
Submitted:
27 November 2024
Posted:
28 November 2024
You are already at the latest version
Abstract
Clavibacter is a phytopathogenic genus that causes severe diseases in economically important crops, yet the role of prophages in their evolution, pathogenicity and adaptation remains poorly understood. In this study, we used PHASTER, Prophage Hunter, and VirSorter2 to identify prophage-like sequences in publicly available Clavibacter genomes. Prophage predictions were checked by hand to make them more accurate. We identified 353 prophages, predominantly in chromosomes, with some detected prophages in plasmids. Most prophages exhibited traits of advanced domestication, such as an unimodal genome length distribution, reduced numbers of integrases, and minimal transposable elements, suggesting long-term interactions with their bacterial hosts. Comparative genomic analyses uncovered high genetic diversity, with distinct prophage clusters showing species-specific and interspecies conservation patterns. Functional annotation revealed prophage-encoded genes were involved in sugar metabolism, heavy metal resistance, virulence factors, and antibiotic resistance, highlighting their contribution to host fitness and environmental adaptation. Defense system analyses revealed that, despite lacking CRISPR-Cas, Clavibacter genomes harbor diverse antiviral systems, including PD-Lambda-1, AbiE and MMB_gp29_gp30, some encoded within prophages. These findings underscore the pervasive presence of prophages in Clavibacter and their role in shaping bacterial adaptability and evolution.
Keywords:
prophages
; domestication
; bacterial immunity
; defense systems
; adaptation
1. Introduction
Phytopathogenic bacteria are one of the main contributors of low agricultural productivity worldwide because they cause devastating diseases in many important crops [1]. Clavibacter is the most economically important gram-positive bacterial plant pathogen, with species that cause severe diseases of maize (C. nebraskensis), wheat (C. tessellarius), tomato (C. michiganensis), potato (C. sepedonicus), bean (C. phaseoli), and pepper (C. capsici) crops [2]. Because they cause significant crop losses, some species are considered quarantine pathogens in different countries [3,4]. Despite considerable efforts to mitigate the impact of Clavibacter-associated diseases through traditional breeding strategies and chemical control measures, their management remains challenging, partly due to the need for a comprehensive understanding of the genetic determinants underlying their pathogenicity and environmental interactions [5,6,7].
Prophages and antiviral defenses represent two interconnected components of bacterial genomes that have received increasing attention in recent years due to their roles in shaping bacterial evolution, pathogenicity and ecological interactions [8,9]. Prophages are viral genomes integrated within bacterial chromosomes or plasmids, which can either enhance or decrease the fitness of the bacteria, impacting their behavior, genetic variability and ecology [10]. Some prophages provide genes encoding toxins that enhance the virulence of pathogenic bacteria, as occurs in Acinetobacter baumannii, Staphylococcus aureus, and Escherichia coli [11,12,13]. Moreover, Bifidobacterium, Lactobacillus, and Streptococcus have been shown to harbor numerous prophages and prophage-like components [14,15,16]. Even though significant research has been conducted on prophages in relation to human bacteria, their significance in the fitness and development of phytopathogenic bacteria is comparable. For example, prophages of some of the most destructive plant pathogens, such as Dickeya, Ralstonia, Xylella, Xanthomonas, Burkholderia, and Pectobacterium, have been implicated in auxiliary genes that encode proteins that inhibit the plant immune response, degradation enzymes and proteins involved in the secretion systems [17,18,19,20]. To make bacterial plant pathogens more competitive, prophages can encode bacteriocins that stop competitors [21], increase the resistance of bacteria to environmental stressors such as metal ions and antibacterial compounds [19,22], or boost metabolic potential to help plants survive when nutrients are limited [23].
On the other hand, bacteria have evolved a plethora of antiviral strategies to resist the invasion of viral and mobile genetic elements (MGEs), thereby influencing their susceptibility to phage predation and horizontal gene transfer [24]. These phage resistance strategies involve diverse mechanisms such as the modification of the surface receptors to prevent phage attachment [25], the action of superinfection exclusion proteins to block phage DNA injection [26], the production of enzymes that degrade phage DNA or modify the bacterial genome [27], the use of cell suicide systems to limit phage spread [28], the DNA interference by the Argonaute system to silence phage DNA [29,30], and the expression of elaborated adaptive immunity mechanisms (e.g. CRISPR-Cas), also called defense systems [31]. In the latter, numerous defense systems with great antiphage power and extensive dispersion in bacteria and archaea have been discovered, but most of their molecular mechanisms are unknown [32,33]. Many of the antiviral systems can be carried by MGEs, such as prophages. In P. aeruginosa, the ϕ297 prophage modifies the structure of O-antigen subunits in lipopolysaccharides, conferring phage resistance[34]. Mycobacteriophages such as Butters, Sbash, and CarolAnn have putative toxin-antitoxin (TA) modules with membrane-associated effectors [35,36]. Some phages encode the stress alarmone guanosine pentaphosphate ((p)ppGpp) for host defense, while others encode Sie membrane proteins or restriction endonucleases [37,38]. The prophage-mediated antiviral systems may play an important role in limiting phage infection and increasing host competence and survival [35].
Despite the well-documented roles of antiviral defenses and prophages in various bacterial species, the distribution, diversity and potential functions in Clavibacter species remain poorly understood. Given the importance of Clavibacter-associated diseases in agriculture and the increasing prevalence of phage-mediated control strategies for bacterial pathogens, the knowledge gap surrounding Clavibacter phages is striking. To date, there have been only three completely sequenced genomes of Clavibacter phages: CMP1 and phage 33, isolated against C. michiganensis, and CN1A phage isolated from C. sepedonicus; and the partially sequenced specific CN77 phage for C. michiganensis [39,40,41]. This emphasizes the necessity to address the impact of the viral elements in Clavibacter biology. In this study, we used bioinformatics and comparative genomics to identify and characterize prophage sequences within publicly available Clavibacter genomes, with the aim to shed light on their genomic features and functional potential. Additionally, we will assess the presence and diversity of antiviral defense mechanisms. Exploring the interplay between Clavibacter and their viral elements will provide valuable insights to understand the impact on pathogenicity, ecology and evolution. Understanding how bacteria defend against viral infections is essential for creating specific strategies to reduce Clavibacter-related diseases and loss to agriculture.
2. Materials and Methods
2.1. Bacterial Genomes and Prophage Identification
A total of 114 genome sequences of the Clavibacter genus were retrieved from GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) (last accessed July 2022), and listed in Table S1. Prophage identification was performed using three tools: the PHASTER (PHAge Search Tool Enhanced Release) [42], the PROPHAGE HUNTER [43], and the command line software VirSorter2 [44]. All potential prophages were manually analyzed to depurate the raw list of prophages. For this, overlapping prophages identified on the same contig were thoroughly analyzed by sequence alignment ClustalW, and they were counted as one, adjusting the prophage length considering the non-overlapping ends. Further analysis depicting prophages type, G+C content, polylysogeny, location, and prophage length was plotted in RStudio (v.4.4.1) using ggplot2 (v.3.5.1). Comparative analysis of the host and the prophage genome sizes were analyzed by correlation with the Kendall method considering significant with a p-value less than or equal to 0.05.
2.2. Annotation of Prophage Genes
Final prophage genomes were annotated using the open-access program RAST: Rapid Annotation using Subsystem Technology [45]. Prophage genomes shorter than 4,000 bp that were not accepted for RAST were annotated with GeneMarkS [46]. Then, prophage genes were evaluated to determine their similarity with bacteriophage genes based on searches with phage-limited BLASTx in the NCBI database. The analysis was restricted to tax IDs relevant to bacteriophages, namely 28883, 10699, 10474, 12333, 38018, 2100421, 186765, 10662, 10744, 79205, 10841, and 102294, using the default settings. Proteins identified as hypothetical in RAST were analyzed with BLASTx and PFAM. For searching integrases on prophage genomes, a database was constructed with phage integrase protein sequences retrieved from UniProt. Subsequently, a BLASTp analysis was performed to identify integrases in the Clavibacter prophages using the integrase database previously constructed. Raw results were filtered based on the following criteria: an E-value threshold of less than 0.0001, a cut-off of 50% identity, and 50% coverage and the presence of integrase-related domains with PFAM, in order to increase the probability of finding divergent integrases.
2.3. Functional Categorization of CDSs
Genes were classified by functional categories using the eggNOG-mapper version 2 [47], to identify the clusters of orthologous genes (COGs) of the CDS of the prophage genes according to eggNOG 5 database (v5). Then, a KEGG pathway enrichment analysis was performed with the advanced clusterProfiler (v4.12.6) package in R. The p-values were adjusted with the Benjamini-Hochberg method, and a p-adjusted ≤ 0.05 was set to identify the significantly enriched pathways. Subsequently, the enhanced terms were displayed through dot plots to show the metabolic pathways significantly enriched in the data.
2.4. Identification of Virulence Factors and Antibiotic Resistance Genes
To identify virulence genes, searches were performed using VirulenceFinder 2.0 [48] on all prophage genomic sequences. An additional survey of antibiotic resistance genes was carried out using the Comprehensive Antibiotic Resistance Database (CARD) with the Resistance Gene Identifier (RGI) option with the default parameters [49]. Detection of Insertion Sequences (IS) in Clavibacter genomes was carried out with MobileElementFinder v1.1.2 using the default parameters [50].
2.5. Comparative Genomic Analyses and Defense System Detection
ANIb (Average Nucleotide Identity using BLAST) was calculated using Pyani (v0.2.12) [51] with default parameters to assess the sequence-level similarity between all prophages. The ANI similarity matrix was used to construct a heatmap using the Pheatmap function (v1.0.12) in R. Hierarchical clustering of rows and columns was performed using the ward.D2/average method, while the distance between rows and columns was calculated using the Euclidean distance. All prophage cluster species levels were aligned, and amino acid synteny was visualized using Clinker [52]. The search for bacterial and phage defense systems was done using Defense Finder (https://defensefinder.mdmlab.fr/) [53].
3. Results
3.1. Identification and Prevalence of Prophages in Clavibacter Strains
To study the prevalence and diversity of prophages, genome sequences of Clavibacter associated with tomato (C. michiganensis n = 71), maize (C. nebraskensis n = 13), lucerne (C. insidious n = 7), and other crops (n = 24) were retrieved from NCBI database and analyzed with three different programs. In the first instance, we identified the presence of prophages in more than 97% (n=111 of 114) of Clavibacter genomes, identifying 123 incomplete prophages with PHASTER, 34 active and 124 ambiguous prophages with Prophage Hunter, and finally, 94 full and 15 partial prophages with Virsorter2, accounting a total of 390 prophages (Figure 1A, Table S2). We performed a manual curation because we observed that several prophages overlapped within their genomic regions and we decided to join all of them in one larger prophage. Manual curation merged several prophages into 21 prophages (highlighted in blue in Table S2), reducing a total of 353 prophages identified in Clavibacter genomes (Figure 1B, Table S3), an observation consistent with previous studies showing that prophages are prevalent in bacteria [54]. We noticed that only 13 out of 353 prophages were detected with two or three programs, strongly supporting their identification.
Further characterization revealed that most Clavibacter strains harbored one prophage (32 out of 114, 28%), followed by two (24 out 114, 21%) and three (21 out 114, 18.4%) prophages, while a higher number of prophages by strain were found less frequently (Figure 2A). In fact, we observed that 17 prophages were detected in CFBP 6488 strain. Because of the diversity of prophage categories, we decided to merge prophages in one category as Complete/Active/Full prophages, resulting in 126 active prophages, and as Incomplete/Ambiguous/Partial prophages in the other grouping 227 cryptic prophages. Then, analysis showed that 281 prophages were located into the chromosome and 72 in plasmids (Figure 2B), where most of the prophages were identified in contigs, indicating that draft genomes are useful for prophage detection. Most of the identified prophages were found in C. michiganensis, as expected due to a higher number of genomes analyzed, and they showed a mean proportion of two phages by genome. In contrast, other Clavibacter species, such as C. californiensis, C. chilensis, and C. insidious, exhibited a higher proportion of 7, 6, and 6 prophages per genome, respectively, although the analyzed genomes were less (Figure 2C-D). Despite many prophages were predicted as active elements (category Complete/Active/Full), further annotation revealed the presence of a few integrases, strongly suggesting that these prophages are cryptic or remanent of domesticated prophages (see below).
Next, the average genome size of prophages was 13.98 kbp, ranging from 1.38 to 123.96 kbp and exhibited an unimodal genome size distribution highly enriched with prophages up to 10 kbp in genome length (Figure 3A-B). This finding is contrary to Bobay et al. [55] in which a bimodal distribution of prophage genome length was observed, suggesting that prophages with larger genomes belong to recently integrated and inactivated prophages, indicating a rapid step of inactivation, while shorter prophages suggest genome degradation due to the phage domestication process. In this context, the prophage genomes frequently accumulate mutations, and those prophage genomic regions that confer beneficial traits to the host are selected at long-term, a mechanism known as phage domestication [56]. Moreover, most of the prophages had an average G+C content of 72.16%, similar to analyzed Clavibacter genomes with 72.56% average GC content, although 145 (41%) prophages showed G+C contents lower than 70% (Figure 3C). Similar G+C content may suggest that most prophage-like elements are the product of long-term interactions with their Clavibacter hosts [57]. Linear regression analysis of host versus prophage genome sizes was very weak (R2=0.065) although was significant (p < 0.001), showing that most prophages were accumulated in genomes between 3.2 and 3.4 Mb in size (Figure 3D). Taken together, these results indicated that Clavibacter genomes exhibit a high prevalence of cryptic prophages, many of them representing domesticated prophages.
All 124 active prophages were analyzed in public databases using BLAST limited to tax ID viruses, showing than only 26 prophages matched to known phage genomes with at least 74.6% identity, although very low coverages were observed. Matched prophages were related to Erwinia phage vB_EamM_Stratton, Gordonia phage BetterKatz, Mycobacterium phage ShrimpFriedEgg, Mycobacterium phage Kykar, and Gordonia phage MagicMan. Because of low coverage, we considered that all active prophages are novel.
3.2. Comparative Genomic Analyses of Clavibacter Prophages
To analyze the genetic diversity of prophages, we created an ANI similarity matrix and constructed a heatmap. This analysis revealed that most of the prophages were grouped into 7 clusters, which shared at least of 90% identity, except for groups B and C, where some pairwise comparisons gave ANI values of at least 80% but were clustered together. The remaining prophage genomes showed identity with a few genomes or remained as single prophages, suggesting they were unique. These results indicated high genetic diversity and revealed novel Clavibacter prophages with unique genomes.
Figure 4.
Clustering and heatmap of prophages regions in Clavibacter species. Similar prophages were classified in A to G groups. .
Figure 4.
Clustering and heatmap of prophages regions in Clavibacter species. Similar prophages were classified in A to G groups. .
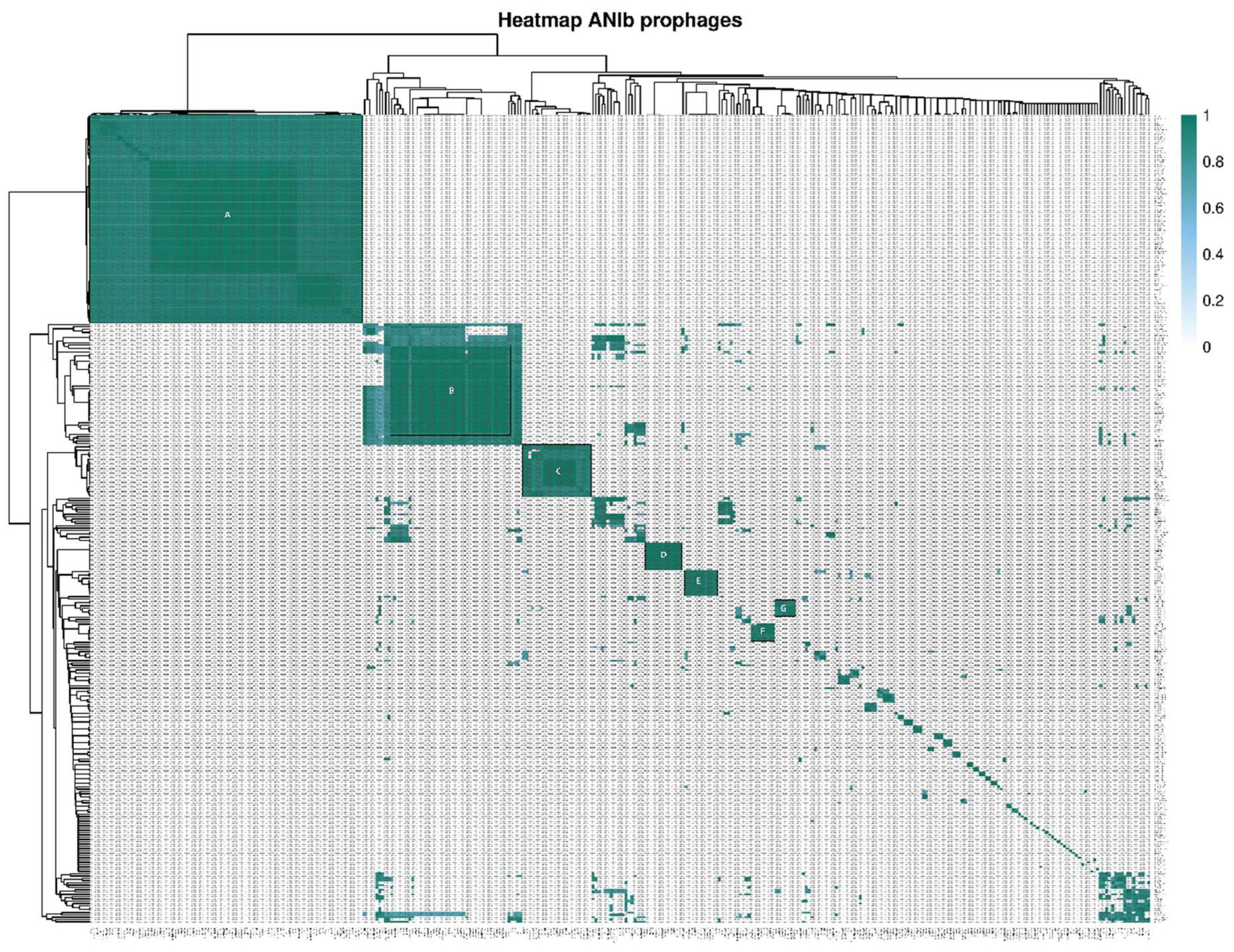
Detailed analysis revealed that most groups of prophages contained at least from two different Clavibacter species, with the exception of cluster D, which was composed of only C. michiganensis prophages. Analysis of C. michiganensis prophages from group A showed low differences in the genomic organization, including some inversions and rearrangements (Figure 5A). A similar pattern was observed for C. michiganensis prophages from cluster D (Figure 5D), showing that the gene organization was conserved with some putative proteins less conserved, reflecting a lower genetic variability among prophages within the group. Strikingly, most of prophages were classified independently of the Clavibacter species, which may suggest these prophages could be acquired by the last ancestor before speciation. In the case of cluster D, only C. michiganensis prophages were grouped together, suggesting a species-specific group of prophages; however, we cannot rule out a potential bias caused by a reduced number of analyzed genomes representing other species than C. michiganensis in our study.
3.3. Functional Annotation of Clavibacter Prophages
In order to gain insights into the nature and function of genes, a total of 6,937 CDS (protein-coding genes) were identified in all the prophage genomes, and 3,858 CDS were filtered after eggNOG analysis representing 55.6% of total CDS. From these, 3,422 CDS were grouped in at least one of the 21 COG functional categories, while 436 out of 3,858 CDS were unclassified, representing 11.3% (Figure 6A, Table S4). Interestingly, after removing redundant descriptions of CDS with COG, we found 594 unique CDS descriptions, indicating that some CDS corresponded to orthologous genes shared among the different prophages. In total, 2,701 CDS were associated with COGs with a defined function, in which the most representative categories were the M group (Cell wall/membrane/envelope biogenesis) alone or in combination with other groups accounting for 26.8% (1,036 out of 3,858 CDS), followed by L group (replication, recombination, and repair) alone or in combination accounting 8.2% (316 out 3,858 CDS). Interestingly, 1,157 CDS (29.9%) were hypothetical proteins (including the S group and the unclassified CDS), which summed up to 3,079 CDS not considered by eggNOG, represented 61% of total proteins with unknown function (4,236 out 6,937 CDS). A complete annotation of prophages was done by combining the results of RAST, BLASTx, CARD, PFAM, and VFDB (Table S5).
Protein-coding genes belonging to the M group and combined categories include several proteins such as glycosyl transferases, cell wall hydrolases, and other enzymes involved in sugar metabolism. Other proteins harbor domains related to dTDP-4-dehydrorhamnose 3,5-epimerase and dTDP-glucose 4,6-dehydratase from the rhamnose pathway, likely playing an important role in the bacterial cell wall structure[58]. In addition, a total of 348 CDS (9% of CDS grouped with COG) are involved in carbohydrate metabolism in G, GK, GM, CG, EG, and EGP categories, which included several ABC-2 type transporters, glycosyl hydrolases, pectate lyases, and cellulases. Interestingly, we found the celA gene, a known virulence factor encoded by the plasmid pCM1 of C. michiganensis [7], was enriched in the G group. Further analysis showed celA was annotated as endoglucanase E1 precursors by RAST and it was present in different prophages, including the phage-plasmids LMG_3663_P4, CFIA_CsR14_P2, 1217_P3, ATCC_10253_P1_3_4, NCPPB382_P1, R1_1_P1_P4, R1_3_P1 and LMG_3663_P5_P1, and the prophages CFBP_7577_P1 and 1106_P1. Other genes related to sugar metabolism were identified, such as UDP-glucose 4-epimerase and glucose-1-phosphate thymidylyltransferase that participate in the production of precursors for the biosynthesis of complex sugars [59,60]. Several CAZymes, mainly of the glycosyltransferase type, were identified in different prophages, such as CASJ001_P1 and CIBA_P1. Furthermore, some Blastx results showed homology with hydrolase proteins from bacteriophages such as Erwinia phage Fifi, suggesting a possible function in the degradation of plant cell wall components [61].
Since the diversity of metabolic functions encoded by prophage genes, we performed a KEGG pathway enrichment analysis. This analysis revealed that metabolic pathways related to carbon, starch, sucrose, and galactose metabolism were enriched in prophage-encoded genes, consistent with findings observed with COG classification. Moreover, biosynthetic pathways involving cofactors, amino acids, nucleotide sugars, and streptomycin were significantly enriched. Interestingly, the streptomycin biosynthesis pathway has only been reported in Streptomyces griseus [62], which belongs to Actinomycetes like Clavibacter. We extracted the genes associated with streptomycin biosynthesis. We found four genes involved in the first metabolic steps of streptomycin biosynthesis (Figure S1), mainly as a unit in the prophage genomes. BLASTn analysis revealed these genes showed high coverages (99%) and at least 70% identity, which suggest these genes might be orthologous acquired by a phage-mediated gene transfer.
On the other hand, prophage genomes were enriched in genes that may confer advantages to adapt to diverse, challenging environments. Several genes related to copper homeostasis and resistance were identified in prophages, such as CopZ, CopG, CopD, and CopC (S group), which are involved in copper translocation. In addition, genes involved in transport and resistance to other metals were identified, such as lead, cadmium, zinc, mercury and arsenic, including transporting P-type ATPases, chaperones, and arsenate-mycothiol transferases, which could confer an advantage to the bacteria in contaminated environments with heavy metals. Other genes encoding ABC (ATP-binding cassette) family transporters related to polysaccharide, cobalt, and Fe3+ transport were identified. For antibiotic resistance, we found genes encoding chloramphenicol acetyltransferases in two prophages (LMG26808_P1_3 and CASJ001_P7) using CARD analysis with perfect cut-off, which was also detected in the H group by COG analysis. Chloramphenicol acetyltransferases are known to inactivate chloramphenicol, thiamphenicol, and azidamfenicol via acetylation, being the most prevalent route for acquiring chloramphenicol resistance in bacteria [63]. Moreover, a putative helix_turn_helix multiple antibiotic resistance protein was enriched in the K group, which harbors a MarR domain (PF01047) that is characteristic of Mar proteins, suggesting a possible role in multiple antibiotic resistance [64]. Other related genes were the metallo-beta-lactamase superfamily proteins, which are often involved in the hydrolysis of β-lactam antibiotics.
Regarding virulence factors, analysis with VFDB revealed several hits associated with functions such as immune modulation, nutritional/ metabolic factors, adherence, biofilm, exotoxin, exoenzyme, accessory secretion factor, effector delivery system, stress survival, motility, invasion and regulation. The most abundant categories were immune modulation, including glycosyltransferases, ABC transporters, capsular proteins, carbohydrate metabolism enzymes, acetyltransferases, antiporters and some kinases; followed by nutritional/metabolic factors, the enriched genes were associated mainly with siderophores biosynthesis and transport, transporter ATPases, sugar transporters and metal iron/manganese transporters. Then, for the effector delivery system were identified several proteins associated to type VI secretion system, ATPases, transcriptional regulators, chaperones and hydrolases. For adherence, Clavibacter prophages were found to encode several proteins identified as C5a peptidases, fimbria and pili biosynthesis and assembly, chaperones, metallopeptidases and adhesins (Table S6).
Based on COG, we found only 16 integrases (L group), 6 structural proteins, and 5 terminases, both belonging to the S group. Combined annotation with RAST, BLASTx (limited to viruses-related tax ids) and PFAM allowed the identification of a total of 109 genes encoding phage-related proteins, such as integrases, major and minor capsid proteins, neck, head closure proteins, fibers, tail proteins and head-to-tail adaptor proteins (Figure 7), which are associated to prophage integration, DNA translocation and virion structure [65,66,67]. Despite their identification, phage-related proteins are few in comparison with all CDS for predicted prophages, thus it is possible that they could be lost under the domestication events.
In addition, we found 37 transposasas grouped in the L group. Previous studies have shown that transposases are highly enriched in prophages with shorter genome lengths, and the mathematical modeling including transposition events, revealed that transposable elements could be relevant primarily during the initial event of domestication by inducing mutagenesis and prophage genome degradation; however, in shorter prophages representing advanced domestication events, transposable elements are lost and only beneficial genes are selected [68]. Thereby, we performed the identification of insertion elements (IS), a kind of transposable element, finding a total of 706 IS in 58 out 114 Clavibacter strains (50.87%), being 107 different IS. From these, the most abundant were IS1121, IS1122, ISCmi2 and ISPfr17 (Table S7). However, after a detailed examination, we found that only 29 prophages contained IS. The presence of IS in the prophage could interfere with the activation of viral genes, influence the ability of these viruses to replicate and spreading, and facilitate horizontal gene transfer [69]. IS may be important in initial events of inactivation and domestication, but the IS are generally lost in advanced genomic decay, similar to the observed pattern in Clavibacter prophages.
Taking together, we hypothesized that the underrepresentation of integrases, transposases, and terminases may indicate an advanced stage of domestication. Some studies have shown that integrases are relatively maintained through evolution, maybe mediating horizontal gene transfer of selfish elements, such as satellite phages, or in the process of recombination [68], meanwhile transposases are present in the first step of prophage inactivation but absent in advanced steps of domestication. Despite Prophage Hunter and Virsorter2 algorithms detected active and complete prophages, our analysis may suggest that most Clavibacter prophages probably are cryptic and domesticated prophages with higher levels of genomic decay.
3.5. Diversity of Defense Systems
Considering that identified prophages seem to be domesticated keeping at long-term interaction with the host, we supposed that Clavibacter probably is infected by prophages at a low rate. This reasoning made us speculate that the diversity of the immunity defense system is poor in this bacteria, considering that phage does not represent selective pressure. To explore this hypothesis, we performed a search of defense systems harbored by Clavibacter genomes, and strikingly, we found that 111 out of 114 genomes contained at least one defense system. Moreover, these strains exhibited 35 types of immunity systems (Figure 8, Table S8). Interestingly, none of the analyzed genomes harbored the CRISPR-Cas system, indicating this system is not found in Clavibacter. Meanwhile, the PD-Lambda-1 was present in 95 out 114 of Clavibacter genomes, followed by Abi (AbiE) in 48 genomes and restriction-modification systems (RM), MMB_gp29_gp30, PD-T7-2 in 131, 34, and 30 genomes, respectively. In bacterial restriction-modification, the restriction endonuclease is crucial in giving tolerance against foreign DNA [70]. The MMB gp29-gp30 system is a defense mechanism identified in the MichelleMyBell (MMB) temperate mycobacteriophage, which consists of a toxic protein (gp29) and a putative membrane protein (gp30). The mechanism is unknown, but it has been determined that this protein complex helps bacterial defense against lytic phages [71].
Since phage predation is a potential threat, prophages may encode anti-phage defense genes to protect its host. Thereby, we searched defense-associated genes in prophages, and we found that only 12 prophages were predicted for the AbiU, RM_Type_IIG, MMB_gp29_gp30, AbiE, CapRel, PD-Lambda-1, Hachiman and Gabija systems within their genomes. Other defense systems were less than 9,000 bp upstream or downstream of four prophage genomes (Table S8). Because Hachiman and Gabija have not been reported in prophages, we performed a survey in GenBank in viral genomes, and we detected the presence of only gajAB genes in phage PfaC02b from Pseudomonas. Genomic surveys from other defense systems revealed the presence of darT in Pseudomonas phage Zuri, kwaAB in phage PfAC02a, and the Thoeris anti-defense 1 protein (ths-1) from Bacillus phage Jack Rabbit (data not shown). Detailed analysis with PFAM, RAST and BLAST revealed that Clavibacter prophages harbored genes encoding proteins related to toxin-antitoxin (TA) systems, such as the HicA toxin and the HicB-like antitoxin from the hicAB system; the yoeB-like toxin of the YoeB/YefM systems; and a complete VapBC TA. Toxin-antitoxin was initially found as selfish genetic elements on plasmids but has been identified in bacterial chromosomes and mobile genetic elements as phages [72,73]. These results lead us to speculate that components of several defense systems, including those described only in bacteria might be encoded by prophages.
4. Discussion
Despite the Clavibacter species are devastating and quarantine pathogens in several important economic crops, very few studies have been conducted to explore the contribution of viral elements in their pathogenesis and evolution. To date, several studies in other relevant phytopathogens, such as Ralstonia and Erwinia, have demonstrated the presence of numerous active and inactive prophages, which contribute to genetic variability and confer diverse properties affecting bacterial adaptation to new environments and competition within microbial communities [74,75]. This study provides novel genomic insights about the diversity of prophage-like elements and defense systems across different Clavibacter species genomes. To improve the detection of prophages, we employed three softwares, each using unique criteria and methods to identify prophages. VirSorter2 uses a viral sequence identification system by examining signature genes, gene density and other genomic features, accurately distinguishing viruses [44]. On the other hand, PHASTER employs searches based on sequence homology by comparison with known phage databases, having limitations in identifying unknown phages or atypical sequences [42]. Lastly, prophage Hunter combines sequence similarity and more than 24 genetic features, such as transcriptional orientation, amino acid composition and Watson-Crick ratio, among others, using a scoring system to detect prophages, although it relies on reference data and it is less effective on highly divergent sequences [43]. Despite this, Prophage Hunter allowed the identification of most prophages, at least in part because it uses machine learning models and detailed comparisons with viral genes, improving the detection of latent or atypical prophages than VirSorter and PHASTER, which may overlook due to bias in the information relying on databases. Nonetheless, the results obtained from this analysis clearly showed that prophage-like elements are pervasive in Clavibacter genomes and encode a repertoire of proteins that may enable survival and fitness.
Detailed analysis of most Clavibacter prophages showed them as inactive or cryptic elements, although some prophages were classified as active or full depending on the detection software. Nevertheless, we speculated that all prophages possibly are inactive based on several criteria: only a few encoded integrases, terminases, transposases; few prophage-associated IS were identified, and genome length exhibited an unimodal distribution enriched in short length genomes. Firstly, the low presence of integrases and terminases may indicate an advanced state of domestication, where prophages are no longer active in the production of complete viral particles and depend on alternative mechanisms for their persistence within the bacterial genome. Moreover, previous studies suggested that in degraded prophages, transposable elements, such as transposases and IS, may play an initial role in mutagenesis and genomic degradation during early domestication events but become less frequent as domestication progresses and only genes of selective benefit to the host are maintained [55]. Despite we found more than 100 IS in half of the analyzed Clavibacter, the fact that only 29 prophages contained IS activity on these elements, may imply they were lost in advanced domestication steps, which would be consistent with an evolutionary trend towards the degradation or domestication of these mobile elements in bacterial genomes [68].
Secondly, the unimodal genome length distribution indicates most prophage-like elements have undergone a process of domestication, where the integrated prophages experienced inactivating mutations, genome reorganization and gene loss due to selective pressures at long-term [55]. Finally, the diverse GC content distribution on prophage-like sequences suggested prophage groups are genetically diverse and that those with similar G+C content might be the product of long-term interactions with their Clavibacter hosts [57], supporting our hypothesis that most prophages are under advanced domestication. Unexpectedly, prophages searches for identification failed using BLAST, which may indicate that that Clavibacter prophages are novel, although bias in the identification process might be due to the scarce genomic information regarding Clavibacter phages in databases.
There was a notable variation in the presence and proportion of prophages among different Clavibacter species. Strikingly, the presence of several orthologus prophages across the different Clavibacter strains may suggest that can be derived from a single prophage present in the last ancestor, indicating vertical transmission [55]. A higher number of prophages in specific genomes could be related to species-specific characteristics, such as their ability to acquire exogenous genetic material through prophages, which could influence their evolution, pathogenicity or adaptation [12,75,76]. Moreover, the presence of prophages in plasmids was identified possibly as representing phage-plasmids, which may offer adaptive advantages since their functions as plasmids and phages represent more pathways for the spreading of genes, improving host fitness [77]. Genomic analysis on phage-plasmids showed that their genes overlapped with plasmids and phages, indicating that phage-plasmids mediate gene exchange among prophages and plasmids, including genes associated with core functions, defense systems and antibiotic resistance [78]. Other studies highlight the interaction and impact of phage-plasmids in phytopathogens, such as the Ralstonia solanacearum species complex [57]. Thereby, prophages on plasmids contribute to host bacteria’s genetic diversity and functionality.
The presence of prophage-like elements in Clavibacter strains indicate that they play important roles in bacterial survival, adaptation and fitness, which is consistent with the fact that prophages harbored a repertoire of genes associated with relevant functions, such as the case of virulence factors, antibiotic resistance, tolerance to metals, siderophores, adhesion, among others functions. In Streptococcus pyogenes, the partial loss of genes in prophages maintains key genes for virulence, allowing an advantage in colonization and persistence in their host [16]. Moreover, the annotation of prophage-encoded genes revealed a remarkable frequency of genes associated with sugar and DNA metabolism, indicating an adaptation towards metabolic functions that could benefit the host, optimizing nutrient availability and facilitating adaptation to adverse environments, such as resistance to heavy metals. The presence of genes related to copper and other metal metabolism suggests that prophages may contribute to homeostasis and resistance to toxic elements, which could confer selective advantages to Clavibacter in agricultural environments. Altogether, these genes may contribute to the ability of Clavibacter strains for adapting to new environments [79]. On the other hand, prophages affect the host in diverse ways upon integration, such as causing mutagenesis and gene disruption, modifying levels of gene expression from the host, facilitating recombination acting as a hot spot or protecting from attacks of similar phages [57]. Nonetheless, some studies propose that acquisition and loss of adaptive prophage genes are constantly occurring in bacterial genomes, favoring the flux of genetic information and their evolution and ecological adaptation [55,80].
On the other hand, the presence of diverse immunity systems suggests that Clavibacter is under selective pressure to keep them, indicating that the bacteria is the target of phages. Unlike other bacteria, Clavibacter do not possess an endogenous CRISPR-cas consistent with previous reports [81]. Instead, other defense systems such as PD-lambda systems, Abi, Hachiman and Gabija, among others, form the repertoire of immunity systems in Clavibacter. Georjon et al. [82] showed that defense systems in Actinobacteria are rare systems, such as gp29-gp30 and Wadjet, and often encoded in mobile genetic elements, which was consistent with our results. Indeed, it was shown that 2% of the defense systems identified in Actinobacteria are encoded within prophages [82], indicating that prophages play a significant role as drivers of host immunity.
In summary, in this study we described the occurrence and diversity of prophage-like elements in Clavibacter spp. We demonstrated that Clavibacter prophages are pervasive and most of them are cryptic and probably with high levels of domestication. Interestingly, many prophages were unique unveiling the diversity of prophages infecting Clavibacter species. Moreover, we identified numerous genes conferring advantageous traits that improve host fitness and adaptation, including some genes associated with defense systems. Taken together, our findings highlight that prophages are relevant mediating phage-host interactions, broadening our understanding of Clavibacter biology and evolution.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: KEGG enrichment of streptomycin biosynthesis in prophage-encoded genes; Table S1: List of Clavibacter strains analyzed in this study; Table S2: Raw list of identified Clavibacter prophages; Table S3: Curated list of identified Clavibacter prophages; Table S4: Results of eggNOG categories; Table S5: Prophage_annotation_Rast_blastx_CARD_PFAM_VFDB; Table S6: VDFB results; Table S7: Results of IS identification; Table S8: Results of defense system searches.
Author Contributions
Conceptualization, L.M.R.-R. and C.V.; methodology, L.M.R.-R. and C.V.; software, L.M.R.-R.; validation, L.M.R.-R. and C.V.; formal analysis, L.M.R.-R.; investigation, L.M.R.-R.; writing—original draft preparation, L.M.R.-R.; writing—review and editing, L.M.R.-R., J.L.-F. and C.V.; visualization, L.M.R.-R.; supervision, C.V.; project administration, C.V.; funding acquisition, C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the CIAD internal project, grant number P00768014, and the Investigadores por México CONAHCYT project number 784.
Data Availability Statement
Part of data are contained within the article and supplementary materials. Additional information can be provided by the authors upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Martins, P.M.M.; Merfa, M. V.; Takita, M.A.; De Souza, A.A. Persistence in Phytopathogenic Bacteria: Do We Know Enough? Front Microbiol 2018, 9. [CrossRef]
- Li, X.; Tambong, J.; Yuan, K. (Xiaoli); Chen, W.; Xu, H.; Lévesque, C.A.; De Boer, S.H. Re-Classification of Clavibacter Michiganensis Subspecies on the Basis of Whole-Genome and Multi-Locus Sequence Analyses. Int J Syst Evol Microbiol 2018, 68, 234–240. [CrossRef]
- EPPO <scp>PM</Scp> 7/42 (3) Clavibacter Michiganensis Subsp. Michiganensis. EPPO Bulletin 2016, 46, 202–225. [CrossRef]
- Bragard, C.; Dehnen-Schmutz, K.; Di Serio, F.; Gonthier, P.; Jaques Miret, J.A.; Justesen, A.F.; MacLeod, A.; Magnusson, C.S.; Milonas, P.; Navas-Cortes, J.A.; et al. Pest Categorisation of Clavibacter Sepedonicus. EFSA Journal 2019, 17. [CrossRef]
- Metzler, M.C.; Laine, M.J.; De Boer, S.H. The Status of Molecular Biological Research on the Plant Pathogenic Genus Clavibacter. FEMS Microbiol Lett 1997, 150.
- Jahr, H.; Bahro, R.; Burger, A.; Ahlemeyer, J.; Eichenlaub, R. Interactions between Clavibacter Michiganensis and Its Host Plants. Environ Microbiol 1999, 1, 113–118. [CrossRef]
- Nandi, M.; Macdonald, J.; Liu, P.; Weselowski, B.; Yuan, Z.C. Clavibacter Michiganensis Ssp. Michiganensis: Bacterial Canker of Tomato, Molecular Interactions and Disease Management. Mol Plant Pathol 2018, 19, 2036–2050.
- Bruneaux, M.; Ashrafi, R.; Kronholm, I.; Laanto, E.; Örmälä-Tiznado, A.; Galarza, J.A.; Zihan, C.; Kubendran Sumathi, M.; Ketola, T. The Effect of a Temperature-sensitive Prophage on the Evolution of Virulence in an Opportunistic Bacterial Pathogen. Mol Ecol 2022, 31, 5402–5418. [CrossRef]
- Tesson, F.; Hervé, A.; Mordret, E.; Touchon, M.; d’Humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. Nat Commun 2022, 13, 2561. [CrossRef]
- Fillol-Salom, A.; Alsaadi, A.; de Sousa, J.A.M.; Zhong, L.; Foster, K.R.; Rocha, E.P.C.; Penadés, J.R.; Ingmer, H.; Haaber, J. Bacteriophages Benefit from Generalized Transduction. PLoS Pathog 2019, 15. [CrossRef]
- Park, D.; Stanton, E.; Ciezki, K.; Parrell, D.; Bozile, M.; Pike, D.; Forst, S.A.; Jeong, K.C.; Ivanek, R.; Döpfer, D.; et al. Evolution of the Stx2-Encoding Prophage in Persistent Bovine Escherichia Coli O157:H7 Strains. Appl Environ Microbiol 2013, 79, 1563–1572. [CrossRef]
- Costa, A.R.; Monteiro, R.; Azeredo, J. Genomic Analysis of Acinetobacter Baumannii Prophages Reveals Remarkable Diversity and Suggests Profound Impact on Bacterial Virulence and Fitness. Sci Rep 2018, 8. [CrossRef]
- Ingmer, H.; Gerlach, D.; Wolz, C. Temperate Phages of Staphylococcus Aureus. Microbiol Spectr 2019, 7. [CrossRef]
- Ventura, M.; Lee, J.H.; Canchaya, C.; Zink, R.; Leahy, S.; Moreno-Munoz, J.A.; O’Connell-Motherway, M.; Higgins, D.; Fitzgerald, G.F.; O’Sullivan, D.J.; et al. Prophage-like Elements in Bifidobacteria: Insights from Genomics, Transcription, Integration, Distribution, and Phylogenetic Analysis. Appl Environ Microbiol 2005, 71. [CrossRef]
- Pei, Z.; Sadiq, F.A.; Han, X.; Zhao, J.; Zhang, H.; Ross, R.P.; Lu, W.; Chen, W. Identification, Characterization, and Phylogenetic Analysis of Eight New Inducible Prophages in Lactobacillus. Virus Res 2020, 286, 198003. [CrossRef]
- Remmington, A.; Haywood, S.; Edgar, J.; Green, L.R.; de Silva, T.; Turner, C.E. Cryptic Prophages within a Streptococcus Pyogenes Genotype Emm4 Lineage. Microb Genom 2021, 7. [CrossRef]
- Slater, S.C.; Goldman, B.S.; Goodner, B.; Setubal, J.C.; Farrand, S.K.; Nester, E.W.; Burr, T.J.; Banta, L.; Dickerman, A.W.; Paulsen, I.; et al. Genome Sequences of Three Agrobacterium Biovars Help Elucidate the Evolution of Multichromosome Genomes in Bacteria. J Bacteriol 2009, 191, 2501–2511. [CrossRef]
- Evans, T.J.; Coulthurst, S.J.; Komitopoulou, E.; Salmond, G.P.C. Two Mobile Pectobacterium Atrosepticum Prophages Modulate Virulence. FEMS Microbiol Lett 2010, 304, 195–202. [CrossRef]
- Czajkowski, R. May the Phage Be With You? Prophage-Like Elements in the Genomes of Soft Rot Pectobacteriaceae: Pectobacterium Spp. and Dickeya Spp. Front Microbiol 2019, 10. [CrossRef]
- Lelis, T.; Peng, J.; Barphagha, I.; Chen, R.; Ham, J.H. The Virulence Function and Regulation of the Metalloprotease Gene PrtA in the Plant-Pathogenic Bacterium Burkholderia Glumae. Molecular Plant-Microbe Interactions 2019, 31, 841–852. [CrossRef]
- Ahmad, A.A.; Stulberg, M.J.; Huang, Q. Prophage Rs551 and Its Repressor Gene Orf14 Reduce Virulence and Increase Competitive Fitness of Its Ralstonia Solanacearum Carrier Strain UW551. Front Microbiol 2017, 8. [CrossRef]
- Roszniowski, B.; McClean, S.; Drulis-Kawa, Z. Burkholderia Cenocepacia Prophages—Prevalence, Chromosome Location and Major Genes Involved. Viruses 2018, 10. [CrossRef]
- Swain, D.M.; Yadav, S.K.; Tyagi, I.; Kumar, R.; Kumar, R.; Ghosh, S.; Das, J.; Jha, G. A Prophage Tail-like Protein Is Deployed by Burkholderia Bacteria to Feed on Fungi. Nat Commun 2017, 8. [CrossRef]
- Baker, E.P.; Barber, M.F. Unearthing the Ancient Origins of Antiviral Immunity. Cell Host Microbe 2020, 28, 629–631. [CrossRef]
- Kim, M.; Ryu, S. Spontaneous and Transient Defence against Bacteriophage by Phase-variable Glucosylation of <scp>O</Scp> -antigen in <scp>S</Scp> Almonella Enterica Serovar <scp>T</Scp> Yphimurium. Mol Microbiol 2012, 86, 411–425. [CrossRef]
- Leavitt, J.C.; Woodbury, B.M.; Gilcrease, E.B.; Bridges, C.M.; Teschke, C.M.; Casjens, S.R. Bacteriophage P22 SieA-Mediated Superinfection Exclusion. mBio 2024, 15. [CrossRef]
- Tock, M.R.; Dryden, D.T. The Biology of Restriction and Anti-Restriction. Curr Opin Microbiol 2005, 8, 466–472. [CrossRef]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.C.; Fineran, P.C. A Widespread Bacteriophage Abortive Infection System Functions through a Type IV Toxin–Antitoxin Mechanism. Nucleic Acids Res 2014, 42, 4590–4605. [CrossRef]
- Swarts, D.C.; Jore, M.M.; Westra, E.R.; Zhu, Y.; Janssen, J.H.; Snijders, A.P.; Wang, Y.; Patel, D.J.; Berenguer, J.; Brouns, S.J.J.; et al. DNA-Guided DNA Interference by a Prokaryotic Argonaute. Nature 2014, 507, 258–261. [CrossRef]
- Kuzmenko, A.; Oguienko, A.; Esyunina, D.; Yudin, D.; Petrova, M.; Kudinova, A.; Maslova, O.; Ninova, M.; Ryazansky, S.; Leach, D.; et al. DNA Targeting and Interference by a Bacterial Argonaute Nuclease. Nature 2020, 587, 632–637. [CrossRef]
- Marraffini, L.A. CRISPR-Cas Immunity in Prokaryotes. Nature 2015, 526, 55–61. [CrossRef]
- Millman, A.; Melamed, S.; Leavitt, A.; Doron, S.; Bernheim, A.; Hör, J.; Garb, J.; Bechon, N.; Brandis, A.; Lopatina, A.; et al. An Expanded Arsenal of Immune Systems That Protect Bacteria from Phages. Cell Host Microbe 2022, 30, 1556-1569.e5. [CrossRef]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic Discovery of Antiphage Defense Systems in the Microbial Pangenome. Science (1979) 2018, 359. [CrossRef]
- Tsao, Y.F.; Taylor, V.L.; Kala, S.; Bondy-Denomy, J.; Khan, A.N.; Bona, D.; Cattoir, V.; Lory, S.; Davidson, A.R.; Maxwell, K.L. Phage Morons Play an Important Role in Pseudomonas Aeruginosa Phenotypes. J Bacteriol 2018, 200. [CrossRef]
- Gentile, G.M.; Wetzel, K.S.; Dedrick, R.M.; Montgomery, M.T.; Garlena, R.A.; Jacobs-Sera, D.; Hatfull, G.F. More Evidence of Collusion: A New Prophage-Mediated Viral Defense System Encoded by Mycobacteriophage Sbash. mBio 2019, 10, 1–20. [CrossRef]
- Mageeney, C.M.; Mohammed, H.T.; Dies, M.; Anbari, S.; Cudkevich, N.; Chen, Y.; Buceta, J.; Ware, V.C. Mycobacterium Phage Butters-Encoded Proteins Contribute to Host Defense against Viral Attack. mSystems 2020, 5. [CrossRef]
- Mahony, J.; McGrath, S.; Fitzgerald, G.F.; van Sinderen, D. Identification and Characterization of Lactococcal-Prophage-Carried Superinfection Exclusion Genes. Appl Environ Microbiol 2008, 74, 6206–6215. [CrossRef]
- Zheng, Z.; Bao, M.; Wu, F.; Van Horn, C.; Chen, J.; Deng, X. A Type 3 Prophage of ‘ Candidatus Liberibacter Asiaticus’ Carrying a Restriction-Modification System. Phytopathology 2018, 108, 454–461. [CrossRef]
- Wittmann, J.; Brancato, C.; Berendzen, K.W.; Dreiseikelmann, B. Development of a Tomato Plant Resistant to Clavibacter Michiganensis Using the Endolysin Gene of Bacteriophage CMP1 as a Transgene. Plant Pathol 2016, 65, 496–502. [CrossRef]
- Kongari, R.R.; Yao, G.W.; Chamakura, K.R.; Kuty Everett, G.F. Complete Genome of Clavibacter Michiganensis Subsp. Sepedonicusis Siphophage CN1A. Genome Announc 2013, 1. [CrossRef]
- Bekircan Eski, D.; Gencer, D.; Darcan, C. Whole-Genome Sequence of a Novel Lytic Bacteriophage Infecting Clavibacter Michiganensis Subsp. Michiganensis from Turkey. Journal of General Virology 2024, 105. [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res 2016, 44, W16–W21. [CrossRef]
- Song, W.; Sun, H.X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An Integrative Hunting Tool for Active Prophages. Nucleic Acids Res 2019, 47, W74–W80. [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genomics 2008, 9, 75. [CrossRef]
- Besemer, J. GeneMarkS: A Self-Training Method for Prediction of Gene Starts in Microbial Genomes. Implications for Finding Sequence Motifs in Regulatory Regions. Nucleic Acids Res 2001, 29, 2607–2618. [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 2021, 38, 5825–5829. [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res 2022, 50, D912–D917. [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res 2023, 51, D690–D699. [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella Enterica Using a Newly Developed Web Tool: MobileElementFinder. Journal of Antimicrobial Chemotherapy 2021, 76, 101–109. [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Analytical Methods 2016, 8, 12–24. [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [CrossRef]
- Tesson, F.; Hervé, A.; Mordret, E.; Touchon, M.; d’Humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. Nat Commun 2022, 13, 2561. [CrossRef]
- López-Leal, G.; Camelo-Valera, L.C.; Hurtado-Ramírez, J.M.; Verleyen, J.; Castillo-Ramírez, S.; Reyes-Muñoz, A. Mining of Thousands of Prokaryotic Genomes Reveals High Abundance of Prophages with a Strictly Narrow Host Range. mSystems 2022, 7. [CrossRef]
- Bobay, L.M.; Touchon, M.; Rocha, E.P.C. Pervasive Domestication of Defective Prophages by Bacteria. Proc Natl Acad Sci U S A 2014, 111, 12127–12132. [CrossRef]
- Touchon, M.; Moura de Sousa, J.A.; Rocha, E.P. Embracing the Enemy: The Diversification of Microbial Gene Repertoires by Phage-Mediated Horizontal Gene Transfer. Curr Opin Microbiol 2017, 38, 66–73.
- Gonçalves, O.S.; Souza, F.D.O.; Bruckner, F.P.; Santana, M.F.; Alfenas-Zerbini, P. Widespread Distribution of Prophages Signaling the Potential for Adaptability and Pathogenicity Evolution of Ralstonia Solanacearum Species Complex. Genomics 2021, 113, 992–1000. [CrossRef]
- Christendat, D.; Saridakis, V.; Dharamsi, A.; Bochkarev, A.; Pai, E.F.; Arrowsmith, C.H.; Edwards, A.M. Crystal Structure of DTDP-4-Keto-6-Deoxy-d-Hexulose 3,5-Epimerase FromMethanobacterium Thermoautotrophicum Complexed with DTDP. Journal of Biological Chemistry 2000, 275, 24608–24612. [CrossRef]
- Frey, P.A.; Hegeman, A.D. Chemical and Stereochemical Actions of UDP–Galactose 4-Epimerase. Acc Chem Res 2013, 46, 1417–1426. [CrossRef]
- Sivaraman, J.; Sauvé, V.; Matte, A.; Cygler, M. Crystal Structure of Escherichia Coli Glucose-1-Phosphate Thymidylyltransferase (RffH) Complexed with DTTP and Mg2+. Journal of Biological Chemistry 2002, 277, 44214–44219. [CrossRef]
- Rodríguez-Rubio, L.; Martínez, B.; Donovan, D.M.; Rodríguez, A.; García, P. Bacteriophage Virion-Associated Peptidoglycan Hydrolases: Potential New Enzybiotics. Crit Rev Microbiol 2013, 39, 427–434. [CrossRef]
- Distler, J.; Mansouri, K.; Mayer, G.; Stockmann, M.; Piepersberg, W. Streptomycin Biosynthesis and Its Regulation in Streptomycetes. Gene 1992, 115, 105–111. [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular Basis of Bacterial Resistance to Chloramphenicol and Florfenicol. FEMS Microbiol Rev 2004, 28, 519–542. [CrossRef]
- Beggs, G.A.; Brennan, R.G.; Arshad, M. MarR Family Proteins Are Important Regulators of Clinically Relevant Antibiotic Resistance. Protein Science 2020, 29, 647–653. [CrossRef]
- Zivanovic, Y.; Confalonieri, F.; Ponchon, L.; Lurz, R.; Chami, M.; Flayhan, A.; Renouard, M.; Huet, A.; Decottignies, P.; Davidson, A.R.; et al. Insights into Bacteriophage T5 Structure from Analysis of Its Morphogenesis Genes and Protein Components. J Virol 2014, 88, 1162–1174. [CrossRef]
- Driedonks, R.A.; Caldentey, J.; Klug, A. Gene 20 Product of Bacteriophage T4. J Mol Biol 1983, 166, 341–360. [CrossRef]
- Smith, M.C.M. Phage-Encoded Serine Integrases and Other Large Serine Recombinases. Microbiol Spectr 2015, 3. [CrossRef]
- Khan, A.; Wahl, L.M. Quantifying the Forces That Maintain Prophages in Bacterial Genomes. Theor Popul Biol 2020, 133, 168–179. [CrossRef]
- Bacciu, D.; Falchi, G.; Spazziani, A.; Bossi, L.; Marogna, G.; Leori, G.S.; Rubino, S.; Uzzau, S. Transposition of the Heat-Stable Toxin AstA Gene into a Gifsy-2-Related Prophage of Salmonella Enterica Serovar Abortusovis. J Bacteriol 2004, 186, 4568–4574. [CrossRef]
- Berg, P.; Baltimore, D.; Boyer, H.W.; Cohen, S.N.; Davis, R.W.; Hogness, D.S.; Nathans, D.; Roblin, R.; Watson, J.D.; Weissman, S.; et al. Potential Biohazards of Recombinant DNA Molecules. Science (1979) 1974, 185, 303–303. [CrossRef]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.G.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-Mediated Defence against Viral Attack and Viral Counter-Defence. Nat Microbiol 2017, 2, 16251. [CrossRef]
- Kamruzzaman, M.; Iredell, J. A ParDE-Family Toxin Antitoxin System in Major Resistance Plasmids of Enterobacteriaceae Confers Antibiotic and Heat Tolerance. Sci Rep 2019, 9, 9872. [CrossRef]
- Li, Y.; Liu, X.; Tang, K.; Wang, W.; Guo, Y.; Wang, X. Prophage Encoding Toxin/Antitoxin System PfiT/PfiA Inhibits Pf4 Production in Pseudomonas Aeruginosa. Microb Biotechnol 2020, 13, 1132–1144. [CrossRef]
- Morgan, T.; Rezende, R.R. de; Lima, T.T.M.; Souza, F. de O.; Alfenas-Zerbini, P. Genomic Analysis Unveils the Pervasiveness and Diversity of Prophages Infecting Erwinia Species. Pathogens 2022, 12, 44. [CrossRef]
- Greenrod, S.T.E.; Stoycheva, M.; Elphinstone, J.; Friman, V.-P. Global Diversity and Distribution of Prophages Are Lineage-Specific within the Ralstonia Solanacearum Species Complex. BMC Genomics 2022, 23, 689. [CrossRef]
- Marshall, C.W.; Gloag, E.S.; Lim, C.; Wozniak, D.J.; Cooper, V.S. Rampant Prophage Movement among Transient Competitors Drives Rapid Adaptation during Infection. Sci Adv 2021, 7. [CrossRef]
- Fong, K.; Lu, Y.T.; Brenner, T.; Falardeau, J.; Wang, S. Prophage Diversity Across Salmonella and Verotoxin-Producing Escherichia Coli in Agricultural Niches of British Columbia, Canada. Front Microbiol 2022, 13. [CrossRef]
- Pfeifer, E.; Rocha, E.P.C. Phage-Plasmids Promote Recombination and Emergence of Phages and Plasmids. Nat Commun 2024, 15, 1545. [CrossRef]
- Valenzuela, M.; Besoain, X.; Durand, K.; Cesbron, S.; Fuentes, S.; Claverías, F.; Jacques, M.A.; Seeger, M. Clavibacter Michiganensis Subsp. Michiganensis Strains from Central Chile Exhibit Low Genetic Diversity and Sequence Types Match Strains in Other Parts of the World. Plant Pathol 2018, 67, 1944–1954. [CrossRef]
- Canchaya, C.; Fournous, G.; Brüssow, H. The Impact of Prophages on Bacterial Chromosomes. Mol Microbiol 2004, 53, 9–18.
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRcompar: A Website to Compare Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res 2008, 36, W145–W148. [CrossRef]
- Georjon, H.; Bernheim, A. The Highly Diverse Antiphage Defence Systems of Bacteria. Nat Rev Microbiol 2023, 21, 686–700. [CrossRef]
Figure 1.
Identification of prophages in Clavibacter genomes using different softwares. A) Raw identification of prophages, B) Identified prophages after manual curation.
Figure 1.
Identification of prophages in Clavibacter genomes using different softwares. A) Raw identification of prophages, B) Identified prophages after manual curation.
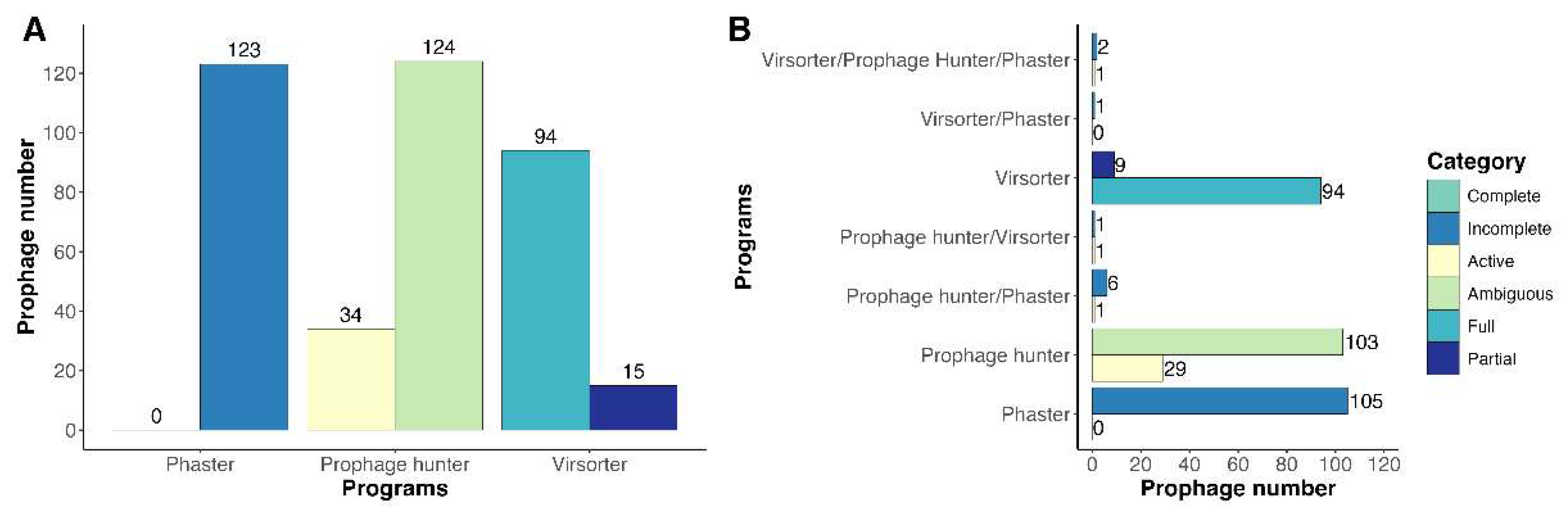
Figure 2.
General features of prophage genomes detected in Clavibacter strains. (A) Frequency of prophages detected on each Clavibacter strain; (B) Distribution of prophages according to the location in the Clavibacter genomes; (C) Presence of prophages in different Clavibacter species; (D) Proportion between the number of strains and the number of prophages present per species.
Figure 2.
General features of prophage genomes detected in Clavibacter strains. (A) Frequency of prophages detected on each Clavibacter strain; (B) Distribution of prophages according to the location in the Clavibacter genomes; (C) Presence of prophages in different Clavibacter species; (D) Proportion between the number of strains and the number of prophages present per species.
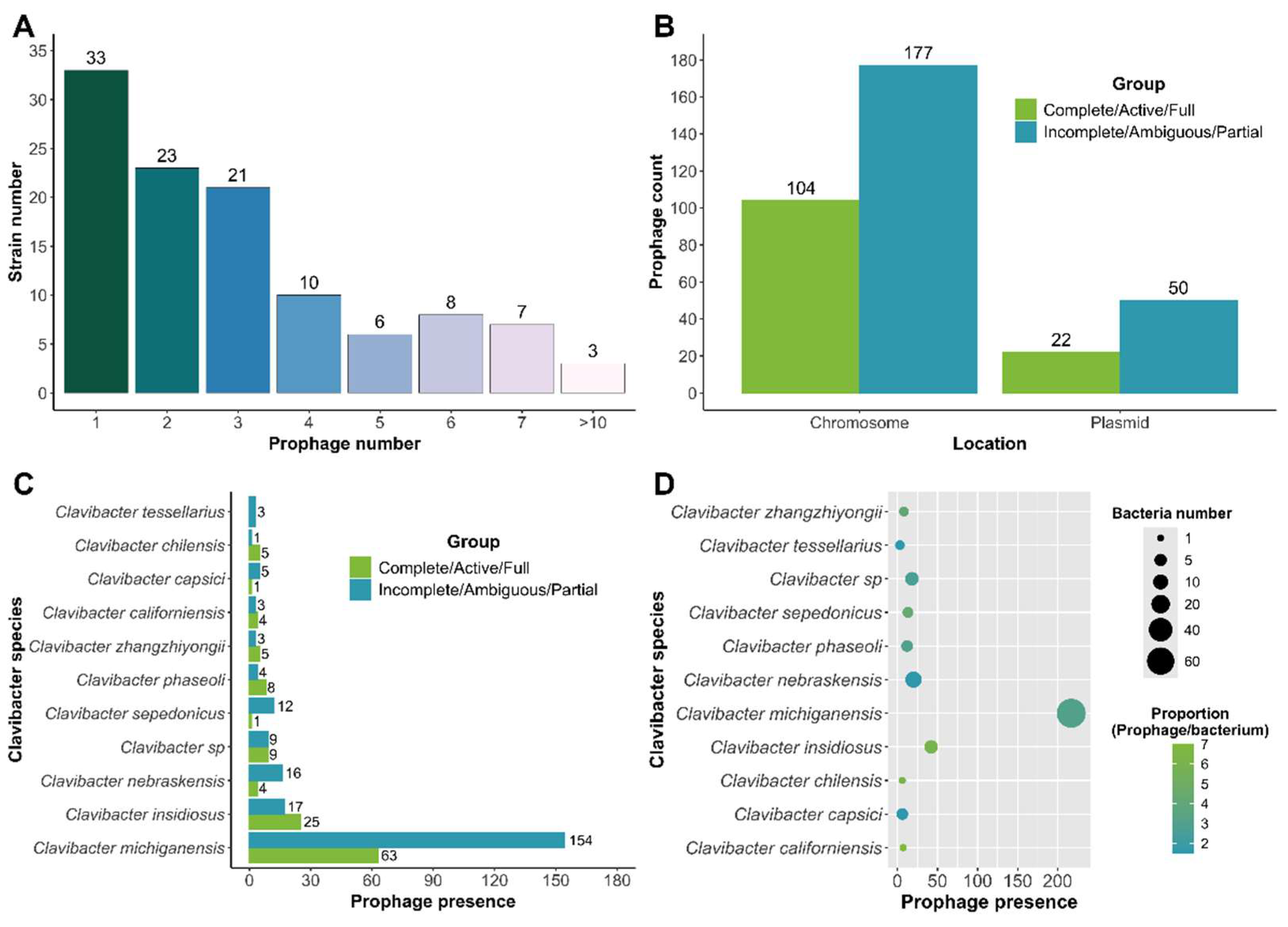
Figure 3.
Genomic characteristics of Clavibacter prophages. (A) Distribution of prophage according to genome size (Kbp). (B) Density of genome length distribution. (C) G+C content (%) of Clavibacter. (D) Correlation between bacterial genome size and prophage genome size.
Figure 3.
Genomic characteristics of Clavibacter prophages. (A) Distribution of prophage according to genome size (Kbp). (B) Density of genome length distribution. (C) G+C content (%) of Clavibacter. (D) Correlation between bacterial genome size and prophage genome size.
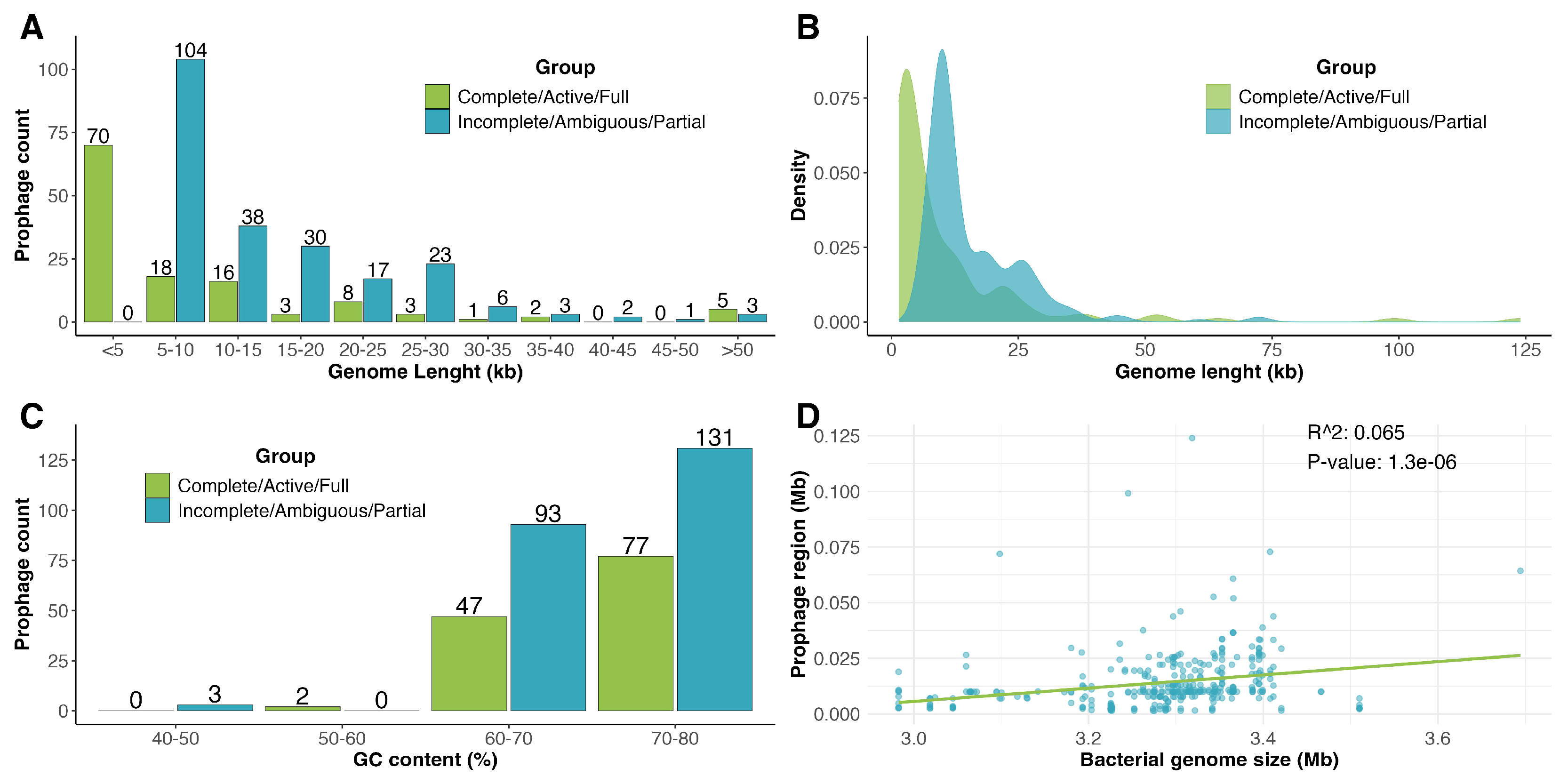
Figure 5.
Genome alignments of clusters of Clavibacter prophages. (A) Analysis of prophages from C. michiganensis from cluster A. (B) Analysis of C. michiganensis prophages from group D. Shared genes across genomes are shown by arrows of the same color. The grey horizontal bar shows the degree of gene similarity between the prophage genomes.
Figure 5.
Genome alignments of clusters of Clavibacter prophages. (A) Analysis of prophages from C. michiganensis from cluster A. (B) Analysis of C. michiganensis prophages from group D. Shared genes across genomes are shown by arrows of the same color. The grey horizontal bar shows the degree of gene similarity between the prophage genomes.
Figure 6.
Functional categorization of prophage-encoded genes. (A) Distribution frequency of prophage-encoded genes classified in COGs. (B) Enrichment analysis of metabolic pathways using KEGG. Abbreviations: -, unclassified; B, Chromatin structure and dynamics; C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; N, Cell motility; O, Postranslational modification, protein turnover, chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; S, Unknown function; T, Signal transduction mechanisms; U, Intracellular trafficking, secretion, and vesicular transport; V, Defense mechanism, Z, Cytoskeleton.
Figure 6.
Functional categorization of prophage-encoded genes. (A) Distribution frequency of prophage-encoded genes classified in COGs. (B) Enrichment analysis of metabolic pathways using KEGG. Abbreviations: -, unclassified; B, Chromatin structure and dynamics; C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; N, Cell motility; O, Postranslational modification, protein turnover, chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; S, Unknown function; T, Signal transduction mechanisms; U, Intracellular trafficking, secretion, and vesicular transport; V, Defense mechanism, Z, Cytoskeleton.
Figure 7.
Distribution of prophage structural proteins identified in Clavibacter prophages. Searches were carried out using COG, BLASTx limited to viruses, RAST and PFAM.
Figure 7.
Distribution of prophage structural proteins identified in Clavibacter prophages. Searches were carried out using COG, BLASTx limited to viruses, RAST and PFAM.
Figure 8.
Diversity of defense systems harbored by Clavibacter strains. Most of defense systems werre enriched in C. michiganensis due to more genomes of this species were analyzed in comparison with the others.
Figure 8.
Diversity of defense systems harbored by Clavibacter strains. Most of defense systems werre enriched in C. michiganensis due to more genomes of this species were analyzed in comparison with the others.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



