Submitted:
11 July 2025
Posted:
14 July 2025
You are already at the latest version
Abstract
Compelling epidemiological evidence suggests that exercise and smoking are modifiable risk factors that are linked to a reduced risk of Parkinson’s disease, possibly because they both exert protective effects on neurodegeneration. Like Yin and Yang, these two risk factors represent opposite ends of a spectrum: exercise is universally embraced, while smoking is rightly eschewed. Yet, intriguingly, preclinical evidence suggests that at their biological cores, exercise and smoking may share strikingly similar working mechanisms in favorably modifying the disease course. Here, we deconstruct these overlapping and putative neuroprotective mechanisms. Our aim is to transform this unexpected overlap into an actionable perspective towards identifying novel targets for disease-modifying therapies that can slow the progression of Parkinson’s disease. We stress that while both factors may theoretically inform disease-modifying strategies for PD, in practice, only exercise should be promoted for its health benefits, whereas smoking is firmly contraindicated due to its known detrimental health effects.
Keywords:
Parkinson's disease
; smoking
; exercise
Introduction
For decades, studies have fairly consistently reported negative associations between both smoking and exercise (or physical activity) and the risk of developing Parkinson’s disease (PD). [1,2] Ever since, smoking has been a hot, intensely debated topic in PD research. [3,4,5,6,7] The inverse association between exercise and PD seems to square with its generally salubrious effects. [8] By contrast, the inverse association between smoking and PD strikes many as paradoxical, as smoking is known to cause or hasten a number of human diseases, including lung cancer and cardiovascular disease. [9] It is therefore critical to underscore that, despite these epidemiological observations, smoking is unequivocally harmful and is not advocated as a preventive or therapeutic strategy for any condition. At its biological core, certain molecular mechanisms engaged by smoking may exert disease-modifying effects that could help explain the reduced risk of PD among smokers. [10,11,12] This notion has held up in several supportive mendelian randomization studies that allow for causal inferences of a true disease-modifying effect even if tempered by their inherent limitations. [13,14,15] Interestingly, various molecular mechanisms of action underlying the cellular effects of smoking overlap with those of exercise, including stabilization of hypoxia inducible factor 1α (HIF-1α) and upregulation of anti-oxidative mechanisms and PGC-1α, an important mitochondrial regulator. [16,17,18,19,20,21,22] As such, we find exercise and smoking to be biological brethren through their putative neuroprotective mechanisms, yet paradoxically situated at opposite extremes of the overall health risk-to-benefit spectrum among potential interventions for PD. Unravelling the shared mechanisms underlying these two putative disease-modifying behaviors for PD might inform further therapeutic pursuits. Notably, several interventions linked to one or more of these mechanisms have been or are now being explored in PD, including several small-molecule compounds, and even controlled exposure to nicotine, low-dose carbon monoxide and moderate hypoxia. [23,24,25,26]
Here, we discuss the presumed neuroprotective mechanisms of smoking and exercise and then demonstrate the commonalities between those mechanisms. Finally, we highlight recent research investigating these pathways in the context of PD, and describe several trials of interventions tailored to these insights that may have potential to prevent or slow the disease. We emphasize that the discussion into putative disease-modifying mechanisms behind smoking is intended solely to inform mechanistic understanding and guide the search for safe and targeted disease-modifying therapies, not to promote a smoking habit in any condition.
Epidemiology of Potentially Protective Effects of Smoking and Exercise
Evidence for an Association
The inverse association between smoking and the subsequent diagnosis of PD is among the most consistent lifestyle links across all of neuroepidemiology, and certainly among risk factors for PD. The first reports about smoking in PD were published over 50 years ago, and a twenty-year old landmark twin study showed a negative correlation between smoking dose and PD within twin pairs, even with a 10-year lag time before PD diagnosis. [10] Several studies report a dose-dependent association between smoking and risk of PD, with heavy smokers having the lowest risk to develop PD [20] and former and current smokers having a lower risk of PD compared to participants who never, or occasionally, smoked. [2,13,27,28,29,30] Whether second-hand smoking is also inversely associated with risk of PD, [31,32,33] and whether smoking behavior is associated with PD phenotype and progression, remain topics of debate. For example, some studies suggest that smokers have a later age of PD onset [34,35,36], lower (better) MDS-UPDRS-III [37] and a different non-motor symptom spectrum [38,39], but other studies report no associations with PD phenotype. [40] Lastly, although smoking is one of the most important risk factors for dementia in the general population, population studies report lower neurology-related mortality in current smokers compared to never-smokers. [41,42]
On the other hand, exercise is a widely advised non-pharmacological intervention in PD management. Exercise offers symptomatic benefits, and may also exert potential disease-modifying effects, although intervention studies addressing the hypothesized preventive effects of exercise in prodromal cohorts are just beginning [NCT06193252]. One synthesis of meta-analyses demonstrated that greater premorbid amounts of physical activity were associated with a dose-dependent reduced risk of developing PD. [1] In one study stratifying for task-related physical activity, only household and commuting-related physical activity were associated with a reduced PD risk, and leisure or occupation-related physical activity were not. [43] Exercise has dose-dependent positive effects on gait and postural stability, independence in activities of daily living and several cognitive symptoms. [44,45] Physical activity levels also correlate inversely with deterioration of dopamine transporter (DAT) imaging. [46] Finally, two randomized controlled trials showed that aerobic exercise resulted in a stabilization of motor progression in PD. [47,48] In one of these studies, this was associated with adaptive plasticity of the brain, which coincided with cognitive improvement. [49]
Limitations
Several limitations exist for observational studies that investigated the association between smoking and PD. First, the association could also be attributed to an intact and higher-than-average dopaminergic state which might make one more prone to addiction-based behaviors like smoking (differences in premorbid personality). [50] This enhanced dopaminergic state might directly protect against PD onset. Similarly, caloric restriction induced by smoking may be a confounding factor, among others, in this association. Evidence suggests caloric restriction may have preventive effects for neurodegenerative disease through reduced oxidative stress, improved autophagy and mitochondrial function, and neurotrophic effects. [51]
On the other hand, the reduced risk of PD among smokers may result from reverse causation, such that in the early, prodromal phase of PD, addictive behaviors such as smoking are less likely to develop or more likely to halt. Indeed, PD patients quit smoking with less difficulty than controls, possibly due to attenuated addictive effects of nicotine due to dopaminergic loss. [52] As noted above, Mendelian randomization studies report comparable risk reductions to observational studies. [13,14,15] Both Mendelian randomization studies and case-control studies remain susceptible to survival bias across groups. For example, putative anti-apoptotic smoking-induced effects on Bcl-2 or p53 lead to higher risk for malignancies and might lead to unbalanced drop-out in the smoking group. However, such problems are not as prominent in prospective cohort studies, where smoking’s association with PD risk reduction remains strong. [2] Similar arguments regarding prodromal behavioral differences and selective survival exist for exercise. Lastly, the lack of mechanistic clarity hinders the identification of putative therapeutic targets, hampering the development of experimental or translational studies of candidate compounds without the associated harm of tobacco use.
Protective Mechanisms in Smoking and Exercise?
Several hypotheses have been proposed for the mechanisms underlying a putative neuroprotective effect of smoking. [53] First, as non-smokers who used snus (smokeless tobacco) have a substantially lower risk of PD, nicotine or other components of tobacco leaves have been suggested to be the mediating factor. [41,54] Briefly, nicotine may act as a stimulant that inhibits the action of striatal DAT, increasing dopamine levels in the synaptic gap [55] and reducing levodopa-induced dyskinesias [56]. However, a recent trial did not show neuroprotective effects of nicotine patches. [24] Second, preclinical studies report neuroprotective effects of carbon monoxide at the low doses seen in smokers, as it activates protective signaling cascades including HIF-1α and heme oxygenase 1 (HO-1), and reduces α-synuclein pathology. [57] Third, perhaps as a result of carbon monoxide exposure, smoking’s benefits on PD risk may be mediated by intermittent hypoxia.[17,58,59] Preclinical evidence demonstrates neuroprotective effects of moderate intermittent hypoxia through activation of the hypoxia response pathway [26]. Fourth, smoking reduces MAO-B activity, which increases dopaminergic activity. Indeed, current smoking is associated with increased DAT binding. [60,61] Lastly, smoking induces cytochrome P450 1A2 (CYP1A2), which plays important roles in detoxification of toxins, whose activity is highest in mitochondria of the striatum. [60,62]
Of note, there is overwhelming evidence that smoking increases the risk of Alzheimer’s disease (AD). [63,64] While this finding might cast doubt about the established relation between smoking and PD as both are usually slowly progressive neurodegenerative diseases occurring in later life, it should be noted that AD and PD have widely differing risk factors, most notable in divergent genetic, lifestyle, environmental and vascular risk factors. [63,65]
Several studies have aimed to elucidate the neuroprotective mechanisms of exercise in the light of PD. These include increase in neuronal resilience through improving adaptive response to energetic stress, primarily through anti-inflammatory control mechanisms and improved mitochondrial turnover, and anti-oxidant response. [8] Furthermore, preclinical evidence substantiates the induction of various growth factor-related mechanisms, including IGF-1, clusterin, irisin, Nrf2, BDNF, VEGF and the hypoxia response pathway (through both HIF-1α and HIF-1α independent mechanisms). [26,66] Such mechanisms seem able to strengthen functional connectivity or alternatively induce compensatory mechanisms, improving function. [8,49]
Overlapping Mechanisms
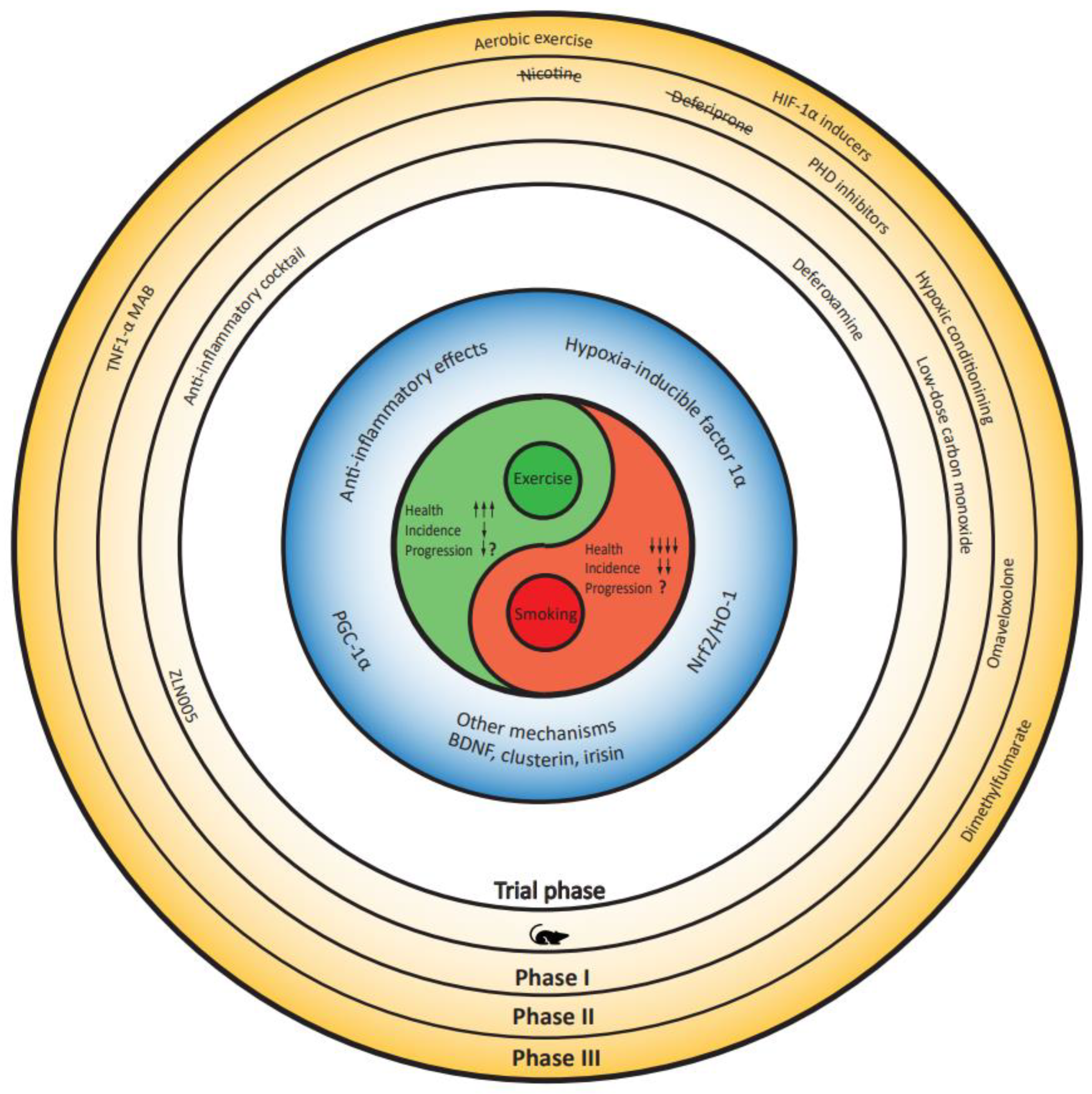
Recent studies suggest a mechanistic overlap between the putative protective effects of smoking and exercise. In Figure 1, we visualized these overlapping mechanisms, which we discuss below.
- Hypoxia response pathway (HIF-1)
The most compelling link between smoking and exercise lies in the innate response to hypoxia, which is mainly mediated by hypoxia-inducible factor (HIF) 1α. [26,67] Exercise leads to increased HIF-1 levels due to increased muscle oxygen consumption, and preclinical studies suggest HIF-1 is essential for the exercise-induced anti-apoptotic effects in neurons. [68,69,70]
Cigarette smoke, carbon monoxide, and nicotine all induce HIF-1 activation, the former in a reactive oxygen species-dependent manner, carbon monoxide via hemoglobin binding and secondary hypoxia, and the latter through the activation of the nicotinic acetylcholine receptor (nAChR)-mediated signaling pathway. [16,17] Preclinical studies suggest that activation of this hypoxia-response pathway through stabilizing HIF-1α has potential beneficial effects on pathophysiological mechanisms in PD, most importantly mitochondrial function, oxidative stress and potentially neuro-inflammation. [26]
- Nrf2/HO-1
The second mechanistic connection between exercise and smoking involves the activation of Nrf2 (nuclear factor erythroid 2-related factor 2), which could control the development of PD by mediation of oxidative stress. [71] Exercise induces a systemic Nrf2-mediated redox stress response in an age- and dose-dependent manner. [72,73,74,75,76,77] However, evidence for the effects of tobacco smoke exposure on Nrf2-ARE activation are conflicting. Some studies demonstrate activation and translocation of Nrf2, potentially as a compensatory protective response to tobacco-induced oxidative stress [18], whereas other studies indicate impaired Nrf2 signaling and the promotion of inflammation in response to tobacco smoke exposure. [78] Both exercise and smoking induce HO-1, an anti-oxidative response element (ARE) downstream from Nrf2. [53,79,80,81,82]
- PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is induced by exercise and promotes mitochondrial biogenesis and neuronal survival. [19,20,21,22] Regarding smoking, preclinical evidence suggests that PGC-1α is downregulated in response to tobacco-induced TNF-α activation. [83] However, active smokers have upregulated PGC-1α expression, possibly as a conditioning response of smoking behavior. [84] Nicotine might be a mediating factor in this longer-term response. [85] Contrarily, asthma-related research suggests adverse effects of nicotine on mitochondrial structural integrity in airway smooth muscle. [86]
- Anti-inflammatory effects
Exercise suppresses pro-inflammatory markers such as TNF-α, IL-6 and IL-1β, while activating anti-inflammatory markers, such as IL-10 and TGF-β. [87,88,89,90,91,92] Evidence for smoking is more conflicting. Whereas carbon monoxide has a similar profile to exercise [93] and nicotine induces inhibition of microglia and reduces TNF-α activation through the activation of α7 nAChR [22,92,94−97], tobacco exposure exerts acute pro-inflammatory effects by inducing the production of IL-1 and TNF-α. [98,99]
- Other mechanisms
Some mechanisms that are considered important for the neuroprotective effects of exercise are minimally explored in smoking. For example, the anti-inflammatory protein clusterin is of recent interest in the neuroinflammatory pathophysiology of PD [66] and increases with exercise. [100] Evidence on its relation with smoking is sparse, although some studies suggest that cigarette smoke induces clusterin expression. [101] One study suggests a relation between smoking intensity and salivary clusterin levels. [71] Similarly, brain-derived neurotrophic factor (BDNF) is potently activated by aerobic exercise in a dose-dependent fashion. Acute effects are unclear, but smokers have higher BDNF than non-smokers, and some studies suggest a dose-dependent increase in BDNF with duration of smoking behavior. [102,103] Irisin, a myokine, is one of the candidate mediators for increased BDNF expression with exercise. [104] Irisin already showed preventative potential in a mouse model of PD. [105]
Translation of Evidence into Therapeutic Opportunities
First, we stress our unequivocal discouragement of smoking and our wholehearted support for exercise, especially aerobic exercise, for all. However, the evidence above warrants further consideration of potential beneficial effects behind smoking that might inform translational or early-phase trials. Despite their behavioral origins, several experimental phase 1 and 2 trials are currently underway or have recently been published that target one or more of smoking’s components and have overlapping pathways with exercise.
Nicotine
Several trials have investigated whether nicotine is the prime mediating mechanism responsible for the neuroprotective effects of smoking. Evidence suggests nicotine is neuroprotective through nicotinic cholinergic activation, which is a proposed mediating pathway for neuroprotective (and neuroplasticity-related) effects of exercise. [106] Inhibition of pro-inflammatory cytokine production, modulation of mitochondrial function through improved mitophagy and increased complex I activity, and stimulation of neurotrophic factors such as BDNF are alternative proposed pathways. However, as recent studies did not show clinical improvement, further initiatives are currently considered futile. [24,107]
Low-Dose Carbon Monoxide
With nicotine trials in PD demonstrating no clear benefit on PD symptoms or progression, carbon monoxide has emerged as a potential novel target. Low-dose carbon monoxide exposure in smokers induces Nrf2/HO-1 and secondary hypoxia causing HIF-1α release, two critical overlapping mechanisms of smoking and exercise, as we mentioned above. Indeed, a recent preclinical study reported neuroprotective effects of low dose carbon monoxide in animal models of PD. They showed protection of dopamine cells and reduced alpha-synuclein pathology in association with engagement of HIF-1α, HO-1 signaling, the lysosomal enzyme cathepsin D, and Polo-like kinase 2, the latter two proteins that contribute to α-synuclein degradation [23]. The application of controlled carbon monoxide exposure as an interesting therapeutic strategy is especially apparent by recent advances in elite sports, that combine carbon monoxide with altitude exposure (see hypoxic conditioning hereafter). [108] An oral liquid formulation of carbon monoxide with completed Phase 1 safety study [NCT03926819] and demonstrated pharmacokinetic and safety markers is slated for evaluation in a pending phase 2a clinical trial in PD.
Hypoxic Conditioning
Recent evidence suggests that evolutionarily preserved adaptive neuronal responses to hypoxia, including the hypoxia-inducible factor (HIF) cascade, impact beneficially on pathophysiological mechanisms in PD. [109] Exercise increases HIF-1 because of increased metabolic demand [110] and HIF-1 is necessary for exercise-induced nigral neuroprotection. [68] This provides a compelling argument for hypoxic conditioning, i.e. repeated exposure to moderate levels of hypoxia, as a non-pharmacological intervention in PD. Exposure to carbon monoxide-induced hypoxia or hypoxia directly reduces oxidative stress and rescues nigrostriatal degeneration and parkinsonism in a preclinical model of complex I deficiency. [111,112] Furthermore, exposure to hypoxia activates HIF-independent pathways that increase mitochondrial volume and decrease oxidative damage. [113] Notably, chronic intermittent hypoxia, for example in obstructive sleep apnea, is associated with accelerated cognitive decline and PD. [114,115,116]
Small-Molecule Approaches
Several small-molecule compounds target mechanisms discussed in the previous section. For the hypoxia response pathway, these mechanisms have been summarized elsewhere. [26] Briefly, stabilization of HIF-1α as the prime mediator of the hypoxia response pathway is effectively exerted by prolyl hydroxylase domain (PHD) inhibitors (e.g. daprodustat, dimethyloxalylglycine or DMOG, roxadustat), HIF-1α inducers (e.g. albendazole, agmatine) and, less specific in its target, iron chelators (deferoxamine or DFO, deferiprone). Out of these, only deferiprone has been investigated in PD. Deferiprone decreased substantia nigra iron levels, [117] but in a phase 2 trial, the participants treated with deferiprone showed worse scores on a gold-standard motor scale for PD. [118] The latter was perhaps caused by a reduction in dopamine synthesis in the drug-naïve participants, so possible beneficial effects in levodopa-treated patients cannot be excluded.
NRF2 stabilizers are small molecules that disrupt Keap1-mediated Nrf2 ubiquitination, decreasing its breakdown and increasing Nrf2 translocation into the nucleus. [119] Several compounds are known and tested in preclinical PD, most prominently sulforaphane [120,121,122] (an anti-oxidant available as a supplement) and dimethylfumarate (commercially available as Tecfidera® in multiple sclerosis) [123,124,125,126]. Omaveloxolone is a KEAP1 inhibitor, currently under investigation in Friedreich’s ataxia and mitochondrial myopathy. [127,128] Comparisons between the potency of inducing downstream effects have yet to be conducted, and safety studies in PD are needed.
Some PGC-1α small molecules have been proposed [129,130], primarily for use in diabetes mellitus as a mediator of glucose metabolism. One of these small molecules (ZLN005) has been explored in preclinical parkinson models and demonstrates increased expression of mitochondrial respiratory chain-associated genes, and reduced loss of α-synuclein- or rotenone-induced dopaminergic neuron loss. [131] Notably, diabetes, as opposed to smoking and exercise, is a medical condition associated with increased PD incidence of 25-40%. [132]
Lastly, given the anti-inflammatory properties of exercise and some components of tobacco smoke described above, a mixture of anti-inflammatory agents could be explored, targeting multiple pathways all at once. Such a pathophysiology-agnostic regimen avoids the single pathway paradigm that might underlie the failure of many disease-modifying efforts in unselected populations. [133] However, what such a cocktail should include, is currently unknown.
Conclusion
Smoking and exercise are two of the most disparate, yet compelling human behaviors associated with reduced incidence of PD. While on the surface these are Yin and Yang, given their overall opposite health effects, at their biological cores these exposures share some striking similarities in their putative neuroprotective mechanisms. Understanding these overlapping mechanisms provides insight into proposed therapeutic strategies using non-pharmacological interventions or small molecules, many of which have already been tested in human trials in other fragile populations such as anemia or cardiovascular disease. Future proof-of-concept endeavors aimed at translating such insights into the PD population will inform larger clinical trials.
References
- Fang, X., et al. Association of Levels of Physical Activity With Risk of Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Netw Open 1, e182421 (2018). [CrossRef]
- Breckenridge, C.B., Berry, C., Chang, E.T., Sielken, R.L., Jr. & Mandel, J.S. Association between Parkinson’s Disease and Cigarette Smoking, Rural Living, Well-Water Consumption, Farming and Pesticide Use: Systematic Review and Meta-Analysis. PLoS One 11, e0151841 (2016). [CrossRef]
- Nefzger, M.D., Quadfasel, F.A. & Karl, V.C. A retrospective study of smoking in Parkinson’s disease. Am J Epidemiol 88, 149-158 (1968). [CrossRef]
- Kessler, II & Diamond, E.L. Epidemiologic studies of Parkinson’s disease. I. Smoking and Parkinson’s disease: a survey and explanatory hypothesis. Am J Epidemiol 94, 16-25 (1971). [CrossRef]
- Baumann, R.J., Jameson, H.D., McKean, H.E., Haack, D.G. & Weisberg, L.M. Cigarette smoking and Parkinson disease: 1. Comparison of cases with matched neighbors. Neurology 30, 839-843 (1980). [CrossRef]
- Marttila, R.J. & Rinne, U.K. Smoking and Parkinson’s disease. Acta Neurol Scand 62, 322-325 (1980). [CrossRef]
- Godwin-Austen, R.B., Lee, P.N., Marmot, M.G. & Stern, G.M. Smoking and Parkinson’s disease. J Neurol Neurosurg Psychiatry 45, 577-581 (1982). [CrossRef]
- Janssen Daalen, J.M., Schootemeijer, S., Richard, E., Darweesh, S.K.L. & Bloem, B.R. Lifestyle Interventions for the Prevention of Parkinson Disease: A Recipe for Action. Neurology 99, 42-51 (2022). [CrossRef]
- Organization, W.H. WHO report on the global tobacco epidemic, 2023: protect people from tobacco smoke. (2023).
- Tanner, C.M., et al. Smoking and Parkinson’s disease in twins. Neurology 58, 581-588 (2002). [CrossRef]
- Gallo, V., et al. Exploring causality of the association between smoking and Parkinson’s disease. Int J Epidemiol 48, 912-925 (2019). [CrossRef]
- Mappin-Kasirer, B., et al. Tobacco smoking and the risk of Parkinson disease: A 65-year follow-up of 30,000 male British doctors. Neurology 94, e2132-e2138 (2020). [CrossRef]
- Domenighetti, C., et al. Mendelian Randomisation Study of Smoking, Alcohol, and Coffee Drinking in Relation to Parkinson’s Disease. J Parkinsons Dis 12, 267-282 (2022). [CrossRef]
- Dominguez-Baleon, C., Ong, J.S., Scherzer, C.R., Renteria, M.E. & Dong, X. Understanding the effect of smoking and drinking behavior on Parkinson’s disease risk: a Mendelian randomization study. Sci Rep 11, 13980 (2021). [CrossRef]
- Ritz, B.R. & Kusters, C.D.J. The Promise of Mendelian Randomization in Parkinson’s Disease: Has the Smoke Cleared Yet for Smoking and Parkinson’s Disease Risk? J Parkinsons Dis 12, 807-812 (2022). [CrossRef]
- Huang, H.K., et al. Nicotine activates HIF-1alpha and regulates acid extruders through the nicotinic acetylcholine receptor to promote the Warburg effect in non-small cell lung cancer cells. Eur J Pharmacol 950, 175778 (2023). [CrossRef]
- Daijo, H., et al. Cigarette smoke reversibly activates hypoxia-inducible factor 1 in a reactive oxygen species-dependent manner. Sci Rep 6, 34424 (2016). [CrossRef]
- Muller, T. & Hengstermann, A. Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem Res Toxicol 25, 1805-1824 (2012). [CrossRef]
- Erlich, A.T., Brownlee, D.M., Beyfuss, K. & Hood, D.A. Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1alpha-dependent manner. Am J Physiol Cell Physiol 314, C62-C72 (2018). [CrossRef]
- Halling, J.F., et al. PGC-1alpha regulates mitochondrial properties beyond biogenesis with aging and exercise training. Am J Physiol Endocrinol Metab 317, E513-E525 (2019). [CrossRef]
- Lira, V.A., Benton, C.R., Yan, Z. & Bonen, A. PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am J Physiol Endocrinol Metab 299, E145-161 (2010). [CrossRef]
- Park, H.J., et al. Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur J Neurosci 26, 79-89 (2007). [CrossRef]
- Rose, K.N., et al. Neuroprotection of low dose carbon monoxide in Parkinson’s disease models commensurate with the reduced risk of Parkinson’s among smokers. bioRxiv (2024). [CrossRef]
- Oertel, W.H., et al. Transdermal Nicotine Treatment and Progression of Early Parkinson’s Disease. NEJM Evidence 2, EVIDoa2200311 (2023). [CrossRef]
- Janssen Daalen, J.M., et al. Multiple N-of-1 trials to investigate hypoxia therapy in Parkinson’s disease: study rationale and protocol. BMC Neurol 22, 262 (2022). [CrossRef]
- Janssen Daalen, J.M., et al. The Hypoxia Response Pathway: A Potential Intervention Target in Parkinson’s Disease? Mov Disord 39, 273-293 (2024). [CrossRef]
- Nie, J., et al. Independent and Joint Associations of Tea Consumption and Smoking with Parkinson’s Disease Risk in Chinese Adults. J Parkinsons Dis 12, 1693-1702 (2022). [CrossRef]
- Gallo, V., et al. Exploring causality of the association between smoking and Parkinson’s disease. Int J Epidemiol 48, 912-925 (2019). [CrossRef]
- Kim, R., Yoo, D., Jung, Y.J., Han, K. & Lee, J.Y. Smoking Cessation, Weight Change, and Risk of Parkinson’s Disease: Analysis of National Cohort Data. J Clin Neurol 16, 455-460 (2020).
- Sieurin, J., Zhan, Y., Pedersen, N.L. & Wirdefeldt, K. Neuroticism, Smoking, and the Risk of Parkinson’s Disease. J Parkinsons Dis 11, 1325-1334 (2021). [CrossRef]
- Liu, W., Wang, B., Xiao, Y., Wang, D. & Chen, W. Secondhand smoking and neurological disease: a meta-analysis of cohort studies. Rev Environ Health 36, 271-277 (2021). [CrossRef]
- Han, C., Lu, Y., Cheng, H., Wang, C. & Chan, P. The impact of long-term exposure to ambient air pollution and second-hand smoke on the onset of Parkinson disease: a review and meta-analysis. Public Health 179, 100-110 (2020). [CrossRef]
- Gatto, N.M., et al. Passive smoking and Parkinson’s disease in California Teachers. Parkinsonism Relat Disord 45, 44-49 (2017). [CrossRef]
- Gigante, A.F., Martino, T., Iliceto, G. & Defazio, G. Smoking and age-at-onset of both motor and non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord 45, 94-96 (2017). [CrossRef]
- Gabbert, C., et al. Coffee, smoking and aspirin are associated with age at onset in idiopathic Parkinson’s disease. J Neurol 269, 4195-4203 (2022). [CrossRef]
- Rosas, I., et al. Smoking is associated with age at disease onset in Parkinson’s disease. Parkinsonism Relat Disord 97, 79-83 (2022). [CrossRef]
- Gigante, A.F., et al. Smoking in Patients with Parkinson’s Disease: preliminary striatal DaT-SPECT findings. Acta Neurol Scand 134, 265-270 (2016). [CrossRef]
- Moccia, M., et al. Non-Motor Correlates of Smoking Habits in de Novo Parkinson’s Disease. J Parkinsons Dis 5, 913-924 (2015). [CrossRef]
- Doiron, M., Dupre, N., Langlois, M., Provencher, P. & Simard, M. Smoking history is associated to cognitive impairment in Parkinson’s disease. Aging Ment Health 21, 322-326 (2017). [CrossRef]
- Neshige, S., Ohshita, T., Neshige, R. & Maruyama, H. Influence of current and previous smoking on current phenotype in Parkinson’s disease. J Neurol Sci 427, 117534 (2021). [CrossRef]
- O’Reilly, E.J., et al. Smokeless tobacco use and the risk of Parkinson’s disease mortality. Mov Disord 20, 1383-1384 (2005). [CrossRef]
- Yoon, S.Y., et al. Association between smoking and all-cause mortality in Parkinson’s disease. NPJ Parkinsons Dis 9, 59 (2023). [CrossRef]
- Yang, F., et al. Physical activity and risk of Parkinson’s disease in the Swedish National March Cohort. Brain 138, 269-275 (2015). [CrossRef]
- Mak, M.K., Wong-Yu, I.S., Shen, X. & Chung, C.L. Long-term effects of exercise and physical therapy in people with Parkinson disease. Nat Rev Neurol 13, 689-703 (2017). [CrossRef]
- Tsukita, K., Sakamaki-Tsukita, H. & Takahashi, R. Long-term Effect of Regular Physical Activity and Exercise Habits in Patients With Early Parkinson Disease. Neurology 98, e859-e871 (2022). [CrossRef]
- Jin, C., Jiang, Y. & Wu, H. Association between regular physical activity and biomarker changes in early Parkinson’s disease patients. Parkinsonism Relat Disord, 105771 (2023). [CrossRef]
- Schenkman, M., et al. Effect of High-Intensity Treadmill Exercise on Motor Symptoms in Patients With De Novo Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol 75, 219-226 (2018). [CrossRef]
- van der Kolk, N.M., et al. Effectiveness of home-based and remotely supervised aerobic exercise in Parkinson’s disease: a double-blind, randomised controlled trial. Lancet Neurol 18, 998-1008 (2019). [CrossRef]
- Johansson, M.E., et al. Aerobic Exercise Alters Brain Function and Structure in Parkinson’s Disease: A Randomized Controlled Trial. Ann Neurol 91, 203-216 (2022). [CrossRef]
- Ben-Shlomo, Y. Smoking and neurogenerative diseases. Lancet 342, 1239 (1993).
- Zhang, L., et al. Beneficial Effects on Brain Micro-Environment by Caloric Restriction in Alleviating Neurodegenerative Diseases and Brain Aging. Front Physiol 12, 715443 (2021). [CrossRef]
- Chuang, Y.H., et al. Genetic variants in nicotinic receptors and smoking cessation in Parkinson’s disease. Parkinsonism Relat Disord 62, 57-61 (2019). [CrossRef]
- Rose, K.N., Schwarzschild, M.A. & Gomperts, S.N. Clearing the Smoke: What Protects Smokers from Parkinson’s Disease? Mov Disord (2024). [CrossRef]
- Yang, F., et al. Moist smokeless tobacco (Snus) use and risk of Parkinson’s disease. Int J Epidemiol 46, 872-880 (2017). [CrossRef]
- Wang, C., et al. Association between cigarette smoking and Parkinson’s disease: a neuroimaging study. Ther Adv Neurol Disord 15, 17562864221092566 (2022). [CrossRef]
- Ma, C., Liu, Y., Neumann, S. & Gao, X. Nicotine from cigarette smoking and diet and Parkinson disease: a review. Transl Neurodegener 6, 18 (2017). [CrossRef]
- Rose, K.N., et al. Neuroprotection of low dose carbon monoxide in Parkinson’s disease models commensurate with the reduced risk of Parkinson’s among smokers. NPJ Parkinsons Dis 10, 152 (2024). [CrossRef]
- Koskinen, L.O., Collin, O. & Bergh, A. Cigarette smoke and hypoxia induce acute changes in the testicular and cerebral microcirculation. Ups J Med Sci 105, 215-226 (2000). [CrossRef]
- Fricker, M., et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight 3(2018). [CrossRef]
- Allam, M.F., Campbell, M.J., Hofman, A., Del Castillo, A.S. & Fernández-Crehuet Navajas, R. Smoking and Parkinson’s disease: systematic review of prospective studies. Mov Disord 19, 614-621 (2004). [CrossRef]
- Lee, Y., et al. Does smoking impact dopamine neuronal loss in de novo Parkinson disease? Ann Neurol 82, 850-854 (2017). [CrossRef]
- Allam, M.F., Del Castillo, A.S. & Navajas, R.F. Parkinson’s disease, smoking and family history: meta-analysis. Eur J Neurol 10, 59-62 (2003). [CrossRef]
- Scheltens, P., et al. Alzheimer’s disease. Lancet 397, 1577-1590 (2021). [CrossRef]
- Ott, A., et al. Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: the Rotterdam Study. Lancet 351, 1840-1843 (1998). [CrossRef]
- Bloem, B.R., Okun, M.S. & Klein, C. Parkinson’s disease. Lancet 397, 2284-2303 (2021).
- Darweesh, S.K.L., et al. Inhibition of Neuroinflammation May Mediate the Disease-Modifying Effects of Exercise: Implications for Parkinson’s Disease. J Parkinsons Dis 12, 1419-1422 (2022). [CrossRef]
- Lee, J.W., Ko, J., Ju, C. & Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 51, 1-13 (2019). [CrossRef]
- Smeyne, M., Sladen, P., Jiao, Y., Dragatsis, I. & Smeyne, R.J. HIF1alpha is necessary for exercise-induced neuroprotection while HIF2alpha is needed for dopaminergic neuron survival in the substantia nigra pars compacta. Neuroscience 295, 23-38 (2015). [CrossRef]
- Nava, R.C., et al. Repeated sprint exercise in hypoxia stimulates HIF-1-dependent gene expression in skeletal muscle. Eur J Appl Physiol 122, 1097-1107 (2022). [CrossRef]
- Lindholm, M.E. & Rundqvist, H. Skeletal muscle hypoxia-inducible factor-1 and exercise. Exp Physiol 101, 28-32 (2016). [CrossRef]
- Pallardo-Fernández, I., Iglesias, V., Rodríguez-Rivera, C., González-Martín, C. & Alguacil, L.F. Salivary clusterin as a biomarker of tobacco consumption in nicotine addicts undergoing smoking cessation therapy. Journal of Smoking Cessation 15, 171-174 (2020). [CrossRef]
- Ostrom, E.L., Valencia, A.P., Marcinek, D.J. & Traustadottir, T. High intensity muscle stimulation activates a systemic Nrf2-mediated redox stress response. Free Radic Biol Med 172, 82-89 (2021). [CrossRef]
- Muthusamy, V.R., et al. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med 52, 366-376 (2012). [CrossRef]
- Monir, D.M., Mahmoud, M.E., Ahmed, O.G., Rehan, I.F. & Abdelrahman, A. Forced exercise activates the NrF2 pathway in the striatum and ameliorates motor and behavioral manifestations of Parkinson’s disease in rotenone-treated rats. Behav Brain Funct 16, 9 (2020). [CrossRef]
- Li, T., et al. Effects of different exercise durations on Keap1-Nrf2-ARE pathway activation in mouse skeletal muscle. Free Radic Res 49, 1269-1274 (2015). [CrossRef]
- Done, A.J., Gage, M.J., Nieto, N.C. & Traustadottir, T. Exercise-induced Nrf2-signaling is impaired in aging. Free Radic Biol Med 96, 130-138 (2016). [CrossRef]
- Aguiar, A.S., Jr., et al. Moderate-Intensity Physical Exercise Protects Against Experimental 6-Hydroxydopamine-Induced Hemiparkinsonism Through Nrf2-Antioxidant Response Element Pathway. Neurochem Res 41, 64-72 (2016). [CrossRef]
- Garbin, U., et al. Cigarette smoking blocks the protective expression of Nrf2/ARE pathway in peripheral mononuclear cells of young heavy smokers favouring inflammation. PLoS One 4, e8225 (2009). [CrossRef]
- Thompson, D., et al. Exercise-induced expression of heme oxygenase-1 in human lymphocytes. Free Radic Res 39, 63-69 (2005). [CrossRef]
- Baglole, C.J., Sime, P.J. & Phipps, R.P. Cigarette smoke-induced expression of heme oxygenase-1 in human lung fibroblasts is regulated by intracellular glutathione. Am J Physiol Lung Cell Mol Physiol 295, L624-636 (2008). [CrossRef]
- Shih, R.H., Cheng, S.E., Hsiao, L.D., Kou, Y.R. & Yang, C.M. Cigarette smoke extract upregulates heme oxygenase-1 via PKC/NADPH oxidase/ROS/PDGFR/PI3K/Akt pathway in mouse brain endothelial cells. J Neuroinflammation 8, 104 (2011). [CrossRef]
- Alves de Souza, R.W., et al. Skeletal muscle heme oxygenase-1 activity regulates aerobic capacity. Cell Rep 35, 109018 (2021). [CrossRef]
- Tang, K., Wagner, P.D. & Breen, E.C. TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol 222, 320-327 (2010).
- Guo, M., et al. Cigarette smoking and mitochondrial dysfunction in peripheral artery disease. Vasc Med 28, 28-35 (2023). [CrossRef]
- Godoy, J.A., Valdivieso, A.G. & Inestrosa, N.C. Nicotine Modulates Mitochondrial Dynamics in Hippocampal Neurons. Mol Neurobiol 55, 8965-8977 (2018). [CrossRef]
- Borkar, N.A., et al. Nicotine affects mitochondrial structure and function in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 325, L803-L818 (2023). [CrossRef]
- Bae, E.J., et al. TNF-alpha promotes alpha-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp Mol Med 54, 788-800 (2022). [CrossRef]
- Jang, Y., et al. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson’s disease mice. Brain Res 1655, 186-193 (2017). [CrossRef]
- Leem, Y.H., Park, J.S., Park, J.E., Kim, D.Y. & Kim, H.S. Suppression of neuroinflammation and alpha-synuclein oligomerization by rotarod walking exercise in subacute MPTP model of Parkinson’s disease. Neurochem Int 165, 105519 (2023). [CrossRef]
- Petersen, A.M. & Pedersen, B.K. The anti-inflammatory effect of exercise. J Appl Physiol (1985) 98, 1154-1162 (2005). [CrossRef]
- Tuon, T., et al. Physical Training Regulates Mitochondrial Parameters and Neuroinflammatory Mechanisms in an Experimental Model of Parkinson’s Disease. Oxid Med Cell Longev 2015, 261809 (2015). [CrossRef]
- Kalkman, H.O. & Feuerbach, D. Modulatory effects of alpha7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol Life Sci 73, 2511-2530 (2016). [CrossRef]
- Otterbein, L.E., et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6, 422-428 (2000). [CrossRef]
- Chen, Y., Guo, Q., Pan, X., Qin, L. & Zhang, P. Smoking and impaired bone healing: will activation of cholinergic anti-inflammatory pathway be the bridge? Int Orthop 35, 1267-1270 (2011). [CrossRef]
- Lakhan, S.E. & Kirchgessner, A. Anti-inflammatory effects of nicotine in obesity and ulcerative colitis. J Transl Med 9, 129 (2011). [CrossRef]
- Stuckenholz, V., et al. The alpha7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J Parkinsons Dis 3, 161-172 (2013). [CrossRef]
- Suzuki, S., et al. 3-[(2,4-Dimethoxy)benzylidene]-anabaseine dihydrochloride protects against 6-hydroxydopamine-induced parkinsonian neurodegeneration through alpha7 nicotinic acetylcholine receptor stimulation in rats. J Neurosci Res 91, 462-471 (2013). [CrossRef]
- El-Zayadi, A.R. Heavy smoking and liver. World J Gastroenterol 12, 6098-6101 (2006).
- Arnson, Y., Shoenfeld, Y. & Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun 34, J258-265 (2010). [CrossRef]
- De Miguel, Z., et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 600, 494-499 (2021). [CrossRef]
- Carnevali, S., et al. Clusterin decreases oxidative stress in lung fibroblasts exposed to cigarette smoke. Am J Respir Crit Care Med 174, 393-399 (2006). [CrossRef]
- Jamal, M., Van der Does, W., Elzinga, B.M., Molendijk, M.L. & Penninx, B.W. Association between smoking, nicotine dependence, and BDNF Val66Met polymorphism with BDNF concentrations in serum. Nicotine Tob Res 17, 323-329 (2015). [CrossRef]
- Shafiee, A., et al. Effect of smoking on Brain-Derived Neurotrophic Factor (BDNF) blood levels: A systematic review and meta-analysis. J Affect Disord 349, 525-533 (2024). [CrossRef]
- Wrann, C.D., et al. Exercise induces hippocampal BDNF through a PGC-1alpha/FNDC5 pathway. Cell Metab. 18, 649-659 (2013).
- Kam, T.I., et al. Amelioration of pathologic alpha-synuclein-induced Parkinson’s disease by irisin. Proc. Natl. Acad. Sci. U. S. A. 119, e2204835119 (2022). [CrossRef]
- Lu, J.Y.D., et al. The neuroprotective effect of nicotine in Parkinson’s disease models is associated with inhibiting PARP-1 and caspase-3 cleavage. PeerJ 5, e3933 (2017). [CrossRef]
- Charbonneau, P.-F. & Damier, P. Nicotine in Parkinson’s Disease — a Therapeutic Track Gone up in Smoke? NEJM Evidence 2, EVIDe2300167 (2023). [CrossRef]
- Urianstad, T., et al. Carbon monoxide supplementation: evaluating its potential to enhance altitude training effects and cycling performance in elite athletes. J Appl Physiol (1985) 137, 1092-1105 (2024). [CrossRef]
- Burtscher, J., Syed, M.M.K., Lashuel, H.A. & Millet, G.P. Hypoxia Conditioning as a Promising Therapeutic Target in Parkinson’s Disease? Mov Disord 36, 857-861 (2021). [CrossRef]
- Halliday, M.R., Abeydeera, D., Lundquist, A.J., Petzinger, G.M. & Jakowec, M.W. Intensive treadmill exercise increases expression of hypoxia-inducible factor 1alpha and its downstream transcript targets: a potential role in neuroplasticity. Neuroreport 30, 619-627 (2019). [CrossRef]
- Jain, I.H., et al. Hypoxia as a therapy for mitochondrial disease. Science 352, 54-61 (2016). [CrossRef]
- Jain, I.H., et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab 30, 824-832 e823 (2019). [CrossRef]
- Gutsaeva, D.R., et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 28, 2015-2024 (2008). [CrossRef]
- Osorio, R.S., et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964-1971 (2015). [CrossRef]
- Leng, Y., McEvoy, C.T., Allen, I.E. & Yaffe, K. Association of Sleep-Disordered Breathing With Cognitive Function and Risk of Cognitive Impairment: A Systematic Review and Meta-analysis. JAMA Neurol 74, 1237-1245 (2017). [CrossRef]
- Yeh, N.C., Tien, K.J., Yang, C.M., Wang, J.J. & Weng, S.F. Increased Risk of Parkinson’s Disease in Patients With Obstructive Sleep Apnea: A Population-Based, Propensity Score-Matched, Longitudinal Follow-Up Study. Medicine (Baltimore) 95, e2293 (2016). [CrossRef]
- Devos, D., et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21, 195-210 (2014). [CrossRef]
- Devos, D., et al. Trial of Deferiprone in Parkinson’s Disease. N Engl J Med 387, 2045-2055 (2022). [CrossRef]
- Robledinos-Anton, N., Fernandez-Gines, R., Manda, G. & Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev 2019, 9372182 (2019). [CrossRef]
- Han, J.M., et al. Protective effect of sulforaphane against dopaminergic cell death. J Pharmacol Exp Ther 321, 249-256 (2007). [CrossRef]
- Deng, C., Tao, R., Yu, S.Z. & Jin, H. Inhibition of 6-hydroxydopamine-induced endoplasmic reticulum stress by sulforaphane through the activation of Nrf2 nuclear translocation. Mol Med Rep 6, 215-219 (2012). [CrossRef]
- Siebert, A., Desai, V., Chandrasekaran, K., Fiskum, G. & Jafri, M.S. Nrf2 activators provide neuroprotection against 6-hydroxydopamine toxicity in rat organotypic nigrostriatal cocultures. J Neurosci Res 87, 1659-1669 (2009). [CrossRef]
- Khot, M., et al. Dimethyl fumarate ameliorates parkinsonian pathology by modulating autophagy and apoptosis via Nrf2-TIGAR-LAMP2/Cathepsin D axis. Brain Res 1815, 148462 (2023). [CrossRef]
- Lastres-Becker, I., et al. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid Redox Signal 25, 61-77 (2016). [CrossRef]
- Ahuja, M., et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J Neurosci 36, 6332-6351 (2016). [CrossRef]
- Jing, X., et al. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience 286, 131-140 (2015). [CrossRef]
- Lynch, D.R., et al. Efficacy of Omaveloxolone in Friedreich’s Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord 38, 313-320 (2023). [CrossRef]
- Madsen, K.L., et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 94, e687-e698 (2020). [CrossRef]
- Pettersson-Klein, A.T., et al. Small molecule PGC-1alpha1 protein stabilizers induce adipocyte Ucp1 expression and uncoupled mitochondrial respiration. Mol Metab 9, 28-42 (2018). [CrossRef]
- Zhang, L.N., et al. Novel small-molecule PGC-1alpha transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 62, 1297-1307 (2013). [CrossRef]
- Zheng, B., et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2, 52ra73 (2010).
- Cheong, J.L.Y., de Pablo-Fernandez, E., Foltynie, T. & Noyce, A.J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J Parkinsons Dis 10, 775-789 (2020). [CrossRef]
- Lang, A.E. & Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov Disord 33, 660-677 (2018). [CrossRef]
Figure 1.
Conceptual framework postulating overlapping mechanisms and therapeutic opportunities shared by - or resulting from - smoking and exercise. Stages of translation are centered around common underlying mechanisms and span from preclinical work (rodent silhouette) to clinical study phases (I, II, and III). BDNF: brain-derived neurotrophic factor, HIF1-α: hypoxia inducible factor 1α, Nrf2: nuclear factor erythroid 2-related factor 2, PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-α, TNFα: Tumor Necrosis Factor-α.
Figure 1.
Conceptual framework postulating overlapping mechanisms and therapeutic opportunities shared by - or resulting from - smoking and exercise. Stages of translation are centered around common underlying mechanisms and span from preclinical work (rodent silhouette) to clinical study phases (I, II, and III). BDNF: brain-derived neurotrophic factor, HIF1-α: hypoxia inducible factor 1α, Nrf2: nuclear factor erythroid 2-related factor 2, PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-α, TNFα: Tumor Necrosis Factor-α.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



