
Submitted:
10 June 2025
Posted:
11 June 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in Wuhan, China, in December 2019, it has evolved, leading to variants that differ in their transmissibility, severity of the disease, and susceptibility to therapy. Our goal was to describe the dynamics of the emergence of SARS-CoV-2 variants among the population of the southern part of Poland (Silesia) in the period from September 2021 to August 2022. Our results showed that, like in the rest of Poland or in neighboring countries (Czech Republic, Slovakia) Delta was replaced by Omicron BA.1 variant, isolated for the first time in December 2021, and subsequent Omicron BA.2 and its derivative subvariants acquiring further mutations. Finally, in August 2022, only the BA.5.2.26 subvariant was present in Silesia. However, we noted differences in the dynamics of emergence and spread of some Omicron subvariants compared to the rest of Poland and the neighboring countries, which may be due to differences in population density or import of the virus from other regions.
Keywords:
SARS-CoV-2 variants
; molecular epidemiology
; whole-genome sequencing
1. Introduction
The continuous monitoring of the spread of the SARS-CoV-2 virus, which causes severe acute respiratory syndrome, started after the first case was reported in late 2019 in Wuhan, China. By analyzing the complete genetic profile of the virus, researchers were able to track the outbreak in real-time and gain valuable insights into its evolution. The COVID-19 pandemic has led to a significant increase in sequencing efforts across Europe, resulting in a larger number of publicly available SARS-CoV-2 genetic sequences [1,2,3,4,5].
As of February 2024, over 16,536,347 complete genomes of SARS-CoV-2 have been recorded in the Global Initiative on Sharing All Influenza Data (GISAID) database [5]. This comprehensive molecular monitoring of the virus necessitates a simple method for categorizing variant genetic variations. Extensive molecular data allows for the real-time tracking of the pandemic's evolution through the examination of outbreaks and surveillance of the emergence of new circulating strains [6,7]. The B.1.617.2+ AY (Delta) variant of the SARS-CoV-2 virus initially appeared during India's second wave than rapidly spread and become the prevailing variant worldwide. However, it still continues to undergo evolutionary changes. On November 26, 2021, the World Health Organization designated a new variant, B.1.1.529, as a cause for concern and named it Omicron. This decision was based on the discovery of several mutations in Omicron, which may potentially impact its behavior [8].
Our previous research showed a genetic analysis of the epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland, focusing on the period from February 2021 to August 2021. At that time, the alpha variant dominated in Silesia, later replaced by the delta variant [9]. The aim of the presented study was to investigate the molecular epidemiology of SARS-CoV-2 lineages in Southern Poland from September 2021 to 31st August 2022. Through comprehensive analysis of 942 viral genomes, including whole-genome sequencing, variant identification, and phylogenetic analysis, we observed the evolutionary trends of these variants. Additionally, we compared our data from Southern Poland with sequences from Poland, as well as neighboring countries such as the Czech Republic and Slovakia.
2. Materials and Methods
2.1. Study Group
Sequential analysis was performed on 942 samples, determined in RT-PCR tests (MediPAN 2G + FAST COVID Kit [Medicofarma], 2019-Novel-Coronavirus [2019-nCOV] Triplex RT-qPCR Detection Kit [Vazyme], MutaPLEX Coronavirus Real-Time-RT -PCR-Kit [Immunodiagnostic] and Xpert Xpress SARS - COV -2 and Xpert Xpress SARS- COV2 / FLU / RSV [GeneXpert]) as positive, from September 2020 to August 2022. Patients mainly come from Southern Poland (Silesian region), from various sources: hospitals (671), hospital diagnostic points (119), other mobile diagnostic points (114). Samples and data for analysis come from 31 institutions/ sources and were analyzed after prior anonymization. Samples with a CT value <32 were qualified for the preparation of libraries used in sequencing analysis.
2.2. SARS-CoV-2 Whole Genome Sequencing (WGS)
RNA SARS-CoV-2 was isolated by Maxwell 48 RSC instrument (Promega) and Maxwell RSC Viral TNA kit (Promega). Qualitative and quantitative analysis of isolates was performed with Quantus fluorymeter (Promega) and Fragment Analyzer 5200 (Agilent). Reactions of RNA rewriting to cDNA or amplification conducted on the Biorad CFX96 Real-Time System (Biorad) and Biorad C1000 Touch Thermal Cycler (Biorad) devices. Sequencing was performed with two library preparation protocols: Respiratory Virus Oligo Panel RVOP (Illumina RNA Prep with Enrichment) for 856 samples and NEBNext ARTIC SARS CoV-2 FS Library Prep Kit (NebNext) for 48 samples. Sequencing of the SARS-CoV-2 virus genome was performed on Illumina devices dedicated to NGS analyzes: miSeq and NextSeq 1000. After sequencing on an Illumina NGS system, data analysis proceeds using the DRAGEN pipelines: DRAGEN COVID Lineage (3.5.3 Version, Pangolin: Max Ambiguous Rate 0,5) and DRAGEN RNA Pathogen Detection (3.5.16 Version, K-mer Reference FASTA SARS-CoV-2_NC_045512.2, Reference Human (hg38)) and Illumina Respiratory Virus Panel, with Human Controls. Samples were qualitatively analyzed with coverage, mapped reads, duplicate reads, row reads, length and% N. Raw data from the sequencing analyzes with information regarding the date of sample collection, location/region in Poland, gender and age of the patient, clinical condition of the patient (where available) were each time deposited in the GISAID database (https://gisaid.org/).
2.3. Phylogenetic Analyses
To complete phylogenetic analyses, we originally compiled a dataset of 135,879 Sars-CoV-2 genomes (covering the period from the first of September 2021, to the thirty-first of August, 2022) based on data available as of July 25, 2023, from GISAID (gisaid.org). For this investigation, we included sequences specifically from the Silesia region (n=942), along with data from three countries: (i) Poland (excluding Silesian sequences, n=62,520), (ii) Czech Republic (n=39,579), and (iii) Slovakia (n=32,838). Given the unknown proportion of delta and omicron cases in those countries, we subsampled these genomes based on the proportion of overall COVID-19 cases reported per epidemiological week in each country, using reports from the "Center for Systems Science and Engineering (CSSE)" Johns Hopkins University (http://github.com/CSSEGISandData/COVID-19). The subsampling process utilized the 'subsampler' pipeline (http://github.com/andersonbrito/subsampler), selecting genomes to simulate a scenario where 0.1% of cases per 'epiweek' per country were sequenced. The final dataset comprised 8,576 virus genomes from three countries: (i) Poland (excluding Silesian sequences, n=3,096), (ii) Czech Republic (n=2,707), and (iii) Slovakia (n=1,831), including the unbiased sequences from Silesia region. All genomes had coverage above 70% and represent the COVID-19 burden as indicated by each country's epidemiological time series data. The complete list of genomes is available in Supplementary Table S1.
Using the augur pipeline [10], we conducted a rigorous multiple sequence alignment (MSA) using MAFFT [11]. We carefully masked the MSA's 5′ and 3′ ends alongside other problematic sites [12] using a script delivered with the pipeline. We then performed a quick maximum likelihood analysis using IQ-Tree [13] under a GTR nucleotide substitution model. Divergence times and ancestral states were inferred using TreeTime 0.8.0 [14]. This preliminary analysis was about determining the placement of the virus genomes and identifying any major molecular clock outliers deviating from more than four interquartile ranges in the root-to-tip regression line. We inferred the final time-scaled tree to identify large clades containing only genomes of Silesian and Polish, Silesian and Czech, and Silesian and Slovakian origin. These clades are significant in our collective understanding of the spread and evolution of the Sars-Cov-2 variants in the investigated regions.
2.4. Epidemiological Data Analysis
Epidemiological data and statistics for the Czech Republic and Slovakia were downloaded from public repository Our World in Data. Data for Poland and the Silesian Voivodeship were downloaded from government reports posted at https://www.gov.pl/web/koronawirus.
3. Results
3.1. The Prevalence of SARS-CoV-2 Variants (hCoV-19) in Southern Poland
From the 942 sequenced samples collected from September 2021 to 31 August 2022 in Southern Poland (Silesia), 939 were analyzed and classified into variants while 3 were rejected due to the large number of missing nucleotides in sequences (>1500). The analyzed period was characterized by a large variety of isolated variants. The B.1.617.2 + AY (Delta) was the dominant variant among the sequenced samples (n = 718, 76%). The single variants of B.1.1.529 + BA (Omicron) were isolated for the first time in December 2021, and it gradually dominated the variants isolated in Silesia in the analyzed months to completely replace the Delta variant in March 2022. In total, this variant appeared with a frequency of 24% (n=221) in the period from December 2021 to August 2022. However, it should be noted that the number of analyzed samples has decreased significantly since February 2022. In February 2022 no sample was collected. Far fewer samples have been isolated since March 2022. It was directly related to the changes in COVID-19 testing policy introduced in Poland on January 28th, where antigen tests for SARS-CoV-2 were allowed to use in a laboratories/mobile collection points [15] and lifting the need for isolation, quarantine, and masks wear in Poland [16]. Changes in the number of samples isolated in individual months in Silesia are presented in Figure 1.
The analysis of the collected data suggests that the diversity of variants circulating in the studied population was very large. The percentage distribution of the main SARS-CoV-2 variants (frequency >5%) isolated in Silesia from September 2021 to August 2022 are presented in Figure 2.
3.2. The Prevalence of SARS-CoV-2 Main Variants (hCoV-19) in Poland, the Czech Republic, and Slovakia
The analysis of whole-genome sequences from Poland, the Slovakia and Czech Republic was carried out based on the data available in GISAID. The diversity of variants in the studied period was very large in all countries. The percentage distribution of the main SARS-CoV-2 variants (frequency >5%) isolated in Poland, Czech Republic and Slovakia from September 2021 to August 2022 are presented in Figure 3, Figure 4 and Figure 5.
An analysis of 65,509 sequences reported in GISAID between September 2021 and 31 August 2022 in Poland was performed. In the Czech Republic, 39,579 samples were collected, while in Slovakia 32,838. The diversity of variants in the studied period was very large in all countries. None of the variants clearly dominated over the others.
3.3. The Phylogenetic Connections of SARS-CoV-2 Clades
The analysis of the genetic history of the samples tested in Southern Poland showed that there were two main groups of the SARS-CoV-2 virus, namely B.1.617.2 + AY and B.1.1.529 + BA. The BA.1 group was derived from B.1.1.529 + BA, similar to BA.2. Figure 6 provides a visual representation of the evolutionary relationships among different clades of the SARS-CoV-2 virus observed in Southern Poland. In addition, we created a diagram showing the evolutionary relationships of Silesia’s clades within the broader context of Poland (Figure 7). Furthermore, a thorough examination comparing Silesia with the Czech Republic and Slovakia was conducted (Figure 8 and Figure 9). The analysis of the trees presented in Figure 7, Figure 8 and Figure 9 allows to conclude that in each case, the dominance of the B.1.617.2 + AY variant was observed, which was gradually replaced by the B.1.1.529 + BA variant.
3.4. COVID-19 Cases in Silesia Compared to the Rest of Poland and Neighboring Countries
According to Our World in Data, during the period covered by present study Poland had 1.2 times more COVID-19 cases than Czechia and approximately 2.3 times more than Slovakia. However, after adjustment for the country’s population, the number of COVID-19 cases per million inhabitants in Poland was 2.8 times fewer than in Czechia and Slovakia. In contrast, we did not observe any differences in the number of COVID-19 cases per 1 million inhabitants between the Silesian Voivodeship and the country-wide area.
4. Discussion
The Silesian region is inhabited by more than 4.3 million people, which is about 12% of the Polish population. The population density in the Silesian Voivodeship is nearly 3 times higher compared to the average population density in Poland (355 vs 121 persons per 1 km2). Moreover, Silesian Voivodeship borders both the Czech Republic and Slovakia and before the pandemic began, thousands of people benefited from the cross-border labor market - many residents of the Silesian region worked in Czech and Slovak companies. After the Polish government introduced mandatory border controls and a two-week quarantine for Poles returning to the country from abroad in March 2020, full border traffic within the internal borders of the European Union was restored on June 13, 2020. Thus, during the period covered by our research, cross-border traffic was no longer restricted, allowing the free circulation of virus variants between neighboring countries.
In the period from September 2021 to January 2022, the Delta variant (B.1.617.2+ AY) dominated in Southern Poland. It was consistently replaced by the Omicron B.1.1.529 + BA variant. Contrasting Silesia with Poland, the Omicron variant appeared in Southern Poland earlier. It was first recorded in December 2021 (BA.1.1), while in the rest of the country it appeared in January 2022 and caused the fifth wave which peaked at the end of January 2022 [17]. The earlier appearance of the Omicron variant in the Silesian province may be responsible for different dynamics of the development of the fifth wave of the pandemic in this region compared to the rest of the country (Figure 10). Namely, during the initial phase of the fifth wave of the pandemic, in the second half of January 2022, the incidence rate of positive cases (per 100 000 inhabitants) was more dynamic in Silesia compared to the rest of Poland. Then, in February, a faster decrease in the number of new infections per 100 000 inhabitants was observed is our region than in Poland. This early and rapid spread of the Omicron variant of SARS-CoV-2, combined with a significant population density, may explain why the presence of the Delta variant of the virus was no longer observed in Silesia in March.
In March and April, the dominant variant both in Silesia and the rest of the country was the BA.2.9 one. The BA.2 variant occurred with a similar frequency. Interestingly, in the given period, the BA.2.7 variant also appeared with high frequency in Poland, but it was not noted in Silesia. Since June 2022, a large variety of variants without any specific dominance has been observed in Southern Poland. A similar situation occurred throughout Poland.
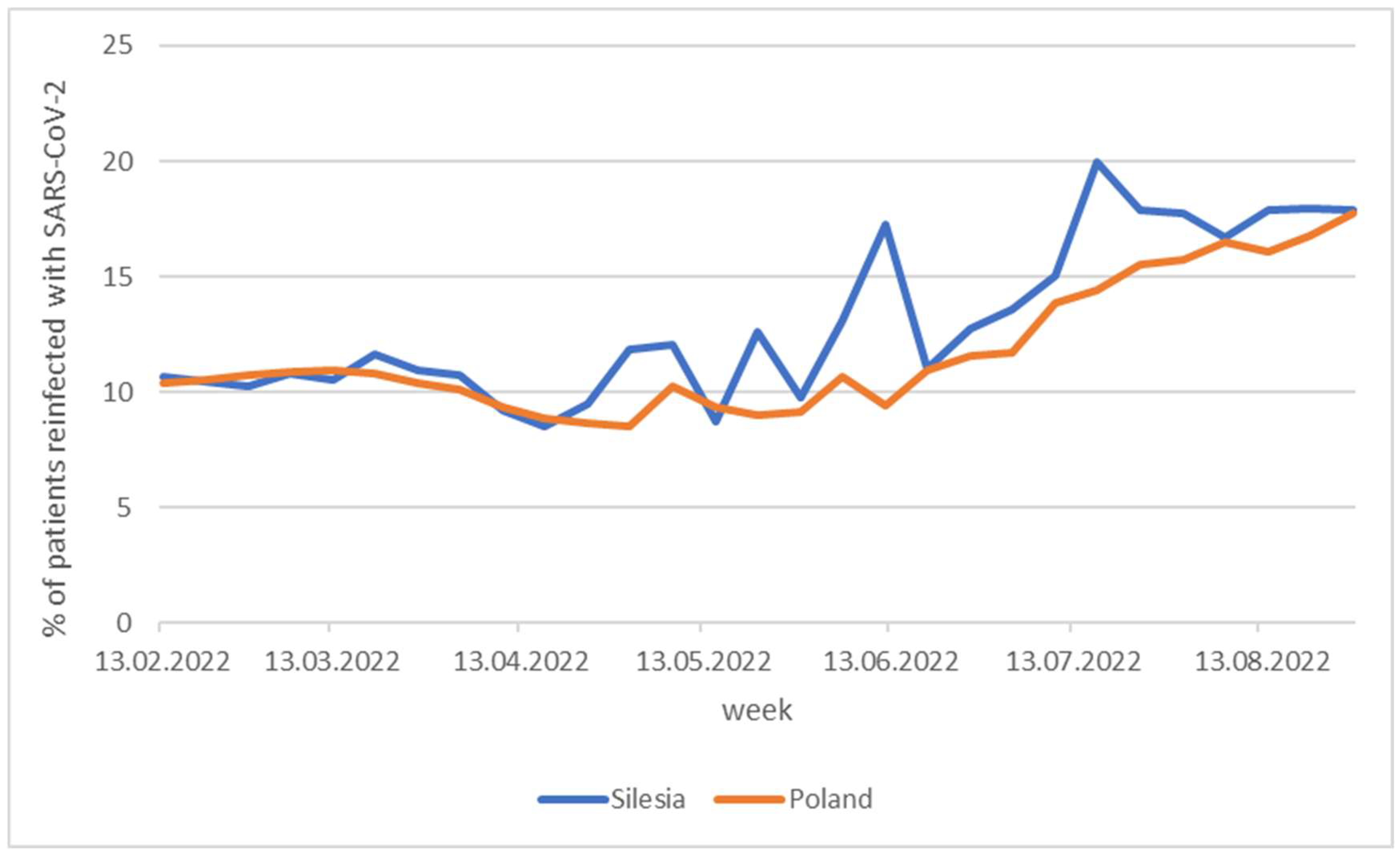
According to the data available on the government website, during the period from early May to mid-July, considerably more cases of reinfection were observed in the region covered by our research compared to the rest of the country (Figure 11), what is especially noteworthy. In Silesia BA.5 subvariants (BA.5.2 and BA.5.32) appeared as early as May, while in the rest of the country presence of BA.5.1, BA.5.2, and BA.5.2.1 subvariants was recorded two months later, in July. The BA.5 clades stem from BA.2 by acquisition of Δ69-70 deletion in N-terminal domain, L452R, F486V, and reversed R493Q mutations in the receptor biding region, S704L mutation located outside the receptor biding region of the spike protein as well as D3N mutation in M protein.
Previously conducted studies have shown that BA.5 competed with BA.2 because of its increased resistance to neutralization [18,19,20] and its higher infectivity resulting from higher replication fitness, transmissibility, and pathogenicity [21,22,23]. The functional examination of impact of mutations characterizing BA.5 S proteins showed that mutations of Δ69-70, L452R, and F486V contribute to the higher infectiousness and fusogenicity of the BA.5 S protein. Moreover, L452R and F486V substitutions were responsible for reduced sensitivity to neutralizing antibodies [24].
The other studies have shown that antibodies isolated from BA.1 infected or vaccinated individuals showed reduced efficacy against L452 mutations causing the most severe escape of variants BA4/BA.5 harboring L452R mutation [20] which may explain the ability of the BA.5 variants to evade the neutralizing effect of antibodies arisen during previous infections. The importance of L452R mutation for impairing antibody binding allowing the B5 variant to escape the immune system was also demonstrated by Tuekprakhon et al. [25]. Moreover, S371F mutation (occurring in Omicron but not Delta variant) was involved in the emergence of conformational changes of the region recognized by antibodies isolated from vaccinated individuals who had recovered from SARS [20].
What is more, in June and July in Silesia region we observed the occurrence of BA.2.12.1, the another derivative of BA.2 variant arising by acquisition of L452Q and S704L mutations in addition to the known mutations in BA.2. Omicron BA 2.12.1 subvariant appeared in December 2021 in the United States and spread very fast in many parts of US, but also in other countries [26]. Many studies showed that the BA.2.12.1 like BA.4 and BA.5 subvariants substantially escape neutralizing antibodies induced by both vaccination and infection. Compared to BA.2 Omicron variant, BA.2.12.1 subvariant shows stronger immune escape and faster transmissibility. In contrary to BA.5, BA.2.12.1 Omicron subvariant showed only modest resistance to sera from vaccinated and boosted individuals compared to BA.2 [19]. The reduced sensitivity of BA.2.12.1 compared to BA.2 S protein to neutralization by sera vaccinated donors was found, however, this effect was donor dependent. On the contrary, S704L mutation increased susceptibility to neutralization [24]. Noteworthy, BA.2.12.1 variant has not been detected either in the rest of Poland or in neighboring countries (Czech Republic, Slovakia), thus, it was probably imported to Silesia from further regions of Europe or the world. In Silesia BA.2.12.1 was completely replaced by BA.5.2.26 in August, which reflects a transmission advantage of BA.5 subvariants. The dominance of previously circulating variants of the virus by more efficient variants has also been observed in other regions, e.g. South Korea, where BA.1 was the dominant variant for 10 weeks starting from the 1st week of 2022, BA.2 was the dominant variant in the 12th and 13th weeks of 2022, and they were subsequently replaced by BA.5 which became the dominant variant in June 2022 [28].
In the Czech Republic, the Omicron variant was first noted in December 2021. An earlier dominance of the Omicron variants was also observed, which replaced the delta variants already in January 2022, when this variant was just appearing in Silesia. Similarly to Silesia and the rest of Poland, the dominance of the BA.2.9 variant has also been observed since March 2022. Since June, the diversity of variants in the Czech Republic has been very large.
Similarly in Slovakia, we observed the earlier displacement of the delta variant by omicron, which dominated already in January 2022. What is more, comparing Slovakia with Silesia, a clear difference can be noticed, consisting in the dominance of the BA.2 variant from March to May 2022 instead of the BA.2.9 variant.
When comparing the molecular diversity of SARS-CoV-2 in Silesia with the above regions, the difference in the occurrence of variants of the BF (BF.5 and BF.7) line locally in Silesia should be also noted What is more, we observed greater diversity among BE line variants (BE.1, BE.1.1 and BE.1).
Analyzing the dynamics of the appearance of individual variants globally, it can be concluded that the Omicron variant appeared earlier in the world than in the regions discussed in this work. It was first sequenced in November 2021 in South Africa, while in Silesia it appeared in December 2021, as well as in the Czech Republic and Slovakia. In other regions of our country, it was detected in January 2022 [29]. An interesting aspect seems to be the earlier appearance of the Omicron variant in Silesia than in the rest of Poland. It seems likely that the variants moved from our southern neighbors into the country. It should be noted, however, that not all samples were reported in GISAID and it cannot be ruled out that Omicron had occurred in other regions of Poland earlier.
Skuza et al. conduct an active monitoring program among military personnel to identify Variants of Concern (VOC) of the SARS-CoV-2 virus, with a particular focus on overseas military operations. Screening of 1699 soldiers using RT-qPCR tests was conducted between November 2021 and May 2022. Out of these, 84 samples tested positive for SARS-CoV-2 and met the criteria for whole genome sequencing analysis for variant identification [30]. Based on analysis of samples from 79 soldiers tested in Poland between November 2021 and March 2022, it can be inferred that the obtained results match the molecular analysis of SARS-CoV-2 in Silesia. Until December 2021, the Delta variant dominated, and then it was replaced by the Omicron lineage from January to March 2022. Similarly to Silesia, the variants BA.1, BA.1.1, BA.1.1.1, and BA.2.9 were dominant in this period. The BA.2.3 variant which was observed in our region in March did not appear in the study by Skuza et al.. Additionally, the population studied by Skuza et al. was characterized by greater molecular diversity. What is more, in March, the Delta variant still appeared there, which had already been completely replaced by Omicron in Silesia during this period [30]. This difference may be due to the fact that Omicron was detected in Silesia as early as December 2021, while in the cohort studied by Skuza this variant was observed only in January 2022. Similar clades were identified in 89 soldiers deployed from Romania in February and March 2022. However, the BA.1.1.13 variant occurred more frequently than among Polish soldiers and samples isolated in Silesia. Similarly, among soldiers returning from France between February and June 2022, the BA.2.9 variant predominated. Additionally, Skuza et al. found a high frequency of the BA.2.56 line, which did not occur in Silesia. Skuza et al. findings indicate that all genetic variants of SARS-CoV-2 found in Polish Armed Forces members had already been circulating in Poland prior to their return from their missions. This suggests that there was minimal transmission of these variants within the national population, or any transmissions that did occur did not significantly impact the SARS-CoV-2 variants present in Poland.
5. Conclusions
In this study we present the alterations in circulating SARS-CoV-2 variants from September 2021 to 31 August 2022 in Southern Poland with reference to the remaining part of the country as well as neighboring countries. We observed the occurrence of new variants of the virus, among which those possessing mutations conferring increased replication efficiency and immune escape spread rapidly and quickly replaced the earlier variants. Tracking the emergence and spread of new SARS-CoV-2 variants is essential for providing information to make decisions on vaccine development, therapy as well as predicting the course of infection with subsequent variants of the virus.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: The complete list of genomes used in phylogenetic analyses nd deposited in GISAID.
Author Contributions
Conceptualization: Tomasz J. Wąsik and Anna Bednarska-Czerwińska; Methodology: Maria Miklasińska-Majdanik, Emilia Morawiec, Karol Serwin and Tomasz J. Wąsik; Software: Karol Serwin, Emilia Morawiec and Adam Pudełko; Validation: Maria Miklasińska-Majdanik, Emilia Morawiec; Jolanta Bratosiewicz-Wąsik, Tomasz J. Wąsik; Formal analysis: Maria Miklasińska-Majdanik, Emilia Morawiec, Jolanta Bratosiewicz-Wąsik Miłosz Parczewski and Tomasz J. Wąsik; Investigation: Emilia Morawiec, Adam Pudełko, Michał Czerwiński and Anna Bednarska-Czerwińska; Resources: Anna Bednarska-Czerwińska and Tomasz J. Wąsik; Data curation: Maria Miklasińska-Majdanik Emilia Morawiec, Jolanta Bratosiewicz-Wąsik and Karol Serwin; Writing—original draft preparation: Maria Miklasińska-Majdanik and Jolanta Bratosiewicz-Wąsik; Writing—review and editing: Tomasz J. Wąsik and Miłosz Parczewski; Wisualization: Karol Serwin, Jolanta Bratosiewicz-Wąsik; Supervision: Tomasz J. Wąsik; Project administration: Anna Bednarska-Czerwińska and Tomasz J. Wąsik; Funding acquisition: Anna Bednarska-Czerwińska and Tomasz J. Wąsik
Funding
This research was funded by Medical University of Silesia in Katowice, Poland, grants number PCN-1-142/N/1/I and PCN-1-005/K/2/I, and Marshal’s Office of the Silesian Voivodeship, EU funds from the Regional Operational Program of the Silesian Voivodeship, grant number UDA-RPSL.10.01.00-24-030H/20-02.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Bioethical Committee at the Medical University in Katowice, Poland, approval number PCN/022/KB1/07/21, approval date March 30, 2021.
Informed Consent Statement
All procedures were performed in accordance with relevant guidelines and regulations. All collected samples and data were anonymous and coded. Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All of the SARS-CoV-2 nucleotide sequences obtained in the study have been deposited in GISAID and are available in Table S1.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript: SARS-CoV-2 - Sever acute respiratory syndrome coronavirus 2; GISAID - Global Initiative on Sharing All Influenza Data; WGS - Whole Genome Sequencing
References
- Serwin, K.; Aksak-Was˛, B.; Parczewski, M. Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; Niu, P.; Zhan, F.; Ma, X.; Wang, D.; Xu, W.; Wu, G.; Gao, G. F.; Tan, W. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B. B.; Nelson, M. I.; Hill, V.; Joy, J. B.; Rambaut, A.; Suchard, M. A.; Wertheim, J. O.; Lemey, P. The Emergence of SARS-CoV-2 in Europe and North America. Science (80-. ). 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S. M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R. A. NextStrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B. B.; Worp, N.; Nieuwenhuijse, D. F.; Sikkema, R. S.; Haagmans, B.; Fouchier, R. A. M.; Koopmans, M. The next Phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef]
- Cairo, A.; Iorio, M. V.; Spena, S.; Tagliabue, E.; Peyvandi, F. Worldwide SARS-CoV-2 Haplotype Distribution in Early Pandemic. PLoS One 2022, 17, 1–12. [Google Scholar] [CrossRef]
- Naqvi, A. A. T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I. K.; Singh, A.; Atif, S. M.; Hariprasad, G.; Hasan, G. M.; Hassan, M. I. Since January 2020 Elsevier Has Created a COVID-19 Resource Centre with Free Information in English and Mandarin on the Novel Coronavirus COVID-19. The COVID-19 Resource Centre Is Hosted on Elsevier Connect, the Company ’ s Public News and Information. BBA - Mol. Basis Dis. 2020, No. January, 1–17.
- Kumar, S.; Thambiraja, T. S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta Variant of SARS-CoV-2: A Comparative Computational Study of Spike Protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef]
- Morawiec, E.; Miklasińska-Majdanik, M.; Bratosiewicz-Wąsik, J.; Wojtyczka, RD.; Swolana, D.; Stolarek, I.; Czerwiński, M.; Skubis-Sikora, A.; Samul, M.; Polak, A.; Kruszniewska-Rajs, C.; Pudełko, A.; Figlerowicz, M.; Bednarska-Czerwińska, A.; Wąsik, TJ. From Alpha to Delta-Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens. 2022, 11 (7), 780. PMID: 35890025; PMCID: PMC9316897. [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S. M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R. A. NextStrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S. M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R. A. NextStrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Available online: https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480 (accessed on 13 February 2025).
- Minh, B. Q.; Nguyen, M. A. T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R. A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.gov.pl/web/zdrowie/komunikat-ministra-zdrowia-w-zwiazku-z-dopuszczeniem-od-dnia-28-stycznia-2022-r-mozliwosci-przeprowadzania-testow-antygenowych-w-kierunku-sars-cov-2-przez-laboratoriummobilny-punkt-pobran (accessed on 13 February 2025).
- Available online: https://www.gov.pl/web/psse-zdunska-wola/zniesienie-obowiazku-noszenia-maseczek-zniesienie-kwarantanny-i-izolacji-domowej (accessed on 13 February 2025).
- Available online: https://koronawirusunas.pl (accessed on 13 February 2025).
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron Lineages BA.4 and BA.5 in South Africa. Nat Med 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Kiso, M.; Iida, S.; Imai, M.; Takashita, E.; Kuroda, M.; Halfmann, P.J.; Loeber, S.; Maemura, T.; Yamayoshi, S.; et al. Characterization and Antiviral Susceptibility of SARS-CoV-2 Omicron BA.2. Nature 2022, 607, 119–127. [Google Scholar] [CrossRef]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2 Subvariants, Including BA.4 and BA.5. Cell 2022, 185, 3992–4007.e16. [Google Scholar] [CrossRef]
- Hoffmann, M.; Wong, L.Y.R.; Arora, P.; Zhang, L.; Rocha, C.; Odle, A.; Nehlmeier, I.; Kempf, A.; Richter, A.; Halwe, N.J.; et al. Omicron Subvariant BA.5 Efficiently Infects Lung Cells. Nat Commun 2023, 14. [Google Scholar] [CrossRef]
- Pastorio, C.; Noettger, S.; Nchioua, R.; Zech, F.; Sparrer, K.M.J.; Kirchhoff, F. Impact of Mutations Defining SARS-CoV-2 Omicron Subvariants BA.2.12.1 and BA.4/5 on Spike Function and Neutralization. iScience 2023, 26. [Google Scholar] [CrossRef]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody Escape of SARS-CoV-2 Omicron BA.4 and BA.5 from Vaccine and BA.1 Serum. Cell 2022, 185, 2422–2433.e13. [Google Scholar] [CrossRef]
- Parums, D. V. Editorial: World Health Organization (WHO) Variants of Concern Lineages Under Monitoring (VOC-LUM) in Response to the Global Spread of Lineages and Sublineages of Omicron, or B.1.1.529, SARS-CoV-2. Medical Science Monitor 2022, 28.
- Tan, T.S.; Toyoda, M.; Ode, H.; Barabona, G.; Hamana, H.; Kitamatsu, M.; Kishi, H.; Motozono, C.; Iwatani, Y.; Ueno, T. Dissecting Naturally Arising Amino Acid Substitutions at Position L452 of SARS-CoV-2 Spike. J Virol 2022, 96. [Google Scholar] [CrossRef]
- Lee, Y.U.; Lee, K.; Lee, H.; Park, J.W.; Cho, S.J.; Park, J.S.; Mun, J.; Park, S.; Lee, C. mi; Lee, J.; et al. Genomic Analysis and Tracking of SARS-CoV-2 Variants in Gwangju, South Korea, From 2020 to 2022. Influenza Other Respir Viruses 2024, 18. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Hodcroft, E. B.; Robertson, D. L. The Evolution and Biology of SARS-CoV-2 Variants. Cold Spring Harb. Perspect. Med. 2022, 12, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Skuza, K.; Rutyna, P.; Krzowski, L.; Rabalski, L.; Lepionka, T. Surveillance of SARS-CoV-2 Genetic Variants in the Polish Armed Forces Using Whole Genome Sequencing Analysis. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Changes in the number of SARS-CoV-2 samples isolated from September 2021 to August 2022 in Silesia.
Figure 1.
Changes in the number of SARS-CoV-2 samples isolated from September 2021 to August 2022 in Silesia.
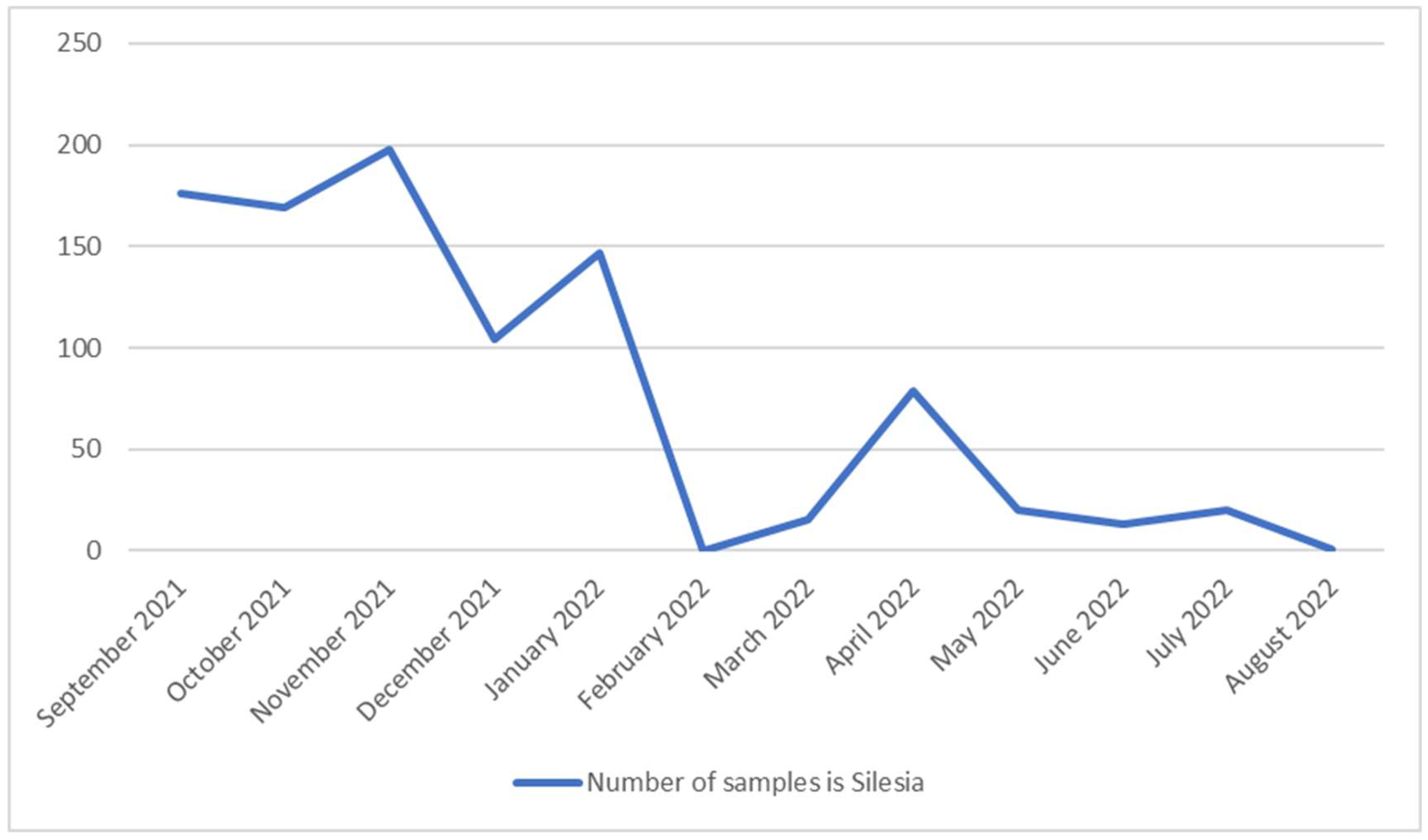
Figure 2.
The percentage distribution of SARS-CoV-2 variants isolated in Southern Poland from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to January 2022, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred in these months with a frequency of >5% were: B.1.617.2, AY.122, AY.4, AY.4.2.3, AY.42. None of these lineages was dominant and they were noted with similar frequency (from 8 to 26%). In addition, several cases of lineages: AY.121.1, AY.126, AY.43, AY.9.2 were recorded during this period. The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In March 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Silesia in the next analyzed months. In February 2022 no sample was isolated. In March and April 2022, the dominated lineage was BA.2.9 (60% and 52%, respectively). In May this lineage was superseded by BA.2 (35%). In June 2022 there were no clearly dominant variant, and lineages: BA.2 (15%), BA.2.9 (15%), BA.2.12.1 (8%), BA.5.1 (8%), BA.5.2 (15%), BA.5.1.10 (7%), BA.5.1.3 (7%), BE.1 (7%), BE.11 (7%), BF.5 (7%) were isolated. In July the BA.5.2 lineage was noted in 30%. Except variants: BA.2.12.1 (5%), BA.5.1 (10%), BA.5.1.3 (5%),.5 (5%) there were also new lineages: BA.5 (5%), BA.5.1.23 (5%), BA.5.2.3 (10%), BA.5.9 (10%), BE.1.1 (15%), BF.5 (5%), BF.7 (5%). One sample was isolated in August 2022 and that was BA.5.2.26 (100%).
Figure 2.
The percentage distribution of SARS-CoV-2 variants isolated in Southern Poland from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to January 2022, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred in these months with a frequency of >5% were: B.1.617.2, AY.122, AY.4, AY.4.2.3, AY.42. None of these lineages was dominant and they were noted with similar frequency (from 8 to 26%). In addition, several cases of lineages: AY.121.1, AY.126, AY.43, AY.9.2 were recorded during this period. The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In March 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Silesia in the next analyzed months. In February 2022 no sample was isolated. In March and April 2022, the dominated lineage was BA.2.9 (60% and 52%, respectively). In May this lineage was superseded by BA.2 (35%). In June 2022 there were no clearly dominant variant, and lineages: BA.2 (15%), BA.2.9 (15%), BA.2.12.1 (8%), BA.5.1 (8%), BA.5.2 (15%), BA.5.1.10 (7%), BA.5.1.3 (7%), BE.1 (7%), BE.11 (7%), BF.5 (7%) were isolated. In July the BA.5.2 lineage was noted in 30%. Except variants: BA.2.12.1 (5%), BA.5.1 (10%), BA.5.1.3 (5%),.5 (5%) there were also new lineages: BA.5 (5%), BA.5.1.23 (5%), BA.5.2.3 (10%), BA.5.9 (10%), BE.1.1 (15%), BF.5 (5%), BF.7 (5%). One sample was isolated in August 2022 and that was BA.5.2.26 (100%).
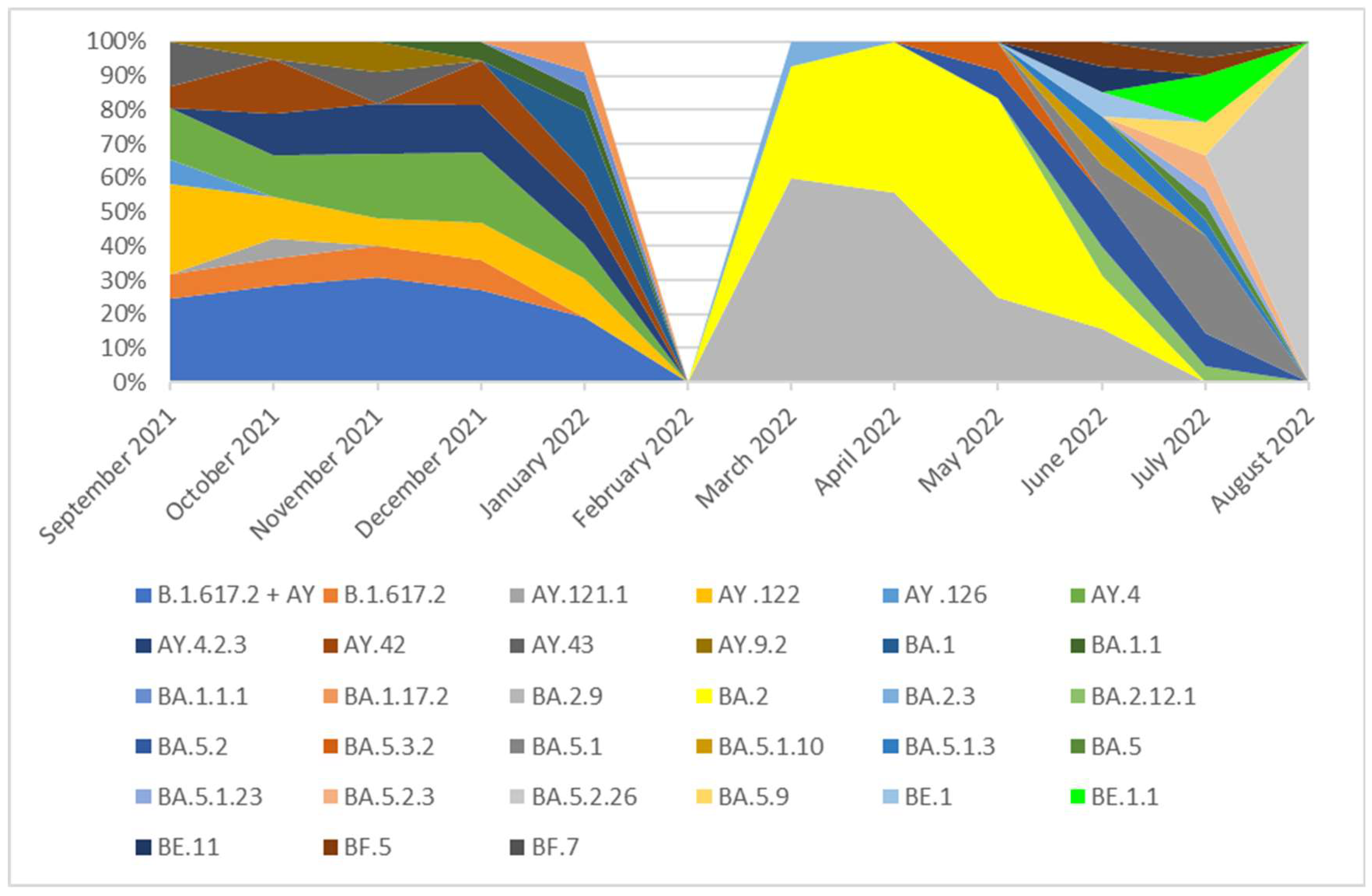
Figure 3.
The percentage distribution of SARS-CoV-2 variants isolated in Poland from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to December 2021, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: B.1.617.2, AY.121.1, AY.122, AY.4, AY.43. None of these lineages was dominant and they were noted with similar frequency (from 6 to 26%). The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2+ AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Poland in the next analyzed months. In January and February 2022, the dominant variant was the BA.1 line and next the BA.1.1 variant. From March to June 2022, the dominated lineages were BA.2 and BA.2.9. The variant BA.2.7 (36%) was also observed with high frequency in March. In July and August 2022 none of these lineages was dominant and they were noted with similar frequency.
Figure 3.
The percentage distribution of SARS-CoV-2 variants isolated in Poland from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to December 2021, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: B.1.617.2, AY.121.1, AY.122, AY.4, AY.43. None of these lineages was dominant and they were noted with similar frequency (from 6 to 26%). The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2+ AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Poland in the next analyzed months. In January and February 2022, the dominant variant was the BA.1 line and next the BA.1.1 variant. From March to June 2022, the dominated lineages were BA.2 and BA.2.9. The variant BA.2.7 (36%) was also observed with high frequency in March. In July and August 2022 none of these lineages was dominant and they were noted with similar frequency.
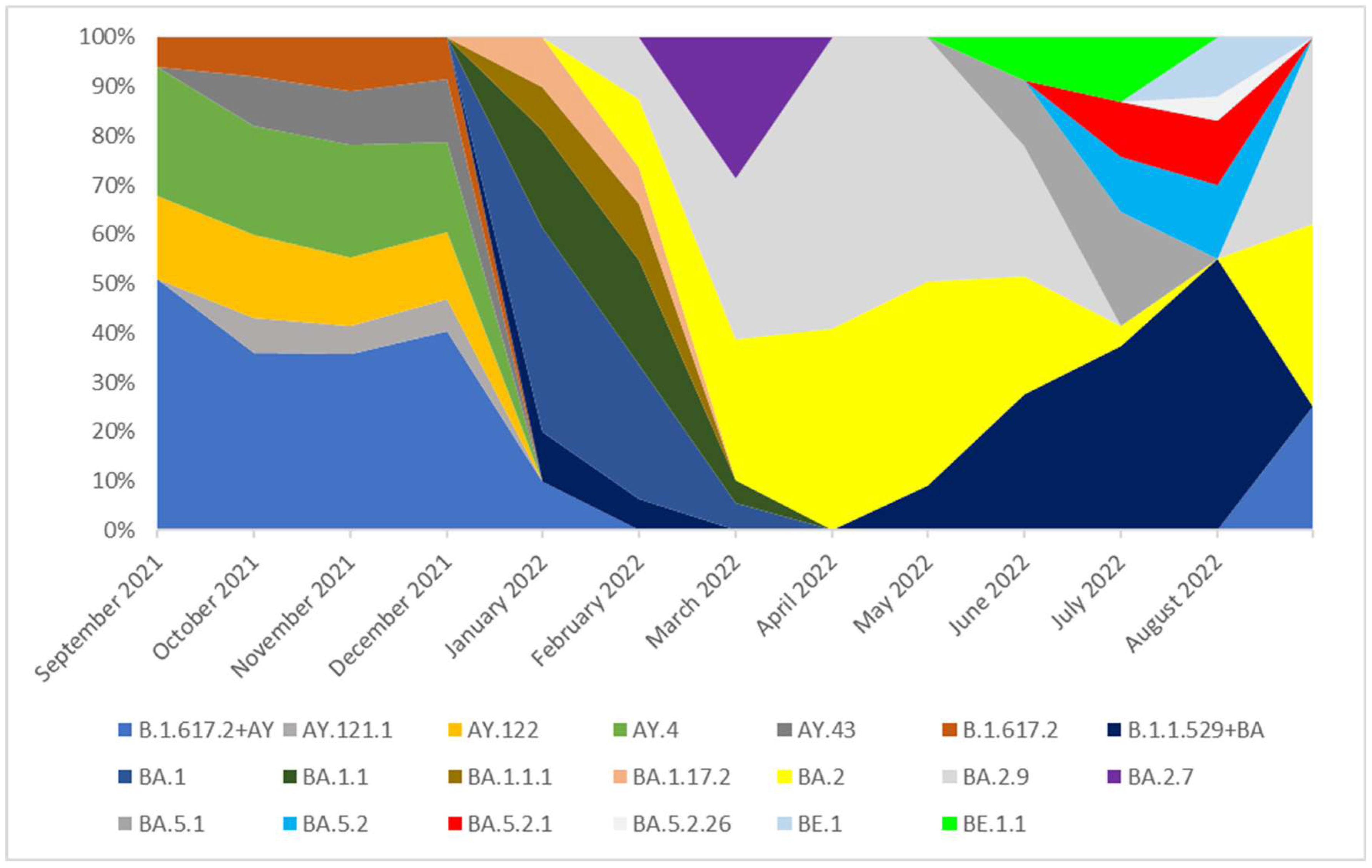
Figure 4.
The percentage distribution of SARS-CoV-2 variants isolated in Czech Republic from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to January 2022, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: AY.122, AY.4, AY.43. None of these lineages was dominant and they were noted with similar frequency (from 13 to 27%). In addition, several cases of lineages: AY.113, AY.13, AY.7.1 were recorded during this period. The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Czech Republic in the next analyzed months. In February 2022, the dominated lineage was BA.1.1 (40%). In March this lineage was superseded by BA.2 one (35%). From March to May 2022 the dominant variants were: BA.2 and BA.2.9. From June to August 2022 none of lineages was dominant and they were noted with similar frequency.
Figure 4.
The percentage distribution of SARS-CoV-2 variants isolated in Czech Republic from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to January 2022, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: AY.122, AY.4, AY.43. None of these lineages was dominant and they were noted with similar frequency (from 13 to 27%). In addition, several cases of lineages: AY.113, AY.13, AY.7.1 were recorded during this period. The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Czech Republic in the next analyzed months. In February 2022, the dominated lineage was BA.1.1 (40%). In March this lineage was superseded by BA.2 one (35%). From March to May 2022 the dominant variants were: BA.2 and BA.2.9. From June to August 2022 none of lineages was dominant and they were noted with similar frequency.
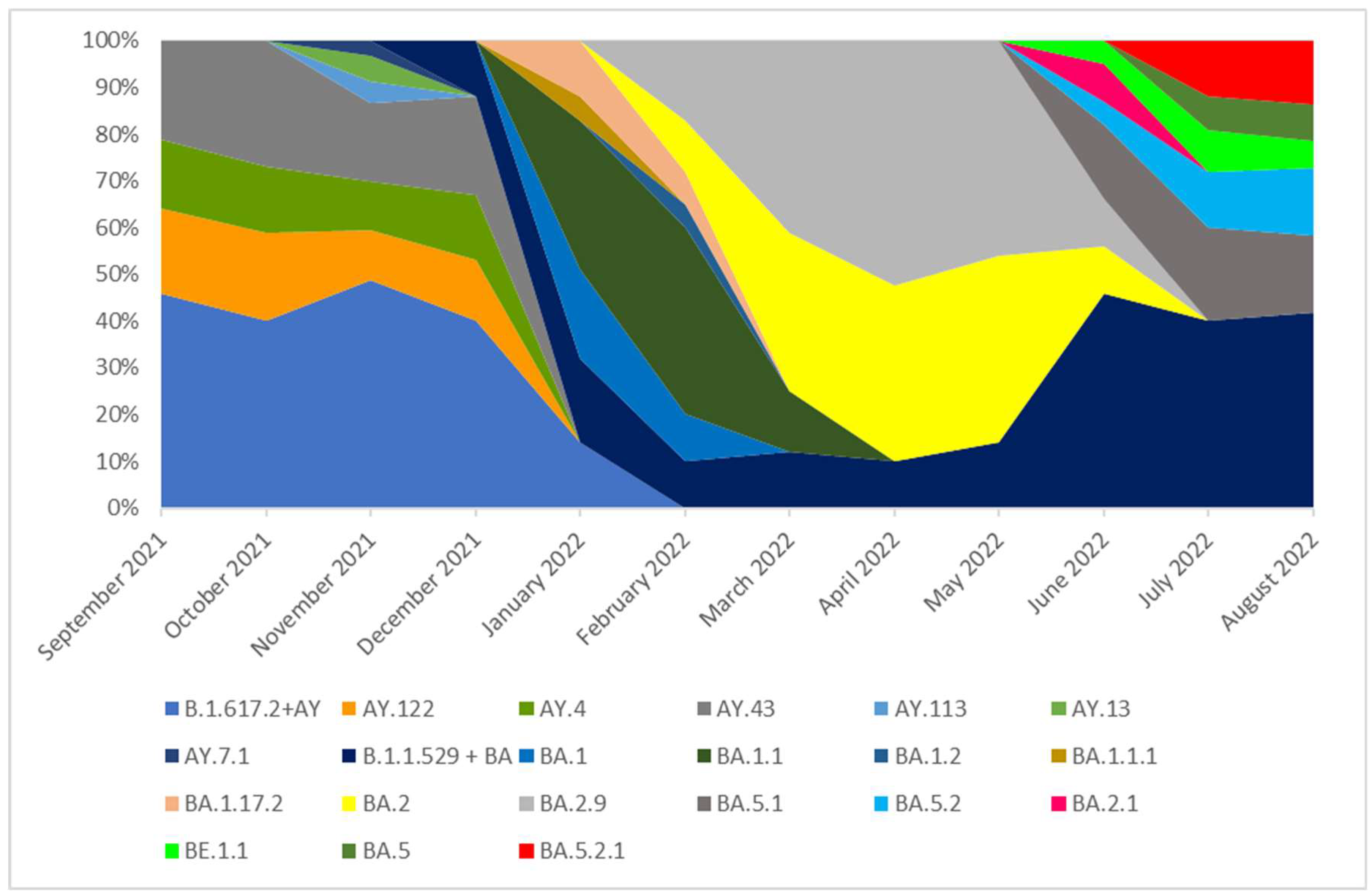
Figure 5.
The percentage distribution of SARS-CoV-2 variants isolated in Slovakia from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to December 2021, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: AY.122, AY.126, AY.4, AY.43, AY.9.2, AY.43.9. None of these lineages was dominant and they were noted with similar frequency (from 5 to 9%). The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Slovakia in the next analyzed months. In February 2022, the dominated lineage was BA.1.1 (36%). In March this lineage was superseded by BA.2 one (50%). From March to May 2022 the dominant variant was BA.2. The BA.2.9 variant also occurred with high frequency. From June to August 2022 none of lineages was dominant and they were noted with similar frequency.
Figure 5.
The percentage distribution of SARS-CoV-2 variants isolated in Slovakia from 1th September 2021 to 31th August 2022 based on whole-genome sequencing. In the period from September 2021 to December 2021, the Delta (B.1.617.2+ AY) variant was dominant and lineages which occurred with these months in a frequency of >5% were: AY.122, AY.126, AY.4, AY.43, AY.9.2, AY.43.9. None of these lineages was dominant and they were noted with similar frequency (from 5 to 9%). The remaining lineages of delta variant, occurring with a frequency of <5%, are marked as B.1.617.2 + AY in the chart. In January 2022, the B.1.617.2 + AY variant was replaced by the B.1.1.529 + BA which completely dominated lineages circulating in Slovakia in the next analyzed months. In February 2022, the dominated lineage was BA.1.1 (36%). In March this lineage was superseded by BA.2 one (50%). From March to May 2022 the dominant variant was BA.2. The BA.2.9 variant also occurred with high frequency. From June to August 2022 none of lineages was dominant and they were noted with similar frequency.
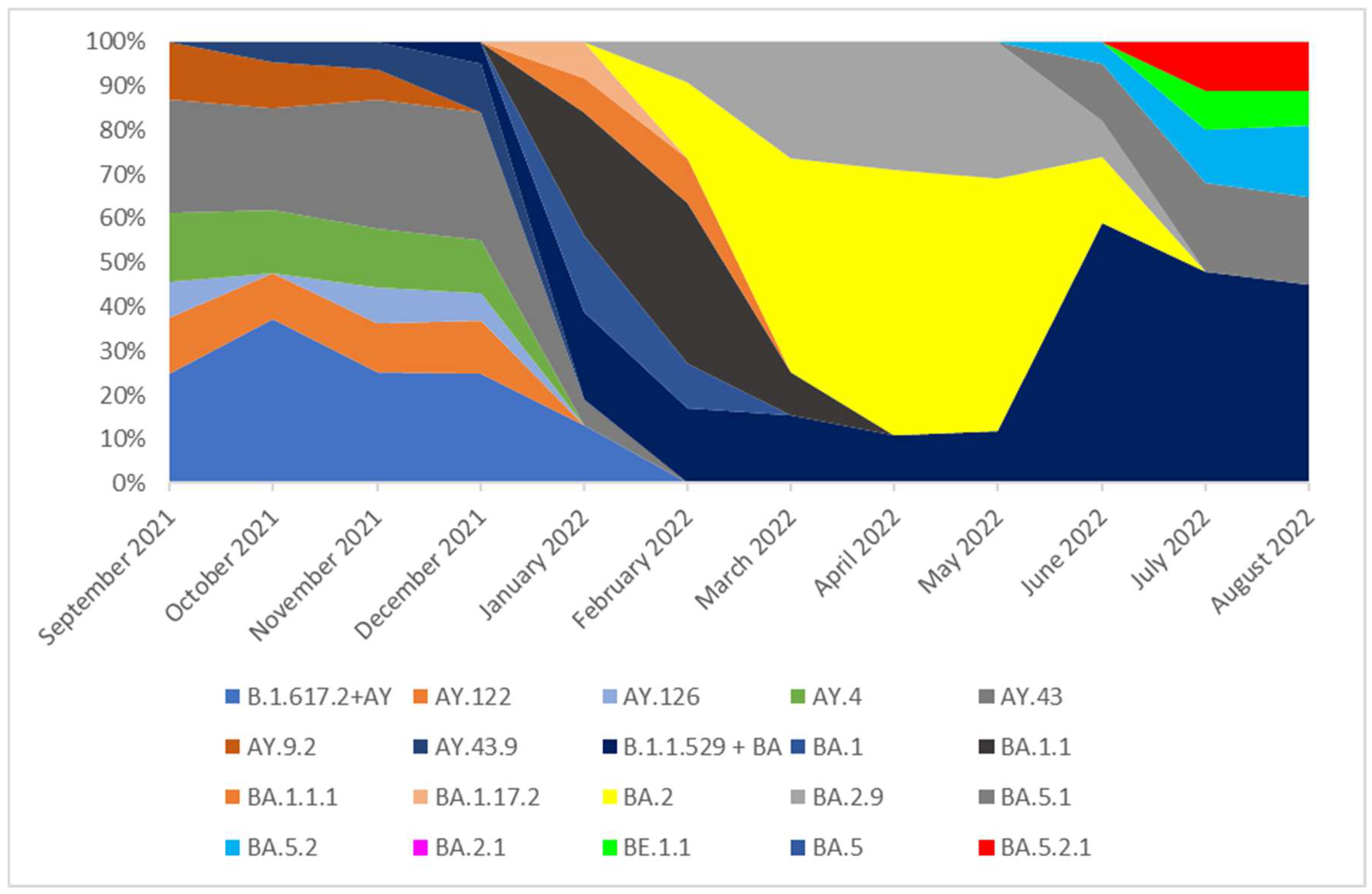
Figure 6.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Southern Poland. The branches corresponding to the five primary clades are highlighted in blue, dark green, light green, orange and red. Virus variants were classified using the Nextstrain lineage systems. The variant 21J (Delta) has definitely dominated in Poland since August 2021, while the 21M (Omicron) variant began to displace the 21J in January 2022. Initially, it was the BA.1 variant gradually replaced by a BA.2 variant and then a BA.5 variant.
Figure 6.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Southern Poland. The branches corresponding to the five primary clades are highlighted in blue, dark green, light green, orange and red. Virus variants were classified using the Nextstrain lineage systems. The variant 21J (Delta) has definitely dominated in Poland since August 2021, while the 21M (Omicron) variant began to displace the 21J in January 2022. Initially, it was the BA.1 variant gradually replaced by a BA.2 variant and then a BA.5 variant.
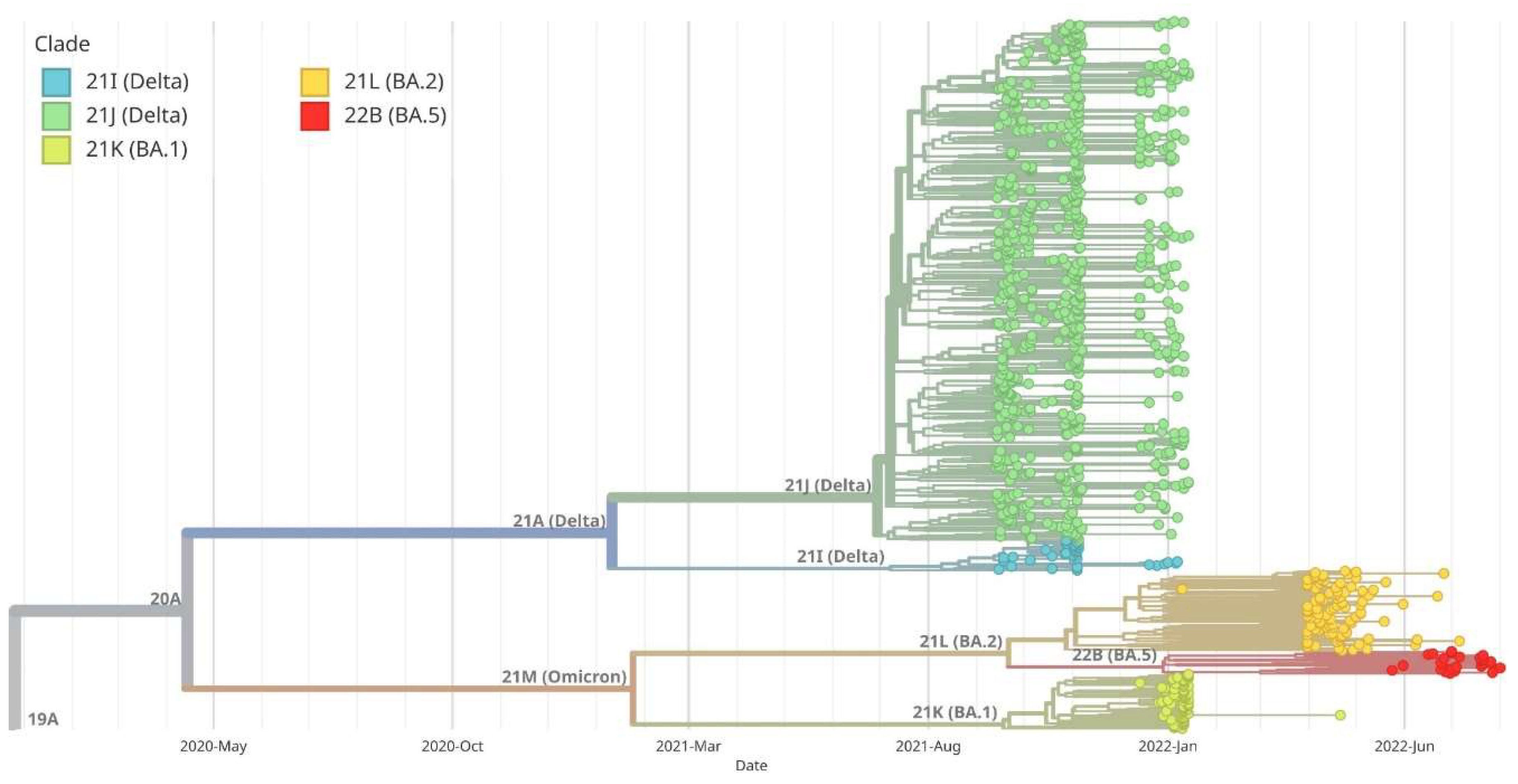
Figure 7.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia within the broader context of Poland. Variants corresponding to Silesia are marked in green, while those corresponding to the rest of the country are marked in red. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared earlier in Silesia than in the rest of the country, while BA.1 variant (Omicron) began to appear at the same time both in Silesia and throughout Poland. On the other hand, BA.2 variant appeared earlier in other regions of Poland, and later in Silesia. The BA.5 variant dominated Silesia earlier than the rest of Poland.
Figure 7.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia within the broader context of Poland. Variants corresponding to Silesia are marked in green, while those corresponding to the rest of the country are marked in red. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared earlier in Silesia than in the rest of the country, while BA.1 variant (Omicron) began to appear at the same time both in Silesia and throughout Poland. On the other hand, BA.2 variant appeared earlier in other regions of Poland, and later in Silesia. The BA.5 variant dominated Silesia earlier than the rest of Poland.
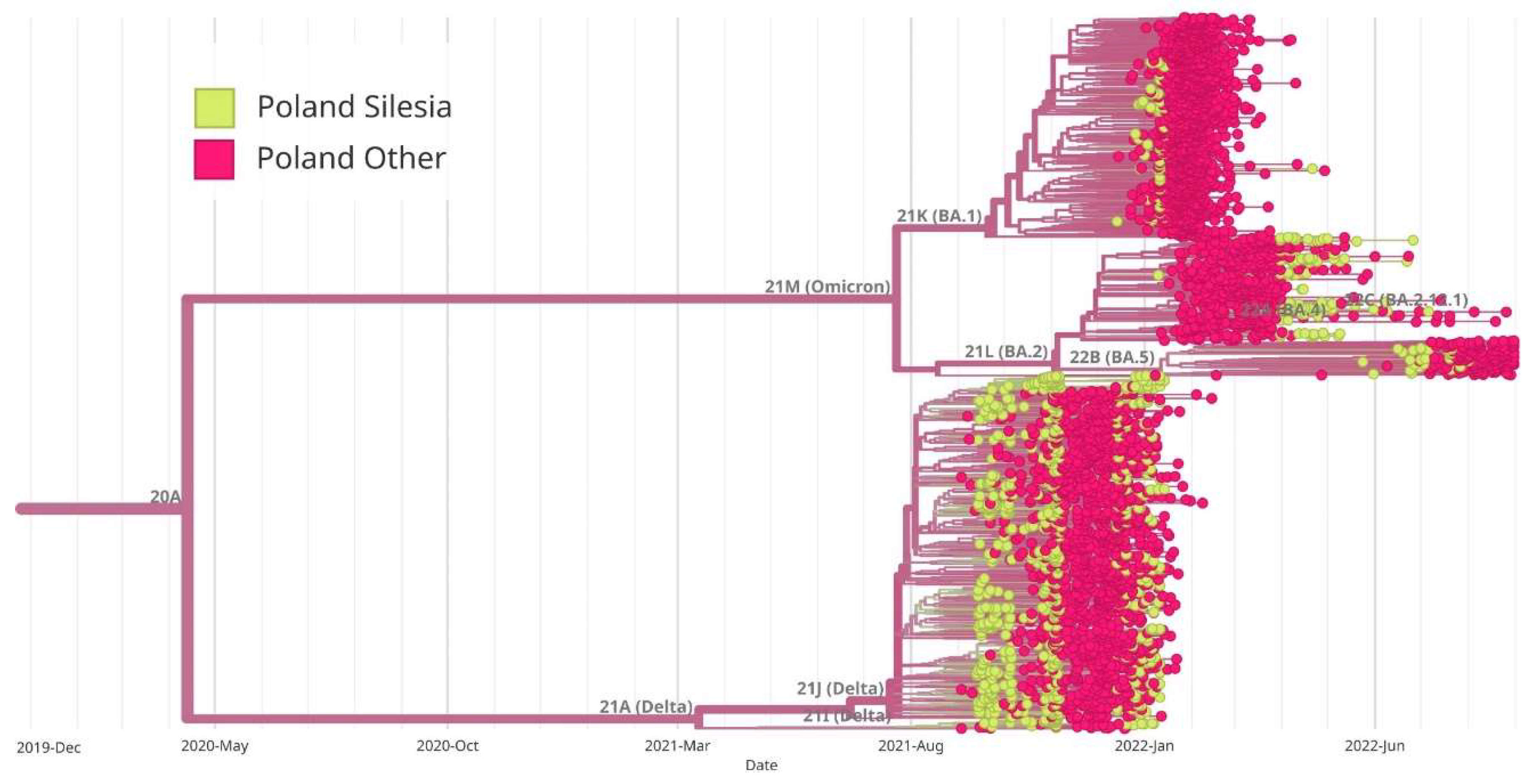
Figure 8.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia comparing to Chech Republic. Variants corresponding to Silesia are marked in yellow, while those corresponding to the Czech Republic are marked in green. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared earlier in Silesia than in the Czech Republic. BA.1 and BA.5 variants began to appear at the same time both in Silesia and Czech Republic, while BA.2 variant (Omicron) dominated the Czech Republic earlier than Silesia.
Figure 8.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia comparing to Chech Republic. Variants corresponding to Silesia are marked in yellow, while those corresponding to the Czech Republic are marked in green. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared earlier in Silesia than in the Czech Republic. BA.1 and BA.5 variants began to appear at the same time both in Silesia and Czech Republic, while BA.2 variant (Omicron) dominated the Czech Republic earlier than Silesia.
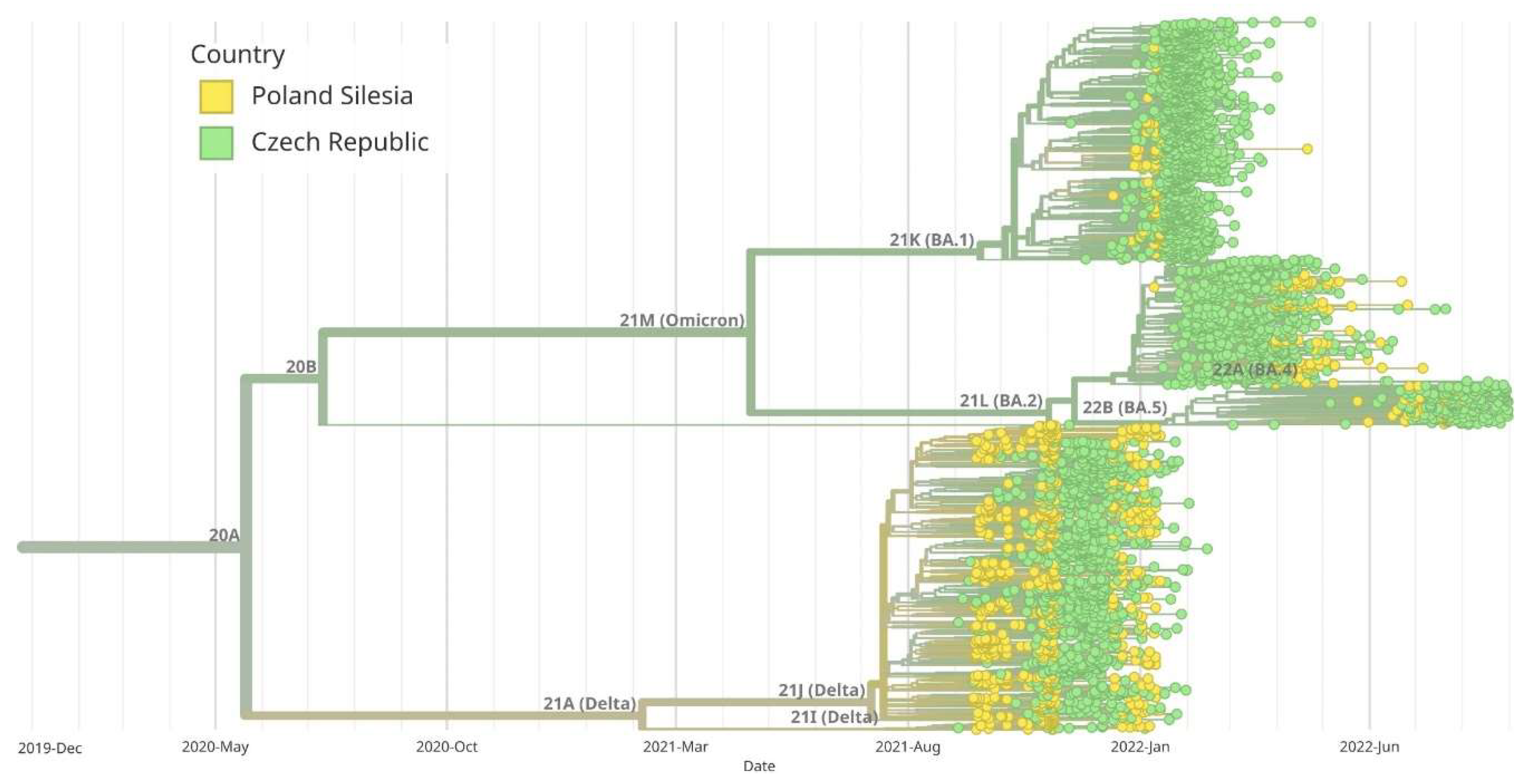
Figure 9.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia comparing to Slovakia. Variants corresponding to Silesia are marked in green, while those corresponding to the Slovakia are marked in yellow. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared at the same time in Silesia and Slovakia. BA.1 and BA.5 variants began to appear earlier in Silesia than in Slovakia, while BA.2 variant dominated the Slovakia earlier than Silesia.
Figure 9.
Maximum likelihood (ML) phylogenetic analysis of SARS-CoV-2 complete genome sequences in Silesia comparing to Slovakia. Variants corresponding to Silesia are marked in green, while those corresponding to the Slovakia are marked in yellow. Virus variants were classified using the Nextstrain lineage systems. Variant 21A (Delta) appeared at the same time in Silesia and Slovakia. BA.1 and BA.5 variants began to appear earlier in Silesia than in Slovakia, while BA.2 variant dominated the Slovakia earlier than Silesia.
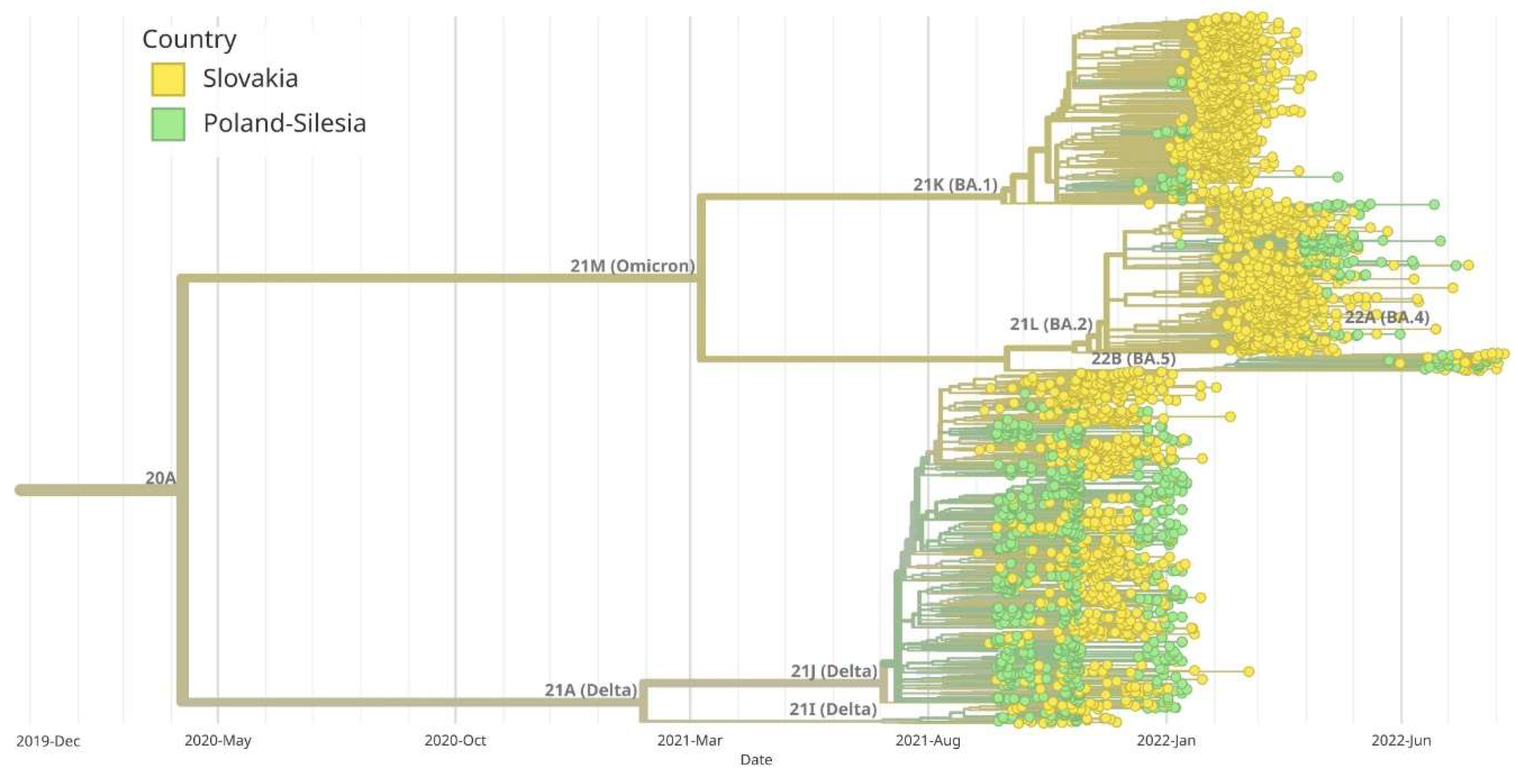
Figure 10.
Daily number of SARS-CoV-2 positive cases per 100 000 inhabitants.
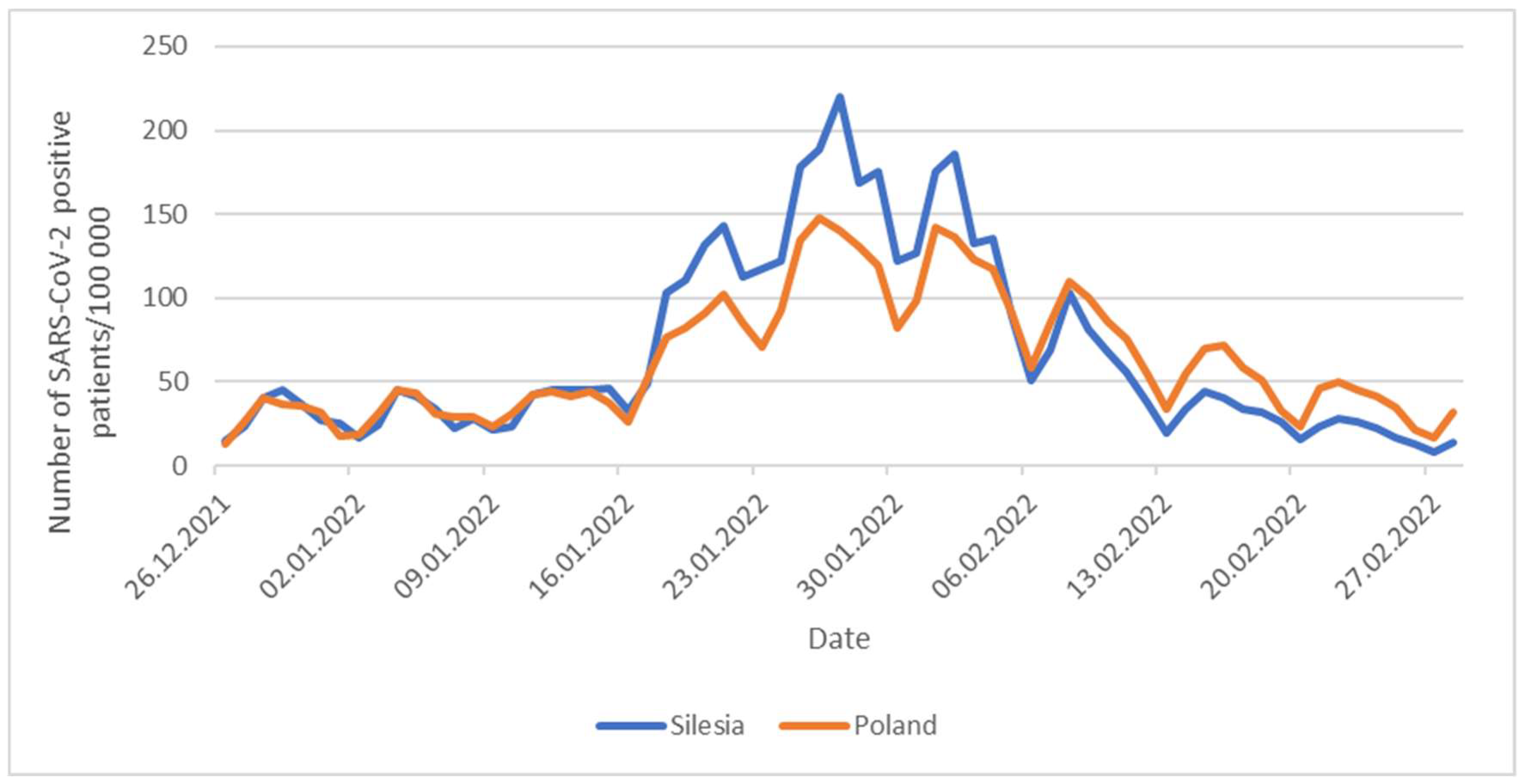
Figure 11.
Weekly percentage of SARS-CoV-2 reinfected patients.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



