
Submitted:
12 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
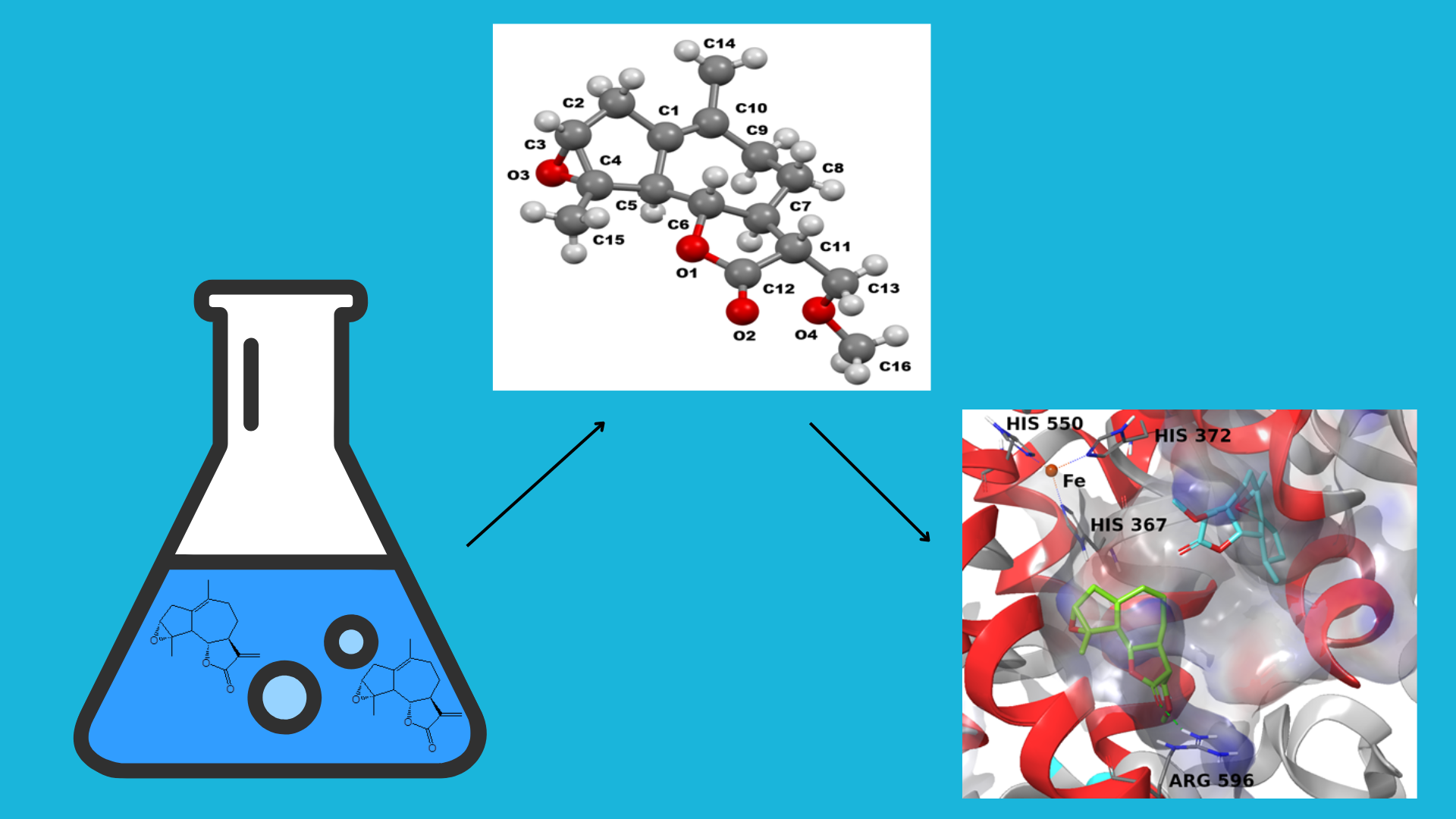
Data on the synthesis of new derivatives of 13-methoxy sesquiterpene lactone from the guiana series, including ludartin and arglabin, are presented. The structures of the synthesized compounds were studied using infrared (IR), ultraviolet (UV), and nuclear magnetic resonance (NMR) spectroscopy (1H and 13C). Chemical shifts, multiplicities, and integral signal intensities in the one-dimensional 1H and 13C NMR spectra were determined. The spatial structure of the new derivatives was determined using X-ray crystallographic analysis. Molecular docking of the obtained compounds with 5-lipoxygenase (PDB ID 6BP2) was performed. It was found that they could theoretically inhibit one of the enzymes of the proinflammatory cascade, 5-lipoxygenase (5-LOX). The molecule of 13-methoxyludartin (2) can participate in the formation of hydrogen bonds with amino acids of the substrate-binding cavity, as well as interact with amino acids of the alpha-helix stabilizing 5-LOX catalytic iron ion. These interactions lead to high stability of the 5-LOX-13-methoxyludartin complex in the first 70 ns of the molecular dynamics simulation.
Keywords:
sesquiterpene lactones
; ludartin
; arglabin
; X-ray
; 5-lipoxygenase
; molecular modeling
1. Introduction
Natural compounds, whether of animal or plant origin, have always attracted the attention of chemists, biologists, and pharmacists, primarily due to their biological activity. It is well known that many plants are used in folk medicine. Many substances of natural origin exhibit a wide range of biological activity and, as a rule, are characterized by low toxicity and do not cause side effects in comparison with similar synthetic drugs.
In this context, sesquiterpene lactones are of particular interest. They are mainly isolated from plants of the Asteraceae family [1,2,3,4,5]. They exhibit pronounced antimicrobial and antifungal activities, as well as antioxidant, attractant, anticancer, bactericidal, and fungicidal properties [6,7,8,9,10]. Today, the list of sesquiterpene lactones and their derivatives used in medical practice totals hundreds [11].
The biological activity of sesquiterpene lactones is primarily associated with the presence of various groups and atoms in their structure, which potentially enhance their pharmacological effects. For example, oxidation of the hydroxyl group in the artemisinin molecule to a keto group in dehydroartemisinin increases antiparasitic activity fourfold. The antiparasitic activity further increases eight times when the hydroxyl group is replaced with an acetyl group in the molecules of grosshemine and austricine. Currently, many sesquiterpene lactones with cytotoxic activity are known.
Chronic inflammation is involved in the pathogenesis of many diseases, including asthma, rheumatoid arthritis, atherosclerosis, and cancer [12]. The arachidonic acid (AA) pathway plays a central role in the inflammatory process, with 5-LOX being a key enzyme in this pathway. 5-LOX catalyzes the conversion of AK into leukotrienes (LT), powerful lipid mediators that promote inflammation, bronchoconstriction, and immune modulation [13].
Several 5-LOX inhibitors have been developed and studied over the past few decades. Among the most well-known is zileuton, a first-generation 5-LOX inhibitor approved for the treatment of asthma. Zileuton works by directly inhibiting the 5-LOX enzyme, thereby reducing the production of leukotrienes [14]. However, its use is limited by potential side effects, including hepatotoxicity, and the need for frequent dosing. Another notable inhibitor is MK-886, which targets the 5-lipoxygenase-activating protein (FLAP), a protein essential for 5-LOX activity. Although MK-886 showed promise in preclinical studies, its clinical development was halted due to insufficient efficacy in human trials [15]. More recently, compounds such as licofelone, a dual 5-LOX/COX inhibitor, have been investigated for their potential to provide broader anti-inflammatory effects by simultaneously targeting multiple pathways in the AA cascade [16].
In addition to synthetic inhibitors, natural products have garnered significant attention as potential 5-LOX inhibitors. Many plant-derived compounds, such as flavonoids, polyphenols, and alkaloids, have demonstrated potent 5-LOX inhibitory activity. For example, quercetin, a flavonoid found in apples, onions, and berries, has been shown to inhibit 5-LOX activity by chelating the iron atom in the enzyme's active site [17]. Similarly, curcumin, the active component of turmeric, exhibits anti-inflammatory properties partly through its ability to inhibit 5-LOX and reduce leukotriene production [18]. Other natural compounds, such as boswellic acids from Boswellia serrata and ginkgolides from Ginkgo biloba, have also shown promising 5-LOX inhibitory effects in preclinical studies [19]. These natural inhibitors often exhibit fewer side effects compared to synthetic compounds, making them attractive candidates for further development.
In connection with the above and continuing the previously initiated research [20], this paper describes the synthesis, structure and molecular modeling of new 13-methoxy derivatives of sesquiterpene lactones of the guaian series ludartin and arglabin.
2. Results and Discussion
Previously, dihydroartemisinin was synthesized by the reduction reaction with sodium tetrahydroborate in methanol, which has high antimalarial activity compared to the parent compound [21] and is an intermediate product for the production of a number of biologically active compounds. In order to study the reduction reaction of sesquiterpene lactones of ludartin guaianolides (3,4α-epoxy-5,7α(H),6,11β(H)-guai-1(10),11(13)- diene-12,6-olide) (1) [22] and arglabin (1,10β -epoxy-5.7α(H),6,11β(H)-guai-3(4),11(13)-diene-12,6-olide) 3 [23] the reaction of compounds 1 and 3 with sodium tetrahydroborate in methanol was carried out.It is known that when sesquiterpene lactones interact with sodium borohydride, the lactone cycle is restored. If the molecule has an exocyclic double bond in conjugation with γ-lactone carbonyl, then it is restored first [1].
In this work, the reduction reaction of sesquiterpene lactones 1 and 3 was studied under conditions of dissolution in methanol and tetrahydrofuran. It has been established that this reaction for compounds 1 and 3 takes place only in methanol.
The interaction of sesquiterpene lactone ludartin (1) with sodium tetrahydroborate in methanol produced a single product 2 with a melting point of 79-81 °C, Rf 0.5 (petroleum ether:EtOAc, 2:1). Absorption bands of the carbonyl group of the γ-lactone cycle at 1777 cm-1 are observed in the IR spectrum of molecule (2). The mass spectrum (2) contains a peak of the molecular ion m/z 278.1 [M]+, corresponding to the gross formula C16H22O4.
Scheme 1.
Synthesis of 13-methoxyludartin (2).
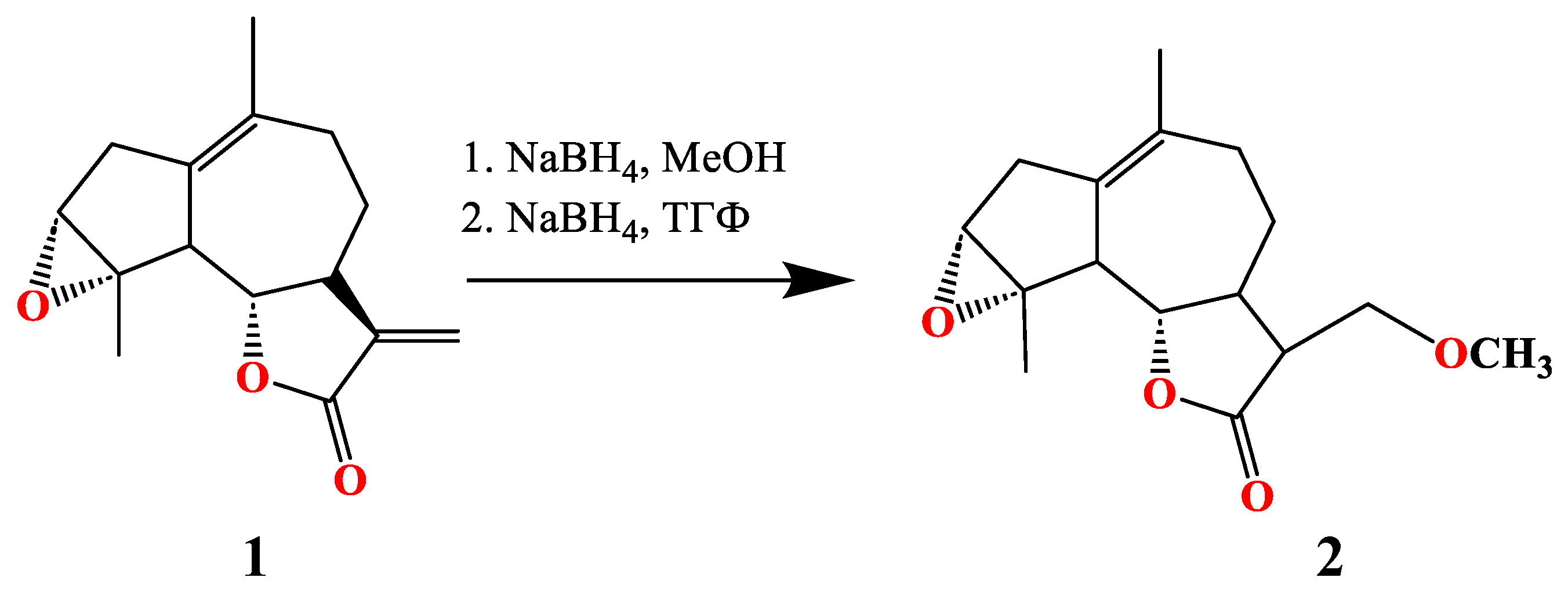
In the 1H NMR spectrum of molecule 2, a signal from the 11th carbon atom appears at 2.28 ppm as a multiplet, while at 3.35 ppm, a singlet corresponds to the methoxy group. The lactone proton signal overlaps with the 13th carbon atom and appears at 3.64 ppm as a multiplet. In the 13C NMR spectrum, an additional signal at 59.23 ppm appears as a quartet, corresponding to the methoxy group's carbon atom. The 11th and 13th carbon signals shift to somewhat weaker fields compared to the original molecule (where these signals were at δС 139.13 as a singlet and 117.72 as a triplet, respectively), now appearing at δС 52.11 as a doublet and δС 68.11 as a triplet.
By reduction of arglabin 3 with sodium borohydride in methanol, a mixture of compounds 4 and 5 was obtained. Through column chromatography separation, two colorless crystalline substances, 4 and 5, were isolated with melting points of 80–84°C and 138–140°C, respectively, and Rf values of 0.67 and 0.53 in petroleum ether:ethyl acetate (2:1).
Scheme 2.
Synthesis of 13α-methoxyarglabin (4) and 13β-methoxyarglabin (5).
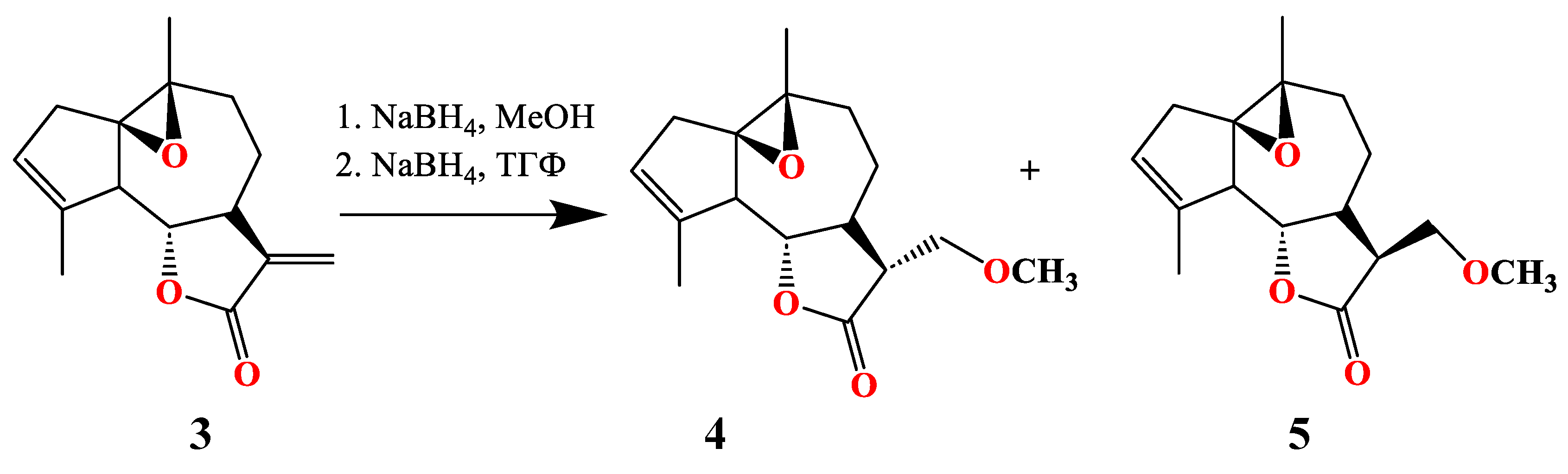
In the IR spectrum of compound 4, absorption bands characteristic of the γ-lactone carbonyl group are observed at 1772 cm⁻¹. The UV spectrum of 4 shows an absorption maximum at 204 nm. In the mass spectrum, compound 4 exhibits a molecular ion peak at m/z 278.1510 [M]+, corresponding to the molecular formula C₁₆H₂₂O₄.
In the 1H NMR spectrum of compound 4, signals corresponding to methyl groups are observed at a singlet of 1.90 ppm, associated with the double bond, and another methyl singlet at 1.30 ppm attached to C-10. The lactone proton appears as a triplet at 3.98 ppm with J = 6.0 Hz, while the olefinic proton resonates as a singlet at 5.52 ppm. The protons attached to C-13 show doublet of doublets at 3.67 ppm and 3.59 ppm, with coupling constants of 4.0 Hz and 3.0 Hz, respectively. The signal at 3.33 ppm, a singlet, is attributed to the methoxy group attached to C-13. In the 13C NMR spectrum, an additional signal at 59.17 ppm is characteristic of the carbon atom of the methoxy group.
In the IR spectrum of compound 5, absorption bands characteristic of the γ-lactone carbonyl group are observed at 1777 cm⁻¹. The mass spectrum shows a molecular ion peak at m/z 278.2 [M]+, corresponding to the molecular formula C₁₆H₂₂O₄. In the 1H NMR spectrum of compound 5, compared to molecule 4, the signal at C-7 is shifted to a more downfield position and appears at 2.55 ppm.
The structures of compounds 2 and 4 have been determined by X-ray diffraction. Their overall molecular conformations are shown in Figure 1.
Figure 1.
Crystal structure of ludartin (2) and arglabin (4) .
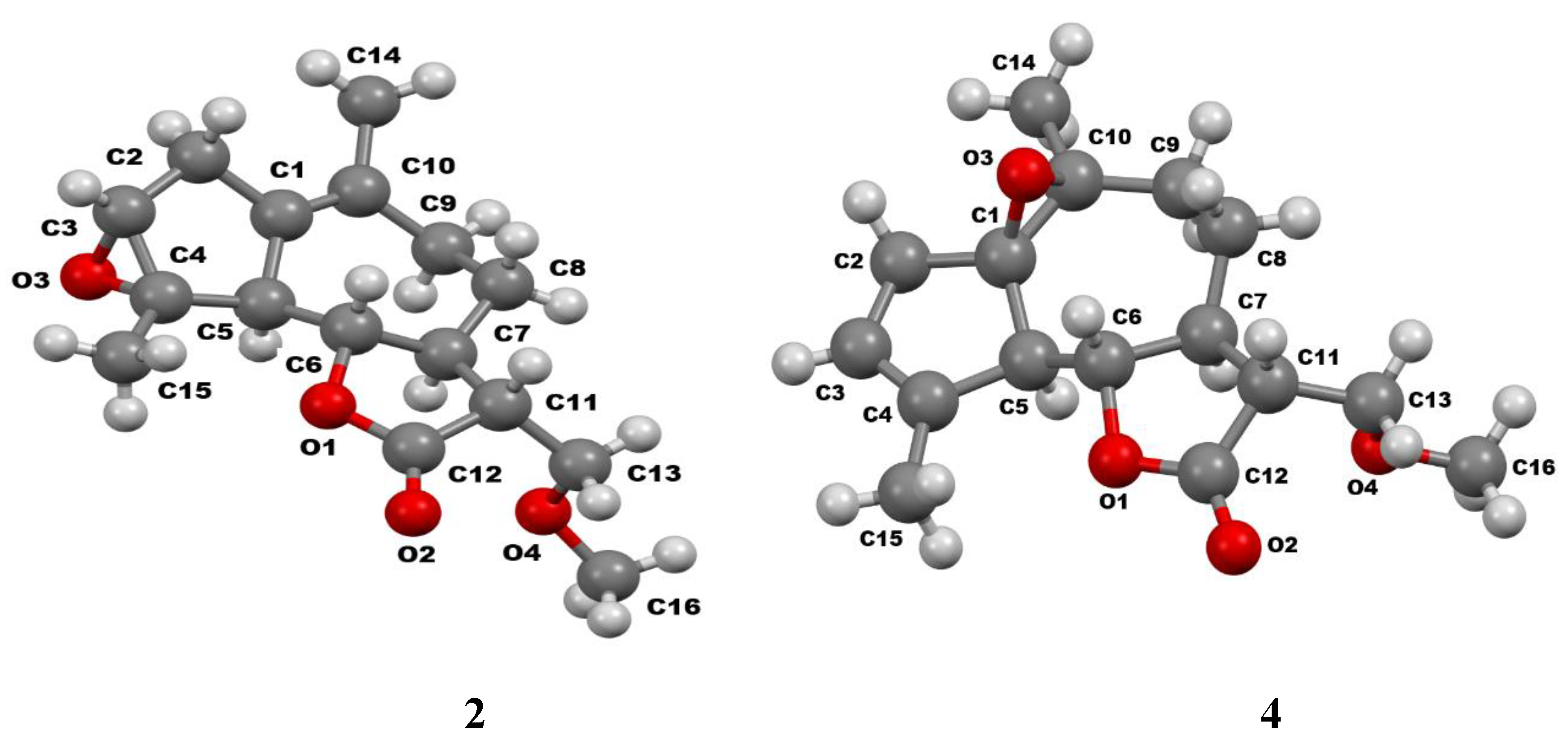
The X-ray data indicate that the bond lengths and valence angles in compounds 2 and 4 are close to the typical values as described in literature [24].
The five-membered (A) and seven-membered (B) carbocycles in molecule 2 are fused in a pseudo-trans configuration, with torsion angles C2–C1–C5–C4 and C10–C1–C5–C6 listed in Table 1. The molecules in compound 4 are fused via a trans configuration, with torsion angles O3–C1–C5–H5 measured at -142° and -144° for the two crystallographically independent molecules, 4a and 4b, respectively. The B-cycle and the lactone cycle (C) are fused in a trans configuration, with torsion angles H6–C6–C7–H7 equal to -172°, -168°, and -169° for compounds 2, 4a, and 4b, respectively.
The five-membered ring A in molecule 2 has a planar structure with an accuracy of ±0.03 Å, while in molecule 4, it adopts the conformation of a slightly distorted 5β envelope (ΔCS5 = 3.3 and 2.8° for 4a and 4b, respectively). The seven-membered ring B in molecules 2 and 4 assumes the conformation of a 7α, 1, 10β chair (ΔCS7=3.2, 2.2, and 1.3° for 2, 4a, and 4b, respectively), which is characteristic of guaianolides with an epoxy group on the C1–C10 bond or a C1 = C10 double bond. This is seen in compounds such as 3,4α-epoxy-4α-hydroxy-5α,7α,6,11β-(N)-gua-6,12-olide and micheliolide, which have a 4α-hydroxyl group at position 4. In compounds 2 and 4, the lactone ring C is in a 7α envelope conformation with varying degrees of distortion (ΔCS7 = 3.7, 5.2, and 3.2° for 2, 4a, and 4b). The intracyclic torsion angles in these molecules are shown in Table 1.
It should be noted that flattening of the five-membered ring in guaianolides with an epoxide group at positions 3,4 and a C1=C10 double bond has not been previously observed. According to the X-ray structural analysis data, the "standard" conformation in such molecules is a 5-envelope, as observed, for example, in arteglasin A (8α-acetoxy-3,4α-epoxy-5α,7α,6β(H)-guaia-1(10),11(13)-dien-6,12-olide) [6] and berlandine 8α-tigloxy-9β-acetoxy-3,4α-epoxy-5α,7α,6β(H)-guaia-1(10),11(13)-dien-6,12-olide) [7].

Table 1.
Intracyclic torsion angles (degrees) in structures 2 and 4.
| Torsion angles | Structure | ||
|---|---|---|---|
| 2 | 4a | 4b | |
| The cycle A | |||
| C5-C1-C2-C3 | -8.1(2) | 21.5(2) | 23.8(2) |
| C1-C2-C3-C4 | 6.7(2) | -11.9(2) | -13.6(3) |
| C2-C3-C4-C5 | -2.8(2) | -2.9(2) | -2.6(3) |
| C3-C4-C5-C1 | -2.2(2) | 16.3(2) | 17.4(2) |
| C2-C1-C5-C4 | 6.4(2) | -23.0(2) | -25.0(2) |
| The cycle В | |||
| C10-C1-C5-C6 | 58.1(2) | 49.2(2) | 47.7(3) |
| C1-C5-C6-C7 | -75.5(2) | -69.7(2) | -68.3(2) |
| C5-C6-C7-C8 | 71.1(2) | 76.0(2) | 76.2(2) |
| C6-C7-C8-C9 | -67.7(2) | -73.4(2) | -74.6(2) |
| C7-C8-C9-C10 | 76.3(2) | 68.4(2) | 69.7(2) |
| C8-C9-C10-C1 | -62.4(2) | -46.7(3) | -46.8(3) |
| C5-C1-C10-C9 | 1.1(3) | -2.1(3) | -2.4(3) |
| The cycle С | |||
| O1-C6-C7-C11 | -39.7(2) | -37.0(2) | -37.9(2) |
| C12-O1-C6-C7 | 27.2(2) | 26.4(2) | 25.9(2) |
| C6-O1-C12-C11 | -2.8(2) | -4.1(2) | -2.3(2) |
| C7-C11-C12-O1 | -22.6(2) | -19.7(2) | -21.8(2) |
| C6-C7-C11-C12 | 37.2(2) | 34.0(2) | 36.0(2) |
Thus, based on spectral data (mass, IR, 1H, 13C, 1H-1H, 1H-13C NMR), physicochemical constants, and X-ray structural analysis, the structure of molecule 2 is assigned as 3,4α-epoxy-13α-methoxy-5,7α(H),6,11β(H)-guaia-1(10)-ene-12,6-olide. Molecule 4 is identified as 1,10β-epoxy-13α-methoxy-5,7α(H),6,11β(H)-guaia-3-ene-12,6-olide, while the structure of compound 5 is established as 1,10β-epoxy-13b-methoxy-5,7α(H),6 β,11α(H)-guaia-3-ene-12,6-olide.
As a result of docking using the IFD algorithm, the values of the estimated parameters were obtained, which are presented in Table 2.
Table 2.
Molecular docking results of new compounds compared to the reference inhibitor NDGA.
| Ligand | Docking parameters, kcal/mol | ||
|---|---|---|---|
| Docking score | LE | Emodel | |
| NDGA | -7.876 | -0.358 | -51.104 |
| compound 4 | -5.892 | -0.295 | -17.585 |
| compound 2 | -5.365 | -0.282 | -37.619 |
The reference inhibitor NDGA demonstrates better docking parameters. The new compounds show similar docking score values, but compound 2 has a significantly higher estimated energy parameter (Emodel), which may indicate a higher probability of forming non-covalent interactions at the enzyme binding site. The distribution of the scoring function over non-hydrogen atoms (LE) of the new compounds is almost identical, reflecting their structural similarity, which leads to similar docking parameters at the binding site.
The reference inhibitor NDGA is located in the hydrophobic active site of 5-LOX in α-helical catalitic domain and blocks the oxidation of the catalytic iron ion Fe2+ to Fe3+, which is required for enzyme activation. Its interaction with the active site causes significant conformational changes, leading to disruption of the coordination of the catalytic iron ion with the surrounding amino acids (Gilbert et al., 2020). The hydroxyl groups of one of the catechol rings of NDGA interact with amino acids Arg596 and His600 of the substrate binding site, while the second catechol ring is located near the catalytic iron ion coordinated with three imidazole rings of histidines. Stacking interactions may occur between the catechol ring of the inhibitor and the imidazole ring of His372 (Figure 2A). The new compound 4 is located in the substrate-binding site of 5-LOX far from amino acids that can form hydrogen bonds with NDGA and does not form non-covalent interactions (Figure 2B). Tne new compound 2 is able to form a hydrogen bond with amino acid Arg596 as well as the NDGA (Figure 2C). The spatial arrangement of the best docking solutions in the binding site for the new compounds differs significantly, characterizing the differences in their molecular structure (Figure 3).
Figure 2.
Features of the spatial arrangement and non-covalent interactions of docking solutions of compounds in the 5-LOX binding site: A - NDGA, B - compound 4, C - compound 2. Interacting amino acids and the catalytic iron ion (orange sphere) are indicated. Non-covalent interactions are shown by dotted lines: green - hydrogen bonds, blue - stacking interactions. The boundaries of the binding site are shown by the surface colored depending on the electrostatic potential.
Figure 2.
Features of the spatial arrangement and non-covalent interactions of docking solutions of compounds in the 5-LOX binding site: A - NDGA, B - compound 4, C - compound 2. Interacting amino acids and the catalytic iron ion (orange sphere) are indicated. Non-covalent interactions are shown by dotted lines: green - hydrogen bonds, blue - stacking interactions. The boundaries of the binding site are shown by the surface colored depending on the electrostatic potential.
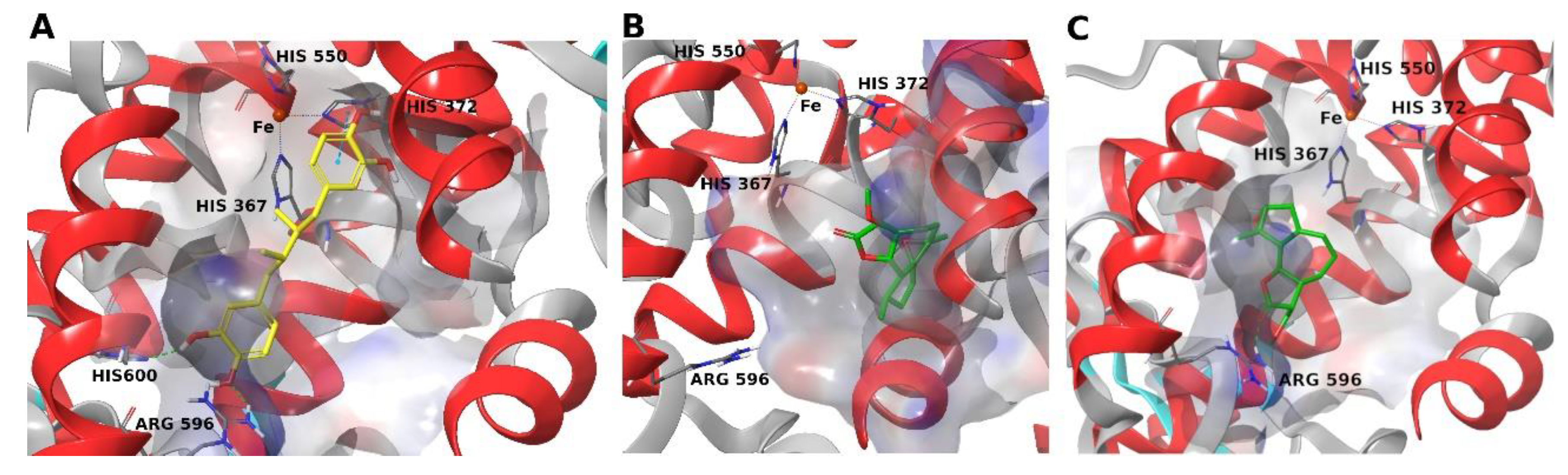
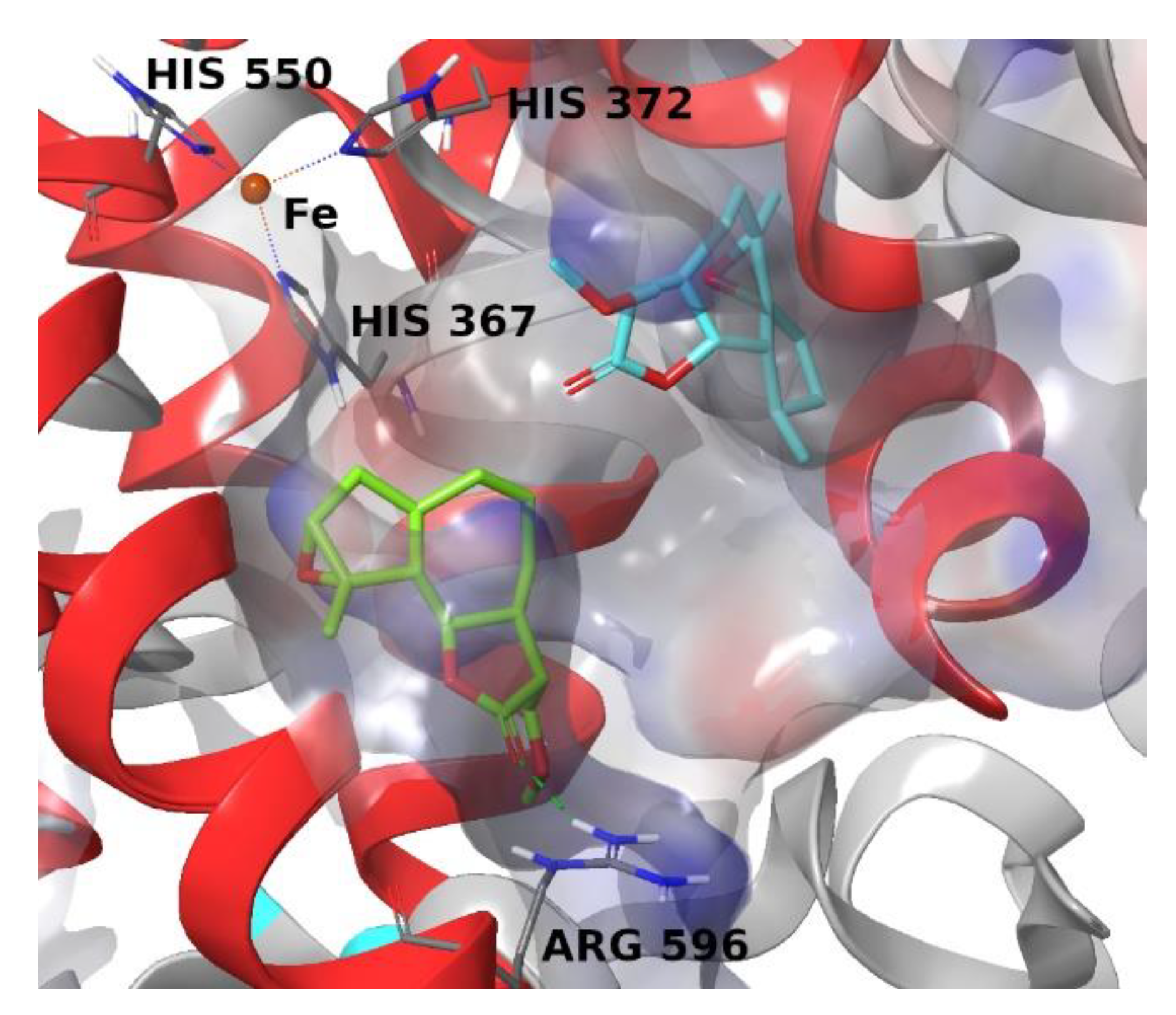
Figure 3.
Superposition of structures of new compounds in the 5-LOX binding site. Blue structure - compound 4, green structure - compound 2.
Figure 3.
Superposition of structures of new compounds in the 5-LOX binding site. Blue structure - compound 4, green structure - compound 2.
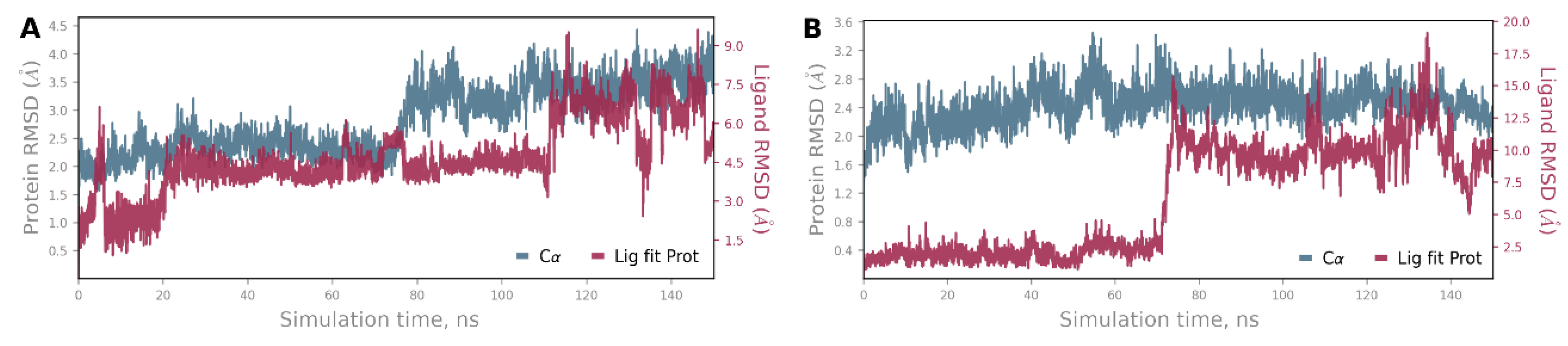
To study the stability of the protein-ligand complexes obtained as a result of molecular docking, as well as the nature and duration of their non-covalent interactions, 150 ns molecular dynamics simulations were performed. Compound 4 is stable for the first 20 ns of the simulation and then remains relatively stable until 110 ns of the molecular dynamics simulation (Figure 4A). After 110 ns of the simulation, compound 4 exhibits significant fluctuations in the enzyme binding site. Compound 2 exhibits strong stability for the first 70 ns of the simulation and then fluctuates markedly (Figure 4B). Interestingly, the enzyme structure remains significantly more stable when compound 2 is bound.
Figure 4.
Protein-ligand RMSD during simulation: A - compound 4, B – compound 2. The blue graph is the RMSD of the protein backbone atoms relative to the simulation starting point, the dark red graph is the RMSD of the ligand atoms relative to the simulation starting point.
Figure 4.
Protein-ligand RMSD during simulation: A - compound 4, B – compound 2. The blue graph is the RMSD of the protein backbone atoms relative to the simulation starting point, the dark red graph is the RMSD of the ligand atoms relative to the simulation starting point.
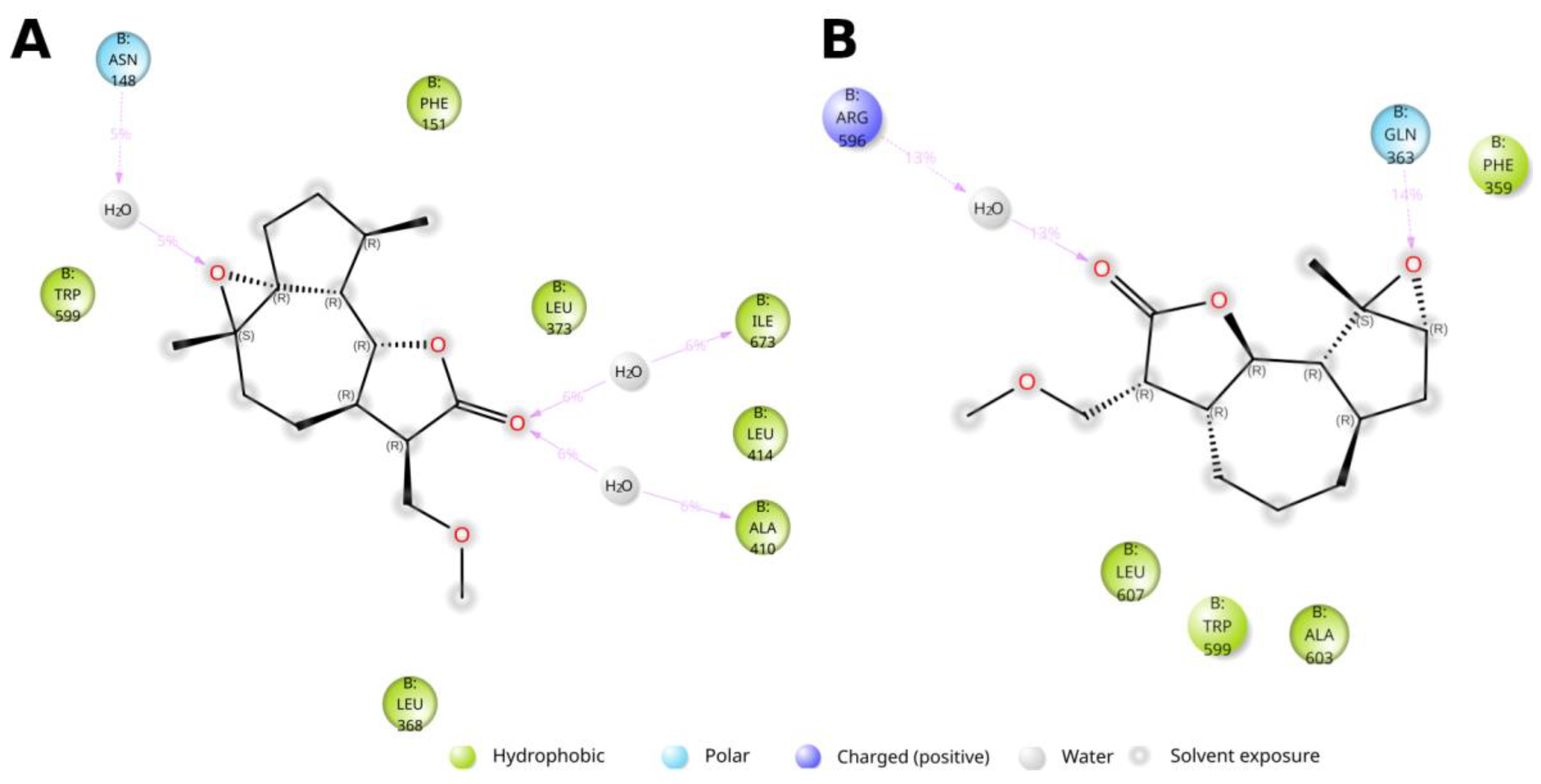
The pronounced exposure of both compounds to the solvent is noteworthy, which is associated with the large size of the binding site, in which the compounds can occupy a wide variety of positions, freely interacting with water molecules. For compound 2, it is possible to form a certain number of water bridges with hydrophobic amino acids and Asn148, which form the boundaries of the binding site, but their duration is not long (Figure 5A). Compound 2 shows longer contacts with the amino acids of the binding site (Figure 5B). Binding to Arg596 is realized through a water bridge. Of interest is the occurrence of a hydrogen bond with Gln363; this amino acid is part of the alpha-helix stabilizing the catalytic iron ion of the enzyme and is located near the histidines that coordinate it. The possibility of such a bond may introduce disturbances into the catalytic center of 5-LOX, inhibiting its function.
Figure 5.
Types and durations of non-covalent interactions of new compounds with amino acids of the enzyme binding site during the molecular dynamics simulation. Hydrogen bonds are indicated by purple arrows with their lifetime as a percentage of the simulation duration.
Figure 5.
Types and durations of non-covalent interactions of new compounds with amino acids of the enzyme binding site during the molecular dynamics simulation. Hydrogen bonds are indicated by purple arrows with their lifetime as a percentage of the simulation duration.
4. Materials and Methods
Melting points were determined using a Boetius apparatus. IR spectra were recorded on an Avatar 360 ESP instrument (Thermo Scientific, Boston, MA, USA) in KBr. UV absorption spectra were obtained with a Helios-β spectrometer (Thermo Scientific, Boston, MA, USA) in the 200–400 nm range. High-resolution mass spectra of the compounds were measured on a DFS (Double Focusing Sector) mass spectrometer by Thermo Electron Corporation with an ionizing voltage of 70 eV. NMR spectra of solutions in CDCl₃ were recorded on Bruker AV-300 (Bruker, Karlsruhe, Germany) (operating frequencies of 300.13 MHz for 1H and 75.47 MHz for 13C) and AV-600 (600.30 MHz for 1H and 150.96 MHz for 13C). The internal standards used were the solvent signals—deuterochloroform (δC 76.90 ppm) and the residual proton signal of chloroform (δH 7.24 ppm).
Reaction progress was monitored by thin-layer chromatography (TLC) on Silufol UV-254 plates using a solvent system of petroleum ether:ethyl acetate (2:1). The spots were visualized by treatment with an aqueous 2% KMnO₄ solution. Reaction products were isolated by column chromatography on silica gel of the "Armosorb" brand.
3.1. Synthesis of of 13-Methoxy Derivatives 2,4,5
3,4α-Epoxy-11α,13-methoxy-5,7α(H),6β(H)-guaia-1(10)-ene-12,6-olide (2). 300 mg (1.2 mmol) of ludartin 1 was dissolved in 10 mL of methanol, then NaOBH₄ (168 mg) was added portionwise over 10 minutes. The reaction was carried out under boiling conditions for 11 hours. The reaction mixture was neutralized with a 30% acetic acid solution. The reaction mixture was further extracted with CHCl₃ (3 × 40 mL). The organic layer was dried over MgSO₄, filtered, and the solvent was removed under reduced pressure on a rotary evaporator. The residue (0.32 g) was purified by column chromatography on 8 g of silica gel. The yield was 78%. The melting point was 83–85°C (petroleum ether:ethyl acetate). IR spectrum (KBr, ν, cm⁻¹): 2980, 2932, 2898, 2853, 2745, 1777 (C=O γ-lactone cycle), 1475, 1439, 1385, 1349, 1316, 1277, 1255, 1240, 1175, 1154, 1123, 1088, 1060, 1045, 1020, 1003, 980, 964, 937, 918, 887, 858, 825, 787, 722, 681, 648, 618, 608, 594, 541, 527, 480, 425. UV spectrum (λ, nm, lgε, EtOH): 202 (0.986). 1H NMR spectrum (δ, ppm, multiplicity, J/Hz): 2.68 (1H, dd, J=18.0, H-2a), 2.41 (1H, dd, J=18.0, H-2b), 3.27 (1H, singlet, H-3), 3.02 (1H, dd, J=10.0, H-5), 3.64 (1H, m, H-6 overlapping with H-13), 2.31 (1H, m, H-7), 1.95 (1H, m, H-8a), 1.20 (1H, m, H-8b), 2.18 (1H, m, H-9a), 2.01 (1H, m, H-9b), 2.28 (1H, m, H-11), 3.69 (1H, dd, J=4.0, H-13a), 3.64 (1H, dd, J=3.0, H-13b), 1.65 (3H, s, H-14), 1.60 (3H, s, H-15), 3.35 (3H, s, OCH₃). 13C NMR spectrum (δ, ppm): 133.28 (s,C-1), 33.38 (t,C-2), 63.67 (d,C-3), 67.06 (s,C-4), 51.72 (t,C-5), 80.25 (t,C-6), 47.06 (t,C-7), 27.35 (t,C-8), 34.13 (t,C-9), 135.27 (s,C-10), 52.11 (d,C-11), 175.22 (s,C-12), 68.11 (t,C-13), 22.42 (q,C-14), 18.95 (q, C-15), 59.23 (q, OCH₃).
1,10β-epoxy-11α,13-methoxy-5,7α(H),6β(H)-guaia-3(4)-ene-12,6-olide (4). 300 mg (1.2 mmol) of arglabin 3 was dissolved in 10 mL of methanol, then 168 mg of sodium borohydride was added in portions over 10 minutes. The reaction was carried out under boiling conditions for 11 hours. The reaction mixture was neutralized with a 30% acetic acid solution. Then, the mixture was extracted three times with CHCl₃ (3 × 40 mL). The organic layer was dried over MgSO₄, filtered, and the solvent was evaporated on a rotary evaporator. The residue (0.37 g) was purified by chromatography on a column with 8 g of silica gel. C₁₆H₂₂O₄, yield 61%. Melting point: 88–91°C (petroleum ether:ethyl acetate). IR spectrum (KBr, ν, cm⁻¹): 3041, 2976, 2929, 2887, 2852, 2833, 2738, 1771 (C=O of γ-lactone ring), 1475, 1443, 1433, 1384, 1353, 1337, 1319, 1288, 1251, 1213, 1185, 1170, 1155, 1130, 1105, 1086, 1058, 1028, 1012, 986, 960, 926, 916, 879, 863, 845, 822, 801, 786, 689, 659, 650, 626, 603, 573, 507, 482, 453, 432. UV spectrum (λ, nm, lgε, E in ethanol): 204 (1.538). 1H NMR spectrum (δ, ppm, J, Hz): 2.73 (1H, d, J=14.0, H-2a), 2.11 (1H, dd, J=14.0, H-2b), 5.52 (1H, s, H-3), 2.84 (1H, d, J=10.0, H-5), 3.98 (1H, t, J1=9.0, J2=19.0, H-6), 2.31 (1H, m, H-7), 1.71 (1H, m, H-8a), 1.43 (1H, m, H-8b), 2.06 (1H, m, H-9a), 1.95 (1H, m, H-9b), 1.79 (1H, m, H-11), 3.67 (1H, dd, J=4.0, H-13a), 3.59 (1H, dd, J=3.0, H-13b), 1.30 (3H, s, H-14), 1.90 (3H, s, H-15), 3.33 (3H, s, OCH₃). 13C NMR spectrum (δ, ppm): 72.36 (s, C-1), 39.45 (t, C-2), 124.55 (d, C-3), 140.55 (s, C-4), 52.16 (d, C-5), 82.55 (d, C-6), 46.74 (d, C-7), 22.57 (t, C-8), 33.34 (t, C-9), 62.51 (s, C-10), 48.55 (d, C-11), 176.18 (s, C-12), 68.04 (t, C-13), 22.60 (q, C-14), 18.13 (q, C-15), 59.17 (q, OCH₃).
1,10β-epoxy-11α,13-methoxy-5,7α(H),6β(H)-guaia-3(4)-ene-12,6-olide (5). C₁₆H₂₂O₄. Yield: 1.5%. Melting point: 138–140°C (petroleum ether:ethyl acetate). IR spectrum (KBr, ν, cm⁻¹): 3042, 3002, 2971, 2952, 2933, 2883, 2827, 2733, 1777, 1650, 1480, 1445, 1385, 1368, 1348, 1317, 1302, 1270, 1235, 1212, 1176, 1140, 1078, 1059, 1110, 1033, 1002, 964, 954, 890, 878, 790, 503, 433. 1H NMR spectrum (δ, ppm, J, Hz): 2.72 (1H, m, H-2a), 2.14 (1H, m, H-2b), 5.52 (1H, m, H-3), 2.76 (1H, s, H-5), 4.35 (1H, t, H-6, J=9.0, 19.0), 2.55 (1H, m, H-7), 1.83 (1H, m, H-8a), 1.64 (1H, m, H-8b), 2.09 (1H, m, H-9a), 1.96 (1H, m, H-9b), 1.91 (1H, m, H-11), 3.70 (1H, dd, J=4.0 Hz, H-13a), 3.53 (1H, dd, J=3.0 Hz, H-13b), 1.31 (3H, s, CH₃-14), 1.54 (3H, s, CH₃-15), 3.27 (3H, s, OCH₃).
3.2. Crystallographic Study:
The cell parameters and reflection intensities of crystals of compounds 2 and 4 were measured using a Bruker Kappa APEX2 CCD (Bruker, Karlsruhe, Germany) diffractometer (with a graphite monochromator, φ-scan mode). The analysis of the raw measured intensity data and absorption corrections were performed using the SAINT [25] and SADABS [26] programs, included in the APEX2 software package.
The structures were solved by direct methods. The positions of nonhydrogen atoms were refined anisotropically using full-matrix least squares. Hydrogen atoms were placed in geometrically calculated positions, and their positions were refined isotropically with fixed positional and thermal parameters (the "rider" model). The structures were solved and refined using the programs SHELXS [27] and SHELXL-2018/3 [28]. The main crystallographic data and characteristics of the X-ray diffraction experiments are presented in Table 3. CCDC 984881 for 2 and 984880 for 4 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk.
Table 3.
Structural information of the crystal of 2 and 4.
| Compound | 2 | 4 |
|---|---|---|
| Empirical formula | C16H22O4 | C16H22O4 |
| Formula weight | 278.34 | 278.34 |
| T, K | 296 | 173 |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | monoclinic | orthorhombic |
| Space group | P21 | P212121 |
| a, Å | 11.2615(4) | 9.4329(8) |
| b, Å | 5.6092(2) | 9.4525(7) |
| c, Å | 12.2180(5) | 33.065(3) |
| β, degree | 106.007(2) | 90 |
| Volume, Å3 | 741.86(5) | 2948.2(4) |
| Z | 2 | 8 |
| Calculated density,g/cm3 | 1.246 | 1.254 |
| Absorption coefficient, mm-1 | 0.088 | 0.089 |
| F(000) | 300 | 1200 |
| Crystal size (mm) | 0.58 × 0.35 × 0.08 | 0.54 × 0.37 × 0.09 |
| Theta range | 1.73 ≤ θ ≤ 27.21 | 1.23 ≤ θ ≤ 25.03 |
| Reflection collected/unique | 15330 / 3269 | 30235 / 5200 |
| R(int) | 0.0216 | 0.0644 |
| Absorption correction | multi-scan | multi-scan |
| Absorption correction, T(min, max) | 0.9535, 0.9920 | 0.9505, 0.9930 |
| Data/restraints/parameters | 3269 / 1 / 184 | 5200 / 0 / 369 |
| Goodness-of-fit on F2 | 1.093 | 1.046 |
| R1, WR2 (I ≥ 2σ(I) | 0.0351, 0.0899 | 0.0356, 0.0752 |
| R1, WR2 (all data) | 0.0456, 0.1046 | 0.0424, 0.0781 |
| Largest diff. peak and hole, е/Å3 | 0.149, -0.118 | 0.261, -0.259 |
3.3. Molecular Modeling
Molecular modeling was performed in the Schrodinger Maestro visualisation environment using the applications from the Schrodinger Small Molecule Drug Discovery Suite 2024-3. The three-dimensional structures of the new derivatives were obtained using quantum chemical optimization in the Jaguar program [29]. The density functional theory method B3LYP-D3 was applied using the basis set 26-31G**. The solvent was taken into account according to the Poisson-Boltzmann solvation model. The X-ray diffraction model of 5-LOX cocrystallised with nordihydroguaiaretic acid (NDGA), PDB ID 6BP2, resolution 2.71 Å, was used for calculations [30]. To simulate a possible mechanism of binding to the selected target, molecular docking was performed using the induced fit docking (IFD) protocol [31], which uses the Glide [32] and Prime [33] programs to predict the positions of ligands in the binding site, taking into account their effect on the target structure. The docking search area was determined automatically based on the size of the reference ligand NDGA. The algorithm of increased docking accuracy XP (extra precision) was used. The following conditions were applied: flexible protein and ligands, the docking area size of 20 Å and amino acids within 5 Å of the ligand were taken into account to optimise its effect. The docking results were ranked by evaluating the following calculated parameters: docking score (based on GlideScore with the exception of penalties that take into account energy parameters that negatively affect binding), ligand efficiency (LE, where the distribution of the evaluation function over the heavy atoms of the ligand is considered), the model energy value parameter (Emodel, including the GlideScore value, the energy of noncovalent interactions and the energy spent on the formation of the stacking of compounds and amino acids in the binding site). Docking was performed in comparison with the docking solution obtained for NDGA which was characterized by the smallest deviation from the coordinates in the X-ray structural model. Molecular dynamics studies were performed in the Desmond program [34] to study the stability of the obtained ligand-protein complexes. Ligand-protein complexes were placed in a virtual cube with a 15 Å buffer filled with a 0.15 M NaCl solution. The aqueous solvent model TIP3P was chosen. The NPT ensemble was utilized to simulate the system at a temperature of 310 K and a pressure of 1.01325 bar. The Nose–Hoover thermostat and a Martyna–Tobias–Klein barostat were used. Preliminary relaxation of the system was carried out for 1 ns. The simulation time was 150 ns, the number of frames was 5000, and the integrator step was 2 fs. Noncovalent interactions of compounds at the binding site were visualised using the Schrodinger Maestro.
4. Conclusions
This study describes the chemical synthesis of new 13-methoxy derivatives of sesquiterpene lactones from the guaianolide series, including ludartin and arglabin. The synthesized compounds were characterized using various spectroscopic methods, such as IR, UV, 1H NMR, and 13C NMR. The structures of the new sesquiterpene lactone derivatives were determined by X-ray diffraction analysis. Compound 2 may participate in hydrogen bonding with amino acids in the substrate-binding pocket, as well as interact with amino acids of the alpha-helix that stabilizes 5-LOX through the catalytic iron ion. This study contains important findings necessary for the identification of new biologically active compounds.
Author Contributions
Conceptualization, K.M.T and D.M.T; methodology A.S.M, D; software, A.S.M; validation, K.M.T, and K.B.K., A.K.; formal analysis, A.S.M; investigation, D.M.T., A.S.K; resources, D.M.T; data curation, K.B.K.; writing—original draft preparation, A.S.M.; writing—review and editing, D.M.T., А.Е.Kh., R.I.J.; visualization, K.B.K.; supervision, K.M.T; project administration, K.M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan (grant "Spatial structure and stereochemistry of derivatives of quinolizidine alkaloids and guaiane sesquiterpenoids", IRN AP23487966)
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available within the article and the Supplementary Materials. Further data are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rybalko, K.S. Natural sesquiterpene lactones, Medicine: Moscow, Russia, 1978; p.320.
- Fischer, N.H.; Olivier, E.J.; Fischer, H.D. The biogenesis and chemistry of sesquiterpene lactones. Fortschr. Chem. Org. Naturst. 1979, 38, 47–390. [Google Scholar] [CrossRef]
- Kagarlitskii, A.D.; Adekenov, S.M.; Kupriyanov, A.N. Sesquiterpene lactones of plants of Central Kazakhstan, Nauka: Almaty, Kazakhstan, 1987; p.240.
- Seaman, F.C. Sesquiterpene Lactones as Taxonomic Characters in the Asteraceae. The Botanical Review. 1982, 48, 121–595. [Google Scholar] [CrossRef]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2006, 23, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; Towers, G.H.N.; Mitchell, J.C. Biological activities of sesquiterpene lactones. Phytochemistry. 1976, 15, 1573–1580. [Google Scholar] [CrossRef]
- Parness, J.; Kingston, D.G.; Powell, R.G.; Harracksingh, C.; Horwitz, S.B. Structure-activity study of cytotoxicity and microtubule assembly in vitro by taxol and related taxanes. Biochem. Biophys. Res. Commun. 1982, 105, 1082–1089. [Google Scholar] [CrossRef]
- Søhoel, H.; Jensen, A.M.L.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Olsen, C.E.; Christensen, S.B. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 2810–2815. [Google Scholar] [CrossRef]
- Ramadan, M.; Goeters, S.; Watzer, B.; Krause, E.; Lohmann, K.; Bauer, R.; Hempel, B.; Imming, P. Chamazulene carboxylic acid and matricin: a natural profen and its natural prodrug, identified through similarity to synthetic drug substances. J. Nat. Prod. 2006, 69, 1041–1045. [Google Scholar] [CrossRef]
- Schmidt, T.; Nour, A.; Khalid, S.; Kaiser, M.; Brun, R. Quantitative structure ‒ antiprotozoal activity relationships of sesquiterpene lactones. Molecules. 2009, 14, 2062–2076. [Google Scholar] [CrossRef]
- Mashkovskii, M.D. Lekarstvennye sredstva. New wave: Moscow, Russia, 2024, p. 1216.
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Drazen, J.M.; Israel, E.; O’Byrne, P.M. Treatment of Asthma with Drugs Modifying the Leukotriene Pathway. New England Journal of Medicine 1999, 340, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Ford-Hutchinson, A.W.; Morton, H.E.; Gillard, J.W. MK886, a potent and specific leukotriene biosynthesis inhibitor blocks and reverses the membrane association of 5-lipoxygenase in ionophore-challenged leukocytes. Journal of Biological Chemistry 1990, 265, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Hornig, M.; Pergola, C.; Meindl, N.; Franke, L.; Tanrikulu, Y.; Dodt, G.; Schneider, G.; Steinhilber, D.; Werz, O. The molecular mechanism of the inhibition by licofelone of the biosynthesis of 5-lipoxygenase products. British Journal of Pharmacology 2007, 152, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Choi, J.S.; Islam, M.N.; Kim, Y.S.; Kim, H.P. Inhibition of 5-lipoxygenase and skin inflammation by the aerial parts of Artemisia capillaris and its constituents. Archives of Pharmacal Research 2011, 34, 1561–1569. [Google Scholar] [CrossRef]
- Pergola, C.; Werz, O. 5-Lipoxygenase inhibitors: A review of recent developments and patents. Expert Opinion on Therapeutic Patents 2010, 20, 355–375. [Google Scholar] [CrossRef]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discovery Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef]
- Adekenov, S.M.; Kagarlitskii, A.D. Chemistry of sesquiterpene lactones, Nauka: Almaty, Kazakhstan, 1990; p.188.
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; Ringwald, P.; Silamut, K.; Imwong, M.; Chotivanich, K.; Lim, P.; Herdman, T.; An, S.S.; Yeung, S.; Singhasivanon, P.; Day, N.P.J.; Lindegardh, N.; Socheat, D.; White, N.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Geissman, T.A.; Griffin, T.S. Sesquiterpene lactones of Artemisia carruthii. Phytochemistry. 1972, 11, 833–835. [Google Scholar] [CrossRef]
- Adekenov, S.M.; Mukhametzhanov, M.N.; Kagarlitskii, A.D.; Kupriyanov, A.N. Arglabin - A new sesquiterpene lactone from Artemisia glabella. Chem. Nat. Compd. 1982, 18, 623–624. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. J. Chem. Soc. Perkin Trans. 2. 1987, S1–S19. [Google Scholar] [CrossRef]
- SMART and SAINT, Area detector control and integration software. 2012, Bruker AXS Inc., Madison, WI-53719, USA.
- Sheldrick, G.M. SADABS. 2012, Bruker AXS Inc., Madison, WI-53719, USA.
- Sheldrick, G.M. A short history of SHELX. Acta Crystaollogr., Sect. A: Found.Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystaollogr., Sect. C: Structural Chemistry. 2015, 714, 3–8. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. International Journal of Quantum Chemistry 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Gilbert, N.C.; Gerstmeier, J.; Schexnaydre, E.E.; Börner, F.; Garscha, U.; Neau, D.B.; Werz, O.; Newcomer, M.E. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nature Chemical Biology 2020, 16, 783–790. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. Journal of Medicinal Chemistry 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. Journal of Medicinal Chemistry 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Structure, Function and Genetics 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06); IEEE; 2006; pp. 43–43. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



