Submitted:
15 May 2025
Posted:
15 May 2025
You are already at the latest version
Abstract
Per- and poly fluoroalkyl substances (PFAS) pose serious health concerns worldwide. Although the use of classical PFAS, such as perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA), is regulated, the toxicological effects of alternative PFASs remain unknown. Cleft palate is a birth defect that is associated with environmental and genetic factors. Although an association between PFOS-induced and cleft palate has been reported, whether alternative PFASs affect cleft palate remains to be fully elucidated. The aim of this study was to clarify the involvement of classical and alternative PFAS (PFHxA and PFHxS) in the proliferation of human embryonic palatal mesenchymal cells (HEPM). Cell viability, apoptosis, and cell cycle-related markers were evaluated after PFASs treatment. In addition, miRNA levels and their downstream genes were measured, and a rescue experiment was performed using the miR-374a-5p inhibitor. Among the four PFASs, PFHxS decreased the number of cells with cyclin- and cyclin-dependent kinase reduction. In addition, PFHxS treatment upregulated miR-374a-5p and its downstream genes. Furthermore, miR-374a-5p inhibitor alleviated the PFHxS-induced inhibition of cell proliferation. These results suggest that PFHxS-induced cleft palate associated with miR-374a-5p and alternative PFAS may have a highly toxic effect on HEPM cells.
Keywords:
Perfluoroalkyl and Polyfluoroalkyl Substance
; cleft palate
; microRNA
; cell cycle arrest
1. Introduction
Gynecological disorders encompass a broad range of conditions that affect the female reproductive system, including the uterus, ovaries, fallopian tubes, uterine cervix, and vagina [1]. These disorders can significantly affect women's health, fertility, and quality of life and range from benign conditions such as polycystic ovary syndrome (PCOS), endometriosis, and uterine fibroids to malignant diseases such as cervical, ovarian, and endometrial cancers [1,2]. Several studies have suggested that gynecological disorders may affect the fetus and the course of pregnancy [3,4]. For example, patients with uterine fibroids have an increased risk of placental abnormalities and preterm birth [5]. Pregnant women with endometriosis have an increased risk of miscarriage and preterm birth [6]. Infants of women with PCOS have an increased risk of congenital anomalies, such as cleft lip (CL) with or without cleft palate (CP) (CL/P) [7].
CL/P is a common congenital anomaly, affecting approximately one in every 500–700 babies born in Asia, and is one of the most frequent birth defects worldwide. Studies have reported that 70% of CL/P cases are non-syndromic, with the remaining 30% linked to syndromic conditions [8,9]. In humans, lip formation begins in the fourth week of gestation and is completed by approximately the seventh week. The development of the secondary palate begins around the sixth week of gestation and is typically completed by the twelfth week. Despite differences in the timing of lip and secondary palate formation, the underlying processes, such as mesenchymal cell proliferation, the fusion of two epithelial cells, and epithelial-mesenchymal transition (EMT), are similar. Disruption of these processes results in CL/P [10,11].
The etiology of CL/P is influenced by a combination of genetic and environmental factors [12,13]. Multiple types of signaling pathway disruptions, resulting from gene deletions or mutations, have been implicated in the development of CL/P through the inhibition of mesenchymal cell proliferation, preventing the fusion of epithelial cells, and impairing EMT [14,15]. For example, the WNT signaling pathway plays an important role in secondary palate formation through by regulating Paired box gene 9 [16,17,18]. A feedback loop between the WNT signaling pathway and Irf6 regulates morphological changes in palatal shelves [19,20]. The bone morphogenetic protein (BMP) signaling plays a critical role in craniofacial development because it regulates key cellular processes including apoptosis, differentiation, and proliferation [21,22,23]. Msx1 knockout mice exhibit mouse CP through the downregulation of WNT and BMP signaling pathways [24,25]. JARID2 exhibits high and specific expression in epithelial cells and is functionally linked to the BMP and transforming growth factor (TGF) β signaling [26,27]. Conditional knock-out of Sirt6 increased mouse embryonic palatal mesenchyme (MEPM) cell proliferation through upregulation of TGF signaling pathway [28]. A systematic review has identified more than 130 genes linked to the occurrence of CP in human [29], whereas more than 190 genes are linked to mouse CP [30]. Suzuki et al. identified 55 genes involved in mouse CL [31] and Yoshioka et al. reported that 173 genes are associated with human CL [32]. While recent studies have identified novel gene mutations linked to orofacial cleft, including CL/P [33], the mechanism by which CL/P is induced remains unknown. Maternal exposure to environmental factors is known to increase the risk of CL/P [34], such as alcohol consumption [35], cigarette smoking [36], and the use of certain medications during pregnancy [37,38]. Exposure to certain environmental factors can lead to CL/P by altering the expression of crucial developmental genes or by perturbing key signaling pathways involved in craniofacial development. Recent studies have identified new environmental factors, including PM2.5, as being associated with the development of CP [39]; therefore, we need to understand how environmental factors are involved in the etiology of CL/P.
Perfluoroalkyl and polyfluoroalkyl substances (PFAS) are a large group of synthetic chemicals that have been manufactured and utilized in various industries since the 1940s [40,41]. There are more than 4700 PFAS compounds in the world [42]. The two most common PFAS compounds are perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), which were produced in large quantities during the 2000s and 2010s [43,44]. The health effects of PFOS and PFOA on adults and fetuses have become serious concerns [45,46,47]. Although the manufacture and import of products containing PFOA and PFOS were banned in Japan in 2010, and 2021, respectively, these substances are still being detected because of their high persistence in the environment [48]. In recent years, perfluorohexanoic acid (PFHxA) and perfluorohexanesulfonic acid (PFHxS) have been employed as alternatives to PFOA and PFOS. However, their toxicological profiles are poorly understood. Environmental exposure to PFAS in drinking water may increase the risk of CL/P [49,50]. In experimental models, exposure to PFOS induces CP in mice [51,52]. However, the molecular mechanisms underlying PFOS-induced CL/P have not yet been reported. Furthermore, it remains unclear whether other PFAS induce CL/P. In this study, we investigated whether two classical PFAS (PFOA and PFOS) and two alternative PFAS (PFHxA and PFHxS) reduce the number of human embryonic palatal mesenchyme (HEPM) cells. Furthermore, we explored the molecular mechanisms focusing on microRNAs (miRNAs).
2. Materials and Methods
2.1. Cell Culture
HEPM cells (JCRB9095) were obtained from the JCRB Cell Bank (Osaka, Japan) and cultured in Minimum Essential Medium Eagle-alpha modification (αMEM) supplemented with 10% fetal bovine serum, and penicillin/streptomycin mixture. HEPM cells were maintained at 37 °C in a humidified incubator with 5% CO2.
2.2. Cell Proliferation Assay
HEPM cells were seeded at a density of 5,000 cells per well in 96-well plates (n=6) and treated 24 h post-seeding with 0–100 μM of PFOA, PFOS, and PFHxS or with 0–2 μM of PFHxA. Following 24 h of treatment (PFOA, PFOS, PFHxA, or PFHxS), cell count was assessed using Alamar Blue, and fluorescence was measured at Ex540/Em600.
2.3. Apoptosis Assay
HEPM cells were seeded at a density of 10,000 cells per chamber and treated with either 100 μM PFHxS or vehicle control (0.1% DMSO). After 48 h of treatment, apoptotic cells were detected using ApoTracker Green [53,54]. The nuclei of the cells were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI).
2.4. Western Blot Analysis
HEPM cells were seeded at a density of 200,000 cells per well in 35 mm dish plates and treated with 100 µM PFHxS or 0.1% DMSO after 24 h of cell seeding. After 48 h of treatment, the cells were washed two times and 100 µL lysis buffer with a protease inhibitor cocktail was added and the cells were incubated at 4°C for five minutes. Subsequently, the cells were collected (20,000 ×g for 20 min at 4°C) [55,56]. Protein samples (10 µg) were separated by 5-20 % gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to the polyvinylidene difluoride membranes. Western blotting was carried out with the antibodies listed below: anti-mouse monoclonal cyclin-D1 (CCND1) antibody (1:750 dilution), anti-mouse monoclonal CCNE (1:1,000 dilution), anti-mouse monoclonal BAX (1:1000 dilution), anti-mouse monoclonal cyclin-dependent kinase-2 (CDK2) antibody (1:1,000 dilution), anti-mouse monoclonal CDK4 antibody (1:1,000 dilution), anti-mouse monoclonal CDK6 antibody (1:2,000 dilution), anti-rabbit polyclonal cleaved CASPASE-3 antibody (1:3,000 dilution), and anti-mouse monoclonal β-actin antibodies (1:4,000 dilution). Peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG were used as secondary antibodies at a dilution of 1:10,000 dilution). Detection of immunoreactive bands was achieved using substrate.
2.5. Bromodeoxyuridine (BrdU) Incorporation Assay
HEPM cells were seeded at a density of 10,000 cells per chamber and treated with either 100 μM PFHxS or vehicle control (0.1% DMSO). After 48 h of treatment, the cells were incubated with 100 μg/mL BrdU for 1 h. The BrdU in the nucleous was detected using an anti-mouse monoclonal BrdU antibody (1:150 dilution) and CoraLite594-conjugated anti-mouse IgG (1:180 dilution). DAPI was used to counterstain the nuclei and the number of BrdU-positive cells was counted across eight fields.
2.6. Quantitative RT-PCR
HEPM cells were seeded at a density of 200,000 cells per well in 35 mm dish plates and treated with 100 µM PFHxS or 0.1% DMSO after 24 h of cell seeding. After 48 h of treatment, the cells were washed two times and total RNA was extracted from the cells using miRNA detectable Kit, following the procedure we previously reported [57,58]. For miRNA detection, total RNA was reverse-transcribed using an miRNA Reverse Transcription Reaction Kit. MiRNA expression was assessed using an all-in-one miRNA qRT-PCR Detection Kit. Information of Probe and PCR conditions were detailed in earlier studies [53]. For normal gene detection, the protocol for reverse transcription and PCR conditions were detailed in earlier studies [55]. Target mRNA levels were normalized to the β-actin levels. The sequences of the primer sets used are as follows: human β-actin (NM_001101), sense 5′-ACCTTCTACAATGAGCTGCGTG-3′ and antisense 5′-TGGGGTGTTGAAGGTCTCAAAC-3′; human cysteine-rich secretory protein LCCL domain containing 1 (CRISPLD1; NM_031461), sense 5′-ATCACAGACAATGACATGCAGAG-3′ and antisense 5′-AGATCTTTCCAGCTCTACATCCC-3′; human fibroblast growth factor receptor 2 (FGFR2; NM_000141), sense 5′-ATGAGGATGACACCGATGGTG-3′ and antisense 5′-TTGACAGTGTTGGCCGCAG-3′; human jumonji and AT-rich interaction domain containing 2 (JARID2; NM_001267040), sense 5′-CTTCATCTTCATGCCAGTCGAC-3′ and antisense 5′-CAGGTTCCTTCTCCCGTGTTG-3′; human msh homeobox 1 (MSX1; NM_002448), sense 5′-GAAGATGCGCTCGTCAAAGC-3′ and antisense 5′-GGTTCGTCTTGTGTTTGCGGAG-3′; and human zinc finger protein 236 (ZNF236; NM_001306089), sense 5′-AGAGTGGCTAGTCTCAAAGCG-3′ and antisense 5′-CAGCTGACTCTGCAGAGTAAAC-3′.
2.7. Rescue Experiments
HEPM cells were seeded at a density of 5,000 cells per well in 96-well plates or a density of 200,000 cells per well in 35 mm dish plates. After 6 h, the cells were treated with 3 or 60 pmol of miR-374a-5p inhibitor or 3 or 60 pmol of control miRNA inhibitor using FuGENE SI Transfection Reagent, following the manufacturer's protocol. Twenty-four hours after transfection, the cells were treated with 100 μM PFHxS or vehicle control (0.1% DMSO) for 48 h. Cell viability and gene expression levels were assessed using the methods described above.
2.8. Statistical Analysis
Comparisons between two groups were performed using Student’s t-test, whereas comparisons among multiple groups were conducted using Tukey’s test. All statistical analyses were performed using SPSS Statistics for Windows (version 27.0). A p-value of <0.05 was considered statistically significant.
3. Results
3.1. PFHxS Reduced Cell Viability in HEPM Cells
Initially, a cell viability assay was conducted to evaluate whether the four PFAS compounds reduced HEPM cell viability. Among the four PFAS, three PFAS (PFOA, PFOS, and PFHxA) did not alter the cell number (Figure 1a, 1b, 1c). By contrast, treatment with 100 μM PFHxS significantly reduced the viability of HEPM cells (40% reduction, Figure 1d). Therefore, we conducted further experiments using 100 μM PFHxS.
3.2. PFHxS Reduced Cell Viability Through G1 Cell Cycle Arrest
It has been reported in several investigations that chemical-induced impaired proliferation of mesenchymal palatal cells can lead to apoptosis and cell cycle arrest [59,60,61]. Therefore, we examined whether PFHxS treatment induces apoptosis and/or cell cycle arrest in HEPM cells. As shown in Figure 2a, treatment with PFHxS did not lead to apoptosis, whereas the positive control treatment did, suggesting that PFHxS did not affect HEPM cell apoptosis. In support of this result, the expression of cleaved CASPASE-3 and BAX, markers of apoptosis, was not detected by treatment with PFHxS (Figure 2b).
- (A)
- HEPM cells treated with 100 µM PFHxS for 48 h and stained with Apotracker. Nuclei were stained with DAPI. P.C.; positive control (CuCl2). Scale bar, 50 μm.
- (B)
- HEPM cells treated with 100 μM PFHxS for 48 h were subjected to western blotting. β-Actin serving as reference control. P.C.; positive control (CuCl2).
Subsequently, cell cycle progression was assessed through a BrdU incorporation assay, revealing a significant reduction in the number of BrdU-positive cells following PFHxS treatment (Fig. 3a). To investigate the molecular mechanisms of PFHxS-induced cell cycle arrest (G1- cell cycle arrest), we analyzed the of cyclins and CDKs protein levels using western blotting. PFHxS treatment downregualted the protein levels of CCND1, CDK4, and CDK6, whereas those of CCNE and CDK4 remained unchanged (Fig. 3b). These results indicated that PFHxS induced G1 cell cycle arrest by inhibiting the CCND1/CDK4 and CCND1/CDK6 pathways in HEPM cells.
- (A)
- HEPM cells were stained for BrdU (green) after 48-hour treatment with 100 μM PFHxS. Nuclei were stained with DAPI (blue). Scale bar, 50 μm. BrdU-positive cell rates are shown. Values are expressed as mean ± standard deviation (SD). **p<0.01 (Student’s t-test) (n=8).
- (B)
- HEPM cells treated with 100 μM PFHxS for 48 h were subjected to western blotting. β-Actin serving as reference control.
3.3. PFHxS Upregulates miR-374a-5p
Several literatures have demonstrated a link between miRNAs and etiology of CL/P [11,62]. Suzuki and Li et al. reported several miRNAs (miR-133b, miR-140-5p, miR-374a-5p, miR-381-3p, and miR-4680-3p) associated with human CP-related genes through a combination of approaches, including systematic reviews, bioinformatics analyses, cell proliferation assays, and qPCR [29,63]. In the present study, we evaluated the expression levels of five miRNAs (miR-133b, miR-140-5p, miR-374a-5p, miR-381-3p, and miR-4680-3p) using qPCR. PFHxS treatment significantly upregulated miR-374a-5p expression, while no changes were observed in the other four miRNAs (Fig. 4).
Expression levels of miR-133b, miR-140-5p, miR-374a-5p, miR-381-3p, and miR-4680-3p were measured using quantitative RT-PCR after treatment of HEPM cells with 100 μM PFHxS for 48 h. Data are shown as the mean ± SD. **p<0.01 (Student’s t-test) (n=3).
3.4. Blocking miR-374a-5p Alleviates PFHxS-Cediated Cell Proliferation Inhibition
To elucidate the role of miR-374a-5p, HEPM cells were transfected with a miR-374a-5p inhibitor to determine whether miR-374a-5p could reverse the PFHxS-induced inhibition of cell proliferation. Under our experimental conditions, expression of miR-374a-5p expression level was reduced by over 85% following transfection with its inhibitor (Fig. 5a). Subsequently, after transfection with miR-374a-5p inhibitor, HEPM cells were treated with PFHxS. We found that miR-374a-5p inhibitor partially restored cell viability, that was inhibited by PFHxS (Fig. 5b). Furthermore, treatment with an miR-374a-5p inhibitor significantly upregulated the gene expression level of CRISPLD1 and FGFR2. The PFHxS-induced downregulation of these genes was significantly alleviated by miR-374a-5p inhibition in HEPM cells (Fig. 5c). These findings imply that miR-374a-5p contributes to PFHxS-induced suppression of HEPM cell proliferation.
- (A)
- Expression level of miR-374a-5p were measured using quantitative RT-PCR after after transfection of HEPM cells with miR-374a-5p inhibitor for 24 h. Data are shown as the mean ± SD. ***p<0.001 (Tukey’s test) (n=3).
- (B)
- HEPM cell proliferation after 48 h of treatment with 100 μM PFHxS, the miR-374a-5p inhibitor, or their combination. Data are shown as the mean ± SD. **p<0.01 and ***p<0.001 (Tukey’s test) (n=6).
- (C)
- Expression level of five predicted genes were evaluated by quantitative RT-PCR following transfection of HEPM cells with the miR-374a-5p inhibitor and/or treatment with 100 μM PFHxS for 48 h. Data are shown as the mean ± SD. *p<0.05, **p<0.01, and ***p<0.001 (Tukey’s test) (n=4).
4. Discussion
In the present study, we examined the effects of four types of PFAS on HEPM cells. Among the four PFAS, PFHxS treatment significantly reduced cell number (Fig. 1d) and the protein levels of CCND1, CDK4, and CDK6 (Figure 3b). A recent report suggested that PFHxS reduces Ccnd1 levels in zebrafish [64], supporting the possibility that PFHxS induces cell cycle arrest (Figure 3a, 3b). PFHxS treatment upregulated miR-374a-5p expression (Figure 4) and decreased its downstream genes expression level (Figure 5c). Importantly, transfection with a miR-374a-5p inhibitor partially alleviated PFHxS-induced cell inhibition (Figure 5b).
Cyclins and CDKs are essential for cell cycle progression [65]. When cells exit the G0 phase and re-enter the cell cycle, CDK4 and CDK6 form active complexes with CCND and other proteins, including the phosphorylated retinoblastoma protein. This interaction facilitates the transition from the G1 phase to the S phase [66]. Moreover, the members of the retinoblastoma protein family are phosphorylated and inactivated by the CDK2–CCNE complex [67], leading to the release of transcription factors, such as E2F, which further drives the G1/S transition. The overexpression of CCND1 represents a central mechanism contributing to therapeutic resistance across multiple cancer types [68,69]. Cdk2 knockout mice has been shown to reduce neural progenitor cell viability [70]. CCNE overexpression also contributes to accelerated G1 phase progression in cancer patients [71]. Targeting these mechanisms, CDK4/6 inhibitors namely abemaciclib, palbociclib, and ribociclib are approved therapies for the epidermal growth factor receptor 2-negative breast cancer [72,73]. Western blot analysis revealed that PFHxS treatment decreased the protein expression of CCND1, CDK4, and CDK6 (Figure 3b). These findings suggest that the PFHxS-induced reduction in cell viability is likely due to G1 phase arrest mediated by the suppression of CCND1/CDK4 and CCND1/CDK6 complexes in HEPM cells.
miRNAs, which are small non-coding RNAs measuring approximately 20–24 nucleotides, are well known for their role as post-transcriptional negative regulators of gene expression. The discovery of the first miRNA in 1993 [74,75] was a landmark event, marking the onset of a new era in RNA biology. These evolutionarily conserved molecules are widely distributed across diverse organisms, and recent research suggests that over 2,500 distinct miRNAs have been identified in the human genome [76]. Recent studies have highlighted the role of miRNAs in the regulation of CL/P [77]. Specifically, the miR-17–92 cluster regulates cell proliferation and cell cycle progression in palatal mesenchymal cells [78]. Mutations in this cluster are associated with severe craniofacial abnormalities [79]. Furthermore, polymorphisms in pre-miR-146a have been shown to influence the expression of tumor necrosis factor receptor-associated factor 6, thereby contributing to the pathogenesis of CP [80]. Li and Suzuki et al. found that the five miRNAs (miR-133b, miR-140-5p, miR-374a-5p, miR-381-3p, and miR-4680-3p) mimic inhibited HEPM cell proliferation [29,63]. Several mouse miRNAs such as miR-27a-3p, miR-27b-3p, miR-124-3p, miR-129-5p, miR-214-3p, miR-340-5p, and miR-486b-5p mimic reduces the number of MEPM cells [38,81,82,83]. Transfection with miR-497-5p and miR-655-3p mimic significantly reduced the viability of cultured human lip fibroblast cells [84]. PFHxS significantly induced the expression of miR-374a-5p in HEPM cells (Figure 4). Moreover, application of a specific inhibitor targeting miR-374a-5p partially mitigated PFHxS-induced inhibition of HEPM cell proliferation (Figure 5b), indicating that miR-374a-5p plays a crucial role in the toxicity associated with PFHxS treatment. Human miR-374a-5p is located on the X chromosome and miR-374a-5p are involved in cell proliferation [85]. miR-374a-5p is associated with WNT/b-catenin singling pathway [86] and overexpression of miR-374a-5p significantly reduced cell viability in human non-small lung carcinoma cell lines A549 and H1299 [87]. Among the five predicted downstream genes (CRISPLD1, FGFR2, JARID2, MSX1, and ZNF236) of miR-374a-5p (Supplementary Figure S1), inhibition of miR-374a-5p significantly upregulated the expression of CRISPLD1 and FGFR2 in HEPM cells (Figure 5c). These findings indicated that CRISPLD1 and FGFR2 play a crucial role in palatal development.
CRISPLD1 is a member of a highly conserved cysteine-rich secretory protein family. This molecule has been implicated in facial morphogenesis, the folate metabolic pathway, and the cellular stress response of chondrocytes upon interleukin-1α stimulation [88]. Single-nucleotide polymorphisms in CRISPLD1/CRISPLD2 have been associated with variations in folate pathway-related genes that contribute to the susceptibility to CL/P [89]. CRISPLD1 inhibition by siRNA was significantly reduced in the human stomach carcinoma cell line HGC-27 through modulation of the PI3K-AKT signaling pathway [90]. FGFR2, including its isoforms FGFR2b and FGFR2c, functions as a receptor for fibroblast growth factors and mediates the RAS/ERK and PI3K/AKT signaling pathways [91,92]. FGFR2 has been reported to regulates cyclin dependent pathways particularly modulation of CCND1 expression [93]. Moreover, CDK4/6 activity has been linked to FGFR2 signaling through the modulation of the MAPK pathway [94]. FGFR2 is essential for palate morphogenesis and plays an important role in CL/P. In Fgfr2 knockout mice, CP was observed, accompanied by a reduction in cell proliferation within both the palatal epithelium and mesenchyme [95]. Given that these genes are involved in multiple signaling pathways associated with cell proliferation, the identified miRNA–mRNA networks may play a critical role in palate development by modulating these pathways.
This study has some limitations. (1) Owing to regulatory restrictions on the purchase of PFOS, we were unable to test concentrations higher than 2 μM in our cell viability assays. Further studies are required to directly compare the toxic effects of PFOS and PFHxS under equivalent experimental conditions. (2) The present study was conducted using an in vitro model. To better understand the in vivo relevance of our findings, it is important to investigate whether PFHxS exposure induces CP in mouse models. However, since the miRNA-gene datasets differ between human and mouse, it remains unknown whether PFHxS induces mesenchymal cell proliferation inhibition through modulation of miRNAs . (3) Our investigation focused on specific miRNAs identified in previous studies by other research groups. To gain a more comprehensive understanding of the miRNA response to PFHxS, future studies should employ miRNA-seq to identify additional miRNAs that may be affected. Further investigations are needed in the future.
5. Conclusions
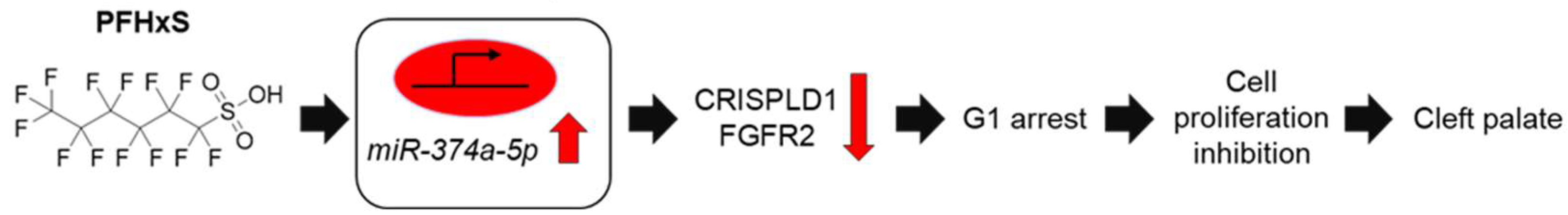
In conclusion, our study demonstrates that PFHxS inhibits cell proliferation by modulating the miR-374a-5p-CRISPLD1 and FGFR2-CCND1/CDK4/CDK6 pathways (Figure 6). To the best of our knowledge, this is the first report to associate PFHxS with miRNAs and inhibit palatal cell growth. Although further research is required to elucidate the specific mechanisms through which miR-374a-5p regulates the G1 phase, our findings provide valuable insights into the potential roles of environmental factors in the etiology of CP.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Identification of putative miR-374a-5p target genes and binding sites associated with cleft palate
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, H.Y. and Y.T.; methodology, H.Y.; software, H.Y. and H.H. (Hanane Horita); validation, H.Y., H.H. (Hanane Horita) and K.O.; formal analysis, H.Y. and H.H. (Hanane Horita); validation, H.Y., H.H. (Hanane Horita) and K.O; investigation, H.Y. and H.H. (Hanane Horita); resources, H.Y., H.H. (Hyogo Horiguchi), and Y.T. ; data curation, H.Y. and H.H. (Hanane Horita); writing—original draft preparation, H.Y.; writing—review and editing, H.H. (Hanane Horita), H.H. (Hyogo Horiguchi), and Y.T.; visualization, H.Y., H.H. (Hanane Horita) and K.O..; supervision, H.Y. and Y.T.; project administration, H.Y.; funding acquisition, H.Y. and Y.T.
Funding
This research was funded by Gifu University of Medical Science research grant A and JSPS KAKENHI Grant Numbers 25K20439.
Data Availability Statement
All relevant data are within the manuscript.
Acknowledgments
The authors thank Aya Ogata (Gifu University, Japan) for her kind suggestions.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BMP | bone morphogenetic protein |
| CCND1 | Cyclin D1 |
| CDK | cyclin-dependent kinases |
| CL | Cleft lip |
| CP | Cleft palate |
| CL/P | Cleft lip with or without cleft palate |
| CRISPLD1 | cysteine-rich secretory protein LCCL domain containing 1 |
| DAPI | 4′,6-diamidine-2′-phenylindole dihydrochloride |
| EMT | epithelial-mesenchymal transition |
| FGFR | fibroblast growth factor receptor |
| HEPM | human embryonic palatal mesenchymal |
| JARID2 | jumonji and AT-rich interaction domain containing 2 |
| IgG | immunoglobulin G |
| miRNA | microRNA |
| MSX1 | msh homeobox 1 |
| PCOS | polycystic ovary syndrome |
| PFAS | perfluoroalkyl substances |
| PFHxA | perfluorohexanoic acid |
| PFHxS | perfluorohexanesulfonic acid |
| PFOA | perfluorooctanoic acid |
| PFOS | perfluorooctanesulfonic acid |
| TGF | transforming growth factor |
| ZNF236 | zinc finger protein 236 |
References
- Tomas, E. J.; Valdes, Y. R.; Davis, J.; Kolendowski, B.; Buensuceso, A.; DiMattia, G. E.; Shepherd, T. G., Exploiting Cancer Dormancy Signaling Mechanisms in Epithelial Ovarian Cancer Through Spheroid and Organoid Analysis. Cells 2025, 14, (2). [CrossRef]
- Zeber-Lubecka, N.; Ciebiera, M.; Hennig, E. E., Polycystic Ovary Syndrome and Oxidative Stress-From Bench to Bedside. Int J Mol Sci 2023, 24, (18). [CrossRef]
- Cheng, C. G.; Su, S. H.; Chien, W. C.; Chen, R.; Chung, C. H.; Cheng, C. A., Diabetes Mellitus and Gynecological and Inflammation Disorders Increased the Risk of Pregnancy Loss in a Population Study. Life (Basel) 2024, 14, (7). [CrossRef]
- MacLean, J. A., 2nd; Hayashi, K., Progesterone Actions and Resistance in Gynecological Disorders. Cells 2022, 11, (4).
- Tirnovanu, M. C.; Lozneanu, L.; Tirnovanu, S. D.; Tirnovanu, V. G.; Onofriescu, M.; Ungureanu, C.; Toma, B. F.; Cojocaru, E., Uterine Fibroids and Pregnancy: A Review of the Challenges from a Romanian Tertiary Level Institution. Healthcare (Basel) 2022, 10, (5). [CrossRef]
- Breintoft, K.; Pinnerup, R.; Henriksen, T. B.; Rytter, D.; Uldbjerg, N.; Forman, A.; Arendt, L. H., Endometriosis and Risk of Adverse Pregnancy Outcome: A Systematic Review and Meta-Analysis. J Clin Med 2021, 10, (4). [CrossRef]
- Witchel, S. F.; Teede, H. J.; Pena, A. S., Curtailing PCOS. Pediatr Res 2020, 87, (2), 353-361.
- Cheng, X.; Du, F.; Long, X.; Huang, J., Genetic Inheritance Models of Non-Syndromic Cleft Lip with or without Palate: From Monogenic to Polygenic. Genes (Basel) 2023, 14, (10). [CrossRef]
- Inchingolo, A. M.; Fatone, M. C.; Malcangi, G.; Avantario, P.; Piras, F.; Patano, A.; Di Pede, C.; Netti, A.; Ciocia, A. M.; De Ruvo, E.; Viapiano, F.; Palmieri, G.; Campanelli, M.; Mancini, A.; Settanni, V.; Carpentiere, V.; Marinelli, G.; Latini, G.; Rapone, B.; Tartaglia, G. M.; Bordea, I. R.; Scarano, A.; Lorusso, F.; Di Venere, D.; Inchingolo, F.; Inchingolo, A. D.; Dipalma, G., Modifiable Risk Factors of Non-Syndromic Orofacial Clefts: A Systematic Review. Children (Basel) 2022, 9, (12). [CrossRef]
- Iwata, J., Gene-Environment Interplay and MicroRNAs in Cleft Lip and Cleft Palate. Oral Sci Int 2021, 18, (1), 3-13. [CrossRef]
- Im, H.; Song, Y.; Kim, J. K.; Park, D. K.; Kim, D. S.; Kim, H.; Shin, J. O., Molecular Regulation of Palatogenesis and Clefting: An Integrative Analysis of Genetic, Epigenetic Networks, and Environmental Interactions. Int J Mol Sci 2025, 26, (3). [CrossRef]
- Gonseth, S.; Shaw, G. M.; Roy, R.; Segal, M. R.; Asrani, K.; Rine, J.; Wiemels, J.; Marini, N. J., Epigenomic profiling of newborns with isolated orofacial clefts reveals widespread DNA methylation changes and implicates metastable epiallele regions in disease risk. Epigenetics 2019, 14, (2), 198-213. [CrossRef]
- Martinelli, M.; Palmieri, A.; Carinci, F.; Scapoli, L., Non-syndromic Cleft Palate: An Overview on Human Genetic and Environmental Risk Factors. Front Cell Dev Biol 2020, 8, 592271. [CrossRef]
- Meng, L.; Bian, Z.; Torensma, R.; Von den Hoff, J. W., Biological mechanisms in palatogenesis and cleft palate. J Dent Res 2009, 88, (1), 22-33. [CrossRef]
- Yoshioka, H.; Tsukiboshi, Y.; Horita, H.; Kurita, H.; Ogata, A.; Ogata, K.; Horiguchi, H., Sasa veitchii extract alleviates phenobarbital-induced cell proliferation inhibition by upregulating transforming growth factor-beta 1. Traditional & Kampo Medicine 2024, 11, (3), 192-199. [CrossRef]
- Jia, S.; Zhou, J.; Fanelli, C.; Wee, Y.; Bonds, J.; Schneider, P.; Mues, G.; D'Souza, R. N., Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development 2017, 144, (20), 3819-3828. [CrossRef]
- Jia, S.; Zhou, J.; Wee, Y.; Mikkola, M. L.; Schneider, P.; D'Souza, R. N., Anti-EDAR Agonist Antibody Therapy Resolves Palate Defects in Pax9(-/-) Mice. J Dent Res 2017, 96, (11), 1282-1289.
- Li, C.; Lan, Y.; Jiang, R., Molecular and Cellular Mechanisms of Palate Development. J Dent Res 2017, 96, (11), 1184-1191. [CrossRef]
- Chu, E. Y.; Tamasas, B.; Fong, H.; Foster, B. L.; LaCourse, M. R.; Tran, A. B.; Martin, J. F.; Schutte, B. C.; Somerman, M. J.; Cox, T. C., Full Spectrum of Postnatal Tooth Phenotypes in a Novel Irf6 Cleft Lip Model. J Dent Res 2016, 95, (11), 1265-73. [CrossRef]
- Tamasas, B.; Cox, T. C., Massively Increased Caries Susceptibility in an Irf6 Cleft Lip/Palate Model. J Dent Res 2017, 96, (3), 315-322. [CrossRef]
- Liu, W.; Sun, X.; Braut, A.; Mishina, Y.; Behringer, R. R.; Mina, M.; Martin, J. F., Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 2005, 132, (6), 1453-61. [CrossRef]
- Graf, D.; Malik, Z.; Hayano, S.; Mishina, Y., Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev 2016, 27, 129-39. [CrossRef]
- Ueharu, H.; Mishina, Y., BMP signaling during craniofacial development: new insights into pathological mechanisms leading to craniofacial anomalies. Front Physiol 2023, 14, 1170511. [CrossRef]
- Satokata, I.; Maas, R., Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 1994, 6, (4), 348-56. [CrossRef]
- Zhang, Z.; Song, Y.; Zhao, X.; Zhang, X.; Fermin, C.; Chen, Y., Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002, 129, (17), 4135-46.
- Scapoli, L.; Martinelli, M.; Pezzetti, F.; Palmieri, A.; Girardi, A.; Savoia, A.; Bianco, A. M.; Carinci, F., Expression and association data strongly support JARID2 involvement in nonsyndromic cleft lip with or without cleft palate. Hum Mutat 2010, 31, (7), 794-800. [CrossRef]
- Hammond, N. L.; Dixon, M. J., Revisiting the embryogenesis of lip and palate development. Oral Dis 2022, 28, (5), 1306-1326.
- Wang, X.; Zhao, X.; Zheng, X.; Peng, X.; Chen, J.; Wang, Y.; Wang, Z.; Meng, M.; Du, J., Sirt6 loss activates Got1 and facilitates cleft palate through abnormal activating glycolysis. Cell Death Dis 2025, 16, (1), 159. [CrossRef]
- Suzuki, A.; Li, A.; Gajera, M.; Abdallah, N.; Zhang, M.; Zhao, Z.; Iwata, J., MicroRNA-374a, -4680, and -133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Med Genomics 2019, 12, (1), 93. [CrossRef]
- Suzuki, A.; Abdallah, N.; Gajera, M.; Jun, G.; Jia, P.; Zhao, Z.; Iwata, J., Genes and microRNAs associated with mouse cleft palate: A systematic review and bioinformatics analysis. Mech Dev 2018, 150, 21-27. [CrossRef]
- Suzuki, A.; Yoshioka, H.; Summakia, D.; Desai, N. G.; Jun, G.; Jia, P.; Loose, D. S.; Ogata, K.; Gajera, M. V.; Zhao, Z.; Iwata, J., MicroRNA-124-3p suppresses mouse lip mesenchymal cell proliferation through the regulation of genes associated with cleft lip in the mouse. BMC Genomics 2019, 20, (1), 852. [CrossRef]
- Yoshioka, H.; Li, A.; Suzuki, A.; Ramakrishnan, S. S.; Zhao, Z.; Iwata, J., Identification of microRNAs and gene regulatory networks in cleft lip common in humans and mice. Hum Mol Genet 2021, 30, (19), 1881-1893. [CrossRef]
- Zhang, S. D.; Lin, Y. S.; Shi, B.; Jia, Z. L., Identifying New Susceptibility Genes of Non-Syndromic Orofacial Cleft Based on Syndromes Accompanied With Craniosynostosis. Cleft Palate Craniofac J 2025, 10556656251313842. [CrossRef]
- Lee, K. S.; Choi, Y. J.; Cho, J.; Lee, H.; Lee, H.; Park, S. J.; Park, J. S.; Hong, Y. C., Environmental and Genetic Risk Factors of Congenital Anomalies: an Umbrella Review of Systematic Reviews and Meta-Analyses. J Korean Med Sci 2021, 36, (28), e183. [CrossRef]
- Yin, X.; Li, J.; Li, Y.; Zou, S., Maternal alcohol consumption and oral clefts: a meta-analysis. Br J Oral Maxillofac Surg 2019, 57, (9), 839-846. [CrossRef]
- Nicoletti, D.; Appel, L. D.; Siedersberger Neto, P.; Guimaraes, G. W.; Zhang, L., Maternal smoking during pregnancy and birth defects in children: a systematic review with meta-analysis. Cad Saude Publica 2014, 30, (12), 2491-529. [CrossRef]
- Puho, E. H.; Szunyogh, M.; Metneki, J.; Czeizel, A. E., Drug treatment during pregnancy and isolated orofacial clefts in hungary. Cleft Palate Craniofac J 2007, 44, (2), 194-202. [CrossRef]
- Yoshioka, H.; Suzuki, A.; Iwaya, C.; Iwata, J., Suppression of microRNA 124-3p and microRNA 340-5p ameliorates retinoic acid-induced cleft palate in mice. Development 2022, 149, (9). [CrossRef]
- Zhou, Y.; Gilboa, S. M.; Herdt, M. L.; Lupo, P. J.; Flanders, W. D.; Liu, Y.; Shin, M.; Canfield, M. A.; Kirby, R. S., Maternal exposure to ozone and PM(2.5) and the prevalence of orofacial clefts in four U.S. states. Environ Res 2017, 153, 35-40.
- Wang, Z.; DeWitt, J. C.; Higgins, C. P.; Cousins, I. T., A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)? Environ Sci Technol 2017, 51, (5), 2508-2518.
- Gluge, J.; Scheringer, M.; Cousins, I. T.; DeWitt, J. C.; Goldenman, G.; Herzke, D.; Lohmann, R.; Ng, C. A.; Trier, X.; Wang, Z., An overview of the uses of per- and polyfluoroalkyl substances (PFAS). Environ Sci Process Impacts 2020, 22, (12), 2345-2373. [CrossRef]
- Vazquez Loureiro, P.; Nguyen, K. H.; Rodriguez Bernaldo de Quiros, A.; Sendon, R.; Granby, K.; Niklas, A. A., Identification and quantification of per- and polyfluorinated alkyl substances (PFAS) migrating from food contact materials (FCM). Chemosphere 2024, 360, 142360. [CrossRef]
- Xie, S.; Wang, T.; Liu, S.; Jones, K. C.; Sweetman, A. J.; Lu, Y., Industrial source identification and emission estimation of perfluorooctane sulfonate in China. Environ Int 2013, 52, 1-8. [CrossRef]
- Li, L.; Zhai, Z.; Liu, J.; Hu, J., Estimating industrial and domestic environmental releases of perfluorooctanoic acid and its salts in China from 2004 to 2012. Chemosphere 2015, 129, 100-9. [CrossRef]
- Das, K. P.; Grey, B. E.; Rosen, M. B.; Wood, C. R.; Tatum-Gibbs, K. R.; Zehr, R. D.; Strynar, M. J.; Lindstrom, A. B.; Lau, C., Developmental toxicity of perfluorononanoic acid in mice. Reprod Toxicol 2015, 51, 133-44. [CrossRef]
- Negri, E.; Metruccio, F.; Guercio, V.; Tosti, L.; Benfenati, E.; Bonzi, R.; La Vecchia, C.; Moretto, A., Exposure to PFOA and PFOS and fetal growth: a critical merging of toxicological and epidemiological data. Crit Rev Toxicol 2017, 47, (6), 482-508. [CrossRef]
- Jane, L. E. L.; Yamada, M.; Ford, J.; Owens, G.; Prow, T.; Juhasz, A., Health-related toxicity of emerging per- and polyfluoroalkyl substances: Comparison to legacy PFOS and PFOA. Environ Res 2022, 212, (Pt C), 113431. [CrossRef]
- Omotola, E. O.; Ohoro, C. R.; Amaku, J. F.; Conradie, J.; Olisah, C.; Akpomie, K. G.; Malloum, A.; Akpotu, S. O.; Adegoke, K. A.; Okeke, E. S., Evidence of the occurrence, detection, and ecotoxicity studies of perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) in aqueous environments. J Environ Health Sci Eng 2025, 23, (1), 10. [CrossRef]
- Chain, E. P. o. C. i. t. F.; Knutsen, H. K.; Alexander, J.; Barregard, L.; Bignami, M.; Bruschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L. R.; Nebbia, C. S.; Oswald, I. P.; Petersen, A.; Rose, M.; Roudot, A. C.; Vleminckx, C.; Vollmer, G.; Wallace, H.; Bodin, L.; Cravedi, J. P.; Halldorsson, T. I.; Haug, L. S.; Johansson, N.; van Loveren, H.; Gergelova, P.; Mackay, K.; Levorato, S.; van Manen, M.; Schwerdtle, T., Risk to human health related to the presence of perfluorooctane sulfonic acid and perfluorooctanoic acid in food. EFSA J 2018, 16, (12), e05194. [CrossRef]
- Save-Soderbergh, M.; Gyllenhammar, I.; Schillemans, T.; Lindfeldt, E.; Vogs, C.; Donat-Vargas, C.; Ankarberg, E. H.; Glynn, A.; Ahrens, L.; Helte, E.; Akesson, A., Fetal exposure to perfluoroalkyl substances (PFAS) in drinking water and congenital malformations: A nation-wide register-based study on PFAS in drinking water. Environ Int 2025, 198, 109381. [CrossRef]
- Thibodeaux, J. R.; Hanson, R. G.; Rogers, J. M.; Grey, B. E.; Barbee, B. D.; Richards, J. H.; Butenhoff, J. L.; Stevenson, L. A.; Lau, C., Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. I: maternal and prenatal evaluations. Toxicol Sci 2003, 74, (2), 369-81.
- Era, S.; Harada, K. H.; Toyoshima, M.; Inoue, K.; Minata, M.; Saito, N.; Takigawa, T.; Shiota, K.; Koizumi, A., Cleft palate caused by perfluorooctane sulfonate is caused mainly by extrinsic factors. Toxicology 2009, 256, (1-2), 42-7. [CrossRef]
- Tsukiboshi, Y.; Horita, H.; Mikami, Y.; Noguchi, A.; Yokota, S.; Ogata, K.; Yoshioka, H., Involvement of microRNA-4680-3p against phenytoin-induced cell proliferation inhibition in human palate cells. J Toxicol Sci 2024, 49, (1), 1-8. [CrossRef]
- Tominaga, S.; Yoshioka, H.; Yokota, S.; Tsukiboshi, Y.; Suzui, M.; Nagai, M.; Hara, H.; Miura, N.; Maeda, T., Copper-induced renal toxicity controlled by period1 through modulation of Atox1 in mice. Biomed Res 2024, 45, (4), 143-149. [CrossRef]
- Yoshioka, H.; Wu, S.; Moriishi, T.; Tsukiboshi, Y.; Yokota, S.; Miura, N.; Yoshikawa, M.; Inagaki, N.; Matsushita, Y.; Nakao, M., Sasa veitchii extract alleviates nonalcoholic steatohepatitis in methionine–choline deficient diet-induced mice by regulating peroxisome proliferator-activated receptor alpha. Traditional & Kampo Medicine 2023, 10, (3), 259-268. [CrossRef]
- Yoshioka, H.; Tominaga, S.; Amano, F.; Wu, S.; Torimoto, S.; Moriishi, T.; Tsukiboshi, Y.; Yokota, S.; Miura, N.; Inagaki, N.; Matsushita, Y.; Maeda, T., Juzentaihoto alleviates cisplatin-induced renal injury in mice. Traditional & Kampo Medicine 2024, 11, (2), 147-155. [CrossRef]
- Yoshioka, H.; Ramakrishnan, S. S.; Shim, J.; Suzuki, A.; Iwata, J., Excessive All-Trans Retinoic Acid Inhibits Cell Proliferation Through Upregulated MicroRNA-4680-3p in Cultured Human Palate Cells. Front Cell Dev Biol 2021, 9, 618876. [CrossRef]
- Tsukiboshi, Y.; Ogata, A.; Noguchi, A.; Mikami, Y.; Yokota, S.; Ogata, K.; Yoshioka, H., Sasa veitchii extracts protect phenytoin-induced cell proliferation inhibition in human lip mesenchymal cells through modulation of miR-27b-5p. Biomed Res 2023, 44, (2), 73-80. [CrossRef]
- Dhulipala, V. C.; Welshons, W. V.; Reddy, C. S., Cell cycle proteins in normal and chemically induced abnormal secondary palate development: a review. Hum Exp Toxicol 2006, 25, (11), 675-82. [CrossRef]
- Yao, Z.; Chen, D.; Wang, A.; Ding, X.; Liu, Z.; Ling, L.; He, Q.; Zhao, T., Folic acid rescue of ATRA-induced cleft palate by restoring the TGF-beta signal and inhibiting apoptosis. J Oral Pathol Med 2011, 40, (5), 433-9. [CrossRef]
- Smane, L.; Pilmane, M.; Akota, I., Apoptosis and MMP-2, TIMP-2 expression in cleft lip and palate. Stomatologija 2013, 15, (4), 129-34.
- Schoen, C.; Aschrafi, A.; Thonissen, M.; Poelmans, G.; Von den Hoff, J. W.; Carels, C. E. L., MicroRNAs in Palatogenesis and Cleft Palate. Front Physiol 2017, 8, 165. [CrossRef]
- Li, A.; Jia, P.; Mallik, S.; Fei, R.; Yoshioka, H.; Suzuki, A.; Iwata, J.; Zhao, Z., Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Brief Bioinform 2020, 21, (4), 1465-1478. [CrossRef]
- Ulhaq, Z. S.; Tse, W. K. F., Perfluorohexanesulfonic acid (PFHxS) induces oxidative stress and causes developmental toxicities in zebrafish embryos. J Hazard Mater 2023, 457, 131722. [CrossRef]
- Sherr, C. J.; Roberts, J. M., Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004, 18, (22), 2699-711. [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P., CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, (6577), eabc1495. [CrossRef]
- Ettl, T.; Schulz, D.; Bauer, R. J., The Renaissance of Cyclin Dependent Kinase Inhibitors. Cancers (Basel) 2022, 14, (2).
- Lukasik, P.; Zaluski, M.; Gutowska, I., Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development-Review. Int J Mol Sci 2021, 22, (6). [CrossRef]
- Alam, S.; Zunic, A.; Venkat, S.; Feigin, M. E.; Atanassov, B. S., Regulation of Cyclin D1 Degradation by Ubiquitin-Specific Protease 27X Is Critical for Cancer Cell Proliferation and Tumor Growth. Mol Cancer Res 2022, 20, (12), 1751-1762.
- Jablonska, B.; Aguirre, A.; Vandenbosch, R.; Belachew, S.; Berthet, C.; Kaldis, P.; Gallo, V., Cdk2 is critical for proliferation and self-renewal of neural progenitor cells in the adult subventricular zone. J Cell Biol 2007, 179, (6), 1231-45. [CrossRef]
- Ohtsubo, M.; Theodoras, A. M.; Schumacher, J.; Roberts, J. M.; Pagano, M., Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol 1995, 15, (5), 2612-24.
- Huang, J.; Zheng, L.; Sun, Z.; Li, J., CDK4/6 inhibitor resistance mechanisms and treatment strategies (Review). Int J Mol Med 2022, 50, (4).
- Purohit, L.; Jones, C.; Gonzalez, T.; Castrellon, A.; Hussein, A., The Role of CD4/6 Inhibitors in Breast Cancer Treatment. Int J Mol Sci 2024, 25, (2).
- Lee, R. C.; Feinbaum, R. L.; Ambros, V., The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, (5), 843-54. [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G., Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, (5), 855-62. [CrossRef]
- Artigas-Arias, M.; Curi, R.; Marzuca-Nassr, G. N., Myogenic microRNAs as Therapeutic Targets for Skeletal Muscle Mass Wasting in Breast Cancer Models. Int J Mol Sci 2024, 25, (12). [CrossRef]
- Shull, L. C.; Artinger, K. B., Epigenetic regulation of craniofacial development and disease. Birth Defects Res 2024, 116, (1), e2271. [CrossRef]
- Shulman, E.; Kargoli, F.; Aagaard, P.; Hoch, E.; Di Biase, L.; Fisher, J.; Gross, J.; Kim, S.; Ferrick, K. J.; Krumerman, A., Socioeconomic status and the development of atrial fibrillation in Hispanics, African Americans and non-Hispanic whites. Clin Cardiol 2017, 40, (9), 770-776.
- Wang, J.; Bai, Y.; Li, H.; Greene, S. B.; Klysik, E.; Yu, W.; Schwartz, R. J.; Williams, T. J.; Martin, J. F., MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet 2013, 9, (9), e1003785.
- Pan, Y.; Li, D.; Lou, S.; Zhang, C.; Du, Y.; Jiang, H.; Zhang, W.; Ma, L.; Wang, L., A functional polymorphism in the pre-miR-146a gene is associated with the risk of nonsyndromic orofacial cleft. Hum Mutat 2018, 39, (5), 742-750. [CrossRef]
- Yoshioka, H.; Jun, G.; Suzuki, A.; Iwata, J., Dexamethasone Suppresses Palatal Cell Proliferation through miR-130a-3p. Int J Mol Sci 2021, 22, (22). [CrossRef]
- Yoshioka, H.; Mikami, Y.; Ramakrishnan, S. S.; Suzuki, A.; Iwata, J., MicroRNA-124-3p Plays a Crucial Role in Cleft Palate Induced by Retinoic Acid. Front Cell Dev Biol 2021, 9, 621045. [CrossRef]
- Dong, X.; Chen, Q.; Du, H.; Qiu, L., 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Suppresses Mesenchymal Cell Proliferation and Migration Through miR-214-3p in Cleft Palate. Cleft Palate Craniofac J 2024, 10556656241286314. [CrossRef]
- Gajera, M.; Desai, N.; Suzuki, A.; Li, A.; Zhang, M.; Jun, G.; Jia, P.; Zhao, Z.; Iwata, J., MicroRNA-655-3p and microRNA-497-5p inhibit cell proliferation in cultured human lip cells through the regulation of genes related to human cleft lip. BMC Med Genomics 2019, 12, (1), 70. [CrossRef]
- Kouroupis, D.; Kaplan, L. D.; Huard, J.; Best, T. M., CD10-Bound Human Mesenchymal Stem/Stromal Cell-Derived Small Extracellular Vesicles Possess Immunomodulatory Cargo and Maintain Cartilage Homeostasis under Inflammatory Conditions. Cells 2023, 12, (14). [CrossRef]
- Fathi, M.; Omrani, M. A.; Kadkhoda, S.; Ghahghaei-Nezamabadi, A.; Ghafouri-Fard, S., Impact of miRNAs in the pathoetiology of recurrent implantation failure. Mol Cell Probes 2024, 74, 101955. [CrossRef]
- Guo, Q.; Wang, H.; Xu, Y.; Wang, M.; Tian, Z., miR-374a-5p inhibits non-small cell lung cancer cell proliferation and migration via targeting NCK1. Exp Ther Med 2021, 22, (3), 943.
- Chiquet, B. T.; Henry, R.; Burt, A.; Mulliken, J. B.; Stal, S.; Blanton, S. H.; Hecht, J. T., Nonsyndromic cleft lip and palate: CRISPLD genes and the folate gene pathway connection. Birth Defects Res A Clin Mol Teratol 2011, 91, (1), 44-9. [CrossRef]
- Creton, M.; Wagener, F.; Massink, M.; Fennis, W.; Bloemen, M.; Schols, J.; Aarts, M.; van der Molen, A. M.; van Haaften, G.; van den Boogaard, M. J., Concurrent de novo ZFHX4 variant and 16q24.1 deletion in a patient with orofacial clefting; a potential role of ZFHX4 and USP10. Am J Med Genet A 2023, 191, (4), 1083-1088. [CrossRef]
- Hu, L.; Shi, J.; Zhu, Z.; Lu, X.; Jiang, H.; Yu, H.; Liu, H.; Chen, W., CRISPLD1 promotes gastric cancer progression by regulating the Ca(2+)/PI3K-AKT signaling pathway. Heliyon 2024, 10, (5), e27569. [CrossRef]
- Ardizzone, A.; Bova, V.; Casili, G.; Repici, A.; Lanza, M.; Giuffrida, R.; Colarossi, C.; Mare, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I., Role of Basic Fibroblast Growth Factor in Cancer: Biological Activity, Targeted Therapies, and Prognostic Value. Cells 2023, 12, (7). [CrossRef]
- Hausott, B.; Pircher, L.; Kind, M.; Park, J. W.; Claus, P.; Obexer, P.; Klimaschewski, L., Sprouty2 Regulates Endocytosis and Degradation of Fibroblast Growth Factor Receptor 1 in Glioblastoma Cells. Cells 2024, 13, (23). [CrossRef]
- Wilson, S. A.; Robertson, S., History of the development of the Confederation of Australian Critical Care Nurses. Confed Aust Crit Care Nurses J 1991, 4, (3), 17. [CrossRef]
- Chang, M. M.; Lai, M. S.; Hong, S. Y.; Pan, B. S.; Huang, H.; Yang, S. H.; Wu, C. C.; Sun, H. S.; Chuang, J. I.; Wang, C. Y.; Huang, B. M., FGF9/FGFR2 increase cell proliferation by activating ERK1/2, Rb/E2F1, and cell cycle pathways in mouse Leydig tumor cells. Cancer Sci 2018, 109, (11), 3503-3518.
- Rice, R.; Spencer-Dene, B.; Connor, E. C.; Gritli-Linde, A.; McMahon, A. P.; Dickson, C.; Thesleff, I.; Rice, D. P., Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest 2004, 113, (12), 1692-700.
Figure 1.
Effect of four PFAS compounds on HEPM cell proliferation after 48 hours of treatment. Panels (A), (B), (C), and (D) show PFOA, PFOS, PFHxA, and PFHxS, respectively. ***p<0.001 versus control (Tukey’s test) (n=6).
Figure 1.
Effect of four PFAS compounds on HEPM cell proliferation after 48 hours of treatment. Panels (A), (B), (C), and (D) show PFOA, PFOS, PFHxA, and PFHxS, respectively. ***p<0.001 versus control (Tukey’s test) (n=6).
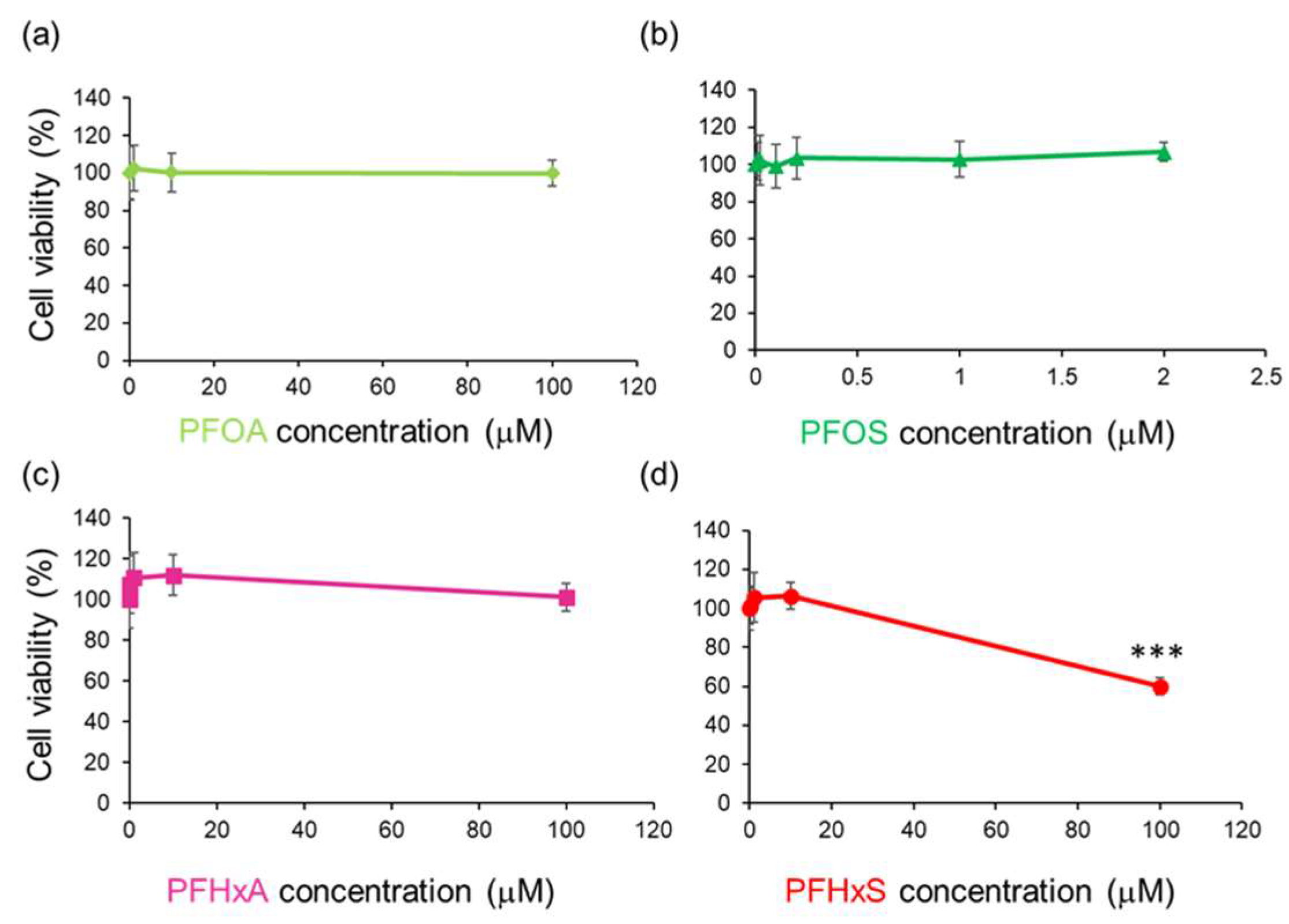
Figure 2.
No Association between PFHxS-Induced cell proliferation and apoptosis.
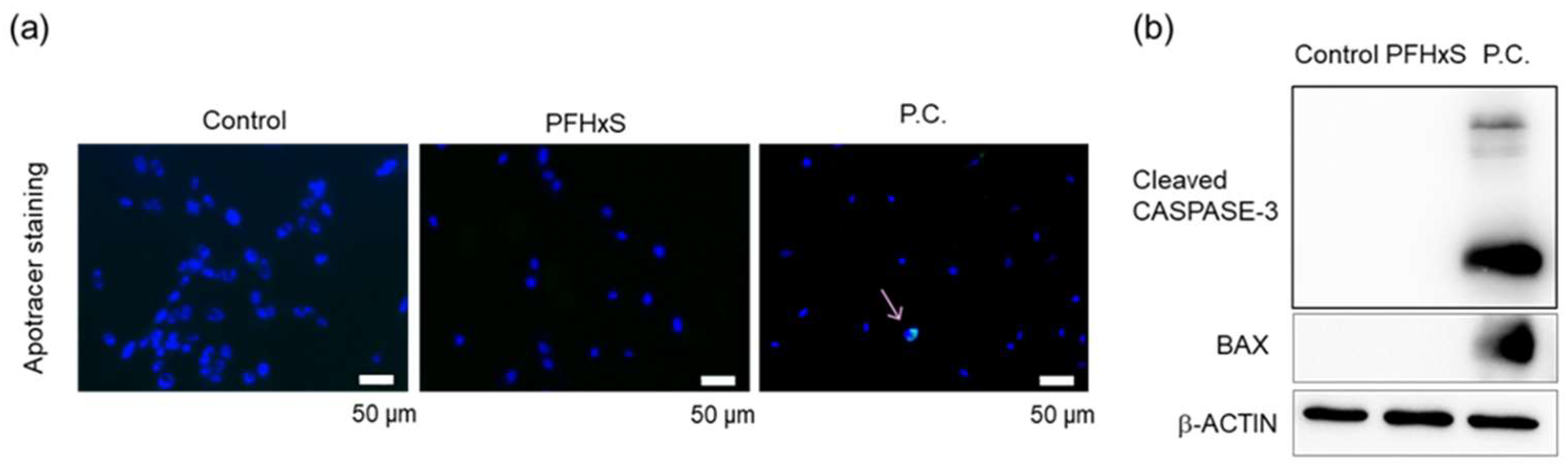
Figure 3.
Association between PFHxS-induced proliferation reduction and G1 cell cycle arrest.
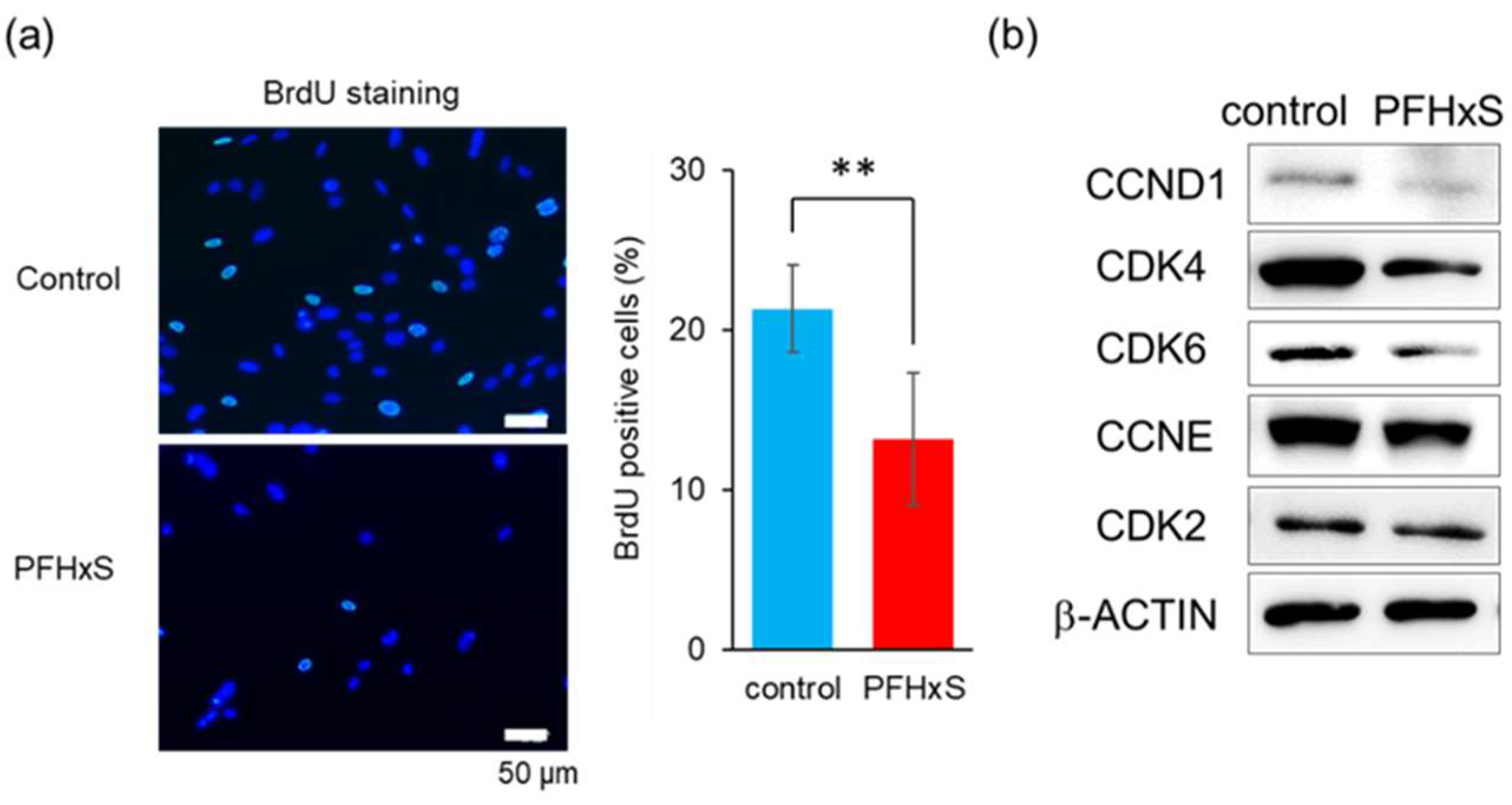
Figure 4.
PFHxS upregulated miR-374a-5p.
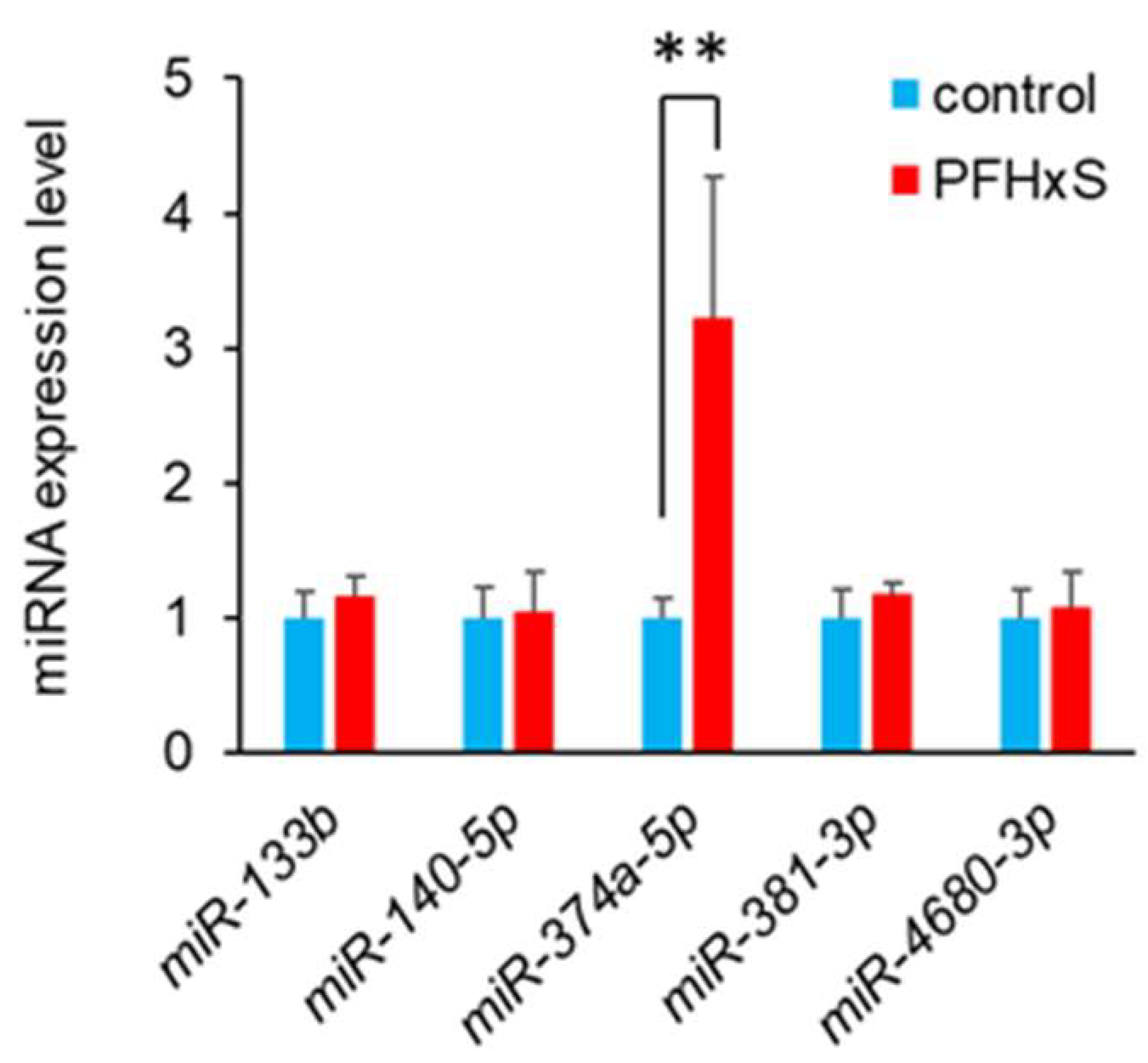
Figure 5.
Inhibition of miR-374a-5p alleviated PFHxS-induced cell proliferation inhibition.
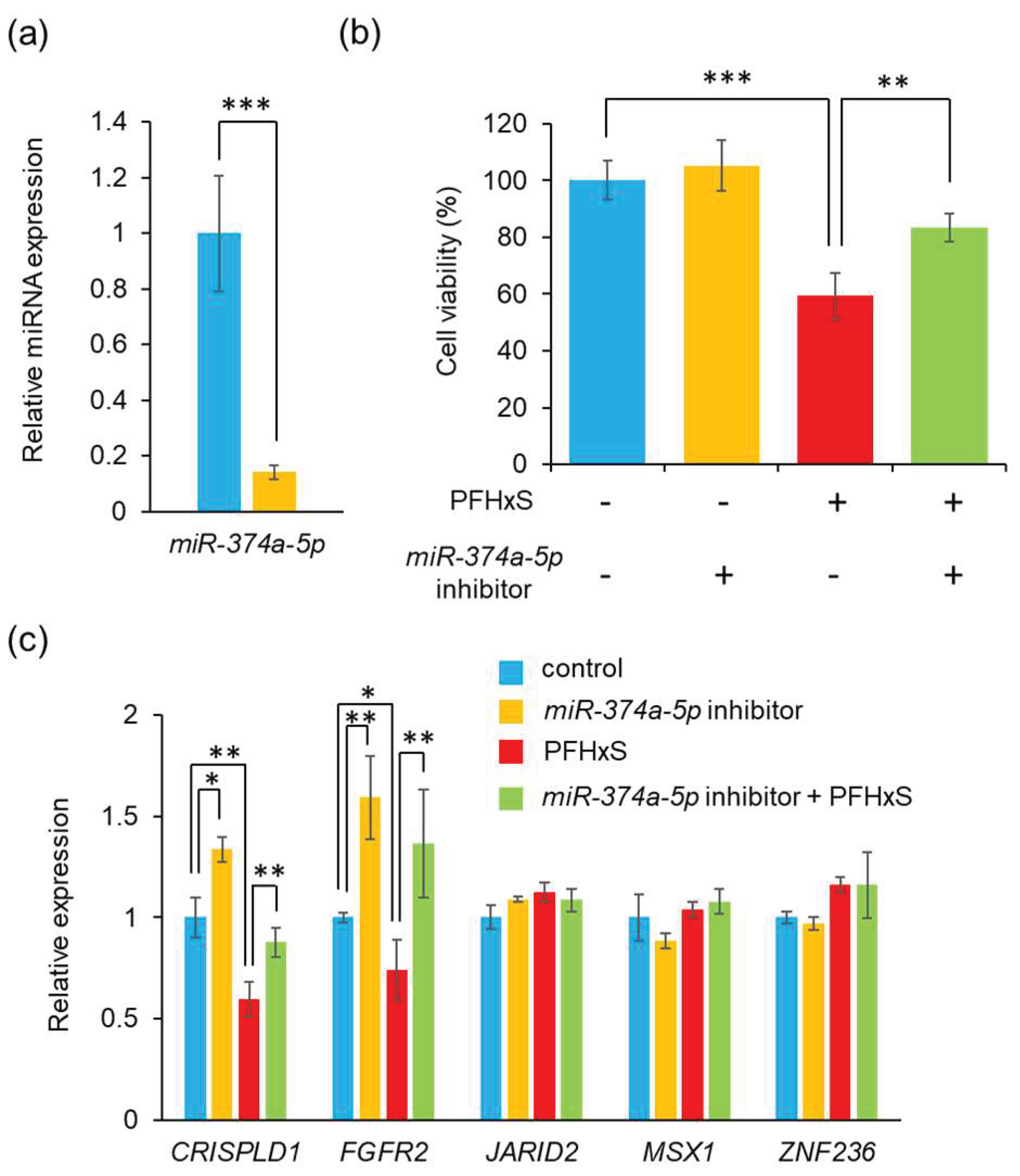
Figure 6.
Proposed mechanism of PFHxS-induced HEPM cell proliferation inhibition.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



