Submitted:
26 June 2025
Posted:
03 July 2025
Read the latest preprint version here
Abstract
The prevailing view for the onset of Multiple Sclerosis is that it is an “outside-in” driven pathology. It's believed that primary infection with Epstein Barr Virus (EBV) in those with a genetic susceptibility to MS, drives the production of auto-reactive B cells that infiltrate the Central Nervous System from the periphery and seed the CNS with EBV. Upon infection of the CNS with EBV, an autoimmune response to the brain from the periphery leads to MS diagnosis and neurodegeneration. We propose an “Inside Out” model of MS genesis where HHV6-A is responsible for the earliest histological findings observed in MS lesions before and during EBV primary infection, EBV primary infection accelerates activation of HHV-6A residual replication, the transactivation of EBV and the development of the Wilkins Lesion where debris from cellular injury induced by HHV6-A replication lead to the development of autoimmune processes to neuronal proteins like myelin, setting a precedent for MS development. Human Herpesvirus 6A (HHV6A) is ubiquitous and undergoes latent and periodic reactivation in the cerebral compartment of young adults. This reactivation may contribute to the development of gliapathy, a dying-back phenomenon characterized by swelling of the inner lamella of the axon, demyelination, apoptosis, or necroptosis of oligodendrocytes (ODC’s) and their precursors as suggested by Theiler’s Murine Encephalomyelitis Virus Infection (TMEV) and Experimental Allergic Encephalomyelitis (EAE) animal models.
Keywords:
human herpes virus–6A
; Epstein Barr-virus
; inside- out model
; citrullination
; MOG
; Molecular Mimicry
; Wilkins Lesion
Introduction
Over recent decades, accumulating evidence has challenged the traditional "outside-in" paradigm of autoimmunity, which posits that external factors initiate immune attacks on the central nervous system (CNS) [1]. The "inside-out" concept, which is complementary to the “outside-in” concept” has gained traction by suggesting that autoimmune responses may originate from within the target organ itself. This perspective is rooted in the primary lesion theory proposed by Terence Wilkin in 1990, which postulates that autoimmunity arises from genetically predisposed immune hyper-reactivity to excessive antigen release caused by primary lesions within specific tissues or organs [2].
According to this theory, autoimmunity is not inherently pathological but represents an exaggerated immune response to internal tissue damage [3]. As further discussed by ‘t Hart et al. [4], the same genetic and environmental risk factors implicated in the outside-in model—such as genetic susceptibility and viral infections—can also contribute to immune hyper-reactivity in the inside-out paradigm. Notably, research using the common marmoset model of experimental autoimmune encephalomyelitis (EAE), a well-established non-human primate model for MS, has highlighted the complex interplay between Epstein-Barr virus (EBV)-infected B cells and auto-aggressive T cells, particularly cytomegalovirus (CMV)-induced natural killer-type cytotoxic T lymphocytes, as central drivers of CNS-targeted autoimmunity [1].
While much attention has been devoted to understanding the activation and progression of autoimmune pathology, the origins and characteristics of the initial, or "primary," lesion remain less well explored. In this review, we introduce the hypothesis that infection of oligodendrocytes with human herpesvirus 6 (HHV-6) may represent a critical, yet underappreciated, viral trigger in MS pathogenesis of the conceptualized primary lesion. This leads us to propose a novel model in which MS could result from a complex "trialogue" of herpesvirus infections—EBV, CMV, and HHV-6—interacting within the CNS to initiate and perpetuate autoimmune pathology.
By examining the interplay between oligodendrocyte dysfunction, viral infections, and immune responses, this review aims to provide new insights into the mechanisms underlying MS and to highlight potential avenues for innovative therapeutic strategies.
2. Multiple Sclerosis
Multiple sclerosis (MS) is a chronic inflammatory disease that mainly affects the brain, and, spinal cord to a variable extent, collectively indicated as CNS [5]. Genetic linkage studies combined with histological analysis and the results of immuno-therapy trials point to a central role of the immune system in the induction of pathology and disease symptoms. However, not with standing decades of intensive research, it is still unclear which factors the immune system is activated by. According to a classical outside-in paradigm exposure of genetically susceptible individuals to infection with certain pathogens, EBV (albeit not exclusively), elicits activation of auto-reactive T- and B-lymphocytes present in the peripheral immune repertoire. Studies in a variety of animal models, including rodents as well as non-human primates, show that these specificities are not innocent, but once appropriately activated, gain the capacity to infiltrate the CNS and to elicit MS pathology [5,6,7].
MS mainly affects the spinal cord and brain via autoimmune inflammation. Histology and genetic linkage studies combined with studies of immuno-therapy trials suggest the central role of the immune system in the induction of disease pathology and symptoms. The classical outside-in paradigm suggests that infection with EBV and other viruses first in the periphery in those with certain genetic profiles induce autoimmunity towards the CNS. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis [8].
3. Epidemiological Studies: Faroe Islands.
The Faroe Islands have provided a unique epidemiological case study for studying MS. The isolated population and literal records have allowed experimenters to probe potential environmental triggers of MS. Specifically, the prevalence of MS in the Faroe Islands increased significantly following the British occupation during World War II, suggesting a possible link between viral infections introduced by those on the islands and the onset of MS in the original population [9].
The MS epidemic in the Faroe Islands offers a compelling example of how viral infections, potentially introduced by outside populations, may interact with local genetic susceptibility factors to trigger multiple sclerosis. The islands present a unique case study not only because of their isolated population but also due to their northern latitude, which results in lower vitamin D levels from reduced sun exposure compared to more southern regions [10,11]. The interplay between viral infections, vitamin D status, and inheritable vulnerability in the Faroe Islands offers investigation of complex etiology of MS and highlights the need for comprehensive approaches in studying and treating this disorder [10]. Epidemiological data from the Faroe Islands suggests an infectious organism may be a precursor to MS development. Animal EAE models have also proven efficacious in understanding the mechanisms behind MS progression [11].
4. The Autoimmune Concept of MS; the Main Immune Players.
Autoimmunity targeting the CNS in MS leads to neurodegeneration primarily through demyelination [12]. This autoimmune process arises from dysregulation of both the innate and adaptive immune responses. CD4+ T cells, particularly the Th1 and Th17 subsets, along with CD8+ cytotoxic T cells, contribute to CNS autoimmunity by secreting pro-inflammatory cytokines that drive tissue damage. B cells also play a critical role by producing oligoclonal bands—antibodies found in the cerebrospinal fluid that serve as markers of CNS-directed autoimmunity—and by functioning as antigen-presenting cells that activate autoreactive T cells [13]. Within the CNS, microglia—the resident immune cells—exhibit plasticity by transitioning between a pro-inflammatory M1 phenotype, which promotes neurodegeneration, and an anti-inflammatory M2 phenotype, which supports tissue repair and remyelination. The balance between these microglial states influences disease progression and recovery, highlighting their dual role in both CNS injury and repair mechanisms in MS [14,15].
5. Earliest Morphological Changes in MS Lesions
Within MS lesions, there is prominent activation of the innate immune system, involving microglia, astrocytes, and nearby oligodendrocytes (ODCs). Activated microglia play a key role in the degradation of myelin, the protective sheath surrounding nerve fibers, by engulfing myelin. The engulfment of myelin is the follow-up of the initial myelin injury via complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC). As demyelination progresses, the resulting tissue damage is gradually replaced by scar tissue. Notably, an iron-containing rim of activated microglia often forms around the edge of chronic lesions, which is thought to mark ongoing inflammation and tissue remodeling [16,17].
Simultaneously, young oligodendrocyte precursor cells (OPCs) are observed attempting to repair the damage by generating new myelin, although this remyelination process is frequently incomplete or unsuccessful in MS. The close spatial relationship between activated microglia and distressed oligodendrocytes suggests that microglia respond directly to signals from damaged or stressed ODCs [17]. Astrocytes, another type of glial cell, also become reactive within and near MS lesions, exhibiting hypertrophy (enlarged processes) that contribute to the formation of the glial scar and influence the local inflammatory environment. In the normal appearing white matter surrounding lesions, microglial nodules—small clusters of activated microglia—are frequently observed. However, only a minority of these nodules appear to develop into full-fledged lesions. Some of these nodules' express interleukin-1 beta (IL-1β), indicating activation of the inflammasome, a key component of the innate immune response that drives inflammation [17,18].
6. Immune Responses
Emerging evidence supports an inside-out model of MS pathogenesis, wherein intrinsic CNS dysfunction, such as oligodendrocyte stress or myelin instability, precedes peripheral immune activation [19]. This model challenges the traditional "outside-in" paradigm by proposing that environmental triggers (e.g., viral infections, vitamin D deficiency) and genetic susceptibility (e.g., HLA-DRB115:01 allele*) synergize to create a pro-inflammatory CNS microenvironment [19,20]. Here, subtle myelin damage releases modified antigens (e.g., citrullinated proteins), which activate auto-reactive T and B cells, (within CNS draining lymph nodes) initiating cycles of immune-mediated neurodegeneration in both white and gray matter [4].
7. Mechanisms of Autoimmunity and Neurodegeneration Antigen Release and Immune
7-1. Activation:
Myelin breakdown exposes myelin proteins (e.g., myelin basic protein, proteolipid protein, myelin oligodendrocyte glycoprotein) that are mistaken for foreign antigens [4]. Innate immune cells (microglia, astrocytes) engulf debris and release pro-inflammatory cytokines (TNF-α, IL-6), recruiting adaptive immune cells from the periphery [21].
7-2. Bystander Toxicity:
Activated CD8+ T cells and macrophages release cytotoxic molecules (e.g., granzyme B, reactive oxygen species), damaging nearby neurons and oligodendrocytes. Failed Repair Mechanisms: Repeated inflammatory episodes impair OPC differentiation, leading to incomplete remyelination and scar formation. Chronic microglial activation perpetuates oxidative stress, accelerating axonal degeneration [4].
8. The Outside-in Pathogenic Concept and Supporting Animal Models
The "outside-in" hypothesis posits that environmental triggers, such as viral infections, play a central role in initiating MS by activating within the peripheral lymphoid compartment auto-reactive lymphocytes that mistakenly target the CNS [22]. According to this model, microbial antigens—like those from viruses—provide not only the initial antigenic stimulus, but also critical co-stimulatory signals required for full T cell activation. Without these secondary signals, auto-reactive T cells may remain in a state of peripheral tolerance, avoiding pathogenic behavior. Mouse models of EAE, the primary animal model of MS, support this theory, as disease induction typically requires both a microbial adjuvant (e.g., complete Freund’s adjuvant) and a myelin-specific antigen. This mirrors the proposed human mechanism where environmental pathogens may "prime" the immune system to breach self-tolerance [23].
Viral infections further exemplify this concept through molecular mimicry, wherein microbial antigens structurally resemble self-antigens like myelin proteins. For instance, Semliki Forest virus (SFV) [24] and Theiler’s murine encephalomyelitis virus (TMEV) [25] are neuro-tropic viruses used in animal studies to demonstrate how viral mimicry of host proteins can trigger cross-reactive immune responses, leading to demyelination. TMEV, in particular, induces chronic CNS inflammation in mice, closely resembling MS pathology. Human studies also implicate viruses such as Epstein-Barr virus (EBV) in MS susceptibility, with evidence suggesting EBV-specific T cells cross-react with myelin basic protein [25,26,27].
These findings underscore the interplay between genetic predisposition and environmental factors in MS. The "outside-in" framework thus emphasizes that microbial encounters may act as essential catalysts, breaking immune homeostasis and enabling autoreactive lymphocytes to infiltrate and damage the CNS.
9. Early changes in NAWM.
Normal-Appearing White Matter (NAWM) exhibits significant abnormalities that are not detectable through conventional MRI but can be revealed using advanced imaging and molecular techniques. Myelin microscopy imaging studies have demonstrated that NAWM is abnormal at a microscopic level, indicating early pathological changes even before the formation of visible MS lesions [28,29].
Immunofluorescence investigations have shown that these myelin abnormalities in NAWM may actually precede the development of overt MS lesions. Despite appearing normal on standard MRI scans, the white matter in the CNS shows subtle but critical changes. For example, myelin water imaging (MWI), a specialized magnetic resonance imaging (MR)I technique, detects a reduced density of myelinated axons within NAWM, highlighting early demyelination processes [30].
The distribution of T2 hyperintense lesions on MRI often follows these initial abnormalities in NAWM, suggesting a progression from microscopic changes to macroscopic lesion formation. At the molecular level, alterations have been observed in lipid composition, the structure of myelin, and the integrity of nodes of ranvier—critical gaps in the myelin sheath essential for rapid nerve signal conduction. Additionally, there is evidence of disrupted iron homeostasis within NAWM. Specifically, a shortage of iron is noted alongside increased activity of ferric oxidases—enzymes that catalyze the oxidation of ferrous iron and facilitate its export from cells. This imbalance may contribute to oxidative stress and further tissue damage [31].
Inflammation and demyelination characterize these NAWM abnormalities, which appear as hyper-intense regions on T2-weighted MRI scans. Advanced brain MRI techniques, including magnetization transfer ratio (MTR) mapping analyzed through brain atlases on a voxel-wise basis, have been used to precisely localize and quantify these abnormalities. The spatial pattern of T2 hyper-intense signals closely matches the areas showing altered MTR values, reinforcing the idea that NAWM changes are an early marker of lesion development [29,31].
10. The Proverbial Primary Lesion
The "inside-out" hypothesis proposes that MS begins with primary degeneration or dysfunction within the CNS, which subsequently triggers an autoimmune response. Research using primate EAE models has identified auto-aggressive effector memory T-cells, including natural killer (NK)-like populations, as part of the normal immune repertoire. Intriguingly, similar T-cell populations have been found in both healthy individuals and those with MS, as demonstrated by the Antel group at McGill University [32,33]. These T-cells can be activated solely through "signal 1"—the interaction between the MHC, antigenic epitope, and T-cell receptor (TCR)—without requiring additional microbial co-stimulation. However, viral infections may still contribute to disease progression. For example, cytomegalovirus (CMV) infection is thought to induce auto-aggressive NK-T cells, while EBV-infected B-cells act as potent antigen-presenting cells (APCs), presenting CNS antigens and sustaining autoimmune activity [34]. Within MS lesions, the innate immune system becomes highly active. Microglia, astrocytes, and nearby ODCs show signs of activation. Myelin degradation occurs, with activated microglia phagocytosing myelin debris. Over time, the loss of myelin is replaced by scar tissue, often surrounded by an iron-containing rim formed by microglia. Young OPCs are observed attempting to remyelinate and repair the damaged tissue [35,36].
Morphological studies suggest that microglia clustering near lesions respond to stressed or damaged oligodendrocytes. Astrocytes near and within lesions exhibit hypertrophy, reflecting a reactive state. Small clusters of activated microglia, known as microglial nodules, are commonly found within lesions. However, only a minority of these nodules progress into full lesions, particularly those expressing interleukin-1 beta (IL-1β), which indicates inflammasome activation and a pro-inflammatory environment [37].
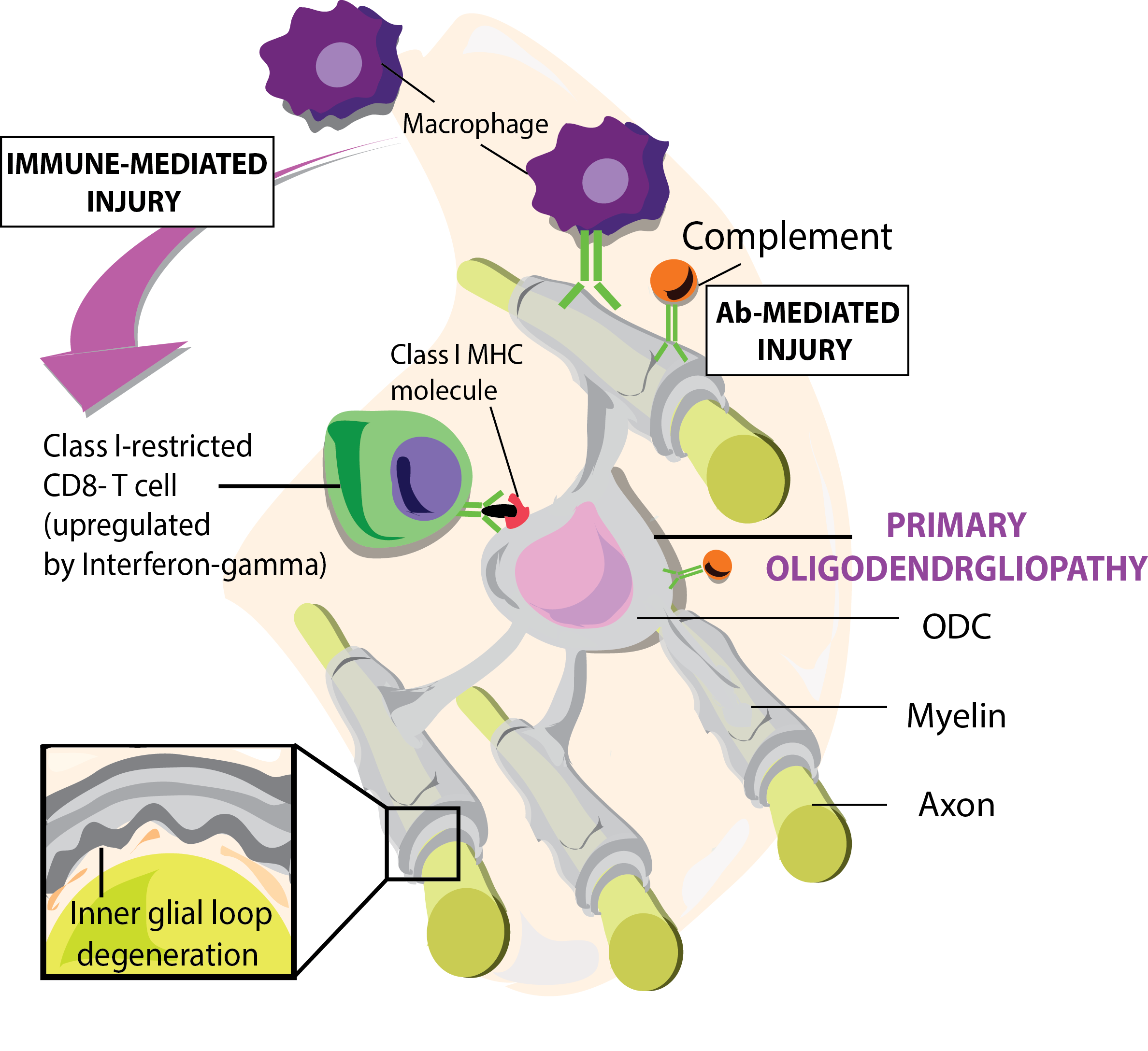
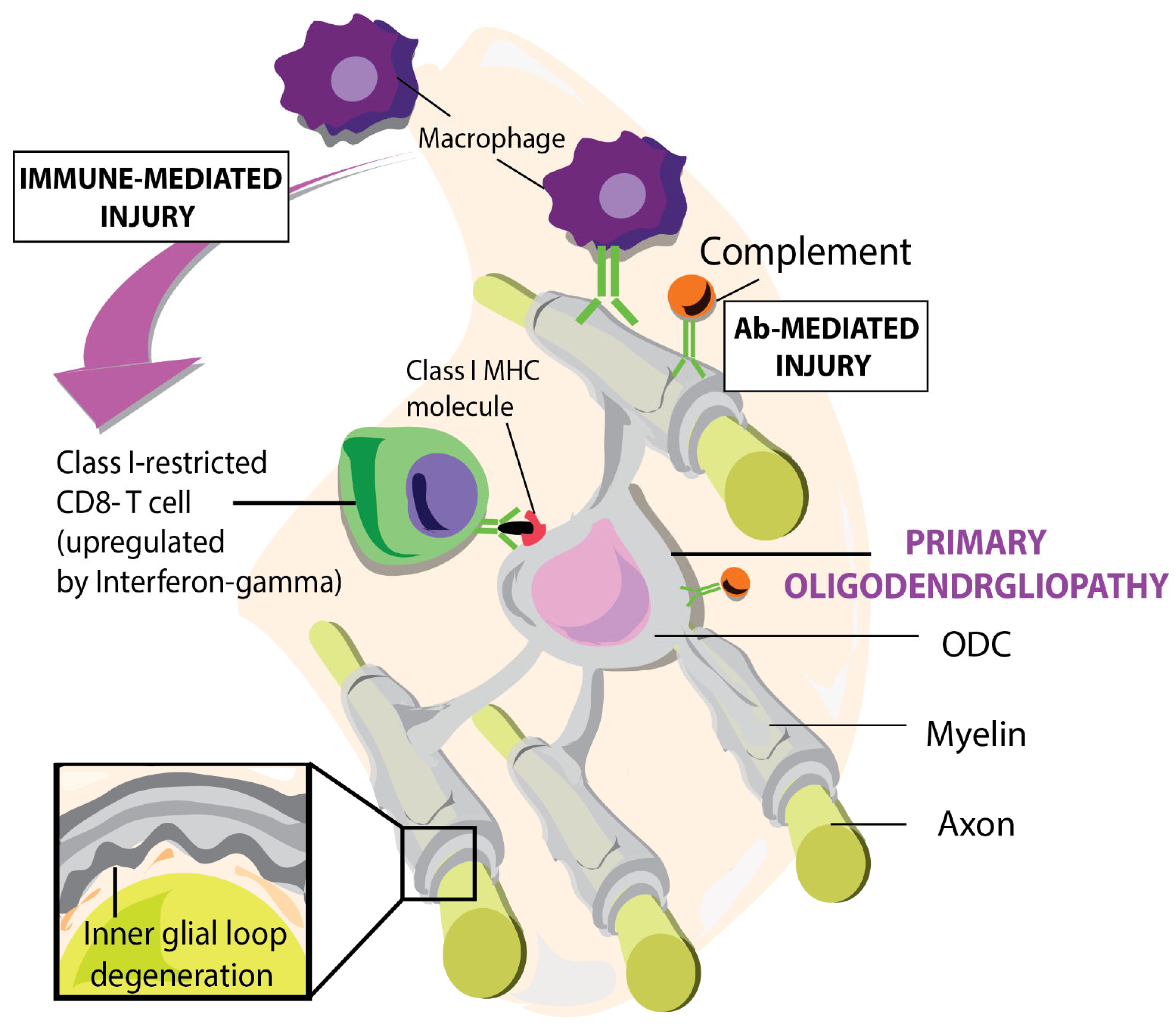
Figure 1.
Graphic represents primary oligodendrogliopathy induced by microglial and CD8 autoimmune responses towards myelin sheaths and other neuronal proteins. Figure from: t’ Hart BA et al. [38].
Figure 1.
Graphic represents primary oligodendrogliopathy induced by microglial and CD8 autoimmune responses towards myelin sheaths and other neuronal proteins. Figure from: t’ Hart BA et al. [38].
Rodriguez and Scheithauer (1994) analyzed the structure of MS lesions via electron microscopy of 11 brain biopsies. Early degenerative changes were observed, including widening of the inner myelin lamellae and disruption of the inner loops. These observations preceded myelin destruction. There was perivascular infiltration of macrophages, lymphocytes, and plasma cells which were interacting with myelin sheaths. It was observed that astrocytes were proliferating even in non-demyelinated areas. There was significant loss of oligodendrocytes in demyelinated areas with denuded axons in proximity to astrocyte processes. There was swelling of the axons in affected areas. Intact were oligodendrocytes at lesion sites. Remyelination was present as thin myelin sheaths at the margins of plaques. There were also “pole-like” crystalline inclusions which may have arisen from the endoplasmic reticulum. The study highlights a "dying-back oligodendrogliopathy" also affecting distal processes. There is heterogeneous lesion activity including acute demyelination and axon loss and gliosis of a chronic nature. There is some nerve regeneration present whereas the macro phage inclusions suggest a protein aggregation of an unknown nature [39].
11. A Case for HHV-6
Infection by HHV-6A disrupts several critical cellular functions of oligodendrocytes. It impairs mitochondrial activity and alters the cytoskeleton, exposing cells to increased oxidative stress, viral byproducts, and chronic inflammatory cytokines. These combined factors can trigger programmed cell death pathways, such as necroptosis or apoptosis, ultimately leading to oligodendrocyte loss and destabilization of the myelin sheath. HHV-6A also interferes with essential cellular processes like mitochondrial fusion and fission, which are vital for maintaining cellular energy balance. Prolonged disruption of these processes can result in cell death. Notably, the earliest pathological damage in MS occurs at the inner glial loop of the myelin sheath, rather than the outer myelin layers [40,41]. Damage to the outer sheath appears only later in the disease progression, highlighting the inner glial loop as the initial site of myelin injury [42].
12. Blistering of Myelin
Recent studies have revealed the blistering of the myelin sheath, particularly at the inner layers closest to the axon. This blistering may represent an early stage of myelin damage that precedes overt demyelination. The mechanism behind ODC blistering is characterized by dislocation of myelin-associated proteins and lipid composition potentially due to viral interference with oligodendrocyte cellular processes [43].
12-1. Citrullination of Myelin
Citrullination is an enzymatic post-translational modification where arginine residues in proteins are converted to citrulline, which plays a significant role in destabilizing myelin and advancing the pathogenesis of MS. In MS patients, there is increased presentation of the citrullinated myelin basic protein (MBP) peptide fragment MBP85-99 to immune cells, contributing to autoimmune responses [44,45]. Pathological changes in oligodendrocytes include swelling of the inner lamella and bleb formation, indicative of cellular degeneration. MBP is essential for maintaining the structural integrity and compaction of the myelin sheath. However, citrullination of MBP disrupts its ability to organize lipid bilayers properly, leading to myelin decompaction. This modification also increases MBP’s susceptibility to proteolytic enzymes such as cathepsins, exacerbating myelin breakdown [44,46].
Calcium signaling dysregulation and glutamate receptor disruption in oligodendrocytes lead to increased intracellular calcium levels, which activate peptidylarginine deiminases (PADs)—the enzymes responsible for citrullination. This cascade results in further myelin destabilization. The loss of myelin compaction exposes the hydrophobic lipid core, making it vulnerable to enzymatic degradation. Additionally, tissues from MS patients show elevated citrullination of cytoskeletal proteins such as vimentin and glial fibrillary acidic protein (GFAP). This modification compromises cytoskeletal integrity, increasing cellular fragility. Mitochondrial dysfunction in oligodendrocytes contributes to myelin decompaction by impairing energy-dependent calcium buffering, which is critical for maintaining axon-myelin interactions [46]. Furthermore, disruption of adhesion molecules, including myelin-associated glycoprotein (MAG), weakens the tethering between axons and myelin, promoting blister formation and further myelin damage. Together, these molecular and cellular alterations highlight the complex mechanisms by which citrullination and MBP dysfunction contribute to myelin instability and MS progression [47,48].
12-2. Citrullination as Result of an Immune Response in the CNS
In MS, tumor necrosis factor (TNF) is prominently expressed by astrocytes and macrophages within active lesions, where it drives neuro-inflammation and oligodendrocyte apoptosis. A key mediator of myelin destabilization in the brain is Peptidyl arginine Deiminase 2 (PAD2), which catalyzes the citrullination of myelin basic protein (MBP). This post-translational modification reduces myelin’s structural stability, rendering it more susceptible to proteolytic degradation and immune targeting. Similarly, PAD4 deaminates histones (e.g., generating citrullinated histone H3, H3cit), altering chromatin compaction and potentially hindering OPC differentiation—a critical barrier to remyelination. While PAD4 is active in the periphery, it can still be found in the CNS. Elevated PAD4 levels correlate with increased H3cit and TNF in normal-appearing white matter (NAWM) of MS patients, a pattern replicated in animal models of demyelination [49,50].
Citrullinated myelin proteins amplify neuro-inflammation by stimulating TNF production in activated microglia. Proteomic studies of MS brain tissue have identified 80 citrullinated proteins, including MBP, GFAP, and vimentin, with citrullinated epitopes enriched in MS patients compared to controls. Intriguingly, CSF-derived T cells from MS patients show no proliferative response to citrullinated myelin antigens in vitro, suggesting citrullination is not a direct T cell activator but rather a consequence of chronic inflammation [51,52,53], however it has been shown in mouse EAE. It is important to note that there is a difference betwen mouse and human findings [54].
12-3. Citrullinated MBP and Astrocyte Dysregulation
Experiments using citrullinated MBP isomers reveal distinct astrocyte responses. The citrullinated isoform (MBP-C8) reduces glutamate release, triggers NF-kB-mediated inflammation (e.g., nitric oxide, IL-17A), and upregulates pro-inflammatory pathways, while unmodified MBP (MBP-C1) enhances glutamate uptake without inducing inflammation. Both isoforms increase expression of PPAR-γ (a regulator of lipid metabolism) and EAAT2 (a glutamate transporter), suggesting a dual role for MBP in modulating astrocyte metabolic and inflammatory states. These findings position citrullinated MBP as a driver of astrocyte-mediated neurotoxicity in MS [53].
13. EAE MS Animal Models
13-1. EAE.
EAE models are widely used to study mechanisms of autoimmune demyelination. In these models, myelin-derived proteins (e.g., proteolipid protein [PLP] or myelin oligodendrocyte glycoprotein [MOG]) are administered with bacterial adjuvants like Complete Freund’s Adjuvant (CFA) to enhance immuno-genicity. For example: SJL/J mice: Immunization with PLP peptides (e.g., PLP139-151) induces a relapsing-remitting EAE course, mimicking some aspects of human MS [55,56]. NOD mice: MOG35-55 immunization triggers a relapsing disease, though it does not fully replicate the chronic progressive phase of MS [57,58]. EAE pathogenesis primarily involves CD4+ T cell-mediated immune responses, while the role of CD8+ T cells in direct neuronal damage remains understudied [59]. In mice, EAE predominantly affects the spinal cord, with limited cortical involvement. In contrast, MS is characterized by extensive subpial cortical demyelination alongside spinal cord lesions [59,60,61]. In the marmoset EAE model, MS pathology in the brain is more closely replicated. Autoimmunity in EAE is driven by co-activation of innate immune receptors (e.g., Toll-like receptors [TLRs] and NOD-like receptors [NLRs]), which break tolerance and activate auto-reactive T and B cells [58,62]. Acute inflammation typically begins 15–20 days post-immunization, followed by symptom relapse and remission [56]
13-2. TMEV EAE Animal Models
Theiler’s Murine Encephalomyelitis Virus (TMEV) models play a crucial role in investigating the involvement of immune cells and viral factors in promoting neuro-inflammation and demyelination. These models are particularly useful for studying the polarization of microglia and macrophages into either pro-inflammatory or anti-inflammatory states, which significantly contribute to the pathogenesis of MS. The relapsing-remitting experimental autoimmune encephalomyelitis (R-EAE) model closely resembles relapsing-remitting MS (RRMS) [63]. In this model, acute inflammation typically begins around 15 days after immunization, followed by a relapse phase occurring between 25 to 30 days, which then resolves. In SJL/J mice, spontaneous relapsing-remitting EAE is induced by myelin oligodendrocyte glycoprotein (MOG)-reactive transgenic T cells that recruit endogenous MOG-specific B cells, amplifying the autoimmune response [64,65]. TMEV infection leads to an upregulation of immunoglobulin-related genes and facilitates the migration of mature B cells across the blood-brain barrier, where they produce intrathecal antibodies and present antigens, thereby sustaining chronic inflammation and autoimmunity. This virus is an excellent model for studying T cell-mediated demyelination in susceptible mouse strains following viral exposure. The disease induced by TMEV is biphasic, with an initial acute phase of viral infection and neuronal damage, followed by a chronic phase characterized by immune-mediated demyelination. This biphasic nature allows researchers to explore viral persistence and chronic inflammation, features that parallel molecular mimicry and bystander activation mechanisms observed in MS [66,67,68]. The autoimmune response is initially dominated by CD4 but taken over later by CD8 [69].
Microscopic examination of TMEV-infected tissue reveals significant myelin abnormalities, including vesiculation and splitting between the myelin layers surrounding axons. Mononuclear cell infiltrates, consisting of lymphocytes, macrophages, and plasma blasts, are found near white matter lesions and in periventricular regions. Vacuolation of oligodendrocyte cytoplasm, a hallmark of demyelination, is also observed. Immuno-peroxidase staining combined with electron microscopy has detected TMEV viral antigens within the inner and outer glial loops of myelin in infected SJL/J mice, indicating primary infection of oligodendrocytes. Structural studies show that axonal damage can occur even when the myelin sheath appears intact, with swollen axons, vacuoles, and abnormal spacing between myelin lamellae. These changes weaken the myelin structure, compromising its insulating function and impairing axonal signal conduction [68,70].
13-3. Non-Human Primate EAE
Callitrichine gammaherpesvirus 3 (CalHV3) is a simian ©1 herpesvirus that, just like it’s human counterpart EBV, causes latent infection of B cells [71]. Complete genomic sequence of the simian EBV-related lymphocryptovirus virus infects marmosets and shares physiological and pathological characteristics with EBV infection of CD20-expressing B lymphocytes and other cell types in humans [72,73]. In EAE models, CalHV3-infected B lymphocytes promote autoimmune responses similarly to EBV’s influence on MS development. In non-human primate EAE models, such as those involving marmosets, MOG and the major capsid protein of CMV become targets of the immune response [74]. This supports the hypothesis that CMV infection generates a repertoire of potentially autoreactive T cells, which, through molecular mimicry, may attack MOG-expressing oligodendrocytes.
Despite the valuable insights gained from non-human primate and murine EAE models, these systems have notable limitations in replicating the full spectrum of human MS pathology. Cortical lesions, which are a hallmark of chronic MS and often correlate with cognitive decline and disease progression, are rarely observed in standard murine models [75]. This discrepancy makes it difficult to translate findings related to myelination abnormalities and remyelination from murine models to human MS. Additionally, rodent models tend to exhibit greater remyelination than typically seen in human patients, which may lead to overestimation of the potential efficacy of candidate therapies targeting repair mechanisms. As noted by Peterson et al. (2001), the rarity of cortical lesions in murine models limits their ability to fully replicate the pathological features of MS [72], while Plemel et al. (2017) highlight that rodent models may provide misleading conclusions regarding remyelination-based treatments [76].
References include Cho et al. (2001) [72] who characterized the genomic sequence of CalHV3 and its relation to EBV, Jagessar et al. (2013) [74] who discussed the role of EBV-infected B cells in primate EAE models, and Axthelm et al. (2011) [75] who explored γ-herpesvirus-induced demyelination in macaques. The virus studied by Axthelm and colleges is a γ2-herpesvirus more closely related to KSHV than to EBV [77]. Studies by Kutzelnigg et al. (2005) [78] and Bevan et al. (2020) [78] provide insights into cortical demyelination in MS and its limited representation in EAE models.
13-4. HHV-6A
Human herpesvirus 6A (HHV-6A) is known to infect humans later in life and exhibits a stronger preference for CNS tissue compared to its closely related counterpart, HHV-6B. Immuno-histochemical studies have detected HHV-6A antigens within oligodendrocytes both inside and adjacent to MS lesions, suggesting a possible involvement of this virus in MS development [79]. Polymerase chain reaction (PCR) analyses have identified HHV-6A DNA in the CSF and serum of individuals with MS, with HHV-6A being detected more frequently in MS lesions than HHV-6B. Evidence of active infection or viral reactivation is indicated by elevated levels of immunoglobulins IgG and IgM against HHV-6A antigens during periods of MS relapse [80]. Additionally, high concentrations of HHV-6A DNA have been found in the serum and urine of MS patients. Although these findings point toward a potential role for HHV-6A in MS pathogenesis, the precise mechanisms remain incompletely understood. It is hypothesized that HHV-6A may contribute to autoimmunity in MS through several mechanisms, such as the release of citrullinated cellular debris following virus-induced cell lysis, aberrant expression of major histocompatibility complex (MHC) molecules, molecular mimicry, or persistent activation of immune cells [80,81]. Interestingly, marmosets have been infected with HHV-6A, inducing neurological disease [82].
While HHV-6B can infect glia, it has a wider cellular tropism is more [83,84]. These viruses establish an initial infection with and the capacity to reactivate later especially in the immune compromised [85]. Treating HHV-6 reactivation infections is a challenge due to difficulties in its diagnosis and shortage of HHV-6 effective anti-antivirals [86]. As in neurons, immunocytochemistry revealed the HHV6-A viral proteins 101K and P41 and proximal to lesions [87]. HHV-6B was detected in the cytoplasm of neurons within gray matter in proximity to lesions and in the NAWM at lower amounts in MS subjects. HHV-6B was also detected in controls, though only MS subjects had HHV-6B nuclear staining in oligodendrocytes [87]. A 1996 study by Sanders and co-workers detected HHV-6 along with other members of the Herpes family in MS subjects as well as in controls via PCR, especially at MS lesion locations [88]. There was no distinction of HHV-6A and HHV-6B variants, due to lack of as variant-specific PCR assays. The observation of HHV-6 in healthy controls implicates HHV-6 as a commensal virus. HHV-6B is known to infect infants at an early age with a tropism for the CNS. A rash known as Roseola Infantum presents itself in toddlers after infection with HHV-6B, and sometimes complications occur [89]. HHV-6A is a plausible candidate for initiation of the first steps in MS development because it is neurotropic and cytopathic [90].
HHV-6 thrives in a host with a deficient immune response towards antigens of the herpes family [91]. The Natural killer cell (NK) immune response has been observed to be reduced in those with MS. HHV-6 IgG levels are also observed to be higher in those presenting with EBV primary infection and during the period before and at MS diagnosis. [92]. Recently, it was also observed that HHV-6A IgG increased preceding an increase in serum neurofilament light chain (sNfL), a marker of axon destruction [93]. HHV-6A u94A alters the ODC cytoskeleton, contributing to ODC dysfunction [94]. These observations suggest that HHV-6A has the capacity to worsen Endoplasmic Reticulum (ER) stress and create abnormalities in UPR pathways in cells of neural origin [95]. HHV-6A has the potential to induce apoptosis by various mechanisms, including Caspase-3 and Caspase 9, enzymes involved in the ODC apoptosis process activation [96,97], in fetal astrocytes. Bax (Bcl-2-associated X protein), a pro-apoptotic protein becomes upregulated while Bcl-2 (B-cell lymphoma 2), anti-apoptotic protein, involved in reduction of apoptosis is down regulated. HHV-6A brings about poly (ADP-ribose) polymerase cleavage. (ADP-ribose) polymerase is a substrate of activated caspase-3 activation [97]. HHV-6A likely induces a "dying back" phenomenon involved in demyelination and the potential apoptosis of ODC’s. HHV-6A can induce caspase independent and dependent pathways of apoptosis in fetal astrocytes [97]. HHV-6A mRNA was expressed at a moderate level in NAWM compared to healthy controls and MS lesion locations having the most HHV-6A mRNA expression [98]. HHV-6A has the capacity to induce apoptosis [99]. A dying back phenomenon characteristic of demyelination may occur before induction of caspase independent apoptosis due to cellular stress [96]. This preferential infection of oligodendrocytes by HHV-6A suggests an underlying medium for the initiation of MS pathology from within the CNS [93]. Myelin debris from apoptotic ODC's undergo citrullination and drain to cervical lymph nodes [100], where putatively auto-reactive T and B cells are activated [101].
HHV-6A DNA and proteins have been found within and in proximity to CNS lesions, including CNS tissue devoid of inflammation. For example, Engdahl et al. (2019) reported that “MS patients showed a significantly higher frequency of anti-HHV-6A antibody responses compared to controls, and these responses were associated with elevated serum neurofilament light chain prior to MS onset.” This finding is supported by Virtanen et al. (2023), who observed that HHV-6A DNA is more commonly detected in active MS plaques than in non-lesional white matter,” implicating viral persistence and injury to neural tissues. suggesting a potential temporal relationship between viral presence and neuroaxonal injury.
Perhaps HHV-6A may also not play a direct role in the development of MS lesions. As reviewed by Opsahl and Kennedy (2005) [98], “the detection of HHV-6 DNA in MS lesions could represent an epiphenomenon, reflecting viral persistence in areas of tissue damage rather than a causal relationship.” Leibovitch and Jacobson (2014) [102], suggest “current evidence is insufficient to distinguish whether HHV-6A is a primary driver of lesion formation or a bystander accumulating in inflamed tissue.” There are also non-consistent findings of anti-HHV-6 antibody levels in CSF and serum. Alvarez-Lafuente et al. (2006) [103], who found “no consistent pattern of HHV-6 antibody elevation in MS patients compared to controls.” Lassmann (2018) [104] states “true causality requires demonstration that targeting the virus can alter disease trajectory, which remains to be shown in human studies.”
14. Citrullination of Myelin and HHV6-A
The link of HHV6-A inducing ODC blebbing via citrullination of myelin is not directly shown in the literature. However, it may be a candidate for such a relationship due to HHV-6A caused molecular mimicry via HHV6-A u24 protein homology with myelin basic protein (MBP), bringing about Th-17 driven autoimmunity towards myelin-related proteins [105]. Caspase-independent ODC death via necroptosis caused by supernatants from HHV-6A-infected ODC's would contribute to the induction of the autoimmune response [99]. The association of very high HHV-6A IgG in those at risk and who develop MS compared to healthy controls suggests a reduced capacity to control transactivated HHV-6A [92]. HHV-6A is often found in a latent state in NAWM. In an active replication state, HHV-6A induced syncytium formation independent of caspase 3 via cell culturing of ODC's [98]. HHV-6 mRNA expression is a tell-tale sign of an active replication cycle [92]. Immune deficiency reduces control of latent HHV-6A and increases the probability of replication mediated toxicity to ODC's. There is a reduction of UL16 Binding Protein 1 (ULBP1), a ligand for NKGD2, which is stress induced. HHV-6B can down regulate NKG2D, an activating receptor which identifies stress induced ligands in virally infected and other dysfunctional cells and NKp30 ligands, cytotoxicity receptors which recognize ligands involved in immune regulation and cellular lysing making it easier for HHV-6 to hide from immune surveillance. HHV-6B can also decrease UL16 Binding Protein 3 (ULBP3) expression which reduces NK cell recognition of virally expressing cells [106]. Th-2 response can be promoted by inducing IL-10, and HHV-6A, modifying signaling inside NK cells involved with sensing external antigens by downregulating MHC-1 [107,108].
15. HHV-6 and Immune Deficiency in MS patients
A medical report presents a female who experienced a mononucleosis-like illness after HHV-6A infection, which resolved. The patient had normal immune parameters with a presence of a potential deficit towards HHV-6A. There was a resolution of the symptoms of the HHV-6A Infection, including encephalomyelitis. A month later, the patient seroconverted to a new EBV infection with symptoms of mononucleosis [109]. This case study shows that HHV-6A has the capacity to induce neuro-inflammation even in adult patients with competent immune responses to antigens other than the herpes family. HHV-6A replication in progenitor and adult ODCs may lead to abnormalities found in NAWM preceding MS lesions.
NK cells and immune deficits towards HHV-6 Killer cell Immunoglobulin-like Receptor (KIR2DL2), an inhibitory receptor on NK cells, can impair the immune response against HHV-6A, leading to persistent viral infection via disrupting the antigen presentation of herpes-related antigens [110]. KIR2DL2 allows for evasion of HHV-6 from the human immune system. People with the KIR2DL2 gene have higher levels of HHV-6A DNA in their blood and cerebrospinal fluid, correlating with increased disease activity and progression in MS patients [111]. The gene's inhibitory effect on NK cells reduces the clearance of HHV-6A-infected cells, leading to sustained viral replication and increased inflammatory responses. The relationship between KIR2DL2 and control of neuro-trophic MS-inducing viruses underscores the importance of genetic and viral factors in MS disease progression. HHV-6A production can occur due to NK cell dysfunction, and poor presentation of antigens of the herpes family [29]. Localized HHV-6A residual replication in those susceptible to MS is correlated with rising neurofilament light chains (NFL) in blood/CSF [93], and the association with KIR2DL2 raises the prospect for reduced control of localized spread of HHV-6A [111]. A study by Goldman (2003) involved a study of live biopsy tissue from five people who developed MS. Patients presented with acute lesions that were initially considered to be tumors, influencing the decision to initiate the biopsy of the patient nervous tissue. The lesions revealed the presence of HHV-6 within inflamed acute lesions as well as areas of the white matter that are not inflamed .HHV-6’s presence in inflammatory lesions and in tissue not inflamed suggests that the virus may also be involved in MS development besides its commensal nature [112]. Recent findings have revealed that HHV-6A could be present in central nervous tissue before symptoms of MS [93]. This finding has reinforced earlier research on potential for HHV-6A involvement in demyelination [102]. HHV-6A can induce cytopathic effects both in a direct and indirect manner [113]. HHV-6A has the capacity to infect ODC’s and precursor cells causing disruption of ODC function and abnormalities in cellular structure [114]. Vesicles between myelin layers can develop [115]. Vesiculation may be a non-specific response due to disruption of normal ODC function [114].
Interestingly, a study from Genuine and Raine in Nat Med 1999 revealed myelin vesicularization in MS enables binding of (cytopathic) anti-MOG antibodies. Both in MS and in marmoset EAE [116]. HHV-6A has the capacity of inducing latency and undergoing reactivation and limited replication in ODC’s inducing cellular stress and perhaps influencing the observed relapsing phenomenon [117].
Residual segments of myelin sheath characterized as short and vacuolated were observed in an immunocompetent 21-year-old female presenting with HHV-6A infection and Fulminant Demyelinating Encephalomyelitis [118] This occurs as demyelination progresses resulting in blebbing of myelin and development of vacuolated short residual segments which are swollen empty spaces within the myelin sheath [119] This is because there is myelin breakdown and phagocytosis of myelin by macrophages [120]. There is also a dysfunction in axon-myelin signaling due to glutamate receptor dysfunction [121].
16. CNS Antigen Drainage in Cervical Lymph Nodes.
The drainage of CNS antigens including CSF, interstitial fluid (ISF) and immune cells into the cervical lymph nodes is facilitated by lymphatic tissues sourced from meningeal tissues [122]. The meningeal vessels allow for immune surveillance via immune cellular traffic including dendritic cells [123]. The meningeal vessels are located beneath the skull in the dorsal and basal positions. CSF enters the lymphatics of the nasal area via drainage via the cribriform plate. The traffic of T cells and antigen-presenting cells is facilitated by basal meningeal vessels [100,123]. There is a correlation between myelin antigens in the cervical lymph nodes and the progression of Multiple Sclerosis [124]. Like CalHV3 in Marmosets, EBV infection of human CD20-bearing B cells delays digestion of citrullinated myelin fragments, which may prolong the opportunity for presentation of neo-antigens, which may set the stage for the generation of autoimmune responses to neural antigens like Myelin Oligodendrocyte Protein (MOG) [125]. Co-repression of EBV and HHV-6A immunoglobulins (IgG), is a marker of reduced control of latent infection of both viruses, which can assist each other in promoting abnormal cellular activation via computer viral transactivation [126]. It has been established that presentation of citrullinated myelin to immune cells in the periphery can induce differentiation of various lymphocytes facilitating autoimmunity towards neural proteins [45]
Louveau et al. (2018) [127] suggests that the surgical removal of meningeal vessels leads to a reduction in the traffic of CNS antigens to the cervical lymph nodes. There is a reduction of T cell activation and a decrease of symptoms related to EAE and a decrease in antigen presentation to other lymphocytes Da Mesquita et al., 2018) [127], Rustenhoven et al. (2021) [128] reported that “myelin-laden antigen-presenting cells are routinely detected in cervical lymph nodes of both EAE mice and MS patients.”
It is unclear if caspase-independent necroptosis of oligodendrocytes occurs. Findings from Ito et al. (2016) [129] suggest that necroptosis may occur due to inflammation. Necroptosis is also found in CNS tissue undergoing demyelination Lloyd et al., 2019) [130]. Ofengeim and Yuan (2013) [131], state “the precise contribution of necroptosis to oligodendrocyte loss in MS lesions awaits further validation in animal models.” As Engdahl et al. (2019) [132] note, “increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis,” suggesting HHV-6A’s association with MS. That viral infection may be an early event in disease development [133], potentially inducing citrullination and blebbing of myelin, central to oligodendro-gliopathy [134], which may precede inflammation among latently infected ODC due to the u94 protein associated with the latent infection of ODC [135].
17. MS as a Tripartite of Herpesviruses (CMV, EBV, HHV6).
It is suggested that MS is a trialogue between 3 herpes viruses: 1. HHV-6A, causing oligodendro-gliopathy and transient neurological dysfunction [82], leading to the development and collection of citrullinated myelin in the cervical lymph nodes [100], 2. EBV transformed B cells into highly effective APC, which acquire the capacity to migrate to the CNS [136], and 3. CMV induces a myelin cross-reactive repertoire of effector memory cytotoxic T cells mediating autoimmune responses, and is capable of eliciting MS-like pathology in a marmoset model [137].
18. Consequences for the Therapeutic Approach to the Disease.
The issue with current and past imaging studies involving HHV-6A and its impact on the dying back phenomenon is that most research groups have used oligodendrocyte culture from neonatal or embryonic tissue in the absence of axons or neurons, complicating observation of a dying back pathology. If co-culture systems are used (adult oligodendrocyte and neuron), HHV-6A's impact on axon function could be better studied. Research using 3-dimensional organotypic nature cultures would be a better representation of in vivo conditions [138]. NFL levels, which highlight axon destruction, give another clue to understanding MS pathogenesis. NFL levels before EBV infection in those who later develop MS are about the same as EBV-positive healthy controls. The NFL levels rise dramatically after EBV exposure in those developing MS [27]. This would support the hypothesis that EBV aggravates axon injury through co-transactivating HHV-6A at the time of primary EBV infection. The concept of the Wilkins lesion was developed by Terrence Wilkins in 1990 [2]. A primary lesion brings about the release of autoantigens, which are detected by and activate a hyperactive immune state. It is postulated that replication from HHV-6A could induce this type of lesion due to its ability to induce cellular toxicity to neural cells. While the inside-out model highlights the primary role of CNS-intrinsic factors in initiation, the importance of the adaptive immune response in disease progression remains evident.
19. Future Research Directions
To further validate and expand upon the inside-out model of MS pathogenesis, several key areas of research should be prioritized: 1. In vivo imaging of early oligodendrocyte dysfunction and myelin changes in preclinical MS models. 2. Specific biomarkers to monitor ODC damage. 3. Understanding viral trans activations between HHV-6A, EBV, and HERVs and how they encourage each other in promoting neurodegeneration. 4. Applying proteomics to increase sensitivity and detection rates of infectious pathogens which encourage neurodegeneration, especially for HHV-6A, HERV's and EBV in MS-related lesions and NAWM. 5. Elucidation of the precise role of citrullinated proteins and amyloid aggregates in triggering and perpetuating MS. 6. Characterizing the degree of induction of UPR in ODCs and glia due to initiation of HHV-6A replication and productivity of the viral life cycle. The effect of anti-HHV-6A pharmaceuticals in halting the replication of HHV-6A’s contribution to furthering MS pathogenesis.
References
- Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). British Journal of Pharmacology. 2011;164(4):1079-106.
- Wilkin TJ. The primary lesion theory of autoimmunity: a speculative hypothesis. Autoimmunity. 1990;7(4):225-35.
- Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125(6):2228-33.
- t Hart BA, Luchicchi A, Schenk GJ, Stys PK, Geurts JJG. Mechanistic underpinning of an inside-out concept for autoimmunity in multiple sclerosis. Ann Clin Transl Neurol. 2021;8(8):1709-19.
- Papiri G, D'Andreamatteo G, Cacchiò G, Alia S, Silvestrini M, Paci C, et al. Multiple Sclerosis: Inflammatory and Neuroglial Aspects. Curr Issues Mol Biol. 2023;45(2):1443-70.
- Pender MP, Burrows SR. Epstein-Barr virus and multiple sclerosis: potential opportunities for immunotherapy. Clin Transl Immunology. 2014;3(10):e27.
- Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9(6):393-407.
- Goris A, Vandebergh M, McCauley JL, Saarela J, Cotsapas C. Genetics of multiple sclerosis: lessons from polygenicity. The Lancet Neurology. 2022;21(9):830-42.
- Kurtzke JF, Hyllested K. Multiple sclerosis in the Faroe Islands. Neurology. 1986;36(3):307-.
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61(4):288-99.
- Simpson S, Jr., Taylor B, Blizzard L, Ponsonby AL, Pittas F, Tremlett H, et al. Higher 25-hydroxyvitamin D is associated with lower relapse risk in multiple sclerosis. Ann Neurol. 2010;68(2):193-203.
- Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14(4):406-19.
- Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67(4):452-61.
- Prinz M, Jung S, Priller J. Microglia Biology: One Century of Evolving Concepts. Cell. 2019;179(2):292-311.
- Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777-83.
- Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225-42.
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, Keeffe S, et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. The Journal of Neuroscience. 2014;34(36):11929.
- Zhang X, Chen F, Sun M, Wu N, Liu B, Yi X, et al. Microglia in the context of multiple sclerosis. Front Neurol. 2023;14:1157287.
- Titus HE, Chen Y, Podojil JR, Robinson AP, Balabanov R, Popko B, Miller SD. Pre-clinical and Clinical Implications of “Inside-Out” vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Frontiers in Cellular Neuroscience. 2020;Volume 14 - 2020.
- Goodin DS, Khankhanian P, Gourraud PA, Vince N. The nature of genetic and environmental susceptibility to multiple sclerosis. PLoS One. 2021;16(3):e0246157.
- Lubetzki C, Stankoff B. Demyelination in multiple sclerosis. Handb Clin Neurol. 2014;122:89-99.
- Sospedra M, Martin R. Immunology of Multiple Sclerosis. Semin Neurol. 2016;36(02):115-27.
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nature Protocols. 2006;1(4):1810-9.
- Fazakerley JK, Webb HE. Semliki Forest Virus-induced, Immune-mediated Demyelination: Adoptive Transfer Studies and Viral Persistence in Nude Mice. Journal of General Virology. 1987;68(2):377-85.
- Oleszak Emilia L, Chang JR, Friedman H, Katsetos Christos D, Platsoucas Chris D. Theiler's Virus Infection: a Model for Multiple Sclerosis. Clinical Microbiology Reviews. 2004;17(1):174-207.
- Lanz TV, Brewer RC, Ho PP, Moon J-S, Jude KM, Fernandez D, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature. 2022;603(7900):321-7.
- Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296-301.
- Allen IV, McQuaid S, Mirakhur M, Nevin G. Pathological abnormalities in the normal-appearing white matter in multiple sclerosis. Neurol Sci. 2001;22(2):141-4.
- Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, et al. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. 2011;70(5):764-73.
- West J, Aalto A, Tisell A, Leinhard OD, Landtblom A-M, Smedby Ö, Lundberg P. Normal Appearing and Diffusely Abnormal White Matter in Patients with Multiple Sclerosis Assessed with Quantitative MR. PLOS ONE. 2014;9(4):e95161.
- Elliott C, Momayyezsiahkal P, Arnold DL, Liu D, Ke J, Zhu L, et al. Abnormalities in normal-appearing white matter from which multiple sclerosis lesions arise. Brain Communications. 2021;3(3):fcab176.
- Sarkar SK, Willson AML, Jordan MA. The Plasticity of Immune Cell Response Complicates Dissecting the Underlying Pathology of Multiple Sclerosis. J Immunol Res. 2024;2024:5383099.
- Ruder J, Rex J, Obahor S, Docampo MJ, Müller AMS, Schanz U, et al. NK Cells and Innate-Like T Cells After Autologous Hematopoietic Stem Cell Transplantation in Multiple Sclerosis. Front Immunol. 2021;12:794077.
- Pender MP. The essential role of Epstein-Barr virus in the pathogenesis of multiple sclerosis. Neuroscientist. 2011;17(4):351-67.
- Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300-12.
- Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131(Pt 7):1749-58.
- Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA. Multiple sclerosis. Nat Rev Dis Primers. 2018;4(1):43.
- t Hart BA, Luchicchi A, Schenk GJ, Killestein J, Geurts JJG. Multiple sclerosis and drug discovery: A work of translation. EBioMedicine. 2021;68:103392.
- Rodriguez M, Scheithauer B. Ultrastructure of multiple sclerosis. Ultrastruct Pathol. 1994;18(1-2):3-13.
- Dietrich J, Blumberg BM, Roshal M, Baker JV, Hurley SD, Mayer-Pröschel M, Mock DJ. Infection with an endemic human herpesvirus disrupts critical glial precursor cell properties. J Neurosci. 2004;24(20):4875-83.
- Wongchitrat P, Chanmee T, Govitrapong P. Molecular Mechanisms Associated with Neurodegeneration of Neurotropic Viral Infection. Molecular Neurobiology. 2024;61(5):2881-903.
- Harberts E, Yao K, Wohler JE, Maric D, Ohayon J, Henkin R, Jacobson S. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc Natl Acad Sci U S A. 2011;108(33):13734-9.
- Luchicchi A, Hart B, Frigerio I, van Dam AM, Perna L, Offerhaus HL, et al. Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Ann Neurol. 2021;89(4):711-25.
- Yang L, Tan D, Piao H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem Res. 2016;41(8):1845-56.
- Martín Monreal MT, Hansen BE, Iversen PF, Enevold C, Ødum N, Sellebjerg F, et al. Citrullination of myelin basic protein induces a Th17-cell response in healthy individuals and enhances the presentation of MBP85-99 in patients with multiple sclerosis. J Autoimmun. 2023;139:103092.
- Faigle W, Cruciani C, Wolski W, Roschitzki B, Puthenparampil M, Tomas-Ojer P, et al. Brain Citrullination Patterns and T Cell Reactivity of Cerebrospinal Fluid-Derived CD4+ T Cells in Multiple Sclerosis. Frontiers in Immunology. 2019;Volume 10 - 2019.
- Tranquill LR, Cao L, Ling NC, Kalbacher H, Martin RM, Whitaker JN. Enhanced T cell responsiveness to citrulline-containing myelin basic protein in multiple sclerosis patients. Multiple Sclerosis Journal. 2000;6(4):220-5.
- Martin R, Whitaker JN, Rhame L, Goodin RR, McFarland HF. Citrulline-containing myelin basic protein is recognized by T-cell lines derived from multiple sclerosis patients and healthy individuals. Neurology. 1994;44(1):123-.
- Musse AA, Li Z, Ackerley CA, Bienzle D, Lei H, Poma R, et al. Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Disease Models & Mechanisms. 2008;1(4-5):229-40.
- Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci. 2006;26(44):11387-96.
- Yang QQ, Zhou JW. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia. 2019;67(6):1017-35.
- Maroto M, Fernández-Morales JC, Padín JF, González JC, Hernández-Guijo JM, Montell E, et al. Chondroitin sulfate, a major component of the perineuronal net, elicits inward currents, cell depolarization, and calcium transients by acting on AMPA and kainate receptors of hippocampal neurons. J Neurochem. 2013;125(2):205-13.
- Tosun D, Schuff N, Rabinovici GD, Ayakta N, Miller BL, Jagust W, et al. Diagnostic utility of ASL-MRI and FDG-PET in the behavioral variant of FTD and AD. Ann Clin Transl Neurol. 2016;3(10):740-51.
- Carrillo-Vico A, Leech MD, Anderton SM. Contribution of Myelin Autoantigen Citrullination to T Cell Autoaggression in the Central Nervous System. The Journal of Immunology. 2010;184(6):2839-46.
- McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38(3):229-40.
- Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol. 2007;Chapter 15:15.1.1-.1.8.
- Baker D, Nutma E, O'Shea H, Cooke A, Orian JM, Amor S. Autoimmune encephalomyelitis in NOD mice is not initially a progressive multiple sclerosis model. Ann Clin Transl Neurol. 2019;6(8):1362-72.
- Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079-106.
- Feizi N, Focaccetti C, Pacella I, Tucci G, Rossi A, Costanza M, et al. CD8+ T cells specific for cryptic apoptosis-associated epitopes exacerbate experimental autoimmune encephalomyelitis. Cell Death & Disease. 2021;12(11):1026.
- Voskuhl RR, MacKenzie-Graham A. Chronic experimental autoimmune encephalomyelitis is an excellent model to study neuroaxonal degeneration in multiple sclerosis. Frontiers in Molecular Neuroscience. 2022;Volume 15 - 2022.
- Lauren G, Richard R, Rhian E, Ryan JB, Mark IR, Djordje G, et al. Substantial subpial cortical demyelination in progressive multiple sclerosis: have we underestimated the extent of cortical pathology? Neuroimmunology and Neuroinflammation. 2020;7(1):51-67.
- Lazarević M, Stanisavljević S, Nikolovski N, Dimitrijević M, Miljković Đ. Complete Freund's adjuvant as a confounding factor in multiple sclerosis research. Front Immunol. 2024;15:1353865.
- Rodriguez M, Leibowitz JL, Lampert PW. Persistent infection of oligodendrocytes in Theiler's virus-induced encephalomyelitis. Ann Neurol. 1983;13(4):426-33.
- Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17(1):174-207.
- Bettelli E, Baeten D, Jäger A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest. 2006;116(9):2393-402.
- Olson JK, Croxford JL, Calenoff MA, Dal Canto MC, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. J Clin Invest. 2001;108(2):311-8.
- Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci. 2014;8:73.
- Lipton HL, Dal Canto MC. Chronic neurologic disease in Theiler's virus infection of SJL/J mice. J Neurol Sci. 1976;30(1):201-7.
- Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 2005;7 Suppl 2(Suppl 2):S4-14.
- Libbey JE, Lane TE, Fujinami RS. Axonal pathology and demyelination in viral models of multiple sclerosis. Discov Med. 2014;18(97):79-89.
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21(23):9246-54.
- Rivailler P, Cho YG, Wang F. Complete genomic sequence of an Epstein-Barr virus-related herpesvirus naturally infecting a new world primate: a defining point in the evolution of oncogenic lymphocryptoviruses. J Virol. 2002;76(23):12055-68.
- Correia S, Bridges R, Wegner F, Venturini C, Palser A, Middeldorp JM, et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J Virol. 2018;92(22).
- t Hart BA, Jagessar SA, Haanstra K, Verschoor E, Laman JD, Kap YS. The Primate EAE Model Points at EBV-Infected B Cells as a Preferential Therapy Target in Multiple Sclerosis. Front Immunol. 2013;4:145.
- Mangiardi M, Crawford DK, Xia X, Du S, Simon-Freeman R, Voskuhl RR, Tiwari-Woodruff SK. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol. 2011;21(3):263-78.
- Packer D, Fresenko EE, Harrington EP. Remyelination in animal models of multiple sclerosis: finding the elusive grail of regeneration. Front Mol Neurosci. 2023;16:1207007.
- Wong SW, Bergquam EP, Swanson RM, Lee FW, Shiigi SM, Avery NA, et al. Induction of B cell hyperplasia in simian immunodeficiency virus-infected rhesus macaques with the simian homologue of Kaposi's sarcoma-associated herpesvirus. J Exp Med. 1999;190(6):827-40.
- Madsen MAJ, Wiggermann V, Bramow S, Christensen JR, Sellebjerg F, Siebner HR. Imaging cortical multiple sclerosis lesions with ultra-high field MRI. medRxiv. 2021:2021.06.25.21259363.
- Ptito M, Moesgaard SM, Gjedde A, Kupers R. Cross-modal plasticity revealed by electrotactile stimulation of the tongue in the congenitally blind. Brain. 2005;128(3):606-14.
- Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol. 2014;159(5):863-70.
- Schreiner P, Harrer T, Scheibenbogen C, Lamer S, Schlosser A, Naviaux RK, Prusty BK. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Immunohorizons. 2020;4(4):201-15.
- Leibovitch E, Wohler JE, Cummings Macri SM, Motanic K, Harberts E, Gaitán MI, et al. Novel marmoset (Callithrix jacchus) model of human Herpesvirus 6A and 6B infections: immunologic, virologic and radiologic characterization. PLoS Pathog. 2013;9(1):e1003138.
- Voumvourakis KI, Fragkou PC, Kitsos DK, Foska K, Chondrogianni M, Tsiodras S. Human herpesvirus 6 infection as a trigger of multiple sclerosis: an update of recent literature. BMC Neurol. 2022;22(1):57.
- Broccolo F, Fusetti L, Ceccherini-Nelli L. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. ScientificWorldJournal. 2013;2013:867389.
- Pantry SN, Medveczky PG. Latency, Integration, and Reactivation of Human Herpesvirus-6. Viruses. 2017;9(7).
- Ljungman P. Is antiviral therapy against HHV-6B beneficial? Blood. 2020;135(17):1413-4.
- Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995;92(16):7440-4.
- Sanders VJ, Felisan S, Waddell A, Tourtellotte WW. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol. 1996;2(4):249-58.
- Hall CB, Long CE, Schnabel KC, Caserta MT, McIntyre KM, Costanzo MA, et al. Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N Engl J Med. 1994;331(7):432-8.
- Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland HF, Jacobson S. Tissue distribution and variant characterization of human herpesvirus (HHV)-6: increased prevalence of HHV-6A in patients with multiple sclerosis. J Infect Dis. 2000;182(5):1321-5.
- Abdel-Haq NM, Asmar BI. Human herpesvirus 6 (HHV6) infection. Indian J Pediatr. 2004;71(1):89-96.
- Lundström W, Gustafsson R. Human Herpesvirus 6A Is a Risk Factor for Multiple Sclerosis. Frontiers in Immunology. 2022;Volume 13 - 2022.
- Grut V, Biström M, Salzer J, Stridh P, Jons D, Gustafsson R, et al. Human herpesvirus 6A and axonal injury before the clinical onset of multiple sclerosis. Brain. 2024;147(1):177-85.
- Lyman MG, Enquist LW. Herpesvirus interactions with the host cytoskeleton. J Virol. 2009;83(5):2058-66.
- Romeo MA, Gilardini Montani MS, Gaeta A, D'Orazi G, Faggioni A, Cirone M. HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 2020;1866(3):165647.
- Li L, Chi J, Zhou F, Guo D, Wang F, Liu G, et al. Human herpesvirus 6A induces apoptosis of HSB-2 cells via a mitochondrion-related caspase pathway. J Biomed Res. 2010;24(6):444-51.
- Gu B, Zhang G-F, Li L-Y, Zhou F, Feng D-J, Ding C-L, et al. Human herpesvirus 6A induces apoptosis of primary human fetal astrocytes via both caspase-dependent and -independent pathways. Virology Journal. 2011;8(1):530.
- Opsahl ML, Kennedy PG. Early and late HHV-6 gene transcripts in multiple sclerosis lesions and normal appearing white matter. Brain. 2005;128(Pt 3):516-27.
- Ahlqvist J, Fotheringham J, Akhyani N, Yao K, Fogdell-Hahn A, Jacobson S. Differential tropism of human herpesvirus 6 (HHV-6) variants and induction of latency by HHV-6A in oligodendrocytes. J Neurovirol. 2005;11(4):384-94.
- Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol. 2013;8(4):840-56.
- de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. 2002;169(10):5415-23.
- Leibovitch EC, Jacobson S. Evidence linking HHV-6 with multiple sclerosis: an update. Curr Opin Virol. 2014;9:127-33.
- Álvarez-Lafuente R, Heras VD, Bartolomé M, García-Montojo M, Arroyo R. Human Herpesvirus 6 and Multiple Sclerosis: A One-Year Follow-up Study. Brain Pathology. 2006;16(1):20-7.
- Lassmann H. Multiple Sclerosis Pathology. Cold Spring Harb Perspect Med. 2018;8(3).
- Fotheringham J, Jacobson S. Human herpesvirus 6 and multiple sclerosis: potential mechanisms for virus-induced disease. Herpes. 2005;12(1):4-9.
- Weaver GC, Schneider CL, Becerra-Artiles A, Clayton KL, Hudson AW, Stern LJ. The HHV-6B U20 glycoprotein binds ULBP1, masking it from recognition by NKG2D and interfering with natural killer cell activation. Frontiers in Immunology. 2024;Volume 15 - 2024.
- Wang F-Z, Pellett PE. HHV-6A, 6B, and 7: immunobiology and host response. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. p. 850-74.
- Hanson DJ, Hill JA, Koelle DM. Advances in the Characterization of the T-Cell Response to Human Herpesvirus-6. Frontiers in Immunology. 2018;Volume 9 - 2018.
- Denes E, Magy L, Pradeau K, Alain S, Weinbreck P, Ranger-Rogez S. Successful treatment of human herpesvirus 6 encephalomyelitis in immunocompetent patient. Emerg Infect Dis. 2004;10(4):729-31.
- Eliassen E, Di Luca D, Rizzo R, Barao I. The Interplay between Natural Killer Cells and Human Herpesvirus-6. Viruses. 2017;9(12).
- Bortolotti D, Gentili V, Bortoluzzi A, Govoni M, Schiuma G, Beltrami S, et al. Herpesvirus Infections in KIR2DL2-Positive Multiple Sclerosis Patients: Mechanisms Triggering Autoimmunity. Microorganisms [Internet]. 2022; 10(3).
- Aarons GA, Goldman MS, Greenbaum PE, Coovert MD. Alcohol expectancies:: Integrating cognitive science and psychometric approaches. Addictive Behaviors. 2003;28(5):947-61.
- Jakhmola S, Upadhyay A, Jain K, Mishra A, Jha HC. Herpesviruses and the hidden links to Multiple Sclerosis neuropathology. Journal of Neuroimmunology. 2021;358:577636.
- Skuja S, Zieda A, Ravina K, Chapenko S, Roga S, Teteris O, et al. Structural and Ultrastructural Alterations in Human Olfactory Pathways and Possible Associations with Herpesvirus 6 Infection. PLOS ONE. 2017;12(1):e0170071.
- Das Sarma J. A mechanism of virus-induced demyelination. Interdiscip Perspect Infect Dis. 2010;2010:109239.
- Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5(2):170-5.
- Komaroff AL, Bateman L. Will COVID-19 Lead to Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Front Med (Lausanne). 2020;7:606824.
- Novoa LJ, Nagra RM, Nakawatase T, Edwards-Lee T, Tourtellotte WW, Cornford ME. Fulminant demyelinating encephalomyelitis associated with productive HHV-6 infection in an immunocompetent adult. J Med Virol. 1997;52(3):301-8.
- Prineas JW, Connell F. Remyelination in multiple sclerosis. Ann Neurol. 1979;5(1):22-31.
- Salzer JL. Schwann cell myelination. Cold Spring Harb Perspect Biol. 2015;7(8):a020529.
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233(1-2):3-13.
- Melloni A, Liu L, Kashinath V, Abdi R, Shah K. Meningeal lymphatics and their role in CNS disorder treatment: moving past misconceptions. Frontiers in Neuroscience. 2023;Volume 17 - 2023.
- Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21(10):1380-91.
- van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med (Berl). 2009;87(3):273-86.
- Morandi E, Jagessar SA, t Hart BA, Gran B. EBV Infection Empowers Human B Cells for Autoimmunity: Role of Autophagy and Relevance to Multiple Sclerosis. J Immunol. 2017;199(2):435-48.
- Meier UC, Cipian RC, Karimi A, Ramasamy R, Middeldorp JM. Cumulative Roles for Epstein-Barr Virus, Human Endogenous Retroviruses, and Human Herpes Virus-6 in Driving an Inflammatory Cascade Underlying MS Pathogenesis. Front Immunol. 2021;12:757302.
- Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185-91.
- Rustenhoven J, Drieu A, Mamuladze T, de Lima KA, Dykstra T, Wall M, et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell. 2021;184(4):1000-16.e27.
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353(6299):603-8.
- Shao H, Wu W, Wang P, Han T, Zhuang C. Role of Necroptosis in Central Nervous System Diseases. ACS Chem Neurosci. 2022;13(23):3213-29.
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nature Reviews Molecular Cell Biology. 2013;14(11):727-36.
- Engdahl E, Gustafsson R, Huang J, Biström M, Lima Bomfim I, Stridh P, et al. Increased Serological Response Against Human Herpesvirus 6A Is Associated With Risk for Multiple Sclerosis. Frontiers in Immunology. 2019;Volume 10 - 2019.
- Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183-93.
- Mastronardi FG, Noor A, Wood DD, Paton T, Moscarello MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. 2007;85(9):2006-16.
- Campbell A, Hogestyn JM, Folts CJ, Lopez B, Pröschel C, Mock D, Mayer-Pröschel M. Expression of the Human Herpesvirus 6A Latency-Associated Transcript U94A Disrupts Human Oligodendrocyte Progenitor Migration. Sci Rep. 2017;7(1):3978.
- Pender MP. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003;24(11):584-8.
- t Hart BA. Experimental autoimmune encephalomyelitis in the common marmoset: a translationally relevant model for the cause and course of multiple sclerosis. Primate Biol. 2019;6(1):17-58.
- Bahramian E, Furr M, Wu JT, Ceballos RM. Differential Impacts of HHV-6A versus HHV-6B Infection in Differentiated Human Neural Stem Cells. Frontiers in Immunology. 2022;Volume 13 - 2022.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



