
Submitted:
09 July 2025
Posted:
09 July 2025
You are already at the latest version
Abstract
Research focusing on targeting the LOX-1 receptor has gained increasing interest because of its established role in the development of cancer and arteriosclerotic diseases, which are among the main causes of human death. In this study, we employed an in silico approach involving molecular docking and molecular dynamics simulations to identify and characterize novel compounds that can target LOX-1 receptors under safe conditions. The results indicated that smilagenin was the compound with the best binding affinity for LOX-1, followed by cannabinol and prasterone. Analysis of protein‒ligand interactions suggested that smilagenin is the most stable of all the selected compounds and that it exhibited a different binding mode to the LOX-1 receptor than the other compounds did. Furthermore, analysis of their pharmacological properties revealed that smilagenin is the safest drug, followed by cannabinol. Moreover, the all-atom molecular dynamics simulation revealed that all the selected compounds are globally stable. These results provide new data on molecules that can target LOX-1 receptors via their respective binding modes. According to this study, all three compounds could be suitable candidates for targeting LOX-1 to treat cancer and arteriosclerosis, especially cannabinol, which showed the most promising results. However, further studies need to be performed to validate these results.
Keywords:
LOX-1 receptor
; Molecular docking
; molecular dynamics simulations
; Interactions
; In silico
1. Introduction
Lectin-like oxidized low-density lipoprotein (LOX-1), discovered in 1997, functions as a membrane scavenger receptor implicated in the internalization of oxLDL by endothelial cells [1]. Its tridimensional structure exists as a disulfide-linked homodimer of 52 kDa found in various cell types, including endothelial cells, macrophages, smooth muscle cells, fibroblasts, platelets, and neurons [2]. LOX-1 is a type II transmembrane protein comprising 273 residues and is characterized by four domains: a short N-terminal cytoplasmic domain (34 aa), a single transmembrane domain (26 aa), an extracellular region containing a coiled-coil domain known as NECK (82 aa), and a C-type lectin-like domain referred to as CTLD (131 aa) at the C-terminus [3,4]. The CTLD, which forms a heart-shaped coiled structure, is composed of antiparallel helix-flanked sheets stabilized by three intrachain disulfide bonds. Six highly conserved cysteine residues within CTLD monomers form an interchain disulfide bond, facilitating the formation of a central hydrophobic tunnel spanning the protein. This tunnel, which is lined with nonpolar amino acids such as tyrosine, aids in lipid transport [3]. In ligand recognition, the CTLD region of Lox-1 plays an important role [5]. This property is attributed to a positively charged CTLD amino acid terminal group involved in the interaction with negatively charged ligands, such as LDL [6]. CTLD inhibition has been shown to disrupt oxLDL binding ability [5]. Furthermore, investigations involving the successive deletion or substitution of basic backbone residues with alanine, which results in the inhibition of LDL association, have highlighted the importance of this terminal chain in the ligand binding mechanism [5,7]. Notably, occasional alterations, including the replacement of uncharged terminal residues for uncharged residues, eliminate LDL binding activity, suggesting that the negative charge of oxLDL cannot be engaged in the absence of positively charged residues in the CTLD (Shi et al., 2001). Moreover, dynamic structural modeling revealed that when arginine residues in the backbone are substituted with alanine, the conformation of CTLD changes, and its affinity for ligand binding is reduced [8].
The NECK domain is another important functional area of LOX-1. The NECK domain has been characterized for the first time as an 80-residue helical structure that links to the transmembrane domain and, independently, the CTLD via a disulfide interchain link [9]. Notably, the third region of the NECK is structurally less stable than the others and has been identified as the target of proteases responsible for cleaving LOX-1 between residues Arg88 and Gln89, releasing its soluble forms with a molecular weight of 34 kDa (sLOX-1) into the bloodstream [9,10] Interleukin-18 (IL-18) causes, at least partially, NECK domain cleavage and LOX-1 release in an ADAM10-dependent manner [11]. The integrity of the CTLD is critical for maintaining the spiral shape of the NECK domain, as indicated by the loss of stability in the NECK domain’s C-terminus caused by mutation of the CTLD residue C140 [9]
LOX-1 expression is increased in both cancer and atherosclerosis, the main cause of cardiovascular disease (CVD) [12,13,14]. Numerous studies have shown that LOX-1 overexpression increases the levels of intracellular reactive oxygen species (ROS), including superoxide anion (O2) and hydrogen peroxide (H2O). Superoxide anions interact with intracellular nitric oxide (NO), which protects against vascular damage, inflammation, and thrombosis. This situation has the potential to promote atherosclerosis and carcinogenesis by connecting ROS, metabolic problems, and cancer [15]. Furthermore, both in vitro and in vivo studies have shown a link between LOX-1 expression and the aggressiveness of different cancer types [13,16]. This protein has also been discovered in cardiomyocytes, where it is linked to the development of cardiac fibrosis and myocyte death, both of which contribute significantly to heart recovery after an ischemic event [17]. As a result, targeting LOX-1 has potential for therapeutic approaches aimed at improving outcomes in atherosclerosis and cancer [15,17]. Considering these findings, modifying or suppressing LOX-1 clearly offers a wide range of therapeutic options, from cancer to cardiovascular illness. To date, only one molecule, 9-chloranyl-5-propyl-11~{H}-pyrido [2,3-b][1,4]benzodiazepin-6-one (BI-0115), has been identified in vitro, characterized and recognized as an inhibitor of the LOX-1 receptor. Indeed, BI-0115 binding inhibits the dimer-forming receptor within the homodimer ligand binding range [18]. Furthermore, the dynamic properties of BI-0115-LOX-1 complexes have never been studied. The discovery of new inhibitors with optimized properties compared with those of BI-0115 could significantly contribute to the development of more effective medications.
Bioinformatics tools are crucial for detecting chemical molecules in various substances, aiding in the identification of novel bioactive molecules, and improving drug design [19]. Structure-based drug design and the quantitative link between macromolecular structure and function are expanding fields in which molecular modeling and docking approaches are becoming increasingly important (Du et al., 2016; Jian et al., 2018). These methods enable in silico evaluation of active compounds; assessment of inhibitory properties; prediction of drug similarity, physicochemical properties, ADME, and toxicological properties; and streamlining of the bioactive compound selection process. Furthermore, in silico evaluation of active compounds can help to predict drug similarity and assess inhibitory, physicochemical, ADME (absorption, distribution, metabolism, and elimination), and toxicological properties, streamlining the bioactive compound selection process (Akhtar et al., 2019; Verma et al., 2022). Molecular dynamics simulations are becoming increasingly important in structure-based drug development to understand the dynamic behavior of receptor–ligand complexes under physiological conditions [20]. Investigating the conformational changes and stability of LOX-1-ligand interactions could be highly important for the identification and validation of new and more effective molecules than BI-0115. The main objective of this research was to use molecular docking and molecular dynamics simulations to identify new inhibitors of the LOX-1 protein receptor. Additionally, the pharmacokinetic properties (ADME), biological activity score, and toxicological properties of the selected compounds were investigated.
2. Methodology
2.1. Protein Preparation
The structure of the LOX-1 receptor (PDB ID: 5w7d) was extracted from the protein database (https://www.rcsb.org/). The Protein Data Bank (PDB) is a database that contains information on experimental protein and nucleic acid structures. The Discovery Studio Visualizer (https://www.3dsbiovia.com/products/collaborative-science/bioviadiscovery studio/visualization-download.php) was used to remove water molecules from the protein. The Discovery Studio Visualizer is free and open-source software that can be used to construct molecular graphs.
2.2. Ligand Preparation
The structures of the twenty-five molecules and those of the recognized inhibitor of the LOX-1 receptor (Table 1) were extracted from the PubChem database. This database contains information on chemical compounds, including their structure, formula, and molecular weight (https://pubchem.ncbi.nlm.nih.gov/). The structures were initially retrieved in SDF format and then converted to PDB format via the software Open Babel (https://sourceforge.net/projects/openbabel/files/openbabel/2.4.0/).
2.3. Molecular Docking
Protein–ligand docking simulations were performed via AutoDock 4 (http://autodock.scripps.edu/downloads/autodock-registration/autodock-4-2-download-page/). AutoDock4 is a molecular docking tool that estimates free binding energy via a scoring formula based on the AMBER force field and linear regression analysis and a large library collection of known protein–ligand interactions and inhibition constants (Ravi and Krishnan 2016). The docking settings were set to 300, 27000 generations, 1000000 evaluations, and 100 genetic algorithm (GA) runs to ensure stability. Following the docking process, a postdocking analysis was performed to determine the best docking pose and associated energy values. Discovery Studio Client 21 was used to generate a three-dimensional representation of the docking data.
2.4. Prediction of Drug-like Properties and Bioactivity Scores
The drug likelihood characteristics of molecules that have the highest affinity for the LOX-1 receptor were determined via the Swiss ADME website [21]. We investigated the drug probability features of these various compounds via Lipinski’s [22] and Hopkins’ [23] methods. Lipinski’s concept is known as the “rule of fives,” and it is a well-known and often-used method for determining whether a substance is well absorbed orally. The “rule of five” is satisfied by a chemical having a molecular weight (MW) ≤500 daltons (Da), an octanol–water partition coefficient (iLOGP) ≤5, a number of hydrogen bond donors ≤ 5, and a number of hydrogen bond acceptors ≤10. Hopkins’s idea is known as the “QED concept,” and it offers four more physicochemical attributes in addition to the four given by the “rule of five” approach. The topological polar surface (20 to 130), the number of heavy atoms (15 to 50), the number of rotational bonds (0 to 5), and the number of alarms (PAINS and Brenk) for subunacceptable structures are additional attributes [20]. Additionally, to evaluate the bioactivity scores of the selected compounds, various molecular properties, such as enzymes, nuclear receptors, kinase inhibitors, GPCR ligands, ion channels, and modulators, were measured via the Molinspiration Toolkit version 2011.06 (www.molinspiration.com).
2.5. ADME Analysis
Before a medication candidate is approved, pharmacokinetic (PK) parameters such as absorption, distribution, metabolism, and excretion should be thoroughly studied [24]. The ADME characteristics of the chosen drug-like compounds were assessed through the utilization of SwissADME (www.swissadme.ch), an openly accessible web tool specifically designed for predicting the pharmacokinetics, physicochemical attributes, and medicinal chemistry of small molecules [21]. In this analysis, we considered the subsequent ADME characteristics, including water solubility, human gastrointestinal absorption, P-glycoprotein substrate, skin permeability, BBB permeability, and the effects of inhibitors of CYP1A2, CYP2D6, CYP2C9 and CYP3A4.
2.6. Toxicological Properties
To evaluate the potential toxicity of a pharmacological ingredient in human organs or cells, the estimated level of toxicity can be utilized. Hence, conducting toxicological tests is crucial during the development of a potential drug. To predict the toxicity levels of our specific compounds, we used the AdmetSAR online toolbox server (http://lmmd.ecust.edu.cn:8000/) and Protox II (https://tox-new.charite.de/protox_II/index.php?site=home) [25].
2.7. Molecular Dynamics Simulation
The best docking poses of smilagenin, DHEA, and cannabinol combined with the LOX-1 receptor were chosen as the initial coordinates for a 100 ns molecular dynamics simulation (MDS). The protein was isolated from the ligand via UCSF Chimera software (https://www.cgl.ucsf.edu/chimera/). GROMACS-2023 [26] was used for the computations, and the visual dynamics interface [27] was used to generate the scripts. Acpype [29], which depends on the ANTECHAMBER module [30], was used to produce the partial charges and ligand topologies after the Amber99 force field [28] was applied. The particle mesh Ewald (PME) technique was used to address electrostatic interactions, with a cutoff distance of 12 Å. A 2 Å margin was added around the outermost protein atoms in all Cartesian directions to automatically determine the dimensions of each system, which was simulated in a cubic box with periodic boundary conditions. Finally, TIP3P water molecules were used to solvate the box [31].
Next, a two-step energy minimization technique was carried out (2000 steps of steepest descent and 2000 steps of the conjugate gradient) until the system reached a resistance force of less than 1000 kJ.mol-1.nm-1. The Maxwell–Boltzmann distribution was then used to assign initial atomic velocities at 300 K. All systems were then equilibrated via two consecutive NVT and NPT equilibration simulations of 200 ps each. Following this phase, all systems were simulated with no restrictions at 300 K in the Gibbs ensemble with a pressure of 1 atm and isotropic coupling. The SHAKE algorithm was used to restrict any chemical linkages that contained hydrogen atoms [32], and the time step was set to 2 fs. Finally, we simulate three independent MD runs of 100 ns for each complex.
The GROMACS package’s utilities were used to process the simulation trajectories [26]. After the heavy atoms were aligned, the RMSD and RMSF were calculated separately for each system, and the starting structure of the production dynamics was used as a guide. Both the intramolecular hydrogen bonds inside the protein and those between the protein and ligand were examined. When the donor angle was greater than 120° and the distance between two polar heavy atoms (at least one of which included a hydrogen atom) was less than 3.5 Å, a hydrogen bond was observed. Furthermore, the protein’s solvent-accessible surface area (SASA), radius of gyration (RG), and secondary structure content were assessed.
3. Results and Discussion
3.1. Molecular Docking Analysis
The crystal structure of the LOX-1 receptor PDB ID: 5wd7d was used for molecular docking with AutoDock 4.0 (Figure 1). AutoDock simplifies this process by allowing the concurrent sampling of multiple configurations. This flexibility is essential for examining side chains, protein active sites, and receptor side chains. AutoDock evaluates the binding affinity of ligand–protein interactions, measuring the binding constant (Kd) and Gibbs free energy value (ΔGL). [33]. Molecular docking has been carried out to study the binding affinity and ligand interaction between LOX 1 and ligands. After performing the docking simulation, only the compounds with the highest binding energy were considered for further analysis. Docking analysis revealed a range from -4.89 to -8.56 kcal/mol, and the highest binding affinity was exhibited by smilagenin, prasterone, and cannabinol, with 8.56 kcal/mol, 8.37 kcal/mol, and 8.14 kcal/mol, respectively, compared with the control (Table 2). Compared with BI-0115, these compounds could function as good inhibitors, as good binding energy is a prerequisite for protein activity inhibition.
To study protein–ligand interactions, the docking poses of our three best-selected compounds with the control were analyzed with Biovia Discover Studio, and the results for protein–ligand interactions are shown in Figure 2 and Figure 3, respectively. The results showed that these interactions are particularly highlighted here by several categories of bonds:
Including hydrogen bonds (conventional hydrogen bonds, carbon hydrogen bonds), van der Waals interactions, hydrophobic interactions (Pi-alkyl and alkyl interactions, Pi-pi T-shaped and amide-Pi stacked interactions, Pi-sigma), electrostatic interactions (Pi-cation, Pi-anion), and some unfavorable bounds (donor-donor, positive-positive). All interactions between amino
The acid residues of the LOX-1 receptor and the selected compounds are described below (Table 3). Overall, we observed that the interactions between LOX-1 and the selected compounds are dominated by weak hydrophobic interactions. However, among the three selected compounds, smilagenin has the highest number of conventional hydrogen bonds when compared with the control BI-0115 and other selected compounds (Table 3). This high number of hydrogen bonds outlined with smilagenin could explain its higher binding affinity than the other compounds, as it has been reported that hydrogen bonds are stronger after covalent bonds and play a key role in binding affinity and molecular recognition in protein–ligand complexes [34]. Moreover, the results revealed that, compared with the two other compounds, only the binding mode of cannabinol was close to that of the control, as we identified only three different residues, namely, Arg124, Ser74, and Gln68. (Table 3).
3.2. Prediction of Drug-like Properties
The selected chemicals, cannabinol, prasterone, and smilagenin, were used to identify their drug likeliness on the basis of the proposals of Lipinski and Hopkins. The results of the analysis revealed that these compounds met Lipinski’s rules, with a molecular weight (MW) ≤500 Da, an octanol–water partition coefficient (iLOGP) ≤5, ≤ 5 hydrogen bond donors, and ≤10 hydrogen bond acceptors (Table 4). These compounds follow both the Lipinski and Hopkins standards, with a TPSA ranging from 20--130, several heavy atoms ranging from 15--50, several rotatable bonds ≤5, and no undesirable structural alerts, except for prasterone, which presented one break alerting an isolated alkene (Table 4). ADMET analysis (absorption, distribution, metabolism, excretion, and toxicity) is the most crucial and challenging step in drug design and development, with approximately 60% of all drugs failing to meet the requirements of the clinical phase [35]. Taken together, these results lead us to conclude that cannabinol and smilagenin are the most promising compounds that could be druggable. Importantly, Smilagenin has previously undergone phase 1 and 2 clinical trials with success, where it was investigated for the treatment of Parkinson’s and Alzheimer’s diseases. It is a neurotrophic factor inducer that works by stopping the depletion of dopamine receptors and neuronal growth factors in the brain as well as the free radical neurotoxicity that 1-ethyl-4-phenylpyridium (MPP+) causes in dopaminergic neurons [36]. It could be a very promising drug for targeting LOX-1 for cardiovascular disease and cancer treatment.
The bioactivity scores anticipated by Molinspiration of the top three compounds are displayed in Table 5. Molecules with higher bioactivity scores are more likely to be effective as medications [37]. A chemical with a bioactivity score greater than 0.00 is more likely to have substantial biological activity, whereas a score between -0.50 and 0.00 is moderately active, and a score less than -0.50 is considered inactive [38]. These results suggest that a compound’s physiological activities may be influenced by diverse mechanisms. It could also be associated with interactions with GCPR ligands, ion channel modulators, nuclear receptor ligands, protease inhibitors, and other enzyme inhibitors. On the basis of bioactivity ratings, the most promising compounds are cannabinol and smilagenin, as they can exhibit five mechanisms compared with those of prasterone and the control, which can achieve only four.
3.3. ADME Property Analysis
One of the key determinants in drug validation and development is the identification of pharmacokinetic (PK) properties, as it helps in understanding the characteristics of an efficient oral drug, such as its rate of absorption from the gastrointestinal tract, its ability to be well transmitted to the site of action, and its metabolism and excretion from the organism without causing any side effects. Many drugs in clinical trials today fail to enter the market because of poor PKs. Today, computational predictions are currently used to analyze the pharmacokinetic profiles of potential drug candidates, with the main objective of selecting only drug-like chemicals with satisfactory PKs [39]. SwissADME and AdmetSAR have been used here to enter the canonical SMILES of our three selected compounds, and the resulting values of the PKs are presented in the table below (Table 6). This analysis revealed that the compound prasterone is more soluble in water than canabinol, the control, and smilagenin, which are moderately soluble and poorly soluble, respectively, in water. All three selected compounds have a high ability to be absorbed through the gastrointestinal tract as controls. In addition, cannabinol is perceived to be a P-gp substrate in contrast to the control, prasterone, and smilagenin. In terms of skin permeability, all three compounds exhibited a high degree of permeability, as their values were greater than -2.5 cm/s. Furthermore, all the selected compounds were able to penetrate the blood–brain barrier (BBB). Cytochrome P450 proteins play an active key role in drug metabolism and subsequently contribute to drug detoxification and pharmacological impact [40]. Only prasterone and smilagenin are noninhibitors of the cytochrome enzymes CYP1A2, CYP2D6, CYP2C9, and CYP3A4. Like the control, cannabinol is an inhibitor of two cytochrome enzymes. This means that, globally, all our selected compounds, especially smilagenin and prasterone, can be metabolized very quickly by the body with reduced toxicity.
3.4. Toxicological Properties
Toxicity prediction is an important phase in drug design since it helps to identify the harmful effects of a potential drug on humans, plants, and the environment [41]. Instead of relying on experimental investigations, several computer tools may now assist in forecasting the toxicological features of specific chemicals in a short period. In this study, AdmetSAR and Protox-II were used to predict the toxicity of our chosen compounds (Table 7). The findings revealed that none of the compounds presented Ames toxicity or carcinogenicity as controls. However, among the selected compounds, only prasterone has been shown to be hepatotoxic and sensitive to the skin. On the other hand, only cannabinol has been revealed to be an inhibitor of the hERG gene. The hERG ion channel is crucial for regulating the electrical activity of the human heart. Disruptions in these channels can cause drug-induced delayed ventricular depolarization and arrhythmia, leading to cardiotoxicity. [42,43]. In terms of the predicted acute oral toxicity values, cannabinol and smilagenin are class III, whereas prasterone is class IV. All the selected compounds have high LD50 values except smilagenin, which has a value of less than 5000 mg/kg and is generally considered suitable from a druggable point of view.
3.5. Molecular Dynamics Simulation Analysis
Molecular dynamics simulations were employed to investigate structural changes and stability within protein–ligand complexes. The examination of structural stability, compactness, and protein expansion volume is essential for interpreting data during molecular dynamics (MD) simulations. This is typically achieved by calculating the RMSD, RMSF, Rg, and SASA [44]. Therefore, we evaluated the structural stability by comparing the RMSD values of the backbone of the initial structure of the three selected ligands with those of the control. The control had an average RMSD of 2.98 Å, whereas prasterone, smilagenin, and cannabinol had average values of 3.08 Å, 1.02 Å, and 2.73 Å, respectively (Figure 4). Differences in RMSD measurements indicated varying levels of stability between the control and the other ligands. Considering these results, it can be inferred that cannabinol is more stable when bound to the LOX-1 receptor, as its RMSD value is lower than that of the control. Smilagenin also has a lower RMSD value than the control. However, the RMSD plot did not converge over the simulation time of 100 ns, as it continues to increase with time, which means that smilagenin could be very unstable. Although prasterone exhibited a higher RMSD value than the control, the difference was not substantial, suggesting that it also presented good stability when complexed with the receptor.
Furthermore, RMSF values were determined to assess residual vibration in the LOX-1 receptor following the binding of various ligands and to better understand how ligand binding affects protein flexibility. Random residual variations were observed between the N-terminus and C-terminus. The control had an RMSF value of 1.64 Å, with a maximum variation of 3.78 Å between (100--110) amino acid residues. The selected compounds have average RMSFs of 1.46 Å, 1.44 Å, and 1.49 Å for prasterone, smilagenin, and cannabinol, respectively (Figure 5). These values are lower than the values of the control, indicating low flexibility of the protein structure and strong interactions in protein–ligand complexes. The chosen compounds had a maximum variation between the 100--110 amino acid residues of 2.67 Å for prasterone, 3.34 Å for smilagenin, and 3.56 Å for cannabinol. These oscillations could be explained by the formation of a beta-sheet secondary structure in that interval (100--110).
The radius of gyration (Rg) was used to assess the compactness and stiffness of the protein–ligand complexes. During simulation, the fluctuation rate has an inverse relationship with a system’s compactness and stiffness. The average Rg for the control was 14.77 Å, whereas the substances prasterone, smilagenin, and cannabinol had average values of 14.99 Å, 14.91 Å, and 14.89 Å, respectively (Figure 6). Overall, we infer that all of the chosen ligands might form a more compact and stiffer complex than the control since there is no significant difference in their Rg values, which remain steady during the simulation time.
The number of hydrogen bonds (H-bonds) generated between the various ligands and the LOX-1 receptor during the 100 ns molecular dynamic simulation was also determined (Figure 7). Our data show that, compared with the two chosen compounds prasterone and cannabinol, the control produced fewer H-bonds with the protein, with an average of ̴ 1H-bonds. Smilagenin establishes an average of 2 H-bonds with the protein, making it more stable and stiffer than the control and the other two compounds. It has been reported that hydrogen bonds are stronger after covalent bonds and play a key role in binding affinity and molecular recognition [34].
SASA analysis was performed to assess how the complex surface area expanded throughout the simulation. Higher SASA values indicate that the protein volume is expanding and that low-level variation is required throughout the simulation. The control SASA value was found to be 7955.53 A2. Figure 8 shows that the system remained constant during the simulation period, with SASA values of 8086.69 A2, 8045.88 A2, and 8133.94 A2 for prasterone, smilagenin, and cannabinol, respectively. These findings demonstrated that all three chosen compounds were stable following interaction with the LOX-1 receptor since their SASA values were closer to those of the control.
The key factor causing conformational changes in proteins is the amount of residual secondary structural dynamics. Therefore, to understand the conformational behavior and folding mechanism of the LOX-1 receptor, we needed to examine the secondary structure dynamics of this protein in the presence of all the selected ligands and the control. As a function of time, the bend, helix, turn, beta-ladder, beta-bridge, loop, helix 3–10, and kappa-helix secondary structural components of the LOX-1 receptor varied from 0.0 to 35% at each time point (Figure 9). Figure 9 shows that, throughout the simulation, the secondary structure of the LOX-1 receptor did not significantly vary in composition, suggesting that, globally, all the complexes are very stable.
4. Conclusions
In the past few years, many studies have been conducted on the LOX-1 receptor, and its role in both cancer progression and atherosclerosis progression, which has been established, has attracted particular attention from the scientific community. Therefore, identification and characterization of molecules that can inhibit this enzyme or reduce its expression level so that it can serve in drug development against these diseases are urgently needed. The present study focused on the identification and characterization of new inhibitors of the LOX-1 protein receptor via molecular docking, molecular dynamics simulation, and pharmacotoxicological property analysis. Autodock 4.0 has been used to process molecular docking data and elucidate protein–ligand interactions. The docking results revealed that, globally, the highest binding affinities were exhibited by smilagenin, prasterone, and cannabinol, with 8.56 kcal/mol, 8.37 kcal/mol, and 8.14 kcal/mol, respectively, compared with that of the control, which was 6.55 kcal/mol. Moreover, the protein–ligand interactions revealed that smilagenin is the most stable, as it established more hydrogen bonds than the other compounds did. It has also been shown that only cannabinol has a binding mode similar to that of the control, with a few differences in three residues, namely, Arg124, Ser74, and Gln68. Furthermore, the analysis of the pharmacotoxicological properties and bioactivity scores was carried out via SwissADME, AdmetSAR, Protox-II, and Molinspiration software. Cannabinol and smilagenin may be the most druggable compounds. Finally, we analyzed the RMSD, RMSF, Rg, H-bonds, SASA, and protein secondary structure content of the LOX-1 receptor during 100 ns and 20 ns molecular dynamic simulations, respectively, with Gromacs 2023 with interface visual dynamics. It has been reported that all the complexes are globally stable and rigid. Together, these results showed that all these compounds can be druggable. The present findings can serve as the basis for new drug development work. However, for our next study, we propose testing our new compounds in vitro and in vivo to further validate their inhibitory efficiency and safety compared with those of the reference LOX-1 inhibitor. Additionally, we recognize the need to investigate the chemical composition and hydrogen function of these compounds to better understand their mechanisms of action. Evaluating other physicochemical factors is also essential for assessing their impact on bioavailability in biological systems.
Funding
This work was not supported by funding or grants.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors confirm that the data supporting the findings are available within the article.
Conflicts of Interest
The authors report that there are no competing interests to declare.
References
- T. Sawamura et al., “An endothelial receptor for oxidized low-density lipoprotein,” Nature, vol. 386, no. 6620, Art. no. 6620, 1997. [CrossRef]
- J. L. Mehta and D. Li, “Identification, regulation and function of a novel lectin-like oxidized low-density lipoprotein receptor,” Journal of the American College of Cardiology, vol. 39, no. 9, Art. no. 9, 2002. [CrossRef]
- H. Park, F. G. Adsit, and J. C. Boyington, “The 1.4 Å Crystal Structure of the Human Oxidized Low Density Lipoprotein Receptor Lox-1,” Journal of Biological Chemistry, vol. 280, no. 14, Art. no. 14, 2005. [CrossRef]
- Q. Xie et al., “Human Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Functions as a Dimer in Living Cells,” DNA and Cell Biology, vol. 23, no. 2, Art. no. 2, 2004. [CrossRef]
- M. Chen, S. Narumiya, T. Masaki, and T. Sawamura, “Conserved C-terminal residues within the lectin-like domain of LOX-1 are essential for oxidized low-density-lipoprotein binding,” Biochemical Journal, vol. 355, no. 2, pp. 289–296, 2001. [CrossRef]
- I. Ohki et al., “Crystal Structure of Human Lectin-like, Oxidized Low-Density Lipoprotein Receptor 1 Ligand Binding Domain and Its Ligand Recognition Mode to OxLDL,” Structure, vol. 13, no. 6, pp. 905–917, 2005. [CrossRef]
- X. Shi, S. Niimi, T. Ohtani, and S. Machida, “Characterization of residues and sequences of the carbohydrate recognition domain required for cell surface localization and ligand binding of human lectin-like oxidized LDL receptor,” Journal of Cell Science, vol. 114, no. 7, Art. no. 7, 2001. [CrossRef]
- M. Falconi, S. Biocca, G. Novelli, and A. Desideri, “Molecular dynamics simulation of human LOX-1 provides an explanation for the lack of OxLDL binding to the Trp150Ala mutant,” BMC Struct Biol, vol. 7, no. 1, Art. no. 1, 2007. [CrossRef]
- T. Ishigaki, I. Ohki, N. Utsunomiya-Tate, and S. -i. Tate, “Chimeric Structural Stabilities in the Coiled-Coil Structure of the NECK Domain in Human Lectin-Like Oxidized Low-Density Lipoprotein Receptor 1 (LOX-1),” Journal of Biochemistry, vol. 141, no. 6, Art. no. 6, 2007. [CrossRef]
- S. Biocca et al., “Simulative and experimental investigation on the cleavage site that generates the soluble human LOX-1,” Archives of Biochemistry and Biophysics, vol. 540, no. 1–2, Art. no. 1–2, 2013. [CrossRef]
- H. Mitsuoka et al., “Interleukin 18 stimulates release of soluble lectin-like oxidized LDL receptor-1 (sLOX-1),” Atherosclerosis, vol. 202, no. 1, Art. no. 1, 2009. [CrossRef]
- M. Ghazi-Khanloosani, A. R. Bandegi, P. Kokhaei, M. Barati, and A. Pakdel, “CRP and LOX-1: a Mechanism for Increasing the Tumorigenic Potential of Colorectal Cancer Carcinoma Cell Line,” Pathol Oncol Res, vol. 25, no. 4, Art. no. 4, 2019. [CrossRef]
- M. Murdocca et al., “LOX-1 and cancer: an indissoluble liaison,” Cancer Gene Ther, vol. 28, no. 10, Art. no. 10, 2021. [CrossRef]
- F. Spaans et al., “Syncytiotrophoblast extracellular vesicles impair rat uterine vascular function via the lectin-like oxidized LDL receptor-1,” PLoS One, vol. 12, no. 7, Art. no. 7, 2017. [CrossRef]
- B. Rizzacasa, E. Morini, S. Pucci, M. Murdocca, G. Novelli, and F. Amati, “LOX-1 and Its Splice Variants: A New Challenge for Atherosclerosis and Cancer-Targeted Therapies,” Int J Mol Sci, vol. 18, no. 2, Art. no. 2, 2017. [CrossRef]
- M. Murdocca et al., “Targeting LOX-1 Inhibits Colorectal Cancer Metastasis in an Animal Model,” Front. Oncol., vol. 9, p. 927, 2019. [CrossRef]
- J. Barreto, S. K. Karathanasis, A. Remaley, and A. C. Sposito, “Role of LOX-1 (Lectin-Like Oxidized Low-Density Lipoprotein Receptor 1) as a Cardiovascular Risk Predictor: Mechanistic Insight and Potential Clinical Use,” ATVB, 2020. [CrossRef]
- G. Schnapp et al., “A small-molecule inhibitor of lectin-like oxidized LDL receptor-1 acts by stabilizing an inactive receptor tetramer state,” Commun Chem, vol. 3, no. 1, Art. no. 1, 2020. [CrossRef]
- M. M. Matin, P. Chakraborty, M. S. Alam, M. M. Islam, and U. Hanee, “Novel mannopyranoside esters as sterol 14α-demethylase inhibitors: Synthesis, PASS predication, molecular docking, and pharmacokinetic studies,” Carbohydrate Research, vol. 496, p. 108130, 2020. [CrossRef]
- M. I. Q. Tonmoy et al., “Identification of novel inhibitors of high affinity iron permease (FTR1) through implementing pharmacokinetics index to fight against black fungus: An in silico approach,” Infection, Genetics and Evolution, vol. 106, p. 105385, 2022. [CrossRef]
- A. Daina, O. Michielin, and V. Zoete, “SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules,” Sci Rep, vol. 7, no. 1, Art. no. 1, 2017. [CrossRef]
- C. A. Lipinski, F. Lombardo, B. W. Dominy, and P. J. Feeney, “Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings,” Advanced Drug Delivery Reviews, vol. 23, no. 1–3, pp. 3–25, Jan. 1997. [CrossRef]
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan, and A. L. Hopkins, “Quantifying the chemical beauty of drugs,” Nature Chem, vol. 4, no. 2, pp. 90–98, 2012. [CrossRef]
- J. A. Pradeepkiran, K. Konidala, N. Yellapu, and Bhaskar, “Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16 M,” DDDT, p. 1897, 2015. [CrossRef]
- F. Cheng et al., “admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties,” J. Chem. Inf. Model., vol. 52, no. 11, pp. 3099–3105, Nov. 2012. [CrossRef]
- M. J. Abraham et al., “GROMACS: High performance molecular simulations through multilevel parallelism from laptops to supercomputers,” SoftwareX, vol. 1–2, pp. 19–25, 2015. [CrossRef]
- I. H. P. Vieira, E. B. Botelho, T. J. de Souza Gomes, R. Kist, R. A. Caceres, and F. B. Zanchi, “Visual dynamics: a WEB application for molecular dynamics simulation using GROMACS,” BMC Bioinformatics, vol. 24, no. 1, Art. no. 1, 2023. [CrossRef]
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, and D. A. Case, “Development and testing of a general amber force field,” J Comput Chem, vol. 25, no. 9, pp. 1157–1174, 2004. [CrossRef]
- L. Kagami, A. Wilter, A. Diaz, and W. Vranken, “The ACPYPE web server for small-molecule MD topology generation,” Bioinformatics, vol. 39, no. 6, p. btad350, 2023. [CrossRef]
- J. Wang, W. Wang, P. A. Kollman, and D. A. Case, “Automatic atom type and bond type perception in molecular mechanical calculations,” Journal of Molecular Graphics and Modeling, vol. 25, no. 2, pp. 247–260, 2006. [CrossRef]
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein, “Comparison of simple potential functions for simulating liquid water,” The Journal of Chemical Physics, vol. 79, no. 2, pp. 926–935, 1983. [CrossRef]
- J.-P. Ryckaert, G. Ciccotti, and H. J. C. Berendsen, “Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes,” Journal of Computational Physics, vol. 23, no. 3, pp. 327–341, 1977. [CrossRef]
- X.-Y. Meng, H.-X. Zhang, M. Mezei, and M. Cui, “Molecular docking: a powerful approach for structure-based drug discovery,” Curr Comput Aided Drug Des, vol. 7, no. 2, pp. 146–157, 2011. [CrossRef]
- G. Bulusu and G. R. Desiraju, “Strong and Weak Hydrogen Bonds in Protein–Ligand Recognition,” J Indian Inst Sci, vol. 100, no. 1, pp. 31–41, 2020. [CrossRef]
- S. Mishra and R. Dahima, “IN VITRO ADME STUDIES OF TUG-891, A GPR-120 INHIBITOR USING SWISS ADME PREDICTOR,” Journal of Drug Delivery and Therapeutics, vol. 9, no. 2-s, Art. no. 2-s, 2019. [CrossRef]
- A. Harvey, “Natural Products in Drug Discovery and Development. 27-28 June 2005, London, UK,” IDrugs, vol. 8, no. 9, pp. 719–721, Sep. 2005.
- A. K. Srivastava, M. Tewari, H. S. Shukla, and B. K. Roy, “In Silico Profiling of the Potentiality of Curcumin and Conventional Drugs for CagA Oncoprotein Inactivation,” Arch Pharm (Weinheim), vol. 348, no. 8, pp. 548–555, 2015. [CrossRef]
- D. Bisht, D. Prakash, and A. Shakya, “Computational Analysis of Pharmacokinetics, Bioactivity and Toxicity Profiling of Some Selected Ovulation-Induction Agents Treating Polycystic Ovary Syndrome (PCOS),” International Journal of Pharmaceutical Sciences and Research, vol. 14, p. 2522, 2023. [CrossRef]
- H. A. Zhong, “ADMET Properties: Overview and Current Topics,” in Drug Design: Principles and Applications, A. Grover, Ed., Singapore: Springer, pp. 113–133, 2017. [CrossRef]
- T. Lynch and A. Price, “The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects,” Cytochrome P, vol. 76, no. 3, 2007.
- N. A. Durán-Iturbide, B. I. Díaz-Eufracio, and J. L. Medina-Franco, “In Silico ADME/Tox Profiling of Natural Products: A Focus on BIOFACQUIM,” ACS Omega, vol. 5, no. 26, pp. 16076–16084, 2020. [CrossRef]
- B. T. Priest, I. M. Bell, and M. L. Garcia, “Role of hERG potassium channel assays in drug development,” Channels (Austin), vol. 2, no. 2, pp. 87–93, 2008. [CrossRef]
- S. Wang, H. Sun, H. Liu, D. Li, Y. Li, and T. Hou, “ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches,” Mol Pharm, vol. 13, no. 8, pp. 2855–2866, 2016. [CrossRef]
- L. Martínez, “Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis,” PLoS One, vol. 10, no. 3, p. e0119264, 2015. [CrossRef]
Figure 1.
a. Representation of the tertiary structure of the homodimer LOX-1 receptor (PDB ID 5w7d) with the BI-0115 inhibitor at the active site. The structural model of the LOX-1 protein is depicted by a rounded ribbon model with the α-helices colored cyan, the β-strands colored yellow, the coils colored magenta, and the red circle delimiting the active site. b. Surface view of the LOX-1 protein after treatment with the BI-0115 inhibitor.
Figure 1.
a. Representation of the tertiary structure of the homodimer LOX-1 receptor (PDB ID 5w7d) with the BI-0115 inhibitor at the active site. The structural model of the LOX-1 protein is depicted by a rounded ribbon model with the α-helices colored cyan, the β-strands colored yellow, the coils colored magenta, and the red circle delimiting the active site. b. Surface view of the LOX-1 protein after treatment with the BI-0115 inhibitor.
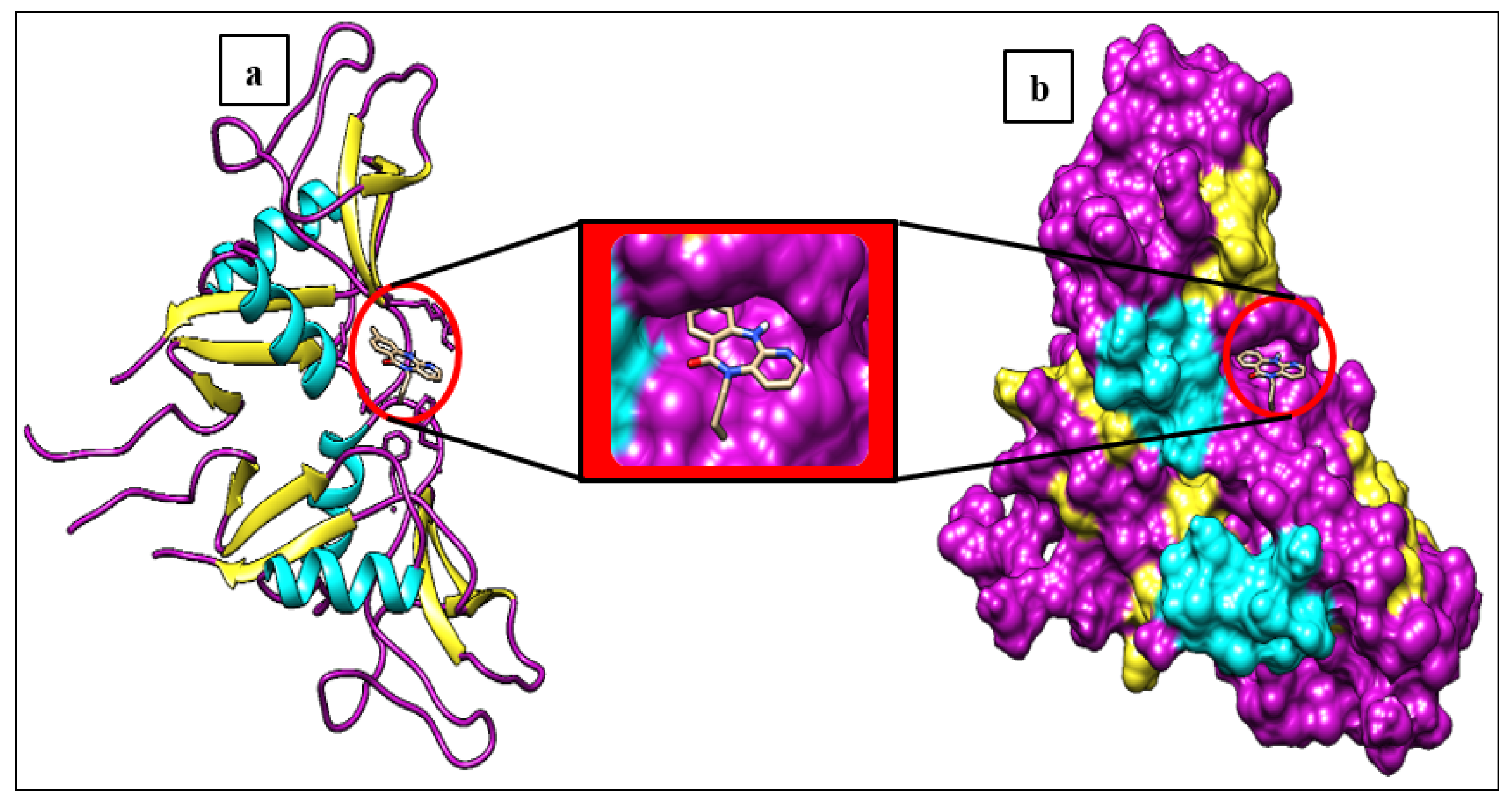
Figure 2.
Protein–ligand interactions of LOX-1 with control BI-0115 and cannabinol. (A) 3D receptor–ligand presentation and (B) 2D receptor–ligand interactions.
Figure 2.
Protein–ligand interactions of LOX-1 with control BI-0115 and cannabinol. (A) 3D receptor–ligand presentation and (B) 2D receptor–ligand interactions.
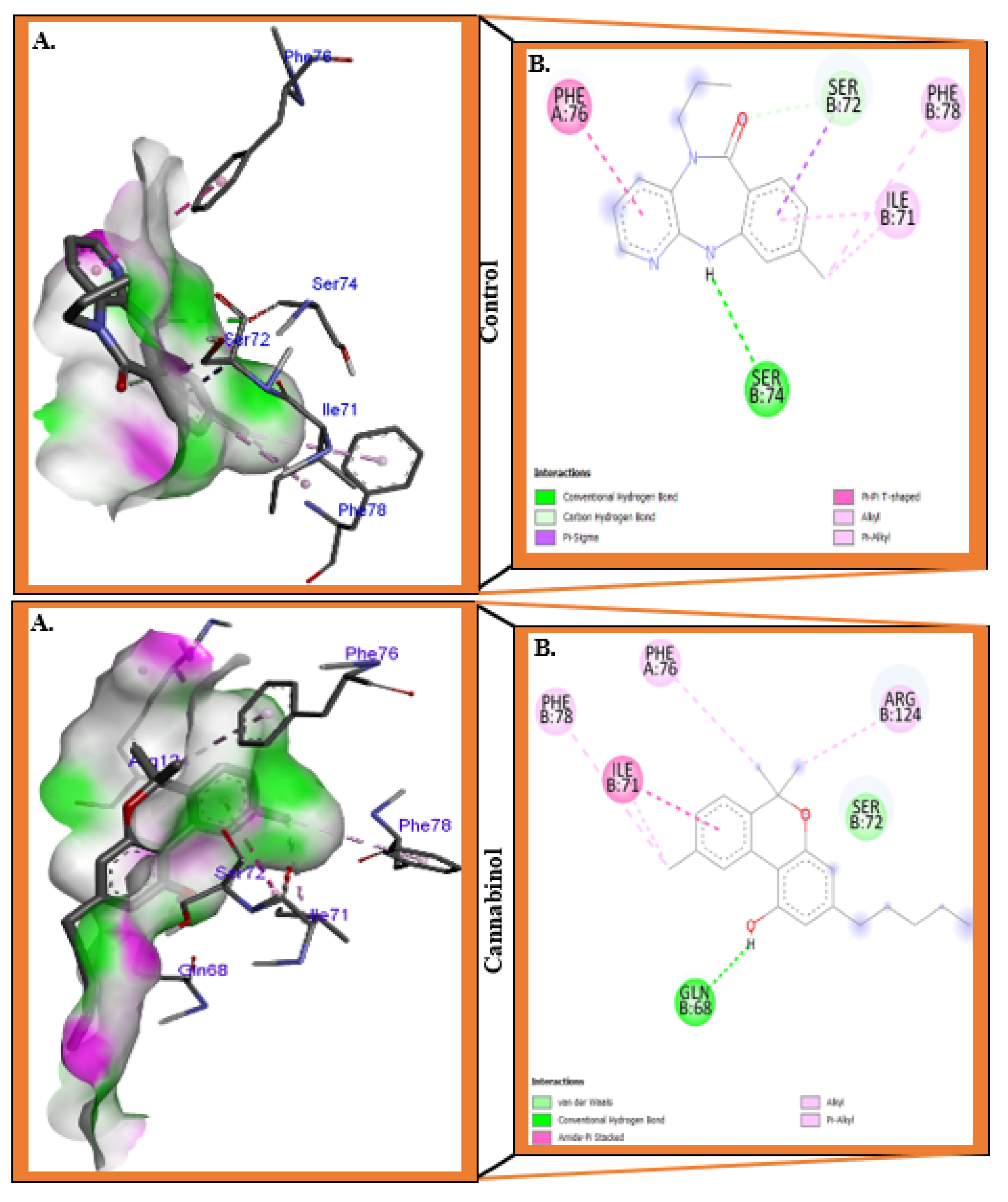
Figure 3.
Protein–ligand interactions of LOX-1 with the molecules DHEA and Smilagenin. (A) 3D receptor–ligand presentation and (B) 2D receptor–ligand interactions.
Figure 3.
Protein–ligand interactions of LOX-1 with the molecules DHEA and Smilagenin. (A) 3D receptor–ligand presentation and (B) 2D receptor–ligand interactions.
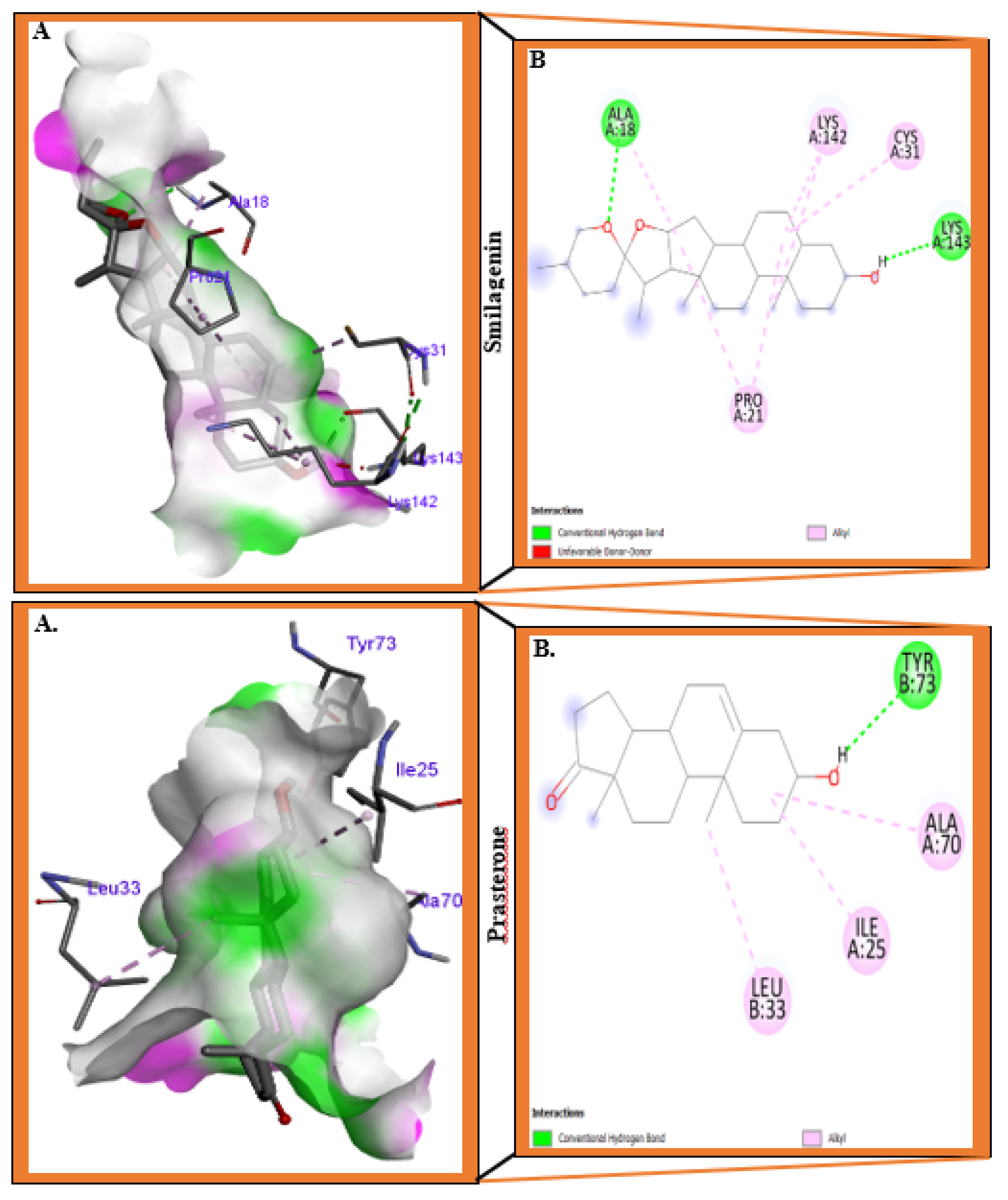
Figure 4.
Root Mean Square Deviation (RMSD) of LOX1-ligand complexes during MDS of 100 ns.
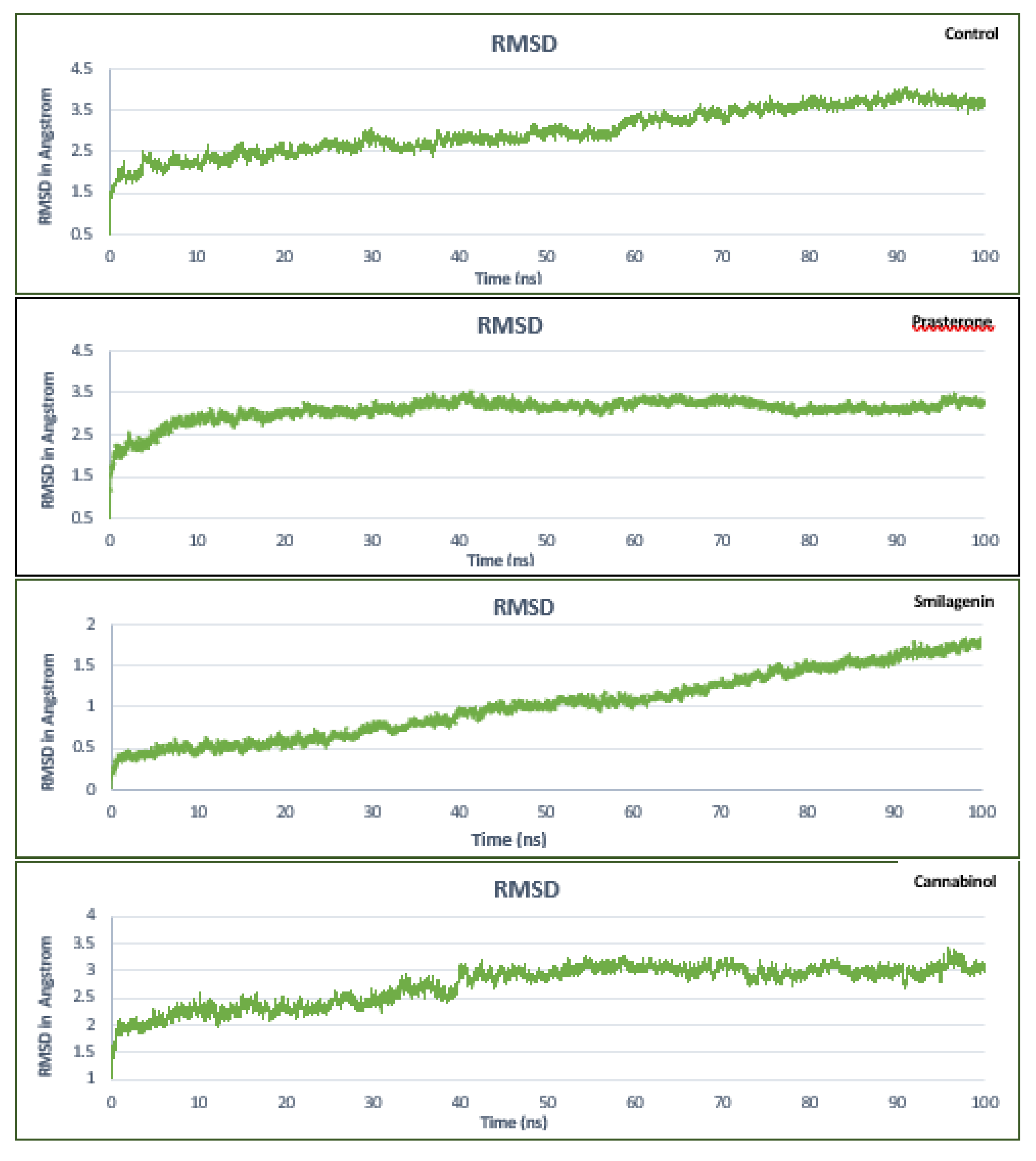
Figure 5.
Root Mean Square Fluctuation (RMSF) of LOX1-ligand complexes during 100 ns of MDS.
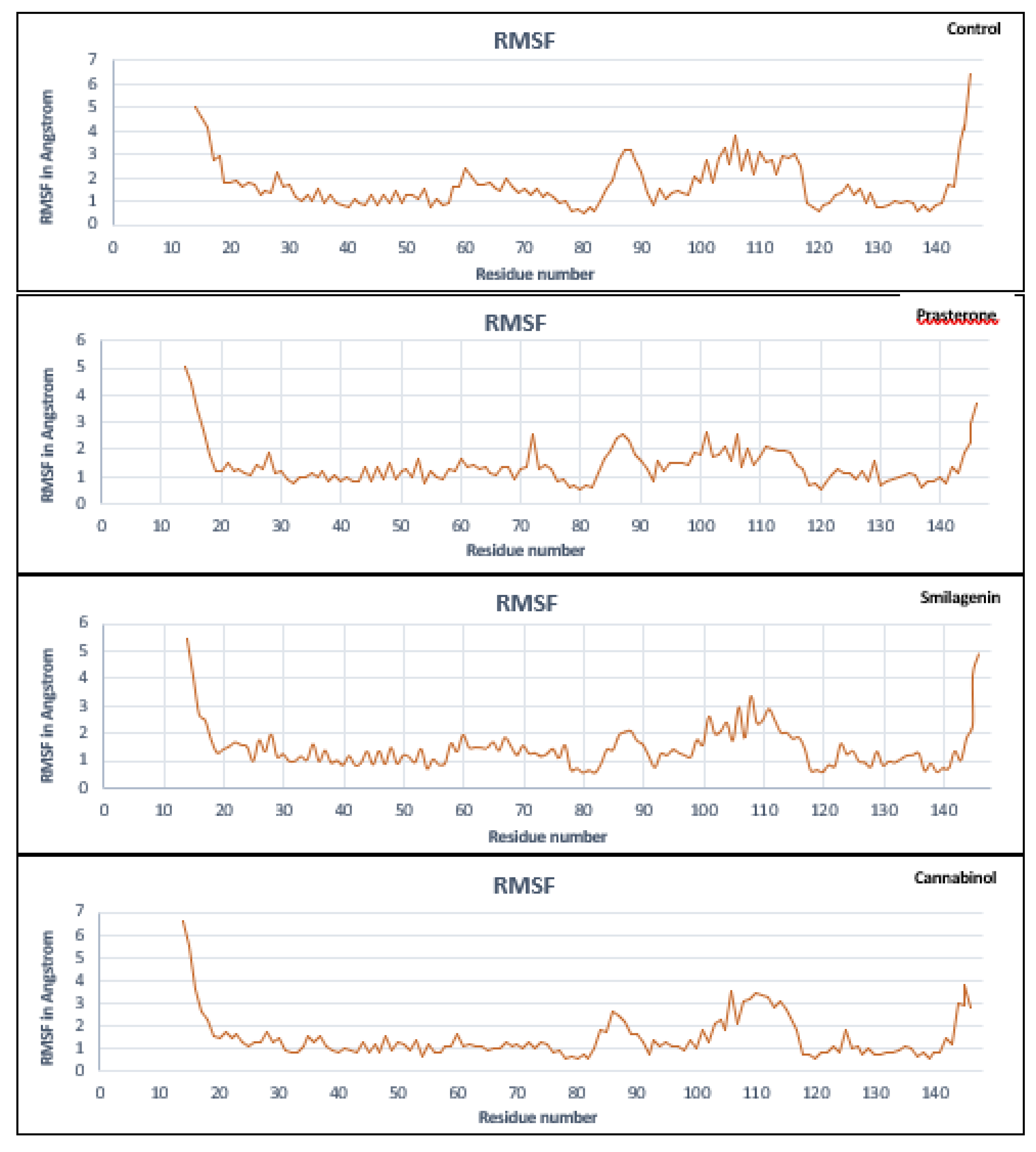
Figure 6.
Radius of gyration (Rg) analysis of LOX1-ligand complexes during a 100 ns MDS time.
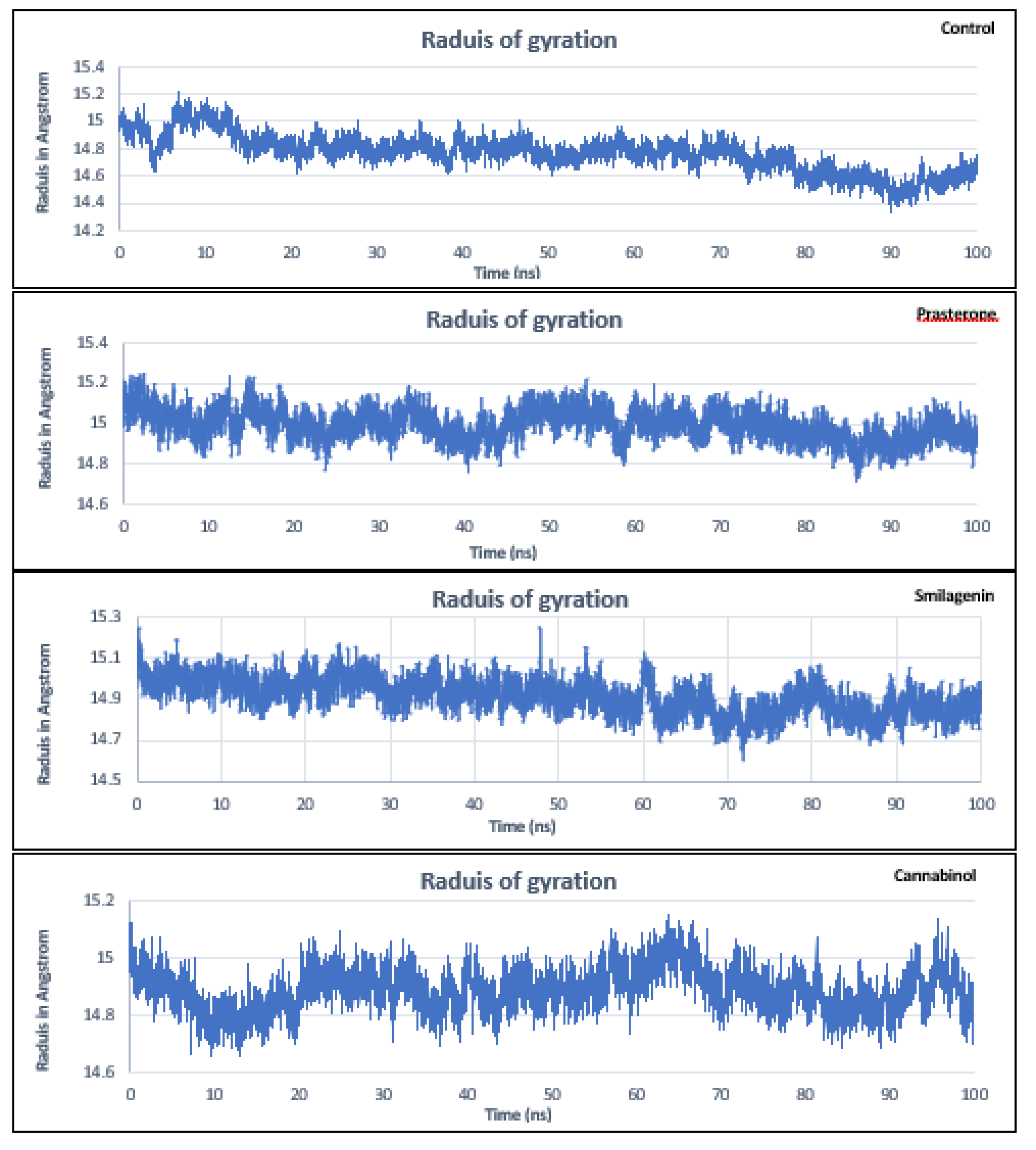
Figure 7.
Hydrogen bonds formed between ligands and LOX-1 receptor during 100 ns of MDS.
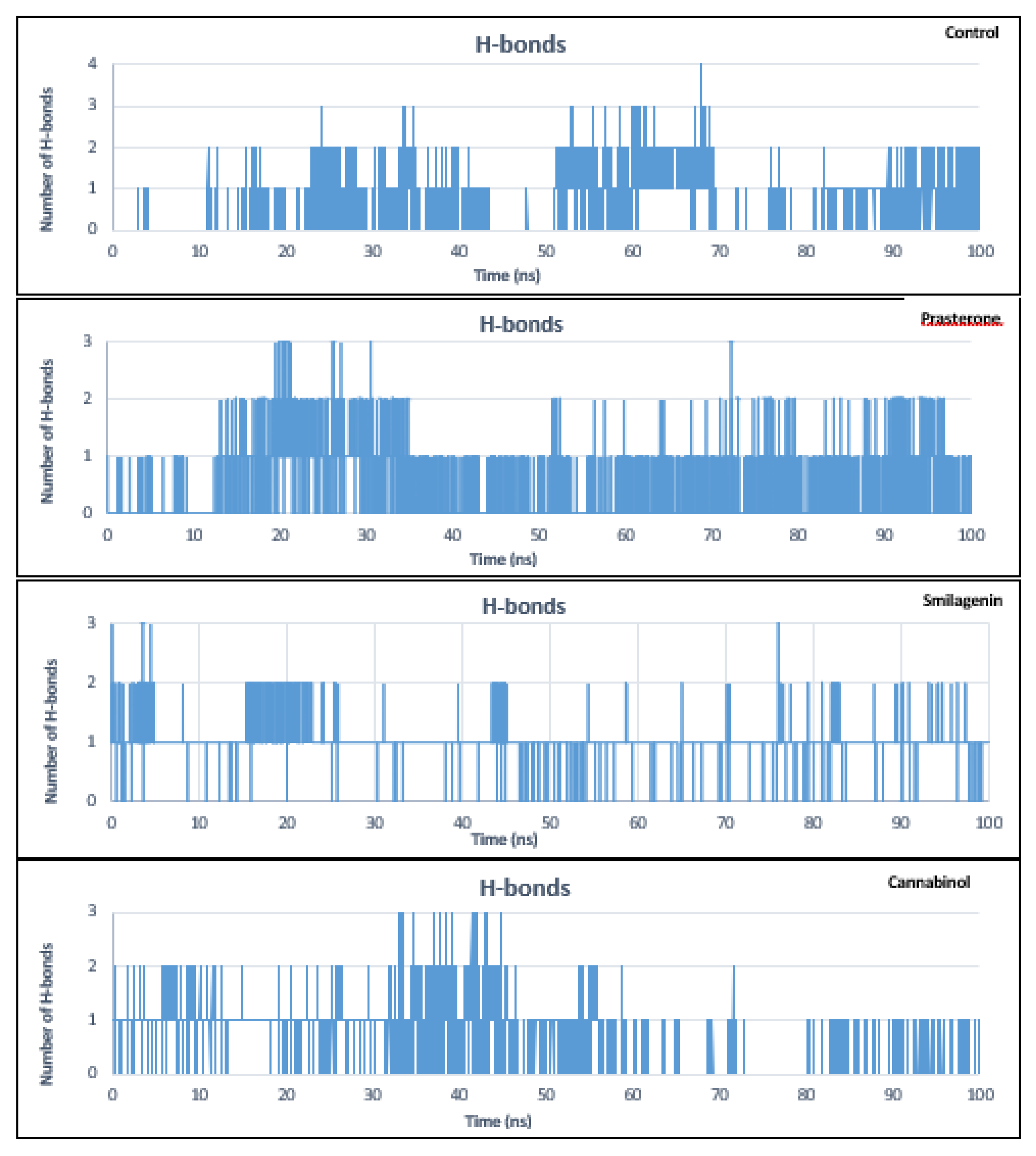
Figure 8.
Solvent Accessible Surface Area (SASA) analysis of the structure of LOX-1-ligands complexes during 100 ns of MDS.
Figure 8.
Solvent Accessible Surface Area (SASA) analysis of the structure of LOX-1-ligands complexes during 100 ns of MDS.
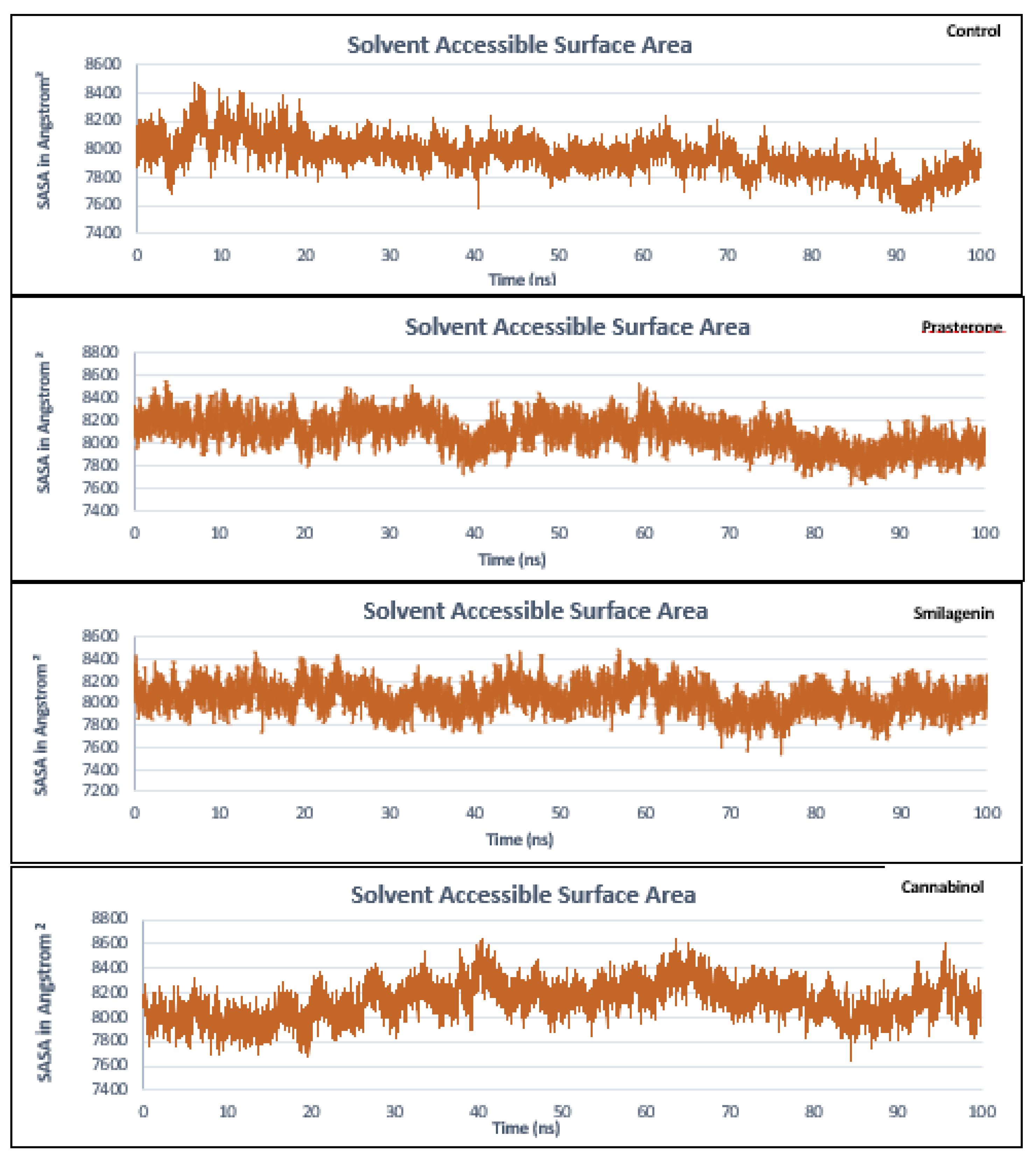
Figure 9.
Protein secondary structure content of LOX-1 receptor with ligands during 20 ns of MDS.
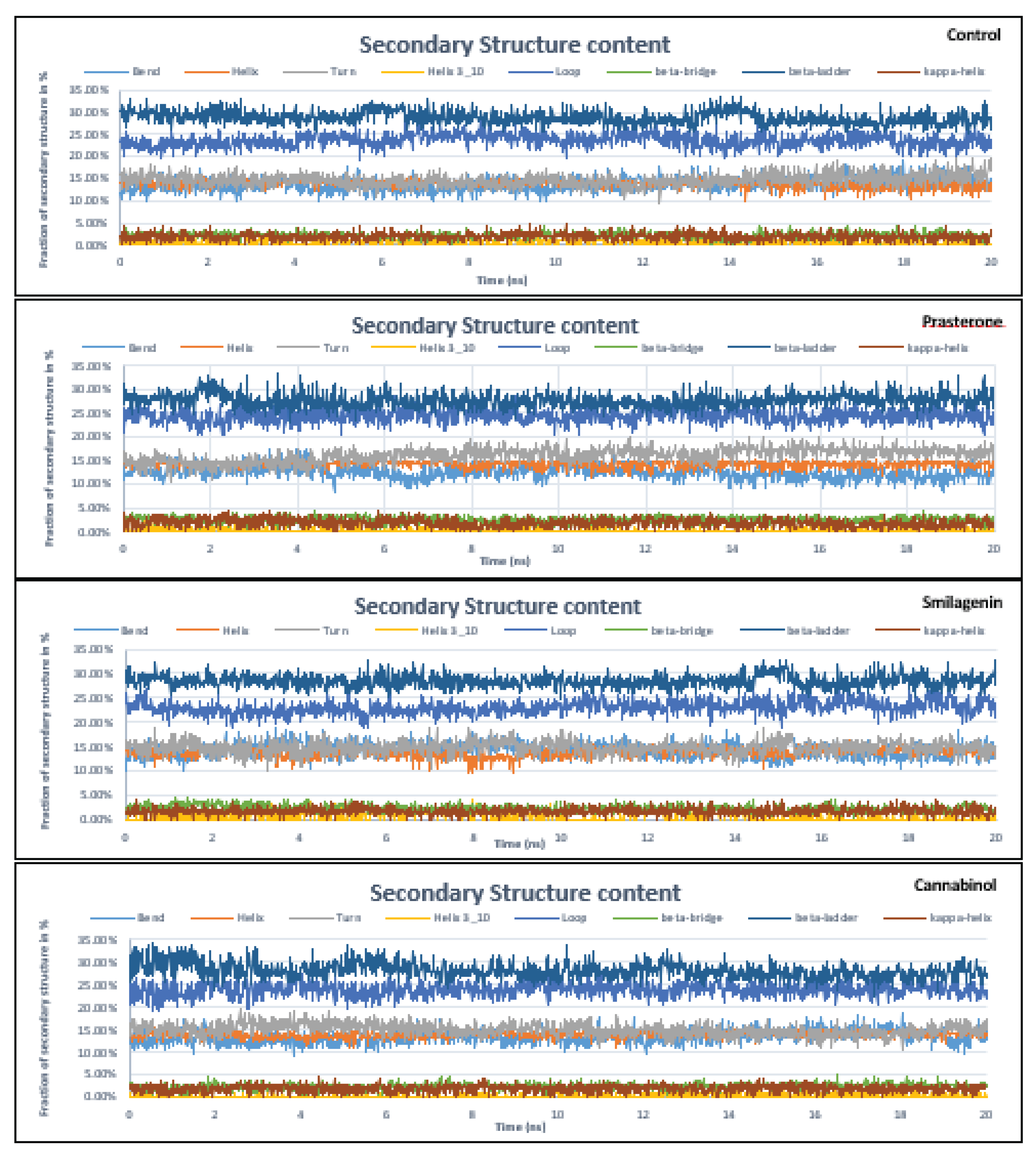

Table 1.
List of screened compounds in the current study along with their PubChem IDs.
| No | PubChem ID | Compounds name |
|---|---|---|
| Control | 146676953 | BI-0115/9-chloranyl-5-propyl-11~{H}-pyrido [2,3-b][1,4]benzodiazepin-6-one |
| 1 | 5281718 | Polydatin |
| 2 | 175265433 | Curcumin |
| 3 | 5881 | Dehydroepiandrosterone/Prasterone |
| 4 | 91439 | Smilagenin |
| 5 | 5742590 | Daucosterol |
| 6 | 16078 | Tetrahydrocannabinol |
| 7 | 30219 | Cannabichromene |
| 8 | 644019 | Cannabidiol |
| 9 | 2543 | Cannabinol |
| 10 | 132279092 | 3,5′-Dimethoxy-resveratrol |
| 11 | 72344 | Nobiletin |
| 12 | 96118 | 4′,5,6,7-Tetramethoxyflavone |
| 13 | 96539 | Gardenin’ B |
| 14 | 96892 | 6-(diethylamino)pyridine-3-carboxamide |
| 15 | 97332 | Quercetin pentamethyl ether |
| 16 | 136417 | 5-Hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone |
| 17 | 145659 | Sinensetin |
| 18 | 261592 | 6-(Pyrrolidin-1-yl)pyridine-3-carboxamide |
| 19 | 629965 | Zapotin |
| 20 | 3010100 | 4′-Hydroxy-5,6,7,8-tetramethoxyflavone |
| 21 | 3031415 | 6-(Cyclohexylamino)pyridine-3-carboxamide |
| 22 | 5315263 | Casticin |
| 23 | 5352005 | Retusin |
| 24 | 132228215 | N-(1-(1-(L-alanyl)piperidine-4-yl)ethyl)-6-aminonicotinamide |
| 25 | 9500 | 6-Aminonicotinamide |
| 26 | 5458896 | scirpusinA |
Table 2.
Docking score (kcal/mol) of the LOX-1 receptor with twenty-six molecules.
| Molecules | Compounds name | Docking score (Kcal/mol) |
|---|---|---|
| Control | BI-0115 | -6.55 |
| 1 | Polydatin | -5.97 |
| 2 | Curcumin | -7.84 |
| 3 | Dehydroepiandrosterone or Prasterone | -8.37 |
| 4 | Smilagenin | -8.56 |
| 5 | Daucosterol | -7.05 |
| 6 | Tetrahydrocannabinol | -7.15 |
| 7 | Cannabichromene | -7.48 |
| 8 | Cannabidiol | -6.78 |
| 9 | Cannabinol | -8.14 |
| 10 | 3,5′-Dimethoxy-resveratrol | -6.59 |
| 11 | Nobiletin | -6.40 |
| 12 | 4′,5,6,7-Tetramethoxyflavone | -6.29 |
| 13 | Gardenin B | -5.81 |
| 14 | 6-(diethylamino) pyridine-3-carboxamide | -4.89 |
| 15 | Quercetin pentamethyl ether | -6.55 |
| 16 | 5-Hydroxy-3,6,7,8,3′,4′-hexamethoxyflavone | -5.80 |
| 17 | Sinensetin | -6.98 |
| 18 | 6-(Pyrrolidin-1-yl) pyridine-3-carboxamide | -5.75 |
| 19 | Zapotin | -6.70 |
| 20 | 4′-Hydroxy-5,6,7,8-tetramethoxyflavone | -5.86 |
| 21 | 6-(Cyclohexylamino)pyridine-3-carboxamide | -6.58 |
| 22 | Casticin | -6.12 |
| 23 | Retusin | -6.27 |
| 24 | N-(1-(1-(L-alanyl) piperidin-4-yl)ethyl)-6-aminonicotinamide | -5.99 |
| 25 | 6-Aminonicotinamide | -5.88 |
| 26 | ScirpusinA | -7.47 |
Table 3.
Nondonding interactions between amino acid residues of LOX1 and our three selected compounds along with the control BI-0115 are given below.
Table 3.
Nondonding interactions between amino acid residues of LOX1 and our three selected compounds along with the control BI-0115 are given below.
| Compounds | Residues | Distance (Å) | Bonds type |
|---|---|---|---|
| Control | Ser72 | 3.89 | Pi-Sigma |
| 3.30 | Carbon | ||
| Ser74 | 3.00 | Conventional hydrogen bond | |
| Phe76 | 5.20 | Pi-Pi T-Shaped | |
| Ile71 | 5.12 | Pi-Alkyl | |
| 3.87 | Alkyl | ||
| Phe78 | 4.99 | Pi-Alkyl | |
| Prasterone | Ile25 | 4.54 | Alkyl |
| Leu33 | 4.52 | Alkyl | |
| Ala70 | 4.58 | Alkyl | |
| Tyr73 | 5.29 | Conventional hydrogen bond | |
| Smilagenin | Cys31 | 5.45 | Alkyl |
| Pro21 | 5.46 | Alkyl | |
| 5.16 | Alkyl | ||
| Lys143 | 2.18 | Conventional hydrogen bond | |
| 2.07 | Donor-donor | ||
| Lys142 | 5.24 | Alkyl | |
| 5.18 | Alkyl | ||
| Ala18 | 3.78 | Alkyl | |
| 2.42 | Conventional hydrogen bond | ||
| Cannabinol | Gln68 | 1.83 | Conventional hydrogen bond |
| Ser72 | 4.47 | Amide Pi-Stacked | |
| Phe78 | 4.99 | Pi-Alkyl | |
| Phe76 | 4.68 | Pi-Alkyl | |
| Arg124 | 4.04 | Alkyl | |
| Ile71 | 4.15 | Alkyl |
Table 4.
Calculated drug-like properties of the three selected compounds.
| Compounds | MW(g/mol) | iLOGP | H-bonds donors | H-bonds acceptors | TPSA(Å2) | Heavy atoms | Rotatable bonds | Alerts | |
| Pains | Brenk | ||||||||
| Control | 287.74 | 2.74 | 1 | 2 | 50.68 | 20 | 2 | 0 | 0 |
| Cannabinol | 310.43 | 3.94 | 1 | 2 | 29.46 | 23 | 4 | 0 | 0 |
| Prasterone | 288.42 | 2.89 | 1 | 2 | 37.30 | 21 | 0 | 0 | 1 |
| Smilagenin | 416.64 | 4.42 | 1 | 3 | 38.69 | 30 | 0 | 0 | 0 |
Table 5.
Bioactivity scores of the three selected compounds.
| Compounds | Molinspiration bioactivity score | GCPR ligand | Ion channel modulator | Kinase inhibitor | Nuclear receptor ligand | Protease inhibitor | Enzyme inhibitor |
| Control | v2022.08 | 0.13 | 0.04 | 0.05 | -0.54 | -0.56 | 0.41 |
| Cannabinol | v2022.08 | 0.50 | 0.02 | -0.08 | 0.60 | 0.11 | 0.22 |
| Prasterone | v2022.08 | 0.07 | 0.01 | -0.60 | 0.90 | -0.04 | 0.82 |
| Smilagenin | v2022.08 | 0.13 | 0.15 | -0.41 | 0.50 | 0.11 | 0.59 |
Table 6.
Absorption, distribution, metabolism, and excretion (ADME) parameters of the three selected compounds.
Table 6.
Absorption, distribution, metabolism, and excretion (ADME) parameters of the three selected compounds.
| Compounds | Water solubility (mg/ml) | GI absorption | P-gp substrate | Skin permeation (cm/s) | BBB permeant | CYP1A2 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | CYP2C9 inhibitor |
| Control | -4.15 | High | No | -5.68 | Yes | Yes | No | No | Yes |
| Cannabinol | -5.74 | High | Yes | -3.86 | Yes | Yes | Yes | No | No |
| Prasterone | -3.66 | High | No | -5.77 | Yes | No | No | No | No |
| Smilagenin | -6.51 | High | No | -4.23 | Yes | No | No | No | No |
Table 7.
Toxicological properties of the three selected compounds.
| Parameters | Molecules | |||
| Control | Cannabinol | Prasterone | Smilagenin | |
| Ames mutagenesis | No | No | No | No |
| LD50 (mg/kg) | 1500 (Class 4) | 13500 (Class 6) | 8800 (class 6) | 2600 (class 5) |
| Acute Oral Toxicity (c) | II | III | IV | III |
| Carcinogenicity (binary) | No | No | No | No |
| Carcinogenicity (trinary) | Nonrequired | Nonrequired | Warning | Nonrequired |
| Hepatotoxicity | Yes | No | Yes | No |
| hERG inhibition | No | Yes | No | No |
| Skin sensitization | No | No | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



