
Submitted:
15 May 2025
Posted:
16 May 2025
You are already at the latest version
Abstract
Pseudomonas aeruginosa is a major pathogen associated with hospital-acquired infections, and the spread of carbapenem-resistant isolates highlights the urgency of developing non-conventional therapies, such as phage therapy. For this alternative to be effective, un-derstanding phage-host interactions is crucial for the selection of candidate phages and offers new insights into these dynamics. Background/Objectives: This study aimed to characterize prophage diversity in clinical P. aeruginosa genomes, assess the relationship between phages and the CRISPR/Cas system, and investigate the potential role of phages in disseminating resistance genes. Methods: A total of 141 genomes from Brazilian hos-pitals were analyzed. Prophage detection was performed using VIBRANT, and in silico analyses were conducted to evaluate taxonomic diversity, presence of resistance genes, phage life cycle, genomic distribution, and the presence of the CRISPR/Cas system. Re-sults: In total, 841 viral sequences were identified, with a predominance of the class Cau-doviricetes and high overall phage diversity. No statistically significant difference was ob-served in the number of prophages between isolates with and without CRISPR/Cas sys-tems. Phages carrying resistance genes were detected in isolates harboring the type I-C CRISPR/Cas system. Additionally, prophages showed no preference for specific insertion sites along the bacterial genome. Conclusions: These findings provide evidence of a well-established phage-host relationship. The dual role of phages—as vectors of antimi-crobial resistance and as potential therapeutic agents—reflects their dynamic impact on bacterial communities and reinforces their importance in developing new strategies to combat antimicrobial resistance.
Keywords:
bacteriophages
; antimicrobial resistance
; CRISPR/Cas system
1. Introduction
Carbapenem resistance in Gram-negative bacteria is a global public health concern due to the increasing spread of resistance to this class of antimicrobials. Carbapenems are known for their broad-spectrum activity and are used as a last-resort treatment for severe infections [1]. In 2024, the WHO updated its list of priority pathogens associated with antimicrobial resistance, highlighting carbapenem-resistant Gram-negative bacteria as a top priority for the development of new therapies, as well as for control and prevention strategies [2].
Pseudomonas aeruginosa has been categorized as a high-priority pathogen on the WHO list (2024) and is considered one of the main causes of healthcare-associated infections (HAIs), particularly affecting immunocompromised patients. Due to its intrinsic and acquired resistance to antimicrobials, as well as its ability to produce biofilms, therapeutic options for treating P. aeruginosa infections are limited [2,3].
One of the promising therapeutic approaches to address antimicrobial resistance is phage therapy, which involves the administration of bacteriophages—viruses that specifically target bacteria and are capable of killing bacterial cells. However, not all phages are suitable candidates for therapeutic use. Ideally, strictly lytic phages are preferred, as they lyse bacterial cells and directly combat the source of infection [4].
Temperate (lysogenic) phages, unlike strictly lytic ones, can integrate their genome into the bacterial chromosome. In this state, they behave as prophages and replicate alongside the host cell. Under stressful conditions, the lysogenic phage may be induced to enter the lytic cycle. During this transition, the phage DNA excises itself from the bacterial genome, initiates replication, and ultimately leads to the production of new phage particles, resulting in the lysis of the host cell and the release of newly formed virions [5,6].
Bacteriophages and bacteria exhibit co-evolutionary dynamics similar to an arms race, in which both species must develop strategies for attack and survival. Within this process of modulation and adaptation, one proposed perspective is that phages, despite being intracellular parasites, may act as symbionts in their hosts due to the presence of phage-carried genes that enhance bacterial fitness [7].
In addition to genetic contributions, prophages can regulate the infection dynamics of related phages through a mechanism known as superinfection exclusion (SIE). This strategy allows prophages integrated into the bacterial genome to block subsequent infections by similar or identical phages, thereby ensuring their own maintenance within the host. SIE not only benefits the prophage but also has biological implications for the host bacterium, as it can provide temporary protection against infections by lytic phages, which might otherwise kill the host cell Therefore, identifying and characterizing prophages is essential for developing effective and safe phage therapies[8].
Moreover, to counter phage infection, bacteria have evolved adaptive immune systems, such as the CRISPR/Cas system (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated genes), one of the most important antiphage defense strategies in bacteria This mechanism provides acquired immunity by capturing fragments of invading phage DNA, which are then used to recognize and neutralize future infections by the same or similar phages. Upon detecting complementary sequences, the system recruits associated proteins, including nucleases, to cleave the foreign genetic material, thereby preventing successful infection [9,10]
For the proper implementation of phage therapy, in addition to understanding the mechanisms underlying phage-bacteria interactions, it is essential to identify the phages that interact with clinical P. aeruginosa isolates, their genomic features, and their role in modulating bacterial responses in the environment.
We analyzed clinical genomes of P. aeruginosa to identify putative phages and explore their diversity. Additionally, we characterized the viral taxonomy, assessed the relationship between phages and CRISPR/Cas systems, mapped their chromosomal distribution and life cycle, and investigated phage-associated resistance genes.
2. Materials and Methods
2.1. Pseudomonas aeruginosa Genome
A total of 141 complete genomes of P. aeruginosa from clinical samples collected in hospitals across various Brazilian states (Pernambuco, Bahia, Ceará, Minas Gerais, Rio de Janeiro, São Paulo, Santa Catarina, Goiás, and Paraná) were selected from the NCBI database for this study. Of the 141 genomes analyzed in this study, 25 (from Recife-Pernambuco) were previously sequenced [11,12] and deposited in the NCBI under BioProject ID PRJNA514718. Additional epidemiological information on the genomes used, such as collection date, isolation source, location, MLST for species confirmation and the corresponding NCBI accession numbers, are available in Supplementary Material 1.
2.2. CRISPR System Identification
The identification of the CRISPR/Cas system in the genomes deposited in the NCBI database was performed using the standalone implementation of the CRISPRCasFinder tool [9]. The strains were then classified according to the type and subtype of CRISPR/Cas system present and categorized as CRISPR/Cas-positive if any type of system was detected, or CRISPR/Cas-negative otherwise.
2.3. Prophage Identification
The tool used for prophage identification was VIBRANT [13]. The results generated by VIBRANT were analyzed to identify phage sequences, their location within the host genome, and their length. Once the sequences were obtained, the identified phage regions were compared against previously deposited genomes in the NCBI Virus database to confirm the presence and identity of the phages. Alignments of unidentifiable regions were performed using the Mugsy tool [14].
The sequences extracted by VIBRANT had their quality assessed using CheckV (https://phage.usegalaxy.eu/), a specific pipeline for evaluating the quality of viral genomes. The sequences were classified as: Complete; High quality (>90% completeness); Medium quality (50–90% completeness); Low quality (<50% completeness); and Not determined.
2.4. Prophage Distribution in Pseudomonas aeruginosa Genomes
The distribution of prophages across the bacterial genomes was analyzed based on the data extracted by VIBRANT. All positions were organized, recording the start and end points of each prophage, as well as their length. Subsequently, the data were mapped in a density plot generated in R using the ggplot2 package, specifically the functions geom_density and aes (https://www.R-project.org/).
2.5. Clustering, Phylogeny and dendogram
We used PhaMMseqs to map and group 841 phages based on sequence similarity. Due to the high volume and diversity, standard clustering failed. To address this, we applied the '--cluster-mode 1' parameter, enabling single-linkage clustering, which is more sensitive to distant homologies
The output file generated by PhaMMseqs was then used as input for the PhamClust software [16]. PhamClust was executed with default parameters, employing the Proteomic Equivalence Quotient (PEQ) metric to calculate the distance matrix.
Phylogenetic analysis was conducted using the VICTOR online server (https://victor.dsmz.de) [17]. All pairwise nucleotide sequence comparisons were performed using the Genome-BLAST Distance Phylogeny (GBDP) method [18] under the settings recommended for prokaryotic viruses.
The resultant intergenomic distances were utilized to generate a balanced minimal evolution tree with branch support using FASTME with SPR post-processing [19]. A total of 100 pseudo-bootstrap replicates were used to determine branch support. Trees were midpoint-rooted [20] and visualizated using ggtree [21].
The OPTSIL program [22] was used to determine taxon boundaries at the species, genus, and family levels, following the recommended clustering thresholds and applying an F value (the fraction of links required for cluster fusion) of 0.5 [17].
The dendrogram of Pseudomonas aeruginosa isolates was constructed based on categorical and quantitative data, including the type of CRISPR/Cas system, MLST profile, and the number of prophages identified per genome. The data matrix was organized in the RStudio environment, and hierarchical clustering analysis was performed using the hclust() function, applied to a dissimilarity matrix generated with the dist() function.
2.6. Identification of Resistance Genes and Virulence Factors in Prophages
The search for antibiotic resistance genes was performed using the Resistance Gene Identifier (RGI) (https://card.mcmaster.ca/analyze/rgi), a tool based on the Comprehensive Antibiotic Resistance Database (CARD) [23]. For the identification of virulence factors, the sequences were submitted to ABRIcate, available on the Galaxy EU (https://usegalaxy.eu/). Viral sequences positive for the presence of resistance and/or virulence genes were annotated using the Pharokka tool (https://phage.usegalaxy.eu/).
2.7. Prophage Life Cycle
To infer whether the bacteriophage life cycle was lytic or lysogenic, the PHACTS tool (PHAge Classification Tool Set) [24] was used. This computational tool is widely applied for classifying and predicting bacteriophage life cycles based on genomic and structural features. The analysis was performed by submitting the phage genomic data to PHACTS, which uses specific algorithms to identify genes and patterns associated with life cycle types. The results provided by PHACTS were interpreted based on the probabilities generated by its predictive model and were used as criteria for the final classification of the phage life cycle.
2.8. Statistical Correlations
Data regarding the number of phage regions per genome, categorized according to the presence or absence of the CRISPR/Cas system, were subjected to exploratory statistical analysis using the Jamovi software (https://www.jamovi.org). To evaluate the possible correlation between the number of viral regions and the presence of the CRISPR/Cas system, Spearman’s correlation test was applied. Only results with a p-value < 0.05 were considered statistically significant.
3. Results
3.1. In Silico Analysis Identifies Prophages in Brazilian Clinical Isolates of Pseudomonas aeruginosa
A total of 141 P. aeruginosa genome sequences were selected and analyzed to investigate the frequency and distribution of prophages in Brazilian clinical isolates.Using the VIBRANT software to search for prophages within the 141 genomes, a total of 841 viral regions were identified, with all genomes containing at least one sequence showing similarity to a phage (Supplementary Material 2).
A wide variation in the length of the viral sequences was observed. The largest sequence was 178,563 bp, and the smallest was 3,028 bp. The average length of the regions was 31,873 bp, with a median of 29,641 bp.
To determine the prevalence of prophages in the P. aeruginosa genomes, the number of prophages per genome was analyzed. From this analysis, it was observed that some phages were rarely found, while others were more commonly present in the isolates’ genomes.
One prophage identified only once showed high sequence similarity to Pseudomonas phage PAJU2. In contrast, Pseudomonas phage phi3 was detected 50 times in 32 analyzed P. aeruginosa genomes. It is worth noting that the phages classified as “Bacteriophage sp.” and “Caudoviricetes sp.” were the most frequently observed. However, it was not possible to determine significant similarity with previously characterized phages (Table 1).
We assessed the completeness and quality of the viral sequences using CheckV. Among the genomes analyzed, 60% were of high or medium quality. In contrast, 39% showed low quality (<50% completeness) (Supplementary Material 3).
Only 1% of the sequences (11 in total) were classified as “not determined.” Despite the low completeness observed in some sequences, all were retained for subsequent analyses, as VIBRANT detected virus-associated proteins, indicating their viral potential. Moreover, host genome contamination exceeded 50% in only 3% of the sequences, suggesting minimal host interference in most cases. After this screening, the sequences were submitted to PhamCluster for group clustering. The clustering results are discussed later.
When classifying the samples as CRISPR/Cas-positive or negative, it was observed that the positive samples contained more viral regions than the negative ones, with an average of six regions compared to an average of five regions in the negative group. However, this difference was not significant, as evidenced by a p-value greater than 0.05 (Table 2).
The number of prophages identified varied markedly among the analyzed genomes. Some strains, such as Pae113 (CRISPR/Cas-positive) and UFMG-H10 (CRISPR/Cas-negative), harbored only a single prophage. In contrast, others contained a much higher number of distinct prophages, with the CRISPR/Cas-positive strain CCBH28612 carrying 17 unique viral sequences and the CRISPR/Cas-negative strain H2-9me containing 16. These findings show that individual P. aeruginosa genomes may harbor multiple distinct prophages, reflecting high variability among strains."
Of the 841 viral sequences observed, 587 were identified in isolates positive for the CRISPR system. Upon analyzing the frequency of these sequences by system type, phages were more frequently detected in hosts harboring the Type I-F system (Figure 1), which may be associated with the higher number of isolates positive for the I-F type. Furthermore, an average of four prophages per isolate was observed in Type I-F positive isolates, three in isolates harboring the I-E system, and seven in those with the I-C system.
A dendrogram was constructed to demonstrate the relationship among P. aeruginosa isolates regarding the type of CRISPR/Cas system, MLST profile, and the number of prophages per genome. Figure 1 shows the distribution of the isolates in relation to these genetic characteristics. It was observed that the clusters were formed in a heterogeneous manner with respect to the isolates, but showed internal homogeneity regarding the analyzed characteristics(Figure 2).
3.2. Caudoviricetes is the Most Commonly Identified Class of Prophages in Pseudomonas aeruginosa Genomes
The recovered phage sequences were subjected to taxonomic classification. Of the 841 regions analyzed, 12 (1.4%) could not be taxonomically assigned due to a lack of significant similarity or because they exhibited low query coverage (<30%) against viral genomes previously deposited in the NCBI viral sequence database (www.ncbi.nlm.nih.gov). Among these 12 sequences, six were identified as three distinct pairs. For each pair of aligned sequences, both corresponded to the same phage and were identified in two different isolates. The remaining six unpaired sequences did not align with any others and were therefore considered as unique phages specific to their respective host genomes.
Among the 829 remaining viral regions, 140 were classified as bacteriophages with no similarity to any previously characterized phage; 136 were identified as Caudoviricetes sp., 14 as Inoviridae sp., and one as Microviridae. The remaining 538 sequences were grouped into 103 distinct phages, resulting in a total of 107 phage types based on their taxonomic classification.
The majority of phage types (104 out of 107, 98%) were identified as belonging to the class Caudoviricetes. This class encompasses all tailed bacteriophages and represents the largest group of phage sequences deposited in the NCBI database. In 2022, with the updated classification system proposed by the International Committee on Taxonomy of Viruses (ICTV), morphology-based taxonomy was abolished, and the former order Caudovirales—along with the families Myoviridae, Podoviridae, and Siphoviridae—was reclassified under the broader class Caudoviricetes [25]. Among the Caudoviricetes phages identified, 44 (42.3%) could not be resolved to the genus level. However, the remaining sequences were distributed across 13 distinct viral genera, with Casadabanvirus being the most frequently represented (Figure 3).
The second most frequently identified class of prophages was Faserviricetes, accounting for approximately 2.6% (n = 4) of the total. Members of the Faserviricetes class are flexible filamentous phages with a positive-sense single-stranded DNA genome, such as those belonging to the family Inoviridae, exemplified by the phage Primolicivirus Pf1 [26]. The third most common class, albeit representing only 0.7% (n = 1) of all identified prophage genomes, was Malgrandaviricetes, which includes phages from the family Microviridae. These viruses possess an icosahedral morphology and ssDNA genomes and primarily infect enterobacteria [26]. Viral sequences that could not be taxonomically classified represented 1.9% of the dataset.
To gain a better understanding of the sequences generically classified as “Bacteriophage” and “Caudoviricetes,” a comparative genomics analysis was performed using the PhamClust software. This analysis resulted in the formation of 91 clusters (Supplementary Material 4). Among the 140 sequences labeled as “Bacteriophage,” 114 were grouped into 15 distinct clusters, suggesting the presence of 15 potential phage types infecting P. aeruginosa. The remaining 26 sequences showed no detectable similarity to each other. Of the 136 sequences identified as “Caudoviricetes,” only 48 were distributed into 16 clusters, while the remaining regions were not clustered.
For each cluster formed, a representative sequence was selected to better understand the phylogenetic distribution of the phages classified as Bacteriophage and Caudoviricetes. This selection was based on the sequence with the highest similarity among the sequences within each cluster.
Based on the phylogenetic analysis, four major branches can be observed, with a distribution of smaller subgroups, highlighting the diversity of the P. aeruginosa phage community (Figure 4).
The sequences classified as Bacteriophage and Caudoviricetes are homogeneously distributed within their respective groups. Clusters corresponding to phages with already-defined taxonomy were grouped more individually and appeared distant from the unclassified sequences. Despite the formation of clusters, it was not possible to infer deeper evolutionary relationships for the unknown sequences.
3.3. Pseudomonas aeruginosa Prophages Do Not Exhibit Unique Insertion Sites
To determine whether prophages, regardless of their taxonomic classification, are integrated into specific regions of the bacterial genome or are randomly distributed throughout the host DNA, all prophage insertion sites were mapped and plotted against a reference genome. This approach allowed for a comprehensive visualization of the genomic distribution of all analyzed prophages
No concentration in specific genomic regions was observed. Based on the density plot (Figure 5A), the start and end positions of the prophages along the bacterial chromosome showed a continuous and dispersed distribution across the genome, suggesting that rather than integrating into preferential loci, prophages may be inserted at various sites throughout the host DNA, indicating a random pattern of genomic dispersion.
Although this result represents the distribution of all prophages, the genomic positions of the most frequently identified prophages were individually analyzed within the bacterial genome. First, the distribution of phage phi3 was assessed (Figure 5B).
The distribution of phage phi3 also shows overlapping density curves, indicating a continuous distribution along the bacterial chromosome. However, notable peaks are observed around 1 Mb and 4 Mb, suggesting a slight preference for insertion in these regions. Overall, the insertions appear to be broadly dispersed throughout the chromosome. The second phage analyzed was phi297. In this case, the distribution of the phage shows peaks at approximately 1 Mb and 3.5 Mb, but similarly exhibits a relatively uniform distribution across the bacterial genome (Figure 5C).
Thus, based on the relative insertions of the phages within the genome, a continuous dispersion was observed, with a less directed or more random pattern.
3.4. Presence of Antibiotic Resistance Genes in Viral sequences
A search for antibiotic resistance genes was conducted in viral genomes, given that these viruses have the ability to mediate the transfer of resistance genes between different bacterial species, thus contributing to the rapid spread of antibiotic resistance. Among the 841 viral sequences analyzed, derived from 141 bacterial genomes, resistance genes were identified in 37 viral sequences (4.4%), which were distributed across 16 bacterial genomes (11.3%) (Table 4).
Although the quality of the sequences carrying resistance genes was considered low (<50% completeness), they were annotated to assist in confirming their identity as viral elements, ensuring that they indeed correspond to viral sequences and not to other genomic regions (Supplementary Material 5).
Upon performing a genomic comparison of a region between two variants of the same phage (DMS3)—one containing the resistance gene rsmA and the other lacking it—it was observed that the overall structure of both phages is highly conserved, especially in the blocks encoding structural proteins and tail-associated proteins (Figure 6). The phage carrying the resistance gene also has an associated tRNA gene, which is possibly linked to rsmA, a gene involved in post-transcriptional modulation in bacteria and, in some contexts, in the regulation of resistance mechanisms [27]. This difference suggests a gene acquisition event, possibly through the integration of bacterial elements during the phage’s lysogenic cycle, highlighting the role of phages as potential gene transfer vectors.
The presence of genes encoding proteins belonging to various classes involved in antimicrobial resistance was observed. Among them, notable examples include β-lactamases (blaOXA-56), genes associated with efflux pump systems (rsmA, mcx, emrE), and sulfonamide resistance genes (sul1). Among the genes analyzed, rsmA, which belongs to the resistance-nodulation-cell division (RND) efflux pump family, and sul1, associated with sulfonamide resistance, showed the highest prevalence. No virulence genes were identified.
The isolates positive for phages carrying the blaOXA-56 gene belong to sequence type ST277 and harbor a type I-C CRISPR-Cas system. This clone is characterized by an enriched resistance profile and the presence of class 1 integron In163, which contains the aacA4, aadA7, and blaOXA-56 genes organized as gene cassettes [28,29]. Interestingly, in isolates LIM1166 and LIM1410, the inserted prophages carried this same association with the aacA4, aadA7, and blaOXA-56 genes (Figure 7).
3.5. The Lytic Cycle Is More Frequent in Clinical Isolates of Pseudomonas aeruginosa
Using PHACTS, a total of 841 prophage sequences from different bacterial genomes were analyzed to infer their potential behavior—whether they are likely to enter the lytic cycle or remain in the lysogenic state. Among these sequences, 474 were classified as prophages with a higher potential to initiate the lytic cycle, while 367 showed a higher tendency to maintain the lysogenic cycle (Supplementary Material 6).
Regarding the prophages carrying resistance genes, 13 exhibited a lysogenic life cycle, while 23 were identified as lytic phages.
The presence of 474 prophages with lytic potential suggests a significant capacity of these genetic elements to excise themselves from the host genome and enter the lytic cycle. This potential enables rapid viral replication in host-rich environments, efficient dispersal through cell lysis, and a selective advantage in competitive conditions. In contrast, the 367 prophages with lysogenic potential indicate an adaptive strategy that favors long-term persistence, horizontal gene transfer, and evasion of host immune defenses.
The coexistence of prophages with both lytic and lysogenic potential reflects a complex interaction between phages and their host populations, influenced by environmental and genetic factors. Variables such as bacterial density, nutrient availability, and the presence of abiotic stressors can modulate the predominance of one cycle over the other, thereby shaping the ecology and evolution of microbial communities [30,31].
4. Discussion
The diversity of phages within bacterial ecosystems is vastly greater than the number of genomes currently deposited in public databases. Studies focused on the identification of prophages and metagenomic analyses can substantially increase the number of available sequences, while also providing deeper insights into this reservoir of phage genetic diversity and their respective hosts [32].
Although the recovery of prophage sequences from bacterial genomes does not allow for the isolation of viable phage particles—as is possible with conventional culture-based methods—the data obtained through in silico analyses enable valuable discoveries and foster meaningful discussions about this viral community.
Despite the vast viral diversity, our taxonomic data support and reinforce the evidence that the class Caudoviricetes is predominant among phages associated with different bacterial communities, underscoring its relevance in regulating bacterial populations and shaping microbial community structure [33,34,35].
In the present analysis, more than 280 prophage sequences showed no homology with previously described phages. Nonetheless, it was possible to group them and analyze the occurrence of uncharacterized phages in Brazilian P. aeruginosa isolates. Most prophages integrate into the host bacterial genome through site-specific recombination at att sites or by transposition into random locations. These insertions usually occur in genomic regions that cause minimal harm to the host, such as areas near non-coding genes like tRNA, regions with few functional genes, or intergenic regions [36].
Phage therapy offers advantages over antibiotics, especially in the phages’ ability to coevolve with bacteria. In fact, unlike antibiotics—which can quickly become ineffective due to bacterial resistance—phages can coevolve in situ with their hosts, potentially making them more effective in the long term. Although the phenomenon of overexpression indeed poses a challenge at this stage, the combination of effective antibiotics with the selection of suitable phages for phage therapy has shown promise in tackling infections caused by pathogens.[8,37]
Phages play a dynamic role in bacterial evolution, going beyond the mere transfer of resistance genes. The interaction between phages and their bacterial hosts can be viewed as a true “biological warfare,” in which bacteria develop defense mechanisms such as CRISPR-Cas systems that provide adaptive immunity against viral infections. In addition, restriction-modification (RM) systems also contribute to bacterial resistance by enabling the recognition and degradation of invading phage DNA through restriction endonucleases [10,38].
Phages themselves can exert control over related phage infections through superinfection exclusion (SIE), a mechanism by which an integrated prophage prevents infection by similar phages. Interestingly, a study based on the genomic analysis of phages using the Anderson phage typing scheme revealed a diversity in phage-host interactions, including the absence of known anti-CRISPR genes, highlighting new opportunities for exploring phage-host-phage relationships [10].
Moreover, it is important to consider intra-host phage diversity, which refers to the presence of multiple phage types coexisting within the same bacterial population inside a host. This diversity can significantly impact the clinical outcome of phage therapy, as different phages may vary in their infectivity and ability to coevolve with bacterial strains. The coexistence of diverse phages enhances the complexity of the phage-bacteria “biological warfare,” potentially driving bacterial populations to develop resistance to some phages while remaining susceptible to others. Clinically, this suggests that phage cocktails, composed of multiple phage types, could be tailored to target a broader spectrum of bacterial variants, improving therapeutic efficacy and reducing the risk of resistance development. Therefore, recognizing and characterizing intra-host phage diversity is crucial for optimizing phage therapy as an adaptive and personalized treatment strategy, as well as for guiding genomic and functional monitoring during therapy to adjust phage combinations in response to bacterial and phage evolution.
Understanding these processes is essential for the development of novel therapeutic approaches involving phages or phage inhibitors, enhancing efforts to combat antimicrobial resistance. Furthermore, the prevalence and diversity of phages in hospital environments—where selective pressure is intense—may amplify the dissemination of resistance genes, thereby compromising the effectiveness of conventional treatments.
Our findings suggest that P. aeruginosa phages integrate in a non-targeted manner into the host genome, or at multiple integration sites throughout the genome. It is important to highlight that each type of insertion may confer a specific advantage to the bacterium. The dispersed presence of prophages enables rapid adaptation of the bacterial host, as they function as genetic buffer zones, promoting genetic variability within the bacterial population.
This variability can accelerate adaptation to environmental changes and selective pressures. On the other hand, such random insertions may also be more deleterious, which is why site-specific insertions tend to confer greater genetic stability to the host. Thus, each bacterium is likely to adopt different strategies for interacting with prophages, depending on its environment, ecological niche, and the selective pressures it encounters [36,39].
Bacteria develop resistance to antimicrobials through intrinsic processes—via the expression of genes located on their own chromosome—or through acquired mechanisms, including mutations or the acquisition of resistance genes via Horizontal Gene Transfer (HGT). Phages are capable of transferring genetic material from one bacterium to another through transduction, including antimicrobial resistance genes, highlighting the role of phages in the acquisition and dissemination of ARGs (Antimicrobial Resistance Genes) [40].
Our results revealed the presence of phages carrying resistance genes. Previous studies involving other clinically relevant bacterial species, such as Staphylococcus aureus and Escherichia coli, have demonstrated that these viral elements can encapsulate fragments of bacterial DNA and, through transduction, transfer this material from donor to recipient cells [41,42].
Although this process may be less efficient—particularly in the presence of virulent phages that lyse co-infected cells—and given that mechanisms such as conjugation are more frequently employed by bacteria, the role of phages in modulating the bacterial genome remains significant and should not be underestimated [41,43].
When evaluating the sequences of prophages, CheckV returned a low completeness quality of the sequences; however, this value does not disregard that prophages can act as vectors. Although the identification of antibiotic resistance genes (ARGs) in phage genomes should be done with caution, here we demonstrate that even in low-quality sequences, it was possible to detect ARGs associated with viral elements.
Most of the viral sequences positive for resistance genes are associated with the genomes of isolates belonging to ST277.This clone is considered endemic among Brazilian strains of Pseudomonas aeruginosa and is characterized by a high-level antibiotic resistance profile and by its association with the type I-C CRISPR/Cas system, which is frequently located within genomic island regions [12,28,44].
Genomic islands are DNA segments acquired through horizontal gene transfer, usually derived from mobile genetic elements or phages. In P. aeruginosa, these segments are known as PAGIs (Pseudomonas aeruginosa Genomic Islands) and commonly harbor genes that provide adaptive advantages to the host, such as antibiotic resistance, virulence factors, and genes associated with metabolism [29,44].
We annotated the genomes of phages carrying resistance genes linked to class 1 integron In163 (aacA4, aadA7, and blaOXA-56) to understand their genetic content. The analysis revealed the presence of typical phage proteins, confirming their viral identity. Additionally, in the phages associated with the aacA4, aadA7, and blaOXA-56 genes, proteins related to integrative conjugative elements were identified. These findings suggest that, during the viral assembly process, these phages may have incorporated bacterial sequences into their genomes, potentially including genes or elements similar to Integrative and Conjugative Elements (ICEs).
Furthermore, Nascimento et al. (2019) characterized PAGIs enriched with transposases, integrases, and phage-associated proteins, along with the prevalence of resistance genes such as blaSPM-1, blaOXA-56, rmtD, cmx, and sul1. Based on the data obtained and the annotation of the genomic sequences, a significant association was observed between the presence of phages and the ability of P. aeruginosa ST277 to acquire and disseminate antimicrobial resistance [45].
The survey of prophages inserted in clinical Pseudomonas aeruginosa genomes in Brazil revealed a prevalence of phages belonging to the class Caudoviricetes, reflecting the high diversity of phages within bacterial communities. The distribution of prophages throughout the host genome, without a preferential integration site, suggests a complex dynamic of viral integration with important implications for bacterial evolution. Furthermore, the presence of antibiotic resistance genes encoded by prophages highlights their role as vectors in the dissemination of resistance determinants, posing a new challenge for the control of bacterial infections.
5. Conclusions
The survey of prophages inserted in clinical Pseudomonas aeruginosa genomes in Brazil revealed a prevalence of phages belonging to the class Caudoviricetes, reflecting the high diversity of phages within bacterial communities. The distribution of prophages throughout the host genome, without a preferential integration site, suggests a complex dynamic of viral integration with important implications for bacterial evolution. Furthermore, the presence of antibiotic resistance genes encoded by prophages highlights their role as vectors in the dissemination of resistance determinants, posing a new challenge for the control of bacterial infections. Prophages coexist with their hosts and play key roles in bacterial evolution and resistance gene dissemination. Understanding this relationship helps explain bacterial genomic plasticity and adaptation. This knowledge also supports better strategies to monitor resistance and develop phage-based therapies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, LEAL-BALBINO.T.C and XAVIER, K.V.M; Methodology, XAVIER, K.V.M ; SILVA.A.M.A; LUZ.A.C.O; SILVA.F.S.C; MELO.B.S.T and PITTA.J.L.L.P; Validation, XAVIER, K.V.M; SILVA.A.M.A and LUZ.A.C.O; Formal analysis, XAVIER, K.V.M ; SILVA.A.M.A; LUZ.A.C.O; SILVA.F.S.C; MELO.B.S.T and PITTA.J.L.L.P; Investigation, XAVIER, K.V.M ; SILVA.A.M.A; Resources, LEAL-BALBINO.T.C; Data curation, LEAL-BALBINO.T.C; XAVIER, K.V.M ; SILVA.A.M.A; Writing—original draft preparation, XAVIER, K.V.M.; Writing—review and editing, LEAL-BALBINO.T.C; XAVIER, K.V.M ; SILVA.A.M.A; Visualization, XAVIER, K.V.M; supervision, LEAL-BALBINO.T.C and LUZ.A.C.O; Funding acquisition, LEAL-BALBINO.T.C.
Funding
This research was funded by Fundação Oswaldo Cruz. Keyla Xavier was financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)-Brazil—Finance Code 001.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| CRISPR/CAS | Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated genes |
| DNA | Deoxyribonucleic acid |
| ICEs | Integrative and Conjugative Elements |
| PAGIs | Pseudomonas aeruginosa Genomic Islands |
References
- Codjoe F, Donkor E. Carbapenem Resistance: A Review. Medical Sciences 2017;6:1. [CrossRef]
- WHO Bacterial Priority Pathogens List 2024: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance. 1st ed. Geneva: World Health Organization; 2024.
- Qin S, Xiao W, Zhou C, Pu Q, Deng X, Lan L, Liang H, Song X, Wu M. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Sig Transduct Target Ther 2022;7:199. [CrossRef]
- Luong T, Salabarria A-C, Roach DR. Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going? Clinical Therapeutics 2020;42:1659–80. [CrossRef]
- Davies EV, Winstanley C, Fothergill JL, James CE. The role of temperate bacteriophages in bacterial infection. FEMS Microbiology Letters 2016;363:fnw015. [CrossRef]
- Chevallereau A, Pons BJ, Van Houte S, Westra ER. Interactions between bacterial and phage communities in natural environments. Nat Rev Microbiol 2022;20:49–62. [CrossRef]
- Tsao Y-F, Taylor VL, Kala S, Bondy-Denomy J, Khan AN, Bona D, Cattoir V, Lory S, Davidson AR, Maxwell KL. Phage Morons Play an Important Role in Pseudomonas aeruginosa Phenotypes. J Bacteriol 2018;200. [CrossRef]
- Bucher MJ, Czyż DM. Phage against the Machine: The SIE-ence of Superinfection Exclusion. Viruses 2024;16:1348. [CrossRef]
- Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research 2018;46:W246–51. [CrossRef]
- Mohammed M, Casjens SR, Millard AD, Harrison C, Gannon L, Chattaway MA. Genomic analysis of Anderson typing phages of Salmonella Typhimrium: towards understanding the basis of bacteria-phage interaction. Sci Rep 2023;13:10484. [CrossRef]
- Luz ACDO, Da Silva JMA, Rezende AM, De Barros MPS, Leal-Balbino TC. Analysis of direct repeats and spacers of CRISPR/Cas systems type I-F in Brazilian clinical strains of Pseudomonas aeruginosa. Mol Genet Genomics 2019;294:1095–105. [CrossRef]
- Xavier KVM, De Oliveira Luz AC, Silva-Junior JW, De Melo BST, De Aragão Batista MV, De Albuquerque Silva AM, De Queiroz Balbino V, Leal-Balbino TC. Molecular epidemiological study of Pseudomonas aeruginosa strains isolated from hospitals in Brazil by MLST and CRISPR/Cas system analysis. Mol Genet Genomics 2025;300:33. [CrossRef]
- Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020;8:90. [CrossRef]
- Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 2011;27:334–42. [CrossRef]
- Gauthier CH, Cresawn SG, Hatfull GF. PhaMMseqs: a new pipeline for constructing phage gene phamilies using MMseqs2. G3 Genes|Genomes|Genetics 2022;12:jkac233. [CrossRef]
- Gauthier CH, Hatfull GF. PhamClust: a phage genome clustering tool using proteomic equivalence. mSystems 2023;8:e00443-23. [CrossRef]
- Meier-Kolthoff JP, Göker M. VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017;33:3396–404. [CrossRef]
- Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013;14:60. [CrossRef]
- Lefort V, Desper R, Gascuel O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program: Table 1. Mol Biol Evol 2015;32:2798–800. [CrossRef]
- Farris JS. Estimating Phylogenetic Trees from Distance Matrices. The American Naturalist 1972;106:645–68. [CrossRef]
- Yu G. Using ggtree to Visualize Data on Tree-Like Structures. CP in Bioinformatics 2020;69:e96. [CrossRef]
- Göker M, García-Blázquez G, Voglmayr H, Tellería MT, Martín MP. Molecular Taxonomy of Phytopathogenic Fungi: A Case Study in Peronospora. PLoS ONE 2009;4:e6319. [CrossRef]
- Alcock BP, Huynh W, Chalil R, Smith KW, Raphenya AR, Wlodarski MA, Edalatmand A, Petkau A, Syed SA, Tsang KK, Baker SJC, Dave M, McCarthy MC, Mukiri KM, Nasir JA, Golbon B, Imtiaz H, Jiang X, Kaur K, Kwong M, Liang ZC, Niu KC, Shan P, Yang JYJ, Gray KL, Hoad GR, Jia B, Bhando T, Carfrae LA, Farha MA, French S, Gordzevich R, Rachwalski K, Tu MM, Bordeleau E, Dooley D, Griffiths E, Zubyk HL, Brown ED, Maguire F, Beiko RG, Hsiao WWL, Brinkman FSL, Van Domselaar G, McArthur AG. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Research 2023;51:D690–9. [CrossRef]
- McNair K, Bailey BA, Edwards RA. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012;28:614–8. [CrossRef]
- Turner D, Shkoporov AN, Lood C, Millard AD, Dutilh BE, Alfenas-Zerbini P, Van Zyl LJ, Aziz RK, Oksanen HM, Poranen MM, Kropinski AM, Barylski J, Brister JR, Chanisvili N, Edwards RA, Enault F, Gillis A, Knezevic P, Krupovic M, Kurtböke I, Kushkina A, Lavigne R, Lehman S, Lobocka M, Moraru C, Moreno Switt A, Morozova V, Nakavuma J, Reyes Muñoz A, Rūmnieks J, Sarkar B, Sullivan MB, Uchiyama J, Wittmann J, Yigang T, Adriaenssens EM. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch Virol 2023;168:74. [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV).
- Allsopp LP, Wood TE, Howard SA, Maggiorelli F, Nolan LM, Wettstadt S, Filloux A. RsmA and AmrZ orchestrate the assembly of all three type VI secretion systems in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 2017;114:7707–12. [CrossRef]
- Van Belkum A, Soriaga LB, LaFave MC, Akella S, Veyrieras J-B, Barbu EM, Shortridge D, Blanc B, Hannum G, Zambardi G, Miller K, Enright MC, Mugnier N, Brami D, Schicklin S, Felderman M, Schwartz AS, Richardson TH, Peterson TC, Hubby B, Cady KC. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio 2015;6:e01796-15. [CrossRef]
- Galetti R, Andrade LN, Varani AM, Darini ALC. SPM-1-producing Pseudomonas aeruginosa ST277 carries a chromosomal pack of acquired resistance genes: An example of high-risk clone associated with ‘intrinsic resistome.’ Journal of Global Antimicrobial Resistance 2019;16:183–6. [CrossRef]
- Argov T, Azulay G, Pasechnek A, Stadnyuk O, Ran-Sapir S, Borovok I, Sigal N, Herskovits AA. Temperate bacteriophages as regulators of host behavior. Current Opinion in Microbiology 2017;38:81–7. [CrossRef]
- Olszak T, Latka A, Roszniowski B, Valvano MA, Drulis-Kawa Z. Phage Life Cycles Behind Bacterial Biodiversity. CMC 2017;24. [CrossRef]
- Dion MB, Oechslin F, Moineau S. Phage diversity, genomics and phylogeny. Nat Rev Microbiol 2020;18:125–38. [CrossRef]
- Shah S, Das R, Chavan B, Bajpai U, Hanif S, Ahmed S. Beyond antibiotics: phage-encoded lysins against Gram-negative pathogens. Front Microbiol 2023;14:1170418. [CrossRef]
- Luo J, Xie L, Yang M, Liu M, Li Q, Wang P, Fan J, Jin J, Luo C. Synergistic Antibacterial Effect of Phage pB3074 in Combination with Antibiotics Targeting Cell Wall against Multidrug-Resistant Acinetobacter baumannii In Vitro and Ex Vivo. Microbiol Spectr 2023;11:e00341-23. [CrossRef]
- Loh B, Chen J, Manohar P, Yu Y, Hua X, Leptihn S. A Biological Inventory of Prophages in A. baumannii Genomes Reveal Distinct Distributions in Classes, Length, and Genomic Positions. Front Microbiol 2020;11:579802. [CrossRef]
- Ramisetty BCM, Sudhakari PA. Bacterial ‘Grounded’ Prophages: Hotspots for Genetic Renovation and Innovation. Front Genet 2019;10:65. [CrossRef]
- Piel D, Bruto M, Labreuche Y, Blanquart F, Goudenège D, Barcia-Cruz R, Chenivesse S, Le Panse S, James A, Dubert J, Petton B, Lieberman E, Wegner KM, Hussain FA, Kauffman KM, Polz MF, Bikard D, Gandon S, Rocha EPC, Le Roux F. Phage–host coevolution in natural populations. Nat Microbiol 2022;7:1075–86. [CrossRef]
- Gogokhia L, Buhrke K, Bell R, Hoffman B, Brown DG, Hanke-Gogokhia C, Ajami NJ, Wong MC, Ghazaryan A, Valentine JF, Porter N, Martens E, O’Connell R, Jacob V, Scherl E, Crawford C, Stephens WZ, Casjens SR, Longman RS, Round JL. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host & Microbe 2019;25:285-299.e8. [CrossRef]
- Naser IB, Hoque MM, Abdullah A, Bari SMN, Ghosh AN, Faruque SM. Environmental bacteriophages active on biofilms and planktonic forms of toxigenic Vibrio cholerae: Potential relevance in cholera epidemiology. PLoS ONE 2017;12:e0180838. [CrossRef]
- Balcázar JL. Implications of bacteriophages on the acquisition and spread of antibiotic resistance in the environment. Int Microbiol 2020;23:475–9. [CrossRef]
- Haaber J, Leisner JJ, Cohn MT, Catalan-Moreno A, Nielsen JB, Westh H, Penadés JR, Ingmer H. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat Commun 2016;7:13333. [CrossRef]
- Hilbert M, Csadek I, Auer U, Hilbert F. Antimicrobial resistance-transducing bacteriophages isolated from surfaces of equine surgery clinics – a pilot study. EuJMI 2017;7:296–302. [CrossRef]
- Da Silva G, Domingues S. Insights on the Horizontal Gene Transfer of Carbapenemase Determinants in the Opportunistic Pathogen Acinetobacter baumannii. Microorganisms 2016;4:29. [CrossRef]
- Silveira MC, Albano RM, Asensi MD, Carvalho-Assef APD. Description of genomic islands associated to the multidrug-resistant Pseudomonas aeruginosa clone ST277. Infection, Genetics and Evolution 2016;42:60–5. [CrossRef]
- Do Nascimento APB, Medeiros Filho F, Pauer H, Antunes LCM, Sousa H, Senger H, Albano RM, Trindade Dos Santos M, Carvalho-Assef APD, Da Silva FAB. Characterization of a SPM-1 metallo-beta-lactamase-producing Pseudomonas aeruginosa by comparative genomics and phenotypic analysis. Sci Rep 2020;10:13192. [CrossRef]
Figure 1.
Distribution of viral regions according to CRISPR/Cas system types in positive bacterial isolates.Viral regions were grouped based on the CRISPR/Cas system type present in each isolate.
Figure 1.
Distribution of viral regions according to CRISPR/Cas system types in positive bacterial isolates.Viral regions were grouped based on the CRISPR/Cas system type present in each isolate.
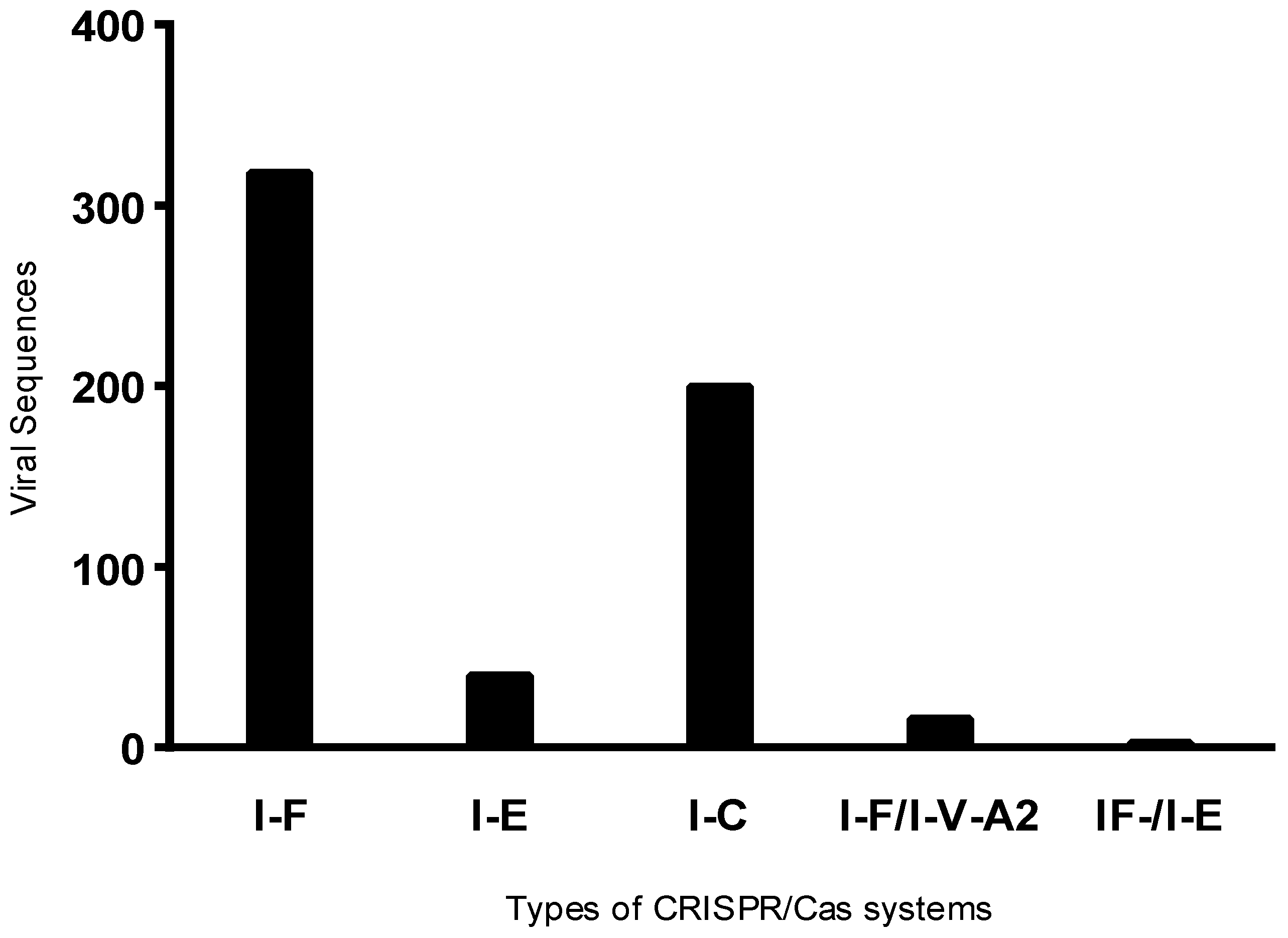
Figure 2.
Dendrogram showing the relationship between Pseudomonas aeruginosa isolates based on the type of CRISPR/Cas system, MLST profile, and number of prophages per genome.
Figure 2.
Dendrogram showing the relationship between Pseudomonas aeruginosa isolates based on the type of CRISPR/Cas system, MLST profile, and number of prophages per genome.
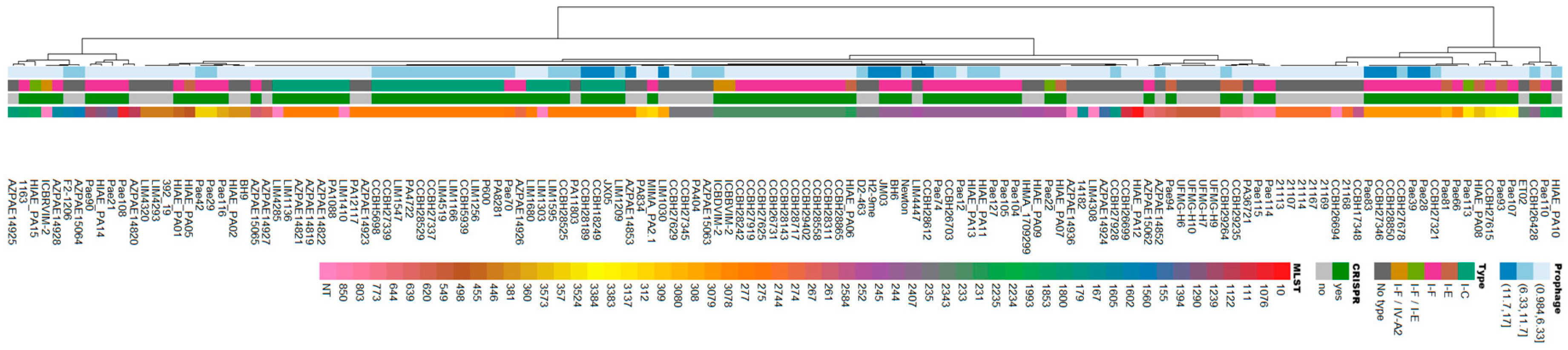
Figure 3.
Prevalence of genera belonging to the class Caudoviricetes.The identified genera belonging to this class were classified according to their frequency among the analyzed viral sequences.
Figure 3.
Prevalence of genera belonging to the class Caudoviricetes.The identified genera belonging to this class were classified according to their frequency among the analyzed viral sequences.
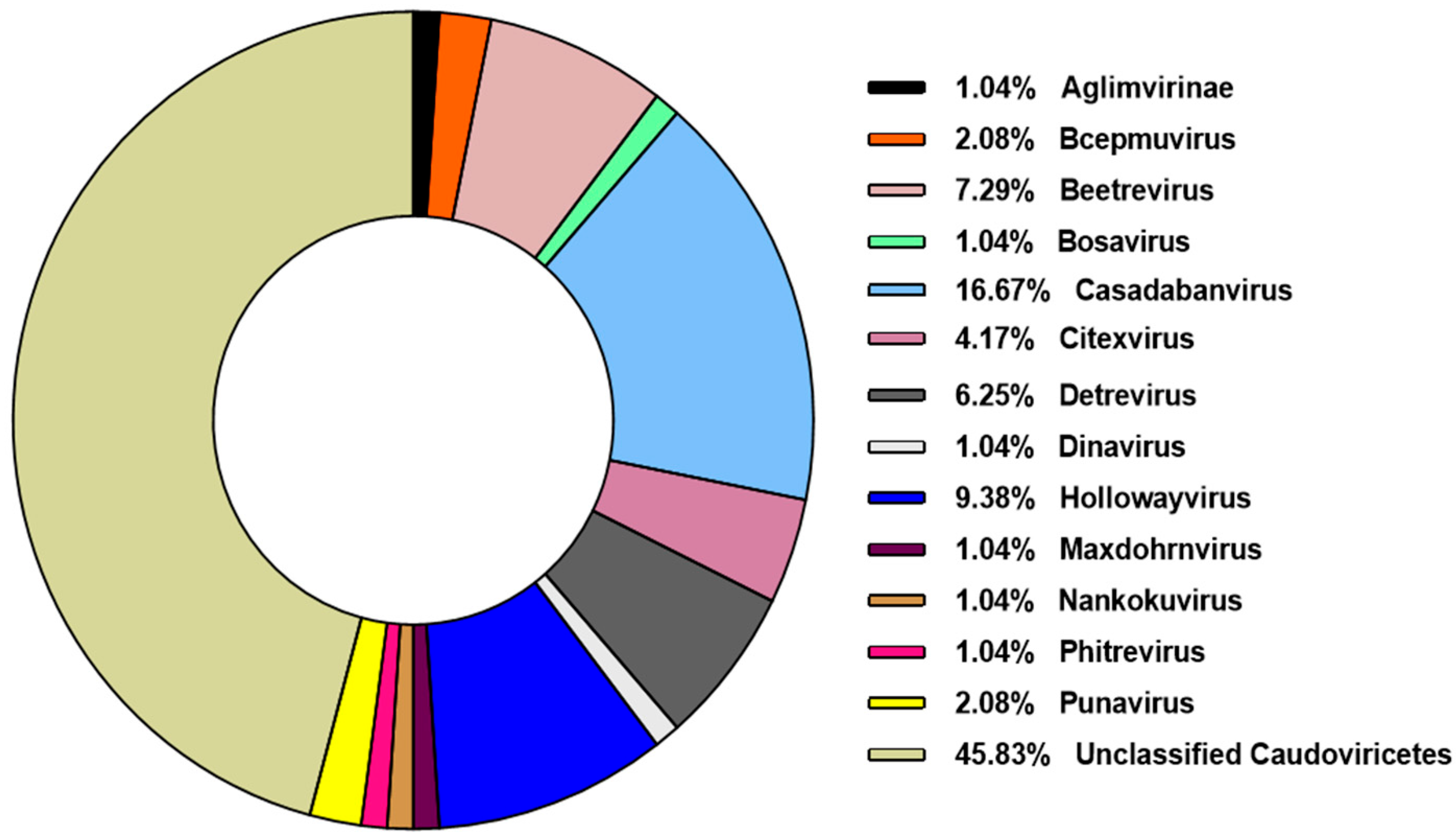
Figure 4.
Phylogenetic analysis of representative prophages identified in clinical isolates of Pseudomonas aeruginosa. The phylogenetic tree was constructed using the Genome-BLAST Distance Phylogeny (GBDP) method through the VICTOR platform, with 100 pseudo-bootstrap replicates to estimate branch support.
Figure 4.
Phylogenetic analysis of representative prophages identified in clinical isolates of Pseudomonas aeruginosa. The phylogenetic tree was constructed using the Genome-BLAST Distance Phylogeny (GBDP) method through the VICTOR platform, with 100 pseudo-bootstrap replicates to estimate branch support.
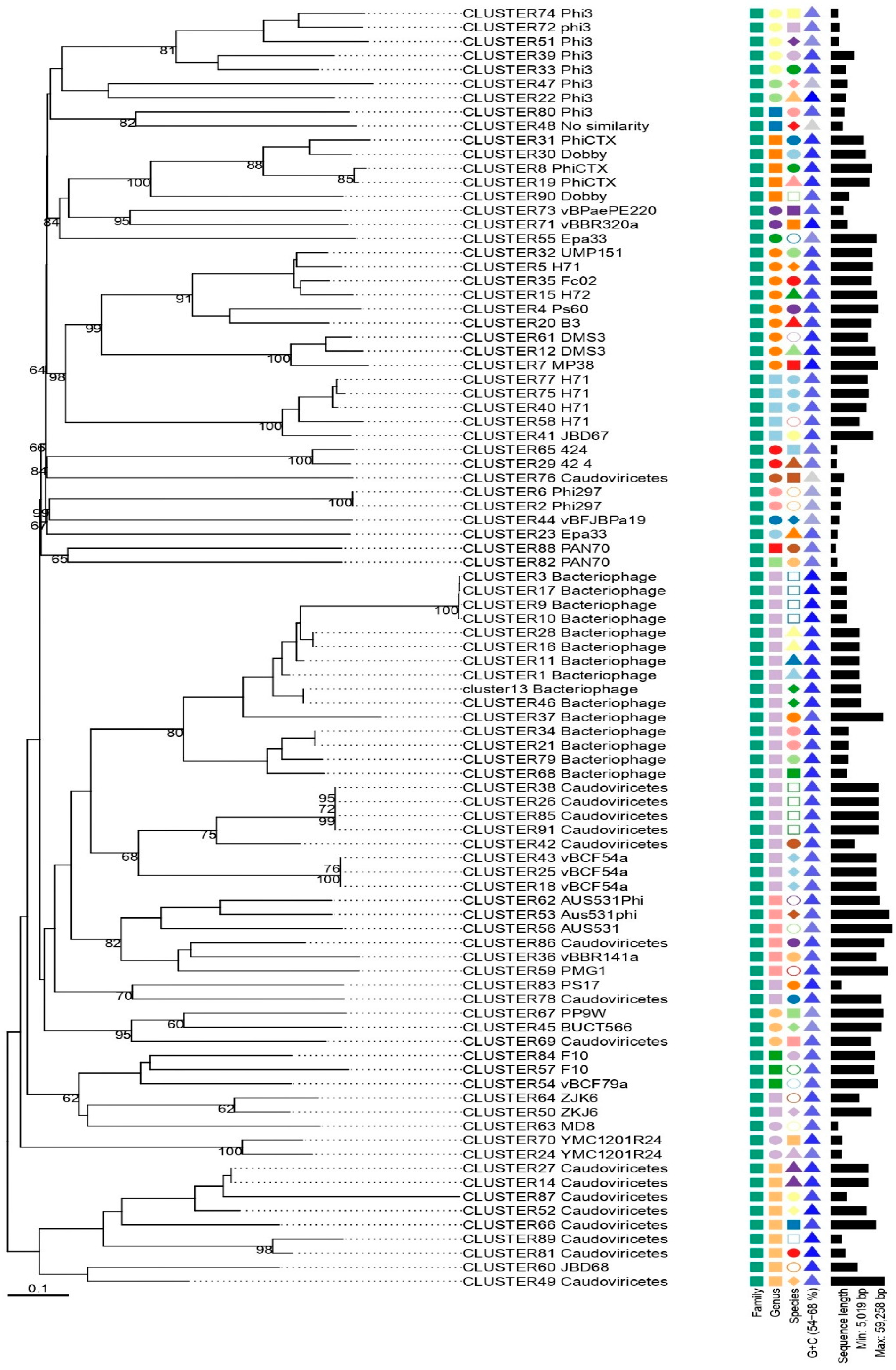
Figure 5.
Distribution of Prophages Identified in Clinical Isolates of Pseudomonas aeruginosa. A) Distribution of All Prophages Extracted from the Genomes of Pseudomonas aeruginosa. B) Distribution of Phage Phi3 along the Chromosome of Pseudomonas aeruginosa. C) Distribution of Phage Phi297 along the Chromosome of Pseudomonas aeruginosa.
Figure 5.
Distribution of Prophages Identified in Clinical Isolates of Pseudomonas aeruginosa. A) Distribution of All Prophages Extracted from the Genomes of Pseudomonas aeruginosa. B) Distribution of Phage Phi3 along the Chromosome of Pseudomonas aeruginosa. C) Distribution of Phage Phi297 along the Chromosome of Pseudomonas aeruginosa.
Figure 6.
Genetic comparison of DMS3 phage variants with and without the presence of a resistance gene. Figure A) shows the structure of a phage region lacking resistance genes, while Figure B) reveals the presence of the rsmA gene, with a highly conserved tail assembly region.
Figure 6.
Genetic comparison of DMS3 phage variants with and without the presence of a resistance gene. Figure A) shows the structure of a phage region lacking resistance genes, while Figure B) reveals the presence of the rsmA gene, with a highly conserved tail assembly region.
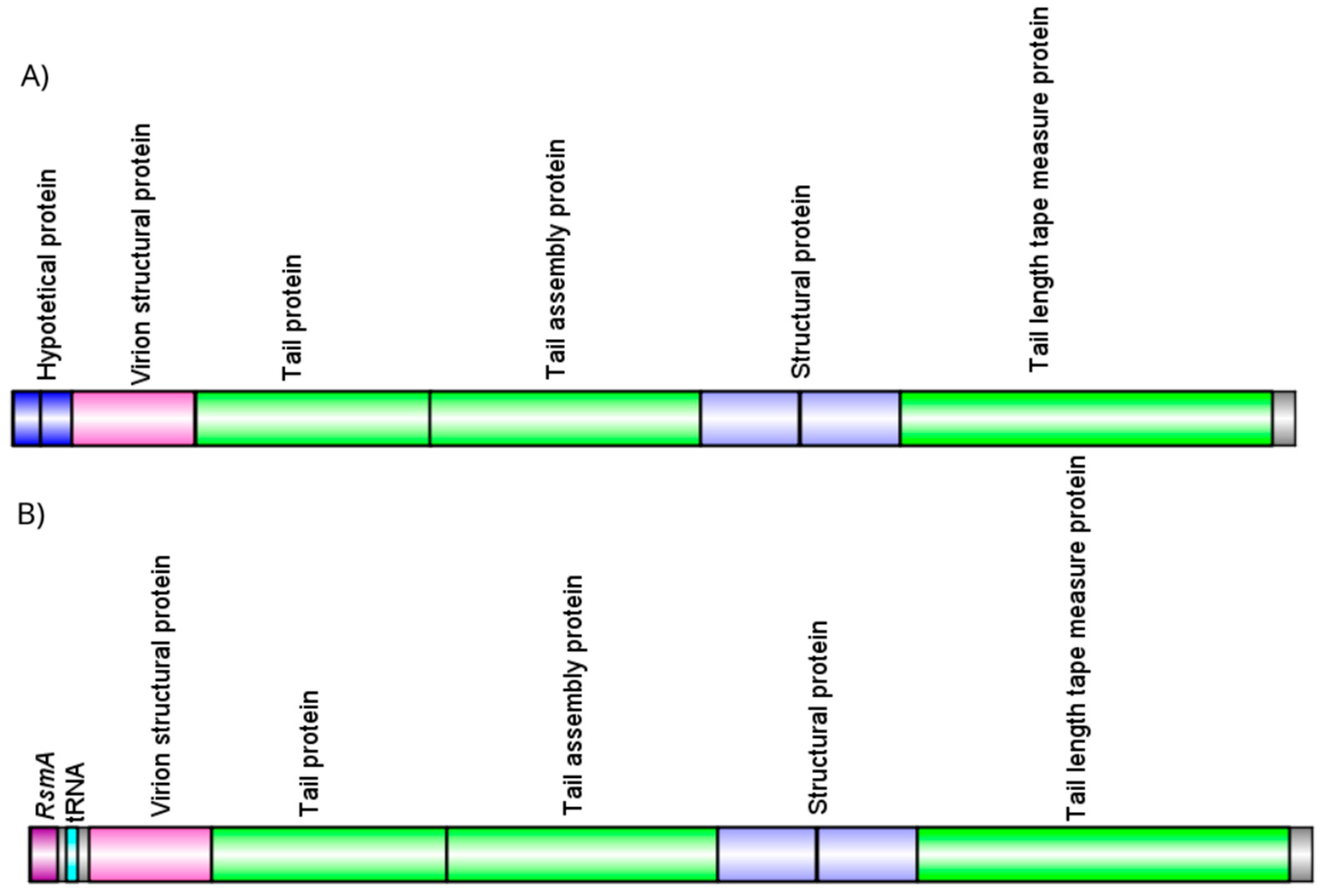
Figure 7.
Schematic representation of resistance genes in gene cassettes (aacA4, aadA7, and blaOXA-56) present in a viral sequence, including their adjacent genes.
Figure 7.
Schematic representation of resistance genes in gene cassettes (aacA4, aadA7, and blaOXA-56) present in a viral sequence, including their adjacent genes.
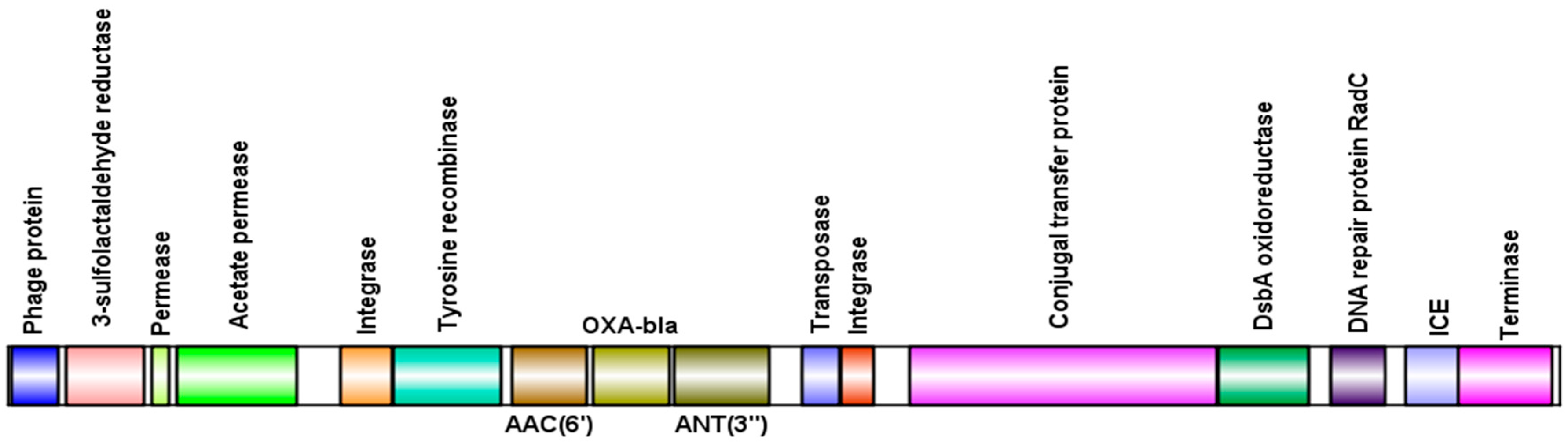

Table 1.
Most and least frequent prophages identified in Pseudomonas aeruginosa strains.
| Most common prophages | Less Common Prophages | |||
| Phage | Total | Phage | Total | |
| Bacteriophage sp. | 147 | Pseudomonas phage PAJU2 | 1 | |
| Caudoviricetes sp. | 136 | Escherichia phage P1 | 1 | |
| Pseudomonas phage phi3 | 50 | Pseudomonas phage JBD26 | 1 | |
| Pseudomonas phage phi297 | 35 | Pseudomonas phage PA8 | 1 | |
| Pseudomonas phage H71 | 32 | Pseudomonas phage JBD5 | 1 | |
| Pseudomonas phage Dobby | 25 | Burkholderia phage phiE255 | 1 | |
| Pseudomonas phage AUS531phi | 24 | Pseudomonas phage vB_Pae_CF125a | 1 | |
| Pseudomonas phage phiCTX | 22 | Ralstonia phage Dina | 1 | |
| Pseudomonas phage vB_Pae_CF54a | 16 | Pseudomonas phage D3112 | 1 | |
| Pseudomonas phage UMP151 | 15 | Pseudomonas phage MP42 | 1 | |
Table 2.
Statistical data on the distribution of phage-designated regions in the studied Pseudomonas aeruginosa genomes.
Table 2.
Statistical data on the distribution of phage-designated regions in the studied Pseudomonas aeruginosa genomes.
| Group Description | ||||||||||
| Group | N | Mean | median | Standard Deviation | Standard Error | |||||
| Phage Region Count | Positive | 93 | 6.31 | 6 | 3.64 | 0.377 | ||||
| Negative | 48 | 5.29 | 5 | 3.64 | 0.525 | |||||
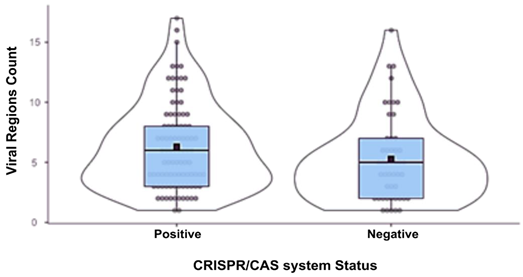 | ||||||||||
| Correlation matrix | ||||||||||
| Viral region count | CRISPR/Cas status | |||||||||
| CRISPR/Cas status | Spearman’s rho | 0,150 | - | |||||||
| p-value | 0,076 | - | ||||||||
| Note. * p < .05** p <.01*** p <.001 | ||||||||||
Table 3.
Pseudomonas aeruginosa isolates with the highest and lowest number of identified prophages.
Table 3.
Pseudomonas aeruginosa isolates with the highest and lowest number of identified prophages.
| Highest number of prophages identified | Fewest number of prophages identified | |||
| Isolate | Nº prophages | Isolate | Nº prophages | |
| CCBH28612 | 17 | AZPAE15065 | 1 | |
| H2-9me | 16 | ET02 | 1 | |
| Pae28 | 16 | Pae113 | 1 | |
| Pae39 | 15 | UFMG-H6 | 1 | |
| AZPAE14853 | 13 | UFMG-H7 | 1 | |
| CCBH27346 | 13 | UFMG-H9 | 1 | |
| JM03 | 13 | UFMG-H10 | 1 | |
| LIM1030 | 13 | Pae93 | 2 | |
| Pae83 | 13 | Pae94 | 2 | |
| BH6 | 12 | Pae110 | 2 | |
Table 4.
Antibiotic resistance genes observed in Pseudomonas aeruginosa genomes.
| Isolate | ST | Phage | AMR gene | Life cycle |
| AZEPAE14852 | 639 | Caudoviricetes sp. | rsmA | Lytic |
| AZEPAE14853 | 277 | Caudoviricetes sp. | rsmA | Lytic |
| CCBH27678 | 3079 | Caudoviricetes sp. | rsmA | Temperate |
| CCBH28189 | 277 | Caudoviricetes sp. | rsmA | Lytic |
| CCBH28529 | 277 | Pseudomonas phage phi3 | rmtD | Lytic |
| CCBH28529 | 277 | Caudoviricetes sp. | sul1 | Lytic |
| CCBH28850 | 3079 | Caudoviricetes sp. | dfrA21 | Lytic |
| CCBH28850 | 3079 | Caudoviricetes sp. | sul1 | Lytic |
| H2-9me | 235 | Pseudomonas phage DMS3 | rsmA | Temperate |
| JX05 | 277 | Pseudomonas phage AUS531phi | sul1 | Lytic |
| JX05 | 277 | Pseudomonas phage AUS531phi | sul1 | Lytic |
| JX05 | 277 | Pseudomonas phage AUS531phi | cmx | Lytic |
| JX05 | 277 | Pseudomonas phage AUS531phi | aadA7 | Lytic |
| JX05 | 277 | Pseudomonas phage AUS531phi | OXA-56 | Lytic |
| LIM1030 | 308 | Bacteriophage sp. | sul1 | Lytic |
| LIM1030 | 308 | Bacteriophage sp. | sul1 | Lytic |
| LIM1166 | 277 | Pseudomonas phage phi3 | cmx | Lytic |
| LIM1166 | 277 | Caudoviricetes sp. | rsmA | Temperate |
| LIM1166 | 277 | Caudoviricetes sp. | rsmA | Temperate |
| LIM1166 | 277 | Pseudomonas phage phi3 | cmx | Lytic |
| LIM1166 | 277 | Caudoviricetes sp. | rsmA | Temperate |
| LIM1256 | 277 | Caudoviricetes sp. | rsmA | Lytic |
| LIM1410 | 277 | Bacteriophage sp. | OXA-56 | Temperate |
| LIM1410 | 277 | Pseudomonas phage phi3 | P. aeruginosa emrE | Lytic |
| LIM1410 | 277 | Bacteriophage sp. | AAC(6')-Ib9 | Temperate |
| LIM1410 | 277 | Bacteriophage sp. | aadA7 | Temperate |
| LIM1547 | 277 | Bacteriophage sp. | sul1 | Temperate |
| LIM1547 | 277 | Bacteriophage sp. | cmx | Temperate |
| LIM1547 | 277 | Bacteriophage sp. | cmx | Temperate |
| LIM1547 | 277 | Bacteriophage sp. | sul1 | Temperate |
| LIM1680 | 277 | Caudoviricetes sp. | SPM-1 | Lytic |
| LIM1680 | 277 | Caudoviricetes sp. | OXA-56 | Lytic |
| LIM1680 | 277 | Caudoviricetes sp. | AAC(6')-Ib9 | Lytic |
| LIM1680 | 277 | Caudoviricetes sp. | aadA7 | Lytic |
| LIM4519 | 277 | Bacteriophage sp. | cmx | Lytic |
| LIM4519 | 277 | Caudoviricetes sp. | rsmA | Temperate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



