
Submitted:
09 June 2025
Posted:
10 June 2025
You are already at the latest version
Abstract
GroE is a chaperonin folding system consisting of GroEL (Cpn60, a 60 kDa chaperonin), and the smaller co-chaperonin GroES (Cpn10). Many “client” proteins require GroE to fold properly, including several that are essential for cell viability. Unsurprisingly then, GroE is found in nearly all bacteria and eukaryotes. Mollicutes are the only microorganisms that lack GroE in almost all cases. Only two clades of Mollicutes have retained the ancestral GroE system, or perhaps reacquired one; these exceptions include the family Acholeplasmataceae (consisting of the genera Acholeplasma and Phytoplasma). The role of GroEL in these “exceptional” Mollicutes is a source of speculation, given how many non-canonical “moonlighting” roles have been ascribed to this protein. GroEL has been suggested to play a role in pathogenesis in plant and animal pathogenic Mollicutes, by binding to host cells and facilitating invasion. However, in one further layer of exception, the phytopathogenic taxon ‘Candidatus Phytoplasma pruni’ (ribosomal group 16SrIII), was reported to lack a GroE system. This study confirms the lack of a functional GroE system in 16SrIII by providing two new, high quality, non-fragmented genome assemblies, as well as a thorough survey of other 16SrIII genomes for genes encoding GroEL/GroES, including those that may not resemble phytoplasma groEL (ie. acquired by horizontal gene transfer, HGT). We discuss the implications of a clearly phytopathogenic, invasive group of Mollicutes that nevertheless lacks GroE, in light of the presumed role of GroEL for these microorganisms. We determined that three groups of genomes of 16SrIII contain short, non-functional groEL pseudogenes, while most of the reported genomes lack any semblance of a GroE system. Examination of the new assemblies allowed us to rule out HGT as a means of GroE acquisition.
Keywords:
pseudogenes
; phytoplasma
; Acholeplasma
; evolution
; genome reduction
1. Introduction
Mollicutes, so named because of their lack of a cell wall (Latin mollis, soft, + cutis, skin), are a major group within the “strong-walled” Gram-positive phylum Firmicutes. This class of bacteria is believed to have evolved around 600 million years ago [1] through a reductive process whereby the microorganisms reduced the metabolic capacities and structural features encoded on their genomes as they adopted an obligately intracellular lifestyle. Extant Mollicutes are universally dependent on eukaryotic host cells to complement their limited metabolic functions, although a few, notably Mycoplasmas, can be cultured in complex artificial medium and are a common contaminant of eukaryotic cell cultures [2]. Mollicutes are commonly associated with pathogenesis in plants (Spiroplasma, Phytoplasma) [3,4] and animals (Mycoplasma) [5]. Due to their reduced genomes, Mollicutes are among the bacteria with the smallest genome size - M. genitalium possesses one of the smallest known bacterial genomes at 580 kbp [6]. Their lack of a cell wall results in a generally pleomorphic shape, with the exception of the helical Spiroplasmas [2]. Other unique features of Mollicute genomes include a low G/C content, and the use of the codon UGA for tryptophan rather than a stop codon as in other bacteria [7]. The single exception to the latter characteristic is the Family Acholeplasmataceae, which consists of the two genera, Acholeplasma and Phytoplasma. Since these taxa use UGA as a stop codon, which is characteristic of non-Mollicute bacteria, they are presumed to be more closely related to the ancestral taxon [1,8].
The protein chaperonin system (GroE) consists of the two proteins GroEL (synonym Cpn60) and GroES (synonym Cpn10) [9]. This system is canonically involved in the prevention of aggregate formation and proper folding of cellular proteins as a part of the basic protein biosynthetic machinery, and for this reason these two genes are anticipated to be present in all prokaryotic and eukaryotic cells [10]. While only a subset of cellular proteins require the GroE system for their tertiary structure formation, these include proteins that are essential for cell viability, so that a loss of this system is thought to result in a cell that is nonviable [11]. Mollicutes, however, are again unique in that they are the only cells known generally to lack a GroE system, although it is present in a subset of these microorganisms [12]. Because the GroE system is an ancestral property (i.e., it is present in all non-Mollicute bacteria), it is thought that Mollicutes lost these genes during genome reduction [11,12]. The retention, or re-acquisition, of GroE is polyphyletic, with two groups of Mollicutes featuring these genes: a smaller clade consisting of Mycoplasma gallisepticum, M. genitalium, and M. pneumoniae, and a much larger group including nearly all of the Acholeplasmas and Phytoplasmas [11]. In addition, there are at least three clear instances of the acquisition of groEL by horizontal gene transfer (HGT) in Mollicutes: the gene of Mycoplasma penetrans is more closely related to that of Helicobacter pylori than other Mycoplasmas [12]; and two species of Spiroplasma, S. turonicium and S. kunkelii, appear to have acquired groEL genes by HGT [11].
These observations have led to speculation regarding the role of these proteins within these intracellular microorganisms, particularly since a very wide array of non-canonical “moonlighting” roles has been ascribed to GroEL in prokaryotic and eukaryotic cells [13]. Notably, Clark and Tillier [12] proposed that, among Mollicutes that encode GroEL in their genomes, the protein may play a role in pathogenesis by acting as an adhesin/invasin, suggesting that the re-acquisition or retention of the GroE chaperonin system may be associated with virulence. This notion is supported by a wide variety of observations that non-Mollicute bacteria can localize GroEL to the cell surface, or even secrete the protein into the extracellular space, clearly indicating a non-canonical, virulence-associated role for the protein in certain bacteria. For example, H. pylori localizes its GroEL to the cell surface, where it interacts with Toll-like receptors to induce interleukin-8 production in epithelial cells, indicating a role in gastric inflammation [14]. In addition, Legionella pneumophila, a non-Mollicute obligate intracellular pathogen, also displays GroEL at the cell surface, and intracellular forms of the pathogen are enriched for GroEL at the cell surface; moreover, L. pneumophila GroEL-coated latex beads are internalized by HeLa cells, where they are maintained as endosomes, mimicking an infective status [15,16]. Brucella abortus, another intracellular pathogen, uses its GroEL to bind to host prion protein and facilitate cell invasion [17]. Other non-Mollicute intracellular pathogens also use the GroEL encoded by one of the paralogous genes to gain access to their host cells and trigger pathogenesis, such as Chlamydia pneumoniae [18] and Mycobacterium tuberculosis [19]. These observations, along with many others in different pathosystems, provide strong reasons to consider carefully the role of GroEL/Cpn60 and the GroE system in pathogenic Mollicutes that have retained or re-acquired these genes.
Phytoplasmas, plant pathogenic microorganisms that are transferred between host plants through an insect vector [20], are among the two groups of Mollicutes that are typically considered to have retained or re-acquired a GroE system [11]. However, an exception to this was noted when the genomes of four members of the ribosomal group 16SrIII, classified as ‘Candidatus Phytoplasma pruni’ were sequenced in 2012 [21]. The genomes of phytoplasmas causing diseases known as Italian clover phyllody, Poinsettia branch-inducing phytoplasma disease, milkweed yellows, and Vaccinium witches’ broom were notably lacking a GroE chaperonin system. The lack of groEL genes in a clearly pathogenic branch of the phytoplasmas calls into question the role that GroEL may play in the pathogenesis caused by these microorganisms. However, all of these genomes are highly fragmented, with at least 150 contigs present in each. Phytoplasma genomes have proven to be difficult to sequence prior to the introduction of long-read sequencing technologies, since the inability to culture them means that the genomes are always metagenome assembled genomes. Despite technological developments for sample preparation including antibody-based enrichment [22], many phytoplasma genomes deposited in public repositories remain fragmented and incomplete. This hinders the search for groEL genes in phytoplasma genomes and can mask the possibility of groEL gene acquisition by HGT. Thus the reported lack of a GroE system in this singular group of phytoplasmas remains an unresolved question.
We address this issue by examining all reported genomes of ‘Candidatus Phytoplasma pruni’ for the presence of groEL/groES genes or pseudogenes, and provide two new high-quality genome assemblies for 16SrIII phytoplasmas in subgroups A and F. We report the pseudogenization of groEL in some of the reported 16SrIII genomes and a complete lack of groES in all of these genomes, and have confirmed through analysis of all reported high-quality phytoplasma genomes that the 16SrIII group constitutes the only phytoplasmas that truly lack a chaperonin system. This observation casts doubt on the previously speculated moonlighting role of GroEL in the attachment and invasion of host cells and suggests that the protein may play a canonical role in the folding of client proteins in these microorganisms.
2. Materials and Methods
2.1. Strain Sources, DNA Isolation, and Genome Sequencing
Syringa (lilac) infected with ‘Ca. P. pruni’ (16SrIII) strain 2A1 was maintained at the Centre for Plant Health, Canadian Food Inspection Agency, North Saanich, British Columbia, Canada. Symptomatic milkweed (Asclepias sp.) was observed at the Canadensis Botanical Garden, Central Experimental Farm of Agriculture and Agri-Food Canada, Ottawa, Ontario, Canada (https://maps.app.goo.gl/urU9SzTiAYX2Zba59). Phytoplasma infection of the milkweed tissue was confirmed using a qPCR assay targeting 16S rRNA genes as described [23]. The phytoplasma was typed using nested PCR amplification of 16S genes (F2nR2) [24,25], and cloned amplicons were sequenced by Sanger (Eurofins). The RFLP type was determined for six clones using the iPhyclassifier [26]. For both plant types, leaf tissue (midrib and petiole) was cut from infected leaves and homogenized in liquid nitrogen using a mortar and pestle that was pre-treated with DNAaway (Thermofisher). DNA was extracted from the plant material using a Wizard High Molecular Weight DNA extraction kit (Promega). DNA yield was determined using a Qubit fluorometer (ThermoFisher), and molecular weight was assessed using agarose gel electrophoresis. This sample was named MYp-CanS4.
Bacterial DNA was enriched from the leaf DNA extract using a NEBNext Microbiome Enrichment Kit (New England Biolabs) as previously described [27]. Both short-read Illumina and long-read Nanopore sequencing technologies were used for genome sequencing. For Illumina, 0.5 μg of enriched DNA was used as the input material for each DNA library preparation. The sequencing libraries were generated using a NEBnext UltraII FS DNA lib prep kit E7805 >100 ng, according to the manufacturer’s instructions, with the following specifications: 10 min fragmentation, size selection using 30 µl / 10 µl beads for 200-475 bp, 5 cycles of amplification, and use of dual index barcodes using NEBnext Multiplex Oligos for Illumina (NEB, USA). The DNA libraries were sequenced on an Illumina NovaSeq 6000 platform (Illumina, San Diego, USA), and 150 bp paired-end reads were generated. For Oxford Nanopore Technology (ONT) sequencing, 0.12 µg for bacterial-enriched lilac and 0.525 µg for bacterial-enriched milkweed DNA were used as the input material for the library preparations. The sequencing library was prepared using an ONT Ligation Sequencing Kit (LSK-SQK114), followed by sequencing on a MinION Mk1C device using an R10.4.1 flowcell (ONT, Oxford, UK).
Illumina reads were quality trimmed using trimmomatic [28], by removing bases from the beginning of reads with a quality score below 3, from the end of reads with a quality score below 20, and cutting any reads with a quality score below 15 over a sliding window of 4 bases. Reads shorter than 36 bases after trimming were removed. The Illumina reads from the lilac-2A1 sample were then mapped (without merging, to minimize reads loss) using a reference database prepared from ‘Ca. P. pruni’ strain PR2021 (GCA_029746895) with bowtie2 [29]. The MYp-CanS4 Illumina reads were mapped using bwa [30] with a reference database prepared from the previously reported milkweed yellows genome (GCA_000309485). Nanopore reads were not mapped but were filtered to exclude reads under 1000 bp using filtlong ( https://github.com/rrwick/Filtlong). The top 90% of reads were retained, based on a composite quality score reflecting the average base quality and read length using filtlong. Illumina and nanopore reads were co-assembled using unicycler [31]. Assemblies were examined for the possible presence of plasmids using plasmer [32], and assembly completeness was assessed using Busco [33] with the default prokaryotic reference database (bacteria_odb12). Assembled genomes were annotated locally using bakta [34], as well as by NCBI using the Prokaryotic Genome Annotation Pipeline (PGAP) [35].
A separate hybrid assembly was prepared for the MYp-CanS4 sample by first removing host reads (including chromosomes, mitochondria, and chloroplast) using bmtagger [36]. Chromosomal sequences for Asclepias syriaca were downloaded from milkweedbase.org, while chloroplast (NC_022432.1) and mitochondrial (NC_022796.1) genomes were downloaded from NCBI. Nanopore reads were filtered to exclude those <1000 bp using filtlong, then mapped to remove host reads using minimap2 [37]. These reads were then used to generate a unicycler Illumina-nanopore co-assembly from all non-host reads.
2.2. Examination of Previously Reported Phytoplasma Genomes
To retrieve all phytoplasma genomes from GenBank, the NCBI Genomes database (https://www.ncbi.nlm.nih.gov/datasets/genome/) was queried using the search term, “phytoplasma”. This search (2025/01/01) retrieved 271 genomes, from which a single example of each unique taxID was selected. In cases where multiple taxID were represented, the genome with the highest completeness score reported was selected. Genomes with a reported CheckM completeness score below 90% were excluded from analysis. This resulted in 65 phytoplasma genomes in the final dataset (Table S1).
Genomes corresponding specifically to group 16SrIII were downloaded from NCBI using the information provided by Fernandez et al. [38] (Table 1). This ensured that no currently reported genomes within this group were missed in our analysis.
Genes annotated as groEL, groES, and 16S rRNA were selected from each genome outside of group 16SrIII. In addition, taxonomic markers secY, secA, tuf, and nusA [39] were retrieved from Peanut Witches’ Broom phytoplasma (PnWB; GCA_000364425.1) and from selected 16SrIII genomes.

Table 1.
‘Candidatus Phytoplasma pruni’ group 16SrIII genomes included in the analysis reported in this work. This table is based on and modified from Fernandez et al. 2024 [38].
Table 1.
‘Candidatus Phytoplasma pruni’ group 16SrIII genomes included in the analysis reported in this work. This table is based on and modified from Fernandez et al. 2024 [38].
| Phytoplasma | strain | 16SrIII- subgroup | Genome level | Host | location | accession | genome size, kb | Reference |
| Cicuta Witches broom | CicWB | 16SrIII-J | contig (16) | Conium maculatum | Argentina | GCA_035853675.1 | 758 | [38] |
| Phytoplasma Vc33 | Vc33 | 16SrIII-J | contig (36) | Catharanthus roseus | Chile | GCA_001623385.2 | 687 | [40] |
| China tree decline | ChTDIII | 16SrIII-B | contig (67) | Melia azedarach | Argentina | GCA_013391955.1 | 791 | [41] |
| Italian clover phyllody | MA | 16SrIII-B | contig (197) | Catharanthus roseus | Italy | GCA_000300695.1 | 597 | [21] |
| Poinsettia branch-inducing | JR | 16SrIII-A | contig (185) | Euphorbia pulcherrima | USA |
GCA_000309465.1 |
631 | [21] |
| Milkweed Yellows Phytoplasma | MW | 16SrIII-F | contig (158) | Catharanthus roseus | Canada | GCA_000309485 | 584 | [21] |
| Vaccinium Witches’ Broom | VAC | 16SrIII-F | contig (272) | Vaccinium myrtillus | Italy |
GCA_000309405.1 |
648 | [21] |
| ‘Candidatus Phytoplasma pruni’ | PR2021 | 16SrIII-A | chromosome (1) | Euphorbia pulcherrima | Taiwan |
GCA_029746895.1 |
710 | [42] |
| ‘Candidatus Phytoplasma pruni’ | CX | 16SrIII-A | contig (46) | Catharanthus roseus | USA | GCA_001277135.1 | 599 | [43] |
| Milkweed Yellows Phytoplasma | MYp- CanS4 | 16SrIII-F | contig (7) | Asclepias syriaca | Canada | GCA_050286905.1 | 694 | this work |
| ‘Candidatus Phytoplasma pruni’ | 2A1 | 16SrIII-A | contig (2) | lilac | Canada | GCA_033391615.1 | 625 | this work |
2.3. Examination of 16SrIII Genomes for Sequences Related to groEL and groES
Each of the downloaded genomes was used to prepare a local BLAST database. The 16SrIII genome 2A1 was queried using tBLASTn, using a GroEL amino acid sequence from ‘Ca. P. asteris’ (strain AYWB; cpnDB ID b8392; 16SrI) as input. This retrieved a short DNA sequence (165 bp) that encoded a predicted protein of 55 amino acids with a BLASTp match to phytoplasma GroEL. This sequence was then used to query all other 16SrIII genomes using BLASTn. This resulted in significant matches in some genomes, but only short, nonspecific matches (under 20 bp) in others. The predicted amino acid sequences of each of the matching regions was compared to the GenBank database using BLASTp, and those with a strongly significant match to GroEL (E values between 10-5 and 10-50) were considered to be potential pseudogenes. A similar approach was used for groES.
2.4. Examination of the Gene Neighborhood of groEL in Group 16SrII and 16SrIII Phytoplasmas
The annotated genomes of ‘Ca. P. pruni’ strain CX (Ga0100078) and ‘Ca. P. aurantifolia strain PnWB (Ga0248296) at the Genomes Online Database (https://gold.jgi.doe.gov/) were used to examine the gene neighborhood of the intact GroE system in PnWB compared to the putative pseudogene identified in strain CX. Genes immediately downstream of the annotated groEL gene or gene fragment from each strain were downloaded and used to identify the corresponding genes in each of the group III phytoplasmas that were suspected to harbor groEL pseudogenes. The locations and annotations of the genes corresponding to the immediate gene neighbors of the GroE system in PnWB were noted in each strain, and intergenic distances calculated. Both gene and predicted amino acid sequences of these genes were downloaded and used for sequence similarity determination.
3. Results and Discussion
3.1. Genome Sequencing of ‘Ca. P. pruni’ Strains 2A1 and MYp-CanS4
The genome sequence of strain 2A1 was 625 kb in length (Table 1), with a G/C content of 27.32%. The genome consisted of two scaffolds with lengths of 336 kb and 289 kb. 594 protein-coding genes were predicted in the genome, 437 of which had function prediction. In addition, the genome contained two copies of 16S rRNA, as is typical for phytoplasmas [44]. Both genes typed at the iPhyclassifier as 16SrIII-A (F=1.0), indicating a lack of the 16S rRNA gene heterogeneity that has been observed in some phytoplasmas in the 16SrIII group [45].
Symptomatic milkweed at the Canadensis Botanical Garden was confirmed to be infected by phytoplasma using qPCR targeting 16S rRNA genes. The 16S rRNA genes were amplified by PCR, sequenced, and examined using RFLP analysis. This showed that strain MYp-CanS4 typed as 16SrIII-F (F=1.0), again with no evidence of gene heterogeneity. This is consistent with previous RFLP typing results of milkweed yellows phytoplasma [21]. The geographic location of the MYp detected in this study is consistent with the original report of MYp in Tichbourne, Ontario in the early 1990s [46], indicating that this phytoplasma has been circulating in Eastern Canada for many years.
The total length of the MYp-CanS4 genome was 694 kb, which is considerably longer than the 584 kb that was previously reported (Table 1). The genome was assembled into 7 contigs with an N50 of 388 kb, which is also an improvement over the N50 of 8 kb in the previously reported MYp genome [21]. The MYp-CanS4 genome, like all phytoplasma genomes, featured a low G/C percentage (26.7%). Annotation of the MYp-CanS4 genome using PGAP revealed the presence of 718 genes in total, including 678 coding sequences, 6 rRNA genes (2 each of 5S, 16S, 23S), and 24 pseudogenes. Both copies of the 16S rRNA-encoding genes typed at the iPhyClassifier as 16SrIII-F, consistent with the PCR results and with the known classification of MYp as a 16SrIII-F phytoplasma [47]. A plasmid of 4.4 kb was predicted by plasmer (contig 6). In accordance with this prediction, BLAST analysis of this contig showed strong similarity (93% sequence identity, e = 0.0) to the annotated plasmid of strain PR2021, pPR2021. Examination of the completeness of all the reported 16SrIII genomes from Table 1 using Busco revealed that the highest completeness score, 67.2%, was observed for the genomes PR2021 (the only chromosome-level assembly), MYp-CanS4, 2A1, and ChTDIII. In contrast, the previously reported MYp assembly had a completeness score of 66.4%, and the lowest score (59.5%) was observed for Vc33. These relatively low Busco completeness scores are due to the reduced genomes of phytoplasmas, which are missing some of the reference genes used by Busco to determine completeness.
3.2. Examination of Phytoplasma Genomes for GroE Chaperonin System
In total, 65 unique taxonomic IDs corresponding to a wide variety of phytoplasmas were examined (Table S1). Phylogenetic analysis of the 16S rRNA genes from these phytoplasmas provided results that were consistent with previous 16S gene-based analysis [11,48], with 3 basal clades represented. One major group consisted mostly of 16Sr groups I and XII, and this group was more closely related to the Acholeplasma taxa compared to the other phytoplasma strains. A second group consisted of a diverse range of phytoplasma taxa, with a complex branching pattern. The third major clade consisted of two main branches, including the 16SrIII group (‘Ca. P. pruni’), and a second branch consisting primarily of group 16SrII strains, along with ‘Ca. P. melaleucae’ (16Sr XXV-A) (Figure 1). The 16SrIII group was most closely related to the 16SrII group, as reported previously [48].
All but 6 of the genomes examined (59) had intact GroE systems, with both groEL and groES found in each genome (Table S2). All of the genomes that lacked a GroE system belonged to the 16SrIII group. The genomes with intact groEL/groES included a wide taxonomic variety, with 31 different phytoplasma species and 16 distinct 16Sr groups represented (Table S2). This analysis indicates that, among all high-quality phytoplasma genomes that have been sequenced to date, ‘Ca. P. pruni’ (16SrIII) is unique in lacking a GroE system. All other phytoplasma taxonomic groups possess both groEL and groES, which are located next to each other in their respective genomes in all cases. While it was previously noted that four phytoplasma genomes from 16SrIII lack a GroE system [21], this observation can now be extended to 11 genomes. Furthermore, while the GroE system was known to be well represented within phytoplasma groups other than 16SrIII [49,50,51,52], it was previously not known if 16SrIII is indeed the only exception. The results shown in Table S1 provide strong evidence that the GroE system is very widely distributed in phytoplasmas other than 16SrIII. Phylogenetic analysis of full-length groEL sequences representing all of the phylogenetic diversity that is currently present in public databases (NCBI) is shown in Figure 2. This analysis demonstrates the very wide sequence divergence of this phylogenetic marker, even in strains that are relatively closely related, underscoring the utility of this marker for differentiation of phytoplasma strains outside of the 16SrIII group [52].
3.3. Pseudogenization of groEL in 16SrIII Phytoplasmas
tBLASTn analysis of the genomes of all group III phytoplasmas showed that, consistent with the initial report, most of the sequenced genomes from this group contained no coding sequences with any significant similarity to amino acid sequences of phytoplasma GroEL. However, 5 of the 11 genomes did contain nucleotide sequences encoding predicted amino acid sequences that were strikingly similar to GroEL from other phytoplasmas (Table 2). These amino acid sequences were short (50, 55, or 62 residues) and could not encode a functional protein. One of these putative pseudogenes, encoded on the genome of ‘Ca. P. pruni’ strain CX (GCA_001277135.1) was annotated as a pseudogene (labelled “hypothetical protein” by the Prokaryotic Genome Annotation Pipeline at GenBank) [35], while the others were identified by local custom BLAST databases. Alignment of these predicted amino acid sequences to the 546 amino acid sequence of GroEL from PnWB phytoplasma (16SrII) revealed that the gene fragments encoded amino acids near the carboxy terminus of the protein (Figure 3). Phylogenetic analysis of the 16SrIII phytoplasma strains based on the sequences of five taxonomic markers (secY; secA; nusA; tuf; rp) revealed that the three types of identified pseudogenes clustered consistently with the phylogenetic placement of the phytoplasmas containing them (Figure 4). The putative pseudogenes encoding these amino acids are considered to be unitary pseudogenes, in that there is no evidence of gene duplication in the genome, so that the loss of these genes is associated with a complete loss of function [55]. No evidence of nucleic acid sequences encoding amino acids with similarity to GroES was found in any of the genomes.
3.4. Conserved Synteny of GroE System in 16SrII and Pseudogenes of 16SrIII
To determine if the pseudogenes were orthologous by synteny, rather than simply a case of coincidental similarity [55], the gene neighborhood of the intact GroE system in a close phylogenetic neighbor of 16SrIII, ‘Ca. P. aurantifolia’ (PnWB; 16SrII) was examined and compared to the locations of the putative pseudogenes.
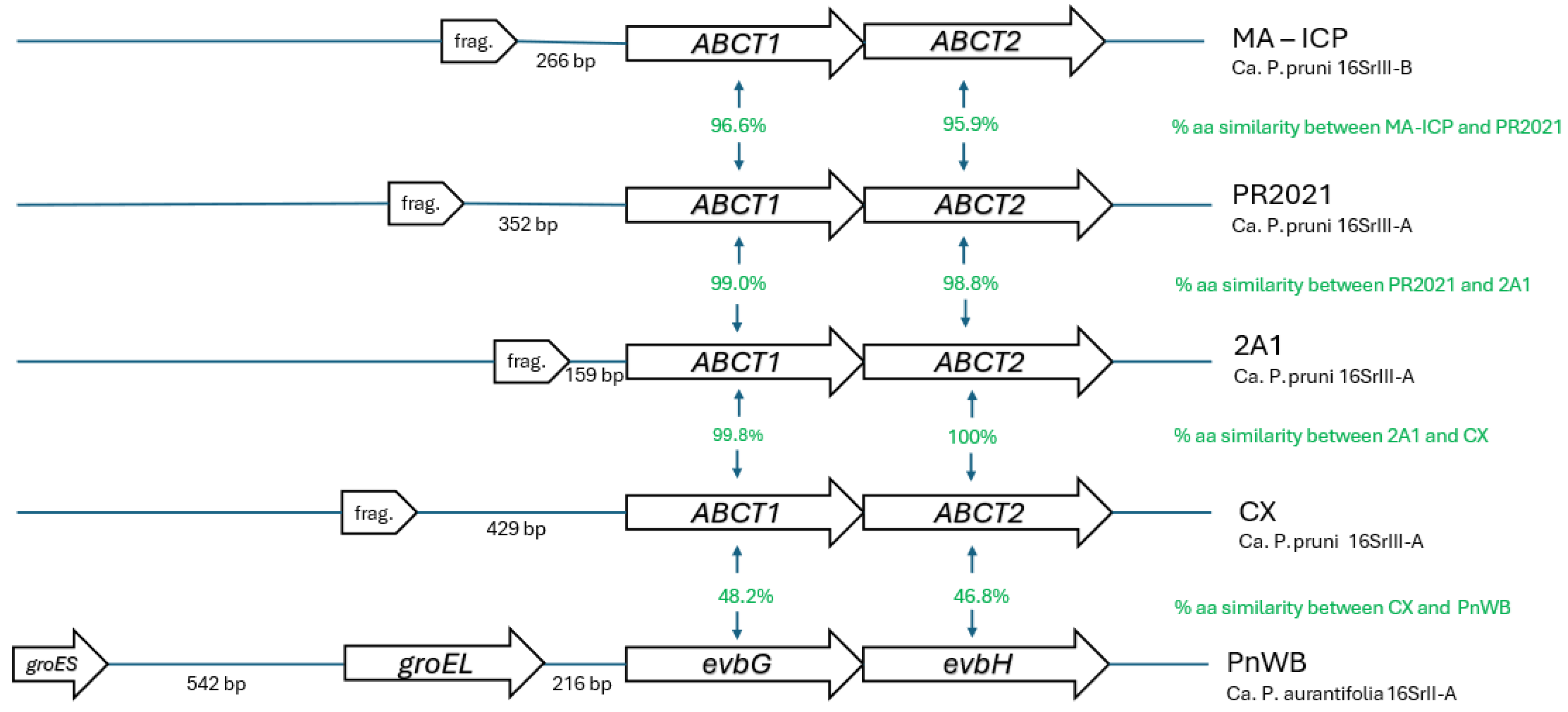
The GroE system (groEL/groES) in PnWB is located immediately adjacent to two genes that are annotated as “multidrug resistance ABC transporter ATP-binding and permease protein” (Figure 5). These genes are identified as evbH (contig 9: 24713-26554, reverse strand) and evbG (contig9: 26551-28284, reverse strand). evbG is located 216 nucleotides downstream of groEL (contig9: 28499-30136, reverse strand), while groES is located 542 nucleotides upstream of groEL (contig9: 30677-30961, reverse strand) (Table 2).
In the 16SrIII phytoplasmas that contain putative groEL pseudogenes, there are two genes located immediately next to each pseudogene (Figure 5). These protein-coding genes are annotated variously at GenBank as “multidrug resistance ABC transporter ATP-binding and permease protein” (strain PR2021), or, “ABC transporter ATP-binding protein” (strains 2A1, CX, MA-ICP). These genes encode proteins that are similar in size to PnWB evbG and evbH – 576/577 amino acids for evbG, and 593/613 amino acids for evbH (Table 2). In each 16SrIII genome, the putative pseudogene is located immediately beside (159-429 nucleotides) the gene that is similar in size to evbG of PnWB, suggesting a similar gene neighborhood of the putative pseudogene in 16SrIII compared to the intact groEL gene in PnWB. (Table 2; Figure 5).
Table 2.
Amino acid lengths and genome coordinates of groEL pseudogenes and genes immediately upstream in 16SrII and 16SrIII.
Table 2.
Amino acid lengths and genome coordinates of groEL pseudogenes and genes immediately upstream in 16SrII and 16SrIII.
| Phytoplasma | strain | 16SrIII-subgroup | groEL (pseudo)gene length, bp | Amino acids | Coordinates (contig:bases) | evbG ortholog length, bp | Amino acids | Coordinates (contig:bases) | groEL-evbG distance, bp | evbH ortholog length, bp | Amino acids | Coordinates (contig:bases) |
| Cicuta Witches broom | CicWB | 16SrIII-J | none | |||||||||
| Phytoplasma Vc33 | Vc33 | 16SrIII-J | none | |||||||||
| China tree decline | ChTDIII | 16SrIII-B | none | |||||||||
| Italian clover phyllody | MA | 16SrIII-B | 153 | 50 | 33:3851-4003 | 1728 | 576 | 33:1856-3586 | 266 | 1782 | 593 | 33:78-1859 |
| Poinsettia branch-inducing | JR | 16SrIII-A | 186 | 62 | 171:1963-2148 | 17031 | 5661 | 5:13448-15150 | ND1 | 1782 | 593 | 5:11670-13451 |
| Milkweed Yellows | MYp-CanS4 | 16SrIII-F | none | |||||||||
| Vaccinium Witches’ Broom | VAC | 16SrIII-F | none | |||||||||
| ‘Candidatus Phytoplasma pruni’ | PR2021 | 16SrIII-A | 186 | 62 | 686562-686747 | 1728 | 576 | 684481-686211 | 352 | 1782 | 593 | 682703-684484 |
| ‘Candidatus Phytoplasma pruni’ | CX | 16SrIII-A | 165 | 55 | 8:22632-22796 | 1728 | 576 | 8:20474-22204 | 429 | 1782 | 593 | 8:18696-20477 |
| ‘Candidatus Phytoplasma pruni’ | 2A1 | 16SrIII-A | 165 | 55 | 1:56157-56321 | 1728 | 576 | 1:56749-58479 | 159 | 1782 | 593 | 1:58476-60257 |
| Peanut witches' broom phytoplasma | PnWB | 16SrII-A | 1638 | 546 | 9:28499-30136 | 1731 | 577 | 9:26551-28284 | 216 | 1842 | 613 | 9:24713-26554 |
| 1minus strand - contig 5 length is 15150 bp. Therefore, the junction of evbG and groEL pseudogene is disrupted and intergenic distance cannot be calculated | ||||||||||||
To determine if these similar genes are likely orthologs, we compared the predicted amino acid sequence similarity of these genes in PnWB and the 16SrIII strains. Within the 16SrIII genomes, the two genes immediately adjacent to the putative groEL pseudogenes had amino acid similarity scores (by clustalw) of 96-99%, suggesting that they are orthologous genes within these strains (Figure 5, Table S2). The amino acid similarity scores of these 16SrIII genes to the PnWB evbG and evbH were 47% and 48% (Figure 5, Table S2). These scores are similar to the amino acid similarities of the orthologous taxonomic markers secY, secA, nusA, and tuf in the 16SrII and 16SrIII genomes, which ranged from 38-56% (Table S3). In contrast, when all other genes in the PnWB chromosome with annotations containing the phrase “ABC transporter ATP-binding” were compared to the genes immediately upstream of the putative groEL pseudogenes in 16SrIII, the amino acid similarity scores were much lower – 15-20% (data not shown). These amino acid sequence similarity comparisons suggest that the two genes encoding ABC transporters in 16SrIII are indeed orthologs of evbG and evbH in PnWB, confirming the syntenic relationship of groEL genes and pseudogenes in these closely related phytoplasmas.
The genomic region upstream of the groEL pseudogenes in 16SrIII strain CX contained no evidence of the shorter groES genes, and displayed a dearth of annotated genes in general compared to the corresponding gene region in PnWB. Only a few short hypothetical proteins are annotated in the 16SrIII genome, on the opposite strand of the groEL pseudogenes and evbG/evbH (Figure S1). The reason for this genomic "desert” is unclear, but this area seems to have been subjected to an ancient pseudogenization that resulted in the loss of the GroE system, with only a small vestige of the groEL gene remaining in this region in extant strains. This was not the case for the 16SrIII strain 2A1, but there was no evidence of a gene with any similarity to groES in the area upstream of the groEL pseudogene (Figure S1).
3.5. No Evidence of HGT in Non-Fragmented 16SrIII Genomes
Due to the clear cases of groEL acquisition by HGT that were previously documented in Mollicutes [11,12], we examined 16SrIII genomes for the presence of any groEL genes, even those more distantly related to phytoplasmas. No evidence of HGT was observed in any of the genomes. Many of the previously reported 16SrIII genomes are highly fragmented (more than 150 contigs) based on short-read but deep and accurate sequencing chemistry (Illumina). In addition, the assembly process commonly involves a mapping step to exclude non-phytoplasma DNA because of the metagenomic nature of phytoplasma DNA samples. To address this possible source of error, we prepared a MYp assembly by only removing host reads (chromosomal, mitochondrial, and chloroplast), leaving all non-host reads (mostly bacterial, but possibly also including other host-associated microorganisms) available for assembly. Removal of host reads left 21,754,990/ 57,879,811 Illumina reads (37.6%), compared to 12,482,861 reads (21.6%) that remained when mapped to the previously reported MYp genome sequence. This suggests that many other microorganisms were associated with the milkweed leaf sample. When these host-depleted, assembled reads were queried for the presence of groEL-like sequences, 9 sequences were retrieved, all from bacteria (Table S2). Examination of the assembled contigs containing these groEL sequences (the chromosomal context) provided taxonomic identifications consistent with the groEL sequences in all cases (Table S4). These results exclude the possibility of a groEL gene being acquired by HGT, as its sequence would then be located within a context of phytoplasma-like DNA sequences.
Here we have provided two additional high-quality, non-fragmented assemblies of 16SrIII phytoplasmas, and we observed no evidence of groEL acquisition by HGT either in the genomes reported here or in previously published assemblies that are less fragmented (eg strain PR2021 is a single contiguous chromosome). Therefore, we conclude that 16SrIII genomes are unique among phytoplasmas in that they contain no functional GroE system.
4. Conclusions
GroEL/Cpn60 is a protein that is essential for cell viability due to its ability to interact with and facilitate the folding of a wide range of client proteins in the cell, including proteins of a variety of sizes (20-80 kDa) with functions in regulatory and structural processes [13]. The protein also possesses a very wide array of non-canonical functions, which are often associated with gene duplication events [13,56,57]. Under this scenario, preservation of the ancestral function of GroEL can be retained while the duplicated gene can accumulate mutations that may provide alternative functions, leading to functional adaptation of the protein [57]. While this model may partially explain the functional diversity of this essential protein, it does not explain how Mollicutes have adapted to genome reduction including the loss of the GroE system. Schwarz et al. concluded that groEL pseudogenization could be overcome by just a few amino acid changes within a limited range of client proteins that are essential for cell viability, implying that the viability barrier caused by the loss of groEL is not particularly high [11].
Nevertheless, a functional GroE system is the ancestral state for bacteria, given that all bacterial taxa except most of the Mollicutes possess it. Since the Acholeplasmataceae are considered to be a basal clade within the Mollicutes due to their phylogenetic position [1,58] and retention of the AUG stop codon [8], it is likely that the single-copy GroE system in the genera Phytoplasma and Acholeplasma represents a retention of the genes from the Mollicute progenitor, rather than a re-acquisition as has been speculated [11,12]. This is evident in the 16SrIII genomes that retain a vestige of the groEL gene, as its predicted amino acid sequence resembles that of other phytoplasmas. If the GroE system in Acholeplasmataceae represents a loss and re-acquisition, then the 16SrIII group would have lost, then re-gained, and lost this system again. While this is possible in the highly plastic genomes of the phytoplasmas [59], it seems unlikely. It is also notable that, while virtually all species of Acholeplasma have retained a GroE system, a single species, A. oculi, has lost these genes [11]. A single taxon within both of these genera seems to have lost the ancestral GroE system, while most members of this group have retained it.
Through genome sequencing and analysis of previously sequenced genomes, we have demonstrated that group 16SrIII phytoplasmas have lost the ancestral GroE system, which is retained in all other taxa of phytoplasmas. No evidence of the presence of groEL/groES genes by horizontal gene transfer was observed, indicating that these bacteria, like most other Mollicutes, have not re-acquired the functionality of GroE. Since groEL is absent in an obviously pathogenic group of phytoplasma, it seems unlikely that the protein is involved in pathogenesis as has been speculated from observations in other Mollicutes [12]. The lack of gene duplication within phytoplasma groEL genes also argues against a moonlighting role for the protein in host cell attachment and invasion and indicates that the protein most likely retains a canonical role in the folding of host cell proteins. Those Mollicutes that have lost the ancestral genes during genome reduction seem to have crossed the low evolutionary barrier identified by Schwarz et al. [11] and adapted their client proteins to be able to fold in the absence of this system.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Genomic context of genes annotated as groEL at JGI-GOLD (https://img.jgi.doe.gov/) in 16SrII and 16SrIII strains. Genes annotated as groEL are shown in red. The two genes immediately adjacent to groEL are shown as follows: evbG (**) and evbH (*) in PnWB (top panel). Putative evbG/evbH orthologs in 16SrIII strains CX (middle panel) and 2A1 (bottom panel) are marked in the same manner. Table S1. List of all phytoplasma genomes examined for the presence of a GroE system. Table S2. Estimates of evolutionary divergence between amino acid sequences encoded by genes next to the groEL gene in PnWB phytoplasma and the putative groEL pseudogenes of the 16SrIII genomes. The number of amino acid substitutions per site between sequences are shown. Analyses were conducted using the Poisson correction model [60] with MEGA X [53]. Table S3. Estimates of evolutionary divergence between amino acid sequences encoded by known orthologous genes secA, secY, nusA, and tuf in PnWB phytoplasma and the corresponding genes from the 16SrIII genomes. The number of amino acid substitutions per site between sequences are shown. Analyses were conducted using the Poisson correction model [60] with MEGA X [53]. Table S4. groEL sequences found in the assembly generated using host-depleted Illumina reads coupled with nanopore reads filtered for reads longer than 1 kb.
Author Contributions
Conceptualization, T.D. and CH.; methodology, C.H.; software, K.M., T.D.; formal analysis, T.D.; investigation, T.D., C.H.; resources, T.D., H.B., D.S.; data curation, T.D., K.M.; writing—original draft preparation, T.D.; writing—review and editing, T.D., C.H., D.S., H.B.; visualization, C.H.; supervision, T.D., H.B.; project administration, T.D., H.B.; funding acquisition, T.D., H.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Canadian Food Inspection Agency grant number N000178.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Whole genome shotgun data has been deposited to NCBI under Bioproject PRJNA958240. The assembly for ‘Ca. P. pruni’ strain 6A1 is deposited under accession number GCA_033391615.1, and for ‘Ca. P. pruni’ MYp-CanS4 is GCA_050286905.1. Sequencing read files are deposited to SRA under accession numbers SRS17436226 (6A1) and SRS24917142 (MYp-CanS4). The code used for genome assembly and analysis is provided at https://github.com/kevmu/milkweed_phytoplasma_genome.git
Acknowledgments
We are grateful to Matthew Osborne, Anna Sierra Heffernan-Wilker, and Heather Kharouba for providing samples of infected milkweed. We also thank Jennifer Town for providing basal code for genome analysis and assembly.
Conflicts of Interest
We declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Stülke, J.; Eilers, H.; Schmidl, S.R. Mycoplasma and Spiroplasma. In Encyclopedia of Microbiology (Third Edition), Schaechter, M., Ed.; Academic Press: Oxford, 2009; pp. 208–219.
- Gasparich, G.E. Spiroplasmas and phytoplasmas: microbes associated with plant hosts. Biologicals 2010, 38, 193–203.
- Kirdat, K.; Tiwarekar, B.; Sathe, S.; Yadav, A. From sequences to species: Charting the phytoplasma classification and taxonomy in the era of taxogenomics. Frontiers in microbiology 2023, 14. [CrossRef]
- You, Y.; Xiao, J.; Chen, J.; Li, Y.; Li, R.; Zhang, S.; Jiang, Q.; Liu, P. Integrated Information for Pathogenicity and Treatment of Spiroplasma. Current Microbiology 2024, 81. [CrossRef]
- Fenta, M.D.; Bazezew, M.; Molla, W.; Kinde, M.Z.; Mengistu, B.A.; Dejene, H. A systematic review and meta-analysis of contagious bovine pleuropneumonia in Ethiopian cattle. Veterinary and Animal Science 2024, 26. [CrossRef]
- Fookes, M.C.; Hadfield, J.; Harris, S.; Parmar, S.; Unemo, M.; Jensen, J.S.; Thomson, N.R. Mycoplasma genitalium: whole genome sequence analysis, recombination and population structure. BMC Genomics 2017, 18, 993. [CrossRef]
- Citti, C.; Baranowski, E.; Dordet-Frisoni, E.; Faucher, M.; Nouvel, L.X. Genomic islands in mycoplasmas. Genes 2020, 11, 1–16. [CrossRef]
- Bove, J.M. Molecular features of mollicutes. Clin Infect Dis 1993, 17 Suppl 1, S10-31.
- Lund, P.A. Multiple chaperonins in bacteria--why so many? FEMS Microbiol. Rev. 2009, 33, 785–800.
- Hemmingsen, S.M.; Woolford, C.; van der Vies, S.M.; Tilly, K.; Dennis, D.T.; Georgopoulos, C.P.; Hendrix, R.W.; Ellis, R.J. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature 1988, 333, 330–334.
- Schwarz, D.; Adato, O.; Horovitz, A.; Unger, R. Comparative genomic analysis of mollicutes with and without a chaperonin system. PLoS One 2018, 13, e0192619. [CrossRef]
- Clark, G.W.; Tillier, E.R.M. Loss and gain of GroEL in the Mollicutes. Biochem. Cell Biol. 2010, 88, 185–194, doi:doi:10.1139/O09-157.
- Henderson, B.; Fares, M.A.; Lund, P.A. Chaperonin 60: a paradoxical, evolutionarily conserved protein family with multiple moonlighting functions. Biological Reviews 2013, 88, 955–987. [CrossRef]
- Takenaka, R.; Yokota, K.; Ayada, K.; Mizuno, M.; Zhao, Y.; Fujinami, Y.; Lin, S.N.; Toyokawa, T.; Okada, H.; Shiratori, Y.; et al. Helicobacter pylori heat-shock protein 60 induces inflammatory responses through the Toll-like receptor-triggered pathway in cultured human gastric epithelial cells. Microbiology 2004, 150, 3913–3922. [CrossRef]
- Hoffman, P.S.; Garduno, R.A. Surface-associated heat shock proteins of Legionella pneumophila and Helicobacter pylori: roles in pathogenesis and immunity. Infect Dis Obstet Gynecol 1999, 7, 58–63.
- Garduno, R.A.; Garduno, E.; Hoffman, P.S. Surface-associated hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa cell model. Infect Immun 1998, 66, 4602–4610.
- Watarai, M.; Kim, S.; Erdenebaatar, J.; Makino, S.; Horiuchi, M.; Shirahata, T.; Sakaguchi, S.; Katamine, S. Cellular prion protein promotes Brucella infection into macrophages. J Exp Med 2003, 198, 5–17. [CrossRef]
- Wuppermann, F.N.; Mölleken, K.; Julien, M.; Jantos, C.A.; Hegemann, J.H. Chlamydia pneumoniae GroEL1 protein is cell surface associated and required for infection of HEp-2 cells. J. Bacteriol. 2008, 190, 3757–3767. [CrossRef]
- Hickey, T.B.M.; Thorson, L.M.; Speert, D.P.; Daffé, M.; Stokes, R.W. Mycobacterium tuberculosis Cpn60.2 and DnaK are located on the bacterial surface, where Cpn60.2 facilitates efficient bacterial association with macrophages. Infect Immun 2009, 77, 3389–3401. [CrossRef]
- Bertaccini, A. Plants and Phytoplasmas: When Bacteria Modify Plants. 2022, 11, 1425.
- Saccardo, F.; Martini, M.; Palmano, S.; Ermacora, P.; Scortichini, M.; Loi, N.; Firrao, G. Genome drafts of four phytoplasma strains of the ribosomal group 16SrIII. Microbiology 2012, 158, 2805–2814. [CrossRef]
- Nijo, T.; Iwabuchi, N.; Tokuda, R.; Suzuki, T.; Matsumoto, O.; Miyazaki, A.; Maejima, K.; Oshima, K.; Namba, S.; Yamaji, Y. Enrichment of phytoplasma genome DNA through a methyl-CpG binding domain-mediated method for efficient genome sequencing. J. Gen. Plant Pathol. 2021, 87, 154–163. [CrossRef]
- Bennypaul, H.; Sanderson, D.; Donaghy, P.; Abdullahi, I. Development of a Real-Time PCR Assay for the Detection and Identification of Rubus Stunt Phytoplasma in Rubus spp. Plant Dis. 2023, 107, 2296–2306. [CrossRef]
- Gundersen, D.E.; Lee, I.M. Ultrasensitive detection of phytoplasmas by nested-PCR assays using two universal primer pairs. Phytopathol. Mediterr. 1996, 35, 144–151.
- Smart, C.D.; Schneider, B.; Blomquist, C.L.; Guerra, L.J.; Harrison, N.A.; Ahrens, U.; Lorenz, K.H.; Seemuller, E.; Kirkpatrick, B.C. Phytoplasma-specific PCR primers based on sequences of the 16S-23S rRNA spacer region. Appl. Environ. Microbiol. 1996, 62, 2988–2993.
- Zhao, Y.; Wei, W.; Lee, I.M.; Shao, J.; Suo, X.; Davis, R.E. The iPhyClassifier, an interactive online tool for phytoplasma classification and taxonomic assignment. Methods Mol Biol 2013, 938, 329–338. [CrossRef]
- Town, J.R.; Wist, T.; Perez-Lopez, E.; Olivier, C.Y.; Dumonceaux, T.J. Genome sequence of a plant-pathogenic bacterium, “Candidatus Phytoplasma asteris” strain TW1. Microbiology Resource Announcements 2018, 7. [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature Meth. 2012, 9, 357–359.
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Computational Biology 2017, 13, e1005595. [CrossRef]
- Zhu, Q.; Gao, S.; Xiao, B.; He, Z.; Hu, S. Plasmer: an Accurate and Sensitive Bacterial Plasmid Prediction Tool Based on Machine Learning of Shared k-mers and Genomic Features. Microbiology spectrum 2023, 11, e04645-04622, doi:doi:10.1128/spectrum.04645-22.
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molec. Biol. Evol. 2021, 38, 4647–4654. [CrossRef]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 2021, 7. [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 2016, 44, 6614–6624. [CrossRef]
- Rotmistrovsky, K.; Agarwala, R. BMTagger: Best Match Tagger for Removing Human Reads from Metagenomics Datasets 2011.
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [CrossRef]
- Fernández, F.D.; Guzmán, F.A.; Conci, L.R. Draft genome sequence of Cicuta witches' broom phytoplasma, subgroup 16SrIII-J: a subgroup with phytopathological relevance in South America. Trop. Plant Pathol. 2024, 49, 558–565. [CrossRef]
- Pusz-Bochenska, K.; Perez-Lopez, E.; Wist, T.J.; Bennypaul, H.; Sanderson, D.; Green, M.; Dumonceaux, T.J. Multilocus sequence typing of diverse phytoplasmas using hybridization probe-based sequence capture provides high resolution strain differentiation. Frontiers in microbiology 2022, 13. [CrossRef]
- Zamorano, A.; Fiore, N. Draft Genome Sequence of 16SrIII-J Phytoplasma, a Plant Pathogenic Bacterium with a Broad Spectrum of Hosts. Genome Announc 2016, 4. [CrossRef]
- Fernández, F.D.; Zübert, C.; Huettel, B.; Kube, M.; Conci, L.R. Draft Genome Sequence of Candidatus Phytoplasma pruni (X-Disease Group, Subgroup 16SrIII-B) Strain ChTDIII from Argentina. Microbiology Resource Announcements 2020, 9, 10.1128/mra.00792-00720, doi:doi:10.1128/mra.00792-20.
- Pei, S.-C.; Chen, A.-P.; Chou, S.-J.; Hung, T.-H.; Kuo, C.-H. Complete genome sequence of Candidatus Phytoplasma pruni PR2021, an uncultivated bacterium associated with poinsettia (Euphorbia pulcherrima). Microbiology Resource Announcements 2023, 12, e00443-00423, doi:doi:10.1128/MRA.00443-23.
- Lee, I.M.; Shao, J.; Bottner-Parker, K.D.; Gundersen-Rindal, D.E.; Zhao, Y.; Davis, R.E. Draft Genome Sequence of "Candidatus Phytoplasma pruni" Strain CX, a Plant-Pathogenic Bacterium. Genome Announc 2015, 3. [CrossRef]
- Wei, W.; Zhao, Y. Phytoplasma taxonomy: nomenclature, classification, and identification. Biology (Basel) 2022, 11. [CrossRef]
- Jomantiene, R.; Davis, R.E.; Valiunas, D.; Alminaite, A. New group 16SrIII phytoplasma lineages in Lithuania exhibit rRNA interoperon sequence heterogeneity. Eur. J. Plant Pathol. 2002, 108, 507–517. [CrossRef]
- Griffiths, H.M.; Gundersen, D.E.; Sinclair, W.A.; Lee, I.M.; Davis, R.E. Mycoplasmalike organisms from milkweed, goldenrod, and spirea represent two new 16S rRNA subgroups and three new strain subclusters related to peach X-disease MLOs. Can. J. Plant Pathol. 1994, 16, 255–260. [CrossRef]
- Valiunas, D.; Samuitiene, M.; Rasomavicius, V.; Navalinskiene, M.; Staniulis, J.; Davis, R.E. Subgroup 16SrIII-F phytoplasma strains in an invasive plant, Heracleum sosnowskyi, and an ornamental, Dictamnus albus. J. Plant Pathol. 2007, 89, 137–140.
- Chung, W.-C.; Chen, L.-L.; Lo, W.-S.; Lin, C.-P.; Kuo, C.-H. Comparative Analysis of the Peanut Witches'-Broom Phytoplasma Genome Reveals Horizontal Transfer of Potential Mobile Units and Effectors. PLoS One 2013, 8, e62770. [CrossRef]
- Mitrović, J.; Smiljković, M.; Seemüller, E.; Reinhardt, R.; Hüttel, B.; Büttner, C.; Bertaccini, A.; Kube, M.; Duduk, B. Differentiation of ‘Candidatus Phytoplasma cynodontis’ Based on 16S rRNA and groEL Genes and Identification of a New Subgroup, 16SrXIV-C. Plant Dis. 2015, 99, 1578–1583. [CrossRef]
- Contaldo, N.; Mejia, J.F.; Paltrinieri, S.; Calari, A.; Bertaccini, A. Identification and GroEL gene characterization of green petal phytoplasma infecting strawberry in Italy. Phytopathogenic Mollicutes 2012, 2, 59–62. [CrossRef]
- Mitrović, J.; Kakizawa, S.; Duduk, B.; Oshima, K.; Namba, S.; Bertaccini, A. The groEL gene as an additional marker for finer differentiation of 'Candidatus Phytoplasma asteris'-related strains. Ann. Appl. Biol. 2011, 159, 41–48. [CrossRef]
- Muirhead, K.; Pérez-López, E.; Bahder, B.W.; Hill, J.E.; Dumonceaux, T. The CpnClassiPhyR is a resource for cpn60 universal target-based classification of phytoplasmas. Plant Dis. 2019, 103, 2494–2497. [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molec. Biol. Evol. 2018, 35, 1547–1549. [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 2021, 49, W293-W296. [CrossRef]
- Dainat, J.; Pontarotti, P. Methods to Identify and Study the Evolution of Pseudogenes Using a Phylogenetic Approach. Methods Mol Biol 2021, 2324, 21–34. [CrossRef]
- Cehovin, A.; Coates, A.R.M.; Hu, Y.; Riffo-Vasquez, Y.; Tormay, P.; Botanch, C.; Altare, F.; Henderson, B. Comparison of the moonlighting actions of the two highly homologous chaperonin 60 proteins of Mycobacterium tuberculosis. Infect Immun 2010, 78, 3196–3206. [CrossRef]
- Wang, G.; Xia, Y.; Cui, J.; Gu, Z.; Song, Y.; Chen, Y.Q.; Chen, H.; Zhang, H.; Chen, W. The Roles of Moonlighting Proteins in Bacteria. Current issues in molecular biology 2014, 16, 15–22.
- Oshima, K.; Maejima, K.; Namba, S. Genomic and evolutionary aspects of phytoplasmas. Frontiers in microbiology 2013, 4, 230. [CrossRef]
- Bai, X.; Zhang, J.; Ewing, A.; Miller, S.A.; Jancso Radek, A.; Shevchenko, D.V.; Tsukerman, K.; Walunas, T.; Lapidus, A.; Campbell, J.W.; et al. Living with genome instability: the adaptation of phytoplasmas to diverse environments of their insect and plant hosts. J Bacteriol 2006, 188, 3682–3696. [CrossRef]
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins, Bryson, V., Vogel, H.J., Eds.; Academic Press: 1965; pp. 97–166.
Figure 1.
Phylogenetic tree (Maximum Likelihood; 100 bootstrap replicates) based on 16S rRNA-encoding gene sequences obtained from publicly available genome sequences. Branch coloring is consistent with Schwarz et al. [11], with red branches indicating taxa with an intact GroE system, and blue branches taxa that lack the GroE system. Circles indicate the bootstrap values, with larger circles denoting higher confidence. The tree was constructed based on clustalw alignments using MEGAX [53] and visualized using the Interactive Tree of Life (itol) [54].
Figure 1.
Phylogenetic tree (Maximum Likelihood; 100 bootstrap replicates) based on 16S rRNA-encoding gene sequences obtained from publicly available genome sequences. Branch coloring is consistent with Schwarz et al. [11], with red branches indicating taxa with an intact GroE system, and blue branches taxa that lack the GroE system. Circles indicate the bootstrap values, with larger circles denoting higher confidence. The tree was constructed based on clustalw alignments using MEGAX [53] and visualized using the Interactive Tree of Life (itol) [54].
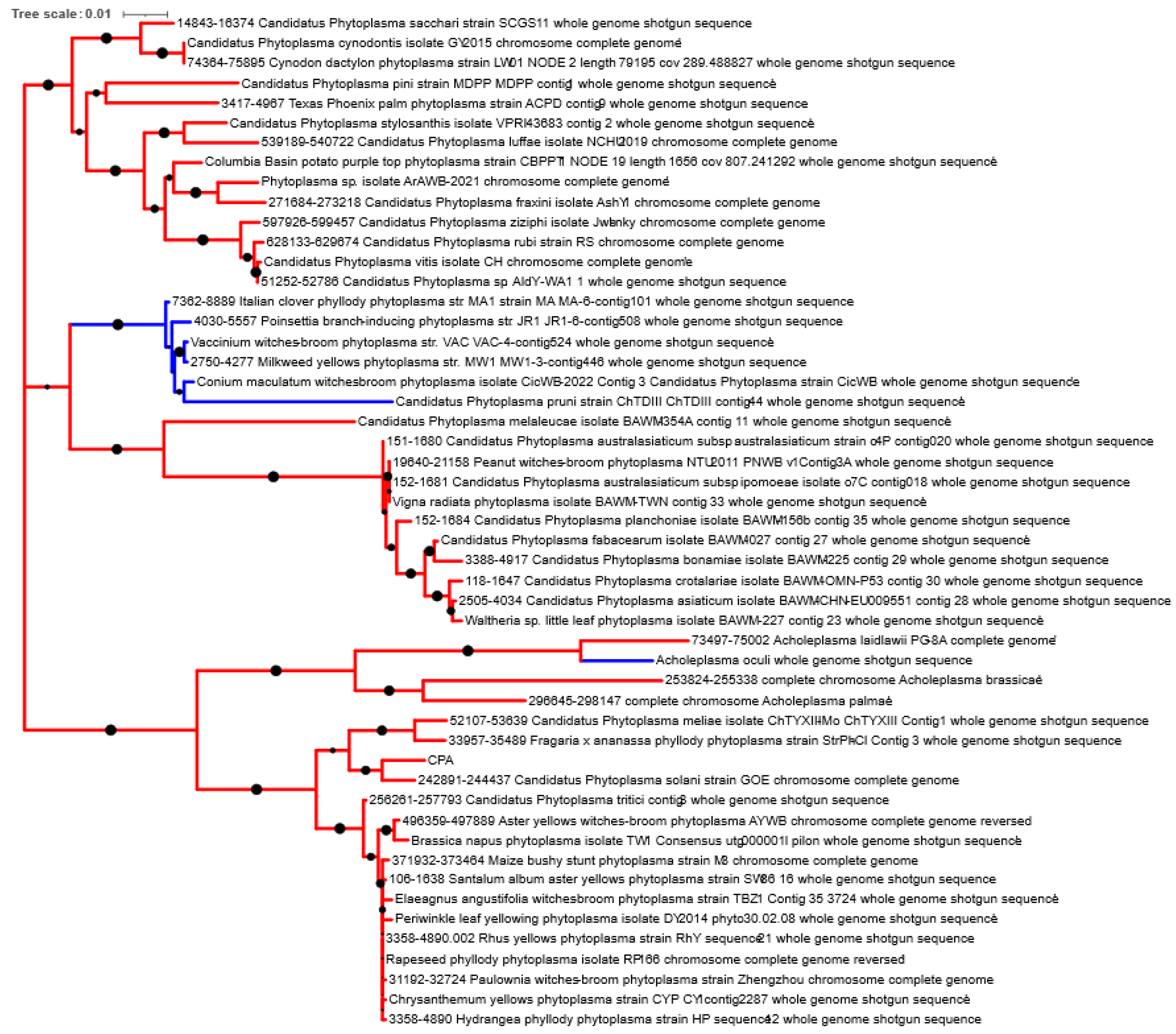
Figure 2.
Phylogenetic tree (Maximum Likelihood; 100 bootstraps) based on full-length groEL (cpn60) sequences obtained from genome sequences deposited in public databases. Circles correspond to bootstrap values, with larger circles denoting higher confidence. The tree was based on clustalw alignments performed using MEGA X [53] and was visualized using the interactive tree of life tool [54].
Figure 2.
Phylogenetic tree (Maximum Likelihood; 100 bootstraps) based on full-length groEL (cpn60) sequences obtained from genome sequences deposited in public databases. Circles correspond to bootstrap values, with larger circles denoting higher confidence. The tree was based on clustalw alignments performed using MEGA X [53] and was visualized using the interactive tree of life tool [54].
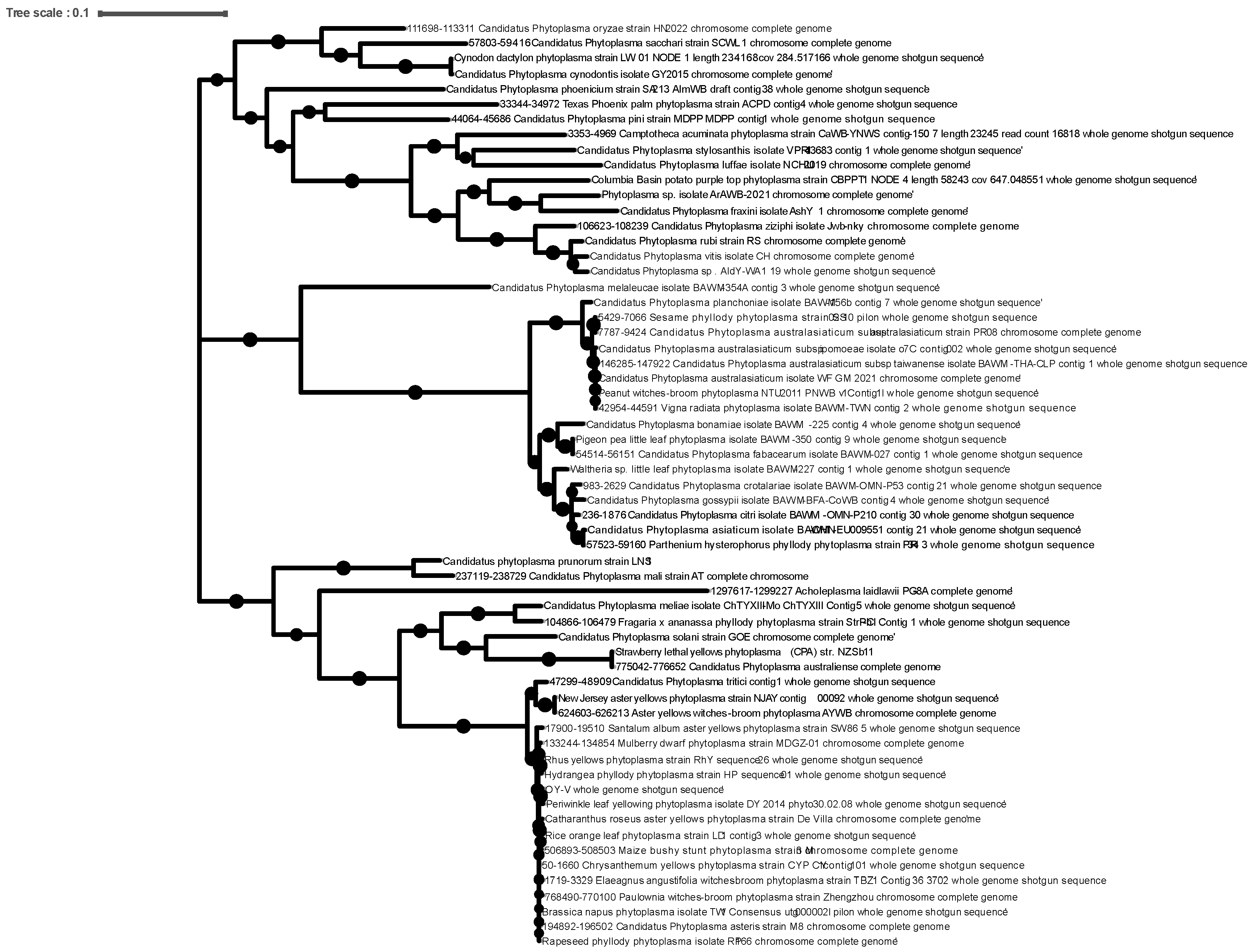
Figure 3.
ClustalW alignment of the predicted amino acid sequences of PnWB (16SrII) and the putative GroEL-encoding pseudogenes in group 16SrIII.
Figure 3.
ClustalW alignment of the predicted amino acid sequences of PnWB (16SrII) and the putative GroEL-encoding pseudogenes in group 16SrIII.

Figure 4.
Phylogenetic tree of the ‘Ca. P. pruni’ (group 16SrIII) based on concatenated taxonomic markers, showing the taxa containing the three different types of pseudogenes. Genomes encoding the three types of pseudogenes are indicated by asterisks: type 1- 55 amino acids, *; type 2 – 62 amino acids, **; type 3 – 50 amino acids, ***.
Figure 4.
Phylogenetic tree of the ‘Ca. P. pruni’ (group 16SrIII) based on concatenated taxonomic markers, showing the taxa containing the three different types of pseudogenes. Genomes encoding the three types of pseudogenes are indicated by asterisks: type 1- 55 amino acids, *; type 2 – 62 amino acids, **; type 3 – 50 amino acids, ***.
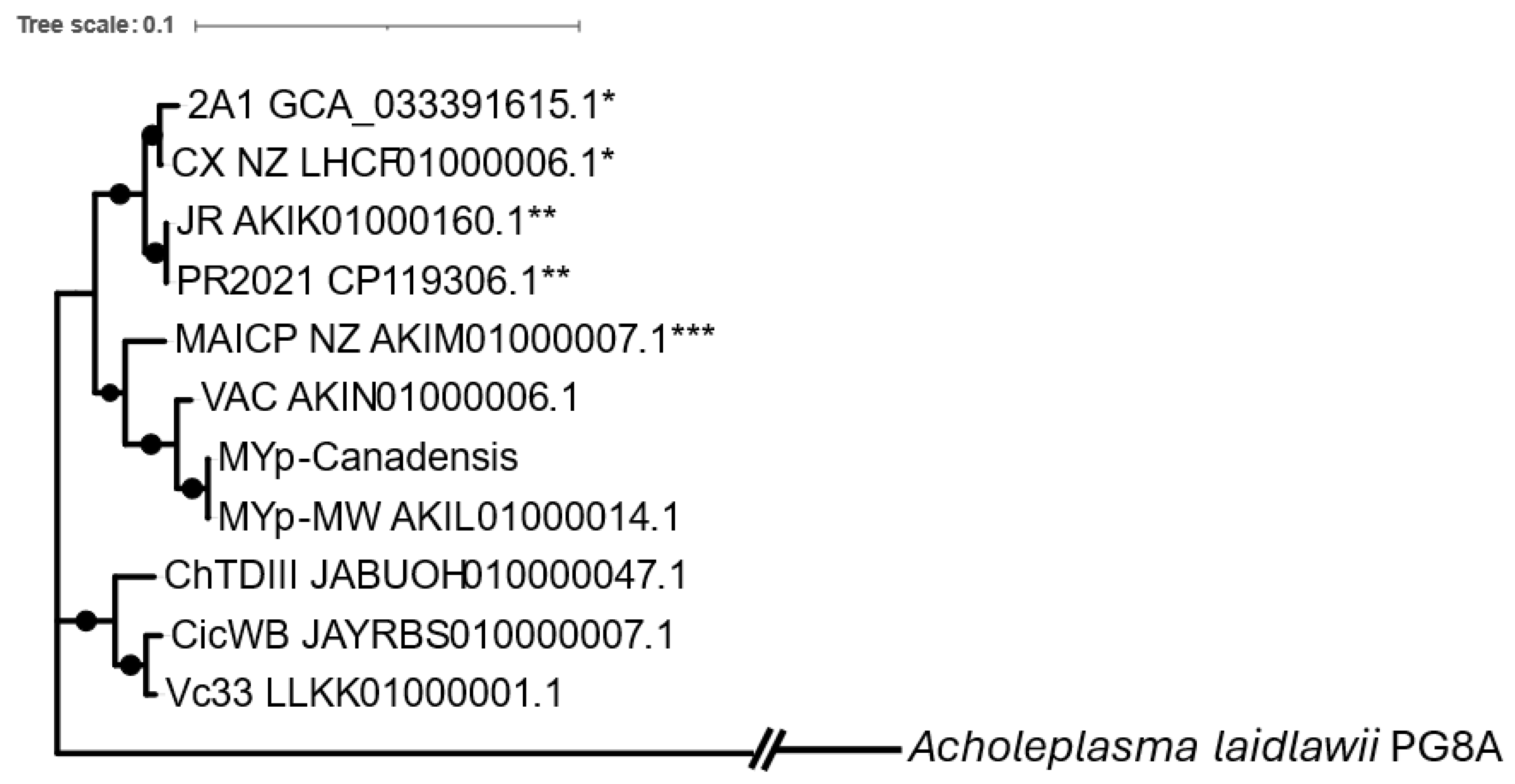
Figure 5.
Gene locations of the predicted pseudogenes (denoted as, “frag.”) and the adjacent genes in 16SrII and 16SrIII. Amino acid similarity scores for all strains are shown in Table S2.
Figure 5.
Gene locations of the predicted pseudogenes (denoted as, “frag.”) and the adjacent genes in 16SrII and 16SrIII. Amino acid similarity scores for all strains are shown in Table S2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



