Submitted:
21 June 2025
Posted:
23 June 2025
Read the latest preprint version here
Abstract
Alzheimers dementia (AD) is a disease of aging brain. It begins in the hippocampal region with the epicentre in the entorhinal cortex, then gradually extends into adjacent brain areas involved in memory and cognition. The events which initiate the damage are unknown and under intense investigation. Localisation to the hippocampus can now be explained by anatomical features of the blood vessels supplying this region. Blood supply and hence oxygen delivery to the area are jeopardized by poor flow through narrowed arteries. In genomic and metabolomic studies, respiratory chain and mitochondrial pathways which generate ATP were leading pathways associated with AD. This review explores the notion that ATP depletion resulting from hippocampal hypoperfusion has a prime role in initiating damage. Sections cover sensing of ATP depletion and protective responses, vulnerable processes with very heavy ATP consumption (the malate shuttle, the glutamate/ glutamine/ GABA (γ-aminobutyric acid) cycle, and axonal transport), phospholipid disturbances and peroxidation by reactive oxygen species, hippocampal perfusion and the effects of hypertension, chronic hypoxia and arterial vasospasm, and an overview of recent relevant genomic studies. The findings demonstrate strong scientific arguments for the proposal with increasing supportive evidence. These lines of enquiry should be pursued.
Keywords:
ATP biosensors
; malate aspartate shuttle
; glutamate/ GABA/ glutamine cycle
; axonal transport
; vasospasm
; cerebral arterial perfusion
; Mitochondrial- derived peptides
; Membrane phospholipids
1. Introduction
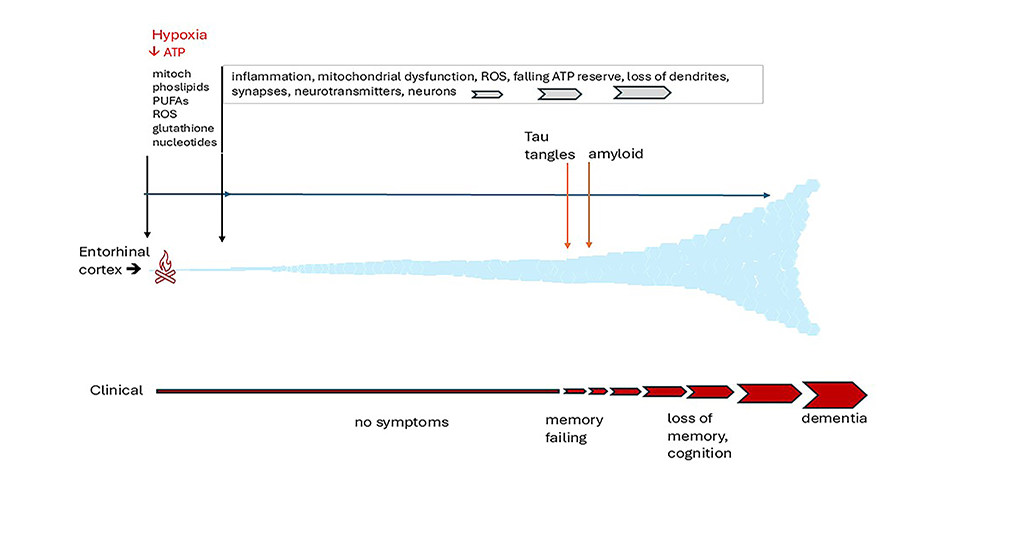
There is a common misconception that Alzheimer’s disease (AD) is merely an exaggeration of the changes which occur normally as brain ages [1]. This is not the case. It is a distinct neurodegenerative disorder which arises within an epicentre in the medial temporal lobe of the brain and leads to characteristic protein accumulations, tau tangles and amyloid plaques, inflammation, destruction of synapses and neuronal loss [2]. These abnormalities are superimposed on changes which occur normally with old age. The earliest changes observed, tau tangles and neuronal loss [1,3] are identifiable in later middle life in the entorhinal cortex (EC), a small region sited deeply in the medial temporal lobe adjoining the hippocampus indicated in Figure 1, which is the main interface between the hippocampus and the brain neocortex [4,5,6].
The EC/ hippocampal system is responsible for creating declarative memories that can be consciously thought of, semantic memory (the ability to recall general facts about the world), memory formation and consolidation, and memory optimization in sleep [3,4,7]. From the EC, the damage extends slowly but relentlessly into the hippocampus, the cingulate gyrus and the brain neocortex, eventually reaching the frontal cortical areas. By this time, there is considerable loss of both grey and white brain matter [8]. When tau tangles are first detectable in the EC, affected individuals may not have had memory problems, or may have only minimal loss of memory and/or cognition (classed as MCI) [9]. However, compared with cognitively normal controls, AD-affected individuals with the mildest clinical dementia had 32% fewer neurons in the EC and 60% and 40% fewer in EC layers II and IV, respectively. In those with severe dementia, neurons in layers II and IV were approximately 90% and 70% fewer than controls. In contrast, cognitively normal subjects had no EC neuronal loss between the sixth and ninth decades of age [1]. Such a dramatic reduction in neurons must have started well before the onset of symptoms [1].
AD is a multifactorial disorder. Table 1 lists some of many factors which have been shown to increase the risk for AD.
Although there is a genetic component to risk, only the 1.5% to 2% of individuals with inherited defects of processing the amyloid precursor protein, APP, have a clear monogenic cause. They present at a young age and are not typical of the 98% of individuals with a later onset of AD (LOAD )[26]. It is apparent from the Table that a high percentage of the world population have these risk factors and yet do not develop the disease [10,11]. To add further confusion, up to one third of community-dwelling older adults have Tau tangles and/or amyloid deposits at autopsy but have experienced negligible cognitive decline in life [2]. Why does AD affect only some of those at risk? Why, of all brain regions, is the hippocampus/EC targeted in AD? Why does this happen in only some individuals? What triggers the process? What is happening during the asymptomatic prodromal phase? These are recurring questions being intensively investigated.
Striking differences in the composition of the brain from other organs increase its vulnerability to pathogenic insult. First, it is wholly reliant on a continuous supply of ATP to function and has a relatively enormous energy requirement for its size, accounting for up 20% of total body energy produced [9]. Hence it is totally dependent on mitochondria which can adapt instantaneously to changing needs. Second, it is a fatty organ. Ten per cent to 20% of the fresh weight and more than 50% of dry weight is composed of lipids [27,28]. Of these, approximately 50% are phospholipids [29] which are the major lipid constituents of membranes [30]; third it has an enormous area of membranes covering the cell surface and internal organelles. Hence the brain is particularly vulnerable to disturbance of lipid turnover; fourth, polyunsaturated fatty acids (PUFAs) are enriched in brain membrane lipids and have an essential structural role. However, they also provide an abundant substrate for cascading free radical attack causing peroxidative membrane damage [31,32]; fifth, it is the most cholesterol-rich body organ. Although accounting for only 2% of body weight it contains around 25% of body cholesterol [33,34]. Most is synthesised in brain and incorporated into membranes [35].
Decades of research studies have shown that many interlinked cellular and molecular disturbances contribute to the AD phenotype, notably neuroinflammation [11], mitochondrial dysfunction [16] and circulatory disturbances [36]. Incredible developments in gene and metabolite analyses, neuroimaging and information technology have enabled deep probing of events in this inaccessible organ. Knowledge is advancing dramatically. It is now widely held that mitochondrial dysfunction has a major role in pathogenesis, and there is growing evidence that it is an impaired blood supply that targets initial damage to the hippocampus.
The review explores the notion that recurrent minor ischaemic events due to reduction in hippocampal blood flow (hypoperfusion) cause episodes of ATP depletion. These are proposed to initiate cellular disturbances which cascade, provoke an inflammatory response, and result in cumulative damage. This combines two concepts which are not new, but it seems timely to review them in the light of recent findings. There are four main sections i) a brief summary of changes which occur normally in aging brain; ii) a large section covering sensing of ATP depletion, processes with very heavy ATP consumption (the malate shuttle, the glutamate/ glutamine/ GABA (γ-aminobutyric acid) cycle and axonal transport), and the consequences of ATP deficiency on lipid turnover, membrane funtion, neurotransmission and synapses; iii) a section covering hippocampal perfusion, responses of cerebral blood flow to hypoxia, neuronal activity and intraluminal pressure, and reduced flow in hypertension and chronic hypoxia; iv) an overview of recent genomic studies relevant to the proposal. From the review, the conclusions are that there are strong scientific arguments for the proposal, that there is supportive evidence, and that these lines of enquiry should be pursued. Studies are already indicating new approaches for preventative interventions, early diagnosis and therapy.
2. Brain Aging
Brain aging is characterized by a progressive loss of grey and white matter volume, a general loss of dendritic spines, loss of synaptic plasticity, increased rates of axonal bouton turnover, and inflammation [reviewed 37]. However, the number of neurons is largely preserved in the neocortex and hippocampus of the aging human brain [1,38].
Brain energy metabolism declines with age, affecting most brain regions [reviewed 39] . Functional brain mitochondrial deficits that occur with age include decreased rates of respiration and of electron transfer, a continuous decrease of the capacity to produce ATP by oxidative phosphorylation [40,41], dynamic changes in shape and size, activation of the permeability transition pore [42] and loss of membrane potential [39]. Increased mitochondrial fusion with elongation was observed with age [43]. Mitochondrial fission factors were reduced in aging mice [44], but were increased in synaptic mitochondria [44]. Expression of OXPHOS proteins in whole brain tissue decreases in advanced age, particularly of complexes I, IV and V [39,40], but inner membrane H+ impermeability and F1-ATP synthase activity are only slightly affected [40]. Expression of TCA cycle proteins was lower in whole brain of elderly mice [45] and rats [46] compared with middle aged rodents. Pathway analysis of expressed genes identified the synaptic vesicle cycle, the GMP-PKG (cGMP-protein kinase G) signaling pathway and oxidative phosphorylation as core gene sets showing highest association with human brain aging [47]. Production of mitochondrial superoxide, oxidative damage and peroxidation of membrane lipids increases with age [48,49]. Significant changes in expression of genes affecting synaptic function were observed in human brain over the age range 20-99 years, often showing progressive down regulation [50]. With a highly sensitive imaging procedure, compared with young animals glutamate levels were significantly lower in elderly lemurs in the hypothalamus and other brain regions, particularly the globus pallidus and nucleus accumbens [51].
3. A Role for ATP Depletion in the Genesis of AD
3.1. Matching ATP Production to Requirement in the Brain
The brain has a continuous high requirement for ATP which oscillates with neuronal activity and cellular stresses. The ATP supply must be adjusted constantly according to need [52] This demands an efficient production system with a generous reserve capacity [53] which can respond immediately to instructions from a wide range of sources. In brain, mitochondria are the main source of ATP which under normal circumstances is generated predominantly from glucose via pyruvate oxidized aerobically, with a smaller proportion from cytoplasmic glycolysis. The production unit comprises the five large complexes of the oxidative phosphorylation (OXPHOS) system tightly configured at the inner mitochondrial membrane. Construction of the system is a highly co-ordinated process. Between 1000-2000 mitochondrial-related genes have been identified in nuclear DNA [54]. Only thirteen of the OXPHOS peptides are encoded by mitochondrial genes. A mitochondrion contains 2-10 copies of mtDNA. The copy number (mtDNA-CN) varies within a cell and correlates to its tissue’s bioenergetic needs. The abundance of mitochondrial transcripts is higher in tissues with high energy demands [55]. Each somatic cell can have upto 1000 mitochondria. Studies have demonstrated a significant association of mtDNA-CN with TFAM (Mitochondrial Transcription Factor A). Other proteins that regulate copy number are TWINKLE, a mitochondrial DNA helicase, and POLG-A, a subunit of DNA polymerase γ. mtDNA is vulnerable to mitochondrial stressors, including oxidative stress, which can disturb the respiratory chain complexes, lead to release of damaging reactive oxygen species (ROS) and reduce ATP production [54]. The tricarboxylic acid (TCA) cycle in the mitochondrial matrix supplies most of the fuel for oxidation as NADH+, but this is supplemented by many other biochemical processes, notably fatty acid oxidation which generates FADH2 [56].
3.2. Biosensors of ATP Status
3.2.1. AMPK
AMPK is a serine/threonine kinase. It is the primary sensor for ATP status in the body through direct interaction with the adenine nucleotides AMP, ADP and ATP [52Treft]. AMPK is heterotrimeric with subunits α, β and γ. The α subunit carries the catalytic domain, β and γ are regulatory [52]. A conserved Thr 172 in the activation loop of the kinase domain is regulated by at least three upstream kinases: LKB1 (liver kinase B1), CaMKK2 (calmodulin-dependent protein kinase kinase β), and TAK1 (TGFβ-activated kinase 1), and dephosphorylated by three phosphatases, PP2A (protein phosphatase 2A, PP2C (protein phosphatase 2C ), and PPM1E (Mg2+/Mn2+ -dependent protein phosphatase 1E) [52,57]. The γ-subunit has 4 tandem repeat motifs (termed CBS1 to CBS4), which assemble to form binding sites for AMP, ADP and ATP. CBS3 seems to be the critical site. In energy replete state (low AMP/ATP and ADP/ATP ratios) phosphatases can access T172 and keep AMPK unphosphorylated and inactive, but phosphatase access is blocked when high levels of AMP or ADP bind to CBS3 in the γ subunit [58].
AMPK maintains and restores ATP when energy levels are falling. It is activated by increasing AMP or ADP coupled with falling ATP. The AMP/ATP ratio is affected by even small changes in AMP [58]. AMPK switches on numerous genes in catabolic pathways which generate ATP, and switches off many in anabolic pathway which use ATP. Up-regulated genes include genes involved in mitochondrial fission, autophagy, mitophagy, mitochondrial biogenesis, glycolysis, glucose uptake, fatty acid uptake, fatty acid catabolism, branched chain amino acid catabolism, and redox regulation, all relevant to this review. Relevant down-regulated genes include genes involved in RNA synthesis, protein synthesis and elongation, and synthesis of triacylglycerol phospholipids, cholesterol, fatty acids and glycogen [Refer to Refs GH,2016,2018, Treft, Jeon for reviews and gene identities]. AMPK also has a role in the metabolic responses to caloric restriction and exercise [52]. It is dysregulated in major chronic diseases including obesity, inflammation and diabetes [58,59].
3.2.2. Sirtuins
Sirtuins (SIRT) are a class of seven NAD+-dependent histone deacetylases that regulate gene transcription in many metabolic pathways [60,61,62,63,64]. Some also remove other acyl groups such as succinyl, malonyl, and long-chain fatty acyl groups [65]. Sirtuins differ in length and sequence in their C- and N-terminal domains. SIRT1 and SIRT2 localize in the nucleus and cytoplasm, and SIRT3, SIRT4 and SIRT5 are mitochondrial [62]. SIRT1 activity is regulated directly by cellular NAD+ levels and indirectly by AMPK activation which increases the intracellular NAD+/NADH ratio [66]. In turn, SIRT1 promotes AMPK activity by deacetylating and thereby activating LKB1 kinase and hence phosphorylation of AMPK [67]. A decrease in the NAD+/NADH ratio when glucose intake is high inhibits AMPK activation [58].
Sirt1 regulates a range of age-related processes including cellular senescence, DNA damage repair, mitochondrial function, and inflammation through histone deacetylation of inflammatory cytokines including NF-kB, HIF1a, AP-1, and P38MAPK [68]. NF-κB associates with accumulating Aβ peptides and activates an inflammatory response via acetylation of its p65 36subunit. Deacetylation of p65 by Sirt1 may limit Aβ-provoked damage. SIRT1/AMPK activity was shown to play a key role in autophagy by inducing mitochondrial fragmentation, which slows progression of neurodegeneration [60,69]. Raising the activity of the AMPK-SIRT1-PGC-1α pathway increases mitochondrial biogenesis. SIRT1 depletion accelerates the ageing process and increases susceptibility to age-associated diseases, [62.One action of the polyphenol resveratrol, which is present in grape skin and red wine, is to activate SIRT1 with neuroprotective effects [70]. SIRT3 regulates enzymes involved in fatty acid oxidation, activates the respiratory succinate dehydrogenase complex flavoprotein subunit A (SDHA), a component of complex II in the respiratory chain [71], and increases activity of the mitochondrial enzyme superoxide dismutase 2 [72]
3.2.3. Phosphofructokinase (PFK)
PFK, the first irreversible step in glucose degradation by glycolysis produces fructose bisphosphate (FBP) which is then committed to pyruvate oxidation. FBP is a good indicator of glucose and energy availability [58].
3.2.4. ATP Regulation by Mitochondrial Nucleotide Transporters
Adenine nucleotide translocase (ANT) is a large mitochondrial solute carrier family of proteins expressed in the inner mitochondrial membrane. In a 1:1 exchange it imports ADP3- into the mitochondrial matrix, for conversion to ATP by ATP synthase and exports ATP4- from matrix to the intermembrane space. It does not alter the mitochondrial adenine nucleotide content. ANT undertakes equimolar exchange of vast amounts of nucleotides daily and is essential for life [73,74,75,76]. Humans express Five ANT genes. ANT1 (SLC25A4) is the most abundant form in brain and in other tissues with high oxidative activity such as heart and skeletal muscle [73,74,75,76]. ANT is incorporated with F1F0-ATP synthase and the phosphate carrier (PiC) proteins in the mitochondrial supercomplex (the ‘ATP synthasome’). It also associates with respiratory chain supercomplexes [74]. AMPK has been implicated in regulation of ANT activity via its interaction with SIRT4 [77], and via the ANT-AMPK-mTORC1 signalling pathway [78]. In addition to roles in energy provision, there is evidence that ANTs have more extensive roles in mitochondrial biology including regulation of opening of the mitochondrial permeability transition pore (MPTP) and mitophagy [74].
ATP- mitochondrial ATP-Mg/Pi carriers are integral proteins of inner mitochondrial membranes which mediate electroneutral exchange of phosphate for adenine nucleotides coupled to magnesium or protons [73,79,80,81]. Their activity enables mitochondria to replenish adenine nucleotide pools depleted by cellular activities [79,81]. They are probably the only transporters responsible for net changes in mitochondrial adenine nucleotide levels. Humans have four carriers. SLC25A23 (Solute Carrier Family 25 Member 23), SLC25A24 and SLC25A25 are calcium-regulated [81,82]. Their N-terminals face the mitochondrial inter-membrane space and hence can transduce cytoplasmic Ca2+ signals. The fourth carrier, SLC25A41, is not regulated by Ca2+ [66]. Fibroblasts from slc25a25-deficient mice embryos had decreased mitochondrial ATP, basal mitochondrial respiration and decreased Ca2+ flux across the sarcoplasmic reticulum. [83]. Glucagon, vasopressin, and epinephrine and a low insulin/glucagon ratio transiently increase cytoplasmic Ca2+by activating their receptors, increase glycolysis and glycogenolysis-derived ATP production and the ATP/ADP ratio. AMPK can influence mitochondrial ATP transporter activity indirectly through activation of membrane Ca2+ transporters. In renal tubular cells SLC25A25 was shown to be activated by Ca2+ entry via the transient receptor potential cation channel PKD2 (polycystin 2) which is activated by AMPK [84,85]. In this study, knock-down of SLC25A25 decreased cellular respiration and significantly reduced ATP concentrations, but had no effect on cell growth or survival. Compared to wild-type cells, there were significant changes in lipids, purine and pyrimidine nucleosides and nucleotides, amino acids, notably with a large decrease in aspartate, and in intermediates of glutathione metabolism [85]. This provided a unique view of the effects of isolated ATP deficiency.
3.3. Hypoxia-Inducible Factor 1 (HIF-1) Mediates the Response to Hypoxia
The transcriptional complex HIF-1 plays an essential role in cellular and systemic oxygen homeostasis [86,87,88]. It induces the transcription of more than 60 neuroprotective proteins which promote erythropoiesis and angiogenesis, thereby increasing oxygen availability, glucose transport and metabolism [86,89].HIF-1 consists of a constitutively expressed HIF-1β subunit (Aryl Hydrocarbon Receptor Nuclear Translocator, ARNT1, HIF1ß ) and one of three subunits (HIF-1α, HIF-2α or HIF-3α). Under normoxic conditions HIF-1α protein is degraded rapidly via the von Hippel-Lindau tumor suppressor gene product (pVHL)-ubiquitin-proteasome pathway [86]. The association of HIF-1α with pVHL is triggered by post-translational hydroxylation of proline residues mediated by prolyl hydroxylase (PHD) or HIF prolyl hydroxylase (HPH). PHD is a dioxygenase. Its activity depends on oxygen concentration and hence PHD has been proposed as the HIF-1α oxygen sensor. In hypoxic conditions, HIF-1α is stabilized, heterodimeric HIF-1α/β translocates into the nucleus and interacts with E1A binding protein p300/ CREB-binding protein (p300/CBP) and other coactivators [91,92,93,94] which synergistically enhance HIF-1α transcription of target genes. Growth factors induce HIF-1α protein translation via PI3K (phosphoinositide 3-kinase)or MAPK (mitogen-activated protein kinase) pathways irrespective of hypoxia.
Genome-wide chromatin immunoprecipitation (ChIP) identified HIF 1-dependent increased or decreased expression levels of hundreds of genes in response to hypoxia [95]. Vascular endothelial cell growth factor (VEGF) and erythropoietin are major HIF1 target genes. Amongst many other transcriptionally activated genes are genes encoding cyclin, IGF2 (Insulin-like Growth Factor 2), IGFBP1 and IGFBP2 (Insulin-like growth factor-binding protein 1 and 2), NOS2 (Nitric Oxide Synthase 2), GLUT1 and GLUT3 (Glucose Transporter 1 and 3), transferrin, the transferrin receptor [86] and caeruloplasmin [96]. VEGF specifically recruits endothelial cells into hypoxic and avascular area and stimulates their proliferation. It is the most potent endothelial-specific mitogen and is known to directly participate in angiogenesis. HIF-1α has also been shown to indirectly contribute to Tau phosphorylation. Because upregulated HIF-1α in chronic hypoxia decreased the activity of protein phosphatase-2A (PP2A), it was proposed that this may mediate Tau hyperphosphorylation with increased risk of resultant cognitive dysfunction [97].
3.3.1. Hypoxia Up-Regulated Mitochondrial Movement Regulator (HUMMR)
HUMMR is expressed in neurons and is markedly induced by HIF-1α [98]. It interacts with Miro-1 and Miro-2, mitochondrial proteins that are critical for mediating mitochondrial transport (refer to section 4.3). Knockdown of HUMMR or HIF-1 function in neurons exposed to hypoxia markedly reduced the mitochondrial content in axons. The percentage of motile mitochondria moving in the anterograde direction decreased and the percentage moving in the retrograde direction was enhanced [98].
3.4. Mitochondrial-Derived Peptides (MDPs) and Nuclear-Encoded Microproteins
The discovery of small bioactive signalling peptides coded by mitochondrial DNA (MDPs) over the last 15 years has radically changed our vision of the roles of mitochondria in directing cell metabolism [reviewed17,99,100]. MDPs are encoded by short open reading frames (ORFs) from noncanonical transcription sites within the known mitochondrial genes [101]. So far eleven have been reported: Seven, humanin and six small humanin-like peptides with 20-35 amino acids (SHLP1 -SHLP6), are encoded within MT-RNA2, the gene for 16S rRNA [102]. Mitochondrial open reading frame of the 12S rRNA-c (MOTS-c) was identified within the 12S rRNA gene and codes for a 16 amino acid peptide [103]. Other MDPs are SHMOOSE (Small Human Mitochondrial ORF Over SErine tRNA) and GAU (gene antisense ubiquitous). In addition, a 99 amino acid polypeptide is translated from an alternative reading frame in the mtDNA gene MT-CYTBmt [104]. In vitro humanin is antiapoptotic, increases mitochondrial respiration, cell proliferation, and cell survival through cell membrane receptors [99,105]. SHLP2 and SHLP3 also decrease apoptosis. SHLP2 has a protective interaction with mitochondrial complex 1 and increases mitochondrial respiration and ATP levels [17,104]. MOTSc however, decreases mitochondrial respiration and increases glycolysis in vitro [104], and MtALTND4, encoded from an alternative open reading frame of the gene for ND4 a subunit of NADH dehydrogenase (Complex 1), decreased the O2 consumption rate, maximum coupled and uncoupled respiration and spare reserve capacity of Hela and HEK-293 cells [104].
Microproteins encoded within nuclear genes which modify mitochondrial function are also being recognized. A uORF located in the 5′ UTR of the nuclear gene for mitochondrial dynamics protein 1 (MID51, Mitochondrial Elongation Factor 1(MIEF1) encodes a mitochondrial microprotein [70 amino acids], named MIEF1-MP (MIEF1-microprotein) that is involved in mitochondrial fission and interacts with the mitochondrial ribosome [106]. Independently, three Groups identified a microprotein (56 aa) encoded by a lncRNA (gene MTLN) which they named mitoregulin [107], MOX1 [108] and MPM [109]. Mitoregulin supported super complexes and modified mitochondrial respiratory efficiency [99,107]. Mitochondrial and nuclear encoded micropeptides /microproteins are likely to have important physiological roles in regulating cellular stress responses, apoptosis, and metabolic processes, and to be under epigenetic control.
A polymorphism of the humanin gene with an incidence of 1-5% in individuals with African ancestry was shown to associate with lower plasma humanin levels and greater cognitive decline [17,110] Surprisingly, this SNP is common among individuals of European descent, with an incidence approaching 50%, and so far has not been associated with dementia, which must reflect the multifactorial nature of the condition. A second humanin variant found in individuals of Ashkenazi descent promoted higher affinity binding to APOE4 than the more common allele in vitro, and in mice expressing human APOE4 the variant reduced AD-related pathology more effectively [111].
3.5. Spectrun of Molecules Involved in ATP Turnover
Bennett, Nguyen and Darch et al. [112,113] developed a fluorescence-activated cell sorting (FACS)-based assay to screen the genome for regulators of cellular ATP levels of K562 leukaemic cells expressing a fluorescent ATP biosensor. They screened the entire genome with CRISPR interference and CRISPR activation libraries and isolated cells with high and low ATP levels under basal conditions or cells dependent on mitochondrial ATP generation (glycolysis inhibited), or on glycolytic production (respiration blocked). They identified numerous gene pathways and ontologies that impacted ATP when knocked down or over-expressed. One of relevance was HSD17B10 (Aβ-binding alcohol dehydrogenase, ABAD) which is involved in isoleucine and neurosteroid metabolism, and is upregulated in Alzheimer’s disease and has been shown to interact with Aβ peptide but it is unclear whether they have concerted or independent roles in AD pathogenesis [114]. Of considerable interest was their demonstration that HIF1α and aryl hydrocarbon nuclear translocator (ARNT1, HIF1ß), which form the functional HIF1 molecule, were at the centre of a network which included the HIF1 targets HK2 (hexokinase 2) and binding partner VDAC1 (voltage dependent anion channel 1) [115], and the HIF1-regulating proteins SENP1 (SUMO specific peptidase 1) and SP1(Sp1 transcription factor) [116] and upstream genes that regulate HIF1.
4. Brain Processes with Very High ATP Consumption/Turnover
There are three intimately linked pathways in brain with very high ATP usage demanding continuous replenishment: the malate-aspartate shuttle, the glutamate/glutamine cycle, and axonal transport / synaptic transmission. These are at high risk of disruption by oxygen depletion with severe consequences for brain function.
4.1. The Malate-Aspartate Shuttle
Intact mitochondria are impermeable to NADH, hence reducing equivalents generated as NADH in the cytosol by NAD+-dependent pathways must enter the mitochondria indirectly. In brain this is via the malate-aspartate shuttle (MAS) which operates in neurons but not in astrocytes. The other major redox shuttle (glycerol-3-phosphate shuttle) has very low activity in brain. Figure 2 shows the shuttle in a presynaptic glutamatergic neuron. The legend explains how it operates.
MDH1 malate dehydrogenase 1, MDH2 malate dehydrogenase 2 (mitochondrial), mAST mitochondrial aspartate amino transferase, alias GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial, cAST cytoplasmic aspartate aminotransferase, alias GOT1 glutamic-oxaloacetic transaminase 1, 2-Oxoglut, 2-oxoglutarate, OGC 2-oxoglutarate carrier
Astrocytes do not express aralar and lack a complete shuttle. The closely related aspartate-glutamate carrier, AGC2 (SLC25A13, citrin) associated with the urea cycle in liver [119] is not expressed in brain. Aralar is regulated by cytosolic Ca2+, and small cytosolic Ca2+ signals activate the Aralar/MAS pathway [120,121].
The MAS is essential for maintaining redox balance in the cytosol and mitochondria, for securing and transferring the energy generated as NADH in the cytosol, and for neuronal use of lactate as fuel. By regenerating NAD+ it enables activities of cytosolic enzyme to continue, and other NAD+ functions such as signaling and regulation of transcription by sirtuins [117], Importantly, it generates aspartate for export from neurons for subsequent uptake by astrocytes where has a central role in glutamine synthesis (refer to Section 4.2). Aspartate is also essential for pyrimidine synthesis [122], and is converted to N-acetylaspartate (NAA) and exported to oligodendrocytes where it is de-acetylated and metabolize, and provides acetate for fatty acid synthesis [118,123,124]. In humans, aralar deficiency presents with severe infantile-onset encephalopathy with epilepsy, global developmental delay, generalized hypotonia loss of cerebral volume, diffuse brain atrophy, and hypomyelination/white matter loss and reduced cerebral NAA on brain imaging [25,118,126]. Infants lacking other shuttle enzymes, GOT2, MDH1, or MDH2, have exhibited similar symptoms [117,127,128,129], which are mirrored in aralar-KO mice [130,131]. Brain Asp levels of KO mice were 80% to 90% lower than controls. Asp and NAA levels of brain and cortical neuronal cell cultures from all brain regions of KO animals were drastically decreased and alanine (Ala) and serine (Ser) were severely reduced [132].
4.2. The Glutamate/GABA/Glutamine Cycle
Glutamine (Gln) and GABA for neurotransmission are synthesised and replenished by interaction of glutamatergic and GABAtergic neurons and astrocytes in a tightly co-ordinated sequence termed the glutamate/GABA/glutamine cycle. Because neurons lack the capacity to synthesise glutamate (Glu) de novo, astrocytes are the primary regulators of Glu and GABA biosynthesis [133,134]. Figure 3 shows the sequence of events. The legend explains how it operates and lists the main transporters and enzymes involved.
Unbound neurotransmitters must be cleared quickly from the synaptic cleft to prevent neurotoxicity from excessive stimulation. There are many members of the solute carrier (SLC) family, and there is current uncertainty about their relative contributions to the cycle processes, particularly in transfer of glutamine from astrocytes to neurons [135]. Glutamine synthetase (GS) is expressed abundantly in the fine astrocytic processes associated with glutamatergic synapses in rat hippocampus [136]. NH3 for glutamine synthesis is supplied by phosphate activated glutaminase (PAG) activity and by other enzyme processes such as inosine monophosphate degradation in the purine nucleotide cycle which is active in brain [137], and from the systemic circulation [137]. Glutamine-glutamate cycling accounts for approximately 80% of total neurotransmitter recycling, and glutamine-GABA cycling for the remainder [133].
By releasing glutamate and GABA , neurons are at the frontline of neurotransmission. However, their capacity to do this is entirely dependent on support from adjoining astrocytes to maintain the glutamine flow [134,138]. This involves complex biochemical interactions. The problem is that the glutamate/GABA/glutamine cycle is a leaky system. Not all of the glutamate from glutamine transferred into neurons is used for neurotransmission. Some 10% to 30% is diverted to cell metabolism [139]. Without replacing this, the neurotransmitter pool would be exhausted very rapidly [133]. Astrocytes bolster supplies by withdrawing 2-oxoglutarate (2OG) from their TCA cycle. Transamination by cytosolic AST yields glutamate for glutamine synthesis. However, this depletes the TCA of an essential component with the risk of rapid malfunction. This is prevented by anaplerotic activities [140] shown in Figure 4 and described in the legend. Pyruvate carboxylase which carboxylates pyruvate to oxaloacetate has a central role in anaplerosis. It is expressed abundantly in astrocytes. (OAA) [141,142].
① SLC1A3, alias excitatory amino acid transporter 1 (EAAT1; GLAST), SLC1A2,alias excitatory amino acid transporter 2 (EAAT2,GLT1), ② SLC6A11 (GAT3), ③ Na+-amino acid cotransporters, SLC38A1 (SNAT1) and SLC38A2 (SNAT2), and Na+-amino acid cotransporters-H+ antiporters, SLC38A3 (SNAT3) and SLC38A5 (SNAT5). cAST cytoplasmic aspartate aminotransferase (alias GOT1 glutamic-oxaloacetic transaminase 1, cytoplasmic), GS glutamine synthetase, PC pyruvate carboxylase, OAA oxaloacetate, 2-oxoglut 2-oxoglutarate, cit citrate, isocit isocitrate, AcCoA acetyl CoA, LCFAs long chain fatty acid, LCFacylCoA , C carnitine shuttle.
Glucose oxidation supplies pyruvate for this pathway according to demand and is closely matched by transport of glucose into the cells, mainly by the glucose transporter GLU1. Astrocyte function is further supported by glycogen which is a local energy reserve [143]. Aspartate generated by the malate aspartate shuttle (Figure 2) is an essential contributor to anaplerosis. Further anaplerotic support is provided by re-uptake of GABA into astrocytes and its conversion to succinate [134](refer to Figure 4).
4.2.1. The Energy Cost of the Glutamate/GABA/Glutamine Cycle
The transport of glutamate into astrocytes places has a high energy requirement which makes significant demand on astrocytic metabolism. For each imported glutamate molecule, three Na+ ions move down its concentration gradient accompanied by the inward movement of one H+ and the counter-transport of a K+ ion [144]. This results in significant depolarisation of the astrocyte membrane [145,146]. The membrane is repolarized by K+ channels and by the Na+/K+ ATPase [reviewed147], which transfers 3 Na+ ions out of the cell and 2 K+ ions inward for each molecule of ATP hydrolyzed [148Kaplan]. In astrocytes, there is a close physical association between glutamate transporters, the Na+/K+ ATPase and mitochondria [149,150].
4.2.2. Disturbances of the Glutamate/GABA/Glutamine Cycle in AD
The activity of GS is vulnerable to mixed-function oxidation which rises exponentially with age [133]. The enzymatic activity of GS was reduced in brain samples of AD patients [151,152,153] and the APP/PS1 mouse model of AD [154,155]. In vivo compared to controls ,GS was found to be significantly oxidized in hippocampus from individuals with MCI or AD [153]. In vitro the Aβ peptide inhibits purified GS [156], as well as GS activity of cortical homogenates [157] and cultured astrocytes [158]. GS was one of the cellular proteins most prone to oxidation after Aβ1–42 treatment in vitro [159]. Expression of the glutamine transporters SNAT1(sodium coupled neutral amino acid transporter1) and SNAT3 was decreased in the APP/PS1 mouse [154,155] and in vitro Aβ exposure leads to downregulation of SNAT1 in cultured cortical neurons [160]. GS expression was reduced in the frontal cortex of 3xTG mice prior to significant Aβ accumulation (1 month of age) [161], indicating that GS dysfunction occurs early in AD development. Glutamine synthesis was reduced in hippocampal slices of 5xFAD mice at an early stage of disease (2 months of age) [162]. With advanced disease at 8 months of age, glutamine synthesis was reduced in both hippocampal and cerebral cortical slices of 5xFAD mice [163]. Reduced glutamine/glutamate levels have been reported in AD brain [reviewed159]. Expression of glutamate transporters, particularly SLC1A2 (EAAT2), is severely reduced in AD brain, resulting in reduced astrocyte glutamate uptake and potential excitotoxicity [164,165,166]. In three independent cohorts the glutamate carrier SLC25A22 was identified as a susceptibility gene for AD, and downregulation was also associated with hippocampal atrophy. It was hypothesised even a small decrease in SLC25A22 could compromise neuronal and mitochondrial glutamate metabolism causing energy deficiency [18].
Hyperammonaemia has been proposed as a pathogenic factor in AD, but the mechanisms are unknown [14,15,167]. Increased NH3 levels could impact on the glutamate/GABA/glutamine cycle by decreasing SNAT3 expression [168] or through increasing ATP consumption during detoxification by GS [137]. Increased serum and CSF levels of ammonia have been reported in AD patients. Asymptomatic women with mild chronic hyperammonaemia due to mutations of X-linked ornithine transcarbamylase (OTC), a urea cycle enzyme, show subtle changes on brain MRI [169]. However, hippocampal changes have not been reported, and neither has an association with AD to date. Mild cognitive impairment may be evident with formal testing [170,171].
4.2.3. Effects of Hypoxia/Ischaemia on the Glutamate/GABA/Glutamine Cycle
Glutamate receptor antagonists have been shown to protect neurons from global and/or focal ischemia which are proposed to increase extracellular glutamate accumulation, leading to excessive activation of glutamate receptors and excitotoxic cell death [164]. Astrocytes are more resistant to hypoxia/ischemia than neurons. Depending on their location relative to a focal ischemic infarct, astrocytes undergo a progressive change in morphology, becoming ‘reactive astrocytes’ with loss of highly branched processes, hypertrophy, and increased expression of glial fibrillary acidic protein (GFAP) [147]. Transient oxygen/glucose deprivation caused relatively rapid fragmentation of mitochondria in the astrocyte processes followed by a gradual decrease in number [172]. Decreased levels of SLC1A2 (EEAT2) and/or SLC1A3 (EEAT1) mRNA and/or protein were observed in models of hypoxia/ischemia [reviewed 173].
4.2.4. Promoting Anaplerosis in Astrocytes to Support Glutamine Synthesis
Triheptanoin, an edible odd-chain fatty acid triglyceride C7:0), can be used as a dietary supplement. The main metabolic product, heptanoate, crosses the blood-brain barrier (BBB) and enters mitochondria to increase succinyl-CoA abundance [174]. Heptanoate can also be converted to five-carbon-ketone bodies or glucose via gluconeogenesis in the liver providing additional substrates for the TCA cycle in the brain [175]. Hence triheptanoin supports the TCA cycle through anaplerosis and by fuelling the cycle hence potentially enhancing ATP production. Triheptanoin has been used in clinical trials for treatment of neurological disorders including glucose transporter type 1 deficiency (Glut1DS) [176]. Treatment of 5×FAD mice with triheptanoin from 3.5m of age for 4.5m, rescued brain ATP content, increased mitochondrial NADH abundance, respiration and redox balance and preserved synaptic density in the hippocampal CA1 region and entorhinal cortex, but did not decrease Aβ load or tau phosphorylation [177].Triheptanoin administration combined with a high-protein ketogenic diet to APP/PS1 mice with AD-like pathology prevented cognitive deficits and astrogliosis [178].
4.3. Axonal Transport Has a High Energy Requirement
Organelles and proteins are generally assembled in the body of neurons and must be transported to synapses in the nerve terminals when required. Material from the synapses requiring neuronal processing must, similarly, be transported in the reverse direction. These functions are carried out by highly co-ordinated events initiated by cell signalling which are tightly regulated by posttranslational modifications of microtubules, and by organelle-specific interactions [179,180]. For each journey, the fundamental requirements are a track, a motor and adaptors [181] to attach the cargo. There are two types of track. One is comprised of actin filaments. These may be used for short-distance transport, as in dendrites, and often associate with actin networks near the cell surface. The others are microtubules. They are composed of α- and β-tubulin molecules which dimerize and then polymerize into parallel protofilaments. These wrap around each other to form a microtubule with a ‘plus’ end orientated toward the distal axon and a ‘minus’ end toward the cell body. They are not permanent structures but assemble and disassemble according to need. Tau protein is an essential binding partner which regulates bundling of the microtubules and stabilizes them [182]. Acetylationof microtubules by α-tubulin N-acetyltransferase (ATAT) may promote stabilization and additionally confer flexibility [183,184,185]. They are deacetylated by histone deacetylase 6 (HDAC6) and sirtuin-2 (SIRT2) [186].
The trafficking system has heavy use, transporting a wide variety of intracellular organelles, including endosomes, lysosomes, autophagosomes, secretory vesicles, mitochondria, proteins and macromolecules. These attach to specific flexible adaptors which bind them to motors on the tracks for transport. Motor proteins are classified into three families, myosins, kinesins and dyneins. The heavy chain of each motor type has a family-specific conserved head domain that binds to the filaments and generates force and motion through cycles of ATP hydrolysis. Organelle movement by myosins can be directed toward the actin filament plus ends, for example by myosin V, or minus ends for example by myosin VI [187].
Kinesin motors generally mediate anterograde axonal transport and dynein drives retrograde axonal transport [179]. The Kinesin-1 family consists of three proteins. Of these, KIF5A, is primarily expressed in neurons. It is composed of two heavy chains and two light chains. The heavy chain binds microtubules with the head domain and hydrolyses ATP near the N-terminus. The head is joined to a long divergent stalk with two coiled-coiled domains, and a C-terminal tail associates with the cargo-binding light chains [180,188,189]. The stalk sequences facilitate homodimerization of the heavy chains that allows the motor to ‘walk’ by alternating cycles of heavy chain to filament binding, such that one head is always attached to the filament [190]. Each ‘step’ consumes ATP. Glycogen synthase kinase-3β (GSK3β) phosphorylates the KIF5A heavy chain to inhibit axonal transport and also phosphorylates the light chain to release cargoes [191,192]. The stress-activated protein kinases c-Jun N-terminal kinase 3 (JNK3) and p38 mitogen-activated protein kinase (p38 MAPK) were also shown to directly phosphorylate the heavy chains and to inhibit anterograde transport [193,194]. Cytoplasmic dynein (referred to as dynein) is a large, 1.4 MDa multimeric complex composed of dimerized heavy chains, two intermediate chains, two light intermediate chains, and additional light chains. The heavy chain binds to the light chains, to a linker connected to the motor, and to cargo via interaction with other dynein subunits at its N-terminal tail [188]. To activate the motor, dynein binds to dynactin, an adaptor comple [180].
4.3.1. Axonal Transport of Mitochondria
Mitochondria in the neuronal cell bodies are transported down axons in response to changes in the local energy state and metabolic demand [179,187].The transport mechanisms are as for other organelles. Increased neuronal Ca2+ released in response to neurotransmitter stimulation inhibited the motility of mitochondria without affecting motion of other organelles [187]. Mitochondrial fusion/fission events, and organelle size have an important influence on mitochondrial motility [187]. Clearly there is close two-way communication between the neuronal cell body and its mitochondria, probably mediated via gene transcription [refer to S3.4]. Microtubule and myosin motors are bound to the mitochondrial surface by a conserved Miro–trafficking kinesin protein (TRAK) adaptor complex [195]. TRAK1 and TRAK2, bind directly to Miro proteins which are anchored to the outer mitochondrial membrane via a C-terminal transmembrane domain [196,197], as shown in Figure 5.
Mammals, express two Miro (Mitochondrial Rho GTPase) proteins, Miro1 and Miro2. Both have two Ca2+-sensing EF-hand domains [196,198,199] and can act as Ca2+ sensors to induce Ca2+-dependent mitochondrial immobilization. Miro-1 and Miro-2 also interact with hypoxia up-regulated mitochondrial movement regulator (HUMMR), which is expressed in neurons and is markedly induced by hypoxia-inducible factor 1 α (HIF-1α) (refer to S ection 3.3). In hypoxic conditions it facilitates anterograde, and represses retrograde, mitochondrial transport. Knockdown of HUMMR or HIF-1 in neurons exposed to hypoxia markedly reduced the mitochondrial content in axons [98,187]. Damaged mitochondria in the axon terminals are transported by dynein to the cell body/soma for elimination by mitophagy [200,201]. Defective retrograde transport of senescent mitochondria results in increased autophagy in axonal swellings [202].
4.3.2. Role of Tau Protein in Axon Transport
Tau (microtubule-associated protein Tau) is encoded by the MAPT gene. Its normal physiological role is to induce tubulin assembly and stabilize microtubules, promote axonal growth and enable axonal transport [203]. In vitro, it also binds to microtubules and actin simultaneously, promoting co-organization and coupled growth of both transport networks [204]. Tau is an intrinsically disordered protein consisting of an N-terminal, a proline-rich ‘projection’ domain, a microtubule-binding domain (MTBD) which incorporates three or four repeated motifs numbered R1 to R4, and a C-terminal tail. The motifs interact with the microtubules. Mutations within the MTBD impair tau-mediated microtubule stabilization [182]. Two of the motifs are conserved hexapeptides, named paired helical filament domains (PHFs). From fluorescence resonance energy transfer (FRET) studies, when not attached to microtubules soluble Tau molecules displayed an unfolded structure. When associated with microtubules, Tau monomers folded, decreasing the distance between the N and C termini [203]. This resulted in formation of hairpin-like structures which stabilized a microtubule-bound conformation [205,294]. The findings strongly suggest that Tau’s capacity to regulate microtubule bundling and stabilizing activities is tightly controlled by its phosphorylation state [182]. Tau is thought to detach from microtubules through hyperphosphorylation of epitopes in the proline-rich domain and C-terminus of the Tau protein [206] by specific kinases such as glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase 5 (CDK5) and phosphokinase A (PKA) [207,208]. AMPK has been linked to the control of mitochondrial anchoring at presynaptic boutons in mature cortical neurons, coupling local metabolic needs with mitochondrial positioning [208,209].
Tau detachment destabilises microtubules and they disaggregate. Hyperphosphorylated tau is consistently associated with pathological lesions in human AD post mortem material and in PET brain imaging [refs], and with pathology and toxicity in animal studies [206,210]. Reactive microglia are often observed near NFTs which are thought to act as danger associated molecular pattern (DAMP) molecules, further activating microglia and triggering an immune response. This could contribute to the chronic inflammation observed in AD [207]. However, p-Tau has been demonstrated in human brain from individuals without AD, and its role in AD pathogenesis is unclear [206]. Tau has additional functions to axonal transport. It localises in the cell nucleus, binds to histones and may be involved in chromatin remodeling, or chromatin compaction and is thought to protect DNA from damage. Misfolding or hyperphosphorylation of Tau would prevent this. Heat or oxidative stress cause nuclear translocation of Tau [211,212].
4.3.3. Disordered Axonal Transport in AD
Dystrophic axons and axonal swellings, areas of expanded axons with accumulation of cargoes and motor proteins, are found in the early stages of AD in brains at autopsy and in an AD mouse model [180,213]. Mouse models with familial AD mutations show axonal pathology before Aβ plaque formation or NFT formation [180] Dysregulation of axonal transport occurs early in neurodegenerative diseases and plays a key role in axonal degeneration [180]. Defective transport of axonal mitochondria is implicated in human neurological disorders and neurodegenerative diseases [213]. Loss of mitochondria from axonal terminals in Drosophila results in impaired synaptic transmission [179]. There is evidence that GSK3β kinase is hyperactive in AD [214]. Synapses are essential for transmitting, processing, and storing information, which all decline in aging and AD [50]. Microarray analysis of brain collected at autopsy from non-AD controls aged 20 to 99 years and individuals with various psychiatric disorders, including AD, identified significant changes in expression of numerous synapse-related genes, with many progressively downregulated across aging [215]. The widespread changes in synaptic gene expression in normal aging suggested that the function of synapses might be impaired, and that aging and AD share a common set of vulnerable synaptic genes [50].
5. Effects of ATP Depletion on Lipid Metabolism
The brain has an exceptionally high lipid content [10,27,28]. In contrast to other fat-laden body organs, only a small lipid fraction is used as an energy source. The majority serves structural and signalling roles in the enormous expanse of membranes covering the organelles and surface of brain cells and their processes. Phospholipids account for approximately 50% of total lipids [29] and the other major contributors are cholesterol and its esters, sphingolipids, glycolipids, and fatty acids. Because of the blood-brain barrier (BBB), most of the cholesterol is synthesized de novo in the brain [216].
Due to liquid-liquid phase separation in cell membranes lipids segregate into ordered (raft) and disordered (non-raft) domains [49,217,218,219,220,221,222]. Rafts are transient and dynamic, heterogeneous with an estimated diameter of 10–200 nm (average 50 nm), and enriched in sphingolipids, cholesterol, and lipids with saturated acyl chains. They harbour most of the proteins involved in synaptic transmission and the amyloidogenic secretases [34,223], and serve as a platform for cellular processes such as cell signaling, pathogen entry, cell adhesion, motility, protein sorting and trafficking. Non-raft domains are enriched in unsaturated and polyunsaturated lipids and other subsets of membrane proteins [34,49,221,224,225]. Changes in the composition of membrane glycerophospholipids, and cholesterol and sphingolipid content impact on the properties of lipid rafts, influencing signal transduction from membrane receptors and activity of membrane transporters [221,226].
5.1. Glycerophospholipids
5.1.1. Synthesis
Graph 3. phosphate. In the initial step, GPAT1 (glycerol phosphate acyltransferase) acylates sn-1 with a preference for saturated stearoyl and palmitoyl fatty acids. Then AGPAT (acylglycerol phosphate acyltransferase) acylates sn-2 with preference for oleoyl-CoA to form phosphatidic acid (1,2-diacylglycerol-3-phosphate) [227]. Further processing via the CDP-DAG (cytidine diphosphate-diacylglycerol) pathway yields glycosylphosphoinositols, phosphoinositides (PIs), phosphatidyl choline (PC), phosphatidyl ethanolamine (PE), phosphatidylserine (PS) and cardiolipin. CDP-DAG is a high energy intermediate synthesised from cytidine triphosphate (CTP). The PI species and plasmalogens synthesised de novo have a miscellany of fatty acyl groups. They may be enriched with arachidonic acid (AA) or docosahexaenoic acid (DHA) by acyl chain remodelling via the Lands cycle (Figure 6) in the ER.
5.1.2. Physiological Functions
Glycerophospholipids are essential to maintain membrane physical bilayer properties for the correct location (in rafts or disordered membrane) of integral proteins and their function. They supply arachidonic acid for eicosanoid production and phosphatidyl inositol PI 4,5 diphosphate, a key signalling molecule with rapid turnover. Approximately 50% of the total inner membrane phospholipids of the inner mitochondrial membrane is comprised of cardiolipin (CL) and phosphatidylethanolamine (PE) [231,232]. Their cone-shape is essential for enabling curvature of the membranes and supporting architecture of the mitochondrial cristae [Ikon], which are the predominant site of OXPHOS assembly and operation [233]. CL also directly interacts with OXPHOS components and is required for formation and stability of Complexes III and IV [234,235].
5.1.3. Pathophysiology
Membrane peroxidation of PUFAs
Lipids containing carbon-carbon double bonds, particularly polyunsaturated fatty acids (PUFAs) undergo free radical attack by oxygen radicals (ROS). The major cell sources are superoxides generated as byproducts of oxygen consumption at the mitochondrial respiratory chain are [236]. Other sources include the activities of NADPH oxidase, cytochrome P450 enzymes and 5-lipoxygenase [90]. ROS may be generated non-enzymically, for example by the Fenton reaction in which hydrogen peroxide (H2O2) reacts with Fe2+,or Cu+ [49,237,238,239], or the Haber-Weiss reaction in which superoxide interacts with H2O2 or another peroxide. Importantly, free radical attack on PUFAs in membranes initiates a self-perpetuating oxidative cascade which generates lipid hydroperoxides, so propagating a rapidly spreading chain reaction [31,240]. The hydroperoxides disrupt membrane function by increasing membrane permeability and perturbing lipid packing, particularly in the disordered membrane regions which have a high PUFA content, and hence alter protein distribution between these domains and rafts [49]. In addition, their degradation produces highly reactive aldehydes, 4-hydroxynonenal (4-HNE) from arachidonic acid, 4-hydroxyhexenal (4-HHE) from docosahexaenoic acid, acrolein and malondialdehyde, which form adducts with lipids, proteins, DNA and other biomolecules [10,49]. Damage to intracellular membranes, particularly of the ER may activate the unfolded protein response [241]. Cells have a high capacity to mount rapid protective measures against free radical attack by recruitment of a host of enzymes including catalase, superoxide dismutase, glutathione peroxidase, and glutathione reductase and non-enzymatic antioxidants such as glutathione, ubiquinone, uric acid and thioredoxin [90,231]. Mitochondrial thioredoxin (TXN2) is expressed ubiquitously, with highest expression in the brain [242] and operates in the mitochondrial thioredoxin system comprising nuclear-encoded peroxiredoxins 3 and 5 (PRDX3 and 5), thioredoxin 2 (TXN2) and thioredoxin reductase 2 (TXNRD2). TXN2-deficiency manifests as an infantile-onset neurodegenerative disorder [231]. Lipid peroxidation is counteracted by several repair systems, especially the system xc−/glutathione/glutathione peroxidase 4 (GPX4), ferroptosis suppressor protein 1 (FSP1)/CoQ10, and GCH1/BH4 pathways [243]. Ferroptosis is a form of non-apoptotic cell death that results from excessive iron-catalyzed peroxidation of membrane phospholipids [244,245] and may contribute to cell death in degenerative diseases, including AD, and acute brain injury [90,243]. Susceptibility to ferroptosis is increased by enrichment of phosphatidylinositol with arachidonate and eicosapentaenoate [243,246].
5.1.4. Potential Role of Disordered Membrane Phospholipids in Promoting Aβ Production from Amyloid Precursor Protein (APP)
The physiological roles of APP are unknown, but proposed functions include regulation of neurite outgrowth, cell adhesion, synaptogenesis and cell survival. APP knockout mice are viable but have impaired spatial learning and long-term potentiation [247]. There are three isoforms produced by alternative splicing, APP695, 751 and 770. APP695 is the major neuronal isoform. APP770 is expressed in most other cell types [34]. APP is synthesized in the endoplasmic reticulum (ER) and trafficked through the secretory pathway. Most is localized in the Golgi apparatus, trans-Golgi network (TGN) and post-TGN vesicles, and only around 10% reaches the plasma membrane. At the cell surface, APP is cleaved enzymically or internalised into endosomes. APP is carried through neuronal axons via the anterograde transport machinery and may be the source of synaptically released Aβ [248,249]. APP is cleaved by two routes, a non-amyloidogenic pathway, and the amyloidogenic Aβ peptide-producing pathway, shown in Figure 7 and explained in the legend. The non-amyloidogenic pathway predominates in non-neuronal cells.
The three secretases are transmembrane proteases. BACE1 is an aspartyl protease [251], α-secretase activity is associated with at least three members of the ADAM (a disintegrin and metalloprotease) family (ADAM9, ADAM10 and ADAM17) [252], and γ-secretase is a complex comprising four core subunits, presenilins (PS1 or PS2), nicastrin, PEN2 (Presenilin Enhancer 2) and APH1 (Anterior Pharynx defective 1) [253] PS1 is the catalytic subunit. Intracellularly, BACE1 and γ-secretase are present in the TGN and endosomes. In cells transfected with APP, Aβ is mainly generated in these organelles. If this also occurs physiologically, it raises the question of the physiological role of APP at these sites [34]. There is good evidence that APP and C99 localise preferentially in the disordered region of cell membranes. Further, both were shown to remain exclusively associated with disordered regions of the membrane following lipid peroxidation [250,254]. The subcellular location of the enzymes is less certain. Variation in bilayer thickness, membrane ordering, and specific interactions with cholesterol affect the structure and orientation of the BACE1 and ADAM10 transmembrane domains. BACE1 can adapt more readily to the wider organised (raft) membranes than ADAM10, suggesting that ADAM10 may be less suited for localization in these domains than BACE1 [254]. γ-Secretase subunits were shown to reside in cholesterol- and sphingolipid-rich detergent-resistant lipid raft microdomains of post-Golgi, TGN and endosome membranes. Both secreted and intracellular Aβ were significantly reduced in neuronal cells when cholesterol transport from late endocytic organelles to the ER was blocked by the cholesterol transport inhibiting drug, U18666A [255]. Similarly, increased cholesterol efflux mediated by ATP-binding cassette transporter A1 (ABCA1) decreased Aβ production by reducing BACE1 and γ-secretase cleavage of APP [256]. BACE1 has many other substrates [257], including low-density lipoprotein receptor-related protein (LRP), β subunits of voltage-gated sodium channels, interleukin-1 receptor II (IL-1R2) and neuregulin1 and 3 [reviewed 34]. If disordered membrane lipids decrease BACE1 activity, would this lead to accumulation of products from its other substrates as well as Aβ which might be implicated in AD pathology?
5.1.5. Disturbances of Membrane Lipids in AD
Studies of brain from patients with AD have observed differences in the lipid content from unaffected controls [reviewed 10]. Concentrations of PUFAs in membrane lipids have been lower in AD compared with controls, including the hippocampus and the entorhinal cortex Low levels of glycerophospholipids including phospholipid-bound arachidonate, sphingomyelin, and the myelin constituents galactosylceramide and sulfatides were observed from the early stages of AD [10,258,259]. The low levels of PUFAs in membrane phospholipids in AD may be explained by decreased synthesis, increased release from sn-2-bound PUFAs, notably arachidonate, by phospholipase A2 which was reported to be increased in AD [260], inadequate replacement by the Lands cycle (Figure 6), or lipid degradation by ROS. In vitro, ATP depletion resulting from slc25a25-knock-down in renal cells had a dramatic effect on intracellular lipids, with increases in unbound PUFAs, lysoplasmalogens and some phospholipids, and decreases in intermediates of phosphatide synthesis, and some lysophospholipids [85] (Section 3.2). These changes could have resulted from a combination of the factors ennumerated above.
Single nucleotide polymorphisms (SNPs) of genes involved in lipid turnover associate with AD. These include ATP-binding cassette subfamily A members 1 and 7 (ABCA1 and ABCA7). ABCA1 initiates the efflux of lipids such as cholesterol and phospholipids by loading them to lipid-free lipoproteins. A loss-of-function mutation in ABCA1 associates with increased risk for AD [19]. ABCA7 is also involved in the transport of cholesterol and phospholipids. Multiple loss of function variants of ABCA7 have been found to associate with altered lipid- and Aβ metabolism and increased AD risks [20,21]. An SNP for the gene for Sterol regulatory-element binding protein-2, SREBP-2, which regulates cholesterol synthesis associated with biomarkers for AD [22].
6. Hypoperfusion of the Hippocampus
Many brain imaging studies have reported decreased cerebral blood flow (CBF) in patients with AD [261,262,263,264,265,266,267,268]. Reduced glucose uptake and perfusion in the hippocampus, parietotemporal cortex and/or posterior cingulate cortex have been demonstrated by FDG-PET in AD in individuals with early AD, MCI or no cognitive impairment prior to progression to AD [265,269,270], and in individuals at genetic risk for AD [271,272]. The primary problem is an inadequate blood supply and not reduced metabolic demand [263]. For decades, the blood supply to the hippocampus has been considered parlous, with limited capacity to meet increased demands [273,274,275]. When coupled with pathological dysfunction of arteries supplying blood to this region, for example atheroma, hypertension or vasospasm after subarachnoid haemorrhage [36,265,276,277] causing arterial constriction, there is a risk of local ischaemia during high neuronal activity. This could trigger a cascade of biochemical events contributory to AD. Comparison of the vasculature of the brain cortex and hippocampus in vivo using neuro imaging has explained the vulnerability.
6.1. Blood Supply to the Brain Cortex and Hippocampus
The brain’s blood supply is provided by three large arteries, the posterior, anterior and middle cerebral arteries, that arise from the Circle of Willis, an arterial hub at the base fed by blood from the internal carotid and vertebral arteries. Those supplying the cortex branch into large pial arteries which run along the surface of the brain, become progressively smaller, and penetrate perpendicularly into the brain substance giving rise to arterioles and capillaries [36,265,273,278]. There are layers of contractile muscle cells in the walls of the arteries and arterioles. In the capillaries, these are replaced by pericytes, small smooth muscle cells which underlie the vascular endothelium and are enclosed within the basal lamina. Astrocytic end feet encase this basal lamina. The astrocytes, pericytes, endothelium and adjacent neurons associate as a neurovascular unit. The pial arteries receive innervation by peripheral nerves, whereas arterioles and micro vessels are innervated intrinsically within the brain substance. Cerebral blood flow (CBF) varies by brain region and adapts constantly to ensure energy supply and waste removal to meet local needs. CBF increases during neuronal activity through dilation of local arterioles in response to the concerted actions of a range of vasoactive agents produced by vascular cells, neurons and astrocytes. Notable among these are nitric oxide, prostacyclin, adenosine and K+ ions [36,265,279,280,281]
6.2. Features of the Hippocampal Vasculature Increase the Risk for Hypoperfusion
The source of blood for the hippocampus is variable. High resolution 7 Tesla time-of-flight MR was used to visualise the brain vasculature of healthy young adults aged 19 – 34 years [273] The most common source (50 % of hemispheres) was a combination from the posterior cerebral artery (PCA) and the anterior choroidal artery (AChA), in agreement with 57% found in an autopsy study [278]. The least common source (3-5%) was the AChA alone. Blood in the PCA was mostly from the vertebrobasilar artery, and not the carotid arteries which are the source in small mammals. Different distribution patterns of the right and left hemispheres were observed (Table 2 [273]). Table 2 summarises studies to investigate CBF, Supplement S1 includes experimental details.
Properties of the hippocampal blood vessels which would increase susceptibility to hypoxia and ischaemia are i) the arteries and veins in the hippocampus have a long tangential course and few anastomoses [36,273], ii) compared with the cortex, there are far fewer capillaries, and these are more widely spaced, iii) they are extremely narrow. The mean diameter of intrahippocampal arteries is 0.09 mm [273]. Delivery of oxygen and glucose may be compromised by low capillary density and red blood cell velocity [273,274,294].
These anatomical features are not confined to individuals with AD but apply to the whole population. Hippocampal perfusion is reduced in healthy older adults aged 60-77 years without dementia (Table2 [284]). In individuals aged around 70y greater blood flow to the hippocampus was positively correlated with memory performance [36]. The vascular reserve of the hippocampus is now considered a primary contributing factor to cognitive performance [295]. Cardiovascular disorders [296,297], and conditions causing chronic hypoxia [298,299] are risk factors for sporadic AD. Vascular dysfunction is a prominent and early feature in prodromal AD [265]. Atherosclerosis of cerebral arteries or hypoxia (Table2 [282,283]), or increased intra-arterial pressure in hypertension (Section 6.4), vasospasm following a subarachnoid haemorrhage [32,269,270] could all reduce the hippocampal blood flow to levels below the safety threshold. At autopsy, atherosclerotic stenosis was significantly greater in circle of Willis arteries and large leptomeningeal arteries from individuals with AD than from nondemented controls [282,283]. The number of stenoses and the index of occlusion were positively correlated (R=0.67; P<0.00001), and the index of stenosis correlated with the scores for total amyloid plaque, neuritic plaque, neurofibrillary tangle, Braak stage, and white matter rarefaction. Severe stenotic lesions consisting of long and continuous stretches of atheroma plaque causing total arterial occlusion were observed in some arterial segments in the AD cases.
6.3. Neurovascular Coupling and the Effects of Hypoxia
The brain is protected from damage due to transient mild oxygen insufficiency during increased neuronal activity by a process termed neurovascular coupling. The neurons signal to local capillaries to dilate, thereby increasing local blood flow and oxygen and glucose delivery. The mechanism which mediates this response is unclear, but HIF may have a central role [36,274,300]. Severe episodic or sustained hypoxia initiates a cascade of pathological events that leads to neuronal degeneration [300,301,302,303].
Shaw, Bell, Boyd et al. [274] investigated neurovascular coupling in vivo in mouse brain cortex through an implanted cranial window. Hippocampal (HC) blood vessels had a blunted response compared with those in the visual cortex (VI), with fewer, smaller dilations. Dilations of HC vessels larger than 7 µm were only half those in the V1. The calculated rate of O2 consumption (VO2) indicated that production of ATP through oxidative phosphorylation in the HC was restricted in tissue furthest from a capillary. A low O2 concentration was estimated to decrease consumption by at least 10% in 30% of HC tissue, and by at least 20% in 10% of tissue. Shaw et al. surmised that, since O2 levels are limiting under physiological conditions, further decreases in O2 availability, as with decreased CBF or local brain ischaemia, would produce a greater reduction in ATP synthesis over a larger volume of tissue in HC than in neocortex.
In healthy young men, hypobaric hypoxia (12% O2 ) reduced blood flow to brain regions with roles in memory functions (Table 2). Hypoxia for approximately 7h significantly reduced O2 delivery of the middle cerebral artery during an executive task and of the posterior cerebral artery during memory tasks but had no effect on cognition [275]. Hypoxia for 2h increased cortical ironment. The decrease resulted from vasoconstriction. After 10 h of hypoxia, decreased blood flow to the major nodes of the default mode network was more pronounced and widespread, involving the posterior cingulate and cuneal cortex, which have roles in declarative and procedural memory [286]. Significantly, decreased flow to the cingulate gyrus was reported in hypertensive individuals [Table 2 [288]]. Ischaemia led to widespread biochemical disturbances in human brain ex vivo and brain from a mouse stroke model (Table 2 [287]). Notably there was a large increase in succinate, but the other TCA intermediates decreased. ATP levels fell dramatically, which probably explains the observed large increase in 5-oxoproline, an intermediate in biosynthesis of glutathione (Figure 8). A fall in glutathione production would dramatically reduce protection from ROS.
Exposure of rodents to hypobaric oxygen also had a significant metabolic impact on brain (Table 2 [290,292]). The hippocampus was most susceptible [292]. Energy production was disturbed (increased lactate dehydrogenas and glutamate dehydrogenase), and ROS generation increased significantly and progressed with time. Protection from ROS decreased strikingly (Table 2 [290,292,293]). The disturbances increased with duration of hypoxia and were more evident in the hippocampus than brain cortex. Exposure of rats to hypoxia for 4 days led to significant morphological changes in the hippocampus. The damage increased between 72h and 144h after hypoxia, suggesting delayed neurotoxicity (Table 2 [291])
Collectively the studies demonstrate that hypoxia causes a significant decrease in hippocampal blood flow. This causes widespread biochemical disturbances with free radical generation which increases with duration of hypoxia and may cause delayed neurotoxicity.
6.4. Effects of Hypertension on Cerebral Blood Flow
Systemic arterial pressure varies during normal activities. The brain self-protects against these oscillations by a process of autoregulation in which the resistance of the cerebral arterioles is adjusted according to intravascular pressure [36,265] By constricting as pressure increases and relaxing as it decreases, the arterioles normally maintain a constant cerebral blood flow over the range 60 to 150mm Hg. This is mediated by depolarisation of arterial muscle cells by rising intraluminal pressure, resulting in influx of Ca2+ and vasoconstriction, together with actions of protein kinase C and Rho kinase which increase the muscle contractile response. Endothelium-derived constrictor factors participate in maintenance of basal arterial tone. Vasodilation is regulated by release of NO, prostacyclin, bradykinin and other agents by endothelial cells [32,36,304].
Hypertension causes changes in arterial structure which can impair blood flow, especially during ischaemic insults or hypotensive episodes. Sustained increases in intraluminal pressure stimulate remodelling of the arterial muscle cells which may undergo hypertrophy and hyperplasia causing thickening of the arterial wall, or rearrangement. Both mechanisms lead to narrowing of the arterial lumen. Arterial resistance is inversely proportional to the luminal diameter, which therefore determines blood flow [36]. In hypertension, myogenic activity (autoregulation) operates at a higher range of pressure (approximately 65 to 190 mm Hg) in cerebral arteries [280]. In addition, hypertension may cause arterial stiffening, promote atherosclerotic plaque formation or cause fibrinoid necrosis of the penetrating arteries which can lead to white matter infarcts or small haemorrhages [305]. The BBB is impaired, with loss of brain protection [36,265]. In the stroke-prone, spontaneously hypertensive rat (SHRSP) model, remodelling increases with age and the lumen diameter decreases progressively [280]. A variety of factors contribute to the muscle hypertrophy, including trophic effects of sympathetic nerves, mechanical effects of increased pressure, increased growth factors, AII (angiotensin II), and oxidative stress [36,265]. Impaired endothelial-mediated vasodilation and regulation of myogenic activity compound the arterial dysfunction. Contributory factors include increased ROS generation via NADPH oxidation, decreases in superoxide dismutase (SOD) and cystathionine b-synthase, deficiency of NO (nitric oxide) due to inactivation by ROS and to decreased activity of eNOS (endothelial nitric oxide synthase), decreased endothelium-derived hyperpolarising factor (EDHF), decreased vasodilatory eicosanoids, prostacyclin and epoxyeicosatrienoic acids, and impaired function of ion channels including store-operated calcium channels, calcium-activated K+ channels and transient receptor potential vanilloid channel 4 (TRPV4) [32,36,304]. Collectively they decrease basal CBF and functional hyperaemia to support increased brain activity.
Hypertension in AD
In a longitudinal study, regional CBF in prefrontal, anterior cingulate, and occipital areas decreased over 6 years in older individuals with hypertension compared with controls (Table 2 [288]). Hypertension in midlife increases the risk of AD in later life. It accelerates progression of AD and is associated with an increase in amyloid deposition [261]. It probably precedes the development of plaques. From observations in SHRSP rats, arterial narrowing may progress with age without effective blood pressure control [306]. Early diagnosis of hypertension and good control is now regarded as a priority for reducing the risk for AD.
7. Genomic, Proteomic, Metabolomic and Imaging Investigations to Identify Causative Genes and Pathways in AD
7.1. Human Studies
The number of studies to explore the genomics of AD using the advanced analytical and information technology now available has escalated. Recent focus has been on identifying disturbances in molecular pathways, rather than associations with individual compounds in order to obtain a broader view to aid reconstruction of events. Many have analysed brain or data from carefully collected samples from cohorts of individuals with AD and controls with normal cognition. The analytical results must be interpreted with caution because of well-recognised problems with the samples analysed. Brain chemistry changes very rapidly ex vivo, and this must also be true of the transcriptome which may not give a true picture of the in vivo situation, despite comparison with controls. Metabolites in CSF reflect production across the whole brain surface and small, deeply seated, regions are poorly represented. Further, normally about 80% of the total protein amount in CSF derives from size-dependent filtration of blood across the blood-brain barrier (BBB) [307]. The human studies documented in Table 3 and presented with more detail in Supplementary Table S2, were selected because they included data for the hippocampus or entorhinal cortex, and/or for early stages of AD, and/or investigated metabolic pathways relevant to the proposal under investigation. Studies of brain rather than CSF and blood were preferred, first because the search was for localised early changes in the hippocampus, second because studies using CSF Tau and Aβ amyloid as inclusion criteria for AD were probably investigating a relatively advanced stage of the AD progression. Readers should refer to the papers reporting the genomic studies. They present a wealth of data which the Tables here cannot convey.
Because the objectives and the research procedures differed, it is impossible to group the data. Of 13 studies tabulated which analysed brain metabolites, six identified significant differences in OXPHOS, mitochondrial, mitochondrial function or energy pathways compared to controls (study number [Ref]: 3[16], 6[47], 7[310],11[313], 13[315],15[2]). This is consistent with the growing view that mitochondrial dysfunction is a major pathogenic factor in AD. Of interest, Study 3 [16] demonstrated increased expression of OXPHOS subunits in young asymptomatic ApoE4 carriers, and study 15 [2] observed increased expression in individuals with AD pathology but normal cognition (AD-resilient) compared to elderly non-demented individuals. This might reflect responses to ATP depletion (refer to discussion in section 8). Other observations from the brain studies to highlight are: Studies 1 [1] and 4 [38], which demonstrated that neuronal cell numbers and hippocampal volume are maintained in older individuals with normal cognition in contrast to AD, Study 2 [308], which observed marked disturbances of the malate-aspartate shuttle, glycerophospholipids and pyrimidines in brain from individuals with AD within 4 hours of death. These are similar to findings for ATP-depleted renal cells with SLC25A25 deficiency [84,85] (Section 3.5 and Section 5.1.5). Study 10 [312] observed decreases in branched chain amino acids with AD progression, probably explained by increased catabolism for energy. Medium and long chain acylcarnitines were increased, which was a surprising finding. These are diagnostic markers for deficiency of very long chain fatty acid dehydrogenase (VLCAD), which is located at the inner mitochondrial membrane (IMM). Reduced enzyme activity might reflect IMM damage. In contrast, expression of the gene for carnitine palmitoyltransferase A (CPTA1) was increased. This enzyme, on the outer mitochondrial membrane, converts acyl-CoA (long-chain fatty acids) into acyl-carnitines for transfer into mitochondria. Study 13 [315] found that expression of 88 proteins was needed to predict progression of asymptomatic AD to AD, Study 14 [18] identified associatons of three mitochondrial solute-linked 25 (SLC25) carriers with AD. One, SLC25A22, which codes for glutamate carrier1 was proposed to participate in the glutamate/GABA/glutamine cycle.
7.2. Animal Studies

Table 4.
Metabolomic, proteomic and genomic studies of AD in animals†.
| Study | Main relevant findings | Reference | |
|---|---|---|---|
| 21 | CSF metabolome of a rabbit model for late onset AD with AD neuropathology induced by a high cholesterol diet | Profiles changed with time; Aβ-like plaques only seen at 12 weeks Four clusters identified in the top 95 metabolites, most at 12 weeks. At 12 weeks, decreased phospholipids, mainly phosphorylated fatty alcohols, akylacyl or dialkyl-glycerophosphates, all potential precursors or degradation products of phospholipids including phosphatidylcholines and plasmalogens. | Liu QY, Bingham EJ, Twine SM, et al., 2012 [26] |
| 22 | Cerebral cortical and glutamine metabolism in a mouse AD model (APPswe/PSEN1dE9) | AD mice: significantly increased lactate and alanine, decreased TCA intermediates, decreased capacity for uptake and oxidative metabolism of glutamine; no change in glial acetate metabolism. | Andersen JV, Christensen SK, Aldana BI, et al., 2017 [320] |
| 23 | Hippocampal proteomic pathways associated with memory status in normal aging and 5FXAD AD mouse model | Normal and AD mice, HDAC4 identified as regulator of memory-related proteins; Top pathways associated with memory deficits in controls: OXPHOS, mitochondrial dysfunction, glutamate receptor signalling; | Neuner SM, Wilmott LA, Hoffmann BR, et al., 2017 [321] |
| 24 | Investigation for overlap in protein expression up to 15m of normal mice following mild traumatic brain injury (TBI) aged 3m; and non-traumatised mice with AD (PSAPP and mice expressing hTau) up to 15m | Impaired in TBI: energy metabolism, clearance, neurotransmitter and intracellular signalling, glial cell function. Little overlap with altered proteins in AD models. TBI and AD damage distinct processes | Ojo JO, Crynen G, Algamal M, et al., 2020 [322] |
| 25 | Characterization of Tg4-42 mouse model for AD [323] | Significant loss of hippocampal CA1 neurons. At 9m caudate, putamen: significant decreases: GABA, glutamine, lactate: increased Aβ42, glutaminase, glutamine decarboxylase, CSF, increased neurofilament light chains (NFL) | Hinteregger B, Loeffler T, Flunkert S, et al., 2021 [323] |
| 26 | Metabolite analyses of cortex and hippocampus of a transgenic AD mouse model with high resolution magic angle spinning NMR. | Controls: changes with age in cortex; at 9m sex differences; at 9m differences from AD mice in hippocampus: glutamate, glutamine, Nacetylaspartate (NAA), glycine, phosphocholine and glycerophosphocholine. | Füzesi MV, Muti IH, Berker Y, et al. 2022 [324] |
| 27 | Investigation of mitochondrial dysfunction and effects of an antibody to a neurotoxic Tau peptide in hippocampus and retina of a mouse AD model | Decreased expression of genes involved in multiple energy generating mitochondrial pathways including OXPHOS pathways; FA oxidation; in the hippocampus and retina of Tg2576 AD mice; GSEA analysis: oxidative phosphorylation the most down-regulated gene set in hippocampus of early symptomatic Tg2576; mitochondrial alterations observed in AD mice significantly reverted by NH2htau antibody. | Morello G, Guarnaccia M, La Cognata V, et al., 2023 [325] |
† For more information refer to Supplement Table S3. HDAC4 Histone Deacetylase 4, GSEA gene set enrichment analysis, Tg25476 AD mice, overexpress a mutated form of APP (the ‘Swedish mutation’); APPswe/PSEN1dE9 (PSAPP) AD mice, carry the Swedish mutation and a presenilin mutation; 5xFAD mice express 5 mutations in two genes (APP and Presenilin-1); Tg4-42 mice carry N-truncated 4- 42 Aβ.
Because of the problems of investigating AD in humans, such models have a central role in AD research. A problem highlighted with transgenic AD models is that they resemble early onset genetic AD rather than the common late onset disorder (LOAD) [26]. Non-transgenic rabbits fed a high cholesterol diet have amyloid deposits and other pathological markers and resemble LOAD more closely. Differences in CSF metabolites from controls were observed in rabbits on a high cholesterol intake for 3 to 12 weeks, with most of the abnormalities at 12 weeks.The majority were glycerophosphates and phospphorylated fatty alcohols. Aβ-like plaques were only observed at 12 weeks (Study 21 [26]). Decreases in TCA intermediates and of oxidative glutamine were recorded in cortex and hippocampus from APPswe/PSEN1dE9 AD mice aged 3m, before amyloid plaque developed 22 [320]. Marked mitochondrial dysfunction was evident in hippocampus and frontal cortex from transgenic 25476 AD mice, a commonly used AD model (Study 27 [325], energy generating mitochondrial pathways including OXPHOS pathways and fatty acid oxidation. From gene set enrichment analysis (GSEA), oxidative phosphorylation was the most down-regulated gene set in hippocampus of early symptomatic Tg2576. An NH2htau antibody produced significant resolution of the abnomalities. In a third mouse model (Study 25 [323]) expressing 4-42 Tau which is truncated at the N-terminal, significant metabolic differences from controls were observed in the caudate and putamen regions at 9m of age. Aβ accumulated intracellularly, but there were no plaques. These mice develop cognitive deficits. 4-42 Tau is not produced physiologically and is more toxic than Aβ1-40 and Aβ1-42. Study 24 [322] demonstrated that proteomes of repetitive mild tramatic injury to the brain and of an AD mouse model showed little overlap, indicating different mechanisms for brain damage.
8. Discussion
The proposal under consideration is that inadequacies of the blood supply to the hippocampus lead to clinically silent focal episodes of hypoperfusion. The resulting ATP depletion disrupts essential neuronal activities, causing a cascade of cumulative damage and inflammation which slowly spreads further into the brain and impacts on memory and learning. This seems a feasible, neat and logical explanation to explain the onset of Alzheimer’s disease. The brain is wholly reliant on a continuous supply of ATP to function and loss of supply can be predicted to have widespread consequences, particularly on processes with heaviest ATP consumption. The high content of PUFAs in membrane phospholipids provide an abundant substrate for cascading free radical attack causing peroxidative damage, which is a common feature of brain ischaemia. This would affect signalling and transport functions throughout the cell, and provoke an inflammatory response at the cell surface. An individual with an unfavourable hippocampal vascular supply would be predicted to have numerous minor ischaemic events over the years whenever the ‘head pressure’ of the main arterial supply falls. These would become more frequent after middle age with cerebral arterial narrowing due to atheroma and/or hypertension, and result in cumulative spreading damage. It is an attractive proposition, but is it tenable? Examination of the scientific basis for ATP depletion and its likely consequences demonstrates that it could indeed have major consequences on mitochondrial and membrane functions, biosynthesis and recycling of glutamate neurotransmitters and axonal transport of organelles. It might possibly explain accumulation of Aβ amyloid and plaque development and Tau tangles. Excessive ROS activity would make a significant contribution to the cascading cell damage. The case for hypoperfusion rests on the parlous blood supply to the hippocampus resulting from variations in its vascular supply, and/or restricted blood flow through narrowed blood vessels. Findings from recent genomic studies are disparate, reflecting differences in the study objectives, but collectively support the proposal and indicate avenues for prevention/intervention. It is interesting that a study of brain from individuals with Parkinson’s disease found that critical processes affected in early disease, before synucleinopathy and neuronal loss were oxidative phosphorylation and ATP synthesis and glutathione and redox regulation [326].
This is an extensive subject, and only the most significant points can be addressed here.
Provision of a ready supply of ATP is paramount for normal brain function. The requirement for ATP is monitored continually by sensors (S2.2) which react immediately to a range of molecular changes, notably a decreased AMP/ATP ratio (AMPK), increased NAD+/NADH ratio (sirtuins), decreased phosphofructokinase activity (fructose bisphosphate) and hypoxia (HFI1 and HUMMR), which indicate a threat to supply. They take corrective action, increasing transcription of numerous genes to activate pathways to increase ATP synthesis, reduce energy expenditure and increase supply of oxygen and/or metabolic substrates. Generation of appropriate amounts of ATP must be controlled by the suppliers-namely the mitochondria. This necessitates very close co-ordination of mitochondrial and extra-mitochondrial activities, particularly to govern the profile of transcribed genes and to continually adjust the production of proteins to metabolic responses. Discovery of the mitochondrial derived peptides (MDPs) over the last 15 years has been an exciting development which provides some insight into how this is achieved (S2.4). Although their function is still not well understood, it is of great interest that four have been shown to impact on mitochondrial respiration. Humanin and SHLP2 increased respiration and SHLP2 had a protective interaction with mitochondrial complex 1 [17]. MOTSc however, decreased mitochondrial respiration and increased glycolysis in vitro and MtALTND4 which decreases complex 1 activity, reduced the O2 consumption rate and maximum coupled and uncoupled respiration [104]. There are probably other MDPs to be discovered. They are likely to be key regulators of mitochondrial function. Microproteins encoded within nuclear genes which modify mitochondrial function have also been identified. MIEF1-microprotein was reported to be involved in mitochondrial fission [106] and mitoregulin to support super complexes and to modify mitochondrial respiratory efficiency [99]. These findings indicate intense cross-communication between the mitochondria and nucleus which likely shapes the responses to significant hypoxic disturbances.
The malate aspartate shuttle (MAS), glutamate/GABA/glutamine cycle and axonal transport of organelles and proteins which have a very high ATP turnover are activities at high risk of disturbance with reduced energy supply (Section 4). The striking feature is the symbiosis of neurons and astrocytes in these processes. Future studies should perhaps investigate them as a single unit, possibly in organ cultures. In addition to essential roles in energy transfer and maintenance of redox balance in neurons, the MAS provides aspartate for export from neurons to adjacent astrocytes for glutamine synthesis and anaplerosis. In turn, astrocytes release glutamine to neurons for glutamate and GABA synthesis and as an energy source. Depletion of ATP, resulting in failure of these three processes, could have a major role in the loss of neurons, dendrites, and synapses, dystrophic axons and axonal swellings, as well as the reduced and disordered neurotransmission observed in AD (Section 1). Tau accumulation appears to be a consequence of reduced axonal trafficking along microtubules. This may be in response to signalling and kinase activation initiated by the neuronal nucleus to reduce energy consumption when ATP is low (Section 4.3.). Phosphorylation of epitopes in the proline-rich domain and C-terminus of Tau leads to detachment of Tau from the microtubules, which are thereby destabilized and disaggregate. Released phosphorylated Tau may form tangles (NFT’s) in the cytosol. Tau tangles induce an inflammatory response in nearby microglia which adds to AD damage. However, Tau has additional functions to axonal transport. It binds to histones in nuclear DNA and may be involved in chromatin remodelling and possibly protection of DNA from damage. This may be relevant to individuals with normal cognition who express Tau.
ATP depletion would be predicted to cause disturbances in brain lipids. Synthesis and remodelling of lipids has high energy dependency (Section 5). Deficiencies of phospholipids result in disordered lipid distribution in cell membranes. Increases or decreases in membrane cholesterol affect compaction of lipid rafts and location and activities of membrane-bound proteins and other molecules. These disturbances impact on signalling. Phospholipids (cardiolipin and phosphatidylethanolamine) have an essential structural function in inner mitochondrial membranes by enabling membrane curvature and providing support for mitochondrial cristae which are the predominant sites of OXPHOS assembly and operation and other super complexes. However, on the downside, the high content of PUFAs in brain membranes are a rich substrate for cascading free radical attack, causing unregulated propagation of damaging lipid peroxidation. This is a common feature of brain damage, particularly with ischaemia. Normal brain cells are well equipped to counteract this with protective antioxidant proteins and enzymes. Studies of brain from patients with AD have reported differences in the brain lipid content from unaffected controls, evidence of increased ROS activity and decreased protective agents (Section 5.1, Section 7). Changes in membrane glycerophospholipids were observed in early AD. Loss of function polymorphisms in two genes involved in cholesterol and phospholipid turnover, ATP-binding cassette subfamily A members 1 (ABCA1 and ABCA7) are associated with AD (Table 1). ABCA1 initiates cellular efflux of lipids by loading them to lipid-free lipoproteins. Is it possible that ApoE4 is among these and, if so, whether this could explain its association with AD?
Amyloid Precursor protein (APP) (Section 5.1.4.) is a trans-membrane protein which is mainly located in intracellular organelles, the Golgi apparatus, trans-Golgi network (TGN) and post-TGN vesicles, with only around 10% at the cell surface. Processing by transmembrane enzymes β-secretase (BACE) and γ-secretase produces Aβ1-40 and Aβ1-42 amyloid which form amyloid plaques. Queries to raise are, first, since BACE1 and γ-secretase are present in the TGN and endosomes, do APP and the Aβ peptides have roles in these organelles?, second, BACE1 has many other substrates. Is it possible that abnormal processing of one or more of these proteins could yield damaging peptides?, third, could misalignment of APP and the cleavage enzymes in ROS-damaged membranes divert APP to the amyloidogenic β-secretase pathway and away from the non-amyloidogenic α-secretase pathway (Figure 7)?
The number of studies to explore the genomics of AD using the advanced analytical and information technology now available has escalated. It was necessary to restrict those presented to studies providing data closely relevant to the proposal under review. The human studies enumerated in Table 3 (Section 7) had varying objectives and used different analytical approaches. Five of 12 brain studies which undertook genomic, proteomic or metabolomic studies on individuals with AD identified significant differences in OXPHOS, mitochondrial, mitochondrial function or energy pathways compared to controls. This is consistent with the growing view that mitochondrial dysfunction is a major pathogenic factor in AD. Three of the 12 studies found lipid disturbances, and one identified marked disturbance in the malate-aspartate shuttle. Studies 3 and 15 in Table 3 unexpectedly observed increased expression of respiratory chain components. This could indicate a response to a low energy status. If this is the case, there might not be enough reserve capacity to restore ATP levels should the blood flow to the hippocampus fall, or neuronal hyperactivity increase ATP consumption. Study 3 investigated young asymptomatic individuals heterozygous for ApoE4 and Study 15, individuals with AD markers but normal cognition (AD resilience). It is clearly important to protect the brains of healthy elderly at times of risk, for example following a cardiac or neurovascular event, surgery or acute urinary infection, by careful control of oxygen intake, blood pressure, prompt antibiotic and antioxidant administration. Short-term administration of protective drugs may be a future option. Cardiologists are exploring ways to prevent memory and cognitive loss after cardiac surgery [274]. Boosting the anaplerotic capacity of astrocytes would give longer protection. Triheptanoin has been used in clinical trials for neurological disorders (Section 4.2.4.). Young carriers of the APOE4 allele have increased medial temporal lobe activity during active encoding tasks compared to non-carriers [327,328,329], which would increase ATP consumption. In addition, there is evidence that APOE4 carriers are less able to regulate cerebral metabolism than APOE4-negative individuals [330]. Adding ApoE4 to well-being checks at middle age, perhaps with follow-up brain imaging to check for cerebrovascular abnormalities, would identify carriers for protective measures as outlined above.
Reduced glucose uptake and perfusion in the hippocampus, parietotemporal cortex and/or posterior cingulate cortex demonstrated by FDG-PET are early features in AD which may precede cognitive impairment [Section 6]. Largely because of recent advances in neuroimaging, this can now be explained by a variable combination of anatomical characteristics of the hippocampal vasculature, and reduced blood flow to the hippocampus, most often because of narrowing of the supplying arteries (Section 6). Atheroma and hypertensive vasoconstriction are the commonest causes. The main source of blood in humans is the vertebrobasilar artery, but the arterial branches from this which supply the hippocampus vary and may differ between hemispheres. Recent in vivo studies of healthy young adults using time-of-flight angiography with very high resolution demonstrated that hippocampal arteries have few anastomoses and that, compared to brain cortex, the hippocampus has relatively few capillaries that are widely spaced. In addition, the protective vasodilatory response of hippocampal blood vessels to low blood O2 concentrations is blunted compared with cortical vessels [274]. O2 delivery to tissues furthest away from the capillaries will be most affected when delivery falls. Collectively, these factors explain why the hippocampus is vulnerable to hypoperfusion and hypoxia and why the vulnerability differs between individuals.
Although low O2 concentrations in the blood due to lung disorders and other hypoxic conditions reduce O2 delivery, by far the commonest cause is reduced blood flow through narrowing of the lumen of hippocampal arteries. Association of AD with cardiovascular diseases has been clearly demonstrated. Arterial narrowing may be caused by atherosclerosis of the cerebral arteries. Although common in the population, in some individuals with AD it may be more extensive than in non-affected subjects. The problem in hypertension is hypertrophy and hyperplasia of arterial muscle cells causing thickening of the arterial wall, sometimes with arterial stiffening, which increases the resistance to flow. Hypertension may also promote arterial plaque formation, damage arteries perforating the brain substance, and decrease the blood brain barrier. The arterial resistance increases with time with a progressive decrease in the arterial lumen. Section 6.4 enumerates some of many factors that contribute to muscle hypertrophy. Clearly to increase protection against development of AD, treatments to prevent progression of atheroma and to control blood pressure must be initiated early and monitored.
Subarachnoid haemorrhage (SAH, a bleed on the surface of the brain due to a ruptured brain aneurysm) leads to cognitive impairment in approximately 50% of patients [269,331]. In around 70% of patients, vasopasm of the cerebral arteries develops from the third day post-bleed, peaks after a week and then subsides [32]. Of interest, expression of apoE is associated with a higher risk of cognitive morbidity and delayed ischemia following SAH [332,333]. In a rat model of SAH, administration of an apoE-mimetic peptide improved functional outcomes and reduced evidence of vasospasm following SAH [334 Mesis]. Vasospasm may be one of several factors that contribute to cognitive loss but it is thought not to be the sole or primary cause [269]. There is no reported association of SAH with AD. The association of vasospasm with apoE is interesting and perhaps merits further exploration.
8.1. Suggestions for Further Study
Investigate neurons and astrocytes together, possibly in organ cultures, in view of their tight symbiosis
Clarify the roles of ApoE4
Consider why ApoE4 increased arterial vasospasm after subarachnoid haemorrhage (SAH) and the beneficial action of an ApoE4-mimetic peptide
Elucidate the roles of mitochondrial derived peptides in regulating ATP synthesis
Clarify the roles of APP/Aβ in the endoplasmic reticulum
Cross-link with cardiologists investigating memory and cognitive impairment following cardiac events and neurosurgeons following SAH
Consider the other BACE substrates and whether any may generate damaging fragments in AD
Investigate the effects of SLC25A25 knock-down on the metabolome and proteome of brain cells to observe the impact of ATP depletion [84,85]
Consider administration of agents to promote the anaplerotic capacity of astrocytes to bolster ATP production
9. Conclusions
The identity of the factor(s) which initiates the brain damage that leads to Alzheimer’s disease remains elusive. The proposal that poor perfusion with reduced oxygen delivery to the hippocampus triggers the process through ATP depletion is attractive. Certainly it would contribute to on-going damage. There is good evidence to implicate hypoperfusion. There is a strong scientific argument that ATP depletion could be a major factor in initiating and propagating the pathological disturbances that characterise AD, and evidence from a variety of sources to support this.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Valerie Walker is the sole Author.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created. Supplementary information to data presented in the Text is provided in Supplementary Tables S1 and S2, with references to the studies reported.
Acknowledgments
Sarah Ennis, Genomics University of Southampton UK, Central and South Genomic Medicine Service Alliance, and Eli Hatchwell, Population Bio UK, Inc, Oxfordshire,UK, for helping me to link recent genomics with brain metabolism, and Paul Cook, University Hospital Southampton UK, for support, encouragement and clinical discussions.
Conflicts of Interest
Valerie Walker declares no conflicts of interest.
Abbreviations
| AMPK 5′ | AMP-activated protein kinase |
| APH1 | (Anterior Pharynx defective 1) a component of the gamma-secretase complex. |
| APP | Amyloid Beta Precursor Protein |
| BACE | Beta-Secretase APP Beta-Secretase |
| BBB | blood brain barrier |
| CBF | cerebral blood flow, |
| rCBF | regional cerebral blood flow |
| FDG-PET | fluorodeoxyglucose (FDG)-positive emission tomography (PET} |
| GSEA | gene set enrichment analysis |
| HDAC4 | Histone Deacetylase |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| HUMMR | hypoxia up-regulated mitochondrial movement regulator |
| LOAD | late onset Alzheimers disease |
| MCI | Mild Cognitive Impairment |
| Miro1 and Miro2 | Mitochondrial Rho GTPase proteins |
| Mitochondrial-derived peptides: | |
| GAU | gene antisense ubiquitous |
| MOTS-c | Mitochondrial ORF of the 12S rRNA Type-C |
| MtALTND4 | protein encoded from an alternative open reading frame of the gene for the NADH-ubiquinone oxidoreductase chain 4 (ND4) protein |
| SHLP1 -SHLP6 | six small humanin-like peptides with 20-35 amino acids |
| SHMOOSE | Small Human Mitochondrial ORF Over SErine tRNA. |
| MRI | Magnetic Resonance Imaging |
| NFTs | neurofibrillary tangles, |
| PEN2 | Gamma-secretase subunit |
| PET | positive emission tomography |
| PS1/PS2 | Presenilin 1/ 2 |
| PUFAs | polyunsaturated fatty acids, |
| SAH | subarachnoid haemorrhage |
| SREBP-2 | Sterol regulatory-element binding protein-2, |
| Transgenic mouse models: | |
| Tg25476 AD mice | overexpress a mutated form of APP (the ‘Swedish mutation’). Develop amyloid plaques and cognitive deficits |
| APPswe/PSEN1dE9 (PSAPP) AD mice | carry two mutations: the Swedish mutation and a presenilin mutation 5xFAD mice express 5 mutations in two genes (APP and Presenilin-1); have increased Aβpeptide, amyloid plaques, cognitive deficits 3xTG mice express the Swedish mutation, a PSEN1 mutation , and a human Tau mutation Tg4-42 mouse model: Mouse model with N-truncated 4- 42 Aβ |
References
- Gómez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996 Jul 15;16(14):4491-500. PMID: 8699259; PMCID: PMC6578866. [CrossRef]
- Morgan GR, Carlyle BC. Interrogation of the human cortical peptidome uncovers cell-type specific signatures of cognitive resilience against Alzheimer’s disease. Sci Rep. 2024 Mar 26;14(1):7161. PMID: 38531951; PMCID: PMC10966065. [CrossRef]
- Thangavel R, Kempuraj D, Stolmeier D, Anantharam P, Khan M, Zaheer A. Glia maturation factor expression in entorhinal cortex of Alzheimer’s disease brain. Neurochem Res. 2013 Sep;38(9):1777-84. Epub 2013 May 29. PMID: 23715664; PMCID: PMC3735652. [CrossRef]
- Kapadia A, Billimoria K, Desai P, Grist JT, Heyn C, Maralani P, et al. Hypoperfusion Precedes Tau Deposition in the Entorhinal Cortex: A Retrospective Evaluation of ADNI-2 Data. J Clin Neurol (2023) 19(2): 131–7. [CrossRef]
- Kahn I, Andrews-Hanna JR, Vincent JL, Snyder AZ, Buckner RL. Distinct cortical anatomy linked to subregions of the medial temporal lobe revealed by intrinsic functional connectivity. J Neurophysiol. 2008 Jul;100(1):129-39. Epub 2008 Apr 2. PMID: 18385483; PMCID: PMC2493488. [CrossRef]
- Torrico TJ, Abdijadid S. Neuroanatomy, Limbic System. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.nlm.nih.gov/books/NBK538491/.
- Patel A, Biso GMNR, Fowler JB. Neuroanatomy, Temporal Lobe. [Updated 2023 Jul 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519512/- accessed 15-06-2025.
- Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. 2006:119–26. [CrossRef]
- Ryzhikova E, Ralbovsky NM, Sikirzhytski V, Kazakov O, Halamkova L, Quinn J, Zimmerman EA, Lednev IK. Raman spectroscopy and machine learning for biomedical applications: Alzheimer’s disease diagnosis based on the analysis of cerebrospinal fluid. Spectrochim Acta A Mol Biomol Spectrosc. 2021 Mar 5; 248:119188. Epub 2020 Nov 13. PMID: 33268033. [CrossRef]
- Yin F. Lipid metabolism and Alzheimer’s disease: clinical evidence, mechanistic link and therapeutic promise. FEBS J. 2023 Mar;290(6):1420-1453. Epub 2022 Jan 18. PMID: 34997690; PMCID: PMC9259766. [CrossRef]
- Li J, Li L, Cai S, Song K, Hu S. Identification of novel risk genes for Alzheimer’s disease by integrating genetics from hippocampus. Sci Rep. 2024 Nov 11;14(1):27484. PMID: 39523385; PMCID: PMC11551212. [CrossRef]
- Wu YY, Lee YS, Liu YL, Hsu WC, Ho WM, Huang YH, Tsai SJ, Kuo PH, Chen YC. Association study of alcohol dehydrogenase and aldehyde dehydrogenase polymorphism with Alzheimer Disease in the Taiwanese Population. Front Neurosci. 2021 Jan 22;15: 625885. PMID: 33551739; PMCID: PMC7862325. [CrossRef]
- Tsai CY, Wu SM, Kuan YC, Lin YT, Hsu CR, Hsu WH, Liu YS, Majumdar A, Stettler M, Yang CM, Lee KY, Wu D, Lee HC, Wu CJ, Kang JH, Liu WT. Associations between risk of Alzheimer’s disease and obstructive sleep apnea, intermittent hypoxia, and arousal responses: A pilot study. Front Neurol. 2022 Nov 30; 13:1038735. PMID: 36530623; PMCID: PMC9747943. [CrossRef]
- Adlimoghaddam A, Sabbir MG, Albensi BC. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front Mol Neurosci. 2016 Aug 8; 9: 57. PMID: 27551259; PMCID: PMC4976099. [CrossRef]
- Seiler N. Ammonia and Alzheimer’s disease. Neurochem Int. 2002 Aug-Sep;41(2-3):189-207. PMID: 12020619. [CrossRef]
- Perkins M, Wolf AB, Chavira B, Shonebarger D, Meckel JP, Leung L, Ballina L, Ly S, Saini A, Jones TB, Vallejo J, Jentarra G, Valla J. Altered Energy Metabolism Pathways in the Posterior Cingulate in Young Adult Apolipoprotein E ɛ4 Carriers. J Alzheimers Dis. 2016 Apr 23;53(1):95-106. PMID: 27128370; PMCID: PMC4942726. [CrossRef]
- Yen K, Miller B, Kumagai H, Silverstein A, Cohen P. Mitochondrial-derived microproteins: from discovery to function. Trends Genet. 2024 Dec 16: S0168-9525(24)00292-0. PMID: 39690001. [CrossRef]
- Tian J, Jia K, Wang T, Guo L, Xuan Z, Michaelis EK, Swerdlow RH; Alzheimer’s Disease Neuroimaging Initiative; Du H. Hippocampal transcriptome-wide association study and pathway analysis of mitochondrial solute carriers in Alzheimer’s disease. Transl Psychiatry. 2024 Jun 10;14(1):250. PMID: 38858380; PMCID: PMC11164935. [CrossRef]
- Nordestgaard LT, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 2015;11: 1430–8.
- Aikawa T, Holm ML, Kanekiyo T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018 Feb 5;8(2):27. PMID: 29401741; PMCID: PMC5836046. [CrossRef]
- De Roeck A, Van Broeckhoven C, Sleegers K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019;138: 201–20. [CrossRef]
- Picard C, Julien C, Frappier J, Miron J, Théroux L, Dea D; United Kingdom Brain Expression Consortium and for the Alzheimer’s Disease Neuroimaging Initiative; Breitner JCS, Poirier J. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol Aging. 2018 Jun; 66: 180.e1-180.e9. Epub 2018 Feb 9. PMID: 29503034. [CrossRef]
- Soleimani Zakeri NS, Pashazadeh S, MotieGhader H. Gene biomarker discovery at different stages of Alzheimer using gene co-expression network approach. Sci Rep. 2020 Jul 22;10(1):12210. PMID: 32699331; PMCID: PMC7376049. [CrossRef]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009; 41:1088–93. [CrossRef]
- Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017 Mar 28;14(3): e1002270. PMID: 28350801; PMCID: PMC5370101. [CrossRef]
- Liu QY, Bingham EJ, Twine SM, Geiger JD, Ghribi O. Metabolomic Identification in cerebrospinal fluid of the effects of high dietary cholesterol in a rabbit model of Alzheimer’s Disease. Metabolomics (Los Angel). 2012 Mar 29;2(3):109. PMID: 24851192; PMCID: PMC4026014.
- Naudi A, Cabre R, Jove M, Ayala V, Gonzalo H, Portero-Otin M, et al. Lipidomics of human brain aging and Alzheimer’s disease pathology. Int Rev Neurobiol. 2015;122: 33–89. [CrossRef]
- Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007 Jan 1;12:2601-15. PMID: 17127266; PMCID: PMC2141543. [CrossRef]
- Sastry PS. Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24(2):69-176. PMID: 3916238. [CrossRef]
- Lamari F, Rossignol F, Mitchell GA. Glycerophospholipids: Roles in Cell Trafficking and Associated Inborn Errors. J Inherit Metab Dis. 2025 Mar;48(2): e70019. PMID: 40101691; PMCID: PMC11919462. [CrossRef]
- Halliwell B, Gutteridge JM. Lipid peroxidation, oxygen radicals, cell damage, and antioxidant therapy. Lancet. 1984 Jun 23;1(8391):1396-7. PMID: 6145845. [CrossRef]
- Walker V, Pickard JD. Prostaglandins, thromboxane, leukotrienes and the cerebral circulation in health and disease. Adv Tech Stand Neurosurg. 1985;12: 3-90. PMID: 3002404. [CrossRef]
- Feringa FM, van der Kant R. Cholesterol and Alzheimer’s Disease; from risk genes to pathological effects. Front Aging Neurosci. 2021 Jun 24;13: 690372. PMID: 34248607; PMCID: PMC8264368. [CrossRef]
- Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim Biophys Acta. 2010 Aug;1801(8): 860-7. Epub 2010 Mar 18. PMID: 20303415; PMCID: PMC2886169. [CrossRef]
- Jin U, Park SJ, Park SM. Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease. Exp Neurobiol. 2019 Oct 31;28(5):554-567. PMID: 31698548; PMCID: PMC6844. [CrossRef]
- Johnson AC. Hippocampal vascular supply and its role in vascular cognitive impairment. Stroke. 2023 Mar;54(3):673-685. Epub 2023 Feb 27. PMID: 36848422; PMCID: PMC9991081. [CrossRef]
- Petralia RS, Mattson MP, Yao PJ. Communication breakdown: the impact of ageing on synapse structure. Ageing Res Rev. 2014 Mar;14: 31-42. Epub 2014 Feb 2. PMID: 24495392; PMCID: PMC4094371. [CrossRef]
- Ledig C, Schuh A, Guerrero R, Heckemann RA, Rueckert D. Structural brain imaging in Alzheimer’s disease and mild cognitive impairment: biomarker analysis and shared morphometry database. Sci Rep. 2018; 8: 11258. [CrossRef]
- Ingram T, Chakrabarti L. Proteomic profiling of mitochondria: what does it tell us about the ageing brain? Aging (Albany NY). 2016 Dec 13;8(12):3161-3179. PMID: 27992860; PMCID: PMC5270661. [CrossRef]
- Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008 May;60(5):308-14. PMID: 18421773. [CrossRef]
- Ferrándiz ML, Martínez M, De Juan E, Díez A, Bustos G, Miquel J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994; 644: 335–38. [CrossRef]
- Mather M, Rottenberg H. Aging enhances the activation of the permeability transition pore in mitochondria. Biochem Biophys Res Commun. 2000; 273:603–08. [CrossRef]
- Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013 Mar;123(3):951-7. Epub 2013 Mar 1. PMID: 23454757; PMCID: PMC3582127. [CrossRef]
- Stauch KL, Purnell PR, Fox HS. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY). 2014 Apr;6(4):320-34. PMID: 24827396; PMCID: PMC4032798. [CrossRef]
- Stauch KL, Purnell PR, Villeneuve LM, Fox HS. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics. 2015;15: 1574–86. [CrossRef]
- Groebe K, Krause F, Kunstmann B, Unterluggauer H, Reifschneider NH, Scheckhuber CQ, Sastri C, Stegmann W, Wozny W, Schwall GP, Poznanović S, Dencher NA, Jansen-Dürr P, et al. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp Gerontol. 2007; 42:887–98. [CrossRef]
- Hu Y, Pan J, Xin Y, Mi X, Wang J, Gao Q, Luo H. Gene expression analysis reveals novel gene signatures between young and old adults in human prefrontal cortex. Front Aging Neurosci. 2018 Aug 27;10: 259. PMID: 30210331; PMCID: PMC6119720. [CrossRef]
- Gomez-Cabrera MC, Ferrando B, Brioche T, Sanchis-Gomar F, Viña J. Exercise and antioxidant supplements in the elderly. J Sport Health Sci. 2013; 2: 94–100. [CrossRef]
- Balakrishnan M, Kenworthy AK. Lipid peroxidation drives liquid-liquid phase separation and disrupts raft protein partitioning in biological membranes. J Am Chem Soc. 2024 Jan 17;146(2):1374-1387. PMID: 37745342; PMCID: PMC10515805. [CrossRef]
- Berchtold NC, Coleman PD, Cribbs DH, Rogers J, Gillen DL, Cotman CW. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol Aging. 2013 Jun;34(6):1653-61. Epub 2012 Dec 27. PMID: 23273601; PMCID: PMC4022280. [CrossRef]
- Garin CM, Nadkarni NA, Pépin J, Flament J, Dhenain M. Whole brain mapping of glutamate distribution in adult and old primates at 11.7T. Neuroimage. 2022 May 1; 251:118984. Epub 2022 Feb 8. PMID: 35149230. [CrossRef]
- Trefts E, Shaw RJ. AMPK: restoring metabolic homeostasis over space and time. Mol Cell. 2021 Sep 16;81(18):3677-3690. PMID: 34547233; PMCID: PMC8549486. [CrossRef]
- Marchetti P, Fovez Q, Germain N, Khamari R, Kluza J. Mitochondrial spare respiratory capacity: mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB j. 2020;34(10):13106–13124. [CrossRef]
- Ferreira T, Rodriguez S. Mitochondrial DNA: Inherent Complexities Relevant to Genetic Analyses. Genes (Basel). 2024 May 12;15(5):617. PMID: 38790246; PMCID: PMC11121663. [CrossRef]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS, The human mitochondrial transcriptome, Cell 146 (2011) 645–658. PubMed: 21854988. [CrossRef]
- Nguyen QL, Corey C, White P, et al. Platelets from pulmonary hypertension patients show increased mitochondrial reserve capacity. JCI Insight. 2018; 2:1-10. [CrossRef]
- Hardie DG, Schaffer BE, Brunet A, 2016. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 26, 190–201. [CrossRef]
- Hardie DG. Keeping the home fires burning: AMP-activated protein kinase. J R Soc Interface. 2018 Jan;15(138):20170774. PMID: 29343628; PMCID: PMC5805978. [CrossRef]
- Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016 Jul 15;48(7): e245. PMID: 27416781; PMCID: PMC4973318. [CrossRef]
- Thapa R, Moglad E, Afzal M, Gupta G, Bhat AA, Hassan Almalki W, Kazmi I, Alzarea SI, Pant K, Singh TG, Singh SK, Ali H. The role of sirtuin 1 in ageing and neurodegenerative disease: A molecular perspective. Ageing Res Rev. 2024 Dec;102: 102545. Epub 2024 Oct 17. PMID: 39423873. [CrossRef]
- Yang JH, Hayano M, Griffin PT, Amorim JA, Bonkowski MS, Apostolides JK, Salfati EL, Blanchette M, Munding EM, Bhakta M, et al. Loss of epigenetic information as a cause of mammalian aging. Cell. 2023 Jan 19;186(2):305-326.e27. Epub 2023 Jan 12. Erratum in: Cell. 2024 Feb 29;187(5):1312-1313. Doi: 10.1016/j.cell.2024.01.049. PMID: 36638792; PMCID: PMC10166133. [CrossRef]
- Razick DI, Akhtar M, Wen J, Alam M, Dean N, Karabala M, Ansari U, Ansari Z, Tabaie E, Siddiqui S. The Role of Sirtuin 1 (SIRT1) in Neurodegeneration. Cureus. 2023 Jun 15;15(6): e40463. PMID: 37456463; PMCID: PMC10349546. [CrossRef]
- Wu QJ, Zhang TN, Chen HH, Yu XF, Lv JL, Liu YY, Liu YS, Zheng G, Zhao JQ, Wei YF, Guo JY, Liu FH, Chang Q, Zhang YX, Liu CG, Zhao YH. The sirtuin family in health and disease. Signal Transduct Target Ther. 2022 Dec 29;7(1):402. PMID: 36581622; PMCID: PMC9797940. [CrossRef]
- Burtscher J, Denti V, Gostner JM, Weiss AK, Strasser B, Hüfner K, Burtscher M, Paglia G, Kopp M, Dünnwald T. The interplay of NAD and hypoxic stress and its relevance for ageing. Ageing Res Rev. 2025 Feb; 104:102646. Epub 2024 Dec 20. PMID: 39710071. [CrossRef]
- Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A, Altucci L. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics. 2016 May 25;8:61. PMID: 27226812; PMCID: PMC4879741. [CrossRef]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009 Apr 23;458(7241):1056-60. PMID: 19262508; PMCID: PMC3616311. [CrossRef]
- Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008 Oct 10;283(41):27628-27635. Epub 2008 Aug 7. PMID: 18687677; PMCID: PMC2562073. [CrossRef]
- Yang Y, Liu Y, Wang Y, Chao Y, Zhang J, Jia Y, Tie J, Hu D. Regulation of SIRT1 and Its Roles in Inflammation. Front Immunol. 2022 Mar 11;13:831168. PMID: 35359990; PMCID: PMC8962665. [CrossRef]
- Song Y, Wu Z, Zhao P. The protective effects of activating Sirt1/NF-κB pathway for neurological disorders. Rev Neurosci. 2021 Nov 8;33(4):427-438. PMID: 34757706. [CrossRef]
- Sarubbo F, Esteban S, Miralles A, Moranta D. Effects of Resveratrol and other Polyphenols on Sirt1: Relevance to Brain Function During Aging. Curr Neuropharmacol. 2018 Jan 30;16(2):126-136. PMID: 28676015; PMCID: PMC5883375. [CrossRef]
- Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010 Jan 19;49(2):304-11. PMID: 20000467; PMCID: PMC2826167. [CrossRef]
- He J, Liu X, Su C, Wu F, Sun J, Zhang J, Yang X, Zhang C, Zhou Z, Zhang X, Lin X, Tao J. Inhibition of Mitochondrial Oxidative Damage Improves Reendothelialization Capacity of Endothelial Progenitor Cells via SIRT3 (Sirtuin 3)-Enhanced SOD2 (Superoxide Dismutase 2) Deacetylation in Hypertension. Arterioscler Thromb Vasc Biol. 2019 Aug;39(8):1682-1698. Epub 2019 Jun 13. PMID: 31189433. [CrossRef]
- Kunji ERS, King MS, Ruprecht JJ, Thangaratnarajah C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology (Bethesda). 2020 Sep 1;35(5):302-327. PMID: 32783608; PMCID: PMC7611780. [CrossRef]
- Bround MJ, Bers DM, Molkentin JD. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J Mol Cell Cardiol. 2020 Jul;144:A3-A13. Epub 2020 May 23. PMID: 32454061; PMCID: PMC7483807. [CrossRef]
- Gavaldà-Navarro A, Mampel T, Viñas O. Changes in the expression of the human adenine nucleotide translocase isoforms condition cellular metabolic/proliferative status. Open Biol. 2016 Feb;6(2):150108. PMID: 26842067; PMCID: PMC4772803. [CrossRef]
- Atlante A, Valenti D. A walk in the memory, from the first functional approach up to its regulatory role of mitochondrial bioenergetic flow in health and disease: focus on the adenine nucleotide translocator. Int J Mol Sci. 2021 Apr 17;22(8):4164. PMID: 33920595; PMCID: PMC8073645. [CrossRef]
- Ho L, Titus AS, Banerjee KK, George S, Lin W, Deota S, Saha AK, Nakamura K, Gut P, Verdin E, Kolthur-Seetharam U. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (Albany NY). 2013 Nov;5(11):835-49. PMID: 24296486; PMCID: PMC38687268. [CrossRef]
- Zhang P, Cheng X, Sun H, Li Y, Mei W, Zeng C. Atractyloside Protect Mice Against Liver Steatosis by Activation of Autophagy via ANT-AMPK-mTORC1 Signaling Pathway. Front Pharmacol. 2021 Sep 21;12:736655. PMID: 34621170; PMCID: PMC8490973. [CrossRef]
- Austin J, Aprille JR. Carboxyatractyloside-insensitive influx and efflux of adenine nucleotides in rat liver mitochondria. J Biol Chem. 1984 Jan 10;259(1):154-60. PMID: 6706925. [CrossRef]
- del Arco A, Satrústegui J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J Biol Chem. 2004;279:24701–24713. [CrossRef]
- Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279: 30722–30730. [CrossRef]
- Aprille JR. Mechanism and regulation of the mitochondrial ATP-Mg/P(i) carrier. J Bioenerg Biomembr. 1993;25:473–481. [CrossRef]
- Anunciado-Koza RP, Zhang J, Ukropec J, Bajpeyi S, Koza RA, Rogers RC, Cefalu WT, Mynatt RL, Kozak LP. Inactivation of the mitochondrial carrier SLC25A25 (ATP-Mg2+/Pi transporter) reduces physical endurance and metabolic efficiency in mice. J Biol Chem. 2011 Apr 1;286(13):11659-71. Epub 2011 Feb 4. PMID: 21296886; PMCID: PMC3064218. [CrossRef]
- Hofherr A, Seger C, Fitzpatrick F, Busch T, Michel E, Luan J, Osterried L, Linden F, Kramer-Zucker A, Wakimoto B, Schütze C, et al. The mitochondrial transporter SLC25A25 links ciliary TRPP2 signaling and cellular metabolism. PLoS Biol. 2018 Aug 6;16(8):e2005651. PMID: 30080851; PMCID: PMC6095617. [CrossRef]
- Hofherr A, Seger C, Fitzpatrick F, Busch T, Michel E, Luan J, Osterried L, Linden F, Kramer-Zucker A, Wakimoto B, Schütze C, et al. The mitochondrial transporter SLC25A25 links ciliary TRPP2 signaling and cellular metabolism, Supplement S2 Data. PLoS Biol 2018; 16(8): e2005651. [CrossRef]
- Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1) alpha: its protein stability and biological functions. Exp Mol Med. 2004 Feb 29;36(1):1-12. PMID: 15031665. [CrossRef]
- Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15: 551-78. PMID: 1061197. [CrossRef]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol (1985). 2000 Apr;88(4):1474-80. PMID: 10749844. [CrossRef]
- Cui C, Jiang X, Wang Y, Li C, Lin Z, Wei Y, Ni Q. Cerebral Hypoxia-Induced Molecular Alterations and Their Impact on the Physiology of Neurons and Dendritic Spines: A Comprehensive Review. Cell Mol Neurobiol. 2024 Aug 6;44(1):58. PMID: 39105862; PMCID: PMC11303443. [CrossRef]
- Lin TK, Huang CR, Lin KJ, Hsieh YH, Chen SD, Lin YC, Chao AC, Yang DI. Potential Roles of Hypoxia-Inducible Factor-1 in Alzheimer’s Disease: Beneficial or Detrimental? Antioxidants (Basel). 2024 Nov 11;13(11):1378. PMID: 39594520; PMCID: PMC11591038. [CrossRef]
- Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 1996; 93:12969-73. [CrossRef]
- Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 1999; 18:1905-14. [CrossRef]
- Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. Hypoxia-induced activation of HIF-1: role of HIF-1alpha-Hsp90 interaction. FEBS Lett 1999; 460: 251-6. [CrossRef]
- Carrero P, Okamoto K, Coumailleau P, O’Brien S, Tanaka H, Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000 Jan;20(1):402-15. PMID: 10594042; PMCID: PMC85095. [CrossRef]
- Mole D.R., Blancher C., Copley R.R., Pollard P.J., Gleadle J.M., Ragoussis J., Ratcliffe P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009; 284:16767–16775. [CrossRef]
- Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999 Aug 20;274(34):24147-52. PMID: 10446188. [CrossRef]
- Lei L., Feng J., Wu G., Wei Z., Wang J.Z., Zhang B., Liu R., Liu F., Wang X., Li H.L. HIF-1alpha causes LCMT1/PP2A deficiency and mediates tau hyperphosphorylation and cognitive dysfunction during chronic hypoxia. Int. J. Mol. Sci. 2022; 23:16140. [CrossRef]
- Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, Rempe DA. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol. 2009 Jun 15;185(6):1065-81. PMID: 19528298; PMCID: PMC2711615. [CrossRef]
- Miller B, Kim SJ, Kumagai H, Mehta HH, Xiang W, Liu J, Yen K, Cohen P. Peptides derived from small mitochondrial open reading frames: Genomic, biological, and therapeutic implications. Exp Cell Res. 2020 Aug 15;393(2):112056. Epub 2020 May 6. PMID: 32387288; PMCID: PMC7778388. [CrossRef]
- Miller B, Kim SJ, Kumagai H, Yen K, Cohen P. Mitochondria-derived peptides in aging and healthspan. J Clin Invest. 2022 May 2;132(9):e158449. PMID: 35499074; PMCID: PMC9057581. [CrossRef]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS, The human mitochondrial transcriptome, Cell 146 (2011) 645–658. PubMed: 21854988. [CrossRef]
- Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY). 2016 Apr;8(4):796-809. PMID: 27070352; PMCID: PMC4925829. [CrossRef]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R,et al.The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015 Mar 3;21(3):443-54. PMID: 25738459; PMCID: PMC4350682. [CrossRef]
- Kienzle L, Bettinazzi S, Choquette T, Brunet M, Khorami HH, Jacques JF, Moreau M, Roucou X, Landry CR, Angers A, Breton S. A small protein coded within the mitochondrial canonical gene nd4 regulates mitochondrial bioenergetics. BMC Biol. 2023 May 18;21(1):111. PMID: 37198654; PMCID: PMC10193809. [CrossRef]
- Yen K, Mehta HH, Kim SJ, Lue Y, Hoang J, Guerrero N, Port J, Bi Q, Navarrete G, Brandhorst S, et al. The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY). 2020 Jun 23;12(12):11185-11199. Epub 2020 Jun 23. PMID: 32575074; PMCID: PMC7343442. [CrossRef]
- Rathore A, Chu Q, Tan D, Martinez TF, Donaldson CJ, Diedrich JK, Yates JR 3rd, Saghatelian A. MIEF1 Microprotein Regulates Mitochondrial Translation. Biochemistry. 2018 Sep 25;57(38):5564-5575. Epub 2018 Sep 14. PMID: 30215512; PMCID: PMC6443411. [CrossRef]
- Stein CS, Jadiya P, Zhang X, McLendon JM, Abouassaly GM, Witmer NH, Anderson EJ, Elrod JW, Boudreau RL. Mitoregulin: A lncRNA-Encoded Microprotein that Supports Mitochondrial Supercomplexes and Respiratory Efficiency. Cell Rep. 2018 Jun 26;23(13):3710-3720.e8. PMID: 29949756; PMCID: PMC6091870. [CrossRef]
- Makarewich CA, Baskin KK, Munir AZ, Bezprozvannaya S, Sharma G, Khemtong C, Shah AM, McAnally JR, Malloy CR, Szweda LI, et al. MOXI is a mitochondrial micropeptide that enhances fatty acid beta-oxidation, Cell Rep. 23 (2018) 3701–3709. [CrossRef]
- Lin YF, Xiao MH, Chen HX, Meng Y, Zhao N, Yang L, Tang H, Wang JL, Liu X, Zhu Y, Zhuang SM. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death Dis. 2019 Jul 11;10(7):528. PMID: 31296841; PMCID: PMC6624212. [CrossRef]
- Yen K, Wan J, Mehta HH, Miller B, Christensen A, Levine ME, Salomon MP, Brandhorst S, Xiao J, Kim SJ,etal. Humanin Prevents Age-Related Cognitive Decline in Mice and is Associated with Improved Cognitive Age in Humans. Sci Rep. 2018 Sep 21;8(1):14212. PMID: 30242290; PMCID: PMC6154958. [CrossRef]
- Miller B, Arpawong TE, Jiao H, Kim SJ, Yen K, Mehta HH, Wan J, Carpten JC, Cohen P. Comparing the Utility of Mitochondrial and Nuclear DNA to Adjust for Genetic Ancestry in Association Studies. Cells. 2019 Apr 3;8(4):306. PMID: 30987182; PMCID: PMC6523867. [CrossRef]
- Bennett NK, Nguyen MK, Darch MA, Nakaoka HJ, Cousineau D, Ten Hoeve J, Graeber TG, Schuelke M, Maltepe E, Kampmann M, Mendelsohn BA, Nakamura JL, Nakamura K. Defining the ATPome reveals cross-optimization of metabolic pathways. Nat Commun. 2020 Aug 28;11(1):4319. PMID: 32859923; PMCID: PMC7455733. [CrossRef]
- Mendelsohn BA, et al. A high-throughput screen of real-time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol. 2018;16: e2004624. [CrossRef]
- Vinklarova L, Schmidt M, Benek O, Kuca K, Gunn-Moore F, Musilek K. Friend or enemy? Review of 17β-HSD10 and its role in human health or disease. J Neurochem. 2020 Nov;155(3):231-249. PMID: 32306391. [CrossRef]
- Zhang D, Yip YM, Li L. In silico construction of HK2-VDAC1 complex and investigating the HK2 binding-induced molecular gating mechanism of VDAC1. Mitochondrion. 2016;30: 222–228. [CrossRef]
- Minet E, Ernest I, Michel G, Roland I, Remacle J, Raes M, Michiels C. HIF1A gene transcription is dependent on a core promoter sequence encompassing activating and inhibiting sequences located upstream from the transcription initiation site and cis elements located within the 5′UTR. Biochem Biophys Res Commun. 1999 Aug 2;261(2):534-40. PMID: 10425220. [CrossRef]
- Broeks MH, Shamseldin HE, Alhashem A, Hashem M, Abdulwahab F, Alshedi T, Alobaid I, Zwartkruis F, Westland D, Fuchs S, Verhoeven-Duif NM, Jans JJM, Alkuraya FS. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum Genet. 2019 Dec;138(11-12):1247-1257. Epub 2019 Sep 19. PMID: 31538237. [CrossRef]
- Pardo B, Herrada-Soler E, Satrústegui J, Contreras L, Del Arco A. AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease. Int J Mol Sci. 2022 Jan 4;23(1):528. PMID: 35008954; PMCID: PMC8745132. [CrossRef]
- Lacabanne D, Sowton AP, Jose B, Kunji ERS, Tavoulari S. Current Understanding of Pathogenic Mechanisms and Disease Models of Citrin Deficiency. J Inherit Metab Dis. 2025 Mar;48(2):e70021. PMID: 40145619; PMCID: PMC11948450. [CrossRef]
- Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrústegui J, et al. Citrin and aralar1 are Ca (2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001 Sep 17;20(18):5060-9. PMID: 11566871; PMCID: PMC125626. [CrossRef]
- Del Arco A, González-Moreno L, Pérez-Liébana I, Juaristi I, González-Sánchez P, Contreras L, Pardo B, Satrústegui J. Regulation of neuronal energy metabolism by calcium: Role of MCU and Aralar/malate-aspartate shuttle. Biochim Biophys Acta Mol Cell Res. 2023 Jun;1870(5):119468. Epub 2023 Mar 28. PMID: 36997074. [CrossRef]
- Cory JG. Purine and Pyrimidine Metabolism in Devlin TM Editor, Textbook of Biochemistry with clinical correlations Fifth Ed Wiley-Liss New York 2002; pp 825-860.
- Amaral A, Hadera MG, Kotter M, Sonnewald U. Oligodendrocytes Do Not Export NAA-Derived Aspartate In Vitro. Neurochem Res. 2017 Mar;42(3):827-837. Epub 2016 Jul 9. PMID: 27394419; PMCID: P. [CrossRef]
- Rosko L, Smith VN, Yamazaki R, Huang JK. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist. 2019 Aug;25(4):334-343. Epub 2018 Aug 20. PMID: 30122106; PMCID: PMC674560MC5357468. [CrossRef]
- Broeks MH, van Karnebeek CDM, Wanders RJA, Jans JJM, Verhoeven-Duif NM. Inborn disorders of the malate aspartate shuttle. J Inherit Metab Dis. 2021 Jul;44(4):792-808. Epub 2021 May 24. PMID: 33990986; PMCID: PMC836216. [CrossRef]
- Wolf NI, Van Der Knaap MS. AGC1 deficiency and cerebral hypomyelination. N Engl J Med. 2009;361(20):1997-1998. [CrossRef]
- Koch J, Broeks MH, Gautschi M, Jans J, Laemmle A. Inborn errors of the malate aspartate shuttle - Update on patients and cellular models. Mol Genet Metab. 2024 Aug;142(4):108520. Epub 2024 Jun 24. PMID: 38945121. [CrossRef]
- Ait-El-Mkadem S., Dayem-Quere M., Gusic M., Chaussenot A., Bannwarth S., Francois B., Genin E.C., Fragaki K., Volker-Touw C.L.M., Vasnier C., et al. Mutations in MDH2, encoding a Krebs cycle enzyme, cause early-onset severe encephalopathy. Am. J. Hum. Genet. 2017;100: 151–159. [CrossRef]
- van Karnebeek CDM, Ramos RJ, Wen XY, Tarailo-Graovac M, Gleeson JG, Skrypnyk C, Brand-Arzamendi K, Karbassi F, Issa MY, van der Lee R,et al. Bi-allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy. Am J Hum Genet. 2019 Sep 5;105(3):534-548. Epub 2019 Aug 15. PMID: 31422819; PMCID: PMC6732527. [CrossRef]
- Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Marmol P, Horiuchi M, Shimotsu K, et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem. 2005 Sep 2;280(35):31333-9. Epub 2005 Jun 29. PMID: 15987682. [CrossRef]
- Sakurai T, Ramoz N, Barreto M, Gazdoiu M, Takahashi N, Gertner M, Dorr N, Gama Sosa MA, De Gasperi R, Perez G, et al. Slc25a12 disruption alters myelination and neurofilaments: a model for a hypomyelination syndrome and childhood neurodevelopmental disorders. Biol Psychiatry. 2010 May 1;67(9):887-94. Epub 2009 Dec 16. PMID: 20015484; PMCID: PMC4067545. [CrossRef]
- Llorente-Folch I, Sahún I, Contreras L, Casarejos MJ, Grau JM, Saheki T, Mena MA, Satrústegui J, Dierssen M, Pardo B. AGC1-malate aspartate shuttle activity is critical for dopamine handling in the nigrostriatal pathway. J Neurochem. 2013 Feb;124(3):347-62. PMID: 23216354. [CrossRef]
- Andersen JV, Schousboe A. Glial Glutamine Homeostasis in Health and Disease. Neurochem Res. 2023 Apr;48(4):1100-1128. Epub 2022 Nov 2. PMID: 363223. [CrossRef]
- Schousboe A, Bak LK, Waagepetersen HS. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front Endocrinol (Lausanne). 2013 Aug 15;4: 102. PMID: 23966981; PMCID: PMC3744088. [CrossRef]
- Sidoryk-Węgrzynowicz M, Adamiak K, Strużyńska L. Astrocyte-Neuron Interaction via the Glutamate-Glutamine Cycle and Its Dysfunction in Tau-Dependent Neurodegeneration. Int J Mol Sci. 2024 Mar 6;25(5):3050. PMID: 38474295; PMCID: PMC10931567. [CrossRef]
- Derouiche A, Frotscher M. Astroglial processes around identified glutamatergic synapses contain glutamine synthetase: evidence for transmitter degradation. Brain Res. 1991 Jun 28;552(2):346-50. PMID: 1680531. [CrossRef]
- Walker V. Ammonia metabolism and hyperammonemic disorders. Adv Clin Chem. 2014;67:73-150. Epub 2014 Nov 4. PMID: 25735860. [CrossRef]
- Marx MC, Billups D, Billups B. Maintaining the presynaptic glutamate supply for excitatory neurotransmission. J Neurosci Res. 2015 Jul;93(7):1031-44. Epub 2015 Feb 3. PMID: 25648608. [CrossRef]
- Rothman DL, Behar KL, Hyder F, Shulman RG. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol. 2003; 65: 401-27. Epub 2002 May 1. PMID: 12524459. [CrossRef]
- Sonnewald U. Glutamate synthesis has to be matched by its degradation - where do all the carbons go? J Neurochem. 2014 Nov;131(4):399-406. Epub 2014 Jul 23. PMID: 24989463. [CrossRef]
- Yu AC, Drejer J, Hertz L, Schousboe A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem. 1983 Nov;41(5):1484-7. PMID: 6619879. [CrossRef]
- Schousboe A, Waagepetersen HS, Sonnewald U. Astrocytic pyruvate carboxylation: Status after 35 years. J Neurosci Res. 2019 Aug;97(8):890-896. Epub 2019 Feb 23. PMID: 30801795. [CrossRef]
- Bak LK, Walls AB, Schousboe A, Waagepetersen HS. Astrocytic glycogen metabolism in the healthy and diseased brain. J Biol Chem. 2018 May 11;293(19):7108-7116. Epub 2018 Mar 23. PMID: 29572349; PMCID: PMC5950001. [CrossRef]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996 Oct 17;383(6601):634-7. PMID: 8857541. [CrossRef]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997 Dec;19(6):1297-308. PMID: 9427252. [CrossRef]
- Mennerick S, Zorumski CF. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature. 1994 Mar 3;368(6466):59-62. PMID: 7906399. [CrossRef]
- Robinson MB, Lee ML, DaSilva S. Glutamate Transporters and Mitochondria: Signaling, Co-compartmentalization, Functional Coupling, and Future Directions. Neurochem Res. 2020 Mar;45(3):526-540. Epub 2020 Jan 30. PMID: 32002773. [CrossRef]
- Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511-35. Epub 2001 Nov 9. PMID: 12045105. [CrossRef]
- Rose EM, Koo JC, Antflick JE, Ahmed SM, Angers S, Hampson DR. Glutamate transporter coupling to Na,K-ATPase. J Neurosci. 2009 Jun 24;29(25):8143-55. PMID: 19553454; PMCID: PMC6666056. [CrossRef]
- Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci. 2011 Dec 14;31(50):18275-88. PMID: 22171032; PMCID: PMC3259858. [CrossRef]
- Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991 Dec 1;88(23):10540-3. PMID: 1683703; PMCID: PMC52964. [CrossRef]
- Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM et al. (1995) Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem 65:2146–2156. [CrossRef]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006 May;22(2):223-32. Epub 2006 Feb 8. PMID: 16466929. [CrossRef]
- Fan S, Xian X, Li L, Yao X, Hu Y, Zhang M, Li W. Ceftriaxone Improves Cognitive Function and Upregulates GLT-1-Related Glutamate-Glutamine Cycle in APP/PS1 Mice. J Alzheimers Dis. 2018;66(4):1731-1743. PMID: 30452416. [CrossRef]
- Fan S, Li L, Xian X, Liu L, Gao J, Li W. Ceftriaxone regulates glutamate production and vesicular assembly in presynaptic terminals through GLT-1 in APP/PS1 mice. Neurobiol Learn Mem. 2021 Sep;183:107480. Epub 2021 Jun 18. PMID: 34153453. [CrossRef]
- Aksenov MY, Aksenova MV, Carney JM, Butterfield DA. Oxidative modification of glutamine synthetase by amyloid beta peptide. Free Radic Res. 1997 Sep;27(3):267-81. PMID: 9350431. [CrossRef]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994 Apr 12;91(8):3270-4. PMID: 8159737; PMCID: PMC43558. [CrossRef]
- Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer’s beta-amyloid peptide (1-40) in cultured hippocampal neurons. Exp Neurol. 1995 Feb;131(2):193-202. PMID: 7895820. [CrossRef]
- Chen J, Herrup K. Chen J, Herrup K. Glutamine acts as a neuroprotectant against DNA damage, beta-amyloid and H2O2-induced stress. PLoS One. 2012;7(3):e33177. Epub 2012 Mar 8. PMID: 22413000; PMCID: PMC3297635. [CrossRef]
- Buntup D, Skare O, Solbu TT, Chaudhry FA, Storm-Mathisen J, Thangnipon W. Beta-amyloid 25-35 peptide reduces the expression of glutamine transporter SAT1 in cultured cortical neurons. Neurochem Res. 2008 Feb;33(2):248-56. Epub 2007 Nov 1. PMID: 18058230; PMCID: PMC2226019. [CrossRef]
- Kulijewicz-Nawrot M, Syková E, Chvátal A, Verkhratsky A, Rodríguez JJ. Astrocytes and glutamate homoeostasis in Alzheimer’s disease: a decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro. 2013 Oct 7;5(4):273-82. PMID: 24059854; PMCID: PMC3791522. [CrossRef]
- Andersen JV, Skotte NH, Christensen SK, Polli FS, Shabani M, Markussen KH, Haukedal H, Westi EW, Diaz-delCastillo M, Sun RC, et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 2021 Oct 16;12(11):954. PMID: 34657143; PMCID: PMC8520528. [CrossRef]
- Andersen JV, Christensen SK, Westi EW, Diaz-delCastillo M, Tanila H, Schousboe A, Aldana BI, Waagepetersen HS. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2021 Jan;148: 105198. Epub 2020 Nov 24. PMID: 33242587. [CrossRef]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996 Nov;40(5):759-66. PMID: 8957017. [CrossRef]
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009 Oct 14;29(41):12957-69. PMID: 19828810; PMCID: PMC2782445. [CrossRef]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grünblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007 Mar;11(1):97-116. PMID: 17361039. [CrossRef]
- Seiler N. Is ammonia a pathogenetic factor in Alzheimer’s disease? Neurochem Res. 1993 Mar;18(3):235-45. PMID: 8479596. [CrossRef]
- Hamdani EH, Popek M, Frontczak-Baniewicz M, Utheim TP, Albrecht J, Zielińska M, Chaudhry FA. Perturbation of astroglial Slc38 glutamine transporters by NH4+ contributes to neurophysiologic manifestations in acute liver failure. FASEB J. 2021 Jul;35(7):e21588. PMID: 34169573. [CrossRef]
- Gropman A. Brain imaging in urea cycle disorders. Mol Genet Metab. 2010;100 Suppl 1(Suppl 1):S20-30. Epub 2010 Feb 13. PMID: 20207564; PMCID: PMC3258295. [CrossRef]
- Maestri NE, Lord C, Glynn M, Bale A, Brusilow SW. The phenotype of ostensibly healthy women who are carriers for ornithine transcarbamylase deficiency. Medicine (Baltimore). 1998 Nov;77(6):389-97. PMID: 9854602. [CrossRef]
- Gyato K, Wray J, Huang ZJ, Yudkoff M, Batshaw ML. Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol. 2004 Jan;55(1):80-6. PMID: 14705115. [CrossRef]
- O’Donnell JC, Jackson JG, Robinson MB. Transient Oxygen/Glucose Deprivation Causes a Delayed Loss of Mitochondria and Increases Spontaneous Calcium Signaling in Astrocytic Processes. J Neurosci. 2016 Jul 6;36(27):7109-27. PMID: 27383588; PMCID: PMC4938859. [CrossRef]
- Malik AR, Willnow TE. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int J Mol Sci. 2019 Nov 12;20(22):5671. PMID: 31726793; PMCID: PMC6888459. [CrossRef]
- Mochel F, Duteil S, Marelli C, Jauffret C, Barles A, Holm J, Sweetman L, Benoist JF, Rabier D, Carlier PG, Durr A. Dietary anaplerotic therapy improves peripheral tissue energy metabolism in patients with Huntington’s disease. Eur J Hum Genet. 2010 Sep;18(9):1057-60. Epub 2010 May 26. PMID: 20512158; PMCID: PMC2987415. [CrossRef]
- Marin-Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab. 2013 Feb;33(2):175-82. Epub 2012 Oct 17. PMID: 23072752; PMCID: PMC3564188. [CrossRef]
- Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, Good LB, Ma Q, Marin-Valencia I, Zhang X, Park JY, Hynan LS, Stavinoha P, Roe CR, Lu H. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014 Oct;71(10):1255-65. PMID: 25110966; PMCID: PMC4376124. [CrossRef]
- Yuan X, Wang L, Tandon N, Sun H, Tian J, Du H, Pascual JM, Guo L. Triheptanoin Mitigates Brain ATP Depletion and Mitochondrial Dysfunction in a Mouse Model of Alzheimer’s Disease. J Alzheimers Dis. 2020;78(1):425-437. PMID: 33016909; PMCID: PMC8502101. [CrossRef]
- Aso E, Semakova J, Joda L, Semak V, Halbaut L, Calpena A, Escolano C, Perales JC, Ferrer I. Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer’s disease. Curr Alzheimer Res. 2013 Mar;10(3):290-7. PMID: 23131121. [CrossRef]
- Cai Q, Sheng ZH. Mitochondrial transport and docking in axons. Exp Neurol. 2009 Aug;218(2):257-67. Epub 2009 Mar 31. PMID: 19341731; PMCID: PMC2710402. [CrossRef]
- Berth SH, Lloyd TE. Disruption of axonal transport in neurodegeneration. J Clin Invest. 2023 Jun 1;133(11):e168554. PMID: 37259916; PMCID: PMC10232001. [CrossRef]
- Cason SE, Holzbaur ELF. Selective motor activation in organelle transport along axons. Nat Rev Mol Cell Biol. 2022;23(11):699–714. [CrossRef]
- Prezel E, Elie A, Delaroche J, Stoppin-Mellet V, Bosc C, Serre L, Fourest-Lieuvin A, Andrieux A, Vantard M, Arnal I. Tau can switch microtubule network organizations: from random networks to dynamic and stable bundles. Mol Biol Cell. 2018 Jan 15;29(2):154-165. Epub 2017 Nov 22. PMID: 29167379; PMCID: PMC5909928. [CrossRef]
- Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Théry M, Nachury MV. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science. 2017 Apr 21;356(6335):328-332. PMID: 28428427; PMCID: PMC5457157. [CrossRef]
- Portran D, Schaedel L, Xu Z, Théry M, Nachury MV. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol. 2017 Apr;19(4):391-398. Epub 2017 Feb 27. PMID: 28250419; PMCID: PMC5376231. [CrossRef]
- Eshun-Wilson L, Zhang R, Portran D, Nachury MV, Toso DB, Löhr T, Vendruscolo M, Bonomi M, Fraser JS, Nogales E. Effects of α-tubulin acetylation on microtubule structure and stability. Proc Natl Acad Sci U S A. 2019 May 21;116(21):10366-10371. Epub 2019 May 9. PMID: 31072936; PMCID: PMC6535015. [CrossRef]
- Janke C, Montagnac G. Causes and Consequences of Microtubule Acetylation. Curr Biol. 2017 Dec 4;27(23):R1287-R1292. PMID: 29207274. [CrossRef]
- Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012 May 1;125(Pt 9):2095-104. Epub 2012 May 22. PMID: 22619228; PMCID: PMC3656622 . [CrossRef]
- Kruppa AJ, Buss F. Motor proteins at the mitochondria-cytoskeleton interface. J Cell Sci. 2021 Apr 1;134(7):jcs226084. Epub 2021 Apr 13. PMID: 33912943; PMCID: PMC8077471 . [CrossRef]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009 Oct;10(10):682-96. PMID: 19773780 . [CrossRef]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42: 39–50. [CrossRef]
- Du J, Wei Y, Liu L, Wang Y, Khairova R, Blumenthal R, Tragon T, Hunsberger JG, Machado-Vieira R, Drevets W, et al. A kinesin signaling complex mediates the ability of GSK-3beta to affect mood-associated behaviors. Proc Natl Acad Sci U S A. 2010 Jun 22;107(25):11573-8. Epub 2010 Jun 7. PMID: 20534517; PMCID: PMC2895136 . [CrossRef]
- Banerjee R, Chakraborty P, Yu MC, Gunawardena S. A stop or go switch: glycogen synthase kinase 3β phosphorylation of the kinesin 1 motor domain at Ser314 halts motility without detaching from microtubules. Development. 2021 Dec 15;148(24):dev199866. Epub 2021 Dec 23. PMID: 34940839; PMCID: PMC8722386 . [CrossRef]
- Padzik A, Deshpande P, Hollos P, Franker M, Rannikko EH, Cai D, Prus P, Mågård M, Westerlund N, Verhey KJ,et al. KIF5C S176 Phosphorylation Regulates Microtubule Binding and Transport Efficiency in Mammalian Neurons. Front Cell Neurosci. 2016 Mar 15;10:57. PMID: 27013971; PMCID: PMC4791394 . [CrossRef]
- Brady ST, Morfini GA. Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol Dis. 2017;105: 273–282. [CrossRef]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013 Jun 1;5(6):a011304. PMID: 23732472; PMCID: PMC3660831 . [CrossRef]
- Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006 Jun 2;344(2):500-10. Epub 2006 Apr 3. PMID: 16630562 . [CrossRef]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006 May 22;173(4):545-57. PMID: 16717129; PMCID: PMC2063864. [CrossRef]
- Klosowiak JL, Focia PJ, Chakravarthy S, Landahl EC, Freymann DM, Rice SE. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013 Nov;14(11):968-74. Epub 2013 Sep 27. PMID: 24071720; PMCID: PMC3818075 . [CrossRef]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009 Feb 26;61(4):541-55. PMID: 19249275; PMCID: PMC2670979 . [CrossRef]
- Cai Q, Zakaria HM, Simone A, Sheng ZH. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012 Mar 20;22(6):545-52. Epub 2012 Feb 16. PMID: 22342752; PMCID: PMC3313683 . [CrossRef]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004 Jun 1;117(Pt 13):2791-804. Epub 2004 May 18. PMID: 15150321 . [CrossRef]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006 Apr;17(4):2057-68. Epub 2006 Feb 8. PMID: 16467387; PMCID: PMC1415296 . [CrossRef]
- Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis. 2012;2012:731526. [CrossRef]
- Elie A, Prezel E, Guérin C, Denarier E, Ramirez-Rios S, Serre L, Andrieux A, Fourest-Lieuvin A, Blanchoin L, Arnal I. Tau co-organizes dynamic microtubule and actin networks. Sci Rep. 2015 May 5;5: 9964. PMID: 25944224; PMCID: PMC4421749 . [CrossRef]
- Kadavath H, Jaremko M, Jaremko Ł, Biernat J, Mandelkow E, Zweckstetter M. Folding of the tau protein on microtubules. Angew Chem Int Ed Engl. 2015;54: 10347–10351. [CrossRef]
- Watamura N, Foiani MS, Bez S, Bourdenx M, Santambrogio A, Frodsham C, Camporesi E, Brinkmalm G, Zetterberg H, Patel S, et al. In vivo hyperphosphorylation of tau is associated with synaptic loss and behavioral abnormalities in the absence of tau seeds. Nat Neurosci. 2025 Feb;28(2):293-307. Epub 2024 Dec 24. PMID: 39719507; PMCID: PMC11802456. [CrossRef]
- Roveta F, Bonino L, Piella EM, Rainero I, Rubino E. Neuroinflammatory Biomarkers in Alzheimer’s Disease: From Pathophysiology to Clinical Implications. Int J Mol Sci. 2024 Nov 6;25(22):11941. PMID: 39596011; PMCID: PMC11593837. [CrossRef]
- Lanfranchi M, Yandiev S, Meyer-Dilhet G, Ellouze S, Kerkhofs M, Dos Reis R, Garcia A, Blondet C, Amar A, Kneppers A, et al. The AMPK-related kinase NUAK1 controls cortical axons branching by locally modulating mitochondrial metabolic functions. Nat Commun. 2024 Mar 21;15(1):2487. PMID: 38514619; PMCID: PMC10958033 . [CrossRef]
- Li S, Xiong G-J, Huang N, Sheng Z-H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020;2: 1077–1095. [CrossRef]
- DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019 Aug 2;14(1):32. PMID: 31375134; PMCID: PMC6679484 . [CrossRef]
- Rico T, Gilles M, Chauderlier A, Comptdaer T, Magnez R, Chwastyniak M, Drobecq H, Pinet F, Thuru X, Buée L, Galas MC, Lefebvre B. Tau Stabilizes Chromatin Compaction. Front Cell Dev Biol. 2021 Oct 14;9: 740550. PMID: 34722523; PMCID: PMC8551707 . [CrossRef]
- Magrin C, Bellafante M, Sola M, Piovesana E, Bolis M, Cascione L, Napoli S, Rinaldi A, Papin S, Paganetti P. Tau protein modulates an epigenetic mechanism of cellular senescence in human SH-SY5Y neuroblastoma cells. Front Cell Dev Biol. 2023 Oct 3;11:1232963. PMID: 37842084; PMCID: PMC10569482 . [CrossRef]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005 Feb 25;307(5713):1282-8. PMID: 15731448 . [CrossRef]
- Lauretti E, Dincer O, Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res. 2020 May;1867(5):118664. Epub 2020 Jan 30. PMID: 32006534; PMCID: PMC7047718 . [CrossRef]
- Lin CW, Chang LC, Ma T, Oh H, French B, Puralewski R, Mathews F, Fang Y, Lewis DA, Kennedy JL, et al. Older molecular brain age in severe mental illness. Mol Psychiatry. 2021 Jul;26(7):3646-3656. Epub 2020 Jul 6. PMID: 32632206; PMCID: PMC7785531 . [CrossRef]
- Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005 Apr;16(2):193-212. PMID: 15797830 . [CrossRef]
- Sych T, Gurdap CO, Wedemann L, Sezgin E. How Does Liquid-Liquid Phase Separation in Model Membranes Reflect Cell Membrane Heterogeneity? Membranes (Basel). 2021 Apr 28;11(5):323. PMID: 33925240; PMCID: PMC8146956 . [CrossRef]
- Brown D A, London E. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 1998, 164 (2), 103–114. [CrossRef]
- London E. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochim. Biophys. Acta, Mol. Cell Res. 2005, 1746 (3), 203–220. [CrossRef]
- Shaw T R, Ghosh S, Veatch SL Critical Phenomena in Plasma Membrane Organization and Function. Annu. Rev. Phys. Chem. 2021, 72, 51–72. [CrossRef]
- Levi M, Gratton E. Visualizing the regulation of SLC34 proteins at the apical membrane. Pflugers Arch. 2019 Apr;471(4):533-542. Epub 2019 Jan 6. PMID: 30613865; PMCID: PMC6436987 . [CrossRef]
- Sebastiao AM, Colino-Oliveira M, Assaife-Lopes N, Dias RB, Ribeiro JA. Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology. 2013;64: 97–107. [CrossRef]
- Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. 2017;18:361–74. [CrossRef]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1: 31–39. [CrossRef]
- Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5: 247–254. [CrossRef]
- Korbecki J, Bosiacki M, Pilarczyk M, Gąssowska-Dobrowolska M, Jarmużek P, Szućko-Kociuba I, Kulik-Sajewicz J, Chlubek D, Baranowska-Bosiacka I. Phospholipid Acyltransferases: Characterization and Involvement of the Enzymes in Metabolic and Cancer Diseases. Cancers (Basel). 2024 May 31;16(11):2115. PMID: 38893234; PMCID: PMC11171337 . [CrossRef]
- Agarwal AK. Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 2012;23: 290–302. [DOI] [PubMed] [Google Scholar]. [CrossRef]
- Maeda Y, Tashima Y, Houjou T, Fujita M, Yoko-o T, Jigami Y, Taguchi R, Kinoshita T. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol Biol Cell. 2007 Apr;18(4):1497-506. Epub 2007 Feb 21. PMID: 17314402 . [CrossRef]
- Kinoshita T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020 Mar;10(3):190290. Epub 2020 Mar 11. PMID: 32156170; PMCID: PMC7125958 . [CrossRef]
- Hishikawa D, Hashidate T, Shimizu T, Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res. 2014 May;55(5):799-807. Epub 2014 Mar 19. Erratum in: J Lipid Res. 2014 Nov;55(11):2444. PMID: 24646950; PMCID: PMC3995458 . [CrossRef]
- Baker MJ, Crameri JJ, Thorburn DR, Frazier AE, Stojanovski D. Mitochondrial biology and dysfunction in secondary mitochondrial disease. Open Biol. 2022 Dec;12(12):220274. Epub 2022 Dec 7. PMID: 36475414; PMCID: PMC9727669 . [CrossRef]
- Ikon N, Ryan RO. Cardiolipin and mitochondrial cristae organization. Biochim Biophys Acta Biomembr. 2017 Jun;1859(6):1156-1163. Epub 2017 Mar 20. PMID: 28336315; PMCID: PMC5426559 . [CrossRef]
- Gilkerson RW, Selker JM, Capaldi RA. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003 Jul 10;546(2-3):355-8. PMID: 12832068 . [CrossRef]
- Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005 Aug 19;280(33):29403-8. Epub 2005 Jun 22. PMID: 15972817; PMCID: PMC4113954 . [CrossRef]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003 Dec 26;278(52):52873-80. Epub 2003 Oct 15. PMID: 14561769 . [CrossRef]
- Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009 Aug 15;47(4):333-43. Epub 2009 May 8. PMID: 19427899 . [CrossRef]
- Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, Jiang F, Peng ZY. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid Med Cell Longev. 2019 Oct 13;2019: 5080843. PMID: 31737171; PMCID: PMC6815535 . [CrossRef]
- Braughler JM, Duncan LA, Chase R. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J. Biol. Chem. 1986, 261 (22), 10282–10289 [DOI] [PubMed] [Google Scholar]. [CrossRef]
- Koppenol WH, Hider RH. Iron and redox cycling. Do’s and don’ts. Free Radic Biol Med. 2019 Mar;133: 3-10. Epub 2018 Sep 17. PMID: 30236787 . [CrossRef]
- Slater TF. Overview of methods used for detecting lipid peroxidation. Methods Enzymol. 1984;105: 283-93. PMID: 6427 . [CrossRef]
- Akagi S, Kono N, Ariyama H, Shindou H, Shimizu T, Arai H. Lysophosphatidylcholine acyltransferase 1 protects against cytotoxicity induced by polyunsaturated fatty acids. FASEB J. 2016;30: 2027–2039. [CrossRef]
- Rybnikova E, Damdimopoulos AE, Gustafsson JA, Spyrou G, Pelto-Huikko M. Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur J Neurosci. 2000 May;12(5):1669-78. PMID: 10792444 . [CrossRef]
- Phadnis VV, Snider J, Varadharajan V, Ramachandiran I, Deik AA, Lai ZW, Kunchok T, Eaton EN, Sebastiany C, Lyakisheva A, et al. MMD collaborates with ACSL4 and MBOAT7 to promote polyunsaturated phosphatidylinositol remodeling and susceptibility to ferroptosis. Cell Rep. 2023 Sep 26;42(9):113023. Epub 2023 Sep 8. PMID: 37691145; PMCID: PMC10591818 . [CrossRef]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM,Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012; 149, 1060–1072. [CrossRef]
- Jiang X, Stockwell BR, and Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. 2021; Cell Biol. 22, 266–282. [CrossRef]
- Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017; 2, e90777. [DOI] [PMC free article] [PubMe. [CrossRef]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008; 283: 29615–29619. [CrossRef]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22: 9785–9793. [CrossRef]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. [CrossRef]
- Capone R, Tiwari A, Hadziselimovic A, Peskova Y, Hutchison JM, Sanders CR, Kenworthy AK. The C99 domain of the amyloid precursor protein resides in the disordered membrane phase. J Biol Chem. 2021 Jan-Jun;296: 100652. Epub 2021 Apr 9. PMID: 33839158; PMCID: PMC8113881 . [CrossRef]
- Vassar R. BACE1: the beta-secretase enzyme in Alzheimer’s disease. J Mol Neurosci. 2004;23 :105–114. [CrossRef]
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74 :342–352. [CrossRef]
- Iwatsubo T. The g-secretase complex: machinery for intramembrane proteolysis. Curr Opin Neurobiol. 2004;14:379–383. [CrossRef]
- Abraham CB, Xu L, Pantelopulos GA, Straub JE. Characterizing the transmembrane domains of ADAM10 and BACE1 and the impact of membrane composition. Biophys J. 2023 Oct 3;122(19):3999-4010. Epub 2023 Sep 1. PMID: 37658602; PMCID: PMC10560698. [CrossRef]
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002; 22:1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]. [CrossRef]
- Sun Y, Yao J, Kim TW, Tall AR. Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J Biol Chem. 2003;278: 27688–27694. [DOI] [PubMed] [Google Scholar]. [CrossRef]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One. 2009;4: e8477. [DOI] [PMC free article] [PubMed] [Google Scholar]. [CrossRef]
- Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 1998 Jan;23(1):81-8. PMID: 9482271. [CrossRef]
- Fabelo N, Martín V, Marín R, Moreno D, Ferrer I, Díaz M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol Aging. 2014 Aug;35(8):1801-12. Epub 2014 Feb 11. PMID: 24613671. [CrossRef]
- Stephenson DT, Lemere CA, Selkoe DJ, Clemens JA. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer’s disease brain. Neurobiol Dis. 1996 Feb;3(1):51-63. PMID: 9173912 . [CrossRef]
- Rubinski A, Tosun D, Franzmeier N, Neitzel J, Frontzkowski L, Weiner M, et al. Lower Cerebral Perfusion Is Associated with Tau-PET in the Entorhinal Cortex across the Alzheimer’s Continuum. Neurobiol Aging 2021; 102:111–8. [CrossRef]
- Chao LL, Buckley ST, Kornak J, Schuff N, Madison C, Yaffe K, Miller BL, Kramer JH, Weiner MW. ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis Assoc Disord. 2010 Jan-Mar;24(1):19-27. PMID: 20220321; PMCID: PMC2865220 . [CrossRef]
- Love S, Miners JS. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016 May;131(5):645-58. Epub 2015 Dec 28. PMID: 26711459; PMCID: PMC4835514 . [CrossRef]
- Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR Jr., Beckett LA, et al. Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain: a journal of neurology 2014; 137(Pt 5): 1550–61. [CrossRef]
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJJ, et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019 Jan;15(1):158-167. doi: 10.1016/j.jalz.2018.07.222. Erratum in: Alzheimers Dement. 2022 Mar;18(3):522. PMID: 30642436; PMCID: PMC6338083. [CrossRef]
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002 Apr;33(4):1152-62. PMID: 11935076. [CrossRef]
- Kalaria RN. Small vessel disease and Alzheimer’s dementia: pathological considerations. Cerebrovasc Dis. 2002; 13: 48–52. [CrossRef]
- Iadecola C, Gorelick PB. Converging pathologic mechanisms in vascular and neurodegenerative dementia. Stroke. 2003; 34: 335–337. [CrossRef]
- Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ; Alzheimer’s Disease Neuroimaging Initiative. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011 Jul;32(7):1207-18. Epub 2009 Aug 5. PMID: 19660834; PMCID: PMC2891865 . [CrossRef]
- Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CR Jr, et al. Alzheimer’s Disease Neuroimaging Initiative. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010 Jul 20;75(3):230-8. Epub 2010 Jun 30. PMID: 20592257; PMCID: PMC2906178 . [CrossRef]
- Ossenkoppele R, van der Flier WM, Zwan MD, Adriaanse SF, Boellaard R, Windhorst AD, Barkhof F, Lammertsma AA, Scheltens P, van Berckel BN. Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology. 2013 Jan 22;80(4):359-65. Epub 2012 Dec 19. PMID: 23255822 . [CrossRef]
- Protas HD, Chen K, Langbaum JB, Fleisher AS, Alexander GE, Lee W, Bandy D, de Leon MJ, Mosconi L, Buckley S, et al. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol. 2013 Mar 1;70(3):320-5. PMID: 23599929; PMCID: PMC3745014 . [CrossRef]
- Spallazzi M, Dobisch L, Becke A, Berron D, Stucht D, Oeltze-Jafra S, Caffarra P, Speck O, Düzel E. Hippocampal vascularization patterns: A high-resolution 7 Tesla time-of-flight magnetic resonance angiography study. Neuroimage Clin. 2019;21: 101609. Epub 2018 Nov 19. PMID: 30581106; PMCID: PMC6413539 . [CrossRef]
- Shaw K, Bell L, Boyd K, Grijseels DM, Clarke D, Bonnar O, Crombag HS, Hall CN. Neurovascular coupling and oxygenation are decreased in hippocampus compared to neocortex because of microvascular differences. Nat Commun. 2021 May 27;12(1):3190. Erratum in: Nat Commun. 2021 Jul 19;12(1):4497. doi: 10.1038/s41467-021-24833-y. PMID: 34045465; PMCID: PMC8160329 . [CrossRef]
- Ando S, Tsukamoto H, Stacey BS, Washio T, Owens TS, Calverley TA, Fall L, Marley CJ, Iannetelli A, Hashimoto T, Ogoh S, Bailey DM. Acute hypoxia impairs posterior cerebral bioenergetics and memory in man. Exp Physiol. 2023 Dec;108(12):1516-1530. Epub 2023 Oct 29. PMID: 37898979; PMCID: PMC10988469 . [CrossRef]
- Geraghty JR, Lara-Angulo MN, Spegar M, Reeh J, Testai FD. Severe cognitive impairment in aneurysmal subarachnoid hemorrhage: Predictors and relationship to functional outcome. J Stroke Cerebrovasc Dis. 2020 Sep;29(9):105027. Epub 2020 Jun 20. PMID: 32807442; PMCID: PMC7438604 . [CrossRef]
- Pickard JD, Walker V, Perry S, Smythe PJ, Eastwood S, Hunt R. Arterial eicosanoid production following chronic exposure to a periarterial haematoma. J Neurol Neurosurg Psychiatry. 1984 Jul;47(7):661-7. PMID: 6589362; PMCID: PMC1027891 . [CrossRef]
- Erdem A, Yaşargil G, Roth P. Microsurgical anatomy of the hippocampal arteries. J Neurosurg. 1993 Aug;79(2):256-65. PMID: 8331410 . [CrossRef]
- den Abeelen AS, Lagro J, van Beek AH, Claassen JA. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res. 2014 Jan;11(1):11-7. PMID: 24251392 . [CrossRef]
- Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol. 2013 Jun 15;304(12):H1598-614. Epub 2013 Apr 12. PMID: 23585139; PMCID: PMC4280158 . [CrossRef]
- Zhang L, Papadopoulos P, Hamel E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br J Pharmacol. 2013 Oct;170(3):661-70. PMID: 23889563; PMCID: PMC3792003 . [CrossRef]
- Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003; 23: 2055–2062. [CrossRef]
- Roher AE, Esh C, Rahman A, Kokjohn TA, Beach TG. Atherosclerosis of cerebr arteries in Alzheimer disease. Stroke. 2004 Nov;35(11 Suppl 1):2623-7. Epub 2004 Sep 16. PMID: 15375298 . [CrossRef]
- Maass A, Düzel S, Goerke M, Becke A, Sobieray U, Neumann K, Lövden M, Lindenberger U, Bäckman L, Braun-Dullaeus R, et al. Vascular hippocampal plasticity after aerobic exercise in older adults. Mol Psychiatry. 2015 May;20(5):585-93. Epub 2014 Oct 14. PMID: 25311366 . [CrossRef]
- Maksimovich IV. Differences in cerebral angioarchitectonics in Alzheimer’s Disease in comparison with other neurodegenerative and ischemic lesions. World Journal of Neuroscience 2018; 8: 454-469. [CrossRef]
- Lawley JS, Macdonald JH, Oliver SJ, Mullins PG. Unexpected reductions in regional cerebral perfusion during prolonged hypoxia. J Physiol. 2017 Feb 1;595(3):935-947. Epub 2016 Sep 24. PMID: 27506309; PMCID: PMC5285718 . [CrossRef]
- Mottahedin A, Prag HA, Dannhorn A, Mair R, Schmidt C, Yang M, Sorby-Adams A, Lee JJ, Burger N, Kulaveerasingam D, et al. Targeting succinate metabolism to decrease brain injury upon mechanical thrombectomy treatment of ischemic stroke. Redox Biol. 2023 Feb;59: 102600. Epub 2023 Jan 2. PMID: 36630820; PMCID: PMC9841348 . [CrossRef]
- Beason-Held LL, Moghekar A, Zonderman AB, Kraut MA, Resnick SM. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke. 2007 Jun;38(6):1766-73. Epub 2007 May 17. PMID: 17510458 . [CrossRef]
- Moskowitz MA, Kiwak KJ, Hekimian K, Levine L. Synthesis of compounds with properties of leukotrienes C4 and D4 in gerbil brains after ischemia and reperfusion. Science.1984;224(4651):886-889. [CrossRef]
- Hota SK, Barhwal K, Singh SB, Ilavazhagan G. Differential temporal response of hippocampus, cortex and cerebellum to hypobaric hypoxia: a biochemical approach. Neurochem Int. 2007 Nov-Dec;51(6-7):384-90. Epub 2007 Apr 8. PMID: 17531352 . [CrossRef]
- Shukitt-Hale B, Kadar T, Marlowe BE, Stillman MJ, Galli RL, Levy A, Devine JA, Lieberman HR. Morphological alterations in the hippocampus following hypobaric hypoxia. Hum Exp Toxicol.1996 Apr;15(4):312-9. PMID: 8845221 . [CrossRef]
- Maiti P, Singh SB, Sharma AK, Muthuraju S, Banerjee PK, Ilavazhagan G. Hypobaric hypoxia induces oxidative stress in rat brain. Neurochem Int. 2006 Dec;49(8):709-16. Epub 2006 Sep 5. PMID: 1691184. [CrossRef]
- Tahir MS, Almezgagi M, Zhang Y, Zhonglei D, Qinfang Z, Ali M, Shouket Z, Zhang W. Experimental brain injury induced by acute hypobaric hypoxia stimulates changes in mRNA expression and stress marker status. International Journal of Research and Review 2021;8 (10): 242-247. [CrossRef]
- Ji X, Ferreira T, Friedman B, Liu R, Liechty H, Bas E, Chandrashekar J, Kleinfeld D. Brain microvasculature has a common topology with local differences in geometry that match metabolic load. Neuron. 2021 Apr 7;109(7):1168-1187.e13. Epub 2021 Mar 2. PMID: 33657412; PMCID: PMC8525211 . [CrossRef]
- Perosa V, Priester A, Ziegler G, Cardenas-Blanco A, Dobisch L, Spallazzi M, Assmann A, Maass A, Speck O, Oltmer J, et al. Hippocampal vascular reserve associated with cognitive performance and hippocampal volume. Brain. 2020;143: 622–634. [CrossRef]
- Purnell C, Gao S, Callahan CM, Hendrie HC. Cardiovascular risk factors and incident Alzheimer disease: a systematic review of the literature. Alzheimer Dis Assoc Disord. 2009; 23(1):1–10. [CrossRef]
- Santos CY, Snyder PJ, Wu WC, Zhang M, Echeverria A, Alber J (2017) Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: a review and synthesis. Alzheimers Dement 2017;7: 69–87. [CrossRef]
- Daulatzai MA. Death by a thousand cuts in Alzheimer’s disease: hypoxia–the prodrome. Neurotox Res 2013; 24(2):216–243. [CrossRef]
- Siachpazidou DI, Stavrou VT, Astara K, Pastaka C, Gogou E, Hatzoglou C, Economou NT, Gourgoulianis KI. Alzheimer’s Disease in Patients with Obstructive Sleep Apnea Syndrome. Tanaffos. 2020 Jul;19(3):176-185. PMID: 33815537; PMCID: PMC8008406.
- Correia S.C., Moreira P.I. Oxygen sensing and signaling in Alzheimer’s disease: A breathtaking story! Cell. Mol. Neurobiol. 2022;42:3–21. [CrossRef]
- Burtscher J, Mallet RT, Burtscher M, Millet. Hypoxia and brain aging: neurodegeneration or neuroprotection? Ageing Res Rev 2021; 68:101343. [CrossRef]
- Lall R, Mohammed R, Ojha U. What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr Dis Treat 2019;151343–1354. [CrossRef]
- Zhang F, Niu L, Li S, Le W. Pathological impacts of chronic hypoxia on Alzheimer’s disease. ACS Chem Neurosci 2019; 10(2):902–909. [CrossRef]
- Zhang L, Papadopoulos P, Hamel E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br J Pharmacol. 2013 Oct;170(3):661-70. PMID: 23889563; PMCID: PMC3792003 . [CrossRef]
- Iadecola C. Dangerous leaks: blood-brain barrier woes in the aging hippocampus. Neuron. 2015 Jan 21;85(2):231-3. PMID: 25611503; PMCID: PMC4731877 . [CrossRef]
- Yamori Y, Horie R. Developmental course of hypertension and regional cerebral blood flow in stroke-prone spontaneously hypertensive rats. Stroke. 1977 Jul-Aug;8(4):456-61. PMID: 898241 . [CrossRef]
- Guldbrandsen A, Vethe H, Farag Y, Oveland E, Garberg H, Berle M, Myhr KM, Opsahl JA, Barsnes H, Berven FS. In-depth characterization of the cerebrospinal fluid (CSF) proteome displayed through the CSF proteome resource (CSF-PR). Mol Cell Proteomics. 2014 Nov;13(11):3152-63. Epub 2014 Jul 18. PMID: 25038066; PMCID: PMC4223498. [CrossRef]
- Paglia G, Stocchero M, Cacciatore S, Lai S, Angel P, Alam MT, Keller M, Ralser M, Astarita G. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J Proteome Res. 2016 Feb 5;15(2):608-18. Epub 2016 Jan 12. PMID: 26717242; PMCID: PMC5751881 . [CrossRef]
- Liang JW, Fang ZY, Huang Y, Liuyang ZY, Zhang XL, Wang JL, Wei H, Wang JZ, Wang XC, Zeng J, Liu R. Application of Weighted Gene Co-Expression Network Analysis to Explore the Key Genes in Alzheimer’s Disease. J Alzheimers Dis. 2018;65(4):1353-1364. PMID: 30124448; PMCID: PMC6218130 . [CrossRef]
- Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron. 2020 Mar 18;105(6):975-991.e7. [CrossRef]
- Liu N, Xu J, Liu H, Zhang S, Li M, Zhou Y, Qin W, Li MJ, Yu C; Alzheimer’s disease Neuroimaging Initiative. Hippocampal transcriptome-wide association study and neurobiological pathway analysis for Alzheimer’s disease. PLoS Genet. 2021 Feb 25;17(2):e1009363. PMID: 33630843; PMCID: PMC7906391 . [CrossRef]
- Horgusluoglu E, Neff R, Song WM, Wang M, Wang Q, Arnold M, Krumsiek J, Galindo-Prieto B, Ming C, Nho K, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI); Alzheimer Disease Metabolomics Consortium. Integrative metabolomics-genomics approach reveals key metabolic pathways and regulators of Alzheimer’s disease. Alzheimers Dement. 2022 Jun;18(6):1260-1278. Epub 2021 Nov 10. PMID: 34757660; PMCID: PMC9085975 . [CrossRef]
- Mullins R, Kapogiannis D. Alzheimer’s Disease-Related Genes Identified by Linking Spatial Patterns of Pathology and Gene Expression. Front Neurosci. 2022 Jun 14;16: 908650. PMID: 35774552; PMCID: PMC9237461 . [CrossRef]
- Luo D, Li J, Liu H, Wang J, Xia Y, Qiu W, Wang N, Wang X, Wang X, Ma C, Ge W. Integrative Transcriptomic Analyses of Hippocampal-Entorhinal System Subfields Identify Key Regulators in Alzheimer’s Disease. Adv Sci (Weinh). 2023 Aug;10(22): e2300876. Epub 2023 May 26. PMID: 37232225; PMCID: PMC10401097 . [CrossRef]
- Tandon R, Levey AI, Lah JJ, Seyfried NT, Mitchell CS. Machine Learning Selection of Most Predictive Brain Proteins Suggests Role of Sugar Metabolism in Alzheimer’s Disease. J Alzheimers Dis. 2023;92(2): 411-424. PMID: 36776048; PMCID: PMC10041447. [CrossRef]
- Czech C, Berndt P, Busch K, Schmitz O, Wiemer J, Most V, Hampel H, Kastler J, Senn H. Metabolite profiling of Alzheimer’s disease cerebrospinal fluid. PLoS One. 2012; 7: e31501. [CrossRef]
- Hajjar I, Liu C, Jones DP, Uppal K. Untargeted metabolomics reveal dysregulations in sugar, methionine, and tyrosine pathways in the prodromal state of AD. Alzheimers Dement (Amst). 2020 Aug 11;12(1): e12064. PMID: 32793799; PMCID: PMC7418891 . [CrossRef]
- de Geus MB, Leslie SN, Lam T, Wang W, Roux-Dalvai F, Droit A, Kivisakk P, Nairn AC, Arnold SE, Carlyle BC. Mass spectrometry in cerebrospinal fluid uncovers association of glycolysis biomarkers with Alzheimer’s disease in a large clinical sample. Sci Rep. 2023 Dec 16;13(1):22406. PMID: 38104170; PMCID: PMC10725469 . [CrossRef]
- Panyard DJ, McKetney J, Deming YK, Morrow AR, Ennis GE, Jonaitis EM, Van Hulle CA, Yang C, Sung YJ, Ali M, et al. Large-scale proteome and metabolome analysis of CSF implicates altered glucose and carbon metabolism and succinylcarnitine in Alzheimer’s disease. Alzheimers Dement. 2023 Dec;19(12):5447-5470. Epub 2023 May 22. PMID: 37218097; PMCID: PMC10663389 . [CrossRef]
- Andersen JV, Christensen SK, Aldana BI, Nissen JD, Tanila H, Waagepetersen HS. Alterations in cerebral cortical glucose and glutamine metabolism precedes amyloid plaques in the APPswe/ PSEN1dE9 mouse model of Alzheimer’s disease. Neurochem Res. 2017;42(6):1589–1598. [CrossRef]
- Neuner SM, Wilmott LA, Hoffmann BR, Mozhui K, Kaczorowski CC. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav Brain Res. 2017 Mar 30;322(Pt B):288-298. Epub 2016 Jun 2. PMID: 27265785; PMCID: PMC5135662 . [CrossRef]
- Ojo JO, Crynen G, Algamal M, Vallabhaneni P, Leary P, Mouzon B, Reed JM, Mullan M, Crawford F. Unbiased Proteomic Approach Identifies Pathobiological Profiles in the Brains of Preclinical Models of Repetitive Mild Traumatic Brain Injury, Tauopathy, and Amyloidosis. ASN Neuro. 2020 Jan-Dec;12:1759091420914768. PMID: 32241177; PMCID: PMC7132820 . [CrossRef]
- Hinteregger B, Loeffler T, Flunkert S, Neddens J, Bayer TA, Madl T, Hutter-Paier B. Metabolic, Phenotypic, and Neuropathological Characterization of the Tg4-42 Mouse Model for Alzheimer’s Disease. J Alzheimers Dis. 2021;80(3):1151-1168. PMID: 33646155; PMCID: PMC8150512 . [CrossRef]
- Füzesi MV, Muti IH, Berker Y, Li W, Sun J, Habbel P, Nowak J, Xie Z, Cheng LL, Zhang Y. High Resolution Magic Angle Spinning Proton NMR Study of Alzheimer’s Disease with Mouse Models. Metabolites. 2022 Mar 17;12(3):253. PMID: 35323696; PMCID: PMC8952313 . [CrossRef]
- Morello G, Guarnaccia M, La Cognata V, Latina V, Calissano P, Amadoro G, Cavallaro S. Transcriptomic Analysis in the Hippocampus and Retina of Tg2576 AD Mice Reveals Defective Mitochondrial Oxidative Phosphorylation and Recovery by Tau 12A12mAb Treatment. Cells. 2023 Sep 12;12(18):2254. PMID: 37759477; PMCID: PMC10527038 . [CrossRef]
- Toomey CE, Heywood WE, Evans JR, Lachica J, Pressey SN, Foti SC, Al Shahrani M, D’Sa K, Hargreaves IP, Heales S,et al. Mitochondrial dysfunction is a key pathological driver of early stage Parkinson’s. Acta Neuropathol Commun. 2022 Sep 8;10(1):134. PMID: 36076304; PMCID: PMC9461181. [CrossRef]
- Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009 Apr 28;106(17):7209-14. Epub 2009 Apr 8. PMID: 19357304; PMCID: PMC2678478 . [CrossRef]
- Dennis NA, Browndyke JN, Stokes J, Need A, Burke JR, Welsh-Bohmer KA, Cabeza R. Temporal lobe functional activity and connectivity in young adult APOE varepsilon4 carriers. Alzheimers Dement. 2010 Jul;6(4):303-11. Epub 2009 Sep 9. PMID: 19744893; PMCID: PMC2891943 . [CrossRef]
- Evans S, Dowell NG, Tabet N, King SL, Hutton SB, Rusted JM. Disrupted neural activity patterns to novelty and effort in young adult APOE-e4 carriers performing a subsequent memory task. Brain Behav. 2017 Jan 5;7(2):e00612. PMID: 28239522; PMCID: PMC5318365 . [CrossRef]
- Brandon JA, Farmer BC, Williams HC, Johnson LA. APOE and Alzheimer’s Disease: Neuroimaging of Metabolic and Cerebrovascular Dysfunction. Front Aging Neurosci. 2018 Jun 14;10:180. PMID: 29962946; PMCID: PMC6010552 . [CrossRef]
- Wenneberg SB, Block L, Sörbo A, Naredi S, Oras J, Hendén PL, Ljungqvist J, Liljencrantz J, Hergès HO. Long-term outcomes after aneurysmal subarachnoid hemorrhage: A prospective observational cohort study. Acta Neurol Scand. 2022 Nov;146(5):525-536. Epub 2022 Jul 18. PMID: 35852005; PMCID: PMC9796482 . [CrossRef]
- Lanterna LA, Rigoldi M, Tredici G, Biroli F, Cesana C, Gaini SM, Dalprà L. APOE influences vasospasm and cognition of noncomatose patients with subarachnoid hemorrhage. Neurology. 2005 Apr 12;64(7):1238-44. PMID: 15824354 . [CrossRef]
- Guo ZD, Sun XC, Zhang JH. The role of apolipoprotein e in the pathological events following subarachnoid hemorrhage: a review. Acta Neurochir Suppl. 2011;110(Pt 2):5-7. PMID: 21125436 . [CrossRef]
- Mesis RG, Wang H, Lombard FW, Yates R, Vitek MP, Borel CO, Warner DS, Laskowitz DT. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg Focus. 2006 Sep 15;21(3):E4. PMID: 17029343 . [CrossRef]
Figure 1.
Location of the entorhinal cortex. The entorhinal cortex is a part of the brain’s limbic system which controls emotional drives and memory formation. It is a collection of structures located deep within the brain which includes the hippocampal formation, amygdala, septal nuclei, cingulate cortex, entorhinal cortex, perirhinal cortex, and parahippocampal cortex. The last three cortical areas comprise different portions of the temporal lobe [6]; image ID HYTTNN hakan çorbac? / Alamy Stock Vector reproduced under license from Alamy Limited, Abingdon, UK https://www.alamy.com .
Figure 1.
Location of the entorhinal cortex. The entorhinal cortex is a part of the brain’s limbic system which controls emotional drives and memory formation. It is a collection of structures located deep within the brain which includes the hippocampal formation, amygdala, septal nuclei, cingulate cortex, entorhinal cortex, perirhinal cortex, and parahippocampal cortex. The last three cortical areas comprise different portions of the temporal lobe [6]; image ID HYTTNN hakan çorbac? / Alamy Stock Vector reproduced under license from Alamy Limited, Abingdon, UK https://www.alamy.com .
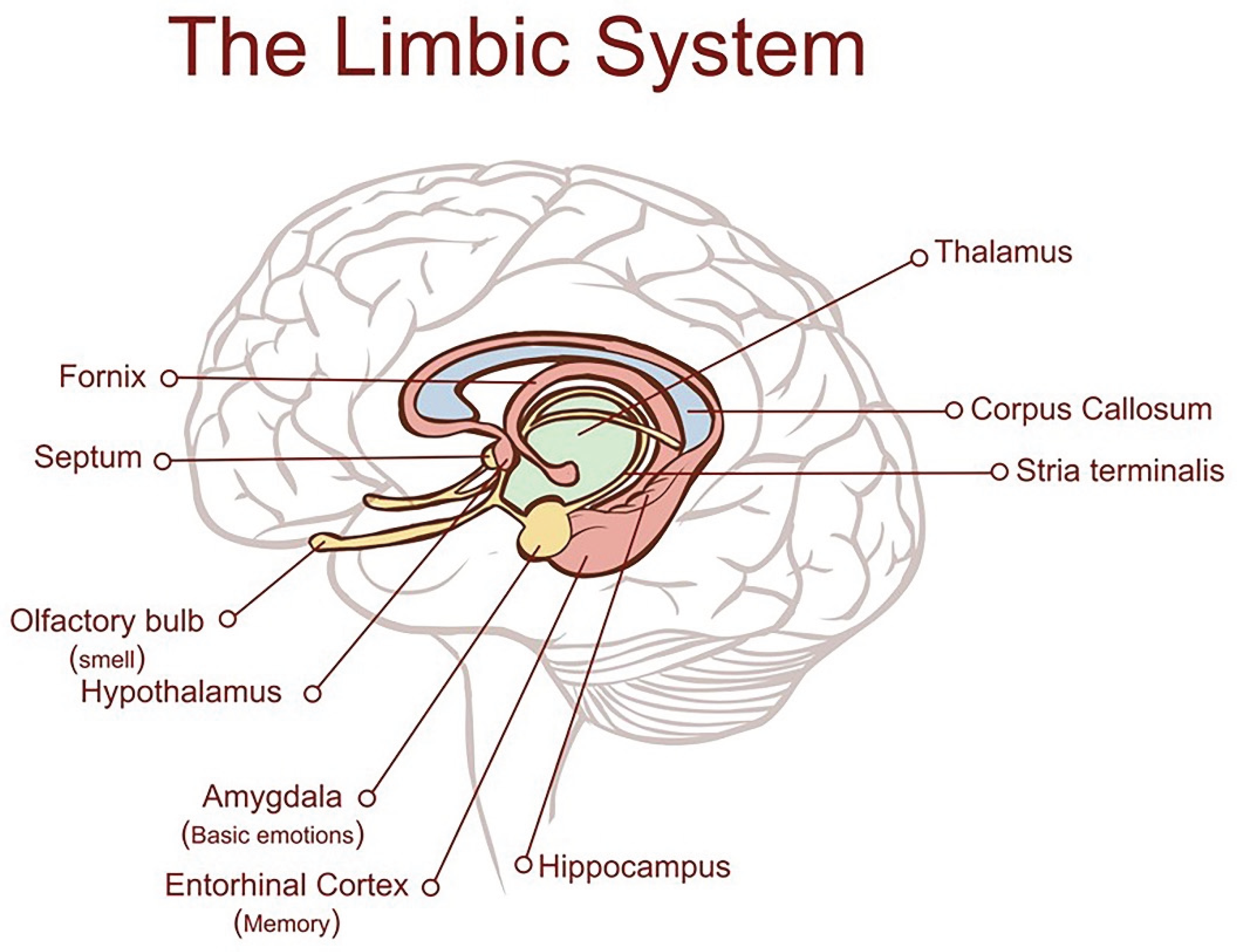
Figure 2.
The malate-aspartate shuttle. NADH cannot be transported from the cytosol into mitochondria. To regenerate NAD from NADH produced during oxidative reactions in the cytosol H+ from NADH is transported into mitochondria via the malate-aspartate shuttle (MAS). MAS requires two cytoplasmic enzymes cAST and MDH1, two mitochondrial enzymes mAST and mMDH2, and two carriers located in the inner mitochondrial membrane, the aspartate-glutamate carrier aralar (SCL25A12) and the 2-oxoglutarate carrier OGC (SLC25A11). i) In the cytoplasm, MDH1 transfers reducing equivalents from NADH to oxaloacetate producing malate ii) OGC transports malate into mitochondrion in exchange for 2-oxoglutarate (2-oxoglut), iii) mMDH2 then oxidizes malate to oxaloacetate (OAA), generating NADH, iv) mAST transfers NH2 from glutamate to OAA producing aspartate and 2-oxoglut, v) aralar transports aspartate out into the cytoplasm in exchange for glutamate and H+ into the mitochondria, vi) finally, cAST transaminates 2-oxoglut forming OAA and glutamate, closing the cycle. After entry into mitochondria electrons are supplied to the electron transport chain in the form of NADH for ATP production, and cytosolic NAD+ is regenerated. [117,118,119]. The Glutamate/H+ symporter, SLCA22 may also contribute to the shuttle activity [18].
Figure 2.
The malate-aspartate shuttle. NADH cannot be transported from the cytosol into mitochondria. To regenerate NAD from NADH produced during oxidative reactions in the cytosol H+ from NADH is transported into mitochondria via the malate-aspartate shuttle (MAS). MAS requires two cytoplasmic enzymes cAST and MDH1, two mitochondrial enzymes mAST and mMDH2, and two carriers located in the inner mitochondrial membrane, the aspartate-glutamate carrier aralar (SCL25A12) and the 2-oxoglutarate carrier OGC (SLC25A11). i) In the cytoplasm, MDH1 transfers reducing equivalents from NADH to oxaloacetate producing malate ii) OGC transports malate into mitochondrion in exchange for 2-oxoglutarate (2-oxoglut), iii) mMDH2 then oxidizes malate to oxaloacetate (OAA), generating NADH, iv) mAST transfers NH2 from glutamate to OAA producing aspartate and 2-oxoglut, v) aralar transports aspartate out into the cytoplasm in exchange for glutamate and H+ into the mitochondria, vi) finally, cAST transaminates 2-oxoglut forming OAA and glutamate, closing the cycle. After entry into mitochondria electrons are supplied to the electron transport chain in the form of NADH for ATP production, and cytosolic NAD+ is regenerated. [117,118,119]. The Glutamate/H+ symporter, SLCA22 may also contribute to the shuttle activity [18].
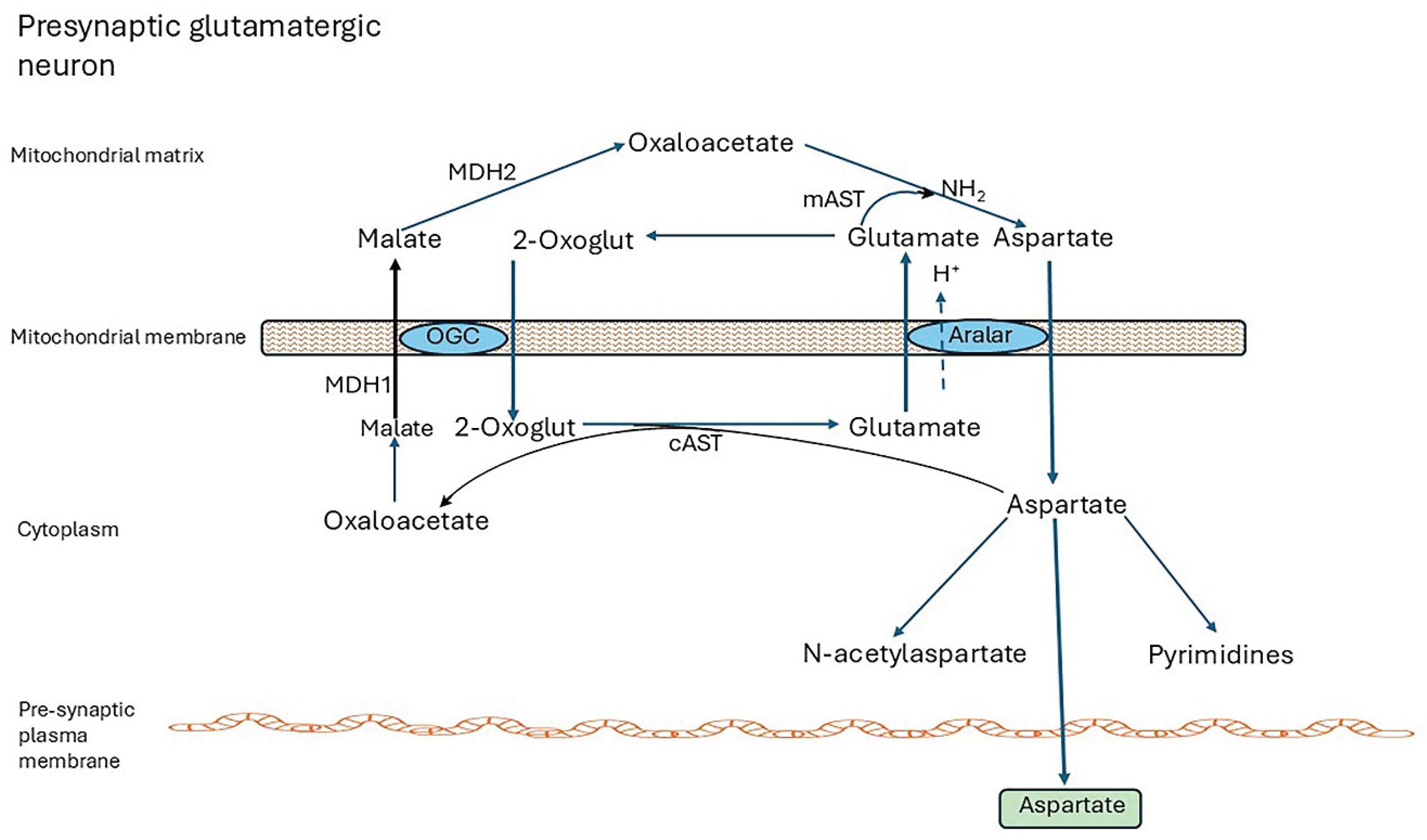
Figure 3.
Overview of the glutamate/GABA/glutamine cycle. The GABA–glutamine–glutamate shuttles replenish the neurotransmitters, L-glutamate and GABA using glutamine generated in astrocytes. After release from glutamatergic neurons into the synaptic clefts, glutamate not bound to postsynaptic receptors is carried with Na+ and H+ into astrocytes by the glutamate–aspartate transporters SLC1A3 (solute carrier 1A3, EAAT1) and SLC1A2 (EAAT2), step ①. Similarly, unbound GABA released from GABAtergic neurons is carried into astrocytes with sodium and chloride by the GABA transporter SLC6A11 (GAT3),②. Within astrocytes, glutamine synthetase (GS) catalyzes the formation of glutamine from glutamate and ammonia in an ATP-dependent reaction. Glutamine is exported with sodium into the extracellular space by Na+-amino acid cotransporters, SLC38A1 (SNAT1) and SLC38A2 (SNAT2), and Na+-amino acid cotransporters-H+ antiporters, SLC38A3 (SNAT3) and SLC38A5 (SNAT5),③. Glutamine is then imported into neurons by one or moreSNAT transporters: SNAT1, 2 and SNAT7 [SLC38A7]),④, where it is hydrolysed by PAG (phosphate activated glutaminase). Glutamate is dehydrogenated by GDH (glutamate dehydrogenase), producing GABA in GABAtergic neurons. The neurotransmitters are then packaged into vesicles, transported to the synapses and released with neuronal stimulation. Most neurotransmitters not bound to postsynaptic receptors are recycled via astrocytes as described above. A fraction is transferred back into neurons: glutamate by SLC1A3,⑤ GABA by SLC6A1 (GAT1),⑥. Mitochondrial glutamate/H+ symporter SLCA22 probably makes a significant contribution to the cycle [18]① SLC1A3, alias excitatory amino acid transporter 1 (EAAT1; GLAST), SLC1A2,alias excitatory amino acid transporter 2 (EAAT2, GLT1), ② SLC6A11 (GAT3), ③ Na+-amino acid cotransporters, SLC38A1 (SNAT1) and SLC38A2 (SNAT2), and Na+-amino acid cotransporters-H+ antiporters, SLC38A3 (SNAT3) and SLC38A5 (SNAT5), ④ SNAT1, SNAT2 and SNAT7 (SLC38A7), ⑤. SLC1 A3 (EAAT2), ⑥ SLC6A1 (GAT1). GS glutamate synthetase, PAG phosphate activated glutaminase, GAD glutamate dehydrogenase; Red lines major pathways, Dotted lines indirect route via TCA cycle.
Figure 3.
Overview of the glutamate/GABA/glutamine cycle. The GABA–glutamine–glutamate shuttles replenish the neurotransmitters, L-glutamate and GABA using glutamine generated in astrocytes. After release from glutamatergic neurons into the synaptic clefts, glutamate not bound to postsynaptic receptors is carried with Na+ and H+ into astrocytes by the glutamate–aspartate transporters SLC1A3 (solute carrier 1A3, EAAT1) and SLC1A2 (EAAT2), step ①. Similarly, unbound GABA released from GABAtergic neurons is carried into astrocytes with sodium and chloride by the GABA transporter SLC6A11 (GAT3),②. Within astrocytes, glutamine synthetase (GS) catalyzes the formation of glutamine from glutamate and ammonia in an ATP-dependent reaction. Glutamine is exported with sodium into the extracellular space by Na+-amino acid cotransporters, SLC38A1 (SNAT1) and SLC38A2 (SNAT2), and Na+-amino acid cotransporters-H+ antiporters, SLC38A3 (SNAT3) and SLC38A5 (SNAT5),③. Glutamine is then imported into neurons by one or moreSNAT transporters: SNAT1, 2 and SNAT7 [SLC38A7]),④, where it is hydrolysed by PAG (phosphate activated glutaminase). Glutamate is dehydrogenated by GDH (glutamate dehydrogenase), producing GABA in GABAtergic neurons. The neurotransmitters are then packaged into vesicles, transported to the synapses and released with neuronal stimulation. Most neurotransmitters not bound to postsynaptic receptors are recycled via astrocytes as described above. A fraction is transferred back into neurons: glutamate by SLC1A3,⑤ GABA by SLC6A1 (GAT1),⑥. Mitochondrial glutamate/H+ symporter SLCA22 probably makes a significant contribution to the cycle [18]① SLC1A3, alias excitatory amino acid transporter 1 (EAAT1; GLAST), SLC1A2,alias excitatory amino acid transporter 2 (EAAT2, GLT1), ② SLC6A11 (GAT3), ③ Na+-amino acid cotransporters, SLC38A1 (SNAT1) and SLC38A2 (SNAT2), and Na+-amino acid cotransporters-H+ antiporters, SLC38A3 (SNAT3) and SLC38A5 (SNAT5), ④ SNAT1, SNAT2 and SNAT7 (SLC38A7), ⑤. SLC1 A3 (EAAT2), ⑥ SLC6A1 (GAT1). GS glutamate synthetase, PAG phosphate activated glutaminase, GAD glutamate dehydrogenase; Red lines major pathways, Dotted lines indirect route via TCA cycle.
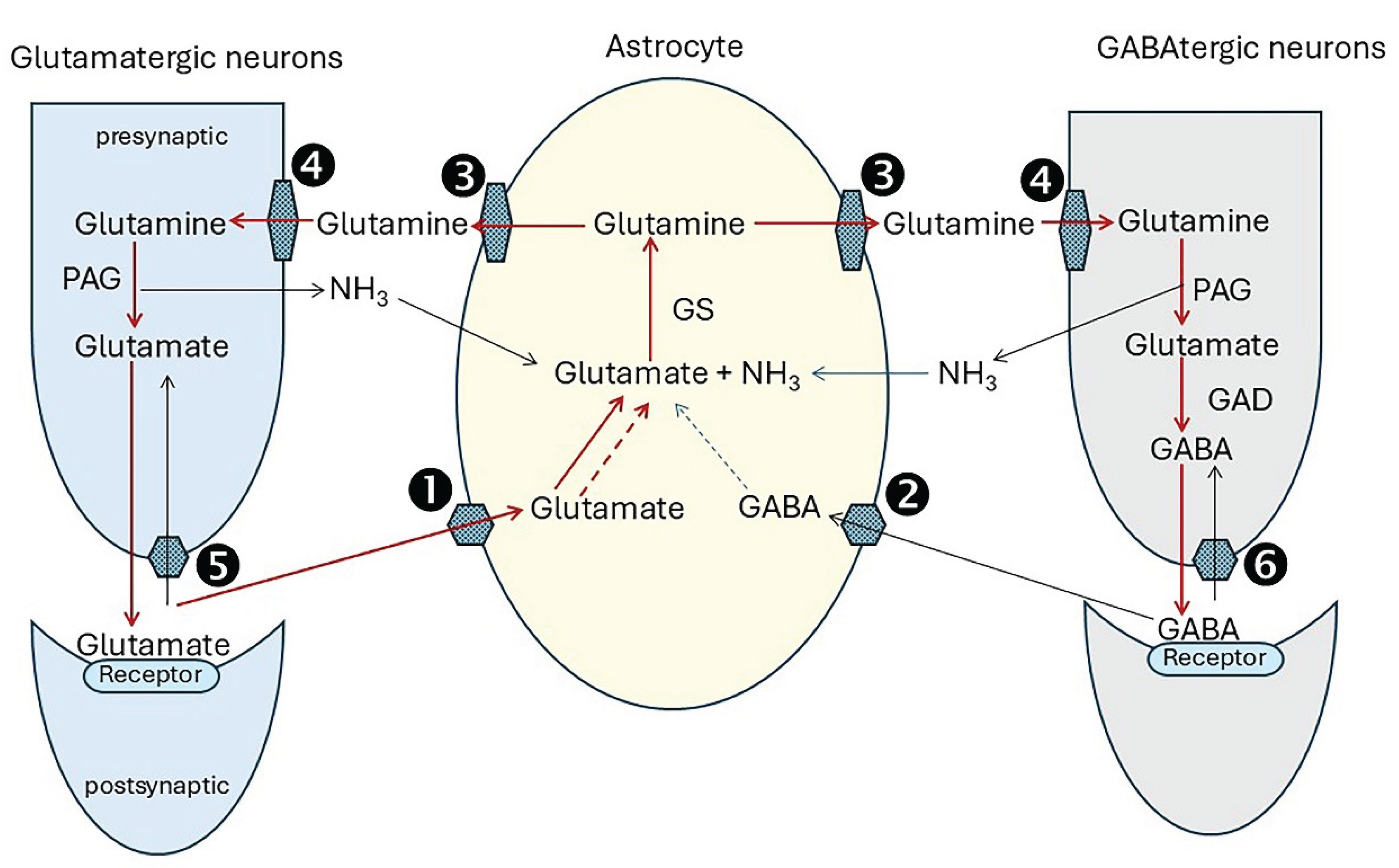
Figure 4.
Astrocytes are central to neurotransmitter turnover of glutamatergic neurons. Glutamine is replenished in astrocytes by transamination of 2-oxoglutarate drawn from the TCA cycle, producing glutamate which GS then converts to glutamine. TCA cycle function is maintained largely by carboxylation of pyruvate (provided by glucose) by cytosolic pyruvate carboxylase forming oxaloacetate. This enters the cycle after hydrogenation to malate and transport into mitochondria. Oxaloacetate is also produced from aspartate generated by the malate aspartate shuttle and makes an essential contribution to anaplerosis. Further anaplerotic support is provided by re-uptake of GABA into astrocytes and its conversion to succinate.
Figure 4.
Astrocytes are central to neurotransmitter turnover of glutamatergic neurons. Glutamine is replenished in astrocytes by transamination of 2-oxoglutarate drawn from the TCA cycle, producing glutamate which GS then converts to glutamine. TCA cycle function is maintained largely by carboxylation of pyruvate (provided by glucose) by cytosolic pyruvate carboxylase forming oxaloacetate. This enters the cycle after hydrogenation to malate and transport into mitochondria. Oxaloacetate is also produced from aspartate generated by the malate aspartate shuttle and makes an essential contribution to anaplerosis. Further anaplerotic support is provided by re-uptake of GABA into astrocytes and its conversion to succinate.

Figure 5.
Axonal transport of mitochondria. For transport down axons mitochondria attach to an adaptor which, in turn, links them to a motor bound to microtubules. Miro proteins anchored to the outer mitochondrial membrane bind to trafficking kinesin protein adaptor complex adaptors (TRAK1 or TRAK 2). For anterograde travel to the nerve terminals, TRAK1/2 binds to the kinesin-1 motor. This has two heavy and two light chains. The heavy chains dimerize and their head domains in alternation bind and then detach from myosin, consuming ATP, and ‘walk’ down the microtubules. Their C-terminals bind to the cargo-carrying light chains. The multimeric complex dynein and its activator dynactin transport damaged mitochondria from the axon terminals to the cell body for elimination by mitophagy [179,180,187,188].
Figure 5.
Axonal transport of mitochondria. For transport down axons mitochondria attach to an adaptor which, in turn, links them to a motor bound to microtubules. Miro proteins anchored to the outer mitochondrial membrane bind to trafficking kinesin protein adaptor complex adaptors (TRAK1 or TRAK 2). For anterograde travel to the nerve terminals, TRAK1/2 binds to the kinesin-1 motor. This has two heavy and two light chains. The heavy chains dimerize and their head domains in alternation bind and then detach from myosin, consuming ATP, and ‘walk’ down the microtubules. Their C-terminals bind to the cargo-carrying light chains. The multimeric complex dynein and its activator dynactin transport damaged mitochondria from the axon terminals to the cell body for elimination by mitophagy [179,180,187,188].
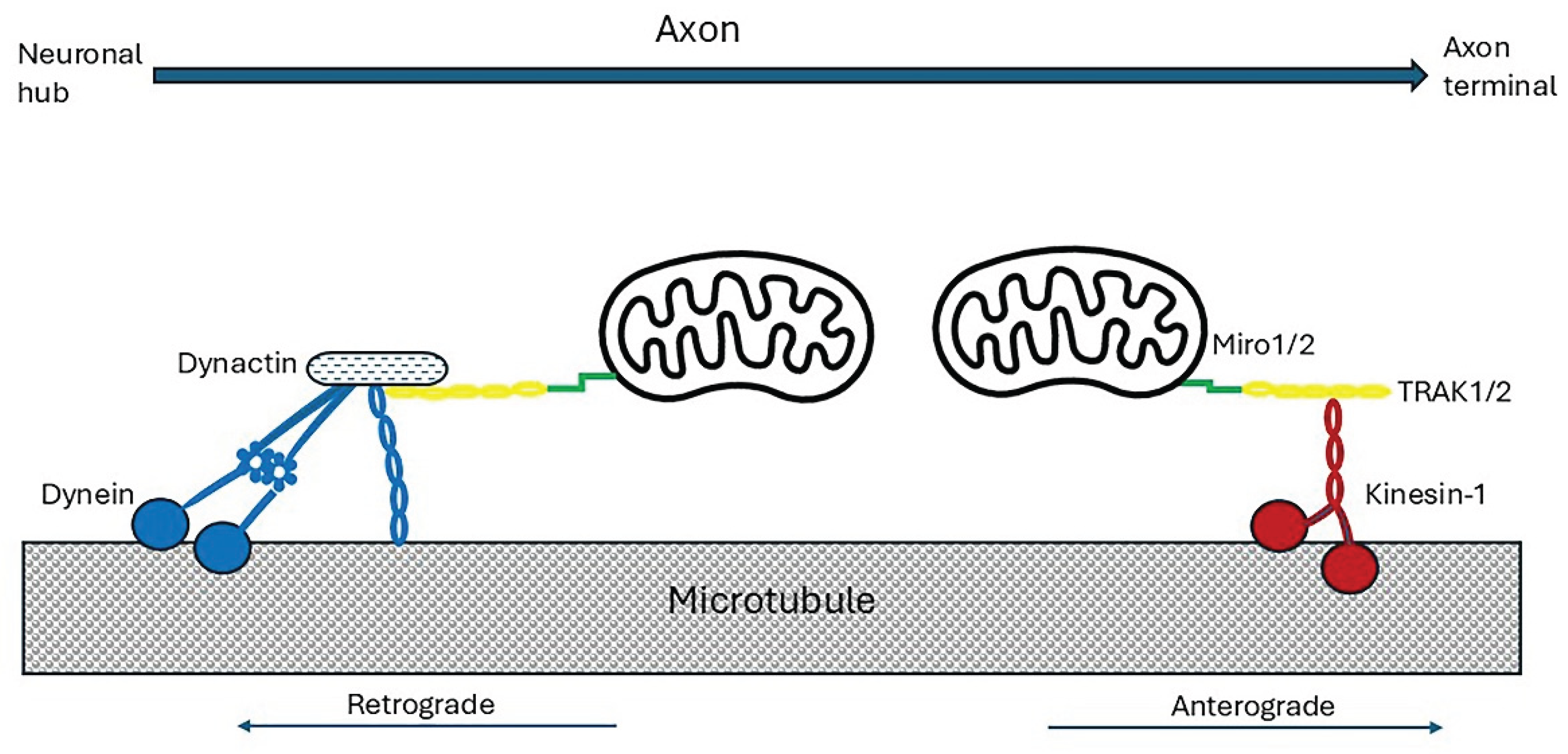
Figure 6.
Remodelling of phosphatidylinositides by the Lands cycle. The phosphatidylinositides (PIs) synthesised de novo have a miscellany of fatty acyl groups. Phospholipase A1 or A2 removes the acyl chains at sn-1 or sn-2, respectively, leaving a lysophosphoinositide. The new, required, acyl groups are transferred to this from stearoyl-CoA or arachidonyl-CoA. PIs incorporated into glycosylphosphatidylinositol (GPI)-anchored proteins are remodelled by the same process, but with replacement of flexible unsaturated fatty acyl chains at sn-2 with rigid stearate chains transferred from stearoyl-CoA. Polyunsaturated acyl groups located at the sn-2 position have a rapid turnover. Synthesis, remodelling and recycling of phospholipids are highly active processes with a high ATP consumption [229,230].
Figure 6.
Remodelling of phosphatidylinositides by the Lands cycle. The phosphatidylinositides (PIs) synthesised de novo have a miscellany of fatty acyl groups. Phospholipase A1 or A2 removes the acyl chains at sn-1 or sn-2, respectively, leaving a lysophosphoinositide. The new, required, acyl groups are transferred to this from stearoyl-CoA or arachidonyl-CoA. PIs incorporated into glycosylphosphatidylinositol (GPI)-anchored proteins are remodelled by the same process, but with replacement of flexible unsaturated fatty acyl chains at sn-2 with rigid stearate chains transferred from stearoyl-CoA. Polyunsaturated acyl groups located at the sn-2 position have a rapid turnover. Synthesis, remodelling and recycling of phospholipids are highly active processes with a high ATP consumption [229,230].
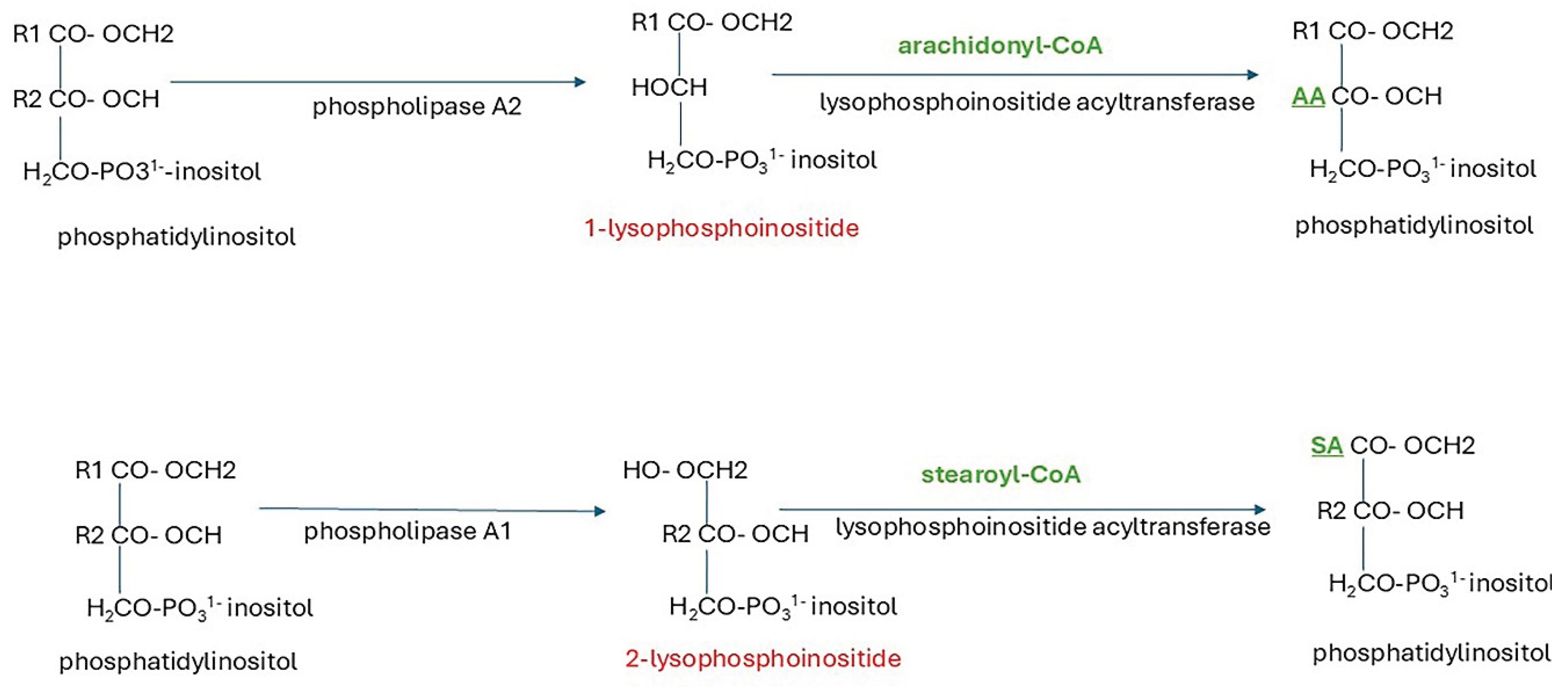
Figure 7.
Processing of amyloid precursor protein (APP). In the non-amyloidogenic pathway (left) APP is first cleaved by α-secretase (α-s) to release a soluble peptide, APPs-α, extracellularly. The 83 residue is then cleaved by γ-secretase (γ-s) in its trans-membrane domain releasing two fragments, a soluble N-terminal fragment P3 peptide, and an intracellular C-terminal fragment (ICTF). In the amyloidogenic pathway (right), β-secretase (β-s, β-APP-cleaving enzyme 1, BACE) cleaves APP proximal to the transmembrane segment releasing an N-terminal ectodomain. The 99 residue C-terminal fragment is then cleaved by γ-secretase, possibly at more than one site. The APP intracellular fragment (ICTF) is released into the cytoplasm, and Aβ peptides with 39 to 42 residues are discharged extracellularly. They may aggregate, oligomerize and form fibrils [34,49,250].
Figure 7.
Processing of amyloid precursor protein (APP). In the non-amyloidogenic pathway (left) APP is first cleaved by α-secretase (α-s) to release a soluble peptide, APPs-α, extracellularly. The 83 residue is then cleaved by γ-secretase (γ-s) in its trans-membrane domain releasing two fragments, a soluble N-terminal fragment P3 peptide, and an intracellular C-terminal fragment (ICTF). In the amyloidogenic pathway (right), β-secretase (β-s, β-APP-cleaving enzyme 1, BACE) cleaves APP proximal to the transmembrane segment releasing an N-terminal ectodomain. The 99 residue C-terminal fragment is then cleaved by γ-secretase, possibly at more than one site. The APP intracellular fragment (ICTF) is released into the cytoplasm, and Aβ peptides with 39 to 42 residues are discharged extracellularly. They may aggregate, oligomerize and form fibrils [34,49,250].
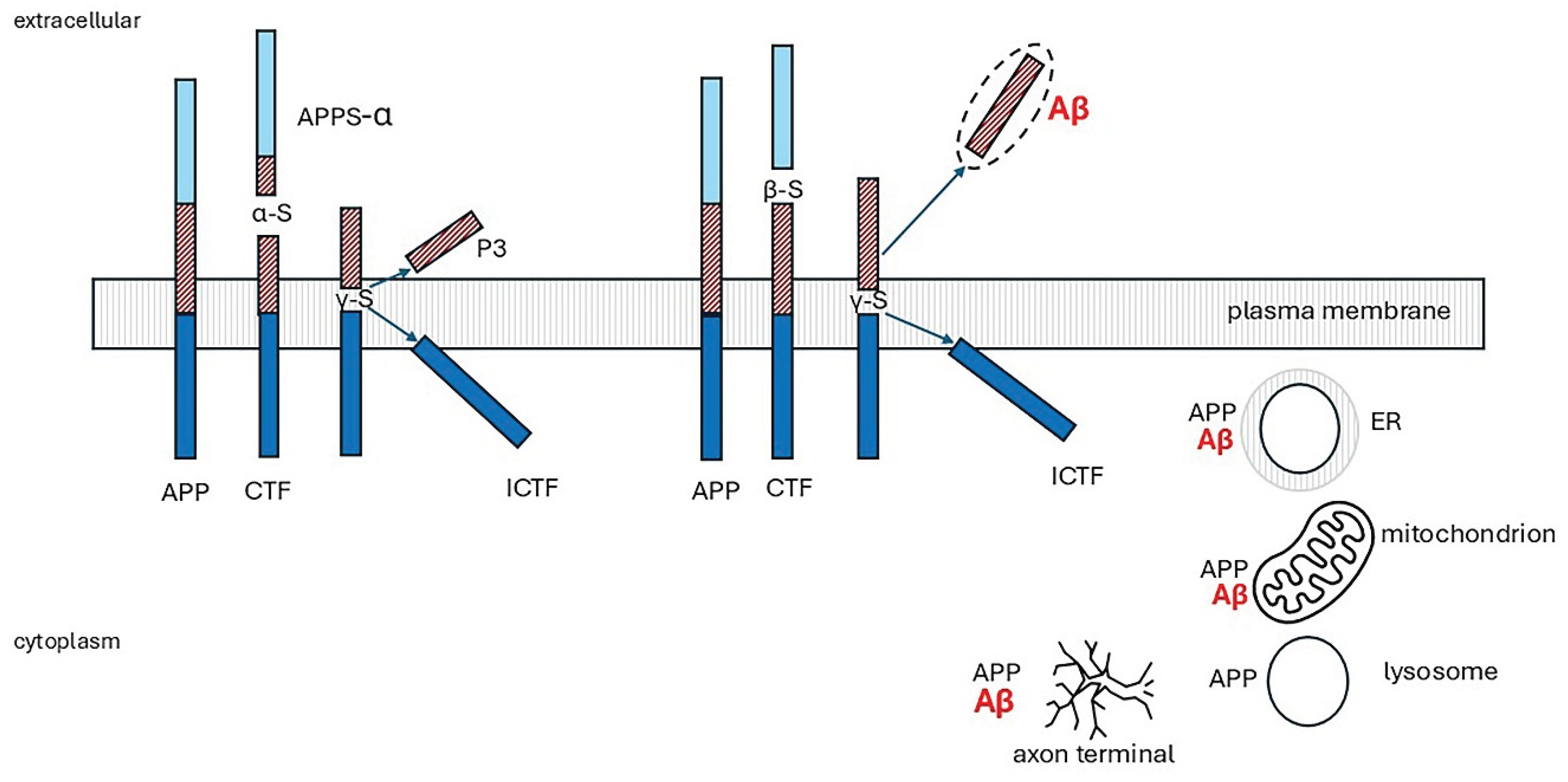
Figure 8.
The γ-glutamyl cycle. 5-oxoproline (pyroglutamic acid) is an intermediate in glutathione metabo279lism. The tripeptide glutathione is synthesised and degraded in the γ-glutamyl cycle. It is synthesised from glutamate by the sequential actions of γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase (GS). γ- glutamine transpeptidase (γ- GT) initiates breakdown by transferring the γ-glutamyl group to amino acid acceptors. γ-glutamylcyclotransferase (γ-GCT) forms 5-oxoproline and releases the amino acids. 5-oxoprolinase (5-OP) converts 5-oxoproline to glutamate. A dipeptidase (not shown) splits the cysteinylglycine moiety of glutathione to cysteine and glycine. The cycle has a heavy ATP consumption.
Figure 8.
The γ-glutamyl cycle. 5-oxoproline (pyroglutamic acid) is an intermediate in glutathione metabo279lism. The tripeptide glutathione is synthesised and degraded in the γ-glutamyl cycle. It is synthesised from glutamate by the sequential actions of γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase (GS). γ- glutamine transpeptidase (γ- GT) initiates breakdown by transferring the γ-glutamyl group to amino acid acceptors. γ-glutamylcyclotransferase (γ-GCT) forms 5-oxoproline and releases the amino acids. 5-oxoprolinase (5-OP) converts 5-oxoproline to glutamate. A dipeptidase (not shown) splits the cysteinylglycine moiety of glutathione to cysteine and glycine. The cycle has a heavy ATP consumption.
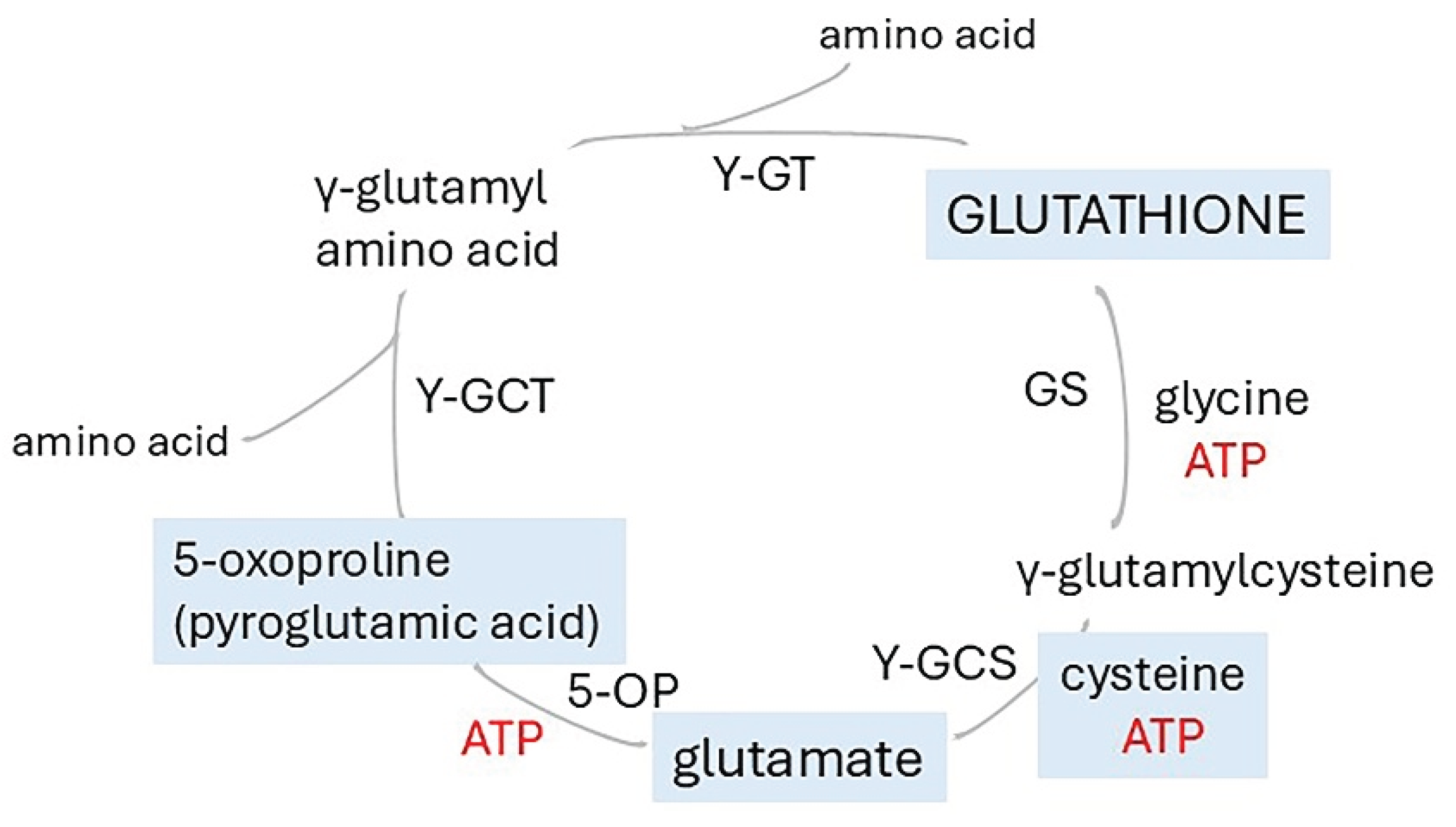
Table 1.
Risk factors for Alzheimer’s disease (AD).
| Factor |
|---|
| Age |
| Overweight-midlife, but not late-life overweight or obesity [10] |
| High blood cholesterol; familial hypercholesterolaemia [10] |
| Low HDL cholesterol [10] |
| Persistent hypertension-midlife [10] |
| Smoking [11] |
| Chronic stress [11] |
| Depression [11] |
| Alcohol-heavy intake or abstinence [12] |
| Poor sleep [11] |
| Recurrent hypoxia-sleep apnoea, COPD [13] |
| Hyperammonaemia (potential risk) [14,15] |
| Genes & polymorphisms |
| ApoE4 [16] |
| TREM2 [11] |
| Humanin rs2854128- African Americans (not Europeans) [17] |
| SLC25A22 [18] |
| ABCA1 (loss of function) [19] |
| ABCA7 numerous polymorphisms [20,21] |
| SREBP-2 [22] |
| MBOAT1 (proposed risk) [23] |
| PICALM [24] |
| Mutations in genes for amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2)-rare early onset AD [25] |
Abbreviations: COPD Chronic obstructive pulmonary disease, TREM2 Triggering Receptor Expressed on Myeloid cells 2, SLC25A22 Solute Carrier Family 25 Member 22 (Glutamate/H(+) Symporter 1), ABC1, ABC7 ATP-binding cassette subfamily A member 1 and member7, SREBP-2 Sterol Regulatory Element Binding Transcription Factor 2, MBOAT1 Membrane Bound Glycerophospholipid O-Acyltransferase 1, PICALM phosphatidylinositol binding clathrin assembly protein rs3851179 .
Table 2.
Investigations of the hippocampal vasculature and blood flow.
| Study | Main findings | Reference | |
|---|---|---|---|
| Human studies | |||
| 1 | Atherosclerosis of Circle of Willis arteries in AD | Number of stenoses and stenosis index in AD> controls; correlated with plaque, NFTs, white matter rarefaction, Braak stage | Roher AE, Esh C, Kokjohn T, et al., 2003 [282] |
| 2 | Atherosclerosis of cerebral arteries in AD | Stenosis of arteries and number of stenoses per individual in AD > controls- highly significant | Roher AE, Esh C, Rahman A, et al., 2004 [283] |
| 3 | Vascular hippocampal plasticity after aerobic exercise in older adults | Fitness improvement correlated with changes in hippocampal perfusion and head volume, but considerable interindividual variability in the response to the physical exercise | Maass A, Düzel S, Goerke M, et al., 2005 [284] |
| 4 | Hippocampal vascularization patterns in vivo | Variable contribution of the anterior choroidal artery, the relationships between hippocampal and posterior cerebral artery patterns, different distribution patterns in the right and left hemispheres. | Spallazzi M, Dobisch L, Becke A, et al., 2019 [273] |
| 5 | Cerebral Angioarchitectonics in AD, compared with other neurodegenerative and ischemic lesions | temporal and fronto-parietal areas of all patients with AD, regardless of disease stage: specific changes in cerebral microcirculation which they named dyscirculatory angiopathy of Alzheimer’s type (DAAT). DAAT was not found in the controls. | Maksimovich IV, 2018 [285] |
| 6 | Effects of acute hypoxia on cerebral bioenergetics and memory. |
In hypoxia, oxygen delivery was reduced in middle cerebral artery during central executive tasks and in posterior cerebral artery during memorization and recall; no effect on cerebral blood flow |
Ando S, Tsukamoto H, Stacey BS, et al., 2023 [275] |
| 7 | Regional cerebral microvascular perfusion in acute and prolonged hypoxia | 2 h of hypoxia: perfusion increased frontal cortex- decreased in ‘default mode’ network; After 10 h decreased blood flow in default mode network more pronounced and widespread, hence reduced local perfusion; Showed related to vasoconstriction | Lawley JS, Macdonald JH, Oliver SJ, 2017 [286] |
| 8 | Effects of brain ischaemia on succinate and other metabolites |
warm ischemia ex vivo: time-dependent accumulation of succinate, other significant changes included increases in purine degradation, PUFAs, 5-oxoproline, decreases in adenosine, acylcarnitines; Stroke model: succinate accumulated, other TCA metabolites decreased, Dramatic decrease in ATP |
Mottahedin A, Prag HA, Dannhorn A, et al., 2023 [287] |
| 9 | Association of regional cerebral perfusion in AD with Tau and amyloid | Tau-PET was associated with lower CBF in the entorhinal cortex, persisted after excluding AD dementia group, was independent of Aβ. APOE genotype and MRI markers for small vessel disease. Amyloid-PET was associated with lower CBF in temporo-parietal regions | Rubinski A, Tosun D, Franzmeier N, et al., 2021 [261] |
| 10 | Tau deposition in entorhinal cortex related to hypoperfusion | baseline CBF was associated with tau deposition at the 6-year follow-up in the left but not the right entorhinal cortex; findings suggest that a reduction in CBF at the entorhinal cortex precedes tau deposition. |
Kapadia A, Billimoria K, Desai P, et al., 2023 [4] |
| 11 | Longitudinal changes in CBF in the older hypertensive brain | Relative to controls, in the hypertensive group rCBF decreased in prefrontal, anterior cingulate and occipital areas over time | Beason-Held LL, Moghekar A, Zonderman AB, et al. 2007 [288] |
| Animal studies | |||
| 12 | Neurovascular coupling in the hippocampus and visual cortex | Compared with visual cortex: hippocampal arteries blunted response: fewer, smaller, dilations. ATP production restricted in tissues furthest from capillaries | Shaw K, Bell L, Boyd K, et al., 2021 [274] |
| 13 | Identification of leukotrienes C4 and D4 in gerbil brains after ischemia and reperfusion. | Significant increases at 5,10, or 15 min of ischaemia, more marked on reperfusion; highest in forebrain grey matter, undetectable in brain regions remote from ischemic zone | Moskowitz MA, Kiwak KJ, Hekimian K, et al.,1984 [289] |
| 14 | Biochemical response to hypobaric oxygen: hippocampus, cortex, cerebellum | Compared with controls, increased lactate dehydrogenase, free radical generation, lipid peroxidation, glutamate dehydrogenase activity, vesicular glutamate transporter expression decreased glutathione reductase, superoxide dismutase activity, reduced glutathione with increased oxidized glutathione |
Hota SK, Barhwal K, Singh SB, et al., 2007 [290] |
| 15 | Hippocampal morphology following hypobaric hypoxia | Significant cell degeneration and death only in the CA3 region; damage more noticeable with longer time following exposure | Shukitt-Hale B, Kadar T, Marlowe BE, et al., 1996 [291] |
| 16 | Oxidative stress in rat brain in hypobaric hypoxia | Significant increase in free radical production, nitric, lipid peroxidation lactate dehydrogenase greater at 7 days than 3 days; reduced glutathione, glutathione peroxidase, glutathione reductase, superoxide dismutase and reduced/oxidized glutathione. Hippocampus most susceptible. | Maiti P, Singh SB, Sharma AK, et al., 2006 [292] |
| 17 | Effect of acute hypobaric hypoxia on SOD and MDA, and mRNA expression of VEGF and HIF1-α in rat brain | Increased expression of HIF1-α and VEGF days1,2,3; significant increased MDA, decreased SOD | Tahir MS, Almezgagi M, Zhang Y, et al. 2021 [293] |
NFTs neurofibrillary tangles, CBF cerebral blood flow, rCBF regional cerebral blood flow, PUFAs polyunsaturated fatty acids, HIF1α, Hypoxia-inducible factor 1-alpha, VEGF vascular endothelial growth factor, MDA malondialdehyde, SOD superoxide dismutase.
Table 3.
Genomic, proteomic, metabolomic and imaging investigations of human brain and CSF in Alzheimer’s Disease (AD) Studies†.
Table 3.
Genomic, proteomic, metabolomic and imaging investigations of human brain and CSF in Alzheimer’s Disease (AD) Studies†.
| Study | Main findings | Reference | |
|---|---|---|---|
| Brain | |||
| 1 | Neuronal loss of entorhinal cortex | Controls: neuronal numbers constant 60y to 90y AD: severe neuronal loss; mainly layers II and IV; loss correlated with NFTs and neuritic but not diffuse or total plaques |
Gómez-Isla T, Price JL, McKeel DW Jr, et al., 1996 [1] |
| 2 | Non-targeted metabolomics to identify pathways altered in AD | Most affected pathway: Ala, Asp, Gln, Asp significant decrease, marked disturbances of malate-aspartate shuttle, glycerophospholipids, pyrimidines; increased S-adenosyl methionine, S-adenosylhomocysteine | Paglia G, Stocchero M, Cacciatore S, et al., 2016 [308] |
| 3 | Brain energy pathways in cingulate cortex of young adult ApoE4 carriers without AD | Carriers: increased expression of subunits of mitochondrial complexes I, II, IV, no change in III or V; qPCR: significant small changes in NDUFB5, NDUF7, ARRDC3 expression | Perkins M, Wolf AB, Chavira B, et al., 2016 [16] |
| 4 | Brain structural changes over 12-24m in MRI scans | Mean annualized hippocampal volume change AD 4.8%, controls 1.1%; AD increased neuronal loss | Ledig C, Schuh A, Guerrero R, et al., 2018 [38] |
| 5 | Investigation of gene pathways enriched in hippocampus in AD | In AD, significant changes in NF-κβ, and cGMP-PKG signalling pathways, MT1, MT2, NOTCH2, ADD3, MSX1, RAB31 key hub genes | Liang JW, Fang ZY, Huang Y, et al., 2018 [309] |
| 6 | Gene pathway analysis to find biomarkers of human brain aging | Modules relevant to brain aging: synaptic vesicle cycle, cGMP-PKG signalling pathway, and oxidative phosphorylation | Hu Y, Pan J, Xin Y, et al., 2018 [47] |
| 7 | Changes in proteome and phosphoproteome in AD progression (7) | Identified three proteome clusters associated with AD progression. Enriched pathways were mitochondria, mitochondrial function, neurotrophic factor signalling | Bai B, Wang X, Li Y, et al., 2020 [310] |
| 8 | Gene pathway analysis to identify new gene and miRNA biomarkers for AD | Identified 8 genes, one of theseMBOAT1 not previously reported, andfive miRNAs | Soleimani Zakeri NS, Pashazadeh S, MotieGhader H, 2020 [23] |
| 9 | Genomic and transcriptomic analyses of hippocampus | Expression of 54 genes associated with AD; 21 were prioritised, including two novel genes Tyrosine-Protein Phosphatase Non-Receptor Type 9 (PTPN9) and Protocadherin Alpha 4 (PCDHA4); QPCTL (glutamyl cyclotransferase, and ERCC2 (excision repair 2) significantly different from elderly controls | Liu N, Xu J, Liu H, et al., 2021 [311] |
| 10 | Investigation of co-expression networks and regulators of metabolism in AD progression | With AD progression, decreased branched chain AAs, and short chain acylcarnitines, increased medium and long chain acyl carnitines, increased expression of adiponectin protein and ATP-Binding Cassette Sub-Family A Member 1 (ABCA1) and Carnitine Palmitoyltransferase 1A (CPT1A) genes in the Hippocampus and para hippocampal gyrus | Horgusluoglu E, Neff R, Song W-M, et al., 2021 [312] |
| 11 | Identification of gene pathways in brain regions with AD pathology identified by use of three different PET scans | Results from Tau scans most relevant. Pathways identified included mitochondrial respiration, electron transport, OXPHOS and metabolism | Mullins R and Kapogiannis D, 2022 [313] |
| 12 | Transcriptomic analyses of hippocampal entorhinal subfields to identify regulators in AD | All 5 subfields positively enriched in AD signalling pathways, extensive neuronal loss in all 5 regardless of AD pathology; most differentially expressed genes in EC and CA4, significant correlation of neuronal and astrocyte profiles, PSP (prosaposin) a key modulator of astrogliosis | Luo D, Li J, Liu H, et al., 2023 [314] |
| 13 | Changes in brain protein expression with AD progression to find proteins to predict progression of MCI to AD, using machine learning | 29 proteins provided best classification of AD and controls; 88 proteins needed to classify AD and asymptomatic AD; predictive proteins of change with disease state were significantly enriched for sugar metabolism supporting dysregulation of energy metabolism | Tandon R, Levey AI, Lah JJ, et al., 2023 [315] |
| 14 | Association of 53 SLC25 carriers with AD | SLC25A10, SLC25A17, and SLC25A22 identified as AD susceptibility genes, down regulation of gene for glutamate carrier1 (SLC25A22) associated with accelerated hippocampal atrophy and increased hazard of dementia. Pathway analysis related SLC25A22 to defects in neuronal function | Tian J, Jia K, Wang T, et al., 2024 [18] |
| 15 | Human cortical peptidome in cognitive resilience against AD | 35 proteins were significantly associated with resilient AD (AD pathology but normal cognition) or with low cognition without AD pathology. In resilient, increased ATP synthase F1 subunit delta (ATP5FLD), cytochrome C oxidase subunit 8A (COX8A). Heterogeneous Nuclear Ribonucleoprotein K (HNRNAP) was enriched in inhibitory neurons | Morgan GR and Carlyle BC, 2024 [2] |
| 16 | Comprehensive hippocampal bio-informatics study using machine learning to identify novel risk genes for AD | 27 down-regulated and 4 up-regulated genes correlated with AD stage. Higher expression of five genes associated with decreased risk and slower progression of AD; 4 with higher risk and faster progression PNMAL1, SLC39A10, GLRB, PTPN3 | Li J, Li L, Cai S, et al., 2024 [11] |
| CSF | |||
| 17 | CSF Metabolite profiles in AD | In mild AD, compared with controls: combination of significantly increased cysteine and decreased uridine 75% predictive of AD, with sensitivity of 75%; Cortisol increased with progression of AD in more advanced AD increased cortisol | Czech C, Berndt P, Busch K, et al., 2012 [316] |
| 18 | Untargeted CSF metabolomics in prodromal AD with mild cognitive impairment | 94 of 294 differentially expressed metabolites were annotated; disturbance in 13 pathways identified. Top four pathways related to bioenergetics and glucose metabolism (N-glycan, sialic acid, amino sugars, galactose); methionine, tyrosine, purine and biopterin metabolism also differentially activated | Hajjar I, Liu C, Jones DP et al., 2020 [317] |
| 19 | Unbiased CSF proteomics in patients with AD | Compared to non-AD groups pyruvate kinase (PKM) and aldolase A (ALDOA) upregulated in AD CSF, glucose increased only in MCI; 33 peptides were differentially abundant between AD with dementia and all nondemented-AD groups, including clusters for glycolytic process or canonical glycolysis, synaptic and immune response markers | de Geus MB, Leslie SN, Lam T, et al., 2023 [318] |
| 20 | CSF Proteome and metabolome of individuals with varying amyloid/taurine (AT) pathology and nine biomarkers of neurodegeneration and neuroinflammation | 61 proteins significantly associated with AT category and 636 proteins with biomarkers. Among amyloid- and tau-associated proteins from glucose and carbon metabolism pathways were enriched, including malate dehydrogenase, aldolase A and succinyl carnitine; Preliminary findings supported association of glucose metabolic dysregulation with alterations in amyloid and tau even before cognitive impairment; preliminary investigations suggested possible abnormalities in insulin signalling | Panyard DJ, McKetney J, Deming YK, et al., 2023 [319] |
† For more information refer to Supplementary Table 2. NDUFB5, NADH:Ubiquinone Oxidoreductase Subunit A5, NDUF7, NADH:Ubiquinone Oxidoreductase Subunit, ARRDC3, Arrestin Domain Containing 3, MT1, A metallothionein 1 gene, MT2, A metallothionein 2 gene, NOTCH2, Notch Receptor 2, ADD3, Adducin 3, MSX1, Msh Homeobox 1, RAB31 RAB31, Member RAS Oncogene Family, SLC25A10,17,22: Solute Carrier Family 25: A10 Mitochondrial Dicarboxylate Carrier, A17 Peroxisomal Membrane Protein 34kD. A17 Glutamate/H (+) Symporter 1, PNMAL1, PNMA8A (Paraneoplastic antigen-like protein 8A, SLC39A10, Solute Carrier Family 39 Member 10 (Zinc-influx transporter), GLRB, Glycine Receptor Beta PTPN3 Protein Tyrosine Phosphatase Non-Receptor Type 3, cGMP-PKG, cGMP-protein kinase G.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



