
Submitted:
07 March 2025
Posted:
11 March 2025
You are already at the latest version
Abstract
(1) Cleft palate is a common birth defect worldwide and is caused by both genetic and environmental factors. Intrauterine drug exposure is one of the environmental factors that can induce cleft palate. Mycophenolate mofetil (MPM) is an immunosuppressant drug with teratogenic effects, including cleft palate. However, the research on MPM-induced cleft palate remains limited. Sasa veitchii extract (SE), a medical plant extract, is commercially available in Asia and has been reported to show the effectiveness on oral diseases. The purpose of the present study is to evaluate whether SE protects against MPM-induced immunosuppression in human embryonic palatal mesenchymal (HEPM) cells. (2) Methods: Cell viability and G1 phase-related cell cycle markers were assessed by cotreatment with MPM and SE. Furthermore, we quantified cleft palate-associated miRNA levels and the expression of its downstream genes. (3) Results: MPM treatment reduced cell viability in a concentration-dependent manner. Co-treatment with SE alleviated MPM-induced inhibition of HEPM cell proliferation. Additionally, SE reduced MPM-induced miR-4680-3p upregulation and the downregulation of its downstream genes (ERBB2 and JADE1). (4) Conclusions: These results suggest that SE alleviated MPM-induced cell proliferation inhibition through modulating miR-4680-3p expression.
Keywords:
cleft palate
; Sasa Veitchii
; mycophenolate mofetil
; microRNA
; cell cycle
1. Introduction
The early stage of pregnancy is a critical period for organogenesis in embryos. Exposure to teratogenic substances during this stage is considered a leading cause of severe congenital anomalies, such as cleft palate (CP) and microphthalmia [1,2]. Cleft lip (CL) with or without CP (CL/P) is a significant congenital defect, posing both functional and aesthetic challenges for children. It is recognized as the most common congenital anomaly globally, with an estimated incidence of approximately 1 in 700 live births [3]. The etiology of CL/P is complex, involving both environmental factors and genetic factors [4]. Given that surgical intervention is frequently the primary treatment for CL/P, it necessitates long-term care and substantial treatment costs. Consequently, preventive strategies, such as avoidance of teratogenic substances and folic acid supplementation, are recommended. Folic acid supplementation in early gestation has been shown to reduce the risk of neural tube defects [5] and has also been reported to decrease the incidence of CL/P, potentially by regulating transforming growth factor (TGF) β3 and mitigating oxidative stress [5,6,7].
Regarding genetic factors, several signaling pathways have been implicated in palate development [8,9]. The Wingless/Integrase-1 (WNT) signaling pathway is crucial for secondary palate formation, notably through Paired box gene (Pax) 9 regulation [10,11]. Mutations in axis inhibition protein 2 have been linked to CL/P, as this protein plays a role in regulating WNT signaling [12,13]. TGFβ3 promotes epithelial cell degradation via epithelial-mesenchymal transition (EMT) by inducing Snail1/2, key transcription factors in EMT [14]. Studies have shown that Snail1/2-deficient mice exhibit reduced apoptosis in epithelial cells, leading to impaired palatal fusion [14]. The bone morphogenic protein signaling pathway is also essential for craniofacial morphogenesis, regulating critical cellular processes such as cell proliferation, differentiation, and apoptosis [15,16]. Recent literature reports that 131 human genes and 252 mouse genes have been associated with CP [17]. Concerning environmental factors, various maternal environmental exposures have been correlated with an elevated risk of CL/P [18]. These include occupational exposure to metals and pesticides [19], infections [20], smoking [21], and medication use during pregnancy [22]. These environmental factors can induce CL/P by disrupting essential genes or signaling pathways. For instance, maternal smoking has been associated with disruptions in TGF-β signaling, specifically through reduced TGF-α expression, thereby increasing the risk of CL/P [23,24].
In recent years, microRNAs (miRNAs), small RNA molecules that regulate gene expression, have attracted significant attention. The first miRNA was discovered in 1993 [25,26], making the inception of a new era in RNA biology. These ubiquitous molecules are present across diverse organisms, and to date, over 2500 miRNAs have been identified in human genome [27]. Recent studies have highlighted the involvement of miRNAs in the epigenetic regulation of CL/P [28]. For instance, the miR-17-92 clusters have been shown to control palatal mesenchyme cell proliferation and cell cycle [29]. Mutation of miR-17-92 induces severe craniofacial abnormalities [30]. The miR-146a rs2910164G allele has been found to regulate tumor necrosis factor receptor associated-factor TRAF6 expression, thereby contributing to the pathogenesis of CP [31]. Suzuki et al. reported that overexpression of miR-374a-5p, miR-4680-3p, and miR-133b suppresses human embryonic palatal mesenchymal (HEPM) cell through downregulation of CP-related genes [32]. Fu et al. demonstrated associations between let-7c-5p-PIGA and miR-193a-3p-TGFB2 signaling pathways and HEPM cell viability [33].
Mycophenolate mofetil (MPM) is an immunosuppressant that selectively inhibits inosine monophosphate dehydrogenase [34]. Compared to conventional immunosuppressants such as azathioprine, MPM is associated with a lower incidence of adverse effects [35]. However, MPM has been reported to induce teratogenic effects, including CL/P and microtia [36]. Lin et al. reported that several proteins, including ribosomal protein 5, mouse double minute protein 2, and tumor suppressor p53, have been associated with MPM-induced CP [37]. We recently demonstrated that MPM reduced cell viability through the upregulation of miR-4680-3p and let-7-5p and the downregulation of their downstream genes in HEPM cells [38].
Sasa veitchii is a member of the Gramineae family and its extract preparation is commercially available as an over-the-counter drug in Japan. In Asia, it has a history of traditional use as herbal medicine and dietary supplements, valued for its health-promoting properties. Furthermore, Sasa veitchii extract (SE) has been shown to possess anti-inflammatory [39,40,41], anti-cancer [40,42], and anti-oxidant effects [39,40,43]. Notably, SE has been demonstrated to reduce the risk of periodontal disease and gingivitis [40,44]. Our previous research indicated that SE alleviated all-trans-retinoic acid (atRA)-induced cell proliferation inhibition through modulation of miR-4680-3p in HEPM cells [45]. Similarly, co-treatment with SE improved phenobarbital-induced cell viability reduction through the upregulation of TGF-β1 in human lip fibroblast cells [46]. Taken together, these findings suggest that SE may also exert protective effects against MPM-induced cell proliferation inhibition.
In this study, we aimed to investigate whether SE could alleviate MPM-induced cell proliferation inhibition using HEPM cells.
2. Results
2.1. MPM Inhibited HEPM Cell Proliferation in a Dose- and Time-Dependent Manner
Firstly, we evaluated the suppressive effect of MPM by treating for 24 and 48 h in HEPM cells. As shown in Figure 1, the number of cells was reduced in a dose- and time-dependent manner and was significantly reduced in the MPM dose of 0.03-30 µM. For the following experiments, we selected 1 and 10 µM MPM for 48 h treatment.
2.2. SE Alleviated MPM-Induced Proliferation Inhibition in HEPM Cells
We examined the protective effects of SE against MPM-induced cell proliferation reduction in HEPM cells. We found that treatment with SE (25, 50, 100 µg/mL) did not affect HEPM cell viability (Supplementary Figure S1). Treatment with 1 and 10 µM MPM reduced cell viability (Supplementary Figure S1). In contrast, cotreatment with SE alleviated MPM-induced cell proliferation inhibition in a dose-dependent manner (Supplementary Figure S1). Of note, we demonstrated that cotreatment of 100 µg/mL (P <0.05) SE significantly alleviated MPM-induced cell viability reduction (Figure 2).
2.3. Cotreatment with Sodium Copper Chlorophyllin (SCC) Failed to Recover MPM-Induced Cell Proliferation Reduction in HEPM Cells
We further focused on the protective effects of SCC against MPM-induced cell proliferation inhibition in HEPM cells since the main component of SE is SCC (0.25%) [46]. We tested the ingredients of SE using 3D HPLC and found that broad peak around 280 nm was detected (Figure 3). These data suggest that SE we used contained many compounds.
We tested that treatment with SCC (0.0003 and 0.001 µg/mL) did not change the HEPM cell number. Cotreatment with SCC failed to recover against MPM-induced cell proliferation inhibition in HEPM cells (Figure 4). This result suggests that the protective effect of SE is contributed by ingredients other than SCC.
2.4. SE Alleviated MPM-Induced Cell Cycle Arrest in HEPM Cells
We tested BrdU incorporation assay since we previously demonstrated that MPM-induced cell number reduction was G1 cell cycle arrest, not apoptosis-induced cell death [38]. We found that BrdU-positive cells significantly decreased by treatment with 1 µM MPM, while cotreatment with SE (100 µg/mL) significantly rescued the MPM-induced inhibition of BrdU incorporation (Figure 5a). To further investigate the molecular mechanism of MPM-induced cell cycle arrest (G1-arrest), we tested cyclins and cyclin-dependent kinases (CDK) by immunoblotting (Figure 5b). We found that MPM treatment reduced CCND1 and CDK6 levels. Moreover, treatment with SE induced these protein levels. These results suggest that SE alleviated MPM-induced cell cycle arrest associated with CCND1/CDK6 upregulation in HEPM cells.
2.5. SE Downregulated miR-4680-3p and Upregulated Its Downstream Genes in HEPM Cells
Finally, we investigated the miRNA expression level by treatment with SE since we recently reported that MPM-induced inhibition of HEPM cell proliferation occurs through upregulation of let-7c-5p and miR-4680-3p expression [38]. We found that the upregulation of let-7c-5p and miR-4680-3p expression was seen by MPM (Figure 6a). Additionally, we revealed that SE significantly downregulated the expression of miR-4680-3p in HEPM cells. Cotreatment with SE significantly alleviated miR-4680-3p expression level in HEPM cells. In contrast, let-7c-5p expression level was not altered by treatment with SE (Figure 6a). To further investigate the effects of miR-4680-3p and let-7c-5p, we conducted a quantitative RT-PCR analysis. We found that MPM treatment significantly suppressed BACH1, PAX3, ERBB2, and JADE1 expression levels (Figure 6b). SE treatment significantly upregulated ERBB2 and JADE1 expression levels, while BACH1 and PAX3 expression levels were not changed. Moreover, cotreatment with SE significantly increased the ERBB2 and JADE1 expression levels compared to MPM treatment. These results indicated that SE exerts the protective effect via modulation of miR-4680-3p-ERBB2/JADE1 expression (Figure 7).
3. Discussion
MPM is a type of immunosuppressant that prevents cell proliferation and autoimmunity. MPM induces G1 cell cycle arrest and results in a loss of the G2/M phase peak, leading to growth inhibition in osteosarcoma U2Os cells [47]. MPM decreases the mesangial cell numbers through the downregulation of CCND1 [48]. MPM induces G1-S phase cell cycle arrest in multiple myeloma cells [49]. We previously reported that MPM reduced human lip fibroblast cell viability associated with CCND1/CDK6 [50]. In addition, we recently showed that cyclin and cyclin-dependent kinase was downregulated by MPM in HEPM cells [38]. In the present study, we demonstrated that MPM-induced CCND1 and CDK6 reduction was recovered by cotreatment with SE. These findings suggest that SE alleviated MPM-induced cell viability reduction through the regulation of CCND1 and CDK6.
Recent reports suggest that miRNA is associated with CL/P [9,51]. Suzuki et al. and Li et al. showed that miRNA was predicted using CP-related genes and bioinformatics analysis. They found that the overexpression of miR-133b, miR-140-5p, miR-374-5p, miR-381a-3p, and miR-4680-3p suppress cell proliferation in HEPM cells by regulating target genes and signaling pathways [17,32]. Fu et al. reported that let-7c-5p and miR-193a-3p were identified in the database of CP patients and overexpression of let-7c-5p and miR-193a-3p reduced HEPM cell viability [33]. As an environmental factor, a relationship between medication intake-induced CP and miRNA was reported. Zhou et al. reported that atRA treatment upregulated miR-470-5p expression and suppressed EMT of mouse embryonic palatal shelf epithelial cells [52]. Zhang et al. showed that upregulation of miR-106a-5p by atRA induced apoptosis through regulation of the TGFb/Smad signaling pathway in mice [53]. Inhibition of miR-4680-3p restored atRA-induced HEPM cell viability reduction [54]. miR-130a-3p significantly contributes to the inhibition of mouse embryonic palatal mesenchymal cell proliferation induced by dexamethasone [55]. miR-4680-3p induction was associated with phenytoin-induced inhibition of cell proliferation in HEPM cells [56]. We recently found that let-7c-5p and miR-4680-3p were upregulated among the above seven miRNAs in HEPM cells and inhibition of let-7c-5p and miR-4680-3p alleviated MPM-induced cell proliferation inhibition [38]. In the present study, we confirmed to upregulate let-7c-5p and miR-4680-3p by treatment with MPM. Among the two miRNAs, we found that miR-4680-3p was significantly reduced by cotreatment with SE, while let-7c-5p expression levels were not changed. These results indicated that the SE-induced protective effect was through the modulation of miR-4680-3p. Since we previously reported that atRA-induced cell proliferation inhibition was attenuated by cotreatment with SE through modulation of miR-4680-3p [45], the present mechanism is reasonable.
The reports related to the miR-4680-3p function were limited compared to let-7c-5p [57,58]. As far as we know, Suzuki et al. first demonstrated that overexpression of miR-4680-3p reduced cell viability in HRPM cells [32] and the same research group found that atRA-induced miR-4680-3p upregulation was associated with HEPM cell proliferation through modulation of downstream genes (ERBB2 and JADE1) [54] (Supplementary Figure S2.). ERBB2 is a part of the ERBB receptor tyrosine kinase family, which also includes the epidermal growth factor receptor [59]. When ligands bind to these receptors, it induces the homo- or heterodimerization, activating the kinase domain. This activation initiates downstream signaling cascades, such as mitogen-activated protein kinase/extracellular signal-regulated kinase and phosphatidylinositol-3 kinase/protein kinase B/mechanism of rapamycin pathways, both of which are crucial for cell proliferation, migration, and differentiation [60,61]. The overexpression of ERBB2 leads to a reduction in the G1 phase of the cell cycle by promoting the levels of CDK6, CCND1, and CCNE [62]. As for the palatal shelf, the bioinformatic analysis suggested that the ERBB signaling pathway may play a significant role in the formation of the palate [63]. In addition, we recently demonstrated that MPM inhibits cell proliferation of HEPM cells by upregulating miR-4680-3p expression, and downregulating ERBB2 expression-induced G1 phase arrest [38]. JADE1, also known as PHF17, is a transcription factor and contains two variants: JADE1-L, which is a long-form with 842 amino acids, and JADE1-S, which is a short form without a C-terminal fragment of 333 amino acids [64,65]. The knockdown of JADE1 (both variants) by siRNA results in inhibition of DNA synthesis in human non-small cell lung carcinoma cell line (h1299 cells) and primary fibroblasts [66]. Although the role of JADE1 remains elusive, the protein exhibits histone acetyltransferase (HAT) activity and acts as a co-factor of the HBO1 complex in histone H4 acetylation during gene regulation, which is essential for regulating the cell cycle [67,68]. JADE1 regulates the WNT/β-catenin signaling pathway [69,70]. Since CCND1 is a downstream gene in this pathway [71], JADE1 may indirectly control CCND1. In the present study, we found that cotreatment with SE recovered MPM-induced ERBB2 and JADE1 expression reduction and CCND1 and CDK6 downregulation. These results suggest that SE-induced ERBB2 and JADE1 upregulation play a crucial role in cell proliferation inhibition against MPM in HEPM cells.
According to the company data, SCC (0.25 %) is the main component of SE [39]. SCC has various potential effects, including antimutagenic [72], anticarcinogenic [73], and antioxidant activities [74]. The total antioxidant status in patients with CL/P was lower than those in the control group (healthy people) [75], and CL/P may be related to oxidation stress [76]. Therefore, we hypothesized that the protective effect of SE is due to the presence of SCC. However, our results failed to alleviate the toxic effect of MPM by cotreatment with SCC in HEPM cells. This result indicated that the protective effect of SE was due to the presence of ingredients other than SCC. This conclusion is corroborated by the results of our previous study since we analyzed SE by 3D-high-performance liquid chromatography and showed several peaks included other than SCC in the chromatogram [46]. Sasa species include various phenolic compounds such as flavone phenol and phenolic acid compounds such as myricetin, vitexin, and luteolin, which have antioxidant capacity [40,77,78]. Moreover, several reports have shown that various SE compounds involve miRNA expression. Chung et al. found that tricin suppresses cell proliferation by increasing miR-7 in C6 glioma cells [79]. Myricetin attenuated hepatic steatosis by regulating miR-146b [80] and inflammatory response by inducing miR-29a-3p [81]. Vitexin-induced apoptosis and oxidative stress were associated with specific miRNAs such as let-7c family, miR-17-5p, and miR-495 [82,83]. Coumaric acid has antitumor and anti-inflammatory effects by regulating miR-7-5p, miR-30a-5p, miR-125a-5p, and miR-146a [84,85]. Luteolin has antitumor effects by miR-34a-5p regulation [86]. Although further investigation is needed, we concluded that compounds in the SE, such as flavones, exert protective effects through miRNA or oxidative regulation. In the future, we need to measure the content of phenolic compounds from SE and identify the active compounds of SE.
While this study provides valuable insights, it is important to acknowledge two limitations. Firstly, the active components within SE remain to be fully elucidated. Further research will focus on identifying these components, initially through fractionation and subsequent bioactivity-guided assays. Secondly, the study is limited to in vivo experiments. In vivo studies are crucial to comprehensively evaluate the protective efficacy of SE in a living system. Notwithstanding these limitations, the present investigation provides a valuable initial assessment of SE’s protective effects against MPM-induced inhibition of cell proliferation in HEPM cells. Given the potential for SE’s use during pregnancy, these findings suggest that it holds promise as a potential preventive agent, pending further rigorous evaluation.
4. Materials and Methods
4.1. Cell Culture
HEPM cells were purchased from the JCRB Cell Bank (JCRB9095, Osaka, Japan) and maintained in Minimum Essential Medium Eagle-alpha modification (αMEM; Fujifilm-Wako Pure Chemical Corporation, Osaka, Japan) supplemented with 10% fetal bovine serum (Millipore-Sigma, St Louis, MO, USA), penicillin (10 U/mL), and streptomycin (10 μg/mL; Nacalai Tesque, Kyoto, Japan). The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
4.2. Preparation of SE
SE was kindly gifted from Sunchlon Co. Ltd. (Nagano, Japan). According to the manufacturer’s data, the SE solution is an extract derived from Sasa veitchii leaves, with 1 ml of the solution containing an equivalent of 2.82 g of the leaves. We Freeze-dried the SE solution. We obtained 9.44 g of powdered SE from 120 mL of SE solution (Sunchlon®, lot# 55222) [42].
4.3. HPLC Analysis
High performance liquid chromatography (HPLC) condition was previously described [42]. We used CAPCELL PAK C18 MGIII (Osaka Soda, Osaka, Japan) and injected 2.0 mg/mL SE and SCC.
4.4. Cell Proliferation Assay
HEPM cells were plated in 96-well plates at a density of 5,000 cells/well (n=6) and treated with various concentrations (0–10 μM) of MPM (Tokyo Kasei Co. Ltd., Tokyo, Japan) after 24 h of cell seeding. After treatment with MPM for 24, or 48 h, the cell viability was evaluated using Alamar Blue (Bio-Rad Laboratories, Hercules, CA, USA). For the rescue experiment, HEPM cells were plated in 96-well plates at a density of 5,000 cells/well (n=6) and treated with 1 or 10 μM MPM and 25, 50, or 100 μg/mL SE after 24 h of cell seeding. After 48h of treatment, cell viability was measured in the presence or absence of SE.
4.5. Bromodeoxyuridine (BrdU) Incorporation Assay
HEPM cells were plated on 8-well chamber slides (Biomedical Sciences Inc.) at a density of 10000 cells/well and treated with 1 μM MPM, 100 μg/mL SE, 1 μM MPM + 100 μg/mL SE, or vehicle. After 48 h of treatment, the cells were incubated with BrdU (100 μg/mL) for 40 min. The incorporated BrdU was stained with an anti-BrdU antibody (1:150, Santacruz Biotechnology, Dallas, TX, USA) and fluorescein (FITC)-conjugated anti-mouse IgG (1:180; MBL, Aichi, Japan). Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, Nacalai Tesque), and BrdU-positive cells were quantified in 6-8 fields.
4.6. Western Blot Analysis
HEPM cells were plated in a 35 mm dish at a density of 2x105 cells per dish and treated with 1 µM MPM, 100 µg/mL SE, 1 µM MPM + 100 µg/mL SE, or vehicle after 24 h cell seeding. After 48 h of treatment, we washed phosphate-buffered saline (PBS) twice and added 100 µL ice-cold RIPA buffer (Nacalai Tesque) containing a protease inhibitor cocktail (Nacalai Tesque) and waited 5 min on ice. Subsequently scraped and centrifuged (20,000 × g for 20 min at 4 °C) as previously described [38,56]. Protein samples (10 µg) were subjected to 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes. anti-mouse cyclin D1 (CCND1) antibody (1:1000 dilution; Santa Cruz Biotechnology), anti-mouse cyclin-dependent kinase 6 (CDK6) antibody (1:2000 dilution; Proteintech Japan, Tokyo, Japan) and anti-mouse b-actin monoclonal antibodies (1:3000 dilution; MBL, Aichi, Japan) were used as primary antibodies for immunoblotting. A peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) and a peroxidase-conjugated anti-mouse IgG (Cell Signaling Technology) were used as secondary antibodies (1:10,000 dilution). The immunoreactive bands were visualized by Western Blot Hyper HRP Substrate (Takara Bio, Shiga, Japan).
4.7. Quantitative RT-PCR
HEPM cells were plated in 35 mm dish at a density of 2x105 cells per dish and treated with 1 μM MPM and/or 100 μg/mL SE or vehicle after 24 h cell seeding. After 48 h of treatment, we washed PBS twice, and total RNA was extracted using a QIAshredder and miRNeasy Mini Kit (QIAGEN, Valencia, CA, USA) as we previously described [56,87]. Total RNA (25 ng) was reverse transcribed using an miRNA Reverse Transcription Reaction Kit (GeneCopoeia, Rockville, MD, USA). miRNA expression was examined using an all-in-one miRNA qRT-PCR Detection Kit (GeneCopoeia). Probe information and PCR conditions were as previously described [38,56].
4.8. Statistical Analyses
Comparisons between more than two groups were performed using Tukey’s test. Cell viability assay for multiple groups were evaluated using two-way analysis of variance with Dunnett’s test. All statistical analyses were performed using SPSS Statistics for Windows (version 26.0; IBM Corp., Armonk, NY). Differences were considered statistically significant at P < 0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Effect of SE and SCC against MPM-induced cell proliferation inhibition in HEPM cells; Figure S2: Putative target site for miR-4680-3p in the ERBB2 and JADE1 3’ UTR.
Author Contributions
Conceptualization, K.O. and H.Y.; methodology, H.Y.; software, S.Y.; validation, H.H., Y.T., A.O., and H.Y.; formal analysis, H.K., H.Y., and H.Y.; investigation, H.H., Y.T., A.O., and H.Y.; resources, A.O., H.K., S.Y., N.I., H.H., and H.Y.; data curation, H.H., and H.Y.; writing—original draft preparation, H.H., and Y.T.; writing—review and editing, K.O., H.K., S. Y., H.Y., N.I., H.H., and H.Y.; visualization, H.H.; supervision, K.O., and H.Y.; project administration, H.Y.; funding acquisition, A.O., and H.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Gifu University of Medical Science research grant A.
Data Availability Statement
All relevant data are within the manuscript.
Acknowledgments
The authors thank Nobuaki Matsui (Gifu University of Medical Science, Japan) for his kind suggestions. We thank Sunchlon Co. Ltd. for providing SE.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| atRA | all-trans-retinoic acid |
| CDK | cyclin-dependent kinases |
| CL | Cleft lip |
| CP | Cleft palate |
| CL/P | Cleft lip with or without cleft palate |
| EMT | epithelial-mesenchymal transition |
| HEPM | human embryonic palatal mesenchymal |
| HPLC | High performance liquid chromatography |
| miRNA | microRNA |
| MPM | Mycophenolate mofetil |
| Pax | Paired box gene |
| SE | Sasa veitchii extract |
| SCC | sodium copper chlorophyllin |
| TGF | transforming growth factor |
| WNT | Wingless/integrase-1 |
References
- Sadler, T.W. Establishing the Embryonic Axes: Prime Time for Teratogenic Insults. J Cardiovasc Dev Dis 2017, 4. [Google Scholar]
- van Gelder, M.M.; van Rooij, I.A.; de Jong-van den Berg, L.T.; Roeleveld, N. Teratogenic mechanisms associated with prenatal medication exposure. Therapie 2014, 69, 13–24. [Google Scholar]
- Babai, A.; Irving, M. Orofacial Clefts: Genetics of Cleft Lip and Palate. Genes (Basel) 2023, 14. [Google Scholar]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat Rev Genet 2011, 12, 167–178. [Google Scholar]
- Blencowe, H.; Cousens, S.; Modell, B.; Lawn, J. Folic acid to reduce neonatal mortality from neural tube disorders. Int J Epidemiol 2010, 39 (Suppl 1), i110–i021. [Google Scholar]
- Maldonado, E.; Murillo, J.; Barrio, C.; del Rio, A.; Perez-Miguelsanz, J.; Lopez-Gordillo, Y.; Partearroyo, T.; Paradas, I.; Maestro, C.; Martinez-Sanz, E.; Varela-Moreiras, G.; Martinez-Alvarez, C. Occurrence of cleft-palate and alteration of Tgf-beta(3) expression and the mechanisms leading to palatal fusion in mice following dietary folic-acid deficiency. Cells Tissues Organs 2011, 194, 406–420. [Google Scholar] [PubMed]
- Zhang, Y.; Kato, H.; Sato, H.; Yamaza, H.; Hirofuji, Y.; Han, X.; Masuda, K.; Nonaka, K. Folic acid-mediated mitochondrial activation for protection against oxidative stress in human dental pulp stem cells derived from deciduous teeth. Biochem Biophys Res Commun 2019, 508, 850–856. [Google Scholar]
- Won, H.J.; Kim, J.W.; Won, H.S.; Shin, J.O. Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review. Cells 2023, 12. [Google Scholar]
- Im, H.; Song, Y.; Kim, J.K.; Park, D.K.; Kim, D.S.; Kim, H.; Shin, J.O. Molecular Regulation of Palatogenesis and Clefting: An Integrative Analysis of Genetic, Epigenetic Networks, and Environmental Interactions. Int J Mol Sci 2025, 26. [Google Scholar]
- Jia, S.; Zhou, J.; Fanelli, C.; Wee, Y.; Bonds, J.; Schneider, P.; Mues, G.; D’Souza, R.N. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development 2017, 144, 3819–3828. [Google Scholar]
- Li, C.; Lan, Y.; Jiang, R. Molecular and Cellular Mechanisms of Palate Development. J Dent Res 2017, 96, 1184–1191. [Google Scholar]
- Letra, A.; Bjork, B.; Cooper, M.E.; Szabo-Rogers, H.; Deleyiannis, F.W.; Field, L.L.; Czeizel, A.E.; Ma, L.; Garlet, G.P.; Poletta, F.A.; Mereb, J.C.; Lopez-Camelo, J.S.; Castilla, E.E.; Orioli, I.M.; Wendell, S.; Blanton, S.H.; Liu, K.; Hecht, J.T.; Marazita, M.L.; Vieira, A.R.; Silva, R.M. Association of AXIN2 with non-syndromic oral clefts in multiple populations. J Dent Res 2012, 91, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Hozyasz, K.K.; Wojcicki, P.; Lasota, A.; Dunin-Wilczynska, I.; Jagodzinski, P.P. Association of DVL2 and AXIN2 gene polymorphisms with cleft lip with or without cleft palate in a Polish population. Birth Defects Res A Clin Mol Teratol 2012, 94, 943–950. [Google Scholar] [PubMed]
- Murray, S.A.; Oram, K.F.; Gridley, T. Multiple functions of Snail family genes during palate development in mice. Development 2007, 134, 1789–1797. [Google Scholar]
- Graf, D.; Malik, Z.; Hayano, S.; Mishina, Y. Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev 2016, 27, 129–139. [Google Scholar]
- Ueharu, H.; Mishina, Y. BMP signaling during craniofacial development: New insights into pathological mechanisms leading to craniofacial anomalies. Front Physiol 2023, 14, 1170511. [Google Scholar] [PubMed]
- Li, A.; Jia, P.; Mallik, S.; Fei, R.; Yoshioka, H.; Suzuki, A.; Iwata, J.; Zhao, Z. Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Brief Bioinform 2020, 21, 1465–1478. [Google Scholar]
- Lee, K.S.; Choi, Y.J.; Cho, J.; Lee, H.; Lee, H.; Park, S.J.; Park, J.S.; Hong, Y.C. Environmental and Genetic Risk Factors of Congenital Anomalies: An Umbrella Review of Systematic Reviews and Meta-Analyses. J Korean Med Sci 2021, 36, e183. [Google Scholar]
- Spinder, N.; Prins, J.R.; Bergman, J.E.H.; Smidt, N.; Kromhout, H.; Boezen, H.M.; de Walle, H.E.K. Congenital anomalies in the offspring of occupationally exposed mothers: A systematic review and meta-analysis of studies using expert assessment for occupational exposures. Hum Reprod 2019, 34, 903–919. [Google Scholar]
- Luteijn, J.M.; Brown, M.J.; Dolk, H. Influenza and congenital anomalies: A systematic review and meta-analysis. Hum Reprod 2014, 29, 809–823. [Google Scholar]
- Nicoletti, D.; Appel, L.D.; Siedersberger Neto, P.; Guimaraes, G.W.; Zhang, L. Maternal smoking during pregnancy and birth defects in children: A systematic review with meta-analysis. Cad Saude Publica 2014, 30, 2491–2529. [Google Scholar]
- Puho, E.H.; Szunyogh, M.; Metneki, J.; Czeizel, A.E. Drug treatment during pregnancy and isolated orofacial clefts in hungary. Cleft Palate Craniofac J 2007, 44, 194–202. [Google Scholar]
- Ebadifar, A.; Hamedi, R.; KhorramKhorshid, H.R.; Kamali, K.; Moghadam, F.A. Parental cigarette smoking, transforming growth factor-alpha gene variant and the risk of orofacial cleft in Iranian infants. Iran J Basic Med Sci 2016, 19, 366–373. [Google Scholar]
- Junaid, M.; Narayanan, M.B.A.; Jayanthi, D.; Kumar, S.G.R.; Selvamary, A.L. Association between maternal exposure to tobacco, presence of TGFA gene, and the occurrence of oral clefts. A case control study. Clin Oral Investig 2018, 22, 217–223. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [PubMed]
- Humphries, B.; Yang, C. The microRNA-200 family: Small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [PubMed]
- Shull, L.C.; Artinger, K.B. Epigenetic regulation of craniofacial development and disease. Birth Defects Res 2024, 116, e2271. [Google Scholar]
- Li, L.; Shi, B.; Chen, J.; Li, C.; Wang, S.; Wang, Z.; Zhu, G. An E2F1/MiR-17-92 Negative Feedback Loop mediates proliferation of Mouse Palatal Mesenchymal Cells. Sci Rep 2017, 7, 5148. [Google Scholar]
- Wang, J.; Bai, Y.; Li, H.; Greene, S.B.; Klysik, E.; Yu, W.; Schwartz, R.J.; Williams, T.J.; Martin, J.F. MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet 2013, 9, e1003785. [Google Scholar] [CrossRef]
- Pan, Y.; Li, D.; Lou, S.; Zhang, C.; Du, Y.; Jiang, H.; Zhang, W.; Ma, L.; Wang, L. A functional polymorphism in the pre-miR-146a gene is associated with the risk of nonsyndromic orofacial cleft. Hum Mutat 2018, 39, 742–750. [Google Scholar]
- Suzuki, A.; Li, A.; Gajera, M.; Abdallah, N.; Zhang, M.; Zhao, Z.; Iwata, J. MicroRNA-374a, -4680, and -133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Med Genomics 2019, 12, 93. [Google Scholar]
- Fu, C.; Lou, S.; Zhu, G.; Fan, L.; Yu, X.; Zhu, W.; Ma, L.; Wang, L.; Pan, Y. Identification of New miRNA-mRNA Networks in the Development of Non-syndromic Cleft Lip With or Without Cleft Palate. Front Cell Dev Biol 2021, 9, 631057. [Google Scholar]
- Chen, H.; Chen, B. Clinical mycophenolic acid monitoring in liver transplant recipients. World J Gastroenterol 2014, 20, 10715–10728. [Google Scholar] [PubMed]
- Narayanaswami, P.; Sanders, D.B.; Thomas, L.; Thibault, D.; Blevins, J.; Desai, R.; Krueger, A.; Bibeau, K.; Liu, B.; Guptill, J.T.; Group, P.-M. S. Comparative effectiveness of azathioprine and mycophenolate mofetil for myasthenia gravis (PROMISE-MG): A prospective cohort study. Lancet Neurol 2024, 23, 267–276. [Google Scholar] [PubMed]
- Perez-Aytes, A.; Ledo, A.; Boso, V.; Saenz, P.; Roma, E.; Poveda, J.L.; Vento, M. In utero exposure to mycophenolate mofetil: A characteristic phenotype? Am J Med Genet A 2008, 146A, 1–7. [Google Scholar]
- Lin, Y.; Song, T.; Ronde, E.M.; Ma, G.; Cui, H.; Xu, M. The important role of MDM2, RPL5, and TP53 in mycophenolic acid-induced cleft lip and palate. Medicine (Baltimore) 2021, 100, e26101. [Google Scholar]
- Yoshioka, H.; Horita, H.; Tsukiboshi, Y.; Kurita, H.; Ogata, A.; Ogata, K. Cleft Palate Induced by Mycophenolate Mofetil Is Associated with miR-4680-3p and let-7c-5p in Human Palate Cells. Noncoding RNA 2025, 11. [Google Scholar]
- Yoshioka, H.; Nonogaki, T.; Fukaya, S.; Ichimaru, Y.; Nagatsu, A.; Yoshikawa, M.; Fujii, H.; Nakao, M. Sasa veitchii extract protects against carbon tetrachloride-induced hepatic fibrosis in mice. Environ Health Prev Med 2018, 23, 49. [Google Scholar]
- Kimura, I.; Kagawa, S.; Tsuneki, H.; Tanaka, K.; Nagashima, F. Multitasking bamboo leaf-derived compounds in prevention of infectious, inflammatory, atherosclerotic, metabolic, and neuropsychiatric diseases. Pharmacol Ther 2022, 235, 108159. [Google Scholar]
- Kojima, S.; Hakamata, M.; Asanuma, T.; Suzuki, R.; Tsuruda, J.I.; Nonoyama, T.; Lin, Y.; Fukatsu, H.; Koide, N.; Umezawa, K. Cellular Anti-Inflammatory and Antioxidant Activities of Bamboo Sasa albomarginata Leaf Extract and Its Constituent Coumaric Acid Methyl Ester. ScientificWorldJournal 2022, 2022, 8454865. [Google Scholar]
- Hamanaka, J.; Mikami, Y.; Horiuchi, A.; Yano, A.; Amano, F.; Shibata, S.; Ogata, A.; Ogata, K.; Nagatsu, A.; Miura, N.; Sano, M.; Suzui, M.; Yoshioka, H. Sasa veitchii extract exhibits antitumor effect against murine pancreatic adenocarcinoma in vivo and in vitro. Traditional & Kampo Medicine n/a, (n/a). [Google Scholar]
- Yoshioka, H.; Wu, S.; Moriishi, T.; Tsukiboshi, Y.; Yokota, S.; Miura, N.; Yoshikawa, M.; Inagaki, N.; Matsushita, Y.; Nakao, M. Sasa veitchii extract alleviates nonalcoholic steatohepatitis in methionine–choline deficient diet-induced mice by regulating peroxisome proliferator-activated receptor alpha. Traditional & Kampo Medicine 2023, 10, 259–268. [Google Scholar]
- Sakagami, H.; Tomomura, M. Dental Application of Natural Products. Medicines (Basel) 2018, 5. [Google Scholar]
- Tsukiboshi, Y.; Mikami, Y.; Horita, H.; Ogata, A.; Noguchi, A.; Yokota, S.; Ogata, K.; Yoshioka, H. Protective effect of Sasa veitchii extract against all-trans-retinoic acid-induced inhibition of proliferation of cultured human palate cells. Nagoya J Med Sci 2024, 86, 223–236. [Google Scholar] [PubMed]
- Yoshioka, H.; Tsukiboshi, Y.; Horita, H.; Kurita, H.; Ogata, A.; Ogata, K.; Horiguchi, H. Sasa veitchii extract alleviates phenobarbital-induced cell proliferation inhibition by upregulating transforming growth factor-beta 1. Traditional & Kampo Medicine 2024, 11, 192–199. [Google Scholar]
- Sun, X.X.; Dai, M.S.; Lu, H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem 2008, 283, 12387–12392. [Google Scholar]
- Gao, Y.; Yang, H.; Wang, Y.; Tian, J.; Li, R.; Zhou, X. Evaluation of the inhibitory effect of tacrolimus combined with mycophenolate mofetil on mesangial cell proliferation based on the cell cycle. Int J Mol Med 2020, 46, 1582–1592. [Google Scholar]
- Takebe, N.; Cheng, X.; Fandy, T.E.; Srivastava, R.K.; Wu, S.; Shankar, S.; Bauer, K.; Shaughnessy, J.; Tricot, G. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol Cancer Ther 2006, 5, 457–466. [Google Scholar]
- Yoshioka, H.; Horita, H.; Tsukiboshi, Y.; Kurita, H.; Mikami, Y.; Ogata, K.; Ogata, A. Mycophenolate mofetil reduces cell viability associated with the miR-205-PAX9 pathway in human lip fibroblast cells. Biomed Res 2025, 46, 1–8. [Google Scholar]
- Schoen, C.; Aschrafi, A.; Thonissen, M.; Poelmans, G.; Von den Hoff, J.W.; Carels, C.E.L. MicroRNAs in Palatogenesis and Cleft Palate. Front Physiol 2017, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, M.; Zhang, M.; Tan, M.; Ji, Y.; Shu, S.; Liang, Y. MiRNA-470-5p suppresses epithelial-mesenchymal transition of embryonic palatal shelf epithelial cells by targeting Fgfr1 during palatogenesis. Exp Biol Med (Maywood) 2023, 248, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shen, Z.; Xing, Y.; Zhao, H.; Liang, Y.; Chen, J.; Zhong, X.; Shi, L.; Wan, X.; Zhou, J.; Tang, S. MiR-106a-5p modulates apoptosis and metabonomics changes by TGF-beta/Smad signaling pathway in cleft palate. Exp Cell Res 2020, 386, 111734. [Google Scholar] [CrossRef]
- Yoshioka, H.; Ramakrishnan, S.S.; Shim, J.; Suzuki, A.; Iwata, J. Excessive All-Trans Retinoic Acid Inhibits Cell Proliferation Through Upregulated MicroRNA-4680-3p in Cultured Human Palate Cells. Front Cell Dev Biol 2021, 9, 618876. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Jun, G.; Suzuki, A.; Iwata, J. Dexamethasone Suppresses Palatal Cell Proliferation through miR-130a-3p. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Tsukiboshi, Y.; Horita, H.; Mikami, Y.; Noguchi, A.; Yokota, S.; Ogata, K.; Yoshioka, H. Involvement of microRNA-4680-3p against phenytoin-induced cell proliferation inhibition in human palate cells. J Toxicol Sci 2024, 49, 1–8. [Google Scholar]
- Roush, S.; Slack, F.J. The let-7 family of microRNAs. Trends Cell Biol 2008, 18, 505–516. [Google Scholar] [CrossRef]
- Su, J.L.; Chen, P.S.; Johansson, G.; Kuo, M.L. Function and regulation of let-7 family microRNAs. Microrna 2012, 1, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat Rev Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat Rev Mol Cell Biol 2011, 12, 104–117. [Google Scholar]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [PubMed]
- Timms, J.F.; White, S.L.; O’Hare, M.J.; Waterfield, M.D. Effects of ErbB-2 overexpression on mitogenic signalling and cell cycle progression in human breast luminal epithelial cells. Oncogene 2002, 21, 6573–6586. [Google Scholar]
- Yang, Y.; Suzuki, A.; Iwata, J.; Jun, G. Secondary Genome-Wide Association Study Using Novel Analytical Strategies Disentangle Genetic Components of Cleft Lip and/or Cleft Palate in 1q32.2. Genes (Basel) 2020, 11. [Google Scholar]
- Borgal, L.; Rinschen, M.M.; Dafinger, C.; Hoff, S.; Reinert, M.J.; Lamkemeyer, T.; Lienkamp, S.S.; Benzing, T.; Schermer, B. Casein kinase 1 alpha phosphorylates the Wnt regulator Jade-1 and modulates its activity. J Biol Chem 2014, 289, 26344–26356. [Google Scholar] [CrossRef]
- Siriwardana, N.S.; Meyer, R.D.; Panchenko, M.V. The novel function of JADE1S in cytokinesis of epithelial cells. Cell Cycle 2015, 14, 2821–2834. [Google Scholar] [PubMed]
- Havasi, A.; Haegele, J.A.; Gall, J.M.; Blackmon, S.; Ichimura, T.; Bonegio, R.G.; Panchenko, M.V. Histone acetyl transferase (HAT) HBO1 and JADE1 in epithelial cell regeneration. Am J Pathol 2013, 182, 152–162. [Google Scholar]
- Panchenko, M.V. Structure, function and regulation of jade family PHD finger 1 (JADE1). Gene 2016, 589, 1–11. [Google Scholar]
- Han, J.; Lachance, C.; Ricketts, M.D.; McCullough, C.E.; Gerace, M.; Black, B.E.; Cote, J.; Marmorstein, R. The scaffolding protein JADE1 physically links the acetyltransferase subunit HBO1 with its histone H3-H4 substrate. J Biol Chem 2018, 293, 4498–4509. [Google Scholar] [PubMed]
- Chitalia, V.C.; Foy, R.L.; Bachschmid, M.M.; Zeng, L.; Panchenko, M.V.; Zhou, M.I.; Bharti, A.; Seldin, D.C.; Lecker, S.H.; Dominguez, I.; Cohen, H.T. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol 2008, 10, 1208–1216. [Google Scholar]
- Tauriello, D.V.; Maurice, M.M. The various roles of ubiquitin in Wnt pathway regulation. Cell Cycle 2010, 9, 3700–3709. [Google Scholar]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 1999, 96, 5522–5527. [Google Scholar] [PubMed]
- Dashwood, R. Chlorophylls as anticarcinogens (review). Int J Oncol 1997, 10, 721–727. [Google Scholar] [PubMed]
- Suparmi, S.; Fasitasari, M.; Martosupono, M.; Mangimbulude, J.C. Comparisons of Curative Effects of Chlorophyll from Sauropus androgynus (L) Merr Leaf Extract and Cu-Chlorophyllin on Sodium Nitrate-Induced Oxidative Stress in Rats. J Toxicol 2016, 2016, 8515089. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Eslava, J.; Gomez-Arroyo, S.; Villalobos-Pietrini, R.; Espinosa-Aguirre, J.J. Antimutagenicity of coriander (Coriandrum sativum) juice on the mutagenesis produced by plant metabolites of aromatic amines. Toxicol Lett 2004, 153, 283–292. [Google Scholar]
- Aizenbud, D.; Peri-Front, Y.; Nagler, R.M. Salivary analysis and antioxidants in cleft lip and palate children. Arch Oral Biol 2008, 53, 517–522. [Google Scholar]
- Li, R.; Huang, C.; Ho, J.C.H.; Leung, C.C.T.; Kong, R.Y.C.; Li, Y.; Liang, X.; Lai, K.P.; Tse, W.K.F. The use of glutathione to reduce oxidative stress status and its potential for modifying the extracellular matrix organization in cleft lip. Free Radic Biol Med 2021, 164, 130–138. [Google Scholar]
- Wang, J.; Yue, Y.D.; Jiang, H.; Tang, F. Rapid Screening for Flavone C-Glycosides in the Leaves of Different Species of Bamboo and Simultaneous Quantitation of Four Marker Compounds by HPLC-UV/DAD. Int J Anal Chem 2012, 2012, 205101. [Google Scholar]
- Wang, J.; Yue, Y.D.; Tang, F.; Sun, J. TLC screening for antioxidant activity of extracts from fifteen bamboo species and identification of antioxidant flavone glycosides from leaves of Bambusa. textilis McClure. Molecules 2012, 17, 12297–12311. [Google Scholar] [CrossRef]
- Chung, D.J.; Wang, C.J.; Yeh, C.W.; Tseng, T.H. Inhibition of the Proliferation and Invasion of C6 Glioma Cells by Tricin via the Upregulation of Focal-Adhesion-Kinase-Targeting MicroRNA-7. J Agric Food Chem 2018, 66, 6708–6716. [Google Scholar]
- Xia, S.F.; Qiu, Y.Y.; Chen, L.M.; Jiang, Y.Y.; Huang, W.; Xie, Z.X.; Tang, X.; Sun, J. Myricetin alleviated hepatic steatosis by acting on microRNA-146b/thyroid hormone receptor b pathway in high-fat diet fed C57BL/6J mice. Food Funct 2019, 10, 1465–1477. [Google Scholar]
- Bai, Y.; Liu, X.; Chen, Q.; Chen, T.; Jiang, N.; Guo, Z. Myricetin ameliorates ox-LDL-induced HUVECs apoptosis and inflammation via lncRNA GAS5 upregulating the expression of miR-29a-3p. Sci Rep 2021, 11, 19637. [Google Scholar] [PubMed]
- Najafipour, R.; Momeni, A.M.; Mirmazloomi, Y.; Moghbelinejad, S. Vitexin Induces Apoptosis in MCF-7 Breast Cancer Cells through the Regulation of Specific miRNAs Expression. Int J Mol Cell Med 2022, 11, 197–206. [Google Scholar]
- Zaki, A.; Mohsin, M.; Khan, S.; Khan, A.; Ahmad, S.; Verma, A.; Ali, S.; Fatma, T.; Syed, M.A. Vitexin mitigates oxidative stress, mitochondrial damage, pyroptosis and regulates small nucleolar RNA host gene 1/DNA methyltransferase 1/microRNA-495 axis in sepsis-associated acute lung injury. Inflammopharmacology 2024. [Google Scholar]
- Jang, M.G.; Ko, H.C.; Kim, S.J. Effects of p-coumaric acid on microRNA expression profiles in SNU-16 human gastric cancer cells. Genes Genomics 2020, 42, 817–825. [Google Scholar] [PubMed]
- Kheiry, M.; Dianat, M.; Badavi, M.; Mard, S.A.; Bayati, V. Does p-coumaric acid improve cardiac injury following LPS-induced lung inflammation through miRNA-146a activity? Avicenna J Phytomed 2020, 10, 50–57. [Google Scholar]
- Jiang, Z.Q.; Li, M.H.; Qin, Y.M.; Jiang, H.Y.; Zhang, X.; Wu, M.H. Luteolin Inhibits Tumorigenesis and Induces Apoptosis of Non-Small Cell Lung Cancer Cells via Regulation of MicroRNA-34a-5p. Int J Mol Sci 2018, 19. [Google Scholar]
- Yoshioka, H.; Tominaga, S.; Amano, F.; Wu, S.; Torimoto, S.; Moriishi, T.; Tsukiboshi, Y.; Yokota, S.; Miura, N.; Inagaki, N.; Matsushita, Y.; Maeda, T. Juzentaihoto alleviates cisplatin-induced renal injury in mice. Traditional & Kampo Medicine 2024, 11, 147–155. [Google Scholar]
Figure 1.
Proliferation of HEPM cells treated with MPM (0.03-10 µM) for 48 h. **p < 0.01, and ***p < 0.001 versus control (n=6).
Figure 1.
Proliferation of HEPM cells treated with MPM (0.03-10 µM) for 48 h. **p < 0.01, and ***p < 0.001 versus control (n=6).
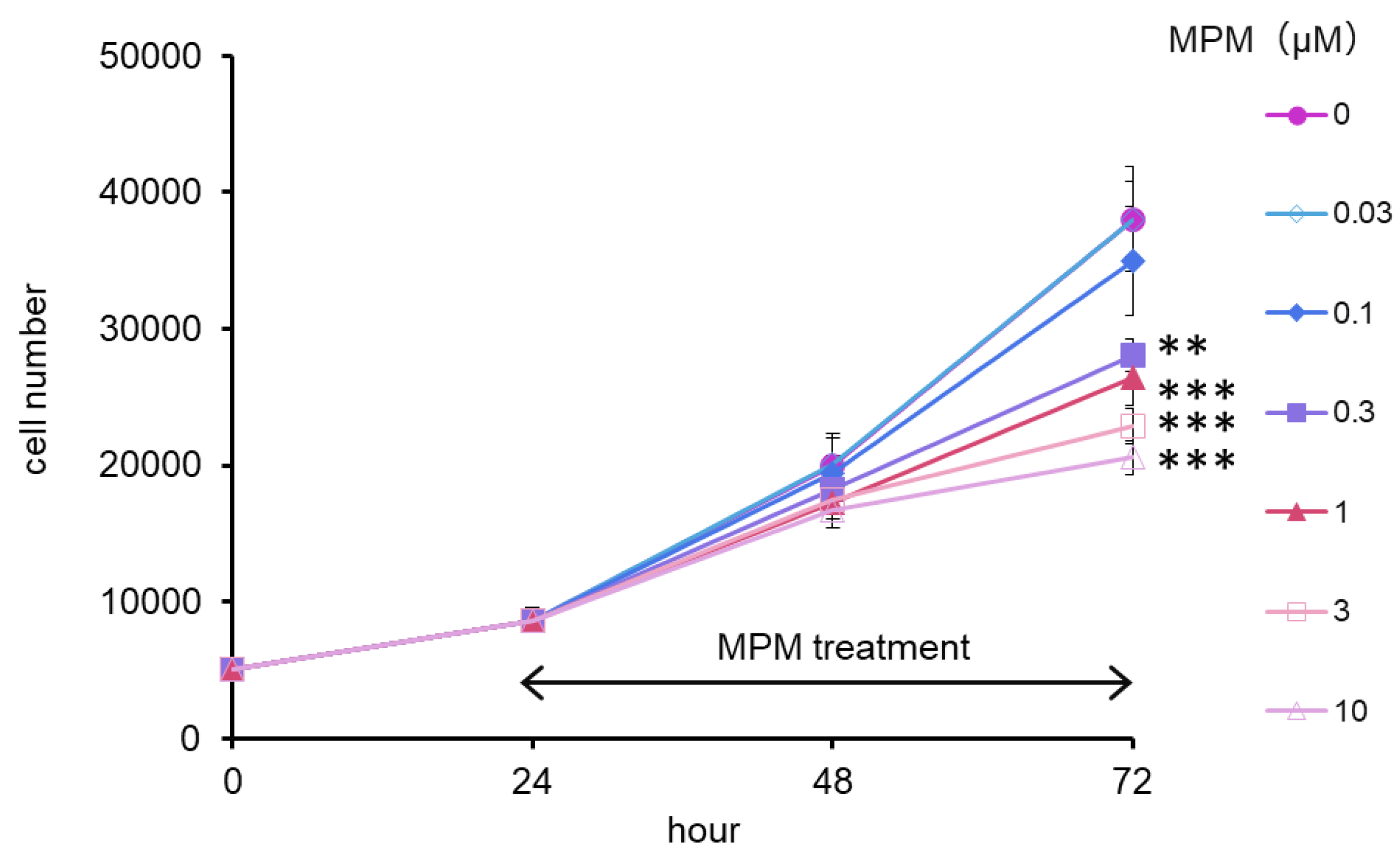
Figure 2.
Protective effect of SE against MPM-induced inhibition of HEPM cell proliferation. 1 µM MPM and 100 µg/ml SE were used. *p < 0.05, and ***p < 0.001 versus control (n=6).
Figure 2.
Protective effect of SE against MPM-induced inhibition of HEPM cell proliferation. 1 µM MPM and 100 µg/ml SE were used. *p < 0.05, and ***p < 0.001 versus control (n=6).
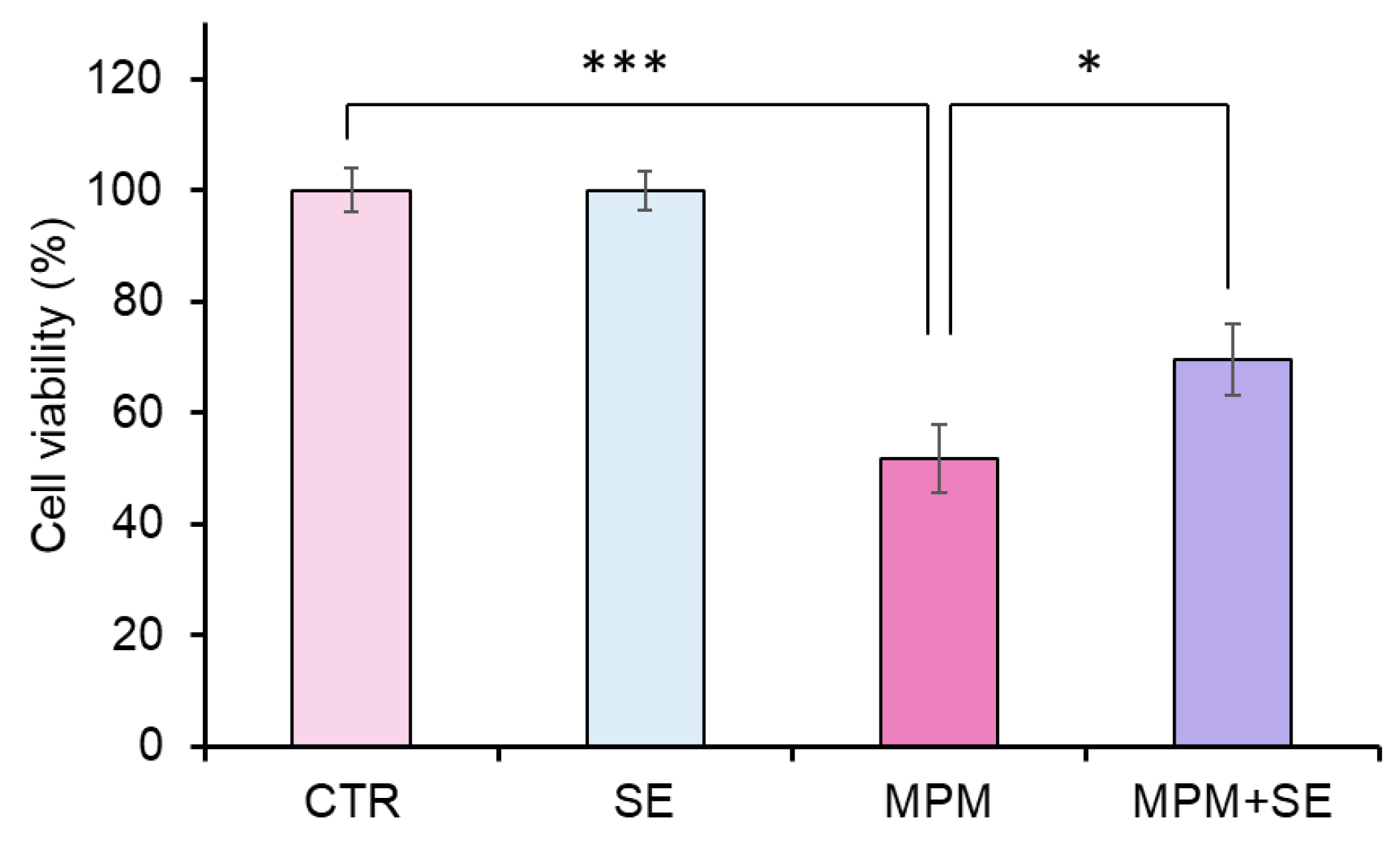
Figure 3.
HPLC analysis results (PDA, UV = 200-650 nm) of SCC and SE. (a) 100 µL injection of SCC (2.0 mg/mL) (b) 100 µL injection of SE (2.0 mg/mL).
Figure 3.
HPLC analysis results (PDA, UV = 200-650 nm) of SCC and SE. (a) 100 µL injection of SCC (2.0 mg/mL) (b) 100 µL injection of SE (2.0 mg/mL).
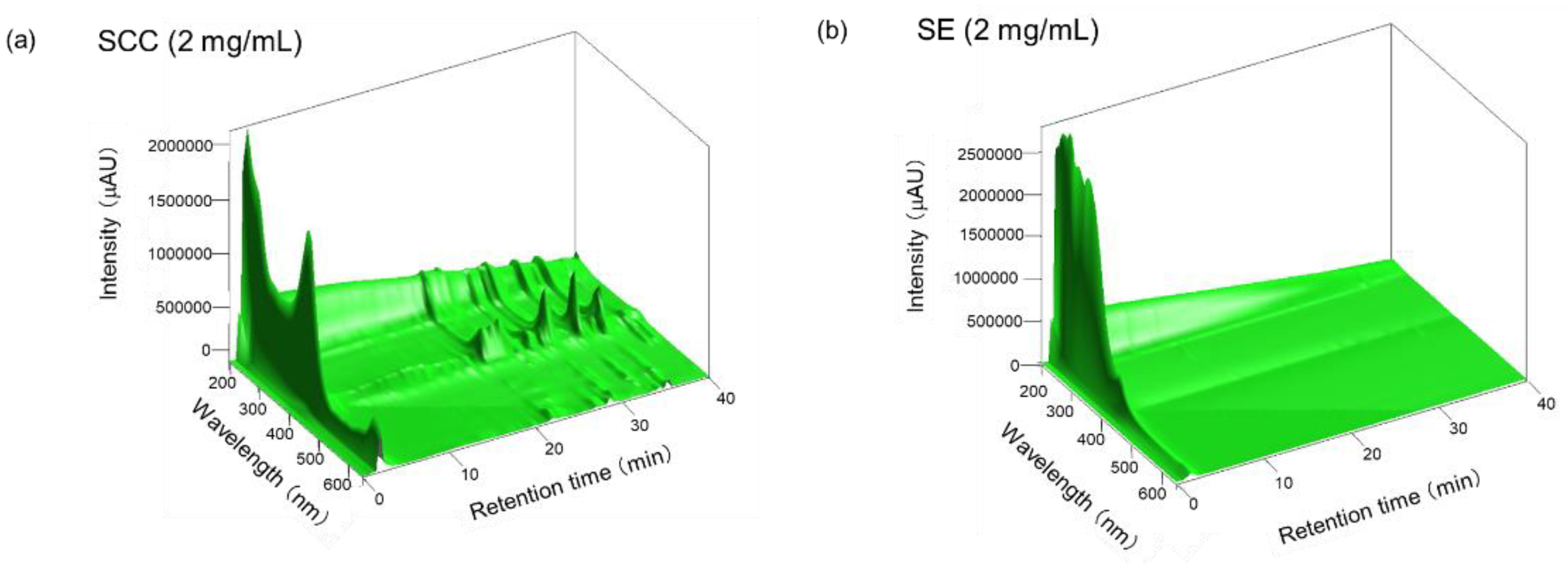
Figure 4.
Sodium copper chlorophyllin failed to alleviate MPM-induced cell proliferation inhibition in HEPM cells. 1 µM MPM and 10×10-4 µg/ml SCC were used. ***p < 0.001 (n=6). N.S.; Not significant.
Figure 4.
Sodium copper chlorophyllin failed to alleviate MPM-induced cell proliferation inhibition in HEPM cells. 1 µM MPM and 10×10-4 µg/ml SCC were used. ***p < 0.001 (n=6). N.S.; Not significant.
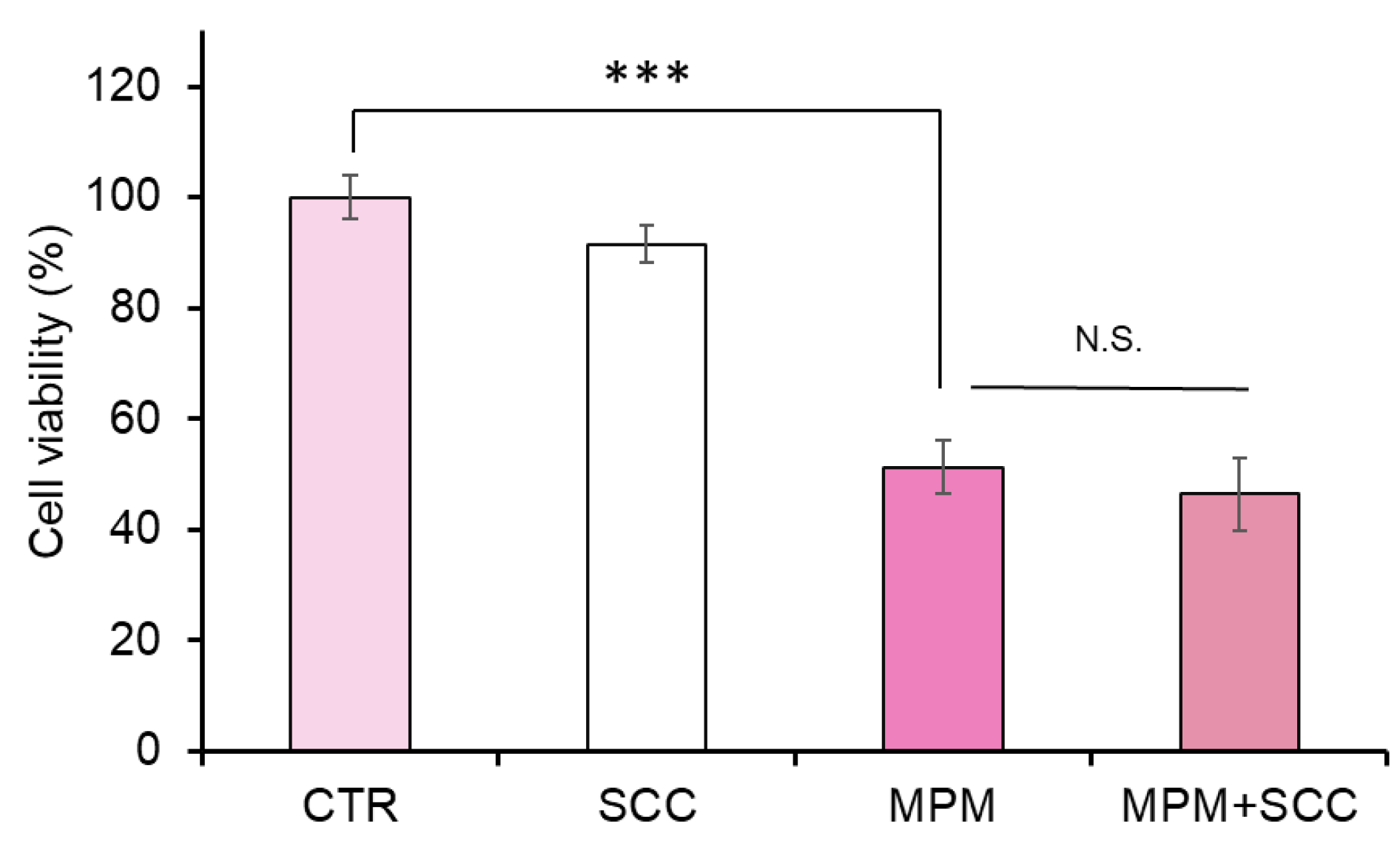
Figure 5.
SE attenuated MPM-induced cell cycle arrest in HEPM cells. (a) BrdU staining (green) of HEPM cells after treatment with 1 µM MPM and/or 100 µg/ml SE for 48 h. BrdU-positive cells were stained green, and nuclei were counterstained with 4’, 6-diamidino-2-phenylindole (blue). Scale bar, 50 µm. The graph shows the quantification of BrdU-positive cells. **p < 0.01 and ***p < 0.001 (n=8-10). (b) Western blotting of HEPM cells treated with 1 µM MPM and/or 100 µg/ml SE for 48 h. β-ACTIN served as an internal control.
Figure 5.
SE attenuated MPM-induced cell cycle arrest in HEPM cells. (a) BrdU staining (green) of HEPM cells after treatment with 1 µM MPM and/or 100 µg/ml SE for 48 h. BrdU-positive cells were stained green, and nuclei were counterstained with 4’, 6-diamidino-2-phenylindole (blue). Scale bar, 50 µm. The graph shows the quantification of BrdU-positive cells. **p < 0.01 and ***p < 0.001 (n=8-10). (b) Western blotting of HEPM cells treated with 1 µM MPM and/or 100 µg/ml SE for 48 h. β-ACTIN served as an internal control.
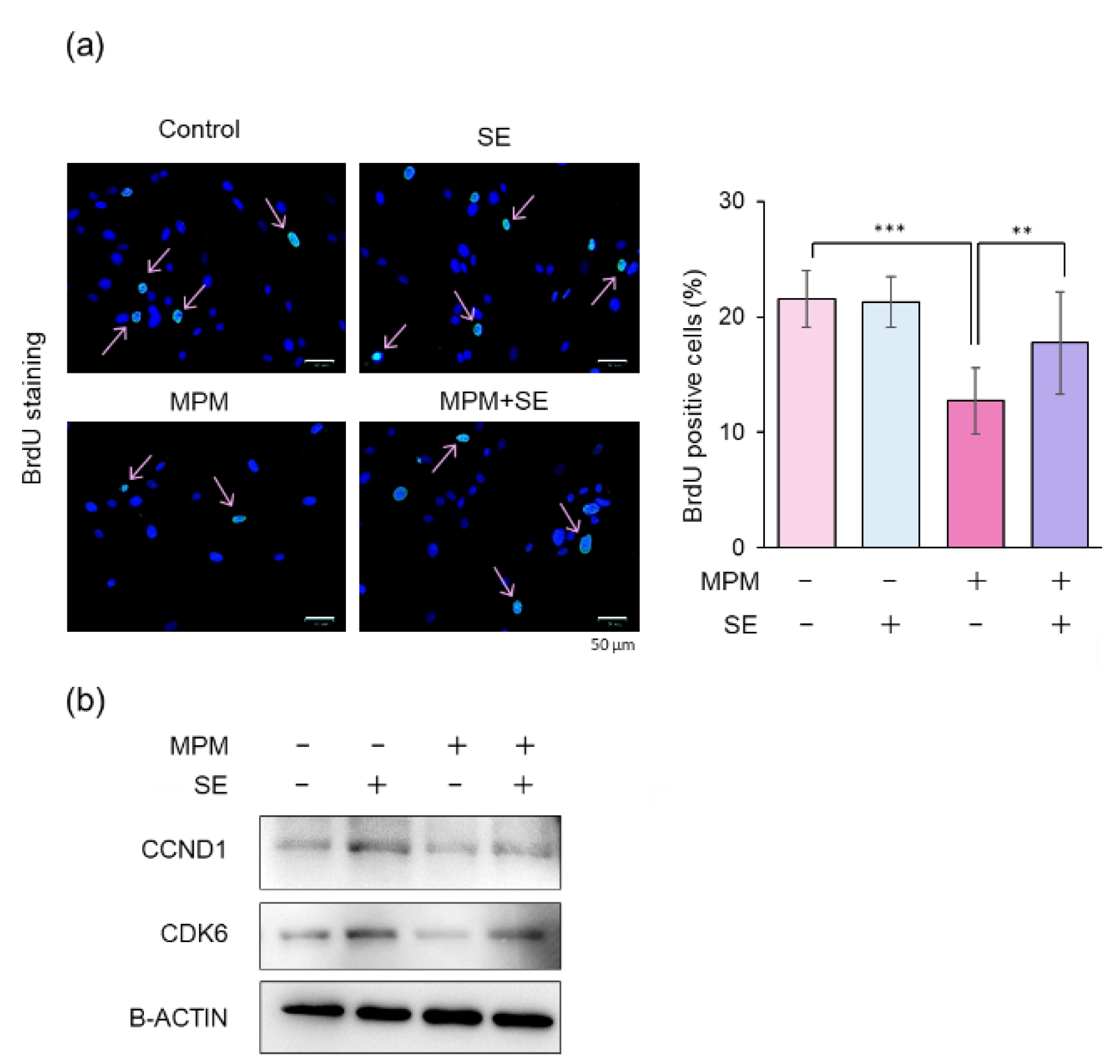
Figure 6.
SE downregulated of miR-4680-3p levels and upregulated of ERBB2 and JADE1 in HEPM cells. (a) Quantitative RT-PCR analysis of Let-7c-5p and miR-4680-3p expression after treatment with 1 µM MPM and/or 100 µg/ml SE in HEPM cells. *p < 0.05, **p < 0.01, and ***p < 0.001. N.S.; Not Significant. (b) Quantitative RT-PCR analysis of BACH1, PAX3, ERBB2, and JADE1 expression after treatment with 1 µM MPM and/or 100 µg/ml SE in HEPM cells. **p < 0.01, and ***p < 0.001. N.S.; Not Significant.
Figure 6.
SE downregulated of miR-4680-3p levels and upregulated of ERBB2 and JADE1 in HEPM cells. (a) Quantitative RT-PCR analysis of Let-7c-5p and miR-4680-3p expression after treatment with 1 µM MPM and/or 100 µg/ml SE in HEPM cells. *p < 0.05, **p < 0.01, and ***p < 0.001. N.S.; Not Significant. (b) Quantitative RT-PCR analysis of BACH1, PAX3, ERBB2, and JADE1 expression after treatment with 1 µM MPM and/or 100 µg/ml SE in HEPM cells. **p < 0.01, and ***p < 0.001. N.S.; Not Significant.
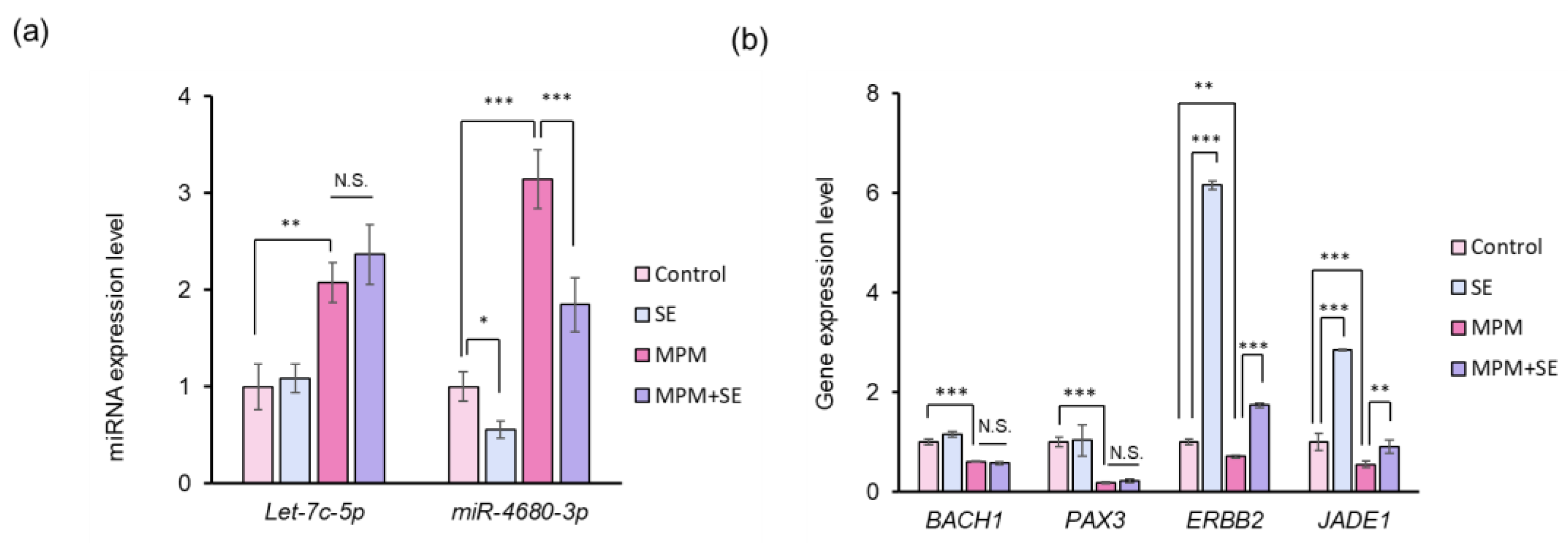
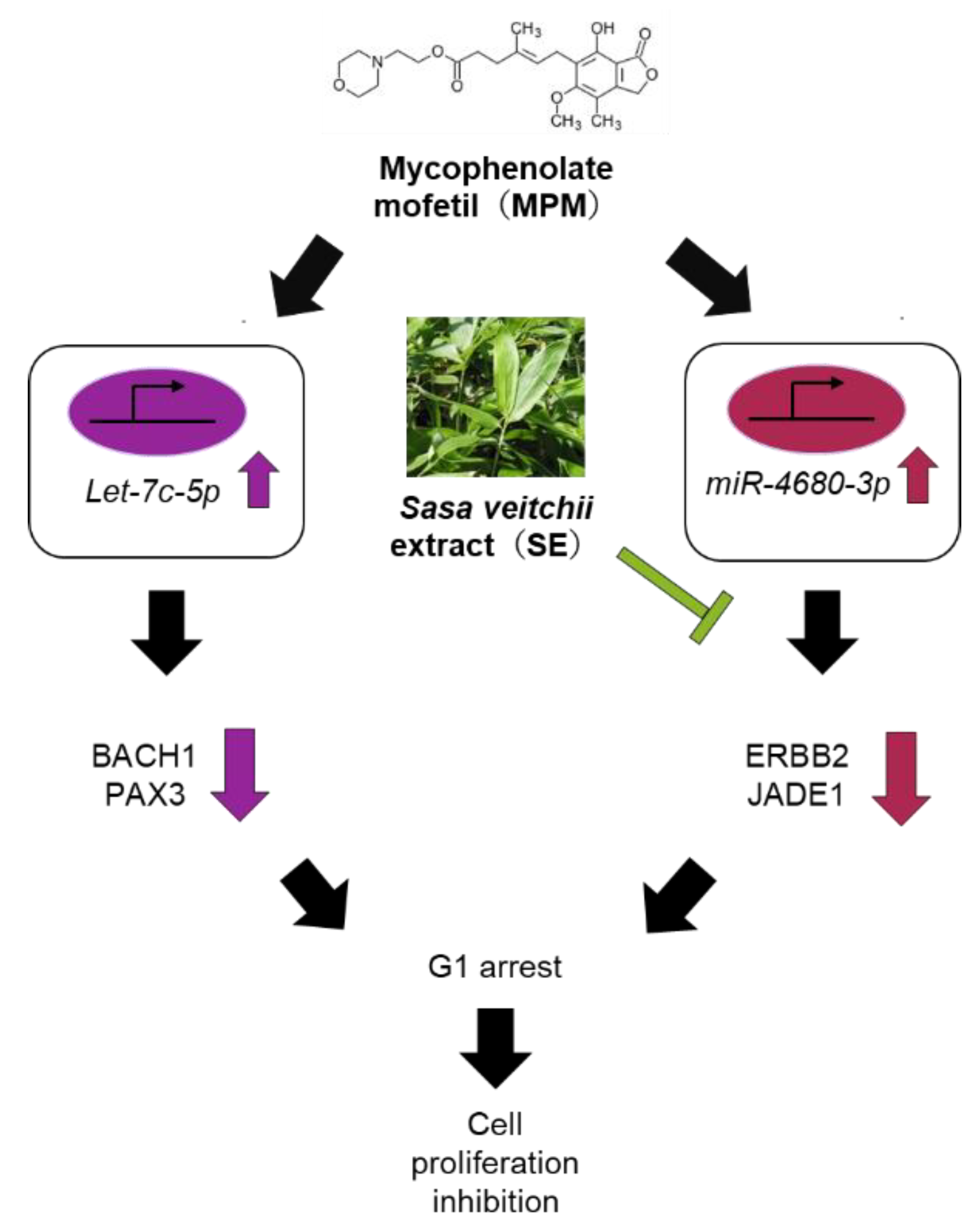
Figure 7.
Proposed mechanism of SE against MPM-induced cell proliferation inhibition.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



