
Submitted:
22 May 2025
Posted:
23 May 2025
You are already at the latest version
Abstract
In this survey, we begin with a concise introduction to information theory within Shannon's framework, focusing on the key concept of Shannon entropy and its related quantities: relative entropy, joint entropy, conditional entropy and mutual information. We then demonstrate how to apply these information-theoretic tools in quantum chemistry, adopting either classical or quantum formalisms based on the choice of information carrier involved.
Keywords:
quantum chemistry
; information theory
; classical information theory
; quantum information theory
; shannon entropy
; von neumann entropy
; relative entropy
; joint entropy
; conditional entropy
; mutual information
; information-theoretic approach
; orbital entanglement
1. Motivation
Information theory was first established in the 1920s through the works of Harry Nyquist and Ralph Hartley [1,2] and propelled to prominence in the 1940s by Claude Shannon [3]. The concept of information theory, which encompasses the quantification, storage, and communication of information [4,5], is too broad to be simply described. Today, information theory serves as a versatile tool in statistics, natural sciences, machine learning, quantum computing, and numerous other fields.
In molecular electronic structure theory, we routinely encounter sets of nonnegative values that sum to unity, corresponding to valid probability distributions. This includes properly normalized electron density distributions, the eigenvalues of the reduced density matrix, the squared modulus of wave function coefficients in an orthonormal basis, and more [6,7]. Information theory can be used to analyze these probability distributions, an approach that has been actively pursued by researchers with remarkable success since the 1970s [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. Incorporating concepts from information theory into quantum chemistry has provided valuable insights into the nature and behavior of electronic systems.
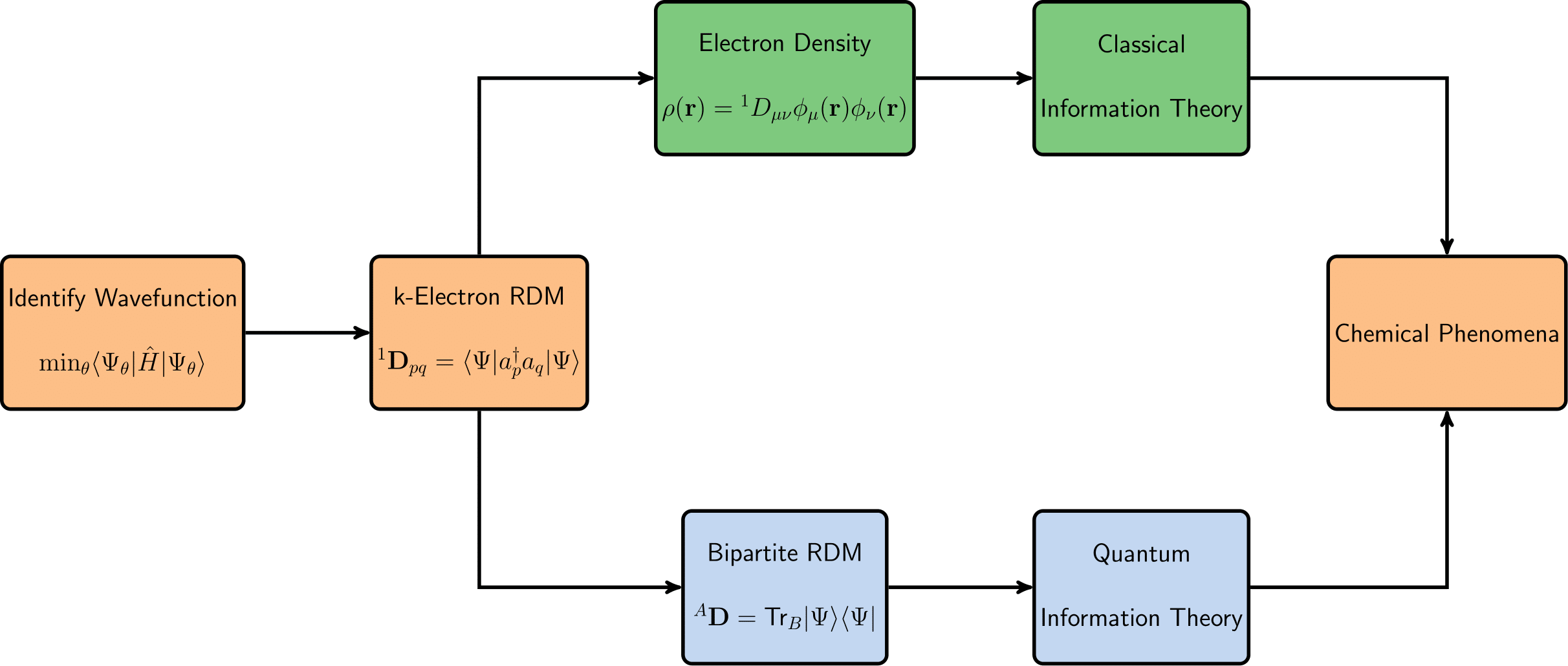
The two most popular approaches to electronic structure theory are quantum many-body theory [37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52] and density functional theory (DFT) [53,54,55,56]. Integrating these strategies with classical information theory (CIT) leads to what is normally called the information-theoretic approach (ITA) [8,9,10,11,12,13,14,15,16,27,28,29,30,31,32,33,34,35,36,57]; integrating these strategies with quantum information theory (QIT) [17,18,19,20,21,22,23,24,25,26] leads to a different set of tools, with different interpretations and utility. Some key concepts of classical and quantum information theory in quantum chemistry that we will discuss in this article are summarized in Figure 1. Although information theory extends far beyond Shannon’s work, other frameworks such as Rényi and Fisher information have their own application domains [58,59,60,61]. Due to space limitations, our discussion in this paper will be confined to Shannon’s framework, although we note that the Rényi formulation and Tsallis formulations, [58,60] in particular, reduce to Shannon’s in appropriate limits and, as such, the following analysis can be generalized to these quantities.
This review concisely introduces some fundamental aspects of information theory and its applications in quantum chemistry. Section 2 discusses the basic concepts of Shannon entropy and its related quantities: relative entropy, joint entropy, conditional entropy, and mutual information. To integrate quantum information theory into quantum chemistry, we introduce the reduced density matrix (RDM) in Section 3, which serves as a powerful tool to simplify the complexity of quantum states while retaining essential information about the state of a subsystem. Section 4 introduces the application of information theory in quantum chemistry; for the classical approach, we convert the reduced density matrix in the position representation to derive the electron density and pair density, which act as information carriers for classical information theory. We also explore the corresponding quantum concepts in Section 5, such as von Neumann entropy and quantum mutual information, which are used to analyze the entanglement in quantum many-body systems.
2. Brief Introduction to Information Theory
2.1. Shannon Entropy
One of the core concepts in information theory is the Shannon entropy, named after Claude Shannon, the founder of information theory [3]. Let X be a discrete random variable with alphabet and probability distribution function for . The Shannon entropy is defined as:
where the expectation is denoted by . Thus, the expected value of the function for is written as:
The Shannon entropy admits multiple interpretations, one of which states that it provides a mathematical framework to measure the uncertainty of a random event. The term entropy originates from the Greek word trope (meaning change) and was first introduced by Clausius in 1854 in the context of the second law of thermodynamics [62]. Shannon himself explained his rationale for adopting this term in a disarming way:
My greatest concern was what to call it. I thought of calling it ‘information’, but the word was overly used, so I decided to call it ‘uncertainty’. When I discussed it with John von Neumann, he had a better idea. Von Neumann told me, “You should call it entropy, for two reasons. In the first place your uncertainty function has been used in statistical mechanics under that name, so it already has a name. In the second place, and more important, nobody knows what entropy really is, so in a debate you will always have an advantage [63].
2.2. Relative Entropy
To understand the geometry of probability distributions, by introducing a second probability distribution , where , we now look for a distinguishability measure of those two probability distributions [64].
By analogy with the concept of distance (norm) in Euclidean space, information theory defines the relative entropy (information divergence) between two probability distributions and . Among the most popular divergence measures include the Bregman divergences [65,66] and f-divergences [67,68,69]. The f-divergence is defined as:
where is a convex function satisfying . The Bregman divergence is defined as:
where is a convex functional.
Most measures of relative entropy fit these frameworks, but the Kullback-Leibler (KL) divergence [70] is both a f-divergence and a Bregman divergence. Therefore, the Kullback-Leibler divergence is the most widely used definition of relative entropy, with its expression given by:
Formally, a metric (such as the Euclidean norm) must satisfy four axioms for all distributions P, Q, and R: [71]
- Non-negativity:
- Identity of indiscernibles:
- Symmetry:
- Triangle inequality:
Figure 2 provides an intuitive illustration of the non-symmetric nature of the Kullback-Leibler divergence. Additionally, counterexamples exist where:
demonstrating that the Kullback-Leibler divergence violates the triangle inequality. Thus, the Kullback-Leibler divergence is considered a premetric but not a full metric.
2.3. Bivariate Entropy
To study correlations between variables, it is useful to extend the Shannon entropy to the multivariate case. For simplicity, consider a pair of variables . Then, bivariate information-theoretic quantities such as joint entropy, conditional entropy, and mutual information can be defined, and their relationships are illustrated in Figure 3.
The most straightforward two-variable extension of Shannon entropy is the joint entropy , which measures the total uncertainty when considering a pair of variables together, and its corresponding expression is:
where is the joint probability distribution function. Since a pair of variables can be treated as a single vector of length two, this extension does not introduce fundamentally new concepts.
Other bivariate entropy measures include conditional entropy and mutual information, which can be expressed in the framework of the Kullback-Leibler divergence we introduced in Equation (5). The conditional entropy of one variable, given another, is defined as the expected value of the entropy of the conditional distributions, averaged over the conditioning variable.
Compare with the concept of entropy, which is a probabilistic measure of uncertainty; information is a measure of a reduction in that uncertainty. The mutual information is a measure of the amount of information that one variable contains about another. It is defined as:
The conditional and mutual information, defined using the Kullback-Leibler divergence, exhibit several important properties, enabling a clear and rigorous analysis.
- The chain rule.
- Subadditivity.
- The relationship between different bivariate entropy,
3. Basic Ingredients of Information Theory in Quantum Chemistry: Reduced Density Matrix
First proposed by Paul Dirac in 1930, [72] the reduced density matrix (RDM) becomes a fundamental tool in quantum chemistry and many-body physics, enabling the analysis of subsystems within larger quantum systems [73,74,75,76,77]. Because the electron-electron repulsion is a two-body operator, the one- and two-electron reduced density matrices (1-RDM and 2-RDM) are the quantities of greatest interest in quantum chemistry, as it suffices to determine most molecular properties, including the electronic energy [78,79,80,81,82]. Similarly, for the application of information theory in quantum chemistry, the reduced density matrix emerges as the natural mathematical framework for elucidating the correlations and entanglement.[83,84]
3.1. Density Matrix and Reduced Density Matrix
For the quantum many-body systems, other than the usual state vectors , quantum states can also be represented by the density matrix (density operator ), which unifies the description of pure and mixed states. If a density matrix is obtained as:
then the quantum state is a pure state. A mixed state cannot be expressed this way and instead requires a statistical mixture,
where is an ensemble of pure states with weights (probability) .
To further elucidate the concepts of pure states and mixed states, we employ a two-state quantum system as an illustrative example. Often implemented by a physical particle with spin with two basis states . As one of the core concepts in quantum information and quantum computing, analogous to the classical bit, it is known as a qubit (quantum bit) [85]. A pure state of such a two-state quantum system is represented by a state vector:
The constraint conditions in Equation (20) indicate that a pure state of a qubit can be equivalently expressed as:
Thus, the pure state of a qubit can be uniquely determined by two parameters and . Here we can establish a geometric representation for this two-state quantum system: every pure state defined in Equation (21) uniquely corresponds to a point (cossin, sinsin, cos) on the surface of the Bloch sphere as shown in Figure 4.
For the mixed states of a qubit, it is evident that the density matrix can be represented by a 2×2 matrix, since.
It can be rigorously proven that every point within the Bloch sphere corresponds to a mixed state. In particular, the center point represents the maximally mixed state, as established in quantum information theory [85].
For concision, henceforth we will assume that the density matrix (DM) and the reduced density matrix (RDM) correspond to pure states, but most of the analysis extends directly to mixed states. The k-electron reduced density matrix (k-RDM) is defined as the partial trace, over dimensions, of an N-electron density matrix; it is usually expanded on the basis of single-particle states (i.e. spin orbitals). Specifically,
where
are elements of the k-RDM, which is Hermitian and positive semidefinite because the density matrix is Hermitian and positive semidefinite. We use the overbar above symbols when we need to explicitly distinguish quantities expressed in the spin-orbital, rather than the spatial orbital, basis.
3.2. 1-RDM and 2-RDM
The quantities of greatest interest to chemists are the one- and two-electron reduced density matrices. The 1-RDM is defined as:
and the 2-RDM is defined as:
In our convention, the trace of the 1-RDM is the number of electrons and the trace of the 2-RDM is the number of electron pairs:
The reader is cautioned that some authors use different normalization conventions (e.g., unit normalization or the number of non-unique electron pairs, ).
The reduced density matrix (RDM) can be represented in a spin block format because the projection of the spin vector onto a specified axis, , commutes with the molecular Hamiltonian.
Thus, the one-particle reduced density matrix (1-RDM) will have a block-diagonal form.
Similarly, the spin-block format of the two-particle reduced density matrix (2-RDM) is:
Note that knowledge of any one of the four opposite-spin blocks, e.g. , suffices to determine the others by (anti)symmetry.
3.3. 3-RDM and 4-RDM
In general, reduced density matrices of order greater than two are essentially redundant for most quantum chemistry problems, as electrons interact only pairwise. However, for certain niche quantum chemistry applications, the information provided by the 3-RDM and 4-RDM is still required. The three-electron reduced density matrix (3-RDM) can be explicitly defined as:
and, similarly, the four-electron reduced density matrix (4-RDM) is defined as:
We can easily determine lower-order reduced density matrices (RDM) by taking the partial trace of a higher-order one. However, using higher-order reduced density matrices than required is undesirable, as the computational cost and memory requirements of high-order reduced density matrices are prohibitively large. For example, the storage required to store the complete 3-RDM and 4-RDM scale as and , respectively, where n is the number of basis functions. Higher-order reduced density matrices can be systematically, but approximately, expressed in terms of lower-order reduced density matrices using diagrammatic and statistical techniques [86,87,88,89,90,91,92,93,94,95,96].
Specifically, using the cumulant expansion one can decompose higher-order RDM into sums of products of lower-order quantities and nonreducible k-order cumulants [86,87,93,97]. As a starting point, the 1-RDM can be expressed as the sum of a mean-field term and a correlated cumulant term:
The two-particle reduced density matrix (2-RDM) can then be expressed as the wedge (or Grassmann) product of two one-particle reduced density matrices (1-RDM) and a cumulant 2-RDM.
the wedge product, denoted as ∧, is defined as an antisymmetric tensor product, such that:
The cumulant expansions of the three-particle and four-particle reduced density matrices (3-RDM and 4-RDM) are given by:
and
The traces of the density matrices in these expressions give the number of distinguishable pairs, triples, and quartets of electrons, respectively. Dividing by , where k is the order of the reduced density matrix, recovers the definition we’ve used elsewhere.
4. Classical Information Theory in Quantum Chemistry
The Hohenberg-Kohn theorems [53,54,98,99,100] imply that all ground state properties are functionals of the electron density; this establishes the theoretical basis for using classical information theory that extracts chemical insights by treating electron densities as probability distributions.
4.1. Electron Density in Position Space
In Section 3, we establish reduced density matrices as the basic ingredients for information-theoretic analysis in quantum chemistry. For molecular systems, the concept of probability distribution in general information theory is specialized as an analytical functional of the electron density , which depends solely on the spatial coordinates, :
By construction,
Another fundamental information carrier in quantum chemistry is the two-electron distribution function, or pair density, defined as:
By definition,
Analogous to density-functional theory, a complete description of electronic structure can be constructed based on the pair density (and also higher-order electron distribution functions) using appropriate generalizations of the Hohenberg-Kohn theorem [101,102,103,104,105,106,107,108,109,110,111,112].
The electron density and pair density can be computed from the reduced density matrices (RDMs) we introduced in Section 3. However, in that section, we considered the spin-resolved reduced density matrices, and it is more convenient in this context to trace out the spin coordinates and obtain a representation of the reduced density matrices in terms of spatial orbitals. I.e.,
Next, we transform the one- and two-electron reduced density matrices from the (second-quantized) spatial-orbital representation into the (first-quantized) position representation,
We use the indices , , , and to label atomic orbitals. The one-electron density (Equation (39)) and the pair-electron density (Equation (41)) represent the diagonal components of the spinless one- and pair-electron reduced density matrix in position space, respectively, which is obtained by setting the un-primed spatial variables equal to the primed spatial variables, . Note that off-diagonal elements of the orbital representation of the RDM contribute to diagonal elements of the spatial representation of the RDM and vice versa. This has significant implications for the N-representable of electron distribution functions [101,113].
4.2. Information-Theoretic Approach Chemical Descriptors
Taking electron density as the information carrier, classical information theory has been applied to DFT since the mid-20th century to study atoms and molecular systems within the framework of the information-theoretic approach (ITA) [8,9,10,11,12,13,14,15,16,27,28,29,30,31,32,33,34,35,36,57].
The Shannon entropy, with as its information carrier, measures the spatial delocalization of the electron density and is defined as:
In the information-theoretic approach, we employ a broader set of formulas beyond Shannon entropy. To maintain notational clarity, we adopt a systematic symbolic convention where Shannon’s formula is specifically denoted by the symbol . When only the one-electron density is considered, the subscript (X) in Equation (47) is typically omitted.
By introducing the reference density , which corresponds to the electron density of the promolecule constructed under the assumption that each atom retains its density as if it were isolated, [114,115,116,117,118,119,120] we can define the relative Shannon entropy . This quantity, also referred to as the Kullback-Leibler divergence, information gain, or information divergence, [16,28,29,120,121,122,123,124] is defined by:
The joint entropy , which measures the localization of the pair of electrons in their respective spaces, is defined as:
Besides joint entropy, other bivariate entropy measures need to maintain the same normalization condition for the state density and the reference state density. Thus, the normalized one- and pair-electron densities and are defined as
and
where N is the number of electrons. Following the definition of Parr and Bartolotti in 1983, [125] these unit-normalized densities are also referred to as shape functions . [125,126,127,128] They exhibit the obvious non-negative properties and .
Using the normalized pair electron density as the distribution functions, the joint entropy is defined as:
The conditional entropy is defined as the Kullback-Leibler divergence of the unit-normalized pair electron density from the unit-normalized electron densities.
The mutual information , is defined as the Kullback-Leibler divergence of the unit-normalized pair electron density from the product of two unit-normalized electron densities. The mutual information then measures the divergence of the pair electron density from the value it would have if the electrons moved entirely independently.
Within the information theory framework introduced in Section 2, which utilizes the one- and pair-electron densities as fundamental information carriers, all the classical information-theoretic quantities we defined in this subsection preserve the essential mathematical properties we introduced in Section 2.3.
4.2.1. Examples and Illustrations
Using the information-theoretic approach, we can systematically interpret chemical concepts such as chemical bonds, chemical reactivity, electron shells, lone electron pairs, and more [8,10,11,13,15,16,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,143,144]. The information-theoretic approach descriptors can be categorized based on the scope of molecular features they capture:
- Global Descriptors: Assign a value to the entire system.
- Local Descriptors: Assign a value to each position in the system.
- Nonlocal Descriptors: Assign a value to each pair of positions in the system.
The global ITA descriptors describe the overall properties of the system as a whole, enabling prediction of properties such as polarizability, aromaticity, acidity/basicity, and reactivity [10,11,13,129,130,131,132,133,134,145,146,147,148,149,148,149]. Figure 5 plots the correlation between Shannon entropy aromaticity() [129] and several established aromaticity indices: the harmonic oscillator model of aromaticity (HOMA) [150,151], the aromatic fluctuation index (FLU) [152], and nucleus-independent chemical shifts (NICS) [153,154]. The strong correlations demonstrate that Shannon entropy effectively characterizes aromaticity.
The local ITA descriptors can serve as regioselectivity indicators, through a coarse-graining process, which involves integrating their values over atoms, functional groups, or fragment regions, condensed descriptors are obtained [145,146,147,148,149,150]. These descriptors help identify the most reactive atoms, functional groups, or bonds. Figure 6 provides an example of how local Shannon entropy is applied to reveal electron shell structures in noble gas atoms. [136] The radial distribution functions of the Shannon entropy densities are defined as:
It shows step-like increases in entropy at shell boundaries.
Non-local ITA descriptors quantify how the properties of a molecule at one location respond to changes at another distant point within the same molecule. These descriptors can also be condensed into response matrices, to quantify the correlation between fragments of the system [156]. Figure 7 illustrates the application of joint entropy, conditional entropy, and the mutual information kernel to analyze electron correlation in the Krypton atom.
5. Quantum Information Theory in Quantum Chemistry
In this section, we transition our discussion of information theory to the quantum realm [23,25,26,49,83,84,85,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,170,171]. As we delve deeper, it will become evident that the quantum case holds far richer possibilities, primarily due to the superposition principle.
5.1. Bipartite Entanglement
In quantum chemistry, for a system of basis set with finite cardinality L, each basis associates with a local Hilbert space . Then the total Hilbert space spanned by the entire basis set is the tensor product of all local Hilbert spaces . [26,159,160,161] For an arbitrary state
When the system is divided into two parts A and B, the composite Hilbert space of the whole system is given as:
where and are the Hilbert spaces of subsystems A and B, respectively. Only when there is no entanglement between the systems can the state be represented as a tensor product of the states of the subsystems and , as shown in Figure 8a.
As a consequence of quantum entanglement among arbitrary subsystems, as shown in Figure 8b, for a generic , it should be represented as a series of tensor products of basis states of subsystems A and B,
The strategy for measuring bipartite entanglement emerges from the concept of partial measurements. [162] In Section 3.1, we have an introduction of the density matrix for both pure and mixed states; for the sake of brevity, here we only consider the case of pure states. A bipartite reduced density matrix of a pure state can be approached by tracing out ("averaging over") one of the subsystems:
thus, and are the reduced density matrices of each subsystem. The quantification of bipartite entanglement can be approached through quantum information theory with the Von Neumann entropy, [158] which quantifies the entanglement of the quantum system as
where is the density matrix. As the quantum counterpart of Shannon entropy we introduced in Equation (1), the Von Neumann entropy can be expressed as the Shannon entropy of the eigenvalues for the density matrix:
To measure the entanglement between the bipartite subsystems, the entanglement entropy (holds for the pure state) is defined as
5.2. Orbital Reduced Density Matrix
For a wavefunction expressed in terms of spatial orbitals, each orbital holds four possible occupations, which can be empty , singly occupied with a spin-up electron , singly occupied with a spin-down electron , and doubly occupied with an electron pair . I.e., the possible orbital occupations are:
As shown in Figure 9, if the system is divided into subsystem A composed of p-orbitals and complement subsystem B (orbital bath) composed of the remaining orbitals, the RDM of subsystem A is then called the p-orbital reduced density matrix (p-orbital RDM), which can be defined in terms of the full, N-electron RDM or, equivalently, the N-electron wavefunction.
The 1-orbital RDM corresponding to the one orbital partition in Figure 9a is expressed in the basis {–, ↑, ,↓, ↑↓}, as shown in Table 1,163,164,165,166] the elements of the matrix can be represented in terms of the spin-dependent 1-electron RDM and 2-electron RDM , where the indices p and indicate spin-up and spin-down electrons of p-th orbital respectively.
The two orbital partition is shown in Figure 9b, elements of the 2-orbital RDM are summarized in Table 2 [163,164,165,166]. Note that requires only some diagonal elements of the 3- and 4-electron reduced density matrices, as well as a few off-diagonal elements of the 1-, 2-, and 3-electron reduced density matrices.
We should note that the 1-orbital RDM can be further simplified for seniority-zero state. Since it excludes singly occupied orbitals and we have the relations , the corresponding basis {–, ↑↓} holds a 2 cardinality, thus the 1-orbital RDM of a seniority-zero state is simplified as a matrix:
Following the same simplification method, the 2-orbital RDM of a seniority-zero state expressed in the basis {—, ↑↓ , ↑↓, ↑↓↑↓} is reduced to a matrix:
5.3. Orbital Entanglement
With the preliminary knowledge of quantum information theory and orbital reduced density matrix, we can define the single-orbital entropy and mutual information from 1- and 2-orbital RDM. The first quantity, single–orbital entropy , measures the entanglement between a given orbital p and the complementary orbital bath, using the eigenvalues of the one–orbital reduced density matrix as information carriers. The single–orbital entropy is defined as:
where and M are the eigenvalues and dimension of the p-th one-orbital reduced density matrix respectively, with for seniority-zero state and for other states. The total quantum information encoded in the system is given by the sum of single-orbital entropy:
Given two states described by the one-orbital reduced density matrices and , one can define the relative orbital entropy by the KL divergence:
It measures how much the entanglement of orbital p deviates from that of orbital q.
If the system is divided into two orbitals and the remaining orbital bath as shown in Figure 9b, the entanglement between them is quantified by the two-orbital entropy defined as
where and M are the eigenvalues and dimension of the two-orbital reduced density matrix respectively, with for seniority-zero state and for other states. The conditional orbital entropy can be written by the KL divergence as
where is the identity matrix with the same dimensions as . The total amount of entanglement between two orbitals p and q can be measured by the orbital pair mutual information, written by the KL divergence as
As an application of the information theory framework introduced in Section 2 where the orbital reduced density matrix serves as the information carrier, those quantum information theory quantities naturally inherit key properties we introduced in Section 2.3, including but not limited to the chain rule and subadditivity.
5.3.1. Examples and Illustrations
The concept of orbital entanglement serves as a complementary tool for interpreting electronic structures, proving particularly valuable in strongly correlated systems. In this subsection, we will present several representative application examples.
As presented in Ref [49,165], the strength of single-orbital entropy to measure orbital entanglement and the orbital-pair mutual information can be associated with different types of electron correlation effects. In Table 3, we map the strength of orbital interactions onto a certain type of correlation effects. Computational investigations demonstrate that orbitals with nondynamic/static electron correlation effects signify substantial multireference character in the system. In contrast, weakly entangled orbitals predominantly exhibit dynamic correlation effects, systems where all orbitals are weakly entangled are can usually be adequately treated by single-reference approaches.
Quantitative visualization of and enables a more intuitive analysis. As shown in Figure 10, the strength of the orbital pair mutual information classified in Table 3 is colour-coded,
- Blue lines: Nondynamic correlated orbital pairs.
- Red lines: Static correlated orbitals.
- Green: Dynamic correlated orbitals.
An important issue for the density matrix renormalization group (DMRG) method [172,173,174] is the order of orbitals in the one-dimensional matrix product state (MPS) wavefunction ansatz; an optimal ordering of orbitals corresponding to maximum entanglement will produce the most efficient results [23,175,176]. Since strong (nondynamic/static) electron correlation is essential for proper molecular dissociation into fragments, orbital entanglement provides both a fundamental framework for understanding bond formation/breaking processes [167] and a practical tool for analyzing chemical reactivity.
For many strongly correlated calculation methods such as complete active space self-consistent field (CASSCF), [43,177] the selection of the complete active space (CAS space) is a crucial prerequisite step to keep computational costs within feasible limits. As shown in Figure 11, for the CAS methodology, all the molecular orbitals are classified into three spaces:
- Inactive space: Always doubly occupied.
- Active space: All the possible configurations are allowed.
- Virtual space: Always empty.
Specifically, the active space, usually denoted as CAS(n,m) where n and m are the number of electrons and orbitals respectively, should encompass orbitals and electrons essential for capturing strong electron correlation effects. Orbital entanglement serve as powerful tools for identifying critical orbitals in active space. Comparing the entanglement diagrams as shown in Figure 10, we can evaluating the quality and convergence behavior of those active-space calculations.
6. Summary and Outlook
This review presents a unified perspective on information theory and its applications in quantum chemistry, integrating both classical and quantum frameworks. Beginning with fundamental concepts like Shannon entropy and its related concepts such as joint entropy, relative entropy, conditional entropy, and mutual information, we demonstrate how information theory can be applied to molecular systems through information carriers such as the electron density and orbital reduced density matrix. The discussion bridges classical concepts with their quantum counterparts, including classical Shannon entropy and quantum correlations to the quantum von Neumann entropy and entanglement. By tracing the historical development from early works by Shannon’s foundational contributions, we highlight how information theory has evolved into a versatile framework with broad applications in quantum chemistry, particularly for analyzing electronic structure and quantum phenomena in chemical systems.
This article only scratches the surface of the vast scope for existing and future applications of information theory in quantum chemistry. Notably, for the application of information theory in quantum chemistry, Shannon’s framework is not the only reasonable formula; the Rényi entropy, the Fisher information, and other f- and Bregman divergences can also be used as a measure of correlation and entanglement. Importantly, these concepts extend far beyond simple pairwise interactions: from single- and pair-electron densities to many-body electron distributions, and from bipartite systems to complex multipartite quantum entanglement. The physical manifestations of information carriers are likewise diverse, ranging from atoms in molecules (AIM) to localized molecular orbitals. By creatively combining the above extended concepts as well as other potential extensions, we can even derive additional novel concepts and tools for advancing our understanding of quantum chemistry.
Furthermore, other domains such as machine learning and quantum computing that represent one of the most active research frontiers in science, also possess profound foundations in information theory. The integration of quantum chemistry with information theory, machine learning, and quantum computing is establishing a transformative new paradigm for quantum chemical research.
Author Contributions
The manuscript was written through contributions of all authors.
Funding
DBZ is supported by the National Natural Science Foundation of China (grant no. 22203071), the High-Level Talent Special Support Plan and the China Scholarship Council. RCY acknowledges support from the National Natural Science Foundation of China (Grant No. 22373034).
Acknowledgments
The authors appreciate financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Research Chairs. The authors also acknowledge computational resources from the Digital Research Alliance of Canada.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Shannon entropy | |
| Relative entropy, Kullback-Leibler divergence | |
| Joint entropy | |
| Conditional entropy | |
| Mutual information | |
| Density matrix, Density operator | |
| k-Electron reduced density matrix with spin and spatial orbital | |
| Electron density at position | |
| Unit-normalized electron density (shape function) at position | |
| Pair-electron density at position and | |
| Unit-normalized pair-electron density at position and | |
| Shannon entropy with electron density | |
| Relative entropy with electron density | |
| Joint entropy with electron density | |
| Conditional entropy with electron density | |
| Mutual information with electron density | |
| k-Orbital Reduced Density Matrix | |
| One-orbital entropy | |
| Orbital relative entropy | |
| Two-orbital entropy | |
| Orbital conditional entropy | |
| Orbital mutual information |
References
- Hartley, R.V.L. Transmission of information. Bell Syst. Tech. J. 1928, 7, 535–563. [CrossRef]
- Nyquist, H. Certain factors affecting telegraph speed. Bell Syst. Tech. J. 1924, 3, 324–346. [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory, 2nd ed.; John Wiley & Sons, Ltd: New York, 2005. [CrossRef]
- Witten, E. A mini-introduction to information theory. Riv. Nuovo Cim. 2020, 43, 187–227. [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press, 1995. [CrossRef]
- Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic-Structure Theory; John Wiley & Sons, Ltd, 2000. [CrossRef]
- Liu, S. Information-Theoretic Approach in Density Functional Reactivity Theory. Acta Phys. -Chim. Sin. 2016, 32, 98–118. [CrossRef]
- Wang, B.; Zhao, D.; Lu, T.; Liu, S.; Rong, C. Quantifications and Applications of Relative Fisher Information in Density Functional Theory. J. Phys. Chem. A 2021, 125, 3802–3811. [CrossRef]
- Zhao, D.; Zhao, Y.; He, X.; Li, Y.; Ayers, P.W.; Liu, S. Accurate and Efficient Prediction of Post-Hartree–Fock Polarizabilities of Condensed-Phase Systems. J. Chem. Theory Comput. 2023, 19, 6461–6470. [CrossRef]
- Zhao, D.; Zhao, Y.; He, X.; Ayers, P.W.; Liu, S. Efficient and accurate density-based prediction of macromolecular polarizabilities. Phys. Chem. Chem. Phys. 2023, 25, 2131–2141. [CrossRef]
- Zhao, D.; Zhao, Y.; Xu, E.; Liu, W.; Ayers, P.W.; Liu, S.; Chen, D. Fragment-Based Deep Learning for Simultaneous Prediction of Polarizabilities and NMR Shieldings of Macromolecules and Their Aggregates. J. Chem. Theory Comput. 2024, pp. 2655–2665. [CrossRef]
- Zhao, Y.; Zhao, D.; Liu, S.; Rong, C.; Ayers, P.W. Why are information-theoretic descriptors powerful predictors of atomic and molecular polarizabilities. J. Mol. Model. 2024, 30, 361. [CrossRef]
- Ayers, P.W.; Fias, S.; Heidar-Zadeh, F. The Axiomatic Approach to Chemical Concepts. Comput. Theor. Chem. 2018, 1142, 83–87. [CrossRef]
- Rong, C.; Zhao, D.; He, X.; Liu, S. Development and Applications of the Density-Based Theory of Chemical Reactivity. J. Phys. Chem. Lett. 2022, 13, 11191–11200. [CrossRef]
- Liu, S. Identity for Kullback-Leibler divergence in density functional reactivity theory. J. Chem. Phys. 2019, 151, 141103. [CrossRef]
- Wu, W.; Scholes, G.D. Foundations of Quantum Information for Physical Chemistry. J. Phys. Chem. Lett. 2024, 15, 4056–4069. [CrossRef]
- Materia, D.; Ratini, L.; Angeli, C.; Guidoni, L. Quantum Information reveals that orbital-wise correlation is essentially classical in Natural Orbitals, 2024. [CrossRef]
- Aliverti-Piuri, D.; Chatterjee, K.; Ding, L.; Liao, K.; Liebert, J.; Schilling, C. What can quantum information theory offer to quantum chemistry? Faraday Discuss. 2024, 254, 76–106. [CrossRef]
- Nowak, A.; Legeza, O.; Boguslawski, K. Orbital entanglement and correlation from pCCD-tailored coupled cluster wave functions. J. Chem Phys. 2021, 154, 084111. [CrossRef]
- Ding, L.; Mardazad, S.; Das, S.; Szalay, S.; Schollwöck, U.; Zimborás, Z.; Schilling, C. Concept of Orbital Entanglement and Correlation in Quantum Chemistry. J. Chem. Theory Comput. 2021, 17, 79–95. [CrossRef]
- Ratini, L.; Capecci, C.; Guidoni, L. Natural Orbitals and Sparsity of Quantum Mutual Information. J. Chem. Theory Comput. 2024, 20, 3535–3542. [CrossRef]
- Legeza, O.; Sòlyom, J. Optimizing the density-matrix renormalization group method using quantum information entropy. Phys. Rev. B 2003, 68, 195116. [CrossRef]
- Convy, I.; Huggins, W.; Liao, H.; Birgitta Whaley, K. Mutual information scaling for tensor network machine learning. Mach. Learn. Sci. Technol. 2022, 3, 015017. [CrossRef]
- Legeza, O.; Sólyom, J. Two-Site Entropy and Quantum Phase Transitions in Low-Dimensional Models. Phys. Rev. Lett. 2006, 96, 116401. [CrossRef]
- Szalay, S.; Pfeffer, M.; Murg, V.; Barcza, G.; Verstraete, F.; Schneider, R.; Legeza, O. Tensor product methods and entanglement optimization for ab initio quantum chemistry. Int. J. Quantum Chem. 2015, 115, 1342–1391. [CrossRef]
- Sears, S.B.; Parr, R.G.; Dinur, U. On the Quantum-Mechanical Kinetic Energy as a Measure of the Information in a Distribution. Isr. J. Chem. 1980, 19, 165–173. [CrossRef]
- Nalewajski, R.F.; Parr, R.G. Information theory, atoms in molecules, and molecular similarity. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8879–8882. [CrossRef]
- Nalewajski, R.F.; Parr, R.G. Information Theory Thermodynamics of Molecules and Their Hirshfeld Fragments. J. Phys. Chem. A 2001, 105, 7391–7400. [CrossRef]
- Levine, R.D.; Bernstein, R.B. Energy disposal and energy consumption in elementary chemical reactions. Information theoretic approach. Acc. Chem. Res. 1974, 7, 393–400. [CrossRef]
- Procaccia, I.; Levine, R.D. The populations time evolution in vibrational disequilibrium: An information theoretic approach with application to HF. J. Chem Phys. 1975, 62, 3819–3820. [CrossRef]
- Dinur, U.; Kosloff, R.; Levine, R.; Berry, M. Analysis of electronically nonadiabatic chemical reactions: An information theoretic approach. Chem. Phys. Lett. 1975, 34, 199–205. [CrossRef]
- Procaccia, I.; Levine, R.D. Vibrational energy transfer in molecular collisions: An information theoretic analysis and synthesis. J. Chem Phys. 1975, 63, 4261–4279. [CrossRef]
- Levine, R.D.; Manz, J. The effect of reagent energy on chemical reaction rates: An information theoretic analysis. J. Chem Phys. 1975, 63, 4280–4303. [CrossRef]
- Levine, R.D. Entropy and macroscopic disequilibrium. II. The information theoretic characterization of Markovian relaxation processes. J. Chem Phys. 1976, 65, 3302–3315. [CrossRef]
- Levine, R.D. Information Theory Approach to Molecular Reaction Dynamics. Ann. Rev. Phys. Chem. 1978, 29, 59–92. [CrossRef]
- Slater, J.C. The Theory of Complex Spectra. Phys. Rev. 1929, 34, 1293–1322. [CrossRef]
- Hartree, D.R. Some Relations between the Optical Spectra of Different Atoms of the same Electronic Structure. II. Aluminium-like and Copper-like Atoms. Math. Proc. Camb. Phil. Soc. 1926, 23, 304–326. [CrossRef]
- Fock, V.A.Z. Näherungsmethode zur Lösung des quantenmechanischen Mehrkörperproblems. Z. Phys. 1930, 61, 126–148.
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [CrossRef]
- Koga, T.; Tatewaki, H.; Thakkar, A.J. Roothaan-Hartree-Fock wave functions for atoms with Z≤54. Phys. Rev. A 1993, 47, 4510–4512. [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [CrossRef]
- Shavitt, I. The history and evolution of configuration interaction. Mol. Phys. 1998, 94, 3–17. [CrossRef]
- Shavitt, I.; Bartlett, R.J. Many-Body Methods in Chemistry and Physics: Theory and Applications. Cambridge University Press 2009. [CrossRef]
- Cooper, N.R.; Leese, M.R. Configuration interaction methods in molecular quantum chemistry. J. Mol. Struct.-THEOCHEM 2000, 94, 71–78.
- Coester, F.; Kümmel, H. Short-range correlations in nuclear wave functions. Nucl. Phys. 1960, 17, 477–485. [CrossRef]
- Ahlrichs, R. Many body perturbation calculations and coupled electron pair models. Comput. Phys. Commun. 1979, 17, 31–45. [CrossRef]
- Bartlett, R.J. Many-body perturbation-theory and coupled cluster theory for electron correlation in molecules. Annu. Rev. Phys. Chem. 1981, 32, 359–401. [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [CrossRef]
- Asadchev, A.; Gordon, M.S. Fast and Flexible Coupled Cluster Implementation. J. Chem. Theory Comput. 2013, 9, 3385–3392. [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [CrossRef]
- Cremer, D. Møller–Plesset perturbation theory: from small molecule methods to methods for thousands of atoms. WIREs Comput. Mol. Sci. 2011, 1, 509–530. [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [CrossRef]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [CrossRef]
- Engel, E.; Dreizler, R.M. Density Functional Theory: An Advanced Course; Theoretical and Mathematical Physics, Springer Berlin Heidelberg: Berlin, Heidelberg, 2011. [CrossRef]
- He, X.; Li, M.; Rong, C.; Zhao, D.; Liu, W.; Ayers, P.W.; Liu, S. Some Recent Advances in Density-Based Reactivity Theory. J. Phys. Chem. A 2024, 128, 1183–1196. [CrossRef]
- Tsallis, C. Possible generalization of Boltzmann-Gibbs statistics. J. Stat. Phys. 1988, 52, 479–487. [CrossRef]
- Onicescu, O. Theorie de l’information energie informationelle. Comptes rendus de l’Academie des Sciences Series AB 1966, 263, 841–842.
- Rényi, A. Probability theory; North-Holland: Amsterdam, 1970.
- Fisher, R.A. Theory of Statistical Estimation. Math. Proc. Cambridge Philos. Soc. 1925, 22, 700–725. [CrossRef]
- Clausius, R. The Mechanical Theory of Heat - Scholar’s Choice Edition; Creative Media Partners, LLC, 2015.
- Accardi, L. Topics in quantum probability. Physics Reports 1981, 77, 169–192. [CrossRef]
- Bengtsson, I.; Zyczkowski, K. Geometry of Quantum States: An Introduction to Quantum Entanglement; Cambridge University Press, 2006.
- Bregman, L.M. The relaxation method of finding the common point of convex sets and its application to the solution of problems in convex programming. USSR Comput. Math. and Math. Phys. 1967, 7, 200–217. [CrossRef]
- Banerjee, A.; Merugu, S.; Dhillon, I.S.; Ghosh, J. Clustering with Bregman divergences. J. Mach. Learn. Res. 2005, 6, 1705–1749.
- Ali, S.M.; Silvey, S.D. A general class of coefficients of divergence of one distribution from another. J. R. Stat. Soc. Ser. B Methodol. 1966, 28, 131–142.
- Csiszár, I. Information-type measures of difference of probability distributions and indirect observations. Stud. Sci. Math. Hung. 1967, 2, 299–318.
- Liese, F.; Vajda, I. On divergences and informations in statistics and information theory. IEEE Trans. Inform. Theory 2006, 52, 4394–4412. [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [CrossRef]
- Burago, D.; Burago, J.D.; Ivanov, S. A Course in Metric Geometry; American mathematical society, 2001.
- Dirac, P.A.M. Note on Exchange Phenomena in the Thomas Atom. Mathematical Proceedings of the Cambridge Philosophical Society 1930, 26, 376–385. [CrossRef]
- Mazziotti, D.A., Ed. Reduced-Density-Matrix Mechanics: With Application to Many-Electron Atoms and Molecules, 1 ed.; Vol. 134, Advances in Chemical Physics, Wiley, 2007. [CrossRef]
- Gidopoulos, N.I.; Wilson, S.; Lipscomb, W.N.; Maruani, J.; Wilson, S., Eds. The Fundamentals of Electron Density, Density Matrix and Density Functional Theory in Atoms, Molecules and the Solid State; Vol. 14, Progress in Theoretical Chemistry and Physics, Springer Netherlands: Dordrecht, 2003. [CrossRef]
- Absar, I. Reduced hamiltonian orbitals. II. Optimal orbital basis sets for the many-electron problem. Int. J. Quantum Chem. 1978, 13, 777–790. [CrossRef]
- Absar, I.; Coleman, A.J. Reduced hamiltonian orbitals. I. a new approach to the many-electron problem. Int. J. Quantum Chem. 2009, 10, 319–330. [CrossRef]
- Coleman, A.J.; Absar, I. Reduced hamiltonian orbitals. III. Unitarily invariant decomposition of hermitian operators. Int. J. Quantum Chem. 1980, 18, 1279–1307. [CrossRef]
- Mazziotti, D.A. Two-Electron Reduced Density Matrix as the Basic Variable in Many-Electron Quantum Chemistry and Physics. Chem. Rev. 2012, 112, 244–262. [CrossRef]
- Mazziotti, D.A. Parametrization of the two-electron reduced density matrix for its direct calculation without the many-electron wave function: Generalizations and applications. Phys. Rev. A 2010, 81, 062515. [CrossRef]
- Verstichel, B.; van Aggelen, H.; Van Neck, D.; Ayers, P.W.; Bultinck, P. Variational Density Matrix Optimization Using Semidefinite Programming. Comput. Phys. Commun. 2011, 182, 2025–2028. [CrossRef]
- Verstichel, B.; van Aggelen, H.; Van Neck, D.; Ayers, P.W.; Bultinck, P. Variational Determination of the Second-Order Density Matrix for the Isoelectronic Series of Beryllium, Neon, and Silicon. Phys. Rev. A 2009, 80, 032508. [CrossRef]
- Eugene DePrince III, A. Variational determination of the two-electron reduced density matrix: A tutorial review. WIREs Comput. Mol. Sci. 2024, 14, e1702. [CrossRef]
- Barcza, G.; Legeza, O.; Marti, K.H.; Reiher, M. Quantum-Information Analysis of Electronic States of Different Molecular Structures. Phys. Rev. A 2011, 83, 012508. [CrossRef]
- Szalay, S.; Barcza, G.; Szilvasi, T.; Veis, L.; Legeza, O. The Correlation Theory of the Chemical Bond. Sci. Rep. 2017, 7, 2237. [CrossRef]
- Nielsen, M.A.; Chuang, I.L. Quantum Computation and Quantum Information: 10th Anniversary Edition; Cambridge University Press, 2010. [CrossRef]
- Kubo, R. Generalized Cumulant Expansion Method. Journal of the Physical Society of Japan 1962, 17, 1100–1120. [CrossRef]
- Ziesche, P. Cumulant Expansions of Reduced Densities, Reduced Density Matrices, and Green’s Functions. In Many-Electron Densities and Reduced Density Matrices; Cioslowski, J., Ed.; Kluwer: New York, 2000; pp. 33–56.
- Alcoba, D.R.; Valdemoro, C. Family of modified-contracted Schrödinger equations. Phys. Rev. A 2001, 64, 062105. [CrossRef]
- Valdemoro, C.; Alcoba, D.R.; Tel, L.M.; Perez-Romero, E. Imposing Bounds on the High-Order Reduced Density Matrices Elements. Int. J. Quantum Chem. 2001, 85, 214–224. [CrossRef]
- Valdemoro, C. Spin-Adapted Reduced Hamiltonians 2. Total Energy and Reduced Density Matrices. Phys. Rev. A 1985, 31, 2123–2128. [CrossRef]
- Valdemoro, C. Spin-Adapted Reduced Hamiltonians. 1. Elementary Excitations. Phys. Rev. A 1985, 31, 2114–2122. [CrossRef]
- Mazziotti, D.A. Contracted Schrödinger equation: Determining quantum energies and two-particle density matrices without wave functions. Phys. Rev. A 1998, 57, 4219–4234. [CrossRef]
- Kutzelnigg, W.; Mukherjee, D. Cumulant expansion of the reduced density matrices. J. Chem Phys. 1999, 110, 2800–2809. [CrossRef]
- Kutzelnigg, W.; Mukherjee, D. Irreducible Brillouin conditions and contracted Schrödinger equations for n-electron systems. II. Spin-free formulation. J. Chem Phys. 2002, 116, 4787–4801. [CrossRef]
- Kutzelnigg, W.; Mukherjee, D. Irreducible Brillouin conditions and contracted Schrödinger equations for n-electron systems. III. Systems of noninteracting electrons. J. Chem Phys. 2004, 120, 7340–7349. [CrossRef]
- Mukherjee, D.; Kutzelnigg, W. Irreducible Brillouin conditions and contracted Schrödinger equations for n-electron systems. I. The equations satisfied by the density cumulants. J. Chem Phys. 2001, 114, 2047–2061. [CrossRef]
- Nooijen, M.; Wladyslawski, M.; Hazra, A. Cumulant approach to the direct calculation of reduced density matrices: A critical analysis. J. Chem. Phys. 2003, 118, 4832–4848. [CrossRef]
- Levy, M. Universal Variational Functionals of Electron-Densities, 1st- Order Density-Matrices, and Natural Spin-Orbitals and Solution of the V-Representability Problem. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 6062–6065. [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [CrossRef]
- Ayers, PW. Axiomatic Formulations of the Hohenberg-Kohn Functional. Phys. Rev. A 2006, 73. [CrossRef]
- Ayers, P.W. Using classical many-body structure to determine electronic structure: An approach using k-electron distribution functions. Phys. Rev. A 2006, 74, 042502. [CrossRef]
- Ziesche, P. Attempts toward a pair density functional theory. International Journal of Quantum Chemistry 1996, 60, 1361–1374. [CrossRef]
- Ziesche, P. Pair density functional theory — a generalized density functional theory. Physics Letters A 1994, 195, 213–220. [CrossRef]
- Nagy, Á. Pair Density Functional Theory. In Proceedings of the The Fundamentals of Electron Density, Density Matrix and Density Functional Theory in Atoms, Molecules and the Solid State; Gidopoulos, N.I.; Wilson, S., Eds., Dordrecht, 2003; pp. 79–87. [CrossRef]
- Nagy, Á. Spherically and system-averaged pair density functional theory. J. Phys. Chem. 2006, 125, 184104. [CrossRef]
- Nagy, Á. Time-Dependent Pair Density from the Principle of Minimum Fisher Information. J. Mol. Model. 2018, 24, 234. [CrossRef]
- Levy, M.; Ziesche, P. The pair density functional of the kinetic energy and its simple scaling property. J. Chem. Phys. 2001, 115, 9110–9112. [CrossRef]
- Ayers, P.W.; Levy, M. Generalized Density-Functional Theory: Conquering the N-representability Problem with Exact Functionals for the Electron Pair Density and the Second-Order Reduced Density Matrix. J. Chem. Sci. 2005, 117, 507–514. [CrossRef]
- Chakraborty, D.; Ayers, P.W. Derivation of Generalized von Weizsäcker Kinetic Energies from Quasiprobability Distribution Functions. In Statistical Complexity: Applications in Electronic Structure; Sen, K.D., Ed.; Springer: New York, 2011; pp. 35–48. [CrossRef]
- Cuevas-Saavedra, R.; Ayers, P.W. Coordinate scaling of the kinetic energy in pair density functional theory: A Legendre transform approach. Int. J. Quantum Chem. 2009, 109, 1699–1705. [CrossRef]
- Ayers, P.W. Generalized Density Functional Theories Using the K-Electron Densities: Development of Kinetic Energy Functionals. J. Math. Phys. 2005, 46, 062107. [CrossRef]
- Ayers, P.W.; Golden, S.; Levy, M. Generalizations of the Hohenberg-Kohn Theorem: I. Legendre Transform Constructions of Variational Principles for Density Matrices and Electron Distribution Functions. J. Chem. Phys. 2006, 124, 054101. [CrossRef]
- Ayers, P.W.; Davidson, E.R., Linear Inequalities for Diagonal Elements of Density Matrices. In Reduced-Density-Matrix Mechanics: With Application to Many-Electron Atoms and Molecules; John Wiley & Sons, Ltd, 2007; chapter 16, pp. 443–483. [CrossRef]
- Keyvani, Z.A.; Shahbazian, S.; Zahedi, M. To What Extent are “Atoms in Molecules” Structures of Hydrocarbons Reproducible from the Promolecule Electron Densities? Chem. Eur. J. 2016, 22, 5003–5009. [CrossRef]
- Spackman, M.A.; Maslen, E.N. Chemical properties from the promolecule. J. Phys. Chem. 1986, 90, 2020–2027. [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Act. 1977, 44, 129–138. [CrossRef]
- Hirshfeld, F.L. XVII. Spatial Partitioning of Charge Density. Isr. J. Chem. 1977, 16, 198–201. [CrossRef]
- Heidar-Zadeh, F.; Ayers, P.W. Generalized Hirshfeld Partitioning with Oriented and Promoted Proatoms. Acta Phys. -Chim. Sin. 2018, 34, 514–518. [CrossRef]
- Nalewajski, R.F.; Switka, E. Information Theoretic Approach to Molecular and Reactive Systems. Phys. Chem. Chem. Phys. 2002, 4, 4952–4958. [CrossRef]
- Nalewajski, R.F. Information Principles in the Theory of Electronic Structure. Chem. Phys. Lett. 2003, 372, 28–34. [CrossRef]
- Nagy, A.; Romera, E. Relative Rényi entropy and fidelity susceptibility. Europhys. Lett. 2015, 109, 60002. [CrossRef]
- Nagy, A. Relative information in excited-state orbital-free density functional theory. Int. J. Quantum Chem. 2020, 120, e26405. [CrossRef]
- Laguna, H.G.; Salazar, S.J.C.; Sagar, R.P. Entropic Kullback-Leibler Type Distance Measures for Quantum Distributions. Int. J. Quantum Chem. 2019, 119, e25984. [CrossRef]
- Borgoo, A.; Jaque, P.; Toro-Labbe, A.; Van Alsenoy, C.; Geerlings, P. Analyzing Kullback-Leibler Information Profiles: An Indication of Their Chemical Relevance. Phys. Chem. Chem. Phys. 2009, 11, 476–482. [CrossRef]
- Parr, R.G.; Bartolotti, L.J. Some remarks on the density functional theory of few-electron systems. J. Phys. Chem. 1983, 87, 2810–2815. [CrossRef]
- Ayers, P.W. Density per particle as a descriptor of Coulombic systems. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 1959–1964. [CrossRef]
- Ayers, P.W. Information Theory, the Shape Function, and the Hirshfeld Atom. Theor. Chem. Acc. 2006, 115, 370–378. [CrossRef]
- Ayers, P.W.; Cedillo, A. The Shape Function. In Chemical Reactivity Theory: A Density Functional View; Chattaraj, P.K., Ed.; Taylor and Francis: Boca Raton, 2009; chapter 19, p. 269. [CrossRef]
- Noorizadeh, S.; Shakerzadeh, E. Shannon entropy as a new measure of aromaticity, Shannon aromaticity. Phys. Chem. Chem. Phys. 2010, 12, 4742–4749. [CrossRef]
- Donghai, Y. Studying on Aromaticity using Information-Theoretic Approach in Density Functional Reactivity Theory. PhD thesis, Hunan Normal University, 2019.
- Li, M.; Wan, X.; Rong, C.; Zhao, D.; Liu, S. Directionality and additivity effects of molecular acidity and aromaticity for substituted benzoic acids under external electric fields. Phys. Chem. Chem. Phys. 2023, 25, 27805–27816. [CrossRef]
- Cao, X.; Rong, C.; Zhong, A.; Lu, T.; Liu, S. Molecular acidity: An accurate description with information-theoretic approach in density functional reactivity theory. J. Comput. Chem. 2018, 39, 117–129. [CrossRef]
- Yu, D.; Rong, C.; Lu, T.; De Proft, F.; Liu, S. Baird’s Rule in Substituted Fulvene Derivatives: An Information-Theoretic Study on Triplet-State Aromaticity and Antiaromaticity. ACS Omega 2018, 3, 18370–18379. [CrossRef]
- Yu, D.; Stuyver, T.; Rong, C.; Alonso, M.; Lu, T.; De Proft, F.; Geerlings, P.; Liu, S. Global and local aromaticity of acenes from the information-theoretic approach in density functional reactivity theory. Phys. Chem. Chem. Phys. 2019, 21, 18195–18210. [CrossRef]
- Rong, C.; Wang, B.; Zhao, D.; Liu, S. Information-theoretic approach in density functional theory and its recent applications to chemical problems. WIREs Comput. Mol. Sci. 2020, 10, e1461. [CrossRef]
- Liu, S. On the relationship between densities of Shannon entropy and Fisher information for atoms and molecules. J. Chem. Phys. 2007, 126, 191107. [CrossRef]
- Heidar Zadeh, F.; Fuentealba, P.; Cárdenas, C.; Ayers, P.W. An information-theoretic resolution of the ambiguity in the local hardness. Phys. Chem. Chem. Phys. 2014, 16, 6019–6026. [CrossRef]
- Heidar-Zadeh, F.; Ayers, P.W.; Verstraelen, T.; Vinogradov, I.; Vöhringer-Martinez, E.; Bultinck, P. Information-Theoretic Approaches to Atoms-in-Molecules: Hirshfeld Family of Partitioning Schemes. J. Phys. Chem. A 2018, 122, 4219–4245. [CrossRef]
- Tehrani, A.; Anderson, J.S.M.; Chakraborty, D.; Rodriguez-Hernandez, J.I.; Thompson, D.C.; Verstraelen, T.; Ayers, P.W.; Heidar-Zadeh, F. An information-theoretic approach to basis-set fitting of electron densities and other non-negative functions. J. Comput. Chem. 2023, 44, 1998–2015. [CrossRef]
- and, R.F.N. On phase/current components of entropy/information descriptors of molecular states. Mol. Phys. 2014, 112, 2587–2601. [CrossRef]
- Nalewajski, R. Phase/current information descriptors and equilibrium states in molecules. Int. J. Quantum Chem. 2014, 115, 1274–1288. [CrossRef]
- Nalewajski, R.F. Resultant Information Description of Electronic States and Chemical Processes. J. Phys. Chem. A 2019, 123, 9737–9752. [CrossRef]
- Nalewajski, R.F. Information-Theoretic Descriptors of Molecular States and Electronic Communications between Reactants. Entropy 2020, 22. [CrossRef]
- Nalewajski, R.F. Information origins of the chemical bond: Bond descriptors from molecular communication channels in orbital resolution. Int. J. Quantum Chem. 2009, 109, 2495–2506. [CrossRef]
- Wang, B.; Rong, C.; Chattaraj, P.K.; Liu, S. A comparative study to predict regioselectivity, electrophilicity and nucleophilicity with Fukui function and Hirshfeld charge. Theor Chem Acc 2019, 138. [CrossRef]
- Liu, S.; Rong, C.; Lu, T. Information Conservation Principle Determines Electrophilicity, Nucleophilicity, and Regioselectivity. J. Phys. Chem. A 2014, 118, 3698–3704. [CrossRef]
- Liu, S. Quantifying Reactivity for Electrophilic Aromatic Substitution Reactions with Hirshfeld Charge. J. Phys. Chem. A 2015, 119, 3107–3111. [CrossRef]
- Zou, X.; Rong, C.; Lu, T.; Liu, S. Hirshfeld Charge as a Quantitative Measure of Electrophilicity and Nucleophilicity: Nitrogen-Containing Systems. Acta Phys. -Chim. Sin. 2014, 30, 2055–2062. [CrossRef]
- Zhou, X.Y.; Rong, C.; Lu, T.; Zhou, P.; Liu, S. Information Functional Theory: Electronic Properties as Functionals of Information for Atoms and Molecules. J. Phys. Chem. A 2016, 120, 3634–3642. [CrossRef]
- Kruszewski, J.; Krygowski, T. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. [CrossRef]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of .pi.-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [CrossRef]
- Matito, E.; Duran, M.; Sola , M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem Phys. 2004, 122, 014109. [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [CrossRef]
- Zhao, Y.; Zhao, D.; Liu, S.; Rong, C.; Ayers, P.W. Extending the information-theoretic approach from the (one) electron density to the pair density. J. Chem Phys. 2025. Accepted for publication.
- Sagar, R.P.; Guevara, N.L. Mutual information and correlation measures in atomic systems. J. Chem Phys. 2005, 123, 044108. [CrossRef]
- Heinosaari, T.; Ziman, M. The Mathematical Language of Quantum Theory: From Uncertainty to Entanglement, 1 ed.; Cambridge University Press, 2011. [CrossRef]
- Bengtsson, I.; Życzkowski, K. Geometry of Quantum States: An Introduction to Quantum Entanglement, 2 ed.; Cambridge University Press: Cambridge, 2017. [CrossRef]
- Ciarlet, P.G.; Lions, J.L. In Computational Chemistry: Reviews of Current Trends. In Computational Chemistry: Reviews of Current Trends; North-Holland, 2003.
- Reed, M.; Simon, B. Methods of Modern Mathematical Physics. IV, Analysis of Operators; Academic Press: London, 1978.
- Yserentant, H. On the regularity of the electronic Schrödinger equation in Hilbert spaces of mixed derivatives. Numer. Math. 2004, 98, 731–759. [CrossRef]
- Islam, R.; Ma, R.; Preiss, P.M.; Eric Tai, M.; Lukin, A.; Rispoli, M.; Greiner, M. Measuring entanglement entropy in a quantum many-body system. Nature 2015, 528, 77–83. [CrossRef]
- Rissler, J.; Noack, R.M.; White, S.R. Measuring orbital interaction using quantum information theory. Chem. Phys. 2006, 323, 519–531. [CrossRef]
- Mazziotti, D.A. Entanglement, Electron Correlation, and Density Matrices, 1 ed.; Vol. 134, Wiley, 2007; pp. 493–535. [CrossRef]
- Boguslawski, K.; Tecmer, P.; Legeza, O.; Reiher, M. Entanglement Measures for Single- and Multireference Correlation Effects. J. Phys. Chem. Lett. 2012, 3, 3129–3135. [CrossRef]
- Boguslawski, K.; Tecmer, P. Orbital entanglement in quantum chemistry. Int. J. Quantum Chem. 2015, 115, 1289–1295. [CrossRef]
- Zhao, Y.; Boguslawski, K.; Tecmer, P.; Duperrouzel, C.; Barcza, G.; Legeza, O.; Ayers, P.W. Dissecting the bond-formation process of d 10-metal–ethene complexes with multireference approaches. Theor. Chem. Acc. 2015, 134, 120. [CrossRef]
- Duperrouzel, C.; Tecmer, P.; Boguslawski, K.; Barcza, G.; Örs Legeza.; Ayers, P.W. A quantum informational approach for dissecting chemical reactions. Chem. Phys. Lett. 2015, 621, 160–164. [CrossRef]
- Boguslawski, K.; Tecmer, P.; Legeza, O. Analysis of two-orbital correlations in wave functions restricted to electron-pair states. Phys. Rev. B 2016, 94, 155126. [CrossRef]
- Boguslawski, K.; Réal, F.; Tecmer, P.; Duperrouzel, C.; Pereira Gomes, A.S.; Legeza, Ö.; W. Ayers, P.; Vallet, V. On the multi-reference nature of plutonium oxides: PuO22+, PuO2, PuO3 and PuO2(OH)2. Phys. Chem. Chem. Phys. 2017, 19, 4317–4329. [CrossRef]
- Brandejs, J.; Veis, L.; Szalay, S.; Barcza, G.; Pittner, J.; Legeza, Ö. Quantum information-based analysis of electron-deficient bonds. J. Chem. Phys. 2019, 150, 204117. [CrossRef]
- White, S.R. Density matrix formulation for quantum renormalization groups. Phys. Rev. Lett. 1992, 69, 2863–2866. [CrossRef]
- White, S.R. Density-matrix algorithms for quantum renormalization groups. Phys. Rev. B 1993, 48, 10345–10356. [CrossRef]
- White, S.R.; Martin, R.L. Ab initio quantum chemistry using the density matrix renormalization group. J. Chem. Phys. 1999, 110, 4127–4130. [CrossRef]
- Fiedler, M. Algebraic connectivity of graphs. Czech. Math. J. 1973, 23, 298–305.
- Fiedler, M. A property of eigenvectors of nonnegative symmetric matrices and its application to graph theory. Czech. Math. J. 1975, 25, 619–633.
- Levine, B.G.; Durden, A.S.; Esch, M.P.; Liang, F.; Shu, Y. CAS without SCF—Why to use CASCI and where to get the orbitals. J. Chem Phys. 2021, 154, 090902. [CrossRef]
Figure 1.
Illustration of some representative concepts of classical information theory (CIT) and quantum information theory (QIT) in quantum chemistry confined to Shannon’s framework.
Figure 1.
Illustration of some representative concepts of classical information theory (CIT) and quantum information theory (QIT) in quantum chemistry confined to Shannon’s framework.
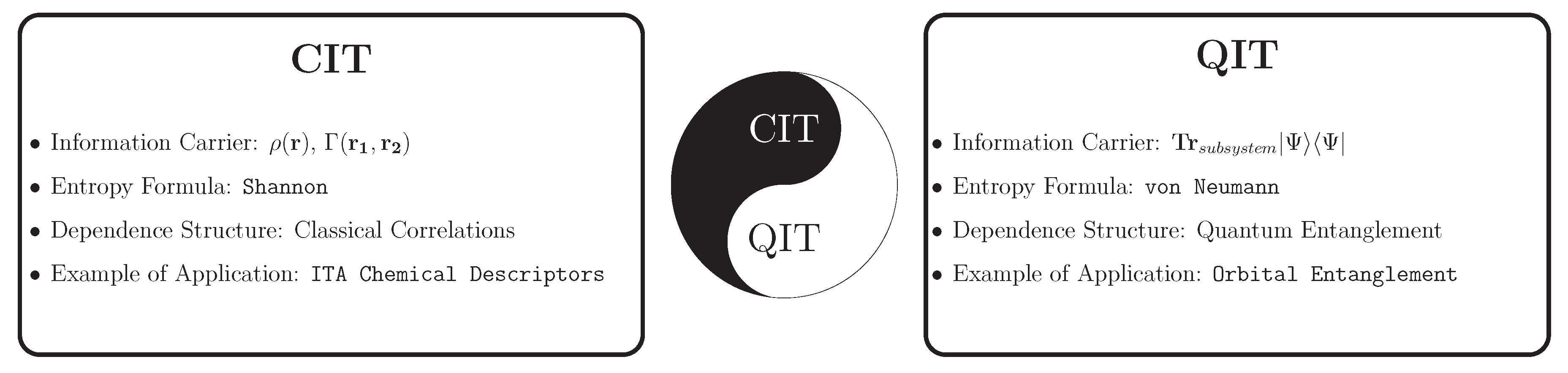
Figure 2.
Different measures of distinguishability between two probability distributions and .
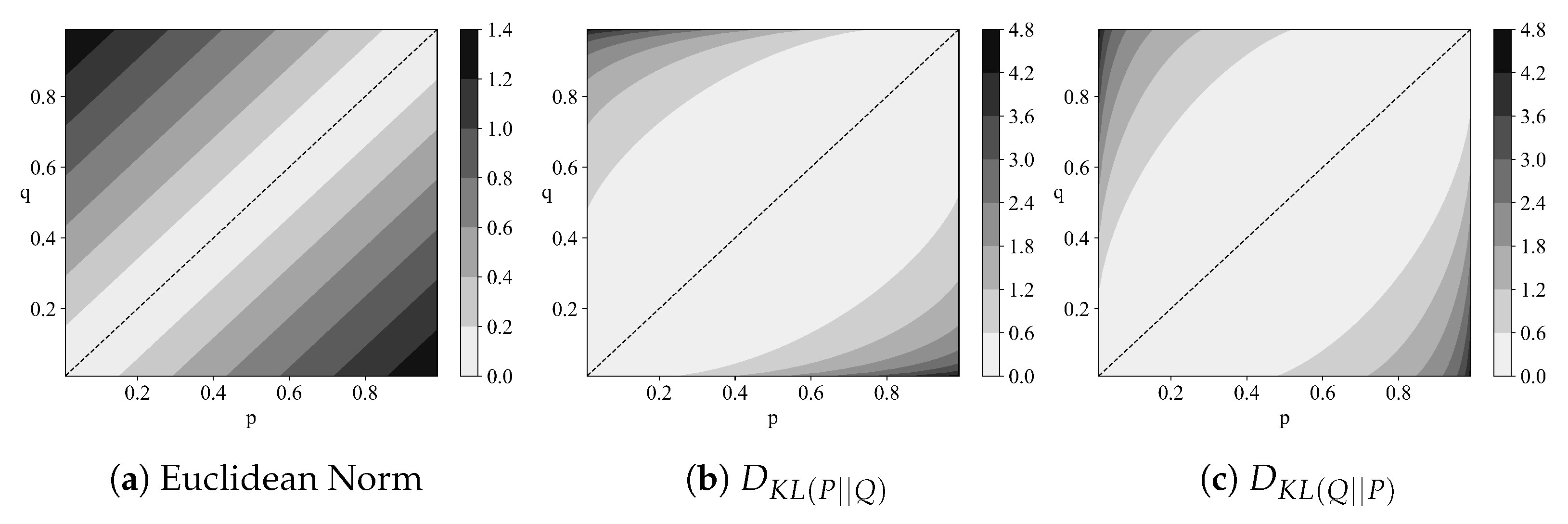
Figure 3.
A pseudo Venn diagram illustrating the relationships between Shannon entropy or along with consequent concepts: joint entropy , conditional entropy or , and mutual information .
Figure 3.
A pseudo Venn diagram illustrating the relationships between Shannon entropy or along with consequent concepts: joint entropy , conditional entropy or , and mutual information .
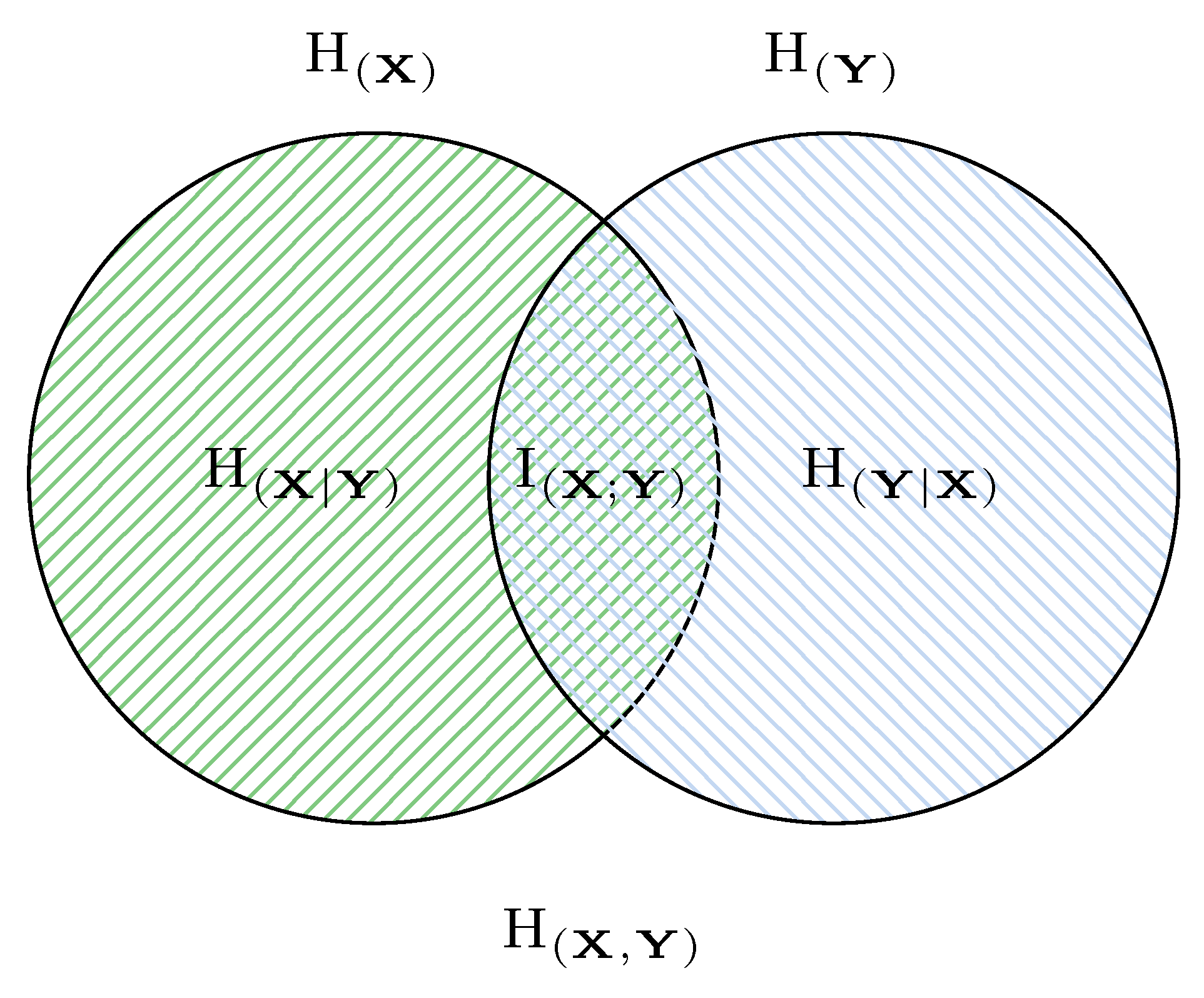
Figure 4.
For the Bloch sphere, every point on the surface corresponds to a pure state, every point inside corresponds to a mixed state, and the point located exact center of the Bloch sphere corresponds to a maximally mixed state.
Figure 4.
For the Bloch sphere, every point on the surface corresponds to a pure state, every point inside corresponds to a mixed state, and the point located exact center of the Bloch sphere corresponds to a maximally mixed state.
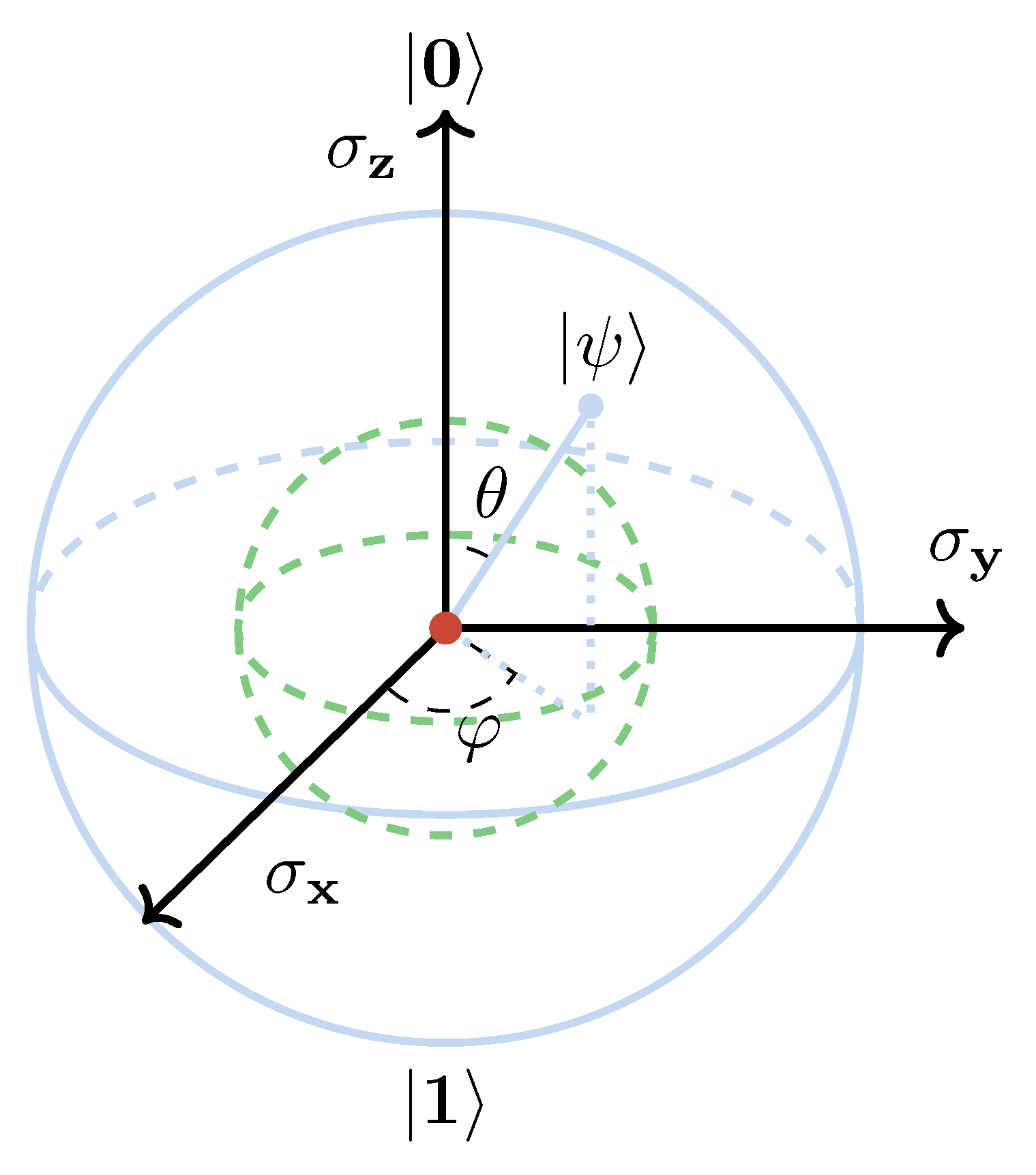
Figure 5.
Linear correlation analysis between Shannon entropy aromaticity and several established aromaticity indices. Reproduced with permission from Ref. [130] Copyright 2019, The Author.
Figure 5.
Linear correlation analysis between Shannon entropy aromaticity and several established aromaticity indices. Reproduced with permission from Ref. [130] Copyright 2019, The Author.
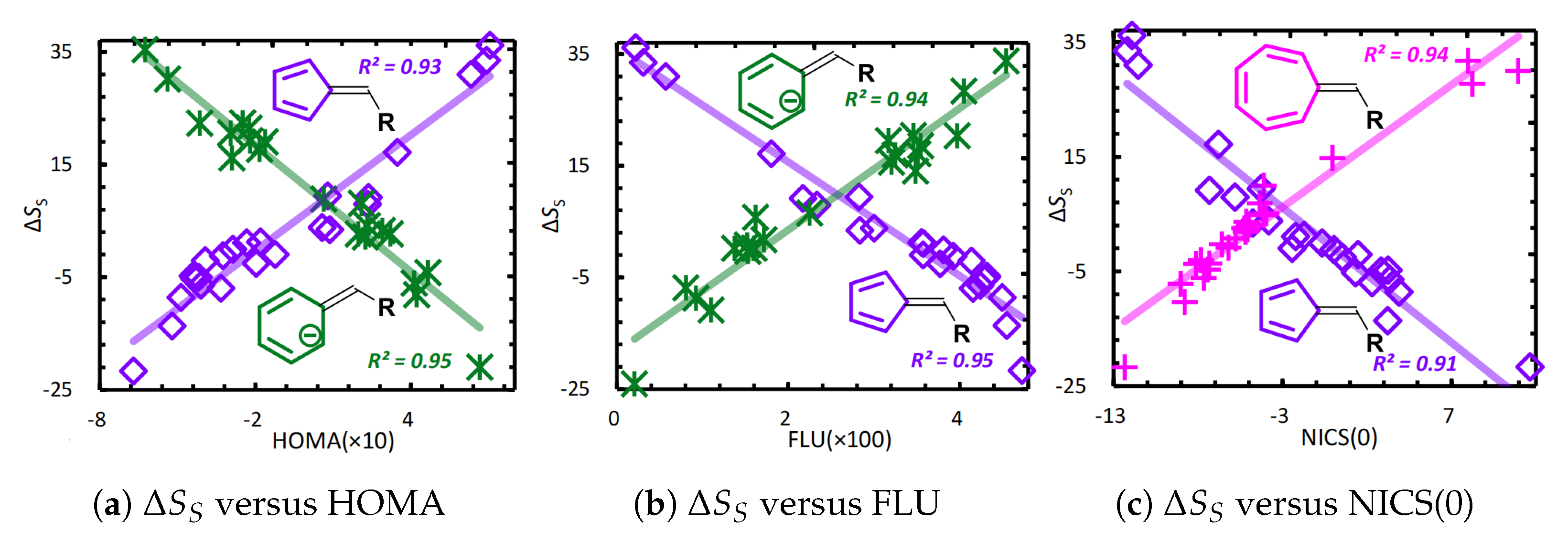
Figure 6.
Radial distribution functions of the Shannon entropy densities , along the r axis for some noble gas atoms Neon, Argon and Krypton. Reproduced with permission from Ref. [155] Copyright 2025, The Author.
Figure 6.
Radial distribution functions of the Shannon entropy densities , along the r axis for some noble gas atoms Neon, Argon and Krypton. Reproduced with permission from Ref. [155] Copyright 2025, The Author.
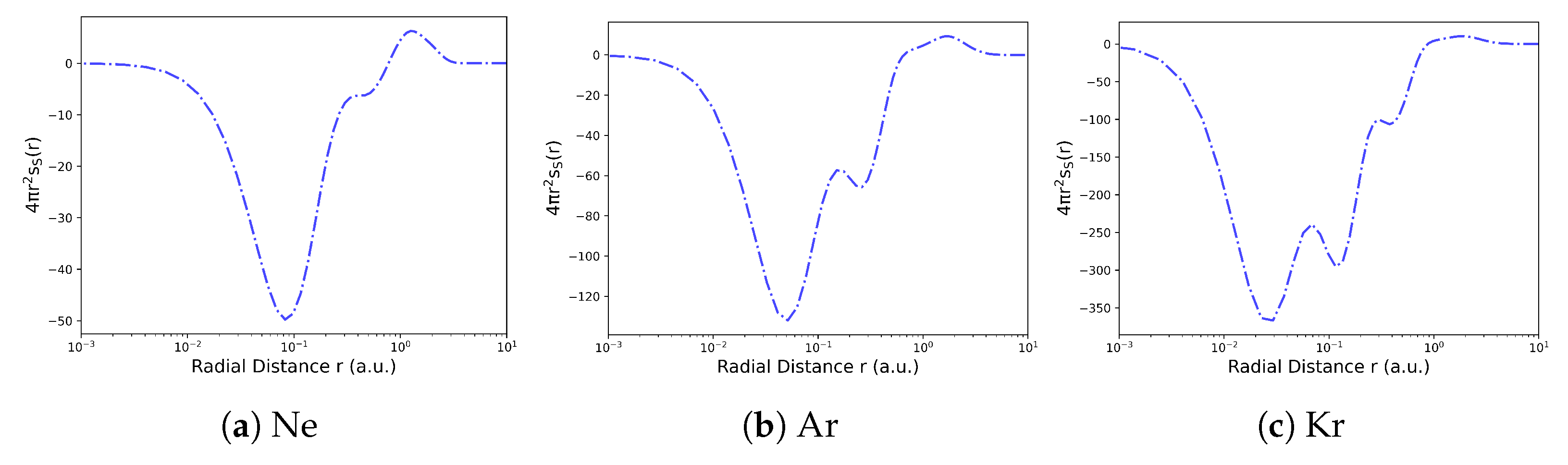
Figure 7.
Contour plots of radial distribution function for joint entropy kernel , conditional entropy kernel and mutual information kernel , for Krypton. Reproduced with permission from Ref. [155] Copyright 2025, The Author.
Figure 7.
Contour plots of radial distribution function for joint entropy kernel , conditional entropy kernel and mutual information kernel , for Krypton. Reproduced with permission from Ref. [155] Copyright 2025, The Author.
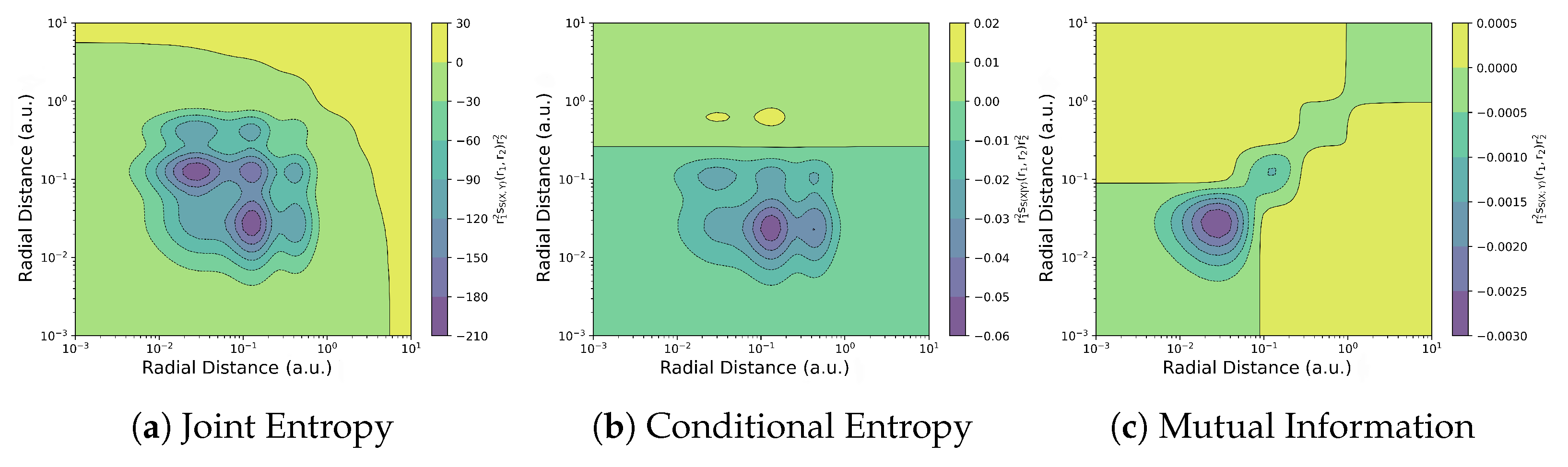
Figure 8.
Bipartite of quantum many-body system for the separable state (product state) and entangled state.
Figure 8.
Bipartite of quantum many-body system for the separable state (product state) and entangled state.
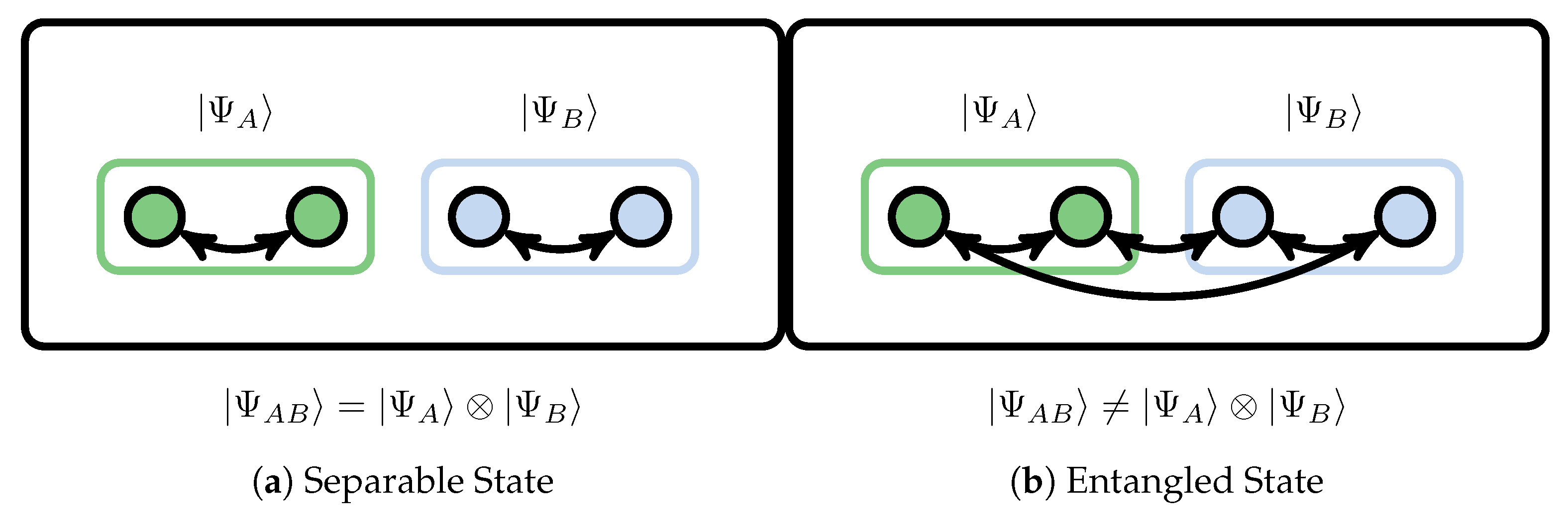
Figure 9.
Graph illustration one- and two- orbital bipartite of the system and the corresponding vector state of the composite system AB is displayed.
Figure 9.
Graph illustration one- and two- orbital bipartite of the system and the corresponding vector state of the composite system AB is displayed.
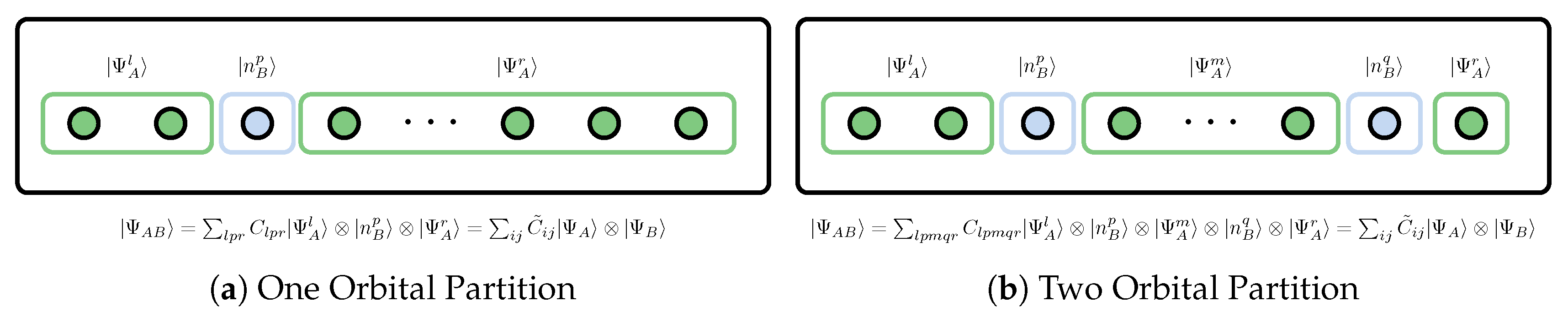
Figure 10.
Single orbital entropy and an alternative definition of orbital mutual information, as proposed in Ref. [165], is given by , specifically applied to Ni(C2H4) through DMRG(36,33) calculations. The classified strengths of mutual information in Table 3 are represented using a color code, with the dynamic entanglement effects being disregarded. Reproduced with permission from Ref. [167] Copyright 2015, The Author.
Figure 10.
Single orbital entropy and an alternative definition of orbital mutual information, as proposed in Ref. [165], is given by , specifically applied to Ni(C2H4) through DMRG(36,33) calculations. The classified strengths of mutual information in Table 3 are represented using a color code, with the dynamic entanglement effects being disregarded. Reproduced with permission from Ref. [167] Copyright 2015, The Author.
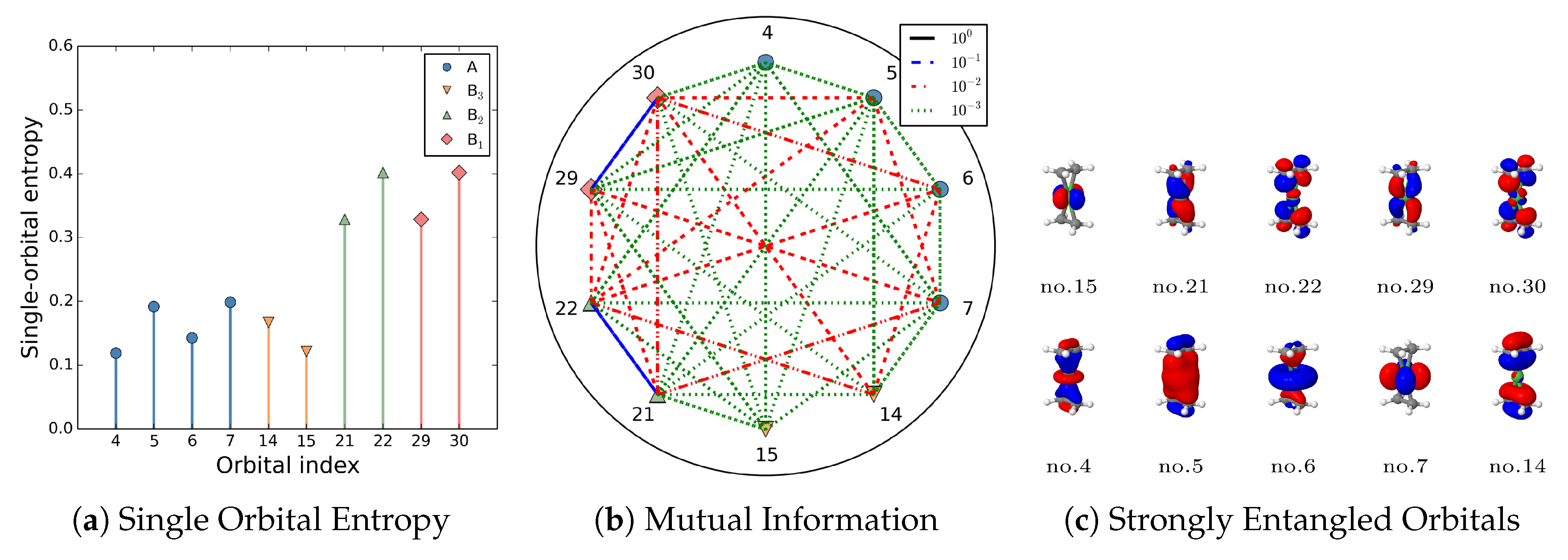
Figure 11.
Orbital classification in CAS Methodology: Inactive space remain doubly occupied, active space allow all possible configurations, and virtual space are unoccupied.
Figure 11.
Orbital classification in CAS Methodology: Inactive space remain doubly occupied, active space allow all possible configurations, and virtual space are unoccupied.
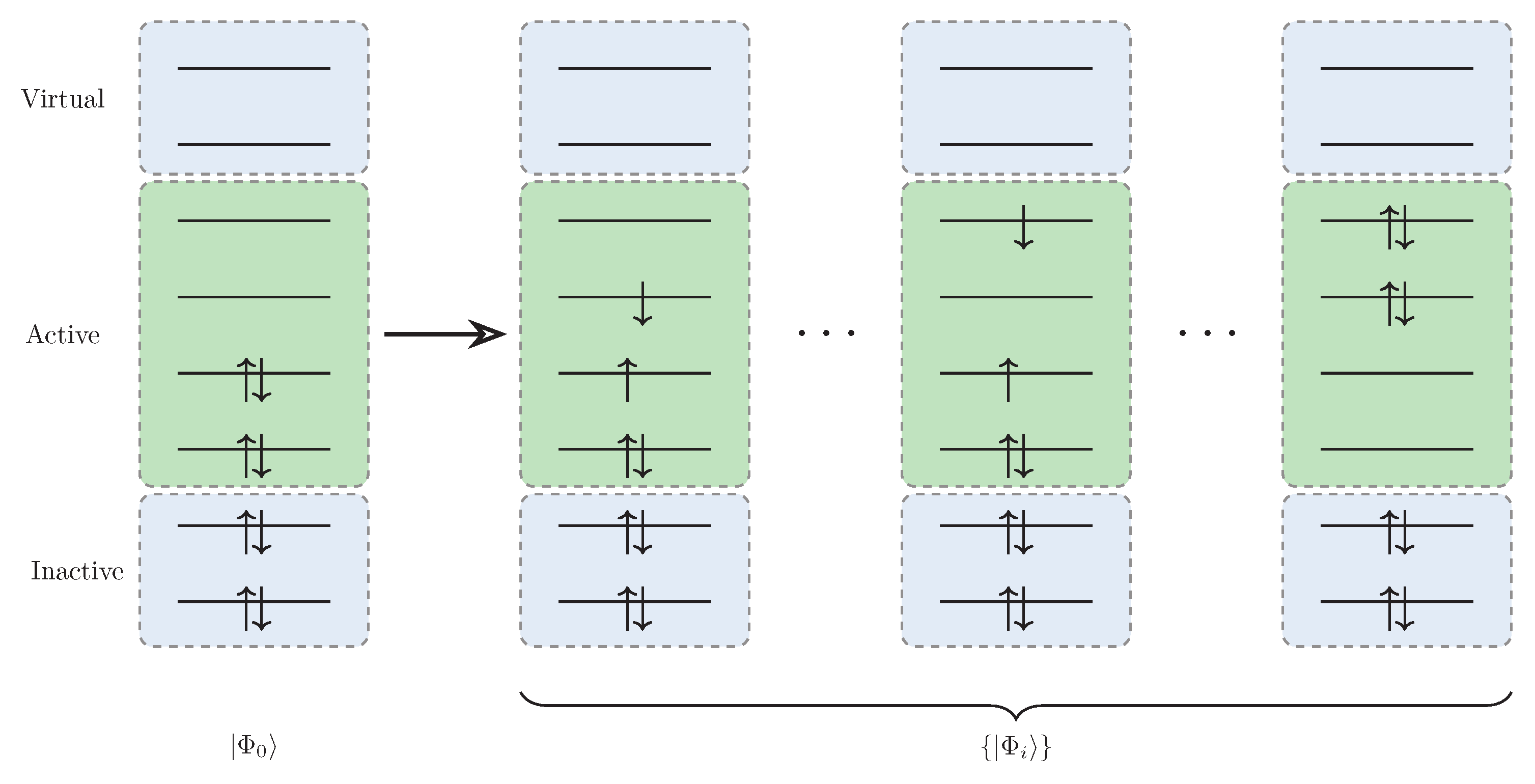

Table 1.
The matrix elements of 1-orbital RDM expressed in terms of 1- and 2-electron RDM.
| - | ||||
|---|---|---|---|---|
| – | 0 | 0 | 0 | |
| 0 | 0 | 0 | ||
| 0 | 0 | 0 | ||
| 0 | 0 | 0 |
Table 2.
The matrix elements of 2-orbital RDM expressed in terms of 1-, 2-, 3-, and 4-electron RDM.
| – |
|
|
|
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| – | 1,1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0 | 2,2 | 2,3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 0 | 3,2 | 3,3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 0 | 0 | 0 | 4,4 | 4,5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 0 | 0 | 0 | 5,4 | 5,5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
|
|
0 | 0 | 0 | 0 | 0 | 6,6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
|
|
0 | 0 | 0 | 0 | 0 | 0 | 7,7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8,8 | 8,9 | 8,10 | 8,11 | 0 | 0 | 0 | 0 | 0 | |
|
|
0 | 0 | 0 | 0 | 0 | 0 | 0 | 9,8 | 9,9 | 9,10 | 9,11 | 0 | 0 | 0 | 0 | 0 |
|
|
0 | 0 | 0 | 0 | 0 | 0 | 0 | 10,8 | 10,9 | 10,10 | 10,11 | 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11,8 | 11,9 | 11,10 | 11,11 | 0 | 0 | 0 | 0 | 0 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12,12 | 12,13 | 0 | 0 | 0 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13,12 | 13,13 | 0 | 0 | 0 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14,14 | 14,15 | 0 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 15,14 | 15,15 | 0 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 16,16 |
Table 3.
Relation between the strength of orbital entanglement and electron correlation effects.
| Correlation Effects | Intensity | ||
|---|---|---|---|
| Nondynamic | Strong | >0.5 | |
| Static | Medium | 0.5-0.1 | |
| Dynamic | Weak | <0.1 |
An alternative definition of orbital mutual information from Ref. [165] is employed: . is the Kronecker delta to ensure when .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



