Submitted:
15 July 2025
Posted:
17 July 2025
You are already at the latest version
Abstract
Cancer remains one of the most challenging diseases to diagnose and treat, requiring the development of innovative therapeutic and diagnostic strategies. Cancer progression includes enzyme dysregulation, which orchestrates key biological processes ranging from metabolic reprogramming, epigenetic modifications to drug metabolism and immune evasion. Enzymes drive and modulate their roles in the tumor microenvironment, influencing how cancer cells adapt to stress, resist treatments, and evade immune surveillance. This review paper examines recent advances in enzyme engineering and its potential in cancer treatment. Computational tools, including artificial intelligence, have contributed significantly to enzyme optimization, enabling improvements in catalytic efficiency, stability, and specificity through structural modeling, functional annotation and generative design. Enzyme engineering can be optimized for targeted therapies, minimizing off-target effects while maximizing therapeutic potential. Challenges such as enzyme stability, delivery mechanisms, and immunogenicity persist, but recent advances offer promising solutions. Therefore, we also review the integration of computational approaches and experimental advances in enzyme engineering, thereby offering insights into future directions for optimizing enzyme engineering strategies in cancer treatment and diagnosis. In essence, this review provides an up-to-date synthesis linking the fundamental biological understanding of enzymes in cancer with the rapidly evolving field of enzyme engineering and the powerful capabilities offered by computational tools, highlighting both promising advances and remaining hurdles such as benchmarking and interpretability of machine learning offered solutions.
Keywords:
enzyme engineering
; cancer
; machine learning
1. Introduction
Cancer remains one of the most significant global health and socioeconomic burdens, accounting for millions of new cases and deaths annually [65]. The persistent rise in cancer incidence underscores the urgent need for innovative and more effective therapeutic strategies that go beyond the limitations of conventional treatments. Among emerging approaches, enzyme engineering has garnered considerable attention for its potential to provide highly selective and efficient therapeutic interventions. Using the catalytic power and substrate specificity of enzymes, researchers aim to develop targeted solutions that minimize off-target effects and enhance therapeutic efficacy.
Catalysis is fundamental to synthetic organic chemistry because it allows reactions to occur more efficiently, selectively, and under milder conditions than would otherwise be possible. Without catalysts, many chemical transformations would require extreme temperatures, pressures, or extended time scales, making them impractical or cost-prohibitive. In a chemical reaction, one or more reactants(substrates) undergo changes that result in one or more products. Catalysts not only speed up reactions without being consumed in the process but can also direct the outcome, such as controlling the orientation or type of product formed (regioselectivity and stereoselectivity). This is especially important in pharmaceutical synthesis, where even small differences in molecular structure can profoundly impact the safety and efficacy of a drug.
Among catalysts, enzymes are in a privileged position. These biological macromolecules operate by stabilizing the transition state, the high energy intermediate between reactants and products. To visualize this, a chemical reaction can be depicted as a hill-climb from reactants to products. The peak of the hill represents the transition state, which is difficult to reach without help. Enzymes act like a tunnel through the hill, allowing the reaction to proceed more easily and quickly without requiring as much energy. This makes enzymatic catalysis not only efficient but also energetically favorable and environmentally sustainable.
Historically, scientists hesitated to use enzymes in synthetic chemistry. Enzymes only worked with limited substrate scope and had low stability. This made them seem limited for large-scale or diverse chemical manufacturing. However, a paradigm shift occurred in the 1990s with the advent of directed evolution, a lab-based process that mimics natural selection. This method involves iterative cycles of mutation (random changes to the genetic code) and selection to evolve enzymes with desired characteristics. Directed evolution revolutionized the field by demonstrating that enzymes could be engineered to catalyze a wide array of reactions, including those requiring high levels of stereoselectivity, where a specific spatial arrangement of atoms (stereochemistry) in the product is preferred[133]. This advancement opened the door to the widespread use of enzymes as biocatalysts in both industrial and biomedical applications, including precision oncology.
Since the initial success of directed evolution, more refined and systematic approaches such as stepwise synergistic random mutations at multiple active-site positions (the combinatorial active-site saturation test) and stepwise one or a few selected positions mutations (the iterative saturation mutagenesis) have emerged to enhance the functional optimization of enzymes. Furthermore, rather than making random changes, rational enzyme design has gained prominence as a targeted, knowledge-driven strategy that introduces specific mutations based on structural insights, in silico predictions, mechanistic understanding, or computational modeling, thereby improving catalytic performance, stability, or substrate specificity. Collectively, these advanced techniques empower scientists to tailor enzymes for a broad range of synthetic transformations, expanding the biocatalytic toolbox available for complex chemical synthesis[133]. As a result, enzymes are now widely recognized as versatile and efficient catalysts in modern synthetic chemistry, particularly in the pharmaceutical industry[159].
Enzymes are increasingly utilized in the synthesis of active pharmaceutical ingredients due to their ability to function under mild, environmentally benign conditions, coupled with their high chemo-, regio-, and stereoselectivity. These attributes not only reduce the environmental footprint but also simplify downstream purification processes, making enzymatic routes attractive alternatives to conventional chemical methods[159].
Importantly, biocatalysis has played a transformative role in the manufacturing of several clinically important drugs. For instance, biocatalysis has contributed to the efficient synthesis of atorvastatin, a widely used statin for lowering cholesterol and preventing cardiovascular disease; montelukast, employed in asthma and allergy management; and duloxetine, prescribed for mood disorders and chronic pain; sitagliptin, used in the treatment of type 2 diabetes; islatravir, an investigational nucleoside analog for HIV therapy; and belzutifan, a therapeutic agent approved for cancers such as von Hippel-Lindau-associated renal cell carcinoma[159,233]. Enzymatic synthesis not only streamlines drug production by reducing the number of synthetic steps, but also enhances overall yield and process efficiency, thereby lowering production costs and improving scalability[233].
The integration of artificial intelligence into biological research has significantly accelerated progress in the field of biocatalysis. A landmark moment in this convergence was recognized by the 2024 Nobel Prize in Chemistry, awarded to Demis Hassabis, John Jumper, and David Baker for their transformative contributions to AI-driven protein structure prediction and computational protein design[32]. Specifically, the development of the AlphaFold model has revolutionized the ability to accurately predict protein three-dimensional structures from amino acid sequences, a longstanding challenge in structural biology. Baker’s complementary work on de novo protein design further underscored the growing capabilities of AI-assisted molecular engineering.
By leveraging deep learning and other machine learning frameworks, researchers can now decipher complex relationships between enzyme sequences, structural motifs, and catalytic functions. This computational power enables the rational design and rapid optimization of enzymes, dramatically reducing the time and experimental cost traditionally associated with wet-lab screening. As a result, the development of next-generation biocatalysts with customized properties for industrial, pharmaceutical, and therapeutic applications has been greatly accelerated.
Recent advances in mechanistic understanding of enzyme-catalyzed reactions have led to the emergence of novel methods for enzyme discovery, engineering, and functional repurposing across various biomedical domains[132], including oncology. These breakthroughs are transforming the landscape of enzyme-based cancer therapeutics, enabling more precise control over catalytic activity and target specificity. Such innovations support the broader goals of precision medicine, where therapies can be tailored to individual patient profiles based on molecular and enzymatic biomarkers.
While artificial intelligence-based models continue to enhance the capacity to design enzymes with tailored functions, it remains essential to rigorously evaluate the real-world performance, safety, and potential risks of these engineered biocatalysts, particularly in clinical contexts. Issues such as off-target effects, immunogenicity, and in vivo stability must be thoroughly addressed before clinical translation.
Enzymes, as the catalytic workhorses of cellular metabolism, are integral to maintaining physiological homeostasis. In the context of cancer, however, this tightly regulated enzymatic environment is frequently subverted. Tumor cells exploit enzymatic networks to promote uncontrolled proliferation, evade immune surveillance, and resist apoptotic signals. The oncogenic rewiring of metabolic and signaling pathways, often mediated by deregulated or overexpressed enzymes, contributes to both disease progression and resistance to standard treatments. Traditional cancer therapies such as chemotherapy and radiotherapy are hampered by non-specific cytotoxicity, resistance mechanisms, and systemic side effects. Consequently, there is an increasing demand for targeted, effective, and less invasive therapeutic and diagnostic modalities.
In this regard, enzymes have emerged not only as functional biomarkers for cancer detection and monitoring but also as actionable molecular targets for therapy. Their roles in critical pathways of tumor biology make them attractive candidates for enzyme-activated prodrugs, enzyme-inhibitor therapies, and enzymatic diagnostics, all of which hold promise for improving clinical outcomes and minimizing collateral damage to healthy tissues.
This review serves as an up-to-date synthesis that bridges the fundamental biological understanding of enzymes in cancer with the rapidly evolving field of enzyme engineering and the powerful capabilities offered by computational tools, including AI and ML. It highlights recent advances in enzyme engineering and its transformative potential in cancer treatment. It also acknowledges persistent challenges, such as enzyme stability, delivery mechanisms, and immunogenicity, while highlighting promising solutions and remaining hurdles in developing enzyme-based cancer therapies.
The next section, Background section, provides fundamental details on enzymes, covering their structure, function, classification using the Enzyme Comission number system, factors influencing their activity, and regulation via post-translational modifications.
Furthermore, the review integrates computational approaches and experimental progress to offer insights into future directions for enzyme engineering.
The review then delves into Cancer and the role of enzymes in cancer study, exploring the cellular transformation process, the impact of oncogenes and tumor suppressor genes, apoptosis, cancer heterogeneity, the hallmarks of cancer (including the Warburg effect), and positioning enzymes as both disease drivers and therapeutic targets. Specific therapeutic applications are detailed in the Enzyme-Aided Drug Delivery section, which outlines various cancer treatment modalities, explains how enzymes such as CYP enzymes activate prodrugs, and describes strategies to stabilize enzymes needed for prodrugs. In parallel, machine learning approaches are discussed to improve understanding of enzymes role in cancer treatment and diagnosis, while also addressing persistent challenges and promising solutions. Finally, the Conclusion summarizes the significance of enzymes as a therapeutic axis, reviewing their roles in key cancer processes and their applications in targeted therapies. The review is supplemented with Appendix A and Appendix B, that list protein design models(Table A1, Table A2) and the benchmark methods(Table B1) used for their evaluation.
2. Background
Proteins are fundamental macromolecules composed of long chains of amino acids that are linked together by peptide bonds through a dehydration synthesis reaction. Each amino acid in the chain contributes to the overall chemical and structural characteristics of the protein. As this linear chain, known as a polypeptide, begins to fold, it adopts local structural motifs such as alpha-helices and beta-sheets, which are stabilized by hydrogen bonds. These elements are referred to as the secondary structure. Regions that lack these regular patterns are described as random coils. As folding progresses, the polypeptide chain assumes a more complex and compact three-dimensional configuration (the tertiary structure), which is stabilized by various interactions including hydrogen bonds, hydrophobic effects, ionic interactions, and disulfide bridges. This three-dimensional conformation is essential because it dictates the biological role the protein can play within the cell.
In this context, amino acids in the protein chain, referred to as residues once linked, are akin to beads on a string, forming a one-dimensional sequence. However, it is their transformation into specific three-dimensional structures that enables biological functionality. This sequence-structure-function relationship is at the core of molecular biology and enzymology. Yet, a major bottleneck persists: the number of protein sequences identified through genomic sequencing technologies far outpaces the number of experimentally determined protein structures. This imbalance, often referred to as the sequence–structure gap, presents a major challenge for understanding protein function at a molecular level[200]. Computational methods, especially those using machine learning, have emerged as crucial tools to bridge this gap by predicting protein structures directly from amino acid sequences, thereby accelerating enzyme characterization and design.
Enzymes are a specialized class of proteins that serve as biological catalysts, significantly increasing the rate of chemical reactions without being consumed in the process. The functionality of an enzyme is intricately linked to its tertiary structure, which determines the configuration of a specific region called the active site. This active site binds target molecules, referred to as substrates, with high specificity and positions them in an optimal environment for the chemical reaction to occur. The catalytic efficiency and specificity of an enzyme are highly dependent on its three-dimensional structure, which is, in turn, determined by the underlying amino acid sequence[41].By engineering changes in the sequence, particularly near or within the active site, scientists can tailor enzymes to enhance their performance, broaden substrate specificity, improve stability under diverse conditions, or develop entirely new catalytic activities, capabilities that are increasingly vital in the design of enzyme-based cancer therapies.
The classical theory of chemical reactions assumes that a reaction must go through a single, well-defined transition-state structure. In enzymatic catalysis, this means that enzymes bind tightly to the transition state, lowering the activation energy and making the reaction proceed more easily. However, this traditional view might be too simplistic[208].
In enzymatic catalysis, the key factor remains transition state stabilization, but this stabilization occurs across a broader range of structures. The catalytic efficiency of enzymes comes from their ability to modulate this entire transition state region, aligning with the complex energy landscape seen in protein folding and molecular binding[208].
Enzymes facilitate this catalytic efficiency through several well-characterized mechanisms:
- Correctly aligning substrates within the active site to ensure effective molecular interactions.
- Inducing strain or distortion in specific substrate bonds, making them more susceptible to cleavage.
- Providing an optimal microenvironment, such as acidic or basic side chains, that stabilizes transition states.
- Forming transient covalent intermediates with substrates, which lowers the energy required to proceed through the reaction pathway.
In microbial genomes, enzymes represent one of the most prevalent categories of functional genes. They are systematically classified by the Enzyme Commission (EC) number system, which provides a hierarchical classification based on the types of reactions they catalyze. For instance, the enzyme alcohol dehydrogenase is designated as EC 1.1.1.1, indicating that it belongs to the oxidoreductases group that acts on the CH-OH group of donors with or as acceptors. The EC classification system not only standardizes the nomenclature, but also facilitates enzyme annotation in large-scale genomic studies[130].
Particularly, EC numbers are crucial for understanding enzyme functions and overall cellular metabolism. EC numbers can be used in tasks such as annotating large amounts of genome sequence data, identifying enzyme functions, linking genes to proteins and reactions, designing new metabolic pathways, and constructing large-scale metabolic networks efficiently[207]. This has important implications for systems biology, metabolic pathway analysis, and synthetic biology applications, fields that benefit immensely from computational models, including machine learning, to predict enzyme functions and guide engineering strategies.
Metabolomics is the comprehensive study of small molecules, commonly referred to as metabolites, within cells, tissues, or biological fluids. Metabolomics provides a functional readout of physiological and pathological states by profiling endogenous metabolites, which are the downstream products of gene expression and enzyme activity. Through the analysis of these low-molecular-weight compounds, metabolomics enables researchers to distinguish metabolic pathways across different tissues, offering insights into disease mechanisms, drug responses, and the impact of environmental factors on cellular metabolism[291].
Metabolic pathways consist of a series of interconnected biochemical reactions that begin with a substrate and proceed through multiple enzymatic steps to generate specific products and intermediates. These pathways include central processes such as glycolysis, the tricarboxylic acid cycle, and lipid metabolism. At the heart of each step lies a specific enzyme—a biological macromolecule that catalyzes the transformation of one metabolite into another by accelerating the rate of the reaction[290].
The activity of enzymes is finely regulated to maintain metabolic homeostasis. One of the key regulators of enzymatic activity is the endocrine system, particularly hormones, which influence enzyme expression levels, catalytic activity, and localization. For example, insulin and glucagon modulate glucose metabolism by activating or inhibiting key metabolic enzymes, thereby directing flux through anabolic or catabolic pathways depending on the organism’s energy demands[290]. Dysregulation of such hormonal controls can result in metabolic imbalances and has been implicated in diseases such as diabetes, obesity, and cancer.
Cofactors are non-protein chemical compounds that are essential for the biological activity of many enzymes. They assist in the catalytic process by stabilizing reaction intermediates, participating in electron transfer, or serving as transient carriers of specific atoms or functional groups. Cofactors are typically categorized into two groups: inorganic ions (such as Mg2+, Fe2+/3+, or Zn2+) and organic molecules known as coenzymes. Coenzymes are a subclass of cofactors that are often derived from dietary vitamins—especially the B-vitamin group—and act as transient carriers of electrons, atoms, or functional groups during enzymatic transformations[289]. Common examples include NAD+/NADH, FAD/FADH2, and coenzyme A, which play pivotal roles in redox reactions and energy metabolism.
The catalytic efficiency and overall functionality of enzymes are influenced by several physicochemical and environmental factors[285]:
- Temperature. Enzyme activity generally increases with temperature due to enhanced molecular motion, reaching a peak at an optimum temperature. Beyond this point, elevated temperatures can lead to enzyme denaturation, a loss of structural integrity that diminishes or abolishes catalytic activity.
- Enzyme concentration. Increasing the amount of enzyme present in a reaction typically raises the reaction rate, provided that substrate availability is not limiting. However, once substrate molecules are fully utilized, further increases in enzyme concentration yield diminishing returns.
- Substrate concentration. At low substrate levels, reaction velocity increases rapidly with substrate concentration. As enzyme active sites become saturated, the reaction rate plateaus, approaching a maximum velocity () as described by Michaelis-Menten kinetics.
- pH levels. Each enzyme exhibits optimal activity within a specific pH range, which reflects the ionization states of amino acid residues in the active site and substrate. Deviation from this range can disrupt ionic interactions or hydrogen bonding, leading to reduced activity or irreversible denaturation.
Enzyme activity can also be modulated through the presence of inhibitors, which reduce or prevent catalytic function via distinct mechanisms:
- Competitive inhibitors. These molecules resemble the enzyme’s natural substrate and bind to the active site, competing with the substrate. This form of inhibition can often be overcome by increasing the substrate concentration.
- Non-competitive inhibitors. These bind to allosteric sites (locations other than the active site), causing conformational changes that diminish the enzyme’s ability to bind its substrate or carry out catalysis, regardless of substrate concentration[285].
In addition to reversible inhibition, certain substances, such as toxins and heavy metals, can act as irreversible inhibitors. These agents often form covalent bonds with amino acid residues in the active site or elsewhere in the protein, resulting in permanent structural alterations and functional loss[285].
To maintain metabolic homeostasis, cells employ a range of regulatory mechanisms involving inhibitors, that ensure enzymes operate at appropriate rates. This allows for fine-tuned, feedback-sensitive control over metabolic fluxes[285].
Disruptions in these tightly regulated systems, whether due to genetic mutations, environmental stressors, or pathological conditions, can affect enzyme function, leading to metabolic disorders, chronic diseases, or even cancer. Such dysfunction highlights the central importance of enzymes in maintaining physiological balance and underscores the relevance of enzyme engineering for therapeutic intervention.
Biocatalysis offers many advantages over conventional chemical methods, including milder reaction conditions, reduced environmental impact, and access to novel chemical functionalities. Natural enzymes often lack the activity, stability, or substrate scope (versatility) required for synthetic applications[95]. Enzymes have evolved over millions of years to meet the needs of their host organisms, which often do not align with industrial requirements. Consequently, enzymes often require tailoring for specific industrial applications [95].
Recent breakthroughs in deep learning, particularly the development of AlphaFold[204] by DeepMind, have significantly transformed our ability to predict protein structures with near-experimental accuracy. AlphaFold demonstrated that it is possible to infer the three-dimensional conformation of a protein solely from its amino acid sequence by modeling long-range interactions and leveraging evolutionary information embedded in multiple sequence alignments. Its performance in the 14th Critical Assessment of Structure Prediction stunned the structural biology community by achieving accuracy comparable to X-ray crystallography for a substantial number of proteins. This advance has profound implications for enzyme engineering, where the precise 3D structure is critical for identifying catalytic residues, understanding substrate binding, and guiding targeted mutations.
Importantly, AlphaFold and similar deep learning tools are helping to close the long-standing sequence–structure gap by providing researchers with high-confidence structural models for millions of proteins whose structures were previously unknown or inaccessible through experimental techniques. These computational models now serve as a foundation for rational enzyme design, enabling in silico exploration of mutational effects on enzyme function, stability, and substrate affinity. In the context of cancer research, this is particularly transformative: the structural elucidation of tumor-associated enzymes allows for the development of novel inhibitors and precision diagnostics.
In summary, enzymes are essential for cellular homeostasis and human health. Understanding their structure-function relationships, regulatory mechanisms, and how they can be designed or modified, particularly through machine learning, is critical for tackling complex diseases such as cancer. As will be demonstrated in the following sections, the convergence of biology, computer science, and synthetic engineering is transforming enzymes from simple natural catalysts into programmable tools for precision medicine.
3. Computational Tools in Enzyme Engineering
Exploring enzyme variants through functional assays and fitness landscape modeling provides a systematic and targeted strategy for enzyme engineering. Functional assays experimentally evaluate the catalytic activity, specificity, and other biochemical properties of enzyme variants, offering empirical insight into how mutations affect performance. Fitness landscapes, on the other hand, enable the identification of mutational hotspots and guide the rational selection of beneficial substitutions during enzyme optimization[46].
The concept of protein fitness refers to the ability of a protein to effectively carry out its biological role, and is influenced by multiple factors, including its structural integrity, thermodynamic stability, and interactions with substrates or cofactors[2]. High-fitness proteins typically retain their function across a broad range of environmental conditions and are less prone to misfolding or degradation. Among these determinants, protein stability is particularly critical, as it ensures the preservation of the active conformation required for enzymatic catalysis. Stability is commonly assessed in terms of the Gibbs free energy difference between folded and unfolded states, or between engineered and wild-type proteins. The lower value indicates a more stable folded structure, which correlates with improved resistance to thermal or chemical denaturation[219].
Computational tools now play a vital role in advancing the selection of enzyme variants by predicting the effects of mutations and enabling the construction of extensive in silico libraries of enzyme variants. These virtual libraries can be rapidly screened using a variety of energy functions, geometric constraints, or machine learning models to assess features such as binding affinity, stability, or catalytic efficiency[83]. By prioritizing the most promising variants computationally, researchers can significantly reduce the experimental burden, focusing validation efforts on a smaller, high-value subset. This integrative approach, which combines in silico modeling, predictive scoring, and targeted functional assays, accelerates the discovery and optimization of enzyme variants with enhanced therapeutic or industrial potential.
Enzyme function can be investigated through two primary frameworks: mechanistic kinetic modeling and machine learning-based predictive models. These complementary approaches differ in their underlying methodologies and data requirements, but both contribute significantly to understanding and optimizing enzymatic activity.
Mechanistic kinetic modeling, rooted in classical enzymology, attempts to explain enzyme function by explicitly characterizing the steps involved in catalysis. This framework is typically structured across three hierarchical levels of enzyme behavior[168]:
- Sequence–structure relationship: The amino acid sequence of the enzyme dictates its folding into a unique three-dimensional structure. This structure, including dynamic conformational states, is essential for the proper positioning of functional groups necessary for catalysis.
- Enzyme as a nanomachine: The enzyme facilitates substrate recognition and binding, correctly orienting the substrate within the active site. Afterward, it assists in the formation and release of the product, while minimizing interference from product inhibition or solvent interactions. This machine-like behavior ensures catalytic efficiency and specificity.
- Catalytic transition state stabilization: Key active-site residues interact with the substrate to stabilize the high-energy transition state, thus lowering the activation energy required for the reaction. This step is crucial in bond breaking and bond formation processes.
Mechanistic models often rely on rate equations derived from Michaelis-Menten kinetics or more complex models when allosteric regulation, inhibition, or multi-step catalysis is involved. Although detailed knowledge of enzyme mechanisms, structure, and kinetics is required, such models are highly informative and can extract critical insights from even limited experimental data. They enable the prediction of enzyme behavior under various conditions, facilitate the identification of rate-limiting steps, and inform strategies for inhibitor design or enzyme reengineering[168].
In contrast, machine learning-based approaches bypass the need for detailed mechanistic information by learning patterns directly from data. These models are trained on large datasets consisting of enzyme sequences, structures, or functional annotations. Once trained, these models can predict enzyme properties such as substrate specificity, catalytic efficiency, thermostability, or mutational effects with remarkable accuracy. Importantly, these models can generalize across enzyme families and are particularly valuable in identifying sequence-function relationships that are non-obvious from traditional biochemical principles.
Current strategies for developing high-performance biocatalysts integrate both mechanistic and data-driven approaches. These strategies include[95]:
- Exploration of natural enzyme diversity, through genome mining and metagenomics, to discover novel catalytic functions.
- Enzyme engineering, which modifies natural enzymes to enhance performance, extend substrate scope, or improve operational stability.
- Mechanism redesign, aimed at introducing entirely new catalytic activities or altering reaction pathways.
- Computational enzyme design, which involves de novo construction of enzymes using structure-guided and machine learning-assisted methods.
Tailored enzymes are especially critical in biocatalysis-driven manufacturing, where specific performance attributes such as catalytic activity, thermal and solvent stability, enantioselectivity, and resistance to inhibition are required for industrial scalability and economic viability.
Two principal strategies for enzyme engineering are directed evolution and rational design. Directed evolution emulates the principles of Darwinian selection by generating diverse libraries of enzyme variants, followed by high-throughput screening or selection for improved functionality. This iterative process has been widely successful in producing enzymes with enhanced properties[159]. In contrast, rational design relies on detailed structural and functional knowledge to introduce specific mutations to achieve a desired outcome. Although more targeted, it often requires reliable structural models and a deep understanding of the enzyme’s catalytic mechanism.
Scientists employ a range of mutagenesis techniques to generate and test genetic variations, enabling the discovery and optimization of proteins with enhanced or novel functions. Key methods include error-prone PCR, which introduces random mutations across the gene sequence; site-saturation mutagenesis, which targets specific amino acid positions to explore all possible substitutions; and DNA shuffling, which recombines gene fragments from related sequences to create chimeric variants. These approaches are frequently coupled with computational modeling and machine learning to guide experimental design, prioritize mutations, and predict functional outcomes[95].
Recent technological innovations in mutagenesis, recombination, and computational biology have made it possible to construct ultra-large gene libraries containing billions of potential variants. A central challenge in directed evolution lies in efficiently identifying functional and improved variants from these massive libraries. This is typically achieved through selection-based or screening-based strategies.
Selection-based approaches, such as phage display or yeast surface display, link protein function to the survival or replication advantage of a host organism, allowing rapid and scalable enrichment of active variants, often capable of handling throughput on the order of clones per round.
In contrast, screening-based approaches provide a more precise assessment of variant function by evaluating individual phenotypes. Techniques such as fluorescence-activated cell sorting and droplet microfluidics allow high-resolution, quantitative screening of enzyme activity, specificity, or binding. Among these, droplet microfluidics has gained prominence due to its ability to compartmentalize and analyze millions of enzyme variants in picoliter volumes, offering reagent efficiency, high throughput, and reduced cross-contamination, which are key features for discovering novel biocatalysts for both industrial and therapeutic applications[110].
Different from the directed evolution, the rational design approach uses computer-based tools to predict how mutations will affect enzyme function to create smaller, but more effective enzyme libraries. The method relies on studying enzyme structures, comparing sequences from different organisms, and analyzing which parts of the enzyme are most important for its function or have remained unchanged through evolution. The goal is to introduce targeted mutations that enhance stability, activity, selectivity, or other desirable properties[83].
When rational design is combined with directed evolution, the result is a powerful, synergistic framework for enzyme engineering. This hybrid strategy enables the development of tailored enzymes capable of exhibiting enhanced thermostability, increased catalytic efficiency, and even non-natural or novel reactivity, traits highly valuable in biocatalysis, pharmaceutical development, and synthetic biology[151].
Despite these advancements, enzyme engineering often faces a significant obstacle known as the "cold-start" problem, a scenario in which little or no experimental sequence–fitness data are available to guide initial library construction[226]. In such cases, it becomes crucial to design libraries that balance diversity with expected functional fitness, thereby maximizing the probability of sampling variants that span multiple peaks in the fitness landscape. These diverse, high-potential starting points help researchers explore evolutionary trajectories that may otherwise be inaccessible to traditional iterative methods[59,63,226].
It is also important to note that mutational effects are not always additive. While some mutations destabilize the protein or abolish function entirely[36], others may interact in epistatic ways, either cooperatively or antagonistically, resulting in non-linear outcomes. Understanding these epistatic interactions is key to mapping rugged fitness landscapes and can reveal mechanistic insights into protein folding, dynamics, and function that are often missed by additive models[230].
Protein engineers face growing challenges in enzyme evolution due to increasing resource demands and slow progress in traditional methods, but emerging technologies offer promising solutions [132]. Innovations such as cell-free protein expression and mRNA display libraries are accelerating evolution cycles and enabling the creation of proteins with enhanced catalytic and binding properties. Additionally, droplet-based microfluidic systems allow ultra-high-throughput screening of enzyme variants, greatly advancing the efficiency and depth of directed evolution [132].
4. Deep Learning Models for Enzyme Engineering
Deep learning approaches are transforming the field of enzyme engineering by enabling sophisticated tasks such as de novo protein design, targeted modification of active or functional regions, and the accurate prediction of structural and functional properties essential for enzymatic activity. These advancements are powered by a new generation of machine learning models capable of capturing complex relationships within protein sequences and structures. Notable model families include protein language models, which learn contextual representations of amino acid sequences; diffusion models, which support structure generation and refinement; graph neural networks, which model proteins as molecular graphs to capture spatial dependencies; and hybrid architectures that integrate multiple modeling strategies to enhance predictive accuracy and generalization.
An overview of state-of-the-art deep learning architectures applied to enzyme engineering is shown in Figure 1. The figure illustrates a comprehensive pipeline that integrates multiple input modalities, including sequence data, structural information (including template-based models), and functional annotations such as Enzyme Commission (EC) numbers. The modality-specific preprocessing phase involves genetic database searches for multiple sequence alignment, structural database queries, and conformer generation using predictive tools such as ESMFold[101] or AlphaFold[204,300]. Following preprocessing, rich molecular representations are constructed through techniques such as autoregression and masked language modeling over amino acid sequences. Additional operations, such as subsampling, substructure extraction, and noise modeling, further enrich the representation space. These representations are refined using a diffusion model which denoises noisy latent encodings to produce structurally and functionally relevant samples. Graph Neural Networks (GNNs) and hybrid architectures are employed to model structural features by extracting edge- and node-level attributes, including spatial distance, direction, orientation, and angular relationships. These features are then passed through contrastive learning modules to produce task-specific embeddings that enhance generalization. The model outputs span a wide range of downstream predictions, including sequence optimization, fitness evaluation, protein structure prediction, physicochemical property estimation, ligand binding affinity, complex formation, and enzyme function classification. Together, these features make this architecture scalable and versatile for data-driven enzyme design and functional analysis.
Large language models (LLMs) have revolutionized natural language processing (NLP) by excelling in tasks such as text generation, translation, and conversational interactions. Their success is driven by extensive training on diverse datasets, allowing them to capture complex language patterns and generate human-like text. Inspired by these advancements, researchers have raised a fundamental question: can LLMs, originally developed for NLP, effectively interpret protein sequences as a form of language[307]? Recent breakthroughs suggest that LLMs can be fine-tuned to predict chemical properties, eliminating the need for extensive domain-specific feature engineering. Open-source models such as GPT-J-6B, Llama-3.1-8B, and Mistral-7B have been fine-tuned on various chemical questions and benchmarked against traditional methods, demonstrating that even with limited datasets, these models outperform conventional approaches in simple classification tasks[191].
Major limitation in applying traditional LLMs to protein understanding lies in the absence of a direct mapping between protein sequences and their textual descriptions, which impairs effective training and evaluation. To overcome this, datasets such as ProteinLMDataset have been introduced to support both self-supervised pretraining and supervised fine-tuning, improving LLMs’ ability to comprehend protein sequences and associated functions [307].
Instead of applying general LLMs, protein language models (pLMs) have emerged as particularly influential tools in computational biology and enzyme engineering. Inspired by techniques from NLP, pLMs conceptualize amino acid sequences as sentences, treating each residue as a “word” to capture contextual relationships analogous to grammatical structures in human language[?]. These models are typically constructed using transformer architectures, which are well-suited for modeling long-range dependencies in sequential data. Transformers have an essential capability for understanding how distant residues within a protein sequence interact during folding and functional site formation.
Transformers accomplish this through two principal training strategies:
- Autoregressive modeling, where the model is trained to predict each successive residue based on all previous residues in the sequence.
- Masked language modeling, where the model reconstructs missing or masked residues using the context of the surrounding sequence.
During training, protein language models learn internal feature representations called embeddings: dense numerical vectors that encode structural, functional, and evolutionary information. These embeddings can be used for a variety of downstream tasks, including prediction of protein structure, novel sequence generation, classification of enzyme functions, prediction of mutation impact, and annotation of active sites.
A key advantage of protein language models lies in their computational efficiency during inference. Although training these models requires substantial computational resources and access to large protein databases, once trained, embeddings can be extracted quickly and efficiently using standard consumer grade hardware, making the technology broadly accessible to the research community[362].
Despite their strengths, transformer-based models exhibit certain limitations, particularly when applied to constraint satisfaction problems (CSPs), which are often encountered in enzyme engineering. CSPs, such as designing active sites to meet specific structural constraints or matching enzymes to substrates with defined physicochemical properties, typically require multi-step logical reasoning. However, standard transformer models have historically struggled with such tasks. For example, solving structured problems such as Sudoku, which demands 20–60 reasoning steps, remains challenging for vanilla transformer architectures[361]. This limitation highlights the need for hybrid or augmented modeling strategies that can bridge the gap between sequence-level learning and logical inference, especially in the context of complex design problems in enzyme engineering.
To overcome the limitations of standard transformer architectures in multi-step reasoning tasks recurrence can be introduced into their design. Discrete constraints such as specifying allowable residue identities at functional positions, enforcing active site geometries, or limiting backbone torsion angles, can be encoded directly into the loss function using differentiable approximations such as Straight-Through Estimators(STEs). STEs allow non-differentiable operations to be integrated into gradient-based learning pipelines. By applying constraint-aware losses to both recurrent layers and attention matrices, Recurrent Transformers can achieve greater predictive accuracy, robustness, and generalization, particularly under semi-supervised learning settings where labeled examples are sparse[361].
Complementing transformer-based approaches, Graph Neural Networks (GNNs) offer a structurally grounded and chemically intuitive method for modeling protein architectures. In GNNs, amino acid residues (or individual atoms) are represented as nodes, while inter-residue interactions such as hydrogen bonds, van der Waals forces, or spatial proximities, are modeled as edges. These networks employ message-passing algorithms, where information is iteratively exchanged between connected nodes, capturing local and global structural dependencies vital for understanding protein folding, stability, and functional site geometry. A prominent example of models that combine transformer-based approaches and GNNs is AlphaFold2, which revolutionized protein structure prediction by combining evolutionary data from multiple sequence alignments with GNN-based geometric modeling[204].
In AlphaFold2, residues are modeled as nodes, and residue-residue relationships as edges, allowing the architecture to encode both sequential and spatial constraints effectively. At the heart of AlphaFold2 lies the Evoformer module, a multi-component architecture that alternates between updating multiple sequence alignments (MSA) embeddings (to capture sequence-level evolution) and pair embeddings (to refine spatial relationships) across several iterative steps, progressively improving the structural prediction fidelity[204]. Evoformer employs two key attention mechanisms[204]:
- MSA Attention, which captures conserved sequence motifs and patterns from MSAs, reflecting evolutionary constraints essential for protein folding.
- Pair Attention, which instead of just considering which residues are close together, uses specialized components such as "Triangle Multiplication" and "Triangle Attention" to model spatial relationships between groups of three residues - building blocks of proteins. These modules help accurately predict inter-residue distances and angles, crucial for constructing the final 3D structure, and allows the model to infer higher-order geometric relationships, surpassing the sequential modeling limitations of traditional transformer layers.
To address more complex biochemical contexts, AlphaFold3 expands upon its structure prediction predecessor AlphaFold2 and reverse structure to sequence models such ProteinMPNN[13] by incorporating additional GNN modules designed to model protein–ligand interactions [206]. In this framework, ligands, including substrates, cofactors, and inhibitors, are also represented as nodes within the graph, allowing the model to predict enzyme-ligand binding poses (how a molecule fits and orients itself inside an enzyme’s binding site), binding affinities (how strongly a ligand attaches to an enzyme), catalytic residues (amino acids that directly participate in the chemical reaction), and allosteric regulatory sites (places on the enzyme where molecules bind to control its activity without touching the active site). This evolution from static structure prediction to interaction modeling marks a critical step for enzyme engineering, especially in drug discovery and therapeutic enzyme design.
Despite these remarkable advances, significant challenges persist in the application of deep learning models to enzyme engineering. One of the main bottlenecks is the labor-intensive nature of curating high-quality datasets, including multiple sequence alignments, structural templates, and enzyme-ligand interaction data. The manual collection and preprocessing of such data require substantial domain expertise and computational resources, limiting the scalability and reproducibility of many structure-based prediction pipelines.
Multiple sequence alignments (MSAs) play a crucial role in AlphaFold by aligning homologous sequences, thereby revealing conserved residues (amino acids preserved through evolution), functional motifs (short, recurring patterns often associated with specific biochemical roles), and domains (structurally and functionally distinct regions of the protein)[204]. These features are key to accurate structure prediction and functional annotation. However, the performance of MSA-based models can degrade when applied to enzymes with low sequence diversity, recently engineered proteins, or orphan sequences lacking known homologs. These limitations hinder the applicability of such models to novel protein design problems, including de novo enzyme engineering. To address this, recent work has proposed hybrid modeling frameworks that dynamically adapt based on the depth of the MSA. For shallow MSAs, models employ autoregressive transformers trained on raw sequence data, while deeper MSAs allow the use of family-specific models enriched with evolutionary priors. This adaptive modeling strategy improves generalization across enzyme families and enhances robustness when dealing with underrepresented or engineered sequences[5].
A promising alternative or complementary approach to MSA, particularly effective for improving enzyme stability, is Ancestral Sequence Reconstruction (ASR). ASR leverages phylogenetic analysis to infer ancient protein sequences by tracing the evolutionary history of extant proteins through phylogenetic trees[57]. These reconstructed ancestral proteins often exhibit enhanced thermostability and robust folding, traits believed to be adaptations to harsher ancient environmental conditions. As such, they serve as valuable scaffolds for enzyme engineering[57]. Unlike traditional directed evolution, which relies on random mutagenesis and high-throughput screening of massive variant libraries, ASR narrows the sequence search space by integrating evolutionary constraints, thereby identifying sequences with innate functional resilience. ASR-generated scaffolds can also serve as pre-trained inputs for machine learning models, particularly those trained on MSA-derived embeddings, providing a stable foundation for further fine-tuning and property optimization in downstream tasks.
In a notable 2022 study, Hie et al.[103] introduced the concept of evo-velocity, utilizing embeddings from transformer-based protein language models such as ESM-1b[299] and the TAPE Transformer[102]. By modeling protein sequences as a flow through an evolutionary vector field, they were able to:
- Reconstruct plausible evolutionary trajectories,
- Detect horizontal gene transfer events,
- Arrange proteins in pseudotime to infer the relative timing of evolutionary divergence.
This evolutionary velocity, which quantifies the rate at which proteins evolve, provides additional insight into enzyme diversification, including phenomena such as epistasis. Epistasis describes how combinations of mutations at different residues produce non-additive functional effects, complicating predictions based on single-residue substitutions.
The study demonstrated that general-purpose protein language models are capable of capturing evolutionary rules solely from raw sequence data, with remarkable generalizability across evolutionary time scales—from short-term viral adaptation over years to long-term eukaryotic protein evolution spanning geologic eons[103]. This long-range evolution is shaped by mechanisms such as:
- Domain shuffling, which recombines existing structural modules to generate new protein functions;
- Gene duplication, which permits the functional divergence of proteins while preserving ancestral roles;
- Natural selection, which filters variants based on functional fitness in changing environments.
Based on these insights, a 2024 study introduced a novel model called AncFlow, which integrates phylo-genetic inference with AlphaFold-based structure prediction to model three-dimensional structures of ancestral proteins[37]. AncFlow enables detailed comparisons between ancestral and extant protein structures, revealing structural adaptations that have driven functional diversification within protein superfamilies, such as acyltransferases and dehydrogenases[37]. By uniting evolutionary modeling with geometric deep learning, AncFlow offers a powerful platform for investigating the molecular basis of enzyme evolution and for guiding the rational design of next-generation biocatalysts.
One notable approach in de novo enzyme design is ProteinGAN, which utilizes Generative Adversarial Networks (GANs). This method has demonstrated a 24% experimental success rate in producing functional enzymes, underscoring its potential to accelerate biocatalyst development [173].
A GAN consists of two neural networks:
- Generator. Creates new protein sequences.
- Discriminator. Acts as a quality control by distinguishing real sequences from generated (fake) ones.
These networks are trained through adversarial competition: the generator aims to "fool" the discriminator by producing sequences that resemble real proteins, while the discriminator improves its ability to detect fakes. Over time, this adversarial process enhances the generator’s capacity to produce realistic sequences that retain key biological properties.
A key disadvantage of ProteinGAN is that while it can generate novel protein sequences, it often lacks fine control over functional properties such as enzymatic activity, stability, or substrate specificity, making experimental validation and screening still essential to identify useful candidates.
Before the advent of deep learning, Rosetta was a leading tool for de novo enzyme engineering [335]. Rosetta employs fragment-based assembly to predict protein structures and refine atomic-level conformations. In this approach, short fragments from known protein structures are assembled using Monte Carlo sampling to generate native-like protein conformations. Each structure prediction involves running multiple short simulations from different random seeds to generate an ensemble of "decoy" structures, which are then clustered by similarity to identify the broadest free energy minima. This strategy helps identify conformations that balance local stability with global protein-like properties. Despite its strengths, Rosetta is computationally intensive, and each simulation requires hours to complete due to the exhaustive conformational sampling involved[335]. To address some of these limitations, modern adaptations such as RoseTTAFold[360] have integrated deep learning-based residue-residue contact predictions into the Rosetta framework, accelerating structure predictions and reducing reliance on fragment libraries. RoseTTAFold was designed as a three-track neural network with attention. RoseTTAFold’s accuracy was comparable to that of AlphaFold in CASP14[360] benchmark. RoseTTAFold’s results showed high consistency with the results of physical experiments and could help solve structures with molecular replacement methods[360].
Rather than relying solely on physics-based models, deep learning models can also be fine-tuned to learn and predict protein physics directly. This shift has led to advances in the prediction of enzyme kinetic parameters, which are crucial to understanding and optimizing enzyme function. The EF-UniKP framework, built on pretrained language models, refines these predictions using sequences, substrate structures, and environmental factors, including pH and temperature. By incorporating these diverse inputs, EF-UniKP has successfully identified high-performance enzyme variants such as Tyrosine ammonia lyase (TAL) mutants with enhanced activity, a 2.6-fold increase in kcat/Km for TrTAL[134]. The catalytic rate constant, or kcat, is a key kinetic parameter in enzymology that represents the number of substrate molecules an enzyme converts into product per second (the turnover number), under saturating substrate conditions. The kcat/Km ratio, often referred to as the enzyme’s catalytic efficiency, reflects how efficiently an enzyme converts substrate into product at low substrate concentrations, where Km represents the substrate concentration at which the enzyme reaches half of its maximum velocity. However, challenges persist in accurately predicting kcat values due to the limited availability of kinetic datasets and the diversity of protein sequences[134].
One of the primary bottlenecks in deep learning-driven enzyme engineering is that models require large amounts of high-quality data to achieve reliable performance. While massive protein sequence datasets, such as UniProt [317,321,324,328,329,330,331], the Big Fantastic Database[325], and structural repositories such as the Protein Data Bank[310], along with curated datasets[3,304,307,323,327,329,369,371,373,377,378,379,380,381], provide a valuable foundation for protein research, they often lack essential functional annotations, detailed mutation effects, and kinetic data, including substrate binding, product formation, and turnover rates. Although some specialized functional datasets and tools [298,301,303,304,306,308,316,319,320,346,372,374,375,376] address these gaps, they remain limited in scope. This limitation hampers the ability of deep learning models to accurately learn enzyme function and generalize across diverse protein families. As a result, generating high-quality labeled data remains a key challenge in advancing machine learning-driven protein engineering[55??].
Inductive biases play a crucial role in guiding machine learning models, shaping the hypothesis space explored during training. In enzyme engineering, these biases emerge from manually defined strategies or representation learning, such as embedding formation, with the chosen encoding method significantly influencing the depth and complexity of captured information [198].
Variations of BERT-based models have been utilized to form embeddings that serve as effective inputs for various tasks. These embeddings were validated across several tasks with impressive results: secondary structure (helix, sheet, coil) prediction (81%-87%), subcellular (nucleus, cytoplasm, mitochondria, etc.) localization (81%), and membrane protein classification (91%)[200]. These models outperformed the state-of-the-art for secondary structure prediction without relying on multiple sequence alignments or evolutionary data, avoiding the need for costly database searches, a significant departure from traditional methods used over the past three decades[200]. However, they did not perform well in identifying Enzyme Commission (EC) numbers [200].
To navigate the rapidly evolving field of enzyme engineering, Harding et al. propose two critical factors for selecting the most effective encoding strategy in machine learning applications: model setup and model objective. The former encompasses dataset size and machine learning architecture, while the latter considers protein properties, mutation effects, and interpretability of model predictions. By focusing on these aspects, researchers can tailor AI-driven approaches for greater accuracy and practical relevance [166].
Scaling transformer models has significantly advanced protein sequence modeling, enabling substantial improvements in sequence generation, secondary structure prediction, and functional annotation. Prominent models like ProGen2 (6.4B parameters) [154] and the ProtTrans family [200], trained on vast protein datasets, have achieved remarkable zero-shot performance across diverse protein engineering tasks, alleviating the need for domain-specific fine-tuning. These models demonstrate how transformer-based architectures can effectively learn evolutionary and structural priors directly from large-scale protein corpora.
Further scaling of protein language models has led to impressive breakthroughs in structure prediction. For instance, ESMfold [101], a 15B-parameter model, improves upon AlphaFold2 in scenarios lacking MSAs or structural templates. OmegaFold [214] similarly excels in predicting the structures of orphan proteins (proteins with few or no homologs) relying solely on primary sequence information to outperform traditional MSA-based methods. Such advancements highlight the versatility of large PLMs in tackling the diversity of protein fitness landscapes.
However, larger PLMs often face diminishing returns in narrow or rugged fitness landscapes, where minor sequence changes can have disproportionately large impacts on functionality [158]. Addressing this challenge, the MODIFY model leverages a dual-objective optimization strategy to balance high fitness with structural diversity, preventing convergence toward local optima. MODIFY integrates zero-shot fitness prediction models, evolutionary strategies, Pareto optimization, and structure-based filtering, allowing the design of structurally diverse proteins with desirable functional properties [36]. Its zero-shot learning capability enables fitness prediction for novel sequences by embedding proteins in a functional space, where similarity to known functional proteins aids in estimating potential fitness.
To further enhance adaptability, test-time training (TTT) has emerged as a dynamic technique that refines model predictions during inference[226]. Unlike conventional models, which remain static after pretraining, TTT enables continued learning on small tasks such as minimizing sequence perplexity or predicting masked amino acids before making final predictions. This adaptability is especially beneficial in protein science, where complex and diverse sequence landscapes demand fine-tuned understanding. In structure prediction, TTT has boosted the performance of ESMFold[101] and ESM3[215] on challenging proteins, while in fitness prediction, TTT-enhanced models like SaProt[7] and ESM2[215] have set new benchmarks in areas such as "Organismal Fitness" and "Binding" [226].
This shift, from simply making models larger to improving their representations and adaptability, marks a positive step forward for protein engineering. By focusing on smaller, more efficient models, it becomes possible to achieve better accuracy and lower computational costs.
For example, Ankh introduces a data-efficient and cost-effective protein language model that achieves superior embedding quality while reducing pretraining and inference costs. Comparing calculation cost to ProtT5-XL-U50[200], ESM-1b[359] and the ESM-2 series[358] (650M, 3B, and 15B parameters), Ankh spent less than 10% for pre-training, less than 7% for inference, and less than 30% for the embedding dimension[223]. By focusing on optimizing representations rather than increasing model size, Ankh predicts missing or masked sequence with evolutionary priors, which biases the model to predict biologically significant positions more accurately[223]. This reduces the need for larger datasets or exhaustive training.
xTrimoPGLM, with its impressive 100 billion parameters, advances protein engineering by integrating both protein understanding and generation through a dual-objective approach of masked language modeling and next-token prediction. By optimizing these complementary tasks, xTrimoPGLM achieves outstanding performance across 13 diverse protein engineering benchmarks. Notably, it surpasses Ankh by 11% in predicting fitness for GB1 protein mutations and outperforms AlphaFold2 in antibody structure prediction, achieving a TM-score of 0.961 [224].
xTrimoPGLM eliminates MSAs and reduces folding blocks and focuses on a proficient encoder, which captures structural information during pretraining. The absence of MSA searches and shallow folding layers drastically reduces runtime. In AlphaFold2, 48 Evoformer blocks are used to refine the embeddings before passing them to the Structure Module[204]. This makes AlphaFold2 highly accurate, but also computationally expensive. xTrimoPGLM reduces the number of Evoformer layers to just 1 without substantial performance losses[224].
Traditional protein language models excel at learning co-evolutionary patterns from sequence data but often lack explicit awareness of protein function, which is the ultimate goal of protein representation learning. To address this, ProtST introduces a multimodal approach by augmenting sequences with textual property descriptions such as function, localization, and family information to guide pretraining with biologically meaningful supervision [9].
Although natural proteins are incredibly diverse, many have subtle structural patterns because of evolutionary pressures. This makes it challenging for generative models that don’t use structural information, as they often fail to produce accurate results for sequences that are different from those they were trained on[7,197].
Built upon the ProtDescribe dataset, ProtST enhances PLMs through a combination of unimodal and multimodal tasks: mask prediction, representation alignment, and multimodal mask prediction. These enable models to learn not just from sequence, but from text-based functional context, improving both whole-protein property prediction and residue-level understanding. For enzyme engineering, this multimodal learning framework offers a powerful tool to bridge the gap between sequence patterns and functional specificity, supporting applications such as active site identification, function annotation, and protein redesign.
OPUS-GO utilizes a modified Multiple Instance Learning (MIL) strategy and outperforms baseline methods in sequence-level classification tasks. MIL, a technique designed to handle weakly labeled data, allows OPUS-GO to improve classification by treating individual residues as instances within a sequence and learning to associate them with the overall sequence label. This approach allows for robust residue-level interpretability while maintaining strong sequence-level accuracy. Furthermore, OPUS-GO accurately identifies residues linked to specific labels. This framework can be integrated into any language model, improving both accuracy and interpretability for downstream tasks[113].
However, OPUS-GO’s interpretability is not designed to identify active sites but rather the residues most directly related to the labels. Analysis of the EC number "5.3.3.2" (Isopentenyl-diphosphate Delta-isomerase) reveals that OPUS-GO identifies consistent, conserved patterns near binding sites, which may be critical for enzyme function, even if they are not active sites [113].
Traditional computational approaches, such as homology-based methods and general machine learning models, often fall short in capturing the full complexity of enzyme active sites and structural features, which are crucial for accurately predicting enzyme function. For example, sequence-based tools like BLAST may misclassify proteins that have similar sequences but different functions, as they typically overlook subtle yet critical structural and functional distinctions [153].
To address these limitations, tools such as GEMME refine sequence conservation analysis by filtering homologous sequences using criteria such as sequence identity (the degree of similarity between amino acid sequences), length coverage (ensuring aligned sequences span most of the target protein), and alignment quality (removing gapped or poorly aligned sequences). This filtering allows GEMME to focus on the most informative homologs, improving the detection of conserved residues important for enzymatic activity [6].
Generative models such as RFDiffusion and RFdiffusion All-Atom (RFdiffusionAA) leverage diffusion models to generate de novo protein backbones and binding interfaces [12]. These models conceptualize structure generation as a denoising process—starting from random coordinates and iteratively refining them into physically plausible protein conformations. However, despite their success in producing realistic protein scaffolds, RFdiffusion models often struggle with the nuanced modeling of protein-ligand interactions and demonstrate limited generalizability across diverse and functionally distinct enzyme families [219].
Similar to GEMME, AlphaProteo [221] leverages structural priors—learned from both experimentally resolved protein structures and AlphaFold-predicted models—and applies sequence filtering to guide de novo protein design. Unlike traditional refinement-based approaches, AlphaProteo directly generates novel protein binders by proposing both the amino acid sequences and their corresponding 3D structures tailored to bind specific target sites. While AlphaFold predicts the structure a given sequence is likely to adopt, AlphaProteo effectively inverts this process by designing sequences that are expected to fold into a desired structure and engage a defined binding region on the target protein [221].
This inversion is particularly transformative in enzyme engineering, where the ability to create custom binders offers precise control over molecular interactions. Traditional binder development methods—such as immunization, directed evolution, or the use of scaffold-based systems like antibodies, nanobodies, and DARPins—are often time-consuming and provide limited control over epitope specificity [221]. In enzymatic contexts, epitopes may include active sites, regulatory loops, or structural motifs essential to catalytic function. In contrast, computational design enables the creation of binders that selectively target user-defined epitopes with high precision, and can yield molecules that are smaller, more thermostable, and easier to express than conventional alternatives.
Expanding on this idea, the design of protein sequences that interact with non-protein molecules such as small molecules, nucleotides, and metals is equally critical in enzyme engineering, particularly for building sensors, inhibitors, and catalytic frameworks. Addressing this need, LigandMPNN introduces a deep learning-based sequence design approach that explicitly models all non-protein components within biomolecular systems [295]. Unlike Rosetta or ProteinMPNN, LigandMPNN achieves significantly higher sequence recovery rates at ligand-contacting sites, including 63.3% for small molecules (vs. 50.4% and 50.5%), 50.5% for nucleotides (vs. 35.2% and 34.0%), and 77.5% for metals (vs. 36.0% and 40.6%). Furthermore, it also predicts sidechain conformations, allowing a detailed evaluation of binding energetics.
To identify and refine functional features such as binding pockets and to distinguish catalytic from non-catalytic residues, the PocketGen model uses an equivariant bilevel graph transformer. The model processes atom-level information (fine-grained details) and residue-/ligand-level information (higher-level, block-based features) separately but in a coordinated way using bilevel attention module, which improves both local precision and global context. At the atom level, a small neural network encodes the distances between atom pairs to guide attention toward closer atom pairs, as closer atoms are more likely to interact. To focus on the most relevant interactions, the model keeps only the top attention scores and sets the rest to zero, encouraging sparsity.Then, the model aggregates atom-level attention to calculate block-level attention between residues or ligands. After computing attention, the model updates each atom’s feature vector and 3D coordinates using specialized update equations that maintain equivariance. PocketGen uses E(3) equivariance, so that the model’s outputs respect the Euclidean transformations in 3D space (translations, rotations, and reflections)[219].
To address the challenges posed by weak or unclear structural signals, contrastive learning has emerged as a powerful approach. By focusing on functional similarities rather than relying solely on structural details, contrastive learning enhances model generalization. For example, the CLEAN model applies contrastive learning to improve enzyme commission (EC) number prediction, a key aspect of enzyme function classification [201]. This is achieved by learning an embedding space where enzyme sequences with the same EC number are positioned closer together, while those with different EC numbers are farther apart. Thus, sequence-function relationships that may be overlooked due to the imbalanced distribution of EC numbers are learned. By refining protein representations from the ESM-1b and the use of contrastive losses, CLEAN achieves superior accuracy, reflected in its 0.865 F1 score [201].
Rather than directly predicting EC numbers, GraphEC enhances enzyme function prediction by first identifying enzyme active sites, which helps guide the prediction of EC numbers for less-studied enzymes. Using a label diffusion algorithm, GraphEC propagates labels through a similarity graph to incorporate homologous information, ensuring that related enzymes share functional annotations. This process improves the accuracy of EC number prediction and even extends to predicting optimal pH values, providing a more comprehensive understanding of enzyme functionality. Importantly, GraphEC’s geometric graph learning framework allows it to capture functional information from protein structures, even when homologous sequences are unavailable, highlighting its versatility and effectiveness in enzyme function prediction [225].
Protein structure predictions from tools such as AlphaFold2 can be integrated into contrastive learning frameworks to enhance structural awareness. The Hierarchical Equivariant Active Learning (HEAL) framework takes this approach by employing a hierarchical graph transformer. This model uses super-nodes to represent functional motifs such as binding pockets or active sites [205].This setup allows HEAL to focus on semantic interactions (how different motifs within the protein relate to each other) both locally (within small regions) and globally (across the entire protein structure). By aggregating the embeddings of these super-nodes with varying emphasis, HEAL generates a comprehensive graph that captures the protein’s overall structure-function relationship[205].
To lessen dependence on homology-based methods, Chai-1, a multi-modal structure prediction model, supports single-sequence input and integrates protein language model embeddings, enabling accurate predictions even when homologs are limited [297]. This is especially valuable for novel or engineered enzymes. One of Chai-1’s key innovations is its ability to incorporate experimental constraints, allowing it to integrate real-world biochemical data into structural predictions. These constraints include:
- Pocket data. Identifies likely active or binding sites within enzymes.
- Contact constraints. Specifies residues that should be spatially close, mimicking atomic contacts.
- Docking data. Provides orientation or distance info between enzyme and ligand or between protein subunits.
Such constraints can be derived from cross-linking mass spectrometry (XL-MS) or hydrogen-deuterium exchange mass spectrometry (HDX-MS), which are experimental techniques that probe spatial proximity and flexibility in enzymes. This integration is especially useful for predicting multi-domain architectures or protein-ligand interactions relevant to catalysis and drug design.
Importantly, Chai-1 uses dropout during training to avoid overfitting to these inputs, allowing it to generalize well even in their absence. The model has been rigorously benchmarked on low-homology multimeric interfaces, where it outperforms AlphaFold-Multimer 2.3 in predicting the orientation and assembly of protein complexes—an essential capability for mapping enzyme-enzyme interactions in metabolic pathways [297].
When evaluating structural prediction accuracy, it’s important to recognize that experimental structure determination itself has inherent variability, which is especially critical in drug discovery and enzyme-target design [341]. Tools such as Chai-1 are therefore judged not only by coordinate error but by their ability to produce results consistent with the range of plausible experimental outcomes.
Table A1, Table A2 in the Appendix A list various models used for protein design, showcasing different approaches and methodologies.
5. Interoperability and Assessment
The "Double-Edged Sword" effect of AI transparency highlights the challenge of balancing its benefits such as fostering trust and enhancing user control with its drawbacks, like cognitive overload, emphasizing the need for user-centric approaches that provide clear, actionable insights without overwhelming users [171].
Neural networks, though highly accurate, often operate as black boxes with unexplained decision-making, highlighting the role of knowledge engineering and the use of knowledge graphs, which organize information, capture relationships, enable semantic queries, and leverage ontologies to ensure interoperability, as demonstrated in applications like gene prioritization in drug discovery and catalysis research analysis[168].
Moreover, knowledge graphs have demonstrated their utility in drug discovery, catalysis research, and prioritizing target genes, but their broader adoption in cancer research is still in its infancy [168]. Leveraging these tools could bridge the gap between raw data and actionable insights, fostering a more robust, ethical, and reproducible framework for future studies.
General-purpose AI models for proteins and large molecules leverage diverse representations, such as residue sequences and 3D structures, to perform tasks such as protein folding prediction, novel protein generation, and functional protein design. These models align with the broader definition of general-purpose AI systems, with advancements enabling the creation of proteins with predictable functions across extensive protein families. Evaluating these systems requires understanding their capabilities and societal impacts, ensuring they meet performance expectations while addressing potential risks such as safety concerns and unintended consequences. Although benchmarks provide valuable insights, they face limitations in fully capturing real-world performance and downstream harms[192].
Large AI models are inherently opaque, with unpredictable[350] and often enigmatic behaviors, and while techniques such as chain-of-thought prompting[349,351,352] and mechanistic interpretability provide some insights, their ability to truly reflect the models’ internal reasoning remains uncertain[222]. These models, lacking specialized training in protein biology, often struggle to interpret conserved motifs, assess the functional impact of mutations, and model the intricate sequence–structure–function relationships that are essential for rational enzyme design [309].
To address these shortcomings, bioinformatics-focused LLMs have been developed and show improved performance on tasks such as protein structure prediction, DNA sequence generation, and functional annotation. However, these domain-specific models still face three major challenges: generalizability to unseen data, scalability across tasks and architectures, and flexibility to adapt to diverse biochemical contexts [193].
Benchmarking studies on tasks from the TAPE benchmark[102] demonstrate that increasing the size of the model and training data can improve scalability. However, achieving the generalizability required to design novel enzymes or predict the effects of distant mutations[102], as well as the flexibility to handle hybrid inputs (e.g., protein-ligand or protein-nucleotide systems), remains an active area of research [295,297,344].
By combining strengths of Bioinformatics LLMs and General-purpose LLMs predictions can be enhanced. LLMs trained on textual descriptions of crystal structures exhibit performance comparable to earlier techniques, such as graph neural networks. However, more accurate predictions can be achieved through the use of a specialized model that directly learns from text representations of the structural data. This specialized model not only improves predictive accuracy but also enables the generation of clear, interpretable explanations regarding the factors that influence the synthesize-ability of a given structure[190].
The choice of performance metrics plays a crucial role in determining model architecture. Likewise, some benchmarks were developed to test model performance[94]. While AlphaFold2[204] has significantly advanced protein modeling, gaps persist between predicted and observed model quality assessment scores, particularly for quaternary structures. For example, Predicted Local Distance Difference Test (plDDT) scores align well with observed lDDT-C scores for tertiary models, but demonstrate lower accuracy for quaternary models, with significant overprediction in low-quality structures [218]. The plDDT is a per-residue confidence metric used by AlphaFold2 to predict the accuracy of atomic positions in the 3D structure compared to the true native structure, making it particularly useful for assessing the reliability of specific regions, such as loops and secondary structures[204]. The lDDT-C score, which compares the distances between alpha carbon atoms (C) in the predicted and reference structures, also provides an indication of structural accuracy, with higher values (ranging from 0 to 100) suggesting a closer match to the native structure and being especially useful for evaluating local accuracy in specific regions[210].
Models can be optimized to favor certain metrics; for instance, pLDDT-Predictor[348], which accelerates protein screening by 250,000 times compared to AlphaFold2, uses pre-trained ESM2[215] embeddings and a Transformer architecture to predict pLDDT scores, achieving a Pearson correlation of 0.7891 with AlphaFold2’s predictions and demonstrating 91.2% accuracy in classifying high-confidence structures, all while also quickly processing proteins in 0.007 seconds [348]. Pearson correlation measures the linear relationship between two continuous variables. It quantifies how strongly and in what direction two variables are related.
Similarly, Predicted Template Modeling (pTM) scores correlate well with Template Modeling scores (TM-scores) for higher-quality models but exhibit overprediction for lower-quality quaternary models, revealing variability in AF2’s confidence metrics [218]. The TM-score is a normalized measure used to assess the topological similarity between two protein structures, independent of protein length[211]. The pTM extends this concept as a global confidence metric, estimating the overall quality of predicted structures and their global fold similarity to reference structures, making it particularly useful for evaluating the reliability of protein models[204].
Benchmarking protein design methods is crucial, though existing benchmarks have notable limitations, as experimental validation of generated sequences remains costly. In silico proxies such as sequence recovery and perplexity metrics provide alternatives but fail to capture real-world foldability, as high sequence similarity does not guarantee correct folding due to potential misfolding caused by single mutations, as seen in diseases such as Alzheimer’s and cystic fibrosis[217]. Perplexity evaluates the uncertainty in a model’s predictions but provides only a point probability mass, rather than a true distribution.
New efforts, such as the PDB-Struct benchmark, specifically designed for structure-based protein design, mark the first comparison of encoder-decoder and structure-prediction-based methods. This helps protein scientists choose the right tools by identifying the strengths and weaknesses of each model, while also introducing two novel metrics: "refoldability," which assesses how well designed structures match input templates using TM score and pLDDT, and a "stability-based metric," which evaluates how accurately methods predict experimental stability using high-throughput datasets and Spearman correlations [217]. Spearman correlation is a non-parametric measure of rank correlation that assesses the strength and direction of the monotonic relationship between two variables. It is based on the ranks of the values rather than their raw scores, making it useful for identifying whether the variables tend to increase or decrease together, even if the relationship is not linear.
To advance the field of enzyme engineering, it is essential to evaluate not only the structural fidelity of designed proteins but also their functional and computational viability. Modern scaffold design workflows like Scaffold-Lab exemplify how benchmark metrics such as novelty, diversity, and efficiency can be integrated to guide the generation and selection of promising protein backbones for downstream engineering tasks [213]. These metrics help optimize protein design for both structure and function, enabling the creation of novel enzymes tailored for specific biochemical tasks.
In Scaffold-Lab, designability measures how easily a generated scaffold can be assigned diverse amino acid sequences that fold correctly under real-world conditions. To ensure a balance between novelty and designability, penalties are applied based on sc-TM scores, with a designability threshold set at 0.5[213]. The sc-TM score quantifies how consistently a predicted protein structure aligns with itself across different iterations, providing insights into self-consistency and robustness [203].
In contrast, novelty is determined by comparing generated backbones to proteins in the protein data bank, using the highest TM-score (pdb-TM) as a reference. A lower pdb-TM score indicates greater deviation from the native fold, which may affect functional viability[213]. To measure novelty, Alphaproteo designers searched each design sequence against Uniref50 using Jackhmmer and considered it novel if its maximum bit-score is less than 50[221].
To assess diversity, protein backbones are clustered using Foldseek-Cluster[347], a computational tool that efficiently compares and groups protein structures based on structural similarity. By applying a TM-score threshold of 0.5, this approach determines the proportion of unique clusters relative to total backbones, reflecting the model’s ability to generate diverse structural topologies—an essential aspect of novel protein design [213].
However, structural fidelity remains a core requirement. Metrics such as Global Distance Test (GDT), Longest Continuous Segment (LCS), and Contact Area Difference score (CAD-score) are used for validating whether generated backbones are not only novel but also physically plausible and functionally relevant[382]:
- RMSD (Root-Mean-Square Deviation) is a foundational metric that calculates the average distance between atoms of superimposed structures. It provides a straightforward measure of structural similarity but is sensitive to outliers and may not accurately reflect functional or local similarities.
- GDT addresses RMSD’s limitations by focusing on the percentage of residues that deviate by less than a set distance threshold (e.g., 1Åor 2Å). It provides a more holistic view of structural similarity, particularly effective for evaluating predictions of multi-domain or flexible proteins.
- LCS complements GDT by identifying the longest uninterrupted stretch of residues that can be superimposed under an RMSD threshold. It emphasizes local consistency, which is crucial in identifying preserved motifs and active sites in enzyme design.
- CAD-score measures the difference in residue-residue contact areas between a model and a reference. It is calculated using Voronoi tessellation to evaluate contact surfaces and is particularly effective in judging physical realism. Unlike RMSD and GDT, CAD-score can directly assess the accuracy of domain interfaces and multimeric assemblies.
These scores, originally developed through rigorous assessments such as CASP and CAMEO, provide insight into local and global structural similarities, enabling robust evaluation of protein candidates regardless of sequence similarity [345,382,384].
In conditional generation tasks, particular emphasis has been placed on motif-scaffolding, where models such as GPDL-H [11] and RFdiffusion [12] have shown high success rates in preserving structural motifs while optimizing scaffold flexibility. These approaches highlight the significance of precise motif incorporation and the need for refined generative strategies in protein design [213]. By integrating machine learning-driven scaffold generation with structural validation metrics, researchers can accelerate the development of customizable protein architectures.
Furthermore, recent tools such as PoseBusters highlight the importance of validating docking predictions through chemical and geometric plausibility checks, which is particularly relevant for enzyme-substrate interaction modeling [344]. These checks go beyond RMSD and help ensure that designed enzymes will exhibit not just structural accuracy, but also realistic active site conformations and ligand binding modes critical for catalysis. Validation becomes even more critical in deep learning-based docking, where models may achieve low RMSD yet generate chemically implausible structures. To address this, PoseBusters introduces a battery of physical and chemical plausibility checks—including planarity, bond lengths, and steric clashes—ensuring that predicted poses are not only accurate in position but also chemically realistic[344]:
- Planarity. Many parts of biomolecules, especially aromatic rings like those in phenylalanine or tyrosine, are expected to be flat due to the nature of their chemical bonds. If a predicted structure distorts this flat shape, it may signal a physically unrealistic conformation that wouldn’t be stable in real conditions.
- Bond lengths. Atoms in proteins are held together by covalent bonds with known average distances. Deviations from these expected values suggest the model may have introduced structural artifacts, which could affect protein stability or activity.
- Steric clashes. In real proteins, atoms are spaced so that they don’t overlap. If two atoms are too close, closer than their physical space allows, it creates a steric clash, indicating an impossible structure. These clashes often arise from poor side-chain packing or inaccurate folding predictions and can disrupt protein function.
Beyond structural fitness, efficiency plays a crucial role in high-throughput applications. It is measured by the average runtime per backbone across identical hardware setups, providing a standardized evaluation of computational resource optimization. This ensures a balance between cost and generative performance, making large-scale protein design more feasible [213].
To enable comprehensive model assessment, benchmark suites such as PEER provide a unified framework for evaluating multiple tasks: structure prediction, functional annotation, and interaction modeling, all in one place [?]. This is particularly valuable for enzyme engineering, where structure and function are deeply intertwined.
At a broader level, platforms such as Aviary are pushing the boundaries of biological reasoning using language-based agents. Aviary treats tasks such as experimental planning, data interpretation, and molecular design as decision-making problems in language-grounded environments [195]. These environments require agents to perform multi-step reasoning under uncertainty, much like a researcher navigating protein optimization strategies. Remarkably, Aviary shows that open source language models can compete with both human experts and advanced proprietary systems, all while maintaining low inference costs. This opens up new avenues for integrating natural language reasoning into the enzyme design pipeline.
Table B1 in the Appendix B list commonly used benchmarks to evaluate protein design and structural prediction methods.
6. Cancer and the Role of Enzymes in Cancer Study
A normal cell transforms into a cancerous cell when the regulatory mechanisms controlling cell division become defective, leading to uncontrolled proliferation. Unlike diseases that result from loss of function, cancer arises due to genetic mutations that activate oncogenes or inactivate tumor suppressor genes, driving the cell cycle beyond normal constraints. Transformed cells exhibit different behaviors, including loss of contact inhibition, the ability to grow in multiple layers, and reduced dependence on growth factors, which allow them to survive and divide indefinitely. These cancerous traits often originate from mutations in proto-oncogenes, which, when altered, become oncogenes that stimulate continuous cell division, much like a car with a stuck accelerator[287].
Oncogenes are mutated or overexpressed versions of normal genes (proto-oncogenes). Activation mechanisms include point mutations, gene amplifications, and chromosomal rearrangements, resulting in increased oncogene expression or function of the oncogene[279].
Retroviruses can induce oncogenesis by integrating their genetic material into the host genome, a process that involves reverse transcription of viral RNA into DNA. The classic dogma of molecular biology (DNA → RNA → Protein) was challenged by the discovery of reverse transcription. David Baltimore’s discovery of reverse transcriptase earned him a Nobel Prize, highlighting the significance of RNA viruses in genetic transformation[277].
This integration can lead to the activation of oncogenes, thereby altering host cell proliferation. For example, the Rous sarcoma virus (RSV) introduces the src gene, which encodes a tyrosine kinase that, when deregulated due to loss of its inhibitory region in viral form (v-Src), drives uncontrolled cell growth and oncogenic transformation[283]. Kinases are crucial enzymes that regulate cellular processes by adding phosphate groups to proteins, a process called phosphorylation, which typically occurs in amino acids such as serine, threonine, or tyrosine. This modification can alter the shape, activity, or interactions of a protein, allowing cells to respond dynamically to various signals, such as turning proteins on or off to regulate processes such as glycogen breakdown for energy. The activity of kinases is tightly regulated by phosphatases, which remove phosphate groups, ensuring a balanced and reversible process that is essential for maintaining proper cellular function and adapting to environmental changes[281].
Tumor suppressor genes regulate cell division, repair DNA errors, and initiate apoptosis. Loss-of-function mutations in these genes remove critical controls on cell growth[271]. A notable example is the TP53 gene, which encodes the p53 protein that prevents the proliferation of cells with damaged DNA[267]. Mutations leading to inactivation of TP53 are found in many types of cancer, allowing cells to grow unchecked[268].
Cells can die through necrosis, caused by acute injury, or apoptosis, a regulated process essential for normal tissue maintenance. Apoptosis occurs when cells do not repair extensive DNA damage, and its inhibition is a key factor in cancer development. Genes such as MYC, BAX, and P53 promote apoptosis, while BCL2, ABL, and RAS inhibit it, altering the balance between cell survival and death[284]. The goal of cancer therapy is to promote the death of cancer cells without causing too much damage to normal cells.[286].
Apoptosis is primarily regulated through two key pathways[280]:
-
Intrinsic Pathway. Triggered by internal cellular stressors such as DNA damage or oxidative stress, this pathway involves the mitochondria releasing cytochrome c into the cytoplasm. This release activates caspases, leading to cell death.Triggered by internal cellular stressors such as DNA damage or oxidative stress, the intrinsic apoptotic pathway is primarily regulated by mitochondria, the energy-producing organelles of the cell. Oxidative stress occurs when an imbalance between reactive oxygen species and antioxidants leads to cellular damage, often compromising mitochondrial function. In response, mitochondria release cytochrome c, a small heme protein that normally facilitates electron transfer in the electron transport chain but, when released into the cytoplasm (the gel-like substance filling the cell and surrounding all organelles), acts as a key apoptotic signal. Once in the cytoplasm, cytochrome c activates initiator caspases. Caspases are a family of proteolytic enzymes - cysteine proteases that cleave target proteins at specific aspartic acid residues, orchestrating a controlled sequence of cellular disassembly. Once activated, executioner caspases systematically degrade cellular components, leading to programmed cell death[280]. This controlled dismantling of the cell ensures minimal damage to surrounding tissues and prevents inflammation, highlighting the tightly regulated nature of apoptosis in maintaining cellular homeostasis.
- Extrinsic Pathway. Initiated by external signals, these signals come from specialized molecules called death ligands, such as Fas ligand (FasL) or tumor necrosis factor (TNF), which bind to death receptors on the cell surface. When a death ligand binds to its matching receptor (e.g., Fas receptor or TNF receptor), it triggers the formation of a protein complex known as the death-inducing signaling complex. This complex directly activates initiator caspases, which function similarly to caspases in the intrinsic pathway[278].
Cancer research faces significant challenges due to extreme interpatient and intratumor heterogeneity, meaning that cancer cells within the same tumor or across different patients can behave unpredictably. A major limitation in understanding cancer biology stems from our scarce knowledge of genome-wide mutational landscapes and epigenetic modifications, which regulate how genes are turned on or off without altering the DNA sequence itself. These mutations and epigenetic changes can profoundly affect transcriptional processes—the way genetic information is copied from DNA to RNA—leading to unpredictable gene expression patterns that drive cancer progression and drug resistance. Additionally, disruptions in signal transduction networks, the molecular pathways that transmit information inside cells, can cause cancer cells to evade normal growth controls and resist therapies. Without a clear grasp of how these genetic and epigenetic alterations regulate cellular processes, it remains difficult to develop precise, long-lasting cancer treatments, highlighting the urgent need for further research in this area[278].
In a landmark review, Hanahan and Weinberg suggested that six essential alterations in cell physiology underlie malignant cell growth [269]. These six hallmarks of cancer include:
- Self-sufficiency in growth signals,
- Insensitivity to growth inhibitory (antigrowth) signals,
- Evasion of programmed cell death (apoptosis),
- Limitless replicative potential,
- Sustained vascularity (angiogenesis),
- Tissue invasion and metastasis.
Genome instability, leading to increased mutability, was considered the essential enabling characteristic for manifesting these hallmarks.
In addition to the six hallmarks, aerobic glycolysis, or the Warburg effect, is recognized as a robust metabolic hallmark of most tumors [293]. Normally, cells use oxidative phosphorylation in the mitochondria when oxygen is present because it is a much more efficient way to produce ATP. If oxygen were completely unavailable, the process would instead be anaerobic glycolysis, which occurs under low oxygen conditions, such as muscle cells during intense exercise. However, cancer cells exhibit aerobic glycolysis, where they rely on substrate-level phosphorylation in the cytoplasm instead of mitochondrial respiration[276]. While glycolysis produces less ATP per glucose molecule than mitochondrial respiration, it provides cancer cells with an advantage: it generates metabolic intermediates that fuel biosynthetic pathways. These intermediates are essential for producing nucleotides, amino acids, and lipids, which are the building blocks needed for cell division and tumor expansion.
A perspective that conceptualizes cancer as a metabolic disease highlights the dual role of enzymes as both drivers of tumor progression and potential therapeutic targets. Enzymes regulate key metabolic pathways that support cancer cell proliferation, immune evasion, and drug resistance. Investigating enzyme expression patterns provides critical insights into their function as cancer biomarkers, facilitating early detection, prognosis assessment, and treatment monitoring. Furthermore, targeted inhibition of specific enzymatic processes represents a promising strategy for suppressing tumor growth and enhancing therapeutic efficacy.
In this context, enzymes can be leveraged in three key ways:
- as drug delivery facilitators,
- as direct therapeutic agents,
- as metabolic regulators influencing cancer proliferation.
Figure 2 shows an overview of the role of enzymes in cancer diagnostics and therapeutics.
The following sections explore these applications, emphasizing their potential to improve cancer treatment outcomes.
6.1. Enzyme-Aided Drug Delivery
Cancer treatment strategies are diverse and tailored to individual patient needs, encompassing:
- Surgery. Optimal for localized tumors, surgical resection aims to remove the tumor entirely, offering the best chance for a cure when the cancer is confined to a specific area [263].
- Chemotherapy. This systemic therapy targets rapidly dividing cells, effectively treating cancers that have spread beyond the primary site. However, chemotherapy can also affect healthy cells, leading to systemic toxicity [263].
- Radiotherapy. Utilizing ionizing radiation, radiotherapy aims to kill cancer cells or inhibit their growth. It is often used in conjunction with other treatments to enhance effectiveness [263].
- Targeted Therapy. This approach uses drugs or other substances to precisely target cancer cells, often by inhibiting specific molecules involved in tumor growth. For example, imatinib is a kinase inhibitor used to treat Philadelphia chromosome-positive leukemia [274].
- Small Molecule Targeted Agents. These therapies inhibit specific molecular targets involved in cancer progression [278].
- Antibody-Drug Conjugates (ADCs). ADCs combine monoclonal antibodies, which are designed to recognize specific proteins on cancer cells, with cytotoxic agents, delivering chemotherapy directly to cancer cells while minimizing damage to normal tissues [274].
- Cell-Based Therapies. Chimeric Antigen Receptor (CAR) T-cell therapies involve modifying a patient’s T cells to attack cancer cells [274].
- Gene Therapy. This approach involves altering the genetic material within a patient’s cells to treat or prevent disease. Gene therapies aim to correct genetic defects or enhance the immune system’s ability to fight cancer [274].
ADCs are often referred to as a "biological missile", where the antibody serves as the “guidance system” and the toxin acts as the “warhead”, allows for precise “strikes” on specific targets [274]. The ADC consists of five core components:
- Target antigen: Must possess high specificity, low exfoliative properties to minimize shedding of the antigen into the bloodstream, endocytosis capabilities to facilitate the internalization of the ADC into the cancer cells.
- Antibody: Binds to the specific antigen on the target cell.
- Linker: Should be stable, specific for tumor conditions, and allow efficient toxin release. Linkers can be divided into cleavable linkers (chemical cleavage linkers, enzyme catalyzed cleavage linkers, photo-cleavable linkers) and non-cleavable linkers (sulfide bond linkers, maleimide bond linkers); cleavable linkers are a prerequisite for exerting bystander killing effect, hence becoming the mainstream trend of ADC linkers.
- Toxin: Must exhibit strong cytotoxicity, high stability, and controlled degradation. Within the cell, the toxin is released after being degraded by lysosomes, ultimately leading to the apoptosis of the target cell.
- Coupling method: Influences drug uniformity and loading for optimized therapeutic effects.
Unlike ADCs that rely on tumor-specific antigen recognition, prodrugs are designed to exploit metabolic differences between cancerous and normal cells to achieve selective activation at the tumor site. This enzyme-substrate specificity is often harnessed in cancer therapies to activate cytotoxic agents directly within the tumor microenvironment (TME) [236].
The TME, comprising abnormal vasculature, acidic pH, hypoxia, and immune evasion, creates a unique biochemical landscape that affects both tumor progression and treatment response. Abnormal vasculature leads to disorganized, leaky blood vessels that hinder drug delivery and oxygen transport. Acidic conditions arise from increased lactate production that promotes survival and resistance in tumor cells. Hypoxia, caused by poor blood flow, contributes to aggressive phenotypes, while immune evasion allows tumors to avoid detection by the host immune system [236].
Stimuli-responsive prodrugs have emerged as a promising strategy that leverages these tumor-specific environmental cues. These systems, including polymer–drug conjugates, are designed to release their therapeutic payloads in response to triggers such as pH, reactive oxygen species, or enzymatic activity. Increasingly sophisticated prodrugs incorporate Boolean logic gates (e.g., "AND" or "OR") to enable multistimuli responsiveness, enhancing targeting precision [235,236].
Earlier designs focused on single-signal activation; however, recent advances in chemical engineering and artificial intelligence have enabled the development of prodrugs that respond to multiple endogenous and exogenous signals simultaneously [50]. AI-driven approaches facilitate this innovation by modeling complex activation patterns and guiding molecular design. Still, the real-time monitoring of in vivo prodrug activation remains a significant challenge. To address this, modern prodrugs can be functionalized with targeting ligands and imaging agents, allowing for enhanced specificity and real-time visualization of drug release [235].
One particularly targeted strategy in enzyme-based cancer therapy is gene-directed enzyme prodrug therapy (GDEPT), which involves delivering genes encoding prodrug-activating enzymes directly into tumor cells. These genes are typically introduced via viral vectors or nanoparticles [129]. Once expressed, the enzymes selectively metabolize systemically administered prodrugs into their active, cytotoxic forms within the tumor, thereby reducing systemic toxicity and improving therapeutic efficacy. For example, the human CYP2B6 gene encodes the cytochrome P450 enzyme CYP2B6, which can convert cyclophosphamide into its active form. When this gene is introduced into tumor cells, the enzyme is produced locally, ensuring that prodrug activation occurs precisely where it’s needed [129]. A major limitation of GDEPT is the efficiency of enzymatic conversion. To address this, enzyme engineering has been applied to improve the catalytic activity, specificity, and stability of these activating enzymes. Engineered enzymes, such as E. coli NfsB mutants, have demonstrated up to a tenfold increase in prodrug activation compared to wild-type forms, showing great promise for enhanced cancer therapy [127].
The enzymatic activation of prodrugs typically involves the cleavage of a linker or chemical transformation of the drug molecule. For instance, matrix metalloproteinase-2 (MMP-2) cleavable linkers release active drugs only in regions where MMP-2 is overexpressed, such as the tumor microenvironment. This approach ensures spatially confined activation, minimizing harm to healthy tissue [236]. Proteases, key enzymes involved in peptide bond cleavage, are widely used in these systems, yet designing selective and efficient substrates for them remains a significant challenge due to the vast design space and limited high-throughput tools. To accelerate this process, machine learning models such as CleaveNet have been developed. CleaveNet is an AI-powered pipeline that enables the in silico design of protease substrates, incorporating features such as biophysical filtering and conditioning tags for customizable cleavage profiles [176]. Experimental validation has shown its ability to produce highly selective substrates, even for difficult targets like MMP13. Furthermore, CleaveNet supports complex use cases where multiple cleavage events by co-expressed proteases are required, opening new possibilities for the rational design of multi-input prodrug activation systems[176].
Unlike cleavage, where enzymes directly break down molecules, instructed mechanisms involve enzymes guiding or triggering changes without cutting a substrate. Instead, they help molecules undergo structural or functional transformations. One example is enzyme-instructed self-assembly (EISA), where an enzyme modifies a molecule, such as removing a phosphate group (dephosphorylation), causing peptides to self-assemble into nanostructures for therapeutic use. Researchers have shown that alkaline phosphatase can dephosphorylate a peptide, leading to the formation of nanofibers that target PD-L1, a protein involved in cancer therapy. Another example is CYP enzymes, which contain a heme iron center and activate prodrugs by adding an oxygen atom, converting them into their active form. While cleaved mechanisms involve direct enzymatic action to activate a substrate, instructed mechanisms rely on enzyme-guided transformations.
In contrast to cleavage, the concept of "instructed" mechanisms involves enzymes guiding or triggering a specific process without directly cleaving a substrate. Instead, enzymes facilitate structural or functional transformations, such as those seen in enzyme-instructed self-assembly (EISA). In EISA, an enzyme catalyzes a modification, such as dephosphorylation, that induces peptides to self-assemble into nanostructures for therapeutic purposes. Wang et al. demonstrated how alkaline phosphatase can dephosphorylate a peptide, prompting its self-assembly into nanofibers targeting PD-L1 for cancer therapy[236].
Another example is CYP enzymes, containing a heme iron center in their active site, activate prodrugs by introducing an oxygen atom into the compound, converting it into its active form[236]. Thus, while "cleaved" mechanisms denote direct enzymatic action for substrate activation, instructed mechanisms rely on enzyme-guided transformations. Both approaches are critical in enzyme-based cancer therapies, enhancing specificity and therapeutic efficacy.
Nanoparticulation of prodrugs is another promising strategy for enhancing the specificity and bioavailability of therapeutic agents. Nanoparticles, which take advantage of the enhanced permeability and retention effect, can shield prodrugs from premature degradation or metabolism and deliver them directly to tumor sites. This strategy, combined with gene-directed enzyme prodrug therapy, forms a "Trojan horse" mechanism that optimizes pharmacokinetics and reduces side effects [177].
Complementing nanoparticulate systems, covalent organic frameworks (COFs) offer a versatile platform for enzyme immobilization and controlled drug delivery. With their open, crystalline structure that support unhindered molecular diffusion, highly tunable porosity and large surface area, COFs can encapsulate enzymes, drugs, and other therapeutic molecules, allowing for precise control over loading capacity and release kinetics. COFs have already been employed to immobilize enzymes such as catalase and glucose oxidase, improving their catalytic efficiency in tumor-targeted catalytic therapies and photodynamic therapy[49]. Furthermore, COFs can be functionalized with specific chemical groups to modulate enzyme activity or facilitate co-factor interactions, effectively mimicking natural enzymatic environments [48].
Within these frameworks, enzymes can be immobilized through noncovalent interactions or in situ biomimetic mineralization, where enzymes are encapsulated during the framework’s formation, emulating natural biomineralization processes. This strategy not only preserves enzymatic activity under harsh physiological conditions but also eliminates size constraints, ultimately enhancing enzyme stability and functionality [49].
While such immobilization strategies improve enzyme performance by modifying external conditions, enzyme stability can also be enhanced at the molecular level through computational design. One example is Stability Oracle, a machine learning model based on a Graph-Transformer architecture, developed to predict stabilizing mutations that improve enzyme robustness[366]. Traditional datasets often suffer from bias toward destabilizing mutations, complicating efforts to train accurate models. To address this, Stability Oracle employs Thermodynamic Permutation, a novel approach that infers stabilizing mutations beyond simple reversions to wild-type sequences, such as substituting alanine at position 50 with leucine or methionine (A50L, A50M), thereby enriching dataset diversity [366].
Unlike conventional physics-based tools such as Rosetta [335] or AlphaFold[204], which depend on computationally intensive structural modeling, Stability Oracle avoids explicit mutant structure generation. Instead, it masks the mutated residue and uses contextual atomic interactions to infer the mutation’s impact on protein stability (). This enables high-throughput prediction of all possible mutations from a single protein structure, dramatically reducing computational overhead. While AlphaFold is tailored for de novo structure prediction using multiple sequence alignments, Stability Oracle is purpose-built for efficiently predicting the thermodynamic consequences of point mutations, offering a powerful tool for enzyme engineering [366].
7. Enzyme-Drug Interactions in Cancer Therapy
Understanding enzyme-drug interactions is essential for improving drug efficacy and safety, especially in personalized medicine and chemotherapy. One critical aspect of this is enzyme-mediated bioactivation, which can lead to diverse outcomes. For instance, a prodrug may be transformed into its active form without structural alteration, resulting in the intended therapeutic effect. However, enzymes can also modify drugs in ways that increase or reduce their activity, and in some cases, even produce toxic effects [65].
These adverse outcomes are often influenced by genetic polymorphisms. Genetic variations in drug-metabolizing enzymes result in distinct metabolic phenotypes. Depending on their enzyme alleles, individuals can be categorized as ultra-rapid, extensive, intermediate, or poor metabolizers. These polymorphisms directly affect how drugs are processed in the body, which may compromise therapeutic efficacy or lead to adverse drug responses [65].
Among the best-characterized enzyme systems is the cytochrome P450 (CYP) superfamily, which is responsible for the oxidative metabolism of a wide range of drugs. Inhibition of CYP enzymes can extend a drug’s half-life, increase its plasma concentration, and elevate toxicity risk. Conversely, enzyme induction may accelerate drug clearance, reducing its therapeutic effect.
A prominent example of enzyme-mediated drug interaction involves furanocoumarins in grapefruit juice, which inhibit CYP3A4—an enzyme that metabolizes over 85 commonly used drugs, including statins and antiretrovirals. This interaction can significantly alter drug bioavailability, potentially leading to severe toxicities such as rhabdomyolysis or nephrotoxicity, and even fatal outcomes, hence the recommendation for patients to avoid grapefruit in certain treatments [246].
CYP enzymes also play central roles in cancer pharmacology, from the metabolism of antineoplastic agents to the activation or deactivation of carcinogens. In response to the complexity of these interactions, deep learning approaches such as the multi-task FP-GNN model have emerged. This model achieved superior predictive performance in identifying CYP inhibitors, validated through metrics such as AUC (0.905), F1 (0.779), BA (0.819), and MCC (0.647). Its interpretability further enabled identification of critical molecular features linked to CYP inhibition. These advancements culminated in the development of DEEPCYPs, an online webserver that facilitates early screening of CYP-interacting compounds, promoting safer drug development [61].
Machine learning models have also been used to predict the bioactivation of CYP enzymes and their substrates [62,63,69] and inhibitors [61,68,115]. These predictions are essential to prevent harmful drug-drug interactions and mitigate toxicity [70,209].
Despite their relevance, comprehensive resources mapping enzyme-drug interactions remain limited. To address this, the INTEDE[237] and DrugMAP[239,240] databases consolidate information on interactions between drug-metabolizing enzymes and microbiome elements, xenobiotics, and host proteins, offering a valuable tool for navigating the landscape of drug metabolism.
7.1. Enzymes as Direct Therapeutic Agents
Beyond prodrug strategies that rely on enzymatic activation for spatial and temporal control of drug release, enzymes can directly modulate cancer cell metabolism. Instead of activating external compounds, some therapeutic enzymes exert their effects by depleting essential nutrients within the tumor microenvironment.
A prime example of this approach is L-asparaginase (L-ASNase), which targets metabolic dependencies in cancer cells to inhibit tumor progression. L-ASNase hydrolyzes L-asparagine into aspartic acid and ammonia, thereby reducing extracellular asparagine (an amino acid essential for protein synthesis and cell viability) levels. While healthy cells can synthesize asparagine through asparagine synthetase (ASNS), certain cancers with low ASNS expression depend on extracellular sources. Depleting asparagine in these contexts inhibits protein synthesis, triggering cell cycle arrest and apoptosis [367]. This enzymatic starvation strategy has proven highly effective in the treatment of acute lymphoblastic leukemia and is under investigation for broader oncological applications, including ovarian, pancreatic, colorectal, and breast cancers, as well as hepatocellular carcinoma and glioblastoma [83]. In addition to its core mechanism, L-ASNase influences several other cancer-associated pathways, including oxidative stress induction, autophagy, and suppression of survival signaling cascades such as Akt/mTOR and Erk [254]. However, despite its clinical success, L-ASNase therapy faces limitations due to adverse effects including hypersensitivity and organ toxicity.
Strategies to mitigate these side effects include enzyme modifications, co-administration of protective agents, and personalized treatment regimens based on asparagine synthetase expression levels [368]. Notably, computational modeling and AI-assisted gene editing have emerged as powerful tools in this effort. CRISPR technology enables precise genetic modifications, while a radial basis function neural network with a specific genetic algorithm predicts the optimal gene edits necessary to enhance enzyme expression, ultimately reducing production costs and optimization time [111].
Furthermore, in microbial production systems, artificial neural networks (ANNs) have outperformed traditional models such as response surface methodology (RSM), particularly in capturing the culture conditions for maximizing enzyme yield. For instance, ANN-guided optimization in Bacillus licheniformis PPD37 has led to a sixfold increase in L-asparaginase yield [255]. While RSM relies on regression models to describe how independent variables (temperature, pH, nutrient concentration) influence a dependent variable (enzyme production), ANNs excel at capturing complex, non-linear bioprocess interactions[255].
7.2. Detection and Modulation of Metabolic Reprogramming of Cancer
Building on the therapeutic potential of enzymes such as L-asparaginase, which directly depletes nutrients vital to certain cancer cells, modern cancer therapies increasingly target the metabolic flexibility of tumor cells, which often rewire key biochemical pathways to support uncontrolled proliferation and resist treatment. Furthermore, some overexpressed enzymes aid cancer in immune evasion and interfere with the metabolism of anticancer agents. This shift has positioned metabolic enzymes, not only as biomarkers but also as intervention points.
7.3. Targeting Cancer Metabolic Reprogramming
One of the hallmark metabolic adaptations in cancer is the Warburg effect, in which tumor cells preferentially rely on glycolysis for energy production, even in the presence of oxygen. This metabolic reprogramming is primarily driven by the upregulation of key glycolytic enzymes, enabling cancer cells to rapidly produce ATP while fueling anabolic processes necessary for tumor proliferation [29,73].
Glycolysis itself is a multi-step process that breaks down glucose to generate energy. Several enzymes act as rate-limiting regulators, controlling the overall pace of this pathway [29]:
- Hexokinase (HK2): Initiates glycolysis by phosphorylating glucose, trapping it inside the cell.
- Phosphofructokinase-1 (PFK-1) functions as a key checkpoint, modulating glucose breakdown based on cellular energy demands.
- Pyruvate Kinase M2 (PKM2) governs the final step of glycolysis and can reroute glucose metabolism toward biosynthetic pathways essential for rapid cell division.
- Lactate Dehydrogenase (LDH) converts pyruvate into lactate, especially under hypoxic conditions, supporting cancer cell survival and contributing to drug resistance.
In the oxygen-deprived tumor microenvironment, LDH sustains energy production via lactate fermentation. This contributes to an acidic extracellular environment that promotes cancer cell invasion, immune evasion, and therapy resistance. High LDH expression correlates with poor prognosis in several malignancies, including breast and colorectal cancers[29].
High expression of glycolytic enzymes, such as Aldolase A (ALDOA), is linked to chemotherapy resistance in cancers such as oral, colorectal, and breast cancer[30,38?]. Validating AI-based model results with Human Protein Atlas (a large database of human tissue samples) and immunohistochemistry (staining specific proteins in cancer cells to confirm gene activity) has confirmed the ALDOA gene expression in various cancer cell lines[38].
Overall, nearly all cancers express aerobic glycolysis, regardless of their tissue or cellular origin. This metabolic phenotype is the basis for tumor imaging using labeled glucose analogs and has become an important diagnostic tool for cancer detection and management [276]. Based on the distinctive glycolytic characteristics of tumor cells, novel imaging technologies have been developed to assess tumor proliferation and metastasis effectively[29]. Histopathology, the gold standard for tumor assessment, now benefits from ML models[112]. For example, computer vision algorithms applied to tissue samples stained for glycolytic enzymes have uncovered metabolic heterogeneity linked to aggressive cancer sub-populations[104].
Alterations in the isocitrate dehydrogenase (IDH) serve as a critical biomarker for the detection and clinical management of gliomas, a type of brain tumor[81]. IDH plays a crucial role in Krebs cycle. It catalyzes the oxidative decarboxylation of isocitrate to -ketoglutarate (-KG) while producing NADH or NADPH, depending on the enzyme isoform([?]. Isoforms of IDH arise from the same gene family but differ in their amino acid sequences or structural configurations, affecting their function in metabolic pathways. Traditionally, detecting IDH mutations required molecular genetic tests that are invasive, expensive, and time-consuming[88]. Cancer tissues are usually examined under a microscope after being stained with hematoxylin and eosin (H&E), producing histopathological images, that contain visual patterns linked to the cancer’s molecular makeup[109]. Histopathology and medical imaging contain hidden clues about IDH status that AI can uncover[108]. Numerous deep learning models have been applied to study status of IDH based on histopathological imaging [79,85,86,87,88,89,90,91,92,108,109,121,125,126], making IDH a valuable cancer marker.
Beyond histopathology,there are methods such as Magnetic Resonance Imaging (MRI), which reveal the physical structure of tissues, while more advanced techniques such as Magnetic Resonance Spectroscopic Imaging (MRSI) provide insight into the molecular composition of cells. It’s akin to adding chemical context to a structural map, allowing us to understand not just the location of a tumor, but also how it’s functioning biologically. Researchers also use MRI and MRSI images to detect IDH mutations using AI, outperforming experienced neuroradiologists in identifying IDH mutation status[106,107,114,122,123,124]. However, more research is needed to refine these models and confirm their effectiveness across different patient populations.
Combining imaging data with genetic data[86], and clinical data such as age and symptoms [125], AI models can provide a more personalized approach to treatment[86]. By using both image and non-image information, their predictions became more reliable.
Furthermore, emerging platforms such as 3D/4D bioprinting now offer physiologically relevant tumor models that recreate the complexity of in vivo environments. These models facilitate real-time observation of metabolic reprogramming and drug responses [98]. While 3D bioprinting creates static structures mimicking tumors, 4D bioprinting incorporates dynamic, stimuli-responsive materials that evolve over time, allowing researchers to simulate tumor progression more realistically.
In a parallel approach, deep reinforcement learning techniques like Double Deep Q-Networks (DDQNs) dynamically simulate tumor microenvironments(glucose, oxygen, and lactate levels, etc.) to optimize therapeutic regimens targeting the Warburg effect [100]. Continuously learning from biochemical interactions, DDQNs offer a powerful strategy to predict optimal drug regimens[100].
Advancements in AI-driven drug discovery have streamlined the search for inhibitors of glycolytic enzymes and other oncogenic targets [340]. When experimental data is limited, generative models using transfer learning can design novel structurally diverse compounds with inhibitory potential[338].
AI-driven screening identified promising p38 Mitogen-Activated Protein Kinase (MAPK) inhibitors, outperforming traditional screening methods[136]. P38 MAPK is well known for its role in metabolic reprogramming. It mediates responses to stress and cytokines and is often hijacked by tumor cells for survival. A prevalent problem with current p38 MAPK inhibitors is their own toxic side effects. Natural products are widely available and have low toxicity, and therefore may be a valuable source for the discovery of p38 MAPK inhibitors.
Small molecule inhibitors selectively target enzymes or receptors to regulate cellular functions with high specificity, making them essential tools in cancer therapy, as, for example, targeting Enolase 1 (ENO1)[53]. ENO1 is another glycolytic enzyme with a dual role in cancer. In addition to its enzymatic role, ENO1 acts as a plasminogen receptor, promoting extracellular matrix remodeling and facilitating tumor invasion and metastasis. Overexpression of ENO1 has been linked to poor prognosis in lung, breast, and liver cancers[53].
Despite promising preclinical findings, targeting metabolic enzymes in cancer therapy remains challenging. Many glycolytic enzymes are essential for normal cellular metabolism, increasing the risk of off-target effects. For example, PKM2 and PFKFB3 inhibitors lack specificity, necessitating the development of highly selective drugs that exploit cancer-specific metabolic dependencies[29].
7.4. Enzymatic Regulation in Cancer Immunotherapy
Cancer cells deploy a variety of mechanisms to evade immune surveillance, including suppressing immune cell activation and reshaping the tumor microenvironment into an immunosuppressive niche.
One critical challenge in cancer therapy is multidrug resistance, often driven by genetic and metabolic adaptations. This resistance commonly arises from the overexpression of drug efflux transporters that expel chemotherapeutic agents from cancer cells, lowering their intracellular concentration and reducing treatment efficacy. While this significantly hampers conventional therapies, immunotherapy, especially immune checkpoint inhibitors (ICIs), has offered new hope by reactivating immune-mediated tumor clearance. However, predicting which patients will respond to ICIs remains difficult, emphasizing the need for reliable biomarkers to guide personalized treatment strategies [19]. Integrating enzyme-based assays with machine learning has accelerated biomarker discovery; a recent study identified glutathione S-transferase A3 as a predictive biomarker for ICI response in melanoma using a supervised learning model [174].
Enzymes also directly mediate immune evasion through metabolic reprogramming in the tumor microenvironment (TME). For instance, arginase and indoleamine 2,3-dioxygenase (IDO) are frequently upregulated in multidrug resistance-associated cancers. These enzymes deplete key nutrients such as L-arginine and tryptophan, respectively, impairing T-cell activation and promoting immunosuppression [1?]. Engineered inhibitors are being developed to target these overexpressed immunosuppressive enzymes. A convolutional neural network recently identified STB-C017, a dual IDO and TDO inhibitor that lowered kynurenine levels and boosted T-cell function [77]. In another example, the ensemble ML model IDO1Stack achieved high accuracy in predicting IDO1 inhibitors, guiding rational drug design with AUC values above 0.9 on both test and external datasets [84]. These examples showcase how AI-driven enzyme-targeting approaches can modulate the TME to enhance immune response. However, enzyme function in the TME is context-dependent. For instance, apoptotic tumor cells can upregulate IDO expression in neighboring cells, reinforcing local immunosuppression. Blocking such enzymatic responses during therapy-induced cell death could amplify inflammation, improve antigen presentation, and reinvigorate immune activation [148].
Tumor-associated adenosine accumulation further inhibits cytotoxic immune cells, but this can be counteracted by adenosine deaminase (ADA), an enzyme currently under investigation for its immunoregulatory potential [186]. A deep learning model AlphaMissense, derived from AlphaFold, has been used to predict pathogenic ADA mutations, enabling the design of more effective ADA variants for immunotherapy. Although highly accurate for severe mutations, the model’s reduced precision for partial-function variants highlights the continued need for biochemical validation alongside computational tools [363].
In addition to their metabolic roles, enzymes are central to epigenetic regulation. Unlike genetic mutations, epigenetic modifications, such as histone methylation, alter gene expression without changing DNA sequence. Histones, the protein components around which DNA is wound, serve as dynamic regulators of gene expression. Enzymatic modifications to histones can either condense chromatin to repress transcription or relax it to permit gene activation, thus functioning as epigenetic switches.
These modifications are orchestrated by specific enzymes and can silence immune-stimulating genes or activate immune checkpoint ligands. One such enzyme, SETD2, is frequently mutated in cancers such as clear cell renal carcinoma. It catalyzes trimethylation of histone H3 on lysine 36, a modification vital for DNA repair and chromatin stability. Loss of SETD2 function contributes to immune evasion by disrupting these regulatory processes [53].
This highlights a broader theme: enzymes are not merely biochemical catalysts, but dynamic regulators of signaling, gene expression, and immunity. Through post-translational modifications (PTMs) such as methylation, phosphorylation, or acetylation, enzymes modulate protein function, localization, and interactions. Despite over 500 types of PTMs being identified, mapping them across the proteome and linking them to specific enzymes remains a formidable challenge.
Machine learning is increasingly pivotal in overcoming this challenge. In a recent study, researchers applied ML to SET8, a histone methyltransferase, and predicted 885 methylation sites, far surpassing conventional techniques in accuracy and throughput. Their work uncovered novel substrate patterns, enabling the mapping of methylation gain and loss across hundreds of proteins [365]. Learning how SET8 “chooses” its substrates, other enzymes and post-translational modifications across the proteome can be studied. Furthermore, enzymes can be designed or reprogrammed with specific target profiles, opening new doors in cancer therapy.
8. Conclusions
Enzymes occupy a pivotal and multifaceted role in cancer biology, functioning both as enablers of malignancy and as strategic intervention points for therapy. This review has highlighted the dualistic nature of enzymes in oncology: on one hand, they drive key cancer hallmarks such as metabolic reprogramming, resistance to chemotherapeutics, and immune evasion, while on the other, they offer potent therapeutic opportunities. For instance, enzyme-assisted prodrug activation systems enable the localized release of cytotoxic agents, minimizing systemic toxicity. Direct enzyme therapeutics, exemplified by L-asparaginase in acute lymphoblastic leukemia, underscore the clinical viability of enzyme-based treatments.
The confluence of enzyme engineering and computational modeling is revolutionizing the development of next-generation enzyme therapeutics. Advances in machine learning, protein language models, and structure prediction algorithms now allow for high-precision prediction of enzyme-substrate interactions, mutational impacts, and inhibitor efficacy. These computational frameworks, especially when integrated with experimental validation, are significantly reducing the time and cost associated with enzyme optimization and drug discovery.
As the field of oncology continues its transition toward precision medicine, enzyme-based strategies are becoming increasingly relevant. Engineered enzymes offer the potential for highly specific, modular, and adaptive treatments that can be tailored to individual patient profiles. Their roles are expanding beyond cytotoxicity to include enzyme-activated imaging, immunomodulation, and synthetic lethality targeting.
However, several challenges must be addressed to facilitate the clinical translation and broad deployment of enzyme-based therapies. Key issues include:
- Immunogenicity. Many therapeutic enzymes, especially those derived from non-human sources, can elicit immune responses that limit their efficacy and safety.
- Stability and bioavailability. Enzymes must maintain catalytic activity under physiological conditions, often requiring stabilization through chemical modification or encapsulation.
- Targeted delivery. Achieving tissue- or tumor-specific localization remains a major hurdle, necessitating advances in delivery vectors such as nanoparticles, antibody conjugates, or gene therapy platforms.
- Manufacturing scalability. Producing large quantities of highly pure, active enzymes in a cost-effective and reproducible manner is essential for clinical and commercial viability.
- Regulatory and translational hurdles. Standardizing protocols for clinical evaluation, safety assessment, and regulatory approval is critical for successful integration into cancer care.
To overcome these challenges, synthetic biology will play central role. Novel engineering strategies including enzyme regulation, switchable activity, and multiplex targeting, are under active exploration to enhance control over enzyme function and context-specificity. Furthermore, the integration of high-throughput screening, AI-guided engineering, and multimodal omics data will further refine enzyme design pipelines.
In summary, enzymes are emerging as powerful and versatile tools in the oncology toolkit. Their ability to act with catalytic precision and biological specificity positions them as ideal agents for next-generation therapeutics. With continued interdisciplinary collaboration across molecular biology, computational science, bioengineering, and clinical oncology, engineered enzymes are poised not only to address unmet clinical needs but also to redefine therapeutic paradigms in the era of precision and personalized cancer treatment.
Author Contributions
Conceptualization, A.A. and S.B.; writing—original draft preparation, A.A.; writing—review and editing, A.A. and Y.A. and S.B.; visualization, A.A and Y.A.; supervision, Y.A. and S.B.; project administration, Y.A. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AI | Artificial Intelligence |
| ML | Machine learning |
Appendix A. Protein engineering machine learning models

Table A1.
2000-2022 Protein Engineering Models.
| No. | Model Name | Year | Input Data Type | Output Type | Datasets Used | Performance Metrics | Performance Results | Limitations |
|---|---|---|---|---|---|---|---|---|
| 1 | ProGen[?] | 2020 | Sequences | Sequences | Uniparc[331], UniProtKB[330], SWISS-PROT[329], TrEMBL[328], Pfam[327], NCBI taxonomic information[326] | Perplexity (PPL), mean per token hard accuracy (HA)1 and soft accuracy (SA)2 primary sequence similarity(SS)3, secondary structure accuracy(SSA)4, conformational energy analysis5 | 8.56 PPL, HA in-distribution 45%, out-of-distribution 22%, Fine-Tuned on OOD-80 50%, SA = 20% HA, SS 25% baseline, SSA 25% baseline | Limited structure integration |
| 2 | ProteinMPNN[13] | 2021 | Protein sequences | 3D structures | Protein Data Bank (PDB)[310], CATH[323] | PDB Test Accuracy (%)6, PDB Test Perplexity7, AlphaFold Model Accuracy (%)8 | with random decoding step: PDB 50.8%, with noise 47.9%, Perplexity 4.74, with noise 5.25, AlphaFold 46.9%, with noise 48.5% | Dependent on high-quality, static backbone inputs. The message-passing framework may not fully capture long-range, non-local interactions, allosteric effects |
| 3 | ProtTrans[200] | 2021 | Sequences | Embeddings, annotations | Uniref50[324], Uniref100[324], BFD(Big Fantastic Database)9[325] | Q3/Q810, Q10 11, Q2 12 | Q3 87%, Q8 77%, Q10 81%, Q2 91% | Requires fine-tuning for specific tasks |
| 4 | AlphaFold 2[204] | 2021 | Sequence, MSA, Homology 3D atom coordinates | 3D protein structures | custom BFD, PDB[310], for MSA Uniref90[324], Uniclust30[321], MGnify[320] | Root Mean Square Deviation (RMSD), pLDDT13, C local-distance difference test (lDDT-C)14, pTM15 | RMSD ∼ 1.46Å, pLDDT 0.76, lDDT-C∼ 80, pTM 0.85 | Predicts static structures |
| 5 | GEMME[6] | 2022 | Protein sequences | Fitness predictions | Mutations dataset16 | Spearman Correlation Coefficient (SCC) | 0.53 | Relies on the quality and diversity of multiple sequence alignments, sensitive to the specific filtering criteria, may struggle to capture complex, non-linear sequence-function relationships. |
| 6 | ProtST[9] | 2022 | Sequences | Secondary structure | ProtDescribe17 | Area Under the Precision-Recall Curve (AUPR)18, Function annotation 19, Binary, subcellular localization accuracy, Fitness landscape Spearman’s 20 | AUPR 0.898, 0.878, Binary 93.04, Subcellular 83.39, Spearman’s 0.895 | Focused on structure tasks |
| 7 | OPUS-GO[113] | 2022 | Protein sequences | Functional annotations | not explicitly mentioned | AUPR, on GO21 term predictions, AUC22 , Calcium ion binding F1-score23, Binary, Subcellular localization Accuracy, AUPR and on two protein EC number prediction datasets | GO AUPR 0.678 , GO 0.690, AUC 0.826 , F1-score 0.327, Bin 0.931, Sub 0.837, Ion 0.827, EC AUPR 0.902, EC 0.881 | Relies on curated graphs |
1 If the predicted amino acid is not exactly the same as the original (ground truth) amino acid, it is counted as incorrect, does not consider whether similar amino acids could replace each other in nature.
2 If an incorrect prediction results in an amino acid that is often substituted in nature(using BLOSUM62[342], a block substitution matrix that specifies which amino acid substitutions are more or less acceptable according to their frequency in known well-formed proteins), it is penalized less than a completely uncommon substitution.
3 Defined by a global, pairwise sequence alignment score with gap(insertions or deletions) open penalty −0.5 and continue penalty −0.1, based on the Needleman-Wunsch algorithm[343], normalized by the length of the protein. The alignment score is influenced by BLOSUM62, which assigns higher scores to substitutions that are more likely to occur in real proteins.
4 Compares PSIPRED (predicts secondary structure based on multiple sequence alignments, generated by PSI-BLAST based on UniRef90 database to find related sequences) predictions with confidence >0.5 to experimentally validated structures from UniProtKB (Universal Protein Knowledgebase).
5 Uses the Rosetta-RelaxBB method, which simulates how proteins fold and calculates their energy. The process involves: keeping the backbone (main structure) fixed while allowing side chains (amino acid-specific parts) to change; performing energy minimization, which adjusts the protein’s shape to reach a lower energy (more stable) state. The lowest energy state is found through Monte Carlo simulations, randomly exploring different folding possibilities. Lower energy means higher stability. Validation restricted to SWISSPROT test samples with experimentally determined 3D structures from RCSB PDB(https://www.rcsb.org).
6 Measures how well the model predicts the correct amino acid sequence from a given protein structure.
7 Represents how "confused" the model is when predicting amino acids.
8 Measures how well the model reconstructs protein sequences when tested on AlphaFold-predicted structures.
9 Merging UniProt[317] and proteins translated from multiple metagenomic sequencing projects.
10 Three/eight-state per-residue accuracy, percentage of residues predicted correctly in either of the 3/8 secondary structure states.
11 Ten-State Accuracy for Subcellular Location Prediction.
12 Two-State Accuracy for Membrane vs. Non-Membrane Classification.
13 Predicted Local Distance Difference Test score. LDDT measures how much a predicted structure deviates from the true (experimental) structure by comparing atomic distances, pLDDT is an AlphaFold-specific version of LDDT that estimates how accurate its own predictions are even without experimental results
14 Compares the prediction with the actual experimental structure at the atomic level.
15 Measures how well AlphaFold captures the overall protein shape (not just local accuracy).
16 High-throughput mutational scans of 33 proteins, 1 protein complex, representing 657,840 mutations.
17 Aligned pairs of protein sequence and property description from SWISS-PROT[329].
18 The area under the Precision-Recall curve, which plots precision vs. recall at different classification thresholds. Since recall is on the x-axis, AUPR prioritizes how well the model finds true positives without over-relying on true negatives. It is suitable for imbalanced data, when positives are much rarer than negatives.
19 The maximum of F1 across thresholds.
20 Monotonic (increasing or decreasing but not necessarily linear) correlation coefficient.
21 Gene Ontology. The most widely used ontology, classifying proteins into molecular function (MF), biological process (BP), and cellular component (CC).
22 Area Under the Curve. It measures the area under the Receiver Operating Characteristic (ROC) curve, which plots the True Positive Rate (TPR, Sensitivity) against the False Positive Rate (FPR, 1 - Specificity) at various threshold settings.
23 The harmonic mean of precision and recall, balancing both measures in a single value.
Table A2.
2023-2024 Protein Engineering Models.
| No. | Model Name | Year | Input Data Type | Output Type | Datasets Used | Performance Metrics | Performance Results | Limitations |
|---|---|---|---|---|---|---|---|---|
| 1 | GPDL-H[11] | 2023 | Protein sequences, structure | Structure optimization | seeding model CATH[323], PDB | Sc-TM-score24, Motif RMSD25, PAE26, pLDDT27 | Sc-TM-score 0.99, Motif RMSD near 0.58 Å, max PAE 2.43, pLDDT 90.66 | Requires large-scale training |
| 2 | RFdiffusion[12] | 2023 | Protein sequences | Structural and functional properties | noising structures from PDB[310] for up to 200 steps | RMSD generated vs AlphaFold2[204], TM-score | RMSD 0.90, 0.98, 1.15, 1.67 Å(increases with protein length), TM-score 100 amino acids 0.85, 200 amino acids 0.75, 300 amino acids 0.65, 400 amino acids 0.55, 600 amino acids 0.50, 800–1000 amino acids 0.40 (decreases with protein length) | Computationally expensive |
| 3 | TranceptEVE[5] | 2023 | Sequences | Fitness landscapes | ProteinGym[318], ClinVar[319], EVE DMS[319] | Protein mutations Spearman correlation of ProteinGym[2]28 data with Low, Medium , High Depth29, AUC of ClinVar[319] data | Spearman correlation Low 0.460, Medium 0.463, High 0.508, All30 0.472, AUC 0.767 | High computational cost |
| 4 | Tranception[94] | 2023 | Sequences | Mutational landscapes | UniProt[317] | Perplexity, log-likelihood | Perplexity 1.2 | Limited scalability for long proteins |
| 5 | SaProt[7] | 2023 | Sequences | Mutational fitness scores | ProteinGym[318], ClinVar[319] | ClinVar AUC, ProteinGym Spearman’s , Thermostability Spearman’s , Human Protein-Protein Interaction (PPI) ACC%, Metal Ion Binding ACC%, EC , GO Molecular function (MF), Biological process(BP), Cellular component(CC) , DeepLoc Subcellular and Binary ACC | ClinVar AUC 0.909, ProteinGym Spearman’s 0.478, Thermostability Spearman’s 0.724, HumanPPI ACC% 86.41, Metal Ion Binding ACC% 75.75, EC 0.882, GO MF 0.682, GO BP 0.486, GO CF 0.479, Subcellular 85.57, Binary 93.55 | Dependent on MSA |
| 6 | TTT[226] | 2023 | Sequences | Task-dependent outputs | MaveDB[346], DeepLoc[316] | Avg. Spearman, TM-score, LDDT | SaProt (650M) + TTT 0.4583, ESM3 + TTT TM-score 0.3954, ESMFold + TTT LDDT 0.5047 | Task-dependent architecture |
| 7 | AlphaProteo[221] | 2024 | Sequences | Protein binders | UniProt[317], PDB[310] | Experimental Success Rate (%)31 tested on BHRF132 SC2RBD 33 IL-7RA34 PD-L135 TrkA36, Binding Affinity ( in nM)37 | BHRF1 88, SC2RBD 8.5, IL-7RA 24.5, PD-L1 9.6, TrkA 9.6, VEGF-A 33, n | Needs fine-tuning |
| 8 | Stability Oracle[366] | 2024 | Structural data | Stability predictions () | C2878, cDNA117K, T2837 from MMseqs2[301] | AUROC, Pearson Correlation Coefficient (PCC), Spearman, precision, RMSE | p53 variations AUROC 0.83, Pearson 0.75, Spearman 0.76, precision 0.55, T2837 RMSE 0.0033 | Requires experimental validation |
| 9 | Chai-1[297] | 2024 | Sequence | Structure prediction | Custom datasets, PDB | The fraction of predictions with ligand root mean square deviation (ligand RMSD) to the ground truth lower than 2 Åfor PoseBusters benchmark set[344] | TM-score > 0.75, RMSD < 1.5Å | Requires large-scale training |
| 10 | LigandMPNN[295] | 2024 | Structural data | Protein-ligand binding predictions | PDB[310]38 | Sequence recovery for residues interacting with small molecules, nucleotides, metals, RMSD, Chi1 fraction39 Chi2, Chi3, and Chi4 fractions40 | Chi1 (small molecules) 86.1%, Chi1 (nucleotides) 71.4%, Chi1 (metals) 79.3% , weighted average Chi1, Chi2, Chi3, and Chi4 fractions for small molecules 84.0%, Chi2 64.0%, Chi3 28.3%, Chi4 18.7%, RMSD near small molecules ∼0.5–1.2Å, RMSD near DNA or RNA in protein-DNA or protein-RNA complexes, RMSD near metal ions ∼0.5–1.1Å | Limited to known binding sites |
| 11 | MODIFY[36] | 2024 | Protein sequences, structural data | Structure, function, stability predictions | For evaluation: ProteinGym[94,318] for zero-shot protein fitness, GB1[315], ParD3[311,313],CreiLOV[314] for high-order mutants | Spearman | all proteins ∼0.42 – 0.65, Low MSA Depth ∼0.35 – 0.50, Medium MSA Depth∼0.40 – 0.55, High MSA Depth ∼0.50 – 0.65 | Still evolving, data-dependent |
| 12 | TourSynbio[309] | 2024 | Protein sequences, textual data | Protein designs, functional annotations | InternLM2-7B[308], ProteinLM-Dataset[307] | ProteinLMBench benchmark set quieries accuracy | 62.18% | High computational requirements |
| 13 | ESM3[8] | 2024 | Sequences | Embeddings, structure | Joint Genome Institute databases[306], UniRef clusters[324], MGnify: the microbiome sequence data analysis resource[304], Observed Antibody Space[303], The Research Collaboratory for Structural Bioinformatics Protein Data Bank[302] | mean pTM41, pLDDT, backbone cRMSD42 by prompting | pTM >0.8, pLDDT >0.8, cRMSD < 1.5Å | Resource-intensive training |
| 14 | AlphaFold 3[300] | 2024 | Sequence data | Multi-protein complexes | PDB[310], Protein monomer distillation43, Disordered protein PDB distillation44, RNA distillation45, JASPAR 9[298] | DockQ score46, DockQ Score for Protein-Protein Interfaces, Predicted Local Distance Difference Test between the predicted structure with the actual structure (pLDDT), Ligand-Protein Binding Accuracy (RMSD-based), lDDT (local Distance Difference Test)47 | DockQ ∼0.7, pLDDT >70, ∼50% of ligand-protein predictions have RMSD ≤ 2Å48, ∼20% of ligand-protein predictions have RMSD < 1Å49, Protein-RNA binding lDDT ∼65%, Protein-DNA (dsDNA) binding lDDT ∼50%, RNA-only (CASP15 RNA benchmark) lDDT ∼85%, Protein-ligand binding lDDT 50-70%, RNA predictions 75-85%, DNA predictions 50-80%, Protein-protein (general) binding lDDT ∼77%, Protein-protein (antibody-involved) binding lDDT 50-60%, Protein-ligand (small molecules) lDDT 40-50% | Requires extensive computational resources |
16 Monotonic (increasing or decreasing but not necessarily linear) correlation coefficient.
17 The largest collection of Deep Mutational Scanning assays for assessing mutation effects predictors. It consists of two different benchmarks measuring mutations made via substitutions and indels.
18 All data.
19 How often a designed protein successfully binds to its target in lab experiments.
20 A viral protein from Epstein-Barr virus (EBV) that helps infected cells avoid immune system detection.
21 The Receptor Binding Domain (RBD) of the SARS-CoV-2 spike protein.
22 Interleukin-7 Receptor Alpha. A receptor protein involved in T-cell development and autoimmune diseases like multiple sclerosis.
23 Programmed Death-Ligand 1. A protein that helps cancer cells evade the immune system by binding to PD-1 on immune cells and turning them off.
24 Tropomyosin receptor kinase A. Mutations in TrkA are linked to neuropathic pain and cancer.
25 PDB (as of Dec 16, 2022) determined by X-ray crystallography or cryo-EM to better than 3.5 Åresolution and with a total length of less than 6,000 residues.
26 Within of the actual crystal structure. Measures how accurately the model predicts the first side-chain torsion angle near small molecules (ligands, inhibitors, or drug molecules), nucleotides, and metals.
27 Additional side-chain torsion angles, which become harder to predict as you go further along the chain.
28 Predicted TM-score, estimates how well the predicted structure resembles a realistic protein fold.
29 Cartesian Root Mean Square Deviation, a structural similarity metric. It quantifies how much the backbone of a predicted protein deviates from an experimental structure.
31 AlphaFold-Multimer v2.3 predictions of PDB proteins from the training set with ground truth nucleic acids and small molecules inserted after the prediction is aligned to the ground truth protein.
32 Clustered Rfam (v14.9) using MMseqs2 with 90% sequence identity and 80% coverage, taking one sequence per cluster (the cluster representative).
33 Shows how well protein-protein docking was predicted.
34 Measures how accurate a predicted 3D structure is compared to the real structure.
35 Moderate binding accuracy.
36 High-precision cases are rarer.
Appendix B. Benchmarks
Table B1.
Benchmarks for Protein Assessment.
| Benchmark | Intended Application | Input | Performance Metric | Datasets Used |
|---|---|---|---|---|
| CASP[14] | Protein structure and folding evaluation | Protein sequences, 3D structures | predicted lDDT, lDDT50, RMSD, TM-score, Pearson correlation | Custom dataset |
| TAPE[102] | Sequence-based tasks | Protein sequences | Perplexity, Accuracy, AUPR, Precision, Spearman’s | Pfam[327] , CB513[369], CASP12, and TS115[370] |
| PEER[4] | Protein function/localization/structure prediction, protein-protein and protein-ligand interaction | Protein sequences | Accuracy, Precision, RMSE, Spearman’s | FLIP[380], Meltome atlas[371], TAPE[102], Envision[379], DeepSol[378], DeepLoc[316], ProteinNet[373], SCOP database[377], Klausen’s dataset[360], CB513[369], Guo’s yeast PPI dataset[376], Pan’s human PPI dataset[375], SKEMPI dataset[374], PDBbind-2019 dataset[372] |
| ProteinGym[2] | Fitness landscape evaluation | Protein sequences and variants | Spearman Correlation, AUC, Matthews Correlation Coefficient (MCC)51, Normalized Discounted Cumulative Gains (NDCG)52, Top K Recall | Deep Mutational Scanning[3], ClinVar[319], FLIP[380], MaveDB[346] |
| Aviary[195] | DNA for molecular cloning, answering research questions, engineering protein stability. | Protein sequences, natural language | Accuracy | LitQA[350], HotpotQA[351], SeqQA[349], GSM8K[352] |
| ProteinLMBench[307] | Protein comprehension | Protein sequences, text pairs | Query Accuracy | ProteinLMDataset[307] |
| DeepLoc[16] | Predicting subcellular protein localization | Protein sequences | Classification Accuracy | DeepLoc dataset |
| PoseBusters[344] | Ligand-protein binding accuracy | The re-docked ligands and the true ligand(s), the protein with any cofactors | Chemical, intramolecular , intermolecular validity, the minimum heavy-atom symmetry-aware root-mean-square deviation, sequence identity, | PDB[310], The Astex Diverse set[381] |
| CAMEO[345] | Continuous protein assessment | Protein sequences, experimental 3D structures | lDDT, Contact Area Difference score (CAD-score)53, TM-score, Global Distance Test(GDT)54, MaxSub score55, Oligomeric state accuracy based on quaternary state scores (QS-score)56, reliability of local model confidence estimates (“model B-factor”)57 | PDB[310] (weekly updated) |
| PDB-Struct[217] | Structural validation | Experimental 3D structures | Clashscore58, MolProbity score59, Ramachandran plot statistics60 | PDB[310] (curated experimental structures) |
1 The average percentage of residues (amino acids) that are within the 1 Å, 2 Å, 4 Å, and 8 Åthresholds when the predicted structure is compared to the real (reference) structure. An angstrom (Å) is a unit of length that measures very small distances, especially at the atomic and molecular scale. Even if some parts of the protein are predicted incorrectly, GDT-TS still gives useful information by considering only correctly positioned residues.
2 Compares model scores with binarized experimental measurements.
3 Up-weights a model if it gives its highest scores to sequences with the highest Deep Mutational Scanning value. For certain goals (e.g., optimizing functional properties of designed proteins), it is more important that a model is able to correctly identify the most functional protein variants, rather than properly capture the overall distribution of all assayed variants.
4 Measures the difference in residue-residue contact areas between a model and a reference. Can directly assess the accuracy of domain interfaces and multimeric assemblies[382].
6 Aims at identifying the largest subset of atoms of a model that superimpose ‘well’ over the experimental structure, and produces a single normalized score that represents the quality of the model.
7 Measures how well the predicted quaternary structure matches the actual arrangement of subunits in the protein complex.
8 Represents the atomic displacement or flexibility of atoms in a protein structure. A higher B-factor suggests a greater uncertainty or flexibility in the local model, while a lower B-factor indicates more confidence and stability in the prediction for that region of the model.
9 The number of clashes per 1000 atoms. A clash is when two atoms come closer than their expected Van der Waals radii (the space atoms naturally occupy). Measures how many atoms are unrealistically close together in a protein structure. Atoms should not overlap, and if they do, it’s a sign of structural errors.
10 Compares structures to high-resolution experimental data to see if they look realistic. A combined quality score that considers: Clashscore (atomic overlaps), Geometry (bond angles, bond lengths), Ramachandran statistics (how well the backbone angles match expected values).
11 Checks if a protein’s backbone angles (, ) are in physically allowed regions. These angles define how a protein folds into its 3D shape. If angles don’t fit expected values, it suggests errors in the structure. The higher the percentage of residues in allowed regions, the better the structure.
References
- Munn, D. H.; Mellor, A. L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. Journal of Clinical Investigation 2007, 117, 2316–2326. [CrossRef]
- Notin, P.; Kollasch, A.; Ritter, D.; Van Niekerk, L.; Paul, S.; Spinner, H.; Rollins, N.; Shaw, A.; Orenbuch, R.; Weitzman, R. Proteingym: Large-scale benchmarks for protein fitness prediction and design. Advances in Neural Information Processing Systems 2024, 36, .
- Hopf, T. A.; Ingraham, J. B.; Poelwijk, F. J.; Schärfe, C. P.; Springer, M.; Sander, C.; Marks, D. S. Mutation effects predicted from sequence co-variation. Nature biotechnology 2017, 35, 128–135. [CrossRef]
- Xu, M.; Zhang, Z.; Lu, J.; Zhu, Z.; Zhang, Y.; Ma, C.; Liu, R.; Tang, J. PEER: A Comprehensive and Multi-Task Benchmark for Protein Sequence Understanding. arXiv preprint arXiv:2206.02096 2022, .
- Notin, P.; Van Niekerk, L.; Kollasch, A. W.; Ritter, D.; Gal, Y.; Marks, D. S. TranceptEVE: Combining family-specific and family-agnostic models of protein sequences for improved fitness prediction. bioRxiv 2022, 2022–12.
- Laine, E.; Karami, Y.; Carbone, A. GEMME: a simple and fast global epistatic model predicting mutational effects. Molecular biology and evolution 2019, 36, 2604–2619. [CrossRef]
- Su, J.; Han, C.; Zhou, Y.; Shan, J.; Zhou, X.; Yuan, F. Saprot: Protein language modeling with structure-aware vocabulary. bioRxiv 2023, 2023–10.
- Hayes, T.; Rao, R.; Akin, H.; Sofroniew, N. J.; Oktay, D.; Lin, Z.; Verkuil, R.; Tran, V. Q.; Deaton, J.; Wiggert, M.; Badkundri R. Simulating 500 million years of evolution with a language model. bioRxiv 2024, 2024–07. [CrossRef]
- Xu, M.; Yuan, X.; Miret, S.; Tang, J. Protst: Multi-modality learning of protein sequences and biomedical texts. International Conference on Machine Learning 2023, 38749–38767.
- Zhuo, L.; Chi, Z.; Xu, M.; Huang, H.; Zheng, H.; He, C.; Mao, X.; Zhang, W. Protllm: An interleaved protein-language llm with protein-as-word pre-training. arXiv preprint arXiv:2403.07920 2024, .
- Zhang, B.; Liu, K.; Zheng, Z.; Zhu, J.; Li, Z.; Liu, Y.; Mu, J.; Wei, T.; Chen, H. Protein Language Model Supervised Scalable Approach for Diverse and Designable Protein Motif-Scaffolding with GPDL. bioRxiv 2023, 2023–10.
- Watson, J. L.; Juergens, D.; Bennett, N. R.; Trippe, B. L.; Yim, J.; Eisenach, H. E.; Ahern, W.; Borst, A. J.; Ragotte, R. J.; Milles, L. F.; Wicky, B.I. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [CrossRef]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R. J.; Milles, L. F.; Wicky, B. I.; Courbet, A.; de Haas, R. J.; Bethel, N.; Leung P. J. Robust deep learning–based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)—Round XV. Proteins: Structure, Function, and Bioinformatics 2023, 91, 1539–1549. [CrossRef]
- van Kempen, M.; Kim, S. S.; Tumescheit, C.; Mirdita, M.; Gilchrist, C. L.; Söding, J.; Steinegger, M. Foldseek: fast and accurate protein structure search. Biorxiv 2022, 2022–02. [CrossRef]
- Almagro Armenteros, J. J.; Sønderby, C. K.; Sønderby, S. K.; Nielsen, H.; Winther, O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nature Reviews Drug Discovery 2017, 16, 315–337. [CrossRef]
- Xing, H.; Cai, P.; Liu, D.; Han, M.; Liu, J.; Le, Y.; Zhang, D; Hu, Q.N. High-throughput prediction of enzyme promiscuity based on substrate–product pairs. Briefings in Bioinformatics 2024, 25, bbab089. [CrossRef]
- Feng, X.; Wang, Z.; Cen, M.; Zheng, Z.; Wang, B.; Zhao, Z.; Zhong, Z.; Zou, Y.; Lv, Q.; Li, S; Huang, L. Deciphering potential molecular mechanisms in clear cell renal cell carcinoma based on the ubiquitin-conjugating enzyme E2 related genes: Identifying UBE2C correlates to infiltration of regulatory T cells. BioFactors 2025, 51, e2143. [CrossRef]
- Moshawih, S.; Goh, H.P.; Kifli, N.; Idris, A.C.; Yassin, H.; Kotra, V.; Goh, K.W.; Liew, K.B; Ming, L.C. Synergy between machine learning and natural products cheminformatics: Application to the lead discovery of anthraquinone derivatives. Chemical Biology and Drug Design 2022, 99, 556–566.
- Hanahan, D; Weinberg, R.A. Weinberg Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [CrossRef]
- Gogoshin, G; Rodin, A.S. Graph neural networks in cancer and oncology research: emerging and future trends. Cancers 2023, 15(24), 5858.
- Avelar, P.H.D.C; Wu, M; Tsoka, S. Incorporating Prior Knowledge in Deep Learning Models via Pathway Activity Autoencoders. arXiv preprint arXiv:2306.05813 2023 .
- Kaur, S.; Xu, K.; Saad, O. M.; Dere, R. C.; Carrasco-Triguero, M. Antibody-drug conjugates for cancer therapy: Design, mechanism, and future applications. Advanced Drug Delivery Reviews 2021, 169, 34–46.
- Jain, R. K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nature Reviews Clinical Oncology 2010, 7, 653–664. [CrossRef]
- Yingying Diao; Yan Zhao; Xinyao Li; Baoyue Li; Ran Huo; Xiaoxu Han A Simplified Machine Learning Model Utilizing Platelet-Related Genes for Predicting Poor Prognosis in Sepsis. Frontiers in Immunology 2023, 14, 1286203. [CrossRef]
- Guojun Lu, W. S.; Yu Zhang Prognostic Implications and Immune Infiltration Analysis of ALDOA in Lung Adenocarcinoma. Frontiers in Genetics 2021, 12, 721021.
- Yang, J.; Virostko, J.; Liu, J.; Jarrett, A. M.; Hormuth, D. A.; Yankeelov, T. E. Comparing mechanism-based and machine learning models for predicting the effects of glucose accessibility on tumor cell proliferation. Scientific Reports 2023, 13, 10387. [CrossRef]
- Wang, Y.; Tang, J.; Liu, Y.; Zhang, Z.; Zhang, H.; Ma, Y.; Wang, X.; Ai, S.; Mao, Y.; Zhang, P.; Chen, S. Targeting ALDOA to Modulate Tumorigenesis and Energy Metabolism in Retinoblastoma. iScience 2024, 27, 10725. [CrossRef]
- Li, Y.N.; Su, J.L.; Tan, S.H.; Chen, X.L.; Cheng, T.L.; Jiang, Z.; Luo, Y.Z.; Zhang, L.M. Machine learning based on metabolomics unveils neutrophil extracellular trap-related metabolic signatures in non-small cell lung cancer patients undergoing chemoimmunotherapy. World Journal of Clinical Cases 2024, 12, 4091. [CrossRef]
- Zhou, C.; Jia, H.; Jiang, N.; Zhao, J.; Nan, X. Establishment of Chemotherapy Prediction Model Based on Hypoxia-Related Genes for Oral Cancer. Journal of Cancer 2024, 15, 5191–5203. [CrossRef]
- Abriata, L.A. The Nobel Prize in Chemistry: past, present, and future of AI in biology. Communications Biology 2024, 7, Article number: 1409. [CrossRef]
- Cooper, S.; Khatib, F.; Treuille, A.; Barbero, J.; Lee, J.; Beenen, M.; Leaver-Fay, A.; Baker, D.; Popović, Z.; Players, F. Predicting protein structures with a multiplayer online game. Nature 2010, 466, 756–760. [CrossRef]
- Leaver-Fay, A.; Tyka, M.; Lewis, S. M.; Lange, O. F.; Thompson, J.; Jacak, R.; Kaufman, K. W.; Renfrew, P. D.; Smith, C. A.; Sheffler, W.; Davis, I. W.; Cooper, S.; Treuille, A.; Mandell, D. J.; Richter, F.; Ban, Y.; Fleishman, S. J.; Corn, J. E.; Kim, D. E.; Lyskov, S.; Berrondo, M.; Mentzer, S.; Popović, Z.; Havranek, J. J.; Karanicolas, J.; Das, R.; Meiler, J.; Kortemme, T.; Gray, J. J.; Kuhlman, B.; Baker, D.; Bradley, P. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods in Enzymology 2011, 487, 545–574.
- Arnold, F. H. Innovation by Evolution: Bringing New Chemistry to Life. Angewandte Chemie International Edition 2019, 58, 14420–14426. [CrossRef]
- Ding, K.; Chin, M.; Zhao, Y.; Huang, W.; Mai, B.K.; Wang, H.; Liu, P.; Yang, Y.; Luo, Y. Machine learning-guided co-optimization of fitness and diversity facilitates combinatorial library design in enzyme engineering. Nature Communications 2024, 15, 6392. [CrossRef]
- Rouzbehani, R.; Kelley, S.T. AncFlow: An Ancestral Sequence Reconstruction Approach for Determining Novel Protein Structural. bioRxiv 2024, 2024-07. [CrossRef]
- Zhou, C.; Jia, H.; Jiang, N.; Zhao, J.; Nan, X. Establishment of chemotherapy prediction model based on hypoxia-related genes for oral cancer. Journal of Cancer 2024, 15(16), 5191. [CrossRef]
- Hollmann, F.; Sanchis, J.; Reetz, M. T. Learning from Protein Engineering by Deconvolution of Multi-Mutational Variants. Angewandte Chemie International Edition 2024, 63, e202404880. [CrossRef]
- Harding-Larsen, D.; Funk, J.; Madsen, N.G.; Gharabli, H.; Acevedo-Rocha, C.G.; Mazurenko, S.; Welner, D.H. Protein representations: Encoding biological information for machine learning in biocatalysis. Biotechnology Advances 2024, 108459. [CrossRef]
- Ahluwalia, V.K.; Kumar, L.S.; Kumar, S. Enzymes. In: Chemistry of Natural Products. Springer International Publishing, 2022.
- Laurent, J. M.; Janizek, J. D.; Ruzo, M.; Hinks, M. M.; Hammerling, M. J.; Narayanan, S.; Ponnapati, M.; White, A. D.; Rodriques, S. G. Lab-bench: Measuring capabilities of language models for biology research. arXiv preprint arXiv:2407.10362 2024, .
- Johnson, S. R.; Fu, X.; Viknander, S.; Goldin, C.; Monaco, S.; Zelezniak, A.; Yang, K. K. Computational scoring and experimental evaluation of enzymes generated by neural networks. Nature biotechnology 2024, 1–10. [CrossRef]
- Lipsh-Sokolik, R.; Fleishman, S. J. Addressing epistasis in the design of protein function. In Proceedings of the National Academy of Sciences 2024, 121, e2314999121. [CrossRef]
- Maynard Smith, J. Natural selection and the concept of a protein space. Nature 1970, 225, 563–564. [CrossRef]
- Ding, X.; Zou, Z.; Brooks III, C. L. Deciphering protein evolution and fitness landscapes with latent space models. Nature communications 2019, 10, 5644. [CrossRef]
- Zhou, Y.; Pan, Q.; Pires, D. E. V.; Rodrigues, C. H. M.; Ascher, D. B. DDMut: predicting effects of mutations on protein stability using deep learning. Nucleic Acids Research 2023, 51, W122-W128. [CrossRef]
- Singh, N.; Won, M.; An, J.; Yoon, C.; Kim, D.; Lee, S. J.; Kang, H.; Kim, J. S. Advances in covalent organic frameworks for cancer phototherapy. Coordination Chemistry Reviews 2024, 506, 215720. [CrossRef]
- Zhou, L.; Guan, Q.; Dong, Y. Covalent Organic Frameworks: Opportunities for Rational Materials Design in Cancer Therapy. Angewandte Chemie International Edition 2024, 63, e202314763. [CrossRef]
- Ding, C.; Chen, C.; Zeng, X.; Chen, H.; Zhao, Y. Emerging strategies in stimuli-responsive prodrug nanosystems for cancer therapy. ACS nano 2022, 16, 13513–13553. [CrossRef]
- Mizera, M.; Lewandowska, K.; Miklaszewski, A.; Cielecka-Piontek, J. Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis. Molecules 2019, 24, 743. [CrossRef]
- Hetrick, K. J.; Raines, R. T. Assessing and utilizing esterase specificity in antimicrobial prodrug development. Methods in enzymology 2022, 664, 199–220.
- Iyer, K. A.; Ivanov, J.; Tenchov, R.; Ralhan, K.; Rodriguez, Y.; Sasso, J. M.; Scott, S.; Zhou, Q. A. Emerging Targets and Therapeutics in Immuno-Oncology: Insights from Landscape Analysis. Journal of medicinal chemistry 2024, 67, 8519-8544. [CrossRef]
- Fu, L.; Li, M.; Lv, J.; Yang, C.; Zhang, Z.; Qin, S.; Li, W.; Wang, X.; Chen, L. Deep neural network for discovering metabolism-related biomarkers for lung adenocarcinoma. Frontiers in Endocrinology 2023, 14, 1270772. [CrossRef]
- Yang, J.; Lal, R. G.; Bowden, J. C.; Astudillo, R.; Hameedi, M. A.; Kaur, S.; Hill, M.; Yue, Y.; Arnold, F. H. Active Learning-Assisted Directed Evolution. bioRxiv 2024, . [CrossRef]
- Long, Y.; Mora, A.; Li, F.Z.; Gürsoy, E.; Johnston, K.E; Arnold, F.H. LevSeq: Rapid Generation of Sequence-Function Data for Directed Evolution and Machine Learning. bioRxiv 2024, .
- Prakinee, K., Phaisan, S., Kongjaroon, S. and Chaiyen, P. Ancestral Sequence Reconstruction for Designing Biocatalysts and Investigating their Functional Mechanisms. Ancestral Sequence Reconstruction for Designing Biocatalysts and Synthetic Biology. JACS Au 2024, 4, 4571–4591. [CrossRef]
- Zou, Z.; Higginson, B.; Ward, T.R. Creation and optimization of artificial metalloenzymes: Harnessing the power of directed evolution and beyond. Chem 2024, 10, 2373-2389. [CrossRef]
- Esteves, F., Rueff, J. and Kranendonk, M., 2021. The central role of cytochrome P450 in xenobiotic metabolism—a brief review on a fascinating enzyme family. Journal of Xenobiotics 2024, 11, pp.94-114. [CrossRef]
- Bundit Boonyarit, N. Y. GraphEGFR: Multi-task and transfer learning based on molecular graph attention mechanism and fingerprints improving inhibitor bioactivity prediction for EGFR family proteins on data scarcity. Journal of Computational Chemistry 2024, 45, Pages. [CrossRef]
- Ai, D., Cai, H., Wei, J., Zhao, D., Chen, Y. and Wang, L., 2023. DEEPCYPs: A deep learning platform for enhanced cytochrome P450 activity prediction. Frontiers in Pharmacology 2023, 14, 1099093. [CrossRef]
- Fang, J.; Tang, Y.; Gong, C.; Huang, Z.; Feng Y.; Liu, G.; Tang, Y.; Li, W. Prediction of Cytochrome P450 Substrates Using the Explainable Multitask Deep Learning Models. Chemical Research in Toxicology 2024, 37, . [CrossRef]
- Li, L.; Lu, Z.; Liu, G.; Tang, Y.; Li, W. Machine Learning Models to Predict Cytochrome P450 2B6 Inhibitors and Substrates. Chemical Research in Toxicology 2023, 36, . [CrossRef]
- Li, H.; Han, Z.; Sun, Y.; Wang, F.; Hu, P.; Gao, Y.; Bai, X.; Peng, S.; Ren, C.; Xu, X.; Liu, Z. CGMega: explainable graph neural network framework with attention mechanisms for cancer gene module dissection. Nature Communications 2024, 15, 5997. [CrossRef]
- Carrera-Pacheco, S.E.; Mueller, A.; Puente-Pineda, J.A.; Zúñiga-Miranda, J.; Guamán, L.P. Designing Cytochrome P450 Enzymes for Use in Cancer Gene Therapy. Frontiers in Bioengineering and Biotechnology 2024, 12, . [CrossRef]
- Mutti, P.; Lio, F.; Hollfelder, F. Microdroplet screening rapidly profiles a biocatalyst to illuminate functional sequence space. bioRxiv 2024, .
- Xing Wan, S. S. Discovery of alkaline laccases from basidiomycete fungi through machine learning-base approach. Biotechnology for Biofuels and Bioproducts 2024, 17, . [CrossRef]
- Changda Gong; Yanjun Feng; Jieyu Zhu; Guixia Liu; Yun Tang; Weihua Li Evaluation of Machine Learning Models for Cytochrome P450 Inhibition Prediction. Journal of Applied Toxicology 2024, 44, .
- Xin-Man Hu; Yan-Yao Hou; Xin-Ru Teng; Yong Liu; Yu Li; Wei Li; Yan Li; Chun-Zhi Ai Prediction of Cytochrome P450-Mediated Bioactivation Using Machine Learning Models and In Vitro Validation. Archives of Toxicology 2024, 98, .
- Kao, D. Prediction of Cytochrome P450-Related Drug-Drug Interactions by Deep Learning. 2022, .
- López-Vidal, E. M.; Schissel, C. K.; Mohapatra, S.; Bellovoda, K.; Wu, C.; Wood, J. A.; Malmberg, A. B.; Loas, A.; Gómez-Bombarelli, R.; Pentelute, B. L. Deep Learning Enables Discovery of a Short Nuclear Targeting Peptide for Efficient Delivery of Antisense Oligomers. JACS Au 2021, 1, 1751–1761. [CrossRef]
- Qin, Y.; Huo, M.; Liu, X.; Li, S. C. Biomarkers and computational models for predicting efficacy to tumor ICI immunotherapy. Frontiers in Immunology 2024, 15, 1368749. [CrossRef]
- Iacobini, C.; Vitale, M.; Pugliese, G.; Menini, S. The “sweet” path to cancer: focus on cellular glucose metabolism. Frontiers in Oncology, 13, p.1202093. Frontiers in Oncology 2024, 13, 1202093. [CrossRef]
- Yin, Q.; Wu, M.; Liu, Q.; Lv, H.; Jiang, R. DeepHistone: a deep learning approach to predicting histone modifications. BMC Genomics 2019, 20, 193. [CrossRef]
- Li, L.; Lu, Z.; Liu, G.; Tang, Y.; Li, W. Machine Learning Models to Predict Cytochrome P450 2B6 Inhibitors and Substrates. Chemical Research in Toxicology 2023, 36, 1227–1236. [CrossRef]
- Neelakandan A. R.; Rajanikant G. K. A deep learning and docking simulation-based virtual screening strategy enables the rapid identification of HIF-1α pathway activators from a marine natural product database. Journal of Biomolecular Structure and Dynamics 2024, 42, 629–651.
- Kim, J. H.; Lee, W. S.; Lee, H. J.; Yang, H.; Lee, S. J.; Kong, S. J.; Je, S.; Yang, H.; Jung, J.; Cheon, J.; Kang, B. Deep learning model enables the discovery of a novel immunotherapeutic agent regulating the kynurenine pathway. Oncoimmunology 2021, 10, 2005280. [CrossRef]
- Boush, M.; Kiaei, A. A.; Mahboubi, H. Trending Drugs Combination to Target Leukemia-associated Proteins/Genes: Using Graph Neural Networks under the RAIN Protocol. medRxiv 2023, 2023–08.
- Liu, X.; Hu, W.; Diao, S.; Abera, D.E.; Racoceanu, D.; Qin, W. Multi-scale feature fusion for prediction of IDH1 mutations in glioma histopathological images. Computer Methods and Programs in Biomedicine 2024, 248, 108116. [CrossRef]
- Cong, C.; Xuan, S.; Liu, S.; Pagnucco, M.; Zhang, S.; Song, Y. Dataset Distillation for Histopathology Image Classification. arXiv preprint arXiv:2408.09709 2024.
- Fang, Z.; Liu, Y.; Wang, Y.; Zhang, X.; Chen, Y.; Cai, C.; Lin, Y.; Han, Y.; Wang, Z.; Zeng, S.; Shen, H. Deep Learning Predicts Biomarker Status and Discovers Related Histomorphology Characteristics for Low-Grade Glioma. arXiv preprint arXiv:2310.07464 2023, 15, .
- Liu, Y.; Xu, W.; Li, M.; Yang, Y.; Sun, D.; Chen, L.; Li, H.; Chen, L. The regulatory mechanisms and inhibitors of isocitrate dehydrogenase 1 in cancer. Acta Pharmaceutica Sinica B 2023, 13, 1438–1466.
- Sonowal, S., Pathak, K., Das, D., Buragohain, K., Gogoi, A., Borah, N., Das, A. and Nath, R. L-Asparaginase Bio-Betters: Insight Into Current Formulations, Optimization Strategies and Future Bioengineering Frontiers in Anti-Cancer Drug Development. Advanced Therapeutics 2024, 7, 2400156. [CrossRef]
- Sun, H.; Yang, Q.; Yu, X.; Huang, M.; Ding, M.; Li, W.; Tang, Y.; Liu, G. Prediction of IDO1 Inhibitors by a Fingerprint-Based Stacking Ensemble Model Named IDO1Stack. ChemMedChem 2023, 18, e202300151. [CrossRef]
- Zhao, Y.; Wang, W.; Ji, Y.; Guo, Y.; Duan, J.; Liu, X.; Yan, D.; Liang, D.; Li, W.; Zhang, Z.; Li, Z. Computational Pathology for Prediction of Isocitrate Dehydrogenase Gene Mutation from Whole Slide Images in Adult Patients with Diffuse Glioma. The American Journal of Pathology 2024, 194, 747–758. [CrossRef]
- Redlich, J.; Feuerhake, F.; Weis, J.; Schaadt, N. S.; Teuber-Hanselmann, S.; Buck, C.; Luttmann, S.; Eberle, A.; Nikolin, S.; Appenzeller, A.; Portmann, A.; Homeyer, A. Applications of artificial intelligence in the analysis of histopathology images of gliomas: a review. npj Imaging 2024.
- Lv, Q.; Liu, Y.; Sun, Y.; Wu, M. Insight into deep learning for glioma IDH medical image analysis: A systematic review. Medicine 2024, 103, e37150.
- Fang, Z.; Liu, Y.; Wang, Y.; Zhang, X.; Chen, Y.; Cai, C.; Lin, Y.; Han, Y.; Wang, Z.; Zeng, S.; Shen, H.; Tan, J.; Zhang, Y. Deep learning predicts biomarker status and discovers related histomorphology characteristics for low-grade glioma. arXiv preprint arXiv:2310.07464 2023. [CrossRef]
- Cong, C.; Xuan, S.; Liu, S.; Pagnucco, M.; Zhang, S.; Zhang, S.; Song, Y. Dataset distillation for histopathology image classification. arXiv preprint arXiv:2408.09709 2024. [CrossRef]
- Guo, J.; Xu, P.; Wu, Y.; Tao, Y.; Han, C.; Lin, J.; Zhao, K.; Liu, Z.; Liu, W.; Lu, C. CroMAM: A Cross-Magnification Attention Feature Fusion Model for Predicting Genetic Status and Survival of Gliomas using Histological Images. IEEE Journal of Biomedical and Health Informatics 2024. [CrossRef]
- Zhang, X.; Shi, X.; Iwamoto, Y.; Chen, Y.W. IDH mutation status prediction by a radiomics associated modality attention network. Visual Computer 2023, 39, 2367–2379. [CrossRef]
- Jiang, S.; Zanazzi G. J.; Hassanpour, S. Predicting prognosis and IDH mutation status for patients with lower-grade gliomas using whole slide images. Scientific Reports 2021, 11, 16849. [CrossRef]
- Basak, K.; Ozyoruk, K.B.; Demir, D. Whole Slide Images in Artificial Intelligence Applications in Digital Pathology: Challenges and Pitfalls. Turkish Journal of Pathology 2023, 39, 101–108.
- Notin, P.; Dias, M.; Frazer, J.; Marchena-Hurtado, J.; Gomez, A.N.; Marks, D.;Gal, Y. Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. ArXiv 2022, abs/2205.13760, .
- Bozkurt, E. U.; rsted, E. C.; Volke, D. C.; Nikel, P. I. Accelerating enzyme discovery and engineering with high-throughput screening. Natural Product Reports 2025, .
- Lu, M. Y.; Williamson, D. F. K.; Chen, T. Y.; Mahmood, F. Data-efficient and weakly supervised computational pathology on whole-slide images. Nature Biomedical Engineering 2021, 5, 555–570.
- Shao, Z.; Bian, H.; Chen, Y.; Wang, Y.; Zhang, J.; Ji, X.; Zhang, Y. TransMIL: Transformer Based Correlated Multiple Instance Learning for Whole Slide Image Classification. In Proceedings of the 35th International Conference on Neural Information Processing Systems (NeurIPS) 2021, 2136–2147.
- Khorsandi, D.; Rezayat, D.; Sezen, S.; Ferrao, R.; Khosravi, A.; Zarepour, A.; Khorsandi, M.; Hashemian, M.; Iravani, S.; Zarrabi, A. Application of 3D, 4D, 5D, and 6D Bioprinting in Cancer Research: What Does the Future Look Like?. Journal of Materials Chemistry B 2024, Issue 19, .
- Laury, A.R.; Zheng, S.; Aho, N.; Fallegger, R.; Hänninen, S.; Saez-Rodriguez, J.; Tanevski, J.; Youssef, O.; Tang, J.; Carpén, O.M. Opening the Black Box: Spatial Transcriptomics and the Relevance of Artificial Intelligence–Detected Prognostic Regions in High-Grade Serous Carcinoma. Modern Pathology 2024, 37, 100508. [CrossRef]
- Nogales, J. M. S.; Parras, J.; Zazo, S. DDQN-based optimal targeted therapy with reversible inhibitors to combat the Warburg effect. Mathematical Biosciences 2023, 363, 109044.
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; Rives, A. Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv 2022, 2022, 500902.
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y; Chen, P; Canny, J; Abbeel, P; Song, Y. TAPE: A Benchmark for Transformer-Based Models of Protein Sequences. arXiv preprint arXiv:2003.11803 2020, .
- Hie, B.L.; Yang, K.K; Kim, P.S. Evolutionary velocity with protein language models predicts evolutionary dynamics of diverse proteins. Cell Systems 2022, 13, 274–285. [CrossRef]
- Howard H.R. Using Machine Vision of Glycolytic Elements to Predict Breast Cancer Recurrences: Design and Implementation. Metabolites 2023, 13, 41.
- Brouwer, B.; Della-Felice, F.; Illies, J.H.; Iglesias-Moncayo, E.; Roelfes, G.; Drienovská, I. Noncanonical Amino Acids: Bringing New-to-Nature Functionalities to Biocatalysis. Chemical Reviews 2024, 124, XX. [CrossRef]
- Moon, H. H.; Jeong, J.; Park, J. E.; Kim, N.; Choi, C.; Kim, Y.; Song, S. W.; Hong, C.; Kim, J. H.; Kim, H. S. Generative AI in glioma: Ensuring diversity in training image phenotypes to improve diagnostic performance for IDH mutation prediction. Neuro-Oncology 2024, 26, 1124-1135. [CrossRef]
- Fayyaz, M., Chaudhry, N. and Choudhary, R. Classification of Isocitrate Dehydrogenase (IDH) Mutation Status in Gliomas Using Transfer Learning. Pakistan Journal of Scientific Research 2024, 3, 224–233. [CrossRef]
- Zhang, X.; Shi, X.; Iwamoto, Y.; Cheng, J.; Bai, J.; Zhao, G.; Han, X. Chen, Y. W. IDH mutation status prediction by a radiomics associated modality attention network. The Visual Computer 2023, 39(6), 2367–2379.
- Krebs, O.; Agarwal, S.; Tiwari, P. Self-supervised deep learning to predict molecular markers from routine histopathology slides for high-grade glioma tumors. Medical Imaging 2023: Digital and Computational Pathology 2023, 12471, 1247102.
- Wang, Y.; Xue, P.; Cao, M.; Yu, T.; Lane, S. T.; Zhao, H. Directed evolution: methodologies and applications. Chemical reviews 2021, 121, 12384–12444.
- Alzaeemi, S. A.; Noman, E. A.; Al-shaibani, M. M.; Al-Gheethi, A.; Mohamed, R. M. S. R.; Almoheer, R.; Seif, M.; Tay, K. G.; Zin, N. M.; El Enshasy, H. A. Improvement of L-asparaginase, an Anticancer Agent of Aspergillus arenarioides EAN603 in Submerged Fermentation Using a Radial Basis Function Neural Network with a Specific Genetic Algorithm (RBFNN-GA). Fermentation 2023, 9, . [CrossRef]
- Madani, A.; McCann, B.; Naik, N.; Keskar, N.S.; Anand, N.; Eguchi, R.R.; Huang, P.S.; Socher, R. ProGen: Language Modeling for Protein Generation. bioRxiv 2020, .
- Xu, G.; Lv, Y.; Zhang, R.; Xia, X.; Wang, Q.; Ma, J. OPUS-GO: An interpretable protein/RNA sequence annotation framework based on biological language model. bioRxiv 2024, 2024–12.
- Safari, M.; Beiki, M.; Ameri, A.; Toudeshki, S.H.; Fatemi, A.; Archambault, L. Shuffle-ResNet: Deep learning for predicting LGG IDH1 mutation from multicenter anatomical MRI sequences. Biomedical Physics and Engineering Express 2022, 8, 065036. [CrossRef]
- Goldwaser, E.; Laurent, C.; Lagarde, N.; Fabrega, S.; Nay, L.; Villoutreix, B.O.; Jelsch, C.; Nicot, A.B.; Loriot, M.A; Miteva, M.A. Machine learning-driven identification of drugs inhibiting cytochrome P450 2C9. PLOS Computational Biology 2022, 18, e1009820. [CrossRef]
- Gao, F.; Huang, Y.; Yang, M.; He, L.; Yu, Q.; Cai, Y.; Shen, J.; Lu, B. Machine learning-based cell death marker for predicting prognosis and identifying tumor immune microenvironment in prostate cancer. Heliyon 2024, 10, e37554. [CrossRef]
- Wang, J.; Lisanza, S.; Juergens, D.; Tischer, D.; Watson, J.L.; Castro, K.M.; Ragotte, R.; Saragovi, A.; Milles, L.F.; Baek, M.; Anishchenko, I. Scaffolding protein functional sites using deep learning. Science 2022, 377, 387–394. [CrossRef]
- Deng, Y.; Li, J.; He, Y.; Du, D.; Hu, Z.; Zhang, C.; Rao, Q.; Xu, Y.; Wang, J.; Xu, K. The deubiquitinating enzymes-related signature predicts the prognosis and immunotherapy response in breast cancer. Aging (Albany NY) 2024, 16, 11553–11567. [CrossRef]
- Zeng, X.; Wang, P. Deep learning for computational biology. BMC Genomics 2019, 20, 1–16.
- Li, Y.; Zeng, M.; Zhang, F.; Wu, F.; Li, M. DeepCellEss: cell line-specific essential protein prediction with attention-based interpretable deep learning. Bioinformatics 2022, 39, btac779. [CrossRef]
- Corpas, F. J.; Barroso, J. B.; Sandalio, L. M.; Palma, J. M.; Lupiánez, J. A.; del Rıo, L. A. Peroxisomal NADP-dependent isocitrate dehydrogenase. Characterization and activity regulation during natural senescence. Plant Physiology 1999, 121, 921–928.
- Li, X.; Strasser, B.; Neuberger, U.; Vollmuth, P.; Bendszus, M.; Wick, W.; Dietrich, J.; Batchelor, T. T.; Cahill, D. P.; Andronesi, O. C. Deep learning super-resolution magnetic resonance spectroscopic imaging of brain metabolism and mutant isocitrate dehydrogenase glioma. Neuro-Oncology Advances 2022, 4, vdac071.
- Tang, W.T.; Su, C.Q.; Lin, J.; Xia, Z.W.; Lu, S.S; Hong, X.N. T2-FLAIR mismatch sign and machine learning-based multiparametric MRI radiomics in predicting IDH mutant 1p/19q non-co-deleted diffuse lower-grade gliomas. Clinical Radiology 2024, 79, e750–e758.
- Zhang, H.; Fan, X.; Zhang, J.; Wei, Z.; Feng, W.; Hu, Y.; Ni, J.; Yao, F.; Zhou, G.; Wan, C.; Zhang, X. Deep-learning and conventional radiomics to predict IDH genotyping status based on magnetic resonance imaging data in adult diffuse glioma. Frontiers in Oncology 2023, 13, 1143688.
- Nakagaki, R.; Debsarkar, S.S.; Kawanaka, H.; Aronow, B.J.; Prasath, V.S. Deep learning-based IDH1 gene mutation prediction using histopathological imaging and clinical data. Computers in Biology and Medicine 2024, 179, 108902.
- Lee, S.H.; Jang, H.J. Deep learning-based prediction of molecular cancer biomarkers from tissue slides: A new tool for precision oncology. Clinical and Molecular Hepatology 2022, 28, 754–772. [CrossRef]
- Sharrock, A.V.; Mumm, J.S.; Williams, E.M.; Čėnas, N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F.; Bagdžiūnas, G.; Arcus, V.L. Structural Evaluation of a Nitroreductase Engineered for Improved Activation of the 5-Nitroimidazole PET Probe SN33623. International Journal of Molecular Sciences 2024, 25, 6593.
- Levinthal, C. Are there pathways for protein folding?Journal de Chimie Physique 1968, 65, 44–45. [CrossRef]
- Xu, G.; McLeod, H.L. Strategies for enzyme/prodrug cancer therapy. Clinical Cancer Research 2001, 7, 3314–3324.
- Kim, G. B.; Kim, J. Y.; Lee, J. A.; Norsigian, C. J.; Palsson, B. O.; Lee, S. Y. Functional annotation of enzyme-encoding genes using deep learning with transformer layers. Nature Communications 2023, 14, 7370. [CrossRef]
- Alam, S.; Pranaw, K.; Tiwari, R.; Khare, S. K. Recent development in the uses of asparaginase as food enzyme. Green bio-processes: enzymes in industrial food processing 2019, 55–81.
- Buller, R.; Lutz, S.; Kazlauskas, R.; Snajdrova, R.; Moore, J.; Bornscheuer, U. From nature to industry: Harnessing enzymes for biocatalysis. Science 2023, 382, eadh8615. [CrossRef]
- Reetz, M. T.; Qu, G.; Sun, Z. Engineered enzymes for the synthesis of pharmaceuticals and other high-value products. Nature Synthesis 2024, 3, 19–32. [CrossRef]
- Yu, H.; Deng, H.; He, J.; Keasling, J. D.; Luo, X. UniKP: a unified framework for the prediction of enzyme kinetic parameters. Nature communications 2023, 14, 8211.
- Ge, F.; Chen, G.; Qian, M.; Xu, C.; Liu, J.; Cao, J.; Li, X.; Hu, D.; Xu, Y.; Xin, Y.; Wang, D. Artificial intelligence aided lipase production and engineering for enzymatic performance improvement. Journal of Agricultural and Food Chemistry 2023, 71, 14911–14930.
- Yang, R.; Zha, X.; Gao, X.; Wang, K.; Cheng, B.; Yan, B. Multi-stage virtual screening of natural products against p38α mitogen-activated protein kinase: Predictive modeling by machine learning, docking study, and molecular dynamics simulation. Heliyon 2022, 8, e10495.
- Kumar, S.; Boehm, J.; Lee, J. C. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nature Reviews Drug Discovery 2003, 2, 717–726.
- Kumar, M.; Anand, S.; Jha, R. K.; Singh, A. Neural Network-Based L-Asparaginase Production Using Acinetobacter baumannii from Pectic Waste: Process Optimization by Response Surface Methodology. Biocatalysis and Agricultural Biotechnology 2016, 7, 173–180.
- Wilkinson, S. P.; Li, Y.; Zhu, Y. Bioinformatic Insights into the Evolutionary Origins of L-Asparaginase Enzymes with Antitumor Properties. Scientific Reports 2022, 12, 4567.
- Smith, J.; Patel, R.; Kim, E. Machine Learning-Assisted Screening of L-Asparaginase Variants for Enhanced Stability and Activity. Computational Biology and Chemistry 2023, 104, 107080.
- Ramirez, D. L.; Hernandez, J. M.; Lopez, M. C. A Novel L-Asparaginase from Thermophilic Bacteria: Biochemical Characterization and Potential Biotechnological Applications. Journal of Molecular Catalysis B: Enzymatic 2021, 180, 105640.
- Jain, P.; Das, S.; Ghosh, K. Deep Learning for Predicting Catalytic Residues in L-Asparaginase Enzymes. BMC Bioinformatics 2024, 25, 89.
- Zhang, H.; Wang, L.; Zhou, J. Enhanced L-Asparaginase Activity through Directed Evolution and Computational Design. Protein Engineering, Design and Selection 2020, 33, 145–152.
- Park, H. J.; Choi, S. W.; Kang, M. J. Cryo-EM Analysis of L-Asparaginase Complex Reveals Molecular Mechanism of Substrate Recognition. Structure 2019, 27, 1023–1031.
- Tanaka, K.; Nakamura, Y.; Ito, A. Synthetic Biology Approaches to Engineer L-Asparaginase for Tailored Therapeutic Applications. Biotechnology Advances 2023, 65, 108068.
- Munn, D. H.; Sharma, M. D.; Baban, B.; Harding, H. P.; Zhang, Y.; Ron, D.; Mellor, A. L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2, 3-dioxygenase. Immunity 2005, 22, 633–642. [CrossRef]
- Muller, A. J.; Manfredi, M. G.; Zakharia, Y.; Prendergast, G. C. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Seminars in Immunopathology 2019, 41, 41–48. [CrossRef]
- Johnson, T. S.; Mcgaha, T.; Munn, D. H. Chemo-immunotherapy: role of indoleamine 2, 3-dioxygenase in defining immunogenic versus tolerogenic cell death in the tumor microenvironment. Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy 2017, 91–104.
- Munn, D. H.; Mellor, A. L. Indoleamine 2, 3 dioxygenase and metabolic control of immune responses. Trends in Immunology 2013, 34, 137–143.
- Niu, B.; Lee, B.; Wang, L.; Chen, W.; Johnson, J. The Accurate Prediction of Antibody Deamidations by Combining High-Throughput Automated Peptide Mapping and Protein Language Model-Based Deep Learning. Antibodies 2024, 13, 74.
- Hua, C.; Lu, J.; Liu, Y.; Zhang, O.; Tang, J.; Ying, R.; Jin, W.; Wolf, G.; Precup, D.; Zheng, S. Reaction-conditioned De Novo Enzyme Design with GENzyme. arXiv preprint arXiv:2411.16694 2024, .
- Wen, Y.; Liu, H.; Cao, C.; Wu, R. Applications of protein engineering in the pharmaceutical industry. Synthetic Biology Journal 2024, 1.
- Yan, Y.; Shi, Z.; Wei, H. ROSes-FINDER: a multi-task deep learning framework for accurate prediction of microorganism reactive oxygen species scavenging enzymes. Frontiers in Microbiology 2023, 14, 1245805. [CrossRef]
- Nijkamp, E.; Ruffolo, J. A.; Weinstein, E. N.; Naik, N.; Madani, A. Progen2: exploring the boundaries of protein language models. Cell Systems 2023, 14, 968–978. [CrossRef]
- Hager, P.; Jungmann, F.; Holland, R.; Bhagat, K.; Hubrecht, I.; Knauer, M.; Vielhauer, J.; Makowski, M.; Braren, R.; Kaissis, G.; Rueckert, D., Evaluation and mitigation of the limitations of large language models in clinical decision-making. Nature Medicine 2024, 30, 2613–2622. [CrossRef]
- Thirunavukarasu, A. J.; Ting, D. S. J.; Elangovan, K.; Gutierrez, L.; Tan, T. F.; Ting, D. S. W. Large language models in medicine. Nature Medicine 2023, 29, 1930–1940. [CrossRef]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T. B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling laws for neural language models. arXiv preprint arXiv:2001.08361 2020, .
- Hoffmann, J.; Borgeaud, S.; Mensch, A.; Buchatskaya, E.; Cai, T.; Rutherford, E.; Casas, D. d. L.; Hendricks, L. A.; Welbl, J.; Clark, A.; Hennigan, T.Training compute-optimal large language models. arXiv preprint arXiv:2203.15556 2022, .
- Huang, C.; Zhang, L.; Tang, T.; Wang, H.; Jiang, Y.; Ren, H.; Zhang, Y.; Fang, J.; Zhang, W.; Jia, X.; You, S. Application of Directed Evolution and Machine Learning to Enhance the Diastereoselectivity of Ketoreductase for Dihydrotetrabenazine Synthesis. JACS Au 2024, 4, 2547–2556.
- López-Cortés, A.; Cabrera-Andrade, A.; Echeverría-Garcés, G.; Echeverría-Espinoza, P.; Pineda-Albán, M.; Elsitdie, N.; Bueno-Miño, J.; Cruz-Segundo, C. M.; Dorado, J.; Pazos, A.; Gonzáles-Díaz, H. Unraveling druggable cancer-driving proteins and targeted drugs using artificial intelligence and multi-omics analyses. Scientific Reports 2024, 14, 19359. [CrossRef]
- Giurini, E. F.; Godla, A.; Gupta, K. H. Redefining bioactive small molecules from microbial metabolites as revolutionary anticancer agents. Cancer Gene Therapy 2024, 31, 187–206. [CrossRef]
- Askari, M.; Kiaei, A. A.; Boush, M.; Aghaei, F. Emerging Drug Combinations for Targeting Tongue Neoplasms Associated Proteins/Genes: Employing Graph Neural Networks within the RAIN Protocol. bioRxiv 2024, 2024–06.
- Dashti, N.; Kiaei, A. A.; Boush, M.; Gholami-Borujeni, B.; Nazari, A. AI-Enhanced RAIN Protocol: A Systematic Approach to Optimize Drug Combinations for Rectal Neoplasm Treatment. bioRxiv 2024, 2024–05.
- Sadeghi, S.; Kiaei, A. A.; Boush, M.; Salari, N.; Mohammadi, M.; Safaei, D.; Mahboubi, M.; Tajfam, A.; Moghadam, S. A graphSAGE discovers synergistic combinations of Gefitinib, paclitaxel, and Icotinib for Lung adenocarcinoma management by targeting human genes and proteins: the RAIN protocol. medRxiv 2024, 2024–04.
- Yang, L.; Lei, S.; Xu, W.; Wang, Z.L. Rising above: exploring the therapeutic potential of natural product-based compounds in human cancer treatment. Tradit Med Res 2025, 10, 18. [CrossRef]
- Harding-Larsen, D.; Funk, J.; Madsen, N. G.; Gharabli, H.; Acevedo-Rocha, C. G.; Mazurenko, S.; Welner, D. H. Protein representations: Encoding biological information for machine learning in biocatalysis. Biotechnology Advances 2024, 108459. [CrossRef]
- Jelassi, S.; Brandfonbrener, D.; Kakade, S. M.; Malach, E. Repeat after me: Transformers are better than state space models at copying. arXiv preprint arXiv:2402.01032 2024, .
- Pleiss, J. Modeling Enzyme Kinetics: Current Challenges and Future Perspectives for Biocatalysis. Biochemistry 2024, 63, 2533–2541.
- Felix A. Döppel; Martin Votsmeier. Robust mechanism discovery with atom conserving chemical reaction neural networks. In Proceedings of the Combustion Institute 2024, 40, 105507.
- Grauman, Å.; Ancillotti, M.; Veldwijk, J.; Mascalzoni, D. Precision cancer medicine and the doctor-patient relationship: a systematic review and narrative synthesis. BMC Medical Informatics and Decision Making 2023, 23, 286. [CrossRef]
- Chen, J.; Jiang, Y.; Zheng, T. Unraveling the Double-Edged Sword Effect of AI Transparency on Algorithmic Acceptance. 2024, .
- Xie, W. J.; Warshel, A. Harnessing generative AI to decode enzyme catalysis and evolution for enhanced engineering. National Science Review 2023, 10, nwad331. [CrossRef]
- Cao, Y.; Zhang, J.; Lee, H. Expanding functional protein sequence spaces using generative adversarial networks. Nature Communications 2023, 14, 2188.
- Ahmed, Y. B.; Al-Bzour, A. N.; Ababneh, O. E.; Abushukair, H. M.; Saeed, A. Genomic and Transcriptomic Predictors of Response to Immune Checkpoint Inhibitors in Melanoma Patients: A Machine Learning Approach. Cancers 2022, 14, 5605. [CrossRef]
- Emran, T.B.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.R.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; Mitra, S. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention, and Therapeutic Approaches. Frontiers in Oncology 2022, 12, 891652.
- Martin Alonso, M. C.; Alamdari, S.; Samad, T. S.; Yang, K. K.; Bhatia, S. N.; Amini, A. P. Deep learning guided design of protease substrates. bioRxiv 2025, 2025–02.
- Zhang, Y.; Cui, H.; Zhang, R.; Zhang, H.; Huang, W. Nanoparticulation of Prodrug into Medicines for Cancer Therapy. Advanced Science 2021, . [CrossRef]
- Bannigan, P.; Bao, Z.; Hickman, R. J.; Aldeghi, M.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning models to accelerate the design of polymeric long-acting injectables. Nature Communications 2023, 14, 35. [CrossRef]
- Guengerich, F. P.; Waxman, D. J.; Mishra, A.; Zhao, X. Designing Cytochrome P450 Enzymes for Use in Cancer Gene Therapy. Frontiers in Bioengineering and Biotechnology 2024, 12, 1405466. [CrossRef]
- Sasidharan, S.; Gosu, V.; Tripathi, T.; Saudagar, P. Molecular Dynamics Simulation to Study Protein Conformation and Ligand Interaction. Protein Folding Dynamics and Stability: Experimental and Computational Methods 2023, 107–127.
- Tandel, G. S.; Biswas, M.; Kakde, O. G.; Tiwari, A.; Suri, H. S.; Turk, M.; Laird, J. R.; Asare, C. K.; Ankrah, A. A.; Khanna, N.; Madhusudhan, B.K. A review on a deep learning perspective in brain cancer classification. Cancers 2019, 11, 111. [CrossRef]
- Nicolás Lefin; Javiera Miranda; Jorge F. Beltrán; Lisandra Herrera Belén; Brian Effer; Adalberto Pessoa Jr; Jorge G. Farias; Zamorano, M. Current state of molecular and metabolic strategies for the improvement of L-asparaginase expression in heterologous systems. Front. Pharmacol. 2023, 14, 1208277.
- Callaghan, R., Luk, F. and Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy?. Drug Metabolism and Disposition 2014, 42, 623–631.
- Wang, X., Zhang, H. and Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2019, 2, 141–160.
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Molecular Cancer 2019, 18, 1-12.
- Jennings, M.R.; Munn, D; Blazeck, J. Immunosuppressive metabolites in tumoral immune evasion: redundancies, clinical efforts, and pathways forward. Journal for ImmunoTherapy of Cancer 2021, 9, e003013.
- Chen, Z.; Liu, Y.; Wang, Y. G.; Shen, Y. Validation of an LLM-based Multi-Agent Framework for Protein Engineering in Dry Lab and Wet Lab. arXiv preprint arXiv:2411.06029 2024, .
- Shen, Y.; Chen, Z.; Mamalakis, M.; He, L.; Xia, H.; Li, T.; Su, Y.; He, J.; Wang, Y. G. A Fine-tuning Dataset and Benchmark for Large Language Models for Protein Understanding. In Proceedings of the 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2024, 2390–2395.
- Pecher, B.; Srba, I.; Bielikova, M. A survey on stability of learning with limited labelled data and its sensitivity to the effects of randomness. ACM Computing Surveys 2024, 57, 1–40.
- Kim, S.; Schrier, J.; Jung, Y. Explainable Synthesizability Prediction of Inorganic Crystal Structures using Large Language Models. Angewandte Chemie International Edition 2024, 64, e202423950. [CrossRef]
- Van Herck, J.; Gil, M. V.; Jablonka, K. M.; Abrudan, A.; Anker, A. S.; Asgari, M.; Blaiszik, B.; Buffo, A.; Choudhury, L.; Corminboeuf, C.; Daglar, H. Assessment of fine-tuned large language models for real-world chemistry and material science applications . Chemical Science 2025, 16, 670–684. [CrossRef]
- Bengio, Y.; Mindermann, S.; Privitera, D.; Besiroglu, T.; Bommasani, R.; Casper, S.; Choi, Y.; Goldfarb, D.; Heidari, H.; Khalatbari, L.; Longpre, S. International Scientific Report on the Safety of Advanced AI (Interim Report). arXiv preprint arXiv:2412.05282 2024 .
- Sarumi, O. A.; Heider, D. Large language models and their applications in bioinformatics. Computational and Structural Biotechnology Journal 2024.
- Yan, K.; Tang, Z. When General-Purpose Large Language Models Meet Bioinformatics. In CS582 ML for bioinformatics workshop , .
- Narayanan, S.; Braza, J. D.; Griffiths, R.; Ponnapati, M.; Bou, A.; Laurent, J.; Kabeli, O.; Wellawatte, G.; Cox, S.; Rodriques, S. G.; White, A.D. Aviary: training language agents on challenging scientific tasks. arXiv preprint arXiv:2412.21154 2024, .
- Brueggemeier, R. W.; Hackett, J. C.; Diaz-Cruz, E. S. Aromatase Inhibitors in the Treatment of Breast Cancer. Endocrine Reviews 2005, 26, 331-345. [CrossRef]
- Qin, Y.; Chen, Z.; Peng, Y.; Xiao, Y.; Zhong, T.; Yu, X. Deep learning methods for protein structure prediction. MedComm–Future Medicine 2024, 3, e96. [CrossRef]
- Gu, Z.; Luo, X.; Chen, J.; Deng, M.; Lai, L. Hierarchical graph transformer with contrastive learning for protein function prediction. Bioinformatics 2023, 39, btad410. [CrossRef]
- Sun, Y.; Li, X.; Dalal, K.; Xu, J.; Vikram, A.; Zhang, G.; Dubois, Y.; Chen, X.; Wang, X.; Koyejo, S.; Hashimoto, T. Learning to (learn at test time): Rnns with expressive hidden states. arXiv preprint arXiv:2407.04620 2024, .
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; Bhowmik, D. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence 2021, 44, 7112–7127. [CrossRef]
- Yu, T., Cui, H., Li, J.C., Luo, Y., Jiang, G. and Zhao, H. Enzyme function prediction using contrastive learning. Science 2023, 379, xx–xx. [CrossRef]
- Sampaio, P.; Fernandes, P. Machine Learning: A Suitable Method for Biocatalysis. Catalysts 2023, 13, 961. [CrossRef]
- Trippe, B. L.; Yim, J.; Tischer, D.; Baker, D.; Broderick, T.; Barzilay, R.; Jaakkola, T. Diffusion probabilistic modeling of protein backbones in 3d for the motif-scaffolding problem. arXiv preprint arXiv:2206.04119 2022, .
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; others Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nature Communications 2022, 13, 4348.
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; Bodenstein, S.W. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500.
- Ryu, J. Y.; Kim, H. U.; Lee, S. Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers. In Proceedings of the National Academy of Sciences 2019, 116, 13996–14001.
- Ma, B.; Kumar, S.; Tsai, C.; Hu, Z.; Nussinov, R. Transition-state ensemble in enzyme catalysis: possibility, reality, or necessity? Journal of theoretical biology 2000, 203, 383–397.
- Guengerich, F.P. Roles of Individual Human Cytochrome P450 Enzymes in Drug Metabolism. Pharmacological Reviews 2024, 76, 1104–1132.
- Huang, Y. J.; Mao, B.; Aramini, J. M.; Montelione, G. T. Assessment of template-based protein structure predictions in CASP10. Proteins: Structure, Function, and Bioinformatics 2014, 82, 43–56. [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics 2004, 57, 702–710. [CrossRef]
- Chae, J.; Wang, Z.; Qin, P. pLDDT-Predictor: High-speed Protein Screening Using Transformer and ESM2. arXiv preprint arXiv:2410.21283 2024 .
- Zheng, Z.; Zhang, B.; Zhong, B.; Liu, K.; Li, Z.; Zhu, J.; Yu, J.; Wei, T.; Chen, H. Scaffold-Lab: Critical Evaluation and Ranking of Protein Backbone Generation Methods in A Unified Framework. bioRxiv 2024, 2024–02. [CrossRef]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; Ma, J. High-resolution de novo structure prediction from primary sequence. BioRxiv 2022, 2022–07.
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; others Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [CrossRef]
- Chen, Z.; Zhao, Y.; Liu, Y. Advanced strategies of enzyme activity regulation for biomedical applications. ChemBioChem 2022, 23, e202200358.
- Wang, C.; Zhong, B.; Zhang, Z.; Chaudhary, N.; Misra, S.; Tang, J. PDB-Struct: A Comprehensive Benchmark for Structure-based Protein Design. arXiv preprint arXiv:2312.00080 2023, .
- Edmunds, N. S.; Genc, A. G.; McGuffin, L. J. Benchmarking of AlphaFold2 accuracy self-estimates as indicators of empirical model quality and ranking: a comparison with independent model quality assessment programmes. Bioinformatics 2024, 40, btae491.
- Zhang, Z.; Shen, W.X.; Liu, Q.; Zitnik, M. Efficient Generation of Protein Pockets with PocketGen. Nature Machine Intelligence 2024, 1–14.
- Hayes, T.; Rao, R.; Akin, H.; Sofroniew, N. J.; Oktay, D.; Lin, Z.; Verkuil, R.; Tran, V. Q.; Deaton, J.; Wiggert, M.; Badkundri, R. Simulating 500 million years of evolution with a language model. Science 2025, eads0018. [CrossRef]
- Zambaldi, V.; La, D.; Chu, A. E.; Patani, H.; Danson, A. E.; Kwan, T. O.; Frerix, T.; Schneider, R. G.; Saxton, D.; Thillaisundaram, A.; Wu, Z. De novo design of high-affinity protein binders with AlphaProteo. arXiv preprint arXiv:2409.08022 2024.
- Qiu, J.; Li, L.; Sun, J.; Peng, J.; Shi, P.; Zhang, R.; Dong, Y.; Lam, K.; Lo, F. P.; Xiao, B.; Yuan, W. Large ai models in health informatics: Applications, challenges, and the future. IEEE Journal of Biomedical and Health Informatics 2023, 27, 6074–6087. [CrossRef]
- Elnaggar, A.; Essam, H.; Salah-Eldin, W.; Moustafa, W.; Elkerdawy, M.; Rochereau, C.; Rost, B. Ankh: Optimized protein language model unlocks general-purpose modelling. arXiv preprint arXiv:2301.06568 2023.
- Chen, B.; Cheng, X.; Li, P.; Geng, Y.; Gong, J.; Li, S.; Bei, Z.; Tan, X.; Wang, B.; Zeng, X.; Liu, C. xTrimoPGLM: unified 100B-scale pre-trained transformer for deciphering the language of protein. arXiv preprint arXiv:2401.06199 2024.
- Song, Y.; Yuan, Q.; Chen, S.; Zeng, Y.; Zhao, H.; Yang, Y. Accurately predicting enzyme functions through geometric graph learning on ESMFold-predicted structures. Nature Communications 2024, 15, 8180. [CrossRef]
- Bushuiev, A.; Bushuiev, R.; Zadorozhny, N.; Samusevich, R.; Stärk, H.; Sedlar, J.; Pluskal, T.; Sivic, J. Training on test proteins improves fitness, structure, and function prediction. arXiv preprint arXiv:2411.02109 2024, .
- Gantz, M.; Mathis, S.V.; Nintzel, F.E.; Zurek, P.J.; Knaus, T.; Patel, E.; Boros, D.; Weberling, F.M.; Kenneth, M.R.; Klein, O.J.; Medcalf, E.J. Microdroplet Screening Rapidly Profiles a Biocatalyst to Enable Its AI-Guided Engineering. bioRxiv 2024, 2024–04.
- Son, A.; Park, J.; Kim, W.; Lee, W.; Yoon, Y.; Ji, J.; Kim, H. Integrating Computational Design and Experimental Approaches for Next-Generation Biologics. Biomolecules 2024, 14, 1073. [CrossRef]
- Zhang, W.; Li, X.; Wang, Y.; Liu, J.; Chen, X.; Zhang, Y.; Wang, X.; Li, H.; Zhang, J. Machine learning-assisted amidase-catalytic enantioselectivity prediction and its application in biocatalyst engineering. Nature Communications 2024, 15, 6392.
- Hollmann, F.; Sanchis, J.; Reetz, M. T. Learning from Protein Engineering by Deconvolution of Multi-Mutational Variants. Angewandte Chemie International Edition 2024, .
- Menke, M. J.; Ao, Y.; Bornscheuer, U. T. Practical Machine Learning-Assisted Design Protocol for Protein Engineering: Transaminase Engineering for the Conversion of Bulky Substrates. ACS Catalysis 2024, .
- Zhou, J.; Huang, M. Navigating the landscape of enzyme design: from molecular simulations to machine learning. Chemical Society Reviews 2024, 53, 8202–8239. [CrossRef]
- Paton, A.; Boiko, D.; Perkins, J.; Cemalovic, N.; Reschützegger, T.; Gomes, G.; Narayan, A. Generation of Connections Between Protein Sequence Space and Chemical Space to Enable a Predictive Model for Biocatalysis. ChemRxiv 2024. [CrossRef]
- Joon, P.; Kadian, M.; Dahiya, M.; Sharma, G.; Sharma, P.; Kumar, A.; Parle, M. Prognosticating Drug Targets and Responses by Analyzing Metastasis-Related Cancer Pathways. Handbook of Oncobiology: From Basic to Clinical Sciences 2023, 1–25.
- Sorrentino, C.; Ciummo, S. L.; Fieni, C.; Di Carlo, E. Nanomedicine for cancer patient-centered care. MedComm 2024, 5, e767.
- Guo, L.; Yang, J.; Wang, H.; Yi, Y. Multistage self-assembled nanomaterials for cancer immunotherapy. Molecules 2023, 28, 7750. [CrossRef]
- Zhang, Y.; Liu, X.; Li, F.; Yin, J.; Yang, H.; Li, X.; Liu, X.; Chai, X.; Niu, T.; Zeng, S.; Jia, Q. INTEDE 2.0: the metabolic roadmap of drugs. Nucleic acids research 2024, 52, D1355–D1364. [CrossRef]
- Yin, J.; Li, F.; Zhou, Y.; Mou, M.; Lu, Y.; Chen, K.; Xue, J.; Luo, Y.; Fu, J.; He, X.; Gao, J. INTEDE: interactome of drug-metabolizing enzymes. Nucleic acids research 2021, 49, D1233–D1243. [CrossRef]
- Li, F.; Yin, J.; Lu, M.; Mou, M.; Li, Z.; Zeng, Z.; Tan, Y.; Wang, S.; Chu, X.; Dai, H.; Hou, T. DrugMAP: molecular atlas and pharma-information of all drugs. Nucleic acids research 2023, 51, D1288–D1299. [CrossRef]
- Li, F.; Mou, M.; Li, X.; Xu, W.; Yin, J.; Zhang, Y.; Zhu, F. DrugMAP 2.0: molecular atlas and pharma-information of all drugs. Nucleic Acids Research 2025, 51, D1372–D1382.
- Srivastava, A.; Vinod, P. A Single-Cell Network Approach to Decode Metabolic Regulation in Gynecologic and Breast Cancers. Systems Biology and Applications 2024, 11(1), 26.
- Jackson, S. E.; Chester, J. D. Personalised cancer medicine. International journal of cancer 2015, 137, 262–266.
- Kearney, T.; Flegg, M. B. Enzyme kinetics simulation at the scale of individual particles. The Journal of Chemical Physics 2024, 161(19).
- Hamdy, N. M.; Basalious, E. B.; El-Sisi, M. G.; Nasr, M.; Kabel, A. M.; Nossier, E. S.; Abadi, A. H. Advancements in current one-size-fits-all therapies compared to future treatment innovations for better improved chemotherapeutic outcomes: a step-toward personalized medicine. Current Medical Research and Opinion 2024, 40, 1943–1961.
- Zhou, L.; Zhu, Z.; Gao, H.; Wang, C.; Khan, M. A.; Ullah, M.; Khan, S. U. Multi-omics graph convolutional networks for digestive system tumour classification and early-late stage diagnosis. CAAI Transactions on Intelligence Technology 2024, 9(6), 1572–1586.
- Bailey, D. G.; Dresser, G.; Arnold, J. M. O. Grapefruit–medication interactions: Forbidden fruit or avoidable consequences? Cmaj 2013, 185(4), 309–316.
- Hackman, G. L.; Collins, M.; Lu, X.; Lodi, A.; DiGiovanni, J.; Tiziani, S. Predicting and quantifying antagonistic effects of natural compounds given with chemotherapeutic agents: Applications for high-throughput screening. Cancers 2020, 12(12), 3714.
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. Journal of Experimental Medicine 2021, 218, e20201606. [CrossRef]
- Qian, L.; Lin, X.; Gao, X.; Khan, R. U.; Liao, J.; Du, S.; Ge, J.; Zeng, S.; Yao, S. Q. The dawn of a new era: targeting the “undruggables” with antibody-based therapeutics. Chemical reviews 2023, 123, 7782–7853. [CrossRef]
- Melton, R. G.; Sherwood, R. F. Antibody-enzyme conjugates for cancer therapy. JNCI: Journal of the National Cancer Institute 1996, 88, 153–165.
- Paiva, S.; Crews, C. M. Targeted protein degradation: elements of PROTAC design. Current opinion in chemical biology 2019, 50, 111–119.
- Xie, L.; Xie, L. Elucidation of genome-wide understudied proteins targeted by PROTAC-induced degradation using interpretable machine learning. PLOS Computational Biology 2023, 19, e1010974.
- Zielezinski, A.; Loch, J. I.; Karlowski, W. M.; Jaskolski, M. Massive annotation of bacterial L-asparaginases reveals their puzzling distribution and frequent gene transfer events. Scientific reports 2022, 12, 15797.
- Shishparenok, A. N.; Gladilina, Y. A.; Zhdanov, D. D. Engineering and expression strategies for optimization of L-Asparaginase development and production. International Journal of Molecular Sciences 2023, 24, 15220. [CrossRef]
- Patel, P.; Gosai, H.; Panseriya, H.; Dave, B. Development of process and data centric inference system for enhanced production of L-asparaginase from halotolerant Bacillus licheniformis PPD37. Applied Biochemistry and Biotechnology 2022, 1–23. [CrossRef]
- Lichtenstein, M.; Zabit, S.; Hauser, N.; Farouz, S.; Melloul, O.; Hirbawi, J.; Lorberboum-Galski, H. TAT for enzyme/protein delivery to restore or destroy cell activity in human diseases. Life 2021, 11, 924. [CrossRef]
- Poongavanam, V.; Kölling, F.; Giese, A.; Göller, A. H.; Lehmann, L.; Meibom, D.; Kihlberg, J. Predictive modeling of PROTAC cell permeability with machine learning. ACS omega 2023, 8, 5901–5916. [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal transduction and targeted therapy 2022, 7, 113. [CrossRef]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 ubiquitin ligases: styles, structures and functions. Molecular biomedicine 2021, 2, 1–17. [CrossRef]
- Smith, B. E.; Wang, S. L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B. D.; Crews, C. M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature communications 2019, 10, 131. [CrossRef]
- Falck, G.; Müller, K. M. Enzyme-based labeling strategies for antibody–drug conjugates and antibody mimetics. Antibodies 2018, 7, 4.
- Ovcharenko, D.; Mukhin, D.; Ovcharenko, G. Alternative Cancer Therapeutics: Unpatentable Compounds and Their Potential in Oncology. Pharmaceutics 2024, 16, 1237.
- Debela, D. T.; Muzazu, S. G.; Heraro, K. D.; Ndalama, M. T.; Mesele, B. W.; Haile, D. C.; Kitui, S. K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE open medicine 2021, 9, 20503121211034366.
- Shi, Z.; Li, C.; Chen, R.; Shi, J.; Liu, Y.; Lu, J.; Yang, G.; Chen, J. The emerging role of deubiquitylating enzyme USP21 as a potential therapeutic target in cancer. Bioorganic Chemistry 2024, 107400.
- Weinberg, R. A.; Weinberg, R. A. The biology of cancer. WW Norton & Company 2006.
- Ruddon, R. W. Cancer biology. Oxford University Press. 2007.
- Marei, H. E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer cell international 2021, 21, 703.
- Joerger, A. C.; Fersht, A. R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annual review of biochemistry 2016, 85, 375–404.
- Hanahan, D.; Weinberg, R. A. The hallmarks of cancer. Cell 2000, 100, 57–70. [CrossRef]
- Mukhopadhyay, R.; Nath, S.; Kumar, D.; Sahana, N.; Mandal, S. Basics of the Molecular Biology: From Genes to Its Function. Genomics Data Analysis for Crop Improvement 2024, 343–374.
- Sherr, C. J. Principles of tumor suppression. Cell 2004, 116, 235–246.
- Alderson, R. F.; Toki, B. E.; Roberge, M.; Geng, W.; Basler, J.; Chin, R.; Liu, A.; Ueda, R.; Hodges, D.; Escandon, E.; Chen, T. Characterization of a CC49-Based Single-Chain Fragment- β-Lactamase Fusion Protein for Antibody-Directed Enzyme Prodrug Therapy (ADEPT). Bioconjugate chemistry 2006, 17, 410–418. [CrossRef]
- Kim, Y.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. In Proceedings of the National Academy of Sciences 1996, 93, 1156–1160.
- Liu, B.; Zhou, H.; Tan, L.; Siu, K. T. H.; Guan, X. Exploring treatment options in cancer: tumor treatment strategies. Signal transduction and targeted therapy 2024, 9, 175.
- Veatch, J. R.; McMurray, M. A.; Nelson, Z. W.; Gottschling, D. E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [CrossRef]
- Seyfried, T. N.; Shelton, L. M. Cancer as a metabolic disease. Nutrition & metabolism 2010, 7, 1–22.
- Outreach, N. P. Press release: The Nobel Prize in Physiology or Medicine 1973. NobelPrize. org 2021.
- Lianos, G. D.; Vlachos, K.; Zoras, O.; Katsios, C.; Cho, W. C.; Roukos, D. H. Potential of antibody-drug conjugates and novel therapeutics in breast cancer management. Onco Targets Ther 2014, 7, 491-500.
- Pierotti, M. A.; Della Porta, G. Mechanisms of oncogene activation. Advances in Oncology 2022, 3–12.
- Jan, R.; others Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Advanced pharmaceutical bulletin 2019, 9, 205. [CrossRef]
- Johnson, J. L.; Yaron, T. M.; Huntsman, E. M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.; Liberatore, K.; Cizin, D. M.; Cohen, B. M.; others An atlas of substrate specificities for the human serine/threonine kinome. Nature 2023, 613, 759–766. [CrossRef]
- Diallo, A.; Prigent, C. The serine/threonine kinases that control cell cycle progression as therapeutic targets. Bulletin du cancer 2011, 98, 1335–1345. [CrossRef]
- Vogt, P. K. Retroviral oncogenes: a historical primer. Nature Reviews Cancer 2012, 12, 639–648. [CrossRef]
- Kontomanolis, E. N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; others Role of oncogenes and tumor-suppressor genes in carcinogenesis: a review. Anticancer research 2020, 40, 6009–6015. [CrossRef]
- Clark, M. A.; Douglas, M.; Choi, J. Biology, 2d ed.; OpenStax: Houston, Texas, 2008; Available online: https://openstax.org/books/biology-2e/pages/6-5-enzymes (accessed on 7 Jul 2025).
- Gerl, R.; Vaux, D. L. Apoptosis in the development and treatment of cancer. Carcinogenesis 2005, 26, 263–270.
- Chow, A. Y. Cell cycle control by oncogenes and tumor suppressors: driving the transformation of normal cells into cancerous cells. Nature Education 2010, 3, 7.
- Kraut, J. A.; Madias, N. E. Metabolic acidosis: pathophysiology, diagnosis and management. Nature Reviews Nephrology 2010, 6, 274–285.
- Robinson, P. K. Enzymes: principles and biotechnological applications. Essays in biochemistry 2015, 59, 1.
- Cooper, G. M. The central role of enzymes as biological catalysts. Sinauer Associates 2000.
- Liu, X.; Locasale, J. W. Metabolomics: a primer. Trends in biochemical sciences 2017, 42, 274–284. [CrossRef]
- Xue, J.; Yang, S.; Seng, S. Mechanisms of cancer induction by tobacco-specific NNK and NNN. Cancers 2014, 6, 1138–1156. [CrossRef]
- Schwartz, L.; T Supuran, C.; O Alfarouk, K. The Warburg effect and the hallmarks of cancer. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) 2017, 17, 164–170.
- Guo, R.; Wang, R.; Wu, R.; Ren, Z.; Li, J.; Luo, S.; Wu, Z.; Liu, Q.; Peng, J.; Ma, J. Enhancing protein mutation effect prediction through a retrieval-augmented framework. Advances in Neural Information Processing Systems 2024, 37, 49130–49153.
- Dauparas, J.; Lee, G. R.; Pecoraro, R.; An, L.; Anishchenko, I.; Glasscock, C.; Baker, D. Atomic context-conditioned protein sequence design using LigandMPNN. Nature Methods 2025, 1–7. [CrossRef]
- Buttenschoen, M.; Morris, G. M.; Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chemical Science 2024, 15, 3130–3139. [CrossRef]
- Chai Discovery team; Boitreaud, J.; Dent, J.; McPartlon, M.; Meier, J.; Reis, V.; Rogozhonikov, A.; Wu, K. Chai-1: Decoding the molecular interactions of life. BioRxiv 2024. [CrossRef]
- Castro-Mondragon, J. A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N.; Mathelier, A. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic acids research 2022, 50, D165–D173.
- Rao, R.; Meier, J.; Sercu, T.; Ovchinnikov, S.; Rives, A. Transformer protein language models are unsupervised structure learners. bioRxiv 2020.
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A. J.; Bambrick, J.; Jumper, J. M. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500.
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature biotechnology 2017, 35, 1026–1028.
- Burley, S. K.; Berman, H. M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J. M.; Dutta, S.; others RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic acids research 2019, 47, D464–D474. [CrossRef]
- Olsen, T. H.; Boyles, F.; Deane, C. M. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Science 2022, 31, 141–146.
- Richardson, L.; Allen, B.; Baldi, G.; Beracochea, M.; Bileschi, M. L.; Burdett, T.; Burgin, J.; Caballero-Pérez, J.; Cochrane, G.; Colwell, L. J.; others MGnify: the microbiome sequence data analysis resource in 2023. Nucleic acids research 2023, 51, D753–D759. [CrossRef]
- Suzek, B. E.; Wang, Y.; Huang, H.; McGarvey, P. B.; Wu, C. H.; UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [CrossRef]
- Grigoriev, I. V.; Nordberg, H.; Shabalov, I.; Aerts, A.; Cantor, M.; Goodstein, D.; Kuo, A.; Minovitsky, S.; Nikitin, R.; Ohm, R. A.; others The genome portal of the department of energy joint genome institute. Nucleic acids research 2012, 40, D26–D32. [CrossRef]
- Shen, Y.; Chen, Z.; Mamalakis, M.; He, L.; Xia, H.; Li, T.; Su, Y.; He, J.; Wang, Y. G. A fine-tuning dataset and benchmark for large language models for protein understanding. 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2024, 2390–2395.
- Cai, Z.; Cao, M.; Chen, H.; Chen, K.; Chen, K.; Chen, X.; Chen, X.; Chen, Z.; Chen, Z.; Chu, P.; others Internlm2 technical report. arXiv preprint arXiv:2403.17297 2024, .
- Shen, Y.; Chen, Z.; Mamalakis, M.; Liu, Y.; Li, T.; Su, Y.; He, J.; Liò, P.; Wang, Y. G. Toursynbio: A multi-modal large model and agent framework to bridge text and protein sequences for protein engineering. 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2024, 2382–2389.
- Bank, P. D. Protein data bank. Nature New Biol 1971, 233, 10–1038.
- Lite, T. V.; Grant, R. A.; Nocedal, I.; Littlehale, M. L.; Guo, M. S.; Laub, M. T. Uncovering the basis of protein-protein interaction specificity with a combinatorially complete library. Elife 2020, 9, e60924. [CrossRef]
- Wu, N. C.; Dai, L.; Olson, C. A.; Lloyd-Smith, J. O.; Sun, R. Adaptation in protein fitness landscapes is facilitated by indirect paths. Elife 2016, 5, e16965. [CrossRef]
- Ding, D.; Green, A. G.; Wang, B.; Lite, T. V.; Weinstein, E. N.; Marks, D. S.; Laub, M. T. Co-evolution of interacting proteins through non-contacting and non-specific mutations. Nature ecology & evolution 2022, 6, 590–603.
- Chen, Y.; Hu, R.; Li, K.; Zhang, Y.; Fu, L.; Zhang, J.; Si, T. Deep mutational scanning of an Oxygen-Independent fluorescent protein CreiLOV for comprehensive profiling of mutational and epistatic effects. ACS Synthetic Biology 2023, 12, 1461–1473. [CrossRef]
- Wu, N. C.; Dai, L.; Olson, C. A.; Lloyd-Smith, J. O.; Sun, R. Adaptation in protein fitness landscapes is facilitated by indirect paths. Elife 2016, 5, e16965. [CrossRef]
- Stärk, H.; Dallago, C.; Heinzinger, M.; Rost, B. Light attention predicts protein location from the language of life. Bioinformatics Advances 2021, 1, vbab035. [CrossRef]
- UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic acids research 2019, 47, D506–D515.
- Notin, P.; Kollasch, A.; Ritter, D.; Van Niekerk, L.; Paul, S.; Spinner, H.; Rollins, N.; Shaw, A.; Orenbuch, R.; Weitzman, R.; Marks, D. Proteingym: Large-scale benchmarks for protein fitness prediction and design. Advances in Neural Information Processing Systems 2023, 36, 64331–64379.
- Landrum, M. J.; Lee, J. M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; Maglott, D. R. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research 2016, 44, D862–D868.
- Mitchell, A. L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M. R.; Kale, V.; Potter, S. C.; Richardson, L. J.; Finn, R. D. MGnify: the microbiome analysis resource in 2020. Nucleic acids research 2020, 48, D570–D578. [CrossRef]
- Mirdita, M.; Von Den Driesch, L.; Galiez, C.; Martin, M. J.; Söding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic acids research 2017, 45, D170–D176. [CrossRef]
- Suzek, B. E.; Wang, Y.; Huang, H.; McGarvey, P. B.; Wu, C. H.; UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932.
- Orengo, C. A.; Michie, A. D.; Jones, S.; Jones, D. T.; Swindells, M. B.; Thornton, J. M. CATH–a hierarchic classification of protein domain structures. Structure 1997, 5, 1093–1109.
- Suzek, B. E.; Wang, Y.; Huang, H.; McGarvey, P. B.; Wu, C. H.; UniProt Consortium UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932.
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nature communications 2018, 9, 2542.
- Federhen, S. The NCBI taxonomy database. Nucleic acids research 2012, 40, D136–D143. [CrossRef]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S. R.; Griffiths-Jones, S.; Howe, K. L.; Marshall, M.; Sonnhammer, E. L. The Pfam protein families database. Nucleic acids research 2002, 30, 276–280. [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.; Estreicher, A.; Gasteiger, E.; Martin, M. J.; Michoud, K.; O’Donovan, C.; Phan, I.; others The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 2003, 31, 365–370. [CrossRef]
- Bairoch, A.; Boeckmann, B.; Ferro, S.; Gasteiger, E. Swiss-Prot: juggling between evolution and stability. Briefings in bioinformatics 2004, 5, 39–55. [CrossRef]
- Bairoch, A.; Apweiler, R.; Wu, C. H.; Barker, W. C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; Yeh, L. S. L. The universal protein resource (UniProt). Nucleic acids research 2005, 33, D154–D159. [CrossRef]
- Leinonen, R.; Diez, F. G.; Binns, D.; Fleischmann, W.; Lopez, R.; Apweiler, R. UniProt archive. Bioinformatics 2004, 20, 3236–3237. [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F. T.; de Beer, T. A. P.; Rempfer, C.; Bordoli, L.; Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research 2018, 46, W296–W303. [CrossRef]
- Dana, J. M.; Gutmanas, A.; Tyagi, N.; Qi, G.; O’Donovan, C.; Martin, M.; Velankar, S. SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic acids research 2019, 47, D482–D489.
- Ramsundar, B. Molecular machine learning with DeepChem. Doctoral dissertation, Stanford University, 2018.
- Rohl, C. A.; Strauss, C. E.; Misura, K. M.; Baker, D. Protein structure prediction using Rosetta. Methods in enzymology 2004, 383, 66–93.
- Dieckhaus, H.; Brocidiacono, M.; Randolph, N. Z.; Kuhlman, B. Transfer learning to leverage larger datasets for improved prediction of protein stability changes. In Proceedings of the national academy of sciences 2024, 121, e2314853121.
- Wang, R.; Albooyeh, M.; Ravanbakhsh, S. Equivariant networks for hierarchical structures. Advances in Neural Information Processing Systems 2020, 33, 13806–13817.
- Kuldeep, J.; Chaturvedi, N.; Gupta, D. Novel molecular inhibitor design for Plasmodium falciparum Lactate dehydrogenase enzyme using machine learning generated library of diverse compounds. Molecular Diversity 2024, 28, 2331–2344. [CrossRef]
- Grunebaum, E.; Loves, R.; Kohn, D. B. Making sense of adenosine deaminase variants and their clinical implications. Journal of Allergy and Clinical Immunology 2025, 155, 92–93. [CrossRef]
- Ibezim, A.; Onah, E.; Osigwe, S. C.; Okoroafor, P. U.; Ukoha, O. P.; de Siqueira-Neto, J. L.; Ntie-Kang, F.; Ramanathan, K. Potential dual inhibitors of Hexokinases and mitochondrial complex I discovered through machine learning approach. Scientific African 2024, 24, e02226. [CrossRef]
- Lane, T. J. Protein structure prediction has reached the single-structure frontier. Nature Methods 2023, 20, 170–173. [CrossRef]
- Henikoff, S.; Henikoff, J. G. Amino acid substitution matrices from protein blocks. In Proceedings of the National Academy of Sciences 1992, 89, 10915–10919.
- Needleman, S. B.; Wunsch, C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology 1970, 48, 443–453.
- Buttenschoen, M.; Morris, G. M.; Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chemical Science 2024, 15, 3130–3139.
- Haas, J.; Barbato, A.; Behringer, D.; Studer, G.; Roth, S.; Bertoni, M.; Mostaguir, K.; Gumienny, R.; Schwede, T. Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins: Structure, Function, and Bioinformatics 2018, 86, 387–398. [CrossRef]
- Esposito, D.; Weile, J.; Shendure, J.; Starita, L. M.; Papenfuss, A. T.; Roth, F. P.; Fowler, D. M.; Rubin, A. F. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome biology 2019, 20, 1–11. [CrossRef]
- Barrio-Hernandez, I.; Yeo, J.; Jänes, J.; Mirdita, M.; Gilchrist, C. L.; Wein, T.; Varadi, M.; Velankar, S.; Beltrao, P.; Steinegger, M. Clustering predicted structures at the scale of the known protein universe. Nature 2023, 622, 637–645. [CrossRef]
- Chae, J.; Wang, Z.; Gul, I.; Ji, J.; Chen, Z.; Qin, P. pLDDT-Predictor: High-speed Protein Screening Using Transformer and ESM2. arXiv preprint arXiv:2410.21283 2024, .
- Laurent, J. M.; Janizek, J. D.; Ruzo, M.; Hinks, M. M.; Hammerling, M. J.; Narayanan, S.; Ponnapati, M.; White, A. D.; Rodriques, S. G. Lab-bench: Measuring capabilities of language models for biology research. arXiv preprint arXiv:2407.10362 2024, .
- Skarlinski, M. D.; Cox, S.; Laurent, J. M.; Braza, J. D.; Hinks, M.; Hammerling, M. J.; Ponnapati, M.; Rodriques, S. G.; White, A. D. Language agents achieve superhuman synthesis of scientific knowledge. arXiv preprint arXiv:2409.13740 2024, .
- Yang, Z.; Qi, P.; Zhang, S.; Bengio, Y.; Cohen, W. W.; Salakhutdinov, R.; Manning, C. D. HotpotQA: A dataset for diverse, explainable multi-hop question answering. arXiv preprint arXiv:1809.09600 2018, .
- Cobbe, K.; Kosaraju, V.; Bavarian, M.; Chen, M.; Jun, H.; Kaiser, L.; Plappert, M.; Tworek, J.; Hilton, J.; Nakano, R.; Schulman, J. Training verifiers to solve math word problems. arXiv preprint arXiv:2110.14168 2021, .
- Touvron, H.; Lavril, T.; Izacard, G.; Martinet, X.; Lachaux, M.; Lacroix, T.; Rozière, B.; Goyal, N.; Hambro, E.; Azhar, F.; Lample, G. Llama: Open and efficient foundation language models. arXiv preprint arXiv:2302.13971 2023, .
- Yıldırım, S.; Asgari-Chenaghlu, M. Mastering Transformers: The Journey from BERT to Large Language Models and Stable Diffusion. Packt Publishing Ltd. 2024, .
- Jeliazkov, J. R.; del Alamo, D.; Karpiak, J. D. ESMFold hallucinates native-like protein sequences. bioRxiv 2023, 2023–05. [CrossRef]
- Wray, R. E.; Kirk, J. R.; Laird, J. E. Language models as a knowledge source for cognitive agents. arXiv preprint arXiv:2109.08270 2021.
- Chen, V.; Yang, M.; Cui, W.; Kim, J. S.; Talwalkar, A.; Ma, J. Applying interpretable machine learning in computational biology—pitfalls, recommendations and opportunities for new developments. Nature methods 2024, 21, 1454–1461. [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; Rives, A. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022.
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C. L.; Ma, J.; Fergus, R. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. In Proceedings of the National Academy of Sciences 2021, 118, e2016239118. [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G. R.; Wang, J.; Cong, Q.; Kinch, L. N.; Schaeffer, R. D.; Baker, D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [CrossRef]
- Yang, Z.; Ishay, A.; Lee, J. Learning to solve constraint satisfaction problems with recurrent transformer. arXiv preprint arXiv:2307.04895 2023, .
- Bordin, N.; Dallago, C.; Heinzinger, M.; Kim, S.; Littmann, M.; Rauer, C.; Steinegger, M.; Rost, B.; Orengo, C. Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences 2023, 48, 345–359.
- Santisteban, I.; Arredondo-Vega, F. X.; Bali, P.; Dalgic, B.; Lee, H. H.; Kim, M.; Hermanson, J.; Tarrant, T. K.; Hershfield, M. S. Evolving spectrum of adenosine deaminase (ADA) deficiency: Assessing genotype pathogenicity according to expressed ADA activity of 46 variants. Journal of Allergy and Clinical Immunology 2025, 155, 166–175. [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L. H.; Zielinski, M.; Sargeant, T.; Avsec, Ž. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492.
- Biggar, K.; Ridgeway, N.; Chopra, A.; Lukinovic, V.; Feldman, M.; Charih, F.; Levy, D.; Green, J. Machine learning-based exploration of enzyme-substrate networks: SET8-mediated methyllysine and its changing impact within cancer proteomes. Research square 2024. [CrossRef]
- Diaz, D. J.; Gong, C.; Ouyang-Zhang, J.; Loy, J. M.; Wells, J.; Yang, D.; Ellington, A. D.; Dimakis, A. G.; Klivans, A. R. Stability Oracle: a structure-based graph-transformer framework for identifying stabilizing mutations. Nature Communications 2024, 15, 6170. [CrossRef]
- El-Naggar, N. E.; El-Shweihy, N. M. Bioprocess development for L-asparaginase production by Streptomyces rochei, purification and in-vitro efficacy against various human carcinoma cell lines. Scientific reports 2020, 10, 7942.
- Touzart, A.; Lengliné, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; Asnafi, V. Epigenetic silencing affects l-asparaginase sensitivity and predicts outcome in T-ALL. Clinical cancer research 2019, 25, 2483–2493. [CrossRef]
- Cuff, J. A.; Barton, G. J. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins: Structure, Function, and Bioinformatics 1999, 34, 508–519.
- Cuff, J. A.; Barton, G. J. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins: Structure, Function, and Bioinformatics 1999, 34, 508–519.
- Jarzab, A.; Kurzawa, N.; Hopf, T.; Moerch, M.; Zecha, J.; Leijten, N.; Bian, Y.; Musiol, E.; Maschberger, M.; Stoehr, G.; Kuster, B. Meltome atlas—thermal proteome stability across the tree of life. Nature methods 2020, 17, 495–503. [CrossRef]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the basis for developing protein–ligand interaction scoring functions. Accounts of chemical research 2017, 50, 302–309.
- AlQuraishi, M. ProteinNet: a standardized data set for machine learning of protein structure. BMC bioinformatics 2019, 20, 1–10. [CrossRef]
- Moal, I. H.; Fernández-Recio, J. SKEMPI: a structural kinetic and energetic database of mutant protein interactions and its use in empirical models. Bioinformatics 2012, 28, 2600–2607.
- Pan, X.; Zhang, Y.; Shen, H. Large-Scale prediction of human protein- protein interactions from amino acid sequence based on latent topic features. Journal of proteome research 2010, 9, 4992–5001.
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic acids research 2008, 36, 3025–3030.
- Fox, N. K.; Brenner, S. E.; Chandonia, J. SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic acids research 2014, 42, D304–D309. [CrossRef]
- Khurana, S.; Rawi, R.; Kunji, K.; Chuang, G.; Bensmail, H.; Mall, R. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics 2018, 34, 2605–2613. [CrossRef]
- Gray, V. E.; Hause, R. J.; Luebeck, J.; Shendure, J.; Fowler, D. M. Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell systems 2018, 6, 116–124. [CrossRef]
- Dallago, C.; Mou, J.; Johnston, K. E.; Wittmann, B. J.; Bhattacharya, N.; Goldman, S.; Madani, A.; Yang, K. K. FLIP: Benchmark tasks in fitness landscape inference for proteins. bioRxiv 2021, 2021–11. [CrossRef]
- Hartshorn, M. J.; Verdonk, M. L.; Chessari, G.; Brewerton, S. C.; Mooij, W. T.; Mortenson, P. N.; Murray, C. W. Diverse, high-quality test set for the validation of protein- ligand docking performance. Journal of medicinal chemistry 2007, 50, 726–741.
- Olechnovič; Kliment; Kulberkytė; Eleonora; Venclovas; Česlovas. CAD-score: a new contact area difference-based function for evaluation of protein structural models. Proteins: Structure, Function, and Bioinformatics 2013, 81, 149–162.
- Kopp, J.; Bordoli, L.; Battey, J. N.; Kiefer, F.; Schwede, T. Assessment of CASP7 predictions for template-based modeling targets. Proteins: Structure, Function, and Bioinformatics 2007, 69, 38–56.
- Zemla, A. LGA: a method for finding 3D similarities in protein structures. Nucleic acids research 2003, 31, 3370–3374. [CrossRef]
Figure 1.
High-level architecture for enzyme engineering using multimodal data integration and advanced modeling techniques.
Figure 1.
High-level architecture for enzyme engineering using multimodal data integration and advanced modeling techniques.
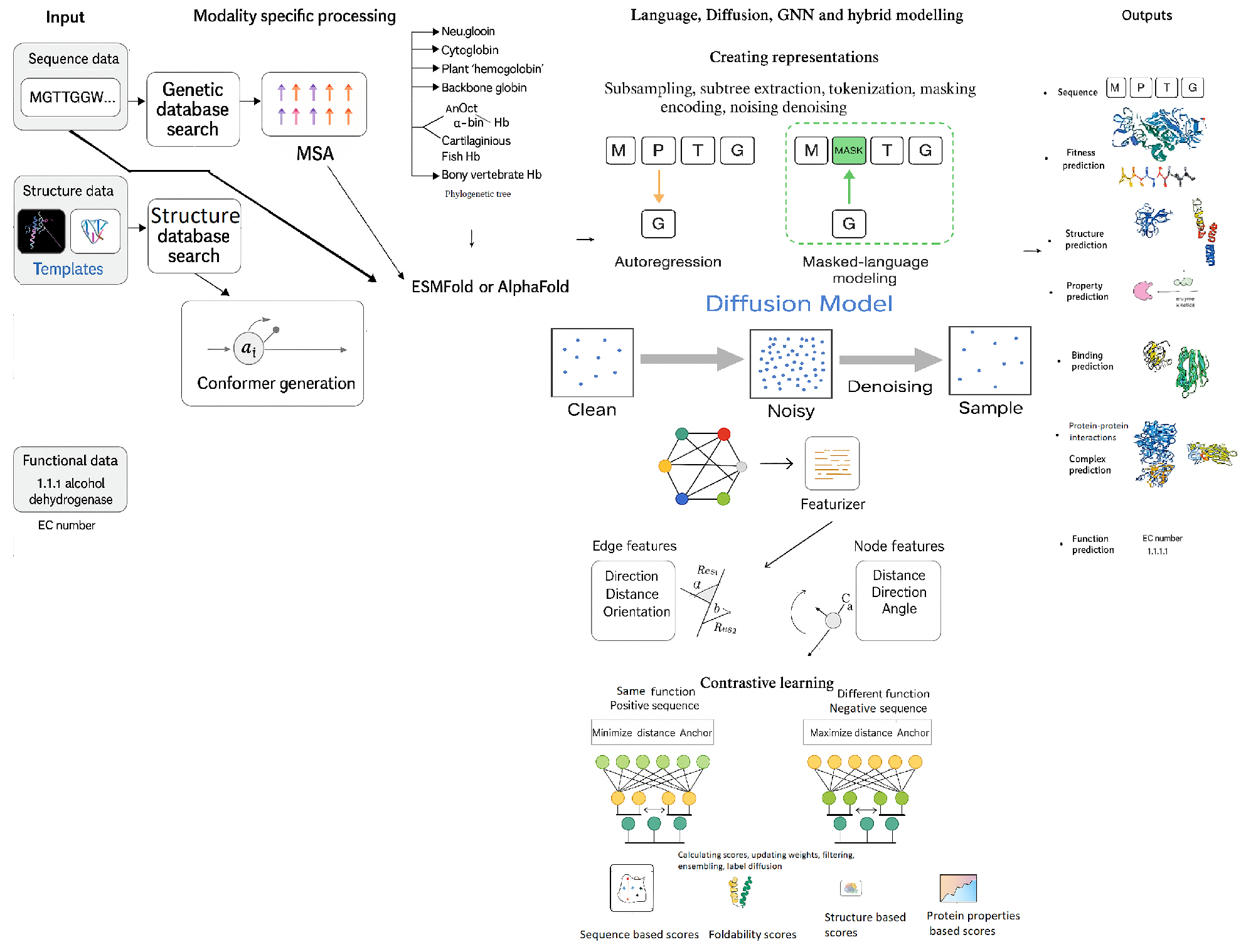
Figure 2.
Enzymes in cancer diagnostics and therapeutics.
Short Biography of Authors
 |
Bolat Sultankulov PhD in Science, Engineering and Technology. Nazarbayev University, School of Engineering and Digital Sciences (2021).CEO&Founder of ARLAN BIOTECH, Inc. StartX alumni startup (Stanford University) developing Generative Physics based models for de novo generation of protein binders. |
 |
Adnan Yazici PhD in Computer Science. Tulane University, Department of EECS, LA, USA (1991). Chair of the Department of Computer Science, Nazarbayev University, Kazakhstan. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



