
Submitted:
22 July 2025
Posted:
22 July 2025
You are already at the latest version
Abstract
The discovery of the structure of DNA and the elucidation of the molecular mechanisms of replication, transcription, and translation are the foundations of modern biology and medicine. However, in the early 80s, the prion hypothesis introduced a new system of biological information transfer that does not rely on DNA; it introduced the concept of conformational propagation through templating. Unlike the molecular biology revolution, which was based on detailed molecular structures and mechanisms, the prion hypothesis was postulated in the absence of clear molecular structures or mechanisms. In this perspective, we highlight 10 points in which the prion hypothesis contradicts the molecular, structural, and mechanistic experimental evidence accrued since its inception four decades ago. Alternatively, we postulate that an extension of the thermodynamic hypothesis of protein folding (Anfinsen’s dogma) to the state of proteins at high concentration (supersaturation) is better suited for explaining the different facets and pathways of protein aggregation.
Keywords:
prion
; amyloid
; seeding
; templating
; propagation
; nucleation
; phase transition
; supersaturation
Article
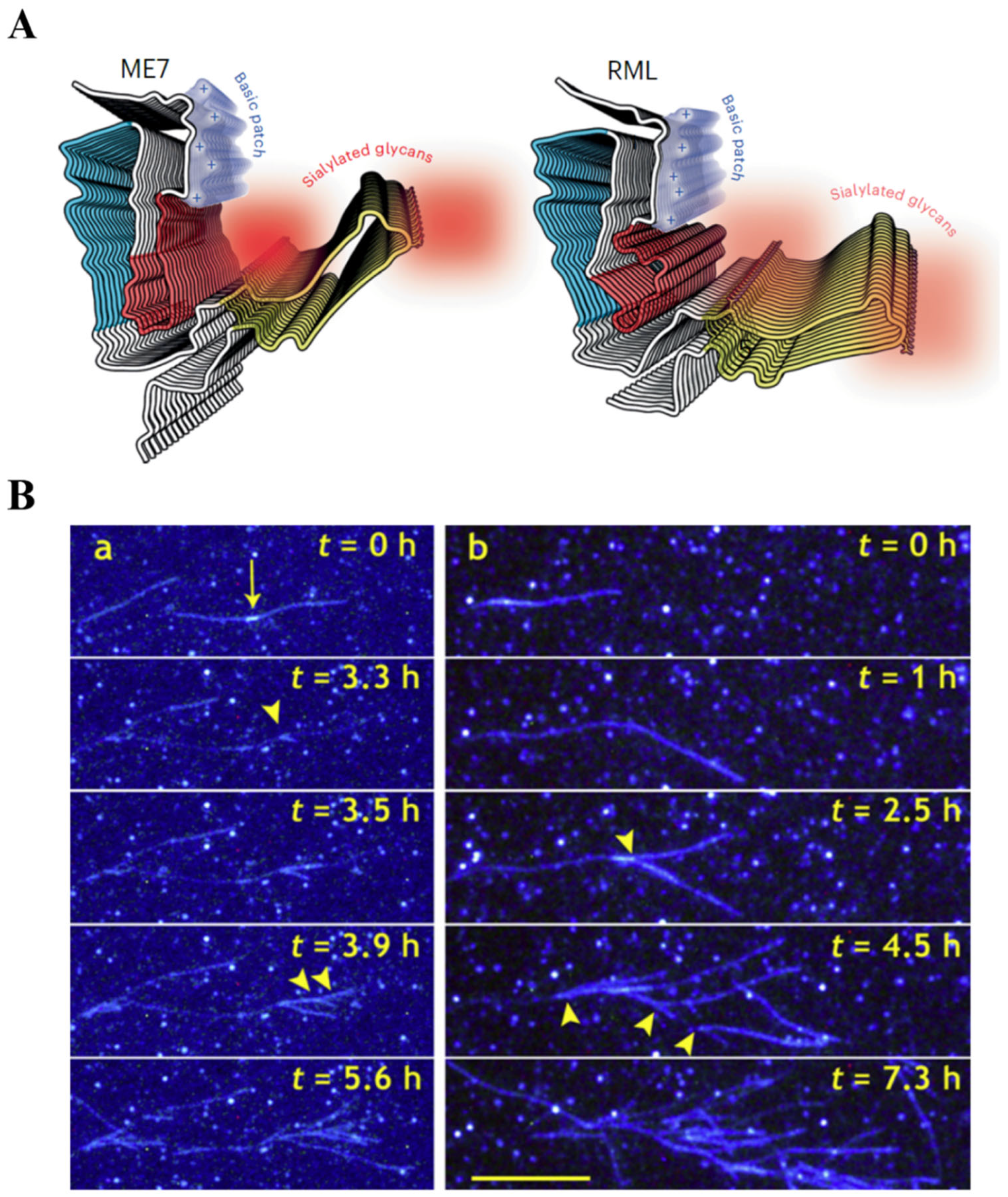
The prion hypothesis introduced a new system of biological information: conformational information transfer through templating. Unlike normal protein folding, which takes place spontaneously based on the primary sequence information of the protein (the thermodynamic hypothesis of protein folding or Anfinsen’s dogma), the amyloid conformation in diseases such as Creutzfeldt-Jakob disease is postulated to require a conformational template, a prion, which acts as a template to “imprint” its corrupt conformation on similar proteins [1]. Experimentally, the prion hypothesis is based on one phenomenon: seeding. This is when an amyloid fibril fragment, a seed, is added to a concentrated solution of other proteins, it catalyzes their transformation into amyloids. However, for decades it has remained unclear what exactly is the conformational information carried by prions and what is the molecular mechanism of templating. More recently, and thanks to a better understanding of the structure of amyloid fibrils, the term “conformational information” has come to designate the specific 2D cross-sectional shape, i.e., the particular cross-sectional pattern of folds and turns of protofilaments and fibrils (Figure 1A) [2]. In this framework, a prion, which is an amyloid fibril fragment, templates its cross-sectional shape on incoming protein molecules binding to its tip during elongation. The incoming molecules must accommodate the cross-sectional shape of the fibril by binding in a parallel, in-register manner (i.e., the N and C termini align in the same direction, and each amino acid of the incoming molecule stacks on top of the identical residue at the tip of the fibril) [3]. This is also the underlying principle behind the concept of prion “strains”, where different cross-sectional seed shapes are postulated to imprint their distinctive pattern of folds and turns on incoming protein molecules, leading to different disease phenotypes [2]. Similarly, prion propagation occurs via breakage or fission of a fibril with a particular cross-sectional shape into smaller fragments or seeds, which then template their shape onto incoming protein molecules. Thus, in its modern formulation, the prion templating and propagation of conformational information is based on elongation and fission.
However, for the elongation and fission mechanism to preserve and propagate specific cross-sectional shape information sustainably across generations of fibrils and across different cells, tissues, and hosts, it must fulfill some basic criteria:
- Incoming proteins must only bind to the fibril tip to accommodate its specific cross-sectional shape.
- Incoming proteins must have the same sequence as the protein in the fibril tip to enable parallel, in-register stacking.
However, there is an overwhelming amount of experimental evidence showing that both criteria are not fulfilled during amyloid growth:
- Amyloid growth cannot maintain elongation even for one generation of fibrils because the process is immediately taken over by branching (Figure 1B), where new fibrils grow as branches on the lateral surface of the parent fibril, a process termed secondary nucleation [4,5]. The lateral surface of the fibril bears no resemblance to its cross-sectional shape (the conformational information) and cannot engage in a parallel, in-register protein binding.
- Seeds of one protein can induce amyloid aggregation of another protein without the sequence homology needed for parallel-in register elongation, a process termed cross-seeding [6]. In this case, heterologous seeds act mainly as catalytic surfaces that do not relay any conformational information [7].
Based on these two points alone, the prion hypothesis as it pertains to sustained conformational templating and propagation via elongation and fission is not supported by experimental evidence. Yet there are additional fundamental problems with the prion hypothesis, including:
- 3.
- There is no thermodynamic incentive for a protein molecule to exit its stable native conformation to mold itself on a fibril’s tip.
- 4.
- There is no mechanism by which seeds can go around in solution “fishing” for similar protein molecules to mold it into their shape.
- 5.
- No machinery has ever been found that could restrict amyloid growth to tip elongation and prevent branching to preserve the cross-sectional template information.
- 6.
- While parallel in-register is the most common β-sheet stacking architecture of amyloids, amyloids with anti-parallel and out-of-register architecture have been experimentally found [8].
- 7.
- 8.
- In a process termed homogenous nucleation, amyloids form spontaneously at high protein concentrations without any template [11].
- 9.
- Amyloid formation is strictly dependent on protein concentration and does not take place under dilute conditions no matter how many seeds or other catalysts are present.
- 10.
The prion hypothesis, postulated long before the mechanistic and structural information outlined above was known, is currently in conflict with the experimental evidence accrued since its inception. It mischaracterized the phenomenon of protein phase transition as replication. What drives amyloid formation, similar to any other phase transition (crystallization, for example), is the increased probability of forming intermolecular bonds due to molecular proximity at higher protein concentrations (supersaturation). From this perspective, Anfinsen principles (the thermodynamic hypothesis) still apply to the amyloid phenomenon, where the amyloid conformation (the cross-β architecture) is spontaneously adopted under supersaturated conditions because it is more thermodynamically favorable than the native conformation, which is dependent on sequence-dependent intramolecular interactions under subsaturated conditions. In this case, no template is needed, just a nucleation catalyst, which can be any surface that lowers the thermodynamic barrier to phase transition [10]. A process that can proceed without a template is by definition a non-templated process. Thus, instead of expanding the prion hypothesis to accommodate even more diseases such as Alzheimer’s and Parkinson’s [1], we suggest extending Anfinsen’s thermodynamic hypothesis to include protein folding into the amyloid conformation under supersaturated conditions. This classical biophysical framework fits better with the experimental evidence as it includes seeding (via homologous or heterologous seeds) without excluding other pathways of amyloid induction that do not involve a proteinaceous seed component (e.g., lipid/microbial catalyzed nucleation, protein overexpression). Such framework frees the phenomenon from the unnecessary constraints of templating, enabling a better understanding of the etiological factors triggering amyloid formation and hopefully opening novel avenues for therapeutic interventions.
Disclosures relevant to this manuscript
The authors have no disclosures relevant to this manuscript.
Disclosures
KE cofounded REGAIN Therapeutics and is co-inventor of the patent “Compositions and methods for treatment and/or prophylaxis of proteinopathies.” AJE has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Mitsubishi Tanabe Pharma America (formerly, Neuroderm), Amneal, Acadia, Avion, Acorda, Bial, Kyowa Kirin, Supernus (formerly, USWorldMeds), NeuroDiagnostics, Inc (SYNAPS Dx), Intrance Medical Systems, Inc., Merz, Praxis Precision Medicines, Citrus Health, and Herantis Pharma; Data Safety Monitoring Board (chair) of AskBio; and publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer. He cofounded REGAIN Therapeutics and is co-inventor of the patent “Compositions and methods for treatment and/or prophylaxis of proteinopathies.” He serves on the editorial boards of the Journal of Parkinson’s Disease, Journal of Alzheimer’s Disease, European Journal of Neurology, Movement Disorders Clinical Practice, and JAMA Neurology.
References
- Condello C, Westaway D, Prusiner SB. Expanding the Prion Paradigm to Include Alzheimer and Parkinson Diseases. JAMA Neurol. 2024;81(10):1023-1024. [CrossRef]
- Manka SW, Wenborn A, Betts J, et al. A structural basis for prion strain diversity. Nat Chem Biol. 2022;(June). [CrossRef]
- Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. BioEssays. 2008;30(10):955-964. [CrossRef]
- Törnquist M, Michaels TCTT, Sanagavarapu K, et al. Secondary nucleation in amyloid formation. Chemical Communications. 2018;54(63):8667-8684. [CrossRef]
- Andersen CB, Yagi H, Manno M, et al. Branching in amyloid fibril growth. Biophys J. 2009;96(4):1529-1536. [CrossRef]
- Subedi S, Sasidharan S, Nag N, Saudagar P, Tripathi T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Published online 2022.
- Koloteva-Levine N, Aubrey LD, Marchante R, et al. Amyloid particles facilitate surface-catalyzed cross-seeding by acting as promiscuous nanoparticles. Proc Natl Acad Sci U S A. 2021;118(36):1-12. [CrossRef]
- Tycko R, Wickner RB. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc Chem Res. 2013;46(7):1487-1496. [CrossRef]
- Jean L, Lee CF, Vaux DJ. Enrichment of amyloidogenesis at an air-water interface. Biophys J. 2012;102(5):1154-1162. [CrossRef]
- Ezzat K, Sturchio A, Espay AJ. Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies. Biology (Basel). 2022;11(4):535. [CrossRef]
- Srivastava AK, Pittman JM, Zerweck J, et al. β-Amyloid Aggregation and Heterogeneous Nucleation. Protein Science. Published online 2019:pro.3674. [CrossRef]
- Peduzzo A, Linse S, Buell A. The Properties of α-Synuclein Secondary Nuclei are Dominated by the Solution Conditions Rather than the Seed Fibril Strain. 2019;(2):1-22. [CrossRef]
- Lövestam S, Schweighauser M, Matsubara T, et al. Seeded assembly in vitro does not replicate the structures of α-synuclein filaments from multiple system atrophy. FEBS Open Bio. 2021;11(4):999-1013. [CrossRef]
- Frey L, Ghosh D, Qureshi BM, et al. On the pH-dependence of α-synuclein amyloid polymorphism and the role of secondary nucleation in seed-based amyloid propagation. Elife. 2024;12. [CrossRef]
Figure 1.
Immediate branching during amyloid growth precludes cross-sectional shape templating even for one generation of fibrils. A. The 2D cross-sectional shape of two PrP amyloid protofilaments that is proposed to denote and propagate different prion strains via elongation. Reproduced with permission from Manka et al. [2]. B. Total internal reflection fluorescence microscopy (TIRFM) images following the growth over time of glucagon amyloid fibrils by branching. The bars represent 10 µm. Reproduced with permission from Andersen et al. [5].
Figure 1.
Immediate branching during amyloid growth precludes cross-sectional shape templating even for one generation of fibrils. A. The 2D cross-sectional shape of two PrP amyloid protofilaments that is proposed to denote and propagate different prion strains via elongation. Reproduced with permission from Manka et al. [2]. B. Total internal reflection fluorescence microscopy (TIRFM) images following the growth over time of glucagon amyloid fibrils by branching. The bars represent 10 µm. Reproduced with permission from Andersen et al. [5].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



