
Submitted:
30 May 2025
Posted:
02 June 2025
You are already at the latest version
Abstract
The emergence of novel viral pathogens, such as SARS-CoV-2, often ignites an urgent quest to pinpoint their precise origins and ancestral lineages. However, the very nature of viral evolution, deeply intertwined with stochastic processes, high mutation rates, genetic exchange, and vast, often unsampled, reservoirs, presents profound challenges to reconstructing deep evolutionary histories with pinpoint accuracy. This essay explores these challenges, arguing that while we can trace recent evolutionary pathways and identify mechanisms of genetic novelty, the quest for a single, identifiable ancestral genotype for viruses that emerged from ancient, diverse reservoirs is often a statistical and biological near-impossibility. Using case studies from Human Immunodeficiency Virus (HIV) and Simian Immunodeficiency Viruses (SIVs), Influenza A viruses, and SARS-CoV-2, we illustrate how stochasticity, sampling effects, and the complexities of viral population dynamics limit our ability to resolve deep ancestral nodes. We propose a conceptual model, visualized through a diagram, to represent the "cloud of unknowing" surrounding ancient viral ancestors, emphasizing a shift from seeking definitive ancestral individuals to understanding the probabilistic nature of viral emergence from diverse populations.
Keywords:
virus evolution
; phylogenetic tree
; genetic recombination
; reassortment
; stochasticity
1. Introduction: The Allure and Difficulty of Origins
The appearance of a new pathogenic virus in the human population invariably triggers intense scientific efforts to understand its origins. This pursuit is driven by critical public health needs: identifying reservoir hosts, understanding pathways of zoonotic transmission, and informing strategies for prevention and control. Genomic sequencing and phylogenetic analysis are primary tools in this endeavor, allowing us to trace the recent evolutionary history of a pathogen with remarkable detail [1]. However, when we attempt to peer deeper into the evolutionary past – to identify the specific ancestral viral lineage within a vast animal reservoir that gave rise to a human pathogen, or to reconstruct the precise sequence of events leading to its emergence – we encounter fundamental limitations imposed by the nature of viral evolution itself.
A central theme often underappreciated in the broader discourse is the profound role of stochasticity – chance events – at every level of viral evolution and emergence. From the random occurrence of mutations and recombination events within a viral quasispecies [2] to the contingent success of a cross-species transmission, chance plays a decisive role. This essay argues that this inherent stochasticity, coupled with the vastness of viral diversity in animal reservoirs and the practical limitations of sampling, makes the precise identification of a single, "pinpoint" ancestral virus for many pathogens a scientifically elusive, if not unattainable, goal. Instead, a more realistic aim is to characterize the properties of the ancestral population or viral cloud from which an emergent lineage likely arose, and to understand the ecological and evolutionary processes that facilitate such emergences.
2. The Challenge of Pinpointing Viral Ancestors: An Analogy
To grasp the difficulty of identifying a specific viral ancestor, an analogy to the human-chimpanzee common ancestor is instructive. We know from overwhelming genetic and fossil evidence that humans and chimpanzees share a common ancestor that lived millions of years ago. We can infer many characteristics of this ancestral population, its approximate timeframe, and even reconstruct parts of its genome with high probability. However, no paleontologist expects to find the fossilized remains of the single individual hominin who was the direct, ultimate common ancestor of all modern humans and all modern chimpanzees. Evolution occurs in populations, not as a linear chain of individuals. The "common ancestor" we refer to is a population whose descendants diverged.
For viruses, the challenge is compounded manyfold. The "fossil record" in the form of ancient viral sequences is virtually non-existent beyond a few decades or, in exceptional cases of DNA viruses preserved in ancient remains, a few centuries or millennia. Most viruses, especially RNA viruses, evolve at rates many orders of magnitude faster than primates. Their populations are vastly larger, their generation times are minuscule, and genetic exchange mechanisms like recombination and reassortment can rapidly create novel genetic combinations, obscuring direct lines of descent [2]. Thus, if finding the "individual" common ancestor of humans and chimps is a practical impossibility, the quest for the specific individual animal virus particle that founded a human epidemic millions of replication cycles later is even more so.
3. Case Studies in Viral Emergence and Deep History
3.1. The Multiplicity of Origins: Lessons from Simian and Human Immunodeficiency Viruses
The story of HIV/SIV provides one of the most compelling illustrations of multiple, independent stochastic emergence events from a diverse animal reservoir. The overwhelming weight of evidence indicates that human immunodeficiency viruses (HIV-1 and HIV-2) arose from cross-species transmissions of simian immunodeficiency viruses (SIVs) naturally infecting various African primate species [3].
- Diverse SIV Reservoir. Dozens of SIV lineages have been identified in many species of African non-human primates. These SIVs are often species-specific and have likely co-evolved with their primate hosts for thousands, if not millions, of years, creating an immense and ancient "reservoir cloud" of viral diversity.
- Multiple Independent Spillovers. Crucially, HIV-1 did not arise from a single SIV transmission. Phylogenetic analyses reveal at least four distinct HIV-1 groups (M, N, O, and P), each reported [3] as an independent cross-species transmission of SIVcpz from chimpanzees (Pan troglodytes troglodytes) or SIVgor from gorillas (Gorilla gorilla gorilla) – gorillas themselves likely acquired their SIV from chimpanzees. HIV-2 also has at least nine distinct groups (A-I), each originating from independent transmissions of SIVsmm from sooty mangabeys (Cercocebus atys). These findings are subject to refinement as other primate populations are sampled.
- Stochastic Nature of Establishment. Each of these successful spillovers represents a rare, stochastic event where a particular SIV variant managed to infect a human, replicate, adapt sufficiently to sustain human-to-human transmission, and establish a new lineage. Countless other SIV exposures to humans likely occurred without leading to sustained epidemics.
- Recombination's Role. Once established in humans, particularly for HIV-1 Group M (responsible for the vast majority of the global pandemic), recombination between different co-infecting subtypes has led to the generation of numerous Circulating Recombinant Forms (CRFs), further diversifying the virus and complicating phylogenetic reconstruction [4,5].
The SIV/HIV paradigm powerfully demonstrates that viral emergence is not necessarily a singular, unique event but can be a repeated process when ecological opportunity (e.g., human-primate contact through hunting) and virological permissiveness (a virus capable of infecting and adapting to a new host) align. While we can identify the primate SIV clades most closely related to each HIV group/type, pinpointing the exact ancestral SIV lineage within the vast diversity of SIVs in primates that founded each human epidemic is beyond the resolution of current data.
3.2. Influenza A Virus: Ancient Reservoirs and Stochastic Reassortment
Influenza A viruses (IAVs) offer another classic example of emergence driven by stochastic genetic exchange, in this case, reassortment, operating against a backdrop of an ancient and vast viral reservoir.
- Avian Reservoir. Wild aquatic birds, particularly waterfowl, are considered the primary natural reservoir for almost all IAV subtypes (HxNy) [6]. Within these bird populations, IAVs exhibit enormous genetic diversity and generally cause asymptomatic infections, co-evolving over long periods.
- Reassortment and Antigenic Shift. IAVs have a segmented genome (eight RNA segments). If two different IAV strains co-infect the same cell, their segments can be "shuffled" during the assembly of new virus particles, producing reassortant viruses with novel combinations of genes. This is the primary mechanism behind antigenic shift, which can lead to the emergence of pandemic influenza strains when a virus with a hemagglutinin (HA) and/or neuraminidase (NA) subtype novel to the human population acquires the ability to transmit efficiently between humans.
- Pandemic Origins:
- The 1957 (H2N2 "Asian flu") and 1968 (H3N2 "Hong Kong flu") pandemics arose from reassortment events where human-adapted IAVs acquired HA/NA and polymerase (PB1) segments from avian influenza viruses.
- The 2009 H1N1 pandemic virus was a particularly complex "quadruple reassortant," possessing segments derived from North American swine, Eurasian swine, human seasonal, and North American avian IAV lineages, likely assembled through multiple reassortment steps in swine, which act as "mixing vessels" [7,8]. Each of these reassortment events is a stochastic process, dependent on co-infection and the viable packaging of a new constellation of segments.
- Challenges in Deep Ancestry. While we can trace the origins of segments in recent pandemic strains to broad avian or swine lineages, identifying the specific ancestral avian virus that contributed, for example, the HA to the 1957 H2N2 pandemic, or the precise sequence of reassortment events in swine leading to the 2009 H1N1, becomes increasingly difficult further back in time due to continuous evolution and extinction of intermediate lineages in animal reservoirs.
Influenza illustrates how a stable, ancient reservoir can periodically "spin off" novel reassortant viruses through stochastic co-infection events, some of which, by chance, possess the right genetic makeup to cross species barriers and cause widespread disease.
3.3. SARS-CoV-2: Reconstructing Recent Emergence and Ongoing Recombination
The COVID-19 pandemic, caused by SARS-CoV-2, has seen unprecedented global efforts to sequence the virus and trace its origins. Despite this, and its very recent emergence, pinpointing its exact proximal origins remains challenging, highlighting the difficulties even for contemporary events.
- Zoonotic Origin. The weight of evidence indicates that SARS-CoV-2 originated from a coronavirus in an animal reservoir, most likely bats, potentially with an intermediate animal host facilitating its spillover to humans [9].
- Role of Recombination. The Spike protein's receptor-binding domain (RBD), crucial for human ACE2 receptor binding, shows evidence suggestive of recombination with coronaviruses from other animal species (e.g., pangolins), although the exact details and timing of such events are still debated and subject to the availability of more comprehensive animal reservoir sampling [10].
- Challenges in Identification. 1) Reservoir Sampling. Despite extensive searching, the exact bat coronavirus population that served as the direct progenitor of SARS-CoV-2 has not been definitively identified. Bats host a vast diversity of coronaviruses, and the specific lineage that jumped to humans (or an intermediate host) may be rare, geographically restricted, or even extinct. 2) Intermediate Host(s). If an intermediate host was involved, identifying it and the specific viral lineage it carried is also challenging. 3) Stochasticity of Spillover. The actual spillover event was likely a stochastic process, possibly one of many attempted jumps that only rarely succeeded.
- Ongoing Recombination in Human Variants. During the pandemic, recombination between circulating SARS-CoV-2 lineages (e.g., between different Omicron sublineages like those forming the XBB lineage) has been documented. The XBB lineage, itself a recombinant of two Omicron BA.2 sublineages, subsequently gave rise to further subvariants like XBB.1.16 ("Arcturus"), which acquired additional mutations impacting transmissibility and immune evasion [11]. These events highlight how recombination can create new genetic backbones for further mutational refinement.
The SARS-CoV-2 case underscores that even with massive resources and a very recent emergence, the stochastic nature of zoonotic spillover from diverse and often undersampled animal reservoirs, potentially compounded by recombination, makes the reconstruction of the precise chain of events leading to human infection extraordinarily difficult.
4. Broader Implications: Sampling, Modeling, and the Nature of Viral Ancestry
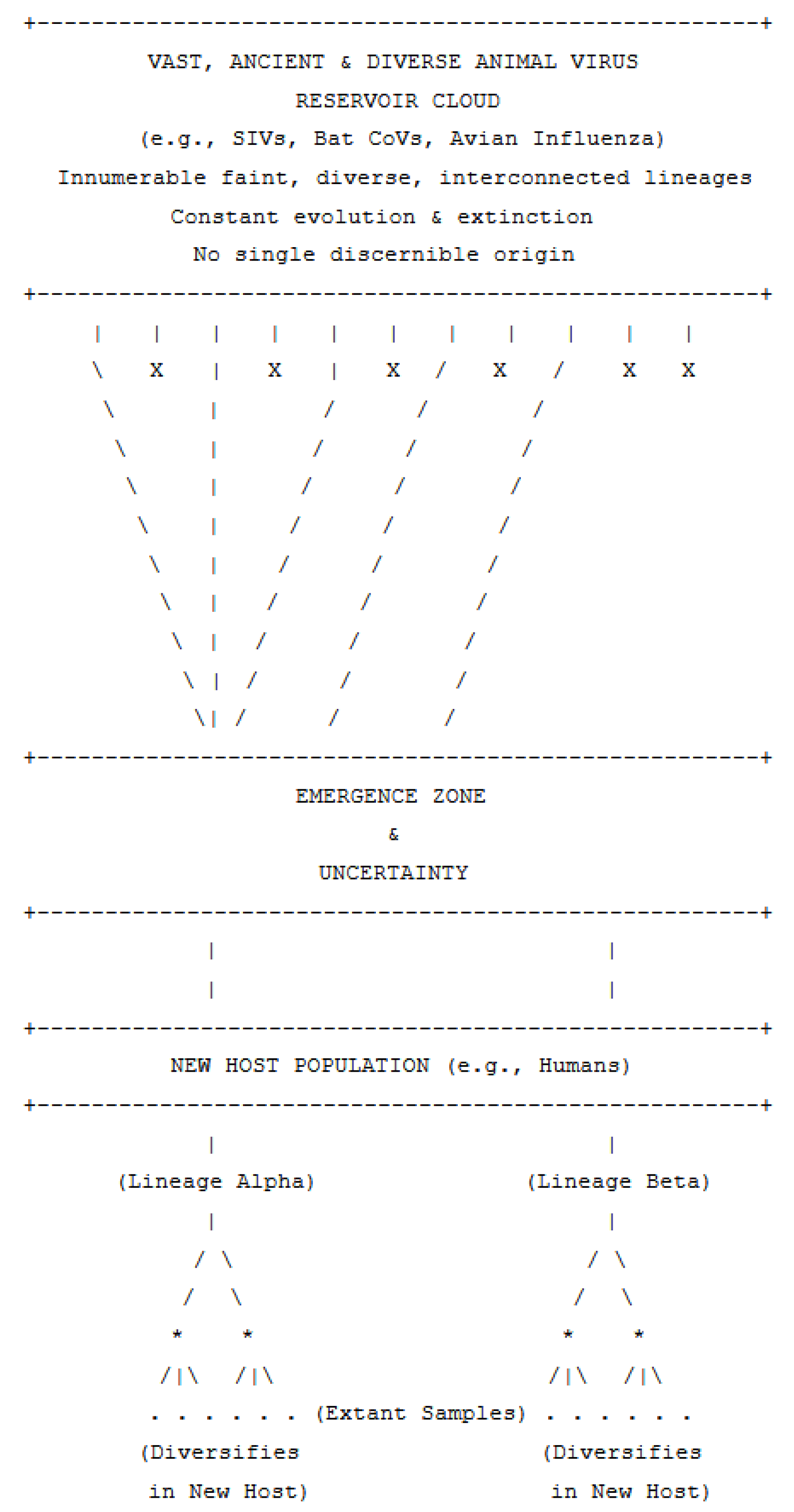
To visually encapsulate the profound challenges in reconstructing deep viral origins and the pivotal role of stochasticity, conceptual model is presented (see Figure 1). This model is not intended as a literal phylogenetic tree but as an abstract representation of the processes and limitations involved.
The case studies and the conceptual model (see Figure 1) highlight several critical implications:
- Sampling Effects are Profound. Our understanding of viral diversity and origins is entirely constrained by what we sample. Wildlife reservoirs are notoriously undersampled, and even within human populations, surveillance is often biased towards symptomatic cases or specific geographic regions. The "viral dark matter" – the vast majority of viruses that exist but have not been sequenced – means our phylogenetic trees are sparse representations of true viral evolutionary history.
- Phylogenetic Reconstruction Limits. While powerful for recent events and well-sampled populations, phylogenetic methods struggle with deep ancestral reconstruction for rapidly evolving viruses with extensive recombination/reassortment. Long branches, homoplasy, and conflicting signals from different genomic regions can obscure deep relationships. Molecular clock estimates for deep viral origins are often highly uncertain due to rate variation and lack of ancient calibration points.
- Ancestral State Reconstruction vs. Pinpointing an Ancestor. It is important to distinguish between inferring the characteristics of an ancestral viral population or node (e.g., likely genetic sequences at certain sites, probable host) and identifying the specific individual ancestral virus. The former is a probabilistic inference based on extant diversity; the latter is generally not feasible for deep time.
- Rethinking "Patient Zero" Narratives. For many viral emergences, the search for a single "patient zero" or a single animal source, while important for initial outbreak control, may be an oversimplification of a more complex ecological and evolutionary process involving a population of viruses in a reservoir and multiple potential spillover opportunities.
5. Conclusion: Embracing Stochasticity and Probabilistic Understanding
The quest to understand viral origins is vital, yet it must be navigated with a profound appreciation for the inherent complexities and limitations imposed by the very nature of viral evolution. The emergence and subsequent diversification of viruses are processes deeply shaped by an intricate interplay of deterministic forces, such as natural selection, and pervasive stochastic events, including the timing and location of mutations, the success of genetic exchange, and the contingent opportunities for cross-species transmission. While our scientific toolkit allows for remarkable insights into recent evolutionary trajectories and the mechanisms of viral adaptation, the precise pinpointing of deep viral ancestors often remains beyond our empirical grasp, lost within the vastness of ancient, unsampled reservoirs and the high tempo of viral change.
Therefore, an essay on this topic, and indeed the broader scientific pursuit, should not be defined by the search for definitive answers to ultimately unanswerable questions about specific ancient ancestral individuals. Instead, its enduring value lies in illuminating the fundamental roles of these core evolutionary processes in shaping the virosphere. It calls for a critical evaluation of the strengths and inherent limitations of our current methodologies for reconstructing viral evolutionary history. Ultimately, this perspective encourages a more nuanced, probabilistic understanding of viral origins. It advocates for a conceptual shift: from the often-publicized search for singular ancestral viruses or "patient zero" narratives, towards a more scientifically grounded endeavor of characterizing the broader ancestral viral populations, the ecological contexts that foster their diversity, and the probabilistic nature of their emergence into new host populations. Such an approach, underpinned by continued efforts in broad viral surveillance, innovative methodological development, and robust interdisciplinary research, will better equip us to understand, anticipate, and mitigate future viral threats, even as we acknowledge and respect the "cloud of unknowing" that inevitably shrouds the deepest recesses of viral history.
Funding
This research received no external funding.
Acknowledgments
The conceptual development and drafting of this essay benefited significantly from discussions and iterative refinement with an AI language model, Gemini 2.5 Pro, (Google, 5/6/2025).
Conflicts of Interest
The author declares no conflict of interest.
References
- Holmes, E. C. , & Drummond, A. J. The evolutionary genetics of viral emergence. Current Topics in Microbiology and Immunology, 2007, 315, 51–66. [Google Scholar] [PubMed]
- Friedman, R. A Hierarchy of Interactions between Pathogenic Virus and Vertebrate Host. Symmetry, 2022, 14, 2274. [Google Scholar] [CrossRef]
- Sharp, P. M. , & Hahn, B. H. Origins of HIV and the AIDS pandemic. Cold Spring Harbor Perspectives in Medicine, 2011, 1, a006841. [Google Scholar] [PubMed]
- Robertson, D. L. , Anderson, J. P., Bradac, J. A., Carr, J. K., Foley, B., Funkhouser, R. K.,... & Korber, B. HIV-1 nomenclature proposal. Science, 2000, 288, 55–56. [Google Scholar] [PubMed]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends in Molecular Medicine, 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Webster, R. G. , Bean, W. J., Gorman, O. T., Chambers, T. M., & Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiological Reviews, 1992, 56, 152–179. [Google Scholar] [PubMed]
- Garten, R. J. , Davis, C. T., Russell, C. A., Shu, B., Lindstrom, S., Balish, A.,... & Cox, N. J. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science, 2009, 325, 197–201. [Google Scholar] [PubMed]
- Smith, G. J. D. , Vijaykrishna, D., Bahl, J., Lycett, S. J., Worobey, M., Pybus, O. G.,... & Rambaut, A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature, 2009, 459, 1122–1125. [Google Scholar] [PubMed]
- Andersen, K. G. , Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. The proximal origin of SARS-CoV-2. Nature Medicine, 2020, 26, 450–452. [Google Scholar] [PubMed]
- Li, X. , Giorgi, E. E., Marichannegowda, M. H., Foley, B., Xiao, C., Kong, X. P.,... & Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Science Advances, 2020, 6, eabb9153. [Google Scholar] [PubMed]
- Uriu, K. , Ito, J., Zahradnik, J., Fujita, S., Kosugi, Y., Schreiber, G. Enhanced transmissibility, infectivity, and immune resistance of the SARS-CoV-2 omicron XBB.1.5 variant. Lancet Infectious Diseases, 2023, 23, 280–281. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Conceptual Model of Viral Emergence and the Limits of Ancestral Reconstruction. The diagram illustrates the probabilistic nature of viral emergence from animal reservoirs. The vertical dimension represents a progression through time: the uppermost section, labeled VAST, ANCIENT & DIVERSE ANIMAL VIRUS RESERVOIR CLOUD, symbolizes events in deep time or the ancient past. Zone #1) This cloud is characterized by innumerable, faint, and interconnected viral lineages, reflecting constant evolution and extinction dynamics with no single discernible origin point. Descending from this reservoir are multiple "Spillover Tendrils"; lines terminating in an 'X' represent the numerous stochastic cross-species transmission attempts that fail to establish sustained infections (Failed Spillovers). Zone #2) Only a very few lines successfully pass through the central box labeled EMERGENCE ZONE & UNCERTAINTY. This zone signifies the conceptual boundary where a lineage from the reservoir successfully transitions to the new host, and also represents a point of profound unknowability: while an emergence event is inferred, the precise, individual ancestral viral genotype that initiated this spillover is considered probabilistically determined and an unknowable pinpoint ancestor within the vastness of the reservoir. Zone #3) The successful lineages (e.g., Lineage Alpha, Lineage Beta) then enter the NEW HOST POPULATION, where they may diversify, leading to the "Extant Samples" (represented by dots) that are typically available for genomic study. The overall depiction underscores the increasing uncertainty and probabilistic nature of reconstructing deep ancestral pathways back into the reservoir cloud.
Figure 1.
Conceptual Model of Viral Emergence and the Limits of Ancestral Reconstruction. The diagram illustrates the probabilistic nature of viral emergence from animal reservoirs. The vertical dimension represents a progression through time: the uppermost section, labeled VAST, ANCIENT & DIVERSE ANIMAL VIRUS RESERVOIR CLOUD, symbolizes events in deep time or the ancient past. Zone #1) This cloud is characterized by innumerable, faint, and interconnected viral lineages, reflecting constant evolution and extinction dynamics with no single discernible origin point. Descending from this reservoir are multiple "Spillover Tendrils"; lines terminating in an 'X' represent the numerous stochastic cross-species transmission attempts that fail to establish sustained infections (Failed Spillovers). Zone #2) Only a very few lines successfully pass through the central box labeled EMERGENCE ZONE & UNCERTAINTY. This zone signifies the conceptual boundary where a lineage from the reservoir successfully transitions to the new host, and also represents a point of profound unknowability: while an emergence event is inferred, the precise, individual ancestral viral genotype that initiated this spillover is considered probabilistically determined and an unknowable pinpoint ancestor within the vastness of the reservoir. Zone #3) The successful lineages (e.g., Lineage Alpha, Lineage Beta) then enter the NEW HOST POPULATION, where they may diversify, leading to the "Extant Samples" (represented by dots) that are typically available for genomic study. The overall depiction underscores the increasing uncertainty and probabilistic nature of reconstructing deep ancestral pathways back into the reservoir cloud.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



