
Submitted:
10 June 2025
Posted:
11 June 2025
You are already at the latest version
Abstract
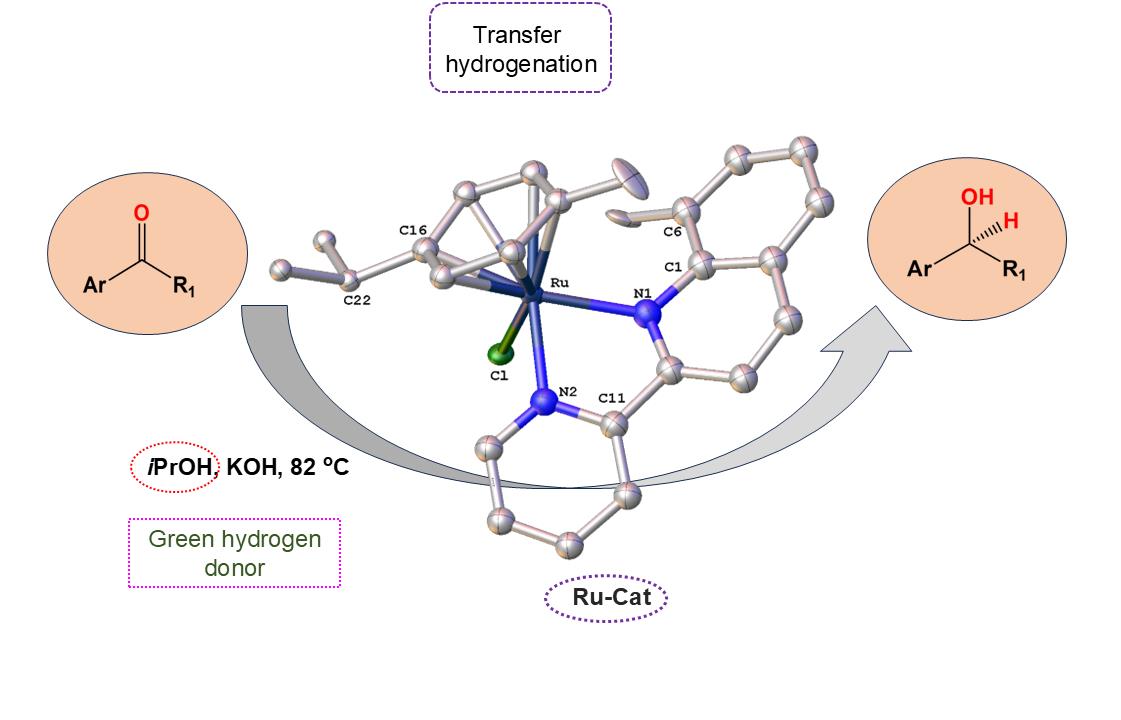
Searching for new and efficient transfer hydrogenation catalysts, a series of new organometallic ruthenium(II)-arene complexes of the formulae [Ru(η6-p-cymene)(L)Cl][PF6] (1–8) and [Ru(η6-p-cymene)(L)Cl][Ru(η6-p-cymene)Cl3] (9–11) were synthesized and fully characterized. These were prepared from the reaction of pyridine-quinoline and biquinoline-based ligands (L) with [Ru(η6-p-cymene)(μ-Cl)Cl]2, in 1:2 and 1:1, metal (M) to ligand (L) molar ratios. Characterization includes a combination of spectroscopic methods (FT-IR, UV-Vis, multi nuclear NMR), elemental analysis and single-crystal X-ray crystallography. The pyridine-quinoline organic entities encountered, were prepared in high yield either via the thermal decarboxylation of the carboxylic acid congeners, namely 2,2′-pyridyl-quinoline-4-carboxylic acid (pqca), 8-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (8-Mepqca), 6′-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (6′-Mepqca) and 8,6′-dimethyl-2,2′-pyridyl- quinoline-4-carboxylic acid (8,6′-Me2pqca), affording the desired ligands pq, 8-Mepq, 6′-Mepq and 8,6′-Me2pq, or by the classical Friedländer condensation, to yield 4,6′-dimethyl-2,2′-pyridyl-quinoline (4,6΄-Me2pq) and 4-methyl-2,2′-pyridyl-quinoline (4-Mepq) respectively. The solid-state structures of complexes 1–4, 6, 8 and 9 were determined showing a distorted octahedral coordination geometry. The unit cell of 3 contains two independent molecules (Ru-3), (Ru′-3) in a 1:1 ratio, due to a slight rotation of the arene ring. All complexes catalyze the transfer hydrogenation of acetophenone, using 2-propanol as a hydrogen donor in the presence of KΟiPr. Among them, complexes 1 and 5 bearing methyl groups at the 8 and 4 position of the quinoline moiety, convert acetophenone to 1-phenylethanol quantitatively, within approximately 10 minutes with final TOFs of 1600 h–1. The catalytic performance of complexes 1–11, towards the transfer hydrogenation of p-substituted acetophenone derivatives and benzophenone, ranges from moderate to excellent. An inner-sphere mechanism has been suggested based on the detection of ruthenium(II) hydride species.
Keywords:
pyridine-quinoline ligands
; p-cymene
; ruthenium complexes
; ketones
; transfer hydrogenation
; hydrogenation
; hydride species
1. Introduction
Hydrogenation (HY) is a fundamental chemical process with a plethora of applications [1], which span from the food (fat hydrogenation) and petrochemical industries to cosmetics and pharmaceutical industry, along with the production of ammonia and other chemical products [2]. Independently, transfer hydrogenation (TH) has been developed as an alternative to classic hydrogenation [3,4]. In this process, 2-propanol or formic acid are used as sacrificial hydrogen donors different to molecular hydrogen. This is a benign and eco-friendly method where no high pressure of molecular hydrogen is required and in general all hydrogen donors are readily available and cheap [5].
Early reports by Henbest and Mitchell [6], Sasson and Blum [7], and Backvall [8], are the cornerstones of this chemical process with potential applications not only in academia but in the chemical industry as well. The pioneering work of Noyori [9], in the field of asymmetric TH, with the development of new enantioselective transfer hydrogenation ruthenium(II) catalysts ([(η6-arene)Ru(Tsdpen)Cl]; (H)Tsdpen = N-p-tosyl-1,2-diphenylethylenediamine), has opened new avenues in homogenous catalysis. Towards this goal a plethora of metal complexes has been synthesized and evaluated as catalysts [10]. Their ability to convert multiple polarized unsaturated substrates to compounds with constantly increasing applications in industry (pharmaceutical, agrochemical, fragrance) demonstrates the high importance of transfer hydrogenation reactions [11]. In this respect we may comment on new potential applications of the TH reactions such as biofuels production [12], along with a new approach for cancer therapy, via intracellular TH reactions [13,14].
This great diversity among catalysts proposed has led to the suggestion of a handy of different mechanistic routes. In general, these can be divided as “inner” and “outer sphere” mechanisms, including their sub-categories as well [15,16]. In this respect, it has been suggested that the structure of the organic ligands attached to the metallic center plays a significant role in the mechanistic route and the overall activity of the catalyst [17].
Among the plethora of metal complexes that served in the transfer hydrogenation reaction of different substrates, organometallic ruthenium(II) complexes comprising the η6-arene ligand, constitute an interesting class of catalyst precursors [3]. The coordination sphere around the metal center can be filled with different organic ligands like NHCs (N-heterocycle carbenes) [18], phosphines [19], thioamides [20], N/P [21], N/O [22], N,N pincer ligands along with N^N ligands (N^N = bidentate), either amino or pyridyl based [23,24,25,26,27], while a series of 2-(2-quinolyl)benzimidazole-based ruthenium(II) complexes containing 8-amino-quinoline derivatives have been used for TH reactions [28,29].
In line with that we have reported previously a series of pyridine-quinoline based ruthenium(II) catalyst precursors bearing triphenyl phosphine ligands, of the formula cis-[RuCl2(PPh3)2(Lx)] (Lx = pq = 2,2′-pyridyl-quinoline; 4-Mepq = 4-methyl-2,2′-pyridyl-quinoline; and pqcame = methyl 2,2′-pyridyl-quinoline-4-carboxylate) for the transfer hydrogenation of polarized unsaturated substrates [30,31,32]. As a continuation of our ongoing research interest in the field, we report herein the synthesis, spectroscopic and structural characterization of a series of ruthenium(II) arene complexes of the general formula [Ru(η6-p-cymene)(L)Cl][PF6] (L = 8-Mepq (1), 6′-Mepq (2), 8,6′-Me2pq (3), 4,6΄-Me2pq (4), 4-Mepq (5), biqcame (6), biq (8) and [Ru(η6-p-cymene)(L)Cl][Ru(η6-p-cymene)Cl3] (L = pqcame (9), 4-Mepq (10), pq (11)). The catalytic activities of all complexes, including those of the known complexes Ru-pqcame [33], 7-Cl [34], and 8 [35], have been examined in the transfer hydrogenation reaction of various aromatic ketones, that are used as substrates. The possible effects related to the position of methyl groups around the ligand periphery, along with that of the different counter anion encountered ([PF6]– or [Ru(η6-p-cymene)Cl3]–), on the catalytic performance of the catalyst precursors, have been also studied.
2. Results and Discussion
2.1. Synthesis, Spectroscopic and Structural Characterization of the Organic Ligands
The organic ligands 2,2′-pyridyl-quinoline (pq) [36], 8-methyl-2,2′-pyridyl-quinoline (8-Mepq), 6′-methyl-2,2′-pyridyl-quinoline (6′-Mepq) and 8,6′-dimethyl-2,2′-pyridyl-quinoline (8,6′-Me2pq) were synthesized from the relevant carboxylic acid congeners pqca, Μepqca, 6′-Mepqca, and 8,6′-Me2pqca with slight modifications of the general route described by Denny et al. for the case of 8-Mepq [37]. In our efforts, thermal decarboxylation occurs without the presence of metallic copper, providing the final products as off-white or light-yellow solids, in higher yields compared to the published procedures. The synthesis of 6′-Mepqca ligand has been reported by Haginiwa et al. [38], and more recently by Fan et al. [39], where in the latter, the preparation of 8,6′-Me2pqca is also included. The 2,2΄-biquinoline (biq) ligand was also synthesized for comparison [40,41,42]. The spectroscopic data (IR and 1H NMR) of the organic ligands prepared are in accord with those of the published procedures.
The 4,6′-Me2pq ligand [38] was reported previously, and characterization was based only on mass spectrometric data. In this study we present a complete spectroscopic characterization of this ligand, including an X-ray structure determination. This compound was prepared according to the procedure described for the synthesis of 4-Mepq [31], following the Friedländer condensation reaction of 2-aminoacetophenone with 2-acetyl-pyridine. The syntheses of the methyl ester analogs, methyl 2,2′-pyridyl-quinoline-4-carboxylate (pqcame) and dimethyl 2,2′-biquinoline-4,4′-dicarboxylate (biqcame) were performed according to the literature [43,44] and are presented in Scheme 1.
Selected NMR data (1H NMR and/or 13C spectra) of 8-Mepq, 6′-Mepq, 8,6′-Me2pq, 4,6′-Mepq and pq, recorded in CDCl3 are included in the Supplementary Materials (Figures S1–S12) and are in accord to the structures shown in Scheme 1. Assignment was based on a combination of 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC methods. Thus, for example, the 1H NMR spectrum of 4,6′-Me2pq (Figure S7) displays eight resonance signals typical for the aromatic ring protons, while the two singlet resonance signals at δ 2.70 and δ 2.80 ppm respectively, are attributed to the methyl group protons.
Additional proof about the nature of the species formed in methanol solution is provided by the high-resolution electrospray ionization mass spectrometry (ESI-HRMS) of the corresponding compounds 8-Mepq, 6′-Mepq and 8,6′-Me2pq (Figures S13–S15). For 8-Mepq and 6′-Mepq, the peak at m/z 221.109 is typical for the protonated [M + H]+ ion, while that at m/z 243.088 can be attributed to the sodium adduct [M + Na]+. For both compounds, the low intensity molecular ion detected at m/z 463.196 can be assigned to the sodium adduct of the dimer [2M + Na]+. Similarly, the mass spectrum of 8,6′-Me2pq displays a basic ion at m/z 235.127 due to the protonated ligand [M + H]+, accompanied by peaks at m/z 257.105 and m/z 273.079, for the sodium [M + Na]+ and potassium adducts [M + K]+ respectively. The low intensity ion at m/z 491.221 is attributable to the sodium adduct of the dimer [2M + Na]+.
The solid-state structures of 8-Mepq, 8,6′-Me2pqca and 4,6′-Me2pq have been unambiguously determined by single-crystal diffraction studies. Colorless single crystals, suitable for X-ray diffraction, were grown upon slow evaporation of a MeOH/H2O solution of the compounds at ambient temperature. Interestingly, a CSD search performed (5/2025), in the crystallographic data center, revealed that the structures of these small molecules had not been reported previously. Their molecular structures are depicted in Figure 1, Figure 2 and Figure 3.
The three molecules 8-Mepq, 4,6′-Me2pq and 8,6′-Me2pq, crystallize in the orthorhombic crystallographic system, space groups Pn21a, P212121 and Pbca respectively. As expected, in the bipyridine domain, the two nitrogen atoms are in the trans-configuration [17,20]. In 8-Mepq the pyridine ring close to that of quinoline, deviates from planarity since the angle between the least-squares planes of the pyridyl and quinoline rings, containing atoms N2 and N1, is 13.69°. For 4,6′-Me2pq this angle drops to 9.11°, while for 8,6′-Me2pq, that is a planar molecule, this angle is 1.74o.
In the asymmetric unit of 8-Mepq there are two crystallographically independent molecules, arranged in a head-to-tail fashion and their bonding parameters differ slightly. The packing in the crystal is such that molecules of 8-Mepq are stabilized by classical intermolecular hydrogen bonding interactions between the nitrogen atom N1 of quinoline moiety and the hydrogen atom H13 of a second molecule (distance of C13–H13ꞏꞏꞏN1΄ = 3.440(4) Å, bond angle = 164.15(4)°; C13΄–H13΄ꞏꞏꞏN1 = 3.561(5) Å, bond angle = 161.84(5)°) as shown in Figure S16 of the Supplementary Materials. For 4,6′-Me2pq the preference of the trans-configuration can be attributed to the characteristic intramolecular hydrogen bonding interactions of N1 and N4 to the adjacent H15 and H4 respectively (distance of N1ꞏꞏꞏH15–C15 = 2.514(5) Å, bond angle = 98.21(5)°and N2ꞏꞏꞏH4–C4 = 2.475(5) Å, bond angle = 100.09(5)°). In the unit cell, stabilization is provided by weak intermolecular non-classical hydrogen bonding interactions (distance of (C10C–H10C)∙∙∙centroid (N1–C1–C2) is 3.529(4) Å, Figure S17) along with typical intermolecular hydrogen bonding interactions via its N2 and N1 atoms respectively (distance of N2…H6–C6 = 2.932(4) Å, bond angle of 131.83°; distance of N1…H14–C14 = 2.581(4) Å, bond angle of 143.94(4)°). Finally, the crystal of 8,6′-Me2pq is stabilized by intramolecular non-classical C–H∙∙∙π contacts, (distance of (C13–H13)∙∙∙centroid (C4–C9) = 3.612(6) Å; distance of (C10C–H10C)∙∙∙centroid (N2–C11–C15) = 3.604(6) Å) and by non-classical intermolecular interactions (C3–H3∙∙∙N2 = 2.954(6) Å; bond angle = 145.19(6)°, Figure S18).
2.2. Synthesis and Characterization of the Ruthenium(II) Complexes 1–8
All the organic ligands reported herein were synthesized aiming to prepare new and efficient ruthenium(II)-based transfer hydrogenation catalysts. Interestingly a literature search revealed that the coordination chemistry of 8-Mepq, 6′-Mepq, 8,6′-Me2pq and 4,6′-Me2pq is currently unknown, while for biqcame only a silver(I) complex has been reported [44]. These findings have prompted us to synthesize new organometallic ruthenium(II) complexes with the aforementioned ligands. The reaction of the ruthenium(II) dinuclear complex [Ru(η6-p-cymene)(μ-Cl)Cl]2, with two molar equivalents (slight excess) of the appropriate ligand L was performed in dry methanol under an argon atmosphere. Subsequent treatment of the non-isolated chlorido intermediates, with a saturated aqueous KPF6 solution, afforded the η6-arene ruthenium(II) complexes of the general type [Ru(η6-p-cymene)(L)Cl][PF6] (L = 8-Mepq (1), 6′-Mepq (2), 8,6′-Me2pq (3), 4,6΄-Me2pq (4), 4-Mepq (5), biqcame (6), biq (8)) as bright yellow (orange for 6) colored solids in high yield. As representative chlorido analogs of the series, complexes 6-Cl and 7-Cl, incorporating the biqcame and pq ligands, were isolated as hygroscopic solids and spectroscopically characterized. In addition the synthesis of 8 was performed with a slight modification of the published procedure [35], using KPF6 instead of NH4PF6, leading in a higher synthetic yield (85 % instead of 71%). All complexes are air stable solids that dissolve in methanol, ethanol, acetone, and dimethylsulfoxide, while remain insoluble in dichloromethane, chloroform, and in water. The synthetic procedure and reaction conditions are depicted in Scheme 2a.
The new complexes were characterized by FT-IR, elemental analysis, multinuclear NMR spectroscopy and UV-Vis spectroscopy. The infrared spectra (Figures S19–S25) recorded in the region of 4000-400 cm–1 are very similar, as expected for compounds with similar molecular structures. The spectra, exception is for 6-Cl and 7-Cl, are dominated by the very strong bands at ∼840 cm−1 and 557 cm−1 that can be assigned to the ν3(P–F) and ν4(P–F) vibration modes of the [PF6]− anion [45]. In addition, the medium intensity band in the region of 800 cm−1 is typical for the stretching vibration mode of ν(Ru–C) [46]. Elemental analyses data of all complexes corroborate with the calculated composition of the proposed structures.
The 1H and 13C NMR spectra of all complexes in Me2CO-d6 (in CDC3 for 6-Cl) were assigned by 2D techniques (Figures S26–S47) and are in accord with the proposed structures depicted in Scheme 2a. The 1H NMR spectra of 1 and 5 are quite similar, displaying the expected resonance signals attributed to the aromatic ring protons of 8-Mepq and 4-Mepq ligands. These are shifted upfield compared to the resonance signals of the free ligands, indicating furthermore coordination to the ruthenium(II) center. The p-cymene ligand displays four separate resonance signals (doublets) in between δ 6.16–5.49 ppm, which are shifted upfield compared to the dinuclear ruthenium(II) precursor. In addition, the two doublets of doublets at δ 1.02, 0.90 (1) and δ 0.95, 0.91 ppm (5) can be assigned to the methyl protons of the isopropyl group. The resonance signals reported above are typical for non-symmetric ruthenium p-cymene complexes [47]. In the aliphatic region, characteristics are the high field signals at δ 3.31 (8-Mepq) and δ 3.00 ppm (4-Mepq), for the methyl groups of the quinoline moieties, respectively. For 6-Cl (CDCl3) and 8 (Me2CO-d6) however, two doublet resonance signals are observed for the p-cymene ligand (δ 5.98/5.81 (6-Cl) and δ 5.98/5.87 (8)), and one doublet resonance for the methyl protons of the isopropyl groups (δ 1.11 (6-Cl) and 0.89 ppm (8)), due to the highly symmetric biqcame and biq ligands. The 1H NMR data of 8 agree to those reported previously for the analogous PF6– salt [35].
The electronic absorption spectra of Me2CO solutions of the ruthenium(II) complexes 1-8 exhibit typical high energy absorption bands in the region of ∼260–350 nm that can be attributed to ligand-centered π–π* transitions and are included in Figures S48–S55. All complexes display a main and broad MLCT absorption band centered between 476 nm and 412 nm, with ε values ranging from 1300–3800 dm3 mol–1 cm–1.
2.3. Structural Characterization of Complexes 1–4 and 6
The molecular structures of complexes 1–4, 6 and 9 in the solid-state have been unambiguously determined by single-crystal X-ray diffraction. For all complexes, suitable single crystals were obtained by slow diffusion of pentane into an acetone solution of the compound. All complexes adopt a pseudo-octahedral geometry also known as three-legged piano stool. The ruthenium(II) center is surrounded by a chlorine atom, two N atoms of the appropriate chelating ligand, while the remaining basal sites are occupied by the η6-p-cymene ligand. Notably, a Cambridge Crystallographic Database search (5/2025) revealed that there are no reports about metal complexes comprising the bidentate ligands reported herein. The solid-state structures of the complex cations of 1 and 4 are depicted in Figure 4 and Figure 5, providing evidence about the nature of the molecules in the crystal.
Complexes 1 and 4 crystallize as clear orange block and plank crystals in orthorhombic and monoclinic Pbca and C2/c space groups. For both complexes, the Ru–Cl (~2.4 Å) and Ru–N (N1 and N2) bond lengths (2.149(15) Å, 2.063(14) Å (1) and 2.102(12) Å, 2.121 (11) Å (4)) are very close to those of similar complexes reported in the literature [26,48,49].
The crystal packing of 1 involves typical intramolecular π…π stacking interactions between ring centroids (C1–N1–C5) and (C16–C21) of adjacent molecules at a separation of 3.742(5) Å. The crystal is stabilized further by non-classical hydrogen bonding interactions (C3–H3…Cl = 2.834(5) Å, bond angle = 177.74(5)°) and C24S–H24D…Cl = 2.751(5) Å, bond angle = 140.80(5)°, Figure S56). Coordinated ligands 8-Mepq and 4,6′-Me2pq in 1 and 4, deviate from planarity as the angle between the planes of the pyridine ring and that of quinoline ring is 9.07(5)o (1) and 7.81(5)o (4) respectively. In the unit cell of 4 the structure is reinforced by non-classical intermolecular hydrogen bonding interactions (C26A–H26A…Cl = 2.886(4) Å; bond angle = 115.14(4)°) and intramolecular hydrogen bonding interactions C12–H12…Cl = 3.074(5) Å, bond angle = 152.70(5)°; C13–H13…Cl = 3.556(5) Å, bond angle = 89.44(5)°, Figure S57).
The mononuclear cationic complex 2 (Figure 4) with a methyl group at the 6′ position, crystallizes in the monoclinic space group P21/c. In this chlorido analog, the Ru–Cl bond length of approximately 2.4 Å and the Ru–N1 and Ru–N2 bond lengths of 2.1129(14) Å and 2.1054(15) Å respectively, are in the same range to other similar ruthenium(II) complexes [49]. In addition, the N1–Ru–Cl and N2–Ru–Cl bond angles are 86.55(4)o and 85.87(4)o respectively. The bond distance between ruthenium atom and p-cymene ring centroid (C16–C21) is 1.694 Å, while the planes defined by the pyridine (N1–C1–C5) and quinoline rings (C1–C2–C6–C9), deviate from planarity by 13.62(14)o. The crystal network is reinforced by characteristic non-classical C–H…π intramolecular interactions (distance of C24B–H24B…centroid (C18–C21 of cymene) = 3.433(4) Å) and by typical intramolecular contacts between the hydrogen atom H24A of the isopropyl group and the quinoline ring centroid from an adjacent molecule (distance of C24–H24A…centroid (C1–C5–N1) = 2.784(4) Å). Moreover, stability is provided by intermolecular hydrogen bonding interactions between molecules of 2 in the crystal (C25–H25A…Cl = 2.769(3) Å, bond angle = 109.68(3)°) along with intramolecular contacts including coordinated Cl atom and H15C for methyl group in pyridine (C15–H15C…Cl = 2.878(4) Å, bond angle = 138.73(4)°). Within the crystal of 2, the hydrogen atom H6 from the quinoline ring, participates in an intermolecular hydrogen bonding interaction with the Cl atom coordinated to ruthenium (C6–H6…Cl = 3.129(4) Å, bond angle = 119.33(4)°, Figure S58).
Clear red plates of 6 were crystallized in monoclinic crystal system and P21/n space group. As expected, the structural features of 6 are quite similar to those of 1, 2 and 4. Within the cell, the crystal is stabilized by intramolecular non-classical hydrogen bonding interactions between O4 and H32 from the isopropyl group of an adjacent molecule (O4(A)…H32(B)-C32(B) = 3.125(4) Å, bond angle 133.30(4)°). Also, stability is provided by non-classical C–H…π intramolecular interactions (distance of C11C–H11C…centroid (C1–C9) = 3.531(4) Å, Figure S59)
Upon crystallization of complex 3, dark red single crystals were afforded which contained two independent molecules of [Ru(η6-p-cymene)(8,6′-Me2pq)Cl][PF6] (Ru-3) and [Ru′(η6-p-cymene)(8,6′-Me2pq)Cl][PF6] (Ru′-3) in the unit cell, in the ratio of 1:1 (Figure 6).
This was the result of a slight rotation of the arene ring, which renders both molecules different. For clarification, a superposition of both independent molecules is shown in Figure 7.
In both molecules the ruthenium metal center adopts a typical pseudo-octahedral coordination geometry, as shown by the N(1)–Ru–N(2), N(1)′–Ru′–N(2)′ bond angles of 78.2(3)° and 77.60(2)°, and by the N(1)–Ru–Cl, N(1)′–Ru–Cl′ bond angles of 93.46(2)° and 94.36(2)° respectively. These structural characteristics are common for η6-arene ruthenium complexes [49]. The bonding parameters of both independent molecules differ slightly. The ruthenium–centroid (C17–C22) and Ru–N1 bond lengths of Ru-3 are slightly elongated compared to those of Ru′-3, while the Ru–N2 bond length slightly decreased, reflecting probably the effect of the slight rotation of the arene ring. Moreover, the relevant N1–Ru–Cl (93.46(2)°) and N2–Ru–Cl (82.90(2)°) bond angles of Ru-3, differ slightly from those of Ru′-3 ((94.36(2)° and (83.37(2)°). The Ru–Cl bond remains practically unaltered at 2.390 Å. In both molecules the aromatic rings comprising the quinoline moieties deviate from planarity by 8.40o and 6.41o respectively. In the unit cell, pairs of molecules of 3 are stabilized by intermolecular π–π stacking interactions including the ring centroids (N2–C11–C15, pyridine) and (C6–C9 quinoline) and the ring centroids (N1–C1–C5, quinoline) and (C11–C15, pyridine) of adjacent molecules, at a separation of 3.980 (4) Å and 3.698(4)Å (Figure S60).
Improved crystallographic data of the known crystal structure of complex 8, are included in the Supplementary Materials (Figure S61 and Table S1). The solid-state structure of 8 reveals a typical pseudo-octahedral geometry considering that the η6-p-cymene ligand occupies three facial coordinated positions. The structural features of 8 reported herein are slightly improved, as shown for example from the bond angles N1–Ru–N2 = 76.49(7)ο (76.6(4)ο published), N1–Ru–Cl = 86.52(5)ο (87.8(3)ο published) and N2–Ru–Cl = 87.59(5)ο (86.4(3)ο published) respectively. In the unit cell, the crystal is stabilized by intramolecular non-classical C–H…π intramolecular interactions (distance of C27A–H27A…centroid (C1–C9) = 3.961(7) Å; distance of C27C–H27C…centroid (C1–C9) = 4.676 (7) Å, Figure S62).
2.4. Synthesis and Characterization of the Ruthenium(II) Complexes 9–11
The cationic ruthenium(II) complexes 9–11 (Scheme 2b), were prepared according to the procedure followed for complexes 1–8, using one mol equivalent of the relevant ligand. As a result, the [Ru(η6-p-cymene)(L)Cl][Ru(η6-p-cymene)Cl3] (L = pqcame (9), 4-Mepq (10), pq (11)) analogs, comprising the [Ru(η6-p-cymene)Cl3]– anion, were isolated as orange solids, in excellent yields (> 90%). They are air stable water-soluble solids, that dissolve in common organic solvents, while they are not soluble in acetone, diethyl ether and pentane. Although there are recent reports of ruthenium(II) complexes consisting of this counter ion [50,51], none of them refer to substituted pyridine – quinoline – based ligands.
The FT-IR spectra of 9–11 (Figures S63–S65) are very similar. The spectra are dominated by the intense ν(C–H) aromatic and aliphatic stretching vibration bands along with those of ν(C–C) stretching vibration modes. The intensity of these vibration modes in 9–11 is significantly higher compared to the relevant congener complexes 3, 5 and 8, which do not contain the [Ru(η6-p-cymene)Cl3]– counter anion.
The 1H and 13C NMR spectra of 9–11 in CDCl3, were assigned by two-dimensional routine techniques (Figures S66–S74). Interestingly, the -CH resonance signal from the -CH(CH3)2 group, corresponding to the [Ru(η6-p-cymene)(pqcame)Cl]+ cation of 9, is split in two resonance signals (septets) at δ 3.20 (CHA) and 2.94 CHB) ppm, in a ratio of 60:40. Accordingly, the two doublet resonances at δ 1.38 (CH3A) and 1.30 (CH3B) ppm are attributed to the methyl group protons of -CH(CH3)2. Moreover, the two singlets at δ 2.31 (CH3A-p-cym) and 2.18 (CH3B-p-cym) ppm (integration for 3H), were assigned to the methyl group hydrogens of p-cymene from the complex cation of 9. By analogy, the septet at δ 2.40 ppm is typical for the -CH' proton of -CH'(CH3')2 group corresponding to the [Ru(η6-p-cymene)Cl3]– counter anion. The doublet resonances at δ 0.96 and 0.91 ppm can be attributed to the methyl group protons of -CH'(CH3')2. Finally, the broad singlet at δ 2.18 ppm overlapped with the resonance signal of CH3B-p-cym, can be assigned to the CH3'-p-cym protons. Analogous behavior has been reported for complexes 10 and 11, with a relevant ratio of approximately 55:45, corresponding to the -CH resonance signals from the -CH(CH3)2 group, which are split.
The absorption spectra of 9–11 recorded in CHCl3 display a relatively broad absorption band in between 415 nm to 450 nm assigned to metal-to-ligand charge transfer (MLCT) transitions (Figures S75, S77, S79). In water, less intense MLCT transitions were observed, but within the same range (Figures S76, S78).
2.5. Structural Characterization of Complex [Ru(η6-p-cymene)(pqcame)Cl][Ru(p-cymene)Cl3]• CH2Cl2 (9)
Additional information about the structure of 9 is provided by single-crystal X-ray crystallography performed on clear red plank crystals, which crystallized in the monoclinic space group P21/n. Suitable crystals of 9•CH2Cl2 were obtained upon the slow diffusion of pentane into a CH2Cl2 solution of the complex (Figure 8). In the unit cell, each [Ru(η6-p-cymene)(pqcame)Cl]+ complex cation is surrounded by a [Ru(η6-p-cymene)Cl3]– complex ion, both displaying a three-legged piano stool geometry, including also a dichloromethane molecule. The bond distance between the ruthenium(II) center and the p-cymene ring centroid (C17–C22) of [Ru(η6-p-cymene)(pqcame)Cl]+ is 1.689(4) Å. All other structural features, (Ru–N, Ru–Cl bond distances and relevant bond angles) comply well with those reported for the analogous [Ru(η6-p-cymene)(pqcame)Cl](PF6) complex [33], and to those of complexes 1–4 and 6, reported herein. Within the [Ru(η6-p-cymene)Cl3]– counter ion, the Ru–Cl bond distances are in the range of 2.429–2.436 Å. Moreover, the bond angles Cl(2)–Ru(2)–Cl(3) = 86.95(4)ο, Cl(2)–Ru(2)–Cl(4) = 88.10(5)ο and Cl(3)–Ru(2)–Cl(4) = 87.91(4)ο respectively, are in agreement with analogous complexes incorporating this complex ion [50]. In the crystal, stabilization is provided by intramolecular non-classical hydrogen bonds (C25A–H25A…Cl2 = 3.817(4) Å, bond angle = 97.87(4)°). Additional short contacts include typical intermolecular C–H…π interactions, between the methyl group of the isopropyl moiety and the pyridine and quinoline rings respectively (C24A–H24A…centroid (C12–C16–N2) = 3.263(4) Å and C24C–H24C…centroid (C1–C5–N1) = 3.351(4) Å, Figure S80).
2.6. Catalytic Transfer Hydrogenation Studies
Complexes 1–11 have been prepared aiming to evaluate their catalytic performance in the transfer hydrogenation reactions of various aromatic ketones (Scheme 3).
All experiments were performed at 82 oC using 2-propanol as a hydrogen donor, in the presence of KOH as a base (KOiPr), according to well-defined protocols [30,52]. From the initial experiments conducted, the optimum conditions were verified by using 0.25 mol% of the ruthenium catalyst and 10 mol% of KOiPr, reaching to a substrate/catalyst/base molar ratio of approximately 400:1:40. The percent conversion of the substrates examined, to the corresponding alcohols was monitored over time by 1H NMR spectroscopy. Acetophenone was used as a model substrate, and the results of the preliminary screening are summarized in Table 1.
All complexes significantly catalyze the transfer hydrogenation of acetophenone, in the presence of a base. At ambient temperature, conversions <10% were observed while the presence of the base proved crucial for the catalytic experiment. Parameters such as (i) the presence of electron donating groups (methyl) either in the pyridine or quinoline ring and/or in both rings (complexes 1–5, 11), (ii) the presence of electron withdrawing groups (COOMe, 6 and Ru-pqcame) and (iii) the effect of the different counter ion used ([PF6]–, 1–8 vs [Ru(η6-p-cymene)Cl3]–, 9–11), were examined.
From Table 1, it becomes evident that the presence of electron donating groups (in certain positions) significantly improves the catalytic properties of complexes 1–5. An exception stands for 3, probably due to steric reasons, as the two methyl groups of the 8,6′-Me2pq ligand are in close proximity to the ruthenium(II) center of the catalyst (entry 5). It seems that an incubation period 15 min is required to activate this catalyst. Within the series, catalysts 1 and 5 bearing methyl groups at positions 8 and 4 of the quinoline moiety exhibit the best activity. The reduction of acetophenone proceeded smoothly to afford quantitatively 1-(phenyl)-ethanol within 15 min (entries 1 and 8). The time dependence of transfer hydrogenation of acetophenone by catalysts 1–5, 7-Cl and 8 is shown in Figure 9.
From a closer view of Figure 9, it is clearly seen that 1 and 5 reduce >80% of acetophenone within the first 5 min, while conversion reaches almost 92%, practically quantitative, after 10 min of reaction. This is in contrast with the reduced catalytic activity of an analogous complex bearing a 2-(5,6-dimethyl-1H-benzimidazol-2-yl)quinoline ligand with methyl groups at the 5,6-positions [53]. For complexes 2 and 4, with methyl groups at positions 6′ and 4,6′ respectively, a two-fold decrease of the activity was observed, since both complexes require approximately 30 min to reduce acetophenone (entries 3 and 7). Catalysts 1, 2, 4 and 5 proved more efficient compared to 7-Cl and 8. The latter containing the non-substituted pq (7-Cl) and biq (8) ligands need almost 60 min to exhibit the highest activity (entries 12 and 13). Notably, for complex 3 comprising the rather bulky 8,6'-Me2pq ligand with two methyl groups close to the ruthenium center, an induction period of 15 min is required (entry 4). However, within the next 5 min of reaction, the conversion rate increases dramatically from 20% to almost 50%, reaching that of 100% after approximately 55 min. On the other hand, the catalytic potency of the methyl ester analogs Ru-pqcame and 6 (electron withdrawing groups) is significantly reduced, since the time required for the hydrogenation of acetophenone is 180 min (entries 10 and 11). For Ru-pqcame, within the first 60 min, hydrogenation reaches a 38% conversion. This result comes to an agreement with that reported for the neutral ruthenium(II) triphenylphosphine complex cis-[RuCl2(PPh3)2(pqcame)] [31,32].
The higher catalytic performance of 1 and 5 is reflected also by the final TOFs of 1600 h–1 achieved. All other complexes display lower TOF values ranging from 360 h–1 to 1440 h–1. To this end we must report that the catalytic activity of complexes 1–5 is comparable to that of other half-sandwich ruthenium(II) complexes bearing 2,2΄-bipyridine and 4,4΄-dimethy-2,2΄-bipyridine, 2-amino-pyridine and 2,2΄-quinoline-benzimidazole ligands [20,24]. Interestingly, their activities are higher than those of the relevant ruthenium(II) arene complexes with N-substituted 3,4-dihydroquinazoline ligands, that need 60 min for conversion of acetophenone [54]. In addition, complexes 1 and 5 exhibit higher conversions in comparison with the [RuCl2(benzene)(PAr3)] complexes (Ar = p-methoxyphenyl, triphenyl, p-trifluoromethylphenyl) [55], and the cationic complex [Ru(ImH)2(p-cym)Cl]Cl; ImH = 1H-Imidazole (33% for acetophenone conversion) [27]. Notably the potency of both catalysts is significantly higher than that reported for the arene ruthenium(II) derivatives using different oximes (11H-indeno [1,2-b]quinoxaline-11-one oximes and tryptanthrin-6-oxime) [26]. On the other hand, catalysts 1 and 5 are less efficient compared to ruthenium(II) complexes that contain bidentate ligands with P,N atoms [17,20].
For complexes 9–11, bearing the same ligands as complexes Ru-pqcame, 5 and 7-Cl respectively, interesting transfer hydrogenation results were obtained. Within the series, complex 11 is the most potent, converting acetophenone by 90% in 15 min (entry 16). In contrast, the structurally similar complex 5 exhibits quantitative conversion, within the same period. Presumably the presence of the less constrained [PF6]– ion, as opposed to the sterically crowded [Ru(η6-p-cymene)Cl3]– analog of 11, may be responsible for the improved catalytic activity of 5. In line with that (entry 17) complex 7-Cl is more active (95%) than 11 (90%). This also may be the case for complex Ru-pqcame when compared to complex 9 (entries 15 and 10), highlighting probably the role of the sterically crowded [Ru(η6-p-cymene)Cl3]– complex anion in 9. For both catalyst precursors, conversion of acetophenone transfer hydrogenation versus reaction time, was compared during the first hour, and the results can be seen in the graph of Figure S81. For the catalyst with the [PF6]– anion, conversion of the sustrate is higher than its [Ru(η6-p-cymene)Cl3]– analog. The TOFs reported for Ru-pqcame and 9 are 120 h–1, somehow lower compared to those of 7-Cl (380 h–1) and 11 (360 h–1) and significantly lower than those of 10 (1440 h–1) and 5 (1600 h–1) respectively.
After an initial screening with acetophenone, the encouraged results obtained by catalysts 1 and 5 prompted us to study transfer hydrogenation reactions with p-substituted derivatives of acetophenone along with benzophenone (Table 2). All complexes showed very good to excellent catalytic potencies. In general, the tendency observed is similar to that reported for the conversion of acetophenone. Notably 4-fluoro-acetophenone and 4-chloro-acetophenone substrates, with electron withdrawing groups at the p-position, were quantitatively converted by 1 and 5 within 15 min. For complexes 3, 6, Ru-pqcame and 11, however the conversion rate is slightly decreased by approximately 5 to 10%. Interestingly, the rate of 4-bromo-acetophenone has decreased (conversion range of 55-73%) compared to the relevant analogs substituted by chlorine and fluorine in the para position. For complex 6 however an 88% conversion was achieved over 180 min. This may be due to the higher mesomeric effect of –Br group that has a result on the reduced electron density of the carbonyl bond of the substrate [31].
In comparison, several catalysts like [RuCp(PN)Br] (PN = N,N-dimethyl-2-diphenylphosphine-ethylamine) [56], [RuCl2(dcype)(bipy)] (dcype = 1,2-di-(dicyclehexaphospine)ethane, bipy = 2,2΄-bipyridine) [57], and [Ru(η6-p-cymene)(L)Cl]+ (L = 4-((Ε)-(4-ethylphenyl)diazenyl)-2((E)-(phenylimino)methyl)phenol) [58], display analogous behavior. Substitution in the para position by a methoxy group decreases the rate of reduction, ranging approximately in between 70–96%, which can be attributed to the electron donating properties of the p-substituted methoxy group.
Finally, we carried out transfer hydrogenation of benzophenone using these catalysts. As seen from Table 2 (entries 49–53), complexes 1–5 exhibited considerable efficiencies ranging from 80 to 99%, though the reduction of benzophenone to benzhydrol is about 2 to 4 times less efficient compared to acetophenone. It should be noted that the cationic ruthenium(II) catalyst 5, is less efficient (60 min) in the transfer hydrogenation of benzophenone, compared to the neutral ruthenium(II) complex cis-[RuCl2(PPh3)2(4-Mepq)]. The latter reached a 96% conversion within 30 min [31]. Under the same conditions, catalysts Ru-pqcame and 6, were less effective over a period of 180 min (entries 54, 55). However, catalyst 7-Cl bearing the non-substituted pq ligand has shown better activity than that of cis-[RuCl2(PPh3)2(pq)], exhibiting 100% conversion, within 150 min. All other complexes hold the same trend followed for the reduction of acetophenone. As an exception, complex 11 reached a 99% conversion that is higher compared to that of acetophenone (90%).
To this end, we performed additional experiments trying to propose key intermediates (active catalysts) that are involved in the TH process reported herein. Thus, we examined the reaction of complex 4 as the catalyst precursor (Cat) with KOiPr (base) in 2-propanol, followed by subsequent heating to reflux for 120 min. The catalyst (Cat) to base molar ratios ranged from 1:3 to 1:10. A characteristic pine green solid was isolated, after evaporation of the solvent, when the Cat to base molar ratio was 1:10. The 1H NMR spectrum of the crude product (in CH3OD), in the hydridic region, revealed the presence of a broad singlet resonance at δ – 0.91 ppm (approximately 12% formation, based on integration, Figure S82), which can be attributed to the formation of Ru-H species [59]. Based on these findings, Ru-hydrides can be considered as the active species, and an inner-sphere mechanism may be proposed. Apparently upon the addition of the base, a ruthenium(II) alkoxide is potentially formed, which then undergoes β-H elimination, leading to the formation of the ruthenium-H species that can be considered as the catalytically active species [60,61].
3. Materials and Methods
The synthesis of the ruthenium(II) complexes was carried out under argon using standard Schlenk techniques, while the organic ligands were prepared under aerobic conditions. All solvents were of analytical grade and were distilled and dried before use, according to standard methods. The distilled solvents were stored over molecular sieves under an argon atmosphere. RuCl3∙H2O was purchased from Riedel de Haën and α-terpinene was purchased from Sigma-Aldrich. Ligand precursors 2,2′-pyridyl-quinoline-4-carboxylic acid (pqca), 8-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (8-Mepqca), 6′-methyl-2,2′-pyridyl-quinoline-4-carboxylic acid (6′-Mepqca), 8,6′-dimethyl-2,2′-pyridyl-quinoline-4-carboxylic acid (8,6′-Me2pqca), 2,2′-pyridyl-quinoline (pq) [36,37,38] and 2,2′-biquinoline (biq) were prepared according to the literature reports [40,41,42]. Ligands 4-methyl-2,2′-pyridyl-quinoline (4-Mepq), methyl 2,2′-pyridyl-quinoline-4-carboxylate (pqcame) and dimethyl 2,2′-biquinoline-4,4′-dicarboxylate (biqcame) [43,44], as well as the ruthenium(II) starting material [Ru(η6-p-cymene)(μ-Cl)Cl]2 [62], were made according to literature procedures. The [Ru(η6-p-cymene)(pqcame)Cl][PF6] (Ru-pqcame) complex was synthesized according to the literature [59]. Infrared spectra (FT-IR) were recorded on IR Affinity-1 SHIMADZU as potassium bromide pellets in the spectral range 4000-400 cm–1. Elemental analyses were obtained from Microanalysis Center of Institut für Anorganische Chemie Universität Bonn. 1H and 13C NMR spectra were recorded on Varian 200 MHz and on Bruker Avance Neo 400 MHz. NMR spectra of metal complexes were assigned using the 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC methods. J values are given in Hz. Absorption spectra were recorded with a CARY 3E UV-vis spectrometer. X-ray diffractions were recorded with Bruker D8-Venture, Bruker APEX-II CCD and Bruker X8-Kappa ApexII.
3.1. Synthesis and Characterization
3.1.1. Synthesis of the Organic Ligands 8-Mepq, 6′-Mepq, 8,6′-Me2pq and pq
In a Pyrex glass tube, 1 mmol of the carboxylic acid precursor (pqca, 8-Mepqca, 6′-Mepqca or 8,6′-Me2pqca) was heated rapidly to reach the melting point temperature. The resulting black solid was left to cool at ambient temperature, treated with hot petroleum ether (25 mL) and subsequently active carbon was added. After filtration, the clear pale-yellow filtrates were evaporated to dryness affording the required ligands.
8-methyl-2,2′-pyridyl-quinoline (8-Mepq). Off-white solid. Yield: (0.140 g, 67%). IR (KBr, νmax/cm−1: 3038 (w, ν(C–H)arom), 2919 (w, νas(C–H)aliph), 1597 (m, ν(C=C)), 1446 (m, ν(C=N)). 1H NMR (CDCl3, 200 MHz, 298 K) δH/ppm 8.74 (2H, m, H5/H6΄), 8.58 (1H, d, J = 6, H4), 8.25 (1H, d, J = 8, H3), 7.86 (1H, t, J = 8, H6), 7.69 (1H, d, J = 8, H3΄), 7.58 (1H, d, J = 6, H5΄), 7.45 (1H, d, J = 6, H4΄), 7.35 (1H, m, H7), 2.92 (3H, s, CH3-quin). 13C{1H} NMR (CDCl3, 50 MHz, 298 K) δC/ppm 156.89 (C8), 154.82 (C2), 149.16 (C8a), 146.96 (C3), 137.83 (C6), 137.13 (C6′), 136.98 (C4΄), 129.72 (C2΄), 128.36 (C3΄), 126.67 (C4a), 125.70 (C4), 124.02 (C5), 121.93 (C5′), 118.53 (C7), 18.00 (CH3-quin). ESI-HRMS (MeOH, positive mode): m/z 221.109 [C15H13N2 + H]+ (calc. 221.107), m/z 243.089 [C15H13N2 + Na]+ (calc. 243.089), m/z 441.201 [C30H26N4 + H ]+ (calc. 441.207), m/z 463.196 [C30H26N4 + Na]+ (calc. 463.189).
6′-methyl-2,2′-pyridyl-quinoline (6′-Mepq). Off-white solid. Yield: (0.135 g, 65%). IR (KBr, νmax/cm−1): 3057 (w, ν(C–H)arom), 2921 (w, νas(C–H)aliph), 1616 (w, ν(C=C)), 1592 (s, ν(C=C)), 1453 (s, ν(C=N)). 1H NMR (CDCl3, 200 MHz) δH/ppm 8.60 (1H, d, J = 8, H5), 8.45 (1H, d, J = 8, H4), 8.26 (1H, d, J = 8, H3), 8.18 (1H, d, J = 8, H8), 7.84 (1H, d, J = 6, H5΄), 7.75 (2H, m, H6/H4΄), 7.54 (1H, t, J = 8, H7), 7.21 (1H, d, J = 6, H3΄), 2.68 (3H, s, CH3-py). 13C{1H} (CDCl3, 50 MHz) δC/ppm 158.07 (C2), 156.69 (C2′), 155.89 (C6΄), 148.09 (C4΄), 137.26 (C5′), 136.82 (C8a), 129.94 (C6), 129.59 (C7), 128.36 (C5), 127.74 (C8), 126.74 (C3), 123.72 (C4), 119.32 (C3΄), 119.01 (C4a), 24.81 (CH3-py). ESI-HRMS (MeOH, positive mode): m/z 221.109 [C15H13N2 + H]+ (calc. 221.107), 243.089 [C15H13N2 + Na]+ (calc. 243.089), 259.063 [C15H13N2 + K ]+ (calc. 259.063), 463.189 [C30H26N4 + Na]+ (calc. 463.189).
8,6′-dimethyl-2,2′-pyridyl-quinoline (8,6′-Me2pq). Off-white solid. Yield: (0.152 g, 72%). IR (KBr, νmax/cm−1): 3054 (w, ν(C–H)arom), 2918 (m, νas(C–H)aliph), 1614 (w, ν(C=C)), 1598 (vs, ν(C=C)), 1453 (s, ν(C=N)). 1H NMR (CDCl3, 400 MHz) δH/ppm 8.64 (1H, d, J = 8, H3), 8.56 (1H, d, J = 8, H3΄), 8.23 (1H, d, J = 8, H4), 7.74 (1H, t, J = 6, H4΄), 7. 68 (1H, d, J = 8, H5), 7.57 (1H, d, J = 8, H7), 7.43 (1H, t, J = 8, H6 ), 7.19 (1H, d, J = 8, H5΄), 2.93 (3H, s, CH3-py), 2.69 (3H, s, CH3-quin). 13C{1H} (CDCl3, 50 MHz) δC/ppm 157.79 (C8), 156.23 (C6΄), 155.09 (C2), 146.94 (C8a), 137.07 (C4′), 136.91 (C4), 129.57 (C8), 128.26 (C7), 126.45 (C6), 125.64 (C5), 123.48 (C5′), 118.88 (C3′), 118.65 (C3), 27.74 (CH3-py), 17.97 (CH3-quin). ESI-HRMS (MeOH, positive mode): 235.128 [C16H15N2 + H]+ (calc. 235.123), 257.106 [C16H15N2 + Na]+ (calc. 257.105), 273.079 [C16H15N2 + K]+ (calc. 273.079), 491.221 [C32H30N4 + N a]+ (calc. 491.221).
2,2′-pyridyl-quinoline (pq). Light yellow solid. Yield: (0.15 g, 60%). IR (KBr, νmax/cm−1): 3056 (w, ν(C–H)arom), 2925 (w, νas(C–H)aliph), 1596 (vs, ν(C=C)), 1450 (m, ν(C=N)), 778(s) and 713 (m) [δ(C-H) out of plane]. 1H NMR (CDCl3, 400 MHz) δH/ppm 8.75 (1H, td, J = 4 and 8, H6’ ), 8.67 (1H, dt, J = 8, H8), 8.57 (1H, d, J = 8, H3), 8.30 (1H, d, J = 8, H4), 8.19 (1H, d, J = 8, H5), 7.90 (2H, m, J = 8, H3’/H4’ ), 7.75 (1H, ddd, J = 4 and 8, H7), 7.57 (1H, ddd, J = 4 and 8, H6), 7.38 (1H, ddd, td, J = 4 and 8, H5΄).
3.1.2. Synthesis of the Organic Ligand 4,6′-Me2pq
In a 50 mL round-bottomed flask and under an argon atmosphere, 2-aminoacetophenone (0.203 g, 1.5 mmol) and 6-methyl-2-acetylpyridine (0.182 g, 1.5 mmol) were dissolved in dry ethanol (5 mL), followed by the addition of 3 drops of 66% (w/w) aqueous KOH solution. The mixture was refluxed for 18 h and then cooled at an ambient temperature. The obtained solution was treated with 1 M HCl(aq), until neutral pH and the organic solvent was rotary evaporated. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL), the organic layer was treated with anhydrous MgSO4, filtered off and the filtrate was evaporated to dryness to yield a yellow oily residue. The residue was treated with a methanol/water mixture (1:9, v/v) to afford an off-white solid, that was dried in a vacuum desiccator under P2O5. Yield: (0.210 g, 60%). IR (KBr, νmax/cm−1): 3054 (w, ν(C–H)arom), 2922 (m, νas(C–H)aliph), 1614 (w, ν(C=C)), 1597 (s, ν(C=C)), 1453 (s, ν(C=N)). 1H NMR (CDCl3, 400 MHz) δH/ppm 8.43 (2H, m, H3/H5΄), 8.18 (1H, d, J = 8, H8), 8.01 (1H, d, J = 8, H5), 7.73 (2H, m, H4΄/H6), 7.56 (1H, t, J = 12, H7), 7.20 (1H, d, J = 8, H3΄), 2.80 (3H, s, CH3-quin), 2.70 (3H, s, CH3-py). 13C{1H} NMR (CDCl3, 100 MHz) δC/ppm 157.96 (C4), 156.22 (C2), 156.12 (C2’), 147.99 (C8α), 144.98 (C6’), 137.19 (C6), 130.52 (C8), 129.24 (C4΄), 128.36 (C4α), 126.46 (C7), 123.88 (C3΄), 123.80 (C5), 119.74 (C5΄), 118.98 (C3), 24.83 (6΄-CH3), 19.05 (4-CH3).
3.1.3. General Synthetic Procedure of Complexes 1–8
In a Schleck tube and under an argon atmosphere, 0.095 g (0.15 mmol) of [Ru(η6-p-cymene)(μ-Cl)Cl]2 and 2.3 mol equivalents (0.36 mmol) of the corresponding organic ligand (L) were added. Upon addition of dry methanol (10 mL) the solution obtained an orange-yellow color, and the mixture was stirred for 18 h at an ambient temperature. The solvent was rotary evaporated, and the oily residue was treated with diethyl ether (2 × 3 mL). The obtained solid was dissolved in water (5 mL) and few drops of a saturated aqueous solution of KPF6 were added. Upon addition, a yellow precipitate appeared, and the mixture was stirred for 30 min to ensure completion of the reaction. After filtration, the yellow solid was washed with water (5 mL), chloroform (3 mL) and finally with diethyl ether (2 × 5 mL) and was dried in vacuo at 50 °C.
3.1.4. Data for Complexes 1–8
[Ru(η6-p-cymene)(8-Mepq)Cl][PF6] (1). Yield: (0.088 g, 92%). Found: C, 46.04; H, 4.15; N, 4.25. C25H26ClF6N2PRu∙H2O requires C, 45.91; H, 4.32; N, 4.28%. IR (KBr, νmax/cm−1): 3051 (w, ν(C–H)arom, 3032 (w, ν(C–H)arom), 2873 (w, νas(C–H)aliph), 1572 (m, ν(C=N)), 1477 (s, ν(C=C)), 1383 (m, ν(C=C)), 841 (vs, ν(P–F)), 803 (s, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4 ): λmax/nm 347 (ε/ dm3 mol–1 cm–1 7600), 453 (1300). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.39 (1H, d, J = 8, H6΄), 8.89 (1H, d, J = 8, H3), 8.64 (1H, d, J = 8, H5), 8.59 (1H, d, J = 8, H4), 8.38 (2H, m, J = 8, H7/H4΄), 8.14 (1H, d, J = 8, H7), 8.02 (1H, d, J = 8, H3΄), 7.89 (2H, t, J = 4, H5΄/H6), 6.01 (1H, d, J = 4, H-cymar), 5.83 (1H, d, J = 4, H-cymar), 5.57 (1H, d, J = 4, H-cymar), 5.49 (1H, d, J = 4, H-cymar), 3.31 (3H, s, CH3-quin), 2.11 (1H, m, CH-(CH3)2), 1.84 (3H, s, CH3-cym), 1.02 (3H, d, J = 8, CH-(CH3)2), 0.90 (3H, d, J = 8, CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 159.24 (C6΄), 156.26 (C2), 155.87 (C8), 152.21 (C8a), 141.91 (C3), 140.23 (C4΄), 136.18 (C3΄), 135.80 (C4a), 130.78 (C6), 129.12 (C5΄), 127.51 (C5), 127.23 (C7), 125.13 (C2΄), 119.60 (C4), 106.57 (CH3-Ccym), 100.51 (Ccym-CH-(CH3)2), 86.89 (C-Ccym-ar), 85.64 (C-Ccym-ar), 84.20 (C-Ccym-ar), 83.29 (C-Ccym-ar), 30.63 (CH-(CH3)2), 21.83 (CH3-quin), 21.27 (CH-(CH3)2), 20.76 (CH-(CH3)2), 16.81 (CH3-cym).
[Ru(η6-p-cymene)(6′-Mepq)Cl][PF6] (2).Yield: (0.085 g, 89%). Found: C, 46.81; H, 4.23; N, 4.36. C25H26ClF6N2PRu requires C, 47.21; H, 4.12; N, 4.40%. IR (KBr, νmax/cm−1): 3099 (w, ν(C–H)arom), 2876 (w, νas(C–H)aliph), 1581 (w, ν(C=N)), 1479 (m, ν(C=C)), 1379 (w, ν(C=C)), 840 (vs, ν(P–F)), 802 (s, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 341 (ε/dm3 mol–1 cm–1 2040), 356 (24305), 427 (3513). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.06 (d, 1H, J= 8 Hz, H8), 8.83 (d, 1H, J= 8Hz, H3), 8.63 (d, 2H, J= 8Hz, H4/H5), 8.23 (m, 2H, H3΄/H4΄), 8.08 (t, 1H, J= 8Hz, H7), 7.89 (m, 2H, H6/H5΄), 6.18 (1H, d, J = 4, H-cymar), 6.05 (1H, d, J = 8, H-cymar), 5.90 (1H, d, J = 8, H-cymar), 5.80 (1H, d, J = 8, H-cymar), 3.34 (3H, s, CH3-py), 2.32 (4H, m, CH3-cym/CH-(CH3)2), 1.00 (3H, d, J = 8, CH-(CH3)2), 0.93 (3H, d, J = 8, CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 166.76 (C6΄), 158.05 (C2΄), 156.69 (C2), 150.73 (C8a), 141.97 (C3), 140.67 (C4΄), 133.44 (C7), 130.62 (C8), 130.42 (C4a), 130.32 (C3΄), 129.86 (C6), 129.72 (C5΄), 123.61 (C5), 120.03 (C4), 106.74 (CH3-Ccym), 104.84 (Ccym-CH-(CH3)2), 86.82 (C-Ccym-ar), 86.47 (C-Ccym-ar), 86.26 (C-Ccym-ar), 86.12 (C-Ccym-ar), 31.44 (CH-(CH3)2), 28.31 (CH3-py), 22.18 (CH-(CH3)2), 22.10 (CH-(CH3)2), 18.32 (CH3-cym).
[Ru(η6-p-cymene)(8,6′-Me2pq)Cl][PF6] (3). Yield: (0.088 g, 90%). Found: C, 47.99; H, 4.57; N, 4.28. C26H28ClF6N2PRu requires C, 48.04; H, 4.34; N, 4.31%. IR (KBr, νmax/cm−1): 3079 (w, ν(C–H)arom), 2928 (m, νas(C–H)aliph), 1571 (w, ν(C=N)), 1472 (m, ν(C=C)), 1383 (w, ν(C=C)), 838 (vs, ν(P–F)), 789 (m, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 339 (ε/ dm3 mol–1 cm–1 12250), 354 (14100), 424 (2100). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 8.88 (1H, d, J = 8, H3), 8.55 (1H, d, J = 8, H4), 8.45 (1H, d, J = 8, H3΄), 8.23 (1H, t, J = 8, H4΄), 8.12 (1H, d, J = 8, H5), 7.99 (1H, d, J = 8, H7), 7.86 (2H, m, H6/H5΄), 6.13 (1H, d, J = 4, H-cymar), 5.71 (1H, d, J = 8, H-cymar), 5.45 (1H, d, J = 8, H-cymar), 4.95 (1H, d, J = 8, H-cymar), 3.30 (3H, s, CH3-py), 3.27 (3H, s, CH3-quin), 2.24 (3H, s, CH3-cym), 2.02 (1H, m, CH-(CH3)2), 0.90 (3 H, d, J = 8, CH-(CH3)2), 0.85 (3H, d, J = 8, CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 165.39, 160.28, 156.77, 152.70, 142.43, 140.12, 136.09, 135.88, 130.37, 129.09, 128.78, 127.41, 123.69, 119.64, 106.0 (CH3-Ccym), 105.0 (Ccym-CH-(CH3)2), 85.63 (C-Ccym-ar), 85.16 (C-Ccym-ar), 82.88 (C-Ccym-ar), 82.11 (C-Ccym-ar), 30.42 (CH-(CH3)2), 29.39 (CH3-py), 22.50 (CH3-quin), 22.04 (CH-(CH3)2), 20.42 (CH-(CH3)2), 17.27 (CH3-cym).
[Ru(η6-p-cymene)(4,6΄-Me2pq)Cl][PF6] (4). Yield: (0.088 g, 90%). Found: C, 47.41; H, 4.50; N, 4.04. C26H28ClF6N2PRu∙0.5H2O requires C, 47.39; H, 4.28; N, 4.25%. IR (KBr, νmax/cm−1): 3087 (w, ν(C–H)arom), 2924 (m, νas(C–H)aliph), 1597 (s, ν(C=N)), 1472 (s, ν(C=C)), 1386 (w, ν(C=C)), 836 (vs, ν(P–F)), 760 (m, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4 ): λmax/nm 338 (ε/ dm3 mol–1 cm–1 13000), 347 (14500), 423 (2200). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.05 (1H, d, J = 4, H8), 8.61 (1H, d, J = 8, H5), 8.55 (1H, s, H3), 8.31 (1H, d, J = 8, H3΄), 8.21 (1H, t, J = 8, H4΄), 8.05 (1H, t, J = 8, H7), 7.91 (1H, t, J = 8, H6), 7.85 (1H, d, J = 8, H5΄), 6.15 (1H, d, J = 8, H-cymar), 6.01 (1H, d, J = 4, H-cymar), 5.86 (1H, d, J = 8, H-cymar), 5.75 (1H, d, J = 8, H-cymar), 3.33 (3H, s, CH3-py), 3.00 (1H, s, CH3-quin), 2.29 (4H, m, CH3-cym/ CH-(CH3)2), 0.99 (3H, d, J = 8, CH-(CH3)2), 0.93 (3H, d, J = 8, CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 166.68(C6’), 157.38 (C2΄), 156.38 (C2), 151.76 (C4), 150.17 (C9), 140.59 (C4′), 132.92 (C8), 131.15 (C6), 130.18 (C9), 129.62 (C5′), 126.11 (C3′), 123.37 (C6), 120.83 (C3), 106.68 (CH3-Ccym), 104.56 (Ccym-CH-(CH3)2), 86.38 (C-Ccym-ar), 86.22 (C-Ccym-ar), 85.92 (C-Ccym-ar), 31.44 (CH-(CH3)2), 28.32 (CH3-py), 22.22 (CH3-quin), 22.06 (CH-(CH3)2), 19.26 (CH-(CH3)2), 18.31 (CH3-cym), 13.26 (4-CH3).
[Ru(η6-p-cymene)(4-Mepq)Cl][PF6] (5). Yield: (0.080 g, 84%). Found: C, 44.01; H, 4.86; N, 3.99. C26H26ClF6N2PRu∙3H2O requires C, 44.48; H, 4.59; N, 3.99%. IR (KBr, νmax/cm−1): 3082 (w, ν(C–H)arom), 2877 (w, νas(C–H)aliph), 1596 (s, ν(C=N)), 1484 (m, ν(C=C)), 1384 (m, ν(C=C)), 839 (vs. br, ν(P–F)), 790 (m, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4): λmax/nm 335 (ε/ dm3 mol–1 cm–1 12600), 348 (13000), 412 (3000). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.59 (1H, d, J = 8, H8), 8.98 (1H, d, J = 8, H6’), 8.74 (1H, d, J = 8, H5), 8.58 (1H, H3), 8.32 (2H, m, J = 8, H3΄/H6), 8.09 (1H, td J = 8, H4’), 7.95(1H, t, J = 8, H5΄), 7.86 (1H, t, J = 8, H7), 6.15 (1H, d, J = 8, H-cymar), 6.06 (1H, d, J = 8, H-cymar), 6.00 (2H, m, H-cymar), 3.00 (3H, s, CH3-quin), 2.41 (1H, sept, J = 5, CH-(CH3)2), 2.29 (3H, s, CH3-cym), 0.95 (3H, d, J = 5, CH-(CH3)2), 0.91 (3H, d, J = 5, CH-(CH3)2).13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 157.12 (2C, C8/C2), 156.51 (C4a), 156.39 (C2΄), 151.71 (C4), 149.75 (C8a), 140.95 (C6), 133.05 (C4΄), 131.58 (C6΄), 130.32 (C5΄), 128.94 (C7), 126.18 (C3΄), 125.80 (C5), 120.83 (C3) 106.46 (CH3-Ccym), 105.18 (Ccym-CH-(CH3)2), 87.78 (C-Ccym-ar), 86.57 (C-Ccym-ar), 85.76 (C-Ccym-ar), 31.75 (CH-(CH3)2), 22.29 (CH-(CH3)2), 21.85 (CH-(CH3)2), 19.22 (CH3-Ccym), 18.71 (CH3-quin).
[Ru(η6-p-cymene)(biqcame)Cl][Cl] (6-Cl). According to the general synthetic procedure 2.4 but under reflux conditions. Yield: (0.108 g, 95%). IR (KBr, νmax/cm−1): 3040 (w, ν(C–H)arom), 2962 (w, νas(C–H)aliph), 1597 (vs, ν(C=N)), 1484 (m, ν(C=C)), 1383 (w, ν(C=C)), 793 (m, ν(Ru–C)). 1H NMR (400 MHz, 298K, CDCl3) δH/ppm 9.14 (2H, d, J = 8, H8/H8΄), 9.06 (H, s, H3/H3΄), 8.95 (2H, d J = 8, H5/H5΄), 8.07 (2H, t, J = 8, H7/H7 ΄), 7.92 (2H, t, J = 8, H6/H6΄), 5.98 (2H, d, J = 8, H-cymar), 5.81 (2H, d, J = 8, H-cymar), 4.19 (6H, s, COOCH3), 2.57 (1H, sept, J = 4, CH-(CH3)2), 2.10 (3H, s, CH3-cym), 1.11 (6H, d, J = 4, CH-(CH3)2). 13C{1H} NMR (100 MHz, CDCl3) δC/ppm 158.58 (CO), 132.91 (C7/C7΄), 131.62 ( C6/C6΄), 130.32 (C8/C8’), 127.09 (C5/C5΄), 124.26 (C3/C3’), 87.62 (2C, C-Ccym), 86.32 (2C, C-Ccym), 53.32 (O-CH3/O-CH3’), 30.68 (CH-(CH3)2), 29.28 (CH-(CH3)2), 22.27 (CH-(CH3)2), 17.74 (CH3-cym).
[Ru(η6-p-cymene)(biqcame)Cl][PF6] (6). As for the synthesis of 6-Cl and then according to the general synthetic procedure 2.4. Yield: (0.115 g, 95%). Found: C, 48.46; H, 4.15; N, 3.36. C32H30ClF6N2O4PRu requires C, 48.77; H, 3.84; N, 3.55%. IR (KBr, νmax/cm−1): 3093 (w, ν(C–H)arom), 3059 (w, ν(C–H)arom), 2926 (m, νas(C–H)aliph), 1726 (s, ν(C=O)), 1583 (m, ν(C=N)), 1474 (w, ν(C=C)), 1381 (w, ν(C=C)), 835 (vs, ν(P-F)), 820 (s, ν(Ru–C)), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4): λmax/nm 371 (ε/ dm3 mol–1 cm–1 21100), 387 (26300), 476 (3800). 1H NMR (400 MHz, 298K, Me2CO-d6) δH/ppm 9.29 (4H, m, H3/H8/H3΄/H8΄), 8.85 (2H, d, J = 8, H5/H5΄), 8.20 (2H, t, J = 8, H7/H7΄), 8.07 (2H, t, J = 8, H6/H6 ΄), 6.07 (2H, d, J = 8, H-cym CH3-Ccym), 5.99 (2H, d, J = 8, H-cymar), 4.17 (6H, s, COOCH3), 2.40 (3H, s, CH3-cym), 2.34 (1H, m, CH-(CH3)2), 0.96 (6H, d, J = 4, CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 164.88 (CO/CO΄), 156.16 (C8a/C8a΄), 151.07 (C2/C2΄), 139.96 (C4/C4΄), 132.95 (C7/C7΄), 131.48 (C6/C6΄), 130.37 (C8/C8΄), 126.53 (C5/C5΄), 126.20 (C4a/C4a΄), 120.93 (C3/C3΄), 107.89 (CH3-Ccym), 103.79 (Ccym-CH-(CH3)2), 87.70(C-Ccym-ar), 86.44 (C-Ccym-ar), 78.42 (C-Ccym-ar), 78.10 (C-Ccym-ar), 53.13 (OCH3/OCH3΄), 30.68 (CH-(CH3)2), 24.90 (CH-(CH3)2), 21.31 (CH-(CH3)2), 17.34 (CH-(CH3)2.
[Ru(η6-p-cymene)(pq)Cl][Cl] (7-Cl). Similar procedure to that of the published synthetic procedure, but without treatment with KPF6. Yield: 70 mg (75%). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.59 (1H, d, J = 8, H6΄), 8.96 (1H, d, J = 8, H8), 8.82 (1H, d, J = 8, H3), 8.74 (1H, d, J = 8, H4), 8.63 (1H, d, J = 8, H3΄), 8.35 (1H, t, J = 8, H4΄), 8.23 (1H, d, J = 8, H5), 8.11 (1H, t, J = 8, H7), 7.92 (1H, t, J = 8, H6), 7.87 (1H, t, J = 4, H5΄), 6.16 (1H, d, J = 8, H-cymar), 6.08 (1H, d, J = 8, H-cymar), 6.01 (2H, m, H-cymar), 2.42 (1H, m, CH-(CH3)2 ), 2.28 (3H, s, CH3-cym), 0.94 (3H, d, J = 4, CH-(CH3)2), 0.90 (3H, d, J = 4, CH-(CH3)2). 13C{1H} NMR (100 MHz, 298K, Me2CO-d6) δC/ppm 157.18 (C2΄), 157.15 (C6΄), 156.16 (C2), 150.30 (C8a), 141.86 (C3), 141.01 (C4΄), 133.57 (C7), 131.00 (C8), 130.53 (C4), 130.49 (C6), 129.92 (C5), 129.06 (C5΄), 119.99 (C3΄), 106.59 (CH3-Ccym), 105.32 (Ccym-CH-(CH3)2), 87.90 (C-Ccym-ar), 86.88 (C-Ccym-ar), 86.48 (C-Ccym-ar), 85.63 (C-Ccym-ar), 31.72 (CH-(CH3)2), 22.30 (CH-(CH3)2), 21.71 (CH-(CH3)2), 18.69 (CH3-cym).
[Ru(η6-p-cymene)(biq)Cl][PF6] (8). Yield: (0.09 g, 85%). Found: C, 49.52; H, 4.21; N, 3.94. C28H26ClF6N2O4PRu requires C, 50.04; H, 4.17; N, 3.55%. IR (KBr, νmax/cm−1): 3069 (w, ν(C–H)arom), 2924 (w, νas(C–H)aliph), 1596 (m, ν(C=N)), 1472 (m, ν(C=C)), 1385 (m, ν(C=C)), 841 (s, ν(P–F), 821 (m, ν(Ru–C), 557 (s, ν(P–F)). UV-vis (Me2CO, 1.0 × 10−4 mol dm−3): λmax/nm 355 (ε/ dm3 mol–1 cm–1 21000), 373 (31100), 450 (2900). 1H NMR (400 MHz, Me2CO-d6) δH/ppm 9.14 (2H, d, J = 8, H8/H8΄), 8.85 (2H, d, J = 8 Hz, H4/H4΄), 8.76 (2H, d, J = 8, H3/H3΄), 8.21 (2H, d, J = 8, H5/H5΄), 8.12 (2H, t, J = 8, H7/H7΄), 7.93 (1H, t, J = 8, H6/H6΄), 5.98 (2H, d, J = 8, H-cymar), 5.87 (2H, d, J = 8, H-cymar), 2.40 (3H, s, CH3-cym), 2.16 (1H, m, CH-(CH3)2), 0.89 (6H, d, J = 8, -CH-(CH3)2). 13C{1H} NMR (100 MHz, Me2CO-d6) δC/ppm 156.72 (C8a/C8a΄), 150.06 (C2/C2΄), 141.27 (C4/C4΄), 136.81 (C7/C7΄), 132.94 (C6/C6΄), 129.71 (C8/C8΄), 129.63 (C5/C5΄), 129.12 (C4a/C4a΄), 120.16 (C3/C3΄), 106.19 (CH3-Ccym), 103.67 (Ccym-CH-(CH3)2), 86.73 (C-Ccym-ar), 86.21 (C-Ccym-ar), 30.55 (CH-(CH3)2), 21.18 (CH-(CH3)2), 17.41 (CH3-cym).
3.2. General Synthetic Procedure of Complexes 9–11
In a Schlenk flask and under an argon atmosphere, 0.095 g (0.15 mmol) of [Ru(η6-p-cymene)(μ-Cl)Cl]2 and an equimolar amount of the appropriate organic ligand (L = pqcame (9); 4-Mepq (10); pq (11) ) were dissolved in dry methanol (10 mL). The obtained orange solution was stirred for 18 h at room temperature, and after filtration the volume of the clear filtrate was diminished to approximately 2 mL and diethyl ether (10 mL) was added. The orange precipitate was washed with diethyl ether (3 × 3 mL) and dried under vacuum to afford the final product.
Data for Complexes 9–11
[Ru(η6-p-cymene)(pqcame)Cl][Ru(η6-p-cymene)Cl3] (9). Yield: (0.130 g, 95%). Found: C, 46.44; H, 4.44; N, 2.92. C36H40Cl4N2O2Ru2∙3H2O requires C, 46.46; H, 4.60; N, 3.20%. IR (KBr, νmax/cm−1): 3077 (w, ν(C–H)arom), 3046 (m, ν(C–H)arom), 2871 (w, νas(C–H)aliph), 1725 (vs, νas(C=O)), 1595 (w, ν(C=N)), 1479 (w, ν(C=C)), 1381 (w, ν(C=C)), 799 (m, ν(Ru–C)). UV-vis (CHCl3, 1.0 × 10−4 mol dm−3): λmax/nm 364 (ε/ dm3 mol–1 cm–1 24 700), 443 (8000). UV-vis (H2O, 1.0 × 10−4 mol dm−3): λmax/nm 358 (27300), 416 (6700). 1H NMR (CDCl3, 400 MHz, 298K) δH/ppm 9.84 (1H, d, J = 4, H8), 9.00 (1H, d, J = 8, H6΄), 8.93 (1H, d, J = 8, H5), 8.67 (1H, s, H3), 8.32 (1H, d, J = 8, H3΄), 8.04 (3H, m, H4΄/H5΄), 7.91 (1H, t, J = 8, H6), 6.33 (2H, d, J = 8, H-cymar), 6.23 (1H, d, J = 4, H-cymar), 6.00 (1H, d, J = 4, H-cymar), 5.77 (1H, d, J = 8, H-cymar), 5.48 (2H, t, J = 4, H-cymar′), 5.35 (1H, d, J = 4, H-cymar′), 5.24 (1H, d, J = 4, H-cymar′), 4.17 (3H, s, COOCH3), 3.20 (0.60H, sept, J = 8, CHA-(CH3)2), 2.94 (0.40H, sept, J = 8, CHΒ-(CH3)2), 2.40 (1H, sept, J = 8, CH'-(CH3)2), 2.31 (1.8H, s, CH3A-cym), 2.18 (4.2H, s, CH3'-cym and CH3Β-cym), 1.38 (3H, d, J = 4, CH-(CH3A)2), 1.30 (3H, d, J = 4, CH-(CH3Β)2), 0.96 (3H, d, J = 8, CH-(CH3')2), 0.91 (3H, d, J = 8, CH-(CH3')2). (assignment marked with a prime refers to the p-cymene protons of the counter anion [Ru(η6-p-cymene)Cl3]–). 13C{1H} NMR (CDCl3, 100 MHz, 298K) δC(ppm) 164.53 (CO), 159.23 (C2΄), 156.32 (C2), 153.21 (C4΄), 150.56 (C6΄), 139.44 (C8), 137.88 (C8a), 132.30 (C4), 130.78 (C3΄), 130.32 (C5΄), 126.75 (C4a), 126.10 (C6), 125.43 (C7), 124.51 (C5), 119.54 (C3), 106.02 (C-Ccym-ar), 103.89 (C΄-Ccym-ar), 100.71 (C-Ccym-ar), 96.82 (C΄-Ccym-ar), 96.36 (C-Ccym-ar), 88.32 (C-Ccym-ar), 87.01 (C-Ccym-ar), 86.20 (C-Ccym-ar), 86.11(C΄-Ccym-ar), 81.83(C΄-Ccym-ar), 81.31 (C΄-Ccym-ar), 80.56 (C΄-Ccym-ar), 79.71 (C΄-Ccym-ar), 53.71 (OCH3), 31.15 (C΄H-(CH3)2), 30.70 (CAH-(CH3)2), 30.64 (CΒH-(CH3)2), 22.40 (2C, CH-(CH3)2), 22.15 (CH-(C΄H3)2), 21.62 (CH-(C'H3)2), 18.92 (CΒH3-cym), 18.84 (CAH3-cym), 18.80 (C΄H3-cym).
[Ru(η6-p-cymene)(4-Mepq)Cl][Ru(η6-p-cymene)Cl3] (10). Yield: (0.121 g, 92%). Found: C, 50.31; H, 5.04; N, 3.33. C35H40Cl4N2Ru2 requires C, 50.49; H, 4.84; N, 3.36%. IR (KBr, νmax/cm−1): 3061 (m, ν(C–H)arom, 3048 (m, ν(C–H)arom), 2920 (m, νas(C–H)aliph), 1600 (s, ν(C=N)), 1486 (s, ν(C=C)), 1383 (m, ν(C=C)), 795 (m, ν(Ru–C)), 773 (m). UV-vis (CHCl3, 1.0 × 10−4 mol dm−3): λmax/nm 258 (ε/ dm3 mol–1 cm–1 30800), 292 (27300), 336 (16400), 350(17700), 414 (3600). UV-Vis (H2O, 1.0 × 10−4 mol dm−3): λmax/nm 253 (17100), 286 (16400), 331 (8600), 345 (11300), 413 (1600). 1H NMR (CDCl3, 400 MHz, 298K) δH/ppm 9.71 (1H, d, J = 4, H8), 8.79 (1H, d, J = 8, H6΄), 8.32 (1H, d, J = 8, H5), 8.25 (1H, s, H3), 8.09 (1H, d, J = 8, H3΄), 7.93 (1H, t, J = 4, H5΄), 7.79 (m, 2H, H7/H4΄), 7.70 (1H, t, J = 8, H6), 6.16 (1H, d, J = 4, H-cymar), 6.11 (1H, d, J =8, H-cymar), 5.78 (1H, d, J = 8, H-cymar), 5.63 (1H, d, J = 4, H-cymar), 5.47 (2H, t, J = 4, H-cymar′), 5.32 (1H, d, J = 4, H-cymar′), 5.24 (1H, d, J = 4, H-cymar′), 3.20 (0.55H, sept, J = 4, CHA-(CH3)2), 2.89 (0.45H, sept, CHΒ-(CH3)2), 2.30 (s, 1.65H, CH3A-cym), 2.24 (sept, J = 8, CH'-(CH3)2), 2.19 (s, 3H, CH3′-cym), 2.15 (s, 1.35H, CH3B-cym), 1.38 (3H, d, J = 4, CH-(CH3A)2), 1.27 (3H, d, J = 4, CH-(CH3Β)2), 0.84 (3H, d, J = 8, CH-(CH3')2), 0.79 (3H, d, J = 8, CH-(CH3')2) (assignment marked with a prime refers to the p-cymene protons of the counter anion [Ru(η6-p-cymene)Cl3]–). 13C{1H} NMR (CDCl3, 100 MHz, 298K) δC/ppm 158.58 (C2΄), 156.45 (C8), 154.45 (C2), 150.56 (C8a), 149.13 (C4), 139.83 (C7), 132.18 (C4a), 130.47 (C4’), 129.85 (C6), 129.54 (C6΄), 125.64 (C5), 125.32 (C3΄), 120.68 (C3), 105.27 (C-Ccym-ar), 104.51 (C΄-Ccym-ar), 101.21 (C-Ccym-ar), 97.19 (C΄-Ccym-ar), 96.75 (C-Ccym-ar), 88.31 (C-Ccym-ar), 86.14 (C-Ccym-ar), 86.11 (C-Ccym-ar), 85.56 (C΄-Ccym-ar), 82.23 (C΄-Ccym-ar), 81.74 (C΄-Ccym-ar), 80.99 (C΄-Ccym-ar), 80.22 (C΄-Ccym-ar), 31.37 (CAH-(CH3)2), 31.34 (CBH-(CH3)2), 31.16 (C΄H-(CH3)2), 31.07 (C΄H-(CH3)2), 22.84 (CH-(C'H3)2), 22.69 (CH-(C'H3)2, 22.58 (CH-(CH3)2), 22.07 (CH-(CH3)2), 19.79 (C΄H3-cym), 19.35 (CΒH3-cym), 19.32 (CAH3-cym), 19.24 (CH3-quin).
[Ru(η6-p-cymene)(pq)Cl][Ru(η6-p-cymene)Cl3] (11). Yield: (0.120 g, 94%). Found: C, 50.28; H, 4.19; N, 3.37. C34H38Cl4N2Ru2 requires C, 50.49; H, 4.84; N, 3.36%. IR (KBr, νmax/cm−1): 3056 (s, ν(C–H)arom), 3031 (s, ν(C–H)arom), 2923 (s, νas(C–H)aliph), 1388 (s, ν(C=C)), 804 (w, ν(Ru–C)). UV-vis (CHCl3, 1.0 × 10−4): λmax/nm 268 (ε/ dm3 mol–1 cm–1 14700), 291 (14300), 337 (8400), 351 (9300), 417 (1900). 1H NMR (CDCl3, 400 MHz, 298K) δH/ppm 9.72 (1H, d, J = 4, H8), 8.79 (1H, d, J = 8, H6΄), 8.52 (1H, d, J = 8, H3), 8.46 (2H, t, J = 8, H4/H5), 7.90 (3H, m, H6/7/H5΄), 7.72 (1H, t, J = 8, H4΄), 6.15 (2H, m, H-cymar), 5.84 (2H, d, J = 8, H-cymar), 5.69 (1H, d, J = 8, H-cymar), 5.48 (1H, t, J = 8, H-cymar′), 5.33 (1H, d, J = 8, H-cymar′), 5.24 (1H, d, J = 8, H-cymar′), 3.20 (0.55H, sept, J = 4, CHA-(CH3)2), 2.92 (0.45H, sept, J = 4, CH′-(CH3)2), 2.30 (4.65H, m, CH3A-cym and CH3′-cym), 2.19 (3H, s, CH3'-cym), 2.15 (1.35H, s, CH3-cym), 1.37 (3H, d, J = 8, CH-(CH3A)2, 1.27 (d, 3H, J = 8, CH-(CH3Β)2), 0.89 (3H, d, J = 4, CH-(CH3)2), 0.83 (3H, d, J = 4, CH-(CH3)2) (assignment marked with a prime refers to the p-cymene protons of the counter anion [Ru(η6-p-cymene)Cl3]–). 13C NMR (CDCl3, 100 MHz, 298K) δC/ppm 158.22 (C2΄), 156.78 (C6΄), 154.21 (C2), 149.51 (C8a), 140.80 (C5), 139.83 (C5΄), 132.43 (C7), 129.65 (C6), 129.62 (C4΄), 129.49 (C8), 129.46 (C4), 129.25 (C4a), 119.67 (C3), 100.90 (C-Ccym-ar), 96.49 (C΄-Ccym-ar), 88.00 (C-Ccym-ar), 86.22 (C΄-Ccym-ar), 85.94 (C-Ccym-ar), 85.47 (C-Ccym-ar), 81.96 (C-Ccym-ar), 81.44 (C΄-Ccym-ar), 80.68 (C΄-Ccym-ar), 79.88 (C΄-Ccym-ar), 31.14 (CAH-(CH3)2), 30.86 (CΒH-(CH3)2), 30.77 (C΄H-(CH3)2), 29.83 (C΄H-(CH3)2), 22.54 (CH-(C'H3)2), 22.52 (CH-(C'H3)2), 22.28 (CH-(CH3)2), 21.68 (CH-(CH3)2), 19.06 (CΒH3-cym), 19.02 (CAH3-cym), 18.93 (C΄H3-cym).
3.3. Single-Crystal X-Ray Structural Determination
The data collections of complexes 1-4, 6, 8 and 9 were performed on a Bruker X8-KappaApexII diffractometer by using graphite monochromated MoKa radiation (λ = 0.7103 Å), generated by a sealed tube. The data collection of 8-Mepq and 4,6′-Me2pq was done using a Bruker D8-Venture diffractometer with Cu Ka radiation (k = 1.5418 Å) generated from an IlsHeliosOptics source. For 8,6′-Me2pq the collection of the data was performed on a Bruker APEX-II CCD diffractometer by using graphite monochromated Mo-Kα radiation (λ = 0.7103 Å), generated by a sealed tube. The diffractometers were equipped with a low-temperature device (Bruker Kryoflex I, and Oxford Cryostream 800er series, both at 100(2) K). Intensities were measured by fine-slicing x and u-scans and corrected for background, polarization, absorption and Lorentz effects. The structures were solved by intrinsic phasing methods implemented in Sheldrick’sXT program and refined anisotropically by the least-square procedure implemented in the SHELX program system [63,64]. Hydrogen atoms were included using the riding model on the bound carbon atoms. The structural data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) with reference numbers 2415190 (8-Mepq), 2415191 (4,6′-Me2pqca), 2415192 (8,6′-Me2pq), 2415193 (1), 2415194 (2), 2415195 (3), 2415196 (4), 2415197 (6), 2415198 (8) and 2415199 (9 ) respectively. Crystallographic data and refinement conditions for 8-Mepq, 4,6′-Me2pqca and 8,6′-Me2pq are summarized in Table S1 while these data for complexes 1-4 and 6, 8 and 9 are presented in Tables S2 and S3.
3.4. Catalytic Transfer Hydrogenation Experiment
A 100 mL two-necked flask was charged with the ruthenium catalyst (0.006 mmol), the appropriate ketone (2.22 mmol) and 15 mL of 2-propanol. The mixture was heated at 82 oC in a preheated oil bath and the appropriate volume of a 0.015 M solution of KOH in 2-propanol solution was added (0.22 mmol) leading to a substrate/catalyst/base ratio of ∼ 400/1/40. Approximately 0.1 mL of the reaction mixture was sampled, the solvent was pumped down and subsequently the oily residue was analyzed by 1H NMR spectroscopy. The percentage conversions during the reaction were determined by means of 1H NMR spectroscopy. Upon completion of the reaction, the solution was cooled at ambient temperature, and the solvent was rotary evaporated to dryness. Hexane (15 mL) was added, and the residue was filtered through a small pad of SiO2. The resulting clear solution was evaporated to dryness affording the relevant alcohol. All experiments were performed twice.
4. Conclusions
In conclusion, a series of new substituted pyridine-quinoline ligands were synthesized as scaffolds to investigate their coordination chemistry. The single-crystal X-ray structures of the organic precursors and relevant ruthenium(II)-p-cymene complexes were determined for the first time. The new catalyst precursors can be easily prepared in high yields, as air stable solids, that can be stored for months without decomposition. The cationic ruthenium(II) p-cymene organometallic complexes catalyze successfully the transfer hydrogenation reaction of various ketone substrates under optimized reaction conditions. Particularly complexes 1 and 5 with methyl groups at positions 8 and 4 of the quinoline moiety, showed the best catalytic activity. Those with electron withdrawing groups (methyl ester) were less efficient, while the presence of the bulkier [Ru(η6-p-cymene)Cl3]– complex counter anion seems to affect, to some extent, the catalytic activity exerted. An inner sphere mechanism can be proposed based on the detection of ruthenium(II)-H species.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figures S1-S12: NMR spectra of the ligand precursors 8-Mepq, 6′-Mepq, 8,6′-Me2pq, 4,6′-Me2pq and pq in CDCl3; Figures S13-S15: ESI-HRMS of 8-Mepq, 6′-Mepq and 8,6′-Me2pq in methanol; Figures S16-S18: Intermolecular interactions in the unit cell of 8-Mepq, 4,6′-Me2pq and 8,6′-Me2pq; Figures S19-S25: FT-IR spectra of complexes 1-6, 8; Figures S26-S47: NMR spectra of complexes 1-5, Ru-pqcame, 6-8 in (CD3)2CO; Figures S48-S55: UV-vis spectra of complexes 1-6, Ru-pqcame and 8 in acetone; Figures S56-S60: Intermolecular interactions in the unit cell of 1-4, 6; Figure S61: Molecular structure of cation of 8; Table S1: Selected improved crystallographic data for 8; Figure S62: Intermolecular interactions in the unit cell of 8; Figures S63-S65: FT-IR spectra of complexes 9-11; Figures S66-S74: NMR spectra of complexes 9-11 in CDCl3; Figures S75-S79: UV-vis spectra of complexes 9-11 in CHCl3 and in H2O; Figure S80: Intermolecular interactions in the unit cell of 9; Figure S81: Conversion versus reaction time for acetophenone transfer hydrogenation by catalysts Ru-pqcame and 10 within 60 min; Figure S82: 1H NMR spectrum (CH3OD) of a sample (pine green solid) of 4, showing the formation of Ru-H species; Tables S2-S4: Crystal and refinement data for ligand precursors 8-Mepq, 4,6′-Me2pqca, 8,6′- Me2pqca and the metal complexes 1-4, 7, 9 and 10.
Author Contributions
Conceptualization, A.Ι.P.; methodology, A.Ι.P., and N.Z.; formal analysis, N.Z., and E.I.P.; investigation, N.Z.; E.I.P.; G.S.; N.S.T., and A.I.P.; resources, A.Ι.P.; N.S.T., and G. S.; data curation, A.Ι.P.; N.Z., and E.I.P; writing—original draft preparation, A.Ι.P. and N.Z; writing—review and editing, A.Ι.P.; supervision, A.Ι.P.; project administration, A.Ι.P.; funding acquisition, A.Ι.P. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded (in part) by The Special Research Account of the National and Kapodistrian University of Athens (NKUA), grant number 15244 (to A.Ι.P.).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
A.Ι.P would like to thank Prof. Dr. A.C. Filippou for access to the elemental analyses center of the University of Bonn, Chemistry Department. We also thank C. Rödde for the X-ray diffraction measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective Hydrogenation for Fine Chemicals: Recent Trends and New Developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Taleb, B.; Jahjah, R.; Cornu, D.; Bechelany, M.; Al Ajami, M.; Kataya, G.; Hijazi, A.; El-Dakdouki, M.H. Exploring Hydrogen Sources in Catalytic Transfer Hydrogenation: A Review of Unsaturated Compound Reduction. Molecules 2023, 28, 7541. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Baidilov, D.; Hayrapetyan, D.; Khalimon, A.Y. Recent advances in homogeneous base-metal-catalyzed transfer hydrogenation reactions. Tetrahedron 2021, 98, 132435. [Google Scholar] [CrossRef]
- Wei, Y.-F.; Liang, Y.; Luo, R.; Ouyang, L. Recent advances of Cp*Ir complexes for transfer hydrogenation: focus on formic acid/formate as hydrogen donors. Org. Biomol. Chem. 2023, 21, 7484. [Google Scholar] [CrossRef]
- Cocker, J.D.; Henbest, H.B.; Phillipps, G.H.; Slater, G.P.; Thomas, D.A. 2. Reactions of ketones with oxidising agents. Part II. Acetoxylation of 11- and 20-oxo-steroids with lead tetra-acetate in the presence of boron trifluoride. J. Chem. Soc 1965, 6–11. [Google Scholar] [CrossRef]
- Sasson, Y.; Albin, P.; Blum, J. Effect of a Ru (II) catalyst on the rate of equilibration of carbinols and ketones. Tetrahedron Lett. 1974, 15, 833–836. [Google Scholar] [CrossRef]
- Chowdhury, R.L.; Bäckvall, J.-E. Efficient ruthenium-catalysed transfer hydrogenation of ketones by propan-2-ol. J.Chem. Soc. Chem. Commun. 1991, 16, 1063–1064. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew Chem. Int Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Morris, R.H. Asymmetric hydrogenation, transfer hydrogenation and hydrosilylation of ketones catalyzed by iron complexes. Chem. Soc. Rev. 2009, 38, 2282–2291. [Google Scholar] [CrossRef]
- Johnson, N.B.; Lennon, I.C.; Moran, P.H.; Ramsden, J.A. Industrial-Scale Synthesis and Applications of Asymmetric Hydrogenation Catalysts. Acc. Chem. Res. 2007, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Fovanna, T.; Campisi, S.; Villa, A.; Kambolis, A.; Peng, G.; Rentsch, D.; .Kröcher, O.; Nachtegaal, M.; Ferri, D. Ruthenium on phosphorous-modified alumina as an effective and stable catalyst for catalytic transfer hydrogenation of furhfural. RSC Adv. 2020, 10, 11507–11516. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015, 6, 6582. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Sadler, P.J. Transfer hydrogenation catalysis in cells. RSC Chem. Biol. 2021, 2, 12–29. [Google Scholar] [CrossRef]
- Comas-Vives, A.; Ujaque, G.; Lledós, A. Inner- and Outer-Sphere Hydrogenation Mechanisms: A Computational Perspective. In Theoretical and Computational Inorganic Chemistry; van Eldik, R., Harvey, J., Eds.; Academic Press, 2010; pp. 231–260. [Google Scholar] [CrossRef]
- Liu, Y.; Yue, X.; Luo, C.; Zhang, L.; Lei, M. Mechanisms of Ketone/Imine Hydrogenation Catalyzed by Transition-Metal Complexes. ENERGY Environ. Mater. 2019, 2, 292–312. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Milstein, D. Metal–Ligand Cooperation. Angew. Chem. Int Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Ma, X.; Guillet, S.G.; Liu, Y.; Cazin, C.S.J.; Nolan, S.P. Simple synthesis of [Ru(CO3)(NHC)(p-cymene)] complexes and their use in transfer hydrogenation catalysis. DaltonTrans. 2021, 50, 13012–13019. [Google Scholar] [CrossRef]
- Mannu, A.; Grabulosa, A.; Baldino, S. Transfer Hydrogenation from 2-propanol to Acetophenone Catalyzed by [RuCl2(η6-arene)P] (P = monophosphine) and [Rh(PP)2]X (PP = diphosphine, X = Cl−, BF4−) Complexes. Catalysts 2020, 10, 162. [Google Scholar] [CrossRef]
- Pandiarajan, D.; Ramesh, R. Ruthenium(II) half-sandwich complexes containing thioamides: Synthesis, structures and catalytic transfer hydrogenation of ketones. J. Organomet. Chem. 2013, 723, 26–35. [Google Scholar] [CrossRef]
- Hounjet, L.J.; Ferguson, M.J.; Cowie, M. Phosphine–Amido Complexes of Ruthenium and Mechanistic Implications for Ketone Transfer Hydrogenation Catalysis. Organometallics 2011, 30, 4108–4114. [Google Scholar] [CrossRef]
- Alonso, D.A.; Brandt, P.; Nordin, S.J.M.; Andersson, P.G. Ru(arene)(amino alcohol)-Catalyzed Transfer Hydrogenation of Ketones: Mechanism and Origin of Enantioselectivity. J. Am. Chem. Soc. 1999, 121, 9580–9588. [Google Scholar] [CrossRef]
- Romain, C.; Gaillard, S.; Elmkaddem, M.K.; et al. New Dipyridylamine Ruthenium Complexes for Transfer Hydrogenation of Aryl Ketones in Water. Organometallics 2010, 29, 1992–1995. [Google Scholar] [CrossRef]
- Türkmen, H.; Kani, İ.; Çetinkaya, B. Transfer Hydrogenation of Aryl Ketones with Half-Sandwich RuII Complexes That Contain Chelating Diamines. Eur. J. Inorg. Chem. 2012, 4494–4499. [Google Scholar] [CrossRef]
- Nieto, I.; Livings, M.S.; Sacci, J.B.; Reuther, L.E.; Zeller, M.; Papish, E.T. Transfer Hydrogenation in Water via a Ruthenium Catalyst with OH Groups near the Metal Center on a bipy Scaffold. Organometallics 2011, 30, 6339–6342. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Sukhikh, T.S.; Gushchin, A.L.; Ivanov, A.Y.; Tennikova, T.B.; Sharoyko, V.V.; Baykov, S.V.; Benassi, E.; Potapov, A. S Arene–Ruthenium(II) Complexes Containing 11H-Indeno[1,2-b]quinoxalin-11-one Derivatives and Tryptanthrin-6-oxime: Synthesis, Characterization, Cytotoxicity, and Catalytic Transfer Hydrogenation of Aryl Ketones. ACS Omega 2020, 5, 11167–11179. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Samsonenko, D.G.; Ermakova, E.A.; Klyushova, L.S.; Baykov, S.V.; Boyarskiy, V.P.; Potapov, A.S. Synthesis and Structural Characterization of Half-Sandwich Arene–Ruthenium(II) Complexes with Bis(imidazol-1-yl)methane, Imidazole and Benzimidazole. Inorganics 2021, 9, 34. [Google Scholar] [CrossRef]
- Erdem, A.; Kılınçarslan, R.; Şahin, Ç.; Dayan, O.; Özdemir, N. Synthesis, thermal, electrochemical and catalytic behavior toward transfer hydrogenation investigations of the half-sandwich RuII complexes with 2-(2′-quinolyl)benzimidazoles. J. Mol. Struct. 2020, 1220, 128556. [Google Scholar] [CrossRef]
- Dayan, S.; Ozpozan Kalaycioglu, N.; Daran, J.-C.; Labande, A.; Poli, R. Synthesis and Characterization of Half-Sandwich Ruthenium Complexes Containing Aromatic Sulfonamides Bearing Pyr id inyl Rings: Catalysts for Transfer Hydrogenation of Acetophenone Derivatives. Eur. J. Inorg. Chem. 2013, 3224–3232. [Google Scholar] [CrossRef]
- Zacharopoulos, N.; Koukoulakis, K.; Bakeas, E.; Philippopoulos, A.I. A 2-(2’-pyridyl)quinoline ruthenium(II) complex as an active catalyst for the transfer hydrogenation of ketones. Open Chem. 2016, 14, 308–315. [Google Scholar] [CrossRef]
- Zacharopoulos, N.; Kolovou, E.; Peppas, A.; Koukoulakis, K.; Bakeas, E.; Schnakenburg, G.; Philippopoulos, A.I. Pyridyl based ruthenium(II) catalyst precursors and their dihydride analogues as the catalytically active species for the transfer hydrogenation of ketones. Polyhedron. 2018, 154, 27–38. [Google Scholar] [CrossRef]
- Kolovou, E.; Peppas, A.; Zacharopoulos, N.; Koukoulakis, K.; Bakeas, E.; Schnakenburg, G.; Philippopoulos, A.I. Facile synthesis of a 2-(2′-pyridyl)-4-(methylcarboxy)quinoline ruthenium (II) based catalyst precursor for transfer hydrogenation of aromatic ketones. Inorg. Chem. Commun. 2018, 92, 64–68. [Google Scholar] [CrossRef]
- Kokkosi, A.; Garofallidou, E.; Zacharopoulos, N.; Tsoureas, N.; Diamanti, K.; Thomaidis, N.S.; Cheilari, A.; Machalia, C.; Emmanouilidou, E.; Philippopoulos, A.I. Ruthenium p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline-Based Ligands: Synthesis, Characterization, and Cytotoxic Properties. Molecules 2024, 29, 3215. [Google Scholar] [CrossRef]
- Tsolis, T.; Ypsilantis, K.; Kourtellaris, A.; Garoufis, A. Synthesis, characterization and interactions with 9-methylguanine of ruthenium(II) η6-arene complexes with aromatic diimines. Polyhedron 2018, 149, 45–53. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Kollipara, M.R. Reactivity studies of η6-arene ruthenium (II) dimers with polypyridyl ligands: isolation of mono, binuclear p-cymene ruthenium (II) complexes and bisterpyridine ruthenium (II) complexes. Polyhedron 2003, 22, 3155–3160. [Google Scholar] [CrossRef]
- Zucca, A.; Cordeschi, D.; Maidich, L.; Pilo, M.I.; Masolo, E.; Stoccoro, S.; Cinellu, M.; Galli, S. Rollover Cyclometalation with 2-(2′-Pyridyl)quinoline. Inorg. Chem. 2013, 52, 7717–7731. [Google Scholar] [CrossRef]
- Atwell, G.J.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 57. 2-Phenylquinoline-8-carboxamides as minimal DNA-intercalating antitumor agents with in vivo solid tumor activity. J. Med. Chem. 1989, 32, 396–401. [Google Scholar] [CrossRef]
- Haginiwa, J.; Higuchi, Y.; Kawashima, T.; Goto, T. Reactions concerned in tertiary amine N-oxides. V. A new method for the preparation of asymmetric 2,2'-dipyridyl, 2,2'-pyridylquinoline and pyridylphenanthroline derivatives (author's transl). Yakugaku Zasshi. 1975, 95, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, F.; He, Y.-M.; Li, C.; Fan, Q.-H. Enantioselective synthesis of tunable chiral pyridine–aminophosphine ligands and their applications in asymmetric hydrogenation. Org. Biomol. Chem. 2019, 17, 50995105. [Google Scholar] [CrossRef]
- Iyoda, M.; Otsuka, H.; Sato, K.; Nisato, N.; Oda, M. Homocoupling of Aryl Halides Using Nickel(II) Complex and Zinc in the Presence of Et4NI. An Efficient Method for the Synthesis of Biaryls and Bipyridines. Bull. Chem. So.c Jpn. 1990, 63, 80–87. [Google Scholar] [CrossRef]
- Verniest, G.; Wang, X.; De Kimpe, N.; Padwa, A. Heteroaryl Cross-Coupling as an Entry toward the Synthesis of Lavendamycin Analogues: A Model Study. J.Org. Chem. 2010, 75, 424–433. [Google Scholar] [CrossRef]
- Martínez, R.; Ramón, D.J.; Yus, M. Transition-Metal-Free Indirect Friedländer Synthesis of Quinolines from Alcohols. J. Org. Chem. 2008, 73, 9778–9780. [Google Scholar] [CrossRef] [PubMed]
- Peppas, A. Synthesis and characterization of homoleptic Copper (I) complexes. Application in third generation solar cells (Gratzel type). Master's thesis, National and Kapodistrian University of Athens, Athens, Greece, 10 October 2015. Available online: https://pergamos.lib.uoa.gr/uoa/dl/object/1320032 (accessed on 10 October 2024).
- Pucci, D.; Crispini, A.; Ghedini, M.; Szerb, E.I.; La Deda, M. 2,2′-Biquinolines as test pilots for tuning the colour emission of luminescent mesomorphic silver(i) complexes. Dalton Trans. 2011, 40, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Milton, A.; Benial, F.; Ramakrishnan, V.; Murugesan, R. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. Spectrochim. Acta Mol. Biomol. Spectrosc. 2002, 58, 1703. [Google Scholar]
- Stanghellini, P.L.; Cognolato, L.; Bor, G.; Kettle, S.F.A. Identification of the IR frequencies of the interstitial carbon atom in some M6C (M = Fe, Ru) carbonyl clusters by 13 C-enrichment. J. Chem. Crystallogr. 1983, 13, 127–133. [Google Scholar] [CrossRef]
- Peacock, A.; Melchart, M.; Deeth, R.; Habtemariam, A.; Parsons, S.; Sadler, P. Osmium(II) and Ruthenium(II) Arene Maltolato Complexes: Rapid Hydrolysis and Nucleobase Binding. Chem. Eur. J. 2007, 13, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Bugarcic, T.; Habtemariam, A.; Stepankova, J.; Heringova, P.; Kasparkova, J.; Deeth, R.J.; Johnstone, R.D.L.; Prescimone, A.; Parkin, A.; Parsons, S.; Brabec, V.; Sadler, P.J. The Contrasting Chemistry and Cancer Cell Cytotoxicity of Bipyridine and Bipyridinediol Ruthenium(II) Arene Complexes. Inorg. Chem. 2008, 47, 11470–11486. [Google Scholar] [CrossRef]
- Fernández, R.; Melchart, M.; Habtemariam, A.; Parsons, S.; Sadler, P.J. Use of chelating ligands to tune the reactive site of half-sandwich ruthenium(II)-arene anticancer complexes. Chem. Eur. J. 2004, 10, 5173–5179. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, P.; Lu, R.; Lu, R.; Qian, Q.; Lei, X.; Huang, S.; Liu, L.; Huang, C.; Su, W. Synthesis, X-ray Diffraction Study, and Cytotoxicity of a Cationic p-Cymene Ruthenium Chloro Complex Containing a Chelating Semicarbazone Ligand. Z. Anorg. Allg. Chem. 2013, 639, 943–946. [Google Scholar] [CrossRef]
- Vock, C.A.; Scolaro, C.; Phillips, A.D.; Scopelliti, R.; Sava, G.; Dyson, P.J. Synthesis, Characterization, and in Vitro Evaluation of Novel Ruthenium(II) η6-Arene Imidazole Complexes. J. Med. Chem. 2006, 49, 5552–5561. [Google Scholar] [CrossRef]
- Zacharopoulos, N. Synthesis and characterization of Ru complexes suitable for transfer hydrogenation and α,β alkylation reactions. Doctoral Dissertation, National and Kapodistrian University of Athens, Athens, Greece, 2023. Persistent URL: https://pergamos.lib.uoa.gr/uoa/dl/object/2973928. [Google Scholar]
- Erdem, A.; Kılınçarslan, R.; Şahin, Ç.; Dayan, O.; Özdemir, N. Synthesis, thermal, electrochemical and catalytic behavior toward transfer hydrogenation investigations of the half-sandwich RuII complexes with 2-(2′-quinolyl)benzimidazoles. J. Mol. Struct. 2020, 1220, 128556. [Google Scholar] [CrossRef]
- Mercan, D.; Çetinkaya, E.; Şahin, E. Ru(II)–arene complexes with N-substituted 3,4-dihydroquinazoline ligands and catalytic activity for transfer hydrogenation reaction. Inorg. Chim. Acta 2013, 400, 74–81. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Q.; Fu, H.-Y.; Chen, H.; Yuan, M.-L.; Li, R.-X. Ru–η6 -benzene–phosphine complex-catalyzed transfer hydrogenation of ketones. Appl. Organometal. Chem. 2011, 25, 626–631. [Google Scholar] [CrossRef]
- Standfest-Hauser, C.; Slugovc, C.; Mereiter, K.; et al. Hydrogen-transfer catalyzed by half-sandwich Ru(ii) aminophosphine complexes. J. Chem. Soc. Dalton Trans. 2001, 20, 2989–2995. [Google Scholar] [CrossRef]
- de Araujo, M.P.; de Figueired, A.T.; Bogado, A.L.; et al. Ruthenium Phosphine/Diimine Complexes: Syntheses, Characterization, Reactivity with Carbon Monoxide, and Catalytic Hydrogenation of Ketones. Organometallics 2005, 24, 6159–6168. [Google Scholar] [CrossRef]
- Çalık, H.S.; Ispir, E.; Karabuga, Ş.; Aslantas, M. Ruthenium (II) complexes of NO ligands: Synthesis, characterization and application in transfer hydrogenation of carbonyl compounds. J. Organomet Chem. 2016, 801, 122–129. [Google Scholar] [CrossRef]
- Dritsopoulos, A.; Zacharopoulos, N.; Peyret, A.-E.; Karampella, E.; Tsoureas, N.; Cheilari, A.; Machalia, C.; Emmanouilidou, E.; Andreopoulou, A.K.; Kallitsis, J.K.; et al. Ruthenium-p-Cymene Complexes Incorporating Substituted Pyridine–Quinoline Ligands with –Br (Br-Qpy) and –Phenoxy (OH-Ph-Qpy) Groups for Cytotoxicity and Catalytic Transfer Hydrogenation Studies: Synthesis and Characterization. Chemistry 2024, 6, 773–793. [Google Scholar] [CrossRef]
- Bäckvall, J.E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002, 652, 105–111. [Google Scholar] [CrossRef]
- Joseph, M.C.; Swarts, A.J.; Mapolie, S.F. Cationic half-sandwich ruthenium (II) complexes ligated by pyridyl-triazole ligands: Transfer hydrogenation and mechanistic studies. Polyhedron 2022, 212, 115579. [Google Scholar] [CrossRef]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, K. Inorganic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 1982. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallorg. C. Struct Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis and structures of organic ligands used in this work; hydrogen atom numbering is included for 1H NMR assignment.
Scheme 1.
Synthesis and structures of organic ligands used in this work; hydrogen atom numbering is included for 1H NMR assignment.
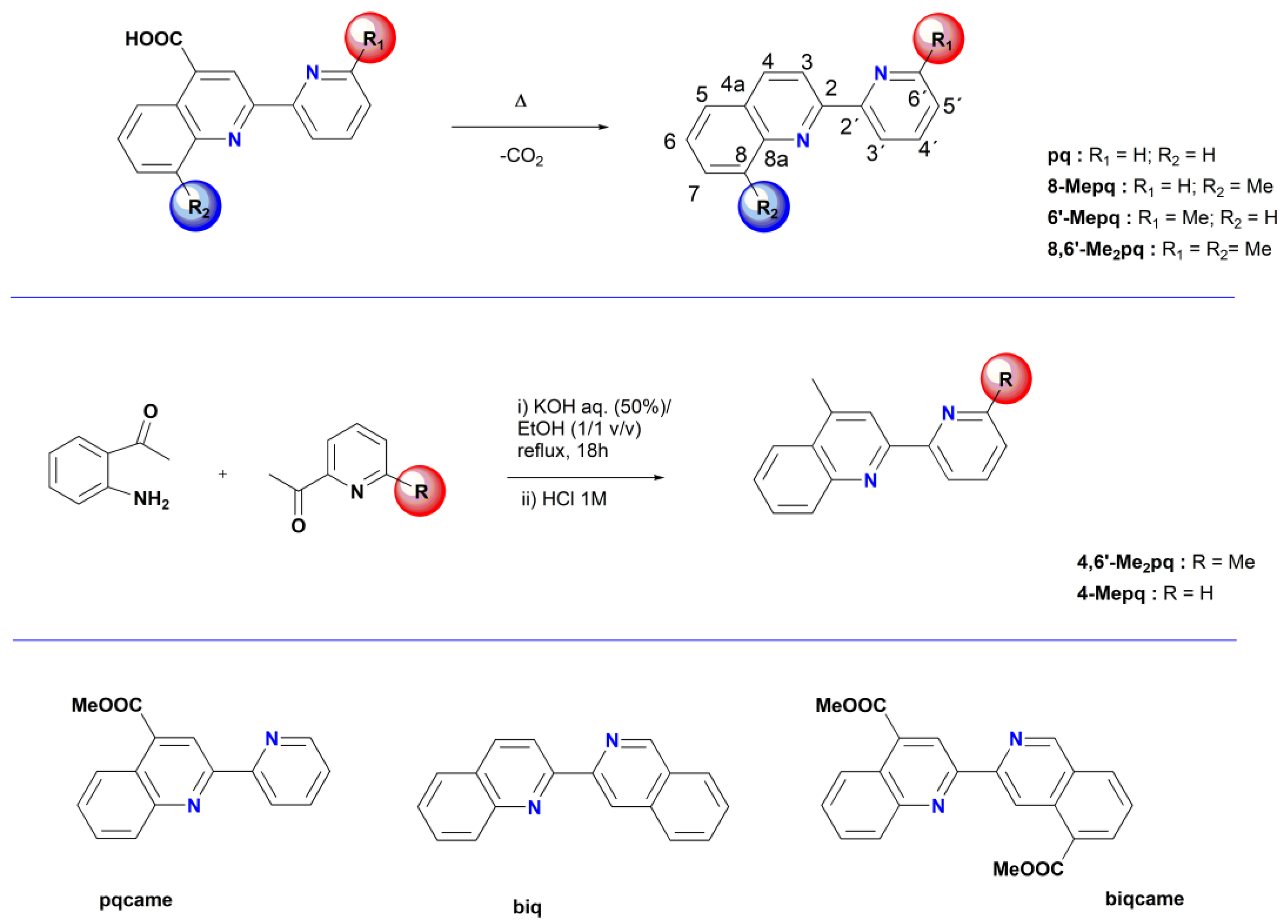
Figure 1.
Molecular structure of 8-Mepq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.376(5); N(1')–C(1') 1.365(5); N(2)–C(11) 1.346(6); N(1)–C(5) 1.321(5); N(1')–C(5') 1.326(5); N(2)–C(11) 1.346(6); C(1)–C(2) 1.416(6); C(1')–C(2') 1.423(6); C(5)–N(1)–C(1) 118.1(3); C(11)–N(2)–C(15) 117.3(4); C(5')– N(1')–C(1') 118(3).
Figure 1.
Molecular structure of 8-Mepq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.376(5); N(1')–C(1') 1.365(5); N(2)–C(11) 1.346(6); N(1)–C(5) 1.321(5); N(1')–C(5') 1.326(5); N(2)–C(11) 1.346(6); C(1)–C(2) 1.416(6); C(1')–C(2') 1.423(6); C(5)–N(1)–C(1) 118.1(3); C(11)–N(2)–C(15) 117.3(4); C(5')– N(1')–C(1') 118(3).
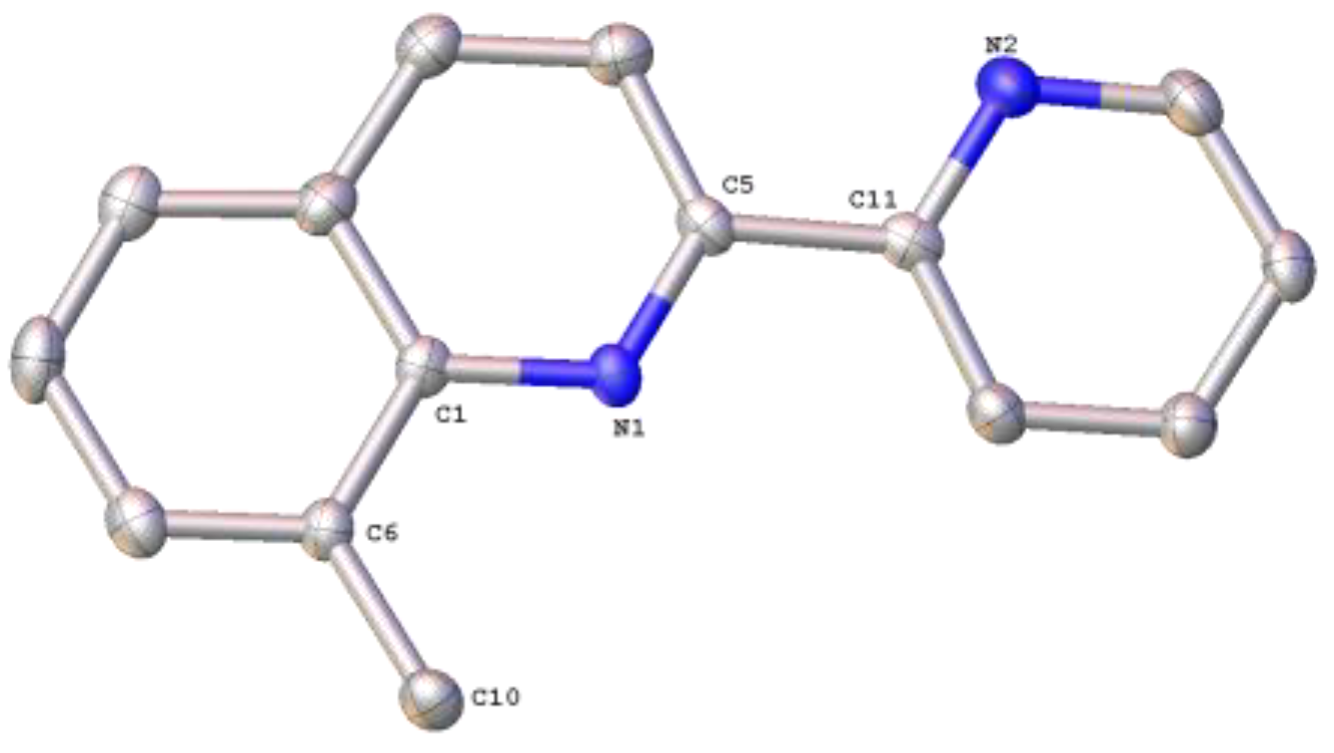
Figure 2.
Molecular structure of 8,6′-Me2pq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.3718(15); N(1)–C(5) 1.372(5); N(2)–C(11) 1.347(5); N(2)–C(15) 1.346(5); C(1)–C(2) 1.415(5); C(5)–N(1)–C(1) 118.6(3); C(11)–N(2)–C(15) 117.7(3); N(1)–C(5)–C(4) 122.5(3).
Figure 2.
Molecular structure of 8,6′-Me2pq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.3718(15); N(1)–C(5) 1.372(5); N(2)–C(11) 1.347(5); N(2)–C(15) 1.346(5); C(1)–C(2) 1.415(5); C(5)–N(1)–C(1) 118.6(3); C(11)–N(2)–C(15) 117.7(3); N(1)–C(5)–C(4) 122.5(3).
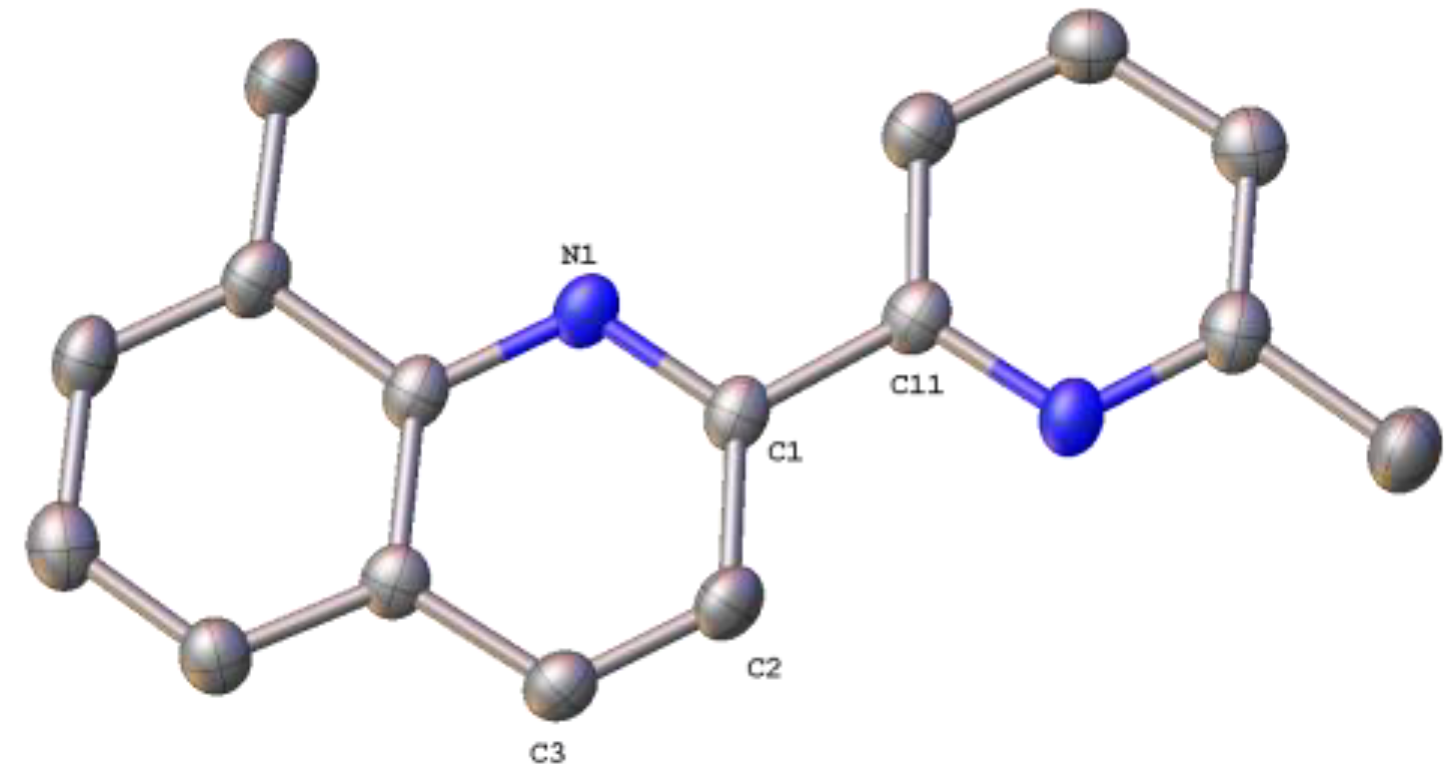
Figure 3.
Molecular structure of 4,6′-Me2pq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.3718(15); N(1)–C(5) 1.3257(16); N(2)–C(11) 1.3476(16); N(2)–C(12) 1.3452(16); C(1)–C(2) 1.4203(17); C(5)–N(1)–C(1) 117.27(10); C(11)–N(2)–C(12) 118.25(11); N(1)–C(5)–C(4) 123.26(10).
Figure 3.
Molecular structure of 4,6′-Me2pq; ellipsoids are plotted at the 50% probability level. H atoms are omitted. Selected bond lengths (Å) and angles (°): N(1)–C(1) 1.3718(15); N(1)–C(5) 1.3257(16); N(2)–C(11) 1.3476(16); N(2)–C(12) 1.3452(16); C(1)–C(2) 1.4203(17); C(5)–N(1)–C(1) 117.27(10); C(11)–N(2)–C(12) 118.25(11); N(1)–C(5)–C(4) 123.26(10).
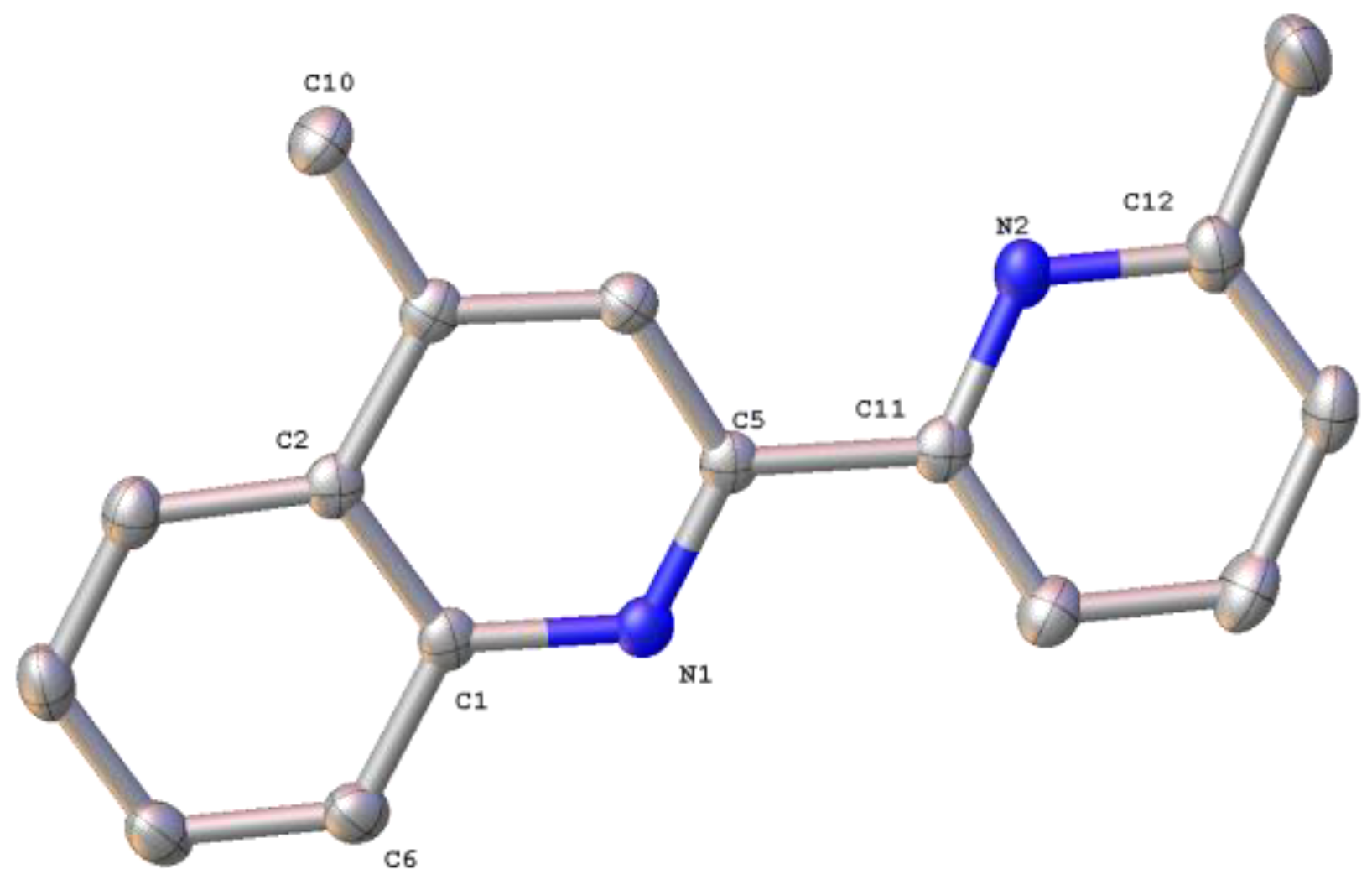
Scheme 2.
General reaction scheme for the synthesis of the ruthenium(II) organometallic complexes 1–8 (a) and 9–11 (b), described in this work.
Scheme 2.
General reaction scheme for the synthesis of the ruthenium(II) organometallic complexes 1–8 (a) and 9–11 (b), described in this work.
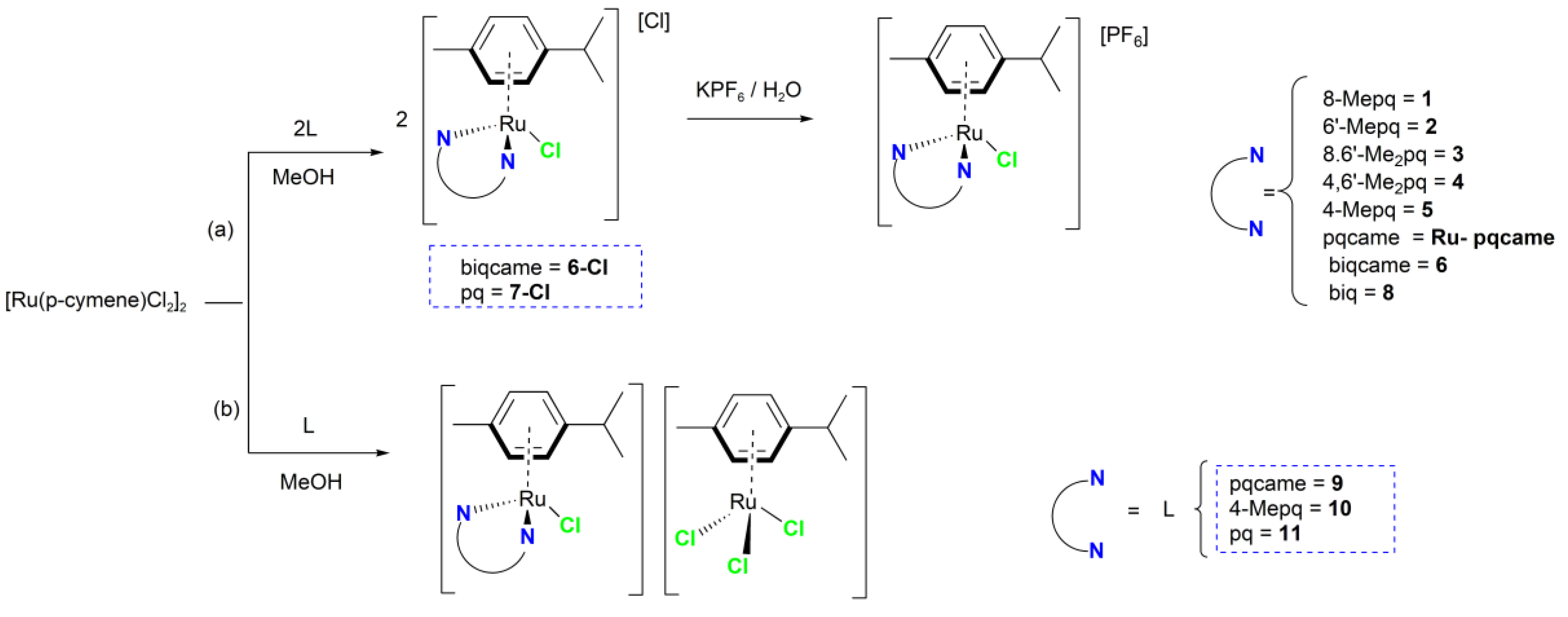
Figure 4.
(a) Molecular structure of the complex cation of 1; (b) Molecular structure of the complex cation of 4. Hydrogen atoms and the PF6 − anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
Figure 4.
(a) Molecular structure of the complex cation of 1; (b) Molecular structure of the complex cation of 4. Hydrogen atoms and the PF6 − anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
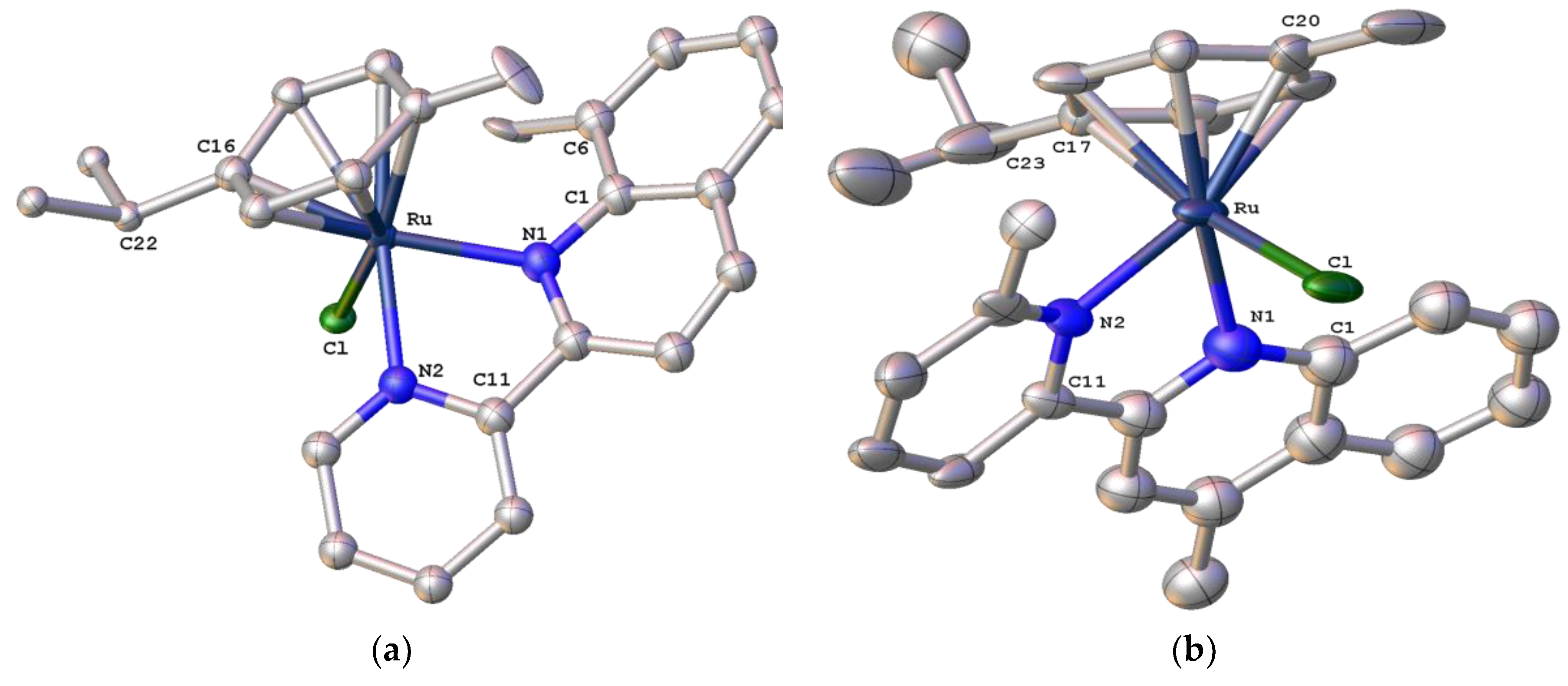
Figure 5.
(a) Molecular structure of the complex cation of 2; (b) Molecular structure of the complex cation of 6. Hydrogen atoms and the PF6 – anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
Figure 5.
(a) Molecular structure of the complex cation of 2; (b) Molecular structure of the complex cation of 6. Hydrogen atoms and the PF6 – anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
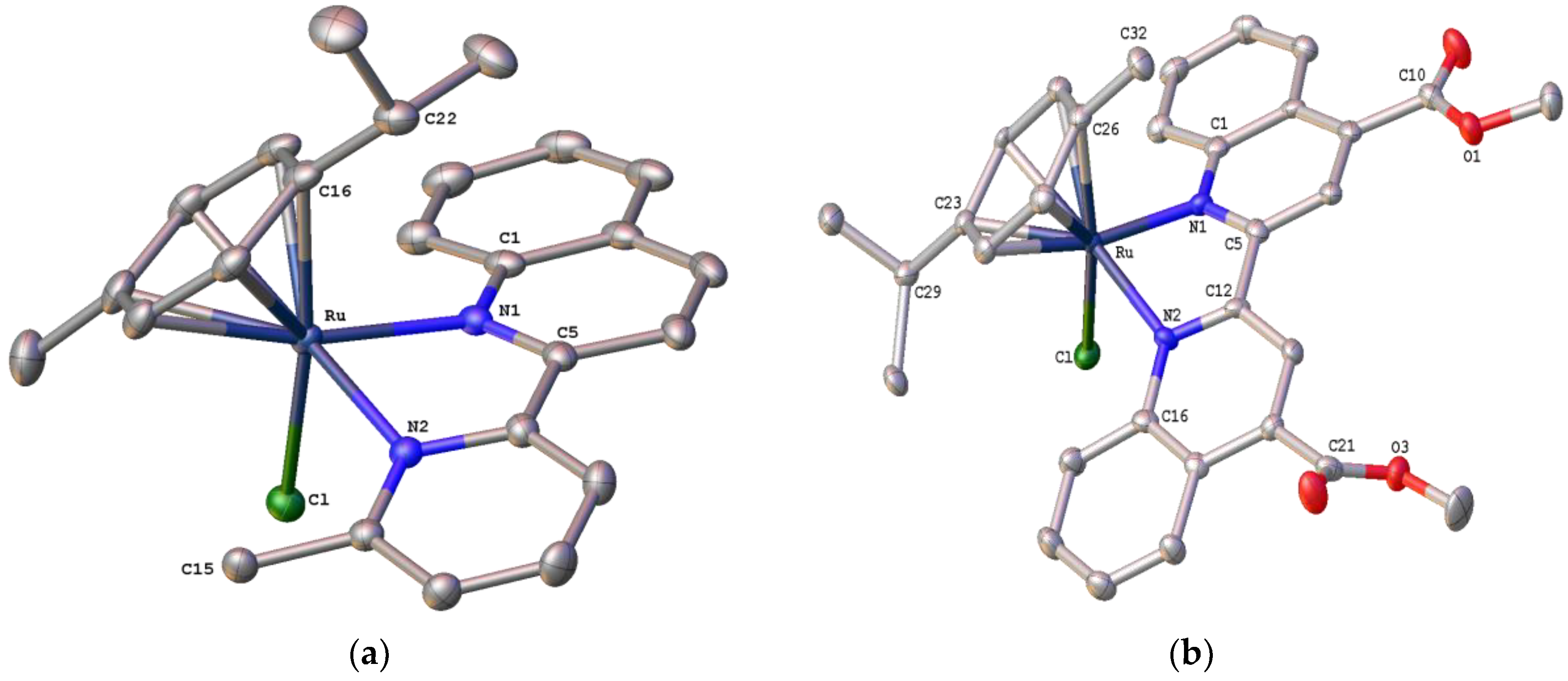
Figure 6.
(a) Molecular structure of the complex cation [Ru(η6-p-cymene)(8,6′-Me2pq)Cl]+ (Ru-3); (b) Molecular structure of the complex cation [Ru′(η6-p-cymene)(8,6′-Me2pq)Cl]+ (Ru′-3). Hydrogen atoms and the PF6 – anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
Figure 6.
(a) Molecular structure of the complex cation [Ru(η6-p-cymene)(8,6′-Me2pq)Cl]+ (Ru-3); (b) Molecular structure of the complex cation [Ru′(η6-p-cymene)(8,6′-Me2pq)Cl]+ (Ru′-3). Hydrogen atoms and the PF6 – anions are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
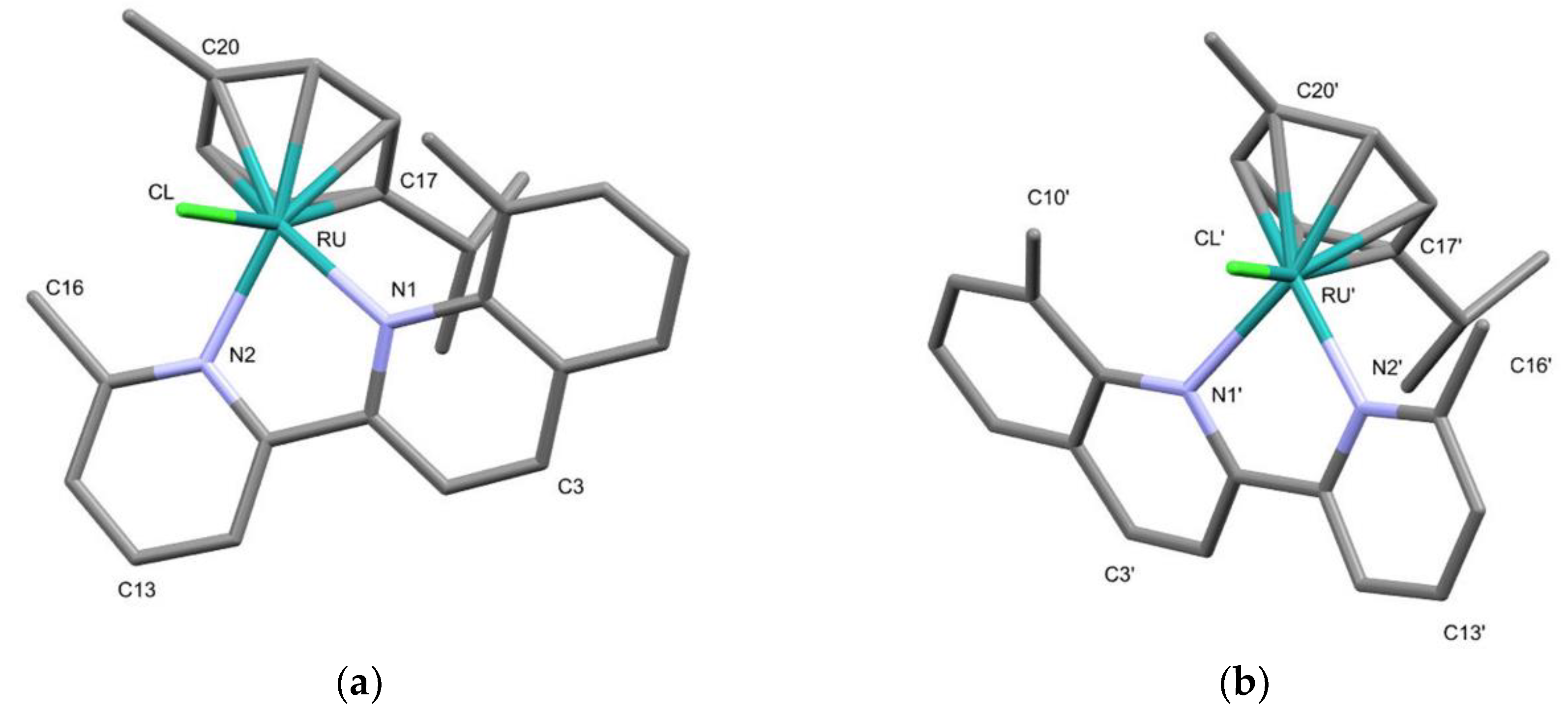
Figure 7.
A superposition of both independent molecules of 3 in the unit cell. Hydrogen atoms are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
Figure 7.
A superposition of both independent molecules of 3 in the unit cell. Hydrogen atoms are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
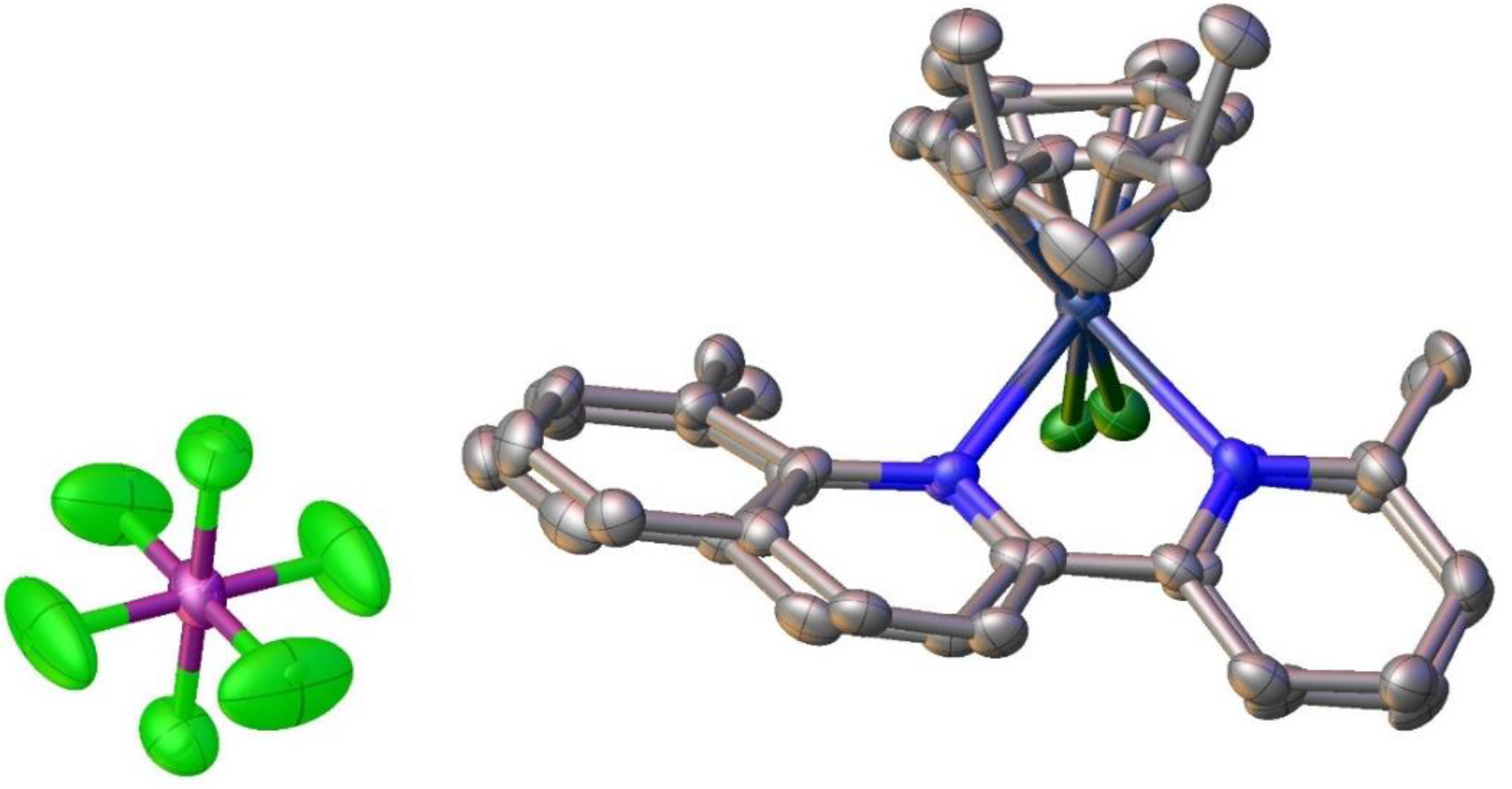
Figure 8.
Molecular structure of the complex cation of 9. Hydrogen atoms, solvent molecules and the [Ru(p-cymene)Cl3]– complex anion are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
Figure 8.
Molecular structure of the complex cation of 9. Hydrogen atoms, solvent molecules and the [Ru(p-cymene)Cl3]– complex anion are omitted for clarity. The ellipsoids were plotted at the 50% probability level.
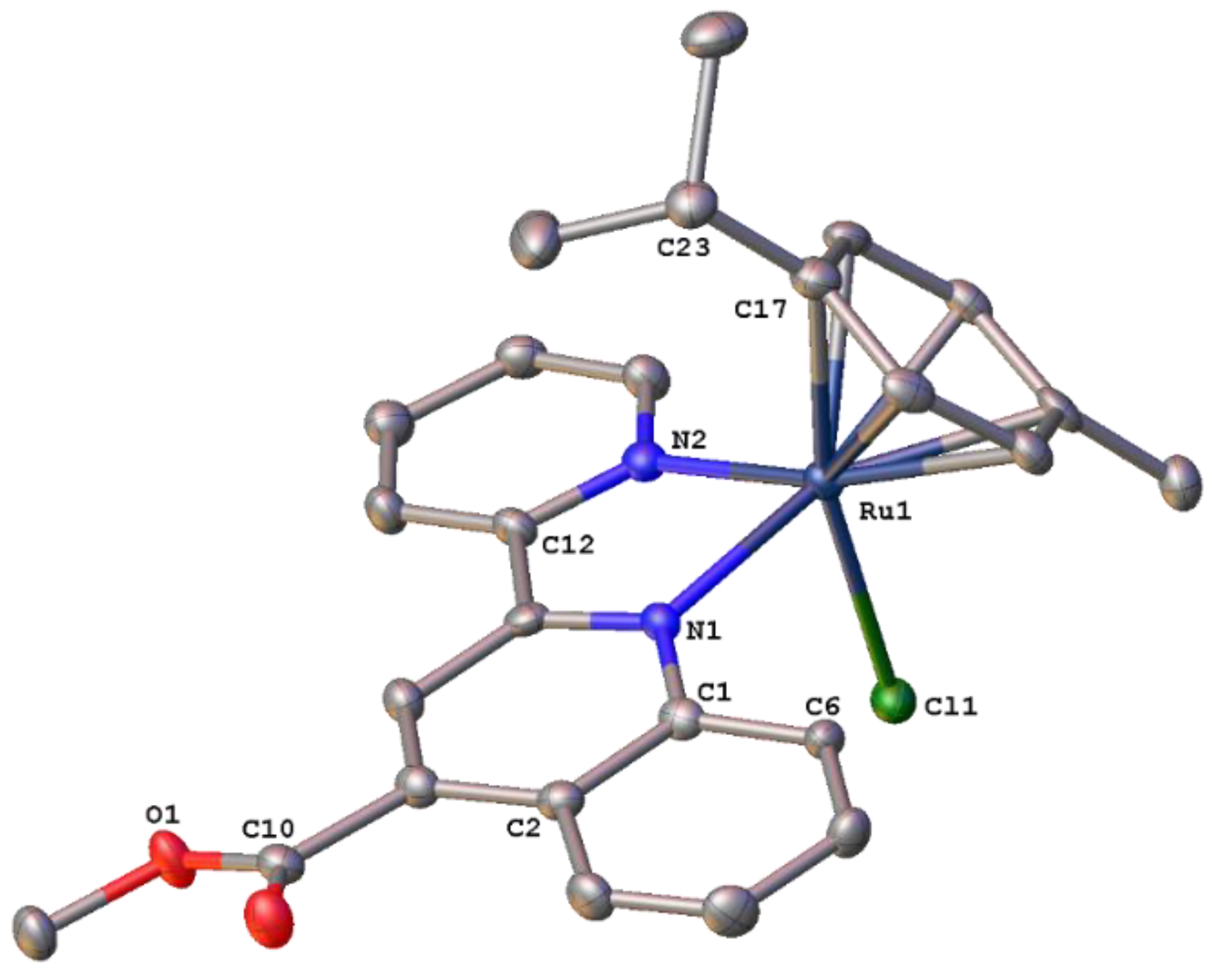
Scheme 3.
Catalytic transfer hydrogenation of benzophenone and acetophenone derivatives by catalysts 1–11, [Ru-Cat].
Scheme 3.
Catalytic transfer hydrogenation of benzophenone and acetophenone derivatives by catalysts 1–11, [Ru-Cat].
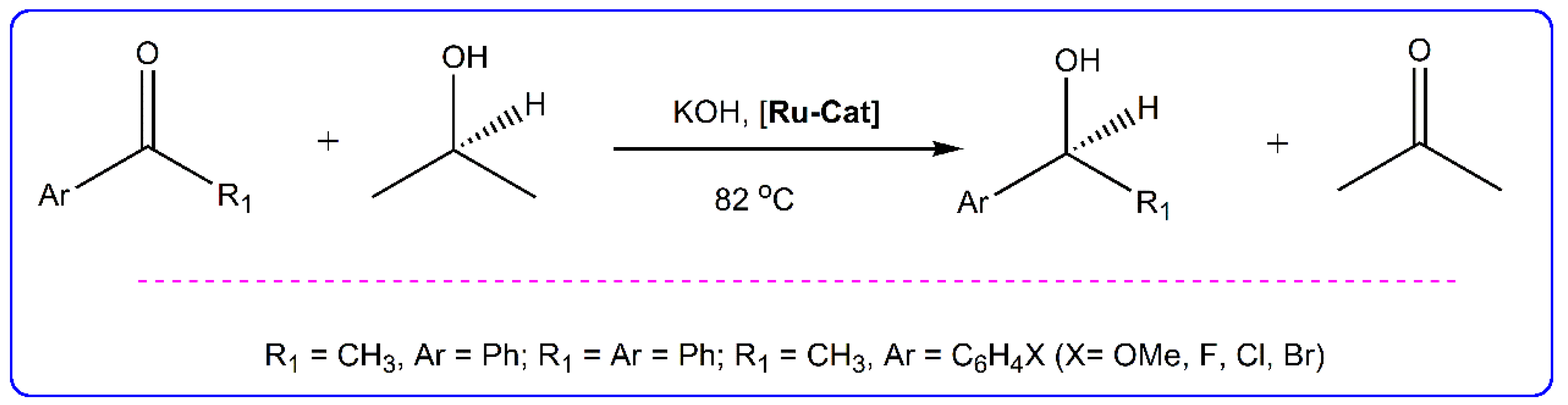
Figure 9.
Time dependence of transfer hydrogenation of acetophenone by catalysts 1–5, 7-Cl and 8.
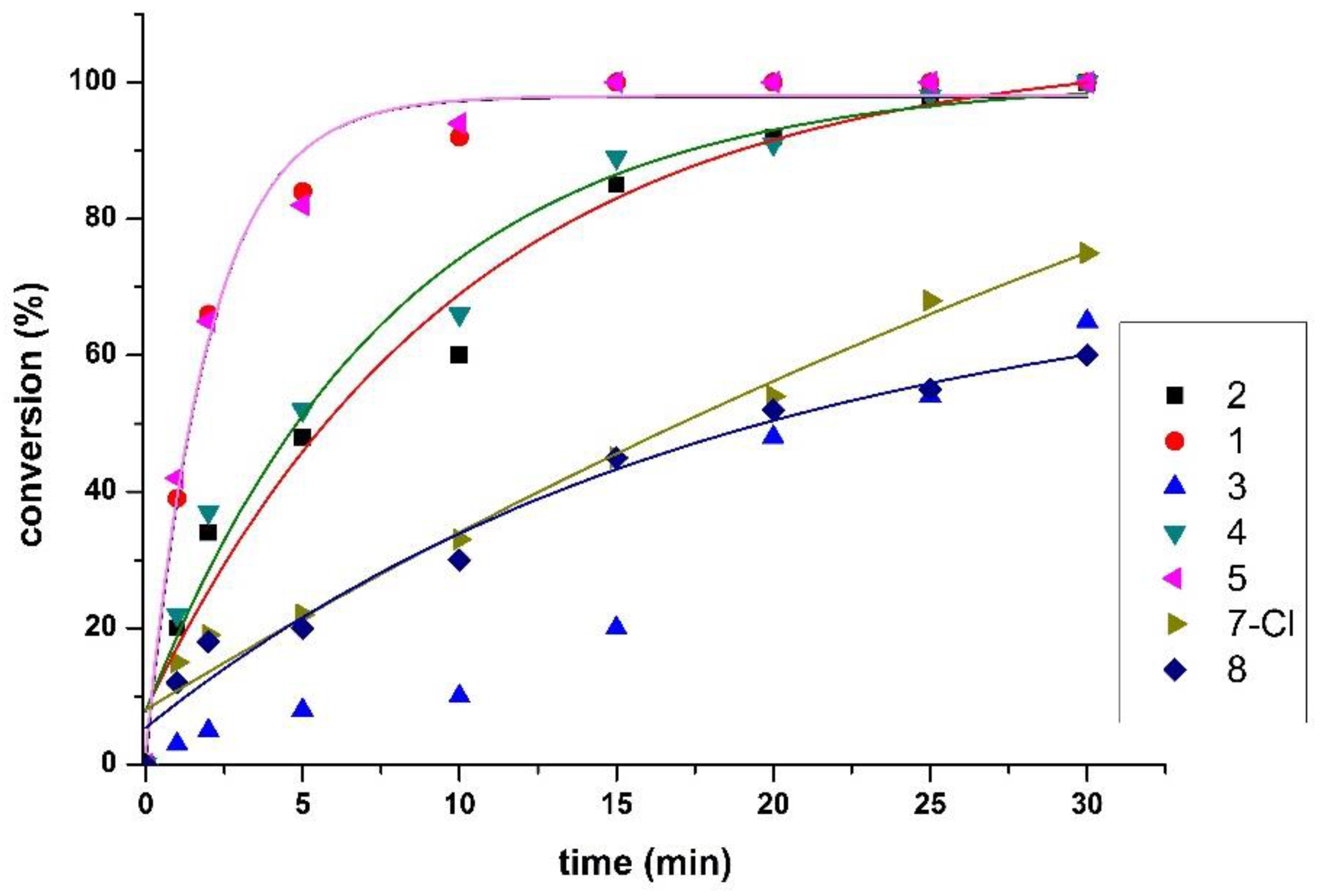

Table 1.
Catalytic transfer hydrogenation of acetophenone using ruthenium(II) complexes 1–11.
| Entry | Complex* | Time (min) | Conversion (%)a | ΤOΝb | TOF (h–1)c |
|---|---|---|---|---|---|
| 1 | 1 | 15 | 100 | 400 | 1600 |
| 2 | 2 | 15 | 85 | – | – |
| 3 | 2 | 30 | 100 | 400 | 800 |
| 4 | 3 | 15 | 20 | – | – |
| 5 | 3 | 60 | 100 | 400 | 400 |
| 6 | 4 | 15 | 90 | – | – |
| 7 | 4 | 30 | 100 | 400 | 800 |
| 8 | 5 | 15 | 100 | 400 | 1600 |
| 9 | Ru-pqcame | 60 | 38 | – | – |
| 10 | Ru-pqcame | 180 | 95 | 380 | 127 |
| 11 | 6 | 180 | 100 | 400 | 133 |
| 12 | 7-Cl | 60 | 95 | 380 | 380 |
| 13 | 8 | 60 | 90 | 360 | 360 |
| 14 | 9 | 60 | 30 | – | – |
| 15 | 9 | 180 | 90 | 360 | 120 |
| 16 | 10 | 15 | 90 | 360 | 1440 |
| 17 | 11 | 60 | 90 | 360 | 360 |
* Reaction conditions: ketone (2 mmol), catalyst (0.25 mol%), KOH (10 mmol%) (Substrate/catalyst/base ratio = 400/1/40). Temperature: 82 oC, hydrogen donor: 2-propanol. aConversion was monitored by 1H NMR and are average of two runs. bTON (Turnover number) = moles of product per moles of ruthenium catalyst. cTOF = TON/h (after completion of the reaction). SD = ±1.0%. For 2, 4 and 6 only for final products.
Table 2.
Catalytic transfer hydrogenation of substituted acetophenone derivatives an benzophenone, using catalysts 1–11.
Table 2.
Catalytic transfer hydrogenation of substituted acetophenone derivatives an benzophenone, using catalysts 1–11.
| Entry | Substrate* | Complex | Time (min) | Conversion (%)a | TONb | TOF (h–1)c |
|---|---|---|---|---|---|---|
| 1 | 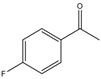 |
1 | 15 | 99 | 396 | 1584 |
| 2 | 2 | 30 | 95 | 380 | 760 | |
| 3 | 3 | 60 | 70 | 280 | 280 | |
| 4 | 4 | 30 | 99 | 392 | 792 | |
| 5 | 5 | 15 | 95 | 380 | 1520 | |
| 6 | Ru-pqcame | 180 | 85 | 340 | 113 | |
| 7 | 6 | 180 | 87 | 348 | 116 | |
| 8 | 7-Cl | 60 | 92 | 368 | 388 | |
| 9 | 8 | 60 | 82 | 328 | 328 | |
| 10 | 9 | 180 | 85 | 340 | 113 | |
| 11 | 10 | 60 | 88 | 352 | 352 | |
| 12 | 11 | 60 | 83 | 332 | 332 | |
| 13 | 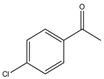 |
1 | 15 | 99 | 396 | 1584 |
| 14 | 2 | 30 | 98 | 392 | 784 | |
| 15 | 3 | 60 | 90 | 360 | 360 | |
| 16 | 4 | 30 | 98 | 392 | 784 | |
| 17 | 5 | 15 | 98 | 392 | 1568 | |
| 18 | Ru-pqcame | 180 | 95 | 380 | 127 | |
| 19 | 6 | 180 | 98 | 392 | 131 | |
| 20 | 7-Cl | 60 | 95 | 380 | 380 | |
| 21 | 8 | 60 | 90 | 360 | 360 | |
| 22 | 9 | 180 | 90 | 360 | 120 | |
| 23 | 10 | 60 | 97 | 388 | 388 | |
| 24 | 11 | 60 | 82 | 328 | 328 | |
| 25 | 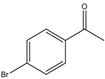 |
1 | 30 | 66 | 264 | 528 |
| 26 | 2 | 30 | 72 | 288 | 576 | |
| 27 | 3 | 60 | 55 | 220 | 220 | |
| 28 | 4 | 30 | 75 | 300 | 600 | |
| 29 | 5 | 30 | 72 | 288 | 676 | |
| 30 | Ru-pqcame | 180 | 72 | 288 | 96 | |
| 31 | 6 | 180 | 88 | 336 | 117 | |
| 32 | 7-Cl | 60 | 65 | 260 | 260 | |
| 33 | 8 | 60 | 66 | 264 | 264 | |
| 34 | 9 | 180 | 60 | 240 | 80 | |
| 35 | 10 | 60 | 73 | 292 | 292 | |
| 36 | 11 | 60 | 66 | 264 | 264 | |
| 37 |  |
1 | 30 | 89 | 356 | 712 |
| 38 | 2 | 30 | 85 | 340 | 680 | |
| 39 | 3 | 60 | 65 | 260 | 260 | |
| 40 | 4 | 30 | 96 | 386 | 772 | |
| 41 | 5 | 30 | 96 | 384 | 768 | |
| 42 | Ru-pqcame | 180 | 78 | 312 | 104 | |
| 43 | 6 | 180 | 94 | 376 | 126 | |
| 44 | 7-Cl | 60 | 75 | 300 | 300 | |
| 45 | 8 | 60 | 70 | 380 | 280 | |
| 46 | 9 | 180 | 75 | 300 | 100 | |
| 47 | 10 | 60 | 79 | 316 | 316 | |
| 48 | 11 | 60 | 78 | 312 | 312 | |
| 49 |  |
1 | 60 | 99 | 396 | 396 |
| 50 | 2 | 60 | 95 | 380 | 380 | |
| 51 | 3 | 60 | 80 | 320 | 320 | |
| 52 | 4 | 60 | 90 | 360 | 360 | |
| 53 | 5 | 60 | 96 | 364 | 364 | |
| 54 | Ru-pqcame | 180 | 85 | 340 | 113 | |
| 55 | 6 | 180 | 82 | 328 | 109 | |
| 56 | 7-Cl | 60 | 96 | 384 | 384 | |
| 57 | 8 | 60 | 88 | 352 | 352 | |
| 58 | 9 | 180 | 82 | 328 | 109 | |
| 59 | 10 | 60 | 88 | 352 | 352 | |
| 60 | 11 | 60 | 99 | 396 | 396 |
* Reaction conditions: ketone (2 mmol), catalyst (0.25 mmol %), KOH (10 mmol %). Temperature: 82 oC, hydrogen donor: 2-propanol; a Conversion was monitored by 1H NMR and are ave-rage of two runs bTON (Turnover number) = moles of product per mole of ruthenium catalyst. c TOF = TON/h (after completion of the reaction). SD = ±1.0%.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



