Submitted:
27 May 2025
Posted:
03 June 2025
You are already at the latest version
Abstract
Objective: The present study aimed to develop and optimize a Cabazitaxel (CTX)-loaded sterically stabilized emulsome (SS-ES) system to enhance drug stability and achieve sustained release, using tristearin as the core lipid via the solvent evaporation method. Methods: Two emulsome formulations were developed: a simple emulsome (P-ES) and a sterically stabilized emulsome (SS-ES). Key formulation variables included the lipid-to-stabilizer ratio (DSPC, CHOL, and DSPE-PEG), the molar ratio of phospholipids to PEG-lipid components, solid lipid content, phospholipid concentration, and the ratio of organic to aqueous phase. The prepared formulations were characterized for particle size, release kinetics, and pH sensitivity. Results: The mean particle size of P-ES was 275 ± 5.52 nm, while SS-ES exhibited a smaller size of 195 ± 6.4 nm, indicating improved nano-sizing with PEGylation and steric stabilization. Both formulations demonstrated pH-sensitive and sustained drug release profiles, with SS-ES showing a slower and more controlled release compared to P-ES. Release kinetics followed a Fickian diffusion mechanism, best fitting the Higuchi model. The SS-ES formulation exhibited enhanced physical stability and potential for prolonged systemic circulation. Conclusion: Sterically stabilized emulsomes represent a promising nanocarrier platform for the delivery of hydrophobic drugs like Cabazitaxel, offering enhanced stability, sustained release, and potential for prolonged bioavailability.
Keywords:
emulsomes
; lipophilic
; cabazitaxel
; pegylation
; drug release kinetics
Introduction
The emergence of nanotechnology has revolutionized drug delivery strategies by providing innovative carrier systems that can be engineered to align with the specific physicochemical properties of therapeutic compounds. Nanocarriers, with their tunable surface characteristics and internal structures, offer enhanced delivery options for both small-molecule drugs and biologics. In recent years, research has emphasized the design of tumour-specific nanocarriers to minimize systemic exposure and reduce unintended side effects1.
One of the critical limitations associated with many chemotherapeutic agents is their poor aqueous solubility, which hampers formulation development and diminishes therapeutic effectiveness. Traditional solubilization techniques, such as the use of organic solvents and surfactants, often provoke adverse immune reactions or toxicity. To address these issues, nanoparticulate systems—especially those based on lipids and polymers—have been explored to enhance solubility, safeguard the drug molecule, and eliminate the need for harmful excipients2.
Cabazitaxel (Ctx), a second-generation taxane used in cancer treatment, is an example of a hydrophobic drug that presents formulation challenges. The commercial formulation, Taxol®, utilizes a combination of Cremophor EL and ethanol, which is linked to significant toxicity. As a safer alternative, emulsome-based delivery systems have gained interest due to their ability to encapsulate hydrophobic drugs without the use of aggressive solubilizers.
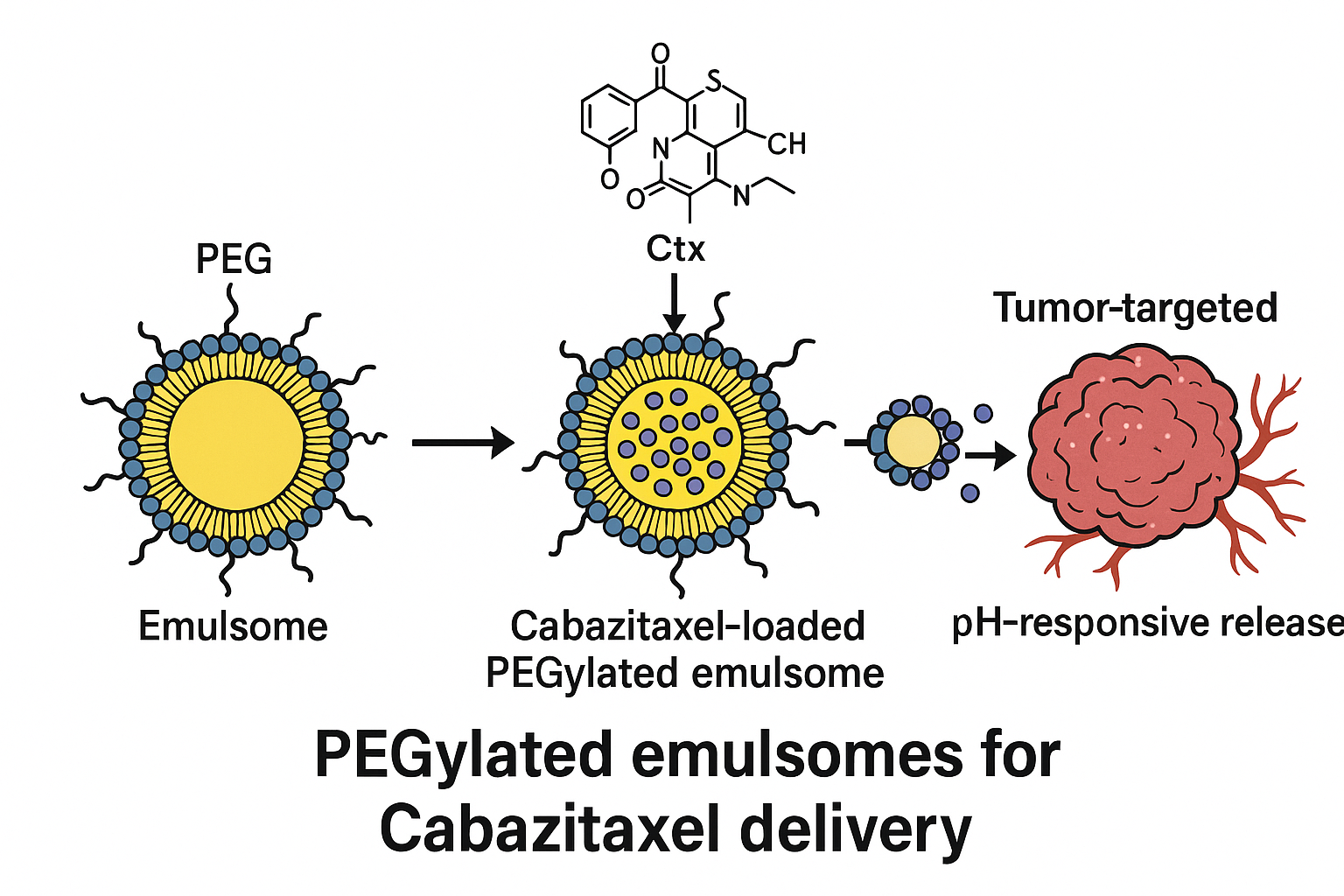
Emulsomes are hybrid systems that integrate the properties of liposomes and solid lipid nanoparticles. They feature a lipid-based solid core surrounded by a phospholipid bilayer, making them suitable for high drug loading and improved physical stability. PEGylation, achieved by incorporating polyethylene glycol (PEG)-linked lipids, further enhances circulation time by reducing recognition and clearance by the immune system. Moreover, these systems can be tailored for controlled and sustained drug release, which can potentially improve therapeutic outcomes and patient adherence3,4.
The present study focuses on the development and optimization of PEGylated emulsomes, also known as sterically stabilized emulsomes (SS-Es), encapsulating Cabazitaxel. The goal was to evaluate their potential as a long-circulating and biocompatible nanocarrier for anticancer therapy. Key physicochemical properties and drug release kinetics were investigated to determine their suitability for clinical application.
Materials and Methods
Materials
Cabazitaxel was generously supplied by Neon Laboratories Ltd., Mumbai, India. Lipid components including DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), cholesterol, and DSPE-PEG (PEGylated phospholipid) were purchased from Lipoid GmbH, Germany. Tristearin was sourced from Sigma-Aldrich, Germany. All other chemicals used were of analytical grade and procured locally.
Preparation of Emulsomes
Both plain emulsomes (P-Es) and PEGylated emulsomes (SS-Es) were prepared using a modified single-step emulsification technique followed by solvent evaporation. Tristearin was utilized as the solid lipid forming the core, while DSPC and cholesterol comprised the lipid bilayer. For steric stabilization, DSPE-PEG was included in selected formulations.
The process began by melting the lipid phase at 80 °C, followed by the addition of Cabazitaxel pre-dissolved in aqueous ethanol. The mixture was stirred at 1000 rpm for 15 minutes to ensure uniform distribution. In parallel, the phospholipids (with or without PEGylation) were dispersed in pre-heated distilled water and mixed at 5000 rpm. The lipid solution was then added gradually (1 ml/min) to the aqueous phase under continuous stirring at 80 °C and homogenized at 10,000 rpm for 30 minutes6,7.
Rapid cooling using an ice bath facilitated the formation of a phospholipid bilayer around the solid lipid core. Residual solvents and unencapsulated drug were removed via dialysis against distilled water using a 10 kDa molecular weight cutoff membrane. The final product was sterile-filtered (0.22 µm), cryoprotected with 5% sucrose, and lyophilized for storage at 4 °C. The composition of the optimised formulation is stated in Table 1.
Optimization Parameters
To achieve optimal characteristics such as particle size and entrapment efficiency, the following formulation parameters were systematically varied:
- Ratio of phospholipids to solid lipid (PL:SL)
- Molar ratio of lipid to PEG-lipid
- Concentration of both lipid and phospholipid components
- Volume ratio of organic to aqueous phase
- Homogenization time
Characterization of Emulsomes
Particle Size and Distribution9,10
Dynamic light scattering (DLS) was employed to measure particle size and polydispersity index (PDI) using a Nanoplus 5.01 Zetasizer. Samples were diluted (1:9) in deionized water. Low PDI values indicated homogeneity and colloidal stability.
Zeta Potential
Surface charge was analyzed to assess colloidal stability and predict in vivo interactions. Zeta potential measurements were performed using the same instrument in flow-through mode.
Morphological Studies
Transmission electron microscopy (TEM) was used to assess the vesicle morphology, including shape and surface texture. A droplet of the formulation was placed on a copper grid, air-dried, and visualized under a Tecnai G2 TEM at 100 kV. Scanning electron microscopy (SEM, NOVA NanoSEM 450) provided additional 3D structural insights.
Entrapment Efficiency11
Drug entrapment was measured using a gel filtration technique involving Sephadex G-50 columns. Sephadex (1.2 g) was pre-swollen in saline for 5 hours and stored at 4 °C. Mini-columns were prepared using disposable syringes and packed with the hydrated gel. A 0.2 ml sample of the emulsomal formulation was applied and centrifuged at 2000 rpm for 3 minutes. The eluted fraction, containing encapsulated drug, was collected. The emulsomes were lysed with 1 ml of 0.1% Triton X-100, and Cabazitaxel concentration was measured at 229 nm using a UV-Vis spectrophotometer.
Entrapment efficiency was calculated using the following formula:
Entrapment Efficiency (%) = (Total drug – Free drug) / Total drug × 100
In Vitro Drug Release Study12,13
Drug release profiles were determined using dialysis in phosphate buffer (pH 4.0) at 37 ± 1 °C. A 1 ml aliquot of the drug-loaded emulsomes was enclosed in a dialysis membrane and placed in 20 ml buffer. Samples were taken at defined intervals for 24 hours, with the buffer volume maintained by replacing each withdrawn sample with fresh medium. Drug concentration was quantified spectrophotometrically at 229 nm.
Drug Release Kinetics14
Release data were analyzed using different kinetic models, including zero-order, first-order, Higuchi, and Korsmeyer-Peppas equations. The best-fitting model was determined based on correlation coefficients (R²), and the diffusion exponent (n) from the Korsmeyer-Peppas model provided insight into the release mechanism, distinguishing between diffusion-controlled and anomalous transport.
Plasma Protein Binding Assay
To assess the stealth characteristics conferred by PEGylation, emulsomes were incubated in a 5% BSA solution. Changes in particle size and PDI were monitored using DLS. Minimal variation in these parameters indicated effective PEG shielding and reduced protein adsorption.
Statistical Analysis
All data were expressed as mean ± standard deviation (n=3). Statistical analysis was conducted using one-way ANOVA followed by Tukey–Kramer post hoc tests via GraphPad Prism (version 3.00). A p-value less than 0.05 indicated statistical significance. (Table 2)
Formulation Variables in Emulsomes Optimization15
To optimize the emulsomal formulation for effective Cabazitaxel delivery, several key formulation parameters were systematically evaluated. These included the molar ratio of phospholipids to DSPE-PEG, the total lipid-to-solid lipid mass ratio (DSPC, cholesterol, and DSPE-PEG to tristearin), the concentrations of tristearin and phospholipids, and the volume ratio between the organic and aqueous phases. Each of these factors was analysed for its influence on particle size (PS) and drug entrapment efficiency (%EE), to develop a stable Nano system capable of efficiently encapsulating Cabazitaxel in a size range suitable for intravenous delivery.
Impact of Phospholipid to DSPE-PEG Molar Ratio on Nanoparticle Characteristics16,17
DSPE-PEG is incorporated into emulsomes to enhance their stability by providing a steric shield that limits particle aggregation and promotes long circulation. In formulations lacking DSPE-PEG (referred to as plain emulsomes), the average particle size was relatively large, measured at 320.5 ± 5.8 nm (Figure 1). Introducing DSPE-PEG led to a progressive decrease in particle size, with the most significant reduction observed at a phospholipid to DSPE-PEG molar ratio of 1:5. At this ratio, the particle size was minimized to 190.5 ± 4.77 nm. A slightly higher size of 195.5 ± 5.6 nm was noted at the 1:3 ratio, which still represented a statistically significant reduction compared to non-PEGylated emulsomes (p < 0.01).
Beyond the 1:3 molar ratio, increasing the DSPE-PEG content further had minimal additional effect on reducing particle size, indicating that 1:3 was the optimal ratio for this formulation. The size reduction is attributed to the steric stabilization effect provided by the hydrophilic PEG chains, which create a repulsive barrier between particles and prevent their agglomeration during formation.
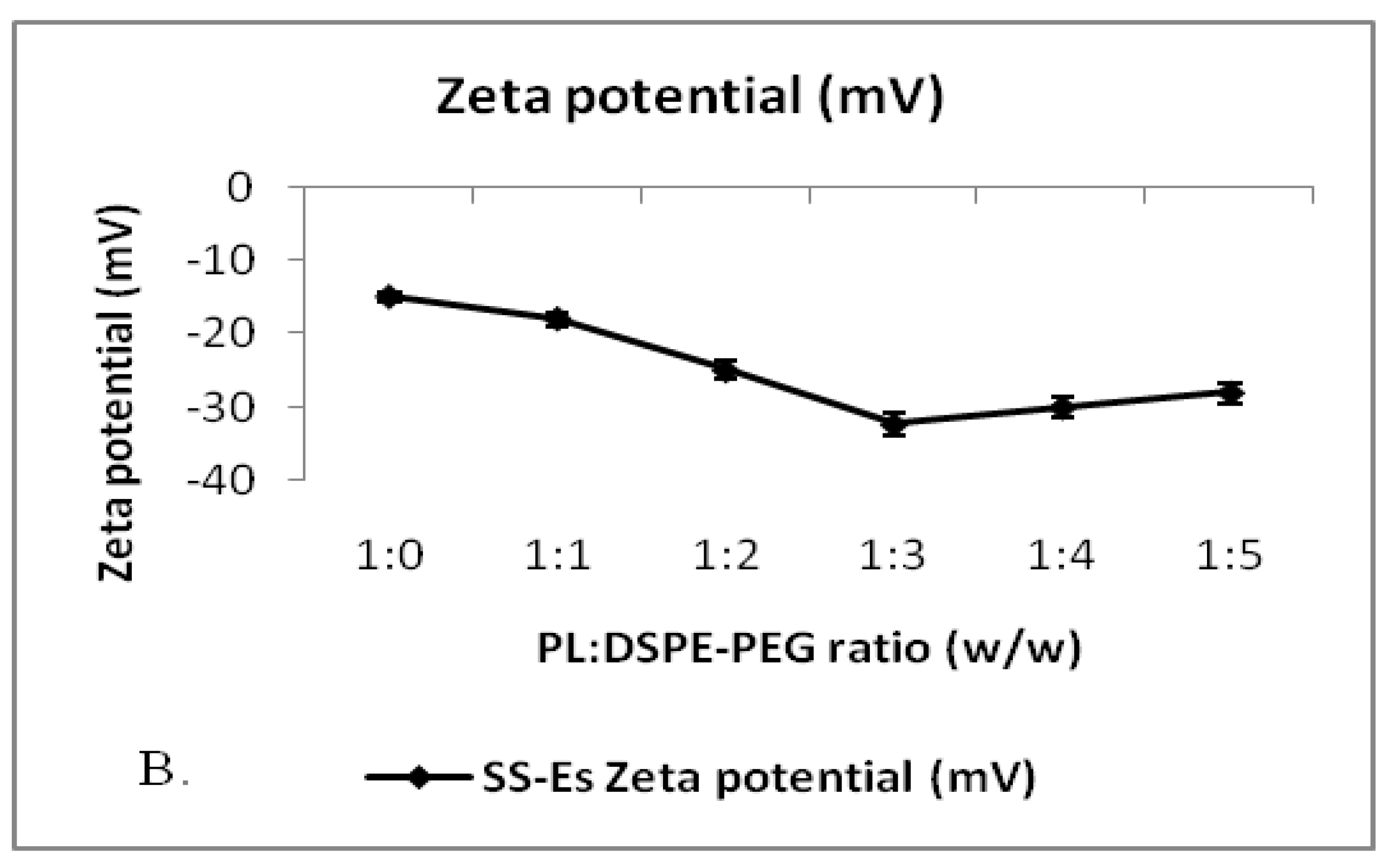
This optimized formulation also achieved a high entrapment efficiency, reaching 78.20 ± 2.8% (Figure 1A, 1B), suggesting that PEGylation not only stabilizes the particles but also promotes effective drug incorporation. A low polydispersity index (PDI) was observed, indicating a narrow size distribution and uniformity of the emulsomes. Zeta potential analysis revealed a surface charge of around –32 mV, which is consistent with stable colloidal systems. The high negative charge contributes to repulsive interactions between particles, preventing aggregation and thereby enhancing the formulation’s shelf life.
Figure 1.
A. Graphs showing the effect of PL to DSPE-PEG ratio on size and entrapment efficiency of emulsomes.
Figure 1.
A. Graphs showing the effect of PL to DSPE-PEG ratio on size and entrapment efficiency of emulsomes.
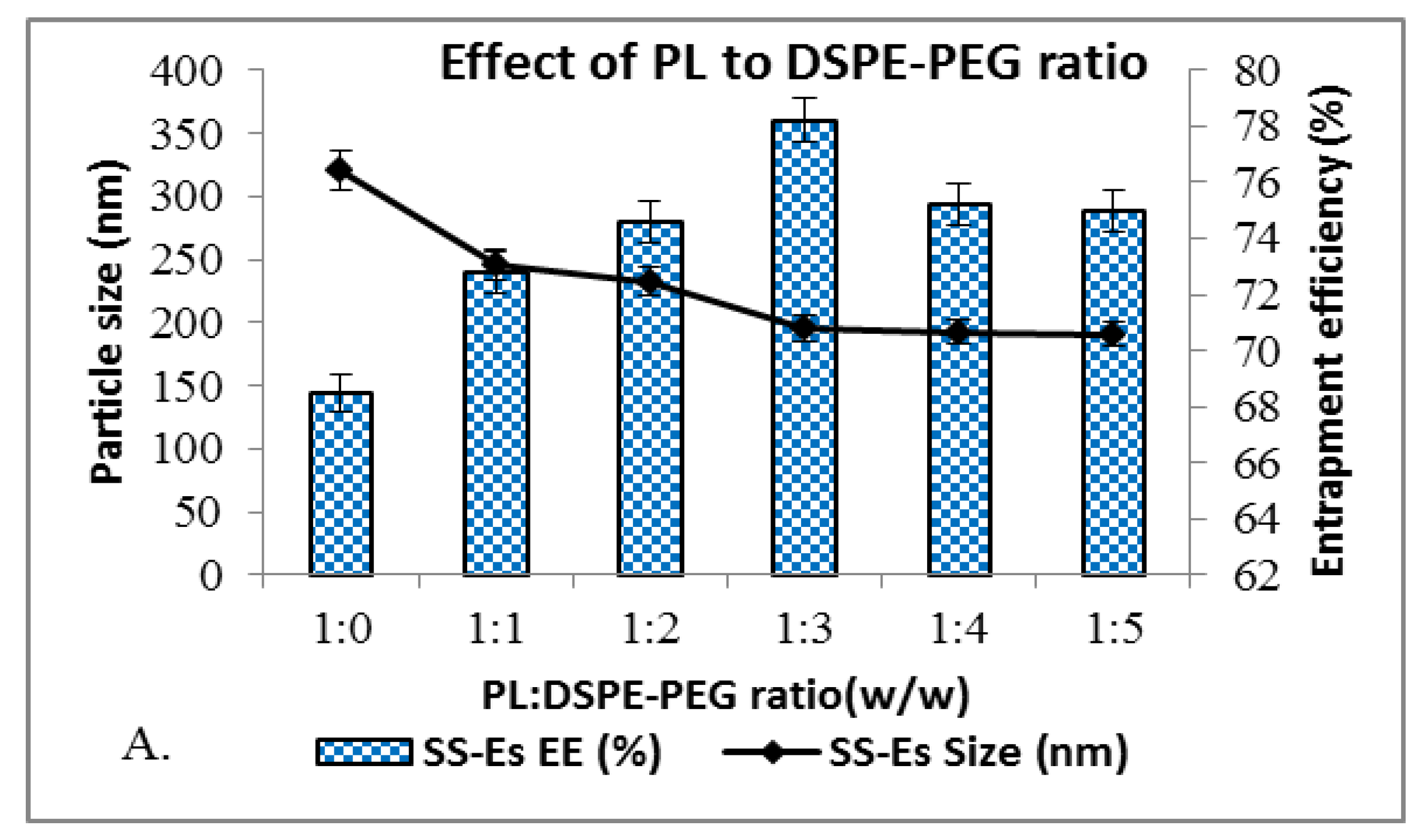
Figure 1.
B. Plot showing the change in zeta potential of emulsomes on the addition of DSPE-PEG to PL. Data represents mean ± SD (n=3).
Figure 1.
B. Plot showing the change in zeta potential of emulsomes on the addition of DSPE-PEG to PL. Data represents mean ± SD (n=3).
Effect of Organic-to-Aqueous Phase Volume Ratio18,19
Another critical variable in emulsomes preparation is the ratio between the volumes of organic and aqueous phases used during emulsification. In this study, the organic phase volume was held constant while the volume of the aqueous phase was varied between 5 and 20 ml. Initial increases in aqueous volume improved dispersion efficiency, leading to a notable reduction in particle size. This effect can be attributed to the enhanced dilution environment, which minimizes droplet collisions and supports better emulsification.
However, when the aqueous phase volume exceeded a certain threshold, an opposite trend was observed—particle size began to increase. This is likely due to a dilution-related reduction in shear energy during homogenization, which diminishes the capacity to break lipid droplets into finer particles. Furthermore, excessive dilution can destabilize emulsification and promote the formation of larger aggregates.
Entrapment efficiency also followed a similar trend. As particle size decreased with increasing aqueous volume, %EE initially improved. However, beyond the optimal point, further increases in aqueous phase volume led to a decline in drug loading. This drop could be due to the increased diffusion of the drug into the surrounding aqueous medium during processing or loss during solvent evaporation, which reduces the proportion of drug retained in the lipid matrix.
When comparing plain emulsomes (P-Es) and sterically stabilized emulsomes (SS-Es), both showed a decrease in particle size as the organic-to-aqueous phase ratio increased from 1:2 to 1:10. For P-Es, the size decreased from 270.5 ± 2.5 nm to 230.2 ± 3.5 nm. The SS-Es exhibited an even greater reduction, from 256.5 ± 2.6 nm to 195.5 ± 3.28 nm (Figure 2). However, beyond this 1:10 ratio, further increases in aqueous phase led to a size increase, likely due to inefficient emulsification at lower energy densities. Based on these observations, an organic-to-aqueous phase ratio of 1:10 was considered optimal for achieving small, stable nanoparticles with desirable drug loading characteristics. At this optimized ratio, the entrapment efficiency was observed to be 70.25 ± 4.4% for P-Es and 75.50 ± 2.5% for SS-Es, indicating effective drug loading in both formulations, with slightly superior performance in the PEGylated emulsomes.
Effect of Phospholipid to Solid Lipid (PL:SL) Ratio on Particle Size and Entrapment Efficiency20,21
The phospholipid-to-solid lipid ratio (PL:SL) plays a pivotal role in determining the physical characteristics and stability of emulsomal formulations. Varying this ratio from 0.25:1 to 1.5:1 led to a significant decrease in particle size across both plain emulsomes (P-Es) and sterically stabilized emulsomes (SS-Es). For P-Es, particle size was reduced from 650.5 ± 1.5 nm to 310.2 ± 2.5 nm, while SS-Es showed a size reduction from 556.4 ± 3.5 nm to 235.5 ± 3.6 nm, indicating improved dispersion and structural uniformity with increasing phospholipid content.
At the extreme of a 0:1 PL:SL ratio—meaning the absence of phospholipids—the lipid-based particles lacked structural integrity, resulting in aggregation and significantly larger particle sizes (approximately 840.5 ± 3.7 nm for P-Es and 650.8 ± 4.5 nm for SS-Es) as shown in Figure 3A. This observation emphasizes the essential role of phospholipids in stabilizing the emulsomal shell and maintaining nanoscale particle dimensions.
In formulations with low PL:SL ratios (0.25:1 to 0.5:1), the produced emulsomes were comparatively larger and exhibited low zeta potential values (Figure 3B), which suggests partial or insufficient encapsulation of the solid lipid core. This incomplete coating likely resulted in unstable particles that were prone to aggregation due to limited steric or electrostatic repulsion and stronger attractive forces between particles.
Improved formulation characteristics were observed near a 1:1 PL: SL ratio. At this ratio, particle sizes were minimized (310.2 ± 2.5 nm for P-Es and 235.5 ± 3.6 nm for SS-Es), and the zeta potential values reached –32 ± 0.12 mV and –35 ± 0.15 mV, respectively. These surface charge values are generally considered ideal for ensuring colloidal stability, as particles with zeta potentials exceeding ±30 mV are less likely to aggregate due to increased electrostatic repulsion.
The enhanced stability at this ratio is likely due to the effective encapsulation of the lipid core by the phospholipid shell, which reduces interfacial tension and supports the formation of uniformly dispersed nanoparticles. As a result, the emulsomes exhibit improved physical properties, including smaller size, better dispersion, and higher entrapment efficiency.
However, exceeding the optimal phospholipid concentration resulted in diminishing returns. With a total lipid content of 30%, excessive phospholipids that do not integrate into the emulsomal matrix may form separate vesicular structures such as liposomes, particularly when the phospholipid concentration surpasses its critical micelle concentration (CMC ~0.4 mg/ml). This leads to an increase in particle size and a decline in zeta potential, signaling reduced formulation stability22.
Conversely, insufficient phospholipid content results in inadequate surface coverage of the lipid core, compromising particle integrity despite potentially high surface charge values. This trade-off highlights the importance of balance in formulation design was shown in graphical data in Figure- 3A, 3B & also in Figure 4.
Impact of Solid Lipid Concentration on Particle Size and Entrapment Efficiency23,24
The concentration of solid lipid was found to significantly influence both particle size and drug entrapment within the emulsomal formulations. As the solid lipid content was increased from 5% to 20% w/w, a notable rise in particle size was observed. In the case of plain emulsomes (P-Es), particle diameter increased from 198.5 ± 4.5 nm to 380.4 ± 2.4 nm, while for sterically stabilized emulsomes (SS-Es), size grew from 182.3 ± 3.5 nm to 358.5 ± 1.5 nm (Figure 4). This size enlargement can be attributed to the elevated viscosity of the organic phase at higher lipid concentrations, which slows down the diffusion of lipids into the aqueous phase during formulation.
When solid lipid concentration exceeded 10% w/w, an imbalance between the lipid core and the phospholipid shell was observed. The available phospholipids were insufficient to fully encapsulate the expanding lipid content, leading to particle coalescence, structural instability, and an associated increase in size. Additionally, these higher lipid levels reduced the system’s ability to efficiently trap the drug, resulting in a drop in entrapment efficiency of 35.8±2.1, respectively (Figure 7C and D).
Conversely, increasing solid lipid content initially led to improved drug entrapment. The greater viscosity of the medium at higher lipid levels slowed the diffusion of Cabazitaxel (Ctx) into the surrounding aqueous phase, facilitating greater retention within the hydrophobic lipid core. These results point to a strong affinity between Ctx and the solid lipid matrix, particularly tristearin, which contributed to enhanced drug loading.
Effect of Homogenization Time on Particle Size and Entrapment Efficiency
Homogenization time is a key processing parameter that directly affects the particle size distribution and entrapment characteristics of emulsomes. This study examined the effect of varying homogenization times between 5 and 15 minutes. It was observed that increasing homogenization time led to a gradual reduction in average particle size and improved uniformity of particle distribution (Figure 5). This can be explained by the higher energy input over time, which facilitates the formation of smaller and more evenly dispersed lipid droplets.
However, extending homogenization beyond the optimal point caused a reversal in this trend. Longer durations introduced excessive turbulence into the system, resulting in increased particle collisions and aggregation. This effect compromised the structural integrity of the particles and reduced the drug’s entrapment efficiency.
Optimal conditions were achieved at a homogenization time that produced particle sizes of 250.5 ± 2.5 nm for P-Es and 215.6 ± 2.3 nm for SS-Es. At this point, entrapment efficiencies reached 76.4 ± 3.2% and 80.2 ± 2.8% for P-Es and SS-Es, respectively, indicating enhanced formulation performance at these conditions.
Physicochemical Characterization of Optimized Emulsomes25,26
Following optimization of formulation and process parameters, the emulsomes were subjected to further physicochemical analysis. Key attributes assessed included particle size, morphology, and surface charge (zeta potential), all of which are critical for evaluating colloidal stability and drug delivery potential.
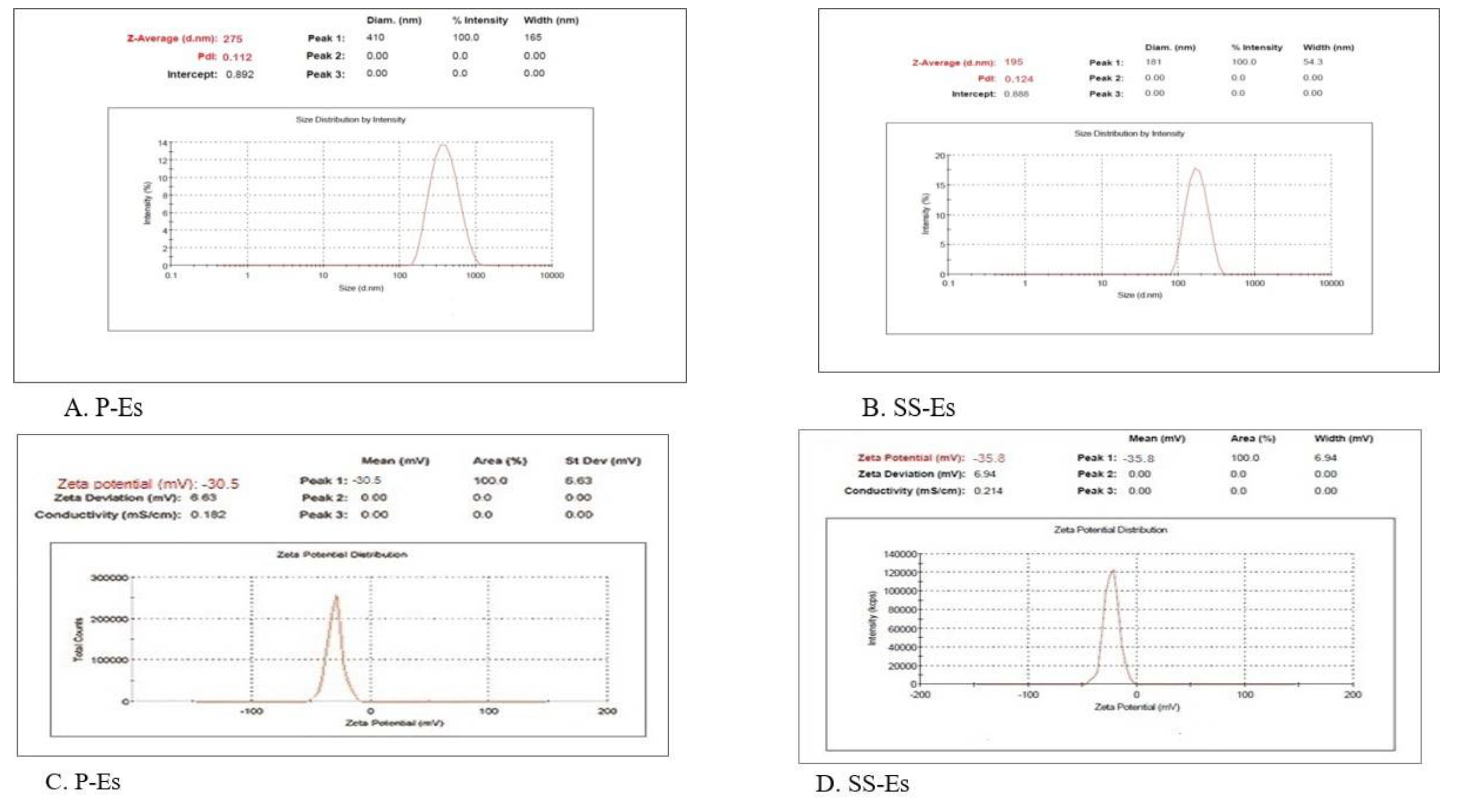
Imaging studies using Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM) confirmed the spherical nature of the emulsomes (Figures 6A and 6B). The uniformity in particle size was further supported by low polydispersity index values, reflecting a narrow size distribution. Mean particle sizes were recorded as 275 ± 5.52 nm for P-Es and 195 ± 6.4 nm for SS-Es (Figures 7A and 7B), consistent with earlier observations.
Surface charge analysis revealed that both P-Es and SS-Es carried a zeta potential of approximately –30.5 ± 2.0 mV. These values indicate strong repulsive forces between particles, minimizing aggregation and supporting the stability of the formulations during storage.
Figure 5.
Graphs showing the effect of homogenization time on the particle size of emulsomes, data represents mean ± SD (n=3).
Figure 5.
Graphs showing the effect of homogenization time on the particle size of emulsomes, data represents mean ± SD (n=3).
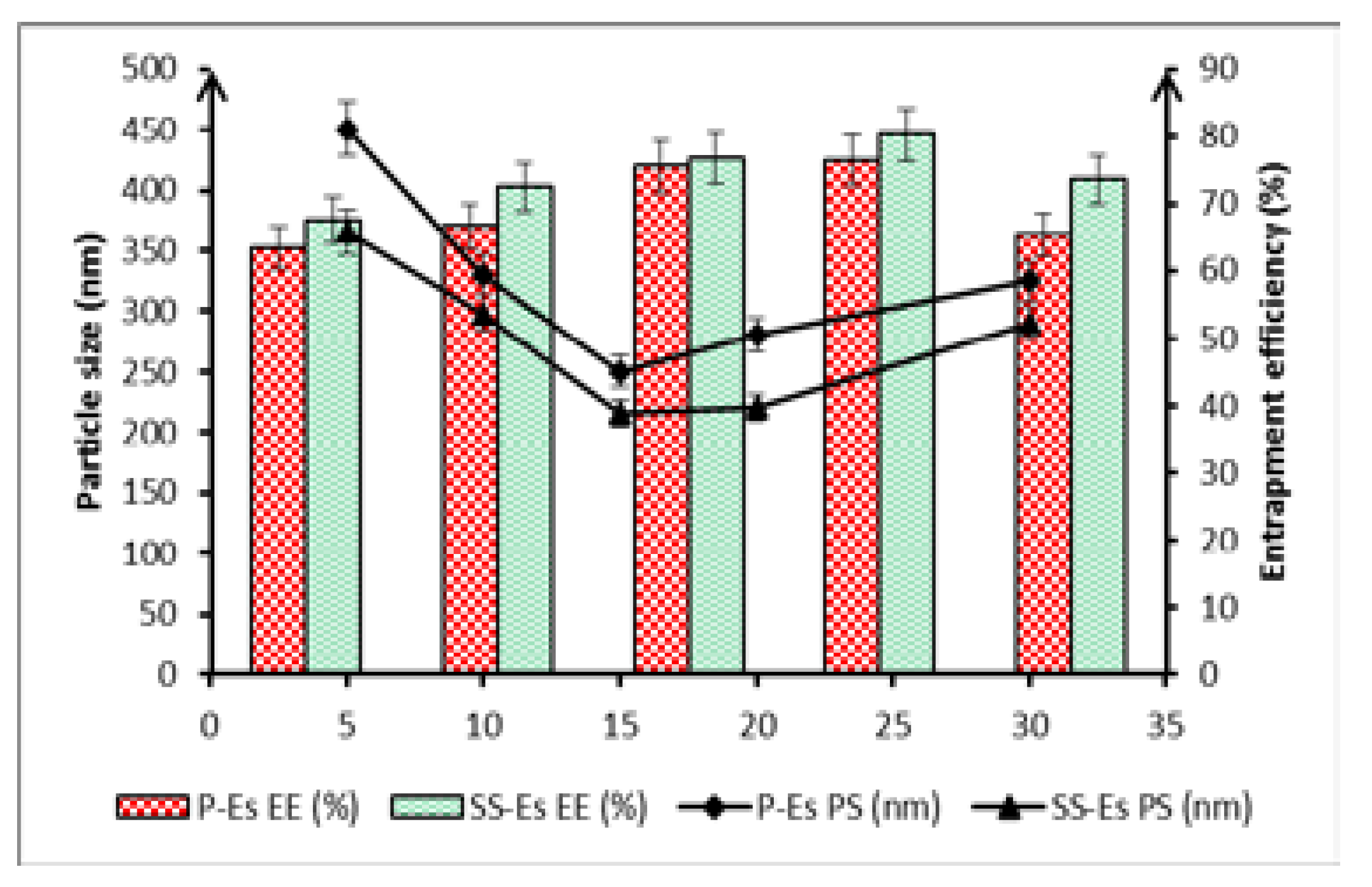
Figure 6.
A. TEM micrograph of SS-Es. The outer light periphery indicated by the arrow shows the presence of PEG on the surface of the emulsomes. B. SEM micrograph of SS-Es.
Figure 6.
A. TEM micrograph of SS-Es. The outer light periphery indicated by the arrow shows the presence of PEG on the surface of the emulsomes. B. SEM micrograph of SS-Es.
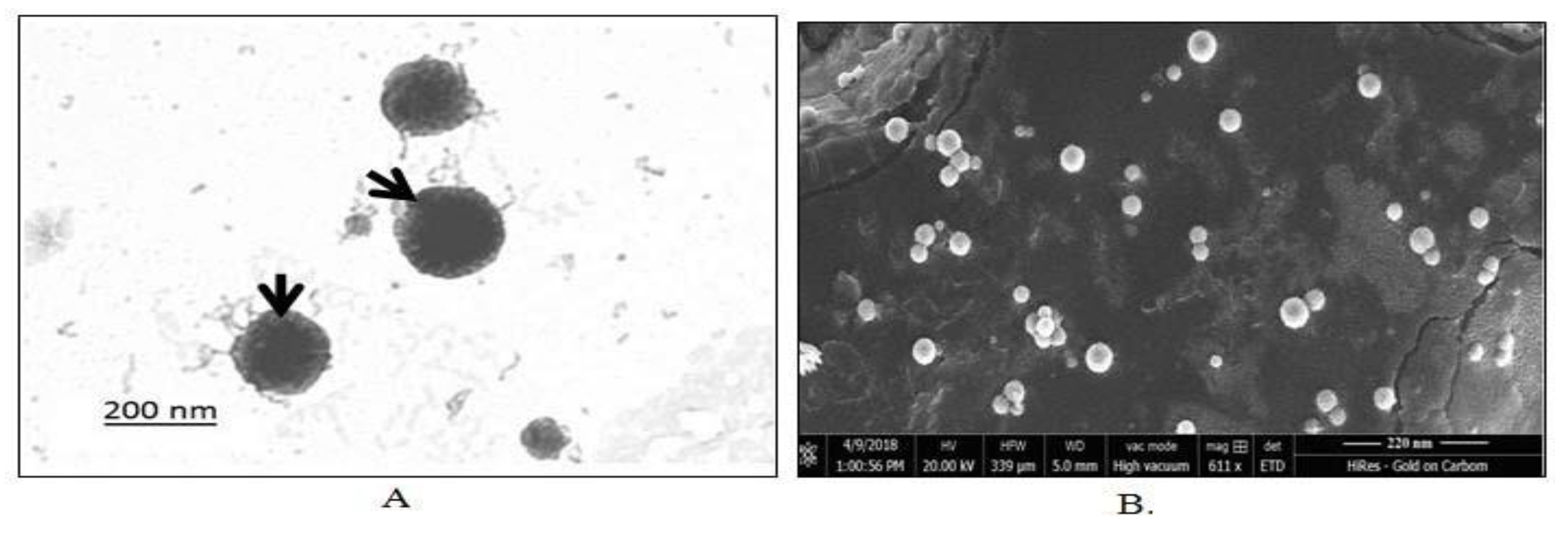
Figure 7.
Plots showing the size distribution of A. P-Es; B. SS-Es and zeta potential value of C. P-Es; D. SS-Es.
Figure 7.
Plots showing the size distribution of A. P-Es; B. SS-Es and zeta potential value of C. P-Es; D. SS-Es.
In Vitro Drug Release27,28
One of the essential attributes of a successful anticancer drug delivery system is its ability to remain stable in physiological conditions while preferentially releasing the drug in the more acidic tumour microenvironment. To examine this, in vitro release studies were performed at two pH levels: pH 7.4 to simulate normal physiological conditions, and pH 4.0 to mimic the acidic conditions typically found in tumour tissues.
Both plain emulsomes (P-Es) and sterically stabilized emulsomes (SS-Es) demonstrated similar release trends under both pH environments, suggesting that the incorporation of lipids and PEGylated components did not drastically alter the drug's release mechanism. At pH 7.4, drug release over the first 24 hours was measured at approximately 40.5 ± 2.7% for P-Es and 32.5 ± 1.5% for SS-Es (Figure 7A). This initial phase of faster release is believed to result from the liberation of the drug associated with the phospholipid layer. Subsequently, a more sustained release phase was observed, extending over the following six days. During this extended period, cumulative release reached 75.50 ± 4.5% for P-Es and 66.80 ± 3.5% for SS-Es..
The slower release rate from SS-Es compared to P-Es is attributed to the presence of the PEG coating, which provides structural reinforcement and acts as a barrier, limiting the diffusion of water and reducing solvent access to the lipid matrix. This barrier effect contributes to a more controlled and prolonged release profile.
When tested at acidic pH (4.0), both formulations exhibited a noticeably faster drug release. Within the first 24 hours, P-Es released 52.5 ± 1.5% of the encapsulated drug, while SS-Es released 48.6 ± 2.5%. This accelerated release behavior in an acidic environment indicates a pH-sensitive release mechanism. The reduced release rate at physiological pH helps maintain the drug within the emulsomal carrier, which could reduce off-target toxicity and enhance safety.
The enhanced drug release under acidic conditions can be explained by the partial disruption of the phospholipid bilayer and increased permeability of the emulsome membrane at lower pH. Acidic conditions may also promote hydrolytic degradation of the lipid matrix, especially tristearin, resulting in faster drug diffusion. This behavior supports the system’s potential for site-specific release in the tumour environment29,30.
Figure 8.
(a). Plot showing the in vitro drug release kinetics as per the various mathematical models. A. Zero-order drug release model. B. First-order drug release model.
Figure 8.
(a). Plot showing the in vitro drug release kinetics as per the various mathematical models. A. Zero-order drug release model. B. First-order drug release model.
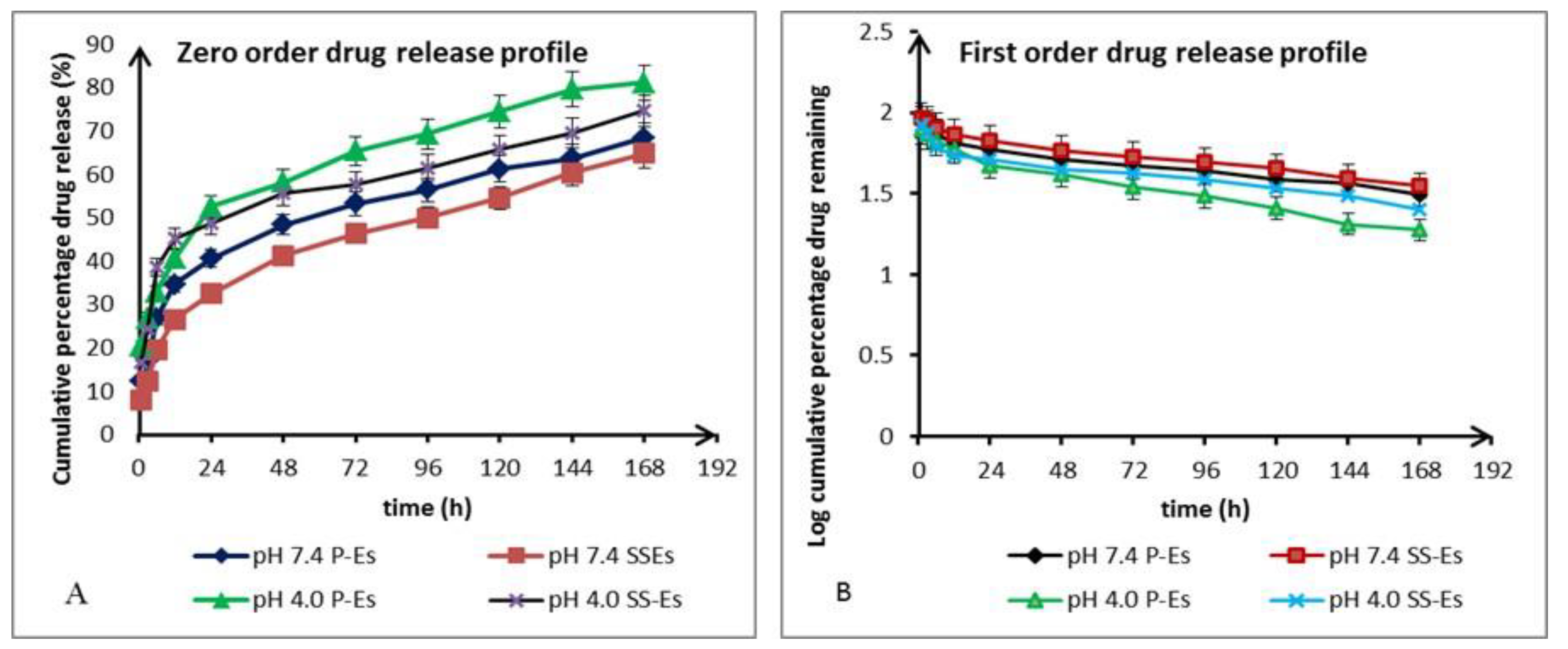
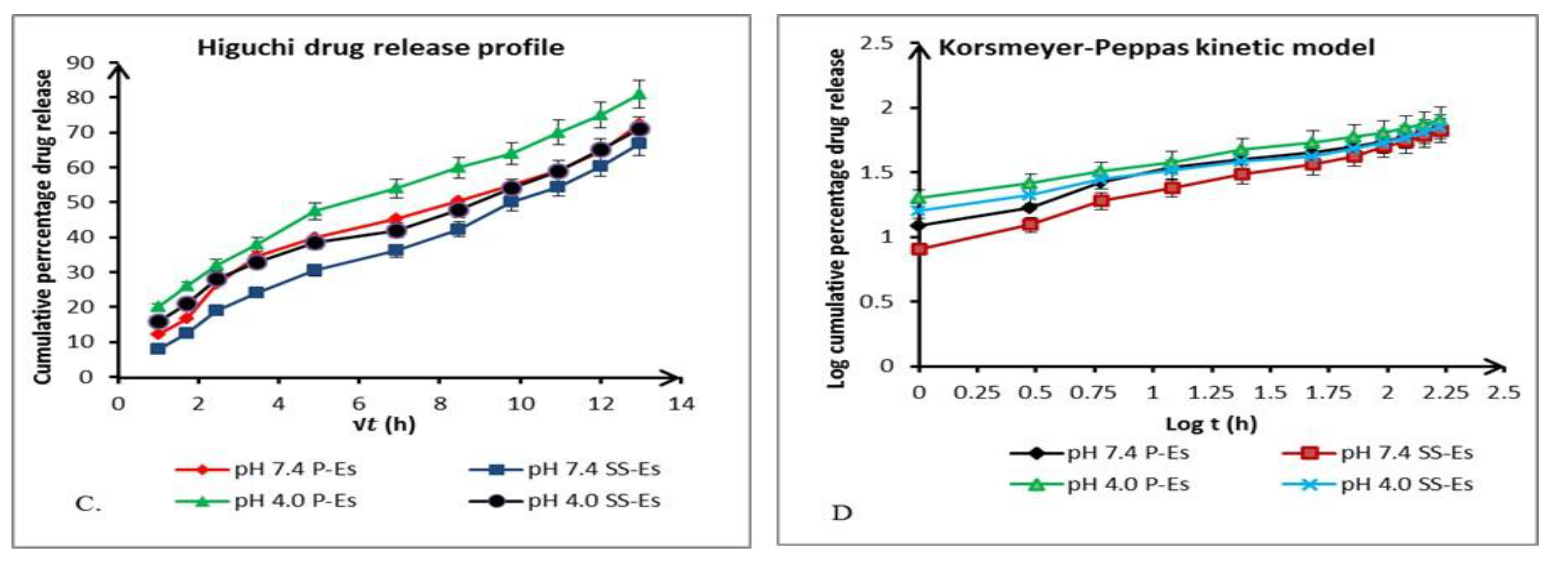
Figure 8.
(b). Plot showing the in vitro drug release kinetics as per the various mathematical models. C. Higuchi model D. Korsmeyer-peppas model. Data represents mean ± SD (n=3).
Figure 8.
(b). Plot showing the in vitro drug release kinetics as per the various mathematical models. C. Higuchi model D. Korsmeyer-peppas model. Data represents mean ± SD (n=3).
Drug Release Kinetics Study31,32
To elucidate the release behaviour of Cabazitaxel (Ctx) from emulsomal formulations, in vitro cumulative release data were analysed using multiple kinetic models. This analysis aimed to determine the underlying mechanism controlling drug release. The models employed included zero-order, first-order, Higuchi, and Korsmeyer-Peppas, each represented by specific plots: cumulative percentage of drug released (Q) versus time (t), log cumulative drug remaining versus time, Q versus the square root of time (√t), and log Q versus log time (log t), respectively (Figure 8A & 8B).
The correlation coefficients (R²) derived from the linear regression of each model were compared to identify the best-fitting kinetic profile (Table 3). Among these, the Higuchi model showed the highest correlation, indicating that the drug release from emulsomes is primarily governed by a diffusion-controlled mechanism.
To further characterize the nature of diffusion, the Korsmeyer-Peppas model was applied. The diffusional exponent ‘n’, determined from the slope of the log-log plot, was found to be less than 0.5. This low ‘n’ value confirms that the drug release conforms to Fickian diffusion, suggesting that the release process is dominated by the passive diffusion of the drug through the emulsomal matrix.
These findings highlight that the emulsomes provide a sustained release platform where the drug diffuses gradually over time, supporting their potential as a controlled drug delivery system for Cabazitaxel with parameters for kinetic models observed given in table-3.
Plasma Protein Absorption Study33,34
Evaluation of Stealth Properties Against Protein Adsorption
One of the primary obstacles in designing long-circulating nanoparticle systems is their rapid clearance by the Reticuloendothelial System (RES), triggered by the adsorption of opsonin proteins onto the nanoparticle surface. This opsonization process flags the particles for uptake by macrophages through phagocytosis. To mitigate this, DSPE-PEG was incorporated into the phospholipid shell of the emulsomes to provide a stealth characteristic, which helps evade immune detection and enhances systemic persistence.
To examine this stealth effect, the emulsomal formulations were incubated in a 5% bovine serum albumin (BSA) solution, with distilled water serving as the control medium. Changes in particle size and distribution were monitored over time to evaluate protein interaction and aggregation behaviour.
Following 24 hours in distilled water, the particle sizes of both plain emulsomes (P-Es) and sterically stabilized emulsomes (SS-Es) remained consistent, indicating good baseline stability. However, when exposed to BSA, a difference in protein interaction became evident. After 12 hours of incubation, both formulations maintained stable sizes, suggesting minimal initial protein adsorption. By 24 hours, P-Es exhibited a significant size increase, reaching approximately 575 ± 2.5 nm, indicative of substantial protein binding and possible aggregation.
In contrast, SS-Es displayed only a slight increase in size, confirming that the PEGylated surface effectively minimized protein adhesion. This resistance to protein adsorption supports the role of the PEG layer as a steric shield that reduces opsonin binding and protects the emulsomes from RES-mediated clearance. Consequently, this stealth behavior contributes to extended circulation time and may enhance the therapeutic potential of the drug delivery system was also shown in Figure 9..
Discussion
The formulation of an effective delivery system for Cabazitaxel (CTX) remains a crucial pharmaceutical challenge due to its low aqueous solubility, high systemic toxicity, and poor oral bioavailability. Traditional formulations, such as those using Cremophor EL, have been associated with significant hypersensitivity reactions, prompting the search for safer and more efficient alternatives. In this context, Compritol-based emulsomes offer a novel and biocompatible approach that addresses many of these limitations.
Rationale for Using Emulsomes
Emulsomes combine the structural advantages of liposomes and solid lipid nanoparticles (SLNs). By encapsulating CTX in a solid lipid core (Compritol or tristearin) and surrounding it with a phospholipid bilayer, the system not only increases the drug’s solubility but also improves its stability and controlled release properties. Unlike conventional systems, these emulsomes were designed without additional surfactants or co-solvents, reducing the risk of toxicity and enhancing patient safety.
Formulation and Physicochemical Characteristics
The use of a modified solvent evaporation technique proved efficient in producing reproducible formulations with uniform particle size, high drug entrapment efficiency, and stable zeta potential. The mean diameters of the simple and stabilized emulsomes-275 ± 5.52 nm (P-ES) and 195 ± 6.4 nm (SS-ES), respectively—fell within the optimal nanoscale range for passive tumor targeting via the enhanced permeability and retention (EPR) effect.
The smaller particle size and tighter distribution of SS-ES suggest that PEGylation and steric stabilization not only enhance physical stability but also contribute to more predictable pharmacokinetics. The phospholipid to PEG-lipid molar ratio and lipid/stabilizer ratios were critical in modulating particle size and stability, underscoring the importance of careful optimization in formulation development.
Drug Release and Mechanism
Both formulations exhibited pH-sensitive, sustained release profiles, which are ideal for cancer therapy. The acidic tumor microenvironment facilitated drug release, while physiological pH conditions maintained drug encapsulation, minimizing off-target toxicity. The release kinetics followed the Higuchi model, indicating a Fickian diffusion-controlled mechanism—suggesting that the drug was primarily released from the matrix through diffusion, a desirable trait for sustained therapeutic action.
The slower release from SS-ES compared to P-ES can be attributed to the stealth characteristics imparted by PEGylation, which may also result in reduced opsonization and extended systemic circulation. This is essential for allowing the drug carrier to accumulate at the tumor site before releasing its payload.
Therapeutic Implications and Biocompatibility
By eliminating harmful excipients like Cremophor EL, the developed emulsome system enhances biocompatibility and reduces the risk of immune-related adverse effects. The PEG layer, aside from extending circulation time, opens up possibilities for active targeting via ligand conjugation (e.g., folate, transferrin, or antibodies), potentially enabling receptor-mediated endocytosis in specific cancer cells.
Additionally, the modular nature of the emulsome design supports the co-encapsulation of multiple drugs, gene delivery components, or imaging agents, making it a multifunctional nanoplatform for combination therapy or theranostic applications.
Comparative Advantage and Future Directions
Compared to other nanoparticle systems like micelles or polymeric nanoparticles, emulsomes offer the unique advantage of a solid lipid matrix with tunable surface characteristics. Their ease of scale-up and reproducibility further support their industrial feasibility. However, the current study is limited to physicochemical characterization and in vitro release. Future investigations should focus on:
- In vivo pharmacokinetic and biodistribution studies
- Toxicological evaluation in animal models
- Comparative studies with conventional CTX formulations
- Ligand-targeted formulations for site-specific delivery
- Evaluation of therapeutic efficacy in cancer-bearing models
The developed sterically stabilized emulsomes present a safe, efficient, and versatile nanocarrier platform for Cabazitaxel delivery. Their physicochemical stability, controlled release, and biocompatible composition mark a significant advancement over traditional formulations. These findings pave the way for further preclinical and clinical exploration, with the potential to transform the delivery of Cabazitaxel and other hydrophobic anticancer agents.
Conclusions
Formulating an effective delivery system for Cabazitaxel (Ctx) remains a significant challenge due to its limited water solubility and poor bioavailability. In response, this study investigated the use of Compritol-based emulsomes as a safer, biocompatible alternative to traditional Cremophor-containing formulations. These emulsomes successfully encapsulated the drug within their solid lipid core, eliminating the need for harmful solubilizers, surfactants, or co-solvents.
A major advantage of this approach lies in the natural stabilization provided by the phospholipid layer surrounding the lipid core, which allowed the development of stable nanoscale formulations without external surfactants. The emulsomes were prepared using a modified solvent evaporation technique, resulting in a reproducible system capable of efficiently encapsulating lipophilic compounds.
The optimized Cabazitaxel-loaded emulsomes demonstrated favourable physicochemical attributes such as appropriate particle size, uniform morphology, stable surface charge, and high entrapment efficiency. Moreover, they exhibited a pH-responsive release profile—remaining stable under physiological conditions with limited drug leakage, while enabling enhanced release in acidic environments mimicking tumour sites. This targeted release behaviour highlights their potential for minimizing systemic toxicity while maximizing therapeutic efficacy at the disease site.
Surface modification with PEG further enhanced the stealth characteristics of the emulsomes, reducing recognition and clearance by the reticuloendothelial system (RES) and extending circulation time. Additionally, the phospholipid shell offers a versatile platform for ligand attachment, paving the way for targeted delivery to specific receptors, thereby broadening their utility for treating a wide range of diseases beyond cancer.
In conclusion, sterically stabilized emulsomes present a promising, non-toxic nanocarrier platform for the delivery of Cabazitaxel and potentially other poorly soluble therapeutic agents. This preliminary work underscores their potential as a next-generation drug delivery system for improved treatment outcomes.
Conflict of Interests: ll authors have contributed equally. The authors have no conflict of interest.
References
- Sawicki, I.E.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat Rev 2016, 50, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V.K. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sinica B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Tarr, B.D.; Yalkowsky, S.H. A new parenteral vehicle for the adminstration of some poorly water soluble anti-cancer drugs. JPST 1987, 41, 31–33. [Google Scholar]
- Patra, J.K.; Das, G.; Fraceto, L.F. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol 2018, 16, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Elgadir, M.A.; Salama, M.; Adam, A. Antia “breast cancer from various natural sources-review. Int J Pharm Pharm Sci 2015, 7, 44–47. [Google Scholar]
- Iwase, K.; Oyama, Y.; Tatsuishi, T.; Yamaguchi, J.Y.; Nishimura, Y.; Kanada, A.; et al. Cremophor EL augments the cytotoxicity of hydrogen peroxide in lymphocytes dissociated from rat thymus glands. Toxicol Lett 2004, 154, 143–148. [Google Scholar] [CrossRef]
- Gupta, R.; Gupta, M.; Mangal, S.; Agrawal, U.; Vyas, S.P. Capsaicin- loaded vesicular systems designed for enhancing localized delivery for psoriasis therapy. Artif Cells Nanomed Biotechnol 2014, 44, 825–834. [Google Scholar] [CrossRef]
- Patravale, V.B.; Mirani, A.G. Preparation and characterization of solid lipid nanoparticles-based gel for topical delivery. Methods in Molecular Biology 2019, 293–302. [Google Scholar]
- Sharma, A.; Mehta, V.; Parashar, A.; Patrwal, R.; Malairaman, U. Solid lipid nanoparticle: fabricated through nanoprecipitation and their physicochemical characterization. Int J Pharm Pharm Sci 2016, 8, 144–148. [Google Scholar] [CrossRef]
- Vyas, S.P.; Gupta, S. Optimizing efficacy of amphotericin B through nanomodification. Int J Nanomed 2006, 1, 417–432. [Google Scholar] [CrossRef]
- Chan, J.M.; Zhang, L.; Yuet, K.; Liao, G. PLGA–lecithin–PEG core– shell nanoparticles for controlled drug delivery. Biomaterials 2009, 30, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- John, A, P. R., S., Priya, s., Raviraj, C., & Ashtekar,. H. (2024). Optimized solid lipid nanoparticles for enhanced oral bioavailability and osteogenic effect of Ipriflavone: formulation, characterization, and in vitro evaluation. International journal of applied pharmaceutics, 16(6), 79–89. [CrossRef]
- Hassan, D. H. , Shohdy, J. M. ( 14(4), 88–93. [CrossRef]
- Aslam, R. , Tiwari, V., Upadhyay, P., & Tiwari, (2024). Revolutionizing therapeutic delivery: diosgenin-loaded solid lipid nanoparticles unleash advanced carriers. International journal of applied pharmaceutics, 16(1), 124–133. [CrossRef]
- Ali, M. , Mohamed, M. I., M. El-say, k., & Megahed, M. A. (2024). Innovative lipidic nanocarriers of flutamide enhancing its in vitro cytotoxicity and in vivo oral bioavailability: design, optimization, characterization, and pharmacokinetic aspects. International journal of applied pharmaceutics, 16(4), 66–77. [CrossRef]
- Konatham, S. , & Patan gay, S. (2023). Abiraterone acetate loaded solid lipid nanoparticles for improved oral bioavailability: design of experiments-based formulation optimization, in vitro, ex vivo, and in vivo characterization. International journal of applied pharmaceutics, 15(2), 131–139. [CrossRef]
- Kumar, J. A. , & Bhikshapathi, D. V. R. N. (2022). Development of Nilotinib-Loaded solid lipid nanoparticles and optimization by central composite design approach. International journal of applied pharmaceutics, 14(2), 172–180. [CrossRef]
- Reddy, M. R. , Gubbiyappa, S. K., Rasheed, S. H., & Parameshwar, k. (2024). Aptamers: Nanomaterials as a potential agent for antiviral therapeutic drug delivery development: a systematic literature review. International journal of applied pharmaceutics, 16(1), 33–50. [CrossRef]
- Sarkar, S., sadhu, S., Roy, R., Tarafdar, S., Mukherjee, N., Sil, M., Goswami, A., & Madhu, n. R. (2023). Contemporary drifts in diabetes management. International journal of applied pharmaceutics, 15(2), 1–9. [CrossRef]
- Raj R., G., S. , Patil, A. B., Jain, V., & Ajmeer, R. (2022). A review of advanced nanotechnologies and drug delivery systems of salinomycin and their role in triple-negative breast cancer. International journal of applied pharmaceutics, 14(4), 103–114. [CrossRef]
- Kumar, H., Rawal, V. B., Ajmeer, R., & Jain, V. (2022). Overview of mitoxantrone potential candidate for the treatment of breast cancer. International journal of applied pharmaceutics, 14(2), 10–22. [CrossRef]
- Gopaiah, K.V.; Shrivastava, B.; Rao, G.S. Lipid-polymer based nanoparticles as a new generation therapeutic delivery platform for ulcerative colitis: in vitro/in vivo evaluation. Int J Innov Technol Explor Eng Sci. 2019, 21, 42–46. [Google Scholar]
- Kumar, R. Characterization and biology of nanomaterials for drug delivery. Lipid-based nanoparticles for drug delivery systems: nanoscience and nanotechnology in drug delivery. In: Micro and Nano Technologies. 1st ed. US: Acdemia Press; 2019. p. 47-76.
- Mainardes, R.M.; Evangelista, R.C. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm 2005, 290, 137–144. [Google Scholar] [CrossRef]
- Song, X.; Zhao, Y.; Wu, W.; Bi, Y.; Cai, Z.; Chen, Q.; et al. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. Int J Pharm 2008, 350, 320–329. [Google Scholar] [CrossRef]
- Hunter, R.J. Applications of the zeta potential. In: Zeta Potential in Colloid Science Principles and Applications. 1st ed. US: Acdemia Press; 1981. p. 219-57.
- Barhoum, A.; Garcia Betancourt, M.L.; Rahier, H.; Assche, G.V. Physicochemical characterization of nanomaterials: polymorph, composition, wettability, and thermal stability. In: Emerging Applications of Nanoparticles and Architecture Nanostructures Current Prospects and Future Trends Micro and Nano Technologies. US: Acdemia Press; 2018. p. 255-78.
- Chirayil, C.J.; Abraham, J.; Mishra, R.K.; George, S.C.; Thomas, S. Instrumental techniques for the characterization of nanoparticles. In: Thermal and Rheological Measurement Techniques for Nanomaterials Characterization. Micro and Nano Technologies. 1st ed. Elsevier; 2017. p. 1-36.
- Venkateswarlu, V.; Manjunath, K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Controlled Release 2004, 95, 627–638. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhia, A.; et al. pH- sensitive nano-systems for drug delivery in cancer therapy. Biotechnol Adv 2014, 32, 693–710. [Google Scholar] [CrossRef]
- Kushwaha, A.K.; Vuddanda, P.R.; Karunanidhi, P.; Singh, S.K. ; SinghS Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. Biomed Res Int 2013, 1–9. [Google Scholar] [CrossRef]
- Gopaiah, K.V.; Mandadapu, G.; Kolli, P.; Medarametla, R.T. Solid lipid nanoparticles of Naftopidil: formulation design and in vitro evaluation for improved oral absorption. Pharma Sci Anal Res J. 2024, 6, 180061. [Google Scholar]
- Gopaiah, K.V.; Mandadapu, G.; Kolli, P.; Medarametla, R.T. Synergistic elegance: harnessing liposomes as advanced cosmetic drug delivery systems with phospholipids and phenolic components. Pharma Sci Anal Res J. 2024, 6, 180077. [Google Scholar]
- Mandadapu, G.; Kolli, P.; Gopaiah, K.V.; Medarametla, R.T. Preparation and evaluation of anti-viral drug entrapped microspheres by using natural gums as rate controlling agent in enhancing bioavailability. Int J Pharm Pharm Res. 2024, 30, 113–126. [Google Scholar]
Figure 2.
Graph showing the effect of organic to aqueous phase volume on size and entrapment efficiency of emulsomes. Data represents mean ± SD (n=3).
Figure 2.
Graph showing the effect of organic to aqueous phase volume on size and entrapment efficiency of emulsomes. Data represents mean ± SD (n=3).
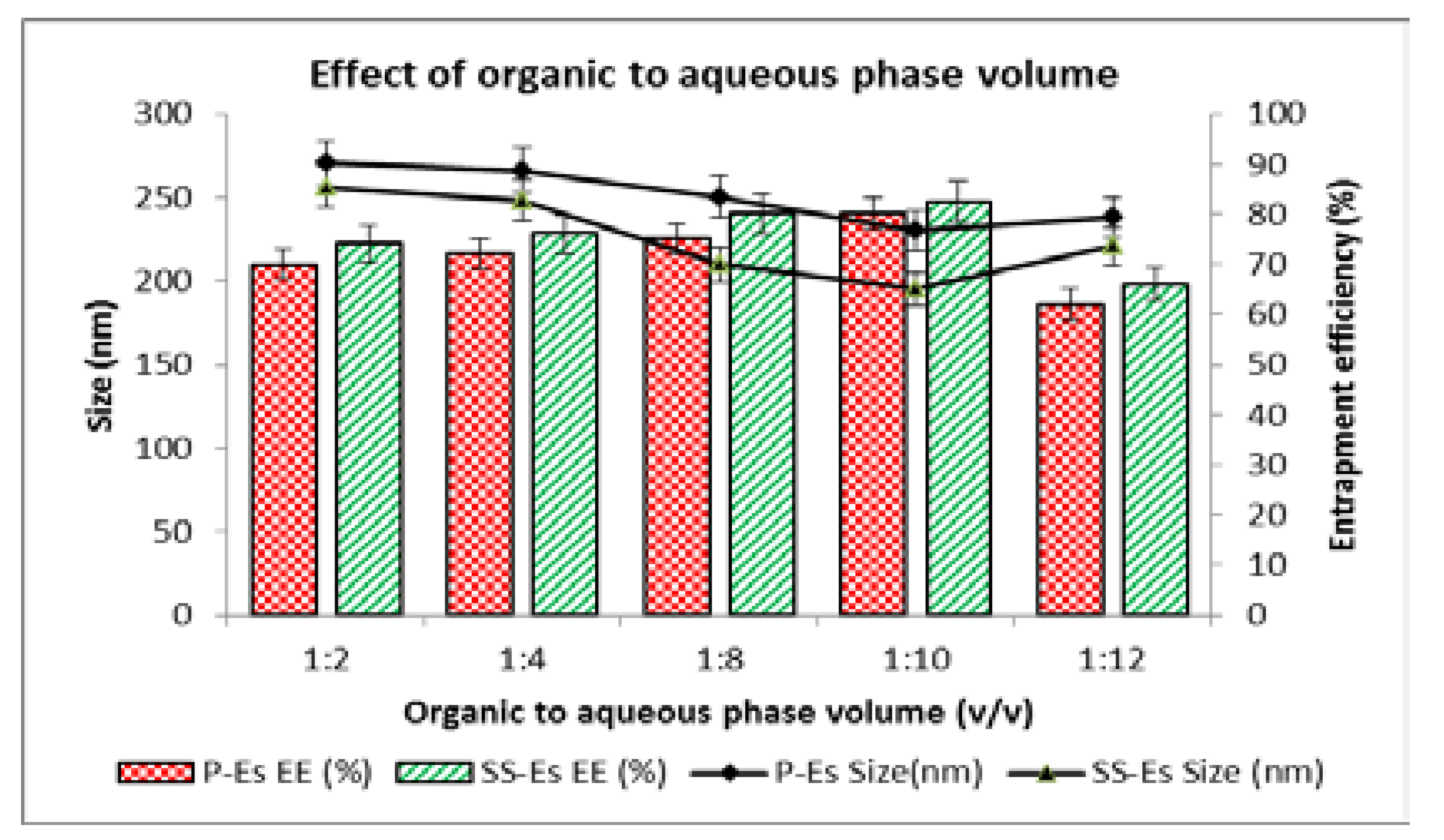
Figure 3.
A. Graphs showing the effect of PL to SL ratio on size and entrapment efficiency of emulsomes. B. Plot showing the change in the zeta potential of emulsomes with PL: SL ratio. Data represents mean ± SD (n=3).
Figure 3.
A. Graphs showing the effect of PL to SL ratio on size and entrapment efficiency of emulsomes. B. Plot showing the change in the zeta potential of emulsomes with PL: SL ratio. Data represents mean ± SD (n=3).
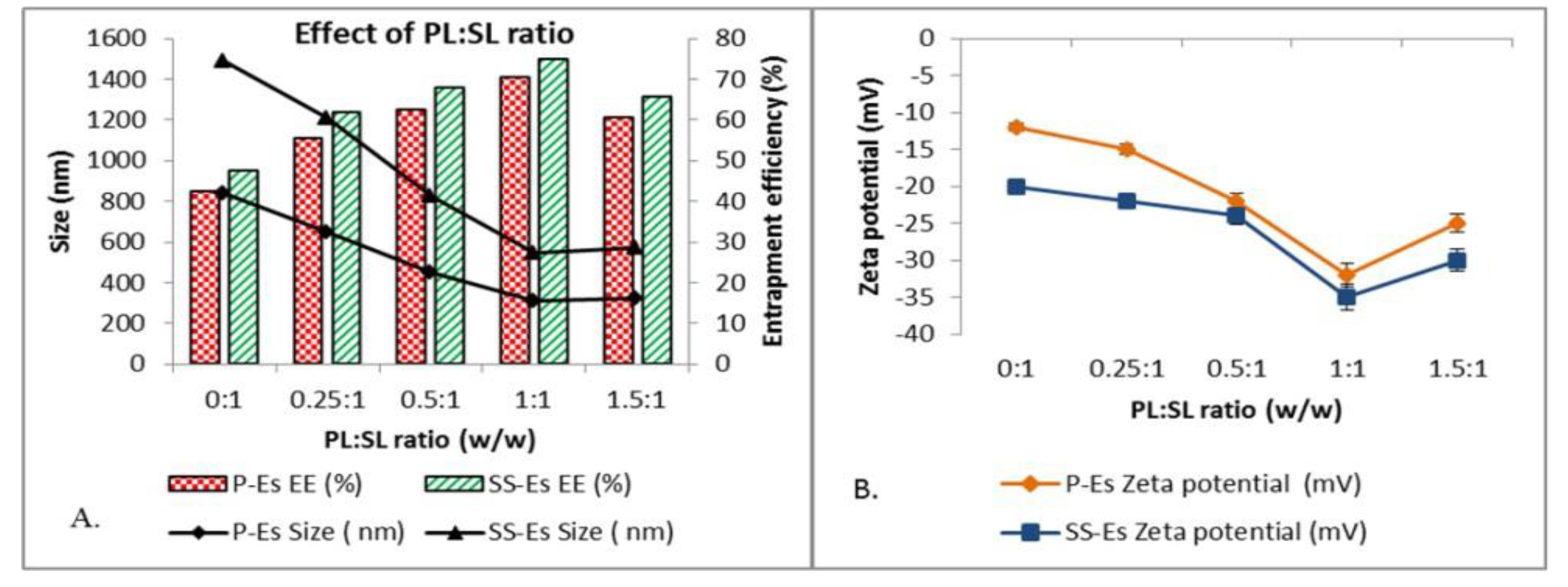
Figure 4.
Graphs showing the effect of SL concentration on particle size and entrapment efficiency of emulsomes. Data represents mean ± SD (n=3).
Figure 4.
Graphs showing the effect of SL concentration on particle size and entrapment efficiency of emulsomes. Data represents mean ± SD (n=3).
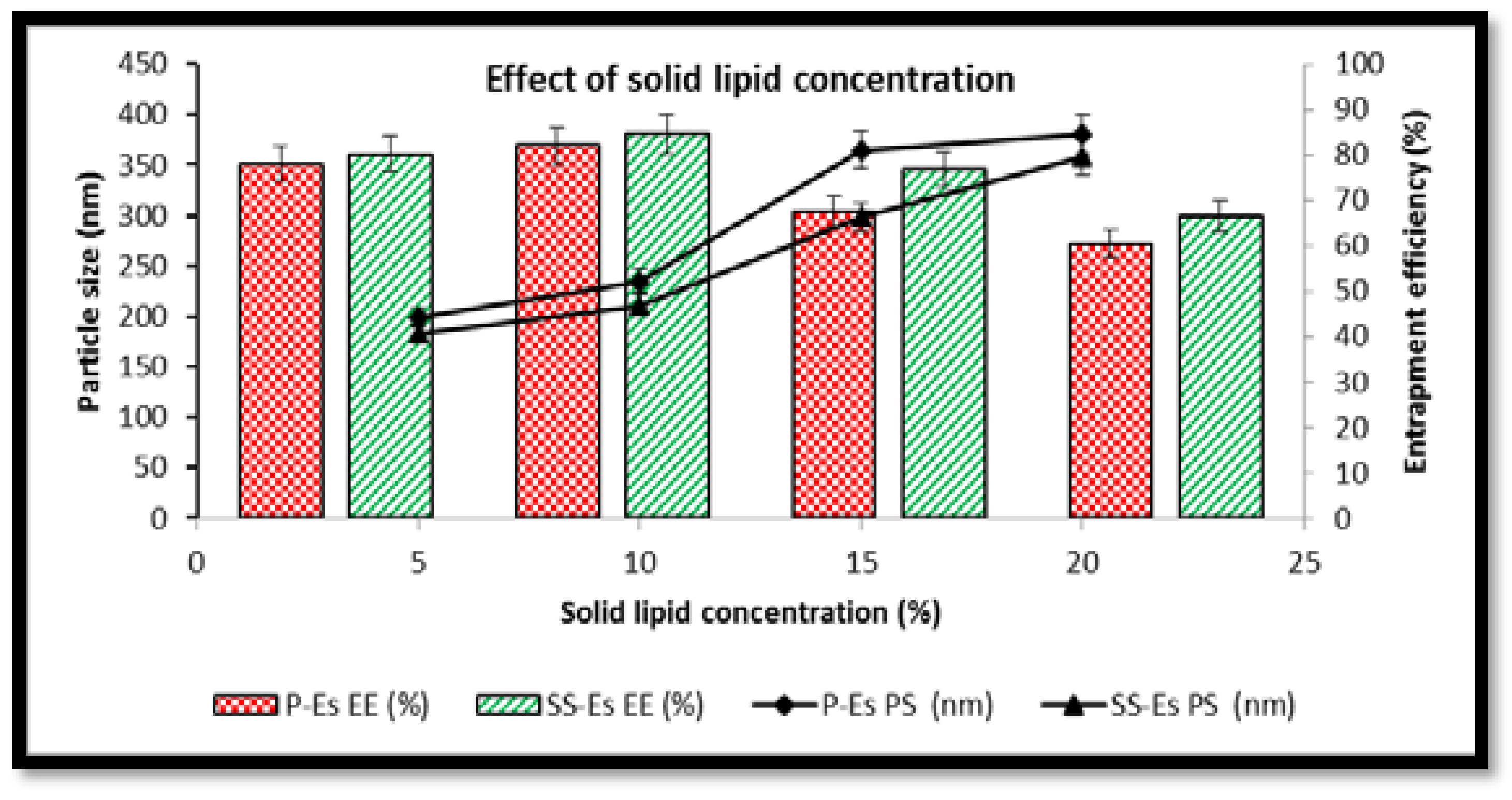
Figure 9.
Change in size of emulsomes on incubation with 5%BSA, data represents mean ± SD (n=3).
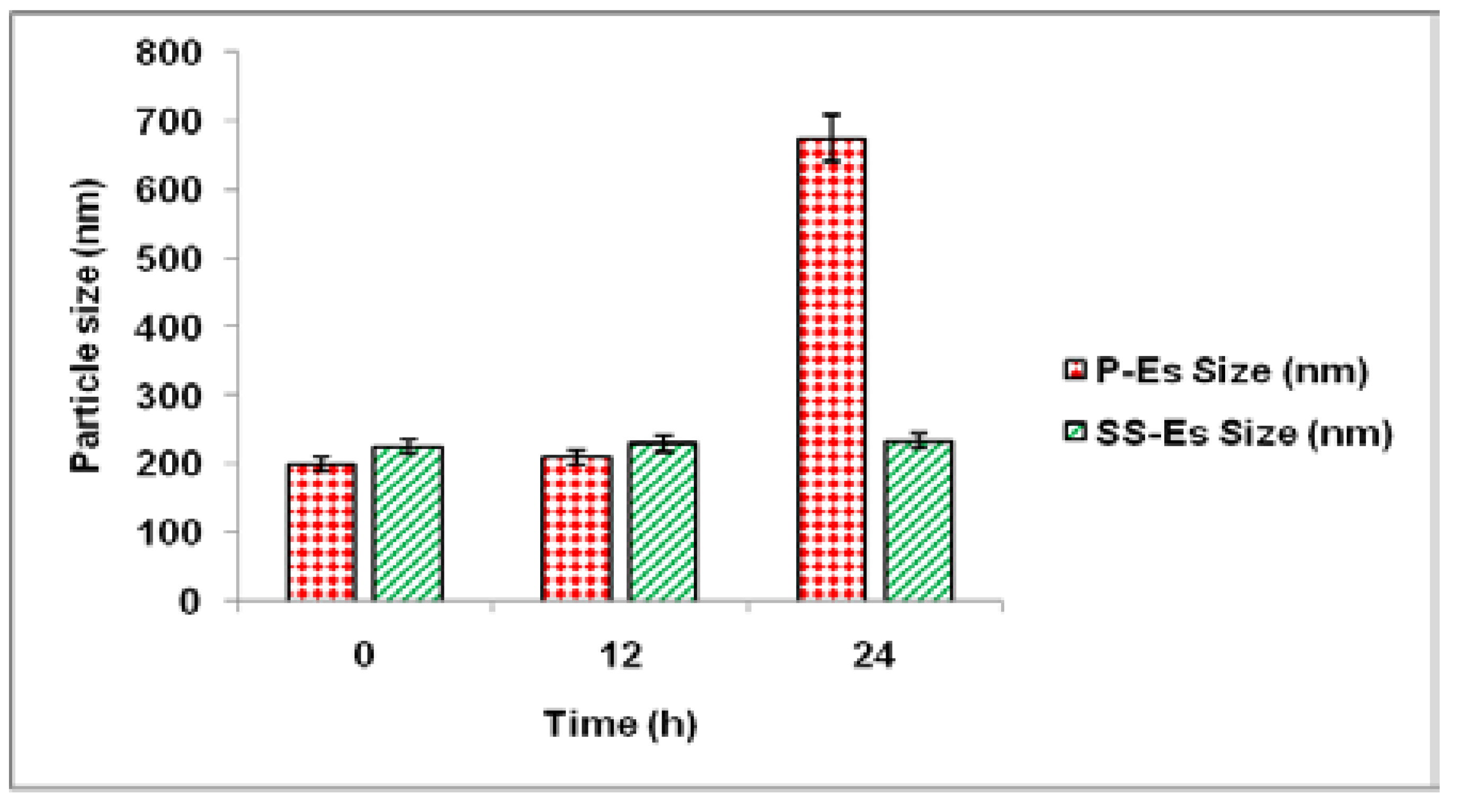

Table 1.
Composition of optimized formulations.
| Formulation code | PL: DSPE-PEG (w/w) |
PL: SL (w/w) |
Organic: aqueous phase volume (v/v) | SL concentration (%) | Homogenization time (min) |
|---|---|---|---|---|---|
| P-Es | - | 1:1 | 1:10 | 10 | 15 |
| SS-Es | 1:3 | 1:1 | 1:10 | 10 | 15 |
P-Es: Plain emulsomes, SS-Es: Sterically stabilized emulsomes, PL: phospholipid, DSPE-PEG: 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine- Poly(ethylene glycol), SL: Solid lipid.
Table 2.
Physicochemical properties of optimized formulations.
| Formulation code | Size (nm) | PI | Zeta potential (mV) | %EE |
|---|---|---|---|---|
| P-Es | 275±5.52* | 0.112±0.011** | -30.5±2.0* | 72.54±3.41* |
| SS-Es | 195±6.4** | 0.124±0.012** | -35.8±2.1* | 75.56±3.25** |
Data represents mean ± SD (n=3), *Statistically significant difference at *p<0.05,** Statistically significant difference at p<0.01.
Table 3.
Parameters for kinetic models applied to in vitro drug release data from emulsomes formulations.
Table 3.
Parameters for kinetic models applied to in vitro drug release data from emulsomes formulations.
| Formulation | Zero order | First order | Higuchi | Korsmeyer-peppas | ||||
|---|---|---|---|---|---|---|---|---|
| R2 | K | R2 | K | R2 | K | R2 | n | |
| P-Es (pH 7.4) | 0.8875 | 0.3011 | 0.9512 | 0.0025 | 0.9947 | 4.4170 | 0.9766 | 0.3252 |
| SS-Es (pH 7.4) | 0.9470 | 0.3166 | 0.9755 | 0.0024 | 0.9912 | 4.5573 | 0.9925 | 0.3942 |
| P-Es (pH 4.0) | 0.9128 | 0.3255 | 0.9766 | 0.0033 | 0.9958 | 5.5072 | 0.9968 | 0.2659 |
| SS-Es (pH 4.0) | 0.9383 | 0.2860 | 0.9755 | 0.0022 | 0.9997 | 4.3476 | 0.9858 | 0.2730 |
R: regression coefficient. K: release constant, n: diffusion coefficient.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



