
Submitted:
22 May 2025
Posted:
22 May 2025
You are already at the latest version
Abstract
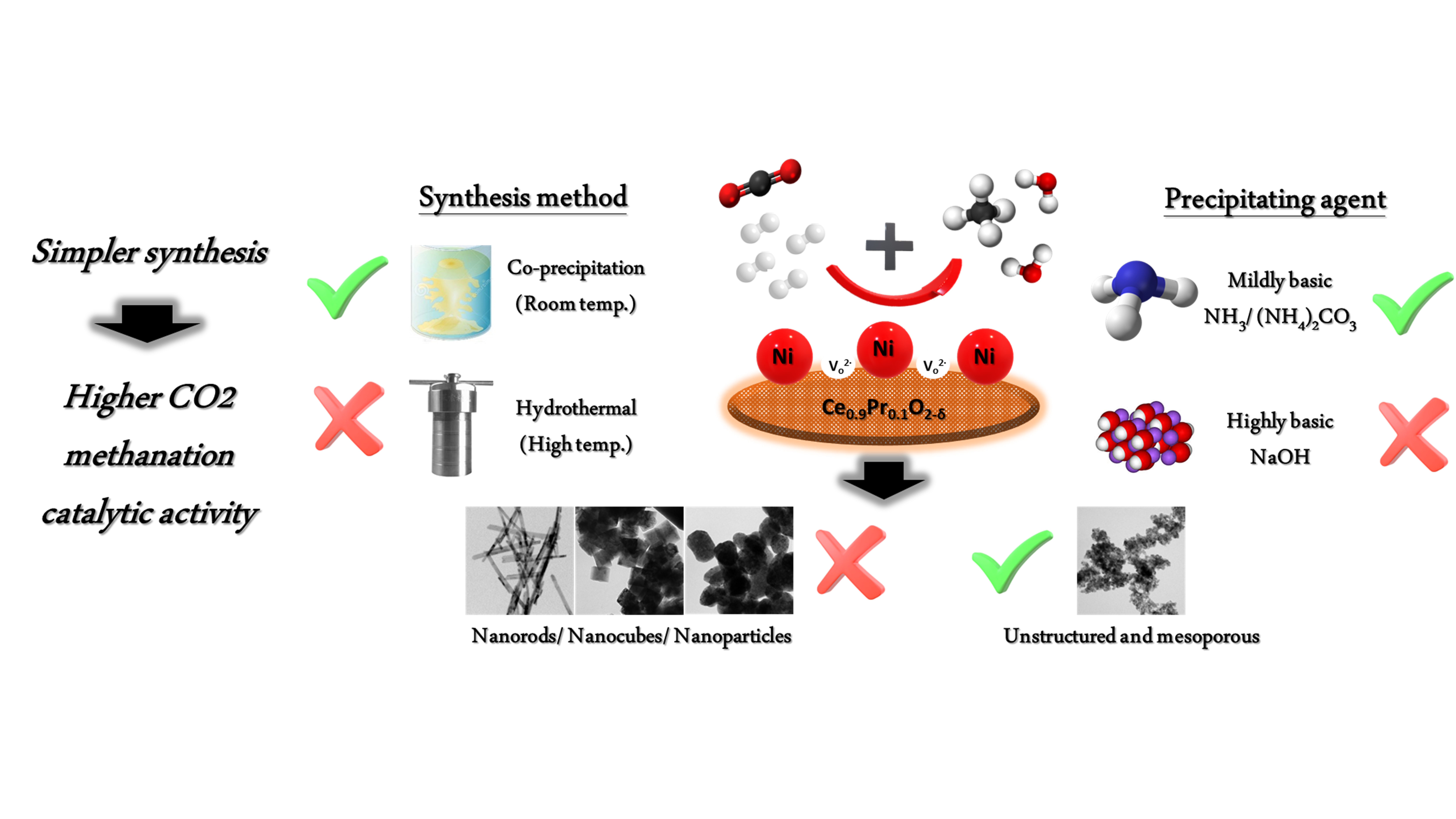
The synthesis method of the Pr-doped CeO2 catalyst support in Ni/Pr-CeO2 CO2 methanation catalysts is varied by changing the type/ basicity of the precipitating solution and the hydrothermal treatment temperature. The use of highly basic NaOH as the precipitating agent and elevated hydrothermal treatment temperature (100 or 180 oC) leads to the formation of structured Pr doped CeO2 nanorods and nanocubes, respectively, whereas the use of a mildly basic NH3 based buffer in the absence of hydrothermal treatment (i.e., co-precipitation) leads to an unstructured mesoporous morphology with medium-sized supported Ni nanoparticles. The latter catalyst (Ni/CP_NH3) displays a high surface area, high population of moderately strong basic sites, and favorable Ni dispersion. These properties lead to a higher catalytic activity for CO2 methanation (75% CO2 conversion and 99% CH4 selectivity at 350 oC) compared to the catalysts with structured nanorod and nanocube support morphologies, which are found to contain a significant amount of leftover Na from the synthesis procedure that can act as a catalyst inhibitor. In addition, the best performing Ni/CP_NH3 catalyst is shown to be highly stable, with minimal deactivation during time on stream operation.
Keywords:
CO2 methanation
; synthesis method
; Pr-doped CeO2
; hydrothermal
; coprecipitation
; nanostructures
1. Introduction
The swift increase in anthropogenic CO2 emissions risks disrupting the Earth's climate, since CO2 functions as a greenhouse gas leading to a steep rise in the atmospheric temperature [1,2]. To keep its concentration in check, it is crucial to develop effective carbon capture and storage, as well as carbon capture and utilization technologies, the latter of which can result in the generation of value-added products from waste CO2 streams [3,4,5]. On the other hand, the fluctuating nature of renewable energy production necessitates long-term energy storage solutions, which can be achieved through chemical energy storage in the form of “green” hydrogen that is produced through electrolysis [6]. However, hydrogen has a low volumetric energy density, presenting challenges for its effective storage and transportation [7]. To this end, the produced green hydrogen can be used to hydrogenate the captured CO2 to generate CH4 (synthetic natural gas), which presents a much higher energy density, as well as easier storage and transportation options, via the CO2 methanation reaction (Eq. 1) [8,9,10].
CO2 + 4H2 → CH4 + 2H2O
Noble metal catalysts like Rh and Ru have demonstrated significant catalytic activity for this particular reaction [9,10,11]. However, their prohibitively high cost remains a considerable limitation, leading to the widespread use of alternative, transition metal catalysts, particularly Ni-based ones [12,13]. Ni catalysts supported on CeO2-based supports are known for their superior CO2 methanation catalytic activity when compared to those supported on other metal oxide supports (e.g., Al2O3, SiO2, or ZrO2), which is ascribed to the rich defect chemistry and high oxygen vacancy population/ oxygen mobility of the CeO2 lattice, facilitating the rapid conversion and removal of intermediate species [13,14,15]. The doping of CeO2 with trivalent cations, like La3+ [16], Sm3+ [17], and Pr3+ [18,19,20], has also been shown to significantly enhance the oxygen vacancy population (to charge-balance the extrinsic substitutional defects) and improve the CO2 methanation catalytic activity. Particularly, in our previous work [20], we found that the Pr-doping of the CeO2 support at 10 mol% can provide the maximum promoting effect for Ni-supported CO2 methanation catalysts.
In recent years, many research studies have focused on the hydrothermal synthesis of CeO2-based nanostructures with varying morphologies and exposed crystalline facets [21,22,23,24,25,26,27]. The hydrothermal synthesis utilizing a highly basic/ concentrated NaOH solution as the precipitating agent has been frequently employed for this purpose, with variations in the hydrothermal treatment temperature typically leading to the formation of nanorods, nanocubes, or other metal oxide support nanostructures [21,22,24,25,26]. For example, Hashimoto et al. [21] demonstrated that a Ni catalyst supported on hydrothermally prepared CeO2 with nanorod morphology exhibited a higher catalytic activity when compared to those supported on CeO2 with nanocube and nanooctahedral morphology, which was attributed to the enhanced surface oxygen reactivity of the (110) facet of crystalline CeO2. Similar findings have also been reported by Bian et al. [22] and Ma et al. [23]. Conversely, according to Jomjaree et al. [24], Ni supported on CeO2 nanopolyhedrons, and according to Bian et al. [25], Ni supported on CeO2 nanoparticles, both prepared using more diluted/ less basic NaOH precipitating solutions, were superior compared to Ni supported on nanorod and nanocube CeO2 support morphologies. The utilization of precipitating solutions with a weaker basicity during hydrothermal synthesis remains considerably less common in the literature. Furthermore, co-precipitation synthesis for the CeO2-based support, which often proceeds similarly to hydrothermal synthesis (but without the hydrothermal treatment step at an autoclave), has also been employed in a number of studies [28,29,30], but it generally receives significantly less attention compared to the hydrothermal synthesis, and these similar preparation techniques are rarely compared with each other.
Therefore, in this work, we perform a comparative study by altering the support synthesis method (Pr-doped CeO2) in Ni-based catalysts via the variation of two synthesis parameters: i) the basicity of the precipitating solution (highly basic NaOH vs mildly basic NH3-based buffer) and ii) the hydrothermal treatment temperature (100 oC, 180 oC, or room temperature, i.e., co-precipitation). The doping of the CeO2 support with 10 mol% Pr was performed to enhance the activity of the corresponding catalysts based on the results of our prior work [20]. It is found that the use of a mildly basic NH3-based buffer in the absence of hydrothermal treatment (co-precipitation) leads to an unstructured mesoporous support morphology that provides a significantly higher catalytic activity during CO2 methanation. This can be attributed to favorable physicochemical properties such as a high surface area, high basic site population of moderate strength, suitable Ni dispersion, and the absence of leftover Na that can act as catalyst inhibitor. As such, the rather simpler co-precipitation support synthesis with a mildly basic precipitating agent can yield better catalytic results when compared to the preparation of structured supports with nanorod and nanocube morphologies using highly basic NaOH.
2. Materials and Methods
2.1. Synthesis Methods
Pr-doped CeO2 nanostructures (10 mol% Pr nominal composition, or Ce0.9Pr0.1O2-δ) were synthesized via several hydrothermal and co-precipitation synthesis methods, by varying the precipitating agent, i.e., the basicity of the precipitating solution during the hydrothermal/ co-precipitation synthesis, and the temperature of the hydrothermal treatment (Table 1).
When NaOH was used as the precipitating agent (high basicity of the precipitating solution, pH > 14), the following procedure was followed: At first, Ce(NO3)3·6H2O (Aldrich, 99%) and Pr(NO3)3·6H2O (Aldrich, 99.9%) in calculated amounts were dissolved in 50 ml of d-H2O in a beaker under stirring. In another beaker, NaOH (Fluka, K ≤0.02%, pellets) was added in 150 ml of d-H2O, so that the final concentration of NaOH (200 ml final solution) would be 10 M. The two solutions were then mixed together, and the final mixture was stirred for another 30 min and then transferred to a 300 ml Teflon-lined stainless-steel autoclave. Two hydrothermal treatment protocols were used: In the first one, the temperature was increased up to 100 oC (NaOH_100), and in the second one up to 180 oC (NaOH_180). In both cases, the mixture remained at that temperature for 24 h. A further co-precipitation protocol was performed, where the final mixture was kept at room temperature for 24 h without undergoing any hydrothermal treatment (CP_NaOH).
When an NH3-based buffer (NH3/(NH4)2CO3) was used as the precipitating agent (low basicity of the precipitating solution, pH = 9), the following procedure was followed: At first, calculated amounts of the metal nitrates of Ce and Pr were dissolved in 100 ml of d-H2O under stirring. The pH was then adjusted to 9 via the dropwise addition of a buffer solution of 3 M NH3/(NH4)2CO3. The volume was then increased to 200 ml (pH remaining at 9) via the further addition of d-H2O and some buffer solution. The final mixture was stirred for 30 min and then transferred to a 300 ml Teflon-lined stainless-steel autoclave. Two hydrothermal treatment protocols were used: In the first one, the temperature was increased up to 100 oC (NH3_100), and in the second one up to 180 oC (NH3_180). In both cases, the mixture remained at that temperature for 24 h. A further co-precipitation protocol was performed, where the final mixture was kept at room temperature for 24 h without undergoing any hydrothermal treatment (CP_NH3).
In all cases, after the 24 h treatment at either room temperature, 100 oC, or 180 oC, the final mixtures were centrifuged, the recovered solids were then washed thoroughly with d-H2O and once with ethanol, then dried at 70 oC overnight and afterwards calcined at 500 oC for 4 h under static air to prepare the corresponding Pr-doped CeO2 support oxides with varying nanostructures.
Wet impregnation was then used to introduce the catalytically active Ni phase. At first, Ni(NO3)2·6H2O (Fluka, 97%), for a final Ni loading of 10 wt%, was added in 100 ml of d-H2O. The metal oxide support powder was then added to the solution under stirring. Afterwards, the water was removed in a rotary evaporator at 72 oC and the leftover slurry was overnight dried at 90 oC, and then calcined at 400 oC for 4h to obtain the calcined catalysts (NiO/ Support). To prepare the corresponding reduced catalysts (Ni/ Support), the calcined ones were reduced under H2 flow at 500 oC for 1 h.
2.2. Characterization Techniques
X-ray diffraction (XRD) was performed on a MiniFlex II Rigaku powder diffractometer, using a Cu-Kα1 radiation source at 30 kV and 20 mA. To calculate the crystallite sizes of each phase, the Scherrer equation was applied on the strongest reflection.
N2 physisorption (adsorption/desorption) was carried out on a 3Flex instrument (Micromeritics) at 77K. The specific surface area (SSA, m2/g) was determined via the multi-point Brunauer-Emmet-Teller (BET) method for 0.05 < P/P◦ < 0.20, and the pore size distribution (PSD) via the Barrett–Joyner–Halenda (BJH) theory.
H2-temperature programmed reduction (H2-TPR) and CO2-temperature programmed desorption (CO2-TPD) were performed on an Autochem 2920 instrument (Micromeritics). For H2-TPR on the calcined catalysts, the samples were first treated under 20% O2/He at 500 oC for 2 h and then cooled down to ambient temperature. Afterwards, 10% H2/Ar was passed through the materials during the temperature ramp (30 oC/min). For CO2-TPD on the reduced catalysts, the samples were first treated under H2 flow at 500 oC for 2 h. Afterwards, 10% CO2/He was flown at room temperature for the CO2 adsorption. The temperature was then increased with a 30 oC/min ramp under He flow. In both cases, the thermal conductivity detector (TCD) signal was continuously recorded during the temperature ramp.
X-ray photoelectron spectroscopy (XPS) was performed on a ThermoFisher Scientific K-Alpha+ instrument using a monochromated Al Ka X-ray source (1486.6 eV), with a 400 μm radius X-ray spot. 200 eV pass energy was employed for the survey spectra and 50 eV for the core level spectra (higher resolution). Instrument modified sensitivity factors were used for quantification. The adventitious carbon C1s peak at 285.0 eV was used for charge referencing.
Lastly, transmission electron microscopy (TEM) was performed on a G2 20 S-Twin Tecnai microscope featuring a LaB6 electron source and a “SuperTwin®” objective lens that allows point-to-point resolution of 0.24 nm. High-angle annular dark field scanning transmission electron microscopy (STEM–HAADF) along with energy dispersive X-ray spectroscopy (EDS) analysis were carried out on an Analytical Titan (FEI) field emission gun microscope (300 kV), featuring a Cs-probe that allows electron probe formation of 0.09 nm (CEOS).
2.3. Catalytic Testing
Catalytic testing was carried out at ambient pressure in a fixed-bed quartz reactor (I.D. = 0.9 cm), with a similar procedure as that described in Ref. [31]. All catalysts were previously reduced in-situ for 1 h at 500 °C under H2 flow. The catalytic performance was evaluated using three experimental protocols (#1, #2, and #3) under a continuous-flow gas feed of: 10% CO2, 40% H2, balance Ar, 100 ml/min total flow.
In short, under Experimental Protocol #1, the catalytic activity was studied as a function of reaction temperature under a relatively low WHSV of 25,000 ml gcat-1 h-1. The temperature of the reactor was gradually increased via 50 oC steps from 200 oC up to 450 oC.
Under Experimental Protocol #2, a higher WHSV of 100,000 ml gcat-1 h-1 was employed, along with additional temperature steps every 10 oC from 250 oC up to 350 oC. The activation energy was calculated via this experimental protocol, making the assumption of pseudo-first order reaction kinetics and for low values of CO2 conversion (< 20%), to negate the influence of mass transfer.
Under Experimental Protocol #3, the catalysts were evaluated regarding their stability during time-on-stream testing for 24 h at a constant temperature of 350 oC (WHSV = 25,000 ml gcat-1 h-1, as in Experimental Protocol #1).
The gases exiting the reactor were analyzed online by a gas chromatography analysis system, as described in Ref. [18]. Besides CH4, CO was the sole hydrogenation by-product detected. Deviations calculated for the carbon balance were limited to ±3%. The following Eqs. 2-4 were used in order to calculate the reaction metrics (CO2 conversion, CH4 selectivity, and CH4 yield):
where Cout is the molar concentration at the reactor outlet for each gas.
Lastly, Eq. 5 was used to calculate the consumption rate of CO2 (mol gcat-1 s-1):
where is the conversion of CO2 (%), is the molar flow rate of CO2 entering the reactor (mol s-1), and Wcat is the catalyst mass (g).
3. Results and Discussion
3.1. Characterization of the Supports and the Ni Catalysts
At first, the prepared Pr-doped CeO2 metal oxide supports and the corresponding Ni-supported catalysts were characterized via XRD (Figure 1a,b). The Pr-doped CeO2 supports (Figure 1a) present the typical diffractions of the fluorite CeO2 lattice with a different peak broadening [18,20]. The calculated average crystallite sizes via the Scherrer equation fall in the range between 8-10 nm (Table 2), except for the HT_NaOH_180 support, which has an average crystallite size of around 20 nm. Therefore, the high basicity of the precipitating agent coupled with a high temperature for the hydrothermal treatment (180 oC) appear to favor the lattice growth of the Pr-doped CeO2 nanocrystallites.
The reduced Ni-supported catalysts were characterized next (Figure 1b). Besides the diffractions for the Pr-doped CeO2 support, diffractions attributed to metallic Ni0 are now also evident due to the presence of the supported Ni nanoparticles [20]. The calculated crystallite sizes for metallic Ni are between 8 and 15 nm (Table 2). The lowest crystallite size was calculated for Ni/HT_NaOH_100 (8 nm) and the highest for Ni/HT_NaOH_180 and Ni/CP_NaOH (15 and 14 nm, respectively). The catalysts whose support was prepared with mildly basic NH3-based buffer as the precipitating agent have a rather similar Ni dispersion and crystallite size (11-13 nm), i.e., medium-sized Ni nanoparticles.
It is also interesting to note, that the diffraction broadening of the Pr-doped CeO2 crystallites changes in some cases from the supports to the reduced catalysts, namely for the Ni/HT_NaOH_100 and especially for the Ni/CP_NaOH catalyst. As a result, all the Ni-supported reduced catalysts prepared via NaOH as the precipitating agent display an almost double average crystallite size for Pr-doped CeO2 compared to those with NH3-based buffer as the precipitating agent. To elucidate the origin of this crystal growth in these cases, the calcined Ni-supported catalysts were also characterized (Figure S1), which display the crystalline diffractions for the support crystallites and the oxidized NiO particles. Since the peak broadening for Pr-doped CeO2 is similar between the bare supports and the calcined catalysts, it can be stated that the Pr-doped CeO2 crystallite growth in Ni/CP_NaOH and Ni/HT_NaOH_100 occurs during the following high-temperature (500 oC) reduction treatment under H2.
Afterwards, N2 physisorption isotherms were collected for the bare supports, as well as for the reduced Ni-supported catalysts (Figure 1c,d). The supports (Figure 1c) display a different porous structure for each material depending on the synthesis method. Most of them are mesoporous with a surface area ranging from 46 up to 93 m2/g (Table 2), with the highest surface area being recorded for the CP_NaOH sample, which also displays the highest pore volume. An outlier is HT_NaOH_180, which has much larger pore sizes and displays a quite small surface area of just 10 m2/g.
Regarding the reduced Ni-supported catalysts (Figure 1d), a general trend is that upon Ni impregnation/ calcination followed by reduction, the pore volume and the surface area both drop, while the pore size distribution is shifted towards larger pore diameters. This is a result of the blocking of the smaller mesopores and pore reconstruction caused by the impregnation of Ni and the formation of supported metallic Ni nanoparticles [31,32]. For the cases of Ni/HT_NaOH_100 and especially Ni/CP_NaOH, a much higher extent of surface area loss occurs (even up to 80%), which is also consistent with the changes in the material crystallinity following Ni impregnation/ calcination and reduction (i.e., significant growth of the Pr-doped CeO2 crystallites) observed during the XRD characterization. Overall, among all of the Ni-supported reduced catalysts, higher porosity is observed for those whose supports were prepared via the mildly basic NH3-based buffer as the precipitating agent.
The catalyst material reducibility was investigated through H2-TPR on the calcined Ni-supported catalysts (Figure 2a). At the first region (I), below 200 oC, the observed reduction peaks could be ascribed to the reduction of highly dispersed NiO species at the catalyst surface, as well as possibly to Ni(OH)2 species, and Ni2+ solubilized in the Pr-doped CeO2 support [31,33]. At the second region (II), up to approx. 400 oC, the main (highest intensity) reduction peak can be assigned to the majority of NiO reduction to metallic Ni0, as well as to the contribution of removal of surface oxygen species from the Pr-doped CeO2 support [31,33,34]. The peak of the main NiO reduction event is located between 290 – 300 oC for all materials, except for the NiO/HT_NaOH_180, whose NiO reduction peak has a maximum at approx. 330 oC. This temperature range of NiO reduction to metallic Ni0 can be attributed to the high reducibility of NiO that is in contact with the defect-rich Pr-doped CeO2 support surface [31,35]. Lastly, the much smaller and quite broad peaks at higher reduction temperatures (region III) can be ascribed to oxygen removal from the bulk of the metal oxide support [31,33,34].
The surface basicity is then evaluated via CO2-TPD for the reduced Ni-supported catalysts (Figure 2b), which shows three types of desorption peaks. The peaks at lower temperatures can be assigned to weakly-bound carbonates and bicarbonates on the weak basic sites of the materials. The CO2 desorption peaks at the intermediate temperature range (approx. between 150 oC and 400 oC) are ascribed to carbonates that are formed over the moderately-strong basic sites, whereas the small and broad peaks at higher temperatures are due to the strong basic sites [31,36,37]. Although some materials present quite intense CO2 desorption peaks at the lower temperature range, Ni/CP_NH3 is found to contain the highest amount of moderately-strong basic sites at intermediate desorption temperatures. Based on the relevant literature [36,38,39], a higher population of moderately-strong basic sites can be associated with a higher catalytic activity during CO2 methanation, since they act to enhance the CO2 chemisorption and activation during the catalytic reaction.
The catalysts’ surface chemistry was studied via XPS, which was conducted ex-situ (Figure 3). The Ni2p spectra (Figure 3a) reveal the following types of surface Ni-species, with increasing binding energy (BE): i) metallic Ni0 surface sites at lower BE, ii) then NiO surface sites which originate following ex-situ oxidation of formerly metallic surface Ni sites and iii) finally, at higher BE, the large peak can be ascribed to the contribution of Ni(OH)2, which can arise following surface Ni oxidation and hydroxylation, Ni2O3 or Ni3+ due to defects in the NiO structure, and Ni species at the Ni-CeO2 interface (Ni-O-Ce sites) [20,34,40]. Regarding the O1s spectra (Figure 3b), these can be separated into surface oxygen from the metal oxide support lattice (Pr-doped CeO2) at lower BE, and adsorbed oxygen species, like hydroxyls and carbonates on the catalyst surface, and also physisorbed H2O, at higher BE [20,41]. The majority of these adsorbed oxygen species at higher BE are also expected to originate following atmospheric exposure [20]. Although differences in the peak shapes are observed for the different catalysts, these can be rather attributed to the exposure of the samples to atmospheric oxygen, and to a different extent of surface oxidation and hydroxylation. In particular, the entirety of the Ni phase is expected to be initially metallic following the reduction treatment at 500 oC, as was verified during H2-TPR characterization (Figure 2a), and thus, the oxidized Ni species in the materials originate during the subsequent atmospheric exposure.
The Ce3d spectra (Figure 3c) show the presence of multiple peaks due to the Ce3d5/2 (peaks labeled as v) and Ce3d3/2 (peaks labeled as u) transitions. Since Ce ions typically exist in both Ce4+ and Ce3+ oxidation states in CeO2-based oxides, the peaks v, v´´, v´´´, u, u´´ and u´´´ can be ascribed to the major Ce4+ oxidation state, whereas the peaks v´ and u´ correspond to the minority Ce3+ ions due to intrinsic defects in the oxide support lattice [18,20]. However, the majority of the oxygen vacancy sites are expected to originate via the extrinsic substitutional defects of aliovalent Pr3+ ions in former Ce4+ sites [20,42]. In the Pr3d spectra (Figure 3d), the position and peak shape of the Pr3d5/2 and Pr3d3/2 transition peaks resemble those of a Pr2O3-like oxide, thus confirming that Pr rather exists in the Pr3+ oxidation state [20,43]. These extrinsic (PrCe´) substitutional defects can generate a large number of oxygen vacancy sites, which can significantly promote the CO2 methanation catalytic activity [14,15,33].
The elemental surface concentrations measured via XPS can be found in Table S2. The differences in adventitious carbon and the surface concentration of some elements can be attributed to atmospheric exposure, although they roughly agree with the expected values. A more pronounced Ni concentration at the surface is expected, since Ni is mainly located as surface metallic nanoparticles, whereas the higher Pr surface concentration can be attributed to a certain extent of PrOx segregation at the support grains [20,44]. An interesting finding is that a significant Na surface concentration was detected for the reduced catalysts whose supports were prepared using NaOH as the precipitating agent (also observed via the intensity of the Na1s XPS peaks in Figure S2), even though the typical washing steps were applied during the material preparation procedures to remove these ions, similarly to other works [22,25,26,45,46,47]. This can be important during the subsequent catalytic evaluation of the materials, since Na can potentially act as a catalyst inhibitor (through electronic modifications and inducing a positive charge on Ni) and has often been reported to impair the CO2 methanation catalytic performance [48,49,50,51].
Lastly, electron microscopy analysis was performed to determine the material nanostructure. TEM images of the reduced Ni-supported catalysts are displayed in Figure 4, whereas the corresponding images of the bare Pr-doped CeO2 metal oxide supports can be found in Figure S3. The reduced catalysts are comprised of the metal oxide support nanostructure alongside the supported spherical-shaped metallic Ni nanoparticles. The majority of the supported Ni nanoparticles are approx. 5 – 20 nm in diameter (medium-sized), although some of them are quite larger, even up to 50 nm. This size range could be consistent with the average crystallite size of Ni that is calculated via XRD (Table 2), assuming predominantly single-crystal particles. A more accurate estimation of the Ni nanoparticle size via TEM is however challenging, due to the low Z-contrast in the TEM images.
From TEM, it is apparent that the metal oxide support nanostructure changes depending on the synthesis method used to prepare it. Regarding the materials prepared using highly basic NaOH as the precipitating agent, the one with no hydrothermal treatment (i.e., co-precipitation, Ni/CP_NaOH) presents small and rather cubic nanoparticles for the metal oxide support (Figure 4a), whereas the ones with the hydrothermal treatment at 100 and 180 oC (Ni/HT_NaOH_100 and Ni/HT_NaOH_180) present the typical nanostructures of nanorods and nanocubes, respectively (Figure 4b), due to the preferred crystal growth along particular crystalline facets, as is also observed in other literature works [21,22,24,25,46]. For the materials prepared using the mildly basic NH3-based buffer as the precipitating agent, the one with no hydrothermal treatment (i.e., co-precipitation, Ni/CP_NH3) presents a rather unstructured mesoporous morphology consisting of aggregates of small crystallites (Figure 4d), while the ones with the hydrothermal treatment at 100 and 180 oC (Ni/HT_NH3_100 and Ni/HT_NH3_180) reveal the formation of large particle aggregates for the metal oxide support with a diameter higher than 100 nm. The hydrothermal treatment at 100 oC (Ni/HT_NH3_100) leads to particle aggregates resembling aggregated nanorods, and the hydrothermal treatment at 180 oC (Ni/HT_NH3_180) leads to rather spherical-shaped and polyhedral-shaped particle aggregates (Figure 4e,f).
The TEM images of the bare metal oxide supports, i.e., prior to Ni impregnation/ calcination and reduction, are shown in Figure S3. Nanorods are observed for HT_NaOH_100, nanocubes for HT_NaOH_180, unstructured mesoporous morphology of aggregated crystallites for CP_NH3, and large aggregated particles resembling aggregated nanorods for HT_NH3_100, and spheres/ polyhedra for HT_NH3_180. The notable exception is CP_NaOH, which presents a quite different morphology for the metal oxide support compared to Ni/CP_NaOH. As mentioned during the XRD characterization results, this change/ consolidation in the nanostructure to small cubic nanoparticles followed by crystal growth for Pr-doped CeO2 occurs during the high-temperature reduction treatment after the Ni impregnation and calcination.
A more precise localization of the metallic Ni supported phase, as well as the determination of the elemental distribution across the materials, are achieved via HAADF-STEM and EDS elemental mapping (Figure 5 and Figure S4). Figure 5 shows the Ni nanoparticle distribution across the catalyst materials, with most of them being medium-sized (5 – 20 nm in diameter), whereas some larger Ni nanoparticles even up to 50 nm in diameter can also be observed. Medium-sized Ni nanoparticles supported over CeO2-based oxides have been previously reported to be quite efficient during CO2 methanation [31,47,52]. The Ni nanoparticles are located between the cubic support grains, nanorods, and nanocubes, for the materials prepared with NaOH as the precipitating agent, throughout the unstructured mesoporous support morphology for Ni/CP_NH3, and for Ni/HT_NH3_100 and Ni/HT_NH3_180 they appear to preferentially reside at the outer surface of the large support particle aggregates. The other elements (O, Ce, and Pr) are found to be evenly distributed across the supports, thereby also verifying that Pr (as Pr3+) is solubilized into the CeO2 lattice. An example of the entire elemental distribution (O, Ce, Pr, and Ni) for Ni/CP_NH3 is shown in Figure S4.
3.2. Catalytic Activity
The CO2 methanation catalytic activity was then evaluated as a function of reaction temperature at two different WHSV values, namely at 25,000 ml gcat-1 h-1 for Experimental Protocol #1, and at 100,000 ml gcat-1 h-1 for Experimental Protocol #2. During Experimental Protocol #1 (Figure 6), a clear observation regarding the catalytic activity can be made that the materials prepared using mildly basic NH3-based buffer as the precipitating agent are significantly more active when compared to the materials prepared using highly basic NaOH as the precipitating agent, being able to reach much higher CO2 conversion and CH4 selectivity values at lower reaction temperatures. The corresponding CH4 yield values are shown in Figure S5a.
Regarding the materials prepared with NaOH as the precipitating agent, the one with no hydrothermal treatment (Ni/CP_NaOH) and the one with the hydrothermal treatment at 100 oC (Ni/HT_NaOH_100) display a similar catalytic activity, whereas the one with the hydrothermal treatment at 180 oC (Ni/HT_NaOH_180) presents a lower one. This is in agreement with other literature works reporting that Ni nanoparticles supported on CeO2 with nanorod morphology are more active than those supported on CeO2 with nanocube morphology, which is typically assigned to differences in the oxygen reactivity of the exposed crystalline facets [21,22,23,24]. Nevertheless, the significantly inferior catalytic activity of all of these three catalysts compared to those prepared with the NH3-based buffer as the precipitating agent can be attributed to: i) the lower surface area accompanied by a higher Pr-doped CeO2 crystallite size, ii) the lower Ni dispersion, and iii) the lower basic site population, especially for the moderately-strong basic sites [31,38]. The significant presence of residual Na on the catalyst surface (Table S1), despite the washing treatments during catalyst preparation, is most probably also responsible for the lower CO2 conversion and CH4 selectivity, in agreement with other literature works [48,49,50,51].
Regarding the materials prepared with the NH3-based buffer as the precipitating agent, the ones prepared following hydrothermal treatment at 100 oC (Ni/HT_NH3_100) and 180 oC (Ni/HT_NH3_180), leading to large particle aggregates for the metal oxide support, have a rather similar catalytic activity between them. Overall however, the best catalytic performance, especially at the low-temperature regime, is obtained for the Ni/CP_NH3 material prepared via co-precipitation, leading to a maximum CO2 conversion of 75% (with 99% CH4 selectivity) at 350 oC (Table 3). The CO2 conversion and CH4 selectivity values then drop at higher temperatures, due to the exothermicity of CO2 methanation and the promotion of the antagonistic reverse water-gas shift reaction [9]. It is thus found herein, that a rather simple catalyst support (Pr-doped CeO2) preparation procedure, with an NH3-based buffer as the precipitating agent and in the absence of hydrothermal treatment, yielding an unstructured mesoporous morphology for the support, leads to an eventually higher CO2 methanation catalytic activity when compared to the materials prepared using highly basic NaOH and hydrothermal treatments that yield specific, well-defined, catalyst support nanostructures such as nanorods and nanocubes. This can be ascribed to the high specific surface area, high population of basic sites (particularly of moderate strength), favorable Ni dispersion and Ni-support interaction, and the absence of catalyst inhibitors such as residual Na [16,31,38,50].
The catalysts were then evaluated at a higher WHSV of 100,000 ml gcat-1 h-1 and with additional temperature steps during Experimental Protocol #2 (Figure 7). The increase in the total flow to catalysts weight (F/W) ratio leads to lower CO2 conversion and CH4 selectivity values, compared to the results obtained via Experimental Protocol #1 (higher F/W ratio and lower WHSV) [31,52]. Again, it is found that Ni/CP_NH3 displays the best catalytic performance, with higher CO2 conversion and CH4 selectivity values especially at the low-temperature regime, followed by Ni/HT_NH3_180 and Ni/HT_NH3_100. On the other hand, the catalysts prepared with NaOH as the precipitating agent display a substantially inferior CO2 methanation catalytic performance. The corresponding CH4 yield values are shown in Figure S5b. The measurements taken for CO2 conversion values below 20%, and thus at the kinetically controlled regime, allow for the calculation of the activation energy values via the Arrhenius plots (Figure 7c), assuming pseudo-first order reaction kinetics [31]. These values can be found in Table 3 and fall in the range between 90 – 110 kJ/mol, as would be expected for Ni/CeO2 type catalysts [22,38,53,54].
3.3. Catalytic Stability and Spent Catalyst Characterization
The stability of the best-performing Ni/CP_NH3 catalyst was then evaluated at a constant temperature of 350 oC and WHSV of 25,000 ml gcat-1 h-1 for a duration of 24 h under Experimental Protocol #3 (Figure 8a). The CO2 conversion remains quite stable, dropping by just 2% during the entire time-on-stream operation, with a final value of 74%. CH4 selectivity also remains constant during the time-on-stream duration, at 99%. It is thus shown, that Ni/CP_NH3 can provide a high and stable catalytic performance under long-term operation.
The spent Ni/CP_NH3 catalyst following the time-on-stream experiment under Experimental Protocol #3 was then characterized via XRD, N2 physisorption, and TEM, to examine potential catalyst deactivation effects. The X-ray diffractograms (Figure 8b) of the fresh (reduced) and the spent catalyst largely overlap, showing no significant changes in the catalyst material crystallinity, although the average crystallite size of Ni is now calculated at 13 nm for the spent catalyst, compared to 11 nm for the fresh one. The N2 physisorption results (Figure 8c) also show similar textural properties for the spent catalyst, with a specific surface area of 29 m2/g, pore volume of 0.13 cm3/g, and average pore diameter of 19 nm, i.e., quite similar values to those obtained for the fresh catalyst (Table 2). Furthermore, TEM characterization of the spent Ni/CP_NH3 catalyst (Figure 8d) displays a similar unstructured mesoporous morphology of aggregated small crystallites for the metal oxide support, with medium-sized (5 – 20 nm) supported Ni nanoparticles. Therefore, it can be stated that Ni/CP_NH3 largely retains its crystallinity, nanomorphology, and Ni dispersion during the time-on-stream operation, with just a potentially minor extent of Ni nanoparticle sintering.
4. Conclusions
This work reports on the co-precipitation and hydrothermal synthesis of Pr-doped CeO2 support nanostructures for Ni/Pr-CeO2 CO2 methanation catalysts by varying the basicity of the precipitating solution and the hydrothermal treatment temperature. It is found that different catalyst support nanostructures can be obtained depending on the support synthesis method, ranging from structured nanorods and nanocubes when using highly basic NaOH and elevated hydrothermal treatment temperature (100 and 180 oC, respectively) to an unstructured mesoporous support morphology consisting of aggregated small crystallites when employing a mildly basic NH3-based buffer as the precipitating solution in the absence of hydrothermal treatment. In all cases, medium-sized Ni nanoparticles are supported on the metal oxide support nanostructures.
Catalytic activity evaluation showed that the catalysts prepared with the mildly basic NH3-based buffer as the precipitating agent performed significantly better during CO2 methanation, with Ni/CP_NH3 synthesized via co-precipitation leading to the best results. On the contrary, the more structured catalyst support nanostructures prepared with highly basic NaOH led to inferior catalytic performance, due to the rather unfavorable physicochemical properties (lower surface area, basic site population of moderate strength, and Ni dispersion), and also due to the catalyst inhibition by residual Na from the synthesis procedure. Furthermore, the best-performing Ni/CP_NH3 catalyst was shown to be highly stable with limited catalyst deactivation during a 24 h time-on-stream experiment.
In short, this study shows that the utilization of rather simpler catalyst support preparation procedures, i.e., co-precipitation in the absence of hydrothermal treatment with the use of a mildly basic precipitating solution, can be more beneficial toward CO2 methanation when compared to the preparation of highly ordered catalyst support nanostructures, such as with a nanorod or nanocube morphology.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org. Figure S1: X-ray diffractograms of the calcined Ni-supported catalysts; Table S1: XPS elemental surface concentrations, given in atomic %, and in parentheses in weight %, for the reduced catalysts; Figure S2: Na1s XPS core level spectra of the reduced catalysts, whose supports were synthesized using NaOH as the precipitating agent; Figure S3: TEM images of the (a) CP_NaOH, (b) HT_NaOH_100, (c) HT_NaOH_180, (d) CP_NH3, (e) HT_NH3_100, and (f) HT_NH3_180 calcined metal oxide supports; Figure S4: HAADF-STEM along with EDS elemental mapping for O, Ce, Pr, and Ni of the Ni/CP_NH3 reduced catalyst; Figure S5: CH4 yield as a function of reaction temperature; (a) Reaction conditions: Experimental Protocol #1; (b) Reaction conditions: Experimental Protocol #2. The thermodynamic equilibrium (dotted lines) is calculated via Aspen Plus (p = 1 atm and H2:CO2 = 4:1).
Author Contributions
Conceptualization, A.I.T.; methodology, A.I.T., A.A.D., A.G.S.H., V.S., S.J.H. and M.A.B.; X.X.; validation, A.I.T.; formal analysis, A.I.T., N.D.C., A.A.D., A.G.S.H., V.S. and S.J.H.; investigation, A.I.T., N.D.C., V.S. and M.A.B.; resources, V.S., M.A.B., K.P. and M.A.G..; data curation, A.I.T., A.A.D., A.G.S.H., V.S. and S.J.H.; writing—original draft preparation, A.I.T.; writing—review and editing, N.D.C, V.S., M.A.B., S.M., K.P. and M.A.G..; supervision, M.A.G.; project administration, M.A.G.; funding acquisition, A.I.T., V.S., K.P. and M.A.G.. All authors have read and agreed to the published version of the manuscript.
Funding
NDC and MAG acknowledge support of this work by the project “Development of new innovative low carbon energy technologies to improve excellence in the Region of Western Macedonia” (MIS 5047197), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure” funded by the Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund). AIT thanks the Hellenic Foundation for Research and Innovation (HFRI) for supporting this research work under the 3rd Call for HFRI PhD Fellowships (Fellowship Number: 6033). KP acknowledges financial support from Khalifa University through the grant RC2–2018-024. VS acknowledges funding from project PID2021-127847OB-I00 MCIN/AEI/10.13039/ 501100011033 CIBER-BBN, an initiative funded by the VI National R&D&i Plan 2008–2011, financed by the Instituto de Salud Carlos III and by Fondo Europeo de Desarrollo Regional (Feder) ‘Una manera de hacer Europa’, with the assistance of the European Regional Development Fund. LMA-ELECMI and NANBIOSIS ICTs are also gratefully acknowledged.
Data Availability Statement
Dataset available on request from the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tong, D.; Zhang, Q.; Zheng, Y.; Caldeira, K.; Shearer, C.; Hong, C.; Qin, Y.; Davis, S.J. Committed emissions from existing energy infrastructure jeopardize 1.5 °C climate target. Nature 2019, 572, 373–377. [Google Scholar] [CrossRef]
- Nong, D.; Simshauser, P.; Binh, D. Greenhouse gas emissions vs CO2 emissions : Comparative analysis of a global carbon tax. Appl. Energy 2021, 298, 117223. [Google Scholar] [CrossRef]
- Zhao, K.; Jia, C.; Li, Z.; Du, X.; Wang, Y.; Li, J.; Yao, Z. Recent Advances and Future Perspectives in Carbon Capture, Transportation, Utilization, and Storage (CCTUS) Technologies : A Comprehensive Review. Fuel 2023, 351, 128913. [Google Scholar] [CrossRef]
- Dziejarski, B.; Krzyżyńska, R.; Andersson, K. Current status of carbon capture, utilization, and storage technologies in the global economy: A survey of technical assessment. Fuel 2023, 342, 127776. [Google Scholar] [CrossRef]
- Latsiou, A.I.; Charisiou, N.D.; Frontistis, Z.; Bansode, A.; Goula, M.A. CO2 hydrogenation for the production of higher alcohols: Trends in catalyst developments, challenges and opportunities. Catal. Today 2023, 420, 114179. [Google Scholar] [CrossRef]
- Ozturk, M.; Dincer, I. A comprehensive review on power-to-gas with hydrogen options for cleaner applications. Int. J. Hydrogen Energy 2021, 46, 31511–31522. [Google Scholar] [CrossRef]
- Abdalla, A.M.; Hossain, S.; Nisfindy, O.B.; Azad, A.T.; Dawood, M.; Azad, A.K. Hydrogen production, storage, transportation and key challenges with applications: A review. Energy Convers. Manag. 2018, 165, 602–627. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, X.; Wang, K.; Liang, J.; Jiang, Y.; Ma, Q.; Zhao, T.; Zhang, J. A short overview of Power-to-Methane : Coupling preparation of feed gas with CO2 methanation. Chem. Eng. Sci. 2023, 274, 118692. [Google Scholar] [CrossRef]
- Tommasi, M.; Naz, S.; Ramis, G.; Rossetti, I. Advancements in CO2 methanation: A comprehensive review of catalysis, reactor design and process optimization. Chem. Eng. Res. Des. 2024, 201, 457–482. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. Bimetallic Ni-based catalysts for CO2 methanation: A review. Nanomaterials 2021, 11, 28. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Lv, C.; Xu, L.; Chen, M.; Cui, Y.; Wen, X.; Li, Y.; Wu, C.E.; Yang, B.; Miao, Z.; Hu, X.; et al. Recent Progresses in Constructing the Highly Efficient Ni Based Catalysts With Advanced Low-Temperature Activity Toward CO2 Methanation. Front. Chem. 2020, 8, 269. [Google Scholar] [CrossRef]
- Alexander, S. Comprehensive review of nickel-based catalysts advancements for CO2 methanation. 2025, 207, 114926. [Google Scholar] [CrossRef]
- Hussain, I.; Tanimu, G.; Ahmed, S.; Aniz, C.U.; Alasiri, H.; Alhooshani, K. A review of the indispensable role of oxygen vacancies for enhanced CO2 methanation activity over CeO2-based catalysts: Uncovering, influencing, and tuning strategies. Int. J. Hydrogen Energy 2023, 48, 24663–24696. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts. Appl. Catal. B Environ. 2020, 265, 118538. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, W.; Gu, F.; Xu, W.; Zhang, J.; Li, Z.; Zhu, T.; Xu, G.; Zhong, Z.; Su, F. Enhancing the low-temperature CO2 methanation over Ni/La-CeO2 catalyst: The effects of surface oxygen vacancy and basic site on the catalytic performance. Appl. Catal. B Environ. 2022, 312, 121385. [Google Scholar] [CrossRef]
- Liu, F.; Park, Y.S.; Diercks, D.; Kazempoor, P.; Duan, C. Enhanced CO2 Methanation Activity of Sm0.25Ce0.75O2-δ-Ni by Modulating the Chelating Agents-to-Metal Cation Ratio and Tuning Metal-Support Interactions. ACS Appl. Mater. Interfaces 2022, 14, 13295–13304. [Google Scholar] [CrossRef]
- Siakavelas, G.I.; Charisiou, N.D.; AlKhoori, S.; AlKhoori, A.A.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Yentekakis, I. V.; Polychronopoulou, K.; Goula, M.A. Highly selective and stable nickel catalysts supported on ceria promoted with Sm2O3, Pr2O3 and MgO for the CO2 methanation reaction. Appl. Catal. B Environ. 2021, 282, 119562. [Google Scholar] [CrossRef]
- Siakavelas, G.I.; Charisiou, N.D.; AlKhoori, A.; AlKhoori, S.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Yentekakis, I. V.; Polychronopoulou, K.; Goula, M.A. Highly selective and stable Ni/La-M (M=Sm, Pr, and Mg)-CeO2 catalysts for CO2 methanation. J. CO2 Util. 2021, 51, 101618. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; AlKhoori, A.; Gaber, S.; Stolojan, V.; Sebastian, V.; van der Linden, B.; Bansode, A.; Hinder, S.J.; Baker, M.A.; et al. Optimizing the oxide support composition in Pr-doped CeO2 towards highly active and selective Ni-based CO2 methanation catalysts. J. Energy Chem. 2022, 71, 547–561. [Google Scholar] [CrossRef]
- Hashimoto, N.; Mori, K.; Asahara, K.; Shibata, S.; Jida, H.; Kuwahara, Y.; Yamashita, H. How the Morphology of NiOx-Decorated CeO2 Nanostructures Affects Catalytic Properties in CO2 Methanation. Langmuir 2021, 37, 5376–5384. [Google Scholar] [CrossRef]
- Bian, Z.; Chan, Y.M.; Yu, Y.; Kawi, S. Morphology dependence of catalytic properties of Ni/CeO2 for CO2 methanation: A kinetic and mechanism study. Catal. Today 2020, 347, 31–38. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, J.; Chu, M.; Yue, J.; Cui, Y.; Xu, G. Enhanced Low-Temperature Activity of CO2 Methanation Over Ni/CeO2 Catalyst. Catal. Letters 2021, 152, 872–882. [Google Scholar] [CrossRef]
- Jomjaree, T.; Sintuya, P.; Srifa, A.; Koo-amornpattana, W.; Kiatphuengporn, S.; Assabumrungrat, S.; Sudoh, M.; Watanabe, R.; Fukuhara, C.; Ratchahat, S. Catalytic performance of Ni catalysts supported on CeO2 with different morphologies for low-temperature CO2 methanation. Catal. Today 2021, 375, 234–244. [Google Scholar] [CrossRef]
- Bian, Y.; Xu, C.; Wen, X.; Xu, L.; Cui, Y.; Wang, S.; Wu, C. e.; Qiu, J.; Cheng, G.; Chen, M. CO2 methanation over the Ni-based catalysts supported on nano-CeO2 with varied morphologies. Fuel 2023, 331, 125755. [Google Scholar] [CrossRef]
- Lin, S.; Li, Z.; Li, M. Tailoring metal-support interactions via tuning CeO2 particle size for enhancing CO2 methanation activity over Ni/CeO2 catalysts. Fuel 2023, 333, 126369. [Google Scholar] [CrossRef]
- Nguyen, T.; Long, B.; Anh, P.; Thuy, T.; Nguyen, V.; Anh, C. Nickel/ceria nanorod catalysts for the synthesis of substitute natural gas from CO2 : Effect of active phase loading and synthesis condition. J. Sci. Adv. Mater. Devices 2024, 9, 100752. [Google Scholar] [CrossRef]
- Yang, W.; Chang, K.; Yang, M.; Yan, X.; Yang, S.; Liu, Y. Facilitating CO2 methanation over oxygen vacancy-rich Ni/CeO2 : Insights into the synergistic effect between oxygen vacancy and metal-support interaction. Chem. Eng. J. 2024, 499, 156493. [Google Scholar] [CrossRef]
- Iglesias, I. Zr promotion effect in CO2 methanation over ceria-supported nickel catalysts. Int. J. Hydrogen Energy 2019, 44, 1710–1719. [Google Scholar] [CrossRef]
- Alcalde-Santiago, V.; Davó-Quiñonero, A.; Lozano-Castelló, D.; Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Ni/LnOx Catalysts (Ln=La, Ce or Pr) for CO2 Methanation. ChemCatChem 2019, 11, 810–819. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Harkou, E.; Hafeez, S.; Manos, G.; Constantinou, A.; Hussien, A.G.S.; Dabbawala, A.A.; Sebastian, V.; Hinder, S.J.; et al. Enhancing CO2 methanation over Ni catalysts supported on sol-gel derived Pr2O3-CeO2: An experimental and theoretical investigation. Appl. Catal. B Environ. 2022, 318, 121836. [Google Scholar] [CrossRef]
- Li, B.; Yuan, X.; Li, B.; Wang, X. Impact of pore structure on hydroxyapatite supported nickel catalysts (Ni/HAP) for dry reforming of methane. Fuel Process. Technol. 2020, 202, 106359. [Google Scholar] [CrossRef]
- Hongmanorom, P.; Ashok, J.; Chirawatkul, P.; Kawi, S. Interfacial synergistic catalysis over Ni nanoparticles encapsulated in mesoporous ceria for CO2 methanation. Appl. Catal. B Environ. 2021, 297, 120454. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Design of active sites in Ni/CeO2 catalysts for the methanation of CO2: tailoring the Ni-CeO2 contact. Appl. Mater. Today 2020, 19, 100591. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Hussien, A.G.S.; Sebastian, V.; Polychronopoulou, K.; Goula, M.A. Integrating capture and methanation of CO2 using physical mixtures of Na-Al2O3 and mono-/ bimetallic (Ru)Ni/Pr-CeO2. Chem. Eng. J. 2024, 491, 151962. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Italiano, C.; Ferrante, G.D.; Pino, L.; Vita, A.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Sharan, A.; et al. Ni-noble metal bimetallic catalysts for improved low-temperature CO2 methanation. Appl. Surf. Sci. 2024, 646, 158945. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Alkhoori, S.; Albedwawi, S.; Alareeqi, S.; Hussien, A.G.S.; Vasiliades, M.A.; Efstathiou, A.M.; Petallidou, K.C.; Singh, N.; Anjum, D.H.; et al. Decoupling the Chemical and Mechanical Strain Effect on Steering the CO2 Activation over CeO2-Based Oxides: An Experimental and DFT Approach. ACS Appl. Mater. Interfaces 2022, 14, 33094–33119. [Google Scholar] [CrossRef]
- Liu, K.; Xu, X.; Xu, J.; Fang, X.; Liu, L.; Wang, X. The distributions of alkaline earth metal oxides and their promotional effects on Ni/CeO2 for CO2 methanation. J. CO2 Util. 2020, 38, 113–124. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, L.; Zheng, Y.; Zhang, S.; Liu, Q.; Gao, G.; Dong, D.; Wang, Y.; Xu, L.; Hu, X. Methanation of CO2 over nickel catalysts: Impacts of acidic/basic sites on formation of the reaction intermediates. Fuel 2020, 262, 116521. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; Villar-Garcia, I.J.; Dieste, V.P.; Calvo, J.A.O.; Velasco, J.R.G.; Bueno-López, A. Monitoring by in situ NAP-XPS of active sites for CO2 methanation on a Ni/CeO2 catalyst. J. CO2 Util. 2022, 60, 101980. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Dou, B.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Ni/Y2O3–ZrO2 catalyst for hydrogen production through the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2020, 45, 10442–10460. [Google Scholar] [CrossRef]
- D’Angelo, A.M.; Chaffee, A.L. Correlations between Oxygen Uptake and Vacancy Concentration in Pr-Doped CeO2. ACS Omega 2017, 2, 2544–2551. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, L.; Feng, J.; Ju, X.; Wang, J.; He, T.; Chen, P. Ru Nanoparticles on Pr2O3 as an Efficient Catalyst for Hydrogen Production from Ammonia Decomposition. Catal. Letters 2022, 152, 1170–1181. [Google Scholar] [CrossRef]
- Borchert, H.; Frolova, Y. V.; Kaichev, V. V.; Prosvirin, I.P.; Alikina, G.M.; Lukashevich, A.I.; Zaikovskii, V.I.; Moroz, E.M.; Trukhan, S.N.; Ivanov, V.P.; et al. Electronic and chemical properties of nanostructured cerium dioxide doped with praseodymium. J. Phys. Chem. B 2005, 109, 5728–5738. [Google Scholar] [CrossRef]
- Li, L.; Jiang, L.; Li, D.; Yuan, J.; Bao, G. Enhanced low-temperature activity of CO2 methanation over Ni/CeO2 catalyst : Influence of preparation methods. Appl. Catal. O Open 2024, 192, 206956. [Google Scholar] [CrossRef]
- Lin, S.; Tang, R.; Liu, X.; Gong, L.; Li, Z. Modulating CO2 methanation activity on Ni/CeO2 catalysts by tuning ceria facet-induced metal-support interaction. Int. J. Hydrogen Energy 2024, 51, 462–475. [Google Scholar] [CrossRef]
- Varvoutis, G.; Lykaki, M.; Stefa, S.; Binas, V.; Marnellos, G.E.; Konsolakis, M. Deciphering the role of Ni particle size and nickel-ceria interfacial perimeter in the low-temperature CO2 methanation reaction over remarkably active Ni/CeO2 nanorods. Appl. Catal. B Environ. 2021, 297, 120401. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The role of alkali and alkaline earth metals in the CO2 methanation reaction and the combined capture and methanation of CO2. Catalysts 2020, 10, 812. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, T.W.; Lee, S.H.; Park, E.D. Effects of Na content in Na/Ni/SiO2 and Na/Ni/CeO2 catalysts for CO and CO2 methanation. Catal. Today 2018, 303, 159–167. [Google Scholar] [CrossRef]
- Wu, H.C.; Chen, T.C.; Wu, J.H.; Pao, C.W.; Chen, C.S. Influence of sodium-modified Ni/SiO2 catalysts on the tunable selectivity of CO2 hydrogenation: Effect of the CH4 selectivity, reaction pathway and mechanism on the catalytic reaction. J. Colloid Interface Sci. 2021, 586, 514–527. [Google Scholar] [CrossRef]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure-sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- Du, Y.; Qin, C.; Xu, Y.; Xu, D.; Bai, J.; Ma, G.; Ding, M. Ni nanoparticles dispersed on oxygen vacancies-rich CeO2 nanoplates for enhanced low-temperature CO2 methanation performance. Chem. Eng. J. 2021, 418. [Google Scholar] [CrossRef]
- Luisetto, I.; Stendardo, S.; Senthil Kumar, S.M.; Selvakumar, K.; Kesavan, J.K.; Iucci, G.; Pasqual Laverdura, U.; Tuti, S. One-pot synthesis of Ni0.05Ce0.95O2−δ catalysts with nanocubes and nanorods morphology for CO2 methanation reaction and in operando DRIFT analysis of intermediate species. Processes 2021, 9, 1899. [Google Scholar] [CrossRef]
- Lin, S.; Hao, Z.; Shen, J.; Chang, X.; Huang, S.; Li, M.; Ma, X. Enhancing the CO2 methanation activity of Ni/CeO2 via activation treatment-determined metal-support interaction. J. Energy Chem. 2021, 59, 334–342. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffractograms of (a) the metal oxide supports and (b) the reduced Ni-supported catalysts. N2 physisorption isotherms along with pore size distribution (inset) of (c) the metal oxide supports and (d) the reduced Ni-supported catalysts.
Figure 1.
X-ray diffractograms of (a) the metal oxide supports and (b) the reduced Ni-supported catalysts. N2 physisorption isotherms along with pore size distribution (inset) of (c) the metal oxide supports and (d) the reduced Ni-supported catalysts.
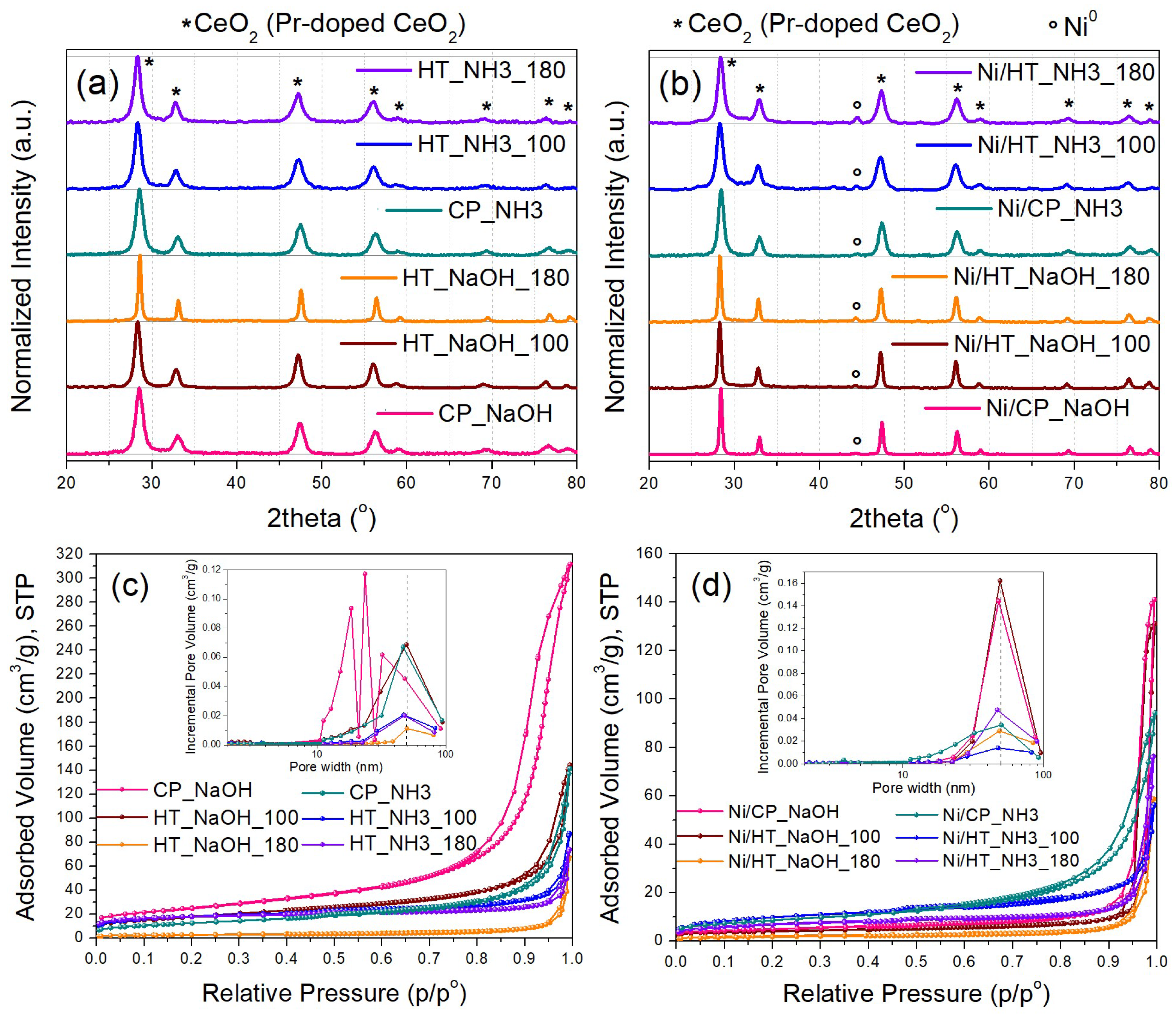
Figure 2.
(a) H2-TPR profiles of the calcined catalysts. (b) CO2-TPD profiles of the reduced catalysts.
Figure 2.
(a) H2-TPR profiles of the calcined catalysts. (b) CO2-TPD profiles of the reduced catalysts.
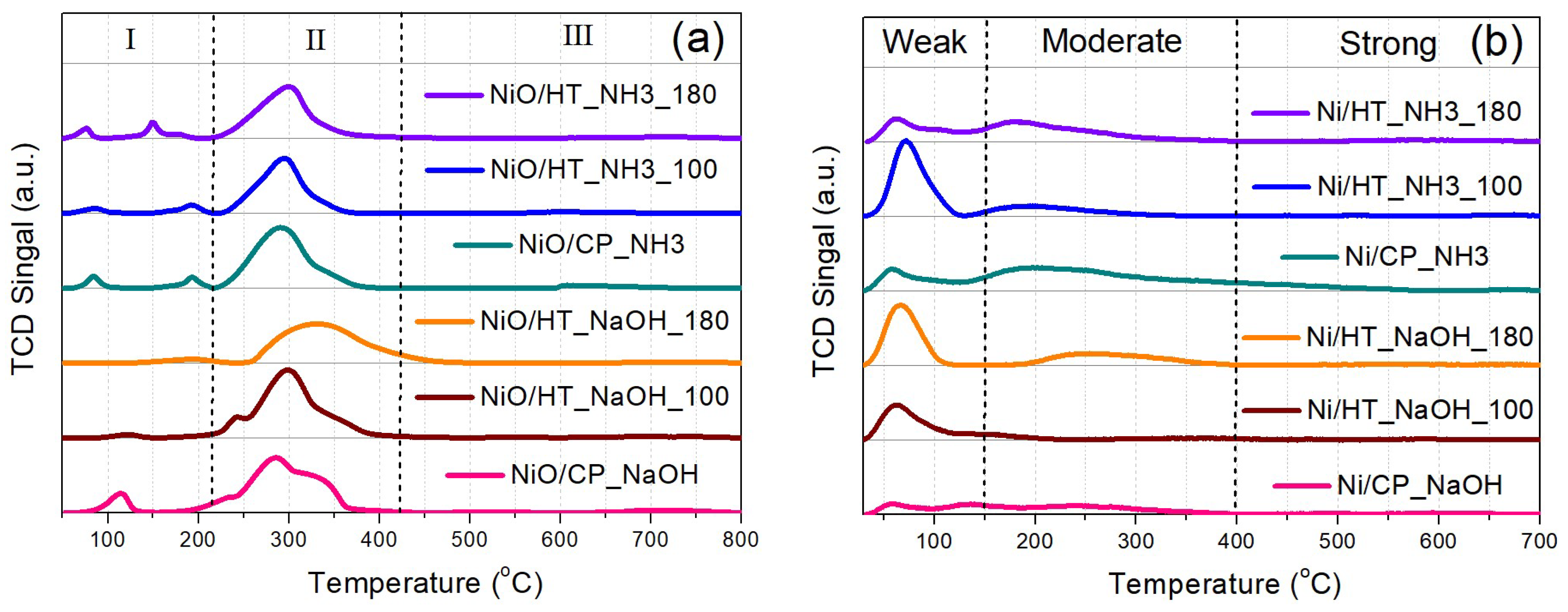
Figure 3.
(a) Ni2p, (b) O1s, (c) Ce3d, and (d) Pr3d XPS core level spectra for the reduced catalysts.
Figure 3.
(a) Ni2p, (b) O1s, (c) Ce3d, and (d) Pr3d XPS core level spectra for the reduced catalysts.
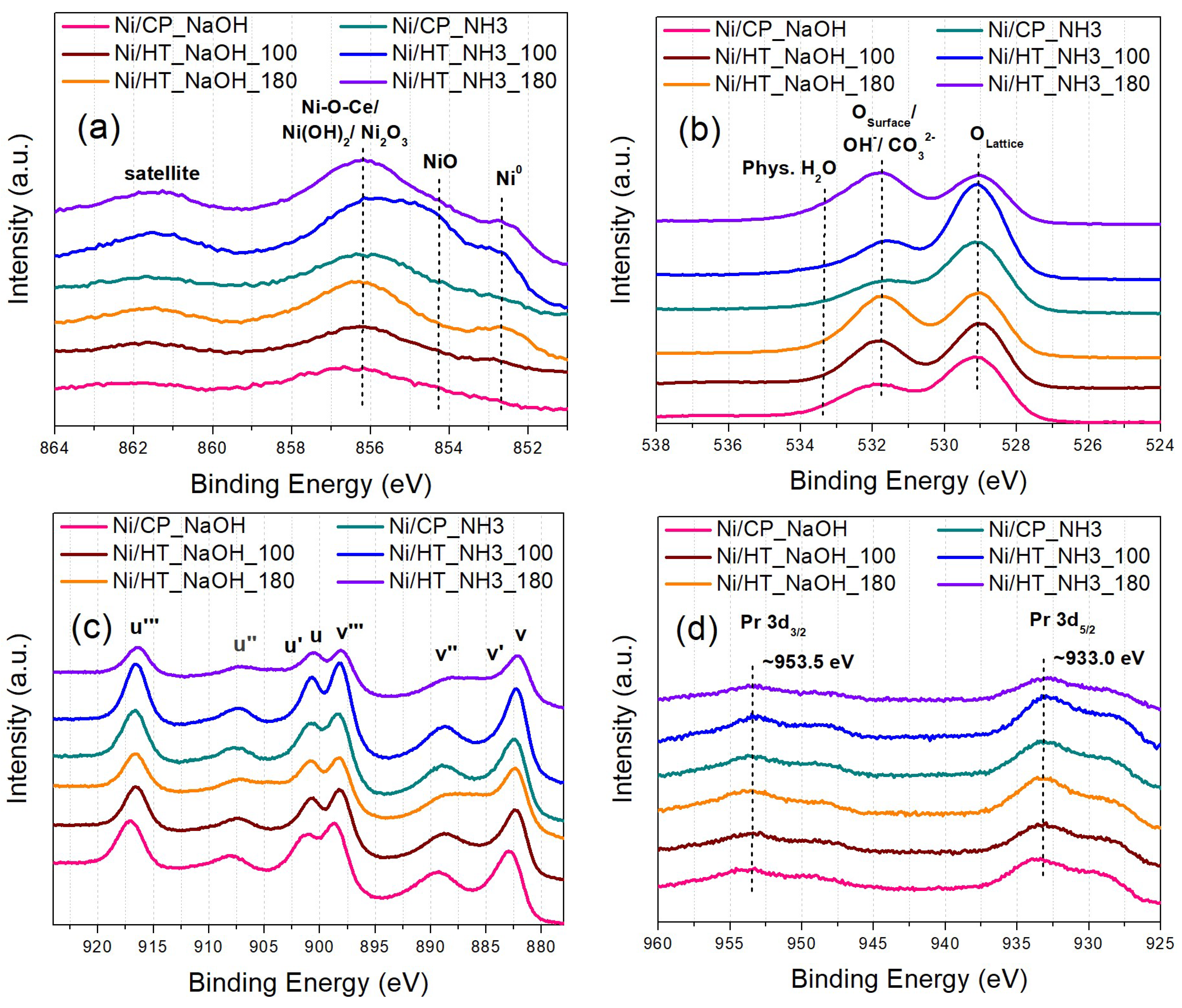
Figure 4.
TEM images of the (a) Ni/CP_NaOH, (b) Ni/HT_NaOH_100, (c) Ni/HT_NaOH_180, (d) Ni/CP_NH3, (e) Ni/HT_NH3_100, and (f) Ni/HT_NH3_180 reduced Ni-supported catalysts. The red circles indicate the location of some Ni nanoparticles.
Figure 4.
TEM images of the (a) Ni/CP_NaOH, (b) Ni/HT_NaOH_100, (c) Ni/HT_NaOH_180, (d) Ni/CP_NH3, (e) Ni/HT_NH3_100, and (f) Ni/HT_NH3_180 reduced Ni-supported catalysts. The red circles indicate the location of some Ni nanoparticles.

Figure 5.
HAADF-STEM EDS elemental mapping images and EDS elemental mapping for Ni of the (a) Ni/CP_NaOH, (b) Ni/HT_NaOH_100, (c) Ni/HT_NaOH_180, (d) Ni/CP_NH3, (e) Ni/HT_NH3_100, and (f) Ni/HT_NH3_180 reduced Ni-supported catalysts.
Figure 5.
HAADF-STEM EDS elemental mapping images and EDS elemental mapping for Ni of the (a) Ni/CP_NaOH, (b) Ni/HT_NaOH_100, (c) Ni/HT_NaOH_180, (d) Ni/CP_NH3, (e) Ni/HT_NH3_100, and (f) Ni/HT_NH3_180 reduced Ni-supported catalysts.
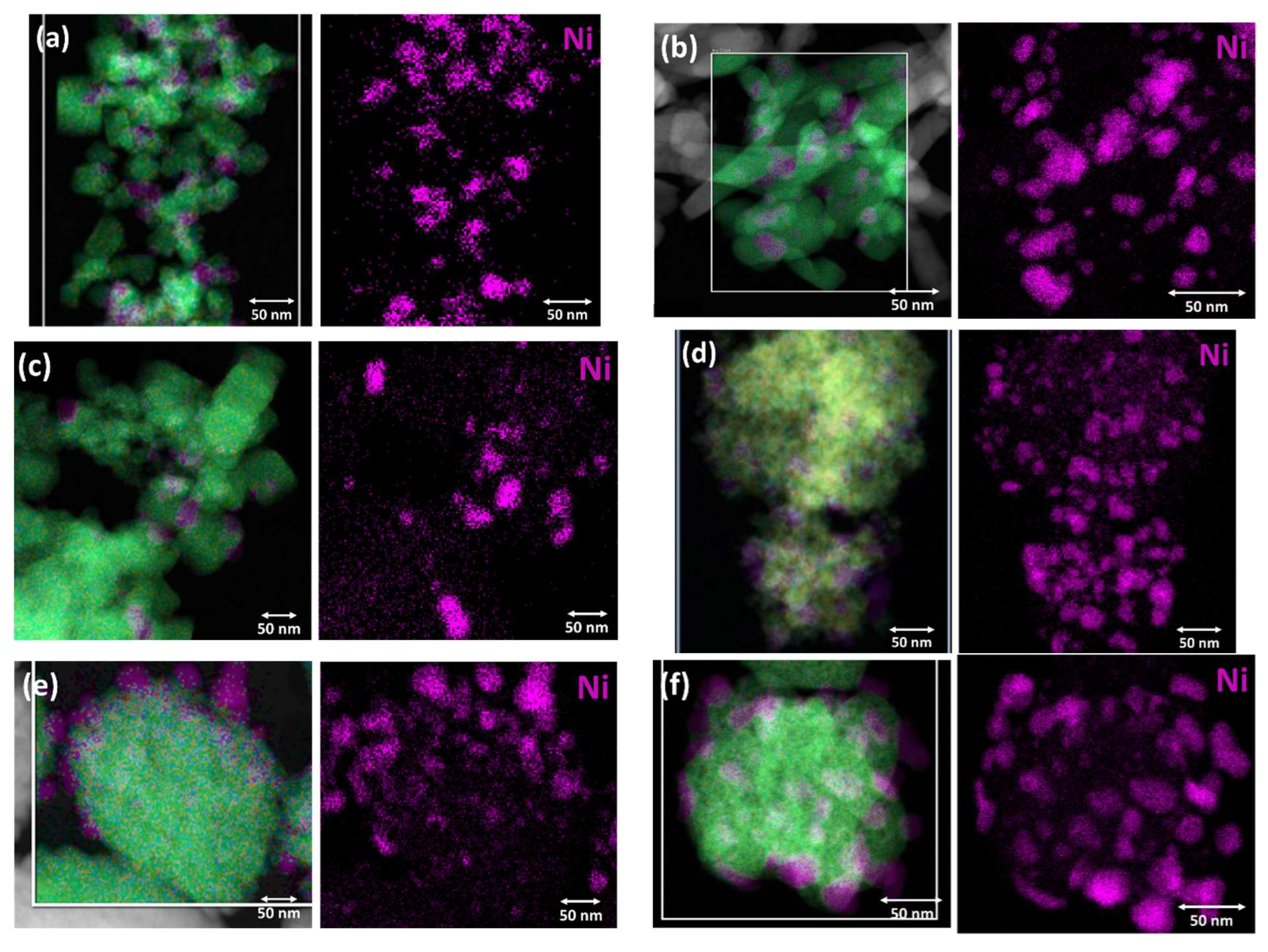
Figure 6.
(a) CO2 conversion and (b) CH4 selectivity as a function of reaction temperature (Experimental Protocol #1). The thermodynamic equilibrium (dotted lines) is calculated via Aspen Plus (p = 1 atm and H2:CO2 = 4:1).
Figure 6.
(a) CO2 conversion and (b) CH4 selectivity as a function of reaction temperature (Experimental Protocol #1). The thermodynamic equilibrium (dotted lines) is calculated via Aspen Plus (p = 1 atm and H2:CO2 = 4:1).
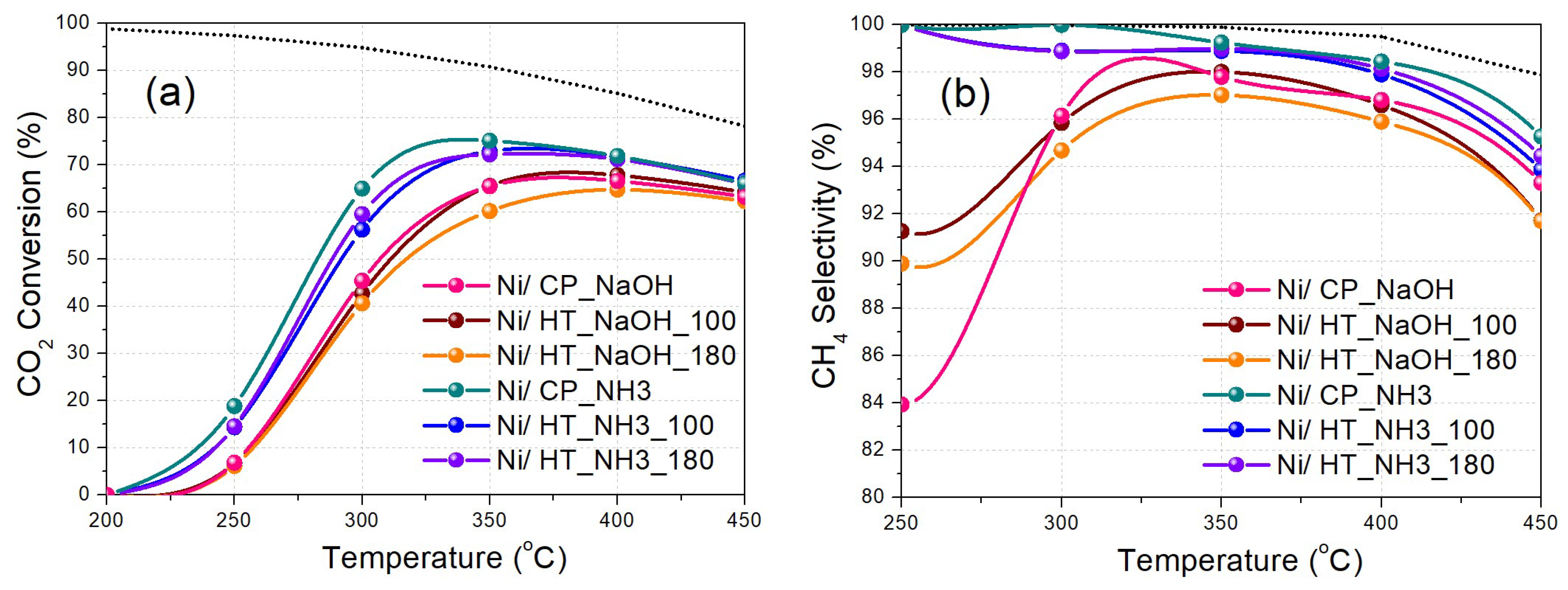
Figure 7.
(a) CO2 conversion and (b) CH4 selectivity as a function of reaction temperature (Experimental Protocol #2). (c) Arrhenius plots (logarithm of the CO2 consumption rate vs 1000/T). The thermodynamic equilibrium (dotted lines) is calculated via Aspen Plus (p = 1 atm and H2:CO2 = 4:1).
Figure 7.
(a) CO2 conversion and (b) CH4 selectivity as a function of reaction temperature (Experimental Protocol #2). (c) Arrhenius plots (logarithm of the CO2 consumption rate vs 1000/T). The thermodynamic equilibrium (dotted lines) is calculated via Aspen Plus (p = 1 atm and H2:CO2 = 4:1).
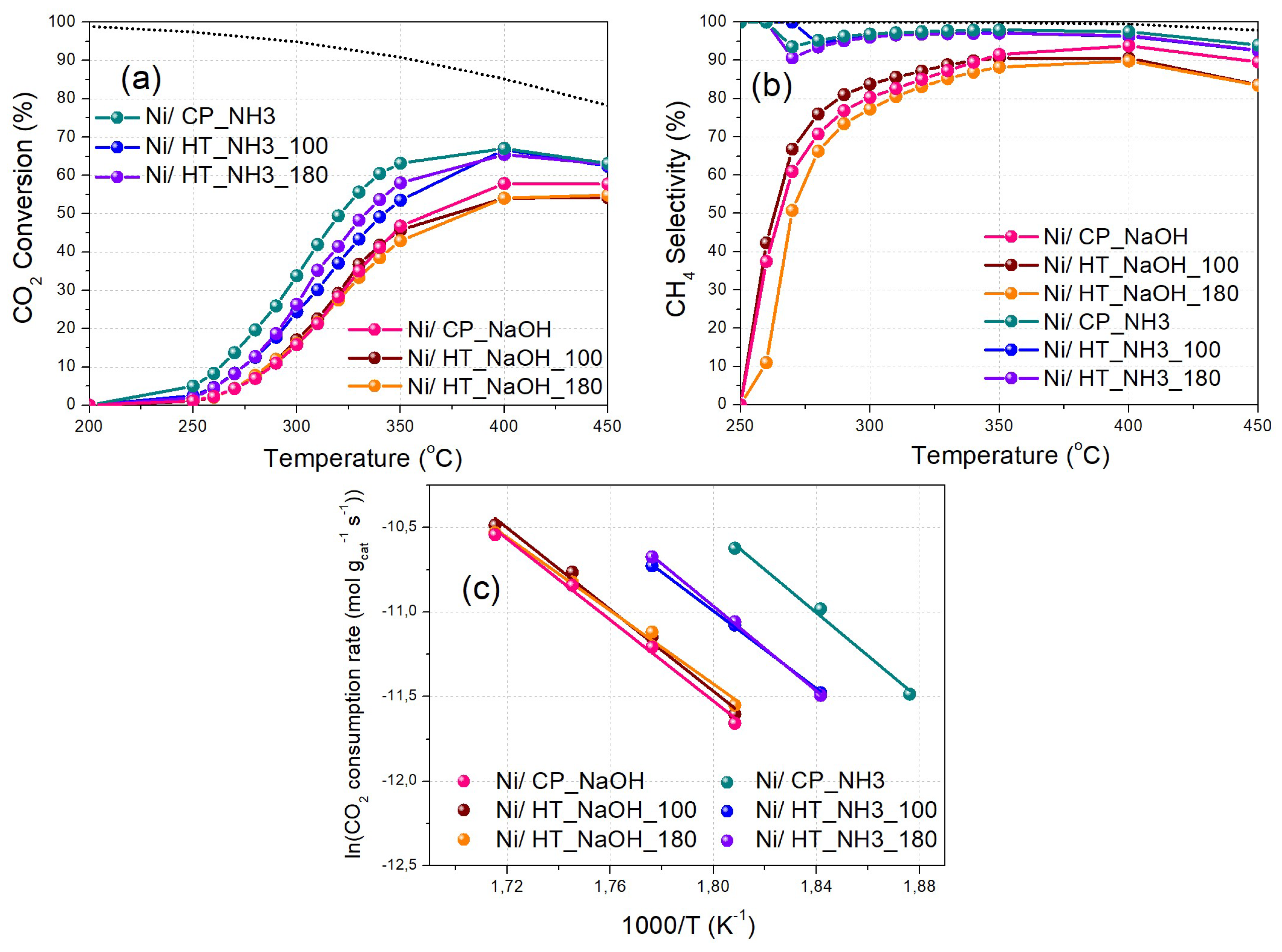
Figure 8.
(a) Time-on-stream catalytic stability for Ni/CP_NH3 at 350 oC for 24 h (Experimental Protocol #3). (b) X-ray diffractograms and (c) N2 physisorption isotherms and pore size distribution graphs (inset) of the fresh (reduced) and spent Ni/CP_NH3 catalysts. (d) TEM image of spent Ni/CP_NH3. The red circles indicate the location of some Ni nanoparticles.
Figure 8.
(a) Time-on-stream catalytic stability for Ni/CP_NH3 at 350 oC for 24 h (Experimental Protocol #3). (b) X-ray diffractograms and (c) N2 physisorption isotherms and pore size distribution graphs (inset) of the fresh (reduced) and spent Ni/CP_NH3 catalysts. (d) TEM image of spent Ni/CP_NH3. The red circles indicate the location of some Ni nanoparticles.
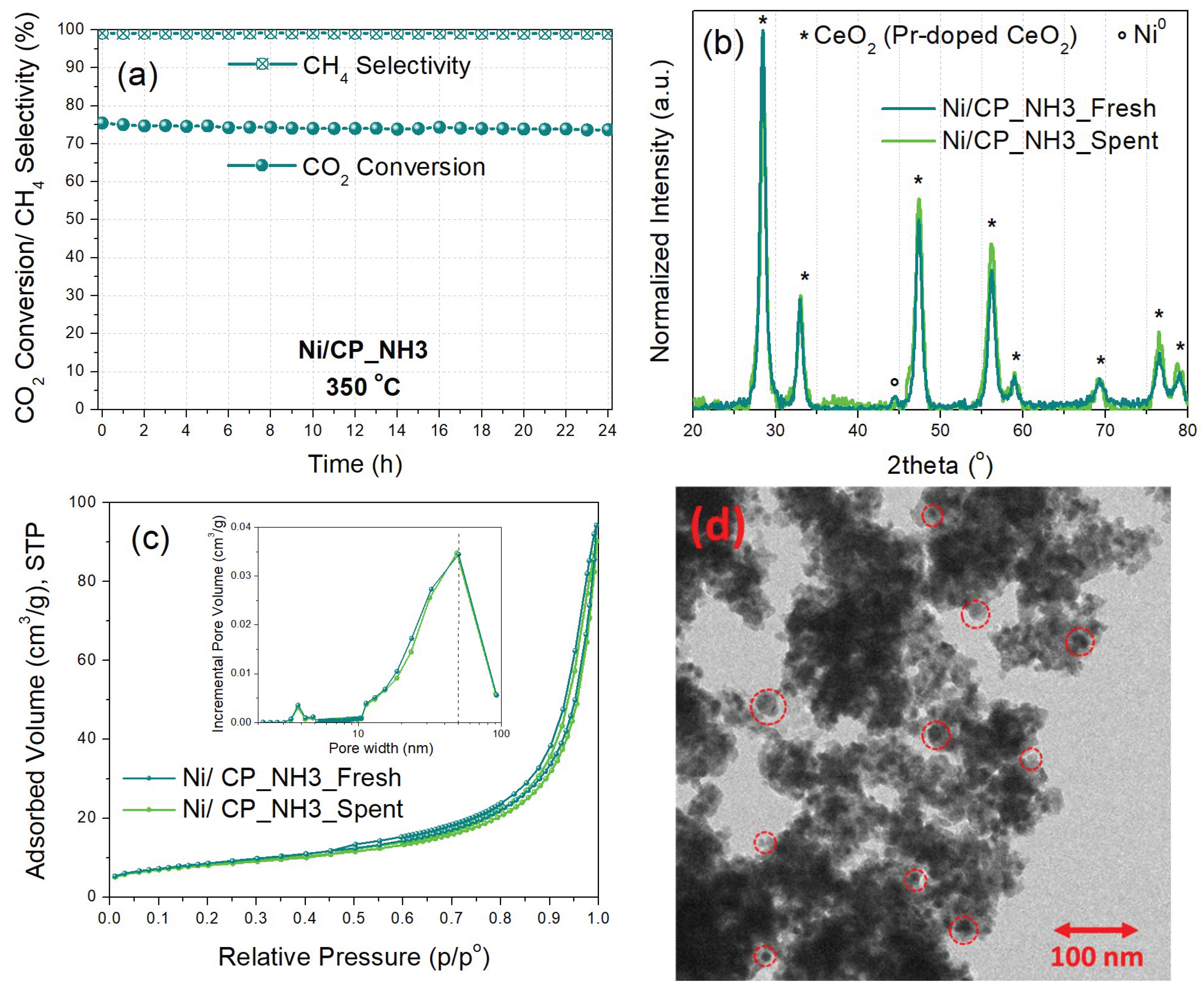

Table 1.
Overview of the different precipitating agents and hydrothermal treatments used to prepare each Pr-doped CeO2 metal oxide support.
Table 1.
Overview of the different precipitating agents and hydrothermal treatments used to prepare each Pr-doped CeO2 metal oxide support.
| Catalyst support | Precipitating agent | Hydrothermal treatment temperature |
|---|---|---|
| CP_NaOH | NaOH | R.T. 1 (Co-precipitation) |
| HT_NaOH_100 | NaOH | 100 oC |
| HT_NaOH_180 | NaOH | 180 oC |
| CP_NH3 | NH3/ (NH4)2CO3 | R.T. 1 (Co-precipitation) |
| HT_NH3_100 | NH3/ (NH4)2CO3 | 100 oC |
| HT_NH3_180 | NH3/ (NH4)2CO3 | 180 oC |
1 R.T. = Room Temperature.
Table 2.
Crystallite sizes of CeO2 (Pr-doped CeO2, ΦCeO2) and Ni0 (ΦNi) calculated from XRD through the Scherrer equation. Specific surface area (SSA), pore volume (VP), and average pore diameter (Dave) determined via N2 physisorption. The values are for the reduced Ni-supported catalysts, while those for the respective metal oxide supports can be found in parentheses.
Table 2.
Crystallite sizes of CeO2 (Pr-doped CeO2, ΦCeO2) and Ni0 (ΦNi) calculated from XRD through the Scherrer equation. Specific surface area (SSA), pore volume (VP), and average pore diameter (Dave) determined via N2 physisorption. The values are for the reduced Ni-supported catalysts, while those for the respective metal oxide supports can be found in parentheses.
| Catalyst | (nm) | (nm) | SSA (m2/g) | (cm3/g) | |
|---|---|---|---|---|---|
| Ni/CP_NaOH | 22 (8) | 15 | 17 (91) | 0.22 (0.48) | 50 (21) |
| Ni/HT_NaOH_100 | 17 (10) | 8 | 13 (63) | 0.20 (0.22) | 61 (14) |
| Ni/HT_NaOH_180 | 20 (20) | 14 | 8 (10) | 0.09 (0.10) | 48 (38) |
| Ni/CP_NH3 | 11 (8) | 11 | 31 (46) | 0.14 (0.21) | 19 (18) |
| Ni/HT_NH3_100 | 9 (9) | 13 | 35 (61) | 0.08 (0.12) | 10 (8) |
| Ni/HT_NH3_180 | 10 (9) | 13 | 24 (58) | 0.12 (0.11) | 19 (7) |
Table 3.
CO2 methanation catalytic performance metrics at 350 oC, and in parenthesis at 300 oC, calculated via Experimental Protocol #1. Activation energy values calculated via Experimental Protocol #2.
Table 3.
CO2 methanation catalytic performance metrics at 350 oC, and in parenthesis at 300 oC, calculated via Experimental Protocol #1. Activation energy values calculated via Experimental Protocol #2.
| Catalyst | CO2 Conversion (%) | CH4 Selectivity (%) | CH4 Yield (%) | Activation Energy (kJ/mol) |
|---|---|---|---|---|
| Ni/CP_NaOH | 65 (45) | 98 (96) | 64 (44) | 101 |
| Ni/HT_NaOH_100 | 66 (43) | 98 (96) | 64 (41) | 100 |
| Ni/HT_NaOH_180 | 60 (41) | 97 (95) | 58 (38) | 91 |
| Ni/CP_NH3 | 75 (65) | 99 (100) | 75 (65) | 106 |
| Ni/HT_NH3_100 | 73 (56) | 99 (99) | 72 (56) | 95 |
| Ni/HT_NH3_180 | 72 (59) | 99 (99) | 72 (59) | 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



