
Submitted:
10 July 2025
Posted:
11 July 2025
You are already at the latest version
Abstract
Cystic fibrosis (CF) is a rare autosomal recessive genetic disease that has a progressive and multisystemic course. The spectrum and frequency of mutations in the gene enco-ding the cystic fibrosis transmembrane conductance regulator (CFTR) varies both in European countries and in other geographical regions. The aim of the our retrospective study was to present the genetic variants identified in a group of 48 patients with CF patients from the Moldova region (Romania), as well as to establish genotype-phenotype correlations. Genetic testing was initially performed for 38 CFTR mutations, and in heterozygous patients or in whom no mutation was detected, CFTR gene sequencing (NGS) was performed.The compound heterozygous genotype was identified in 26 (54.16%) of the patients (one of the alleles being F508del), while 22 (45.83%) patients had the homozygous F508del genotype. The F508del variant was the most frequent (69.79%), followed by: G542X (6.25%, 6/96), c.621 +1G>T (3.12%, 3/96), 1677delTA (3.12%, 3/96), 185+1G->T (3.12%, 3/96), 2184insA (2.08%, 2/96), c.917dupA (2.08%, 2/96) and 3849G>A (2.08%, 2/96). Several new variants were also identified, which had not been reported in other studies from Romania (R1158X, K598*, R347H, c.2589_2599del, R496H, and CFTRdele2). We compared the results obtained with data from the literature and correlated the detected CFTR variant (genotype) with the phenotypic manifestations, highlighting certain particularities present in some patients. Genetic testing allows for early diagnosis and adapted management, including personalized treatment for each patient. Identification of novel unclassified CFTR variants still remains a challenge for clinicians. NGS-based screening of heterozygous healthy carriers is important for both genetic counseling and prenatal diagnosis.
Keywords:
CFTR
; variant
; genotype
; homozygous
; heterozygous
; NGS
1. Introduction
Cystic fibrosis (CF) (ORPHA: 586; OMIM: 219700) is a rare autosomal recessive genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on chromosome 7q31.2 (OMIM, 602421) [https://omim.org/entry/602421?search=CFTR&highlight=cftr] [1].
CF is a progressive multisystem disease with respiratory tract involvement (recurrent sinusitis and bronchitis, progressive obstructive pulmonary disease with bronchiectasis), exocrine pancreatic insufficiency (EPI), diabetes mellitus, gastrointestinal manifestations (meconium ileus, intestinal obstructive syndrome, rectal prolapse), hepatobiliary disease, and male infertility (Congenital absence of the vas deferens) (CAVD). Pulmonary disease is the major cause of morbidity and mortality in CF [2,3]. CF is the most common potentially lethal genetic disease in Caucasians with an incidence of approximately 1 in 3,000–4,000 live births, less common in African Americans (incidence 1 in 15,000–20,000) and even less commonly in Asian Americans [1]. Approximately 1 in 25–30 Caucasians are carriers of a CFTR pathogenic variant [4].
Mutations in the CFTR gene lead to a modification of the chloride and bicarbonate transport channel regulated by cyclic adenosine monophosphate (cAMP). Functional deficiency of CFTR at the apical membrane of the secretory epithelium also causes increased Na+ absorption and mucus secretion. Thus, impaired mucociliary clearance predisposes to inflammation and recurrent respiratory infections, with lung disease representing the main cause of morbidity and mortality in CF [5].
To date, 2121 CFTR variants have been reported in the Cystic Fibrosis Mutations Database, whose distribution and frequency vary in different regions and ethnic groups [http://www.genet.sickkids.on.ca/StatisticsPage.html, accessed on March 2025 ] [6].
Worldwide, the most common variant is c.1521_1523delCTT (F508del) (66,8%), with the highest frequency in northern European countries (eg, 87.2% in Denmark) and the lowest in Algeria (26.3%) [7,8,9]. In some populations, the prevalence of F508del is lower than other variants, and some CFTR mutations have been identified only in certain geographical regions [9].
Along with the F508del, the most common variants are G542X (2.6%), N1303K (1.6%), G551D (1.5%), W1282X (1.0%), 1717-1G->A (0.83%), R553X (0.75%), 621+1G->T (0.54%), and R1162X (0.51%) [10,11,12].
In the Mediterranean area, the most common variant is G542X (6.1%). W1282X is common in the Mediterranean region and North Africa, with the highest frequency in Ashkenazi Jews in Israel (up to 40%) [12,13]. G551D is common in northwestern and central Europe [10,11]. The highest frequency (17.2%) of the N1303K variant is in Tunisia, but it is found in most western and Mediterranean countries [11,12,13,14].
Depending on their effect on the function, quantity or stability of CFTR in the cell membrane, CFTR variants were initially classified into 6 classes [15,16].
Kris De Boeck and Margarida Amaral extended initial classification to 7 classes: the initial class I mutations (absence of CFTR protein synthesis) were divided into two groups: class I mutations, which are characterized by the lack of CFTR protein and are susceptible to pharmacological rescue by read-through compounds, and class VII mutations, which are characterized by the lack of CFTR mRNA transcript and which “cannot be rescued pharmacologically” [17]. Phenotypic variability in CF correlates with genetic heterogeneity. Depending on the phenotypic consequences they generate, a classification of CFTR mutations has been proposed into “minimal function variants” (classes I, II, and VII), which are considered high-risk mutations, associated with a more severe phenotype and early complications, and “residual function variants” or low-risk mutations (classes IV, V, and VI), which retain residual function of CFTR and are associated with milder phenotypic manifestations with late onset [7,18].
Most class III mutations (channel opening defect) are minimal function variants, but they can be found in both classifications [19].
It is known that the phenotypic variability and severity of clinical manifestations in CF is correlated with the age of the patients, the progression of the disease, different environmental factors or the intervention of modifier genes [20,21]. Many studies have described genotype-phenotype correlations in CF using different methods of classifying CFTR variants, especially in the case of pancreatic, gastrointestinal and reproductive manifestations [22,23].
Confirming the diagnosis in patients with characteristic cystic fibrosis symptoms through molecular genetic testing (DNA test for 38 mutations and/or CFTR gene sequencing) is essential for identifying the gene variant and the class of mutations to which it belongs, both in order to assess the likely evolution and for the therapeutic approach.
The aim of our retrospective study was to present the genetic variants identified in a group of 48 patients with cystic fibrosis, as well as to establish genotype-phenotype correlations. We also compared the results obtained with data from the literature and correlated the detected CFTR variant (genotype) with the phenotypic manifestations, highlighting certain particularities present in some patients.
2. Materials and Methods
We retrospectively analyzed a group of 48 patients diagnosed with cystic fibrosis, registered at Children’s Emergency Clinical Hospital, St. Maria Iași, Romania, during the period 1990-2025. All patients came from the geographical region of Moldova. The clinical diagnosis of CF was made based on the clinical picture that included symptoms of chronic sinopulmonary disease and / or gastrointestinal abnormalities / nutritional disorders associated with sweat test values higher than 80 mmol/L, performed by Nanoduct systems (Wescor, USA). The modern Nanoduct sweat analysis system updates the classic method (Macroduct/ Gibson-Cook methods) of inducing sweating by iontophoresis with pilocarpine. The Nanoduct system represents a new, simple and rapid method that allows the collection of sweat and the measurement of sodium chloride levels by conductivity analysis. The normal values of this method are different from the classic Gibson-Cooke test; according to the Nanoduct user guide, the reference ranges for children under 16 years of age are: normal - below 50 mmol/L NaCl, equivocal - 50 to 80 mmol/L NaCl and diagnostic of FC above 80 mmol/L NaCl (1 mmol is equal to 1 mEq NaCl).
In patients with sweat chloride values greater than 80 mmol/L NaCl (performed twice to confirm results), but also in the case of patients who presented clinical manifestations suggestive of cystic fibrosis, but in whom sweat chloride values revealed equivocal values between 50-80 mmol/L, genetic testing was also performed (because we were unable to confirm the results obtained by Nanoduct by another method, e.g. Macroduct / Gibson- Cooke methods).
Genetic testing was initially performed for 38 CFTR gene mutations, and in cases where a single mutation was detected (heterozygous status) or in whom no mutation was detected, CFTR gene sequencing (NGS) was performed. The latter was performed in laboratories abroad, with the cost of the analysis being borne by the patients’ families, since in Romania testing cannot be performed in state hospitals through the National Rare Diseases Program.
For the analysis of 38 CFTR mutations, DNA extraction was performed with the Invitrogen™ PureLink™ Genomic DNA Mini Kit (lot 1691968), from peripheral blood samples collected on EDTA according to the manufacturer’s instructions. Genotyping was performed using the IVD Cystic Fibrosis Genetic Assay kit (Nuclear Laser Medicine). The kit detects 38 mutations in the CFTR gene using a multiplex PCR amplification with biotinylated primers, followed by reverse hybridization on the strip and colorimetric detection (mutant allele/wild-type allele). Equipment used: Corbett Research thermal cycler (Corbett Life Science).
CFTR gene sequencing was performed at laboratories abroad (Blueprint genetics and Invitae), using Next Generation Sequencing (NGS) (Illumina technology).
The total genomic DNA was extracted from the biological sample using a bead-based method. Quantity of DNA was assessed using the fluorometric method. After assessment of DNA quantity, qualified genomic DNA sample was randomly fragmented using non-contact, isothermal sonochemistry processing. Sequencing library was prepared by ligating sequencing adapters to both ends of DNA fragments. Ready sequencing libraries that passed the quality control were sequenced using Illumina’s sequencing-by-synthesis method using paired-end sequencing (150 by 150 bases). Primary data analysis converting images into base calls and associated quality scores was carried out by the sequencing instrument using Illumina’s proprietary software, generating CBCL files as the final output. The pathogenicity of the identified gene variants was assessed according to the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG/AMP) guidelines. For the interpretation of the variants identified in patients with CF, we also used the HGMD Professional and ClinVar databases.
After the molecular classification of the mutations, for each patient and his family we analyzed the data available in the observation files, including those obtained through family history and genealogical tree. We analyzed the correlation between the patient’s phenotype and the detected gene variant (genotype) and highlighted the particularity present in some of the patients.
We also compared the obtained results with the data from the literature, highlighting both the similarities and the differences identified in the patients in the study group.
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the St. Mary’s Emergency Children Hospital, Iasi (certificate no. 31600/2023) and by the Research Ethics Committee of the University of Medicine and Pharmacy Grigore T. Popa Iasi (Certificate no. 434 /2024).
For all children, informed consent was obtained from the parents, as well as from all adults who were clinically evaluated and who underwent genetic testing.
3. Results
The retrospective study included 48 (13.15%) patients with CF confirmed by molecular testing (DNA test for 38 CFTR mutation and/or CFTR gene sequencing) out of a total of 365 patients with suspected CF based on clinical manifestations correlated with iontophoresis values. The compound heterozygous genotype was identified in 26 (54.16%) of the patients (one of the alleles being F508del), while 22 (45.83%) patients had the homozygous F508del genotype (Table 1).
In the 48 patients, 96 CFTR gene variants were identified. The F508del variant was the most frequently detected (69.79%, 67/96), being present in 93.75% (45/48) of the patients, followed by nonsense (6.96%, 6/96) and intronic variants (4.16%, 4/96) (Figure 1). We also identified two non-F508del deletions (2.08%, 2/96), two frameshift mutations (2.08%, 2/96) and two missense variants (2.08%, 2/96) (Table 1 and Figure 1).
Apart from the F508del variant, the most frequent variants detected were: G542X (6.25%, 6/96), 621 +1G>T (c.489+1 G>T) (3.12%, 3/96), 1677delTA (3.12%, 3/96), 185+1G->T (c.53+1G>T) (3.12%, 3/96), 2184insA (2.08%, 2/96), c.917dupA p.(Asn306Lysfs*2) (2.08%, 2/96) and 3849G>A (c.3717G>A) (p.Arg1239=) (2.08%, 2/96) (Table 2).
Phenotypic manifestations detected in the 48 patients with CF are presented in Table 3 .
Male patients (26/48, 54.16%) were more numerous than female (22/48, 45.83%) (Table 2). Respiratory tract involvement was present in 77.08% (37/48) of the patients, being more frequent in patients with compound heterozygous genotype (22/26, 84.61%), compared to homozygous for F508del (15/22, 68.18%). Recurrent upper respiratory tract infections (R-URTIs) (23/48, 47.91%) were more common than bronchiectasis (14/48, 29.16%). The most common colonization of patients was with Staphylococcus aureus (23/48, 47.91%) and Pseudomonas aeruginosa (18/48, 37.5%) (Table 3).
Exocrine pancreatic insufficiency (EPI) was detected in 41/48 patients (85.41%) with CF (22 homozygotes and 19 compound heterozygotes) (Table 3). Other CF-Related gastrointestinal (GI) manifestations included hepatocytolysis (32/48, 66.66%), and biliary cirrhosis (5/48, 10.41%) (Table 3). In 93.75% (45/48) of the cases there was growth failure, and the most frequent metabolic disorders were 25-OH Vitamin D deficiency (27/48, 56.25%), hepatic steatosis (11/48, 22.91%) and Cystic Fibrosis-Related Diabetes (CFRD) (6/48, 12.5%) (Table 3). Meconium ileus was present in 18.75% (9/48) of the cases, subocclusive syndrome was detected in 6.25% (3/48) of the cases, and 12.5% (6/48) patients presented with rectal prolapse (Table 3).The most frequent nonspecific, atypical manifestations detected in patients with CF were neuropsychiatric disorders (11/48, 22.91%) and cardiac involvement (10/48, 20.83%). Other manifestations were congenital adrenal hyperplasia (CAH) (2/48, 4.16%), Congenital anomalies of the kidney and urinary tract (CAKUT) (2/48, 4.16%), Gluten-related disorders (GRDs) (2/48, 4.16%) and Growth hormone deficiency (GHD) (1/48, 2.08%).
4. Discussions
Molecular testing in the 48 patients identified 96 CFTR variants, the most frequent being F508del (67/96, 69.79%). F508del (class I mutation) was identified in 93.75% (45/48) of the patients: 22 (45.83%) cases with homozygous genotype and 26 (54.16%) patients with compound heterozygous genotype (Table 1 and Figure 1). In 3 patients (6.25%) with compound heterozygous genotype, 2 different allelic variants, other than F508del, were identified (Table 1).
To date, there are few studies that attest to the prevalence of CFTR mutations in the Romanian population. The frequency of the F508del variant varied in different studies.
Frentescu et al. [24] analyzed 128 Romanian patients with CF, of whom 56.3% carried at least one F508del allele (35.93% with homozygous genotype) [24].
In another study, Apostol et al. [29] identified the F508del variant in 89.5% of the 19 patients with CF, all of whom came from southern region of Romania [29].
In 203 of the 355 Romanian children with CF included in the database of the Romanian Cystic Fibrosis, Dobre et al. [30] identified a frequency of 62% for the F508del variant [30]. The authors showed that most of the patients were F508del homozygotes (44.3%), while 35.9% of the patients were compound heterozygotes with at least one F508del mutation [30]. The majority of patients came from Timis, Iasi (Moldova) and Bucharest counties [30]. In a 1997 study, Popa et al. [31] reported a frequency of the F508del variant in the Romanian population of 25% [31].
We believe that the frequency of the F508del variant in the Romanian population is underestimated in some studies, and the differences that exist between the data reported by different authors could have several causes. These could include the small number of patients analyzed in some studies, the variation of CFTR mutations in different geographical regions of the country, including the lack of uniform criteria for patient selection or clinical diagnostic difficulties.
Also, access to genetic testing plays a key role in confirming the diagnosis of CF. In Romania, free testing through the National Rare Diseases Program only includes screening for 38 mutations of the CFTR gene, while the cost of CFTR gene sequencing is borne by the patient’s family. Thus, many of the patients heterozygous for one of the 38 tested mutations, who have clinical manifestations characteristic of CF and positive iontophoresis, remain undiagnosed, because the parents do not afford the cost of CFTR gene sequencing.
The prevalence of the F508del variant in Europe is between 65-70%, with large variations between southern Europe (30-35%) compared to northern Europe (87%) [https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf] [32]. We consider that the frequency of the F508del variant in patients in our study (69.79%) is consistent with European data.
In Table 2 we have compared the frequency of CFTR variants identified in our study with those presented in other European studies.
In a 2024 study that analyzed CFTR variant diversity across ancestries characterized using 454,727 UK biobank whole exome sequence, Ideozu et al. [33] identified 3192 (76.1%) CFTR variants in the European population. The F508del variant represented 90% of the CF-causing variant, compared to other ancestries, where the frequency of this variant was lower [33]. We believe that the results of our study (in which we identified 96 CFTR-causing variants, of which the F508del variant represented 69.79% (67/96), being present in 93.75% (45/48) of CF patients) are consistent with those reported by Ideozu et al.
In a study that included 61 patients with CF from the Republic of Moldova, Sciuca et al. [25] identified a frequency of the F508del variant of 54.71% (70/122), similar to those reported for the Romanian population in the studies of Frentescu et al. (56.3%) [24] and Dobre et al. (62%) [30].
In another study that included 192 CF patients from the Murcia region (Spain) (southwestern Europe), Rueda-Nieto et al. [7] identified the F508del variant in 58.3% of patients (27% homozygotes and 73% compound heterozygotes), representing 37% of all allelic variants identified [7].
The frequency of the F508del variant was 83.19% in the study by Kasmi et al. [26] in 116 patients from Albania [26].The results of our study and the Albanian study (both countries from south-eastern Europe) showed (a fact already known) that there are variations in the prevalence of the F508del variant in south-eastern Europe [26,27].
Genotype-Phenotype Correlations in Patients with Cystic Fibrosis
Genotype-phenotype correlations in CF, the intervention of modifier genes, the interaction with environmental and epigenetic factors facilitate the understanding of the wide spectrum of disease manifestations, which can vary from single to multisystemic involvement and between mild to severe disease [15,20,34].
Phenotypic manifestations in patients with CFTR class I, II, III and VII variants (homozygous and compound heterozygous) were more severe compared to those of patients with CFTR class IV, Vand VI variants, the data obtained being consistent with those in the literature.
Meconium ileus was present in 9/48 patients (18,75%), of which 6 (12.5%) were homozygous F508del/F508del and 3 (6,25%) were compound heterozygous: F508del/1677delTA, F508del/c.1792A>T and F508del/R347H (Figure 2).
The prevalence of meconium ileus in CF patients in our study (18,75%), is consistent with data from the literature (the prevalence of meconium ileus varies in different registries between 13-21%). In their study, Dupui et al. [23] showed that the presence of meconium ileus correlates with severe forms of the disease that evolve with EPI in patients who presented the G542X and F508del variants [23].
Bronchiectasis was present in 5/22 (22.72%) homozygous F508del/F508del patients and in 9/26 (34.61%) compound heterozygous: F508del/2184insA – 2 cases, F508del/c.53+1G>T – 1 case; F508del/R347H – 1 case, F508del/c.917dupA – 1 case, F508del/ G542X – 2 cases and F508del/ 621 +1G>T – 2 cases (Figure 2).
The majority of patients (41/48) with CF had exocrine pancreatic insufficiency (EPI) (85.41%).This finding aligns with data from the literature according to which over 85% of CF patients manifest EPI (European Cystic Fibrosis Society Patient Registry) [32].
The evaluation of pancreatic function was done by determining fecal pancreatic elastase-1 (FE-1) in two stool samples, considering Pancreatic insufficiency at FE-1 values <200 µg/g (European Cystic Fibrosis Society (ECFS) standards for the care of people with CF) [32]. In the case of our patients, most had severe pancreatic insufficiency, with FE-1 values below 100 µg/g [32].
EPI was identified in 22 homozygotes F508del/F508del and 19 compound heterozygotes: F508del/1677delTA – 3 cases, F508del/c.917dupA - 2 cases, F508del/c.2589_2599delAATTTGGTGCT - 1 case, F508del/2184insA - 2 cases, F508del/ R1158X - 1 case, F508del/1717-1G>A - 1 case, F508del/c.545940_273_10250del (CFTRdele2,3) - 1 case, F508del/G542X - 3 cases, F508del/c.621+1 G>T - 3 cases, G542X/185+1G->T (c.53+1G>T) – 1 case, F508del/c.(53+1_54-1)_(164+1_165-1)del (CFTRdele2) – 1 case, F508del/c.1487G>A, p.(Trp496Ter) (W496*) – 1 case (Figure 2).
Biliary cirrhosis was identified in 3 (6.25%) patients: 2 patients with compound heterozygous genotypes: F508del/c.917dupA p.(Asn306Lysfs*2) and F508del/ 621 +1G>T and 3 patients homozygous F508del/F508del (Figure 2).
Five (10.41%) patients died due to complications (age of death ranging between 2 and 8 months of life): 3 (6.25%) with homozygous genotype F508del/F508del and 2 (4.16%) patients compound heterozygous F508del/1677del TA. The main cause of death was sepsis with multiple organ dysfunction, and one patient was diagnosed with intestinal volvulus with ileal loop necrosis, meconium peritonitis, common mesenteric thrombosis, and severe sepsis with cloacal Enterobacter and P. aeruginosa.
The G542X variant (class I, nonsense) was detected in 6 (6.25%) compound heterozygous patients: F508del/G542X – 3 cases and G542X/185+1G->T (c.53+1G>T) – 3 cases, being the most frequent mutation detected in our study, after the F508del variant (Table 1).
G542X is frequent in Mediterranean countries (6.1- 8%) [11,12]. The phenotype of patients with the F508del/G542X compound heterozygous mutation is severe, involving meconium ileus, EPI, hepatobiliary involvement and congenital absence of the vas deferens (CAVD) [36,37,38]. In our study, in the case of the three patients with CF and the F508del/G542X genotype, the phenotype was severe and included growth and development delay, recurrent upper respiratory tract infections (R-URTIs), recurrent pneumonia, bronchiectasis, chronic coinfection with P.aeruginosa and Staphylococcus aureus, hepatocytolysis, hepatic steatosis and vitamin D deficiency. Two of the patients presented with rectal prolapse and EPI. CFRD was detected in one of the patients.
In other three patients with CF, the compound heterozygous genotype G542X/185+1G->T (c.53+1G>T) was detected (Table 1). All three patients had a similar severe phenotype manifested by recurrent pneumonia, chronic coinfections with P.aeruginosa and Staphylococcus aureus, anemia and Cystic fibrosis-associated liver disease (CFLD). Only one of the patients manifested EPI and rectal prolapse associated with immune deficiency and atopic dermatitis. The CFTR c.53+1G>T variant (class V, splice donor) is located in a canonical splice site and is predicted to affect mRNA splicing, resulting in an abnormal protein (either by exon skipping or by inclusion of intronic material).
The CFTR c.53+1G>T variant is classified as pathogenic and is present in the ClinVar database (Variation ID: 53988) [39], having been reported in patients with CF in various studies in the literature (PMID: 22658665, 16596947, 23276700, 31126253) [28,40,41,42].
Three other patients with CF in the study group were compound heterozygous F508del/621 +1G>T (c.489+1 G>T) (Table 1). In all three patients the phenotype was severe, with clinical manifestations including recurrent pneumonia, EPI, growth delay and vitamin D deficiency. Other manifestations were inflammatory bowel disease (IBD) (1 case), hepatocytolysis, pituitary dwarfism (1 case) and intellectual disability (1 case).
The CFTR variant 621+1G>T (c.489+1 G>T) (class I, splice donor) is present in the ClinVar database (Variation ID: 38799) [39] and is classified as pathogenic. It has been reported in the literature in patients whose phenotype included bronchiectasis, hereditary pancreatitis and CFTR-related disorders (CFTR – RD) [https://cftr2.org/mutations_history, accessed on 29 March 2025] [43,44,45,46,47].
The 1677delTA variant (class II, nonsense) was detected in three compound heterozygous patients, the other allele being F508del (Table 1). The 1677delTA variant (a 2bp deletion in exon 10 of the CFTR gene) is common in the Black Sea basin (the highest frequency being detected in Georgia) and is associated with a severe CF phenotype, with a high rate of early mortality in homozygotes and possibly with an increased risk of meconium ileus [48,49].
Our results are consistent with those in the literature, the phenotype of the three patients with the compound heterozygous genotype F508del/1677delTA being severe, two of them dying in the first months of life, while the other patient manifested meconium ileus, the subsequent evolution being severe. The F508del/1677delTA variant was previously reported in a patient from Romania by Frentescu et al. [24] and by Sciuca et al [Sciuca-25] in a patient from the Republic of Moldova.
The compound heterozygous genotype F508del/2184insA was identified in two of the patients with CF (Table 1) and was associated with a severe disease phenotype. Thus, one of the patients associated growth failure, recurrent respiratory infections, bronchiectasis, chronic colonization with P.aeruginosa and EPI, and the other two patients also presented chronic coinfection with Moraxella catarrhalis, Burkholderia cepacia, renal lithiasis, dyslipidemia, immune deficiency and CFRD.
The 2184insA c.2052dup (p.Gln685fs) (Q685fs) variant (class I, frameshift) was previously reported by Frentescu et al. [24] in a patient with CF from other geographical region of Romania (Cluj) than Moldova and in four patients from the Republic of Moldova, in the study by Sciuca et al. [25].
The pathogenic CFTR c.2052dupA (p.Gln685ThrfsTer4) (Q685Tfs*4, or 2184insA) variant (most likely originating from the former Galicia) is present in the ClinVar database (Variation ID: 35838) [39] and has been reported in the literature in several individuals affected by FC with exocrine pancreatic insufficiency (EPI) [40,50,51,52,53,54,55,56,57].
In other two patients with CF, the compound heterozygous genotype F508del/c.917dupA was detected (Table 1). In both cases, the phenotype was severe and included EPI associated with CFLD (hepatocytolysis and liver cirrhosis), history of recurrent pneumonia, bronchiectasis, immune deficiency and vitamin D deficiency. Both patients had iontophoresis values above 100 mmol/L NaCl. The CFTR variant (NM_000492.4) c.917dupA (p.Asn306LysfsTer2) (p.N306Kfs*2) is a missense mutation (class I) that is not found in the ClinVar, GnomAD and other databases, and has not been reported in previous studies in patients from Romania, nor in other studies in the literature.
The compound heterozygous genotype F508del/3849G->A (c.3717G>A) (p.Arg1239=) was detected in two patients in the study group, in whom clinical manifestations included growth failure, R-URTIs, recurrent pneumonia, chronic colonization with P.aeruginosa and Staphylococcus aureus, hepatocytolysis, dyslipidemia and immune deficiency, without pancreatic involvement.
The CFTR variant NM_000492.4: c.3717G>A (3849G->A) (p.Arg1239=) (class V, splice donor ) is present in the ClinVar (Variation ID: 53791) [39] and GnomAD databases (allele frequency in the general population 0.00001) being classified as pathogenic [52]. This variant has been reported in several other studies in multiple individuals with CF and pancreatic insufficiency [58,59,60,61,62]. It has also been reported in an individual with unilateral CAVD by Akinsal et al. [63] and also reported as a lower risk variant to develop CFRD by Adler et al. [64].
The CFTR variant c.2589_2599del (p.Ile864fs) (I864fs) (class I, frameshift) was detected in a patient with compound heterozygous genotype (the other allele being F508del). He presented with severe clinical manifestations that included growth failure, recurrent pneumonia, Staphylococcus aureus chronic infection, rectal prolapse, and EPI. This mutation is classified as pathogenic and is present in the ClinVar database (Variation ID: 53516) [39] and absent in GnomAD and is reported in several studies in the literature (PMID: 30979683, 30888834, 30548586) [65,66,67].
Two other novel variants (not previously reported in the Romanian population) associated with meconium ileus were K598*(c.1792A>T) (p.Lys598Ter) and R347H. Both patients had compound heterozygous genotype, the other allele being F508del (Table 1).
The patient with the K598*(c.1792A>T) (p.Lys598Ter) variant (class V, nonsense) came from a family with consanguineous parents, who had a history of another child with CF, who died at the age of 5 weeks. The clinical evolution was with recurrent respiratory infections, growth failure, maldigestion with secondary anemia, hypoproteinemia and severe deficiency rickets. The variant is present in the ClinVar (Variation ID: 1705874) [39] and GnomAD databases and is reported as pathogenic in a single study by Claustres et al. [68].
The patient in whom the c.1040G>A (p.Arg347His) (R347H) variant (class III, missense) was identified presented, in addition to meconium ileus, recurrent respiratory infections, colonization with Staphylococcus aureus, bronchiectasis, and vitamin D deficiency. This missense variant is present in the ClinVar database (Variation ID: 7182) [39] and is classified as pathogenic. It has been reported in several studies in patients with CF who developed pancreatitis (PMID: 30726326, 22658665, 27086061) [36,42,69].
In two other patients who developed EPI, the variants R1158X (c.3472C>T) (class I, nonsense) and 1717-1G>A (class VII, splice acceptor) were identified, both being novel variants, not previously reported in the population of Romania or the Republic of Moldova (Table 2).
Both patients had compound heterozygous genotype, the other allele being F508del (Table 1). Clinical manifestations in the case of the R1158X variant included, in addition to EPI, growth failure, R-URTIs, recurrent pneumonia, hepatocytolysis, immune deficiency and adrenogenital syndrome (CAH).
The nonsense variant R1158X c.3472C>T (p.Arg1158Ter) is present in the ClinVar (Variation ID: 7144) [39] and GnomAD databases and is classified as pathogenic and has been reported in several studies in patients with CF (PMID: 22658665, 23974870, 31672438) [40,47,70].
In addition to EPI, clinical manifestations in the patient with the 1717-1G>A (c.1585-1G>A) variant (IVS10, G-A, -1) included growth failure, chronic diarrhea, recurrent pneumonia, and vitamin D deficiency. The intronic variant 1717-1G>A (c.1585-1G>A) is present in the ClinVar (Variation ID: 7112) [39] and GnomAD databases and is classified as pathogenic. It is considered one of the most common variants associated with CF and has been reported in several studies in the literature [40,71,72,73].
In two patients with CF who presented with neonatal cholestasis syndrome, the CFTR variants c.(53+1_54-1)_(164+1_165-1)del (CFTRdele2) and c.54-5940_273+10250del (CFTRdele2,3 (21kb) were identified, in both cases with compound heterozygous genotype, the other allele being F508del (Table 1).
In the case of the first patient, clinical manifestations included growth failure, EPI, IBD (chronic diarrhea), CFLD (hepatocytolysis, acute liver failure), hyper IgE syndrome and immune deficiency. The c.(53+1_54-1)_(164+1_165-1)del variant (class I) has not been reported in previous studies in the Romanian and is present in the ClinVar database (Variation ID: 1705984) [39], being classified as pathogenic [74].
The second patient in whom the variant c.54-5940_273+10250del (CFTRdele2,3 (21kb) (class VII) was identified, clinical manifestations included, in addition to delayed meconium elimination, hemolytic anemia and hypoproteinemia. The variant was previously reported in Romania by Frentescu et al. [24], as well as in the Republic of Moldova by Sciuca et al. [25] (Table 2). This variant (CFTRdele2,3) involves the deletion of exons 2 and 3 of the CFTR gene and is found in the ClinVar database (Variation ID: 66105) [39] and is classified as pathogenic, having been reported in several studies in the literature (PMID: 24624459, 23974870) [75,76].
In a patient with CF whose phenotype included short stature, chronic diarrhea, chronic rhinosinusitis, chronic infection with P.aeruginosa and Staphylococcus aureus, EPI, hepatocytolysis, CFRD, dyslipidemia, immune deficiency and vitamin D deficiency, the compound heterozygous genotype F508del/c.1487G>A, p.(Trp496Ter) (W496*) was detected. The CFTR variant c.1487G>A, p.(Trp496Ter) (W496*) (class V, nonsense) is present in the ClinVar database (Variation ID: 53265) [34], is classified as pathogenic and is reported in the literature in several studies of patients with CF [77,78].
5. Conclusions
Although it included a relatively small number of patients, our retrospective study revealed that genetic heterogeneity correlates with phenotypic variability in cystic fibrosis.
The F508del variant was the most frequent (69.79%) detected , a similar frequency being reported in the literature for other European countries (65-70%). New variants were identified, as well as variants already reported in other studies both in the Romanian population, and in other European countries or other regions of the world.
Genetic testing has allowed early diagnosis and adapted management (including personalized treatment) for each patient.
The particular phenotypic manifestations of certain patients could be explained by the intervention of phenotype-modifying factors (genes or environmental factors), including epigenetic regulation of CFTR gene expression.
The identification of a large number of unclassified CFTR variants by molecular testing (NGS) can be challenging and, at the same time, of interest and relevance to clinicians. NGS-based screening for the identification of healthy heterozygous carriers in affected families is important for genetic counseling and calculating the risk of recurrence in offspring, as well as for prenatal diagnosis.
Author Contributions
Conceptualization, M.A.D., L.I.B. and L.M.T.; methodology, L.I.B, L.M.T., D.A.P.; software, M.A.D., L.I.B. and A.M.M.; validation, L.I.B., L.M.T and E.C.; formal analysis, E.Ț., A.M.M. and G.G.; investigation, M.A.D.; resources, C.R., R.P., and D.A.P.; data curation, M.C.P., G.G., and E.C.; writing—original draft preparation, L.I.B.; writing—review and editing, L.I.B., L.M.T and M.A.D.; visualization, E.C., G.G. and E.Ț.; supervision, L.I.B. and L.M.T.; project administration, L.I.B and L.M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the St. Mary’s Emergency Children Hospital, Iasi, approved number 31600/26.10.2023 and by the Research Ethics Committee of the University of Medicine and Pharmacy Grigore T. Popa Iasi, approved number 434 /24.04.2024.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
CF: Cystic fibrosis; CFTR: cystic fibrosis transmembrane conductance regulator; R-URTIs: Recurrent upper respiratory tract infections; CFLD: Cystic Fibrosis-associated liver disease; CFRD: Cystic Fibrosis-Related Diabetes; IBD: Inflammatory bowel disease; CFTR – RD: CFTR-related disorders; EPI; Exocrine pancreatic insufficiency;
References
- Online Inherithance of Man (OMIM). Available online: https://omim.org/entry/602421?search=CFTR&highlight=cftr, (accessed on 6 March 2025).
- Diab Cáceres, L.; Zamarrón de Lucas, E. Cystic fibrosis: Epidemiology, clinical manifestations, diagnosis and treatment. Med Clin (Barc). 2023, 161(9),389-396.
- Savant, A.; Lyman, B.; Bojanowsk, C.; Upadia, J.Cystic Fibrosis. 2001 Mar 26 [Updated 2024 Aug 8]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1250/, (accessed on 7 March 2025).
- Crespo-Lessmann, A.; Bernal, S.; Del Río, E.; Rojas, E.; Martínez-Rivera, C.; Marina, N.; Pallarés-Sanmartín, A.; Pascual, S.; García-Rivero, J.L.; ,Padilla-Galo A. Association of the CFTR gene with asthma and airway mucus hypersecretion. PLoS One. 2021, 16(6), e0251881.
- Yu, C.; Kotsimbos, T. Respiratory Infection and Inflammation in Cystic Fibrosis: A Dynamic Interplay among the Host, Microbes, and Environment for the Ages. Int J Mol Sci. 2023, 24(4),4052. [CrossRef]
- Cystic Fibrosis Mutations Database. Available online: http://www.genet.sickkids.on.ca/StatisticsPage.html, accessed on 6 March 2025.
- Rueda-Nieto, S.; Mondejar-Lopez, P.; Mira-Escolano, M.P.; Cutillas-Tolín, A.; Maceda-Roldán, L.A.; Arense-Gonzalo, J.J.; Palomar-Rodríguez, J.A. Analysis of the genotypic profile and its relationship with the clinical manifestations in people with cystic fibrosis: study from a rare disease registry. Orphanet J Rare Dis. 2022,17(1):222. [CrossRef]
- Van Rens, J.; Fox, A.; Krasnyk, M.; Orenti, A.; Zolin, A.; Jung, A.; Naehrlich, L. The European Cystic Fibrosis Society Patient Registry’s Data Quality programme. In 10th European Conference on Rare Diseases & Orphan Products (ECRD 2020). Orphanet J Rare Dis. 2020, 15 (Suppl 1), 310 .
- Mei-Zahav, M., Orenti, A.; Jung, A.; Kerem, E. Variability in disease severity among cystic fibrosis patients carrying residual-function variants: data from the European Cystic Fibrosis Society Patient Registry. ERJ Open Res. 2025 , 11(1), 00587-2024. [CrossRef]
- Petrova, N.; Balinova, N.; Marakhonov, A.; Vasilyeva, T.; Kashirskaya, N.; Galkina, V.; Ginter, E.; Kutsev, S.; Zinchenko, R. Ethnic Differences in the Frequency of CFTR Gene Mutations in Populations of the European and North Caucasian Part of the Russian Federation. Front Genet. 2021,12, 678374. [CrossRef]
- Estivill, X.; Bancells, C.; Ramos, C. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. The Biomed CF Mutation Analysis Consortium. Hum Mutat. 1997, 10(2),135-54.
- WHO Human Genetics Programme. (2004). The molecular genetic epidemiology of cystic fibrosis : report of a joint meeting of WHO/IECFTN/ICF(M)A/ECFS, Genoa, Italy, 19 June 2002. World Health Organization. Available online: https://iris.who.int/handle/10665/68702, (accessed on 19 March 2025).
- Abeliovich, D.; Lavon, I.P.; Lerer, I.; Cohen, T.; Springer, C.; Avital, A.; Cutting, G.R. Screening for five mutations detects 97% of cystic fibrosis (CF) chromosomes and predicts a carrier frequency of 1:29 in the Jewish Ashkenazi population. Am J Hum Genet. 1992, 51(5), 951-6.
- Messaoud, T.; Bel Haj Fredj, S.; Bibi, A.; Elion, J.; Férec, C.; Fattoum, S. Epidémiologie moléculaire de la mucoviscidose en Tunisie [Molecular epidemiology of cystic fibrosis in Tunisia]. Ann Biol Clin (Paris). 2005, 63(6), 627-30.
- Marson, F.A.L.; ,Bertuzzo C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir Med. 2016, 4(8), e37-e38.
- Stanke, F.; Tümmler, B. Classification of CFTR mutation classes. Lancet Respir Med. 2016, 4(8), e36. [CrossRef]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016, 4(8), 662-674.
- Zemanick, E.T.; Emerman, I.; McCreary, M.; Mayer-Hamblett, N.; Warden, M.N.; Odem-Davis, K.; VanDevanter, D.R.; Ren, C.L.; Young, J.; Konstan, M.W.; et al. Heterogeneity of CFTR modulator-induced sweat chloride concentrations in people with cystic fibrosis. J Cyst Fibros. 2024, 23(4), 676-684. [CrossRef]
- Lopes-Pacheco, M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2019, 10, 1662. [CrossRef]
- Butnariu, L.I.; Țarcă, E.; Cojocaru, E.; Rusu, C.; Moisă, Ș.M.; ,Leon Constantin M.M.; Gorduza, E.V.;Trandafir, L.M. Genetic Modifying Factors of Cystic Fibrosis Phenotype: A Challenge for Modern Medicine. J Clin Med. 2021, 10(24), 5821.
- Mésinèle, J.; Ruffin, M.; Guillot, L.; Corvol, H. Modifier Factors of Cystic Fibrosis Phenotypes: A Focus on Modifier Genes. Int J Mol Sci. 2022, 23(22),14205. [CrossRef]
- Varkki, S.D.; Aaron, R.; Chapla, A.; Danda, S.; Medhi, P.; Jansi Rani, N.; Paul, G.R. CFTR mutations and phenotypic correlations in people with cystic fibrosis: a retrospective study from a single centre in south India. Lancet Reg Health Southeast Asia. 2024, 27, 100434. [CrossRef]
- Dupuis, A.; Keenan, K.; Ooi, C.Y.; Dorfman, R.; Sontag, M.K.; Naehrlich, L:, Castellani, C.; Strug, L.J.; Rommens, J.M.; Gonska, T. Prevalence of meconium ileus marks the severity of mutations of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Genet Med. 2016, 18(4), 333-40. [CrossRef]
- Frenţescu, L.; Brownsell, E.; Hinks, J.; Malone, G.; Shaw, H.; Budişan, L.; Bulman, M.; Schwarz, M.; Pop, L.; Filip, M et al. The study of cystic fibrosis transmembrane conductance regulator gene mutations in a group of patients from Romania. J Cyst Fibros. 2008, 7(5), 423-8. [CrossRef]
- Sciuca, S.; Turcu, O.; Posselt, H.G.; Fergelot, P.; Hedtfeld, S.; Sylvie Labatut, S.; Oberkanins, C.; Pühringer, H.; Reboul, M. P.; Tümmler, B. CFTR mutations of patients with cystic fibrosis from Republic of Moldova., MJHS, 2015, 3, 27-30. Available online:https://repository.usmf.md/bitstream/20.500.12710/2445/1/Mutatiile_CFTR_la_pacientii_cu_fibroza_chistica_din_RM.pdf, accesed on 13 March 2025.
- Kasmi, I.; Kasmi, G.; Basholli, B.; Sefa, H.S.; ,Vevecka E. The Spectrum and Frequency of Cystic Fibrosis Mutations in Albanian Patients. Balkan J Med Genet. 2024, 27(1), 31-36.
- Petrova, G.; Yaneva, N.; Hrbková, J.; Libik, M.; Savov, A.; Macek, M. Jr. Identification of 99% of CFTR gene mutations in Bulgarian,Bulgarian Turk, and Roma cystic fibrosis patients. Mol Genet Genomic Med. 2019, 7(8), e696.
- Křenková, P.; Piskáčková, T.; Holubová, A.; Balaščaková, M.; Krulišová, V.; Čamajová, J.; Turnovec, M.; Libik, M.; Norambuena, P.; Štambergová A et al. Distribution of CFTR mutations in the Czech population: positive impact of integrated clinical and laboratory expertise, detection of novel/de novo alleles and relevance for related/derived populations. J Cyst Fibros. 2013, 12(5), 532-7. [CrossRef]
- Apostol, P.; Cimponeriu, D.; Radu, I.; Gavrila, L. The analysis of some CFTR gene mutations in a small group of cf patients from southern part of Romania. Analele Univ din Oradea Fasc Biol [Internet]. 2009, TOM XVI(1), 8-11. Available online: https://bioresearch.ro/2009-1/008-11-APOSTOL.P.-An.U.O.Bio.2009.1.pdf.
- Dobre, M.; Chesaru, B.; Romila, A.; Tutunaru, D.; Gurău, G. Cystic fibrosis in Romanian children [Internet]. 2015 [cited 2020 Apr 3]. Available online: https://www.re searchgate.net/publication/279202066_Cystic_fibrosis_in_Romanian_children, accessed on 19 March 2025.
- Popa, I.; Pop, L.; Popa, Z.; Schwarz, M.J.; Hambleton, G.; Malone, G.M.; Haworth, A.; Super, M. Cystic fibrosis mutations in Romania. Eur J Pediatr. 1997, 156(3), 212-3. [CrossRef]
- European Cystic Fibrosis Society Pacient Registry. ECFSPR Annual Report 2021. Zolin A, Orenti A, Jung A, van Rens J et al, 2023. Available online: https://www.ecfs.eu/sites/default/files/Annual%20Report_2021_09Jun2023.pdf, (accessed on 12 March 2025).
- Ideozu, J.E.; Liu, M.; Riley-Gillis, B.M.; Paladugu, S.R.; Rahimov, F.; Krishnan, P.; Tripathi, R.; Dorr, P, Levy, H.; ,Singh A.; et al. Diversity of CFTR variants across ancestries characterized using 454,727 UK biobank whole exome sequences. Genome Med. 2024,16(1), 43.
- Brennan, M.L.; Schrijver, I. Cystic Fibrosis: A Review of Associated Phenotypes, Use of Molecular Diagnostic Approaches, Genetic Characteristics, Progress, and Dilemmas. J Mol Diagn. 2016, 18(1), 3-14.
- Casals, T.; Nunes, V.; Palacio, A.; Giménez, J.; Gaona, A.; Ibáñez, N.; Morral, N.; Estivill, X. Cystic fibrosis in Spain: high frequency of mutation G542X in the Mediterranean coastal area. Hum Genet. 1993, 91(1), 66-70.
- Rosa, K.M.D.; Lima, E.D.S.; Machado, C.C.; Rispoli, T.; Silveira, V.D.; Ongaratto, R.; Comaru, T.; Pinto, L.A. Genetic and phenotypic traits of children and adolescents with cystic fibrosis in Southern Brazil. J Bras Pneumol. 2018, 44(6), 498-504. [CrossRef]
- McHugh, D.R.; Steele, M.S.; Valerio, D.M.;Miron, A.; Mann, R.J.; LePage, D.F.; Conlon, R.A.; Cotton, C.U.; Drumm, M.L.; Hodges, C.A. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS One. 2018, 13(6), e0199573. [CrossRef]
- Viotti Perisse, I.; Fan, Z.; Van Wettere, A.; Liu, Y.; Leir, S.H.; Keim, J.; Regouski, M.; Wilson, M.D.; Cholewa, K.M.; Mansbach, S.N.; et al. Sheep models of F508del and G542X cystic fibrosis mutations show cellular responses to human therapeutics. FASEB Bioadv. 2021, 3(10), 841-854. [CrossRef]
- ClinVar database. Available online; https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 29 March 2025.
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J Cyst Fibros. 2012, 11(5), 355-62. [CrossRef]
- Bienvenu, T.; Viel, M.; Leroy, C.; Cartault, F.; Lesure, J.F.; Renouil, M. Spectrum of CFTR mutations on Réunion Island: impact on neonatal screening. Hum Biol. 2005, 77(5), 705-14. [CrossRef]
- Indika, N.L.R.; Vidanapathirana, D.M.; Dilanthi, H.W.; Kularatnam, G.A.M.; Chandrasiri, N.D.P.D.; Jasinge ,E. Phenotypic spectrum and genetic heterogeneity of cystic fibrosis in Sri Lanka. BMC Med Genet. 2019, 20(1), 89. [CrossRef]
- https://cftr2.org/mutations_history, (accessed on 29 March 2025).
- Zielenski, J.; Bozon, D.; Markiewicz, D.; Aubin, G.; Simard, F.; Rommens, J.M.; Tsui, L.C. Analysis of CFTR transcripts in nasal epithelial cells and lymphoblasts of a cystic fibrosis patient with 621 + 1G-->T and 711 + 1G-->T mutations. Hum Mol Genet. 1993, 2(6), 683-7. [CrossRef]
- De Braekeleer, M.; Allard, C.; Leblanc, J.P.; Simard, F.; Aubin, G. Genotype-phenotype correlation in five cystic fibrosis patients homozygous for the 621 + 1G-->T mutation. J Med Genet. 1997, 34(9), 788-9. [CrossRef]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Kondratyeva, E.I.; Zhekaite, E.K.; Voronkova, A.Y.; Sherman, V.D.; Galkina, V.A.; Ginter, E.K.; Kutsev, S.I.; et al. Analysis of CFTR Mutation Spectrum in Ethnic Russian Cystic Fibrosis Patients. Genes (Basel). 2020,11(5), 554. [CrossRef]
- Sosnay, P.R.; Raraigh, K.S.; Gibson, R.L. Molecular Genetics of Cystic Fibrosis Transmembrane Conductance Regulator: Genotype and Phenotype. Pediatr Clin North Am. 2016, 63(4), 585-98.
- Angelicheva, D.; Boteva, K.; Jordanova, A.; Savov, A.; Kufardjieva, A.; Tolun, A.; Telatar, M.; Akarsubaşi, A.; Köprübaşi, F.; Aydoğdu, S.; et al. Cystic fibrosis patients from the Black Sea region: the 1677delTA mutation. Hum Mutat. 1994, 3(4), 353-7. [CrossRef]
- Tkemaladze, T.; Kvaratskhelia, E.; Ghughunishvili, M.; Rtskhiladze, I.; Zaalishvili, Z.; Nakaidze, N.; Lentze, M.J.; Abzianidze, E.; Skrahina, V.; Rolfs, A. Additional evidence on the phenotype produced by combination of CFTR 1677delTA alleles and their relevance in causing CFTR-related disease. SAGE Open Med Case Rep. 2023, 11. 2050313X231177163. [CrossRef]
- Amato, F.; Bellia, C.; Cardillo, G.; Castaldo, G.; Ciaccio, M.; Elce, A.; Lembo, F.; Tomaiuolo, R. Extensive molecular analysis of patients bearing CFTR-related disorders. J Mol Diagn. 2012, 14(1), 81-9. [CrossRef]
- Cohn, J.A.; Friedman, K.J.; Noone, P.G.; Knowles, M.R.; Silverman, L.M.; Jowell, P.S. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998, 339(10), 653-8. [CrossRef]
- Genome Aggregation Database. Available on: https://gnomad.broadinstitute.org/, (accessed on 25 March 2025).
- Makukh, H.; Krenková, P.; Tyrkus, M.; Bober, L.; Hancárová, M.; Hnateyko, O.; Macek, M. Jr. A high frequency of the Cystic Fibrosis 2184insA mutation in Western Ukraine: genotype-phenotype correlations, relevance for newborn screening and genetic testing. J Cyst Fibros. 2010 , 9(5), 371-5.
- Dörk, T.; Mekus, F.; Schmidt, K.; Bosshammer, J.; Fislage, R.; Heuer, T.; Dziadek, V.; Neumann, T.; Kälin ,N.; Wulbrand, U.; et al. Detection of more than 50 different CFTR mutations in a large group of German cystic fibrosis patients. Hum Genet. 1994, 94(5), 533-42. [CrossRef]
- Stuhrmann, M.; Dörk, T.; Frühwirth, M.; Golla, A.; Skawran, B.; Antonin, W.; Ebhardt, M.; Loos, A.; Ellemunter, H.; Schmidtke, J. Detection of 100% of the CFTR mutations in 63 CF families from Tyrol. Clin Genet. 1997, 52(4), 240-6. [CrossRef]
- Ivady, G.; Madar, L.; Nagy, B.; Gonczi, F.; Ajzner, E.; Dzsudzsak, E.; Dvořáková, L.; Gombos, E.; Kappelmayer, J.; Macek, M.; et al. Distribution of CFTR mutations in Eastern Hungarians: relevance to genetic testing and to the introduction of newborn screening for cystic fibrosis. J Cyst Fibros. 2011, 10(3), 217-20. [CrossRef]
- Kolesar, P.; Minarik, G.; Baldovic, M.; Ficek, A.; Kovacs, L.; Kadasi, L. Mutation analysis of the CFTR gene in Slovak cystic fibrosis patients by DHPLC and subsequent sequencing: identification of four novel mutations. Gen Physiol Biophys 2008, 27, 299–305.
- Buratti, E.; Chivers, M.; Královicová, J.; Romano, M.; Baralle, M.; Krainer, A.R.; Vorechovsky, I. Aberrant 5’ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007, 35(13), 4250-63. [CrossRef]
- Zhang, M.Q. Statistical features of human exons and their flanking regions. Hum Mol Genet. 1998, 7(5), 919-32. [CrossRef]
- Terzic, M.; Jakimovska, M.; Fustik, S.; Jakovska, T.; Sukarova-Stefanovska, E.; Plaseska-Karanfilska D. Cystic Fibrosis Mutation Spectrum in North Macedonia: A Step Toward Personalized Therapy. Balkan J Med Genet. 2019, 22(1), 35-40. [CrossRef]
- Yang, B.;,Wang .; Zhang, W.; Li, H.; Wang, B. Compound heterozygous mutations in CFTR causing CBAVD in Chinese pedigrees. Mol Genet Genomic Med. 2018, 6(6), 1097-1103.
- Kilinç, M.O.; Ninis, V.N.; Dağli, E.; Demirkol, M.; Ozkinay, F.; Arikan, Z.; Coğulu, O.; Hüner, G.; Karakoç, F.; Tolun, A. Highest heterogeneity for cystic fibrosis: 36 mutations account for 75% of all CF chromosomes in Turkish patients. Am J Med Genet. 2002, 113(3), 250-7. [CrossRef]
- Akinsal, E.C.; Baydilli, N.; Dogan, M.E.; Ekmekcioglu, O. Comorbidity of the congenital absence of the vas deferens. Andrologia. 2018, 50, e12994. [CrossRef]
- Adler, A.I.; Shine, B.S.; Chamnan, P.; Haworth, C.S.; Bilton, D. Genetic determinants and epidemiology of cystic fibrosis-related diabetes: results from a British cohort of children and adults. Diabetes Care. 2008, 31(9),1789-94.
- Nowak, J.K.; Szczepanik, M.; Wojsyk-Banaszak, I.; Mądry, E.; Wykrętowicz, A.; Krzyżanowska-Jankowska, P.; Drzymała-Czyż, S; Nowicka, A.; Pogorzelski, A, Sapiejka E, et al. Cystic fibrosis dyslipidaemia: A cross-sectional study. J Cyst Fibros. 2019, 18(4), 566-571. [CrossRef]
- McCague, A.F.; Raraigh, K.S.; Pellicore, M.J.; Davis-Marcisak, E.F.; Evans, T.A.; Han, S.T.; Lu, Z.; Joynt, A.T.; Sharma, N.; Castellani, C.; et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. Am J Respir Crit Care Med. 2019, 199(9), 1116-1126. [CrossRef]
- Petrova, N.V.; Marakhonov, A.V.; Vasilyeva, T.A.; Kashirskaya, N.Y.; Ginter, E.K., Kutsev, S.I.; Zinchenko, R.A. Comprehensive genotyping reveals novel CFTR variants in cystic fibrosis patients from the Russian Federation. Clin Genet. 2019, 95(3), 444-447. [CrossRef]
- Claustres, M.; Thèze, C.; des Georges, M.; Baux, D.; Girodon, E.; Bienvenu, T.; Audrezet, M.P.; Dugueperoux, I.; Férec, C.; Lalau, G.; et al. CFTR-France, a national relational patient database for sharing genetic and phenotypic data associated with rare CFTR variants. Hum Mutat. 2017, 38(10), 1297-1315.
- Gaitch, N.; Hubert, D.; Gameiro, C.; Burgel, P.R.; Houriez, F., Martinez, B.; Honoré, I.; Chapron, J.; Kanaan, R.; Dusser, D.; et al. CFTR and/or pancreatitis susceptibility genes mutations as risk factors of pancreatitis in cystic fibrosis patients? Pancreatology. 2016, 16(4), 515-22. [CrossRef]
- Smits, R.M.; Oud, M.S.; Vissers L.E.L.M.; Lugtenberg, D.; Braat, D.D.M.; Fleischer, K.; Ramos, L.; D’Hauwers, K.W.M. Improved detection of CFTR variants by targeted next-generation sequencing in male infertility: a case series. Reprod Biomed Online. 2019, 39(6), 963-968. [CrossRef]
- Kerem, B.S.; Zielenski, J.; Markiewicz, D.; Bozon, D.; Gazit, E.; Yahav, J.; Kennedy, D.; Riordan, J.R.; Collins, F.S.; Rommens, J.M.; et al. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc Natl Acad Sci U S A. 1990, 7(21):8447-51. [CrossRef]
- Hull, J.; Shackleton, S.; Harris, A. Abnormal mRNA splicing resulting from three different mutations in the CFTR gene. Hum Mol Genet. 1993, 2(6), 689-92. [CrossRef]
- Sharma, N.; Sosnay, P.R.; Ramalho, A.S.; Douville, C.; Franca, A.; Gottschalk, L.B.; Park, J.; Lee, M.; Vecchio-Pagan, B.; Raraigh, K.S.; et al. Experimental assessment of splicing variants using expression minigenes and comparison with in silico predictions. Hum Mutat. 2014, 35(10), 1249-59. [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17(5), 405-24. [CrossRef]
- Shelton, C.; LaRusch, J.; Whitcomb, D.C. Pancreatitis Overview. 2014 Mar 13 [updated 2020 Jul 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK190101/, (accessed on 25 March 2025).
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013, 45(10), 1160-7. [CrossRef]
- Kanavakis, E.; Efthymiadou, A.; Strofalis, S.; Doudounakis, S.; Traeger-Synodinos, J.; Tzetis, M. Cystic fibrosis in Greece: molecular diagnosis, haplotypes, prenatal diagnosis and carrier identification amongst high-risk individuals. Clin Genet. 2003, 63(5), 400-9.
- Cohn, J.A.; Friedman, K.J.; Noone, P.G.; Knowles, M.R.; Silverman, L.M.; Jowell, P.S. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998, 339(10), 653-8. [CrossRef]
Figure 1.
Types of CFTR variants detected in the group of patients with cystic fibrosis. * the percentages expressed are related to the total number of 96 CFTR variants detected in the 48 patients with cystic fibrosis
Figure 1.
Types of CFTR variants detected in the group of patients with cystic fibrosis. * the percentages expressed are related to the total number of 96 CFTR variants detected in the 48 patients with cystic fibrosis
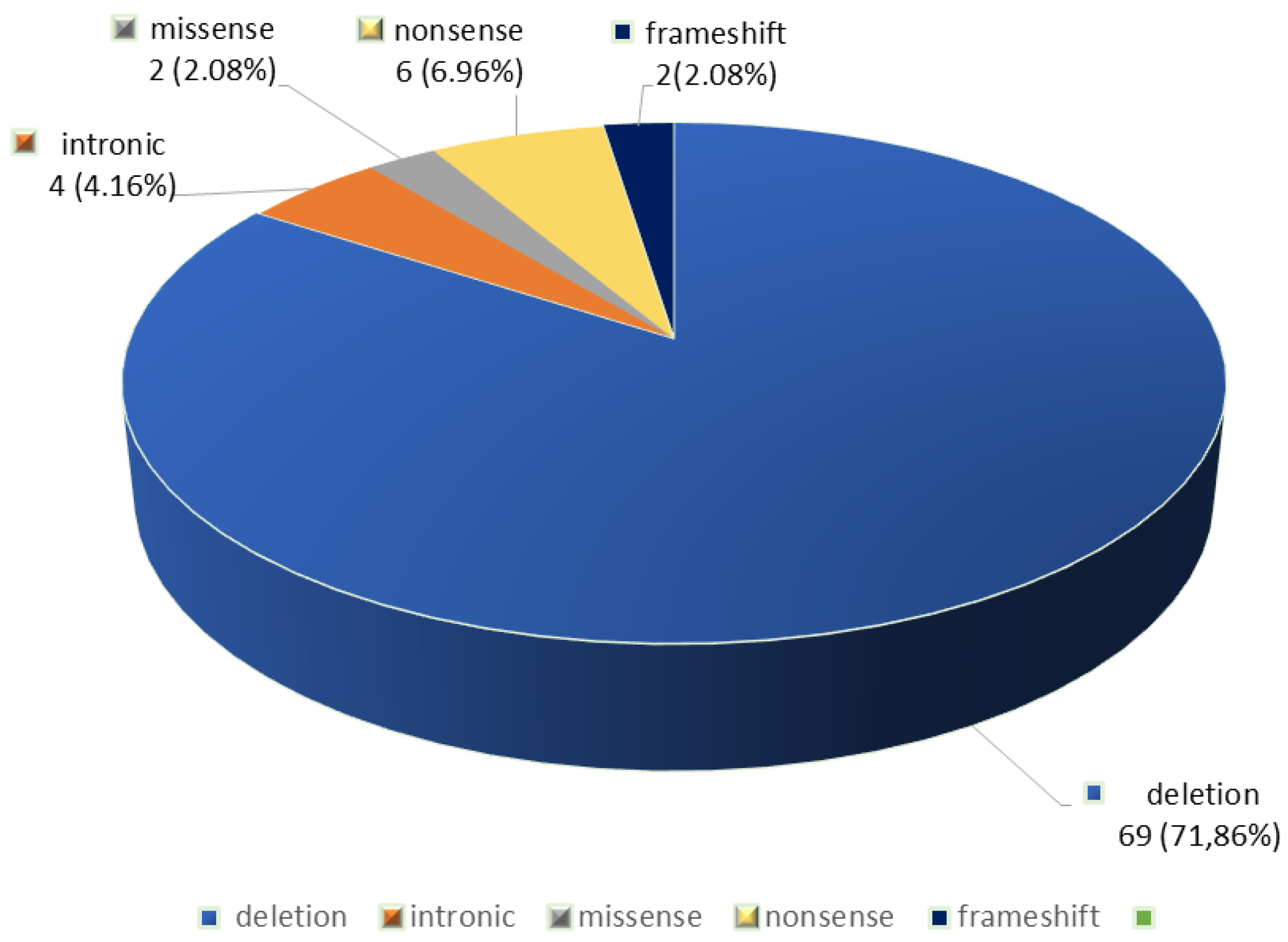
Figure 2.
Genotype - phenotype correlation in 48 patients with cystic fibrosis.
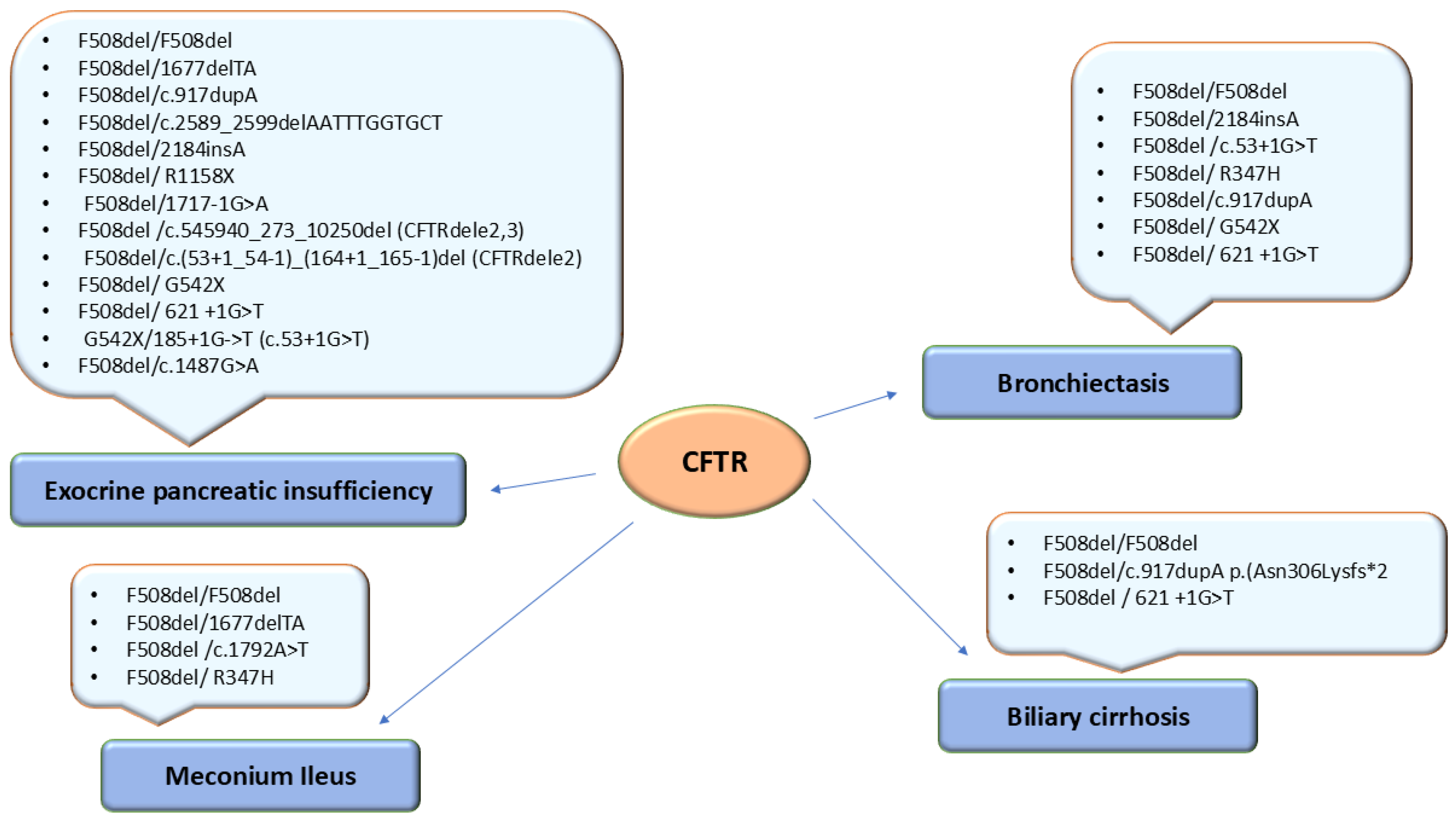

Table 1.
CFTR variant detected in the group of 48 patients with cystic fibrosis.
| Allele1 | Allele 2 | No of cases |
|---|---|---|
| F508del | F508del | 22 |
| F508del | 621 +1G>T (c.489+1 G>T) | 3 |
| F508del | G542X | 3 |
| F508del | 1677delTA | 3 |
| G542X | 185+1G->T (c.53+1G>T) | 3 |
| F508del | 2184insA (c.2052dup) (p.Gln685fs ) (Q685fs) | 2 |
| F508del | c.917dupA p.(Asn306Lysfs*2) | 2 |
| F508del | c.3849G->A (c.3717G>A) (p.Arg1239=) | 2 |
| F508del | R1158X | 1 |
| F508del | K598*(c.1792A>T ) (p.Lys598Ter) | 1 |
| F508del | c.1040G>A (p.Arg347His) (R347H) | 1 |
| F508del | c.54-5940_273+10250del (CFTRdele2,3 (21kb) | 1 |
| F508del | 1717-1G>A | 1 |
| F508del | c2589_2599delAATTTGGTGCT c.2589_2599del (p.Ile864fs)(I864fs) |
1 |
| F508del | c.1487G>A, p.(Trp496Ter) | 1 |
| F508del | c.(53+1_54-1)_(164+1_165-1)del (CFTRdele2) | 1 |
Table 2.
The spectrum of CFTR variants identified in CF patients in our study and their frequency, compared to the results of other European studies.
Table 2.
The spectrum of CFTR variants identified in CF patients in our study and their frequency, compared to the results of other European studies.
| CFTR variant | No. allele (%)(our study) | Frentescu et al. [24](Romania) | Sciucaet al. [25](Rep. Moldova) | Kasmi et al. [26](Albania) | Petrova et al. [27](Bulgaria | Křenková et al.[28]Czech Rep.) | Ruenda -Nieto et al. [7](Spain) |
|---|---|---|---|---|---|---|---|
| F508del | 67 (69.79%) | 144 (56.3%) |
79 (57.4%) | 203 (83.19%) | 154 (55%) | 809 (67.42%) | 142 (37.0%) |
| G542X | 6 (6.25%) | 10 (3.9%) | 4 ( 3.3%) | 4(1.63%) | 11 (3.93%) | 24 (2%) | 31 (8.1%) |
| 621 +1G>T (c.489+1G>T) | 3 (3.12%) | 2 ( 0.8%) |
1 (0.8%) | 6(2.45%) | 4 (1.43%) | 5 (0.43%) | 1 (0.3%) |
| 1677delTA | 3 (3.12%) | 1 (0.4%) | 1 (0.8%) | - | 3 (1.07%) | - | - |
| 185+1G ->T (c.53+1G>T) | 3 (3.12%) | - | 1 (0.8%) | - | - | 2 (0.17%) | - |
| 2184insA | 2 (2.08%) | - | 4 ( 3.3%) | - | 8 (2.89%) | 5 (0.42%) | - |
| c.917dupA p.(Asn306Lysfs*2) | 2 (2.08%) | - | - | - | - | - | |
| 3849G>A (c.3717G>A) (p.Arg1239=) |
2 (2.08%) | - | 2 ( 1.6%) | - | - | - | 1 (0.3%) |
| R1158X (c.3472C>T) | 1 (1.04%) | - | - | 1(0.40%) | 1 (0.36%) | 1 (0.08) | 1 (0.3%) |
| K598* (c.1792A>T ) (p.Lys598Ter) | 1 (1.04 %) | - | - | - | - | - | - |
| R347H (c.1040G>A) | 1 (1.04%) | - | - | - | - | 1 (0.08%) | - |
| c.54-5940_273+10250del (CFTRdele2,3 (21kb) |
1 (1.04%) | 4 (1.6%) | 2 ( 1.6%) | 1(0.40%) | 2 (0.71) | 69 (5.75%) | - |
| 1717-1G>A (c.1585-2A>T) | 1 (1.04%) | 1 (0.4%) | - | - | - | 4 (0.33) | 1 (0.3%) |
| c2589_2599delAATTTGGTGCT c.2589_2599del (p.Ile864fs) |
1 (1.04%) | - | - | - | - | 1 (0.08) | - |
| R496H (c.1487G>A) p.(Trp496Ter) | 1 (1.04%) | - | - | - | - | - | |
| c.(53+1_54-1)_(164+1_165-1)del (CFTRdele2) | 1 (1.04%) | - | - | - | - | - | - |
| No total allele | 96 | 256 | 122 | 244 | 277 | 1200 | 384 |
Table 3.
Phenotypic manifestations in the 48 patients with CF correlated with the homozygous and compound heterozygous genotypes.
Table 3.
Phenotypic manifestations in the 48 patients with CF correlated with the homozygous and compound heterozygous genotypes.
| Criteria | Homozygous no. (%) | Compound Heterozygous no. (%) |
|---|---|---|
| Female | 12 / 22 (54.5%) | 12 /26 (46.15%) |
| Male | 10 / 22 (45.5%) | 14 /26 (53.84%) |
| Respiratory Manifestations | ||
| Bronchiectasis | 5 /22 (22.72%) | 9 /26 (34.61%) |
| R-URTIs a | 10 /22 (45.45%) | 13 /26 (50%) |
|
Gastrointestinal and nutritional manifestations |
||
| Hepatocytolysis | 14 /22 (63.63%) | 18 /26 (69.23%) |
| Biliary cirrhosis | 3 /22 (13.63%) | 2 /26 (7.69%) |
| Liver fibrosis | 1 /22 (4.54%) | - |
| Gallbladder stones | 2 /22 (9.09%) | 1 /26 (3.84%) |
| EPIb | 22/22 (100%) | 19/26 (73.07%) |
| CF-related pancreatitis | 1/22 (4.54%) | - |
| CF-Related GI manifestations | - | 1 /26 (3.84%) |
| Growth Failure | 19/22 (86.36%) | 26/26 (100%) |
| Metabolic manifestations | ||
| Dyslipidemia | 2 /22 (9.09%) | 3 /26 (11.53%) |
| Hepatic steatosis | 5 /22 (22.72%) | 6 /26 (23.07%) |
| CFRD | 3 /22 (13.63%) | 3 /26 (11.53%) |
| 25-OH Vitamin D Deficiency | 13/22 (59.09%) | 14 /26 (53.84%) |
| Surgical manifestations | ||
| Meconium ileus | 6 /22 (27.27%) | 3 /26 (11.53%) |
| Subocclusive syndrome | 2 /22 (9.09%) | 1 /26 (3.84%) |
| Rectal prolapse | 2 /22 (9.09%) | 4 /26 (15.38%) |
| Renal and urological manifestations | ||
| Urolithiasis | - | 1 /26(3.84%) |
a It includes chronic colonization with Staphylococcus aureus, Pseudomonas aeruginosa and Burkholderia cepacia, Moraxella catarrhalis, Acinetobacter; R-URTIs: Recurrent upper respiratory tract infections; CF-Related GI manifestations: Cystic fibrosis gastrointestinal manifestations; CFRD: Cystic Fibrosis-Related Diabetes; FE: Fecal elastase; EPIb: Exocrine pancreatic insufficiency defined by FE-1 levels < 200 μg/g (twice); 95% of patients diagnosed with EPI had levels of FE-1 <100μg/g (widely considered as severe EPI).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



