Submitted:
05 June 2025
Posted:
06 June 2025
You are already at the latest version
Abstract
Metastatic dissemination defines a complex phenomenon driven by genetic forces and importantly determined by interaction between cancer cells and the surrounding stroma. Although the biologic and immune reactions which characterize the process have been widely and extensively evaluated, fewer data are available regarding the mechanic and physical forces to which circulating neoplastic clones are exposed. It should be hypothesized that this interaction can be modified in case of concomitant pathologic conditions, such as chronic vasculopathy which frequently occurs in lung cancer patients. We here aim at analysing and discussing the complex interplay between lung malignant transformation and artheriopathy, mainly focusing on the immune-inflammatory sistemic reaction. Notably - in most instances – smoking history and a consequent chronic obstructive pulmonary disease cohexists. A main attention is payed to the analysis of the role of immunecheckpoint inhibitors and their interaction with triple bronchodilation and antiaggreagants. Understanding the biomechanical and molecular dynamics of lung cancer progression in altered vascular territories has several translational implications in defining risk stratification, in surgical planning and therapeutic targeting: Moreover, computational modelling of the physical forces which regulate the transit and extravasation of metastatic clones in altered contexts could be of help in deciphering the whole process and in determining more effective blockade strategies.
Keywords:
lung cancer
; COPD
; artheriopathy
; inflammation
; atherosclerosis
; imunecheckpoint inhibitors
; triple therapy
1. Introduction
Although the scientific knowledge on the onset and progression of lung cancer (LC) has significantly increased in recent decades with favourable implications on the outcome of affected patients, it is confirmed, worldwide, as one of the big killers primarily due to its high metastatic potential and the frequent occurrence of advanced-stage diagnosis [1,2,3,4].
Unequivocal and consolidated epidemiological and experimental evidence confirms tobacco smoking as the most important risk factor for the development of lung cancer [5,6,7,8,9], although individual susceptibility [10,11,12,13,14,15,16]and other concomitant lung pathologies [17,18,19,20] impact on LC incidence. There is a close correlation between the onset of the disease and the number of cigarettes/day smoked, with the duration of the habit in years, the depth with which the smoke is inhaled, the tar and nicotine content of the cigarettes smoked [21]. The relative risk of smokers of getting LC is 14 times higher than that of non-smokers, while for heavy smokers (for those who consume more than 25 cigarettes/day), it rises to 30 times. For those who quit smoking, the risk progressively reduces over the following 7-10 years, after which it returns to overlap that of non-smokers [22]. Exposure to passive smoking is recognised as risk factor, mainly if it occurred at a young age (under 25 years) [23]. It is more complicated to define the pathogenetic role of air pollution given the complexity and heterogeneity of the toxicants that can be detected; however, in residents of urban areas the risk of having LC is 1.2-2.3 times greater than in residents of rural areas [24,25,26,27,28]. Within respect the clinical presentation and molecular features of smoke-associated LC, most data actually regard non-small-cell LC (NSCLC) and in this review the term LC refers to NSCLC.
The complex natural history of LC implies that distant metastases have most often detectable at time of clinical diagnosis. Metastatic progression is driven by genetic programmes based on the dynamic crosstalk between genes and distant microenvironment [29,30,31]: it is generally a late event but in some cases, it is starts early and prevales over the growth of the primary lesion [32]. Overall metastatic clones need to acquire the biological properties required to detach from the mass, invade blood (or lymphatic) vessels, survive into the flow in absence of anchorage and then extravasate and colonize distant organs. Moreover, it is now well known thar the immune system interacts closely with tumors throughout the entire process of malignant transformation and the last decade has seen the rapid development of immunotherapy and its role as a crucial strategy in the treatment of cancer, especially LC.
Smoking habit acts as risk factor for chronic vascular disease as well. Thus, LC patients are frequently co-affected by chronic obstructive pulmonary disease (COPD) and arteriopathy. Chronic obstructive pulmonary disease (COPD), a common consequence of long-term tobacco exposure, is associated with profound changes in vascular structure and function, systemic inflammation, and immune dysregulation [33,34,35]. Smoke-related vasculopathy may profoundly alter the mechanical and immunologic microenvironment [36], potentially modifying the success of metastatic colonization at distant sites [37,38]. Although the both conditions are well characterized and treated, few data are available regarding the complex interaction between them and LC in terms of mechanistic and therapeutic reciprocal interferences and effects.
Thus, the aim of this work is to point out from the available literature data, the interplay between the two chronic conditions and LC progression, which shares with them smoke as risk factor with signifcant therapeutic implications. This knowledge gap becomes even more relevant in the context of chronic comorbidities frequently observed in lung cancer patients, especially those with a history of heavy smoking. Moreover, a deeper understanding of the fate of LC metastatic cells in the blood flow in a real-life context should be of help in designing and modulating innovative systemic diagnostic tools, surgical strategies and drug delivery technologies.
2. Methods
We performed an extensive search using the following biomedical databases: Medline using the PubMed interface, Web of Science and Embase. Search terms were as follows: “lung cancer” and “chronic pulmonary obstructive disease -COPD” and “vascular disease”, “aneurysm”, “arteriopathy”, “metastases”, “immunotheraphy”, “triple broncodilation”; “antiaggregants” and “circulating tumor cells” and “metastatic niche”. No restriction on publication date was applied and the last search was performed on 21 April 2025. Only articles written in or translated into English were included. The search for each topic was performed independently by pneumo-oncologists, pulmunologists, radiologist and vascular surgeons focusing on their area of interest.
3. Lung Cancer in Smokers
Cigarette smoking has a direct carcinogenic effect (genotoxic damage) due to the various substances produced during combustion. Among the components of cigarette smoke, the main carcinogenic action is attributed to polycyclic aromatic hydrocarbons, indirect carcinogens that require transformation into active intermediates by microsomal enzymes present at the level of bronchial cells [39,40,41]. Cigarette devices and vaping fluids as well have been shown to contain a number of carcinogens classified as both definite and probable, including nicotine derivatives (e.g., nitrosonornicotine, nitrosamine ketone), polycyclic aromatic hydrocarbons, heavy metals (including organometallic compounds) and aldehydes/other complex organic compounds. These molecules are present both in the e-liquid (with many aldehydes and other complex organic compounds used as flavors) and as a result of pyrolysis/complex organic reactions in the e-cigarette device (including clear carcinogens such as formaldehyde, formed by the pyrolysis of glycerol). Various studies demonstrate the transforming and cytotoxic activity of these derivatives in vitro and in vivo. The use of e-cigarette devices is significantly increasing, particularly among younger people and previously non-smokers. Considering the latency times (extrapolated from data on tobacco smoking) which reach up to 20 years, it is highlighted how this type of exposure can have very significant future implications on public health [42,43,44,45]. Moreover, many substances demonstrate an additive and sometimes synergistic effect with tobacco smoke. This is true, for example, for exposure to asbestos fibers since it has been shown that non-smoking workers in the asbestos industry display a risk of developing LC 5 times higher than non-exposed and non-smokers, while the risk of exposed workers and even current smokers rises to 95 times [46,47]. Lung cancer cells express the nicotinic acetylcholine receptors (nAChRs) [48] and through the α7-nAChRs, nicotine can increase the invasive potential of LC cells whereas it also promotes the expression of genes involved in epithelial-to-mesenchymal transition (EMT) in several cancer types [49,50,51,52,53]. Overall, experimental evidence suggests that smoke inhibits vascular collagen by reducing local production of prolyl-4-hydroxylase [54] and impairs matrix homeostasis by altering production of metalloprotease and increasing T cell infiltrates in response to the smoke-induced injury [55,56]. Smoke seems also to promote hyperplasia of the tunica intima as well as senescence of vascular smooth cells [57]. Nicotine promotes aneurysms formation by activating AMP-activated protein kinase α2 (AMPK-α2) and miR-21 probably in response to cellular stress and hypoxia [58]. Smoke exposure is also associated with the induction of a number of indirect genotoxic effects [59,60,61,62,63,64]: i) alteration of DNA repair mechanisms and the consequent failure to repair to direct genotoxic damage: ii) expression of polymorphisms of genes coding for enzymes involved in the metabolization process with increased exposure to genotoxic and carcinogenic agents: iii) induction of the expression of genes involved in the pathogenetic mechanisms of inflammatory processes, oxidative stress and tissue repair which ultimately leads to chronic tissue damage and activation of molecular mechanisms promoting neoplastic progression and inflammatory infiltrates [65].
Although a deep analysis of the molecular profile of LC in smokers goes beyond the scope of this review, some issues deserve to be reminded. Chromosomal alterations (e.g., promoter hypermethylation and loss-of-heterozigosity) are more frequent in tumors of smokers and among these in squamous cell cancers than in adenocarcinomas. The genetic asset of LC in smokers is mainly addicted to somatic mutations in KRAS gene (mainly affecting exon 12) correlating with poor survival. KRAS mutational frequency is allele/tissue-specific and associated to smoking habit although they are not described in small-cell lung cancers (SCLCs). KRAS alleles are non-uniformly distributed across cancers; they have different mutagenic origins related to the exposure of tobacco smoke. The KRAS alleles have distinct co-mutation networks with mutual exclusion with EGFR mutations [66,67]. Once defined as undruggable genetic driver, more recently the activated G12C KRAS has become actionable via a newly identified switch II pocket and an innovative class of small molecules have been developed. Thus, sotorasib and adagrasib are now approved in locally advanced or metastatic KRASG12C NSCLC [68,69,70,71]. Moreover, p53 mutations are more common in NSCLCs (SCC) in smokers, independently of KRAS and EGFR status [72]. Smoking also affects epigenetic status and more than 2600 cytosine-phosphate-guanine sites (CpGs) are statistically significantly differentially methylated in smokers [73]. Exposure to cigarette smoke on the human airway epithelial cell is known to affect transcriptome profile as well. A number of data on hierarchical clustering analysis performed on samples derived from current- and never-smoker samples have documented that in smokers some genes are overexpressed such as those in: regulation of drug metabolism and oxidation-reduction reactions, mucous secretion and some oncogenes (RAS pathway); tumor growth and inflammation (cystatin, IL-8, CD55), fibroblast activation and proliferation (HBP17), cell cycle progression and control (TOB1, DUSP6, BRD2). Unexpressed genes are those involved in negative regulation of inflammatory processes (antioxidant defence gene BACH2, COX5B), tumor suppressor genes, ubiquitinization (UBE2D2), DNA repair (GTF2H3). In particular, aberrant transcripts (deletions, LOH, allelic loss) of FHIT (Fragile Histidine Triad) have been described in NSCLCs (SCC), directly related to smoking exposure. FHIT acts as a tumor suppressor and is localized in the chromosomal region 3p14.2, close to the most common fragile site of the human genome FRA3B: loss of function of the FHIT protein is associated with increased proliferation and reduction in the apoptosis index and is an independent indicator of clinical outcome [74,75,76,77,78]. It should be also remarked that even upon cessation of exposure to smoking, in some instances gene expression is irreversible altered, thus justifying the persistence upon cessation of the risk of lung disease as cancer [79,80,81,82].
Solid tumors consist of multiple cell types that differ in their state of differentiation and contain a cellular subset with phenotypic stemness characteristics, namely cancer stem cells (CSC) which are characterized by the capacity of self-renewal and differentiation and which constitutively express non-specific molecular markers of multi-drug and radio- resistance; they retain the exclusive ability to support tumorigenesis (for a review see [83,84,85,86,87]). It is conceivable that anatomically distinct CSC niches exist in the lung: in proximal tract elements carrying stem-like potential can be identified in basal cells (expressing keratins K5/K14+) [88] whereas in the peripheral one the morphological identification is more difficult, based on the expression of TTF1- thyroid transcription factor 1 [89] and correspond to Club cells (CC10+) in bronchioles, alveolar type II pneumocytes (SP-C +) and Club cell variants co-expressing CC10 and SP-C [90,91,92,93]. Chronic cigarette smoking results in lung inflammation and epithelial damage that ultimately activate a chronic wound repair programme. It induces, as widely reported in upper airways [94,95], the formation of multifocal epithelial lesions carrying genetic alterations as LOH in 17p,3p,9p,8p,18q and 11q13 amplification. Thus, is likely that, in smokers, the phenomenon of “field cancerization” [96,97,98] culminates in malignant transformation starting from precursor stem cells which define pre-neoplastic cyto-histologic settings. This model is coherent with the risk of lung recurrence after radical surgery and the arousal of second primary or synchronous tumors and justifies the rationale for monitoring pre-neoplastic “field” lesions [99,100]. Moreover, recent studies have demonstrated the ability of bone-marrow derived stem cell to respond to epithelial wounding and contribute to epithelial repair through directly crossing lineage or fusion with lung epithelial cells and sustaining malignant transformation induced by the carcinogens in cigarette smokes [101,102].
Cigarette smoke contributes not only to malignant transformation but also to tumor progression by promoting (seed) and creating a favourable substrate (soil) to the process of neoplastic spreading and metastasis growth (Figure 1) [103,104,105,106,107,108,109,110,111,112,113,114,115]. Overall, the strong immunogenicity associated with cigarette smoking is a reliable predictor of response to immune checkpoint inhibitors in LC. Increased number of mutations associated to the cellular damage exerted by smoke toxics is associated to the increased release of neo-antigens which may improve response to immunotherapy. In this perspective, smoke and smoke-induced mutational load are a proxy for tumor response [116]
4. Smoke-Induced Vascular Pathology
The arterial wall are multi-layered systems with each layer helping to maintain arterial physiology: the inner-most tunica intima, middle tunica media, and outer-most tunica adventitia. The intima consists of an endothelial cell (EC) monolayer, contains smooth muscle fibrocells and free cells, and is directly exposed to the hemodynamic environment; it anchored to the basal membrane through a pool of elastic fibers defined as internal elastic lamina. The EC layer regulates biomacromolecule permeability and acts as hemodynamic mechanosensors transducing fluid force into biochemical signals. The tunica media is made primarily of vascular smooth muscle cells (vSMCs), elastic fibers and laminae, collagen fibers and proteoglycans which contribute in the molecule trafficking and in orchestrating both physiological and pathological vascular functions, such as remodeling after injuries. The VSMCs regulate vessel contraction/dilation, modulating blood pressure in response to flow and also produce a significant portion of extracellular matrix (ECM) molecules, such as elastin, collagen, fibrillin and proteoglycans which maintain arterial biomechanical and contractile integrity. The vasa vasorum may also be missing in the tunica media of the largest arteries and the supply of oxygen probably occurs much more effectively from the lumen than from them thanks to the high partial pressure of oxygen. The adventitia is mainly made of collagen and continues into the perivascular connective tissue. The external elastic lamina is between the media and adventitia which is mainly composed of type I collagen fibrils, organised to form collagen fibers and and fibroblasts, providing strength and support [121]. Notably, in largest arteries in the media, elastic fibers are arranged concentrically and joined together by elastic connection bands. This allow high distensibility and recoil capacity of the vascular wall, whereas the frame of fibrillar collagen assure the length-tension properties. The anatomic composition and mechanical performances vary near vascular bifurcation, where the adventitia increases its thickness. The aorta, the largest artery, is defined an elastic or conducting vessel, based on its relatively high elastin content which allow to maintain constant blood pressure gradient throughout the cardiac cycle. This phenomenon is termed Windkessel’s effect [122], the physiological phenomenon that allows, based on resistance on compliance variations, at the level of the large elastic arteries, to modify the discontinuous flow of the cardiac output into a more continuous flow, transforming the kinetic energy of the blood coming from the left ventricle into elastic potential energy.
Blood circulation induces into vessels 3 different types of mechanic energy: i) tangential shear stress (τ) which exerts a direct action of the flow on endothelial cells; ii) perpendicular or radial shear stress (γ) which is defined as the radial thrust of blood pressure; iii) stress related to parietal stretching which is associated with the parietal wall deformation. A fluid is said to be Newtonian if the tangential stress (τ) is directly proportional to deformation rate (shear stress, γ), according to the formula τ = γ where is the dynamic viscosity: a thermophysical property of the fluid, which depends only on the temperature and not on the flow. A fluid that does not present this property is defined as non-Newtonian and is described by the same equation replacing the viscosity with the apparent viscosity (app), τ = app γ [123].
The apparent viscosity is not constant and is no longer a thermophysical property of the fluid, but depends on the flow properties. Blood isn’t a Newtonian fluid but it is a complex suspension in which cells are immersed into a colloidal solution, the plasma [124]. Non-Newtonian effects of blood are considered negligible when examining the motion of blood in large vessels, but they can become relevant on those areas of flow reversal and/or separation, characterized by low shear rate values. The main differences with the Newtonian model for blood are mainly found at low speeds, on the distribution of WSS, on recirculation zones and on secondary flows [125].
It is well known that smoking is a potent risk factor for clinically relevant peripheral artery disease (PAD), with a significant dose-response association [126,127,128]. Arterial vasculature is subjected to morphological and functional alterations due to cigarette smoking. Pathologic lesions involve the arterial wall and blood cells as well. The effects of smoke on microcirculation encompass alterations in endothelium, platelet aggregation and adhesiveness, nervous system and metabolic changes which overall alter the flow and tissue perfusion [129]. Moreover, two major compounds of cigarette smoke are capable of determining vascular damage: nicotine acts preferably on large arteries and carbon monoxide on small arteries, although both compounds damage the vascular system [130]. In animal models, smoke exposure induces the formation of lipid plaques by impacting on total cholesterol content, ultimately leading to atherosclerosis [131,132]. The most relevant role in generation of atherosclerotic lesions is played by nicotine [133]. In addition to alter lipid metabolisms, cigarette smoke extracts are known to induce oxidative stress and inflammation which contribute to atherosclerotic process through dysfunction of macrophages, smooth cells and endothelial elements which are implicated in the development of PAD [134,135,136].
4.1. Circulating Tumor Cells Dynamics in Vasculopathy
Circulating tumor cells (CTCs) face a hostile intravascular environment characterized by hemodynamic stress, endothelial barriers, and immune surveillance [137], yet the interplay between these biomechanical constraints and cancer cell survival has not been adequately dissected in lung cancer. Recent studies have begun to elucidate how mechanical factors such as fluid shear stress (FSS) influence CTC survival. FSS has been shown to trigger epithelial-mesenchymal transition (EMT) through JNK signalling, promoting resistance to apoptosis and enhancing metastatic fitness [138]. Moreover, shear stress can induce nuclear expansion via histone acetylation, a process that appears essential for CTC viability during circulation [139]. The mechanical resilience of CTCs, including their ability to deform through capillary-sized vessels and interact dynamically with blood components such as platelets, further contributes to their metastatic potential [140]. Platelets in particular play a protective role by shielding CTCs from immune recognition and facilitating vascular adhesion and extravasation [141]. These findings highlight the underappreciated contribution of mechanobiological forces in lung cancer metastasis and suggest novel avenues for therapeutic intervention. The process of metastatic dissemination involves a series of tightly regulated and highly selective steps, among which the intravascular survival and migration of CTCs represent a major bottleneck [142]. Once detached from the primary tumor mass, CTCs must enter the bloodstream, withstand shear stress from blood flow, evade immune detection, and ultimately extravasate at distant sites [143]. Although the molecular characteristics of CTCs have been extensively studied, the physical and mechanical challenges encountered during their journey through the circulation have received comparatively limited attention [144]. Blood flow exposes CTCs to varying degrees of shear stress depending on the vessel type and the local hemodynamic conditions. In large arteries, high shear forces can induce cell deformation, membrane disruption, and mechanical apoptosis, while in capillary beds or venous systems, lower shear may facilitate cellular arrest and adhesion [145,146]. Moreover, the shape, stiffness and deformability of CTCs influence their ability to navigate narrow capillaries and avoid entrapment or destruction [147]. These biomechanical parameters are further complicated by the interaction of CTCs with circulating platelets, leukocytes, and endothelial cells, forming heterotypic aggregates that may either protect ttumor cells or contribute to their clearance [148,149].
Importantly, in patients with chronic vascular conditions such as smoking-induced vasculopathy, the intravascular landscape may be profoundly altered. Endothelial dysfunction, vessel wall thickening, and microvascular rarefaction can all change the physical forces acting on CTCs and the mechanical properties of the vascular niche [150,151]. Such alterations may impair the regular laminar flow and induce turbulent patterns that influence CTC arrest, rolling, and extravasation [152]. Moreover, stiffened and inflamed endothelium may facilitate abnormal adhesion molecule expression, allowing neoplastic clones to interact more readily with the vessel wall and initiate the metastatic cascade [153]. A particularly overlooked and clinically relevant scenario is the presence of arterial aneurysms, especially in major vessels such as the aorta, iliac, and femoral arteries, which are not infrequent in heavy smokers or patients with coexisting COPD [154,155]. These aneurysmal dilatations represent zones of altered wall compliance and disturbed flow dynamics, often characterized by low and oscillatory shear stress, vortex formation, and prolonged blood residence times [156]. Such hemodynamic profiles are known to promote thrombus formation and endothelial activation but may also enhance the mechanical trapping and adhesion of CTCs [157,158]. From a pathophysiological standpoint, the interface between intraluminal thrombi, endothelial dysfunction, and turbulent flow within aneurysms could create a permissive microenvironment for metastatic cell docking and transmigration [159]. Intraluminal thrombus may act as a scaffold where platelets and neutrophils release cytokines and extracellular traps (NETs), contributing to local immunosuppression and promoting CTC survival [160,161]. Furthermore, the transition zones between aneurysmal and normal-caliber segments may represent focal points of flow deceleration and mechanical impaction, further amplifying the risk of CTC arrest [162].
These observations are not only of theoretical interest but hold significant clinical implications with valuable insights into the anatomic distribution and hemodynamic profiles of aneurysms in LC patients, potentially identifying anatomical “hot spots” for metastatic seeding [163]. In selected cases, vascular imaging data could be integrated with circulating tumor DNA (ctDNA) or CTC analysis to correlate vascular abnormalities with metastatic burden [164]. Moreover, in the future, prophylactic or therapeutic vascular interventions aimed at modulating aneurysmal flow (e.g., endograft reshaping) could become part of a multidisciplinary strategy to limit systemic dissemination in high-risk oncologic patients [165]. Recent advances in computational modelling and in vitro flow-based systems have begun to shed light on these biomechanical interactions. Microfluidic devices mimicking capillary networks and blood flow conditions have been used to quantify the deformability and adhesive behaviour of tumor cells under dynamic shear conditions [166,167]. Such platforms may be instrumental in deciphering how altered hemodynamics in vasculopathic patients affect the efficiency of metastatic spread and could lead to the identification of novel mechanical vulnerabilities in circulating cancer cells [168]. In patients with atherosclerotic disease, vessel wall stiffness and plaque formation lead to disturbed flow profiles characterized by low shear stress zones and vortex formation [28]. These microenvironments favor CTC margination, rolling, and eventual adhesion to the endothelial surface. Similarly, arterial aneurysm, which are increasingly detected in lung cancer patients, especially smokers, create localized areas of low velocity and high recirculation, acting as potential mechanical traps for CTCs [151]. Preliminary computational models suggest that such aneurysmal niches could serve as “metastatic filters,” wherein tumor cells are temporarily retained, exposed to altered oxygen gradients and cytokine profiles, and eventually driven to extravasate under local inflammatory and mechanical cues [169]. These regions may also sustain platelet-rich microthrombi, further enhancing tumor–endothelial interactions [170]. Advanced computational fluid dynamics (CFD), integrated with patient-specific vascular geometries obtained via CT-angiography or MRI, allows precise simulation of hemodynamic patterns in both normal and diseased vessels. In silico studies have successfully modelled flow and shear profiles within thoracic aneurysms, CTC trajectory and deformation under pulsatile pressure waves and tumor cell adhesion under pro-inflammatory endothelial activation [171]. Recent hybrid models also incorporate multi-physics elements, combining mechanical stress with biochemical gradients (e.g., oxygen tension, cytokine fields) to predict the likelihood of metastatic seeding in given vascular districts [172]. These platforms may help identify high-risk vascular phenotypes, especially in patients with combined COPD and systemic vasculopathy [173].
4.2. Apolipoproteins
A growing body of evidence implicates apolipoproteins (particularly ApoA-I and ApoE) as key modulators of lung cancer metastasis through both vascular and immune pathways. ApoA-I, the principal protein of high-density lipoprotein (HDL), exerts potent anti-oxidative and anti-inflammatory effects while promoting cholesterol efflux from macrophages and endothelial cells. In murine models, ApoA-I mimetic peptides have been shown to reprogram tumor-associated macrophages toward an M1-like phenotype, reduce secretion of pro-metastatic cytokines (e.g., IL-6, TNF-α), and ultimately could suppress pulmonary metastatic burden [174]. By contrast, ApoE appears to facilitate neoplastic dissemination: elevated ApoE expression in lung cancer cells enhances adhesion to LRP1-expressing endothelium, promotes transendothelial migration under disturbed flow, and increases metastatic colonization in distal organs [175].
Importantly, patients with COPD and smoking-related vasculopathy often exhibit dysregulated lipoprotein profiles, characterized by reduced ApoA-I/HDL levels and increased ApoE/LDL ratios [176]; this may exacerbate endothelial dysfunction and alter local shear stress patterns. Such lipid imbalances can impair endothelial barrier integrity, promote low-shear niches in aneurysmal segments, and synergize with the inflammatory milieu to create “metastatic traps” for circulating tumor cells. Taken together, these findings position apolipoproteins at the crossroads of lipid metabolism, vascular mechanics, and immune modulation, suggesting their potential as both biomarkers of metastatic risk and targets for novel interventions within the immune-vascular axis.
5. Smoking, COPD and Systemic Vasculopathy in Lung Cancer
The intricate interplay between chronic smoking, COPD and systemic vasculopathy forms a complex and pro-inflammatory background that influences both carcinogenesis and cancer progression in the lung. Cigarette smoke represents a multifaceted insult, rich in oxidants, reactive nitrogen species, and carcinogens, which chronically activates epithelial cells, fibroblasts, macrophages, and endothelial structures. The result is a persistent inflammatory milieu that extends well beyond the airway architecture, involving the pulmonary vasculature and systemic endothelium [177]. COPD itself is now recognized not only as a localized respiratory disorder but as a systemic inflammatory syndrome, often associated with endothelial dysfunction, arterial stiffness, and accelerated atherosclerosis. Structural and functional alterations in the vascular tree (including reduced nitric oxide bioavailability, increased endothelin-1 expression, and pro-coagulant status) are frequently observed in patients with moderate to severe COPD. These changes predispose to vascular remodelling, aneurysm formation, and chronic hypoperfusion, and may facilitate a microenvironment permissive for tumor growth and dissemination [34]. Furthermore, hypoxia (a common feature in advanced COPD) plays a dual role in modulating tumor biology. Hypoxic stress in the lung activates hypoxia-inducible factors (HIFs) that promote angiogenesis, epithelial–mesenchymal transition (EMT), and immune evasion in neoplastic clones. Simultaneously, hypoxia exacerbates endothelial injury and promotes local and systemic vasoconstriction, further impairing vascular homeostasis. The end result is a “double-hit” phenomenon, where the vasculature is simultaneously primed for neoplastic invasion and incapable of mounting an effective barrier [38]. Another critical dimension is the profound impact of smoking and COPD on the immune-inflammatory landscape. Both are associated with systemic neutrophilic activation, elevated levels of TNF-α, IL-6, and IL-1β, and reduced regulatory T cell activity. These alterations skew immune responses toward a pro-tumorigenic, tolerogenic profile. Moreover, smoking is a major inducer of clonal hematopoiesis and immunosenescence, which could further impair antitumor immunity and impact the efficacy of immune checkpoint inhibitors (ICIs) [33].
Interestingly, vascular damage in COPD patients is not limited to atherosclerosis. Medial thinning, elastin degradation, and focal aneurysmal dilatations have been documented in postmortem analyses of patients with advanced COPD, particularly in smokers with concurrent pulmonary hypertension. These vascular abnormalities may alter the distribution of pulmonary and systemic blood flow, enhancing the retention and survival of CTCs and contributing to the peculiar metastatic patterns observed in lung cancer patients with significant smoking history [178].
These observations could potentially have implications for metastasis, therapeutic delivery, and immune modulation. This complex scenario calls for integrated therapeutic strategies capable of targeting both the neoplastic process and the underlying vascular-inflammatory substrate [179].
6. The Immune-Inflammatory Axis in Vasculopathy and Lung Cancer Dissemination.
The chronic immune-inflammatory state observed in both smoking-induced vasculopathy and lung malignancy creates a biologically fertile ground for tumor progression and systemic dissemination (Tab.1). In this setting, the immune system, rather than acting as a barrier, may paradoxically facilitate metastatic spread through a series of dysfunctional responses involving both innate and adaptive components [180,181,182,183,184]. Central to this phenomenon is the endothelial cell, which serves as both a target and a mediator of inflammatory injury. Activated endothelium in vasculopathic patients expresses increased levels of adhesion molecules such as ICAM-1, VCAM-1, and E-selectin, promoting leukocyte trafficking and, inadvertently, facilitating the adhesion and transmigration of circulating tumor cells (CTCs). Simultaneously, endothelial permeability is increased under inflammatory conditions, easing the paracellular passage of tumor cells into distant tissues [185,186,187].

Table 1.
Point for clinical practice. Risk factors, mechanisms and potential therapeutic targets of metastatic dissemination in smoke-induced pathologic blood vessels. TNF: tumor necrosis factor; CSF-1R: Receptor of the Colony-Stimulating Factor-1; ROS: Reactive Oxygen Species, NO: Nitric Oxide; IFP: Interstitial Fluid Pressure; MMP: Matrix MetalloProteinases; ECM: Extracellular Matrix; WSS: Wall Shear Stress; DNTM: DNA Methyltransferase; HDAC: Histone Deacetylase.
Table 1.
Point for clinical practice. Risk factors, mechanisms and potential therapeutic targets of metastatic dissemination in smoke-induced pathologic blood vessels. TNF: tumor necrosis factor; CSF-1R: Receptor of the Colony-Stimulating Factor-1; ROS: Reactive Oxygen Species, NO: Nitric Oxide; IFP: Interstitial Fluid Pressure; MMP: Matrix MetalloProteinases; ECM: Extracellular Matrix; WSS: Wall Shear Stress; DNTM: DNA Methyltransferase; HDAC: Histone Deacetylase.
| l | Factor | Mechanism | Potential actionable target |
| Immune-Inflammatory | IL-1β, TNF-α, IL-6 | Pro-inflammatory cytokines promoting chronic inflammation, tumor promotion, angiogenesis | Anti-cytokine therapies (e.g., IL-1β inhibitors, anti-TNF agents) |
| NF-κB, STAT3, HIF-1α signalling | Sustained pro-survival and inflammatory signalling pathways | NF-κB/STAT3 inhibitors, HIF-1α modulators | |
| T-reg depletion, CD8+ T cell predominance | Immune imbalance, reduced immunosurveillance | T-reg restoration, immune checkpoint modulation | |
| Macrophage polarization (M1 dominance, tumor-associated macrophages) | Pro-tumor inflammation, matrix remodeling | CSF-1R inhibitors, macrophage reprogramming | |
| Oxidative stress | ROS, mitochondrial dysfunction | DNA damage, impaired apoptosis | Antioxidants, mitochondrial protective agents |
| Vascular dysfunction | Endothelial adhesion molecules (VCAM-1, ICAM-1, E-selectin) | Promotes leukocyte adhesion, CTC arrest | Anti-adhesion therapies (e.g., selectin blockers) |
| Reduced NO bioavailability | Endothelial dysfunction, impaired vasodilation | NO donors, endothelial stabilizers | |
| Pathologic angiogenesis (VEGF, Angiopoietin-2) | Abnormal, leaky vessels facilitating metastasis | Anti-VEGF therapies, angiopoietin pathway inhibitors | |
| Biomechanical | Low shear stress, turbulent flow, flow stagnation | Facilitates CTC adhesion, extravasation | Vascular normalization, flow modulation |
| Elevated IFP | Drives outward migration of tumor cells | Anti-VEGF, normalization of tumor IFP | |
| Extracellular matrix | MMP-2, MMP-9 | ECM degradation enabling invasion | MMP inhibitors |
| Epigenetic/Genetic | DNA methylation, histone modifications | Silencing of tumor suppressor genes | Epigenetic drugs (e.g., DNMT inhibitors, HDAC inhibitors) |
| Apolipoproteins/Lipids | Oxidized LDL, ApoB/ApoE dysregulation | Endothelial activation, macrophage recruitment | Lipid-lowering agents, ApoE modulators |
| Aneurysmal niche | MMP overexpression, inflammatory cell infiltration | Vessel wall degradation, permissive microenvironment | MMP inhibitors, anti-inflammatory therapies |
| Hemodynamic abnormalities in aneurysm (low WSS, recirculation zones) | Increased CTC residence time and adhesion | Flow-altering endovascular interventions | |
| Pharmacologic interactions | Corticosteroids, ICS | Immune suppression, impaired antigen presentation | Tapering strategies, ICS alternatives |
| Aspirin, P2Y12 inhibitors | Reduce platelet cloaking of CTCs, inhibit thrombosis | Consider as adjunct to anti-metastatic therapy | |
| Triple inhaled therapy (ICS/LABA/LAMA) | Reduces inflammation, improves oxygenation | May indirectly mitigate hypoxia-driven tumor progression | |
| PD-L1 expression | Immune evasion by inhibiting T-cell-mediated cytotoxicity | Enhances tumor immune escape and metastatic spread | Anti-PD-1/PD-L1 therapy (e.g., pembrolizumab) |
| Tumor-stromal cross-talk | EMT promotion (TGF-β, matrix degradation) | Increases invasiveness, resistance to apoptosis | TGF-β inhibitors, EMT blockers |
Among innate immune players, neutrophils have a prominent role. Chronic inflammation leads to persistent neutrophil activation and degranulation, with release of reactive oxygen species (ROS), matrix metalloproteinases (MMPs), and the formation of neutrophil extracellular traps (NETs). NETs have been shown to ensnare CTCs, protect them from immune clearance, and even promote their extravasation and metastatic outgrowth. In murine models, NET-rich microenvironments were associated with enhanced metastatic colonization of the lung and liver [188]. Monocyte/macrophage polarization is also critical. In the vasculopathic lung, macrophages are often turned toward an M2-like phenotype, promoting tissue remodelling, immunosuppression, and tumor angiogenesis. These macrophages can release VEGF, TGF-β, and IL-10, factors that not only support tumor growth but also modulate endothelial behaviour and vascular permeability [189]. The adaptive immune system is not spared. Chronic smoking and inflammation drive T cell exhaustion, reduce cytotoxic CD8+ T cell activity, and promote the expansion of regulatory T cells (Tregs). This shift in immune balance may reduce immune surveillance against disseminated tumor cells and promote their survival and proliferation at secondary sites. Moreover, altered dendritic cell function in vasculopathic tissues limits effective antigen presentation, further weakening anti-tumor immunity [190].
At the molecular level, the crosstalk between inflammatory cytokines (IL-6, IL-1β, TNF-α), chemokines (CXCL12, CCL2), and proangiogenic signals (VEGF-A, Ang2) orchestrates a permissive metastatic niche in vasculopathic environments. This network sustains the pre-metastatic conditioning of distant organs, paving the way for efficient seeding of tumor clones [191]. A particularly relevant concept is the “inflammatory pre-metastatic niche”, wherein systemic inflammation and vascular pathology create specific molecular and cellular landscapes in organs distant from the primary tumor, which become more receptive to metastatic colonization. As above discussed, this niche is characterized by endothelial activation, extracellular matrix remodelling, stromal cell recruitment, and immune suppression, all of which are exacerbated in patients with underlying vasculopathy. In this scenario, the vasculature is no longer a passive conduit but becomes an active facilitator of metastatic spread, both as a dysfunctional barrier and as a pro-inflammatory signal amplifier [138].
Immune Checkpoint Inhibitors, Bronchodilation, and Anti-Platelet Therapy
The advent of immune checkpoint inhibitors (ICIs) has transformed the therapeutic landscape of non-small cell lung cancer (NSCLC), offering prolonged survival even in advanced disease stages [192]. However, the efficacy and safety profile of ICIs can be significantly influenced by the underlying cardio-pulmonary and vascular status of the patient, including co-administered medications like bronchodilators and antiplatelet agents. Indeed, ICIs act by restoring cytotoxic T cell function, primarily via blockade of the PD-1/PD-L1 and CTLA-4 axes [9,37,162,193]. However, vascular inflammation can modulate the tumor microenvironment in ways that interfere with ICI efficacy. Platelets are emerging as crucial players in cancer biology. and secrete growth factors such as PDGF and TGF-β that enhance tumor cell survival. In vasculopathic patients, low-dose aspirin or platelet receptor P2Y₁₂ inhibitors are frequently prescribed, and their anti-metastatic potential is receiving growing attention [194]. Triple bronchodilation (typically combining a long-acting β₂-agonist (LABA), a long-acting muscarinic antagonist (LAMA), and an inhaled corticosteroid (ICS) is the cornerstone of COPD management in patients with severe airflow limitation. These drugs, beyond their airway-specific effects, can have systemic implications relevant to tumor-immune dynamics [8,33,195]. Taken together, this pharmacologic triad, immunotherapy, bronchodilation, and antiplatelet therapy represents a highly dynamic system, in which reciprocal interactions may profoundly shape lung cancer outcomes. However, some relevant issues need to be implemented and clarified (Tab.2). Understanding these interdependencies is essential for designing personalized treatment strategies in patients with complex vascular and respiratory comorbidities.
Table 2.
Known key points and gap of knowledge regarding synergy and mutual interferences between immune checkpoint inhibitors, triple bronchodilators and antiplatelet agents.
Table 2.
Known key points and gap of knowledge regarding synergy and mutual interferences between immune checkpoint inhibitors, triple bronchodilators and antiplatelet agents.
| Key points | Knowledge gap |
|
LUNG CANCER →Immune checkpoint inhibitors Inflamed or remodelled vasculature in COPD or systemic vasculopathy may be a critical determinant of response to immunotherapy | |
|
|
|
COPD → Triple bronchodilation Beyond airway-specific effects, it can have systemic implications relevant to tumor-immune dynamics | |
|
|
|
VASCULOPATHY→Antiplatelet therapy Platelets can shield CTCs from immune recognition and promote their extravasation | |
|
|
7. Conclusion
Smoking habit is known be strictly related and interconnected to the arousal of both arteriopathy and cancer and a large amount of experimental reports points out the biologic basis linking smoke-induced malignant transformation and peripheral arterial disease. Although smokers most often develop cancer and chronic arteriopathy, these conditions are usually evaluated as distinct nosologic entities. While substantial research has focused on the genetic, epigenetic, and immunologic drivers of LC spreading progression, the role of mechanical forces governing metastatic dissemination remains largely unexplored. Understanding the biomechanical and molecular dynamics of LC progression in altered vascular territories offers several translational opportunities: i) risk stratification: CFD models could be used to predict regions at higher metastatic risk based on vascular topology and flow conditions [196]; ii) surgical planning: in patients with aneurysms or localized vascular remodelling, identifying pro-metastatic hemodynamic niches may guide endovascular interventions aimed not only at preventing rupture, but also at disrupting potential metastatic highways [197]; iii) therapeutic targeting: modulation of flow dynamics via antithrombotic therapy, endothelial normalization agents, or even implantable flow regulators could theoretically reduce CTC retention and dissemination .
Ultimately, bridging vascular surgery, oncology, and computational biology may open novel avenues in the prevention of metastatic disease, particularly in patients with complex comorbid profiles.
Conflicts of Interest
The authors have nothing to disclose.
Support Statement
Ricerca corrente 5x1000-2020 (cod. 090000X121—progetto 08050122) to G.M. Stella.
References
- Cancer Facts & Figures 2023. Atlanta: American Cancer Society, Inc.; 2022. Accessed January 9, 2024. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf.
- Kızılırmak D, Yılmaz Z, Havlucu Y, Çelik P. Impact of the COVID-19 Pandemic on Diagnosis of Lung Cancer. SN Compr Clin Med. 2023;5(1):23. [CrossRef] [PubMed]
- Cantini L, Mentrasti G, Russo GL, Signorelli D, Pasello G, Rijavec E, Russano M, Antonuzzo L, Rocco D, Giusti R, Adamo V, Genova C, Tuzi A, Morabito A, Gori S, Verde N, Chiari R, Cortellini A, Cognigni V, Pecci F, Indini A, De Toma A, Zattarin E, Oresti S, Pizzutilo EG, Frega S, Erbetta E, Galletti A, Citarella F, Fancelli S, Caliman E, Della Gravara L, Malapelle U, Filetti M, Piras M, Toscano G, Zullo L, De Tursi M, Di Marino P, D'Emilio V, Cona MS, Guida A, Caglio A, Salerno F, Spinelli G, Bennati C, Morgillo F, Russo A, Dellepiane C, Vallini I, Sforza V, Inno A, Rastelli F, Tassi V, Nicolardi L, Pensieri V, Emili R, Roca E, Migliore A, Galassi T, Rocchi MLB, Berardi R. Evaluation of COVID-19 impact on DELAYing diagnostic-therapeutic pathways of lung cancer patients in Italy (COVID-DELAY study): fewer cases and higher stages from a real-world scenario. ESMO Open. 2022 Apr;7(2):100406. [CrossRef] [PubMed]
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024 Jan-Feb;74(1):12-49. [CrossRef]
- Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454.
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non–small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594.
- Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495–509.
- Tsoukalas N, Aravantinou-Fatorou E, Baxevanos P, Tolia M, Tsapakidis K, Galanopoulos M, et al. Molecular biomarkers in non-small cell lung cancer: a review. Curr Pharm Biotechnol. 2018;19(8):586–597.
- O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–167.
- Brennan P, Hainaut P, Boffetta P. Genetics of lung-cancer susceptibility. Lancet Oncol. 2011 Apr;12(4):399-408. [CrossRef]
- e Alencar VTL, Formiga MN, de Lima VCC. Inherited lung cancer: a review. Ecancermedicalscience. 2020 Jan 29;14:1008. [CrossRef]
- Kanwal M, Ding XJ, Cao Y. Familial risk for lung cancer. Oncol Lett. 2017 Feb;13(2):535-542. [CrossRef]
- Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008 Jul;9(7):676-82. [CrossRef]
- Stella GM, Luisetti M, Pozzi E, Comoglio PM. Oncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamics. Lancet Respir Med. 2013 May;1(3):251-61. [CrossRef]
- Benusiglio PR, Fallet V, Sanchis-Borja M, Coulet F, Cadranel J. Lung cancer is also a hereditary disease. Eur Respir Rev. 2021 Oct 20;30(162):210045. [CrossRef]
- Smolle E, Pichler M. Non-Smoking-Associated Lung Cancer: A distinct Entity in Terms of Tumor Biology, Patient Characteristics and Impact of Hereditary Cancer Predisposition. Cancers (Basel). 2019 Feb 10;11(2):204. [CrossRef]
- Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010 Mar;35(3):496-504. [CrossRef]
- Stella GM, D'Agnano V, Piloni D, Saracino L, Lettieri S, Mariani F, Lancia A, Bortolotto C, Rinaldi P, Falanga F, Primiceri C, Corsico AG, Bianco A. The oncogenic landscape of the idiopathic pulmonary fibrosis: a narrative review. Transl Lung Cancer Res. 2022 Mar;11(3):472-496. [CrossRef]
- Lettieri S, Oggionni T, Lancia A, Bortolotto C, Stella GM. Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? Int J Mol Sci. 2021 Mar 12;22(6):2882. [CrossRef]
- Perrotta F, Chino V, Allocca V, D'Agnano V, Bortolotto C, Bianco A, Corsico AG, Stella GM. Idiopathic pulmonary fibrosis and lung cancer: targeting the complexity of the pharmacological interconnection. Expert Rev Respir Med. 2022 Oct;16(10):1043-1055. [CrossRef]
- CDC https://www.cdc.gov/lung-cancer/risk-factors/index.html#:~:text=People%20who%20smoke%20cigarettes%20are,the%20risk%20of%20lung%20cancer.
- Crispo A, Brennan P, Jöckel KH, Schaffrath-Rosario A, Wichmann HE, Nyberg F, Simonato L, Merletti F, Forastiere F, Boffetta P, Darby S. The cumulative risk of lung cancer among current, ex- and never-smokers in European men. Br J Cancer. 2004 Oct 4;91(7):1280-6. [CrossRef]
- Asomaning K, Miller DP, Liu G, Wain JC, Lynch TJ, Su L, Christiani DC. Second hand smoke, age of exposure and lung cancer risk. Lung Cancer. 2008 Jul;61(1):13-20. [CrossRef]
- Berg CD, Schiller JH, Boffetta P, Cai J, Connolly C, Kerpel-Fronius A, Kitts AB, Lam DCL, Mohan A, Myers R, Suri T, Tammemagi MC, Yang D, Lam S; International Association for the Study of Lung Cancer (IASLC) Early Detection and Screening Committee. Air Pollution and Lung Cancer: A Review by International Association for the Study of Lung Cancer Early Detection and Screening Committee. J Thorac Oncol. 2023 Oct;18(10):1277-1289. [CrossRef]
- Loomis D, Huang W, Chen G. The International Agency for Research on Cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: focus on China. Chin J Cancer. 2014 Apr;33(4):189-96. [CrossRef]
- Myers R, Brauer M, Dummer T, Atkar-Khattra S, Yee J, Melosky B, Ho C, McGuire AL, Sun S, Grant K, Lee A, Lee M, Yuchi W, Tammemagi M, Lam S. High-Ambient Air Pollution Exposure Among Never Smokers Versus Ever Smokers With Lung Cancer. J Thorac Oncol. 2021 Nov;16(11):1850-1858. [CrossRef]
- Brunekreef B, Beelen R, Hoek G, Schouten L, Bausch-Goldbohm S, Fischer P, Armstrong B, Hughes E, Jerrett M, van den Brandt P. Effects of long-term exposure to traffic-related air pollution on respiratory and cardiovascular mortality in the Netherlands: the NLCS-AIR study. Res Rep Health Eff Inst. 2009 Mar;(139):5-71; discussion 73-89. [PubMed]
- Shahadin MS, Ab Mutalib NS, Latif MT, Greene CM, Hassan T. Challenges and future direction of molecular research in air pollution-related lung cancers. Lung Cancer. 2018 Apr;118:69-75. [CrossRef]
- Celià-Terrassa T, Kang Y. Metastatic niche functions and therapeutic opportunities. Nat Cell Biol. 2018 Aug;20(8):868-877. [CrossRef]
- Patel SA, Rodrigues P, Wesolowski L, Vanharanta S. Genomic control of metastasis. Br J Cancer. 2021 Jan;124(1):3-12. [CrossRef]
- Kiri S, Ryba T. Cancer, metastasis, and the epigenome. Mol Cancer. 2024 Aug 2;23(1):154. [CrossRef]
- Stella GM, Senetta R, Cassenti A, Ronco M, Cassoni P. Cancers of unknown primary origin: current perspectives and future therapeutic strategies. J Transl Med. 2012 Jan 24;10:12. [CrossRef]
- Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27.
- Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459.
- Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026.
- Mazzuca P, Carloni A, Cazzato F, et al. Tobacco smoke and the vascular endothelium: from inflammation to transformation. Int J Mol Sci. 2020;21(21):7755.
- Yang IA, Relan V, Wright CM, Davidson MR, Sriram KB, Savarimuthu Francis SM, et al. Common pathogenic mechanisms and epigenetic changes in lung cancer and COPD. Am J Pathol. 2011;178(3):1353–1360.
- Wang Y, Zhang Y, Ma J, et al. The role of COPD in lung cancer: insights from human and murine studies. Cancer Lett. 2022;543:215797.
- Moorthy B, Chu C, Carlin DJ. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol Sci. 2015 May;145(1):5-15. [CrossRef]
- Goldman R, Enewold L, Pellizzari E, Beach JB, Bowman ED, Krishnan SS, Shields PG. Smoking increases carcinogenic polycyclic aromatic hydrocarbons in human lung tissue. Cancer Res. 2001 Sep 1;61(17):6367-71.
- Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005 Sep;289(3):L391-9. [CrossRef]
- Shehata SA, Toraih EA, Ismail EA, Hagras AM, Elmorsy E, Fawzy MS. Vaping, Environmental Toxicants Exposure, and Lung Cancer Risk. Cancers (Basel). 2023 Sep 12;15(18):4525. [CrossRef]
- Wang Q, Jiang C, Hsu ML, Wisnivesky J, Dowlati A, Boffetta P, Kong CY. E-Cigarette Use and Lung Cancer Screening Uptake. JAMA Netw Open. 2024 Jul 1;7(7):e2419648. [CrossRef]
- Allbright K, Villandre J, Crotty Alexander LE, Zhang M, Benam KH, Evankovich J, Königshoff M, Chandra D. The paradox of the safer cigarette: understanding the pulmonary effects of electronic cigarettes. Eur Respir J. 2024 Jun 28;63(6):2301494. [CrossRef]
- https://www.cdc.gov/tobacco/e-cigarettes/youth.
- Klebe S, Leigh J, Henderson DW, Nurminen M. Asbestos, Smoking and Lung Cancer: An Update. Int J Environ Res Public Health. 2019 Dec 30;17(1):258. [CrossRef]
- van Zandwijk N, Frank AL, Reid G, Dimitri Røe O, Amos CI. Asbestos-Related lung Cancer: An underappreciated oncological issue. Lung Cancer. 2024 Aug;194:107861. [CrossRef]
- Saracino L, Zorzetto M, Inghilleri S, Pozzi E, Stella GM. Non-neuronal cholinergic system in airways and lung cancer susceptibility. Transl Lung Cancer Res. 2013 Aug;2(4):284-94. [CrossRef]
- Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009 Jan 1;124(1):36-45. [CrossRef]
- Di Cello F, Flowers VL, Li H, Vecchio-Pagán B, Gordon B, Harbom K, Shin J, Beaty R, Wang W, Brayton C, Baylin SB, Zahnow CA. Cigarette smoke induces epithelial to mesenchymal transition and increases the metastatic ability of breast cancer cells. Mol Cancer. 2013 Aug 6;12:90. [CrossRef]
- Prashanth N, Meghana P, Sandeep Kumar Jain R, Pooja S Rajaput, Satyanarayan N D, Raja Naika H, Kumaraswamy H M. Nicotine promotes epithelial to mesenchymal transition and gemcitabine resistance via hENT1/RRM1 signalling in pancreatic cancer and chemosensitizing effects of Embelin-a naturally occurring benzoquinone. Sci Total Environ. 2024 Mar 1;914:169727. [CrossRef]
- Chen PC, Lee WY, Ling HH, Cheng CH, Chen KC, Lin CW. Activation of fibroblasts by nicotine promotes the epithelial-mesenchymal transition and motility of breast cancer cells. J Cell Physiol. 2018 Jun;233(6):4972-4980. [CrossRef]
- Dinicola S, Masiello MG, Proietti S, Coluccia P, Fabrizi G, Catizone A, Ricci G, de Toma G, Bizzarri M, Cucina A. Nicotine increases colon cancer cell migration and invasion through epithelial to mesenchymal transition (EMT): COX-2 involvement. J Cell Physiol. 2018 Jun;233(6):4935-4948.
- Raveendran M, Senthil D, Utama B, Shen Y, Dudley D, Wang J, Zhang Y, Wang XL. Cigarette suppresses the expression of P4H[alpha] and vascular collagen production. Biochem Biophys Res Com. 2004;323:592–598. [CrossRef]
- Lemaître V, Dabo AJ, D'Armiento J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicological Sciences. 2011;123:542–549. [CrossRef]
- Arif B, Garcia-Fernandez F, Ennis TL, Jin J, Davis EC, Thompson RW, Curci JA. Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol. 2012;32:2901–2909. [CrossRef]
- Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res. 2000;92:85–95. [CrossRef]
- Polytarchou C, Iliopoulos D, Hatziapostolou M, Kottakis F, Maroulakou I, Struhl K, Tsichlis PN. Akt2 regulates all Akt isoforms and promotes resistance to hypoxia through induction of miR-21 upon oxygen deprivation. Cancer Research. 2011;71:4720–4731. [CrossRef]
- Husgafvel-Pursiainen, K. Genotoxicity of environmental tobacco smoke: a review. Mutat Res. 2004 Nov;567(2-3):427-45. [CrossRef]
- Stannard L, Doak SH, Doherty A, Jenkins GJ. Is Nickel Chloride really a Non-Genotoxic Carcinogen? Basic Clin Pharmacol Toxicol. 2017 Sep;121 Suppl 3:10-15. [CrossRef]
- Jianlin L, Guohai C, Guojun Z, Jian J, Fangfang H, Juanjuan X, Shu Z, Zhijian C, Wei J, Yezhen L, Xiaoxue L, Jiliang H. Assessing cytogenotoxicity of cigarette smoke condensates using three in vitro assays. Mutat Res. 2009 Jun-Jul;677(1-2):21-6. [CrossRef]
- Messner B, Frotschnig S, Steinacher-Nigisch A, Winter B, Eichmair E, Gebetsberger J, Schwaiger S, Ploner C, Laufer G, Bernhard D. Apoptosis and necrosis: two different outcomes of cigarette smoke condensate-induced endothelial cell death. Cell Death Dis. 2012 Nov 15;3(11):e424. [CrossRef]
- Khulan B, Ye K, Shi MK, Waldman S, Marsh A, Siddiqui T, Okorozo A, Desai A, Patel D, Dobkin J, Sadoughi A, Shah C, Gera S, Peter Y, Liao W, Vijg J, Spivack SD. Normal bronchial field basal cells show persistent methylome-wide impact of tobacco smoking, including in known cancer genes. Epigenetics. 2025 Dec;20(1):2466382. [CrossRef]
- Herzog C, Jones A, Evans I, Raut JR, Zikan M, Cibula D, Wong A, Brenner H, Richmond RC, Widschwendter M. Cigarette Smoking and E-cigarette Use Induce Shared DNA Methylation Changes Linked to Carcinogenesis. Cancer Res. 2024 Jun 4;84(11):1898-1914. [CrossRef]
- Essogmo FE, Zhilenkova AV, Tchawe YSN, Owoicho AM, Rusanov AS, Boroda A, Pirogova YN, Sangadzhieva ZD, Sanikovich VD, Bagmet NN, Sekacheva MI. Cytokine Profile in Lung Cancer Patients: Anti-Tumor and Oncogenic Cytokines. Cancers (Basel). 2023 Nov 13;15(22):5383. [CrossRef]
- Cook JH, Melloni GEM, Gulhan DC, Park PJ, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun. 2021 Mar 22;12(1):1808. [CrossRef]
- O'Sullivan É, Keogh A, Henderson B, Finn SP, Gray SG, Gately K. Treatment Strategies for KRAS-Mutated Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 Mar 7;15(6):1635. [CrossRef]
- Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, Kato T, Curioni-Fontecedro A, Sacher A, Spira A, Ramalingam SS, Takahashi T, Besse B, Anderson A, Ang A, Tran Q, Mather O, Henary H, Ngarmchamnanrith G, Friberg G, Velcheti V, Govindan R. Sotorasib for Lung Cancers with KRASp.G12C Mutation. N Engl J Med. 2021 Jun 24;384(25):2371-2381. [CrossRef]
- Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, Johnson ML, Sabari JK, Leventakos K, Yau E, Bazhenova L, Negrao MV, Pennell NA, Zhang J, Anderes K, Der-Torossian H, Kheoh T, Velastegui K, Yan X, Christensen JG, Chao RC, Spira AI. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N Engl J Med. 2022 Jul 14;387(2):120-131. [CrossRef]
- Sidaway, P. Sotorasib effective in KRAS-mutant NSCLC. Nat Rev Clin Oncol. 2021 Aug;18(8):470. [CrossRef]
- Longo DL, Rosen N. Targeting Oncogenic RAS Protein. N Engl J Med. 2022 Jul 14;387(2):184-186. [CrossRef]
- Le Calvez F, Mukeria A, Hunt JD, Kelm O, Hung RJ, Tanière P, Brennan P, Boffetta P, Zaridze DG, Hainaut P. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005 Jun 15;65(12):5076-83. [CrossRef]
- Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S, Moreno-Macias H, Smith JA, Brody JA, Dhingra R, Yousefi P, Pankow JS, Kunze S, Shah SH, McRae AF, Lohman K, Sha J, Absher DM, Ferrucci L, Zhao W, Demerath EW, Bressler J, Grove ML, Huan T, Liu C, Mendelson MM, Yao C, Kiel DP, Peters A, Wang-Sattler R, Visscher PM, Wray NR, Starr JM, Ding J, Rodriguez CJ, Wareham NJ, Irvin MR, Zhi D, Barrdahl M, Vineis P, Ambatipudi S, Uitterlinden AG, Hofman A, Schwartz J, Colicino E, Hou L, Vokonas PS, Hernandez DG, Singleton AB, Bandinelli S, Turner ST, Ware EB, Smith AK, Klengel T, Binder EB, Psaty BM, Taylor KD, Gharib SA, Swenson BR, Liang L, DeMeo DL, O'Connor GT, Herceg Z, Ressler KJ, Conneely KN, Sotoodehnia N, Kardia SL, Melzer D, Baccarelli AA, van Meurs JB, Romieu I, Arnett DK, Ong KK, Liu Y, Waldenberger M, Deary IJ, Fornage M, Levy D, London SJ. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet. 2016 Oct;9(5):436-447. [CrossRef]
- Raso MG, Wistuba II. Molecular pathogenesis of early-stage non-small cell lung cancer and a proposal for tissue banking to facilitate identification of new biomarkers. J Thorac Oncol. 2007 Jul;2(7 Suppl 3):S128-35. [CrossRef]
- Okamoto T, Suzuki Y, Fujishita T, Kitahara H, Shimamatsu S, Kohno M, Morodomi Y, Kawano D, Maehara Y. The prognostic impact of the amount of tobacco smoking in non-small cell lung cancer--differences between adenocarcinoma and squamous cell carcinoma. Lung Cancer. 2014 Aug;85(2):125-30. [CrossRef]
- Maeda R, Yoshida J, Ishii G, Hishida T, Nishimura M, Nagai K. The prognostic impact of cigarette smoking on patients with non-small cell lung cancer. J Thorac Oncol. 2011 Apr;6(4):735-42. [CrossRef]
- Lee JY, Bhandare RR, Boddu SHS, Shaik AB, Saktivel LP, Gupta G, Negi P, Barakat M, Singh SK, Dua K, Chellappan DK. Molecular mechanisms underlying the regulation of tumour suppressor genes in lung cancer. Biomed Pharmacother. 2024 Apr;173:116275. [CrossRef]
- Tao S, Pu Y, Yang EJ, Ren G, Shi C, Chen LJ, Chen L, Shim JS. Inhibition of GSK3β is synthetic lethal with FHIT loss in lung cancer by blocking homologous recombination repair. Exp Mol Med. 2025 Feb;57(1):167-183. [CrossRef]
- Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004 Jul 6;101(27):10143-8. [CrossRef]
- Spira A, Beane JE, Shah V, Steiling K, Liu G, Schembri F, Gilman S, Dumas YM, Calner P, Sebastiani P, Sridhar S, Beamis J, Lamb C, Anderson T, Gerry N, Keane J, Lenburg ME, Brody JS. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007 Mar;13(3):361-6. [CrossRef]
- Castaldi P, Sauler M. Cigarette Smoking and the Airway Epithelium: Characterizing Changes in Gene Expression over Time. Am J Respir Crit Care Med. 2023 Oct 1;208(7):749-750. [CrossRef]
- Lin BC, Li QY, Tian L, Liu HL, Liu XH, Shi Y, He C, Ding SS, Yan J, Li K, Bian LP, Lai WQ, Zhang W, Li X, Xi ZG. Identification of apoptosis-associated protein factors distinctly expressed in cigarette smoke condensate-exposed airway bronchial epithelial cells. J Biochem Mol Toxicol. 2020 Mar;34(3):e22444. [CrossRef]
- Alamgeer M, Peacock CD, Matsui W, Ganju V, Watkins DN. Cancer stem cells in lung cancer: Evidence and controversies. Respirology. 2013 Jul;18(5):757-64. [CrossRef]
- Chu X, Tian W, Ning J, Xiao G, Zhou Y, Wang Z, Zhai Z, Tanzhu G, Yang J, Zhou R. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal Transduct Target Ther. 2024 Jul 5;9(1):170. [CrossRef]
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006 Sep 21;355(12):1253-61. [CrossRef]
- Bomken S, Fiser K, Heidenreich O, Vormoor J. Understanding the cancer stem cell. Br J Cancer. 2010 Aug 10;103(4):439-45. [CrossRef]
- Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells--old concepts, new insights. Cell Death Differ. 2008 Jun;15(6):947-58. [CrossRef]
- Alam H, Sehgal L, Kundu ST, Dalal SN, Vaidya MM. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol Biol Cell. 2011 Nov;22(21):4068-78. [CrossRef]
- Tanaka Y, Yamaguchi M, Hirai S, Sumi T, Tada M, Saito A, Chiba H, Kojima T, Watanabe A, Takahashi H, Sakuma Y. Characterization of distal airway stem-like cells expressing N-terminally truncated p63 and thyroid transcription factor-1 in the human lung. Exp Cell Res. 2018 Nov 15;372(2):141-149. [CrossRef]
- Hiemstra PS, Bourdin A. Club cells, CC10 and self-control at the epithelial surface. Eur Respir J. 2014 Oct;44(4):831-2. [CrossRef]
- Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell. 2020 Mar 5;26(3):346-358.e4. [CrossRef]
- Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell. 2020 Mar 5;26(3):346-358.e4. [CrossRef]
- Salwig I, Spitznagel B, Vazquez-Armendariz AI, Khalooghi K, Guenther S, Herold S, Szibor M, Braun T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019 Jun 17;38(12):e102099. [CrossRef]
- Campo-Trapero J, Cano-Sánchez J, Palacios-Sánchez B, Sánchez-Gutierrez JJ, González-Moles MA, Bascones-Martínez A. Update on molecular pathology in oral cancer and precancer. Anticancer Res. 2008 Mar-Apr;28(2B):1197-205.
- Vairaktaris E, Spyridonidou S, Papakosta V, Vylliotis A, Lazaris A, Perrea D, Yapijakis C, Patsouris E. The hamster model of sequential oral oncogenesis. Oral Oncol. 2008 Apr;44(4):315-24. [CrossRef]
- Willenbrink TJ, Ruiz ES, Cornejo CM, Schmults CD, Arron ST, Jambusaria-Pahlajani A. Field cancerization: Definition, epidemiology, risk factors, and outcomes. J Am Acad Dermatol. 2020 Sep;83(3):709-717. [CrossRef]
- SLAUGHTER DP, SOUTHWICK HW, SMEJKAL W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953 Sep;6(5):963-8. [CrossRef]
- Simple M, Suresh A, Das D, Kuriakose MA. Cancer stem cells and field cancerization of oral squamous cell carcinoma. Oral Oncol. 2015 Jul;51(7):643-51. [CrossRef]
- Aramini B, Masciale V, Grisendi G, Bertolini F, Maur M, Guaitoli G, Chrystel I, Morandi U, Stella F, Dominici M, Haider KH. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers (Basel). 2022 Feb 15;14(4):976. [CrossRef]
- Kitamura H, Okudela K, Yazawa T, Sato H, Shimoyamada H. Cancer stem cell: implications in cancer biology and therapy with special reference to lung cancer. Lung Cancer. 2009 Dec;66(3):275-81. [CrossRef]
- Tura-Ceide O, Lobo B, Paul T, Puig-Pey R, Coll-Bonfill N, García-Lucio J, Smolders V, Blanco I, Barberà JA, Peinado VI. Cigarette smoke challenges bone marrow mesenchymal stem cell capacities in guinea pig. Respir Res. 2017 Mar 23;18(1):50. [CrossRef]
- Zhu F, Guo GH, Chen W, Wang NY. Effects of bone marrow-derived mesenchymal stem cells engraftment on vascular endothelial cell growth factor in lung tissue and plasma at early stage of smoke inhalation injury. World J Emerg Med. 2010;1(3):224-8.
- Chen JT, Lin TS, Chow KC, Huang HH, Chiou SH, Chiang SF, Chen HC, Chuang TL, Lin TY, Chen CY. Cigarette smoking induces overexpression of hepatocyte growth factor in type II pneumocytes and lung cancer cells. Am J Respir Cell Mol Biol. 2006 Mar;34(3):264-73. [CrossRef]
- Yoneyama R, Aoshiba K, Furukawa K, Saito M, Kataba H, Nakamura H, Ikeda N. Nicotine enhances hepatocyte growth factor-mediated lung cancer cell migration by activating the α7 nicotine acetylcholine receptor and phosphoinositide kinase-3-dependent pathway. Oncol Lett. 2016 Jan;11(1):673-677. [CrossRef]
- Du B, Leung H, Khan KM, Miller CG, Subbaramaiah K, Falcone DJ, Dannenberg AJ. Tobacco smoke induces urokinase-type plasminogen activator and cell invasiveness: evidence for an epidermal growth factor receptor dependent mechanism. Cancer Res. 2007 Sep 15;67(18):8966-72. [CrossRef]
- Jiang YJ, Chao CC, Chang AC, Chen PC, Cheng FJ, Liu JF, Liu PI, Huang CL, Guo JH, Huang WC, Tang CH. Cigarette smoke-promoted increases in osteopontin expression attract mesenchymal stem cell recruitment and facilitate lung cancer metastasis. J Adv Res. 2022 Nov;41:77-87. [CrossRef]
- Jung YS, Park JH, Park DI, Sohn CI, Lee JM, Kim TI. Impact of Smoking on Human Natural Killer Cell Activity: A Large Cohort Study. J Cancer Prev. 2020 Mar 30;25(1):13-20. [CrossRef]
- Dinicola S, Morini V, Coluccia P, Proietti S, D'Anselmi F, Pasqualato A, Masiello MG, Palombo A, De Toma G, Bizzarri M, Cucina A. Nicotine increases survival in human colon cancer cells treated with chemotherapeutic drugs. Toxicol In Vitro. 2013 Dec;27(8):2256-63. [CrossRef]
- Liu X, Zhang J, Sun W, Cao J, Ma Z. COX-2 in lung cancer: Mechanisms, development, and targeted therapies. Chronic Dis Transl Med. 2024 Mar 12;10(4):281-292. [CrossRef]
- Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005 Nov;1056:218-33. [CrossRef]
- Guo JH, Thuong LHH, Jiang YJ, Huang CL, Huang YW, Cheng FJ, Liu PI, Liu CL, Huang WC, Tang CH. Cigarette smoke promotes IL-6-dependent lung cancer migration and osteolytic bone metastasis. Int J Biol Sci. 2024 Jun 3;20(9):3257-3268. [CrossRef]
- Zheng L, Sun R, Zhu Y, Li Z, She X, Jian X, Yu F, Deng X, Sai B, Wang L, Zhou W, Wu M, Li G, Tang J, Jia W, Xiang J. Lung microbiome alterations in NSCLC patients. Sci Rep. 2021 Jun 3;11(1):11736. [CrossRef]
- Wu F, Yin Z, Yang L, Fan J, Xu J, Jin Y, Yu J, Zhang D, Yang G. Smoking Induced Extracellular Vesicles Release and Their Distinct Properties in Non-Small Cell Lung Cancer. J Cancer. 2019 Jun 9;10(15):3435-3443. [CrossRef]
- Jiang C, Zhang N, Hu X, Wang H. Tumor-associated exosomes promote lung cancer metastasis through multiple mechanisms. Mol Cancer. 2021 Sep 13;20(1):117. [CrossRef]
- Liang Z, Fang S, Zhang Y, Zhang X, Xu Y, Qian H, Geng H. Cigarette Smoke-Induced Gastric Cancer Cell Exosomes Affected the Fate of Surrounding Normal Cells via the Circ0000670/Wnt/β-Catenin Axis. Toxics. 2023 May 17;11(5):465. [CrossRef]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015 Apr 3;348(6230):124-8. [CrossRef]
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer. 2007 Oct;7(10):778-90. [CrossRef]
- Lam DC, Liam CK, Andarini S, Park S, Tan DSW, Singh N, Jang SH, Vardhanabhuti V, Ramos AB, Nakayama T, Nhung NV, Ashizawa K, Chang YC, Tscheikuna J, Van CC, Chan WY, Lai YH, Yang PC. Lung Cancer Screening in Asia: An Expert Consensus Report. J Thorac Oncol. 2023 Oct;18(10):1303-1322. [CrossRef]
- Luo G, Zhang Y, Rumgay H, Morgan E, Langselius O, Vignat J, Colombet M, Bray F. Estimated worldwide variation and trends in incidence of lung cancer by histological subtype in 2022 and over time: a population-based study. Lancet Respir Med. 2025 Apr;13(4):348-363. [CrossRef]
- Tang FH, Wong HYT, Tsang PSW, Yau M, Tam SY, Law L, Yau K, Wong J, Farah FHM, Wong J. Recent advancements in lung cancer research: a narrative review. Transl Lung Cancer Res. 2025 Mar 31;14(3):975-990. [CrossRef]
- Thaxton C, Kano M, Mendes-Pinto D, Navarro TP, Nishibe T, Dardik A. Implications of preoperative arterial stiffness for patients treated with endovascular repair of abdominal aortic aneurysms. JVS Vasc Sci. 2024 May 21;5:100209. [CrossRef]
- Westerhof N, Lankhaar JW, Westerhof BE. The arterial Windkessel. Med Biol Eng Comput. 2009 Feb;47(2):131-41. [CrossRef]
- Schwartz JA, Keagy BA, Johnson G Jr. Determination of whole blood apparent viscosity: experience with a new hemorheologic technique. J Surg Res. 1988 Aug;45(2):238-47. [CrossRef]
- Baieth, HE. Physical parameters of blood as a non - newtonian fluid. Int J Biomed Sci. 2008 Dec;4(4):323-9.
- Lee HJ, Kim YW, Shin SY, Lee SL, Kim CH, Chung KS, Lee JS. A Physics-Integrated Deep Learning Approach for Patient-Specific Non-Newtonian Blood Viscosity Assessment using PPG. Comput Methods Programs Biomed. 2025 Mar 23;265:108740. [CrossRef]
- Willigendael EM, Teijink JA, Bartelink ML, Kuiken BW, Boiten J, Moll FL, Büller HR, Prins MH. Influence of smoking on incidence and prevalence of peripheral arterial disease. J Vasc Surg. 2004 Dec;40(6):1158-65. [CrossRef]
- Lu L, Mackay DF, Pell JP. Meta-analysis of the association between cigarette smoking and peripheral arterial disease. Heart. 2014 Mar;100(5):414-23. [CrossRef]
- Wang W, Zhao T, Geng K, Yuan G, Chen Y, Xu Y. Smoking and the Pathophysiology of Peripheral Artery Disease. Front Cardiovasc Med. 2021 Aug 27;8:704106. [CrossRef]
- Leone A, Landini L. Vascular pathology from smoking: look at the microcirculation! Curr Vasc Pharmacol. 2013 Jul;11(4):524-30. [CrossRef]
- Whitehead AK, Erwin AP, Yue X. Nicotine and vascular dysfunction. Acta Physiol (Oxf). 2021 Apr;231(4):e13631. [CrossRef]
- Lietz M, Berges A, Lebrun S, Meurrens K, Steffen Y, Stolle K, Schueller J, Boue S, Vuillaume G, Vanscheeuwijck P, Moehring M, Schlage W, De Leon H, Hoeng J, Peitsch M. Cigarette-smoke-induced atherogenic lipid profiles in plasma and vascular tissue of apolipoprotein E-deficient mice are attenuated by smoking cessation. Atherosclerosis. 2013 Jul;229(1):86-93. [CrossRef]
- Yamaguchi Y, Matsuno S, Kagota S, Haginaka J, Kunitomo M. Oxidants in cigarette smoke extract modify low-density lipoprotein in the plasma and facilitate atherogenesis in the aorta of Watanabe heritable hyperlipidemic rabbits. Atherosclerosis. 2001 May;156(1):109-17. [CrossRef]
- Centner AM, Bhide PG, Salazar G. Nicotine in Senescence and Atherosclerosis. Cells. 2020 Apr 22;9(4):1035. [CrossRef]
- Whitehead AK, Erwin AP, Yue X. Nicotine and vascular dysfunction. Acta Physiol (Oxf). 2021 Apr;231(4):e13631. [CrossRef]
- Siasos G, Tsigkou V, Kokkou E, Oikonomou E, Vavuranakis M, Vlachopoulos C, Verveniotis A, Limperi M, Genimata V, Papavassiliou AG, Stefanadis C, Tousoulis D. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr Med Chem. 2014;21(34):3936-48. [CrossRef]
- Gimbrone MA Jr, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016 Feb 19;118(4):620-36. [CrossRef]
- Aceto, N. Aceto N. Bring it on: blood-based analysis of circulating tumor cells in lung cancer. EMBO Mol Med. 2020;12(12):e12802.
- Li F, Tiede C, Massi D, Campaner E, Calorini L, Kohlhammer H, et al. Fluid shear stress regulates epithelial–mesenchymal transition via JNK signaling pathway in circulating tumor cells. Biochem Biophys Res Commun. 2020;533(4):1095–1100.
- Zhang J, Qin Y, Fan J, Zhou J, Zhang Y, Zhong W, et al. Shear stress-induced nuclear expansion through histone acetylation promotes survival of suspended tumor cells. Nat Commun. 2022;13(1):2494.
- Krog BL, Henry MD. Biomechanical regulation of CTC metastasis in lung cancer. Biophys J. 2018;115(2):186–195.
- Kurma K, Alix-Panabières C. Platelet–CTC interactions: mechanisms and implications in metastasis. Cancer Cell. 2023;41(4):439–452.
- Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292.
- Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458.
- Szczerba BM, Castro-Giner F, Vetter M, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553–557.
- Mitchell MJ, King MR. Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via trimeric death receptors. New J Phys. 2013;15:015008.
- Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292(3):H1209–H1224.
- Au SH, Storey BD, Moore JC, et al. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc Natl Acad Sci U S A. 2016;113(18):4947–4952.
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590.
- Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Cell. 2014;158(5):1045–1059.
- Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636.
- Pries AR, Secomb TW. Microvascular blood viscosity in vivo and the endothelial surface layer. Am J Physiol Heart Circ Physiol. 2005;289(6):H2657–H2664.
- Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327–387.
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689.
- Lederle FA, Johnson GR, Wilson SE, et al. The prevalence and associated factors of abdominal aortic aneurysm detected through screening. Ann Intern Med. 1997;126(6):441–449.
- Rzucidlo EM, Powell RJ. Arterial aneurysms in smokers and patients with chronic obstructive pulmonary disease: a common pathophysiologic link. Vasc Med. 2009;14(3):195–204.
- Bluestein D, Chandran KB, Manning KB. Towards non-thrombogenic performance of blood recirculating devices. Ann Biomed Eng. 2010;38(3):1236–1256.
- Chatzizisis YS, Coskun AU, Jonas M, et al. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling. J Am Coll Cardiol. 2007;49(25):2379–2393.
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219.
- Vorp DA, Lee PC, Wang DH, Makaroun MS, Nemoto EM, Ogawa S, Webster MW. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J Vasc Surg. 2001;34(2):291–299.
- Demers M, Wagner DD. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost. 2014;40(3):277–283.
- Park J, Wysocki RW, Amoozgar Z, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138.
- Les AS, Shadden SC, Figueroa CA, et al. Quantification of hemodynamic wall shear stress in patients with abdominal aortic aneurysms using computational fluid dynamics. Ann Biomed Eng. 2010;38(4):1040–1053.
- Fillinger MF, Marra SP, Raghavan ML, Kennedy FE. Prediction of rupture risk in abdominal aortic aneurysm during observation: wall stress versus diameter. J Vasc Surg. 2003;37(4):724–732.
- Alix-Panabières C, Pantel K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 2021;11(4):858–873.
- Pfister G, Wang Y, Mehta K, et al. Vascular-targeted therapies: a new approach to fighting metastasis? Trends Mol Med. 2020;26(4):401–414.
- Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX, Konstantopoulos K. Water permeation drives tumor cell migration in confined microenvironments. Cell. 2014;157(3):611–623.
- Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc Natl Acad Sci U S A. 2011;108(27):11115–11120.
- Ma H, Wells A, Wei L, Li Y, Yu F. Emerging microfluidic platforms for cancer metastasis research. Trends Biotechnol. 2020;38(6):556–572.
- Les AS, Shadden SC, Figueroa CA, et al. Quantification of hemodynamic wall shear stress in patients with abdominal aortic aneurysms using computational fluid dynamics. Ann Biomed Eng. 2010;38(4):1040–1053.
- Vorp DA, Lee PC, Wang DH, Makaroun MS, Nemoto EM, Ogawa S, Webster MW. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J Vasc Surg. 2001;34(2):291–299.
- Les AS, Shadden SC, Figueroa CA, et al. Quantification of hemodynamic wall shear stress in patients with abdominal aortic aneurysms using computational fluid dynamics. Ann Biomed Eng. 2010;38(4):1040–1053.
- Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc Natl Acad Sci U S A. 2011;108(27):11115–11120.
- Ma H, Wells A, Wei L, Li Y, Yu F. Emerging microfluidic platforms for cancer metastasis research. Trends Biotechnol. 2020;38(6):556–572.
- Pan C, Kumar C, Bohl M, Wilkins DE, Gupta N, Bazan NG. Apolipoprotein A-I mimetic peptides suppress lung tumor growth by reprogramming tumor-associated macrophages. Mol Med. 2017;23(1):79–88. [CrossRef]
- Dong Z, Wang Y, Weng H, Wang B. Apolipoprotein E enhances lung cancer metastasis by interacting with endothelial LRP1 receptors. Cancer Lett. 2018;431:157–167. [CrossRef]
- Kotlyarov, S. High-Density Lipoproteins: A Role in Inflammation in COPD. Int J Mol Sci. 2022 Jul 23;23(15):8128. [CrossRef]
- Mazzuca P, Carloni A, Cazzato F, et al. Tobacco smoke and the vascular endothelium: from inflammation to transformation. Int J Mol Sci. 2020;21(21):7755.
- Rzucidlo EM, Powell RJ. Arterial aneurysms in smokers and patients with chronic obstructive pulmonary disease: a common pathophysiologic link. Vasc Med. 2009;14(3):195–204.
- Pfister G, Wang Y, Mehta K, et al. Vascular-targeted therapies: a new approach to fighting metastasis? Trends Mol Med. 2020;26(4):401–414.
- Tibuakuu M, Kamimura D, Kianoush S, DeFilippis AP, Al Rifai M, Reynolds LM, White WB, Butler KR, Mosley TH, Turner ST, Kullo IJ, Hall ME, Blaha MJ. The association between cigarette smoking and inflammation: The Genetic Epidemiology Network of Arteriopathy (GENOA) study. PLoS One. 2017 Sep 18;12(9):e0184914. [CrossRef]
- Elisia I, Lam V, Cho B, Hay M, Li MY, Yeung M, Bu L, Jia W, Norton N, Lam S, Krystal G. The effect of smoking on chronic inflammation, immune function and blood cell composition. Sci Rep. 2020 Nov 10;10(1):19480. [CrossRef]
- Leduc C, Antoni D, Charloux A, Falcoz PE, Quoix E. Comorbidities in the management of patients with lung cancer. Eur Respir J. 2017 Mar 29;49(3):1601721. [CrossRef]
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016 Nov 4;354(6312):618-622. [CrossRef]
- Warren GW, Sobus S, Gritz ER. The biological and clinical effects of smoking by patients with cancer and strategies to implement evidence-based tobacco cessation support. Lancet Oncol. 2014 Nov;15(12):e568-80. [CrossRef]
- Zhang J, Huang S, Zhu Z, Gatt A, Liu J. E-selectin in vascular pathophysiology. Front Immunol. 2024 Jul 19;15:1401399. [CrossRef]
- Turhan H, Saydam GS, Erbay AR, Ayaz S, Yasar AS, Aksoy Y, Basar N, Yetkin E. Increased plasma soluble adhesion molecules; ICAM-1, VCAM-1, and E-selectin levels in patients with slow coronary flow. Int J Cardiol. 2006 Apr 4;108(2):224-30. [CrossRef]
- Park-Windhol C, D'Amore PA. Disorders of Vascular Permeability. Annu Rev Pathol. 2016 May 23;11:251-81. [CrossRef]
- Demers M, Wagner DD. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost. 2014;40(3):277–283.
- Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Cell. 2014;158(5):1045–1059.
- Hargadon, KM. Tumor-altered dendritic cell function: implications for anti-tumor immunity. Front Immunol. 2013 Jul 11;4:192. [CrossRef]
- Geindreau M, Bruchard M, Vegran F. Role of Cytokines and Chemokines in Angiogenesis in a Tumor Context. Cancers (Basel). 2022 May 16;14(10):2446. [CrossRef]
- Ramakrishnan R, Subramanian J. Long-Term Survival by Number of Immune Checkpoint Inhibitors in PD-L1-Negative Metastatic NSCLC: A Systematic Review and Meta-Analysis. JAMA Netw Open. 2025 Feb 3;8(2):e2457357. [CrossRef]
- Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018 Sep;8(9):1069-1086. [CrossRef]
- Braun A, Anders HJ, Gudermann T, Mammadova-Bach E. Platelet-Cancer Interplay: Molecular Mechanisms and New Therapeutic Avenues. Front Oncol. 2021 Jul 12;11:665534. [CrossRef]
- Lycan TW Jr, Norton DL, Ohar JA. COPD and Immune Checkpoint Inhibitors for Cancer: A Literature Review. Int J Chron Obstruct Pulmon Dis. 2024 Dec 9;19:2689-2703. [CrossRef]
- Pfister G, Wang Y, Mehta K, et al. Vascular-targeted therapies: a new approach to fighting metastasis? Trends Mol Med. 2020;26(4):401–414.
- Fillinger MF, Marra SP, Raghavan ML, Kennedy FE. Prediction of rupture risk in abdominal aortic aneurysm during observation: wall stress versus diameter. J Vasc Surg. 2003;37(4):724–732.
Figure 1.
Figure 1. Pro-metastatic mechanisms induced by smoke. Smoke affects erays phases of LC onset in most cases in a COPD context. It is known that LC arouses earlier and with multifocal lesions in Asian women and never-smokers. Adenocarcinoma has been the predominant oncotype in non-smokers, but currently its prevalence is also increasing in smokers (changes in the composition of cigarettes). The parenchymal site of growth of neoplastic lesions in smokers is both peripheral and central lung; in non-smokers the sub-mantle areas are generally involved. Survival data (corrected for known prognostic factors) are more favourable for non-smokers [117,118,119,120]. Circulating tumor cells (CTCs) are constantly exposed to a complex set of mechanical forces during their transit through the bloodstream, including shear stress, pressure gradients, turbulence, and mechanical deformation. These forces are not uniformly distributed across the vascular system, and in pathological conditions such as arteriopathy and aneurysms the biomechanical landscape is profoundly altered, affecting tumor cell survival, adhesion, and extravasation. A number of biomolecular transducers activated by smoke promote their pro-invasive phenotype. Smoke-related vasculopathy and aneurysmal niche promote profoundly influence the invasive CTC capacity. uPA: urokinase plasminogen activator; OPN: osteopontin. Create in BioRender.com.
Figure 1.
Figure 1. Pro-metastatic mechanisms induced by smoke. Smoke affects erays phases of LC onset in most cases in a COPD context. It is known that LC arouses earlier and with multifocal lesions in Asian women and never-smokers. Adenocarcinoma has been the predominant oncotype in non-smokers, but currently its prevalence is also increasing in smokers (changes in the composition of cigarettes). The parenchymal site of growth of neoplastic lesions in smokers is both peripheral and central lung; in non-smokers the sub-mantle areas are generally involved. Survival data (corrected for known prognostic factors) are more favourable for non-smokers [117,118,119,120]. Circulating tumor cells (CTCs) are constantly exposed to a complex set of mechanical forces during their transit through the bloodstream, including shear stress, pressure gradients, turbulence, and mechanical deformation. These forces are not uniformly distributed across the vascular system, and in pathological conditions such as arteriopathy and aneurysms the biomechanical landscape is profoundly altered, affecting tumor cell survival, adhesion, and extravasation. A number of biomolecular transducers activated by smoke promote their pro-invasive phenotype. Smoke-related vasculopathy and aneurysmal niche promote profoundly influence the invasive CTC capacity. uPA: urokinase plasminogen activator; OPN: osteopontin. Create in BioRender.com.
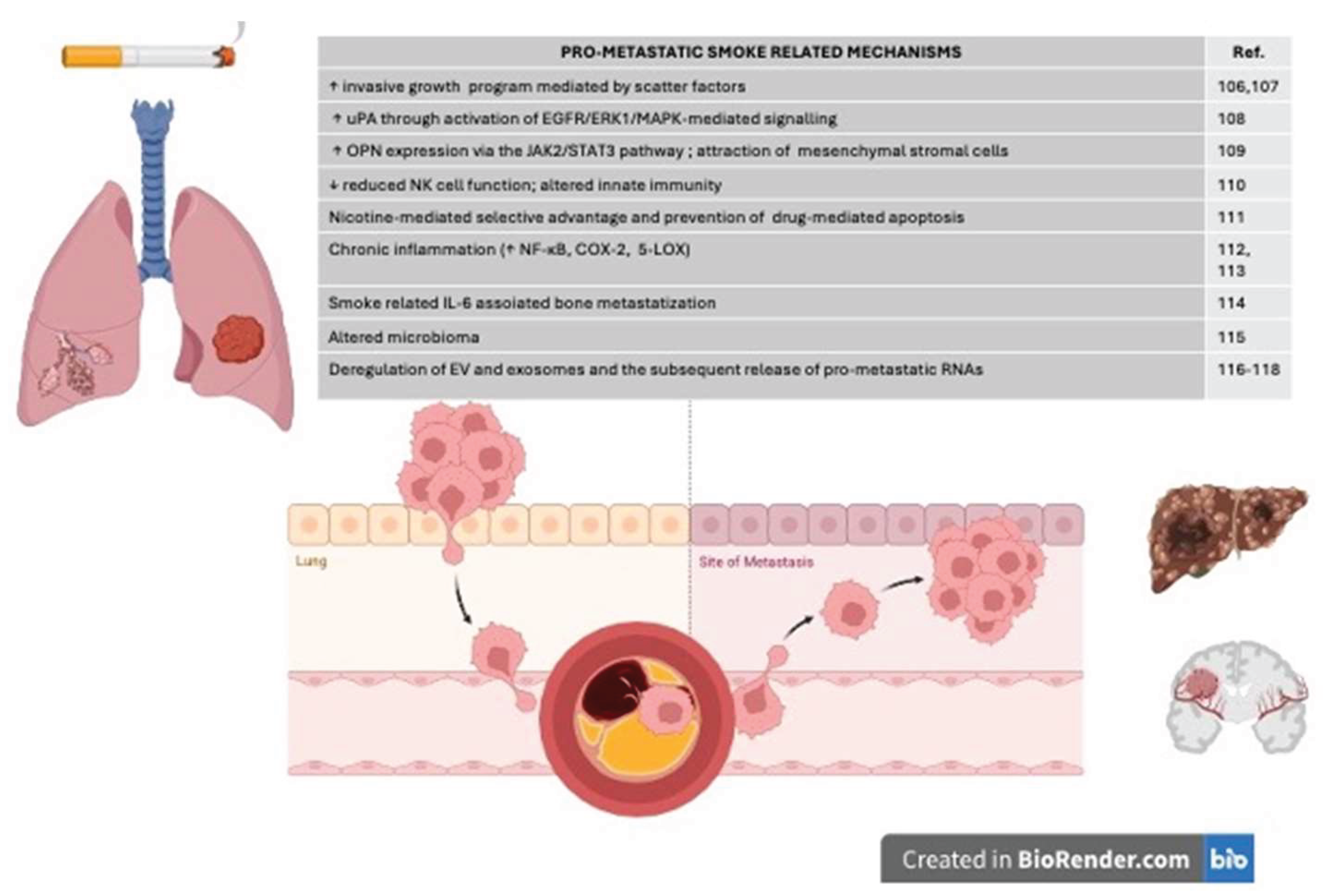
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



