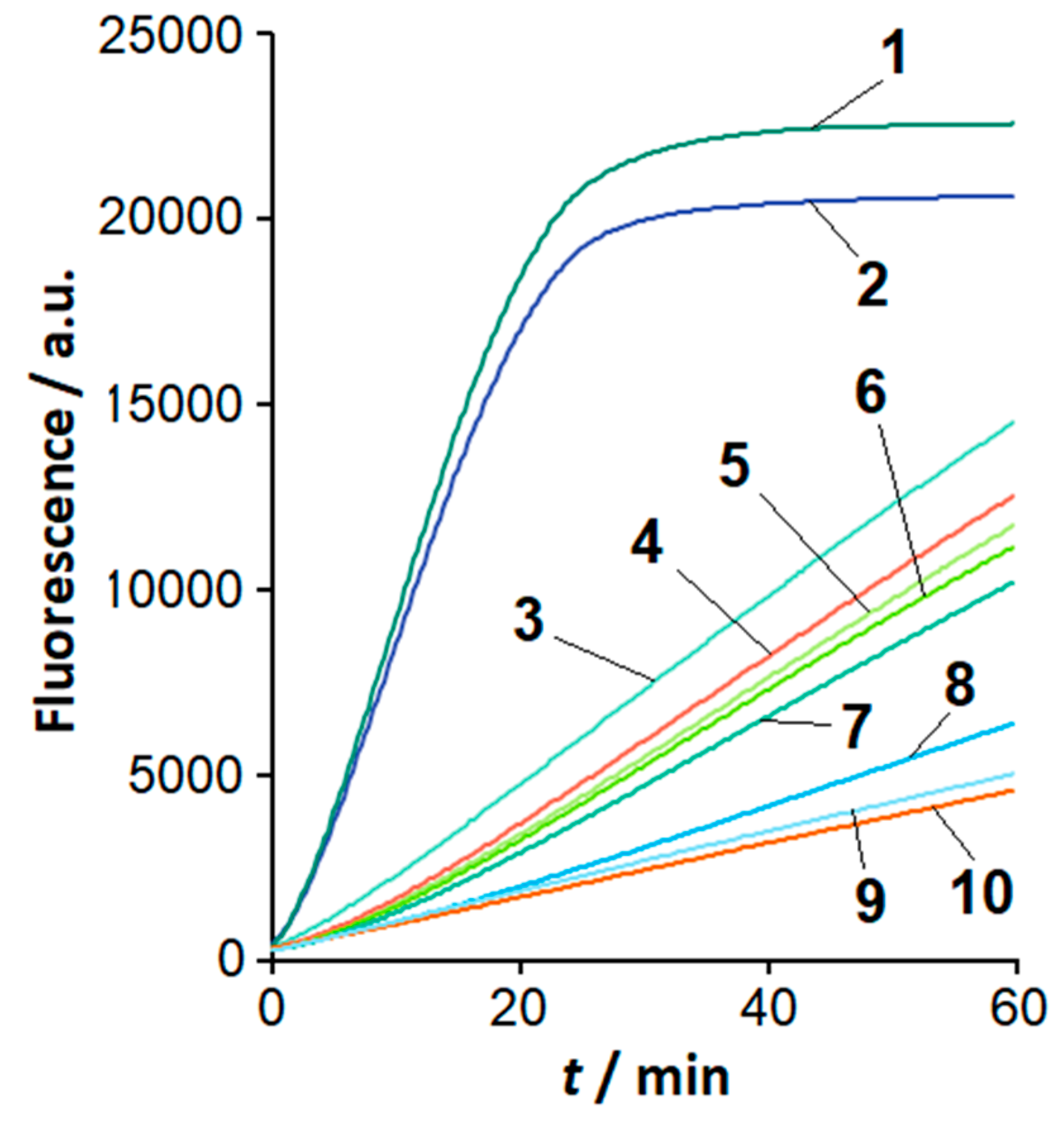
Figure 4.
The initial velocity, V0, of FAM-MR cleavage by Cas12a nuclease for different Clavibacter species and gRNA-F variants. The LAMP and Cas12a analysis conditions as in Figure 3, except for various gRNA-F. Numbers 1 to 10 indicate Cs (Ac-2753 and Ac-1405), Cm (Ac-1403), Cn, Ci, Cp, Cm (Ac-1144), Ct, NTC LAMP, and control, respectively. (a) – gRNA-F-16, (b) – gRNA-F-18, (c) – gRNA-F-20, (d) – gRNA-F-22, (e) – gRNA-F-24.
Figure 4.
The initial velocity, V0, of FAM-MR cleavage by Cas12a nuclease for different Clavibacter species and gRNA-F variants. The LAMP and Cas12a analysis conditions as in Figure 3, except for various gRNA-F. Numbers 1 to 10 indicate Cs (Ac-2753 and Ac-1405), Cm (Ac-1403), Cn, Ci, Cp, Cm (Ac-1144), Ct, NTC LAMP, and control, respectively. (a) – gRNA-F-16, (b) – gRNA-F-18, (c) – gRNA-F-20, (d) – gRNA-F-22, (e) – gRNA-F-24.
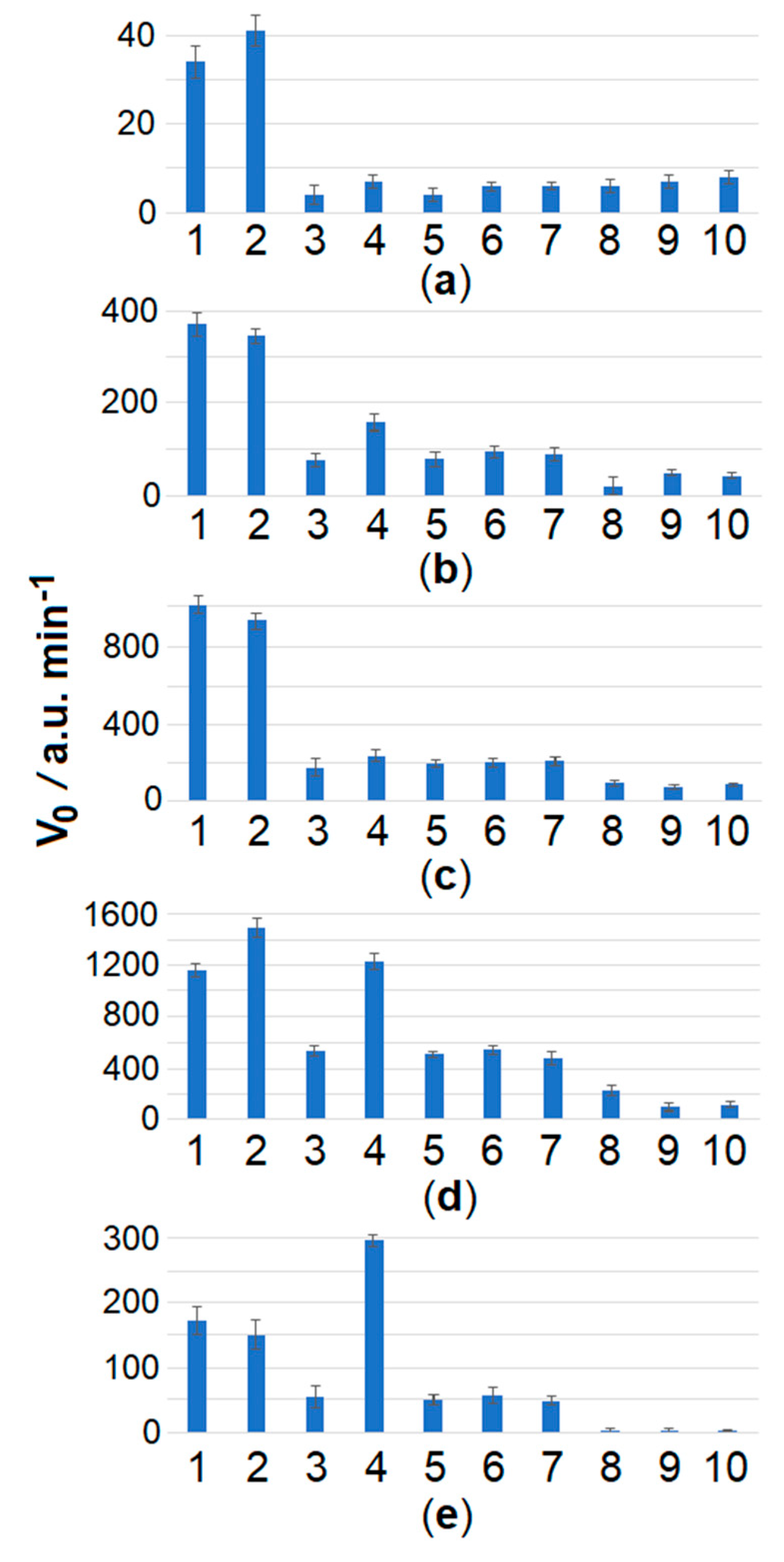
Figure 5.
The color development at various ROX-MR-5 concentrations. The single test tube assay with the naked-eye detection. Various concentrations of ROX-MR-5: 1 µM – tubes 1 and 2, 10 µM – tubes 3 and 4, 18 µM – tubes 5 and 6. LAMP with 37 fg of Cs (Ac-1405) genomic DNA for 45 min; FnCas12a/gRNA-B complex. Tubes 2, 4, and 6 – NTC LAMP.
Figure 5.
The color development at various ROX-MR-5 concentrations. The single test tube assay with the naked-eye detection. Various concentrations of ROX-MR-5: 1 µM – tubes 1 and 2, 10 µM – tubes 3 and 4, 18 µM – tubes 5 and 6. LAMP with 37 fg of Cs (Ac-1405) genomic DNA for 45 min; FnCas12a/gRNA-B complex. Tubes 2, 4, and 6 – NTC LAMP.
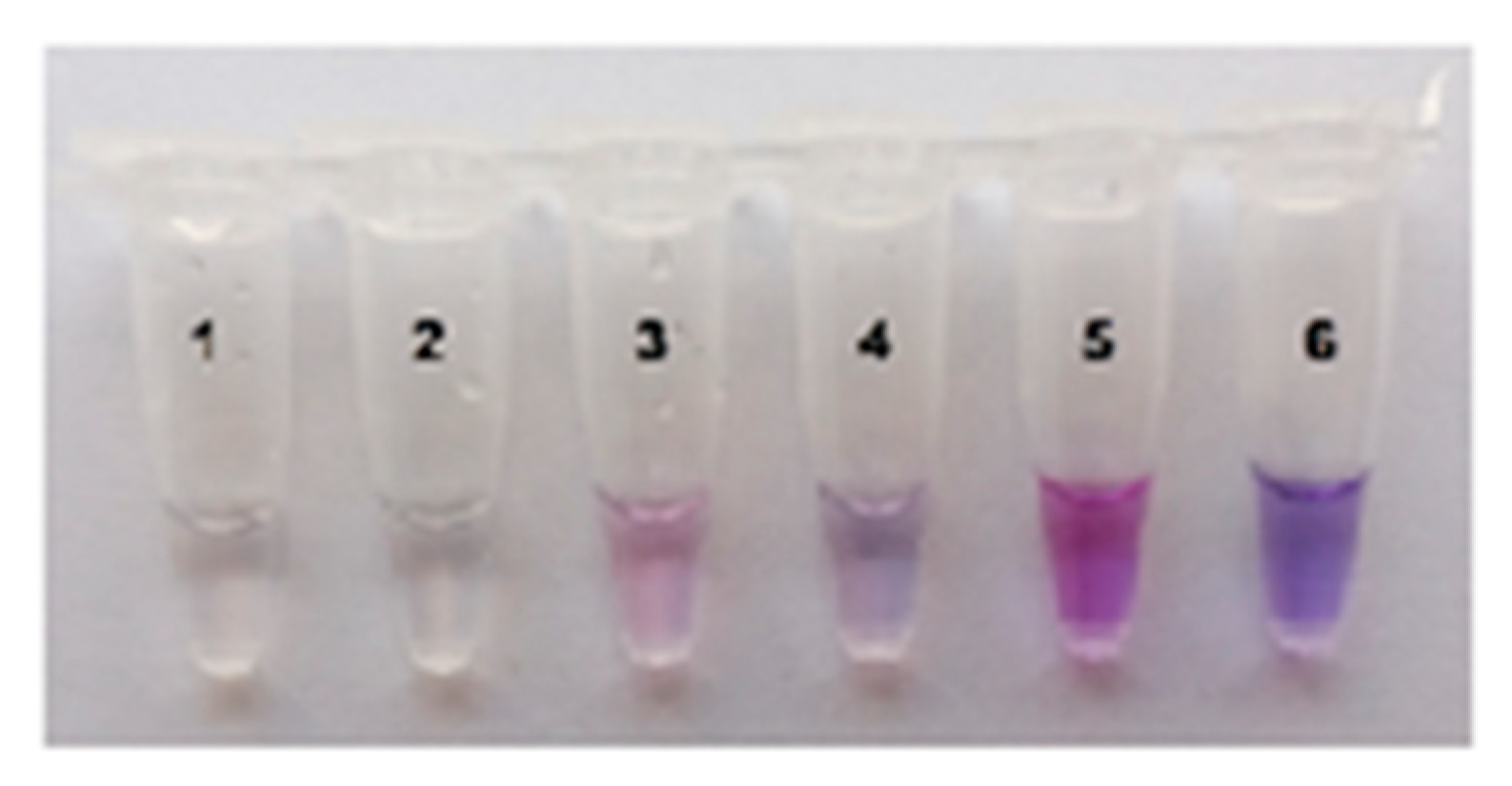
Figure 6.
The color development with time. The single test tube assay with ROX-MR-5 or ROX-MR-8 and naked-eye detection. The incubation time is indicated on the left. ROX-MR concentrations – 18 µM. Cas12a/gRNA-B complex. Cas12a and Mg2+ ions concentrations – 180 nM and 18 mM, respectively. Tubes 1 and 2 – LAMP with 37 fg of Cs (Ac-1405) genomic DNA for 45 min and NTC LAMP, respectively.
Figure 6.
The color development with time. The single test tube assay with ROX-MR-5 or ROX-MR-8 and naked-eye detection. The incubation time is indicated on the left. ROX-MR concentrations – 18 µM. Cas12a/gRNA-B complex. Cas12a and Mg2+ ions concentrations – 180 nM and 18 mM, respectively. Tubes 1 and 2 – LAMP with 37 fg of Cs (Ac-1405) genomic DNA for 45 min and NTC LAMP, respectively.

Figure 7.
Differential detection of Cs with the single test tube assay. LAMP is as in Figure 1b. The other conditions as in Figure 6, except for that the incubation time was 5 min. Test tubes 1 to 11 – Ci, Cm (Ac-1403), Cm (Ac-1144), Cs (Ac-1405), Ct, Cp, Cn, repeat of Cn, Cs (Ac-2753), NTC LAMP, and control, respectively.
Figure 7.
Differential detection of Cs with the single test tube assay. LAMP is as in Figure 1b. The other conditions as in Figure 6, except for that the incubation time was 5 min. Test tubes 1 to 11 – Ci, Cm (Ac-1403), Cm (Ac-1144), Cs (Ac-1405), Ct, Cp, Cn, repeat of Cn, Cs (Ac-2753), NTC LAMP, and control, respectively.
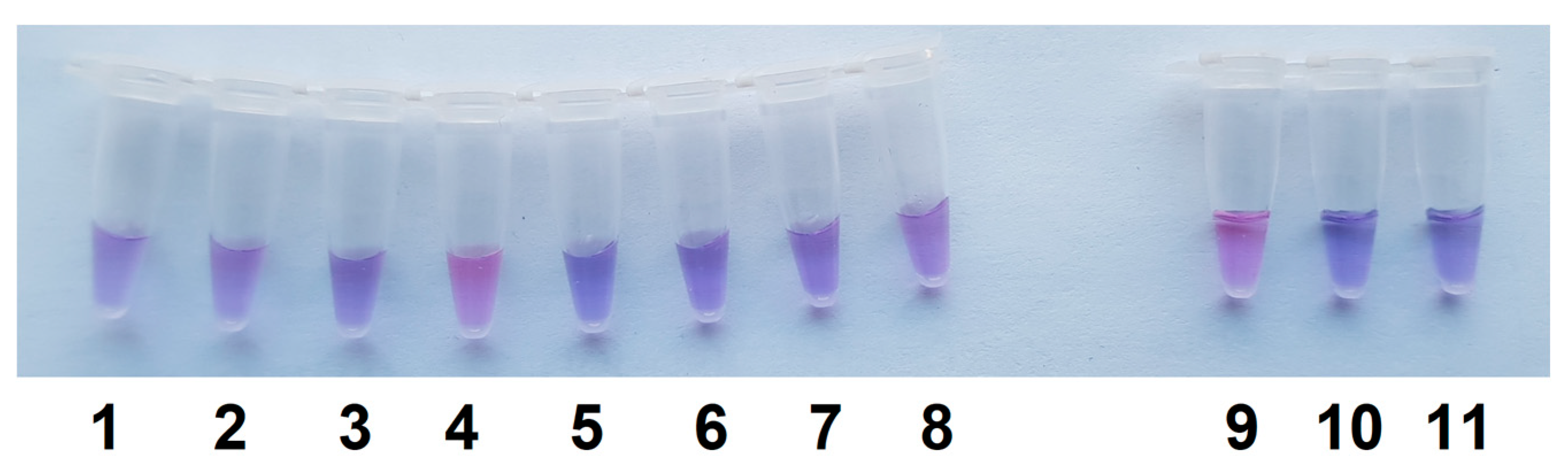

Table 1.
Sequences of loops B and F of LAMPlicons. The sequences were retrieved from NCBI databases for Clavibacter species, based on the sequences of the inner LAMP primers (FIP and BIP, Table S2). Underlined sections are targeted by gRNA spacers. Annealing sites for primers LF and LB are shown in bold. The single nucleotide substitutions in the loop F are indicated in red.
Table 1.
Sequences of loops B and F of LAMPlicons. The sequences were retrieved from NCBI databases for Clavibacter species, based on the sequences of the inner LAMP primers (FIP and BIP, Table S2). Underlined sections are targeted by gRNA spacers. Annealing sites for primers LF and LB are shown in bold. The single nucleotide substitutions in the loop F are indicated in red.
| Loop designation | Species* | Loop sequence (5' → 3') |
|---|---|---|
| Loop B | All | GGCGTCTCGCTCCTGAGCCTCCTGCTCGGGCAGCTCGTCTTCA |
| Loop F | Cs | GACTTGCGCACGTTCTCCACGATGATGCGCG |
| Ct | GACTTCCGCACGTGCTCGACGATGATGCGCG | |
| Cm | GGACTTCCGCACGTTCTCGACGATGATGCGCG | |
| Cn | GACTTCCGCACGTTCTCGACGATGATGCGCG | |
| Ci | GACTTCCGCACGTTCTCGACGATGATGCGCG | |
| Cp | GGACTTCCGCACGTTCTCGACGATGATGCGCG |
* Cs – C. sepedonicus, Ct – C. tesselarius, Cm – C. michiganensis, Cn – C. nebraskensis, Ci – C. in-sidiosus, Cp – C. phaseoli.
Table 2.
The list of gRNAs used. The spacer sequence is underlined.
| Name | Sequence (5' → 3') |
|---|---|
| gRNA-B | GGGAAUUUCUACUGUUGUAGAUCAGGAGGCUCAGGAGCGAGA |
| gRNA-F-14 | GGGAAUUUCUACUGUUGUAGAUGGAGAACGUGCGCA |
| gRNA-F-16 | GGGAAUUUCUACUGUUGUAGAUGUGGAGAACGUGCGCA |
| gRNA-F-18 | GGGAAUUUCUACUGUUGUAGAUUCGUGGAGAACGUGCGCA |
| gRNA-F-20 | GGGAAUUUCUACUGUUGUAGAUCAUCGUGGAGAACGUGCGCA |
| gRNA-F-22 | GGGAAUUUCUACUGUUGUAGAUAUCAUCGUGGAGAACGUGCGCA |
| gRNA-F-24 | GGGAAUUUCUACUGUUGUAGAUGCAUCAUCGUGGAGAACGUGCGCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



