Submitted:
18 May 2025
Posted:
19 May 2025
Read the latest preprint version here
Abstract
The investigation of alternatives for the management of breast cancer (BC), has led to the study of extracts of plant origin and their compounds, within these, the extracts of oregano variants and carvacrol (Cv) have shown promising results, however, the Mexican oregano (L. graveolens) infusion (MoI) has not been studied, so the aims of this work are; to characterize the plant used, and to compare against Cv, the impact on the metabolism and cytotoxicity of BC cell lines. Within the characterization of the plant, moisture, ash, protein, available carbohydrates, ethereal extraction and dietary fiber were determined, as well as antioxidant capacity analysis; ABTS, DPPH, phenols, flavonoids and Ferric reducing antioxidant power (FRAP) and Fourier-transform infrared spectroscopy (FTIR), as well as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays (MTT) and lactate dehydrogenase (LDH) determination. The results demonstrated that MoI possesses potent antioxidant activity and antiproliferative effect against BC cell lines, with lower cytotoxicity on non-cancer cells compared to Cv. However, both the infusion and Cv caused a reduction in cell metabolism, with Cv having the most acute effect. Thus, the present investigation provides evidence on the efficacy of these alternative treatments in BC cell lines, including aggressive subtypes such as triple negative (TN).
Keywords:
Mexican oregano
; anticancer agents
; alternative treatments
; breast cancer triple negative
; antioxidant capacity
; plant extracts
; isolated compounds
1. Introduction
Cancer is a broad term encompassing a group of diseases characterized by uncontrolled growth and proliferation of abnormal cells, resulting from genetic or epigenetic alterations that disrupt cell cycle regulation. These malignant cells can invade adjacent tissues and organs and, in advanced stages, metastasize to distant sites through the bloodstream or lymphatic system. BC originates in mammary tissues and primarily affects the lobules, responsible for milk production, the ducts that transport milk to the nipple, or the connective tissue that provides structural support. Most cases arise in the ducts or lobules and have the potential to spread via blood and lymphatic vessels. According to the Global Cancer Observatory, BC accounted for 2,296,840 cases worldwide in 2022, representing 11.5% of all cancer diagnoses. It was also the leading cause of cancer-related mortality among women, with 666,103 deaths (15.4%). These statistics underscore its significant global burden [1,2,3].
In 2000, Perou et al. proposed a molecular classification of BC based on the expression of three key receptors critical for conventional treatment efficacy: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). This classification defines four molecular subtypes with distinct therapeutic responses: Luminal A (ER+ and PR+/-), Luminal B (ER+, PR+/- and HER2+), HER2-enriched (HER2+), and TN (ER-, PR- and HER2-) [4]. Among these, TN is clinically recognized as the most aggressive subtype due to its poor response to conventional therapies, leading to lower overall survival and disease-free survival rates [4,5,6].
Natural products have played a fundamental role in traditional medicine throughout history. Since the World Health Organization endorsed the use of complementary therapies in 2004, interest in herbal medicine and the development of phytochemical-based anticancer agents has grown significantly. Bioactive compounds derived from plants have gained recognition for their potential in cancer treatment [7,8,9]. Cv is a monoterpenoid phenol synthesized via the mevalonate pathway from acetyl-coenzyme A, which has attracted attention for its anticancer properties. It one of the predominant compounds in oregano extracts [10,11,12]. Lippia graveolens, commonly known as Mexican oregano, is used as condiment of many traditional Mexican dishes as well as raw material to produce cosmetics, drugs, and liquors. Oregano variants have been reported to have antioxidant potential and anti-inflammatory activity; these properties are attributed to their phytochemical profile, mainly to its phenolic compounds [13, 14].
However, limited evidence exists regarding the composition, antioxidant capacity, and anticancer potential of aqueous extracts from oregano variants, leaving a gap in knowledge about the bioactivity of MoI. Therefore, the aims of this study was, first, to characterize an endemic species of Mexican oregano (Lippia graveolens) from Totatiche, Jalisco, Mexico, as well as to evaluate the antioxidant capacity of the MoI prepared with this plant sample with different tests (ABTS, DPPH, total phenols, total flavonoids and FRAP) and second, to compare the anticancer effects of Cv and the MoI at the level of cell metabolism and cytotoxicity against BC cell lines, luminal A (MCF-7), HER2-enriched (HCC-1954) and TN (MDA-MB-231), also, these tests were performed on HaCaT cells (immortalized keratinocytes that do not express oncogenes) to evaluate the effects of both treatments on non-cancerous cells.
2. Results
2.1. Bromatological analysis of L. graveolens
Table 1 shows the results obtained from the bromatological analysis of the L. graveolens sample.
2.2. Antioxidant capability of MoI
We found an important antioxidant capacity in the Mol, the data shows in the Table 2.
2.3. FTIR Measurement
The FTIR spectrum of MoI exhibits characteristic absorption bands corresponding to its diverse phytochemical constituents. The broad absorption centered around 3300 cm⁻¹ is attributed to O-H stretching vibrations, confirming the presence of hydroxyl groups, which are commonly associated with polyphenols, flavonoids, and other bioactive compounds. The peak at ~2900 cm⁻¹ corresponds to C-H stretching, indicative of aliphatic hydrocarbons. A sharp and well-defined band around ~1700 cm⁻¹ suggests the presence of carbonyl (C=O) groups, likely from carboxylic acids, aldehydes, or ketones, which are essential components of organic acids and flavonoids.
The region between 1500–1200 cm⁻¹ displays peaks that correspond to aromatic C=C stretching, suggesting the presence of phenolic compounds. Additionally, intense peaks in the 1200–1000 cm⁻¹ range confirm the presence of C-O stretching, typically found in esters, alcohols, and glycosides. Below 1000 cm⁻¹, the complex peaks in the fingerprint region suggest contributions from terpenoids and polysaccharides. A notable absorption around 950 cm⁻¹ may correspond to out-of-plane bending of =C-H, which is characteristic of aromatic and conjugated systems, reinforcing the presence of flavonoids and other bioactive molecules in the L. graveolens extract. These results confirm that MoI contains a combination of phenols, terpenoids and carboxylic acids, which reinforces its potential in pharmaceutical, antioxidant and antimicrobial applications (Figure 1). The stretching and bending vibrations characteristic of the L. graveolens secondary metabolites is observed as reported by Soto et al. 2024 [15].
2.4. MoI dose response curve
Treatment with MoI for 24 hours does not modify the metabolic activity of MDA-MB-231 cells. Instead, the infusion significantly decreases formazan production after 48 hours (0.08 mg/mL, 83.09% ± 5.17%; 0.09 mg/mL, 88.20% ± 1.81%; and 0.1 mg/mL, 82.66% ± 5.68%) and this effect is similar at 72 hours (0.08 mg/mL, 86.23% ± 3.48%; 0.09 mg/mL, 90.65% ± 1.31% and 0.10 mg/mL, 89.88% ± 2.63%). These results are shown in Figure 2.
2.5. Cell metabolism assay
Treatment of HaCaT cells with MoI for 48 hours resulted in a significant reduction in formazan production starting at 0.10 mg/mL (84.51% ± 4.50%), following a dose-dependent trend (0.15 mg/mL, 78.58% ± 6.06%; 0.20 mg/mL, 36.65% ± 6.38%; 0.30 mg/mL, 9.50% ± 3.82%). In a similar manner, MoI treatment of MCF-7 cells also led to a significant decrease in formazan production from the 0.10 mg/mL dose (34.67% ± 6.31%), exhibiting a dose-dependent effect (0.15 mg/mL, 15.49% ± 3.91%; 0.20 mg/mL, 15.90% ± 4.02%; 0.30 mg/mL, 13.70% ± 3.91%). Moreover, in HCC-1954 cells, MoI significantly decreased formazan production after 48 hours, starting from the second dose (85.90% ± 4.10%), in a dose-dependent manner (0.15 mg/mL, 74.56% ± 3.59%; 0.20 mg/mL, 47.60% ± 6.92%; 0.30 mg/mL, 48.48% ± 5.91%). Finally, treatment of MDA-MB-231 cells with MoI for 48 hours also resulted in a significant reduction in formazan production from the second dose onwards (74.71% ± 5.36%), following a dose-dependent pattern (0.15 mg/mL, 73.66% ± 8.59%; 0.20 mg/mL, 49.67% ± 6.01%; 0.30 mg/mL, 41.56% ± 5.89%). These results are shown in Figure 3.
Treatment of HaCaT cells with Cv for 24 hours resulted in a significant reduction in formazan production from the first dose (54.49% ± 6.42%), exhibiting a dose-dependent response (100 µM, 44.70% ± 4.83%; 200 µM, 25.45% ± 3.59%; 300 µM, 22.61% ± 4.15%; 500 µM, 26.85% ± 4.05%). Similarly, MCF-7 cells demonstrated a significant decrease in formazan production starting at the initial dose (81.00% ± 5.86%), following a dose-dependent pattern (100 µM, 81.13% ± 6.04%; 200 µM, 60.85% ± 4.94%; 300 µM, 32.54% ± 3.37%; 500 µM, 27.55% ± 2.77%). In HCC-1954 cells, Cv treatment for 24 hours significantly reduced formazan production from the first dose onward (85.18% ± 5.59%), with a clear dose-dependent effect (100 µM, 57.04% ± 7.61%; 200 µM, 40.95% ± 3.04%; 300 µM, 40.46% ± 2.68%; 500 µM, 40.76% ± 3.23%). Likewise, MDA-MB-231 cells treated under the same conditions exhibited a significant reduction from the first dose (85.76% ± 4.14%), maintaining a dose-dependent trend (100 µM, 80.21% ± 4.17%; 200 µM, 44.52% ± 4.62%; 300 µM, 25.78% ± 4.51%; 500 µM, 25.15% ± 3.25%). These results are shown in Figure 4.
2.6. IC50 calculation
The IC50 values calculated for each cell line under each treatment are presented in Table 3.
2.7. Cytotoxicity of MoI and Cv (IC50)
As shown in Figure 5 the IC50 Cv concentrations induced approximately 50% cytotoxicity, with values of 49.85% ± 6.53% in MDA-MB-231 cells, 51.26% ± 9.42% in HCC-1954 cells, 53.84% ± 8.59% in MCF-7 cells, and 57.66% ± 10.05% in HaCaT cells. In contrast, the IC50 concentrations of MoI resulted in a lower cytotoxicity of approximately 15%, with specific values of 11.68% ± 1.97% in MDA-MB-231 cells, 21.94% ± 3.25% in HCC-1954 cells, 15.43% ± 0.15% in MCF-7 cells, and 12.07% ± 2.08% in HaCaT cells.
3. Discussion
Both oregano extracts and Cv have been extensively studied due to their diverse biological activities, which postulates them as potential therapeutic agents for the management of different diseases, including cancer. Among their properties, their antioxidant activity stands out, as well as their antiproliferative and cytotoxic effects, characteristics that make them promising options for oncological treatment [16,17,18].
The bromatological analysis performed on the plant sample used in this study can be compared with the data reported by the U.S. Department of Agriculture (USDA) for dried oregano. Compared to this reference, our sample presented 12.3 % less moisture, 15.3 % less ash, 24 % less protein, 2.3 % less lipids, but an increase of 51.9 % in available carbohydrates and 70.1 % in total sugars, with a reduction of 9.1 % in dietary fiber. These differences can be attributed to several factors, such as soil type and fertility, pH, growing conditions (temperature, light, humidity), irrigation regime, fertilization, pest control, harvest timing and drying methods. Each of these factors significantly influences the chemical composition of the plant sample and, consequently, its biological activity [19].
Regarding the antioxidant capacity of L. graveolens, it has been demonstrated that both the plant matrix and its extracts are rich in antioxidant compounds, which confers a high capacity to neutralize free radicals [17, 20]. The results obtained in the present study support this claim, since it was observed that MoI exhibited potent total antioxidant activity in ABTS and DPPH radical neutralization assays, as well as in the quantification of total phenols, total flavonoids and FRAP. Similar results have been reported in the literature. Mahomoodally et al. (2018) evaluated the antioxidant capacity of aqueous and methanolic extracts of Origanum onites (Greek oregano), finding that the aqueous extract presented a higher antioxidant capacity compared to the methanolic one, which was attributed to a higher concentration of phenolic compounds [21]. In contrast, Simirgiotis et al. (2020) analyzed a lipid extract of Origanum vulgare (oregano species native to the mediterranean), finding a lower amount of phenolic compounds and, consequently, a lower antioxidant capacity against ABTS and DPPH radicals [22]. Similarly, Kogiannou et al. (2013) compared the antioxidant capacity of 6 herbal infusions of plants originating from Greece, including a species of oregano, where they observed that this infusion had the best antioxidant capacity in all the techniques they used (total phenols, total flavonoids, DPPH and FRAP), compared to other plants with recognized antioxidant capacity such as chalk marjoram, pink savory, mountain tea, pennyroyal and chamomile [23]. These findings reinforce the evidence that extracts from oregano variants, especially in infusion form, have a high antioxidant potential. The antioxidant potential of MoI could be related to its anticancer activity, since there is a correlation between the amount and type of phenolic compounds and their cytotoxicity. It has been proposed that certain functional groups, such as carbonyls and free hydroxyls, facilitate the formation of ortho-diphenolic radicals (two hydroxyl groups attached to a benzene ring in adjacent positions), which could chelate transition metals involved in redox processes, thus affecting the viability of cancer cells [24, 25]. This suggests the need to continue investigating their role in the management of chronic degenerative diseases, such as BC.
On the other hand, treatment with Cv showed a differential impact on the metabolic activity of the different cell lines used in this study. It was observed that the MDA-MB-231 cell line (TN BC) was more susceptible to treatment compared to the HCC-1954 (HER2+) and MCF-7 (luminal A) lines, with IC50 values of 121 µM, 123 µM and 211 µM, respectively. However, the control cell line (HaCaT) presented higher susceptibility to Cv, with an IC50 of 91 µM, highlighting the importance of including non-cancer cell models to assess treatment specificity. These findings are consistent with previous studies. Elshafie et al. (2017) evaluated the metabolic activity of a hepatocellular carcinoma cell line (HepG2) treated with the Origanum vulgare lead compounds (Cv, thymol, and citral). To evaluate this activity, they performed MTT assays and found that Cv was the compound with the highest potential to reduce metabolic activity, with an IC50 of 48 mg/L. Furthermore, they observed that the metabolic activity of the control line (HEK293; non-cancerous human kidney cells) was considerably higher (38%) than that exhibited by HepG2 cells (12%). These findings support the idea that Cv, besides being the most abundant compound in oregano variants, also exhibits a remarkable cytotoxic potential against cancer cells, even becoming the most effective compound among those present in the plant against the HepG2 line [16]. Both previous research and the results of this study agree that the ability of Cv to decrease cell viability in cancer cells is significant in both hepatocellular carcinoma cells and aggressive BC subtypes. Further support for the cytotoxic potential of Cv against cancer cells was provided by Elbe et al. in 2020, who subjected ovarian cancer cells (Skov-3) to different doses of thymol and Cv. The results, analyzed by MTT assays, revealed that both compounds exhibit a potent effect and that this occurs in a dose-dependent manner, with IC50 values of 316.08 µM for thymol and 322.50 µM for Cv after 24 hours of treatment. Although the authors reported that thymol showed a higher cytotoxic potential than Cv in this cell line, both compounds are considered interesting options for the development of adjuvant cancer treatments [26]. These results contrast with those presented by Elshafie et al. in 2017, who found that Cv exhibited greater cytotoxic potential compared to other major Origanum vulgare compounds, including thymol [16]. The differences may be attributed to the different cell lines used in each study. Despite this, Cv demonstrated significantly high cytotoxic potential, supported by the results presented in the present study, which evidences its efficacy in reducing the metabolic activity of different cancer cell lines, including BC, and consequently supports the importance of continuing its study to assess the possibility of its use in the management of neoplastic diseases.
Regarding MoI, it was found that as the aggressiveness of the molecular subtype of the cell line increases, so does the dose required to reach IC-50. Furthermore, the control cell line (HaCaT) required the same infusion dose as the triple-negative BC cells, suggesting that MoI has a less aggressive effect on non-cancerous cells compared to the isolated Cv compound. These findings are consistent with that reported by Tuncer et al. in 2013. In their study, they treated three BC cell lines (MCF-7, MDA-MB-468 and MDA-MB-231), using an aqueous extract of Origanum acutidens (Turkish oregano) and assessed the impact on metabolic activity using MTT assays as in our research, treatment efficacy is presented in a dose-dependent manner and together, these reports and our results support that oregano extracts represents a good alternative treatment against BC cell lines, including the most aggressive subtypes [27]. Furthermore, in 2018, Makrane et al. investigated the effect of infusion of a Moroccan species of oregano in HT-29 colon cancer cells and MDA-MB-231 BC cells and evaluated its impact on metabolic activity by WST1 assay, finding that the treatment reduced metabolic activity in a dose-dependent manner in both cell lines. They observed that triple-negative BC cells were more susceptible to treatment, with an IC50 ranging from 30.90 to 87.09 µg/mL, while in HT-29 cells this range ranged from 50.11 to 158.48 µg/mL [28]. These results are relevant to our study, as we also employed an infusion as a treatment in a TN BC cell line, providing a close reference to contrast our results. Based on this study and our findings, we can conclude that oregano extracts, including MoI, significantly reduce the metabolic activity of BC cell lines, even in the TN subtype, and that this effect occurs in a dose-dependent manner.
The results obtained in this study indicate that Cv exhibits greater cytotoxicity compared to MoI (~50 % and ~15 %, respectively), which suggests that while infusion may reduce cellular metabolism, the isolated Cv compound exhibits greater cytotoxicity. Despite the limited availability of studies on oregano cytotoxicity, the existing evidence concurs that its extracts, regardless of extraction method, possess activity against BC cells, including the most aggressive subtypes.
4. Materials and Methods
4.1. Chemical and reagents
Cv ≥98% purity (499-75-2) and dimethyl sulfoxide (DMSO, D8418), was purchased from Sigma Aldrich (St Louis, MO, USA). Dulbecco’s Modified Eagle Medium (DMEM, 11966025) Roswell Park Memorial Institute (RPMI-1640, 11875119), phosphate buffered saline (PBS, 10010023), fetal bovine serum (FBS,16140071), penicillin/ streptomycin (15140122), MTT (6494), and trypsin (25300054) were purchased from Thermo Fisher ScientificTM (Waltham, MA, USA). All solvents and chemicals were of an analytical grade.
4.2. Cell lines and culture conditions
Cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Human keratinocyte transformed and immortalized HaCaT cells and TN BC MDA-MB-231 cells were maintained in RPMI-1640, luminal A MCF-7 and HER2 HCC-1954 BC cell lines were maintained in DMEM, both mediums were supplemented with 10% FBS and 1% of penicillin/ streptomycin. The cell cultures were kept in incubation under physiological conditions (37 °C, 95% humidity and 5% CO2 saturation).
4.3. Plant material
The oregano used in this study is an endemic species from the region of Totatiche, Jalisco, Mexico.
4.4. Bromatological analysis of L. graveolens
Bromatological analyses were performed with the dried leaves of L. graveolens. The analyses performed were humidity, ash, proteins by the micro-Kjeldahl method, direct reducing sugars, total reducing sugars, ethereal extraction by Soxhlet and dietary fiber.
4.5. MoI preparation
230 mg of dry leaves were ground to a fine powder using a porcelain mortar. This powder was suspended in 10mL of sterile boiling water for 10 minutes, and the resulting infusion was filtered with a 0.22 µM sterile filter into a laminar flow hood.
4.6. Preparation of Cv solution
Cv isolate was mixed with DMSO (<0.05% final concentration) and culture medium to obtain a 1000µM solution.
4.7. Antioxidant activity protocols (DPPH and ABTS) in MoI
In brief, a total of 225 μL of ABTS (2,2’-azinobis-3-ethylbenzothiazoline-6-sulfonic acid) solution was placed in each microplate well, and an initial reading was taken at 734 nm, which served as the initial absorbance datum. Subsequently, 75 μL of 1:1000 diluted MoI (with deionized water) was added, and the microplate was incubated for 5 minutes with stirring. After which, the absorbance was measured at 734 nm. A Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) standard curve was performed to express the results as mMEQ Trolox per gram of dried sample.
For the DPPH (1,1diphenyl-2-picrylhydrazyl) method, 150 µL of the 1:1000 diluted MoI (with deionized water) was placed in a 96-well plate, and 150µL of 1010 µM methanolic solution of DPPH was added and allowed to react in the dark at room temperature for 30 minutes. A Trolox standard curve was performed to express the results as mMEQ Trolox per gram of dried sample. The absorbance was measured at 517 nm (BioTek-SynergyHTX, Winooski, VE, USA).
4.8. Determination of total phenolic content in MoI
This was performed using the Folin–Ciocalteu reaction. Total phenolic content of MoI was determined by the Folin–Ciocalteu method. 200 μL of distilled water were mixed with 250 μL of Folin-Ciocalteu solution (1 N) and 50 μL of MoI (1:10) or different concentrations (20–180 g/mL) of gallic acid (used as standard) were tested. Further, after incubation for 5 minutes at room temperature, 500 µL of sodium carbonate solution (15% Na2CO3) was added and the solution was mixed thoroughly and incubated for 15 minutes in the dark at 45ºC. 200 μL of each reaction were measured at 760 nm (BioTek-SynergyHTX, Winooski, VE, USA). All results were expressed as milligram of Gallic Acid Equivalent per gram of dried sample (mgGAE/g).
4.9. Determination of total flavonoids in MoI
Briefly, 100 µL of MoI was mixed with 200 µL of 1M potassium acetate, 200 µL of aluminum chloride solution (10% AlCl₃) and 500 µL of 80% ethanol. The mixture was shaken vigorously and incubated for 40 minutes at room temperature. The absorbance was measured at a wavelength of 415 nm (BioTek-SynergyHTX, Winooski, VE, USA). A calibration curve was performed with quercetin as standard. The result was expressed as mg Eq quercetin/g dry weight of oregano.
4.10. FRAP in MoI
40 μL of MoI or standard was mixed with 250 μL of FRAP reagent (25 mL 300 mM sodium acetate, 1.6% acetic acid; 2.5 mL TPTZ (2, 4, 6-tri (2-pyridyl)-s-triazine), 40 mM hydrochloric acid). 240 μL of distilled water was used as blank and FeSO₄ as standard for the calibration curve. The absorbance was determined at 593 nm (BioTek-SynergyHTX, Winooski, VE, USA). Results were expressed as millimoles of Fe2+.
To prepare the MoI, 1 g of dried oregano leaves was heated in 100 mL of distilled water at 60°C for 10 minutes with continuous stirring. After cooling to room temperature, the mixture was centrifuged at 10,000 rpm for 15 minutes. The supernatant was carefully poured through Whatman No. 1 filter paper into a clean glass container to remove any solid residues. The filtered extract was stored at 4°C for further use.
To obtain a concentrated extract, the filtered solution was transferred into a round-bottom flask connected to a rotary evaporator. The flask was immersed in a 40–50°C water bath, and a gradual vacuum was applied. The rotation speed was set to approximately 100 rpm to enhance evaporation. Once most of the solvent had evaporated, the concentrated extract was collected and further dried in a desiccator until a solid or semi-solid form was obtained. The final dried extract was stored in a sealed container at 4°C for analysis.
4.11. FTIR Measurement
The MoI was centrifuged at 10,000 rpm for 15 minutes. The supernatant was carefully filtered through Whatman No. 1 filter paper into a clean glass container to remove any solid residues. To obtain a concentrated extract, the filtered solution was transferred to a round-bottom flask connected to a rotary evaporator. The flask was immersed in a water bath maintained at 40–50°C, and a gradual vacuum was applied. The rotation speed was set to approximately 100 rpm to facilitate solvent evaporation. Once most of the solvent had evaporated, the concentrated extract was collected and further dried in a desiccator until a solid or semi-solid form was achieved. The final dried extract was stored in a sealed container at 4°C for analysis. The MoI was analyzed using FTIR spectroscopy within the 4000–400 cm⁻¹ range, utilizing a PerkinElmer 400 FTIR spectrometer (Waltham, MA, USA). A background spectrum was recorded prior to measurement to eliminate environmental interference. For analysis, a small amount (1–2 mg) of the extract was placed on the sample holder of the ATR (Attenuated Total Reflectance) crystal, forming a thin film. The instrument performed 32 scans, which were averaged to generate the final spectrum. The results were displayed as Transmittance vs. Wavenumber (cm⁻¹) for functional group identification.
4.12. MoI time response curve
Since there are no previous reports on the use of MoI on cancer cells, we evaluated the effect at the metabolic level of different doses of the infusion for 24, 48 and 72 hours in MDMA-MB-231 cells to establish an exposure time for further testing, because for Cv treatment we started from the background of Arunasree et al. in MDA-MB-231 cells [16]. Briefly, 1.5 × 10⁴ cells/well were seeded in 96 well-plates, after 24 hours of incubation MoI was added in different concentrations, untreated groups received culture medium, and cells were incubated for the corresponding time (24, 48 or 72 hours). Next, MTT (5 mg/mL in PBS) was added, and cells were incubated for 4 hours to allow formazan production, these crystals were dissolved using a solution prepared with sodium dodecyl sulfate (Thermo Fisher, 28312) and dimethylformamide (Sigma-Aldrich, 227056) at pH 4.6, followed by a final incubation of 18–24 hours. Absorbance was measured at 570 nm (BioTek-800TS, Winooski, VE, USA), with untreated cells considered as 100% formazan production.
4.13. Evaluation of cell metabolism (MTT assay)
Cells were seeded in 96-well plates at a density of 1.5 × 10⁴ cells/well. After 24 hours of incubation increasing concentrations of MoI or Cv solution were added, while untreated groups received culture medium. Cells were incubated 24 hours with Cv and 48 hours with MoI. Subsequently, MTT (5 mg/mL in PBS) was added, and cells were incubated for 4 hours. Formazan crystals were dissolved using a solution prepared with sodium dodecyl sulfate (Thermo Fisher, 28312) and dimethylformamide (Sigma-Aldrich, 227056) at pH 4.6, followed by a final incubation of 18–24 hours. Absorbance was measured at 570 nm (BioTek-800TS, Winooski, VE, USA), with untreated cells considered as 100% formazan production. A dose-response curve was generated for each cell line under both treatments independently, and half maximal inhibitory concentration (IC50) values were determined using the four-parameter logistic model (J. A. Nelder and R. W. M. Wedderburn, Journal of the Royal Statistical Society, 1972). The IC50 values obtained were subsequently used for the cytotoxicity assay.
4.14. Cytotoxicity assay (LDH in supernatant)
Cytotoxicity was assessed using the CyQUANT™ LDH Cytotoxicity Assay Kit (Invitrogen, C20300), following the manufacturer’s instructions. Briefly, 1.5 × 10⁴ cells were seeded in a 96-well plate and treated with MoI, Cv, or ultrapure water (for the spontaneous LDH activity control) in a final volume of 100 µL. Cells were incubated for 24 hours in the Cv group and 48 hours in the MoI group. Subsequently, 10 µL of 10X Lysis Buffer was added to the maximum LDH control group and incubated for 45 minutes. Then, 50 µL of supernatant from each well was transferred to a new 96-well plate, followed by the addition and mixing of 50 µL of Reaction Mixture. After 30 minutes of incubation at room temperature, 50 µL of Stop Solution was added and mixed. Finally, absorbance was measured at 490 nm with correction at 690 nm (BioTek-SynergyHTX, Winooski, VE, USA). Cytotoxicity was calculated using the following formula:
4.15. Statistical analysis
Results were expressed as means ± SD, T-student, ANOVA and Dunnet as post-hoc test was performed using R version 4.4.3, and a p value of <0.05 was considered statistically significant.
5. Conclusions
In conclusion, our findings confirm that both MoI and Cv represent promising therapeutic alternatives for the treatment of BC, particularly the most aggressive subtypes. It is essential to continue investigating how treatments rich in antioxidant compounds, such as MoI or Cv, affect the redox status of cancer cells, given that this mechanism is emerging as a promising therapeutic target for the management of cancers with poor prognosis, such as TN BC.
Author Contributions
Conceptualization, B.R.R., T.G.I. and M.M.G.; methodology, B.R.R., T.G.I., M.M.G, G.C.R, E.V.M., J.G.L., T.G.C., P.S.H., A.M.S., R.L.R. and I.B.L; investigation, B.R.R., T.G.I., M.M.G. and G.C.R.; writing—original draft preparation, B.R.R., and T.G.I., writing—review and editing, B.R.R., T.G.I., M.M.G, G.C.R, E.V.M., J.G.L., T.G.C., P.S.H., A.M.S., R.L.R. and I.B.L; supervision, T.G.I. and M.M.G.; funding acquisition, T.G.I. All authors have read and agreed to the published version of the manuscript.
Funding
The author(s) declared financial support was received for the research, authorship, and/or publication of this article. The study was supported by Fondo de Proyectos de Impulso a la Investigación (PIN 2022-III), and PRO-SNI 2022-2024 to the Universidad de Guadalajara.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to express our gratitude to CONAHCYT for awarding B.E.R.R (CVU number: 1182751) a doctoral scholarship through the National Postgraduate Program (Sistema Nacional de Posgrados SNP-CONAHCYT). Additionally, we extend our thanks to Doctorado en Ciencias en Biología Molecular en Medicina (DCBMM) from Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BC | Breast cancer |
| Cv | Carvacrol |
| MoI | Mexican oregano infusion |
| FRAP | Ferric reducing antioxidant power |
| FTIR | Fourier-transform infrared spectroscopy |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| LDH | Lactate dehydrogenase |
| TN | Triple negative |
| ER | Estrogen receptor |
| PR | Progesterone receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| HER2+ | HER2-enriched |
| DMSO | Dimethyl sulfoxide |
| DMEM | Dulbecco’s Modified Eagle Medium |
| RPMI | Roswell Park Memorial Institute |
| PBS | Phosphate buffered saline |
| FBS | Fetal bovine serum |
| ATCC | American Type Culture Collection |
| IC50 | Half maximal inhibitory concentration |
| USDA | U.S. Department of Agriculture |
References
- World Health Organization Cancer. https://www.who.int/health-topics/cancer#tab=tab_1. 2025.
- Cilibrasi C, Papanastasopoulos P, Samuels M, Giamas G (2021) Reconstituting Immune Surveillance in Breast Cancer: Molecular Pathophysiology and Current Immunotherapy Strategies. Int J Mol Sci 22:12015. [CrossRef]
- Global Cancer Observatory Breast cancer. https://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf. 2025.
- Perou CM, Sørile T, Eisen MB, et al (2000) Molecular portraits of human breast tumours. Nature 406:747–752. [CrossRef]
- Tsang JYS, Tse GM (2019) Molecular Classification of Breast Cancer.
- Nolan E, Lindeman GJ, Visvader JE (2023) Deciphering breast cancer: from biology to the clinic. Cell 186:1708–1728. [CrossRef]
- Amin ARMR, Kucuk O, Khuri FR, Shin DM (2009) Perspectives for cancer prevention with natural compounds. Journal of Clinical Oncology 27:2712–2725. [CrossRef]
- Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M MM (2015) The Global Burden of Cancer 2013. JAMA Oncol 1:505–527. [CrossRef]
- Dias DA, Urban S, Roessner U (2012) A Historical overview of natural products in drug discovery. Metabolites 2:303–336. [CrossRef]
- Arigesavan K, Sudhandiran G (2015) Carvacrol exhibits anti-oxidant and anti-inflammatory effects against 1, 2-dimethyl hydrazine plus dextran sodium sulfate induced inflammation associated carcinogenicity in the colon of Fischer 344 rats. Biochem Biophys Res Commun 461:314–320. [CrossRef]
- Lim W, Ham J, Bazer FW, Song G (2019) Carvacrol induces mitochondria-mediated apoptosis via disruption of calcium homeostasis in human choriocarcinoma cells. J Cell Physiol 234:1803–1815. [CrossRef]
- Khan F, Khan I, Farooqui A, Ansari IA (2017) Carvacrol Induces Reactive Oxygen Species (ROS)-mediated Apoptosis Along with Cell Cycle Arrest at G0/G1 in Human Prostate Cancer Cells. Nutr Cancer 69:1075–1087. [CrossRef]
- Martínez-Natarén DA, Parra-Tabla V, Dzib G, et al (2012) Essential oil yield variation within and among wild populations of mexican oregano (lippia graveolens h.b.k.-verbenaceae), and its relation to climatic and edaphic conditions. Journal of Essential Oil-Bearing Plants 15:589–601. [CrossRef]
- Nieto-Ramírez MI, Feregrino-Pérez AA, Aguirre Becerra H, et al (2023) Response of Phenolic Compounds in Lippia graveolens Kunth Irrigated with Aquaculture Wastewater and Steiner Solution. International Journal of Plant Biology 14:483–492. [CrossRef]
- Soto KM, Hernández-Iturriaga M, Cárdenas A, Mendoza S (2024) Biosynthesis of Silver Nanoparticles Mediated by Lippia graveolens Aqueous Extract. J Mex Chem Soc 68:402–411. [CrossRef]
- Elshafie HS, Armentano MF, Carmosino M, et al (2017) Cytotoxic activity of origanum vulgare L. on Hepatocellular carcinoma cell line HepG2 and evaluation of its biological activity. Molecules 22:. [CrossRef]
- Oniga I, Pus C, Silaghi-Dumitrescu R, et al (2018) Origanum vulgare ssp. vulgare: Chemical composition and biological studies. Molecules 23:. [CrossRef]
- Sharifi-Rad M, Varoni EM, Iriti M, et al (2018) Carvacrol and human health: A comprehensive review. Phytotherapy Research 32:1675–1687. [CrossRef]
- USDA Spices, oregano, dried. In: 2019. https://fdc.nal.usda.gov/fdc-app.html#/food-details/171328/nutrients. 2025.
- Zhang XL, Guo YS, Wang CH, et al (2014) Phenolic compounds from Origanum vulgare and their antioxidant and antiviral activities. Food Chem 152:300–306. [CrossRef]
- Mahomoodally MF, Zengin G, Aladag MO, et al (2019) HPLC-MS/MS chemical characterization and biological properties of Origanum onites extracts: a recent insight. Int J Environ Health Res 29:607–621. [CrossRef]
- Simirgiotis MJ, Burton D, Parra F, et al (2020) Antioxidant and antibacterial capacities of origanum vulgare l. Essential oil from the arid andean region of chile and its chemical characterization by gc-ms. Metabolites 10:1–12. [CrossRef]
- Kogiannou DAA, Kalogeropoulos N, Kefalas P, et al (2013) Herbal infusions; their phenolic profile, antioxidant and anti-inflammatory effects in HT29 and PC3 cells. Food and Chemical Toxicology 61:152–159. [CrossRef]
- Rice-Evans CA, Miller NJ, Paganga G (1996) Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med 20:933–956. [CrossRef]
- Berdowska I, Zieliński B, Fecka I, et al (2013) Cytotoxic impact of phenolics from Lamiaceae species on human breast cancer cells. Food Chem 141:1313–1321. [CrossRef]
- Elbe H, Yigitturk G, Cavusoglu T, et al (2020) Comparison of ultrastructural changes and the anticarcinogenic effects of thymol and carvacrol on ovarian cancer cells: which is more effective? Ultrastruct Pathol 44:193–202. [CrossRef]
- Tuncer E, Unver-Saraydin S, Tepe B, et al (2013) Antitumor effects of Origanum acutidens extracts on human breast cancer. 7: Journal of BUON 18.
- Makrane H, El Messaoudi M, Melhaoui A, et al (2018) Cytotoxicity of the Aqueous Extract and Organic Fractions from Origanum majorana on Human Breast Cell Line MDA-MB-231 and Human Colon Cell Line HT-29. Adv Pharmacol Sci 2018. [CrossRef]
Figure 1.
The FTIR spectrum of MoI shows characteristic bands of various functional groups, indicating the presence of bioactive compounds. The broad band between 3200-3600 cm-¹ is attributed to O-H stretches of alcohols and phenols, suggesting a high presence of polyphenolic compounds. The signals at 2800-3000 cm-¹ correspond to C-H stretches of methyl and methylene groups. The intense band at 1700-1750 cm-¹ confirms the presence of carbonyls (C=O), characteristic of carboxylic acids, ketones or aldehydes. Absorptions at 1500-1650 cm-¹ can be associated with C=C vibrations of aromatic rings, while bands at 1000-1300 cm-¹ indicate C-O stretches, typical of esters and phenolic compounds.
Figure 1.
The FTIR spectrum of MoI shows characteristic bands of various functional groups, indicating the presence of bioactive compounds. The broad band between 3200-3600 cm-¹ is attributed to O-H stretches of alcohols and phenols, suggesting a high presence of polyphenolic compounds. The signals at 2800-3000 cm-¹ correspond to C-H stretches of methyl and methylene groups. The intense band at 1700-1750 cm-¹ confirms the presence of carbonyls (C=O), characteristic of carboxylic acids, ketones or aldehydes. Absorptions at 1500-1650 cm-¹ can be associated with C=C vibrations of aromatic rings, while bands at 1000-1300 cm-¹ indicate C-O stretches, typical of esters and phenolic compounds.
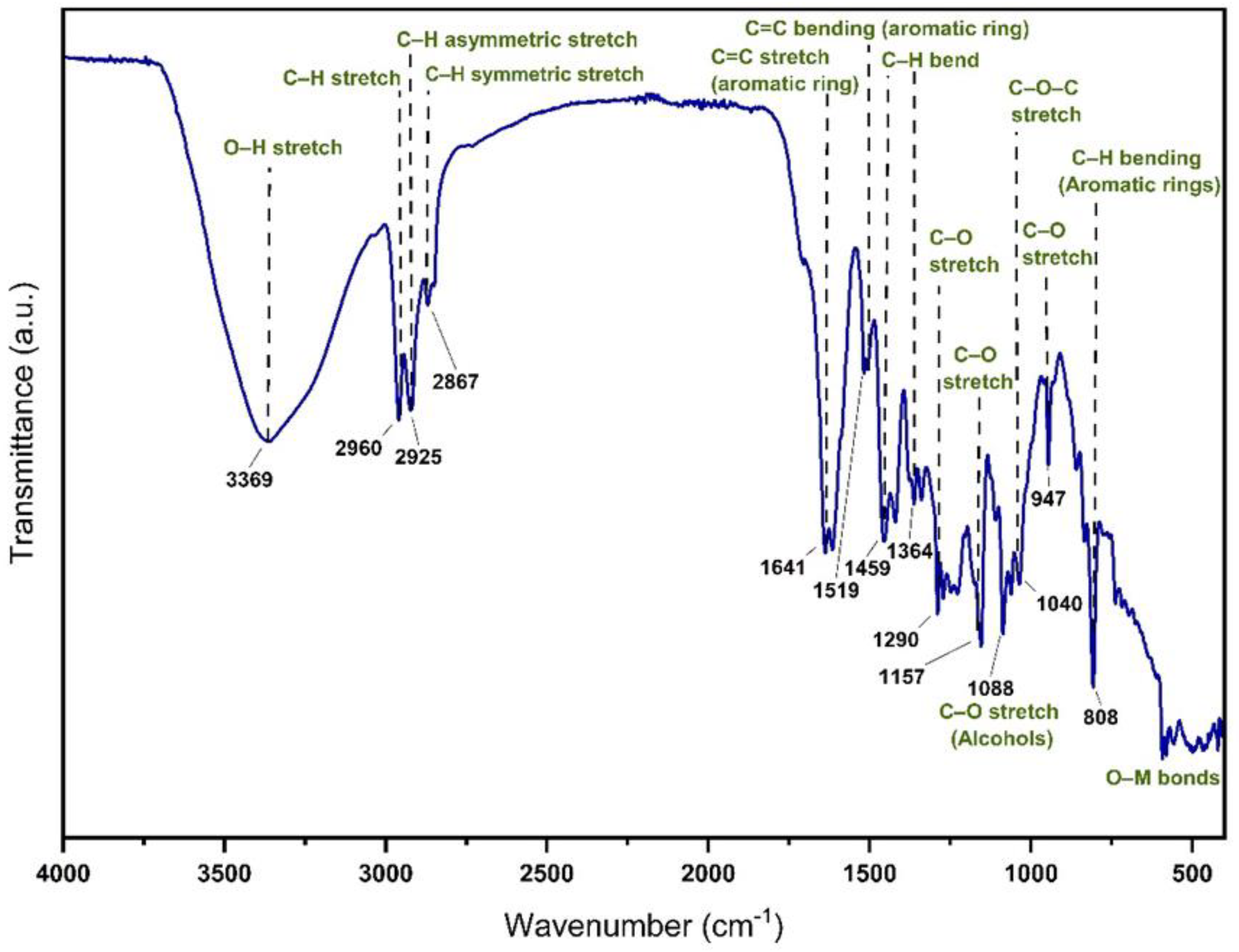
Figure 2.
Comparison of formazan production in MDA-MB-231 cells after exposure to MoI for 24, 48 and 72 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (* = p<0.05; *** = p<0.001, compared with control).
Figure 2.
Comparison of formazan production in MDA-MB-231 cells after exposure to MoI for 24, 48 and 72 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (* = p<0.05; *** = p<0.001, compared with control).
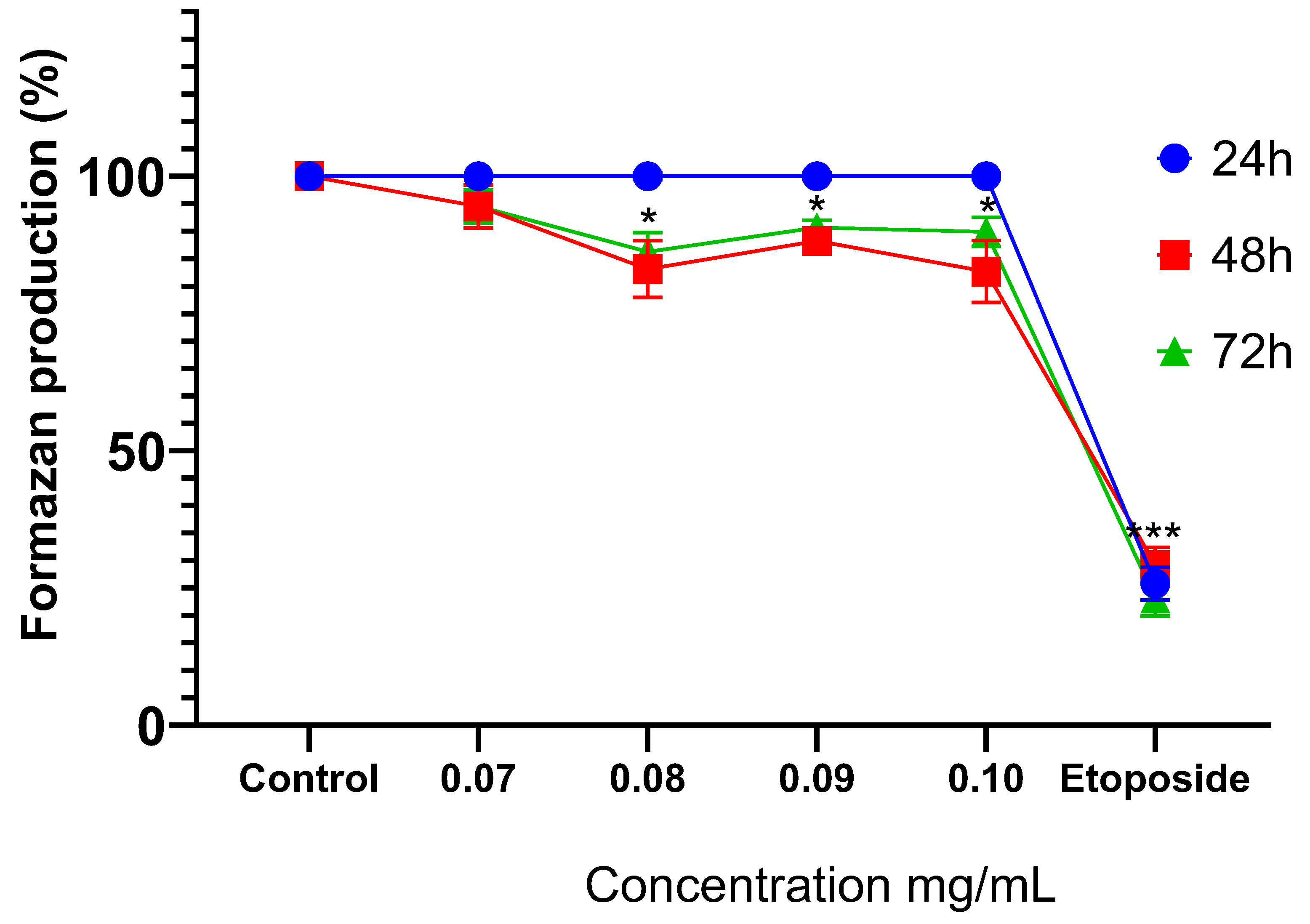
Figure 3.
Percentage of formazan produced after treatment with MoI for 48 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (*** = p<0.001, compared with control). .
Figure 3.
Percentage of formazan produced after treatment with MoI for 48 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (*** = p<0.001, compared with control). .
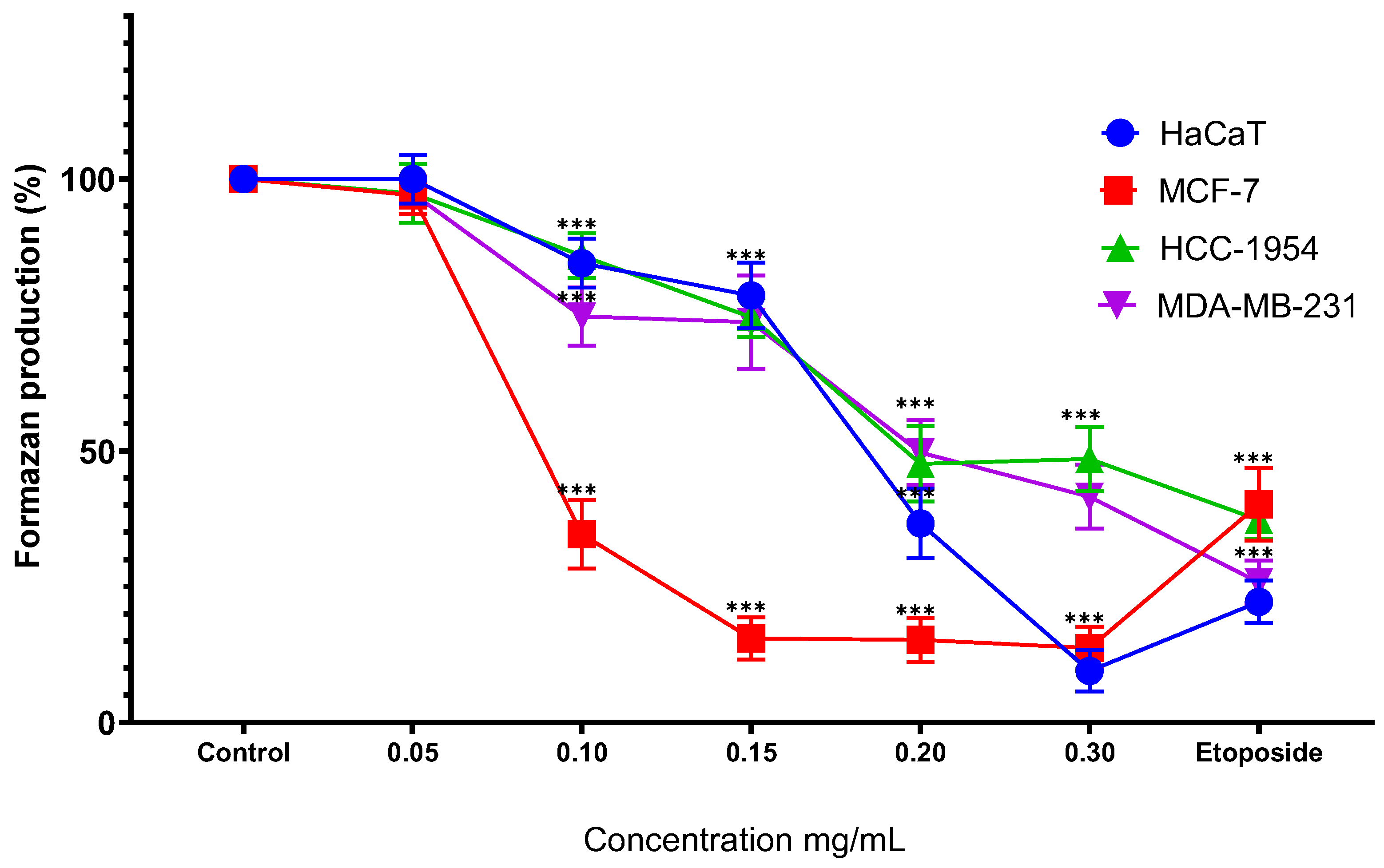
Figure 4.
Percentage of formazan produced after treatment with Cv for 24 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (*** = p<0.001, compared with control). .
Figure 4.
Percentage of formazan produced after treatment with Cv for 24 hours, etoposide was used as cell death-control, the determination was performed by MTT assay, the percentage was calculated in comparison to the untreated group taking it as 100% formazan production. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Analysis of variance between groups was performed using ANOVA and Dunnett’s post-hoc test (*** = p<0.001, compared with control). .
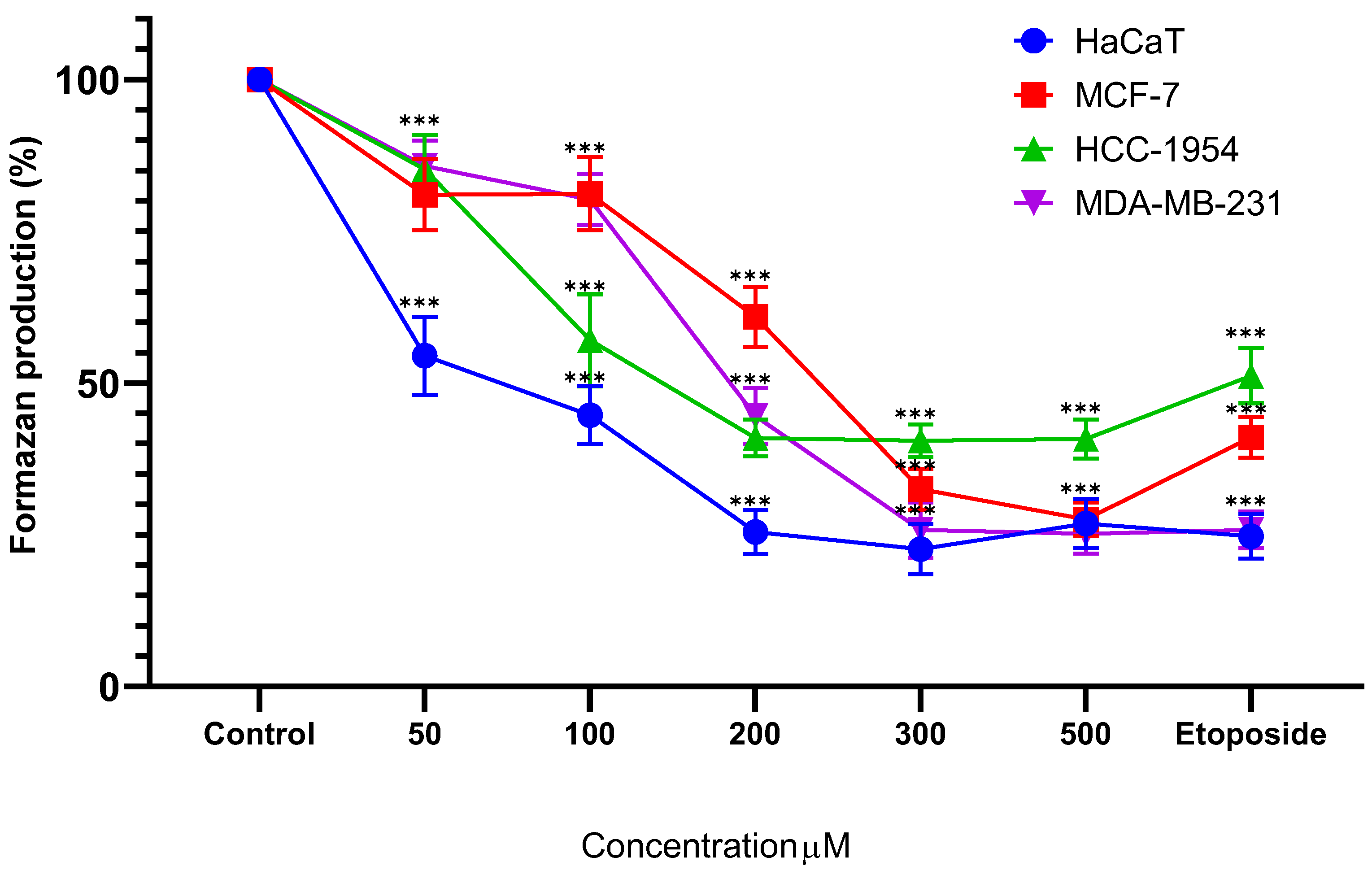
Figure 5.
Cytotoxicity of MoI and Cv was assessed by quantifying LDH release in the supernatant of treated cells. Cells were exposed to the IC50 of both treatments. A control group treated with the lysis buffer included in the kit was used to define 100% cytotoxicity. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Mean comparisons between groups were conducted using Student’s t-test (*** = p < 0.001).
Figure 5.
Cytotoxicity of MoI and Cv was assessed by quantifying LDH release in the supernatant of treated cells. Cells were exposed to the IC50 of both treatments. A control group treated with the lysis buffer included in the kit was used to define 100% cytotoxicity. Results are expressed as mean ± standard deviation of three independent experiments performed in triplicate. Mean comparisons between groups were conducted using Student’s t-test (*** = p < 0.001).
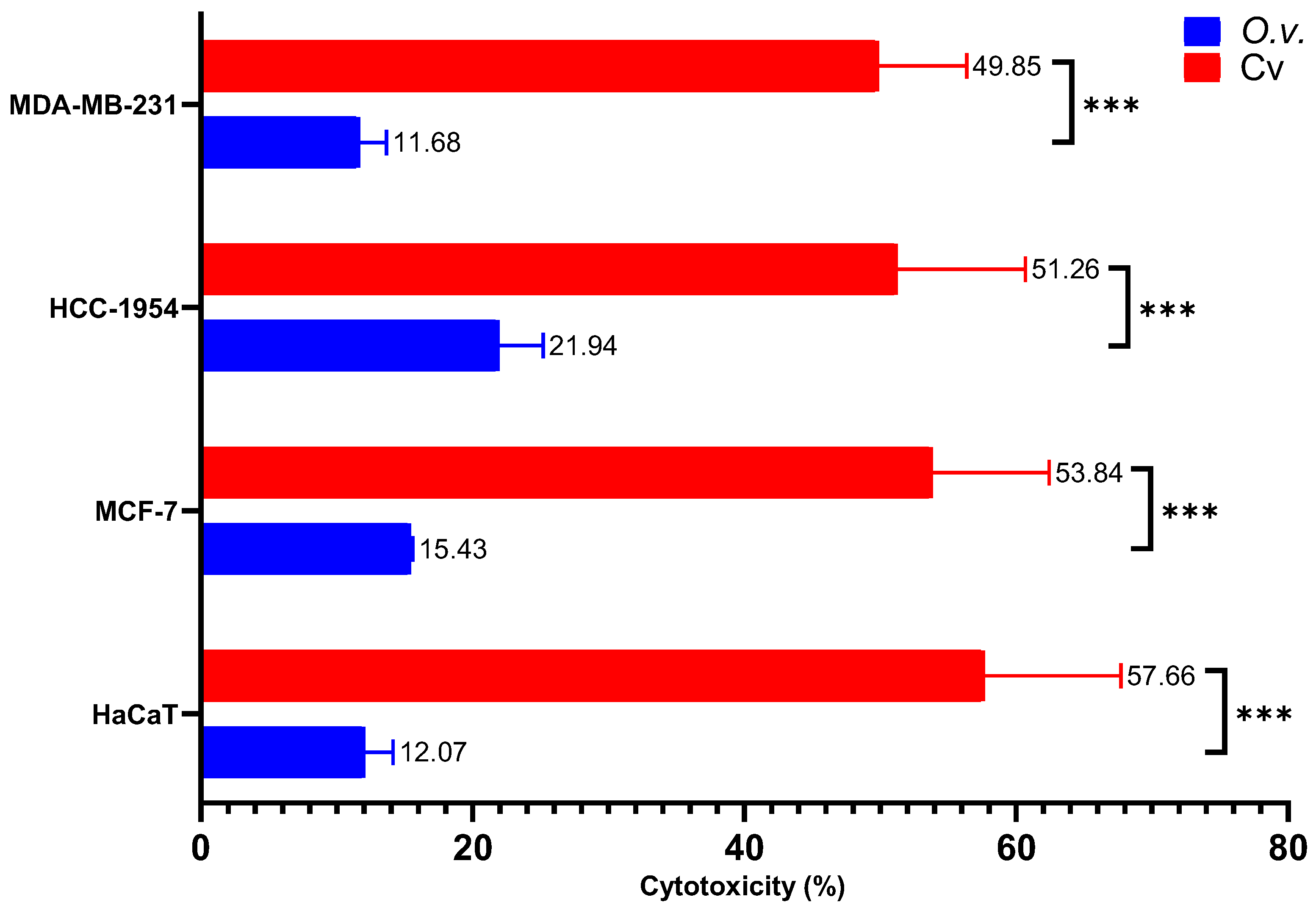

Table 1.
Bromatological analysis of the L. graveolens.
| Nutritional Content | Per 100g |
|---|---|
| Energy content | 363.4 Kcal (1539.2 KJ) |
| Moisture | 7.8% |
| Ash | 6.6% |
| Protein | 6.8g |
| Total fat | 4.2g |
| Available carbohydrates | 36g |
| Dietary fiber | 38.6g |
Table 2.
Total phenolic and flavonoids content and antioxidant capacity (DPPH, ABTS and FRAP) of MoI.
Table 2.
Total phenolic and flavonoids content and antioxidant capacity (DPPH, ABTS and FRAP) of MoI.
| ABTS | DPPH | Total phenolics | Total Flavonoids | FRAP |
|---|---|---|---|---|
| 42.837 ± 0.175 mMEQ Trolox/ g | 66.303 ± 0.228 mMEQ Trolox/ g | 27.765 ± 1.095§mgGAE/g | 22.343 ± 0.096 mg Eq quercetin/g | 109.85 ± 0.51 mMEQ FeSO4/ g |
Table 3.
IC50 of MoI and Cv in each cell line.
| Cell line | MoI IC50 | Cv IC50 |
|---|---|---|
| HaCaT | 0.18mg/mL | 110µM |
| MCF-7 | 0.08mg/mL | 211µM |
| HCC-1954 | 0.15mg/mL | 123µM |
| MDA-MB-231 | 0.17mg/mL | 121µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



