
Submitted:
24 June 2025
Posted:
25 June 2025
You are already at the latest version
Abstract
Nitrogen-containing bisphosphonates (N-BPs) are commonly used drugs in the treatment of bone diseases due to their potent inhibition of the mevalonate pathway, leading to disrupted protein prenylation and reduced osteoclast activity. Although N-BPs are effective in reducing bone resorption, increasing evidence indicates their side effects on various non-skeletal cells. At the cellular level, N-BPs may reduce viability, modulate inflammatory responses, trigger apoptosis, disrupt cytoskeletal organization and influence signaling and energy metabolism. N-BPs may also impair prenylation of proteins essential for mitochondrial dynamics and quality control, and may disrupt Ca2+ homeostasis. By inhibiting the mevalonate pathway, N-BPs may lead to the reduction of key components of the mitochondrial respiratory chain, such as coenzyme Q (CoQ) and a-heme. These effects can contribute to impaired mitochondrial respiratory function, increased oxidative stress and mitochondria-dependent apoptosis, effecting cellular energy metabolism and viability. These findings underscore the multifaceted impact of N-BPs beyond bone, emphasizing the importance of mitochondrial health and energy metabolism in understanding their broader biological effects and potential adverse outcomes.
Keywords:
mevalonate pathway
; mitochondria
; nitrogen-containing bisphosphonates
1. Introduction
1.1. History of Bisphosphonates
Osteoporosis is the most common metabolic bone disease, particularly prevalent among postmenopausal women. As the world's population ages, the incidence of osteoporosis is increasing, with over 200 million people now suffering from the disease [1]. Bone health is maintained by a delicate balance between bone resorption by osteoclasts and bone formation by osteoblasts. This balance can be disturbed by many factors, including aging, genetic disease, lifestyle and endocrine disorders [2]. The primary goal of osteoporosis treatment is the prevention of fractures, which can be achieved either by stimulating bone formation or, more commonly, by inhibiting bone resorption. Bisphosphonates (BPs) are the most commonly prescribed antiresorptive drugs, approved as first-line treatment for osteoporosis, Paget's disease and bone diseases in cancer patients [3]. Their clinical use has also been extended to the treatment of bone metastases and other diseases associated with increased bone turnover.
BPs are stable synthetic pyrophosphate (PPi) analogues, in which the central carbon atom replaces the PPi oxygen bridge and carries two variable side chains, R1 and R2 (Figure 1). Historically, before their potential in preventing bone resorption was discovered in 1969 [4,5], they were first used industrially for their calcium-chelating properties [6,7] as water softeners, corrosion inhibitors and fertilizers. Manipulation of BP side chains has led to the discovery of new compounds with different potencies, while the synthesis of BPs containing a nitrogen atom in the R2 alkyl chain has created a new type of drugs with significantly improved antiresorptive properties [8].
1.2. Types of BPs
BPs are divided into two main groups based on their R2 side chain – non-nitrogen-containing bisphosphonates and nitrogen-containing bisphosphonates (N-BPs) [9]. N-BPs can be further divided into second-generation (including alendronate) and more efficient third-generation (including ibandronate and zoledronate) drugs. First-generation BPs ‒ non-nitrogen-containing compounds such as etidronate and clodronate ‒ can interfere with ATP-dependent enzymes by generating non-hydrolyzable ATP analogues [10]. These analogues compete with ATP, ultimately inducing apoptosis in osteoclasts. Most often, these types of BPs contain a simple hydroxyl group in the R1 chain, while the R2 chain may contain a chlorine atom (in clodronate) or an alkyl group (in etidronate). Clodronate is metabolized by class II aminoacyl-tRNA synthases to adenosine-5′-(β,γ-dichloromethylene) triphosphate (AppCCl₂p), a toxic ATP analogue that accumulates and disrupts cellular energetics and other cellular processes [11,12]. Non-hydrolyzable ATP analogues may reduce cytokine release, offering potential in treating inflammatory conditions. They reach mitochondria, changing oxygen consumption and depolarizing the mitochondrial membrane potential [13].
In contrast, N-BPs act by inhibiting farnesyl diphosphate synthase (FPP synthase) in the mevalonate pathway, preventing prenylation of proteins essential for osteoclast survival and function (e.g., membrane ruffling and stress fibre assembly) [13,14,15]. Greater efficacy of N-BPs compared to first-generation BPs is attributed to their greater affinity for bone mineral [14]. Bone-targeting properties of N-BPs provide selective accumulation at resorption sites, where they effectively inhibit osteoclast recruitment, activity and induce apoptosis. The most commonly used N-BPs are alendronate, prescribed primarily to prevent bone fractures, and zoledronate, a more potent agent used to prevent pathological fractures or treat bone loss associated with cancer [15].
1.3. Side Effects of N-BPs
N-BPs can be administered intravenously or orally. However, the oral administration is linked with poor absorption and has many contradictions. The negative charge of N-BPs impedes their transport across the lipophilic membrane. A similar effect is achieved by complexing with calcium ions, to which they owe their high affinity to sites of intense bone resorption. Most of the contradictions of N-BPs use are connected to the frequent comorbidities of osteoporosis, e.g., inflammatory bowel diseases (Crohn’s disease, ulcerative colitis, celiac disease), small bowel resection or gastojejunostomies [16]. Intravenous treatment with N-BPs, particularly zoledronate, shows greater drug adhesion to the bone surface, with approximately 50–70% of the dose binding to bone minerals within 6–10 hours. Compared to the low oral bioavailability (~5%), intravenous treatment demonstrates its superiority [17,18].
Use of alendronate, a second-generation bisphosphonate, has tripled from 2.7% in the 1990s to 7.8% in less than a decade [19]. The global market of N-BPs is expected to be 9.8 billion dollars by 2032 [20]. Given this, the side effects of these medications have been extensively studied (Table 1). Upper gastrointestinal side effects are most commonly reported with oral N-BPs. Failure to maintain an upright position for 60 minutes after taking the drug caused esophagitis in patients [21].
The most feared side effect of intravenous N-BPs is osteonecrosis of the jaw, which is likely due to the antiangiogenic properties of N-BPs. Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is thought to be related to the use of high doses of the drugs, but the condition also affects patients taking lower doses. More recently, BRONJ has been proposed to be caused by the presence of Actinomyces infection, decreased bone turnover, inhibition of angiogenesis and immune dysregulation [23]. Ziebart et al. suggested that the effect of N-BPs on the onset of BRONJ could be reversed [24]. By studying the products of the mevalonate pathway, they found that one of them, geranylgeraniol (GGOH), when added to in vitro cultures of HUVEC cells, reversed the effects of clodronate, zoledronate, pamidronate and ibandronate.
2. Mevalonate Pathway – A Biological Role and Products
The mevalonate pathway is a key metabolic pathway that supplies isoprenoid units and plays a central role in the biosynthesis of important molecules, including cholesterol, steroid hormones, heme a and coenzyme Q (CoQ) (Figure 2) [26,27]. The mevalonate pathway has been the subject of in-depth research due to its importance in the pathophysiological processes of cancer, cardiovascular and neurodegenerative diseases [27].
2.1. Cholesterol
Cholesterol, the major sterol in mammals, is found in all cell membranes and forms lipid rafts that are an important part of signaling complexes. It constitutes about one third of lipids in the plasma membrane, packaging phospholipids into membranes and insuring their fluidity and barrier function [28]. The sterols present in cell membranes are mainly produced endogenously. If cholesterol production is upregulated, sterol regulatory element-binding proteins (SREBP) step in, step in, regulating the expression of genes involved in lipid synthesis and maintaining a narrow, nontoxic range of this molecule [29]. Cholesterol is also essential as a precursor for the production of steroid hormones. Increasing evidence suggests that cancer cells are highly dependent on high cholesterol levels, moderating the antiviral response of type I interferon (IFN) in macrophages while avoiding detection by the immune system [30].
2.2. Dolichol
Dolichol, a polyprenol, is an another product of the mevalonate pathway which plays an important role in protein glycosylation. As an essential oligosaccharide carrier, it is involved in the synthesis of C-mannosylation, glycosylphosphatidylinositol (GPI)-anchored protein and N-glycans [31]. It has been described over 30 years ago that the level of dolichol increases with age in the human brain. Although in neurodegenerative diseases, such as Alzheimer’s, the situation is opposite showing possible increases in dolichyl phosphate glycosylation as the part of the pathophysiological processes of this disease [32].
2.3. CoQ
CoQ is a lipid-soluble molecule found in cell membranes. Although the human body is able to synthesize it, it is also abundant in our diet. CoQ is an important electron transport carrier of the respiratory chain involved in producing ATP by mitochondrial oxidative phosphorylation system (Figure 3) [33]. CoQ moves across the inner mitochondrial membrane, transporting electrons from substrate dehydrogenases (complexes I and II), where it is reduced, to complex III, facilitating electron transport in the respiratory chain. In addition, CoQ is an antioxidant and can replenish the pool of other antioxidants, such as vitamin C or E, by reducing their oxidized form. The antioxidant properties of CoQ are largely dependent on processes that renew the reduced pool of CoQH2 [34]. However, if the molecule is not fully reduced, a semiquinone radical can be formed, which reacts with oxygen to produce a precursor of various reactive oxygen species (ROS) – the superoxide anion. CoQ levels naturally decline with age, and decreased concentrations have also been observed in various pathological conditions, including diabetes, cancer and neurodegenerative disorders [35].
2.4. Heme a
Heme a is an essential molecule in organisms that depend on aerobic respiration. It is embedded in electron transport chains as a prosthetic group in respiratory oxidases containing cytochrome a [36]. In mammals, hemes a + a3 are components of cytochrome c oxidase (complex IV of the mitochondrial respiratory chain) (Figure 3). Mutations in the COX15 gene encoding heme a synthase cause fatal infantile hypertrophic cardiomyopathy [36]. On the other hand, increased levels of heme a were found in Alzheimer's disease patients [37]. Furthermore, the accumulation of Cox-1 protein and other mitochondrial markers in autophagocytic vesicles is associated with a higher level of mitochondrial turnover.
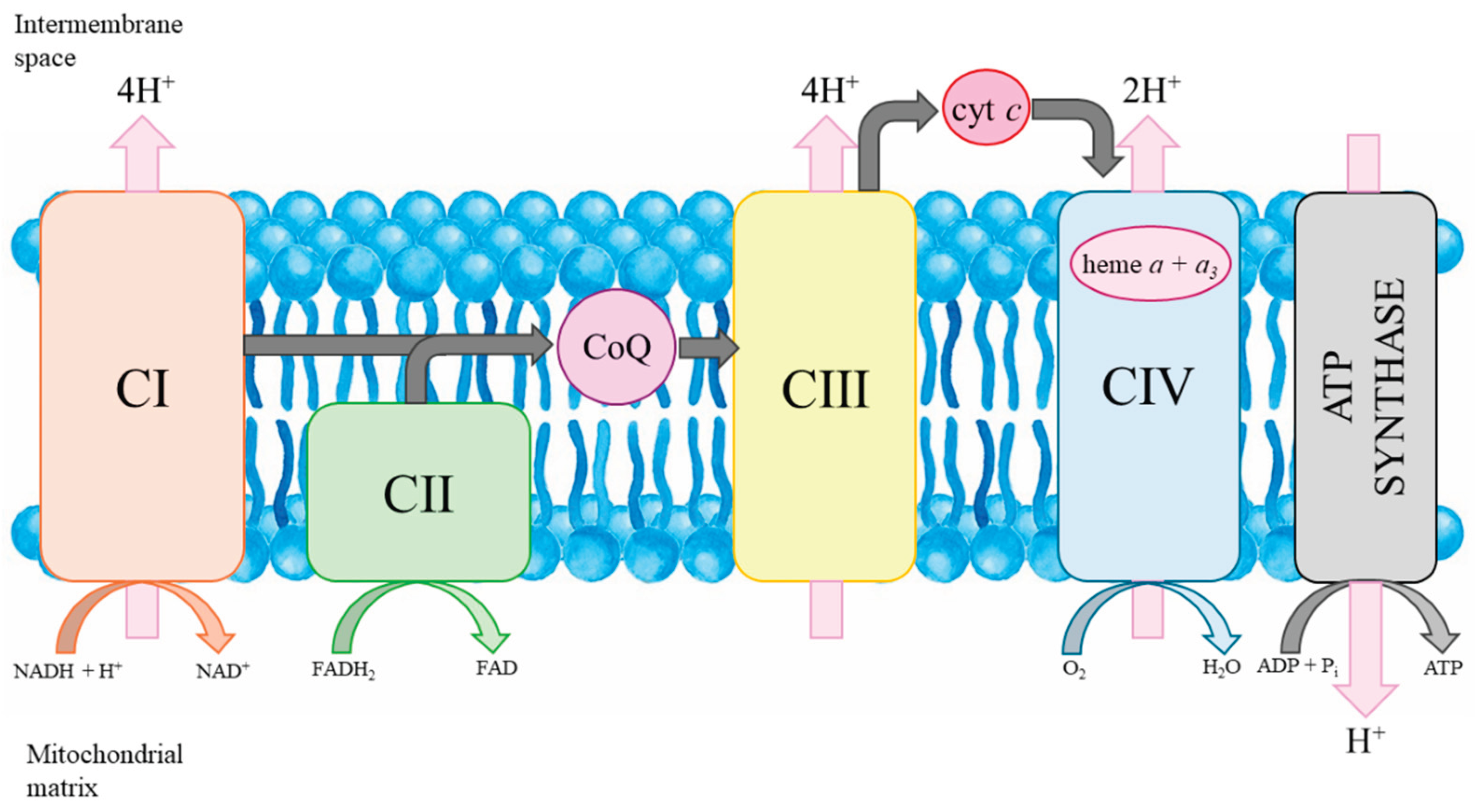
Figure 3.
Mitochondrial oxidative phosphorylation system. cyt c, cytochrome c; CI-CIV, respiratory chain complexes.
Figure 3.
Mitochondrial oxidative phosphorylation system. cyt c, cytochrome c; CI-CIV, respiratory chain complexes.
2.5. Protein Prenylation
Protein prenylation is essential for cell growth, cell division, receptor trafficking and cell polarization. It is the first step in targeting and binding to the cell membrane and also mediates protein-protein interactions. Interest in the process of protein prenylation increased following the description of protein prenylation as being required for the malignant activity of oncogenic Ras proteins [38].
Two main isoprenoid molecules involved in protein prenylation, which are produced in mevalonate pathway, are farnesyl-diphosphate (FPP) and geranylgeranyldiphosphate (GGPP) [39]. GGPP is crucial for membrane localization of small GTP-binding proteins such as Ras, Rho, Rac and Rap. Ras and Rho GTPases require post-translational modifications such as farnesylation or geranylgeranylation. Prenylation allows them to associate with lipid membranes and act as a molecular switch that toggles between an inactive GDP-bound state and an active GTP-bound state. N-BPs inhibit the post-translational modifications of small GTPases by inhibiting the FPP synthase [39]. The concomitant use of N-BPs and statins, other mevalonate pathway inhibitors that block 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCoA reductase), has been proposed as an effective strategy for the treatment of malignant tumors due to the inhibition of FPP and GGPP biosynthesis from two points in the pathway [40].
To inhibit tumor growth and induce apoptosis of cancer cells, a new treatment method was proposed in which zoledronate was placed in a liposome, which allowed to bypass the high affinity of the drug for bone [41]. Interestingly, this technique led to the inhibition of FPP synthase activity, loss of protein prenylation, and also allowed zoledronate to cross the blood-brain barrier, as detected in the brains of the tested mice. It has been suggested that prenylation inhibitors should be further investigated as a potential new class of anti-cancer drugs.
3. Inhibition of the Mevalonate Pathway by N-BPs
3.1. Effects on the Cellular Level
3.1.1. Effects on Bone Cells
The main target of N-BPs are osteoclasts. Selective absorption of N-BP to sites with high bone resorption rates causes the drugs to come into close contact with osteoclasts, which translates into their high safety and efficacy [42]. N-BPs reduce bone resorption by inhibiting osteoclast recruitment, resorption activity and inducing apoptosis. N-BPs, by inhibiting FPP synthase, prevent protein prenylation, which is crucial for obtaining resorption activity by osteoclasts. The correlation between FPP synthase inhibition and antiresorptive activity of N-BPs was shown over 20 years ago by Dunford et al. [43].
Despite the expected effect of reducing the rate of bone resorption, N-BPs also act on other cells with which they come into contact [44]. It has been shown that these drugs can alter the expression of osteoprotegerin (OPG) and macrophage colony stimulating factor (M-CSF) in osteoblasts, affecting osteoblastogenesis. Variable effects on the proliferation and differentiation of osteogenic cells have been observed when different concentrations of N-BPs were administered. The anti-apoptotic properties of N-BPs towards osteoblasts are observed when using concentrations ranging from 10-6 to 10-8 M. The pro-apoptotic events in osteoblasts and osteocytes were observed after increasing the dose to 10-4 -10-5M.
N-BPs inhibit bone resorption leading to lower serum calcium levels [15,45,46]. The P-C-P moiety in N-BPs is responsible for quick binding to the bone mineral. However, the different substitutions in the R1 and R2 positions in the carbon atom allow them to chelate Ca2+ more effectively [47]. Most studies focus on N-BPs effects within osteoclasts or their precursors, particularly concerning calcium-related ion channels. For example, Sheng-Nan et al. investigated the effects of ibandronate on intermediate-conductance Ca²⁺-activated K⁺ (IKCa) channels in the osteoclast precursor cell line RAW 264.7 [48]. They found that ibandronate inhibited IKCa channel activity, resulting in membrane depolarization and reduced cell migration.
3.1.2. N-BP Effects on off-Target Cells
The way in which BP treatment alters cellular processes and leads to apoptosis depends on their molecular structure [10]. Non-nitrogen containing BPs bind to ATP, leading to the production of non-hydrolyzable ATP analogues that compete with the nucleotide, resulting in impaired energy metabolism and apoptosis. N-BPs inhibit FPP synthase, a key enzyme of the mevalonate pathway, thereby preventing the prenylation and activation of small GTPases that are essential for cytoskeletal organization, vesicular trafficking, cell survival and other cellular processes. In vitro studies demonstrating the toxicity of both types of BPs (N-BPs and non-nitrogen containing BPs) do not contain information allowing comparison of the concentrations of these drugs with the concentrations observed in the plasma of patients undergoing BP therapy. In the case of studies with N-BPs, depending on the cell line, their concentrations range from 1 µM in the human endothelial cell line (EA.hy926), 5 µM in the human epithelial cell line (HaCaT), to even 100 µM in the umbilical vein endothelial cell line (HUVEC) [49,50,51].
Anti-Cancer Effects, Cytoskeleton Alterations
Relatively little is known about the effects of N-BPs on tissues other than bone. Most of the existing knowledge comes from studies of cancer cells, as N-BPs are used in anti-cancer therapies [52]. N-BPs, such as zoledronic acid, are widely used to prevent skeletal-related events in cancer patients with bone metastases. Beyond their bone-targeting properties, preclinical studies have demonstrated that N-BPs exert direct anti-cancer effects. These effects include the inhibition of tumor cell proliferation, induction of apoptosis, and modulation of the tumor microenvironment. For instance, N-BPs have been shown to stimulate the expansion and cytotoxic activity of human T cells, which possess potent anti-tumor capabilities. The anti-tumor effect of N-BPs by blocking the prenylation of small GTPases has been explained by the disruption of cytoskeletal organization [53]. After adding 10-5 M alendronate to human prostate cancer (PC-3) cells, Virtanen at al. observed significant changes in the organization of F-actin and cofilin level. Phosphorylation of cofilin is crucial for actin filament elongation, which is carried out by Rho GTPases. This observation led to the hypothesis that N-BPs, which cause loss of prenylation of small GTPases, may be associated with lower levels of phosphophilin, which disrupts the balance between phosphorylated and unphosphorylated cofilin, affecting actin dynamics [54,55]. Morphological changes caused by disturbances in the polymerization of the actin cytoskeleton were observed during zoledronate treatment in fibroblasts, vascular smooth muscle cells and tumor cells [56,57,58].
Effects Related to Nephrotoxicity
In human embryonic kidney (HEK-293) cells, zoledronic acid increase ROS production and induce oxidative damage, as evidenced by altered expression of genes related to oxidative stress and apoptosis [59]. Additionally, endoplasmic reticulum (ER) stress-mediated apoptosis has been observed. Studies in zoledronate-treated rats and renal tubular HK-2 cells suggest that zoledronate-induced nephrotoxicity is linked to disruptions in glutathione biosynthesis and the tricarboxylic acid (TCA) cycle, leading to excessive ROS production, oxidative stress, and inflammation, thereby leading to nephrotoxicity [60].
Effects on Endothelial Cell Viability, Inflammation and Apoptosis
N-BPs have been shown in vitro to reduce the viability of human endothelial cells, induce their inflammatory response and impair the endothelial differentiation potential of human mesenchymal stem cells [51,61,62]. One of the signaling pathways involved in endothelial cell proliferation, apoptosis and angiogenesis is the EGFR/Akt/PI3K pathway, which begins with activation of the epidermal growth factor receptor (EGFR) by ligands, which then triggers activation of phosphatidylinositol 3-kinase (PI3K) and Akt, releasing the Bcl2-related cell death antagonist [63]. The Akt pathway is responsible for the activation of nuclear factor κB (NFκB), reprograming the expression of genes whose products are involved in stress responses, inflammation and inhibition of apoptosis. N-BPs are considered good candidates for anti-cancer activity because they inhibit the key cell survival pathway by inhibiting the activation of Akt and NFκB [64]. Moreover, zoledronate has been shown to impair endothelial cell function and survival by inhibiting the extracellular signal regulated protein kinase 1/2 (ERK1/2) pathway, other prenylation-dependent signaling cascade [65]. Importantly, ERK1/2 also serves as an anti-inflammatory signal to prevent inflammatory signaling in endothelial cells [66]. Endothelial cells treated with zoledronate but not alendronate exhibited a significant decrease in active phospho-ERK1/2 levels, along with increased expression of inflammatory markers [intercellular adhesion molecule 1 (ICAM1) and cytokine interleukin-6 (IL6)], indicating endothelial cell activation and the initiation of local inflammation through the upregulation of inflammatory cytokines and adhesion molecules [51]. In osteoblasts and fibroblasts, zoledronate, unlike alendronate, also induced an increased level of inflammatory cytokines [67]. These observations indicate that zoledronate induces a more potent inflammatory response than alendronate, although both N-BPs have a similar effect on ERK1/2 phosphorylation [51].
Effects on Endothelial Cell CoQ Homeostasis and Energy Metabolism
N-BPs, by inhibiting the mevalonate pathway, lead to the inhibition of synthesis of CoQ, an important intracellular antioxidant that is present in all cell membrane [68,69,70]. Reduced CoQ (CoQH2), by binding free radicals, inhibits lipid peroxidation processes and prevents oxidative modifications of DNA and proteins. Decreased CoQ levels due to age or blockade of the mevalonate pathway by drugs (e.g., statins) may result in oxidative stress and damage. For example, patients with coronary heart disease have lower levels of plasma CoQ, which contributes to lipid peroxidation and DNA damage [71]. In 2014, for the first time (and for a long time the only) an association was observed between N-BP therapy and reduced levels of CoQ10 in the plasma of postmenopausal women [72]. However, to date, CoQ10 supplementation is rarely recommended [72,73]. Reductions in CoQ levels in tissues or organs during N-BP treatment have not yet been investigated, apart from our findings in endothelial cells [51,73]. We recently showed that both alendronate and zoledronate significantly reduced CoQ levels in EA.hy926 human endothelial cells [51]. In particular, in vitro treatment with 2.5 µM zoledronate caused a 60% decrease in cellular CoQ content. N-BPs disrupted the cellular CoQ redox homeostasis by decreasing the CoQ redox state (CoQH2/CoQ). The significant N-BP-induced CoQ deficiency seems to contribute to metabolic reprogramming, including reduced mitochondrial respiratory function and reduced ATP production. A general decline in mitochondrial respiration has been observed in N-BP-treated endothelial cells, particularly with stronger reducing substrates such as pyruvate and glutamate. However, the oxidative capacity as well as aerobic metabolism in N-BP-treated endothelial cells were increased. Impairment of the oxidative phosphorylation system in N-BP-treated endothelial cells was evidenced by reduced ATP-linked respiration with stronger reducing substrates and decreased cellular ATP levels. These alterations in oxidative metabolism resulted in elevated intracellular oxygen levels. N-BP-treated endothelial cells showed significant increases in histone lysine-specific demethylase 6A (KDM6A), a direct oxygen sensor involved in the regulation of gene transcription, along with significant reductions in hypoxia-inducible factor 1α (HIF1α), a key marker of cellular hypoxia. These observations suggest the presence of elevated oxygen levels in N-BP-treated cells, which may enhance reactive oxygen species (ROS) production and contribute to oxidative stress. In N-BP-treated endothelial cells, a decrease in CoQ level, particularly its antioxidant-active reduced form, was accompanied by increased total and mitochondrial ROS production, resulting in oxidative stress, as evidenced by elevated levels of the antioxidant enzymes glutathione reductase and superoxide dismutase (SOD1). These studies represent the first comprehensive investigation into the effects of N-BPs on cellular energy metabolism, using endothelial cells as a model system.
Effects on Lipid Metabolism
In vivo, zoledronate treatment reduced hepatic lipid accumulation in mice fed a high-fat diet, suggesting a potential regulatory effect on lipid metabolism in liver cells [74]. Additionally, zoledronate increased the expression of genes involved in fatty acid oxidation and reduced the size of lipid droplets in hepatocytes of treated mice. An increase in palmitate oxidation was observed in EA.hy926 endothelial cells treated with zoledronate or alendronate, indicating a metabolic shift towards fatty acid catabolism [51]. In contrast to these findings, Cheng et al. showed that zoledronate induces fatty acid accumulation in renal HK-2 cells [75]. It has been proposed that, although β-oxidation of fatty acids is the primary energy source for renal tubular epithelial cells, excessive fatty acid accumulation may contribute to nephrotoxicity and the development of tissue fibrosis or renal disease.
Effects on Authophagy, Mitochondrial Dynamics and Turnover
Defective autophagy has also been linked to a severe reduction in protein prenylation, which may lead to inflammasome activation and subsequent cell death [76]. However, the effects of NBPs, as inhibitors of the mevalonate pathway, on autophagy in non-bone cells remain largely unexplored.
The mevalonate pathway supports the prenylation of small GTPases, including Rab proteins, which are essential for mitochondrial quality control processes such as fission and mitophagy [76,77,78]. However, this aspect remains poorly investigated in the context of N-BP inhibition of the pathway and so far only one study shows that N-BPs can affect mitochondrial dynamics and turnover [51]. In EA.hy926 human endothelial cells treated with N-BPs, no changes were observed in the levels of mitochondrial biogenesis markers, including peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) and nuclear factor erythroid 2–related factor 2 (NRF2) [51]. However, the reduction in mitochondrial fission markers − mitochondrial fission factor (MFF) and active phospho-dynamin-related protein 1 (phospho-DRP1) − along with unchanged levels of the fusion marker optic atrophy protein 1 (OPA1), suggests reduced mitochondrial clearance via mitophagy in N-BP-treated endothelial cells, likely as a consequence of ERK1/2 signaling downregulation. These studies provide the first evidence that N-BPs alter mitochondrial dynamics and turnover.
Effects on Calcium Homeostasis
N-BPs can lower plasma calcium levels by inhibiting bone resorption, thereby reducing the release of calcium into the bloodstream [15,45,46]. Currently, there is limited evidence that N-BPs disrupt calcium homeostasis in tissues beyond bone and blood. For example alendronate has been shown to affect cellular calcium dynamics in cardiomyocytes in vitro [79].
3.2. N-BP Effects on the Mitochondrial Level
Mitochondria are the site of oxidative phosphorylation, the process by which cells produce ATP from nutrients [80,81]. Oxidative phosphorylation occurs across the inner mitochondrial membrane via the electron transport chain and ATP synthase (Figure 3). Mitochondria are also key guardians of cellular homeostasis. In addition to synthesizing ATP via oxidative phosphorylation, mitochondria are also major sources of ROS. While ROS at the physiological level function as critical signaling molecules involved in maintaining cellular homeostasis, excessive ROS production becomes detrimental. When the balance is disturbed, elevated levels of ROS can damage lipids, proteins, and DNA, contributing to oxidative stress and playing a central role in the pathogenesis of various diseases, including neurodegeneration, cancer, and cardiovascular disorders. In addition, impaired mitochondria release apoptotic factors that act as signals that induce cell death. Mitochondria also play an important role in Ca2+ homeostasis by buffering intracellular calcium levels, thereby supporting proper cell function and survival [82].
Effects on Mitochondria-Induced Apoptosis
Mitrofan et al. investigated the mechanism of zoledronate-induced apoptosis in the HF28RA human follicular lymphoma cells and identified key mitochondrial events involved in this process [83]. Zoledronate treatment led to mitochondrial membrane permeabilization, which resulted in the release of cytochrome c into the cytosol, activation of caspase-3, and DNA fragmentation, i.e., hallmarks of mitochondrial-dependent apoptosis. The study identified inhibition of the mevalonate pathway as a major trigger of this response. Geranylgeraniol (GGOH) supplementation, which restores prenylation of proteins downstream of the blocked pathway, effectively rescued cells from zoledronate-induced apoptosis, underscoring the importance of prenylated proteins in maintaining mitochondrial integrity and cell survival. Thus, N-BPs can activate the intrinsic pathway of apoptosis by interfering with mitochondrial function through loss of membrane potential, oxidative stress, and impaired protein prenylation. Although the exact molecular link between N-BP and mitochondrial apoptosis signaling remains unclear, the results support potential therapeutic strategies combining N-BPs with agents that modulate mitochondrial sensitivity or protein prenylation to enhance their efficacy, particularly in resistant cancer cells.
Effects on Mitochondrial Calcium Homeostasis
Some evidence suggests that N-BPs may influence mitochondrial calcium handling in kidney cells. For instance, studies have reported reduced calcium release from renal mitochondria in vitro [84] as well as increased mitochondrial calcium levels in vivo [85]. Interestingly, etidronate has been shown to protect the kidney from ischemic injury [86]. This protective effect is proposed to result from enhanced mitochondrial calcium sequestration, which helps prevent intracellular Ca2+ overload.
N-BP-Induced Mitochondrial Heme a Decrease
Mevalonic acid serves as a precursor for the synthesis of isoprenoid units-containing heme a, an essential prosthetic group of cytochrome c oxidase (complex IV) [87]. In EA.hy926 endothelial cells, treatment with inhibitors of the mevalonate pathway, zoledronate or alendronate, led to a reduction in cytochromes a + a3, suggesting a reduction in the heme a content of complex IV [73]. Despite this reduction, the maximum enzymatic activity of complex IV in intact endothelial mitochondria remained unchanged compared with control untreated cells. Interestingly, this is the only study to date to directly examine the effect of N-BPs on mitochondrial heme a levels, emphasizing the need for further studies to elucidate the broader implications of N-BP-induced disruption of mitochondrial heme a biosynthesis and respiratory chain integrity.
N-BP-Induced Changes in Mitochondrial Respiratory Function
Mitochondrial dysfunction induced by N-BPs remains an underexplored area and relatively few studies have addressed this aspect. In mitochondria isolated from the kidneys of zoledronate-treated rats, significant decreases in mitochondrial dehydrogenase activities, mitochondrial membrane depolarization and permeabilization as well as ATP depletion were observed [88]. In mitochondria isolated from N-BP-treated endothelial cells, an overall reduction in the oxidation of respiratory substrates was observed, with the exception of increased fatty acid oxidation [73]. These mitochondria exhibited reduced respiratory rates, diminished membrane potential and lower efficiency of ATP synthesis during the oxidation of complex I and II substrates. Moreover, in the mitochondria of endothelial cells treated with N-BPs, a reorganization of respiratory chain supercomplexes was observed, notably a downregulation of the complex III2 + IV supercomplex and III2 dimer . This changes were accompanied by reduced protein levels and enzymatic activities of complexes II, III, and V, indicating impaired structural and functional integrity of the electron transport chain.
N-BP-Induced mtCoQ Deficiency
N-BPs as inhibitors of the mevalonate pathway may disturb the synthesis of isoprenoid-derived molecules, including mitochondrial CoQ (mtCoQ), a key electron carrier in the mitochondrial electron transport chain (Figure 3) [69,70]. Reduced mtCoQ (CoQH2) participates in the production of mitochondrial ROS through the formation of superoxide/H2O2 due to electron leakage from the semiubiquinone radical at the mtCoQ-biding sites of the respiratory chain. mtCoQ redox homeostasis, maintained through its cycling between oxidized and reduced forms, is crucial for efficient electron transport and the regulation of oxidative stress [69].
Recently, we demonstrated for the first time that treatment with N-BPs leads to a significant reduction in mtCoQ level. In EA.hy926 endothelial cells treated with alendronate or zoledronate, a 45–55% reduction in total mtCoQ was observed [73]. Importantly, the pool of reduced mtCoQ (mtCoQH2), which is essential for its antioxidant function, was lost. The disrupted mtCoQ redox homeostasis induced a compensatory response characterized by increased expression of mitochondrial antioxidant proteins, including superoxide dismutase 2 (SOD2) and uncoupling protein 2 (UCP2). Moreover, N-BP treatment resulted in a significantly increase in mitochondrial H2O2 production, which has been attributed to increased mtCoQ reduction level (elevated mtCoQH2/mtCoQtot ratio). These findings suggest that decreased mitochondrial CoQ levels, due to mevalonate pathway inhibition by N-BPs, may impair mitochondrial respiratory function and disrupt CoQ redox balance, leading to elevated mitochondrial ROS production. These results underscore the need for further studies on mtCoQ deficiency induced by N-BP treatment. Severe mtCoQ depletion, coupled with excessive mitochondrial ROS production, can disrupt mitochondrial integrity and cellular homeostasis. Such dysfunctions are known to contribute to the pathogenesis of several diseases, including cardiovascular and neurodegenerative diseases, and may be associated with accelerated aging and reduced lifespan [89]. Elucidation of the mechanisms underlying mtCoQ depletion and its downstream effects may point to new strategies to alleviate N-BP-induced mitochondrial toxicity, particularly through CoQ10 supplementation, which may help restore mitochondrial function and redox balance.
4. Conclusions and Future Perspectives
Although widely used N-BPs are clinically effective in treating bone resorption disorders, they are increasingly recognized as drugs with a broad spectrum of adverse effects resulting from inhibition of the mevalonate pathway in non-skeletal cells. The cellular consequences of N-BP exposure include anti-tumor activity, cytoskeletal disruption, renal toxicity, endothelial dysfunction, and alterations in lipid and energy metabolism. Many of these effects may be related to mitochondrial dysfunction, including impaired respiratory function, impaired calcium homeostasis, deficiencies in CoQ and heme a in mitochondria and activation of mitochondria-dependent apoptosis. Despite the growing awareness of these off-target effects, the impact of N-BPs at the mitochondrial level remains a largely unexplored area of research. In particular, the role of mitochondrial CoQ deficiency as a contributor to N-BP toxicity is understudied, and the potential for CoQ supplementation to ameliorate these effects has not been investigated.
Future perspectives should include an in-depth exploration of N-BP-induced mitochondrial dysfunction, with a focus on respiratory chain function and mitochondrial quality control mechanisms. Particular attention should be paid to the evaluation of CoQ supplementation as a potential protective strategy to ameliorate mitochondrial impairment and disruption of cellular energy metabolism. Further studies on the complex interplay between the mevalonate pathway, mitochondrial function, and overall cellular homeostasis will be important to optimize the clinical use of N-BPs while reducing unintended effects on non-target tissues.
Author Contributions
Writing original draft (A.B.), review and editing (W.J.), Funding (W.J.). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre, Poland, grant OPUS 2020/37/B/NZ1/01188.
References
- Cooper, C.; Campion, G.; Melton, L.J. 3rd Hip fractures in the elderly: a world-wide projection. Osteoporos. Int. a J. Establ. as result Coop. between Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 1992, 2, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Sözen, T.; Özışık, L.; Başaran, N.Ç. An overview and management of osteoporosis. Eur. J. Rheumatol. 2017, 4, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.N.; Lie, J.D.; Wan, C.K.V.; Cameron, M.; Austel, A.G.; Nguyen, J.K.; Van, K.; Hyun, D. Osteoporosis: A Review of Treatment Options. P T 2018, 43, 92–104. [Google Scholar]
- Barbosa, J.S.; Braga, S.S.; Almeida Paz, F.A. Empowering the Medicinal Applications of Bisphosphonates by Unveiling their Synthesis Details. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Russell, R.G.G. Bisphosphonates: The first 40years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H.; Russell, R.G.; Francis, M.D. Diphosphonates inhibit hydroxyapatite dissolution in vitro and bone resorption in tissue culture and in vivo. Science 1969, 165, 1262–1264. [Google Scholar] [CrossRef]
- Francis, M.D.; Russell, R.G.; Fleisch, H. Diphosphonates inhibit formation of calcium phosphate crystals in vitro and pathological calcification in vivo. Science 1969, 165, 1264–1266. [Google Scholar] [CrossRef]
- Russell, R.G.; Croucher, P.I.; Rogers, M.J. Bisphosphonates: pharmacology, mechanisms of action and clinical uses. Osteoporos. Int. a J. Establ. as result Coop. between Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 1999, 9 Suppl 2, S66–80. [Google Scholar] [CrossRef]
- Cremers, S.; Drake, M.T.; Ebetino, F.H.; Bilezikian, J.P.; Russell, R.G.G. Pharmacology of bisphosphonates. Br. J. Clin. Pharmacol. 2019, 85, 1052–1062. [Google Scholar] [CrossRef]
- Russell, G.R. Bisphosphonates: mode of action and pharmacology. Pediatrics 2007, 119, 150–162. [Google Scholar] [CrossRef]
- Frith, J.C.; Mönkkönen, J.; Blackburn, G.M.; Russell, R.G.; Rogers, M.J. Clodronate and liposome-encapsulated clodronate are metabolized to a toxic ATP analog, adenosine 5’-(beta, gamma-dichloromethylene) triphosphate, by mammalian cells in vitro. J. bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1997, 12, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Mönkkönen, H.; Rogers, M.J.; Makkonen, N.; Niva, S.; Auriola, S.; Mönkkönen, J. The cellular uptake and metabolism of clodronate in RAW 264 macrophages. Pharm. Res. 2001, 18, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Lehenkari, P.P.; Kellinsalmi, M.; Näpänkangas, J.P.; Ylitalo, K. V; Mönkkönen, J.; Rogers, M.J.; Azhayev, A.; Väänänen, H.K.; Hassinen, I.E. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol. Pharmacol. 2002, 61, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.L.; Guo, K.; Dunford, J.E.; Wu, X.; Knapp, S.; Ebetino, F.H.; Rogers, M.J.; Russell, R.G.G.; Oppermann, U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. 2006, 103, 7829–7834. [Google Scholar] [CrossRef]
- Giannasi, C.; Niada, S.; Farronato, D.; Lombardi, G.; Manfredi, B.; Farronato, G.; Brini, A.T. Nitrogen containing bisphosphonates impair the release of bone homeostasis mediators and matrix production by human primary pre-osteoblasts. Int. J. Med. Sci. 2019, 16, 23–32. [Google Scholar] [CrossRef]
- Diab, D.L.; Watts, N.B.; Miller, P.D. Chapter 80 - Bisphosphonates: Pharmacology and Use in the Treatment of Osteoporosis. In Osteoporosis (Fourth Edition); Marcus, R., Feldman, D., Dempster, D.W., Luckey, M., Cauley, J.A., Eds.; Academic Press: San Diego, 2013; ISBN 978-0-12-415853-5. [Google Scholar]
- Coleman, R.E. Risks and benefits of bisphosphonates. Br. J. Cancer 2008, 98, 1736–1740. [Google Scholar] [CrossRef]
- Jagpal, A.; Saag, K.G. How to use bisphosphonates safely and optimally. Rheumatology 2018, 57, 1875–1876. [Google Scholar] [CrossRef]
- Jha, S.; Wang, Z.; Laucis, N.; Bhattacharyya, T. Trends in Media Reports, Oral Bisphosphonate Prescriptions, and Hip Fractures 1996–2012: An Ecological Analysis. J. Bone Miner. Res. 2015, 30, 2179–2187. [Google Scholar] [CrossRef]
- market.us Global Osteoporosis Drug Market By Drug Class (Bisphosphonates, Parathyroid Hormone Therapy Drugs), By Route Administration (Oral and injectable), and By Distribution Channel (Retail Pharmacies, Hospital Pharmacies), By Region and Companies - Industry Seg; 2023;
- Kennel, K.A.; Drake, M.T. Adverse effects of bisphosphonates: implications for osteoporosis management. Mayo Clin. Proc. 2009, 84, 632–7, quiz 638. [Google Scholar] [CrossRef]
- Dunstan, C.R.; Felsenberg, D.; Seibel, M.J. Therapy insight: the risks and benefits of bisphosphonates for the treatment of tumor-induced bone disease. Nat. Clin. Pract. Oncol. 2007, 4, 42–55. [Google Scholar] [CrossRef]
- Kishimoto, H.; Noguchi, K.; Takaoka, K. Novel insight into the management of bisphosphonate-related osteonecrosis of the jaw (BRONJ). Jpn. Dent. Sci. Rev. 2019, 55, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Ziebart, T.; Koch, F.; Klein, M.O.; Guth, J.; Adler, J.; Pabst, A.; Al-Nawas, B.; Walter, C. Geranylgeraniol – A new potential therapeutic approach to bisphosphonate associated osteonecrosis of the jaw. Oral Oncol. 2011, 47, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef]
- Hashemi, M.; Hoshyar, R.; Ande, S.R.; Chen, Q.M.; Solomon, C.; Zuse, A.; Naderi, M. Mevalonate Cascade and its Regulation in Cholesterol Metabolism in Different Tissues in Health and Disease. Curr. Mol. Pharmacol. 2017, 10, 13–26. [Google Scholar] [CrossRef]
- Lasunción, M.A.; Martínez-Botas, J.; Martín-Sánchez, C.; Busto, R.; Gómez-Coronado, D. Cell cycle dependence on the mevalonate pathway: Role of cholesterol and non-sterol isoprenoids. Biochem. Pharmacol. 2022, 196, 114623. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: convergent physiology — divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Denecke, J.; Kranz, C. Hypoglycosylation due to dolichol metabolism defects. Biochim. Biophys. Acta - Mol. Basis Dis. 2009, 1792, 888–895. [Google Scholar] [CrossRef]
- Edlund, C.; Söderberg, M.; Kristensson, K. Isoprenoids in aging and neurodegeneration. Neurochem. Int. 1994, 25, 35–38. [Google Scholar] [CrossRef]
- Sood, B.; Patel, P.; Keenaghan, M. Coenzyme Q10. In; Treasure Island (FL), 2024.
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- de la Bella-Garzón, R.; Fernández-Portero, C.; Alarcón, D.; Amián, J.G.; López-Lluch, G. Levels of Plasma Coenzyme Q(10) Are Associated with Physical Capacity and Cardiovascular Risk in the Elderly. Antioxidants (Basel, Switzerland) 2022, 11. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 Produce a Defect in the Mitochondrial Heme Biosynthetic Pathway, Causing Early-Onset Fatal Hypertrophic Cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Dwyer, B.E.; Stone, M.L.; Gorman, N.; Sinclair, P.R.; Perry, G.; Smith, M.A.; Zhu, X. Heme-a, the heme prosthetic group of cytochrome c oxidase, is increased in Alzheimer’s disease. Neurosci. Lett. 2009, 461, 302–305. [Google Scholar] [CrossRef]
- Palsuledesai, C.C.; Distefano, M.D. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef]
- Walker, K.; Olson, M.F. Targeting Ras and Rho GTPases as opportunities for cancer therapeutics. Curr. Opin. Genet. Dev. 2005, 15, 62–68. [Google Scholar] [CrossRef]
- Vincenzi, B.; Santini, D.; Avvisati, G.; Baldi, A.; Cesa, A. La; Tonini, G. Statins may potentiate bisphosphonates anticancer properties: a new pharmacological approach? Med. Hypotheses 2003, 61, 98–101. [Google Scholar] [CrossRef]
- Rondeau, J.-M.; Bitsch, F.; Bourgier, E.; Geiser, M.; Hemmig, R.; Kroemer, M.; Lehmann, S.; Ramage, P.; Rieffel, S.; Strauss, A.; et al. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem 2006, 1, 267–273. [Google Scholar] [CrossRef]
- Russell, R.G.G. Determinants of structure–function relationships among bisphosphonates. Bone 2007, 40, S21–S25. [Google Scholar] [CrossRef]
- Dunford, J.E.; Thompson, K.; Coxon, F.P.; Luckman, S.P.; Hahn, F.M.; Poulter, C.D.; Ebetino, F.H.; Rogers, M.J. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 2001, 296, 235–242. [Google Scholar] [CrossRef]
- Xu, X.-L.; Gou, W.-L.; Wang, A.-Y.; Wang, Y.; Guo, Q.-Y.; Lu, Q.; Lu, S.-B.; Peng, J. Basic research and clinical applications of bisphosphonates in bone disease: what have we learned over the last 40 years? J. Transl. Med. 2013, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A. A patent review of bisphosphonates in treating bone disease. Expert Opin. Ther. Pat. 2019, 29, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Shahi, A.K. Nitrogen containing bisphosphonates associated osteonecrosis of the jaws: A review for past 10 year literature. Dent. Res. J. (Isfahan). 2014, 11, 147–153. [Google Scholar] [PubMed]
- Russell, R.G.G.; Rogers, M.J. Bisphosphonates: from the laboratory to the clinic and back again. Bone 1999, 25, 97–106. [Google Scholar] [CrossRef]
- Wu, S.-N.; Huang, Y.-M.; Liao, Y.-K. Effects of Ibandronate Sodium, a Nitrogen-Containing Bisphosphonate, on Intermediate-Conductance Calcium-Activated Potassium Channels in Osteoclast Precursor Cells (RAW 264.7). J. Membr. Biol. 2015, 248, 103–115. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.; Han, W.; Li, H. Zoledronate induces autophagic cell death in human umbilical vein endothelial cells via Beclin-1 dependent pathway activation. Mol. Med. Rep. 2016, 14, 4747–4754. [Google Scholar] [CrossRef]
- Dong, X.; He, Y.; An, J.; He, L.; Zheng, Y.; Wang, X.; Wang, J.; Chen, S.; Zhang, Y. Increased apoptosis of gingival epithelium is associated with impaired autophagic flux in medication-related osteonecrosis of the jaw. Autophagy 2023, 19, 2899–2911. [Google Scholar] [CrossRef]
- Budzinska, A.; Galganski, L.; Jarmuszkiewicz, W. The bisphosphonates alendronate and zoledronate induce adaptations of aerobic metabolism in permanent human endothelial cells. Sci. Rep. 2023, 13, 16205. [Google Scholar] [CrossRef]
- Clézardin, P.; Massaia, M. Nitrogen-containing bisphosphonates and cancer immunotherapy. Curr. Pharm. Des. 2010, 16, 2014–3007. [Google Scholar] [CrossRef]
- Virtanen, S.S.; Ishizu, T.; Sandholm, J.A.; Löyttyniemi, E.; Väänänen, H.K.; Tuomela, J.M.; Härkönen, P.L. Alendronate-induced disruption of actin cytoskeleton and inhibition of migration/invasion are associated with cofilin downregulation in PC-3 prostate cancer cells. Oncotarget 2018, 9, 32593–32608. [Google Scholar] [CrossRef]
- Lauterborn, J.C.; Gall, C.M. Chapter 14 - Defects in Rho GTPase Signaling to the Spine Actin Cytoskeleton in FMR1 Knockout Mice. In; Willemsen, R., Kooy, R.F.B.T.-F.X.S., Eds.; Academic Press, 2017; pp. 277–299 ISBN 978-0-12-804461-2.
- Oleinik, N. V; Helke, K.L.; Kistner-Griffin, E.; Krupenko, N.I.; Krupenko, S.A. Rho GTPases RhoA and Rac1 mediate effects of dietary folate on metastatic potential of A549 cancer cells through the control of cofilin phosphorylation. J. Biol. Chem. 2014, 289, 26383–26394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shen, W.; Zhu, H.; Lin, L.; Jiang, G.; Zhu, Y.; Song, H.; Wu, L. Zoledronate inhibits fibroblasts’ proliferation and activation via targeting TGF-β signaling pathway. Drug Des. Devel. Ther. 2018, 12, 3021–3031. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, L.; Shi, W.-H.; Zhang, J.; Ma, D.; Yu, B. Zoledronate inhibits the proliferation, adhesion and migration of vascular smooth muscle cells. Eur. J. Pharmacol. 2009, 602, 124–131. [Google Scholar] [CrossRef]
- Tassone, P.; Tagliaferri, P.; Viscomi, C.; Palmieri, C.; Caraglia, M.; D’Alessandro, A.; Galea, E.; Goel, A.; Abbruzzese, A.; Boland, C.R.; et al. Zoledronic acid induces antiproliferative and apoptotic effects in human pancreatic cancer cells in vitro. Br. J. Cancer 2003, 88, 1971–1978. [Google Scholar] [CrossRef]
- Kara, M.; Boran, T.; Öztaş, E.; Jannuzzi, A.T.; Özden, S.; Özhan, G. Zoledronic acid-induced oxidative damage and endoplasmic reticulum stress-mediated apoptosis in human embryonic kidney (HEK-293) cells. J. Biochem. Mol. Toxicol. 2022, 36, e23083. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Chai, K.; Jiang, Y.; Liu, X. Characterization of urinary biomarkers and their relevant mechanisms of zoledronate-induced nephrotoxicity using rats and HK-2 cells. Hum. Exp. Toxicol. 2019, 38, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, J.; Guo, T.; Liu, D.; Pan, J. Epidermal Growth Factor Reverses the Inhibitory Effects of the Bisphosphonate, Zoledronic Acid, on Human Oral Keratinocytes and Human Vascular Endothelial Cells In Vitro via the Epidermal Growth Factor Receptor (EGFR)/Akt/Phosphoinositide 3-Kinase (PI3K). Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 700–710. [Google Scholar] [CrossRef]
- Sharma, D.; Hamlet, S.M.; Petcu, E.B.; Ivanovski, S. The effect of bisphosphonates on the endothelial differentiation of mesenchymal stem cells. Sci. Rep. 2016, 6, 20580. [Google Scholar] [CrossRef]
- Treda, C.; Popeda, M.; Ksiazkiewicz, M.; Grzela, D.P.; Walczak, M.P.; Banaszczyk, M.; Peciak, J.; Stoczynska-Fidelus, E.; Rieske, P. EGFR Activation Leads to Cell Death Independent of PI3K/AKT/mTOR in an AD293 Cell Line. PLoS One 2016, 11, e0155230. [Google Scholar] [CrossRef]
- Inoue, R.; Matsuki, N.; Jing, G.; Kanematsu, T.; Abe, K.; Hirata, M. The inhibitory effect of alendronate, a nitrogen-containing bisphosphonate on the PI3K-Akt-NFkappaB pathway in osteosarcoma cells. Br. J. Pharmacol. 2005, 146, 633–641. [Google Scholar] [CrossRef]
- Hasmim, M.; Bieler, G.; Rüegg, C. Zoledronate inhibits endothelial cell adhesion, migration and survival through the suppression of multiple, prenylation-dependent signaling pathways. J. Thromb. Haemost. 2007, 5, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.-S.; Min, J.-K.; Kim, J.H.; Yamagishi, A.; Mochizuki, N.; Kwon, J.-Y.; Park, Y.-W.; Kim, Y.-M.; Kwon, Y.-G. ERK is an anti-inflammatory signal that suppresses expression of NF-kappaB-dependent inflammatory genes by inhibiting IKK activity in endothelial cells. Cell. Signal. 2006, 18, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Açil, Y.; Arndt, M.L.; Gülses, A.; Wieker, H.; Naujokat, H.; Ayna, M.; Wiltfang, J. Cytotoxic and inflammatory effects of alendronate and zolendronate on human osteoblasts, gingival fibroblasts and osteosarcoma cells. J. cranio-maxillo-facial Surg. Off. Publ. Eur. Assoc. Cranio-Maxillo-Facial Surg. 2018, 46, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Broniarek, I.; Jarmuszkiewicz, W. [Statins and mitochondria]. Postepy Biochem. 2016, 62, 77–84. [Google Scholar]
- Jarmuszkiewicz, W.; Dominiak, K.; Budzinska, A.; Wojcicki, K.; Galganski, L. Mitochondrial coenzyme Q redox homeostasis and reactive oxygen species production. Front. Biosci. (Landmark Ed. 2023, 28, 61. [Google Scholar] [CrossRef]
- James, A.M.; Smith, R.A.J.; Murphy, M.P. Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch. Biochem. Biophys. 2004, 423, 47–56. [Google Scholar] [CrossRef]
- Kaya, Y.; Çebı, A.; Söylemez, N.; Demır, H.; Alp, H.H.; Bakan, E. Correlations between oxidative DNA damage, oxidative stress and coenzyme Q10 in patients with coronary artery disease. Int. J. Med. Sci. 2012, 9, 621–626. [Google Scholar] [CrossRef]
- Kalyan, S.; Huebbe, P.; Esatbeyoglu, T.; Niklowitz, P.; Côté, H.C.F.; Rimbach, G.; Kabelitz, D. Nitrogen-bisphosphonate therapy is linked to compromised coenzyme Q10 and vitamin E status in postmenopausal women. J. Clin. Endocrinol. Metab. 2014, 99, 1307–1313. [Google Scholar] [CrossRef]
- Budzinska, A.; Galganski, L.; Wojcicki, K.; Jarmuszkiewicz, W. Adaptation of mitochondrial bioenergetics to coenzyme Q deficiency in human endothelial cells after chronic exposure to bisphosphonates. Sci. Rep. 2025, 15, 17734. [Google Scholar] [CrossRef]
- Tang, Q.; Jiang, S.; Jia, W.; Shen, D.; Qiu, Y.; Zhao, Y.; Xue, B.; Li, C. Zoledronic acid, an FPPS inhibitor, ameliorates liver steatosis through inhibiting hepatic de novo lipogenesis. Eur. J. Pharmacol. 2017, 814, 169–177. [Google Scholar] [CrossRef]
- Cheng, L.; Ge, M.; Lan, Z.; Ma, Z.; Chi, W.; Kuang, W.; Sun, K.; Zhao, X.; Liu, Y.; Feng, Y.; et al. Zoledronate dysregulates fatty acid metabolism in renal tubular epithelial cells to induce nephrotoxicity. Arch. Toxicol. 2018, 92, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Yang, Q.; Guo, Y.; Liu, X.; Shen, D.; Jiang, C.; Wang, X.; Li, K.; Wang, H.; Yang, C.; et al. Oocytes orchestrate protein prenylation for mitochondrial function through selective inactivation of cholesterol biosynthesis in murine species. J. Biol. Chem. 2023, 299, 105183. [Google Scholar] [CrossRef]
- Kemeny-Suss, N.; Kasneci, A.; Rivas, D.; Afilalo, J.; Komarova, S. V; Chalifour, L.E.; Duque, G. Alendronate affects calcium dynamics in cardiomyocytes in vitro. Vascul. Pharmacol. 2009, 51, 350–358. [Google Scholar] [CrossRef]
- San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Mitrofan, L.M.; Castells, F.B.; Pelkonen, J.; Mönkkönen, J. Lysosomal-mitochondrial axis in zoledronic acid-induced apoptosis in human follicular lymphoma cells. J. Biol. Chem. 2010, 285, 1967–1979. [Google Scholar] [CrossRef]
- Guilland, D.F.; Sallis, J.D.; Fleisch, H. The effect of two diphosphonates on the handling of calcium by rat kidney mitochondria in vitro. Calcif. Tissue Res. 1974, 15, 303–314. [Google Scholar] [CrossRef]
- Guilland, D.F.; Fleisch, H. The effect of in vivo treatment with EHDP and/or 1,25-DHCC on calcium uptake and release in isolated kidney mitochondria. Biochem. Biophys. Res. Commun. 1974, 61, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Greif, F.; Anais, D.; Frei, L.; Arbeit, L.; Sorroff, H.S. Blocking the calcium cascade in experimental acute renal failure. Isr. J. Med. Sci. 1990, 26, 301–305. [Google Scholar] [PubMed]
- Keyhani, J.; Keyhani, E. Mevalonic acid as a precursor of the alkyl sidechain of heme a of cytochrome c oxidase in yeast Saccharomyces cerevisiae. FEBS Lett. 1978, 93, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, H.; Rouhani, A.; Yüzügülen, J.; Ghaderi, F.; Fazlinezhad, R.; Kiafar, M.R.; Honarpishefard, Z.; Matinpour, P.; Arjmand, A.; Azarpira, N.; et al. Zoledronic acid-induced mitochondrial impairment, inflammation, and oxidative stress in the rat kidney. Trends Pharm. Sci. 2023, 9, 243–252. [Google Scholar] [CrossRef]
- Díaz-Casado, M.E.; Quiles, J.L.; Barriocanal-Casado, E.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The Paradox of Coenzyme Q(10) in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef]
Figure 1.
General structure of pyrophosphate and bisphosphonate.
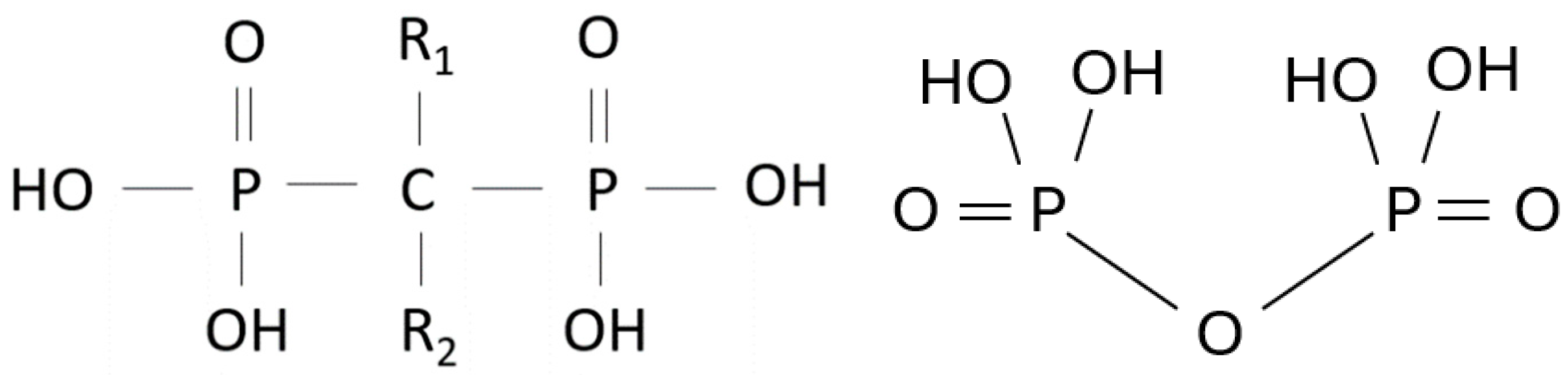
Figure 2.
Mevalonate pathway diagram. HMGCS1 ‒ hydroxymethylglutaryl-CoA synthase, HMG CoA ‒ 3-hydroxy-3-methylglutaryl coenzyme A, HMGCR ‒ 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, FPPS ‒ farnesyldiphosphate synthase.
Figure 2.
Mevalonate pathway diagram. HMGCS1 ‒ hydroxymethylglutaryl-CoA synthase, HMG CoA ‒ 3-hydroxy-3-methylglutaryl coenzyme A, HMGCR ‒ 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, FPPS ‒ farnesyldiphosphate synthase.
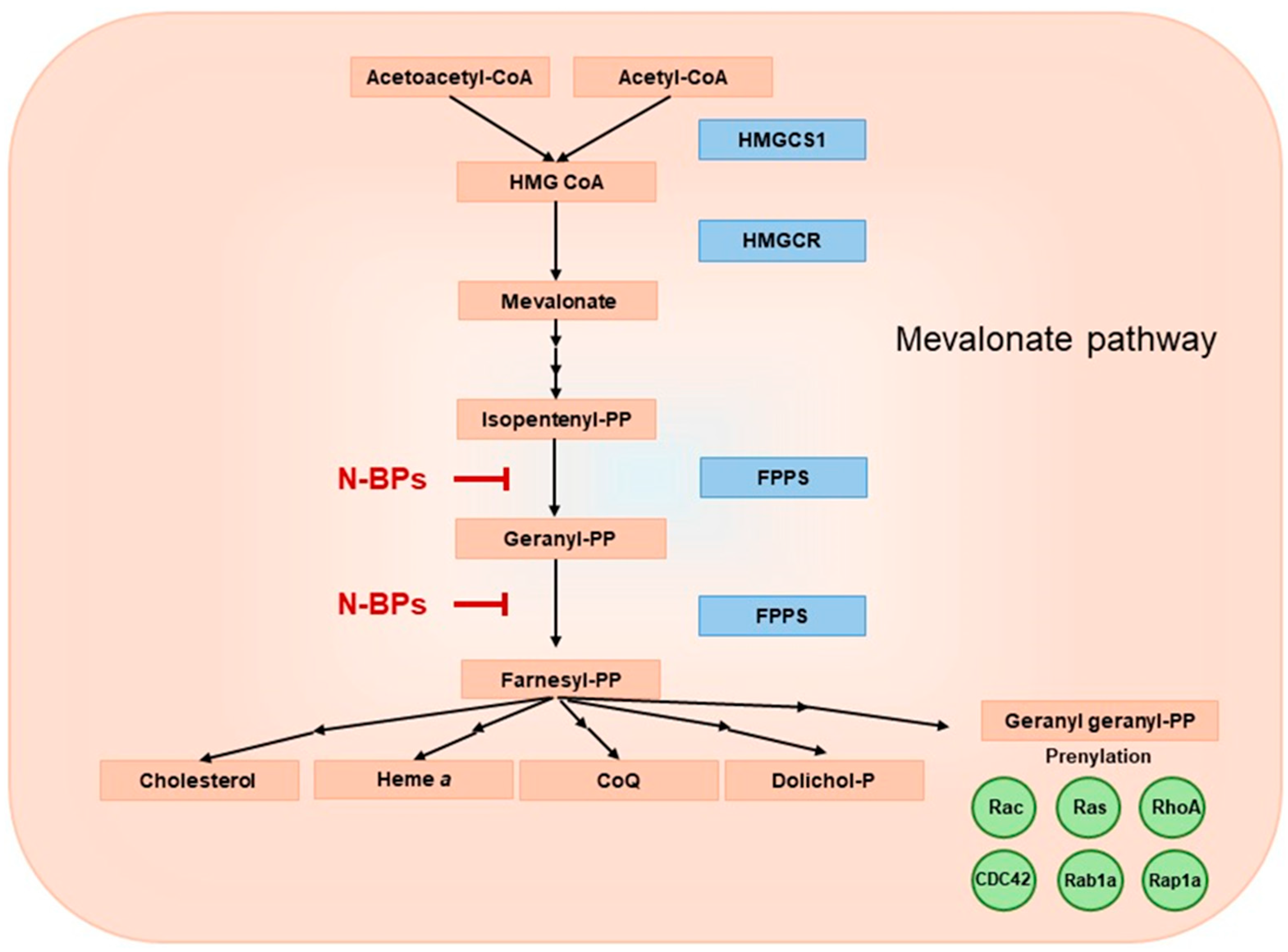

Table 1.
Documented major side effects of BP therapy [22].
Table 1.
Documented major side effects of BP therapy [22].
| Side effects | BPs |
| Osteomalacia | Etidronate |
| Hypocalcemia | Zoledronate, pamidronate, clodronate |
| Acute phase reaction | Zoledronate, ibandronate |
| Gastrointestinal side effects | Orally administered N-BPs, alendronate |
| Nephrotoxicity, renal failure | Zoledronate, pamidronate, ibandronate, alendronate |
| Ocular side effects | Pamidronate, zoledronate |
| Bisphosphonate-related osteonecrosis of the jaw (BRONJ) | Zoledronate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



