
Submitted:
08 July 2025
Posted:
08 July 2025
You are already at the latest version
Abstract
Background: Lipoprotein(a) [Lp(a)]-induced inflammation contributes to coronary artery spasm (CAS) by contraction of vascular smooth muscle cells. However, the interaction between Lp(a) and soluble CD36 (sCD36)/interleukin(IL)-6/RAS Homolog Family Member A(RhoA)-GTP signaling pathway has not been evaluated. Methods: We investigated the relevance of Lp(a)/CD36 signaling in CAS patient monocyte-derived macrophages (PMDMs) and human coronary artery smooth muscle cell (HCASMC) line using expression profile correlation analyses, molecular docking, RNA sequencing, flow cytometry, immunoblotting and quantitative reverse transcription polymerase chain reaction. Results: Plasma Lp(a) and sCD36 levels in 41 CAS patients were significantly higher (p = 0.001) and positively correlated (r2 = 0.3145, p < 0.001), a trend not observed in 36 non-CAS controls. RNA-sequencing indicated a significant co-overexpression of CD36 and RhoA in Lp(a)-treated CAS PMDMs and HCASMCs, of which the mRNA and protein expression of CD36 and RhoA were significantly enhanced (p < 0.001) dose-dependently. Lp(a) rather than LDL preferentially induced CD80+ PMDM (M1) polarization. In HCASMCs, the CD36 knockdown using either short hairpin RNA or natural biflavonoid amentoflavone suppressed Lp(a)-upregulated protein expression of CD36, RhoA-GTP, IL-6, tumor necrosis factor(TNF)-α, nuclear factor(NF)-κB and CD80; however, overexpressed CD36 increased their levels. Lp(a) decreased and amentoflavone increased the epigenetic expression of CD36 inhibitors, miR-335-5p and miR-448, respectively. Reciprocally, miRNA inhibitor or mimic could magnify or diminish Lp(a)-induced CD36, TNF-α, NF-κB and IL-6 expressions on HCASMCs, respectively. Conclusions: Elevated Lp(a) levels upregulate the CD36-dependent TNF-α/NF-κB/IL-6/RhoA-GTP signaling pathway in CAS PMDMs and HCASMCs, indicating that Lp(a)/CD36 inflammatory signaling, HCASMC activation and macrophage M1 polarization mediate CAS development.
Keywords:
biomarker
; coronary artery spasm
; cluster of differentiation 36
; lipoprotein(a)
; molecular docking
; ras homolog family member A
; RhoA GTPase
; Rho-kinase
; soluble CD36
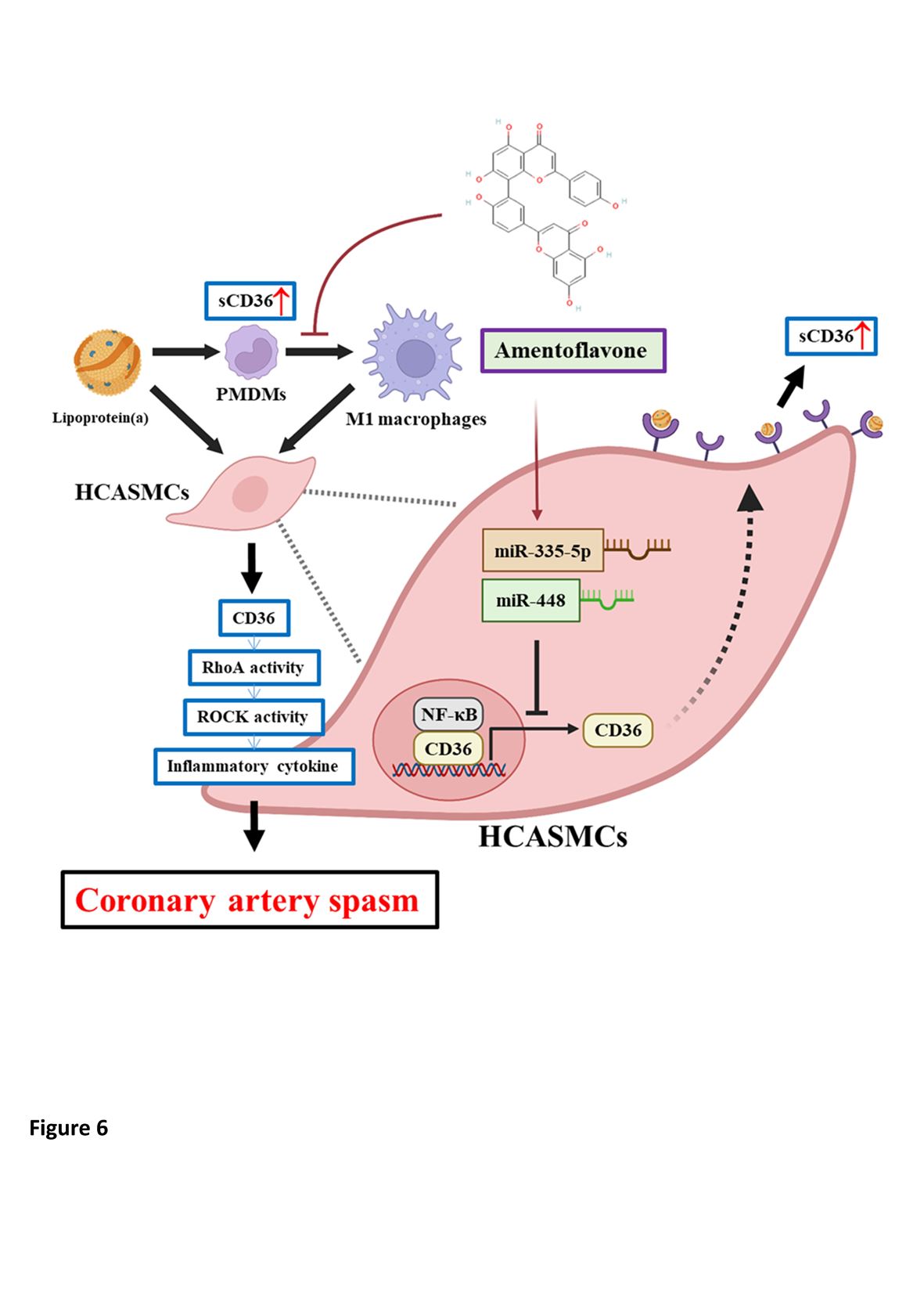
Graphical Abstract. Graphical abstract depicting how Lp(a)-triggered HCASMC dysfunction drives CAS development through a novel pathway of CD36-dependent TNF-α/NF-κB/IL-6/RhoA-GTP signaling induction, epigenetic regulation of miRNA expression and macrophage M1 polarization, which makes CD36 and its downstream effectors promising therapeutic targets.
1. Introduction
Rest- and/or effort-associated coronary artery spasm (CAS), an excessive vasoconstriction of vascular smooth muscle cell (VSMC) resulting in total or subtotal vessel obstruction, plays a key role in acute coronary syndrome (ACS), such as unstable angina, ischemia or myocardial infarction but without obstructive coronary artery disease, and sudden cardiac death [1,2]. CAS has been linked to inflammation marked by elevated peripheral monocyte count [3], levels of C-reactive protein (CRP) [4] and interleukin (IL)-6 [5], among them the increased IL-6 and CRP levels reduce endothelial nitric oxide production [6,7], leading to CAS development. Among lipoproteins, lipoprotein(a) [Lp(a)] is a risk factor for CAS [8]. The key proinflammatory oxidized phospholipids is predominantly found on apolipoprotein(a) isoforms of Lp(a), with only small amounts on low-density lipoprotein (LDL) and high-density lipoprotein [9]. Moreover, in vitro transfer studies show that oxidized LDL preferentially donates oxidized phospholipids to Lp[a], as opposed to LDL, in a time- and temperature-dependent manner [10]. We previously demonstrated that Lp(a)-triggered inflammation mediated CAS through α7-nicotinic acetylcholine receptor (α7-nAChR)/p38 MAPK/IL-6/RAS Homolog Family Member A (RhoA)-GTP signaling induction, CAS patient monocyte-derived macrophages (PMDMs) M1 polarization, and activation of human coronary artery smooth muscle cells (HCASMCs) [11]. In addition, the uncontrolled VSMC contraction in CAS is related to RhoA GTPase/Rho-kinase (ROCK1, ROCK2) pathway, which further induces inflammation and oxidative stress [12]. Although these observations highlight the important role of Lp(a) in modulating the inflammation-associated development of CAS, and while scavenger receptors’ recognition of oxidized lipoproteins on phagocytic cells evolve with the innate immune system, the interactions between Lp(a) and scavenger receptors have not been evaluated in CAS.
The scavenger receptor cluster of differentiation 36 (CD36), a class B protein (SR-B2) with dual functions as both an oxidized LDL receptor and a fatty acid transporter in a tissue-specific manner [13], has been widely expressed in many immune and non-immune cells [14]. While CD36 is preferentially expressed in tissues performing very active fatty acid metabolism, such as heart, the binding of modified lipoproteins to CD36 might regulate CAS development [14]. CD36 promotes inflammation and has been associated with ACS, among which the CD36 gene expression is ≥1.91-fold in all clinical spectrum of ACS in comparison with non-ACS control group [15]; however, whether circulating soluble CD36 (sCD36) is connected with CAS development is unknown [15]. Notably, the activation of CD36 by hexarelin, a growth hormone-releasing peptide, induces a dose-dependent increase in coronary perfusion pressure and CAS in isolated rat/mice perfused hearts [16], suggesting CD36 may mediate CAS evolution. Nonetheless, scarce data are available on Lp(a) in relation to CD36 in CAS. On the other hand, smoking and CRP are 2 important risk factors for CAS [17] and strongly correlated with insulin resistance [18]. Moreover, considering the significance of CD36 in prediabetes [19], the redistribution of CD36 in cardiomyocytes from intracellular compartments to the sarcolemma is one of the earliest changes in insulin resistance [20], which has a significant positive association with CAS [21]. Reciprocally, CAS is a risk factor for incident diabetes regardless of sex [22]. In addition, in glucose-intolerant subjects, sCD36 is positively associated with insulin resistance, IL-6, fasting triglycerides and platelet count [23]. In early molecular events of ischemic brain injury, CD36 leads to nuclear factor-κB (NF-κB) activation and postischemic inflammation [24]. Moreover, RhoGTPases play a context-dependent positive or negative regulatory role on NF-κB activation, as the RhoA–NF-κB interaction has been shown to be important in tumor necrosis factor-α (TNF-α)-activated NF-κB processes [25]. NF-κB can activate the myosin light chain kinase/myosin light chain-2 pathway and upregulate endothelin-1 expression by binding to their promoter regions, thus inducing VSMC contraction in an in-vitro model of CAS [26]. Notably, Lp(a) inhibits lipopolysaccharide-induced TNF-α production by human mononuclear cells [27]. Collectively, these studies indicate the effects of CD36 as well as sCD36 on TNF-α/NF-κB/IL-6/RhoA-GTP signaling pathway may mediate CAS development.
Clinically, CAS-induced repeat transient myocardial ischemia-reperfusion can stimulate proinflammatory responses from macrophages and coronary VSMCs [11,28], which can reciprocally activate CAS [29]. As a result, the dysregulated immunometabolism [30] may have a crucial role in the occurrence of CAS. Recent studies indicate that the dual functions of CD36 can influence innate and adaptive immune cell activation and differentiation fates [31]. In monocytes, CD36 is upregulated by peroxisome proliferator-activated receptor gamma and cytokines, such as macrophage colony-stimulating factor, IL-4, and IL-10, which are involved in differentiation into dendritic cells and reparative M2 phenotypes [32,33]. In contrast, lipopolysaccharide and dexamethasone downregulate CD36 expression [34]. However, whether Lp(a) up- or down-regulate CD36 expression in CAS remains unclear. On the other hand, the regulation of CD36 expression at both the transcriptional and posttranslational levels vary among different cell types, the differentiation states as well as exposure to soluble mediators [31]. Under chronic inflammation, oxidant stress, hyperlipidemia, or diabetes, CD36 deficiency alleviates atherosclerosis and thrombosis to maintain homeostasis [35]. We, therefore, analyzed (1) the Lp(a)-associated mRNA and protein expression levels of CD36 in the PMDMs of CAS patients and HCASMCs; (2) the relationship of CD36 and monocyte-to-macrophage differentiation and polarization based on CD80 or CD206 positivity in CAS PMDMs; (3) the probable positive interaction and modulatory loop between Lp(a), CD36 and TNF-α/NF-κB/IL-6/RhoA-GTP signaling pathway in HCASMCs; (4) the role of CD36-dependent epigenetic regulation of anti-inflammatory microRNAs (miRNAs) expression in HCASMCs.
2. Results
2.1. Study Cohort Baseline Characteristics
A total of 77 patients were enrolled in the study (median age = 59.0 years; interquartile range, 48–66 years). The CAS group compared with the control group are more likely to be male, current smokers (p < 0.05), had significantly lower high-density lipoprotein, higher sCD36 and Lp(a) levels (p = 0.001, respectively), as well as significantly higher monocyte, macrophage counts and hemoglobin values (all p < 0.05) (Table 1). Single-vessel spasm was the most common finding in the CAS patients, and spasm was provoked mostly in the right coronary artery. No between-group difference in medication use before coronary angiography was observed. However, after coronary angiography, the numbers of patients being treated with beta and calcium channel blockers were significantly lower and higher in the CAS group than in the control group, respectively (Table 1).
2.2. Correlation of Lp(a) with sCD36
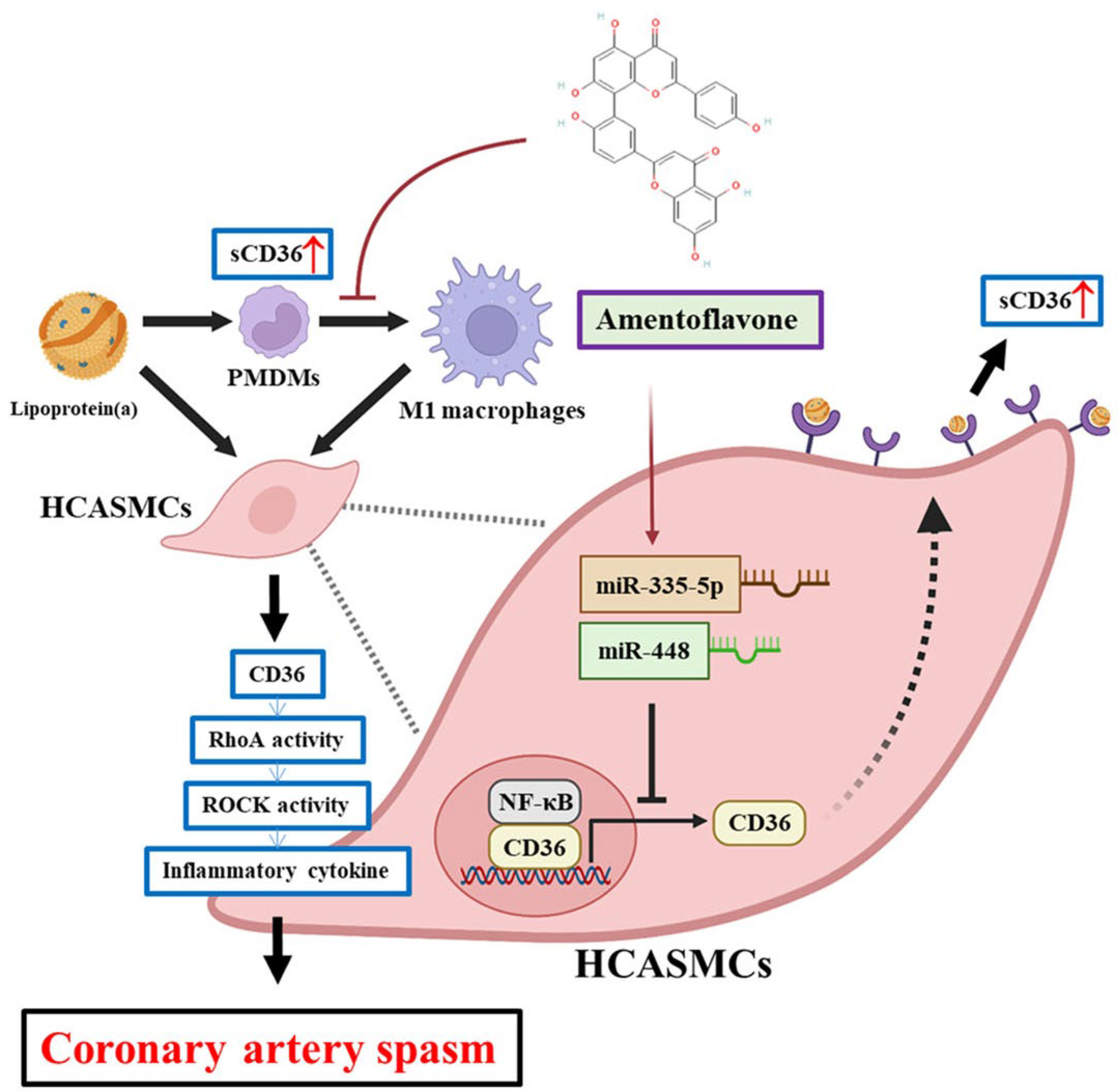
The microarray sequencing of Lp(a)-stimulated PMDMs of CAS patients (n = 3), and HCASMCs (n = 3) were analyzed to examine expression correlation between them (Figure 1A). In the differential expression genes (DEGs) analysis under Lp(a) treatment, distinct upregulated or downregulated patterns of gene expression between CAS PMDMs and HCASMCs were observed and based on the cut-off criteria log2FC>1.5 and p < 0.05, including a total of 1955 DEGs consisting of 749 upregulated and 1206 downregulated DEGs from stimulated CAS PMDMs, and a total of 1955 DEGs consisting of 749 upregulated and 1206 downregulated DEGs from HCASMCs cells, as shown in the heatmap displaying sample cluster (Figure 1B) and a volcano plot (Figure 1C). The Venn diagram, which displayed the differences in DEGs between Lp(a)-treated CAS PMDMs and HCASMCs, demonstrated that CD36 and RhoA genes were among the overlapping and overexpressed DEGs (Figure 1D). This comparison identified the genes with expression changes affected by Lp(a) in CAS PMDMs and HCASMCs. Furthermore, Lp(a) levels were positively correlated with sCD36 levels in CAS (r2 = 0.3145, p < 0.001) but not in non-CAS patients. (Figure 1E)
2.3. Lp(a) Stimulated CD36 and RhoA Expression in PMDMs, HCASMCs, and Preferentially Induced PMDM M1 Polarization
Commercially available native human Lp(a) was used in all functional assays. Under phorbol-12-myristate-13-acetate treatment of PMDMs together with 500 nM of LDL or Lp(a), Lp(a) up-regulated CD36 and RhoA mRNA by 2.5- and 3.5-fold, respectively, compared to both untreated and LDL-treated controls (Figure 2A). In parallel assays, exposure to 100 nM-1 μM, corresponding to 2.5-25 mg/dL, Lp(a) increased dose-dependently (p < 0:001) CD36 and RhoA mRNA, as well as sCD36 and RhoA protein expression level (Figure 2B-C). A dose-dependent induction of CD36 and RhoA mRNA expression was also observed in HCASMCs incubated with 0.5 to 2 μM, Lp(a) for 48 hours (Figure 2D). In our CAS PMDMs, 500 nM Lp(a) as compared to 500 nM LDL elicited a 3.2-fold stronger shift in fluorescence intensity of the CD80+ macrophage population (p < 0.001) (Figure 2E, left side). However, exposure to neither LDL nor Lp(a) had any obvious effect on the median fluorescence intensity of the CD206+ macrophage population (Figure 2E, right side), suggesting that Lp(a) preferentially induced M1 macrophage polarization in patients with CAS.
2.4. In Silico Molecular Docking to Examine Lp(a)/sCD36 Binding Interactions
By using AlphaFold models of the CD36 ectodomain (AF-Q07954-F1, residues 30–439) and apolipoprotein(a) on Lp(a), we ran a 2-step workflow: 70,000 rigid-body poses were generated with ClusPro and the 30 best clusters were flexibly refined in HADDOCK (online server: http://hdock.phys.hust.edu.cn/). Cartoon view of the best-ranked complex (Figure 3A). The top ClusPro solution scored -273.47 kcal mol−1 (confidence 0.922) and all 10 highest poses laid within -225 to -273 kcal mol−1 and ≤1.7 Å ligand Root Mean Square Deviation (Figure 3B). HADDOCK water refinement kept 128 near-identical poses averaging a score of -122 kcal mol−1, which was about 20 kcal mol−1 better than the next cluster and placed the lysine-rich groove of apolipoprotein(a) squarely on the Asp238–Glu242 acidic patch of CD36, burying ~1.6 × 103 Å2 of surface through a tight Lys70/Lys74 ↔ Asp238/Glu240 salt-bridge network (Figure 3C). A 100-ns explicit-solvent molecular dynamics simulation (ff19SB/OPC) confirmed stability, in which the complex Root Mean Square Deviation rose only to 2.6 ± 0.2 Å after equilibration, the interface held ~12 H-bonds and 5 salt bridges, and Molecular Mechanics Generalized Born Surface Area binding energy settled at -54 ± 5 kcal mol−1. Altogether, these data support a single, high-affinity pose in which Lp(a) clamped onto sCD36 through a lysine-acid “Velcro” interface to drive the conformational changes linked to outside-in signalling, offering a structural rationale for Lp(a)-induced CD36-dependent macrophage activation and providing clear targets for cross-link validation.
2.5. CD36 Knockdown Reduced Lp(a)-Induced Proinflammatory Signaling in HCASMCs
The short hairpin RNA for CD36 (shCD36) knockdown remarkably suppressed Lp(a)-induced CD36 protein expression in HCASMCs by 81% compared with Lp(a) treatment alone (Figure 3A). Additionally, shCD36 significantly decreased both protein and mRNA expression levels of RhoA-GTP, RhoA, IL-6, TNF-α, NF-κB and CD80 (Figure 4A-B). In Lp(a)-treated HCASMCs, shCD36 and a natural biflavone CD36 antagonist amentoflavone significantly reduced the RhoA-GTP/RhoA ratio using a pull-down assay with the glutathione-S-transferase-Rhotekin-Rho-binding domain (Figure 4C). On the other hand, while HCASMCs transfected with CD36 overexpression (OE-CD36) vector, cDNA-CD36, showed a significantly higher RhoA-GTP/RhoA ratio than CD36 knockout cells and the vehicle vector control, addition of amentoflavone to OE-CD36 medium significantly decreased the CD36 expression to the level similar to that observed with shCD36 (Figure 4D). In 1 μM Lp(a)-treated HCASMCs, addition of shCD36, OE-CD36, or OE-CD36+amentoflavone for 60 min, compared with controls, significantly decreased, increased or decreased ROCK activity, which was the downstream effector of Rho GTPase and RhoA-GTP, respectively (Figure 4E).
2.6. Epigenetic Regulation of Lp(a)-Triggered CD36, IL-6, TNF-α and NF-κB Expression in HCASMCs
Amentoflavone has a molecular formula of C30H18O10 (Figure 5A). Treatment with 12.5 μM–100 μM amentoflavone did not affect cell viability in CAS PMDMs and HCASMCs (Figure 5B). In HCASMCs, exposure to 10μM amentoflavone significantly reversed the 1 μM Lp(a)-induced downregulation of hsa-miR-335-5p and hsa-miR-448 expression levels to the extent that were also significantly higher than non-Lp(a)-treated controls (Figure 5C). In HCASMCs, dual luciferase reporter gene assay to examine the interaction between the miRNAs and their targeting sites demonstrated that the targeting sequence for miR-335-5p and miR-448 were identified in the 3′-UTR of CD36 mRNA (Figure 5D). In HCASMCs, the miR-335-5p’s and miR-448’s inhibitors largely increased, and mimics decreased, the Lp(a)-induced target mRNA translation, respectively, such as CD36, cytokines IL-6, TNF-α and transcription factor NF-κB (Figure 5E), suggesting miR-335-5p and miR-448 mediated the Lp(a)/CD36 interaction.
3. Discussion
We demonstrated that, compared with controls, CAS patients had higher serum Lp(a) levels, which were positively correlated with sCD36 levels in CAS but not in controls. Lp(a) preferentially induced PMDM M1 polarization in CAS and increased expression of RhoA and CD36 in CAS PMDMs and HCASMCs, and the subsequent elevated sCD36 protein expression in PMDMs. Our molecular docking predicted a good binding mode and affinity of sCD36 within Lp(a) binding site in a stable and favorable manner. In Lp(a)-treated HCASMCs, shCD36 significantly decreased both protein and mRNA expression levels of CD36, RhoA-GTP, RhoA, IL-6, TNF-α, NF-κB and CD80. In addition, shCD36 and amentoflavone significantly reduced the RhoA-GTP/RhoA ratio. On the other hand, while Lp(a)-treated HCASMCs showed a significantly higher RhoA-GTP/RhoA ratio in cells transfected with OE-CD36 than shCD36, the addition of amentoflavone to OE-CD36 medium significantly downregulated CD36 expression to the level similar to that of shCD36. Amentoflavone exerted epigenetic regulation of CD36, IL-6, TNF-α and NF-κB mRNA expression through miR-335-5p and miR-448, which mediated the Lp(a)/CD36 interaction in HCASMCs. Our novel pathways of Lp(a)-triggered CD36-dependent TNF-α/NF-κB/IL-6/RhoA-GTP signaling induction, macrophage M1 polarization, and HCASMC activation may be one of the important mechanisms contributing to CAS development.
Lp(a) is a well-established genetic risk factor for coronary artery disease (CAD), while its pathogenic mechanism in CAS has remained unclear [36]. Lp(a) can be upregulated in chronic inflammatory disorders, such as rheumatoid arthritis, Crohn’s disease, and among patients receiving hemodialysis, further implicating its role in immune-mediated cardiovascular risk [37,38,39]. Lp(a) levels also increase under acute stress conditions such as surgery or myocardial infarction, highlighting its involvement in innate immunity [40]. While Lp(a) only exists in monkeys, apes, and humans [41], and some species lack KV, human apolipoprotein (a) kringles are specialized domains to mediate ligand interactions [42], often with lysine-containing substrates, as it contains both KV and an intact lysine binding site in KIV10. Therefore, human Lp(a) is exceptionally pathogenic. Though its exact receptor remains unknown, Lp(a) interacts with several receptors, including scavenger receptors like CD36 [43]; however, the roles of these receptors, including lipoprotein receptors, toll-like and scavenger receptors, lectins, and plasminogen receptors, remain unclear [43].
Our previous studies showed that Lp(a) correlates with IL-6 in CAS, contrary to the lack of such correlation in CAD [11,44], suggesting a distinct inflammatory mechanism from that of CAD. The current study builded upon these findings, showing that Lp(a)-induced CD36 expression contributes to CAS pathogenesis by driving inflammation and VSMC dysfunction [45,46]. Despite CD36 being associated with lipid uptake and foam cell formation in atherosclerosis, our findings highlight a contrasting role in CAS where it mediates proinflammatory signaling and HCASMC hyperreactivity [13,14,47,48]. Nonetheless, although CD36 deficiency is associated with reduced risks for cardiovascular diseases, the morbidity of CAD is significantly higher in CD36 deficiency patients suffering from severe atherosclerosis [49], implying that the status of CD36 deficiency might be atherogenic rather than VSMC contraction-prone. These observations imply that CD36 is a context-dependent multifunctional receptor that both up-regulation and deficiency of this protein may increase the cardiovascular risk.
Additionally, we observed that Lp(a) increased RhoA and CD36 expression in a dose-dependent manner in CAS-derived PMDMs and HCASMCs, resulting in elevated sCD36 protein expression in PMDMs. Despite limited understanding of the RhoA signaling network within the cardiovascular context, especially given its ambiguous outcomes in both in vitro and in vivo models [50], our findings align with prior observations linking RhoA/ROCK activity to NF-κB activation in intestinal inflammation [51] and increased RhoA and ROCK mRNA expression at CAS-affected coronary sites [52]. Though Lp(a) is known to activate HCASMCs via α7-nAChR/p38 MAPK/IL-6/RhoA-GTP signaling [11], the interplay between Lp(a), CD36, and RhoA in humans remains underexplored. Our data suggest a positive feedback mechanism among these 3 markers. Notably, HCASMCs demonstrated increased CD80 expression following Lp(a) stimulation. Although CD80 is predominantly found on immune cells like B cells, monocytes, T cells, and dendritic cells, this upregulation in HCASMCs may point to an immunological checkpoint role during CAS development [53,54].
Data regarding human monocyte-to-macrophage differentiation and polarization upon Lp(a) exposure remain limited. However, interspecies differences in gene expression profiles during macrophage polarization have been documented [55]. Immunometabolism, which is the study of how the competing cellular aerobic and anaerobic metabolisms influences immune cell differentiation and function [56], particularly in myocardial ischemia, is emerging as a promising therapeutic concept [57]. CD36 stands out as a central regulator within this framework. Immune modulation holds promise across diverse pathological contexts [58], particularly as M1 macrophages exacerbate inflammation. In CAD, high Lp(a) levels are associated with increased CD36 expression on monocytes without corresponding changes in receptors such as CD163 and CD206 [59]. Similarly, in atherosclerosis, enhanced M1 marker expression (e.g., CD40/CD86) strengthens adaptive immunity [60]. Our findings indicate a comparable upregulation of CD80 in CAS PMDMs, linked to pathogenic eicosanoid production. As IL-6 and TNF-α are hallmark cytokines of M1 polarization [61,62], it is plausible that Lp(a) fosters a proinflammatory cellular environment through CD36-mediated signaling in CAS PMDMs.
This study is the first to delineate how Lp(a)-triggered CD36-dependent monocyte/HCASMC interactions upregulate the expression of TNF-α, NF-κB, IL-6, and RhoA-GTP in VSMC dysfunction in CAS development. MiRNAs regulate gene expression by targeting mRNAs for gene silencing or translational repression [63]. Expression levels of certain miRNAs change in pathological states, including malignancies, acute kidney injury, and CAD [64]; hence, they might potentially control the inflammatory cascade in CAS. Through databases like miRTarBase and MirTarget, we identified miR-335-5p and miR-448 as novel regulators of CD36. MiR-335-5p has been identified as a possible biomarker of non-alcoholic fatty liver disease, modulating genes involved in lipid metabolism, such as peroxisome proliferator-activated receptor-γ (PPAR-γ) and sterol regulatory element-binding protein1c (SREBP1c) [65]. PPAR-γ agonists added on calcium channel blockers significantly reduce CAS compared with calcium channel blockers alone after 6 months of treatment [66]. In non-alcoholic fatty liver disease, CD36 influences lipogenesis via SREBP1 processing [67], which is also elevated in lipopolysaccharide-induced M1 macrophages [68]. Additionally, miR-448 is overexpressed in VSMCs from atherosclerotic plaques compared to normal arteries [69]. Our study confirmed that both miR-335-5p and miR-448 target key inflammatory mediators, including NF-κB, IL-6, and TNF-α, linking them to CAS development. Evidence indicates that hypercontraction of VSMC is caused by enhanced Ca2+ sensitization through the activation of the RhoA/ROCK pathway and that inflammatory cytokines, such as TNF-α and IL-1β, enhance the expression and activity of RhoA [70]. In rat CAS models, NF-κB triggers the myosin light chain kinase/myosin light chain-2/endothelin-1 axis, further exacerbating VSMC contraction [26]. Taken together, these findings highlight miR-335-5p and miR-448 as regulators of Lp(a)/CD36-driven inflammation in CAS.
Amentoflavone, a biflavonoid with anti-inflammatory and antioxidant properties, is derived from traditional Chinese medicinal plants such as ginkgo biloba in the treatment of cardiovascular diseases [71]. It suppresses oxidized-LDL-induced lipid accumulation by downregulating the PPAR-γ/CD36 signaling axis [71]. Our findings revealed that amentoflavone mimicked shCD36 effects by attenuating Lp(a)-induced CD36 and RhoA-GTP expression, even in the presence of OE-CD36. It also epigenetically decreased CD36 expression and suppressed the downstream TNF-α/NF-κB/IL-6/RhoA-GTP/CD80 signaling via miR-335-5p and miR-448 in HCASMCs. These findings suggest that miRNA-based therapeutics hold great promise for treating inflammation-associated CAS.
Our study has some limitations. Firstly, the relatively small sample size (n = 77) may restrict causal inferences. Secondly, the use of certain medications, including beta blockers, statins, Ca2+-channel blockers, or nitrates, is known to affect IL-6 expression and/or activity [11]. Thirdly, the presence of confounders that could have affected the accurate measurement of patients’ cytokine or Lp(a) level is probable.
4. Materials and Methods
4.1. Cells, Compounds, and Reagents
The HCASMCs (ATCC® PCS-100-021™, American Type Culture Collection, Manassas, VA, USA) were cultured in smooth muscle cell growth medium 2 (#C-22062, PromoCell GmbH, Heidelberg, Germany), and all PMDMs were cultured in the RPMI-1640 culture media (Sigma-Aldrich Corporation, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 50 µg/ml streptomycin and 50 U/ml penicillin in 5% carbon dioxide humidified atmosphere incubator at 37°C to 98% - 100% confluence. Cells were sub-cultured and culture media changed every 48 h. Human IL-6 (Sigma-Aldrich, Cat #407652, ≥95% purity, Lot #SLCL0481), LDL (Sigma-Aldrich, Cat #LP2, ≥95% purity, Lot #SLBP8776V), and amentoflavone (Sigma-Aldrich, HPLC grade ≥98%, Cat #A9034, Lot #MKBT9052V) were sourced from Sigma-Aldrich. Amentoflavone was prepared in DMSO (Sigma-Aldrich, Cat #D2650, Lot #BCBL1917V) and diluted in medium to obtain the concentrations for assays. Methylergonovine (Methergin®, Novartis, Basel, Switzerland, Batch #MTG-2109) and nitroglycerin (Millisrol®, G. Pohl-Boskamp, Hohenlockstedt, Germany, Lot #NTG-3459) were obtained from the respective manufacturers.
4.2. Study Population
This prospective cohort study was approved by the Taipei Medical University Joint Institutional Review Board (approval number: TMU-JIRB 201503007). All patients provided signed informed consent regarding use of their blood in scientific research, and the study was compliant with the guidance of the Declaration of Helsinki for biomedical research involving human subjects. A total of 77 patients (35 men and 42 women), who had chest pain and suspected ischemic heart disease on noninvasive tests, undergoing diagnostic coronary angiography with or without established CAS, but without obstructive stenosis, from September 2019 to February 2021 were enrolled in this study. Study subjects were stratified into control (n = 36) and CAS (n = 41) groups. Among them, 4 from the control and 15 CAS subjects were active smokers. Inclusion criteria for patients with CAS included spontaneous chest pain at rest associated with ST-segment elevation or depression on electrocardiogram that was relieved by sublingual administration of nitroglycerin, no angiographic evidence of obstructive CAD after intracoronary nitroglycerin administration, and a positive result on intracoronary methylergonovine provocation testing. CAS was not induced in the remaining 36 patients (non-CAS, control), which consisted of patients who presented with atypical chest pain, no angiographic evidence of obstructive CAD, and negative results on intracoronary methylergonovine provocation testing (no CAS). Atypical chest pain was defined as spontaneous chest pain at rest and/or provoked by exertion that was eased by sublingual administration of nitroglycerin [72] but not linked with ST-segment change on resting electrocardiogram. Exclusion criteria included the presence of obstructive CAD, coronary microvascular spasm [73], inflammatory manifestations probably associated with noncardiac diseases (e.g., infections and autoimmune disorders), liver disease/renal failure (serum creatinine level >2.5 mg/dL), collagen disease, malignancy, and loss of blood samples. None of our patients had allergic or hypersensitivity conditions.
4.3. Patient Data Collection
Patients’ demographic, anthropometric, and laboratory data as well as details of their comorbidities, medicine use, habits, and number of functional units were collected. Current smoking was defined as having smoked a cigarette within 3 weeks of cardiac catheterization. Diabetes mellitus was diagnosed when the fasting glucose level was ≥126 mg/dL on >2 occasions or was defined from dietary treatment and/or medical therapy. Baseline seated blood pressure was derived from the mean of 6 readings obtained during the first 2 office visits at 2 weeks apart. Hypertension was defined as a blood pressure >140/90 mmHg on >2 occasions or receiving antihypertensive treatment.
4.4. Spasm Provocation Test Protocol
Coronary angiography was done using the Judkins method, with prior withdrawal of nitrates and calcium channel blockers for at least 24 hours [74]. Obstructive CAD was defined as a ≥50% decrease in luminal diameter after administration of intracoronary nitroglycerin [6]. If no obstructive CAD was discovered, intracoronary methylergonovine was given stepwise (1, 5, 10, 30 μg) first into the right coronary artery and subsequently into the left coronary artery. CAS was defined as a >70% reduction in luminal diameter compared with postintracoronary nitroglycerin, with associated angina and/or ST depression or elevation [6]. Provocation testing was stopped with an intracoronary injection of 50–200 μg of nitroglycerin.
4.5. Monocyte Isolation from Human Peripheral Blood
Following overnight fasting just before coronary angiography, blood was collected in BD Vacutainer® CPT™ mononuclear cell preparation tubes (#362753, BD Diagnostics, Sparks Glencoe, MD, USA) and centrifuged at 1800× g at room temperature for 20 min. After removing the upper layer containing plasma and Ficoll™Hypaque™, and without disturbance of the red lowest layer, the opaque interface containing the mononuclear cells was carefully transferred to a new 50 mL conical tube. The mononuclear cells were washed twice with phosphate buffered saline (PBS). Subsequently, monocytes were isolated using Invitrogen™ Dynabeads® CD14 superparamagnetic beads (#11149D, Thermo Fisher Scientific Inc., Waltham, MA, USA) and magnetic activated cell sorting. Isolated monocyte purity was assessed by flow-cytometry of fluorescin-labeled CD14 postive cells. Finally, isolated monocytes were re-suspended in Invitrogen™ TRIzol™ reagent (#15596026, Thermo Fisher Scientific Inc., Waltham, MA, USA), and the total RNA extract was stored at −80°C until use.
4.6. Monocyte Differentiation into Macrophages
Monocytes were allowed to adhere at 37°C and 5% CO2 for 2 hours. While nonadherent cells with the supernatant were carefully discarded, adherent monocytes were carefully washed with prewarmed 15mL phosphate buffered saline (PBS) and washing solution aspirated. Thereafter, the ImmunoCult™-SF macrophage medium (#10961, STEMCELL Technologies Inc., Kent, WA, USA) was used for monocyte differentiation to macrophages as previously described [11].
4.7. HCASMCs Culture
HCASMC cells was obtained from Lonza (Lonza Walkersville, Cat #CC-2583, Lot #HC2938) and cultured in DMEM (Gibco, Cat #11965092, Lot #DMEM2244) supplemented with 10% FBS (Gibco, Cat #16000044, Lot #2297076). Cells were kept at 37°C in 5% CO2, with medium changes every 3 days to ensure the cells remained healthy and active. For assays, cells in their 5th to 7th passages underwent 24-hour serum starvation with 0.5% FBS, followed by stimulation using 20 ng/ml Platelet-Derived Growth Factor-BB (BioVision, Cat #4577-50, Lot #PDGF1021) at 37°C for 24 hours, which readied the cells for the subsequent stages of our experimental analysis.
4.8. Lp(a) Assay
Native human Lp(a) was purchased from Cell Sciences (www.cellsciences.com; Newburyport, MA, USA; Cat. No. CRA169B). The supplier specifies a concentration of 1 mg mL−1 in PBS (no preservative), a buoyant density of 1.063–1.10 g mL−1, and ≥ 95 % purity confirmed by SDS-PAGE; electrophoretic profiling shows negligible contamination with other plasma lipoproteins, with only a minor Apo-A1 band visible. The molecular-weight distribution of the preparation is 280–700 kDa. and quantified using a Human Lp(a) ELISA kit (Abcam, Cat #ab212165, Lot #ELISA9083), with a detection limit of 17.2 ng/mL.
4.9. CD36 Expression Analysis in HCASMCs
To investigate the effects of Lp(a) on CD36-mediated signaling pathways, we employed both gene silencing and overexpression strategies. For knockdown, we utilized a commercially available short hairpin RNA targeting CD36 (Origene, Cat # TL314090). For overexpression, a human CD36 plasmid (NM_001001548) with a tagged open reading frame and its corresponding empty vector were obtained from Origene (Cat #RC221976). HCASMCs were transfected with either the shCD36 construct or the CD36 overexpression (OE-CD36) plasmid using the Neon™ electroporation system (Invitrogen Neon Transfection System, Cat. No. MPK5000), achieving an approximate transfection efficiency of 80%. Scrambled shRNA and empty vector plasmids served as negative controls in the knockdown and overexpression experiments, respectively. To establish a stable OE-CD36 cell line, transfected HCASMCs were subjected to selection with puromycin (5 μg/ml). The efficacy of CD36 knockdown and overexpression was confirmed by Western blot analysis.
4.10. RNA Processing and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) for CD36, RhoA and miRNA
Total RNA was extracted using a guanidinium isothiocyanate buffer followed by one step of phenol-chloroform-isoamyl alcohol extraction. For reverse transcription, 1 μg of this RNA was incubated with 200 U of reverse transcriptase in a 20 μL reaction buffer. This buffer included 0.25 μg of random primers and 0.8 mM dNTPs, and the incubation occurred at 42°C for 1 hour. The resulting first-strand cDNA served as the template for the PCR; thereafter, we used 20 ng aliquots of cDNA in each 20-μL reaction mix containing 10 μM primer, 2× Real Quantitative PCR Master Mix, and 6 μL DEPC H2O. The PCR was conducted using the SYBR Green method on a MxPro-Mx3000P Quantitative PCR machine (Stratagene, La Jolla, CA). Each sample underwent triplicate runs, with GAPDH as the reference gene. For miRNA quantification, total RNA from both cells and tissue samples was extracted using Trizol reagents (Invitrogen, Life Technologies). We then synthesized the first-strand cDNA using the PrimeScript RT Reagents Kit (Takara). The miRNA levels in the cells were quantified using qRT-PCR, with U6 miRNA as the normalization control. This qRT-PCR was performed using an SYBR Premix Ex Taq Kit (Takara) on a QIAGEN rotor real-time PCR machine (QIAGEN, Valencia, CA, USA).
4.11. Molecular Docking
The soluble ectodomain of CD36 (AlphaFold model AF-Q07954-F1, residues 30–439, Protein Data Bank, downloaded from https://alphafold.ebi.ac.uk/) and apolipoprotein(a) crystal structure Protein Data Bank file downloaded from https://www.rcsb.org/structure/1AV1 were trimmed, protonated at pH 7.4, and energy-minimised with AMBER ff19SB. Rigid-body sampling was carried out on the ClusPro 2.0 server (balanced energy set-up; ~70,000 fast Fourier transform rotations, 1 Å grid). The 1,000 best poses were clustered at 9 Å Cα Root Mean Square Deviation and ranked by cluster size; the 10 highest-scoring centers (docking scores −273.47 to −225.09 kcal mol−1; confidence 0.82–0.92) were forwarded to HADDOCK 2.4 (Guru mode, web server http://hdock.phys.hust.edu.cn). In HADDOCK, Lys70/Lys74 of apolipoprotein(a) and Asp238/Glu240/Glu242 of CD36 were defined as active residues; default passive residues were automatically assigned. Each run produced 2,000 rigid-body (it0), 200 semi-flexible (it1) and 20 water-refined models, followed by fraction of common contacts clustering at 1.0 Å. The top cluster (128 models) yielded an average HADDOCK score of −122 ± 8 kcal mol−1 and buried ~1,600 Å2 of interface surface, defining the final docked complex used for subsequent analyses.
4.12. Western Blot Analysis
Western blotting was performed according to standard protocol [75] using antibodies against CD36 (Cell Signaling Technology, Cat #74002, Lot #CD362031), IL-6 (Cell Signaling Technology, Cat #12153, Lot #IL61213), TNF-α (Cell Signaling Technology, Cat #3707, Lot #TNF7081), NF-κB (Cell Signaling Technology, Cat #8242, Lot #NFkB1093), CD80 (Abcam, Cat #ab134120, Lot #CD801209), GAPDH (Abcam, Cat #ab9484, Lot #GAP2203), and RhoA (Cell Signaling Technology, Cat #2117), RhoA-GTP (Cat #8820), ROCK1 (Cat #4035), ROCK2 (Cat #9029), t-MBS (Cat #2634), and p-MBS (Cat #3040). Protein bands were detected using the Amersham ECL system (GE Healthcare, NJ, USA) and quantified via ImageJ software (https://imagej.nih.gov/ij/).
4.13. Flow Cytometry Analysis
Approximately 2 million cells were fixed using 4% formaldehyde (Sigma-Aldrich, Cat #F8775, pH 7.5, Lot #FA9081) for 15 minutes at ambient temperature. Cells were then incubated in a blocking buffer containing 1% bovine serum albumin (Sigma-Aldrich, Cat #A7906, Lot #BSA2023) and 1% goat serum (Gibco, Cat #16210072, Lot #GS12345) in PBS for 30 minutes. Cells were then treated for 2 hours with primary antibodies targeting CD36 (Abcam, Cat #ab133625, Lot #CD36988) and RhoA (Abcam, Cat #ab54835, Lot #RHA6521), each diluted 1:100. After washing, cells were incubated for 1 hour in PBS containing fluorescein isothiocyanate-conjugated IgG secondary antibody (Sigma-Aldrich, Cat #F6257, Lot #FITC7742). Surface marker expression was quantified using a BD fluorescence-activated cell sortingCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
4.14. Statistical Analysis
All assays were performed at least 3 times in triplicates. Continuous variables are expressed as the mean ± standard deviation or median (2-quartile), and positively skewed variables were log-transformed for subsequent intergroup Student’s t-test. Discrete variables were expressed as numbers and percentages (%) of the total sample and comparisons made using the two-tailed Fisher’s exact test, while categorical variables were analyzed using the χ2 test. Correlation between the levels of serum Lp(a) and sCD36 expression in patients with CAS were determined by Spearman correlation. The coefficient of determination (r2) and associated p-value were calculated using linear regression analysis. All statistical analyses were performed with SPSS (IBM Corp. Released 2017. IBM SPSS Statistics for Windows, Version 25.0 Armonk, NY: IBM Corp.). A two-tailed p-value <0.05 was considered statistically significant.
5. Conclusions
In CAS, higher serum Lp(a) levels were positively correlated with the levels of sCD36, which was not observed in controls. Lp(a) activated macrophage M1 polarization and, increasedd RhoA and CD36 expression in PMDMs of CAS patients and HCASMCs, resulting in elevated sCD36 protein in PMDMs. In Lp(a)-treated HCASMCs, shCD36 significantly decreased both protein and mRNA expression levels of CD36, RhoA-GTP, RhoA, IL-6, TNF-α, NF-κB and CD80. In addition, shCD36 and amentoflavone significantly reduced the RhoA-GTP/RhoA ratio. Conversely, OE-CD36+Lp(a) induced a significantly higher RhoA-GTP/RhoA ratio than Lp(a) alone does. The addition of amentoflavone to OE-CD36 significantly downregulated the CD36 expression level to the extent similar to that with shCD36. Amentoflavone mediated Lp(a)/CD36 interaction in HCASMCs through epigenetic regulation of CD36, IL-6, TNF-α and NF-κB mRNA expression via miR-335-5p and miR-448. Our study provides a novel Lp(a)-triggered CD36-dependent TNF-α/NF-κB/IL-6/RhoA-GTP signaling pathway mediating HCASMC dysfunction in CAS (Graphical Abstract), which makes CD36 and its downstream effectors promising therapeutic targets.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Y.-K.L. and T.-H.H. contributed equally to this work. Conceptualization and design of the research: Y.-K.L., T.-H.H. and M.-Y.H.; acquisition of data: C.-T.Y., V.K.Y., I.-H.F. and K.-T.K.; analysis and interpretation of data: C.-T.Y., V.K.Y., I.-H.F.; statistical analysis: Y.-K.L; funding acquisition: C.-T.Y., and M.-Y.H.; drafting of the manuscript: Y.-K.L., T.-H.H. and C.-T.Y.; revision of the manuscript for important intellectual content: N.G.K., P.H. and M.-Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Council of Taiwan under grants awarded to Ming-Yow Hung (MOST 111-2314-B-038-028 and NSTC 112-2314-B-038-104-MY3). Additional funding support was provided by Taipei Medical University under project numbers 111TMU-SHH-01 and 112TMU-SHH-01.
Institutional Review Board Statement
The study was conducted in accordance with the guidelines of the Declaration of Helsinki, and the protocol was approved by the Joint Institutional Review Board of Taipei Medical University (Approval number: TMU-JIRB 201503007, approved on 8 July 2015).
Informed Consent Statement
Informed consent for participation was obtained from all subjects involved in the study.
Data Availability Statement
Data are contained within the article and the Supplementary Materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
We gratefully acknowledge the technical assistance provided by the staff of the Cancer Translational Research Laboratory and the Core Facility Center at Taipei Medical University – Shuang Ho Hospital, and the intellectual support from Taiwan Society of Coronary Artery Spasm. Their support with molecular docking, flow cytometry, molecular assays, and cell-based experiments was invaluable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Writing Committee Members; Gulati, M. ; Levy, P.D.; Mukherjee, D.; Amsterdam, E.; Bhatt, D.L.; Birtcher, K.K.; Blankstein, R.; Boyd, J.; Bullock-Palmer, R.P.; Conejo, T.; et al. 2021 AHA/ACC/ASE/CHEST/SAEM/SCCT/SCMR Guideline for the Evaluation and Diagnosis of Chest Pain: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Cardiovasc Comput Tomogr. 2022, 16, 54–122. [Google Scholar] [CrossRef]
- Hung, M.-J.; Hsu, K.-H.; Chang, N.-C.; Hung, M.-Y. Increased Numbers of Coronary Events in Winter and Spring Due to Coronary Artery Spasm: Effect of Age, Sex, Smoking, and Inflammation. J Am Coll Cardiol. 2015, 65, 2047–2048. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-J. , Kuo, L.-T.; Cheng, C.W.; Chang, C.-P; Cherng, W.J. Comparison of peripheral monocyte counts in patients with and without coronary spasm and without fixed coronary narrowing. Am J Cardiol. 2004, 93, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-J.; Hsu, K.-H.; Hu, W.-S.; Chang, N.-C.; Hung, M.-Y. C-reactive protein for predicting prognosis and its gender-specific associations with diabetes mellitus and hypertension in the development of coronary artery spasm. PLoS One. 2013, 8, e77655. [Google Scholar] [CrossRef]
- Hung, M.-J.; Cherng, W.-J.; Cheng, C.-W.; Li, L.-F. Comparison of serum levels of inflammatory markers in patients with coronary vasospasm without significant fixed coronary artery disease versus patients with stable angina pectoris and acute coronary syndromes with significant fixed coronary artery disease. Am J Cardiol. 2006, 97, 1429–1434. [Google Scholar] [CrossRef]
- Hung, M.-Y.; Wu, Y.H.; Bamodu OA, Chen X, Lin YK, Hu P, Chang NC, Pang JS, Yeh CT. Activation of the monocytic α7 nicotinic acetylcholine receptor modulates oxidative stress and inflammation-associated development of coronary artery spasm via a p38 MAP-kinase signaling-dependent pathway. Free Radic Biol Med. 2018, 120, 266–276. [Google Scholar] [CrossRef]
- Kugiyama, K.; Yasue, H.; Okumura, K.; Ogawa, H.; Fujimoto, K.; Nakao, K.; Yoshimura, M.; Motoyama, T; Inobe, Y. ; Kawano, H. Nitric oxide activity is deficient in spasm arteries of patients with coronary spastic angina. Circulation. 1996, 94, 266–271. [Google Scholar] [CrossRef]
- Miwa, K.; Nakagawa, K.; Yoshida, N.; Taguchi, Y.; Inoue, H. Lipoprotein(a) is a risk factor for occurrence of acute myocardial infarction in patients with coronary vasospasm. J Am Coll Cardiol. 2000, 35, 1200–1205. [Google Scholar] [CrossRef]
- Hung, M.-Y.; Witztum, J.L.; Tsimikas, S. New therapeutic targets for calcific aortic valve stenosis: the lipoprotein(a)-lipoprotein-associated phospholipase A2-oxidized phospholipid axis. J Am Coll Cardiol. 2014, 63, 478–480. [Google Scholar] [CrossRef]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.R.; Shin, M.J.; Binder, C.J.; Hörkkö, S.; Krauss, R.M.; Chapman, M.J.; et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008, 49, 2230–2239. [Google Scholar]
- Lin, Y.-K.; Yeh, C.-T.; Kuo, K.-T.; Fong, I.-H.; Yadav, V.-K.; Kounis, N.G.; Hu, P.; Hung, M.-Y. Apolipoprotein (a)/Lipoprotein(a)-Induced Oxidative-Inflammatory α7-nAChR/p38 MAPK/IL-6/RhoA-GTP Signaling Axis and M1 Macrophage Polarization Modulate Inflammation-Associated Development of Coronary Artery Spasm. Oxid Med Cell Longev. 2022, 2022, 9964689. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-J.; Cherng, W.-J.; Hung, M.-Y.; Kuo, L.-T.; Cheng, C.-W.; Wang, C.-H.; Yang, N.-I.; Liao, J.K. Increased leukocyte Rho-associated coiled-coil containing protein kinase activity predicts the presence and severity of coronary vasospastic angina. Atherosclerosis. 2012, 221, 521–526. [Google Scholar] [CrossRef]
- Puchałowicz, K.; Rać, M.E. The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications-Update in Pathogenesis, Treatment and Monitoring. Cells. 2020, 9, 1877. [Google Scholar] [CrossRef]
- Kaewmalee, J.; Ontawong, A.; Duangjai, A.; Tansakul, C.; Rukachaisirikul, V.; Muanprasat, C.; Srimaroeng, C. High-Efficacy α,β-Dehydromonacolin S Improves Hepatic Steatosis and Suppresses Gluconeogenesis Pathway in High-Fat Diet-Induced Obese Rats. Pharmaceuticals (Basel). 2021, 14, 375. [Google Scholar] [CrossRef]
- Parra-Reyna, B.; Padilla-Gutiérrez, J.R.; Aceves-Ramírez, M.; García-Garduño, T.C.; Martínez-Fernández, D.E.; Jacobo-García, J.J.; Valdés-Alvarado, E.; Valle, Y. Genetic variants, gene expression, and soluble CD36 analysis in acute coronary syndrome: Differential protein concentration between ST-segment elevation myocardial infarction and unstable angina. J Clin Lab Anal. 2022, 36, e24529. [Google Scholar] [CrossRef] [PubMed]
- Bodart, V.; Febbraio, M.; Demers, A.; McNicoll, N.; Pohankova, P.; Perreault, A.; Sejlitz, T.; Escher, E.; Silverstein, R.L.; Lamontagne, D.; et al. CD36 mediates the cardiovascular action of growth hormone-releasing peptides in the heart. Circ Res. 2002, 90, 844–849. [Google Scholar] [CrossRef]
- Hung, M.-Y.; Kounis, N.G.; Lu, M.-Y.; Hu, P. Myocardial Ischemic Syndromes, Heart Failure Syndromes, Electrocardiographic Abnormalities, Arrhythmic Syndromes and Angiographic Diagnosis of Coronary Artery Spasm: Literature Review. Int J Med Sci. 2020, 17, 1071–1082. [Google Scholar] [CrossRef]
- Thorand, B.; Löwel, H.; Schneider, A.; Kolb, H.; Meisinger, C.; Fröhlich, M.; Koenig, W. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: results from the MONICA Augsburg cohort study, 1984-1998. Arch Intern Med. 2003, 163, 93–99. [Google Scholar] [CrossRef]
- Mansor, L.S.; Sousa Fialho, M.D.L.; Yea, G.; Coumans, W.A.; West, J.A.; Kerr, M.; Carr, C.A.; Luiken, J.J.F.P.; Glatz, J.F.C.; Evans, R.D.; et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017, 113, 737–748. [Google Scholar] [CrossRef]
- Glatz, J.F.; Angin, Y.; Steinbusch, L.K.; Schwenk, R.W.; Luiken, J.J. CD36 as a target to prevent cardiac lipotoxicity and insulin resistance. Prostaglandins Leukot Essent Fatty Acids. 2013, 88, 71–77. [Google Scholar] [CrossRef]
- Kang, K.W.; Choi, B.G.; Rha, S.W. Impact of Insulin Resistance on Acetylcholine-Induced Coronary Artery Spasm in Non-Diabetic Patients. Yonsei Med J. 2018, 59, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-J.; Chang, N.-C.; Hu, P.; Chen, T.-H.; Mao, C.-T.; Yeh, C.-T.; Hung, M.-Y. Association between Coronary Artery Spasm and the risk of incident Diabetes: A Nationwide population-based Cohort Study. Int J Med Sci. 2021, 18, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Handberg, A.; Lopez-Bermejo, A.; Bassols, J.; Vendrell, J.; Ricart, W.; Fernandez-Real, J.M. Circulating soluble CD36 is associated with glucose metabolism and interleukin-6 in glucose-intolerant men. Diab Vasc Dis Res. 2009, 6, 15–20. [Google Scholar] [CrossRef]
- Kunz, A.; Abe, T.; Hochrainer, K.; Shimamura, M.; Anrather, J.; Racchumi, G.; Zhou, P.; Iadecola, C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008, 28, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Tergaonkar, V. Rho protein GTPases and their interactions with NFκB: crossroads of inflaowiojldflmmation and matrix biology. Biosci Rep. 2014, 34, e00115. [Google Scholar] [CrossRef]
- Wu, B.-W.; Wu, M.-S.; Liu, Y.; Lu, M.; Guo, J.-D.; Meng, Y.-H.; Zhou, Y.-H. SIRT1-mediated deacetylation of NF-κB inhibits the MLCK/MLC2 pathway and the expression of ET-1, thus alleviating the development of coronary artery spasm. Am J Physiol Heart Circ Physiol. 2021, 320, H458–H468. [Google Scholar] [CrossRef]
- Netea, M.G.; de Bont, N.; Demacker, P.N.; Kullberg, B.J.; Jacobs, L.E.; Verver-Jansen, T.J.; Stalenhoef, A.F.; Van der Meer, J.W. Lipoprotein(a) inhibits lipopolysaccharide-induced tumor necrosis factor alpha production by human mononuclear cells. Infect Immun. 1998, 66, 2365–2367. [Google Scholar] [CrossRef]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med. 2012, 52, 556–592. [Google Scholar] [CrossRef]
- Li, C.; Ma, Q.; Toan, S.; Wang, J.; Zhou, H.; Liang, J. SERCA overexpression reduces reperfusion-mediated cardiac microvascular damage through inhibition of the calcium/MCU/mPTP/necroptosis signaling pathways. Redox Biol. 2020, 36, 101659. [Google Scholar] [CrossRef]
- Mathis, D.; Shoelson, S.E. Immunometabolism: an emerging frontier. Nat Rev Immunol. 2011, 11, 81. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J Exp Med. 2022, 219, e20211314. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.Y.; Pearce, S.F.; Yesner, L.M.; Schindler, J.L.; Silverstein, R.L. Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood. 1996, 87, 2020–2028. [Google Scholar] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998, 93, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Yesner, L.M.; Huh, H.Y.; Pearce, S.F.; Silverstein, R.L. Regulation of monocyte CD36 and thrombospondin-1 expression by soluble mediators. Arterioscler Thromb Vasc Biol. 1996, 16, 1019–1025. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis. 2014, 234, 95–101. [Google Scholar] [CrossRef]
- Missala, I.; Kassner, U.; Steinhagen-Thiessen, E. A Systematic Literature Review of the Association of Lipoprotein(a) and Autoimmune Diseases and Atherosclerosis. Int J Rheumatol. 2012, 2012, 480784. [Google Scholar] [CrossRef]
- Koutroubakis, I.E. , Malliaraki, N.; Vardas, E.; Ganotakis, E.; Margioris, A.N.; Manousos, O.N.; Kouroumalis, E.A. Increased levels of lipoprotein (a) in Crohn’s disease: a relation to thrombosis? Eur J Gastroenterol Hepatol. 2001, 13, 1415–1419. [Google Scholar] [CrossRef]
- Zimmermann, J.; Herrlinger, S.; Pruy, A.; Metzger, T.; Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999, 55, 648–658. [Google Scholar] [CrossRef]
- Maeda, S.; Abe, A.; Seishima, M.; Makino, K.; Noma, A.; Kawade, M. Transient changes of serum lipoprotein(a) as an acute phase protein. Atherosclerosis. 1989, 78, 145–150. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Patthy, L.; Trexler, M.; Váli, Z.; Bányai, L.; Váradi, A. Kringles: modules specialized for protein binding. Homology of the gelatin-binding region of fibronectin with the kringle structures of proteases. FEBS Lett. 1984, 171, 131–136. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P.A.; Schneider, W.J. Lipoprotein(a) catabolism: a case of multiple receptors. Pathology. 2019, 51, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Holm, S.; Oma, I.; Hagve, T.A.; Saatvedt, K.; Brosstad, F.; Mikkelsen, K.; Rydningen, H.; Risnes, I.; Almdahl, S.M.; Ueland, T.; et al. Levels of Lipoprotein (a) in patients with coronary artery disease with and without inflammatory rheumatic disease: a cross-sectional study. BMJ Open. 2019, 9, e030651. [Google Scholar] [CrossRef]
- Berthold, H.K.; Laudes, M.; Krone, W.; Gouni-Berthold, I. Association between the interleukin-6 promoter polymorphism -174G/C and serum lipoprotein(a) concentrations in humans. PLoS One. 2011, 6, e24719. [Google Scholar] [CrossRef]
- Müller, N.; Schulte, D.M.; Türk, K.; Freitag-Wolf, S.; Hampe, J.; Zeuner, R.; Schröder, J.O.; Gouni-Berthold, I.; Berthold, H.K.; Krone, W.; et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. 2015, 56, 1034–1042. [Google Scholar] [CrossRef]
- Sun, Y.; Scavini, M.; Orlando, R.A.; Murata, G.H.; Servilla, K.S.; Tzamaloukas, A.H.; Schrader, R.; Bedrick, E.J.; Burge, M.R.; Abumrad, N.A.; et al. Increased CD36 expression signals monocyte activation among patients with type 2 diabetes. Diabetes Care. 2010, 33, 2065–2067. [Google Scholar] [CrossRef]
- Yue, H.; Febbraio, M.; Klenotic, P.A.; Kennedy, D.J.; Wu, Y.; Chen, S.; Gohara, A.F.; Li, O.; Belcher, A.; Kuang, B.; et al. CD36 Enhances Vascular Smooth Muscle Cell Proliferation and Development of Neointimal Hyperplasia. Arterioscler Thromb Vasc Biol. 2019, 39, 263–275. [Google Scholar] [CrossRef]
- Yuasa-Kawase, M.; Masuda, D.; Yamashita, T.; Kawase, R.; Nakaoka, H.; Inagaki, M.; Nakatani, K.; Tsubakio-Yamamoto, K.; Ohama, T.; Matsuyama, A.; et al. Patients with CD36 deficiency are associated with enhanced atherosclerotic cardiovascular diseases. J Atheroscler Thromb. 2012, 19, 263–275. [Google Scholar] [CrossRef]
- Kilian, L.S.; Voran, J.; Frank, D.; Rangrez, A.Y. RhoA: a dubious molecule in cardiac pathophysiology. J Biomed Sci. 2021, 28, 33. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Blétière, D.; Sauzeau, V.; Bourreille, A.; Hilaret, G.; Cario-Toumaniantz, C.; Pacaud, P.; Galmiche, J.P.; Loirand, G. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: evidence in Crohn’s disease and experimental colitis. Gastroenterology. 2003, 124, 1180–1187. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ Res. 2016, 118, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Lochmüller, H.; Melms, A.; Weller, M. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: a novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J. 2003, 17, 1892–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Manes, T.D.; Pober, J.S.; Tellides, G. Human vascular smooth muscle cells lack essential costimulatory molecules to activate allogeneic memory T cells. Arterioscler Thromb Vasc Biol. 2010, 30, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.J.; Martinez, F.O.; Smelt, M.J.; Sironi, M.; Locati, M.; Mantovani, A.; Sozzani, S. Transcriptional profiling reveals complex regulation of the monocyte IL-1 beta system by IL-13. J Immunol. 2005, 174, 834–845. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Voss, K.; Hong, H.S.; Bader, J.E.; Sugiura, A.; Lyssiotis, C.A.; Rathmell, J.C. A guide to interrogating immunometabolism. Nat Rev Immunol. 2021, 21, 637–652. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation. 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Aicher, A.; Heeschen, C.; Mohaupt, M.; Cooke, J.P.; Zeiher, A.M.; Dimmeler, S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003, 107, 604–611. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity. 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Lai, C.-Y.; Yeh, D.-W.; Liu, Y.-L.; Su, Y.-W.; Hsu, L.-C.; Chang, C.-H.; Catherine Jin, S.-L.; Chuang, T.-H. Involvement of M1 Macrophage Polarization in Endosomal Toll-Like Receptors Activated Psoriatic Inflammation. Mediators Inflamm. 2018, 2018, 3523642. [Google Scholar] [CrossRef] [PubMed]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J Med Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Park, C.S.; Kim, I.; Oh, G.C.; Han, J.K.; Yang, H.M.; Park, K.W.; Cho, H.J.; Kang, H.J.; Koo, B.K.; Chung, W.Y.; et al. Diagnostic Utility and Pathogenic Role of Circulating MicroRNAs in Vasospastic Angina. J Clin Med. 2020, 9, 1313. [Google Scholar] [CrossRef]
- Zhang, J.W.; Pan, H.T. microRNA profiles of serum exosomes derived from children with nonalcoholic fatty liver. Genes Genomics. 2022, 44, 879–888. [Google Scholar] [CrossRef]
- Morita, S.; Mizuno, Y.; Harada, E.; Kashiwagi, Y.; Yoshimura, M.; Murohara, T.; Yasue, H. Pioglitazone, a peroxisome proliferator-activated receptor γ activator, suppresses coronary spasm. Coron Artery Dis. 2014, 25, 671–677. [Google Scholar] [CrossRef]
- Zeng, H.; Qin, H.; Liao, M.; Zheng, E.; Luo, X.; Xiao, A.; Li, Y.; Chen, L.; Wei, L.; Zhao, L.; et al. CD36 promotes de novo lipogenesis in hepatocytes through INSIG2-dependent SREBP1 processing. Mol Metab. 2022, 57, 101428. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, G.; Lou, Z. Role of the Sterol Regulatory Element Binding Protein Pathway in Tumorigenesis. Front Oncol. 2020, 10, 1788. [Google Scholar] [CrossRef]
- Zhang, R.; Sui, L.; Hong, X.; Yang, M.; Li, W. MiR-448 promotes vascular smooth muscle cell proliferation and migration in through directly targeting MEF2C. Environ Sci Pollut Res Int. 2017, 24, 22294–22300. [Google Scholar] [CrossRef]
- Sakai, H.; Otogoto, S.; Chiba, Y.; Abe, K.; Misawa, M. Involvement of p42/44 MAPK and RhoA protein in augmentation of ACh-induced bronchial smooth muscle contraction by TNF-alpha in rats. J Appl Physiol (1985). 2004, 97, 2154–2159. [Google Scholar] [CrossRef]
- Zhuang, J.L.; Liu, Y.Y.; Li, Z.Z.; Zhuang, Q.Z.; Tang, W.Z.; Xiong, Y.; Huang, X.Z. Amentoflavone prevents ox-LDL-induced lipid accumulation by suppressing the PPARγ/CD36 signal pathway. Toxicol Appl Pharmacol. 2021, 431, 115733. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; Chatterjee, K.; Daley, J.; Douglas, J.S.; Fihn, S.D.; Gardin, J.M.; Grunwald, M.A.; Levy, D.; Lytle, B.W.; O’Rourke, R.A.; et al. ACC/AHA/ACP-ASIM guidelines for the management of patients with chronic stable angina: executive summary and recommendations. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients with Chronic Stable Angina). Circulation. 1999, 99, 2829–2848. [Google Scholar] [CrossRef] [PubMed]
- JCS Joint Working Group. Guidelines for diagnosis and treatment of patients with vasospastic angina (coronary spastic angina) (JCS 2008): digest version. Circ J. 2010, 74, 1745–1762. [Google Scholar] [CrossRef]
- Fichtlscherer, S.; Breuer, S.; Schächinger, V.; Dimmeler, S.; Zeiher, A.M. C-reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. Eur Heart J. 2004, 25, 1412–1418. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Mahmood, T.; Yang, P.C. Western blot: technique, theory and trouble shooting. N Am J Med Sci. 2014, 6, 160. [Google Scholar] [CrossRef]
Figure 1.
Lipoprotein(a) (Lp(a) upregulated CD36 and RhoA expression, and were positively correlated with soluble cluster of differentiation 36 (sCD36) levels in coronary artery spasm (CAS). (A) RNA sequencing after isolation and treatment of CAS patient monocyte-derived macrophages (PMDMs) and human coronary artery smooth muscle cell (HCASMC) line. (B) Heatmap of differentially expressed genes (DEGs) from CAS PMDMs and HCASMCs. The colored codes from blue to yellow indicated expression levels from low (in blue) to high (in yellow). (C) Volcano plot of the expression levels of the PMDMs and HCASMCs compared with that of the control groups. The blue and red dots indicated the lowest level and the highest level of expression, respectively. (D) Up-regulated CD36 and RAS Homolog Family Member A (RhoA)-GTP/Rho-kinase (ROCK) DEGs (at the center) from PMDMs and HCASMCs were identified in Venn Diagram. (E) Lp(a) levels were positively correlated with sCD36 in CAS patients but not in non-CAS controls.
Figure 1.
Lipoprotein(a) (Lp(a) upregulated CD36 and RhoA expression, and were positively correlated with soluble cluster of differentiation 36 (sCD36) levels in coronary artery spasm (CAS). (A) RNA sequencing after isolation and treatment of CAS patient monocyte-derived macrophages (PMDMs) and human coronary artery smooth muscle cell (HCASMC) line. (B) Heatmap of differentially expressed genes (DEGs) from CAS PMDMs and HCASMCs. The colored codes from blue to yellow indicated expression levels from low (in blue) to high (in yellow). (C) Volcano plot of the expression levels of the PMDMs and HCASMCs compared with that of the control groups. The blue and red dots indicated the lowest level and the highest level of expression, respectively. (D) Up-regulated CD36 and RAS Homolog Family Member A (RhoA)-GTP/Rho-kinase (ROCK) DEGs (at the center) from PMDMs and HCASMCs were identified in Venn Diagram. (E) Lp(a) levels were positively correlated with sCD36 in CAS patients but not in non-CAS controls.
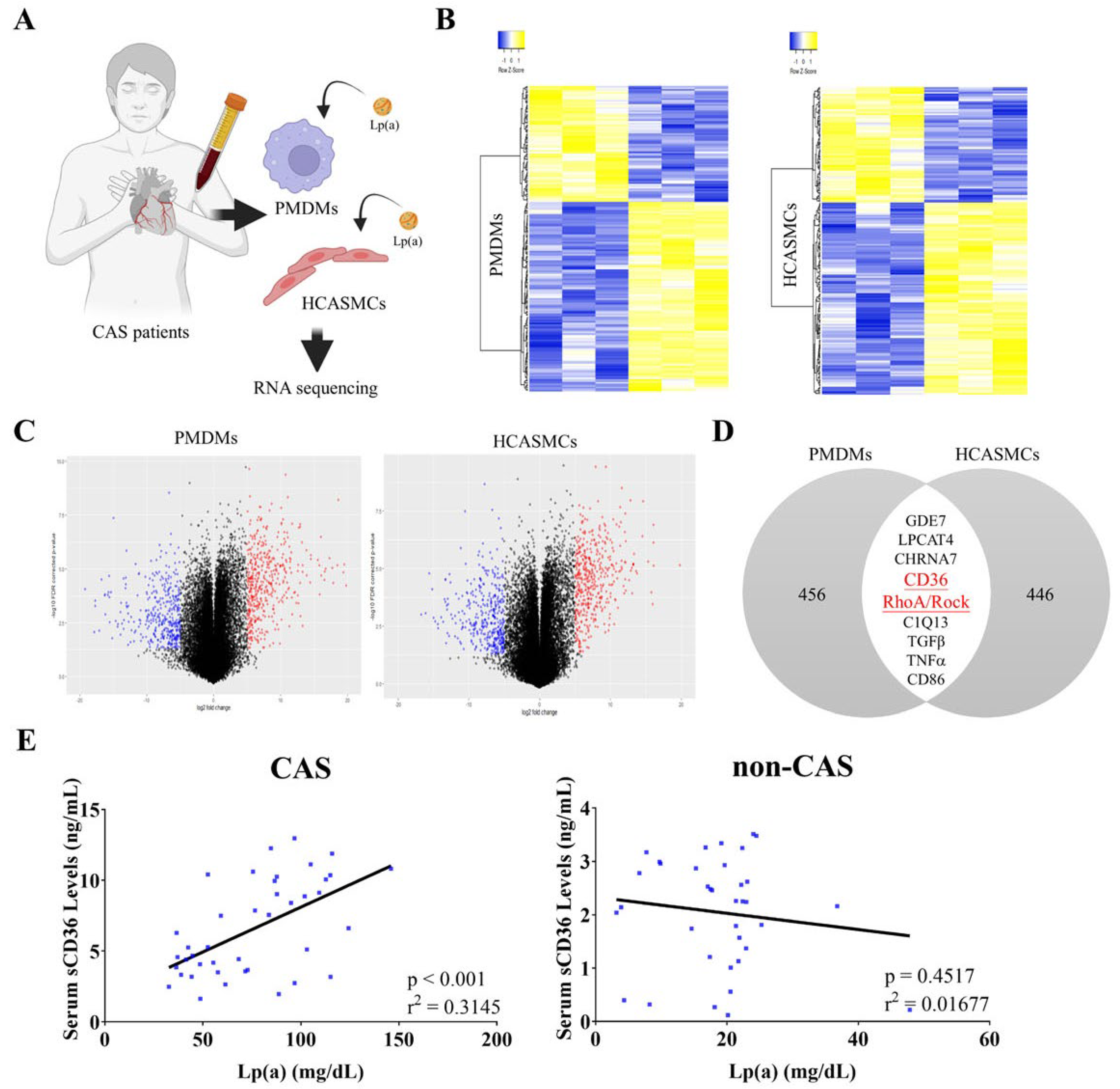
Figure 2.
Lp(a)-induced CD36 and RhoA expression in CAS PMDMs and HCASMCs, and M1 polarization. (A) Graphical representation of the effect of 500 nM low-density lipoprotein (LDL) or Lp(a) on the relative expression of RhoA or CD36 in the CAS PMDMs. (B, C) Histograms showing the dose-dependent effect of 100 nM-1000 nM Lp(a) on the relative expression of CD36/RhoA mRNA or relative luciferase reporter activity of sCD36/RhoA in the CAS PMDMs, while (D) 0.5 nM–2 nM Lp(a) were investigated for the dose-dependent relative expression of CD36/RhoA mRNA in HCASMCs. (E) Flow cytometry cell count histograms depicting the effect of 500 nM LDL or Lp(a) treatment for 24 hours on the CD80 or CD206 median fluorescence intensity of CAS PMDMs, in which colors are blue for control antibody and red for target antibody. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 2.
Lp(a)-induced CD36 and RhoA expression in CAS PMDMs and HCASMCs, and M1 polarization. (A) Graphical representation of the effect of 500 nM low-density lipoprotein (LDL) or Lp(a) on the relative expression of RhoA or CD36 in the CAS PMDMs. (B, C) Histograms showing the dose-dependent effect of 100 nM-1000 nM Lp(a) on the relative expression of CD36/RhoA mRNA or relative luciferase reporter activity of sCD36/RhoA in the CAS PMDMs, while (D) 0.5 nM–2 nM Lp(a) were investigated for the dose-dependent relative expression of CD36/RhoA mRNA in HCASMCs. (E) Flow cytometry cell count histograms depicting the effect of 500 nM LDL or Lp(a) treatment for 24 hours on the CD80 or CD206 median fluorescence intensity of CAS PMDMs, in which colors are blue for control antibody and red for target antibody. *p < 0.05, **p < 0.01, ***p < 0.001.
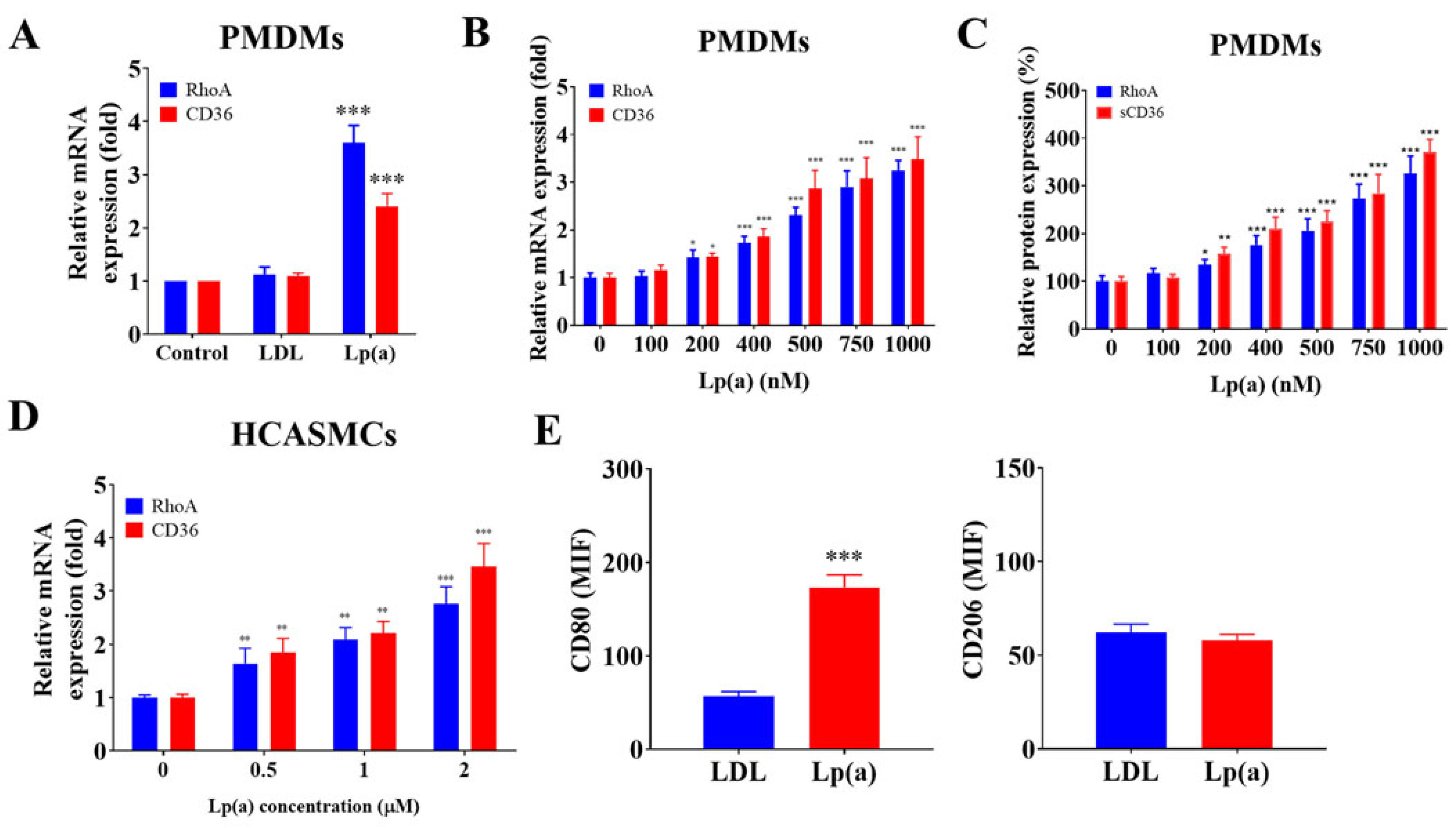
Figure 3.
Docking and interface analysis of apolipoprotein(a) on Lp(a) with the sCD36 ectodomain. (A) Best-ranked docked complex: rainbow-coloured apolipoprotein(a) bond the Asp238–Glu242 acidic patch of sCD36 (orange), blocking the lipid tunnel. (B) Score table for the 10 top models showed a leading docking score of –273.47 kcal mol−1 with all poses clustering within ≤1.7 Å ligand Root Mean Square Deviation. (C) Interface map highlighted the dominant Lys70/Lys74 ↔ Asp238/Glu240 salt bridges that underlay the favorable –122 kcal mol−1 HADDOCK score and the 2.6 Å backbone Root Mean Square Deviation maintained during 100-ns molecular dynamics.
Figure 3.
Docking and interface analysis of apolipoprotein(a) on Lp(a) with the sCD36 ectodomain. (A) Best-ranked docked complex: rainbow-coloured apolipoprotein(a) bond the Asp238–Glu242 acidic patch of sCD36 (orange), blocking the lipid tunnel. (B) Score table for the 10 top models showed a leading docking score of –273.47 kcal mol−1 with all poses clustering within ≤1.7 Å ligand Root Mean Square Deviation. (C) Interface map highlighted the dominant Lys70/Lys74 ↔ Asp238/Glu240 salt bridges that underlay the favorable –122 kcal mol−1 HADDOCK score and the 2.6 Å backbone Root Mean Square Deviation maintained during 100-ns molecular dynamics.
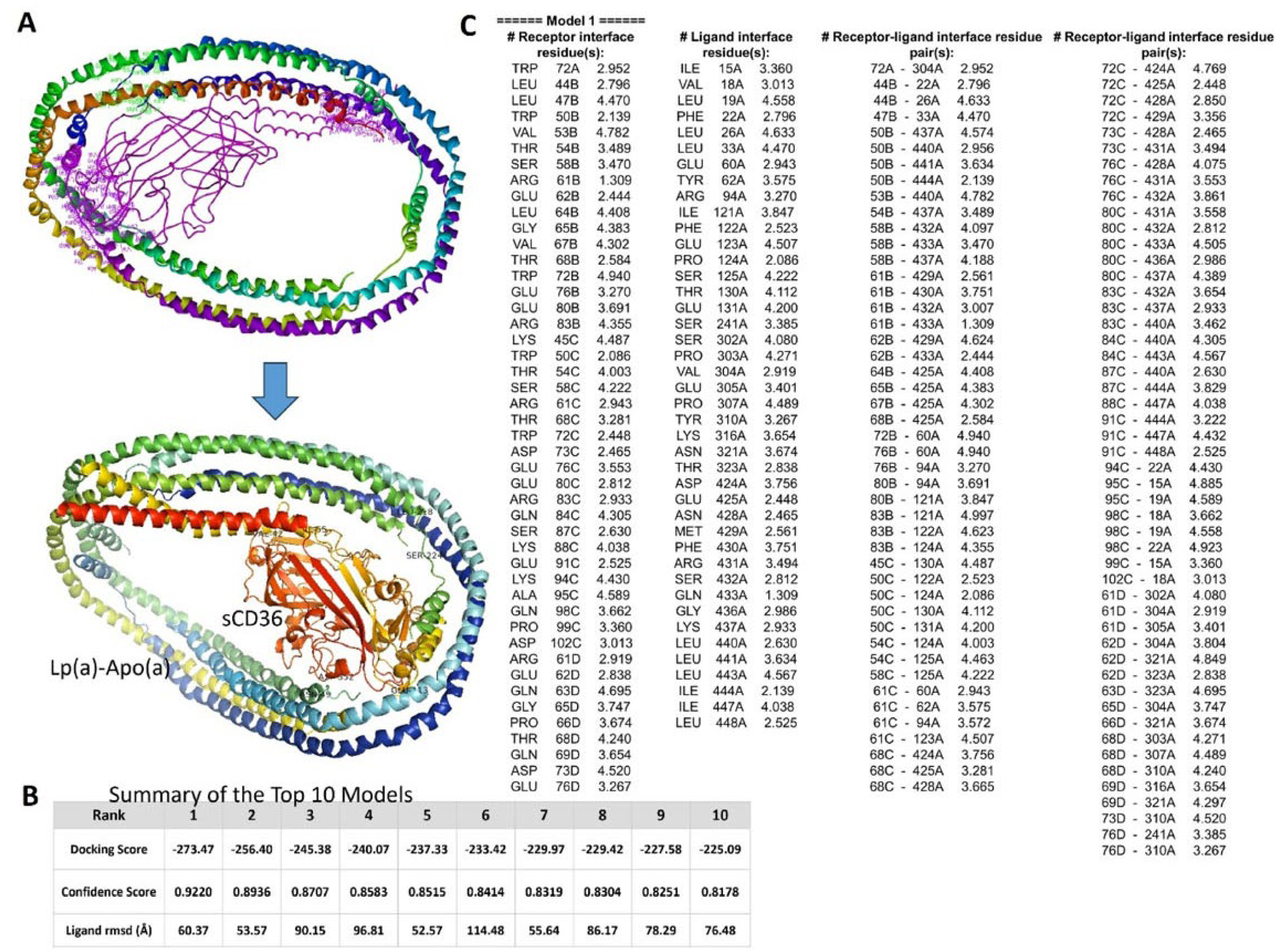
Figure 4.
The impact of short hairpin RNA for CD36 (shCD36), CD36 overexpression (OE-CD36) vector and amentoflavone on Lp(a)-induced cellular signaling pathways in HCASMCs. (A, B) Immunoblotting and quantitative reverse transcription polymerase chain reaction investigating the effects of Lp(a) alone or with shCD36 knockdown demonstrated that reduction in CD36 mRNA and protein expression levels could significantly downregulate tumor necrosis factor(TNF)-α/nuclear factor kappa B(NF-κB)/interleukin(IL)-6/RhoA-GTP signaling pathways. (C) In Lp(a)-treated HCASMCs, shCD36 and amentoflavone, in contrast with control, significantly reduced the RhoA-GTP/RhoA ratio using a pull-down assay with the glutathione-S-transferase-Rhotekin-Rho-binding domain. (D) While HCASMCs transfected with OE-CD36 vector showed a significantly higher RhoA-GTP/RhoA ratio than CD36 knockdown cells and the vehicle vector control, addition of amentoflavone to OE-CD36 medium significantly decreased the CD36 expression level similar to that of shCD36. (E) Representative western blot photo images showing that in 1 μM Lp(a)-treated HCASMCs, addition of shCD36, OE-CD36, or OE-CD36+amentoflavone for 60 min, compared with controls, significantly decreased, increased or decreased ROCK activity, respectively. The ROCK activity was expressed as a relative blot density ratio, which was the expression ratio of phosphorylation level of myosin-binding subunit of myosin light chain phosphatase (pMBS) sample density/total MBS (tMBS) sample density. *p < 0.05, **p < 0.01, ***p < 0.001; GAPDH served as loading control.
Figure 4.
The impact of short hairpin RNA for CD36 (shCD36), CD36 overexpression (OE-CD36) vector and amentoflavone on Lp(a)-induced cellular signaling pathways in HCASMCs. (A, B) Immunoblotting and quantitative reverse transcription polymerase chain reaction investigating the effects of Lp(a) alone or with shCD36 knockdown demonstrated that reduction in CD36 mRNA and protein expression levels could significantly downregulate tumor necrosis factor(TNF)-α/nuclear factor kappa B(NF-κB)/interleukin(IL)-6/RhoA-GTP signaling pathways. (C) In Lp(a)-treated HCASMCs, shCD36 and amentoflavone, in contrast with control, significantly reduced the RhoA-GTP/RhoA ratio using a pull-down assay with the glutathione-S-transferase-Rhotekin-Rho-binding domain. (D) While HCASMCs transfected with OE-CD36 vector showed a significantly higher RhoA-GTP/RhoA ratio than CD36 knockdown cells and the vehicle vector control, addition of amentoflavone to OE-CD36 medium significantly decreased the CD36 expression level similar to that of shCD36. (E) Representative western blot photo images showing that in 1 μM Lp(a)-treated HCASMCs, addition of shCD36, OE-CD36, or OE-CD36+amentoflavone for 60 min, compared with controls, significantly decreased, increased or decreased ROCK activity, respectively. The ROCK activity was expressed as a relative blot density ratio, which was the expression ratio of phosphorylation level of myosin-binding subunit of myosin light chain phosphatase (pMBS) sample density/total MBS (tMBS) sample density. *p < 0.05, **p < 0.01, ***p < 0.001; GAPDH served as loading control.
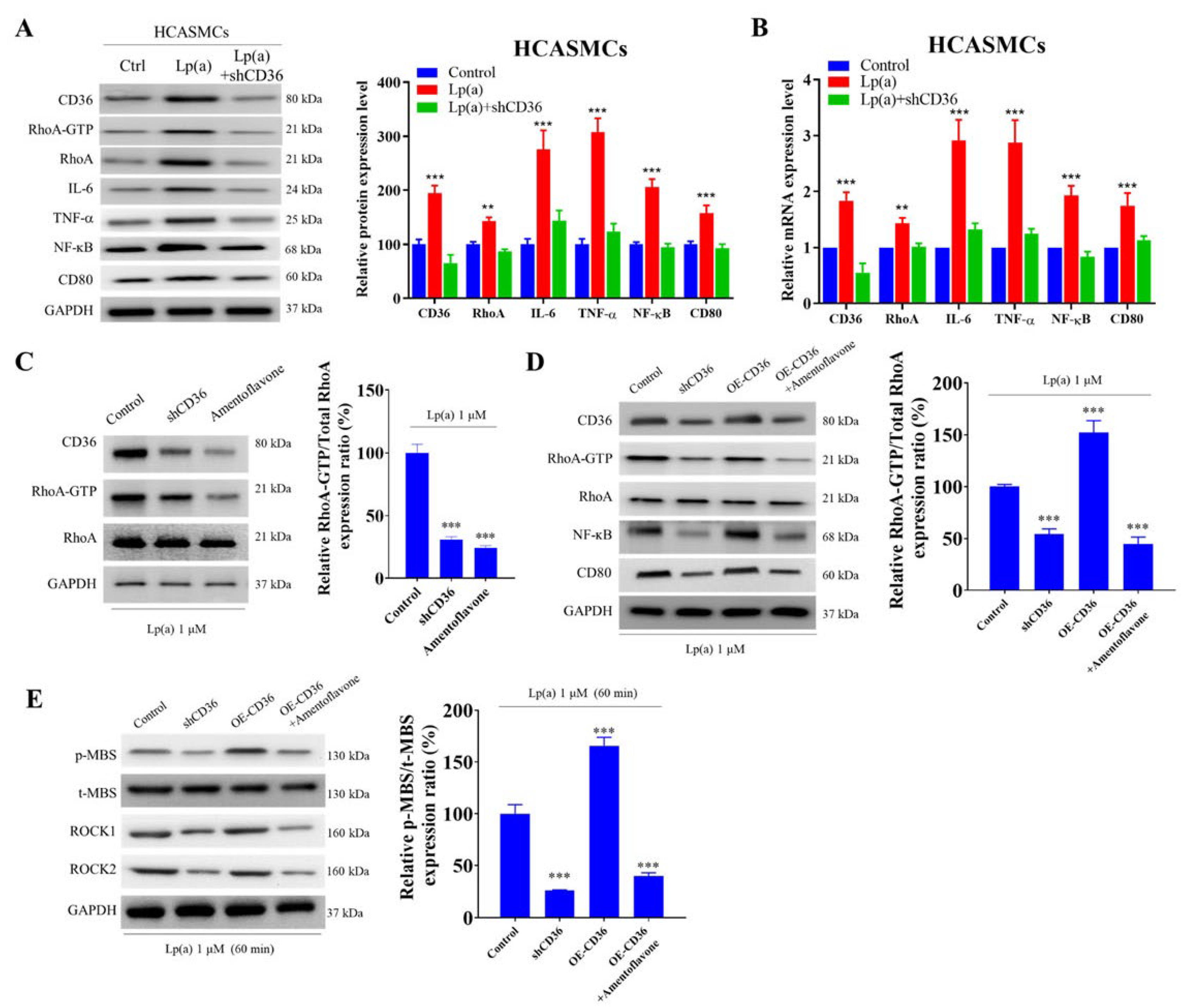
Figure 5.
Amentoflavone exerted epigenetic regulation of CD36, IL-6, TNF-α and NF-κB mRNA expression through miR-335-5p and miR-448, which mediated the Lp(a)/CD36 interaction in HCASMCs. (A) Amentoflavone has a molecular formula of C30H18O10 and molar mass 538.45 g/mol. (B) Graphical representation of the effect of 12.5 μM–100 μM amentoflavone on the viability of HCASMCs or CAS PMDMs. (C) Exposure to 10μM amentoflavone significantly reversed the 1 μM Lp(a)-induced downregulation of hsa-miR-335-5p and hsa-miR-448 expression levels in HCASMCs, which were also significantly higher than non-Lp(a)-treated controls. (D) In HCASMCs, dual luciferase reporter gene assay demonstrated that the targeting sequence for miR-335-5p and miR-448 was identified in the 3′-UTR of CD36 mRNA using constructs containing the predicted targeting sequence (CD36 3’UTR) and mutated targeting sequence (CD36 3’UTR mut) cloned into the 3′-UTR of the reporter gene. (E) In HCASMCs, the miR-335-5p’s and miR-448’s inhibitors largely increased, and mimics decreased, the 1 μM Lp(a)-induced target mRNA translation, respectively, such as CD36, IL-6, TNF-α and NF-κB. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 5.
Amentoflavone exerted epigenetic regulation of CD36, IL-6, TNF-α and NF-κB mRNA expression through miR-335-5p and miR-448, which mediated the Lp(a)/CD36 interaction in HCASMCs. (A) Amentoflavone has a molecular formula of C30H18O10 and molar mass 538.45 g/mol. (B) Graphical representation of the effect of 12.5 μM–100 μM amentoflavone on the viability of HCASMCs or CAS PMDMs. (C) Exposure to 10μM amentoflavone significantly reversed the 1 μM Lp(a)-induced downregulation of hsa-miR-335-5p and hsa-miR-448 expression levels in HCASMCs, which were also significantly higher than non-Lp(a)-treated controls. (D) In HCASMCs, dual luciferase reporter gene assay demonstrated that the targeting sequence for miR-335-5p and miR-448 was identified in the 3′-UTR of CD36 mRNA using constructs containing the predicted targeting sequence (CD36 3’UTR) and mutated targeting sequence (CD36 3’UTR mut) cloned into the 3′-UTR of the reporter gene. (E) In HCASMCs, the miR-335-5p’s and miR-448’s inhibitors largely increased, and mimics decreased, the 1 μM Lp(a)-induced target mRNA translation, respectively, such as CD36, IL-6, TNF-α and NF-κB. *p < 0.05, **p < 0.01, ***p < 0.001.
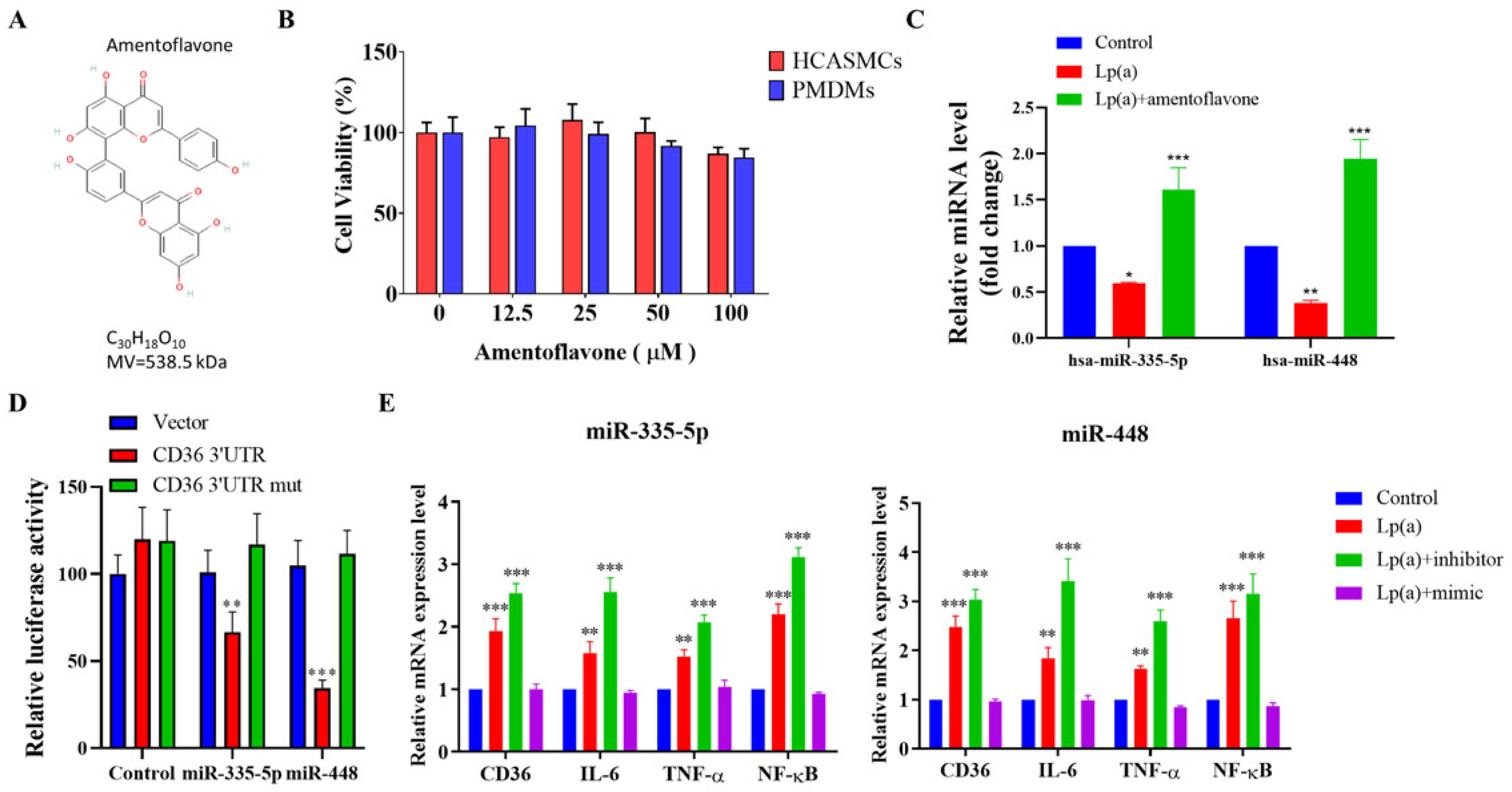

Table 1.
Baseline characteristics of the study cohort.
| Controls (n = 36) | CAS (n = 41) | p value | ||||
|---|---|---|---|---|---|---|
| Age, years | 56.6 ± 15.4 | 56.3 ± 11.9 | 0.94 | |||
| Male sex, n (%) | 12 (33) | 23 (56) | 0.045 | |||
| Body mass index, kg/m2 | 24.5 ± 4.1 | 26.0 ± 4.5 | 0.12 | |||
| Current smoker, n (%) | 4 (11) | 15 (41) | 0.004 | |||
| Diabetes mellitus, n (%) | 4 (11) | 2 (5) | 0.38 | |||
| Hypertension, n (%) | 5 (14) | 9 (24) | 0.26 | |||
| Systolic blood pressure, mmHg | 113 ± 14 | 115 ± 17 | 0.56 | |||
| Diastolic blood pressure, mmHg | 67 ± 10 | 71 ± 10 | 0.10 | |||
| Heart rate, beats/min | 67 ± 9 | 71 ± 14 | 0.24 | |||
| Left ventricular ejection fraction, % | 65 ± 5 | 65 ± 7 | 0.98 | |||
| Total cholesterol, mg/dL | 171 ± 35 | 166 ± 33 | 0.56 | |||
| Triglyceride, mg/dL | 85 ± 51 | 99 ± 54 | 0.26 | |||
| HDL cholesterol, mg/dL | 53 ± 12 | 47 ± 12 | 0.034 | |||
| LDL cholesterol, mg/dL | 95 ± 29 | 98 ± 27 | 0.66 | |||
| Lipoprotein (a), mg/dL | 18.6 ± 8.7 | 75.9 ± 29.4 | 0.001 | |||
| sCD36 | 2.1 ± 1 | 6.6 ± 3.3 | 0.001 | |||
| Peripheral leukocytes, /mm3 | 6006 ± 1459 | 6786 ± 2182 | 0.088 | |||
| Monocytes, /mm3 | 458 ± 159 | 551 ± 178 | 0.027 | |||
| Macrophage, /mm3 | 113 ± 36 | 411 ± 113 | 0.001 | |||
| Lymphocytes, /mm3 | 1725 ± 643 | 1669 ± 866 | 0.76 | |||
| Hemoglobin, g/dL | 13.4 ± 1.5 | 14.1 ± 1.3 | 0.03 | |||
| Hematocrit, % | 39.0 ± 5.0 | 41.3 ± 3.6 | 0.026 | |||
| Platelet, ×103/mm3 | 222 ± 57 | 241 ± 61 | 0.18 | |||
| hs-CRP, mg/L* | 0.63 (0.26–1.40) | 0.70 (0.21–0.98) | 0.31 | |||
| Provoked coronary artery | ||||||
| Left anterior descending artery, n (%) | 10 (24) | |||||
| Left circumflex artery, n (%) | 1 (2) | |||||
| Right coronary artery, n (%) | 32 (78) | |||||
| Number of spastic arteries | ||||||
| One-vessel spasm, n (%) | 35 (90) | |||||
| Two-vessel spasm, n (%) | 4 (10) | |||||
| Three-vessel spasm, n (%) | 0 (0) | |||||
| Medications | A | D | A | D | A | D |
| Aspirin, n (%) | 31 (86) | 33 (81) | 31 (89) | 36 (88) | 0.51 | 0.92 |
| β-blockers, n (%) | 34 (94) | 34 (83) | 18 (51) | 7 (17) | 0.12 | 0.001 |
| Calcium channel blockers, n (%) | 4 (11) | 11 (27) | 17 (49) | 39 (95) | 0.08 | <0.001 |
| Diuretics, n (%) | 0 (0) | 1 (2) | 1 (3) | 1 (2) | 0.35 | 0.91 |
| Angiotensin receptor blocker, n (%) | 7 (19) | 12 (29) | 7 (20) | 11 (27) | 0.32 | 0.49 |
| Nitrates, n (%) | 1 (3) | 1 (2) | 1 (3) | 0 (0) | 0.93 | 0.28 |
| Statins, n (%) | 10 (28) | 20 (49) | 11 (31) | 22 (54) | 0.06 | 0.05 |
Values are expressed as mean ± SD or median (interquartile range). A, before angiography; CAS, coronary artery spasm; D, at discharge; hs-CRP, high-sensitivity C-reactive protein. CAS, coronary artery spasm; hs-CRP: high-sensitivity C-reactive protein. *Log-transformed values were used for the analyses.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



