
Submitted:
14 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
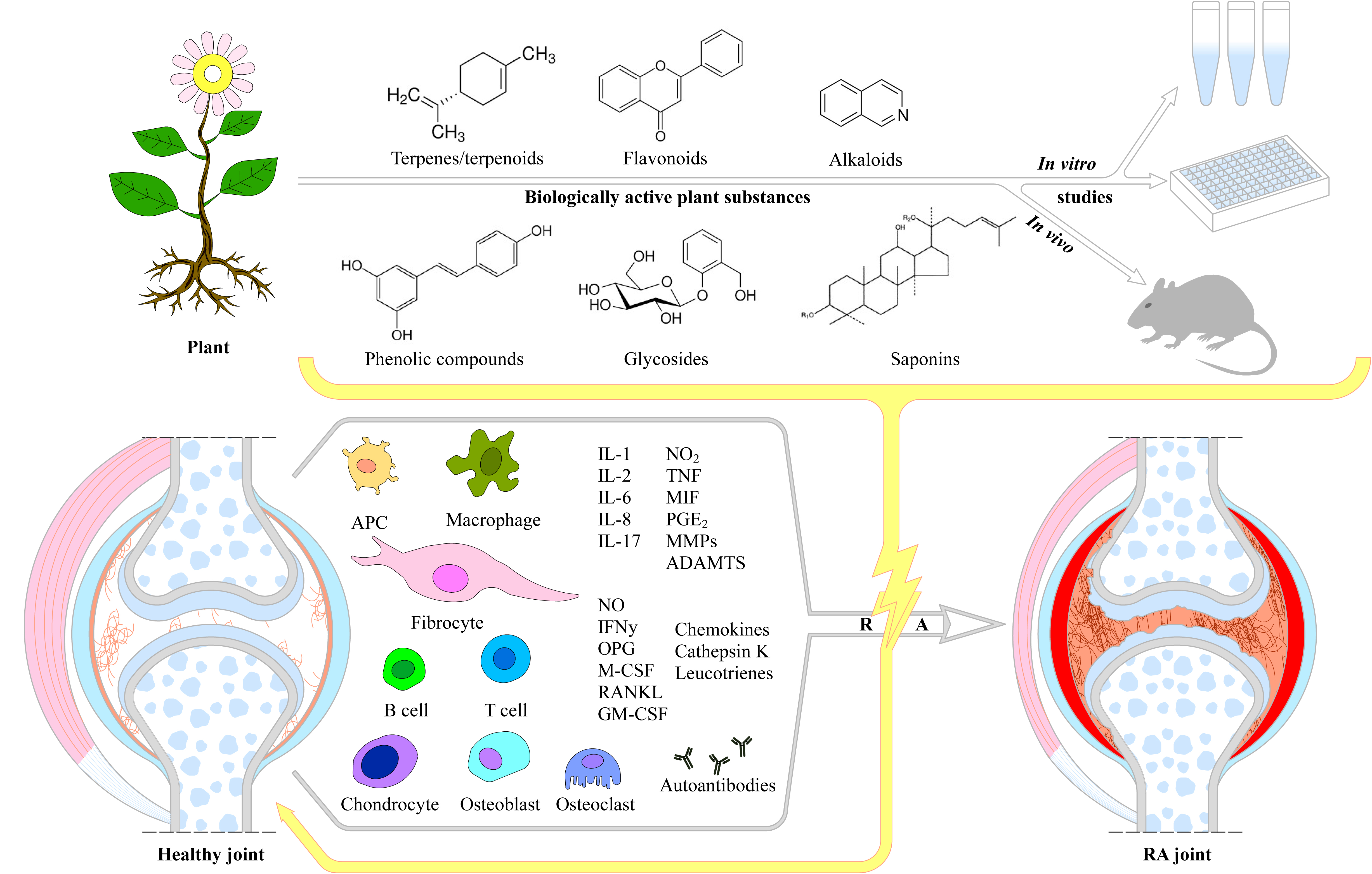
Rheumatoid arthritis (RA) is a progressive and systemic autoimmune disease, characterized with a chronic inflammatory process, affecting the lining of the synovial joints, many body organs/systems and blood vessels. Its pathological hallmarks are hyperplasic synovium, bone erosion, and progressive join destruction. Rheumatoid arthritis affects over 20 million people with a worldwide prevalence of 0.5-1.0%, exhibiting gender, ethnic and geographical differences. The progressive disability severely impairs physical motion, quality of life, and is finally leading to a shortened life span. The pathogenesis of RA is a complex and still poorly understood process in which genetic and environment factors are principally associated. The current treatment mostly relies on conventional/non-biological disease modifying anti-rheumatic drugs (cDMARDs), analgesics, non-steroidal anti-inflammatory drugs, glucocorticoids, steroids, immunosuppresants and biologic DMARDs, which only control inflammation and pain. Along with the side effects (drug toxicity and intolerance), these anti-rheumatic drugs possess limited efficacy. Therefore, the discovery of novel multi-target therapeutics with no side effects that function as inhibitors of RA-linked signaling systems are in high demand and this is in interest in both, patients and clinicians. Plant-derived extracts, nutritional supplements, dietary medicine and molecules with anti-inflammatory activity represent promising adjuvant agents or alternatives for RA therapeutics. This review aims not only to discuss the basic features of RA pathogenesis, risk factors and signaling pathways, but highlights the research progress in pre-clinical RA in vitro and in vivo models revealing new avenues in management of the disease in terms of comprehensive multidisciplinary strategies originating from medicinal plants and plant-derived molecules.
Keywords:
Rheumatoid arthritis
; risk factors
; pathogenesis
; signaling pathways
; medicinal plants
; natural products
; in vitro and in vivo models
1. Introduction
Rheumatoid arthritis (RA) is the most common progressive and systemic autoimmune disease, characterized with a chronic inflammatory process, which predominantly affects the lining of the synovial joints (articular damage), as well as, a variety of other body organs/systems and blood vessels (extra-articular manifestations) [1,2,3]. The pathological hallmarks of RA are defined by hyperplasic synovium, bone erosion, and progressive join destruction due to autoantibodies production towards immunoglobulin G (IgG, named rheumatoid factor (RF)) and citurllinated proteins (anti-citrullinated protein antibodies (ACPAs)), carbamylated proteins (anti-carbamylated protein antibodies (anti-CarP)) and lately acetylated proteins (anti-acetylated protein antibodies). [4,5,6]. Both, RF and ACPAs have approximately equal sensitivity and specificity to RA. The disease is also marked up by elevated levels of C-creative protein (CRP), and erythrocyte sedimentation rate (ESR) [7]. The most common serum prognostic biomarkers for RA are RF and ACPA, since their appearance is observed approximately 4.5 years prior to clinical onset of the disease. A relatively new potential biomarker for RA, revealing high specificity is the oncoprotein survivin detected in 50.7% of RA patients and only 5.6% in controls [8]. Nevertheless, 50-80% of RA patients harbor autoantibodies are seropositive, while other patients are seronegative for these auto-antibodies. For that reason to identify RA patients, a set of clinical diagnosis e.g. physical examination, clinical symptoms, antibodies present in the blood and imaging findings are necessary to be applied [9,10,11].
1.1. Epidemiology Overview and Global Prevalence
Although RA can occur at any time, it is mainly manifested in elderly population peaking between the ages of 40 to 70 [12]. Rheumatoid arthritis affects over 20 million people and its average worldwide prevalence is about 0.5-1.0%, exhibiting gender (prevalence of female to male is in ratio 3:1), ethnic and geographical differences, therefore significantly varying among different populations [7,12]. Along with joint damage, extra-articular multiple co-morbidities affect cardiovascular, endocrine, neurological, ocular and pulmonary systems, including hematologic, renal and hepatic disorders [13]. The progressive disability severely impairs physical motion and the quality of life, finally leading to a shortened life span [6], reporting mortality rates twice higher in patients with RA and this number is increasing [14]. Based on analyses for the period 1990-2020, it was reported that in 2020 the RA patients were 17.6 million worldwide and the age-standardized global prevalence rate increased 14.1% since 1990. The age-standardized death rate decreased with 23.8% from 1990 to 2020, while the disability-adjusted life-years (DALYs) increased with 76.4%. In spite of that, RA is still a key public health concern worldwide. The global burden of RA has increased during the past decades and will continue to increase, forecasting that 31.7 million individuals will be living with RA until 2050 [15]. Rheumatoid arthritis has a negative impact on the most productive and active years (30-50 years) of an individual’s lifespan [16], severely affecting the quality of daily life, including high disability rates and potential loss of labor [17]. Socioeconomic surveys indicated that people with severe (60%) or moderate (48%) pain experience additional work obstacles compared with dose with mild (34%) or no (19%) pain and significant correlation was found between severity, pain, disability, and early retirement [18]. The major economic costs for RA patients are based on both healthcare expenditures and loss of productivity [19]. For example the average annual cost of medical care for RA depending on the medication may vary between $12 500 to $36 000, while the Rheumatoid Arthritis Support Network estimates that low productivity, absenteeism, and lost wages can cost $1,500 to $22,000 per year, per patient [20]. In addition to the joint, many advanced patients develop systemic and comorbid extra-joint diseases, such as hypertension, diabetes mellitus, ischemic heart disease, chronic obstructive pulmonary disease, asthma, tuberculosis, chronic liver disease, hypothyroidism, hyperthyroidism, active malignancy etc., that significantly reduces life expectancy [7,21].
The clinical manifestation of RA differs in its early and untreated/inadequately treated late stage. The typical symptoms for the early stage are fatigue, swollen and tender joints, redness, stiffness not only in the morning, but also after a period of inactivity. On the other hand, the late stage is characterized with more severe manifestations, such as Swan neck deformity, Ulnar deviation and subcutaneous nodules formation [6,22], also bone corrosion, muscle atrophy, synovitis invasion of articular cartilage, sub-cartilage bone erosion, damage to ligaments and tendons [17], which may finally result in a premature death [15]. Rheumatoid arthritis has a multi-synovial form, which primary clinical manifestation is the repeated and symmetrical multiple micro arthritis [17]. It is a form of polyarthritis due to its involvement of multiple joints (typically six or more), mainly affecting hand, wrists, knees, feet, metacarpophalangeal, proximal interphalangeal and metatarsophalangeal joints. Erosions of the feet and hands are observed radiographically [17,23]. The pathogenesis of RA is a complex and still poorly understood process in which genetic and environment factors are principally associated with the disease onset and progression [21,23]. Joint inflammation is being triggered and maintained by invasion and interaction between several types of immune cells, including neutrophils, antigen-presenting cells (B-cells, macrophages and dendritic cells), as well as, T-cells and fibroblast-like synoviocytes (FLS) [16,24]. These chain events contribute to the releasing of pro-inflammatory cytokines, such as interleukin-1 (IL-1), -6, -17, prostaglandins and tumor necrosis factor alpha (TNF-α), which mediators activate further additional signaling pathways, including the upregulation of cyclooxygenase-2 (COX-2), and matrix metalloproteinases (MMPs), Janus kinase-signal transducers and activators of transcription (induced by IL-6), and nuclear factor kappa-B (NF-κβ) pathway (induced by IL-1 and TNF-α) that continue to generate and accumulate inflammation [16,24]. The above indexed cells and cytokines pattern the RA microenvironment (consisting mainly of extracellular matrix (ECM) and stromal cells), where the generated inflammation creates hypoxic environment and subsequently triggers the generation of reactive oxygen species (ROS), angiogenesis and as a consequence the formed microvessels cause the synovial membrane to invade the cartilage surface and to form pannus, destroying the structure and function of bone and cartilage [25].
Rheumatoid arthritis is often regarded as incurable and currently no adequate treatment is available [6]. The current treatments aim to control inflammation and pain, contributing to patients’ symptoms relief and therefore avoiding or minimizing joint destructive processes [7]. The current treatment of RA mostly relies on conventional/non-biological disease modifying anti-rheumatic drugs (cDMARDs), analgesics, non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids (GC), steroids, immunosuppresants and biologic DMARDs (bDMARDs) [7,12,26]. However, in most cases the drugs need to be accepted weeks or month before take any effect and may cause liver and bone marrow toxicity, certain malignance and/or opportunistic effects [12,27]. The patients are considered as “difficult to treat” or “refractory” to RA and although the introduction of targeted synthetic DMARDs, RA treatment is still a great therapeutic challenge and considerable part of patients remain symptomatic [28]. Along with the side effects, such as drug toxicity and intolerance, these anti-rheumatic drugs possess limited efficacy, therefore the discovery of novel multi-target therapeutics with no side effects are in high demand and this is in interest in both, patients and clinicians [12]. Plant-derived extracts, nutritional supplements, dietary medicine and molecules with anti-inflammatory activity represent promising adjuvant agents or alternatives for RA therapeutics [12]. The pathogenesis of RA is tightly related to many characterized signaling pathways and the research attention has focused on discovering plant-derived molecules that function as inhibitors of RA-linked signaling systems [17].
This review attempts to recapitulate first of all the dynamics of RA pathogenesis, highlighting the role of risk factors, effector cells involved, the role of metalloproteinases, oxidative and nitrosative stress, angiogenesis, cell migration and invasion, as well as, the production of cytokines and chemokines. Next, we discuss the existing conventional therapies for RA and their limitations. A summary of various signaling pathways involved in RA development has been performed outlining the efficacy and mechanism of plants and their phytoconstituents in RA management supported by preclinical data based on state-of-the-art in vitro and in vivo models. Finally, we analyze the existing theoretical and practical challenges, and unanswered questions in the field aiming to improve the RA therapy by natural products.
1.2. Risk factors
Unlike osteoarthritis, RA does not develop due to age, but its onset and development is rather as a result of a combination between immune dysfunction, hereditary (genetic factors) and environmental factors. Because of the complexity, it has not been thoroughly clarified yet, therefore research into the genes, pathways, and pathogenic immune cell subsets in RA has advanced the understanding of mechanisms involved in pathogenesis [16,27]. The major differences between healthy and RA joint, as well as its process of establishment has been illustrated on Figure 1.
Genetic factors
The contribution of genetic predisposition is thought to be about 50 to 60%, which has the most significant impact on the vulnerability to RA [29]. Genetic studies have been successful in determining the heritable component of RA susceptibility and outcome, of which RA severity and response to treatment are important [30]. The presence or absence of RF and ACPAs classifies RA into seropositive and seronegative and differences between the risk factors involved exists. Tyrosine phosphatase non-receptor type 22 (PTPN22) risk alleles [31,32], human leukocyte antigen D-related (HLA-DR) alleles [33], and tumors necrosis factor-receptor associated factor 1 (TRAF1) and complement component 5 (TRAF1/C5) related genes are the main genetic factors associated with an ACPA-positive subtype [34], while interferon regulatory factor 5 (IRF-5) is confined to the ACPA-negative subtype [35]. Despite the lack of concrete evidence supporting a direct role in RA pathology, some genetic markers regulate immune responses and account for the variations in RA susceptibility [21]. The strongest genetic association with RA susceptibility is located at the HLA locus. This is a specific HLA class II gene also known as a major histocompatibility complex (MHC) loci, encoding MHC molecules that may contain the shared epitope [4]. Individuals carrying a single HLA allele, have a five-fold higher risk of developing RA than individuals without this gene [36]. The susceptibility and outcome of RA may be related to specific HLA-DR alleles, however, these alleles vary by ethnicity and geographic region [37,38]. The HLA-DRB1 gene constitutes the strongest genetic association linked to RA, and the allele associated with the disease is named as a shared epitope, which is a conserved sequence of five amino acids at positions 70-74 of the HLA-DRB1 gene [17] and this concept has been highly correlated with the ACPA-positive RA [30]. The shared epitope hypothesis indicates that some alleles with this conserved sequence are linked with the pathogenesis of RA since they allow antigen-presenting cells to incorrectly present their antigens to T cells, which results in T cell mediated autoimmune responses that directly contribute to the RA pathogenesis [39].
On the other hand, single nucleotide polymorphisms (SNPs) are variations in the DNA code at a specific position in the genome and these variations have been associated with susceptibility to diseases, including RA [30]. A SNPs has been detected in T cells regulating the HLA-gene. In the presence of SNPs, the T cells induce tissue damage and inflammation linked to RA during the rapid resolution of the joint defects and therefore its detection may enable assessment of the risk of developing RA and hereditary autoimmune disorders in general [40]. The specific RA-related gene HLA-DR4 is found in 60% to 70% of patients diagnosed with RA, while it is detected in only 20% of the general population [41]. The HLA-DRB1 gene has been associated with the susceptibility of this disease, especially with the shared epitope coding alleles (HLA-DRB1*0401, *0404, *0405, *0408, *0101, *0102, *1402, and *1001) [42]. In addition, genetic differences between ACPA-positive and ACPA-negative RA have been demonstrated, e.g. variants in HLA-DRB1, PTPN22, BLK, Ankyrin Repeat Domain 55 (ANKRD55) and IL6ST associate with RA regardless of serological status, whereas AFF3, CD28 and TNFAIP3 are found only in seropositive RA and PRL and NFIA are found only in seronegative RA [43,44]. All these variants demonstrate very well the RA susceptibility in both positive and negative type of RA. However, equally important is to identify markers of disease severity. In this regard it has been demonstrated that several markers for susceptibility are also associated with severity, e.g. HLA-DRB1, IL2RA, DKK1, GRZB, MMP9 and SPAG16 [43,45], although some of them, such as FOXO3 are associated with severity alone [43,46]. There are also other HLA loci independently associated with RA susceptibility: amino acid position 9 of HLA-DPB1 (another HLA class II gene) [47] and two associations within HLA class I genes, such as amino acid position 9 of HLA-B [47] and position 77 of HLA-A [48]. Along with the HLA gene, there are many genetic variations outside the HLA complex, related to RA, such as PTPN22, STAT4, TRAF1-C5, CTLA4 and PADI4 [49]. The PTPN22 gene encodes for the cytoplasmic lymphoid specific tyrosine phosphatase (Lyp). This enzyme has gained enormous interest due to a genetic SNP of its gene PTPN22 rs2476601 (R620W) which has been associated with several human autoimmune diseases, including rheumatoid arthritis (RA). The Lyp has been shown to be a powerful inhibitor of T-cell and B-cell activation by binding to the SH3 domain of the Csk tyrosine kinase, an important negative regulator of T-cell and B-cell antigen receptor signaling [50,51,52,53]. Along with that, the role of Lyp in TNFα-induced priming of neutrophil ROS production has been investigated during RA development. It has been demonstrated that Lyp-selective inhibitors, inhibited TNFα-induced priming of neutrophil superoxide anion production, through inhibition of key pathways involved in neutrophil priming, such as inhibition of p47phox phosphorylation on Ser345, ERK1/2 phosphorylation and Pin1 [54].
Epigenetic factors
Genetic heterogeneity can not explain all aspects of RA, therefore examination of epigenetic factors and mechanisms might be of ultimate importance for the provision of novel therapeutic factors [55]. Epigenetics are heritable changes of gene expression without altering the DNA sequence, but epigenetics determines which genes are turned on and/or off, which mainly include histone modification, DNA methylation, and non-coding RNA mechanisms and these processes can be affected by different genetics and environmental factors. The epigenetic modifications are reversed processes, and the corresponding enzymes which control histone modification or DNA methylation could be proposed as drug targets for RA [17]. DNA methylation is the most commonly occurring postreplication DNA modification under the activity of DNA methyltransferases (DNMT), which transfers the methyl group from S-adenosine methionine (SAM) to the DNA sequence. DNA methylation occurs at the cytosine of CpG (cytosine-phosphoric acid guanine) islands to produce 5MC, most of which are located in the promoter region. Hypermethylation in the promoter region is related to gene silencing or gene inactivation, while its hypomethylation activates transcriptional activity and promotes gene expression of extracellular matrix proteins, growth factors/receptors, matrix-degrading enzymes, and adhesion molecules [56]. For example, several studies show that fibroblast-like synoviocytes (FLS) and peripheral blood mononuclear cells (PBMCs) in RA patients are characterized by extensively hypomethylated genomic DNA. Rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) have a unique, non-random methylation pattern-methylome, which is specifically reorganized during the disease progression and varies depending on the joint localization [55], and DNA methylation reduction is often found in highly proliferative tissues and is associated with a relative lack of methyl groups’ donor S-adenosylmethionine (SAM) [17]. T-box transcription factor 5 (TBX5) regulates expression of pro-inflammatory cytokines and chemokines in SF including CXCL12 (chemokine C-X-C motif ligand 12) chemokine. Upon hypomethylation TBX5 increases its own expression and that of CXCL12 which is associated with protein accumulation in RA patients and contributes to chronic inflammation [57]. The promoter demethylation of IL-6 and IL-10 genes in a single CpG sequence contribute to the increase in cytokine levels as the disease progresses [58]. Comprehensive analysis of DNA methylation in RA-SFs identified 1 859 differently methylated loci, of which the hypomethylated ones such as CHI3L1, CASP1, STAT3, MAP3K5, MEFV, and WISP3 were of critical importance and found to be involved in cell migration, adhesion, transendothelial penetration and interactions in the extracellular matrix [59]. It has been reported that the PBMCs from RA patients have a significant overall DNA hypomethylation state compared to healthy people [60]. Analyses of the whole-genome DNA methylation and mRNA expression profiles of PBMCs from patients with RA revealed that approximately 1,046 DNA methylation sites were closely associated with the pathogenesis of RA. Among the identified differentially methylated positions (DMPs) and genes formed an interferon-inducible gene interaction network (such as MX1, IFI44L, DTX3L and PARP9). Methylation of PARP9 was correlated with mRNA level in Jurkat cells and T lymphocytes isolated from patients with RA. The PARP9 gene exerted significant effects on Jurkat cells (eg, cell cycle, cell proliferation, cell activation and expression of inflammatory factor IL-2) [61]. Epigenetic modification in immune cells is also critical for the development of RA. The differentially methylated patterns of B lymphocytes in RA and systemic lupus erythematosus (SLE) patients and in the control group CpG sites were located in the CD1C, TNFSF10, PARVG, NID1, DHRS12, ITPK1, ACSF3, and TNFRSF13C genes [62]. On the other hand, hypermethylation of 4 CpG dinucleotides in exon 7 of LRPAP1 has been correlated with patients who demonstrated no response to therapy by TNF inhibitors compared to responders. The LRPAP1 gene is expressed in PBMCs and encodes the chaperone of low density lipoprotein receptor-related protein 1, that affects the activity of transforming growth factor beta (TGF-β) [63]. The aberrant function of regulatory T cells (Treg) in RA patients was associated with the hypermethylation of a specific region in the promoter of the cytotoxic T-lymphocyte associated protein 4 (CTLA-4;−658 CpG) in comparison with healthy controls. DNA hypermethylation prevents binding of the nuclear factor of activated T cells (NF-AT) with cytoplasmic one, called NF-ATc2, which leads to decrease of CTLA-4 expression. As a consequence, Treg cells were unable to induce expression and activation of the tryptophan-degrading enzyme indoleamine 2.3-dioxygenase (IDO), which in turn resulted in a failure to activate the immunomodulatory kynurenine pathway [64]. Furthermore, treatment with methotrexate induced DNA hypomethylation of FoxP3 locus in Treg. This results in the gene upregulation with consequent increase of CTLA-4 concentration and normalization of Treg function in RA. These studies clearly illustrate how aberrant DNA methylation can affect cell functions and how epigenetic mechanisms can be used in therapy [65].
Histone modification is a post-translational modification of a specific site on histones in chromatin. Acetylation, methylation, phosphorylation, and ubiquitination are all included within the modifications of the histone tails and among them acetylation is the most common. Histone deacetylases (HDAC) have a significant role in the activation or silent regulation of pro-inflammatory genes, and their inhibitors are often used to study the pathogenesis of RA. The HDAC activity and histone H3 acetylation status in PBMCs are potential biomarkers for evaluating the disease activity [56]. For example, the hyperacetylation state caused by the decreased activity and expression of HDACs promotes the pro-inflammatory processes and ultimately leads to RA in PBMCs [66]. Mice with a T cell-specific deficiency of HDAC1 (HDAC1-cKO) were found to be resistant to the development of collagen-induced arthritis (CIA), while its activation produces pro-inflammatory factors. The HDAC1 is a promising target for the treatment of RA patients, since the inflammatory cytokines, IL-17 and IL-6, were significantly reduced in the serum of HDAC1-cKO mice. Along with that, selective HDAC inhibitors restrained chemokine receptor 6 (CCR6) upregulation in a mouse model and human CD4+ T-cells [67]. Another potential target for RA treatment is SIRT1 (a type 3 histone deacetylase that possesses anti-inflammatory properties) and can reduce the inflammatory responses in RA by regulating M1/M2 macrophages polarization. For example, activated SIRT1 promotes the phosphorylation of adenosine monophosphate-activated protein kinase α (AMPKα)/acetyl-CoA carboxylase in macrophages through upregulation of the M2 genes, such as MDC, FcϵRII, MrC1, and IL-10 expression. Simultaneously, activated SIRT1 downregulates the LPS/γ interferon mediated NF-κB activity by inhibiting p65 acetylation and M1 genes (including CCL2, iNOS, IL-12p35, and IL-12p40) expression [68].
MicroRNAs (miRNAs) widely present in all organisms and present endogenous non-coding single-stranded small RNAs of about 22 nucleotides in length [56]. Initially, the miRNA gene is transcribed to form a primary miRNA in the nucleus, cleaved by Drosha to form a precursor-miRNA. Exportin-5 transfers miRNA to the cytoplasm where it is cleaved by Dicer to form mature miRNA duplexes, which are then unfolded, and a miRNA strand is added to the RNA-induced silencing complex and act as a post-translational repressor of the gene [17]. MicroRNAs have been implicated in the occurrence and progression of many diseases, including RA. For example, hsa-miR-132-3p, hsa-miR-146a-5p, and hsa-miR-155-5p are potential biomarkers of responsiveness to methotrexate (MTX) therapy, which levels were lower in responders [69]. However, the downregulation of miR-10a in RA-FLs accelerated NF-κB activation and significantly promoted the production of various inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-8 and MCP-1 and matrix metalloproteinase (MMP)-1 and MMP-13 [70]. Some miRNAs have been regulated in RA and it has been demonstrated that DNA methylation increased the expression of miR-203, which lead to increased secretion of MMP-1 and IL-6 via the NF-κB pathway and contributed to the activated phenotype of RA-FLs [71]. The upregulation of miR-203 induces RA through promoting the generation of MMP-1 and IL-6 [71]. Depletion of miR-19b regulates positively NF-κB signaling through suppression of a regulon of negative regulators of the same pathway [72].
Environmental factors
Many environmental factors, including smoking, alcohol, air pollution, and exposure to insecticides, viruses, bacteria, toxic minerals or mineral oil increase the risk of RA [21]. Most of the listed factors here support the hypothesis that the aetiopathogenesis of RA is of ‘mucosal origin’, in which the autoimmune processes that lead to the development of RA are triggered in the mucosa-associated lymphoid tissues in the lung, the oral cavity and the gut, prior to systemic spread [73]. Rheumatoid arthritis related autoantibodies may be generated in the lung mucosa and in draining lymph nodes prior to the onset of clinically apparent RA [74,75,76]. Smoking is the strongest environmental factor in RA development and raises the risk of RA in a gradient fashion, with double risk among smokers with a 20-year history of tobacco use compared with nonsmokers. The correlation between tobacco use and RA is strongest or almost limited to RF- or ACPA-positive individuals [77] with at least one copy of the shared epitope alleles HLA-DR Beta 1 [78] and has no or very little effect on ACPA-negative [79]. The risk for developing RA between shared epitope and smoking can be increased by 20-fold or more compared with nonsmokers who do not carry the shared epitope [80]. Actually, no association between passive smokers and risk of developing RA has been observed [81]. In addition, the increased risk associated with smoking might be mediated by epigenetic modifications, as smoking was significantly associated with hypomethylation of certain DNA regions [82]. Population-based studies revealed that smoking is a strong risk factor for RA in men rather in women, which effect might be due to hormonal differences in the modulation of the immune cascade activated by smoking [83]. Air pollution and the inhaled particulars, such as pulverized cement, silica, asbestos, glass fibers, textile dust and many others are all associated with RA development [84,85]. For example, there is correlation between silicosis and RA, which mainly affects patients with ACPA-positive RA and is caused due to silica dust inhalation [86]. Chronic exposure to silica can lead to rheumatoid pneumoconiosis, also known as Caplan’s syndrome, a rare disease of RA patients who have developed silicosis [87].
Lifestyle factors (nutrition and gut microbiota)
It has been suggested that some healthy diets, e.g. Mediterranean diet has modest protective effect in individuals with seropositive RA [88]. The consumption of fruits and fish, including omega-3 fatty acids has a protective effect related to RA-associated autoimmunity [89,90,91]. Long-term supplementation of omega-3 and vitamin D resulted in a 25-30% lower incidence of RA [92]. Coffee consumption may be a risk factor for RA, a possible explanation being the involvement in the production of RF [93]. The intake of raw meat and sugar-sweetened sodas is associated with increased risk of RA [94,95,96]. Microbiota (periodontal and gut) and infection microorganisms are associated with increased risk of RA development as well [97]. For example the oral microbiota Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans causing periodontal disease, also related with RA development [98,99]. The dysregulation many intestinal bacteria are associated with RA onset. For example, expansion of Prevotella species is associated with RA-related autoimmunity and a marker for early disease preclinical development [100]. Other genus, such as Bacteroides, Eggerthella and Collinsella have also been related to RA [101,102,103]. Several infectious agents have been considered as possible causes of RA, including rubella virus, Epstein–Barr virus, and mycoplasma organisms [21].
Personal factors
Although both, males and females are at risk of RA development, RA is more prevalent in women with a with a female-to-male sex ratio ranging from 4:1 in younger individuals to less than 2:1 in older populations with the disease [100]. This might be due to hormonal changes (such as from contraceptive use) or a sudden decline of oestrogenic function (seen in menopause, or with the use of anti-oestrogenic therapies) [104,105]. The manifestations of RA improve or even disappear during pregnancy [106]. In women, RA most commonly becomes symptomatic around middle age or at the time of menopause. Men have a later disease onset, are more likely to be positive for RF and have higher titers of ACPAs [107]. Epidemiologic studies have shown a complex interaction between RA and obesity. Obesity would contribute to the development of seronegative RA, especially in younger women, although it is suggested a decreased risk of RA among men [108]. Adipose tissue secretes different pro-inflammatory and anti-inflammatory factors, including the adipokines leptin, adiponectin, resistin, and visfatin as well as cytokines and chemokines, such as TNF-α, IL-6, which participates in RA development [109].
1.3. Mechanisms and (pato)etiology of Rheumatoid Arthritis Initiation, Development and Progression
The pathogenesis of RA is a complex and poorly understood in terms of causes, initial triggers and progression process [16]. However, the initiation of RA involves an interplay among components of the innate and adaptive immune responses, where immune cells (T-cells, B-cells, mast cells, and dendritic cells) and synoviocytes (macrophages and fibroblasts) are the key players [8]. In general, the RA pathophysiology goes through three distinct stages: 1) occurrence of immune abnormalities; 2) synovial inflammation and extensive proliferation; and 3) joint deterioration and bone erosion [8]. The mechanisms involved in initiation and progression of RA are presented on Figure 2.
The initiation of RA has multifactorial origin including many genetic and environmental factors. Collectively, all these factors initiate the early development of RA, including post-translational modifications of a wide range of cellular (collagen) and nuclear (histones) proteins, including the conversion of the amino acid arginine to citrulline (a process called citrullination) through peptidyl arginine deiminase, type IV enzyme (PADI4; EC: 3.5.3.15) [1,110]. After citrullination or other post-translational modifications (acetylation or carbamylation) the altered modified self-proteins are recognized by immune system through binding to major histocompatibility complex (MHC) protein heterodimers, especially those containing the shared epitope and produce antigen presenting cells (APCs), such as dendritic cells and macrophages, which carry antigen to lymph node [4,22,111]. These APCs activate CD4 T-helper (Th) cells causing stimulation and proliferation of B-cells, which in turn distinguish into plasma cells and produce autoantibodies such as ACPA (targeting citrullinated proteins) and RF (targeting IgGs). The presence of these and other antibodies can be detected up to 10 years before the clinical disease onset [4,22,112]. The antibodies bind with its fixed complement to form immune complex and release chemotactic factors such as C5a and consequently due to chemotactic gradient, inflammatory cells are subsequently recruited to the rheumatoid joint where they are activated and cause localized inflammation [22,113]. The APCs such as plasmacytoid dendritic cells (PDC) and myeloid dendritic cell (MDC) in response to modified protein, express HLA class II molecules, cytokines (such as IL-12, -15, -18, and -23), and co-stimulatory molecules leading to activation of synovial T cells. Dendritic-cell–derived TGF-β and interleukins (IL-1β, -6, -21, and -23) supports Th17 cells (pathogenic cells) differentiation and suppress the differentiation of regulatory T cells (Treg) which shifts T-cell homeostasis toward inflammation. Activated Th17 cells release cytokines such as IL-17, IFN-γ to provoke expansion of the intimal lining and to recruit macrophages, which release interleukins (IL-1, -6, -12, -15, -18, and-23), TNF-α, TGF-β, granulocyte–macrophage colony-stimulating factor (GM-CSF), macrophage migration inhibitory factor (MIF), MMPs, disintegrin, ADAMTs, prostaglandins, leukotrienes, and reactive nitric oxide. They all together cause FLSs proliferation known as synovial hyperplasia [1,22,30]. Treg cells produce anti-inflammatory cytokines IL-10 and TGF-β and inhibit autoimmunity, therefore the balance between Th17 and Treg is important in the pathology of RA [114]. The increase in mass of synovial cells and immune cells in joints leads to pannus formation (thick, swollen synovial membrane with granulating tissue consisted of myofibroblast, fibroblast and inflammatory cells) [22,115].
This hyperplastic pannus tissue contributes to cartilage damage but may also be responsible for propagation and systemic spreading of inflammation by migrating between joints or other organs [22,26]. The synovial immune cell infiltration transform the paucicellular synovium into chronically inflamed tissue [1]. Due to the resulting local hypoxia, new vessels are formed, which facilitate the inflammatory process by increasing the amount of adaptive immune cells, and especially CD4+ Th cells infiltrating the synovial sublining. Lymphocyte infiltrates accumulate and form aggregates, which may develop germinal centers that facilitate local T cell - B cell interactions. In these ectopic germinal structures are specific pathologic follicular helper T cells (Tfh), which promote B-cell responses and (auto)antibody production within pathologically-inflamed non-lymphoid tissues [1,116]. It has been established that specific pathogenic infiltrating immune cell subsets, such as IL-1β positive pro-inflammatory monocytes, autoimmune-associated B cells, and peripheral helper T (Tph) cells sharing similarities with Tfh cells, distinct subsets of CD8+ T cells, as well as mast cells, contribute to the inflammatory pattern of the RA synovial lining/sublining [117,118,119,120,121,122]. Finally, the invasive and destructive FLS-front of synovial tissue (called the pannus), attaches to the articular surface and contributes to local matrix destruction and cartilage degradation. The chondrocytes of the damaged articular cartilage contribute to the vicious cycle of cartilage degeneration by inducing inflammatory cytokines, such as IL-1β and TNF-α, as well as MMPs and nitric oxide (NO). Additionally, FLSs negatively affect the subchondral bone by activation and maturation of bone-resorbing osteoclasts. Osteoclasts are highly responsive to autoantibodies; pro-inflammatory cytokines, in particular TNF, IL-1, and IL-6; and more importantly, receptor activator of nuclear factor kappa B ligand (RANKL), which is the key regulator of osteoclastogenesis. RANKL binds to its receptor, the receptor activator of nuclear factor-B (RANK), and activates osteoclasts, leading to an enhancement of bone resorption and destruction. Conversely, osteoblasts that play a key role in the regulation of anabolic bone metabolism produce bone matrix constituents, induce bone matrix mineralization, and modulate osteoclasts through the production of osteoprotegerin (OPG) [32]. Although osteoblasts producing OPG, which is a decoy receptor for RANKL, results in protection from bone destruction by osteoclasts, they also generate RANKL and M-CSF, both of which contribute to osteoclastogenesis. Imbalanced bone remodeling both in the subchondral and periarticular bone of joints leads to bone erosions and periarticular osteopenia; generalized bone loss is a general feature of established RA [1,123].
2. Effectors Cells Involved in Rheumatoid Arthritis Pathology
Rheumatoid arthritis has a complex pathogenesis and the activation of cells of both, innate and adaptive immunity and aberrant cytokine production are the main events responsible for the generation and development of pathological immune response. Consequently, synovial inflammation leading to massive destruction of cartilage and bone structures is observed. The pathogenesis of RA involves a diverse array of effector cells, including T cells, B cells, macrophages, and fibroblasts, each contributing to the inflammatory milieu and tissue damage observed in affected individuals [124,125].
The association of the particular MHC-II alleles in RA chronic inflammation supported the role of CD4+ T-cells. Disease-associated HLA-DR alleles, and particularly those with the ”shaped epitope” may present arthritis-related peptides, such as those modified by citrullination, leading to the stimulation and expansion of autoantigen-specific CD4+ T cells in the joints and lymph nodes [126].
As in the regular immune response, the autoimmune process starts with antigen presentation of the processed antigen on APCs, and triggering the activation of naïve T lymphocytes. This activation includes proliferation and secretion of cytokines as IL2, IFN-γ, TNF, and IL-4 that leads to modulation of the immune system through differentiation of various subsets of cells like T helper (Th)1, Th2, T follicular helper (Tfh), Th17 and regulatory T (Treg) cells. The Th1/Th2 imbalance is thought to exacerbate the inflammatory processes in RA, as a lack of Th2-mediated regulation allows for unchecked Th1-driven inflammation. Particularly CD4+ T helper cells and CD8+ cytotoxic T cells, play pivotal roles in RA pathology. A notable increase in the ratio of CD4+ to CD8+ T cells has been documented in the blood of RA patients, suggesting a skewed immune response favoring CD4+Tcell activation [127]. Furthermore, studies have shown that effector memory CD8+ Tcells are significantly altered in RA, with a decrease in their numbers in peripheral blood, potentially due to their migration to inflamed tissues [128]. This migration is indicative for their pathogenic role because they can produce pro-inflammatory cytokines such as IFN-γ, contributing to the inflammatory environment within the synovium [129].
Th17 cells are characterized by their production of IL17 and have emerged as critical player in RA pathology. These cells promote the recruitment of neutrophils and enhance the production of pro-inflammatory cytokines, contributing to joint inflammation and damage [130,131]. Studies have shown that Th17 cells are enriched in the synovial fluid of RA patients, correlating with disease severity and joint destruction [132,133]. The differentiation of Th17 cells is often favored in the inflammatory milieu of RA witch further perpetuates the cycle of inflammation and tissue damage [134]. T Follicular Helper (Tfh) cells are essential for B cells in the germinal centers, promoting their differentiation into plasma cells that produce of high-affinity antibodies. In RA, the frequency of circulating Tfh cells is significantly elevated, and these cells are associated with increased levels of autoantibodies, such as anti-citrullinated protein antibodies [135,136]. The presence of Tfh cells in the synovial tissue of RA patients suggests that they play a direct role in sustaining the autoimmune response and contributing to the chronicity of the disease.
Regulatory T (Treg) cells are typically involved in maintaining tolerance and preventing excessive inflammation. In the RA, their functional capacity is often compromised. This dysfunction leads to inadequate suppression of effector T cell responses, allowing for the persistence of inflammation and autoimmunity [137].
Macrophages are central to the inflammatory processes in RA, acting as a major producers of pro-inflammatory cytokines such as TNF-α and IL-1β. They are found in increased numbers within the synovial tissue, correlating with disease severity and joint erosion [138,139]. Additionally, macrophages expressing heparin-binding EGF-like growth factor (HB-EGF) have been shown to enhance fibroblast invasiveness, further exacerbating joint destruction [140,141].
B cells contribute significantly to RA through the production of autoantibodies and pro-inflammatory cytokines. Dysregulation of B cells subsets, including the expansion of RANKL+ effector B cells, has been observed in RA patients, indicating their involvement in osteoclastogenesis and joint destruction [142]. The efficacy of B cell depletion therapies, such as Rituximab, reinforces the pathogenic role of B cells in RA, as these cells not only produce antibodies but also act as antigen-presenting cells, perpetuating the inflammatory cycle [143]. Furthermore, the presence of FcRL4+ B cells in the synovial fluid suggests a unique subset that may contribute to the inflammatory process through enhanced activation and cytokine production [144].
Fibroblast-like synoviocytes are critical effector cells in RA, contributing to the formation of the invasive pannus that characterizes the disease. These cells are activated by various inflammatory mediators and exhibit increased production of matrix metalloproteinases (MMPs) and cytokines, facilitating tissue remodeling and joint destruction [145,146]. The interaction between FLSs and infiltrating immune cells, particularly macrophages, creates a feedback loop that sustains inflammation and joint damage [147,148]. Recent studies have identified functionally district subsets of FLSs that correlate with disease activity, suggesting that targeting these cells may offer therapeutic potential [145].
2.1. Cytokines and the Impact on Effector Cells
Experimental studies on RA patients as well as on different mouse models point to the role of interactions between cytokines and effector cells in the disease progression [149,150]. Genetically modified TNF-α transgenic and IL-1Ra knockout mouse models of RA showed the importance of TNF-α and Th17 in the activation of the immune cells [151,152]. RA can also be induced and by the administration of different agents generating local or systemic inflammation. In the most frequently used model of RA - collagen-induced arthritis, the presence of collagen type II specific CD4+ T cells leads to the activation of collagen-specific B lymphocytes together with the secretion of various pro-inflammatory cytokines (TNF-α, IL-6), although the anti-inflammatory IL-10 and IL-1 receptor antagonist can also be detected in the affected tissues [150,153]. In antigen-induced model of RA, the antigen binds directly to the cartilage and induce inflammation with CD4+CCR6+ T cells via IL23 secretion [154]. Another mouse model of RA is the proteoglycan-induced arthritis in which scheduled injections of human cartilage proteoglycan resulted in synovial inflammation and infiltration of immune cells. CD4+ T cell immune response is observed with high levels of TNF-α, IL-1β, -6 and -12 in the experimental mice [155]. Finally, RA can be induced in various mouse strains by the administration of specific anti-collagen type II monoclonal antibodies. In this model the major effector cells appeared to be macrophages and neutrophils, although B- and T-lymphocytes also participated [156].
In RA patients, the role of effector immune cells and cytokine secretion has also been studied. Recent study reported that accumulation of B lymphocytes in synovium correlates positively with the severity of the disease [157]. B cells are the major source of autoantibodies to various antigens as some antibodies can be detected before the onset of the disease, whereas the presence of others indicates more severe form of RA [158]. In addition, being effective antigen-presenting cells [159] and producers of a number of pro-inflammatory cytokines [160,161], B lymphocytes stimulate autoreactive T-lymphocyte differentiation and proliferation [162]. RA pathogenesis is also caused by activated T cells and massive infiltration of CD4+ T lymphocytes can be observed in inflamed synovial tissues [157]. One particular population – peripheral helper PD-1+CD4+ T cells has been shown to have a central place in B cell co-stimulation and secretion of pro-inflammatory cytokines [163]. Although part of the innate immune system, another important players in RA pathogenesis are the neutrophils. Abnormal neutrophil activation due to aberrations in gene expression or in metabolic pathways, can lead to inappropriate local degranulation with subsequent chronic inflammation [164]. Finally, macrophages in synovium participate in the development of the inflammatory process by secretion of pro-inflammatory cytokines, chemokines and metabolites [165]. In addition, Zec et. al. have recently reported that synovial inflammation is initiated by lining macrophages in the niches and attract preferentially neutrophils in the inflamed tissues [166].
All these data demonstrated the complexity of RA pathogenesis and proposed interactions between the immune cells as well as particular cell populations as new targets for the forthcoming therapies.
2.2. The Role of Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases belonging to the superfamily of metzincin proteases, involved in the extracellular matrix (ECM) degradation and remodeling. The MMPs have a critical role in RA, since they degrade ECM to destroy the integrity of synovial, cartilage and bone tissue through selectively cleavage of many non-matrix components present in the extracellular environment, such as cell surface receptor, cytokines, chemokines, cell-cell adhesion molecules, clotting factors and other proteinases like binding proteins to involve in inflammation and immunity in RA [167,168]. The key role of MMPs in immune cell development, function and inflammatory response turned them as attractive target therapy in RA disease [169]. Tissue inhibitors of metalloproteinases (TIMPs) are produced by all connective tissue cells and are firmly and irreversibly bound to the active MMPs and pro-MMPs to form a 1:1 non-selective complex to take part in ECM homeostasis [170]. These TIMPs control tissue breakdown by blocking the activities of MMPs and when imbalance occurs due to increased production of MMPs or a lack of TIMPs adequate regulation, the pathogenesis of RA is being initiated due to dysregulated tissue remodeling (the fine balance between ECM degradation and production) [171]. The ECM is typically composed of the interstitial connective tissue matrix and a basement membrane (BM). The major hydrolases responsible for tissue breakdown are MMP1, 8, 13 and membrane-type I metalloproteinase (MT1-MMP) and further MMP9 degrade type II collagen fragments to generate immunodominant epitope [172]. The MMPs 1 and 13 predominate in RA due to their capacity to restrict collagen degradation rate, while MMP13 is considered to have a dual role in RA since their capability of degrading aggrecan and proteoglycan, suggesting it has a dual role in ECM destruction in RA [173,174]. In RA mice model, during joint inflammation has been observed down-regulation of collagens (COL1A1, COL3A1 and COL5A1) accompanied by increase in metalloproteases (MMP3 and MMP11), and down-regulation of enzymes involved in matrix degradation, such as ADAMTS1 and ADAMTS2 (a disintegrin and metalloprotease with thrombospondin motifs) [175]. Similarly, in TNFα-treated decellularized chondrocyte matrix many different collagens (COL1A1, COL1A2, COL2A1, COL5A1, COL5A2, COL12A1, COL14A1), but also aggrecan and chondroadherin were strongly downregulated in the damaged matrix, while matrix-degrading enzymes including MMP2, -3, -13 and 19, as well as, ADAMTS5 and ADAMTSL4 were upregulated [176]. The MMPs also participate in bone destruction processes, cell migration and invasion, as well as, in cytokine and chemokine processing [177]. The MMPs participation in joint destruction has related through three mechanisms: 1) Adhesion to chondrocytes, leading to degradation of collagen and cartilage damage; 2) Provoking of imbalance in affected join homeostasis (through regulation of inflammatory cytokines and chemokines) and activation of inflammatory signaling pathways that promote osteoclast differentiation and bone resorption; 3) Promotion of cell migration and invasive angiogenesis [169]. The FLs are involved in both, synovial inflammation and bone erosion in RA through production of various factors such as TNFα, IL-1β, -6, -8, MMP-1, and MMP-13. Stimulated chondrocytes are able to express various proteases, including MMP-1, -2, -3, -7, -8, -9, -10, -13, -14, ADAM9, 10, 17 and ADAMTS4, all related to cartilage destruction [70,178]. Interleukin-1β is inducing a variety of MMPs, including MMP-1, -3, -8, -13, -14, -29 and activating osteoclasts to breakdown the cartilaginous matrix [168]. Bone destruction can be effectively ameliorated by inhibiting osteoclast differentiation and regulating multiple signaling pathways including osteoclast differentiation, IL-17, and TNF, as well as, MMP-9 and protein kinase B (AKT) signaling pathway [179]. In collagen induced arthritis (CIA) in mice bone destruction was correlated with the significantly elevated levels of MMP-2 and MMP-9 in serum, as well as, elevated levels of serum TNF-α, IL-6, and IL-1β [180]. In RA mice, the cartilage destruction process has been slowed down through downregulation of the expression of AKT1, VEGFA, IL-1β, IL-6, MMP-9, ICAM1, VCAM1, MMP-3, MMP-13 and TNF-α [181].
The destruction of the BM of synovial cells enable them to migrate through the tissue and permeate interior of the joint. It has been reported that MMP2 and MT1-MMP are essential for instigating invadopodia generation, cell migration and invasion [182]. In the CIA induced arthritis cartilage destruction and bone erosion was reduced by decreased protein levels of TNF-α , IL-1β, IL-6, iNOS, COX2, MMP-1, and MMP-3 in ankle joint tissue [183]. Inhibitors of epithelial-mesenchymal transition (EMT) significantly reduced the excessive proliferation, migration and invasive behavior of RA-FLs by reduction of MMP1, -3 and -13 secretion. In addition, the expression of N-Cadherin (an EMT marker) has been also reduced [184]. The RA-FLs migration and invasion mechanisms were significantly inhibited through inhibiting the classical TLR4-NF-κB inflammatory pathway and regulating the dynamic balance of MMP-2/TIMP-2, MMP-9/TIMP-1 [185]. Inhibition of the extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling pathways (ERK/JNK) significantly attenuated the migration and invasion n RA-FLs through Leukocyte Ig-like receptor A3 (LILRA3), which resulted in decreased expression of expression of IL-6, IL-8 and MMP3 [186]. Similarly, the decreased expression of MMP-1 and -3, as well as, the pro-inflammatory factors (IL-1 and IL-6) and the elevated level of the pro-apoptotic factor (Bax nd cleaved caspase 3) inhibited the RA-FLs invasion via inactivation of WNT/β-catenin signaling pathway [187].
2.3. The Role of Angiogenesis
An intensive and important process in early stage of RA is angiogenesis (the development of new blood vessels), which is regulated by many inducers and inhibitors along with the involvement of pro-angiogenic factors like acidic and basic fibroblast growth factors (FGFs), transforming growth factor (TGF)-β, angiopoietin, placental growth factor, and vascular endothelial growth factor (VEGF) [188,189]. A previously unknown mechanism based on inhibition of the angiogenic functional module of circHIPK3/miR-149-5p/FOXO1/VEGF, demonstrated to have a significant protective effect on RA-FLS and CIA synovium, thus confirming the cross-talk between circular RNAs (circRNAs) and RA [190]. A broad investigation in RA patients revealed extensive correlation between pro-inflammatory and proangiogenic profile of the disease activity. The most important angiogenic markers were angiopoietin-1, neuropilin-1, Tie-2, endostatin, platelet factor 4 (CXCL4), interleukin-8 (IL-8, CXCL8), vascular cell adhesion molecule 1 (VCAM-1), VEGF and placenta growth factor (PlGF) [191]. Another mechanism involved in RA angiogenesis suppression is the hypoxia inducible factor-1α/ vascular endothelial growth factor/ angiopoietin 2 (HIF-1α/VEGF/ANG2) axis [192]. Interestingly, the release of heat shock protein 70 kDa (HSP70) can induce angiogenesis. However, the inhibited proteins in the sphingosine Kinases 1/sphingosine-1-phosphate/sphingosine-1-phosphate receptors/Gα protein subunit (SphK1/S1P/S1PRs/Gαi) pathway prevent HSP70 release and therefore producing anti-angiogenic effect [193].
2.4. The Role of Free Radicals
Free radicals and especially reactive oxygen species (ROS) play vital roles in RA pathogenesis and are key modulators of join inflammation [194]. When ROS are over-generated and/or the antioxidant system is in imbalance, ROS and its related metabolites are accumulated excessively and cause oxidative stress. Hypoxia and oxidative stress might be important drivers of the inflammatory processes in arthritic joints. Excessive ROS accumulation might contribute to somatic mitochondrial DNA (mtDNA) alterations, including mtDNA mutations in synovial tissue and somatic cells of RA patients and increased mitochondrial mutagenesis associated with reduced oxygen tension (pO2) in synovial membranes, which induced a pro-inflammatory mitochondrial phenotype [195,196,197]. It has been reported that ROS production increased lipid peroxidation, protein oxidation and DNA damage in the peripheral blood of RA patients [198,199]. Some markers of DNA damage, such as 8-Hydroxyl-2-deoxyguanosine (8-oxodG) has been found in synovial fluids and blood of RA patients [200].
3. Current Rheumatoid Arthritis Drug Treatment
The treatment for rheumatoid arthritis is focused on controlling inflammation and relieving pain with the end goal being low disease activity or remission. To achieve this, the European League Against Rheumatism (EULAR) has listed 10 recommendations for the management of RA with disease-modifying antirheumatic drugs (DMARDs). Furthermore, based on those recommendations an initiative called treat-to-target (T2T) was developed with the idea that RA treatment shouldn’t be universal but rather individual for each patient [117,201].
The overarching principle of the T2T approach is the shared decision making between patient and rheumatologist on the next step in the treatment plan based on the efficacy of previous treatments and the treatment goals. Creating an individual therapeutic approach follows an algorithm adapted from the 2016 update on the EULAR recommendations. The treatment guideline is divided into 3 phases with each phase focusing on a different class of DMARDs based on the stage of the disease upon first diagnosis. Failure to achieve the treatment goal within 6 months of starting the therapy moves the patient to the next phase [202].
3.1. Conventional Synthetic DMARDs (csDMARDs)
Glucocorticoids (GCs) are the most widely used anti-inflammatory drugs in the field of rheumatology. Intra-articular injections are used to treat acute disease flares, but this type of treatment has been associated with severe adverse reactions. To prevent this, low-dose oral GCs should be used only for short-term pain relief [203]. For long-term control of the inflammation, csDMARDs treatment should be started as soon as rheumatoid arthritis is diagnosed.
Methotrexate
Methotrexate (MTX, Trexall) is usually the first choice for RA treatment as it has been proven to be effective as low-dose monotherapy. Oral administration is usually starts at a dose of 15 mg/week but can be increased up to 25 mg. The mechanism of action of low-dose MTX is thought to be the inhibition of aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase (ATIC). AICAR accumulates in the cell which leads to increased adenosine release (adenosine is a potent immunosuppressant) [204,205]. Common side effects include hematologic abnormalities, gastrointestinal problems, elevated liver enzymes, fatigue and nausea [206].
Leflunomide
Leflunomide (Arava) is administered orally 50 mg/week. Its effect is lymphocyte-specific immunomodulation. Leflunomide inhibits the mitochondrial enzyme dihydroorotate dehydrogenase, which plays a key role in the de novo synthesis of the pyrimidine ribonucleotides which in term prevents clonal expansion of activated lymphocytes [207]. The most common adverse effects are diarrhoea, elevated liver enzymes, rashes, and hypertension [208].
Sulfasalazine
Sulfasalazine (SSZ, Azulfidine) was designed specifically for the treatment of rheumatoid arthritis. It combines an antibiotic, sulphapyridine, with an anti-inflammatory, salicylic acid, linked via an azo bond. The treatment of RA with SSZ is effective with the main effects being on the gut microbiota and inflammatory cell functions although the exact mechanism of action is not completely understood. Adverse reactions include gastrointestinal problems, rashes and nausea [209].
3.2. Biologic DMARDs (bDMARDs)
If the patient continues to display moderate disease activity without poor prognostic markers the therapy should be adjusted to a combination of csDMARDs instead of monotherapy. At the appearance of poor prognostic markers and failure to achieve low disease activity due to csDMARDs’ lack of efficacy and/or toxicity the therapy should move forward to phase Ⅱ – using biologic DMARDs. Those agents are highly specific and target pathways of the immune system so screening, and treatment of any latent infections is highly advised before starting any bDMARDs [210].
Tumor Necrosis Factor-alpha inhibitors (TNFi)
TNF-α is a cytokine with both pro-inflammatory and immunoregulatory functions. In the context of rheumatoid arthritis, dysregulated TNF-α was the first cytokine to be proven to directly cause tissue destruction as well as lead to the overproduction of other pro-inflammatory cytokines. Hence, blocking TNF-α signalling is one of the most effective ways to slow down the disease progression [211,212].
Since 2000, five TNFi are available. Each antibody has different molecular structure, administration and dose but all have the same effect – binding soluble and/or membrane-bound TNF-α. This can be summarized in Table 1.
Interleukin-1 inhibitor
Anakinra (Kineret) is a recombinant human IL-1 receptor antagonist administered subcutaneously 100 mg once a day. Studies on the effectiveness of Anakinra suggest that it might be dependent on the cytokine profile of the patient and that the drug works better in combination with other DMARDs [214,215].
Interleukin-6 receptor inhibitor
Besides TNF-α, IL-6 is the other cytokine proven to have a key role in the pathogenesis of rheumatoid arthritis. Tocilizumab (TCZ, Actemra) is a humanized recombinant IgG monoclonal antibody that binds to the soluble and membrane-bound IL-6 receptor. It’s injected intravenously 8 mg/kg every 4 weeks. Studies have shown that Tocilizumab is effective and safe as monotherapy or in combination with other DMARDs with both short-term and long-term effects regardless of the stage of the disease [216,217].
Anti-CD20 antibody
CD20 is a membrane-bound molecule on the surface of B-cells, which plays a role in their development and differentiation into plasma cells. Rituximab (RTX, Rituxan) is a chimeric monoclonal antibody originally created to bind to the CD20 molecule on the surface of peripheral B-cells in blood cancer patients but since then has been proven effective for treating RA by depleting B-cells in the synovium as well. Administration is intravenous, two 1000 mg doses 2 weeks apart [218,219].
3.3. Mesenchymal Stem Cells (MSCs)
Mesenchymal stem cells, also known as multipotent mesenchymal stromal cells, are a heterogeneous group of cells with a plastic-adherent, fibroblast-like morphology. In vitro they can differentiate into osteocytes, chondrocytes, and adipocytes. According to the criteria established by the International Society for Cellular Therapy (ISCT), MSCs are identified based on the expression of surface markers such as CD73, CD90, CD105, CD44, CD71, and CD106, while lacking markers associated with hematopoietic and endothelial cells, including CD34, CD45, CD11b, CD14, and CD31 [220,221].
The MSCs’ main role in physiology is supporting the function and maintenance of hematopoietic stem cells (HSCs) within the bone marrow. From MSCs, the structural elements of the specialized HSC microenvironment are formed; those are referred to as the "niche," which is essential for regulating HSC behaviour, including their self-renewal, differentiation, and survival. Through direct cell-to-cell interactions and the secretion of various cytokines, growth factors, and extracellular matrix components MSCs modulate the balance between hematopoietic stem cell quiescence and activation, ensuring a steady supply of blood cells while preventing exhaustion of the stem cell pool [222,223].
The MSCs’ therapeutic potential in autoimmune diseases and inflammatory disorders is due to their critical role in suppressing T-cell proliferation. Several mechanisms for MSC-mediated inhibition of proliferation have been described, primarily secreting immunosuppressive cytokines like TGF-β, IL-10, which induce a state of energy or functional inactivity in T cells. In addition, MSCs express surface molecules such as PD-L1, which bind PD-1 receptors on T cells, promoting T-cell apoptosis [224,225]. Lastly, it has been described that MSCs also influence the polarization of T cells, promoting the expansion of regulatory T cells, which further supress the immune response and maintain immune tolerance [226].
Preclinical studies in animal models of rheumatoid arthritis have demonstrated that MSCs can effectively reduce inflammation, inhibit immune activation, and promote tissue repair in damaged joints. MSCs migrate to inflamed tissues where they secrete both anti-inflammatory cytokines, such as TGF-β and IL-10, and pro-inflammatory cytokines, such as TNF-α and IL-6. In models with acute joint damage, MSCs have been shown to differentiate into chondrocytes and osteoblasts, restoring the structural integrity to the affected joints, decreasing bone erosion, and reducing cartilage degeneration [227,228].
A key focus of all preclinical studies is to understand the mechanisms behind MSC homing in inflamed tissues and their interaction with the immune system. Research has been done to identify factors that influence the effectiveness of MSC therapy, including the source of MSCs (e.g., bone marrow, adipose tissue, or umbilical cord), culture conditions, and delivery methods (e.g., intra-articular injection or systemic administration) [229].
4. In Vitro Studies Using Plant-Derived Natural Products for the Management of Rheumatoid Arthritis and Signaling Pathways
Multiple transcription factors and signaling pathways are involved in the pathogenesis of RA, the most important and key pathways are the MAPK (mitogen-activated protein kinase), NF-κB (nuclear factor kappa B), PI3/AKT (phosphatidylinositol 3 kinase-AKT, also known as PKB), JAK/STAT (Janus-activated kinase signal transduction and activator of transcription), Wnt/β-catenin (Wingless/Integrated), SYK/BTK (spleen tyrosine kinase)/Bruton’s tyrosine kinase), and Notch [16,17]. The major signaling pathways and their possible modulation by plant-derived molecules has been presented on Figure 3.
The NF-κB signaling pathway controls many biological processes, mainly inflammation that is associated with RA. Inflammation has been intensively observed in the early and late stage of RA and is triggered by NF-κB activation, in both T cells and antigens- presenting cells directly or indirectly by extracellular and/or intracellular stimuli (e.g. IL-1β, -6, TNF-α, MMPs etc.). The NF-κB signaling may be activated by two different ways: direct activation (canonical and noncanonical pathways) mediated by inhibitor of kappa B (IkB) kinase (IKK) and NF-kB-inducing kinase (NIK), respectively and indirect, which is interconnected with other cellular pathways, including MAPK, Rho, and phosphoinositide3-kinase (PI3-K) [24]. The NF-kB regulates more than 150 genes involved in anti-apoptosis, cell proliferation, inflammation and plays a key role in regulating the activation, survival, and differentiation of innate and adaptive immune cells. In RA pathogenesis, dysregulated NF-kB signaling contributes to activation of both immune and non-immune cells through transcriptional regulation of inflammatory mediators, including TNF-α, IL-1, -2, -6, -8, -9, -12, -18, -23, GM-CSF, VEGF, RANKL, MCP-1, MIP-2, CXCL1, CXCL10, RANTES, ICAM-1, VCAM-1, MMPs, and COX-2 [24]. On the other hand, inflammatory cytokines also modulate NF-kB through positive feedback, forming a vicious loop, which intensifies RA development [17]. At the same time, excessive NF-κB activation induces apoptosis of abnormal FLS cells in RA, which further accumulates in joint tissues and debris adheres to cartilage and bone, exacerbating the articular cartilage and bone destruction [17]. Along with that NF-κB dysregulation activates different types of T cells and also differentiate Th1 and Th17 cells by inducing IL-12 production and promotes IL-17 synthesis in Th17 cells, and thereby recruiting neutrophils and monocytes to sites of inflammation. Th17 cells contribute to inflammation by regulating expression of TNF, IL-1b, IL17, IL-21, and IL-22 [24]. The SYK (spleen tyrosine kinase) is a central molecule of B-cell receptor signaling and the level of phosphorylated SYK in peripheral blood B cells of RA patients has been dramatically increased. Strong positive autoantibodies against citrullinated peptides has been also established [230]. Relevant examples of plant derived molecules demonstrating their effect in in vitro RA models have been presented in Table 2.
The 70% ethanol extract of Periploca forrestii Schltr. rich of flavonoids revealed notable anti-RA activity. At concentrations from 25 to 500 ng/mL the extract mitigated the cell injury, reduced cell apoptosis and inflammation through reduced mRNA expression of COX-2, iNOS, IL-6 and IL-1β in TNF-α stimulated L929, HEK293T and MH7A human RA-FLS by inhibiting the activation of NF-κB signaling [110]. The sesquiterpene lactones-enriched fraction from Xanthium mongolicum Kitag exhibited the strongest anti-RA, which dose dependently (from 1 to 640 μg/mL) decreased the expression of M1-related genes IL-6, -1β, TNF-α, -12b and iNOS through suppression of NF-kB signaling [111]. The ethanol extract of Achyranthes aspera L (doses from 100 to 300 µg/mL) revealed its potential as natural anti-inflammatory remedy in treatment of RA by downregulation of the mRNA expression levels of inflammatory genes, blocking the NF-kB promoter activity induced by TNF-α and inactivation of two upstream signaling molecules, such as Src and Syk kinases [246]. The mechanism of anti-RA activity of Siweixizangmaoru decoction (dose from 12.5 to 400 µg/mL) from Tibet revealed to include modulation of TNF-α, IL-1β, -6, -4, and -10, MMP-2, -3, -9, and MMP-13 through suppression of JAK2/STAT3 and NF-κB signaling [112]. The Chinese herb Kadsura coccinea (Lem.) A. C. Smith also revealed its anti-RA activity through inhibition of NF-κB and STAT1 signaling [113], while Nyctanthes arbortristis L from India reduced the production of various inflammatory mediators and factors in vitro, such as NO, ROS, iNOS, COX-2, TNF-α, IL-6 and -1β by inhibiting the activation of NF-kB [247]. Investigating the effect of Geranium Wilfordii Maxim. in MH7A cells was established that its anti-RA activity was due to modulation of the expression of Bax, Bcl-2, IL-6, -8, MMP-1, MMP-2, -3, -9, COX-2, and iNOS through inhibition of NF-kB and MAPK pathways [238]. The JAK/STAT pathway is another crucial signaling pathway deregulated in RA and governing cell differentiation, proliferation, with special emphasis of inflammation and immune functions [24].
The JAK family has four members, JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), while the STAT family of TFs consists of seven members, namely STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. Upon receptor ligation, JAKs are autophosphorylated, and recruit and phosphorylate members of the STAT family and many of the proinflammatory cytokines, such as TNF-α, IL-1β, -6, -7, -8, -12, -15, -17, -23, -32, IFN-γ and GM-CSF, that are highly expressed in RA are known to be regulated by JAK/STAT signaling pathway [17,24].
Tinospora cordifolia (Thunb.) Miers extract, rich of polyphenols effectively downregulated the level of pro-inflammatory mediators (IL-6, TNF-α, PGE2, COX-2, iNOs) and angiogenic factor VGEF through targeted the upstream kinases of the JAK/STAT pathway [248]. The acidified methanol extract of Pennisetum glaucum (L.) R.Br., rich of polyphenols demonstrated strong anti-RA activity in vitro by inhibiting MMP-9 and PTGS2 via suppression of JAK2 signaling pathway [249]. The water extract of Pueraria montana (Lour.) Merr. suppressed the inflammation, migration and invasion in human rheumatoid fibroblast-like synoviocyte line, MH7A through modulating the expression of Bcl-2, Cas-3, Cas-9, MMP-1, -9, IL-6, -8, -1β, TNF-α and SOCS1 [116]. In single osteoclasts cell culture, the classic Chinese herbal compound Yi Shen Juan Bi Pill relieved the symptoms of RA through regulating the bone immune microenvironment via JAK2/STAT3. In vitro, this compound decreased the number of TRAP+ cells, the areas of bone resorption and inhibited the expression of RANK, NFATc1, c-fos, JAK2, and STAT3, while promoting the expression of IL-10 [250]. Similarly, another traditional Tibetan medicine (Shi-Wei-Ru-Xiang pills). In a co-culture system of IL-1β-stimulated synoviocytes or chondrocytes exhibited inhibition of chondrocytes apoptosis, associated with attenuation of inflammation by inhibition of the phosphorylation of p38, Erk1/2, and STAT3 [123]. Jolkinolide B (an ent-abietane-type diterpenoid found in Euphorbia plants) revealed its anti-RA potential at 1 µM concentration through suppression of TNF-α and IL-6 by decreasing the protein expression level of JAK2/STAT3 pathway in vitro [251].

Table 2.
Plant-derived molecules targeting the major cytokines, TFs and signaling pathways in RA directly or indirectly in vitro.
Table 2.
Plant-derived molecules targeting the major cytokines, TFs and signaling pathways in RA directly or indirectly in vitro.
| Molecule | Dose, µM | Cell line | Targets | Main findings | Modulated pathway | Reference |
| Curcumin | 50 | MH7A | TNF-α, IL-6, IL-17 | Inhibition of migration, invasion and inflammation | PI3K/AKT | [231] |
| Emodin | 15 | L929 | IL-6, IL-1β, COX-2 | Inhibition of inflammation | NF-κB | [232] |
| Ginsenoside compound K | 30 | Isolated FLS | FLUT1, HK2, PKM1, PKM2 | Inhibition of glycolysis | NF-κB | [233] |
| Glytabastan B | 3 and 6 | SW982 | TNF-α, IL-6, IL-8, COX-2 and MMP-1 | Inhibition of inflammation and invasion | MAPK, PI3K/AKT, NF-κB | [234] |
| Isobavachalcone | 20 | MH7A | TNF-α, MAPK13, EGFR, PTGS2, MMP-3 | Inhibition of migration, invasion and inflammation | PI3K/AKT, JAK/STAT | [235] |
| Kaempferol | 10 | HFLS-RA | IL-1β, MMP-2, -9, N-cadherin, vimentin | Inhibition of inflammation and abnormal proliferation | MAPK | [236] |
| Leocarpinolide B | 20 | SW982 | IL-6, IL-8, IL-1β, | Inhibition of proliferation, migration, invasion and inflammation | NF-κB | [237] |
| Magnoflorine | 10 | MH7A | iNOS, COX-2, IL-6, IL-8, MMP-1, -2, -3, -9 and -13 | Inhibition of proliferation, migration, invasion | PI3K/AKT, NF-κB, Nrf-2, | [238] |
| Nimbolide | 1 | HIG-82 | MMP-2, IL-6, iNOS, COX-2 | Reduction of inflammation | MAPK, NF-κB, Nrf-2 | [239] |
| Quercetin | 1.5 | L929, HEK293T, MH7A | COX-2, iNOS, IL-6, IL-1β | Reduction of cell apoptosis, improvement of cell injury | NF-κB | [110] |
| Sappanone A | 40 | HFLS-RA | TNF-α, IL-1β, IL-6, IL-10, IL-17A | Inhibition of inflammation | JAK2/STAT3, PI3K/AKT, NF-κB | [240] |
| Shikonin | 1x10-7 | MH7A | VEGF, VEGFR2, TNF-α, IL-1β, PDGF and TGF-β | Inhibition of migration, invasion and adhesion | MAPK (ERK1/2, JNK, and p38) | [241] |
| Scopoletin | 30 | HFLS-RA | IL-1β, TNF-α, MMP-3, MMP-9, COX-2, Bcl-2 | Inhibition of proliferation, migration and invasion | NF-κB | [242] |
| Suberosin | 5 | RA-FLS | IL-6, IL-1β, TNF-α, IL-8, MMP-1, MMP-3, MMP-9, MMP-13 | Inhibition of inflammation | JAK/STAT | [243] |
| Tectoridin | 50 | HFLS-RA | IL-1β, IL-6, COX-2, iNOS | Inhibition of inflammation | MAPK (ERK1/2, JNK, and p38) | [244] |
| Umbelliferone | 20 | HFLS-RA | IL-1β, TNF-α, MMP-3, MMP-9, COX-2, Bcl-2 | Inhibition of proliferation, migration and invasion | NF-κB | [242] |
| Wilforine | 0.4 | Isolated FLS | IL-1β, IL-6, TNF-α, CCND1, GSK-3β, and c-Myc, MMP-3 | Inhibition of inflammation and abnormal proliferation | Wnt11/β-catenin | [245] |
The PI3K/AKT pathway regulates proliferation, metabolism, angiogenesis, and cell survival and is correlated with the occurrence and development of RA [17]. This pathway participate in the abnormal proliferation of FLS cells and synovial inflammation by stimulating the expression of inflammatory molecules like IL-1β, -6, -17, -21, -22, and TNF-α [17]. Abnormal PI3K/AKT pathway activation stimulate the expression of VEGF and HIF-1α to promoting angiogenesis, which not only disturbs the nutrition processes in the synovium, but also promotes glycolysis [27] and release diverse inflammatory mediators [17]. The PI3K/AKT activates mammalian target of rapamycin (mTOR) and further inhibiting autophagy in FLS, promoting continuous abnormal proliferation of synovial cells, and is also critical for the survival and differentiation of osteoclasts, aggravating RA [17].
Hedyotis diffusa Willd modulated its anti-inflammatory targets (TNF-α, IL-6, IL-17 and IL-10) in vitro through suppression of PI3K/AKT signaling pathway, thus revealing its anti-RA capacity at varying concentration between 0.5 and 2.0 mg/mL [17]. The ethanolic extract of Ammopiptanthus nanus (M. Pop.) Cheng f. inhibited the pathways PI3K/AKT/NF-κB closely related to RA by suppressing the expression of inflammatory cytokines IL-1β, COX-2 and iNOS in vitro used in concentration between 1 to 30 µg/mL [252].
The MAPK signaling pathway plays a key role in the pathological process of RA in terms of regulation of various cellular activities, including gene expression, metabolism, migration, survival, cell cycle progression, apoptosis, and differentiation and its over activation is closely correlated to the articular cartilage destruction and inflammatory hyperplasia of the synovial tissues. P38 MAPK, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK) are the three main subfamilies of the MAPK pathway. The ERK1 and ERK2 are important for the regulation of cell differentiation, proliferation, and survival. On the other hand, the main effect of JNK MAPKs in RA is cartilage destruction mediated by MMPs. Similarly, P38 is linked to the inflammatory response in RA and activates many protein kinases and transcription factors that play key roles in the regulation of humoral and cellular autoimmune responses [17].
The dried roots of Lithospermum erythrorhizon Sieb containing shikonin as a major compound inhibited the migration, invasion and inflammation process related to RA in vitro through inhibition of angiogenic and inflammation mediators, such as VEGF, TNF-α and IL-1β [241]. The phenolic compound 5-hydroxyconiferaldehyde, isolated from the Campanula takesimana revealed its anti-RA potential by inhibiting the inflammatory response in vitro (inhibition of PGE2, iNOS, TNF-α, COX-2, IL-6, -1β) through suppression of several signaling pathways, such as MAPK, NF-κB, Nrf-2 [253]. The saponins rich fraction from Rhizoma Panacis Majoris exhibited its anti-RA potential through decreasing the expression of autophagy-related indicators (LC3II/LC3I, Beclin-1) and the corresponding signaling pathways, such as MAPK and PI3K/AKT [254].
The Wnt (Wingless/Integrated) signaling pathway takes part in a variety of pathological symptoms such as maintenance, differentiation, proliferation, and self-renewal in RA. The Wnt pathway also plays a key role in synovial inflammation and in the regulation of bone metabolism in RA [17]. A traditional Chinese Medicine Er Miao San (a mixture of Atractylodis Rhizoma and Phellodendri Cortex) revealed a promising anti-RA activity in vitro by decreasing angiogenesis and inflammatory microenvironment in MH7A cells by inhibiting the Wnt/β-catenin signaling [255]. The total saponins fraction of Radix clematidis have a pronounced anti-RA activity inhibiting the excessive proliferation of FLS during in vitro studies. At concentrations ranging from 0.5 to 1562.5 µg/mL inhibited c-Myc, cyclin D1, GSK-3β, and SFRP4 markers through Wnt7b/β-catenin [256].
The Notch signaling affects numerous processes of normal cell morphogenesis, including cell proliferation, the differentiation of pluripotent progenitors, apoptosis, and the formation of cell boundaries [17]. The Qi-Sai-Er-Sang-Dang-Song decoction (concentrations from 0.25 to 4%) inhibited the RA symptoms with focus on inflammation by inhibiting IL-6, -18 and -1β that were regulated by Notch/NF-κB axis [257]. In the range of 1 to 30 µM norisoboldine (an alkaloid compound isolated from Radix Linderae) inhibited synovial angiogenesis in vitro by decreasing VEGF expression and Notch1 signaling pathway [258].
5. In Vivo Studies Using Plant-Derived Natural Products for the Management of Rheumatoid Arthritis
Rheumatoid arthritis is a chronic autoimmune disorder characterized by various inflammatory processes causing pain, swelling, redness and malfunctioning of joints [259]. The ethiology of RA represents a complex combination of genetic and epigenetic predispositions as well as of environmental factors [260]. Despite the long list of medications for RA, most of them bring only partial relief to the patients and new therapeutic agents for treating the sources of disease and not just the symptoms are still needed [261,262].
The most frequently applied drugs for RA treatment are conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs), among which methotrexate (MTX) is the recommended first-line therapy for RA; biological DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs) [263]. The treatment with csDMARDs ideally comprise MTX plus low-dose glucocorticoids. However, response to MTX varies and only after 6 months of therapy, 43% of the patients are classed as non-responders to MTX [264]. The bDMARDs include targeting monoclonal antibodies against TNF-α, IL-6, soluble receptor for TNF and T-cell co-stimulation. The tsDMARDs are inhibitors of the Janus tyrosine kinase family (JAK), which targets intracellular signalling of type I and II cytokines [263]. In addition to poor response, many patients often experience adverse events (AEs) such as gastrointestinal AEs and/or elevated liver enzymes [264], liver fibrosis, myelosuppression, pneumotitis [265] and are at a higher chance of developing infections, tuberculosis and certain malignancies, such as lung, breast, skin cancer and lymphoma [266].
Therefore, there is not only a clinical need to identify patients at high risk of non-response to DMARDs, but also to identify novel effective RA therapeutics without any side effects. In this regard, natural products with anti-inflammatory activity represent promising adjuvant agents or alternatives to RA therapeutics [4].
Experimental therapy with newly developed drugs in humans is limited for technical and ethical reasons. In this case, the rodent models are very useful because of low cost, homogeneity of the genetic background, and ease of handling. Although the animal models mimic a part of a disease and never duplicates or reproduces the entire human pathology, they are very useful to test the hypothesis of the global effect of a molecule.
The animal models of RA can be classified into two broad categories of: (A) induced animal models for RA, (B) genetic models of RA. The induced models vary according to the used chemical agent with arthritogenic properties for instance, collagen, pristane, complete Freund’s adjuvant (CFA), Cartilage oligomeric matrix protein (COMP). In the case of the genetic models, the animals are either deficient in (knockout) or transgenic for a specific gene of interest [267]. The genetic models have a valuable role in the arthritis research. The information regarding the role of depleted or introduced particular gene, gives a valuable insight to the process of inflammation and regulation. These models serve as a tool to study the effect of therapeutics in mice prone to developing joint inflammation spontaneously and to understand varying manifestations of autoimmunity in general. In the present report, we summarized the widely used rodent models of RA for testing the therapeutic potential of the biologically-active compounds.
5.1. Collagen Induced Arthritis Model
Known as a gold standard of the in vivo models, the collagen induced arthritis (CIA) model sharing many similarities with the pathological and immunological features of the human RA. The clinical signs of this polyarthritis model are inflammation of the synovium, cartilage destruction and bone erosion [268]. The antibodies play an important role in the inflammatory phase of CIA. The immunization with heterologous type II collagen in complete Freund’s adjuvant (CFA) to genetically susceptible mice strains with MHC haplotypes H-2q or H-2r (DBA/1, B10.Q, and B10.RIII) leads to generation of autoantibodies against self-antigens and collagen [153]. It was also shown that C57BL/6 (B6; H-2b) [269] can also develop CIA with a high incidence (60–70%) and sustained severity dependently on B and CD4 + T cells. The susceptibility to CIA requires more than MHC class II haplotypes restriction but also TCR, and non-MHC susceptibility genes. This is also confirmed by the fact that the passive transfer of collagen type II specific T cells doesn`t induce severe inflammation compare to the transfer of collagen type II specific antibodies. The dominant pathological role of Th1 and Th17 cells is well known, but the antibodies against collagen type II seems to have primary role in the immunopathogenesis of this model. CIA is an important rodent model for the analysis of non-MHC genes and their role in RA development [153].
5.2. Collagen Antibody Induced Arthritis Model
The passive transfer of commercially available monoclonal antibody cocktail to collagen type II presents an alternative to the CIA, significantly reducing the study duration and cost while increasing disease induction and synchronicity among individual animals. This model can be induced in MHC haplotypes non- susceptible to CIA. Terato et al. [270] have shown that by i.v. injection of a combination of 4 different monoclonal antibodies can be induced antibody-mediated CIA. Further research by Nutty Nandakumar and Rikard Holmdahl [271] has improved the antibody cocktail by selecting epitope specific antibodies, which is critical for their pathogenicity. The combination of monoclonal antibody cocktail and LPS leads to the development of severe and persistent arthritis to nearly 100% of the mice. As advantages of the CAIA model can be pointed out: length of the development (typically within days instead of weeks as in the classic CIA model); synchronization between the animals into the group (the onset of the disease is 2 days after LPS injection); Susceptibility (not only CIA-susceptible mice strains, but also some CIA-resistant mice, such as Balb/c and C57BL/6) [272]. The pathology of this model and the induction of the inflammation is expressed in the formation of immune complexes with the collagen type II on the cartilage or synovium, and activation of the classical and alternative complement` pathways. Additionally, the antibody complexes may also activate the monocytes in the joint via Fc receptors, and the releasing of pro-inflammatory cytokines (i.e. TNF-a and IL-1) recruit neutrophils and macrophages [273]. This model of RA can be used to studying inflammatory mechanisms in arthritis and to screen candidate therapeutic agents.
5.3. Adjuvant Induced Arthritis Model
The adjuvant induced arthritis model (AIA) present an efficient way to enhance both the cell-mediated and humoral immune responses towards the antigens by emulsified them in Complete Freund’s adjuvant (CFA) [274]. AIA is severe, sub-chronic arthritis, characterized by persistent inflammation with numerous systemic alterations as synovial hyperplasia, cartilage and bone damage, as well as edema and deformation. The massive leucocyte infiltration leads to increased chemokine and cytokine levels (as IL-1 and TNF-α), and release of reactive oxygen species (ROS) [275]. AIA is suitable as a model for screening and testing anti-arthritic drugs. Its manifestations are very similar to those in the human RA [276].
5.4. Pristane Induced Arthritis Model
Pristane induced arthritis (PIA) is severe and chronic inflammatory model. The introduction of Pristane, a synthetic mineral oil (2,6,10,14-tetramethylpentadecane), induce an acute arthritic, massive formation of osteoclasts, bone erosion and new bone formation, and also an inflammatory cell infiltration. It`s characterized with prolonged and delayed clinical features (from weeks to months). Serological parameters as rheumatoid factor (RF), antibodies against heat shock proteins and collagen type I and II, and the elevated levels of cartilage oligomeric matrix protein can be analyzed. The development of PIA is joint-specifically regulated by T cells and by MHC genes and fulfill the clinical criteria for RA. This model is useful for validation of new drug candidates [276,277,278,279].
In the recent years, different herbal extracts have been reported to have beneficial effect on RA development in animal models of the disease. The rhizome of Acorus gramineus Sol. ex Aiton (a widely used plant in traditional Chinese medicine) has shown therapeutic effect in CIA mouse model of RA – it decreases IL-6 and TNF-α and ameliorates swelling of the hind limb [280]. The fruit peel extract of Annona squamosa L contains various immunomodulating chemical substances that have anti-RA functions - decreased leucocyte levels in the serum and decreased necrosis in the paws are observed after its administration in CFA-injected mice [281]. Another report pointed to the beneficial effect of Saururus chinensis (Lour.) Baill leaves extract on type II collagen-induced arthritis in mice [282]. Authors concluded that S. chinensis extract administration has positive effect on different RA manifestations – increased serum levels of IL-6 and TNF-alpha, swelling of hind limbs and accumulation of inflammatory cells in the synovial membrane. In their research, Kim et al. investigated the therapeutic potential in RA of extract mixture of two folk remedies - Cudrania tricuspidata (Carrière) Bureau and Stewartia koreana Maxim. The results showed that treatment with the extract mixture has strong anti-inflammatory effect as it reduced nitric oxide levels, tumor necrosis factor, IL-6 and IL-1 levels [283]. Another extract, rich in antioxidants, catechins and proanthocyanidins is the grape seed proanthocyanidin extract. This extract expresses therapeutic effect in CIA mouse model of RA by regulating the TLR4-MyD88-NF-κB signaling pathway which further resulted in improvement of clinical features and joint histology [284]. Thorn extract of Gleditsia sinensis in combination with Lactobacillus casei has anti-inflammatory effects in mouse model of type II collagen-induced RA [285]. The amelioration was observed in decrease in serum nitrite and total cholesterol as well as in pro-inflammatory cytokine (TNF-αand IL 6) levels. Another interesting observation is the report of Allam et al. concerning the therapeutic effect of the phenolic substance ellagic acid in CFA-injected mice [286]. Authors clearly demonstrated that treatment with ellagic acid attenuated paw swelling and bone dysfunction by modulating pro- and anti-inflammatory cytokines.
In addition, the beneficial effect of different plants has been reported and in rat models of RA. Wood bark extract from the fruit Durian (Durio zibethinus Murr.) has shown therapeutic effect in CFA injected-rat rats - improvement of hind limb swelling and of histopathological changes and suppression of iNOS expression due to the presence of various phenols, alkaloids, tannins, terpenes, saponins, and flavonoids in the extract [287]. Root and leaf extracts of Chloranthus serratus (Thunb.) Roem. & Schult. (a traditional Chinese medicine plant) also showed beneficial effect in RA treatment. The administration of extract in CFA-injected rats resulted in inhibition of secretion of pro-inflammatory cytokines [288]. Finally, ethyl-acetate extract from the root of Caragana sinica (Buc'hoz) Rehder (herbal product in traditional Chinese medicine) has been reported to have therapeutic efficacy for RA treatment in adjuvant-induced arthritis in rats. The administration of extract resulted in reduced paw swelling and protected bone structures due to negative regulation of the NF-κB pathway [289].
All these results pointed herbal extracts as promising therapeutic agents in experimental mouse and rat models of RA with unexploited potential for further clinical implications (Table 3).
6. Conclusions and Future Perspectives
Rheumatoid arthritis is a chronic autoimmune disorder characterized by inflammation of the joints, resulting in pain, joint swelling and finally joint damage and bone erosion. Profound insight into RA etiology reveals that many genetic, epigenetic changes, posttranslational modifications and environmental factors, including complex interactions at mucosal levels between microbiome and host immune cells provoke the pathogenesis and progression of RA. In spite of the numerous drugs and novel biological therapies, they have several limitations and drawbacks, including disease recurrence and adverse effects due to long-term use. Rheumatoid arthritis has turned from a highly disabling disease for which no effective remedies existed to a disorder that can be controlled well, with many patients reaching remission. However, still many unmet needs remain, since not all patients reach sustained clinical remission and even about 25% still suffer from moderate or even high disease activity; even some patients still do not reach low disease activity; great similarity of response rates to the various targeted therapies has been frequently observed. Therefore, it is still needed to uncover the cause(s) for RA in order to assure cure or at least prevention; novel therapies need to be established; different and yet unidentified signaling pathways in non-responders need to be identified.
Currently, although still underestimated plant-based natural extracts and molecules, including flavonoids, terpenoids, alkaloids and many others possessing anti-inflammatory, immunomodulatory, anti-oxidant and anti-apoptotic activity have demonstrated excellent curative effect on autoimmune diseases, including RA and even some of them have already been used in treating RA patients. Major challenges using medicinal plants are their complexity related to identification of the bioactive components, understanding the mechanisms of the phytopharmaceutical activity and their poor availability. The inclusion of plant-derived extracts and molecules into novel delivery systems, such as nanoparticles improves their pharmacological and therapeutic properties. Nanoencapsulation seems to be a promising strategy to enhance the permeability, solubility of the molecules protecting them from degradation and improving their bioavailability. Therefore, this encapsulation allows precise dosage, prolonged and controlled release of the phytochemicals, as well as, reducing dosage regimen and drug toxicity, and improving patient compliance. The use of nanocariers offer an innovative platform for the topical delivery of plant-derived molecules in the treatment of RA. Using different “omics” technologies, such metabolomics, as well as, 2- and 3-D in vitro models and different panels of in vivo models is also a possibility to overcome some of the challenges associated using plant-derived products and finally decrease the failure of potential new therapies in clinical trials. Combining different 3D tissue models with microfluidic devices could be the next generation in vitro approach to study the complex cross talk between tissues/organs and immune system, including the spreading of (auto)immune reactions across different organs. Although, the broad diversity of conventional RA mice models, they are partially suited to preclinical testing of cell-based therapies. For example, spontaneous mice models are suitable to study autoimmunity, while induced models are more focused on the effector phases of immune-mediated arthritis. For that reason, the translational research in humanized mice models that accurately mirrors autoimmune processes of human RA and permits its modulation by the transfer of human immunoregulatory cells are expected to be a powerful tool for preclinical evaluation of cell-based immunotherapies. Human-based approach will provide opportunities to identify objective patient-related biomarkers to elucidate disease subtypes and treatment response, but will also enable strategies for the management of patients who are ‘refractory’ or resistant to available treatments.
As future outlook, plant-derived products also contain prebiotic components, whose interaction with the host microbiome might have a significant impact on health and disease. A future task for researchers in the field will be to identify how these parameters interact to trigger an autoimmune inflammatory response and the development of RA and would further help to optimize the selection of plant-derived products for therapy and define their mechanisms of activity. In the near future additional research efforts might be invested in combining phytotherapy with stem-cell research, clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-Cas9) genome editing or gene therapy that would provide a long-term therapeutic advantage for RA patients following evaluation of safety, ethical, and medical concerns.
Author Contributions
Conceptualization, A.S.M., N.M.M., K.A.N.; writing – original draft preparation – A.S.M., N.M.M., K.A.N., L.A.K, K.I.I., A.S.Z., L.K., N.L.P. and M.M.I.; writing – reviewer and editing – A.S.M., N.M.M., K.A.N.; Supervision, A.S.M. and N.M.M.; project administration, A.S.M; funding, A.S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the Bulgarian National Science Fund (contract number КП-06-Н61/4) and from Bulgaria’s Recovery and Resilience Plan, a part of European Union’s NextGenerationEU instrument for the implementation of an investment under C2I2 "Enhancing the innovation capacity of the Bulgarian Academy of Sciences (BAS) in the field of green and digital technologies" (contract number BG-RRP-2.017-0046-C01).
Data Availability Statement
Not applicable.
Acknowledgements
The authors are thankful to: Senior Assist. Prof. Aleksandar K. Slavov, Ph.D. (Department of Ecological Engineering, University of Food Technologies Plovdiv) for the courtesy provision of all figures used in the paper and Dr. Krasimir Kosev (Director of University Botanic Gardens, Sofia University "St. Kliment Ohridski", Sofia, Bulgaria) for providing biological material from plants of the genus Lespedeza.
Abbreviations
| ACPAs | Anti-citrullinated protein antibodies |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin Motifs |
| Anti-CarP | Anti-carbamylated protein antibodies |
| CCR6 | Chemokine receptor 6 |
| cDMARDs | Conventional disease modifying anti-rheumatic drugs |
| COX-2 | Cyclooxygenase-2 |
| CRP | C-creative protein |
| CTLA-4 | T-lymphocyte associated protein 4 |
| CXCL12 | Chemokine C-X-C motif ligand 12 |
| DALYs | Disability-adjusted life-years |
| DNMT | DNA methyltransferases |
| ECM | Extracellular matrix |
| ESR | Erythrocyte sedimentation rate |
| FGFs | Fibroblast growth factors |
| FLS | Fibroblast-like synoviocytes |
| GC | Glucocorticoids |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HDAC | Histone deacetylases |
| HLA-DR | Human leukocyte antigen D-related |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| IRF-5 | Interferon regulatory factor 5 |
| JAK/STAT | Janus-activated kinase signal transduction and activator of transcription |
| MAPK | Mitogen-activated protein kinase |
| M-CSF | Macrophage colony-stimulating factor |
| MDC | Myeloid dendritic cells |
| MHC | Major histocompatibility complex |
| MMPs | Matrix metalloproteinases |
| MSCs | Mesenchymal stem cells |
| MTX | Methotrexate |
| NF-κB | Nuclear factor kappa B |
| NF-κβ | Nuclear factor kappa-B |
| NO | Nitric oxide |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| OPG | Osteoprotegerin |
| PADI4 | Peptidyl arginine deiminase, type IV enzyme |
| PBMCs | Peripheral blood mononuclear cells |
| PDC | Plasmacytoid dendritic cells |
| PI3/AKT | Phosphatidylinositol 3 kinase-AKT |
| PlGF | Placenta growth factor |
| RA | Rheumatoid arthritis |
| RANKL | Receptor activator of nuclear factor-B ligand |
| RF | Rheumatoid factor |
| ROS | Reactive oxygen species |
| SAM | S-adenosine methionine |
| SLE | Systemic lupus erythematosus |
| SNPs | Single nucleotide polymorphisms |
| SYK/BTK | Spleen tyrosine kinase)/Bruton’s tyrosine kinase |
| TBX5 | T-box transcription factor 5 |
| Tfh | T follicular helper |
| TGF-β | Transforming growth factor beta |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TNF-α | Tumor necrosis factor alpha |
| TRAF1 | Tumors necrosis factor-receptor associated factor 1 |
| Treg | Regulatory T cells |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| Wnt/β-catenin | Wingless/Integrated |
References
- Damerau, A.; Gaber, T. Modeling Rheumatoid Arthritis In Vitro: From Experimental Feasibility to Physiological Proximity. IJMS 2020, 21, 7916. [CrossRef]
- Kour, G.; Choudhary, R.; Anjum, S.; Bhagat, A.; Bajaj, B.K.; Ahmed, Z. Phytochemicals Targeting JAK/STAT Pathway in the Treatment of Rheumatoid Arthritis: Is There a Future? Biochemical Pharmacology 2022, 197, 114929. [CrossRef]
- Hosseinikhah, S.; Barani, M.; Rahdar, A.; Madry, H.; Arshad, R.; Mohammadzadeh, V.; Cucchiarini, M. Nanomaterials for the Diagnosis and Treatment of Inflammatory Arthritis. IJMS 2021, 22, 3092. [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat Rev Dis Primers 2018, 4, 18001. [CrossRef]
- Jannat, A.; John, P.; Bhatti, A.; Hayat, M.Q. Tomorou Attenuates Progression of Rheumatoid Arthritis through Alteration in ULK-1 Independent Autophagy Pathway in Collagen Induced Arthritis Mice Model. Cell Death Discov. 2019, 5, 142. [CrossRef]
- Kour, G.; Haq, S.A.; Bajaj, B.K.; Gupta, P.N.; Ahmed, Z. Phytochemical Add-on Therapy to DMARDs Therapy in Rheumatoid Arthritis: In Vitro and in Vivo Bases, Clinical Evidence and Future Trends. Pharmacological Research 2021, 169, 105618. [CrossRef]
- Li, P.; Wang, C.; Huo, H.; Xu, C.; Sun, H.; Wang, X.; Wang, L.; Li, L. Prodrug-Based Nanomedicines for Rheumatoid Arthritis. Discover Nano 2024, 19, 9. [CrossRef]
- Dudics, S.; Langan, D.; Meka, R.R.; Venkatesha, S.H.; Berman, B.M.; Che, C.-T.; Moudgil, K.D. Natural Products for the Treatment of Autoimmune Arthritis: Their Mechanisms of Action, Targeted Delivery, and Interplay with the Host Microbiome. IJMS 2018, 19, 2508. [CrossRef]
- Juarez, M.; Bang, H.; Hammar, F.; Reimer, U.; Dyke, B.; Sahbudin, I.; Buckley, C.D.; Fisher, B.; Filer, A.; Raza, K. Identification of Novel Antiacetylated Vimentin Antibodies in Patients with Early Inflammatory Arthritis. Annals of the Rheumatic Diseases 2016, 75, 1099–1107. [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid Arthritis. The Lancet 2010, 376, 1094–1108. [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; Van Der Linden, M.P.; Janssen, G.M.C.; Van Veelen, P.A.; Levarht, N.E.W.; Van Der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17372–17377. [CrossRef]
- Gandhi, G.R.; Jothi, G.; Mohana, T.; Vasconcelos, A.B.S.; Montalvão, M.M.; Hariharan, G.; Sridharan, G.; Kumar, P.M.; Gurgel, R.Q.; Li, H.-B.; et al. Anti-Inflammatory Natural Products as Potential Therapeutic Agents of Rheumatoid Arthritis: A Systematic Review. Phytomedicine 2021, 93, 153766. [CrossRef]
- Han, Y.; Huang, S. Nanomedicine Is More than a Supporting Role in Rheumatoid Arthritis Therapy. Journal of Controlled Release 2023, 356, 142–161. [CrossRef]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.-C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global Epidemiology of Rheumatoid Arthritis. Nat Rev Rheumatol 2022. [CrossRef]
- Black, R.J.; Cross, M.; Haile, L.M.; Culbreth, G.T.; Steinmetz, J.D.; Hagins, H.; Kopec, J.A.; Brooks, P.M.; Woolf, A.D.; Ong, K.L.; et al. Global, Regional, and National Burden of Rheumatoid Arthritis, 1990–2020, and Projections to 2050: A Systematic Analysis of the Global Burden of Disease Study 2021. The Lancet Rheumatology 2023, 5, e594–e610. [CrossRef]
- Bahuguna, R.; Awasthi, R. Unlocking New Dimensions in Rheumatoid Arthritis Therapy: Harnessing the Power of Lipid Based Vesicles beyond Traditional Therapies. Journal of Drug Delivery Science and Technology 2023, 89, 105106. [CrossRef]
- Ding, Q.; Hu, W.; Wang, R.; Yang, Q.; Zhu, M.; Li, M.; Cai, J.; Rose, P.; Mao, J.; Zhu, Y.Z. Signaling Pathways in Rheumatoid Arthritis: Implications for Targeted Therapy. Sig Transduct Target Ther 2023, 8, 68. [CrossRef]
- Galloway, J.; Capron, J.-P.; De Leonardis, F.; Fakhouri, W.; Rose, A.; Kouris, I.; Burke, T. The Impact of Disease Severity and Duration on Cost, Early Retirement and Ability to Work in Rheumatoid Arthritis in Europe: An Economic Modelling Study. Rheumatology Advances in Practice 2020, 4, rkaa041. [CrossRef]
- Fischer, B.D.; Adeyemo, A.; O’Leary, M.E.; Bottaro, A. Animal Models of Rheumatoid Pain: Experimental Systems and Insights. Arthritis Res Ther 2017, 19, 146. [CrossRef]
- Huang, Y.; Li, J.; Agarwal, S.K. Economic and Humanistic Burden of Rheumatoid Arthritis: Results From the US National Survey Data 2018–2020. ACR Open Rheumatology 2024, 6, 746–754. [CrossRef]
- Akram, M.; Daniyal, M.; Sultana, S.; Owais, A.; Akhtar, N.; Zahid, R.; Said, F.; Bouyahya, A.; Ponomarev, E.; Ali Shariat, M.; et al. Traditional and Modern Management Strategies for Rheumatoid Arthritis. Clinica Chimica Acta 2021, 512, 142–155. [CrossRef]
- Patidar, V.; Shah, S.; Kumar, R.; Singh, P.K.; Singh, S.B.; Khatri, D.K. A Molecular Insight of Inflammatory Cascades in Rheumatoid Arthritis and Anti-Arthritic Potential of Phytoconstituents. Mol Biol Rep 2022, 49, 2375–2391. [CrossRef]
- Liu, X.; Tao, T.; Yao, H.; Zheng, H.; Wang, F.; Gao, Y. Mechanism of Action of Quercetin in Rheumatoid Arthritis Models: Meta-Analysis and Systematic Review of Animal Studies. Inflammopharmacol 2023, 31, 1629–1645. [CrossRef]
- Balendran, T.; Lim, K.; Hamilton, J.A.; Achuthan, A.A. Targeting Transcription Factors for Therapeutic Benefit in Rheumatoid Arthritis. Front. Immunol. 2023, 14, 1196931. [CrossRef]
- Miao, C.; Bai, L.; Yang, Y.; Huang, J. Dysregulation of lncRNAs in Rheumatoid Arthritis: Biomarkers, Pathogenesis and Potential Therapeutic Targets. Front. Pharmacol. 2021, 12, 652751. [CrossRef]
- George, G.; Shyni, G.L.; Raghu, K.G. Current and Novel Therapeutic Targets in the Treatment of Rheumatoid Arthritis. Inflammopharmacol 2020, 28, 1457–1476. [CrossRef]
- Luo, T.-T.; Wu, Y.-J.; Yin, Q.; Chen, W.-G.; Zuo, J. The Involvement of Glucose and Lipid Metabolism Alteration in Rheumatoid Arthritis and Its Clinical Implication. JIR 2023, Volume 16, 1837–1852. [CrossRef]
- Németh, T.; Nagy, G.; Pap, T. Synovial Fibroblasts as Potential Drug Targets in Rheumatoid Arthritis, Where Do We Stand and Where Shall We Go? Annals of the Rheumatic Diseases 2022, 81, 1055–1064. [CrossRef]
- Mueller, A.-L.; Payandeh, Z.; Mohammadkhani, N.; Mubarak, S.M.H.; Zakeri, A.; Alagheband Bahrami, A.; Brockmueller, A.; Shakibaei, M. Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies. Cells 2021, 10, 3017. [CrossRef]
- Sharma, S.D.; Leung, S.H.; Viatte, S. Genetics of Rheumatoid Arthritis. Best Practice & Research Clinical Rheumatology 2024, 38, 101968. [CrossRef]
- Källberg, H.; Padyukov, L.; Plenge, R.M.; Rönnelid, J.; Gregersen, P.K.; Van Der Helm-van Mil, A.H.M.; Toes, R.E.M.; Huizinga, T.W.; Klareskog, L.; Alfredsson, L. Gene-Gene and Gene-Environment Interactions Involving HLA-DRB1, PTPN22, and Smoking in Two Subsets of Rheumatoid Arthritis. The American Journal of Human Genetics 2007, 80, 867–875. [CrossRef]
- Kokkonen, H.; Johansson, M.; Innala, L.; Jidell, E.; Rantapää-Dahlqvist, S. The PTPN221858C/T Polymorphism Is Associated with Anti-Cyclic Citrullinated Peptide Antibody-Positive Early Rheumatoid Arthritis in Northern Sweden. Arthritis Res Ther 2007, 9, R56. [CrossRef]
- Van Der Helm-van Mil, A.H.M.; Verpoort, K.N.; Breedveld, F.C.; Huizinga, T.W.J.; Toes, R.E.M.; De Vries, R.R.P. The HLA–DRB1 Shared Epitope Alleles Are Primarily a Risk Factor for Anti–Cyclic Citrullinated Peptide Antibodies and Are Not an Independent Risk Factor for Development of Rheumatoid Arthritis. Arthritis & Rheumatism 2006, 54, 1117–1121. [CrossRef]
- Zhang, X.; Li, W.; Zhang, X.; Zhang, X.; Jiang, L.; Guo, Y.; Wang, X. Association between Polymorphism in TRAF1/C5 Gene and Risk of Rheumatoid Arthritis: A Meta-Analysis. Mol Biol Rep 2014, 41, 317–324. [CrossRef]
- Sigurdsson, S.; Padyukov, L.; Kurreeman, F.A.S.; Liljedahl, U.; Wiman, A.; Alfredsson, L.; Toes, R.; Rönnelid, J.; Klareskog, L.; Huizinga, T.W.J.; et al. Association of a Haplotype in the Promoter Region of the Interferon Regulatory Factor 5 Gene with Rheumatoid Arthritis. Arthritis & Rheumatism 2007, 56, 2202–2210. [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and Genetics of Rheumatoid Arthritis. Arthritis Res 2002, 4, S265. [CrossRef]
- Kapitány, A.; Zilahi, E.; Szántó, S.; Szücs, G.; Szabó, Z.; Végvári, A.; Rass, P.; Sipka, S.; Szegedi, G.; Szekanecz, Z. Association of Rheumatoid Arthritis with HLA-DR1 and HLA-DR4 in Hungary. Annals of the New York Academy of Sciences 2005, 1051, 263–270. [CrossRef]
- Becart, S.; Whittington, K.B.; Prislovsky, A.; Rao, N.L.; Rosloniec, E.F. The Role of Posttranslational Modifications in Generating Neo-Epitopes That Bind to Rheumatoid Arthritis-Associated HLA-DR Alleles and Promote Autoimmune T Cell Responses. PLoS ONE 2021, 16, e0245541. [CrossRef]
- Van Der Helm-van Mil, A.H.M.; Huizinga, T.W.J.; Schreuder, G.M.Th.; Breedveld, F.C.; De Vries, R.R.P.; Toes, R.E.M. An Independent Role of Protective HLA Class II Alleles in Rheumatoid Arthritis Severity and Susceptibility. Arthritis & Rheumatism 2005, 52, 2637–2644. [CrossRef]
- Kanaan, S.B.; Sensoy, O.; Yan, Z.; Gadi, V.K.; Richardson, M.L.; Nelson, J.L. Immunogenicity of a Rheumatoid Arthritis Protective Sequence When Acquired through Microchimerism. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 19600–19608. [CrossRef]
- Okada, E.; Daimon, K.; Fujiwara, H.; Nishiwaki, Y.; Nojiri, K.; Watanabe, M.; Katoh, H.; Shimizu, K.; Ishihama, H.; Fujita, N.; et al. Twenty-Year Longitudinal Follow-up MRI Study of Asymptomatic Volunteers: The Impact of Cervical Alignment on Disk Degeneration. Clinical Spine Surgery: A Spine Publication 2018, 31, 446–451. [CrossRef]
- Holoshitz, J. The Rheumatoid Arthritis HLA–DRB1 Shared Epitope. Current Opinion in Rheumatology 2010, 22, 293–298. [CrossRef]
- Viatte, S.; Massey, J.; Bowes, J.; Duffus, K.; arcOGEN Consortium; Eyre, S.; Barton, A.; Worthington, J. Replication of Associations of Genetic Loci Outside the HLA Region With Susceptibility to Anti–Cyclic Citrullinated Peptide–Negative Rheumatoid Arthritis. Arthritis & Rheumatology 2016, 68, 1603–1613. [CrossRef]
- Viatte, S.; Plant, D.; Bowes, J.; Lunt, M.; Eyre, S.; Barton, A.; Worthington, J. Genetic Markers of Rheumatoid Arthritis Susceptibility in Anti-Citrullinated Peptide Antibody Negative Patients. Annals of the Rheumatic Diseases 2012, 71, 1984–1990. [CrossRef]
- Krabben, A.; Huizinga, T.W.J.; Mil, A.H.M. Biomarkers for Radiographic Progression in Rheumatoid Arthritis. CPD 2014, 21, 147–169. [CrossRef]
- Lee, J.C.; Espéli, M.; Anderson, C.A.; Linterman, M.A.; Pocock, J.M.; Williams, N.J.; Roberts, R.; Viatte, S.; Fu, B.; Peshu, N.; et al. Human SNP Links Differential Outcomes in Inflammatory and Infectious Disease to a FOXO3-Regulated Pathway. Cell 2013, 155, 57–69. [CrossRef]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.-S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five Amino Acids in Three HLA Proteins Explain Most of the Association between MHC and Seropositive Rheumatoid Arthritis. Nat Genet 2012, 44, 291–296. [CrossRef]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; González-Gay, M.A.; Rantapää-Dahlqvist, S.; et al. Fine Mapping Seronegative and Seropositive Rheumatoid Arthritis to Shared and Distinct HLA Alleles by Adjusting for the Effects of Heterogeneity. The American Journal of Human Genetics 2014, 94, 522–532. [CrossRef]
- Ishigaki, K.; Sakaue, S.; Terao, C.; Luo, Y.; Sonehara, K.; Yamaguchi, K.; Amariuta, T.; Too, C.L.; Laufer, V.A.; Scott, I.C.; et al. Multi-Ancestry Genome-Wide Association Analyses Identify Novel Genetic Mechanisms in Rheumatoid Arthritis. Nat Genet 2022, 54, 1640–1651. [CrossRef]
- Hill, R.J.; Zozulya, S.; Lu, Y.-L.; Ward, K.; Gishizky, M.; Jallal, B. The Lymphoid Protein Tyrosine Phosphatase Lyp Interacts with the Adaptor Molecule Grb2 and Functions as a Negative Regulator of T-Cell Activation. Experimental Hematology 2002, 30, 237–244. [CrossRef]
- Bottini, N.; Peterson, E.J. Tyrosine Phosphatase PTPN22: Multifunctional Regulator of Immune Signaling, Development, and Disease. Annu. Rev. Immunol. 2014, 32, 83–119. [CrossRef]
- Vang, T.; Liu, W.H.; Delacroix, L.; Wu, S.; Vasile, S.; Dahl, R.; Yang, L.; Musumeci, L.; Francis, D.; Landskron, J.; et al. LYP Inhibits T-Cell Activation When Dissociated from CSK. Nat Chem Biol 2012, 8, 437–446. [CrossRef]
- Arechiga, A.F.; Habib, T.; He, Y.; Zhang, X.; Zhang, Z.-Y.; Funk, A.; Buckner, J.H. Cutting Edge: The PTPN22 Allelic Variant Associated with Autoimmunity Impairs B Cell Signaling. The Journal of Immunology 2009, 182, 3343–3347. [CrossRef]
- Gardette, A.; Marzaioli, V.; Bedouhene, S.; Hayem, G.; Hurtado-Nedelec, M.; He, Y.; Dang, P.M.-C.; Dieudé, P.; Zhang, Z.-Y.; Marie, J.-C.; et al. The Protein Tyrosine Phosphatase Lyp/PTPN22 Drives TNFα-Induced Priming of Superoxide Anions Production by Neutrophils and Arthritis. Free Radical Biology and Medicine 2025, 228, 68–78. [CrossRef]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A. Epigenetic Changes in the Pathogenesis of Rheumatoid Arthritis. Front. Genet. 2019, 10, 570. [CrossRef]
- Yang, C.; Li, D.; Teng, D.; Zhou, Y.; Zhang, L.; Zhong, Z.; Yang, G.-J. Epigenetic Regulation in the Pathogenesis of Rheumatoid Arthritis. Front. Immunol. 2022, 13, 859400. [CrossRef]
- Karouzakis, E.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. Epigenome Analysis Reveals TBX5 as a Novel Transcription Factor Involved in the Activation of Rheumatoid Arthritis Synovial Fibroblasts. The Journal of Immunology 2014, 193, 4945–4951. [CrossRef]
- Li, X.-F.; Wu, S.; Yan, Q.; Wu, Y.-Y.; Chen, H.; Yin, S.-Q.; Chen, X.; Wang, H.; Li, J. PTEN Methylation Promotes Inflammation and Activation of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Front. Pharmacol. 2021, 12, 700373. [CrossRef]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA Methylome Signature in Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2013, 72, 110–117. [CrossRef]
- Liebold, I.; Grützkau, A.; Göckeritz, A.; Gerl, V.; Lindquist, R.; Feist, E.; Zänker, M.; Häupl, T.; Poddubnyy, D.; Zernicke, J.; et al. Peripheral Blood Mononuclear Cells Are Hypomethylated in Active Rheumatoid Arthritis and Methylation Correlates with Disease Activity. Rheumatology 2021, 60, 1984–1995. [CrossRef]
- Zhu, H.; Wu, L.-F.; Mo, X.-B.; Lu, X.; Tang, H.; Zhu, X.-W.; Xia, W.; Guo, Y.-F.; Wang, M.-J.; Zeng, K.-Q.; et al. Rheumatoid Arthritis–Associated DNA Methylation Sites in Peripheral Blood Mononuclear Cells. Annals of the Rheumatic Diseases 2019, 78, 36–42. [CrossRef]
- Julià, A.; Absher, D.; López-Lasanta, M.; Palau, N.; Pluma, A.; Waite Jones, L.; Glossop, J.R.; Farrell, W.E.; Myers, R.M.; Marsal, S. Epigenome-Wide Association Study of Rheumatoid Arthritis Identifies Differentially Methylated Loci in B Cells. Human Molecular Genetics 2017, 26, 2803–2811. [CrossRef]
- Lev Maor, G.; Yearim, A.; Ast, G. The Alternative Role of DNA Methylation in Splicing Regulation. Trends in Genetics 2015, 31, 274–280. [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by CTLA-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis & Rheumatology 2014, 66, 2344–2354. [CrossRef]
- Cribbs, A.; Feldmann, M.; Oppermann, U. Towards an Understanding of the Role of DNA Methylation in Rheumatoid Arthritis: Therapeutic and Diagnostic Implications. Therapeutic Advances in Musculoskeletal 2015, 7, 206–219. [CrossRef]
- Li, Y.; Zhou, M.; Lv, X.; Song, L.; Zhang, D.; He, Y.; Wang, M.; Zhao, X.; Yuan, X.; Shi, G.; et al. Reduced Activity of HDAC3 and Increased Acetylation of Histones H3 in Peripheral Blood Mononuclear Cells of Patients with Rheumatoid Arthritis. Journal of Immunology Research 2018, 2018, 1–10. [CrossRef]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.-Y.; Backlund, J.; Matthias, P.; et al. Histone Deacetylase 1 (HDAC1): A Key Player of T Cell-Mediated Arthritis. Journal of Autoimmunity 2020, 108, 102379. [CrossRef]
- Park, S.Y.; Lee, S.W.; Lee, S.Y.; Hong, K.W.; Bae, S.S.; Kim, K.; Kim, C.D. SIRT1/Adenosine Monophosphate-Activated Protein Kinase α Signaling Enhances Macrophage Polarization to an Anti-Inflammatory Phenotype in Rheumatoid Arthritis. Front. Immunol. 2017, 8, 1135. [CrossRef]
- Li, M.; Hu, W.; Wang, R.; Li, Z.; Yu, Y.; Zhuo, Y.; Zhang, Y.; Wang, Z.; Qiu, Y.; Chen, K.; et al. Sp1 S-Sulfhydration Induced by Hydrogen Sulfide Inhibits Inflammation via HDAC6/MyD88/NF-κB Signaling Pathway in Adjuvant-Induced Arthritis. Antioxidants 2022, 11, 732. [CrossRef]
- Mu, N.; Gu, J.; Huang, T.; Zhang, C.; Shu, Z.; Li, M.; Hao, Q.; Li, W.; Zhang, W.; Zhao, J.; et al. A Novel NF-κB/YY1/microRNA-10a Regulatory Circuit in Fibroblast-like Synoviocytes Regulates Inflammation in Rheumatoid Arthritis. Sci Rep 2016, 6, 20059. [CrossRef]
- Stanczyk, J.; Ospelt, C.; Karouzakis, E.; Filer, A.; Raza, K.; Kolling, C.; Gay, R.; Buckley, C.D.; Tak, P.P.; Gay, S.; et al. Altered Expression of microRNA-203 in Rheumatoid Arthritis Synovial Fibroblasts and Its Role in Fibroblast Activation. Arthritis & Rheumatism 2011, 63, 373–381. [CrossRef]
- Gantier, M.P.; Stunden, H.J.; McCoy, C.E.; Behlke, M.A.; Wang, D.; Kaparakis-Liaskos, M.; Sarvestani, S.T.; Yang, Y.H.; Xu, D.; Corr, S.C.; et al. A miR-19 Regulon That Controls NF-κB Signaling. Nucleic Acids Research 2012, 40, 8048–8058. [CrossRef]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid Arthritis and the Mucosal Origins Hypothesis: Protection Turns to Destruction. Nat Rev Rheumatol 2018, 14, 542–557. [CrossRef]
- Willis, V.C.; Demoruelle, M.K.; Derber, L.A.; Chartier-Logan, C.J.; Parish, M.C.; Pedraza, I.F.; Weisman, M.H.; Norris, J.M.; Holers, V.M.; Deane, K.D. Sputum Autoantibodies in Patients With Established Rheumatoid Arthritis and Subjects at Risk of Future Clinically Apparent Disease. Arthritis & Rheumatism 2013, 65, 2545–2554. [CrossRef]
- Demoruelle, M.K.; Bowers, E.; Lahey, L.J.; Sokolove, J.; Purmalek, M.; Seto, N.L.; Weisman, M.H.; Norris, J.M.; Kaplan, M.J.; Holers, V.M.; et al. Antibody Responses to Citrullinated and Noncitrullinated Antigens in the Sputum of Subjects With Rheumatoid Arthritis and Subjects at Risk for Development of Rheumatoid Arthritis. Arthritis & Rheumatology 2018, 70, 516–527. [CrossRef]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural Changes and Antibody Enrichment in the Lungs Are Early Features of Anti–Citrullinated Protein Antibody–Positive Rheumatoid Arthritis. Arthritis & Rheumatology 2014, 66, 31–39. [CrossRef]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [CrossRef]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A Gene–Environment Interaction between Smoking and Shared Epitope Genes in HLA–DR Provides a High Risk of Seropositive Rheumatoid Arthritis. Arthritis & Rheumatism 2004, 50, 3085–3092. [CrossRef]
- Linn-Rasker, S.P.; Van Der Helm-van Mil, A.H.M.; Van Gaalen, F.A.; Kloppenburg, M.; De Vries, R.R.P.; Le Cessie, S.; Breedveld, F.C.; Toes, R.E.M.; Huizinga, T.W.J. Smoking Is a Risk Factor for Anti-CCP Antibodies Only in Rheumatoid Arthritis Patients Who Carry HLA-DRB1 Shared Epitope Alleles. Annals of the Rheumatic Diseases 2006, 65, 366–371. [CrossRef]
- Källberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Smoking Is a Major Preventable Risk Factor for Rheumatoid Arthritis: Estimations of Risks after Various Exposures to Cigarette Smoke. Annals of the Rheumatic Diseases 2011, 70, 508–511. [CrossRef]
- Hedström, A.K.; Klareskog, L.; Alfredsson, L. Exposure to Passive Smoking and Rheumatoid Arthritis Risk: Results from the Swedish EIRA Study. Annals of the Rheumatic Diseases 2018, 77, 970–972. [CrossRef]
- Svendsen, A.J.; Gervin, K.; Lyle, R.; Christiansen, L.; Kyvik, K.; Junker, P.; Nielsen, C.; Houen, G.; Tan, Q. Differentially Methylated DNA Regions in Monozygotic Twin Pairs Discordant for Rheumatoid Arthritis: An Epigenome-Wide Study. Front. Immunol. 2016, 7. [CrossRef]
- Krishnan, E. Smoking, Gender and Rheumatoid Arthritis–Epidemiological Clues to Etiology. Joint Bone Spine 2003, 70, 496–502. [CrossRef]
- Too, C.L.; Muhamad, N.A.; Ilar, A.; Padyukov, L.; Alfredsson, L.; Klareskog, L.; Murad, S.; Bengtsson, C. Occupational Exposure to Textile Dust Increases the Risk of Rheumatoid Arthritis: Results from a Malaysian Population-Based Case–Control Study. Annals of the Rheumatic Diseases 2016, 75, 997–1002. [CrossRef]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L. Silica Exposure among Male Current Smokers Is Associated with a High Risk of Developing ACPA-Positive Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2010, 69, 1072–1076. [CrossRef]
- Mehri, F.; Jenabi, E.; Bashirian, S.; Shahna, F.G.; Khazaei, S. The Association Between Occupational Exposure to Silica and Risk of Developing Rheumatoid Arthritis: A Meta-Analysis. Safety and Health at Work 2020, 11, 136–142. [CrossRef]
- Alaya, Z.; Braham, M.; Aissa, S.; Kalboussi, H.; Bouajina, E. A Case of Caplan Syndrome in a Recently Diagnosed Patient with Silicosis: A Case Report. Radiology Case Reports 2018, 13, 663–666. [CrossRef]
- on behalf of the EIRA study group; Johansson, K.; Askling, J.; Alfredsson, L.; Di Giuseppe, D. Mediterranean Diet and Risk of Rheumatoid Arthritis: A Population-Based Case-Control Study. Arthritis Res Ther 2018, 20. [CrossRef]
- Jin, J.; Li, J.; Gan, Y.; Liu, J.; Zhao, X.; Chen, J.; Zhang, R.; Zhong, Y.; Chen, X.; Wu, L.; et al. Red Meat Intake Is Associated with Early Onset of Rheumatoid Arthritis: A Cross-Sectional Study. Sci Rep 2021, 11, 5681. [CrossRef]
- Gan, R.W.; Demoruelle, M.K.; Deane, K.D.; Weisman, M.H.; Buckner, J.H.; Gregersen, P.K.; Mikuls, T.R.; O’Dell, J.R.; Keating, R.M.; Fingerlin, T.E.; et al. Omega-3 Fatty Acids Are Associated with a Lower Prevalence of Autoantibodies in Shared Epitope-Positive Subjects at Risk for Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2017, 76, 147–152. [CrossRef]
- Gan, R.W.; Young, K.A.; Zerbe, G.O.; Demoruelle, M.K.; Weisman, M.H.; Buckner, J.H.; Gregersen, P.K.; Mikuls, T.R.; O’Dell, J.R.; Keating, R.M.; et al. Lower Omega-3 Fatty Acids Are Associated with the Presence of Anti-Cyclic Citrullinated Peptide Autoantibodies in a Population at Risk for Future Rheumatoid Arthritis: A Nested Case-Control Study. Rheumatology 2016, 55, 367–376. [CrossRef]
- Costenbader, K.H.; Cook, N.R.; Lee, I.; Hahn, J.; Walter, J.; Bubes, V.; Kotler, G.; Yang, N.; Friedman, S.; Alexander, E.K.; et al. Vitamin D and Marine N-3 Fatty Acids for Autoimmune Disease Prevention: Outcomes Two Years After Completion of a Double-Blind , Placebo-Controlled Trial. Arthritis & Rheumatology 2024, 76, 973–983. [CrossRef]
- Karlson, E.W.; Mandl, L.A.; Aweh, G.N.; Grodstein, F. Coffee Consumption and Risk of Rheumatoid Arthritis. Arthritis & Rheumatism 2003, 48, 3055–3060. [CrossRef]
- Pattison, D.J.; Symmons, D.P.M.; Lunt, M.; Welch, A.; Luben, R.; Bingham, S.A.; Khaw, K.; Day, N.E.; Silman, A.J. Dietary Risk Factors for the Development of Inflammatory Polyarthritis: Evidence for a Role of High Level of Red Meat Consumption. Arthritis & Rheumatism 2004, 50, 3804–3812. [CrossRef]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intake of High-Fructose Corn Syrup Sweetened Soft Drinks, Fruit Drinks and Apple Juice Is Associated with Prevalent Arthritis in US Adults, Aged 20–30 Years. Nutr & Diabetes 2016, 6, e199–e199. [CrossRef]
- Hu, Y.; Costenbader, K.H.; Gao, X.; Al-Daabil, M.; Sparks, J.A.; Solomon, D.H.; Hu, F.B.; Karlson, E.W.; Lu, B. Sugar-Sweetened Soda Consumption and Risk of Developing Rheumatoid Arthritis in Women , , ,. The American Journal of Clinical Nutrition 2014, 100, 959–967. [CrossRef]
- Radu, A.-F.; Bungau, S.G. Management of Rheumatoid Arthritis: An Overview. Cells 2021, 10, 2857. [CrossRef]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas Gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis & Rheumatology 2016, 68, 604–613. [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter Actinomycetemcomitans –Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8. [CrossRef]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella Copri in Individuals at Risk for Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2019, 78, 590–593. [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The Oral and Gut Microbiomes Are Perturbed in Rheumatoid Arthritis and Partly Normalized after Treatment. Nat Med 2015, 21, 895–905. [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of Intestinal Prevotella Copri Correlates with Enhanced Susceptibility to Arthritis. eLife 2013, 2, e01202. [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An Expansion of Rare Lineage Intestinal Microbes Characterizes Rheumatoid Arthritis. Genome Med 2016, 8, 43. [CrossRef]
- Bengtsson, C.; Malspeis, S.; Orellana, C.; Sparks, J.A.; Costenbader, K.H.; Karlson, E.W. Association Between Menopausal Factors and the Risk of Seronegative and Seropositive Rheumatoid Arthritis: Results From the Nurses’ Health Studies. Arthritis Care & Research 2017, 69, 1676–1684. [CrossRef]
- Beydoun, H.A.; el-Amin, R.; McNeal, M.; Perry, C.; Archer, D.F. Reproductive History and Postmenopausal Rheumatoid Arthritis among Women 60 Years or Older: Third National Health and Nutrition Examination Survey. Menopause 2013, 20, 930–935. [CrossRef]
- Kobak, S.; Bes, C. An Autumn Tale: Geriatric Rheumatoid Arthritis. Therapeutic Advances in Musculoskeletal 2018, 10, 3–11. [CrossRef]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and Prevalence of Rheumatoid Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review. Seminars in Arthritis and Rheumatism 2006, 36, 182–188. [CrossRef]
- George, M.D.; Baker, J.F. The Obesity Epidemic and Consequences for Rheumatoid Arthritis Care. Curr Rheumatol Rep 2016, 18, 6. [CrossRef]
- Alunno, A.; Carubbi, F.; Giacomelli, R.; Gerli, R. Cytokines in the Pathogenesis of Rheumatoid Arthritis: New Players and Therapeutic Targets. BMC Rheumatol 2017, 1, 3. [CrossRef]
- Chen, S.; Xue, W.; Wu, Z.; Lu, D.; Zheng, L.; Zhou, M.; Li, Y.; Wang, Y.; Liu, T. Quercetin, a Compound of the Total Flavonoids of Periploca Forrestii Schltr., Ameliorates Rheumatoid Arthritis by Targeting TNF-α. JIR 2025, Volume 18, 2879–2898. [CrossRef]
- Han, J.; Wang, J.; Wang, Y.; Zhu, Z.; Zhang, S.; Wu, B.; Meng, M.; Zhao, J.; Wang, D. Sesquiterpene Lactones-Enriched Fractions from Xanthium Mongolicum Kitag Alleviate RA by Regulating M1 Macrophage Polarization via NF-κB and MAPK Signaling Pathway. Front. Pharmacol. 2023, 14, 1104153. [CrossRef]
- Niu, Y.; Feng, Q.; Cui, M.; Fan, C.; Wang, T.; Yuan, R.; Tsering, D.; Huang, S.; Li, B. Siweixizangmaoru Decoction Ameliorated Type II Collagen-Induced Arthritis in Rats via Regulating JAK2–STAT3 and NF-κB Signaling Pathway. Biological & Pharmaceutical Bulletin 2024, 47, 1511–1524. [CrossRef]
- Peng, W.; Yang, Y.; Lu, H.; Shi, H.; Jiang, L.; Liao, X.; Zhao, H.; Wang, W.; Liu, J. Network Pharmacology Combines Machine Learning, Molecular Simulation Dynamics and Experimental Validation to Explore the Mechanism of Acetylbinankadsurin A in the Treatment of Liver Fibrosis. Journal of Ethnopharmacology 2024, 323, 117682. [CrossRef]
- Cao, F.; Cheng, M.-H.; Hu, L.-Q.; Shen, H.-H.; Tao, J.-H.; Li, X.-M.; Pan, H.-F.; Gao, J. Natural Products Action on Pathogenic Cues in Autoimmunity: Efficacy in Systemic Lupus Erythematosus and Rheumatoid Arthritis as Compared to Classical Treatments. Pharmacological Research 2020, 160, 105054. [CrossRef]
- Shen, Y.; Fan, X.; Qu, Y.; Tang, M.; Huang, Y.; Peng, Y.; Fu, Q. Magnoflorine Attenuates Inflammatory Responses in RA by Regulating the PI3K/Akt/NF-κB and Keap1-Nrf2/HO-1 Signalling Pathways in Vivo and in Vitro. Phytomedicine 2022, 104, 154339. [CrossRef]
- Wang, F.; Liu, M.; Tang, Q.; Sun, H.; Yang, G.; Sun, J. Anti-Rheumatic Arthritis Efficacy of Pueraria Montana Extract against Type-II Collagen-Induced Rheumatoid Arthritis Rat Model an in Vitro and in Vivo Assessment. Journal of Ethnopharmacology 2025, 340, 119175. [CrossRef]
- Smolen, J.S.; Landewé, R.; Breedveld, F.C.; Buch, M.; Burmester, G.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gossec, L.; Nam, J.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2013 Update. Annals of the Rheumatic Diseases 2014, 73, 492–509. [CrossRef]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically Expanded Peripheral T Helper Cell Subset Drives B Cells in Rheumatoid Arthritis. Nature 2017, 542, 110–114. [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat Immunol 2019, 20, 928–942. [CrossRef]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends in Molecular Medicine 2019, 25, 215–227. [CrossRef]
- Rivellese, F.; Mauro, D.; Nerviani, A.; Pagani, S.; Fossati-Jimack, L.; Messemaker, T.; Kurreeman, F.A.S.; Toes, R.E.M.; Ramming, A.; Rauber, S.; et al. Mast Cells in Early Rheumatoid Arthritis Associate with Disease Severity and Support B Cell Autoantibody Production. Annals of the Rheumatic Diseases 2018, 77, 1773–1781. [CrossRef]
- Schubert, N.; Dudeck, J.; Liu, P.; Karutz, A.; Speier, S.; Maurer, M.; Tuckermann, J.; Dudeck, A. Mast Cell Promotion of T Cell–Driven Antigen-Induced Arthritis Despite Being Dispensable for Antibody-Induced Arthritis in Which T Cells Are Bypassed. Arthritis & Rheumatology 2015, 67, 903–913. [CrossRef]
- Xiong, H.; Meng, F.; Luo, M.; Chen, W.; Tian, J.; Chen, L.; Ju, Y.; Mei, Z. Anti-Inflammatory and Osteoprotective Effects of Shi-Wei-Ru-Xiang Pills on Collagen-Induced Arthritis in Rats via Inhibiting MAPK and STAT3 Pathways. Journal of Ethnopharmacology 2023, 300, 115693. [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N Engl J Med 2011, 365, 2205–2219. [CrossRef]
- Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. IJMS 2021, 22, 10922. [CrossRef]
- Cope, A.P. T Cells in Rheumatoid Arthritis. Arthritis Res Ther 2008, 10, S1. [CrossRef]
- Choi, E.W.; Lee, K.W.; Park, H.; Kim, H.; Lee, J.H.; Song, J.W.; Yang, J.; Kwon, Y.; Kim, T.M.; Park, J.B.; et al. Therapeutic Effects of Anti-CD154 Antibody in Cynomolgus Monkeys with Advanced Rheumatoid Arthritis. Sci Rep 2018, 8, 2135. [CrossRef]
- Cho, B.-A.; Sim, J.H.; Park, J.A.; Kim, H.W.; Yoo, W.-H.; Lee, S.-H.; Lee, D.-S.; Kang, J.S.; Hwang, Y.-I.; Lee, W.J.; et al. Characterization of Effector Memory CD8+ T Cells in the Synovial Fluid of Rheumatoid Arthritis. J Clin Immunol 2012, 32, 709–720. [CrossRef]
- Jonsson, A.H.; Zhang, F.; Dunlap, G.; Gomez-Rivas, E.; Watts, G.F.M.; Faust, H.J.; Rupani, K.V.; Mears, J.R.; Meednu, N.; Wang, R.; et al. Granzyme K+ CD8 T Cells Form a Core Population in Inflamed Human Tissue. Sci. Transl. Med. 2022, 14, eabo0686. [CrossRef]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 Cells in Human Autoimmune Arthritis. Arthritis & Rheumatism 2010, 62, 2876–2885. [CrossRef]
- Van Baarsen, L.G.; Lebre, M.C.; Van Der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous Expression Pattern of Interleukin 17A (IL-17A), IL-17F and Their Receptors in Synovium of Rheumatoid Arthritis, Psoriatic Arthritis and Osteoarthritis: Possible Explanation for Nonresponse to Anti-IL-17 Therapy? Arthritis Res Ther 2014, 16, 426. [CrossRef]
- Li, S.; Yin, H.; Zhang, K.; Wang, T.; Yang, Y.; Liu, X.; Chang, X.; Zhang, M.; Yan, X.; Ren, Y.; et al. Effector T Helper Cell Populations Are Elevated in the Bone Marrow of Rheumatoid Arthritis Patients and Correlate with Disease Severity. Sci Rep 2017, 7, 4776. [CrossRef]
- Edavalath, S.; Singh, A.; Soni, N.; Mohindra, N.; Kumar, S.; Misra, R. Peripheral Blood T Helper Type 17 Frequency Shows an Inverse Correlation with Disease Activity and Magnetic Resonance Imaging-Based Osteitis and Erosions in Disease-Modifying Anti-Rheumatic Drug- and Steroid-Naive Established Rheumatoid Arthritis. Clinical and Experimental Immunology 2016, 186, 313–320. [CrossRef]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central Role of T Helper 17 Cells in Chronic Hypoxia-Induced Pulmonary Hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017, 312, L609–L624. [CrossRef]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.; Tong, J.; Yang, M.; et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients with Rheumatoid Arthritis. Clinical and Developmental Immunology 2012, 2012, 1–7. [CrossRef]
- Kondo, Y.; Yokosawa, M.; Kaneko, S.; Furuyama, K.; Segawa, S.; Tsuboi, H.; Matsumoto, I.; Sumida, T. Review: Transcriptional Regulation of CD 4+ T Cell Differentiation in Experimentally Induced Arthritis and Rheumatoid Arthritis. Arthritis & Rheumatology 2018, 70, 653–661. [CrossRef]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [CrossRef]
- Ambarus, C.A.; Noordenbos, T.; De Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal Lining Layer Macrophages but Not Synovial Sublining Macrophages Display an IL-10 Polarized-like Phenotype in Chronic Synovitis. Arthritis Res Ther 2012, 14, R74. [CrossRef]
- Soler Palacios, B.; Estrada-Capetillo, L.; Izquierdo, E.; Criado, G.; Nieto, C.; Municio, C.; González-Alvaro, I.; Sánchez-Mateos, P.; Pablos, J.L.; Corbí, A.L.; et al. Macrophages from the Synovium of Active Rheumatoid Arthritis Exhibit an Activin A-dependent Pro-inflammatory Profile. The Journal of Pathology 2015, 235, 515–526. [CrossRef]
- Kuo, D.; Ding, J.; Cohn, I.S.; Zhang, F.; Wei, K.; Rao, D.A.; Rozo, C.; Sokhi, U.K.; Shanaj, S.; Oliver, D.J.; et al. HBEGF+ Macrophages in Rheumatoid Arthritis Induce Fibroblast Invasiveness. Sci. Transl. Med. 2019, 11, eaau8587. [CrossRef]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct Synovial Tissue Macrophage Subsets Regulate Inflammation and Remission in Rheumatoid Arthritis. Nat Med 2020, 26, 1295–1306. [CrossRef]
- Ota, Y.; Niiro, H.; Ota, S.; Ueki, N.; Tsuzuki, H.; Nakayama, T.; Mishima, K.; Higashioka, K.; Jabbarzadeh-Tabrizi, S.; Mitoma, H.; et al. Generation Mechanism of RANKL+ Effector Memory B Cells: Relevance to the Pathogenesis of Rheumatoid Arthritis. Arthritis Res Ther 2016, 18, 67. [CrossRef]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus Tocilizumab in Anti-TNF Inadequate Responder Patients with Rheumatoid Arthritis (R4RA): 16-Week Outcomes of a Stratified, Biopsy-Driven, Multicentre, Open-Label, Phase 4 Randomised Controlled Trial. The Lancet 2021, 397, 305–317. [CrossRef]
- Yeo, L.; Lom, H.; Juarez, M.; Snow, M.; Buckley, C.D.; Filer, A.; Raza, K.; Scheel-Toellner, D. Expression of FcRL4 Defines a Pro-Inflammatory, RANKL-Producing B Cell Subset in Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2015, 74, 928–935. [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally Distinct Disease-Associated Fibroblast Subsets in Rheumatoid Arthritis. Nat Commun 2018, 9, 789. [CrossRef]
- Yin, G.; Li, Y.; Yang, M.; Cen, X.; Xie, Q. Pim-2/mTORC1 Pathway Shapes Inflammatory Capacity in Rheumatoid Arthritis Synovial Cells Exposed to Lipid Peroxidations. BioMed Research International 2015, 2015, 1–8. [CrossRef]
- Kuo, D.; Ding, J.; Cohn, I.S.; Zhang, F.; Wei, K.; Rao, D.A.; Rozo, C.; Sokhi, U.K.; Shanaj, S.; Oliver, D.J.; et al. HBEGF+ Macrophages in Rheumatoid Arthritis Induce Fibroblast Invasiveness. Sci. Transl. Med. 2019, 11, eaau8587. [CrossRef]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct Synovial Tissue Macrophage Subsets Regulate Inflammation and Remission in Rheumatoid Arthritis. Nat Med 2020, 26, 1295–1306. [CrossRef]
- Gao, Y.; Zhang, Y.; Liu, X. Rheumatoid Arthritis: Pathogenesis and Therapeutic Advances. MedComm 2024, 5, e509. [CrossRef]
- Kong, J.-S.; Jeong, G.H.; Yoo, S.-A. The Use of Animal Models in Rheumatoid Arthritis Research. J Yeungnam Med Sci 2023, 40, 23–29. [CrossRef]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Cartilage Destruction and Bone Erosion in Arthritis: The Role of Tumour Necrosis Factor α. Annals of the Rheumatic Diseases 2000, 59, i75–i80. [CrossRef]
- Horai, R.; Saijo, S.; Tanioka, H.; Nakae, S.; Sudo, K.; Okahara, A.; Ikuse, T.; Asano, M.; Iwakura, Y. Development of Chronic Inflammatory Arthropathy Resembling Rheumatoid Arthritis in Interleukin 1 Receptor Antagonist–Deficient Mice. The Journal of Experimental Medicine 2000, 191, 313–320. [CrossRef]
- Kannan, K.; Ortmann, R.A.; Kimpel, D. Animal Models of Rheumatoid Arthritis and Their Relevance to Human Disease. Pathophysiology 2005, 12, 167–181. [CrossRef]
- Razawy, W.; Asmawidjaja, P.S.; Mus, A.; Salioska, N.; Davelaar, N.; Kops, N.; Oukka, M.; Alves, C.H.; Lubberts, E. CD4+ CCR6+ T Cells, but Not Γδ T Cells, Are Important for the IL-23R-dependent Progression of Antigen-induced Inflammatory Arthritis in Mice. Eur J Immunol 2020, 50, 245–255. [CrossRef]
- Giant, T.T.; Mikecz, K.; Bartlett, R.R.; Deák, F.; Thonar, E.J.-M.A.; Williams, J.M.; Mattar, T.; Kuettner, K.E.; Schleyerbach, R. Immunomodulation of Proteoglycan-Induced Progressive Polyarthritis by Lefluflomide. Immunopharmacology 1992, 23, 105–116. [CrossRef]
- Nandakumar, K.S.; Bäcklund, J.; Vestberg, M.; Holmdahl, R. Collagen Type II (CII)-Specific Antibodies Induce Arthritis in the Absence of T or B Cells but the Arthritis Progression Is Enhanced by CII-Reactive T Cells. Arthritis Res Ther 2004, 6, R544. [CrossRef]
- Yamada, H. Adaptive Immunity in the Joint of Rheumatoid Arthritis. Immunological Medicine 2022, 45, 1–11. [CrossRef]
- Nandakumar, K.S.; Fang, Q.; Wingbro Ågren, I.; Bejmo, Z.F. Aberrant Activation of Immune and Non-Immune Cells Contributes to Joint Inflammation and Bone Degradation in Rheumatoid Arthritis. IJMS 2023, 24, 15883. [CrossRef]
- Takemura, S.; Klimiuk, P.A.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T Cell Activation in Rheumatoid Synovium Is B Cell Dependent. The Journal of Immunology 2001, 167, 4710–4718. [CrossRef]
- Schlegel, P.M.; Steiert, I.; Kötter, I.; Müller, C.A. B Cells Contribute to Heterogeneity of IL-17 Producing Cells in Rheumatoid Arthritis and Healthy Controls. PLoS ONE 2013, 8, e82580. [CrossRef]
- Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium; Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat Immunol 2019, 20, 928–942. [CrossRef]
- Aarvak, T.; Natvig, J.B. Cell-Cell Interactions in Synovitis: Antigen Presenting Cells and T Cell Interaction in Rheumatoid Arthritis. Arthritis Res 2001, 3, 13–17. [CrossRef]
- Takeshita, M.; Suzuki, K.; Kondo, Y.; Morita, R.; Okuzono, Y.; Koga, K.; Kassai, Y.; Gamo, K.; Takiguchi, M.; Kurisu, R.; et al. Multi-Dimensional Analysis Identified Rheumatoid Arthritis-Driving Pathway in Human T Cell. Annals of the Rheumatic Diseases 2019, 78, 1346–1356. [CrossRef]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [CrossRef]
- Yang, X.; Chang, Y.; Wei, W. Emerging Role of Targeting Macrophages in Rheumatoid Arthritis: Focus on Polarization, Metabolism and Apoptosis. Cell Proliferation 2020, 53, e12854. [CrossRef]
- Zec, K.; Schonfeldova, B.; Ai, Z.; Van Grinsven, E.; Pirgova, G.; Eames, H.L.; Berthold, D.L.; Attar, M.; Compeer, E.B.; Arnon, T.I.; et al. Macrophages in the Synovial Lining Niche Initiate Neutrophil Recruitment and Articular Inflammation. Journal of Experimental Medicine 2023, 220, e20220595. [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360. [CrossRef]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw 2018, 18, e8. [CrossRef]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De La Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. IJMS 2020, 21, 9739. [CrossRef]
- Yamamoto, K. Targeting Dysregulation of Metalloproteinase Activity in Osteoarthritis.
- Del Buono, A.; Oliva, F.; Osti, L.; Maffulli, N. Metalloproteases and Tendinopathy. Muscle Ligaments and Tendons J 2019, 03, 51. [CrossRef]
- Van Den Steen, P.E.; Proost, P.; Brand, D.D.; Kang, A.H.; Van Damme, J.; Opdenakker, G. Generation of Glycosylated Remnant Epitopes from Human Collagen Type II by Gelatinase B. Biochemistry 2004, 43, 10809–10816. [CrossRef]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint Diseases and Matrix Metalloproteinases: A Role for MMP-13.
- Burrage, P., S. Matrix Metalloproteinases: Role In Arthritis. Front Biosci 2006, 11, 529. [CrossRef]
- Montero-Melendez, T. Therapeutic Senescence via GPCR Activation in Synovial Fibroblasts Facilitates Resolution of Arthritis.
- Zeinert, I. Matrix-Mediated Activation of Murine Fibroblast-like Synoviocytes. Experimental Cell Research 2025.
- Bian, Y.; Xiang, Z.; Wang, Y.; Ren, Q.; Chen, G.; Xiang, B.; Wang, J.; Zhang, C.; Pei, S.; Guo, S.; et al. Immunomodulatory Roles of Metalloproteinases in Rheumatoid Arthritis. Front. Pharmacol. 2023, 14, 1285455. [CrossRef]
- Wang, J. et al. Functional Analysis of Discoidin Domain Receptor 2 in Synovial Fibroblasts in Rheumatoid Arthritis.
- Wang, Y.; Hao, Z.; Lu, D.; Naseem, A.; Sun, Y.; Sun, Y.; Li, J.; Kuang, H.; Liu, Y.; Yang, B. Effects of Viscum Coloratum (Kom.) Nakai on Collagen-Induced Rheumatoid Arthritis. Journal of Ethnopharmacology 2024, 327, 118026. [CrossRef]
- Guo, X.; Zhang, J.; Feng, Z.; Ji, J.; Shen, X.; Hou, X.; Mei, Z. The Antiangiogenic Effect of Total Saponins of Panax Japonicus C.A. Meyer in Rheumatoid Arthritis Is Mediated by Targeting the HIF-1α/VEGF/ANG-1 Axis. Journal of Ethnopharmacology 2024, 333, 118422. [CrossRef]
- Duan, Z.; Jin, C.; Deng, Y.; Liu, J.; Gu, C.; Wang, J.; Cai, X.; Li, S.; Zhou, Y. Exploring the Chondroprotective Effect of Chaenomeles Speciosa on Glucose-6-Phosphate Isomerase Model Mice Using an Integrated Approach of Network Pharmacology and Experimental Validation. Journal of Ethnopharmacology 2023, 314, 116553. [CrossRef]
- Maybee, D.V.; Ink, N.L.; Ali, M.A.M. Novel Roles of MT1-MMP and MMP-2: Beyond the Extracellular Milieu. Int. J. Mol. Sci. 2022.
- Yeon, K.Y. Role of Activating Transcription Factor 3 as a Mediator of the Protective Effects of Berberine against Lipopolysaccharide-Stimulated SW982 Cells and in Rheumatoid Arthritis Animal Models. Toxicology and Applied Pharmacology 2025.
- Zhou, W.; Cheng, H.; Fan, C.; Zhou, X.; Chen, W.; Xie, C.; Hu, Y.; Chen, Y.; Wang, X.; Wu, J. LAMP3-Mediated Epithelial-Mesenchymal Transition Promotes the Invasion and Excessive Proliferation of Fibroblast-like Synoviocytes in Rheumatoid Arthritis. Journal of Autoimmunity 2025, 151, 103359. [CrossRef]
- Fu, Y.; Gao, C.; Sun, X.; Zhao, Y.; Zhang, H. Study on the Mechanism of Action of Wu Mei Pill in Inhibiting Rheumatoid Arthritis through TLR4-NF-κB Pathway. J Orthop Surg Res 2024, 19, 65. [CrossRef]
- Liu, M.; Tang, Y.; Du, Y.; Zhang, J.; Hu, F.; Zou, Y.; Li, Y.; Zhu, L.; He, J.; Guo, J.; et al. Leukocyte Ig-like Receptor A3 Facilitates Inflammation, Migration and Invasion of Synovial Tissue-Derived Fibroblasts via ERK/JNK Activation.
- Jin, Y.; Chang, C.; Zhou, X.; Zhang, R.; Jiang, P.; Wei, K.; Xu, L.; Shi, Y.; Yang, G.; Lv, X.; et al. LncRNA NONHSAT042241 Inhibits Rheumatoid Synovial Proliferation, Inflammation and Aggression via Ina.
- Lin, F.-J.; Wei, X.-L.; Liu, H.-Y.; Li, H.; Xia, Y.; Wu, D.-T.; Zhang, P.-Z.; Gandhi, G.R.; Hua-Bin Li; Gan, R.-Y. State-of-the-Art Review of Dark Tea: From Chemistry to Health Benefits. Trends in Food Science & Technology 2021, 109, 126–138. [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019.
- Bruno, A.; Wang, G.; Chen, Y.; Zhang, Z. Angiogenesis Is Inhibited by Arsenic Trioxide Through Downregulation of the CircHIPK3/miR-149-5p/FOXO1/ VEGF Functional Module in Rheumatoid Arthritis. Frontiers in Pharmacology 2021, 12.
- Lesturgie-Talarek, M.; Gonzalez, V.; Combier, A.; Thomas, M.; Boisson, M.; Poiroux, L.; Wanono, S.; Hecquet, S.; Carves, S.; Cauvet, A.; et al. Inflammatory and Angiogenic Serum Profile of Refractory Rheumatoid Arthritis. Sci Rep 2025, 15, 7159. [CrossRef]
- Yuan, M. Clematichinenoside AR Alleviates Rheumatoid Arthritis by Inhibiting Synovial Angiogenesis through the HIF-1α/VEGFA/ANG2 Axis. 2025.
- Wei, Y. The New Anti-Angiogenesis Perspective of Rheumatoid Arthritis with Geniposide: Reducing the Extracellular Release of HSP70 in HUVECs. International Immunopharmacology 2025.
- Wang, X.; Fan, D.; Cao, X.; Ye, Q.; Wang, Q.; Zhang, M.; Xiao, C. The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment. 2022.
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia Induces Mitochondrial Mutagenesis and Dysfunction in Inflammatory Arthritis.
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.J.; Fearon, U. Mitochondrial Mutagenesis Correlates with the Local Inflammatory Environment in Arthritis. Annals of the Rheumatic Diseases 2012, 71, 582–588. [CrossRef]
- Du, J.; Yu, S.; Wang, D.; Chen, S.; Chen, S.; Zheng, Y.; Wang, N.; Chen, S.; Li, J.; Shen, B. Germline and Somatic mtDNA Mutation Spectrum of Rheumatoid Arthritis Patients in the Taizhou Area, China.
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [CrossRef]
- Kardeş, S.; Karagülle, M.; Durak, İ.; Avcı, A.; Karagülle, M.Z. Association of Oxidative Stress with Clinical Characteristics in Patients with Rheumatoid Arthritis. Eur J Clin Investigation 2018, 48. [CrossRef]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular Mitochondrial DNA and Oxidatively Damaged DNA in Synovial Fluid of Patients with Rheumatoid Arthritis. Arthritis Res Ther 2003, 5. [CrossRef]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compán, M.V.; Oliver, S.; Schoels, M.; et al. Treating Rheumatoid Arthritis to Target: 2014 Update of the Recommendations of an International Task Force. Annals of the Rheumatic Diseases 2016, 75, 3–15. [CrossRef]
- Combe, B.; Landewe, R.; Daien, C.I.; Hua, C.; Aletaha, D.; Álvaro-Gracia, J.M.; Bakkers, M.; Brodin, N.; Burmester, G.R.; Codreanu, C.; et al. 2016 Update of the EULAR Recommendations for the Management of Early Arthritis. Annals of the Rheumatic Diseases 2017, 76, 948–959. [CrossRef]
- Hoes, J.N.; Jacobs, J.W.G.; Boers, M.; Boumpas, D.; Buttgereit, F.; Caeyers, N.; Choy, E.H.; Cutolo, M.; Da Silva, J.A.P.; Esselens, G.; et al. EULAR Evidence-Based Recommendations on the Management of Systemic Glucocorticoid Therapy in Rheumatic Diseases. Annals of the Rheumatic Diseases 2007, 66, 1560–1567. [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and Its Mechanisms of Action in Inflammatory Arthritis. Nat Rev Rheumatol 2020, 16, 145–154. [CrossRef]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of Action of Methotrexate in Rheumatoid Arthritis, and the Search for Biomarkers. Nat Rev Rheumatol 2016, 12, 731–742. [CrossRef]
- Solomon, D.H.; Glynn, R.J.; Karlson, E.W.; Lu, F.; Corrigan, C.; Colls, J.; Xu, C.; MacFadyen, J.; Barbhaiya, M.; Berliner, N.; et al. Adverse Effects of Low-Dose Methotrexate: A Randomized Trial. Ann Intern Med 2020, 172, 369. [CrossRef]
- Fox, R.I.; Herrmann, M.L.; Frangou, C.G.; Wahl, G.M.; Morris, R.E.; Strand, V.; Kirschbaum, B.J. Mechanism of Action for Leflunomide in Rheumatoid Arthritis. Clinical Immunology 1999, 93, 198–208. [CrossRef]
- Li, E. Leflunomide in the Treatment of Rheumatoid Arthritis. Clinical Therapeutics 2004, 26, 447–459. [CrossRef]
- Pullar, T.; Hunter, J.A.; Capell, H.A. SULPHASALAZINE IN THE TREATMENT OF RHEUMATOID ARTHRITIS: RELATIONSHIP OF DOSE AND SERUM LEVELS TO EFFICACY. Rheumatology 1985, 24, 269–276. [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360. [CrossRef]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic Mice Expressing Human Tumour Necrosis Factor: A Predictive Genetic Model of Arthritis. The EMBO Journal 1991, 10, 4025–4031. [CrossRef]
- Taylor, P.C.; Feldmann, M. Anti-TNF Biologic Agents: Still the Therapy of Choice for Rheumatoid Arthritis. Nat Rev Rheumatol 2009, 5, 578–582. [CrossRef]
- Ma, X.; Xu, S. TNF Inhibitor Therapy for Rheumatoid Arthritis. Biomedical Reports 2013, 1, 177–184. [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for Rheumatoid Arthritis: A Systematic Review. J Rheumatol 2009, 36, 1118–1125. [CrossRef]
- Konttinen, L.; Kankaanpää, E.; Luosujärvi, R.; Blåfield, H.; Vuori, K.; Hakala, M.; Rantalaiho, V.; Savolainen, E.; Uutela, T.; Nordström, D.; et al. Effectiveness of Anakinra in Rheumatic Disease in Patients Naive to Biological Drugs or Previously on TNF Blocking Drugs: An Observational Study. Clin Rheumatol 2006, 25, 882–884. [CrossRef]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [CrossRef]
- Zhang, X.; Chen, Y.-C.; Fettner, S.; Rowell, L.; Gott, T.; Grimsey, P.; Unsworth, A. Pharmacokinetics and Pharmacodynamics of Tocilizumab after Subcutaneous Administration in Patients with Rheumatoid Arthritis. CP 2013, 51, 620–630. [CrossRef]
- Cohen, M.D.; Keystone, E. Rituximab for Rheumatoid Arthritis. Rheumatol Ther 2015, 2, 99–111. [CrossRef]
- Lee, Y.H.; Bae, S.-C.; Song, G.G. The Efficacy and Safety of Rituximab for the Treatment of Active Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Rheumatol Int 2011, 31, 1493–1499. [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the Nomenclature for MSC: The International Society for Cellular Therapy Position Statement. Cytotherapy 2005, 7, 393–395. [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [CrossRef]
- Muguruma, Y.; Yahata, T.; Miyatake, H.; Sato, T.; Uno, T.; Itoh, J.; Kato, S.; Ito, M.; Hotta, T.; Ando, K. Reconstitution of the Functional Human Hematopoietic Microenvironment Derived from Human Mesenchymal Stem Cells in the Murine Bone Marrow Compartment. Blood 2006, 107, 1878–1887. [CrossRef]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal Stem Cells: Revisiting History, Concepts, and Assays. Cell Stem Cell 2008, 2, 313–319. [CrossRef]
- Nasef, A.; Chapel, A.; Mazurier, C.; Bouchet, S.; Lopez, M.; Mathieu, N.; Sensebé, L.; Zhang, Y.; Gorin, N.-C.; Thierry, D.; et al. Identification of IL-10 and TGF-β Transcripts Involved in the Inhibition of T-Lymphocyte Proliferation During Cell Contact With Human Mesenchymal Stem Cells. gene expr 2006, 13, 217–226. [CrossRef]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells 2017, 35, 766–776. [CrossRef]
- Naserian, S.; Shamdani, S.; Arouche, N.; Uzan, G. Regulatory T Cell Induction by Mesenchymal Stem Cells Depends on the Expression of TNFR2 by T Cells. Stem Cell Res Ther 2020, 11, 534. [CrossRef]
- Ansboro, S.; Roelofs, A.J.; De Bari, C. Mesenchymal Stem Cells for the Management of Rheumatoid Arthritis: Immune Modulation, Repair or Both? Current Opinion in Rheumatology 2017, 29, 201–207. [CrossRef]
- Maumus, M.; Guérit, D.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal Stem Cell-Based Therapies in Regenerative Medicine: Applications in Rheumatology. Stem Cell Res Ther 2011, 2, 14. [CrossRef]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [CrossRef]
- Iwata, S.; Nakayamada, S.; Fukuyo, S.; Kubo, S.; Yunoue, N.; Wang, S.; Yoshikawa, M.; Saito, K.; Tanaka, Y. Activation of Syk in Peripheral Blood B Cells in Patients With Rheumatoid Arthritis: A Potential Target for Abatacept Therapy. Arthritis & Rheumatology 2015, 67, 63–73. [CrossRef]
- Xu, Z.; Shang, W.; Zhao, Z.; Zhang, B.; Liu, C.; Cai, H. Curcumin Alleviates Rheumatoid Arthritis Progression through the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway: An in Vitro and in Vivo Study. Bioengineered 2022, 13, 12899–12911. [CrossRef]
- Lu, D.; Tian, X.; Cao, T.; Chen, S.; Liu, C.; Zheng, L.; Zhou, M.; Peng, X.; Li, Y.; Liu, T. Emodin Mitigates Rheumatoid Arthritis through Direct Binding to TNF-α. Front. Pharmacol. 2025, 16. [CrossRef]
- Wang, Y.; Bao, X.; Xian, H.; Wei, F.; Song, Y.; Zhao, S.; Zhang, Y.; Wang, Y.; Wang, Y. Glucocorticoid Receptors Involved in Ginsenoside Compound K Ameliorate Adjuvant Arthritis by Inhibiting the Glycolysis of Fibroblast-like Synoviocytes via the NF-κB/HIF-1α Pathway. Pharmaceutical Biology 2023, 61, 1162–1174. [CrossRef]
- Tu, Y.; Tan, L.; Lu, T.; Wang, K.; Wang, H.; Han, B.; Zhao, Y.; Chen, H.; Li, Y.; Chen, H.; et al. Glytabastan B, a Coumestan Isolated from Glycine Tabacina, Alleviated Synovial Inflammation, Osteoclastogenesis and Collagen-Induced Arthritis through Inhibiting MAPK and PI3K/AKT Pathways. Biochemical Pharmacology 2022, 197, 114912. [CrossRef]
- Wang, S.; Du, Q.; Sun, J.; Geng, S.; Zhang, Y. Investigation of the Mechanism of Isobavachalcone in Treating Rheumatoid Arthritis through a Combination Strategy of Network Pharmacology and Experimental Verification. Journal of Ethnopharmacology 2022, 294, 115342. [CrossRef]
- Liu, H.; Lu, H.; Fan, X.; Chen, S.; Chen, X.; Gao, W. Probing the Molecular Mechanism of Kaempferol in Relieving Rheumatoid Arthritis Based on Network Pharmacology. Sci Rep 2025, 15. [CrossRef]
- Linghu, K.-G.; Zhao, G.-D.; Zhang, D.-Y.; Xiong, S.-H.; Wu, G.-P.; Shen, L.-Y.; Cui, W.-Q.; Zhang, T.; Hu, Y.-J.; Guo, B.; et al. Leocarpinolide B Attenuates Collagen Type II-Induced Arthritis by Inhibiting DNA Binding Activity of NF-κB. Molecules 2023, 28, 4241. [CrossRef]
- Shen, Y.; Teng, L.; Qu, Y.; Liu, J.; Zhu, X.; Chen, S.; Yang, L.; Huang, Y.; Song, Q.; Fu, Q. Anti-Proliferation and Anti-Inflammation Effects of Corilagin in Rheumatoid Arthritis by Downregulating NF-κB and MAPK Signaling Pathways. Journal of Ethnopharmacology 2022, 284, 114791. [CrossRef]
- Anchi, P.; Swamy, V.; Godugu, C. Nimbolide Exerts Protective Effects in Complete Freund’s Adjuvant Induced Inflammatory Arthritis via Abrogation of STAT-3/NF-κB/Notch-1 Signaling. Life Sciences 2021, 266, 118911. [CrossRef]
- Deng, C.; Sun, S.; Zhang, H.; Liu, S.; Xu, X.; Hu, Y.; Ma, H.; Xin, P. Sappanone A Attenuates Rheumatoid Arthritis via Inhibiting PI3K/AKT/NF-κB and JAK2/STAT3 Signaling Pathways in Vivo and in Vitro. International Immunopharmacology 2024, 143, 113560. [CrossRef]
- Liu, C.; He, L.; Wang, J.; Wang, Q.; Sun, C.; Li, Y.; Jia, K.; Wang, J.; Xu, T.; Ming, R.; et al. Anti-Angiogenic Effect of Shikonin in Rheumatoid Arthritis by Downregulating PI3K/AKT and MAPKs Signaling Pathways. Journal of Ethnopharmacology 2020, 260, 113039. [CrossRef]
- Chen, Q.; Zhou, W.; Huang, Y.; Tian, Y.; Wong, S.Y.; Lam, W.K.; Ying, K.Y.; Zhang, J.; Chen, H. Umbelliferone and Scopoletin Target Tyrosine Kinases on Fibroblast-like Synoviocytes to Block NF-κB Signaling to Combat Rheumatoid Arthritis. Front. Pharmacol. 2022, 13. [CrossRef]
- Liu, H.; Li, Q.; Chen, Y.; Dong, M.; Liu, H.; Zhang, J.; Yang, L.; Yin, G.; Xie, Q. Suberosin Attenuates Rheumatoid Arthritis by Repolarizing Macrophages and Inhibiting Synovitis via the JAK/STAT Signaling Pathway. Arthritis Res Ther 2025, 27. [CrossRef]
- Huang, Q.; Xiao, X.; Yu, J.; Yang, Y.; Yu, J.; Liu, Y.; Song, H.; Han, T.; Zhang, D.; Niu, X.; et al. Tectoridin Exhibits Anti-Rheumatoid Arthritis Activity through the Inhibition of the Inflammatory Response and the MAPK Pathway in Vivo and in Vitro. Archives of Biochemistry and Biophysics 2022, 727, 109328. [CrossRef]
- Huang, Y.; Peng, Y.; Li, H.; Li, C.; Wu, Y.; Wang, X.; Chang, J.; Miao, C. Wilforine Inhibits Rheumatoid Arthritis Pathology through the Wnt11/β-Catenin Signaling Pathway Axis. Arthritis Res Ther 2023, 25. [CrossRef]
- Lee, J.-O.; Yang, W.S.; Park, J.G.; Jeong, D.; Kim, H.G.; Yoon, K.D.; Aravinthan, A.; Kim, J.-H.; Kim, E.; Cho, J.Y. Src and Syk Contribute to the Anti-Inflammatory Activities of Achyranthes Aspera Ethanolic Extract. Journal of Ethnopharmacology 2017, 206, 1–7. [CrossRef]
- Sharma, V.K.; Prateeksha, P.; Singh, S.P.; Rao, C.V.; Singh, B.N. Nyctanthes Arbor-Tristis Bioactive Extract Ameliorates LPS-Induced Inflammation through the Inhibition of NF-κB Signalling Pathway. Journal of Ethnopharmacology 2024, 320, 117382. [CrossRef]
- George, G.; Shyni, G.L.; Mohan, S.; Abraham, B.; Nisha, P.; Ranjith, S.; Rajankutty, K.; Raghu, K.G. In Vitro and in Vivo Anti-Inflammatory and Anti-Arthritic Effect of Tinospora Cordifolia via Modulation of JAK/STAT Pathway. Inflammopharmacol 2023, 31, 1009–1025. [CrossRef]
- Sharif, M.; John, P.; Bhatti, A.; Paracha, R.Z.; Majeed, A. Evaluation of the Inhibitory Mechanism of Pennisetum Glaucum (Pearl Millet) Bioactive Compounds for Rheumatoid Arthritis: An in Vitro and Computational Approach. Front. Pharmacol. 2024, 15. [CrossRef]
- Xia, Y.; Fan, D.; Li, X.; Lu, X.; Ye, Q.; Xi, X.; Wang, Q.; Zhao, H.; Xiao, C. Yi Shen Juan Bi Pill Regulates the Bone Immune Microenvironment via the JAK2/STAT3 Signaling Pathway in Vitro. Front. Pharmacol. 2021, 12. [CrossRef]
- Yan, Y.; Zhang, L.-B.; Ma, R.; Wang, M.-N.; He, J.; Wang, P.-P.; Tao, Q.-W.; Xu, Y. Jolkinolide B Ameliorates Rheumatoid Arthritis by Regulating the JAK2/STAT3 Signaling Pathway. Phytomedicine 2024, 124, 155311. [CrossRef]
- Yao, Y.; Wang, J.; Zhang, H.; Peng, T.; Sun, Y.; Zhang, R.; Meng, X.; Lu, X.; Gao, Y.; Jin, Y.; et al. Ammopiptanthus Nanus (M. Pop.) Cheng f. Stem Ethanolic Extract Ameliorates Rheumatoid Arthritis by Inhibiting PI3K/AKT/NF-κB Pathway-Mediated Macrophage Infiltration. Journal of Ethnopharmacology 2025, 338, 118974. [CrossRef]
- Kim, S.-Y.; Kim, J.-M.; Chung, K.-S.; Jang, D.S.; Lee, J.-Y.; Kim, C.; Lee, J.Y.; Lee, J.K.; Lee, K.-T. In Vitro and in Vivo Anti-Inflammatory Effects of 5-Hydroxyconiferaldehyde via NF-κB, MAPK/AP-1, and Nrf2 Modulation. Chemico-Biological Interactions 2025, 409, 111427. [CrossRef]
- Ren, M.; Ma, K.; Pang, X.; Liu, Y.; Song, Z.; Zhou, R.; Tang, Z. Anti-Rheumatoid Arthritis Effects of Total Saponins from Rhizoma Panacis Majoris on Adjuvant-Induced Arthritis in Rats and Rheumatoid Arthritis Fibroblast-like Synoviocytes. Phytomedicine 2023, 119, 155021. [CrossRef]
- Chen, S.; Wang, J.; Wang, J.; Jia, X.; Xuan, Z.; Cheng, Z.; Meng, X.; Su, W. Wnt/β-Catenin Signaling Pathway Promotes Abnormal Activation of Fibroblast-like Synoviocytes and Angiogenesis in Rheumatoid Arthritis and the Intervention of Er Miao San. Phytomedicine 2023, 120, 155064. [CrossRef]
- Pan, L.; Sun, Y.; Jiang, H.; Chen, Y.; Jiang, Y.; Han, Y.; Wang, Y. Total Saponins of Radix Clematis Regulate Fibroblast-Like Synoviocyte Proliferation in Rheumatoid Arthritis via the LncRNA OIP5-AS1/MiR-410-3p/Wnt7b Signaling Pathway. Evidence-Based Complementary and Alternative Medicine 2022, 2022, 1–12. [CrossRef]
- Su, J.; Tao, Y.; Liu, J.; Sun, J.; Zeng, Y.; Meng, X.; Fan, G.; Zhang, Y. Tibetan Medicine Qi-Sai-Er-Sang-Dang-Song Decoction Inhibits TNF-α-Induced Rheumatoid Arthritis in Human Fibroblast-like Synoviocytes via Regulating NOTCH1/NF-κB/NLRP3 Pathway. Journal of Ethnopharmacology 2023, 310, 116402. [CrossRef]
- Lu, Q.; Lu, S.; Gao, X.; Luo, Y.; Tong, B.; Wei, Z.; Lu, T.; Xia, Y.; Chou, G.; Wang, Z.; et al. Norisoboldine, an Alkaloid Compound Isolated from Radix Linderae, Inhibits Synovial Angiogenesis in Adjuvant-Induced Arthritis Rats by Moderating Notch1 Pathway-Related Endothelial Tip Cell Phenotype. Exp Biol Med (Maywood) 2012, 237, 919–932. [CrossRef]
- Deshmukh, R. Rheumatoid Arthritis: Pathophysiology, Current Therapeutic Strategies and Recent Advances in Targeted Drug Delivery System. Materials Today Communications 2023, 35, 105877. [CrossRef]
- Kim, Y.; Yang, H.-I.; Kim, K.-S. Etiology and Pathogenesis of Rheumatoid Arthritis-Interstitial Lung Disease. IJMS 2023, 24, 14509. [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St.Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care & Research 2021, 73, 924–939. [CrossRef]
- Hein, T.R.; Peterson, L.; Bartikoski, B.J.; Portes, J.; Espírito Santo, R.C.; Xavier, R.M. The Effect of Disease-Modifying Anti-Rheumatic Drugs on Skeletal Muscle Mass in Rheumatoid Arthritis Patients: A Systematic Review with Meta-Analysis. Arthritis Res Ther 2022, 24, 171. [CrossRef]
- Gehringer, C.K.; Martin, G.P.; Hyrich, K.L.; Verstappen, S.M.M.; Sergeant, J.C. Clinical Prediction Models for Methotrexate Treatment Outcomes in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Seminars in Arthritis and Rheumatism 2022, 56, 152076. [CrossRef]
- Tamura, T.; Higuchi, Y.; Kitamura, H.; Murao, N.; Saitoh, R.; Morikawa, T.; Sato, H. Novel Hyaluronic Acid–Methotrexate Conjugate Suppresses Joint Inflammation in the Rat Knee: Efficacy and Safety Evaluation in Two Rat Arthritis Models. Arthritis Res Ther 2016, 18, 79. [CrossRef]
- Simon, T.A.; Boers, M.; Hochberg, M.; Baker, N.; Skovron, M.L.; Ray, N.; Singhal, S.; Suissa, S.; Gomez-Caminero, A. Comparative Risk of Malignancies and Infections in Patients with Rheumatoid Arthritis Initiating Abatacept versus Other Biologics: A Multi-Database Real-World Study. Arthritis Res Ther 2019, 21, 228. [CrossRef]
- Choudhary, N.; Bhatt, L.K.; Prabhavalkar, K.S. Experimental Animal Models for Rheumatoid Arthritis. Immunopharmacology and Immunotoxicology 2018, 40, 193–200. [CrossRef]
- Bevaart, L.; Vervoordeldonk, M.J.; Tak, P.P. Evaluation of Therapeutic Targets in Animal Models of Arthritis: How Does It Relate to Rheumatoid Arthritis? Arthritis & Rheumatism 2010, 62, 2192–2205. [CrossRef]
- Campbell, I.K.; Hamilton, J.A.; Wicks, I.P. Collagen-Induced Arthritis in C57BL/6 (H-2b) Mice: New Insights into an Important Disease Model of Rheumatoid Arthritis. Eur. J. Immunol. 2000, 30, 1568–1575. [CrossRef]
- Terato, K.; Hasty, K.A.; Reife, R.A.; Cremer, M.A.; Kang, A.H.; Stuart, J.M. Induction of Arthritis with Monoclonal Antibodies to Collagen. J Immunol 1992, 148, 2103–2108.
- Nandakumar, K.S.; Holmdahl, R. Efficient Promotion of Collagen Antibody Induced Arthritis (CAIA) Using Four Monoclonal Antibodies Specific for the Major Epitopes Recognized in Both Collagen Induced Arthritis and Rheumatoid Arthritis. Journal of Immunological Methods 2005, 304, 126–136. [CrossRef]
- Khachigian, L.M. Collagen Antibody-Induced Arthritis. Nat Protoc 2006, 1, 2512–2516. [CrossRef]
- Nandakumar, K.S.; Holmdahl, R. Collagen Antibody Induced Arthritis. In Arthritis Research; Cope, A.P., Ed.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, 2007; Vol. 136, pp. 215–223 ISBN 978-1-58829-918-5.
- Pearson, C.M. Development of Arthritis, Periarthritis and Periostitis in Rats Given Adjuvants. Experimental Biology and Medicine 1956, 91, 95–101. [CrossRef]
- Negi, P.; Agarwal, S.; Garg, P.; Ali, A.; Kulshrestha, S. In Vivo Models of Understanding Inflammation (in Vivo Methods for Inflammation). In Recent Developments in Anti-Inflammatory Therapy; Elsevier, 2023; pp. 315–330 ISBN 978-0-323-99988-5.
- Ye, L.; Mingyue, H.; Feng, Z.; Zongshun, D.; Ying, X.; Xiong, C.; Liang, L. Systematic Review of Robust Experimental Models of Rheumatoid Arthritis for Basic Research. Digital Chinese Medicine 2021, 4, 262–272. [CrossRef]
- Hopkins, S.J.; Freemont, A.J.; Jayson, M.I.V. Pristane-Induced Arthritis in Balb/c Mice: I. Clinical and Histological Features of the Arthropathy. Rheumatol Int 1984, 5, 21–28. [CrossRef]
- Holmdahl, R. Experimental Models for Rheumatoid Arthritis. In Kelley and Firestein’s Textbook of Rheumatology; Elsevier, 2017; pp. 449–460 ISBN 978-0-323-31696-5.
- van den Berg, W.B. Animal Models of Arthritis. What Have We Learned? J Rheumatol Suppl 2005, 72, 7–9.
- Nho, J.-H.; Kim, A.-H.; Jung, H.-K.; Lee, M.-J.; Jang, J.-H.; Yang, B.-D.; Lee, H.-J.; Lee, K.-H.; Woo, K.-W.; Cho, H.-W. Water Extract of Acori Graminei Rhizoma Attenuates Features of Rheumatoid Arthritis in DBA/1 Mice. Evidence-Based Complementary and Alternative Medicine 2019, 2019, 1–12. [CrossRef]
- Phan, H.T.; Nguyen, T.T.; Tran, P.N.T. EVALUATION OF ANTI-INFLAMMATORY EFFECT OF FRUIT PEEL EXTRACTS OF ANNONA SQUAMOSA L. ON MOUSE MODELS OF RHEUMATOID ARTHRITIS. J microb biotech food sci 2021, 11, e2075. [CrossRef]
- Nho, J.-H.; Lee, H.-J.; Jung, H.-K.; Jang, J.-H.; Lee, K.-H.; Kim, A.-H.; Sung, T.-K.; Cho, H.-W. Effect of Saururus Chinensis Leaves Extract on Type II Collagen-Induced Arthritis Mouse Model. BMC Complement Altern Med 2019, 19, 2. [CrossRef]
- Kim, I.; Kim, H.; Lee, E.H.; Jo, G.; Na, C.S.; Kang, K.; Lee, T.H. Anti-Inflammatory Effect of Cudrania Tricuspidata Extract and Stewartia Koreana Extract Mixture in a Collagen-Induced Arthritis Mouse Model. Applied Sciences 2021, 11, 6660. [CrossRef]
- Kim, S.-H.; Bang, J.; Son, C.-N.; Baek, W.-K.; Kim, J.-M. Grape Seed Proanthocyanidin Extract Ameliorates Murine Autoimmune Arthritis through Regulation of TLR4/MyD88/NF-κB Signaling Pathway. Korean J Intern Med 2018, 33, 612–621. [CrossRef]
- Eor, J.Y.; Park, N.; Son, Y.J.; Kim, S.H. Therapeutic Effects of Gleditsia Sinensis Thorn Extract Fermented by Lactobacillus Casei 3260 in a Type II Collagen-Induced Rheumatoid Arthritis Mouse Model. Food Sci Anim Resour 2021, 41, 497–508. [CrossRef]
- Allam, G.; Mahdi, E.A.; Alzahrani, A.M.; Abuelsaad, A.S. Ellagic Acid Alleviates Adjuvant Induced Arthritis by Modulation of Pro- and Anti-Inflammatory Cytokines. cejoi 2016, 4, 339–349. [CrossRef]
- Ulhaq, Z.S.; Hendyatama, T.H.; Alluza, H.H.D.; Aulanni’am, A. Therapeutic Effects of Durian Wood Bark Extract on a Rat Model of Rheumatoid Arthritis. Revista Colombiana de Reumatología 2021, 28, 118–123. [CrossRef]
- Sun, S.; Li, S.; Du, Y.; Wu, C.; Zhang, M.; Li, J.; Zhang, X. Anti-Inflammatory Effects of the Root, Stem and Leaf Extracts of Chloranthus Serratus on Adjuvant-Induced Arthritis in Rats. Pharmaceutical Biology 2020, 58, 528–537. [CrossRef]
- Qu, B.; Wang, S.; Zhu, H.; Yin, T.; Zhou, R.; Hu, W.; Lu, C. Core Constituents of Caragana Sinica Root for Rheumatoid Arthritis Treatment and the Potential Mechanism. ACS Omega 2023, 8, 2586–2595. [CrossRef]
Figure 1.
Comparison between healthy and RA joint and RA establishment. (A): due to immune activation, join swelling is observed reflecting in synovial inflammation, synovial hypertrophy (pannus) formation and osteoclast activation, extensive angiogenesis, bone and cartilage erosion, join space narrowing, joint structure destruction and muscle loss. Increased levels of metalloproteinases (MMPs) and reactive oxygen species (ROS) and decrease in pH is observed as well. The key players are accumulated cells of innate and adaptive immune systems, including T cells, dendritic cells, B cells, macrophages, and osteoclasts; Establishment of RA (B): multiple risk factors (genetic and non-genetic) are required to achieve the threshold for triggering RA. Before the first subclinical synovitis (inflammation of the synovium) and clinical symptoms, years before, the disease progression involves initiation and propagation of autoimmunity against modified self-proteins. Therefore, once established, RA can be classified according to the clinical symptoms.
Figure 1.
Comparison between healthy and RA joint and RA establishment. (A): due to immune activation, join swelling is observed reflecting in synovial inflammation, synovial hypertrophy (pannus) formation and osteoclast activation, extensive angiogenesis, bone and cartilage erosion, join space narrowing, joint structure destruction and muscle loss. Increased levels of metalloproteinases (MMPs) and reactive oxygen species (ROS) and decrease in pH is observed as well. The key players are accumulated cells of innate and adaptive immune systems, including T cells, dendritic cells, B cells, macrophages, and osteoclasts; Establishment of RA (B): multiple risk factors (genetic and non-genetic) are required to achieve the threshold for triggering RA. Before the first subclinical synovitis (inflammation of the synovium) and clinical symptoms, years before, the disease progression involves initiation and propagation of autoimmunity against modified self-proteins. Therefore, once established, RA can be classified according to the clinical symptoms.
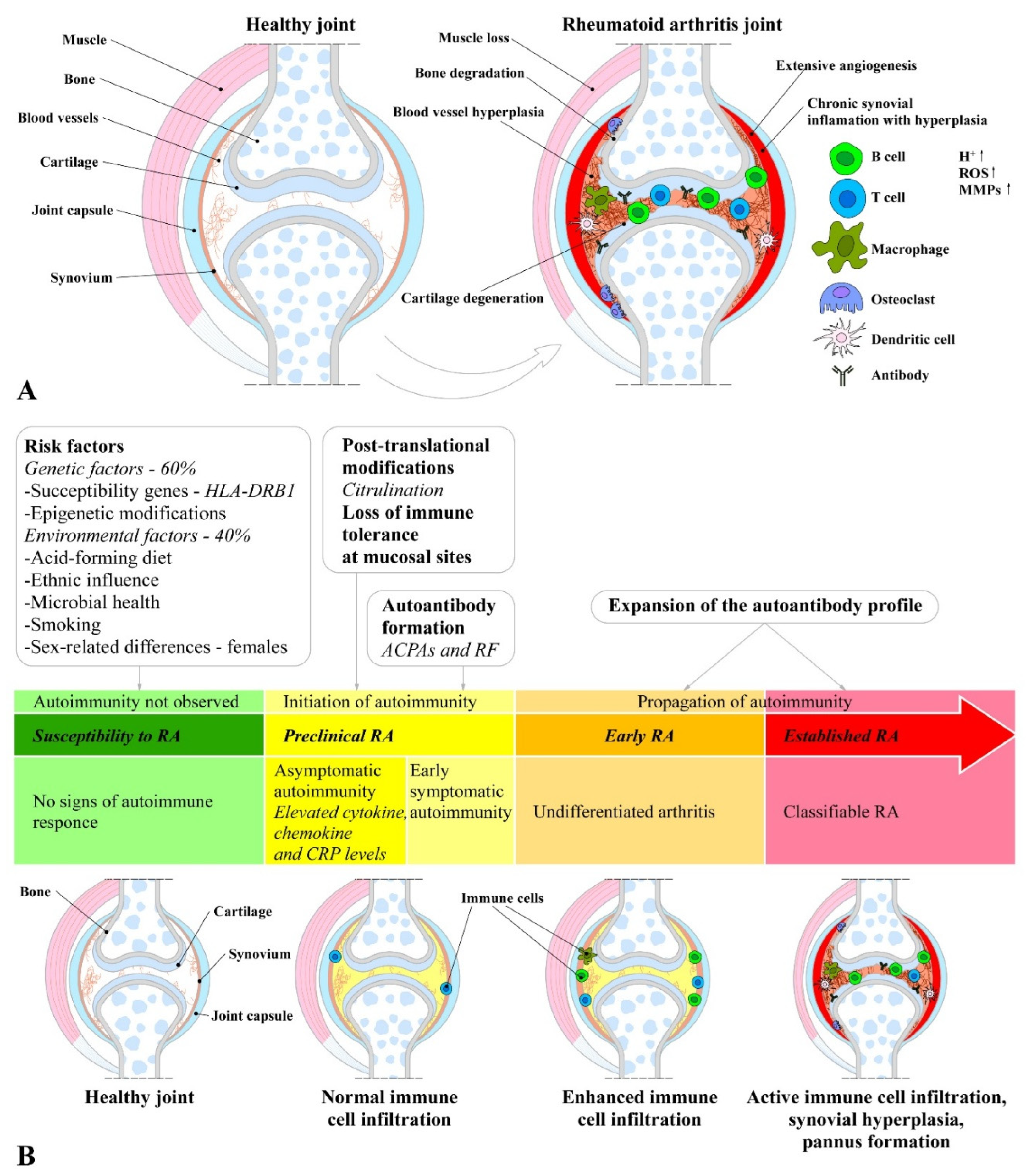
Figure 2.
Mechanisms involved in initiation and progression of RA. (A): the onset of autoimmunity is supposed to occur in the mucosa (e.g. mouth, lung and gut) by the production of neo-epitopes resulting of post-translational modifications, such as by citrullination or carbamylation; (B): the altered peptides (neo-peptides) are recognized by antigen-presenting cells (APCs) of the adaptive immune system and (C): are presented to adaptive immune cells in lymphoid tissues activating an immune response, and induce autoantibody formation (e.g. ACPA and RF); (C): activated immune cells and complexes activate synovial cells, such as fibroblast-like synoviocytes (FLS) and macrophage-like synoviocytes of the intimal lining and APCs in the sublining area, thus producing a range of inflammatory factors and resulting in expansion and formation of the cartilage- and bone-invasive pannus. The activation and infiltration of immune cells (T cells, B cells and macrophages) of the sublining area further contribute to the excessive production of inflammatory factors, auto-antibodies, and synovial vascular leakage, resulting in articular cartilage and subchondral bone destruction due to matrix-degrading enzymes and a de-balanced bone homeostasis characterized by an imbalanced RANKL/RANK/OPG system and activated osteoclasts.
Figure 2.
Mechanisms involved in initiation and progression of RA. (A): the onset of autoimmunity is supposed to occur in the mucosa (e.g. mouth, lung and gut) by the production of neo-epitopes resulting of post-translational modifications, such as by citrullination or carbamylation; (B): the altered peptides (neo-peptides) are recognized by antigen-presenting cells (APCs) of the adaptive immune system and (C): are presented to adaptive immune cells in lymphoid tissues activating an immune response, and induce autoantibody formation (e.g. ACPA and RF); (C): activated immune cells and complexes activate synovial cells, such as fibroblast-like synoviocytes (FLS) and macrophage-like synoviocytes of the intimal lining and APCs in the sublining area, thus producing a range of inflammatory factors and resulting in expansion and formation of the cartilage- and bone-invasive pannus. The activation and infiltration of immune cells (T cells, B cells and macrophages) of the sublining area further contribute to the excessive production of inflammatory factors, auto-antibodies, and synovial vascular leakage, resulting in articular cartilage and subchondral bone destruction due to matrix-degrading enzymes and a de-balanced bone homeostasis characterized by an imbalanced RANKL/RANK/OPG system and activated osteoclasts.
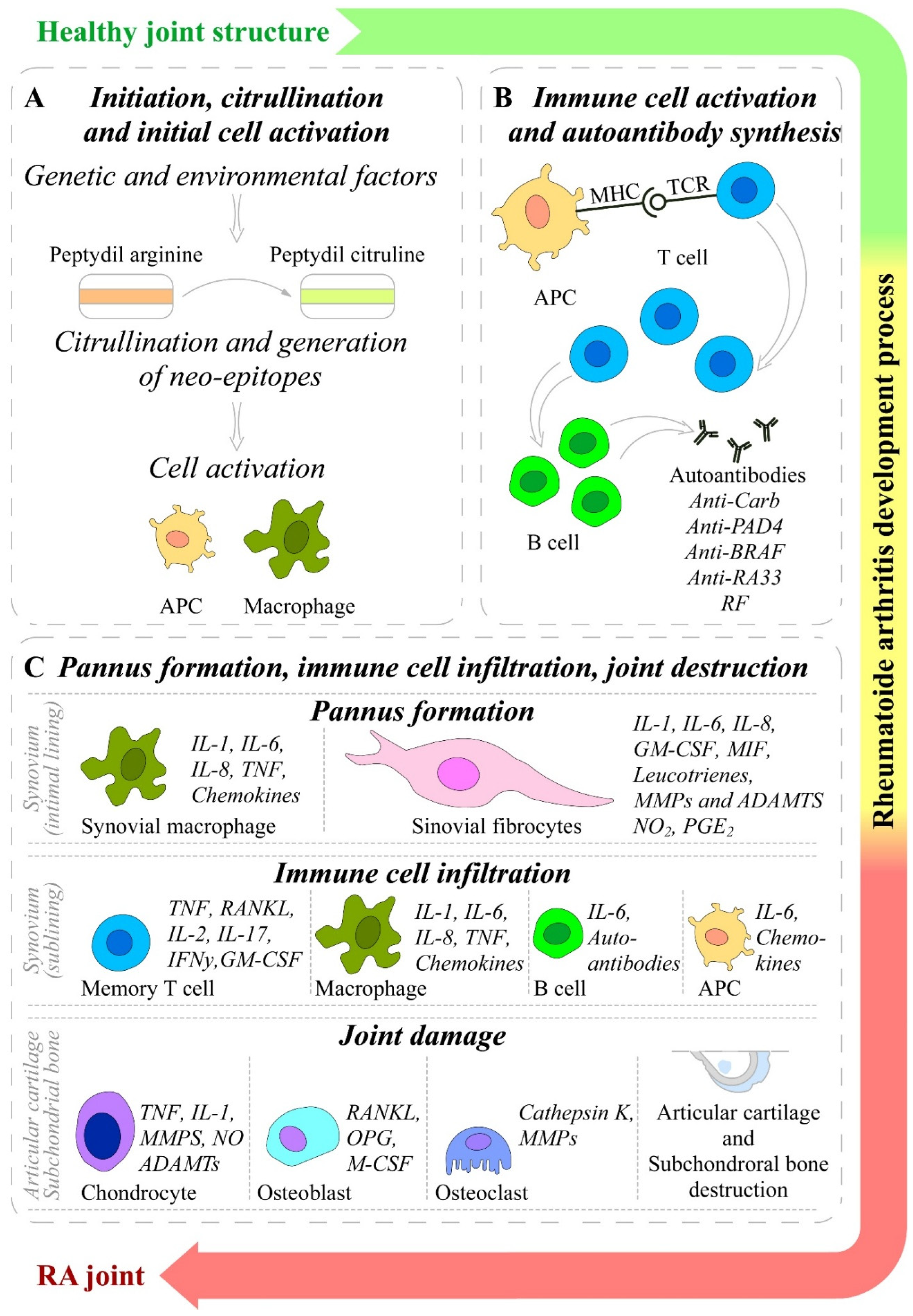
Figure 3.
Signaling network in RA and its possible inhibition by plant-derived molecules. The red lines indicate the blocked signaling pathways.
Figure 3.
Signaling network in RA and its possible inhibition by plant-derived molecules. The red lines indicate the blocked signaling pathways.
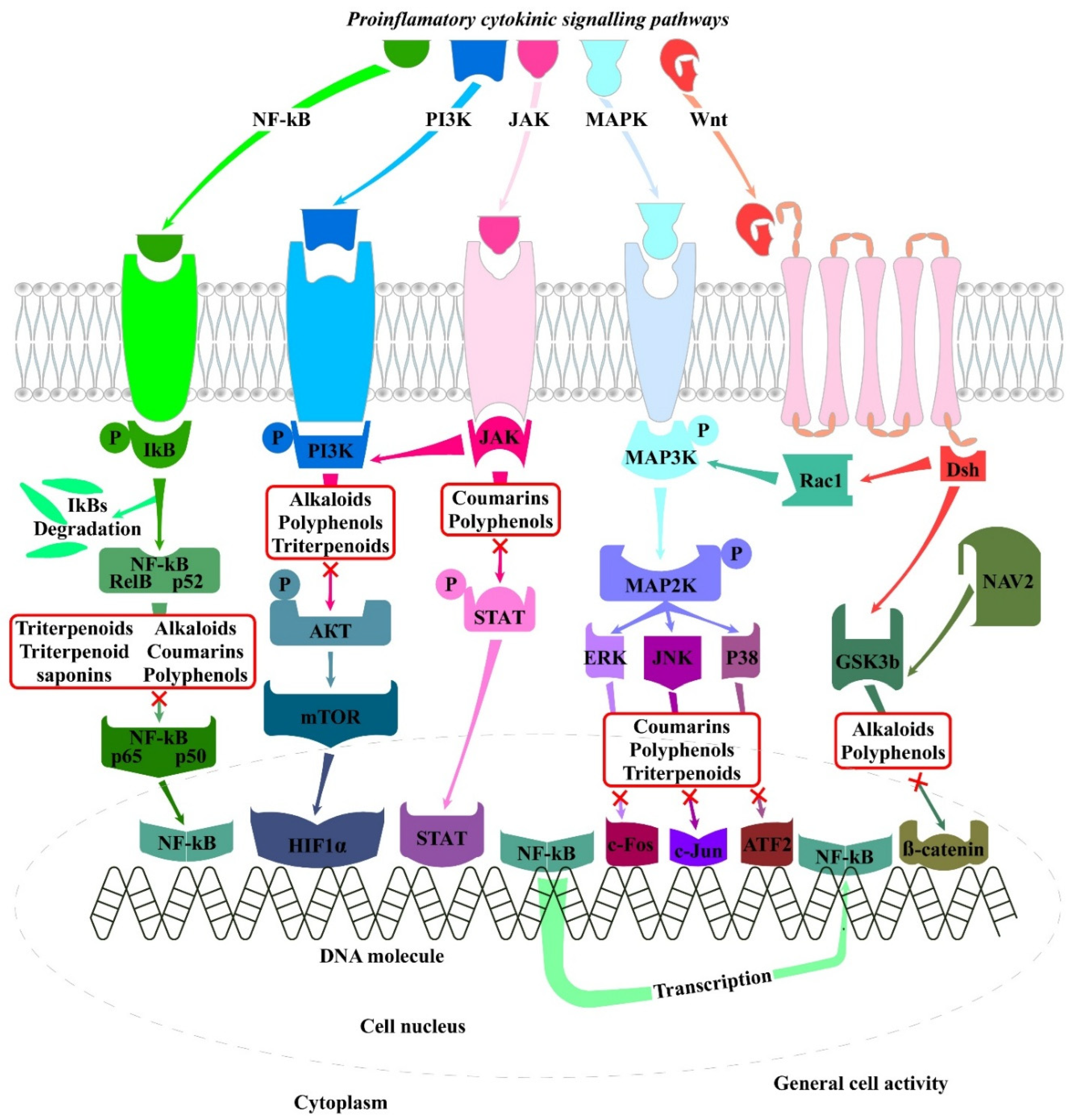
Table 1.
Molecular structure, administration and dose of the five FDA approved TNFi antibodies [213].
Table 1.
Molecular structure, administration and dose of the five FDA approved TNFi antibodies [213].
| Antibody | Molecular structure | Administration and dose |
| Infliximab (IFX, Remicade) | Chimeric IgG1 monoclonal antibody | Intravenous injections 3-10 mg/kg every 4-8 weeks |
| Etanercept (ETN, Enbrel) | Recombinant human fusion protein (TNF-α receptor bound to Fc fragment) | Subcutaneous injections 50 mg/week or 25 mg/twice a week |
| Adalimumab (ADA, Humira) | Recombinant human IgG monoclonal antibody | Subcutaneous injections 25 mg/twice a week |
| Golimumab (GOL, Simponi) | Human IgG monoclonal antibody | Subcutaneous injections 50 mg/month |
| Certolizumab Pegol (CZP, Cimzia) | Recombinant humanized Fab fragment |
Subcutaneous injections 400 mg at weeks 0, 2 and 4 followed by 200 mg/ every 2 weeks |
Table 3.
Mode of action of different herbal extracts reported to have beneficial effect on RA in animal models.
Table 3.
Mode of action of different herbal extracts reported to have beneficial effect on RA in animal models.
| Host | Biological agent | Mode of action | Reference |
| Mouse | Acori Graminei | Reduction of inflammation indicators including IL-6 and TNF-α | 96 |
| Mouse | Fruit peel extracts of Annona Squamosa L. |
Decrease in the leukocytes in the serum | 97 |
| Mouse | Saururus chinensis | Reduction of inflammatory cytokines | 98 |
| Mouse | Cudrania tricuspidata and Stewartia koreana | Decrease in inflammatory cytokine levels, NOS inhibitors | 99 |
| Mouse | Grape seed proanthocyanidin extract | Inhibition of TLR4/ MyD88/NF-κB signaling pathway. | 100 |
| Mouse |
Gleditsia sinensis thorn extract fermented by Lactobacillus |
Reduction of inflammatory cytokine levels | 101 |
| Mouse | Ellagic acid | Downregulation of pro-inflammatory cytokines and upregulation of anti-inflammatory cytokines |
102 |
| Rat | Duran wood bark extract | iNOS suppression/NOS inhibitor | 103 |
| Rat | Chloranthus serratus | Inhibition of the releases of inflammatory cytokines and amelioration of antioxidant capacity |
104 |
| Rat | Caragana sinica | Negative regulation of the NF-κB pathway |
105 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



