Submitted:
01 May 2025
Posted:
02 May 2025
You are already at the latest version
Abstract
This manuscript reports the synthesis and characterization of 19 novel heterostilbene carbamates, designed as selective butyrylcholinesterase (BChE) inhibitors with potential application in the treatment of neurodegenerative disorders, particularly Alzheimer's disease. The compounds were synthesized from resveratrol analogs, and their structures were confirmed by NMR spectroscopy, high-resolution mass spectrometry (HRMS), and single-crystal X-ray diffraction for selected derivatives (compounds 1 and 4). In vitro assays demonstrated high selectivity toward BChE over acetylcholinesterase (AChE), with compound 16 exhibiting exceptional inhibitory activity (IC₅₀ = 26.5 nM). Furthermore, compound 16 showed moderate anti-inflammatory effects by inhibiting LPS-stimulated TNF-α production in peripheral blood mononuclear cells. In silico ADME(T) profiling revealed favorable pharmacokinetic properties and low mutagenic potential for the majority of compounds. Molecular docking and molecular dynamics simulations confirmed stable binding interactions within the BChE active site. These results highlight heterostilbene carbamates as promising lead structures for developing novel therapeutic agents targeting neurodegenerative diseases.
Keywords:
anti-inflammatory activity
; butyrylcholinesterase inhibition
; carbamates
; docking
; molecular dynamics
; synthesis
1. Introduction
Carbamates are a group of compounds that have garnered increasing interest as potential therapeutic agents for treating neurodegenerative diseases. These compounds primarily function by inhibiting acetylcholinesterase (AChE), but they can also target butyrylcholinesterase (BChE), another cholinesterase enzyme involved in the hydrolysis of acetylcholine. While AChE is the primary enzyme responsible for acetylcholine breakdown in the central nervous system under normal conditions, BChE becomes upregulated in several neurodegenerative conditions, including Alzheimer's disease (AD), where it is often found in higher concentrations in the brain [1]. Notably, BChE is thought to play a more significant role in the pathogenesis of AD than previously appreciated, and this has led to increased interest in selective BChE inhibitors as potential treatments. Selective inhibition of BChE using carbamates is being actively explored for its therapeutic benefits, particularly in AD. In contrast to traditional carbamates, which inhibit both AChE and BChE, selective BChE inhibitors aim to specifically target BChE while leaving AChE activity relatively intact. This selective inhibition may offer several advantages. For instance, while both AChE and BChE contribute to cholinergic dysfunction in AD, BChE is particularly important in the later stages of the disease, where its levels increase and correlate with the severity of cognitive decline [2,3]. By selectively inhibiting BChE, carbamates can help restore a more balanced cholinergic tone, potentially improving cognitive function with fewer side effects than dual inhibitors of AChE and BChE. Moreover, BChE is implicated in the aggregation of amyloid-beta peptides, a hallmark of AD pathology, and selective inhibition of BChE may reduce amyloid-beta toxicity, providing an additional therapeutic benefit [4].
One notable example of a carbamate selective for BChE is Phenserine, which has been studied for its ability to inhibit BChE without significantly affecting AChE activity at lower doses [5]. Phenserine has shown promise in preclinical studies and clinical trials by improving cognitive function in patients with AD while reducing the risk of side effects such as nausea and diarrhea, which are common with non-selective cholinesterase inhibitors [6]. Additionally, Phenserine’s selective inhibition of BChE has been linked to a reduction in amyloid-beta aggregation, an effect that has significant therapeutic implications for slowing the progression of AD. This dual action—restoring cholinergic function and reducing amyloid-beta toxicity—highlights the potential advantages of selective BChE inhibitors over more traditional, non-selective carbamates.
Other selective BChE inhibitors have also been synthesized and tested for their effects on both cholinergic transmission and amyloid-beta pathology. For example, Rivastigmine, although initially developed as a dual AChE and BChE inhibitor, has been shown to have a higher affinity for BChE in certain doses, suggesting that at optimal concentrations, it may primarily target BChE in the treatment of AD [7,8]. Similarly, carbamates like Donepezil have been shown to exert some degree of selective BChE inhibition under specific conditions, making them viable candidates for future exploration in diseases with pronounced BChE activity, such as AD and Parkinson’s disease [8]. These findings suggest that carbamates with selective BChE inhibitory properties could not only help alleviate symptoms of cognitive decline but may also have disease-modifying effects by interacting with the neurodegenerative processes associated with amyloid plaque formation. The role of BChE in amyloid-beta aggregation has driven much of the research into selective BChE inhibitors. In AD, amyloid plaques accumulate and disrupt normal neuronal function. Research results suggest that BChE can interact with amyloid-beta peptides, promoting their aggregation and exacerbating neurodegeneration [4]. By inhibiting BChE specifically, carbamates may reduce amyloid-beta aggregation, thus providing a dual benefit: enhancing cholinergic transmission and potentially mitigating the pathological effects of amyloid plaques.
Selective BChE inhibitors may also offer therapeutic benefits in other neurodegenerative diseases. For example, Parkinson’s disease (PD), which involves both dopaminergic and cholinergic dysfunction, may benefit from the modulation of BChE activity. In Parkinson's disease, BChE contributes to the imbalance between the cholinergic and dopaminergic systems. The selective inhibition of BChE could potentially restore this balance, alleviating cognitive and motor symptoms [9]. Given the high prevalence of cognitive decline in PD and other conditions like Lewy body dementia, selective BChE inhibition is a promising strategy for improving both cognitive and motor function. In conclusion, selective BChE inhibition by carbamates represents a promising strategy in the treatment of neurodegenerative disorders, particularly AD, where BChE activity is upregulated and linked to disease progression. By specifically targeting BChE, these compounds may enhance cholinergic transmission, reduce amyloid-beta aggregation, and provide neuroprotective effects, all while minimizing the side effects commonly seen with non-selective cholinesterase inhibitors. Research into the design and testing of selective BChE inhibitors continues to expand the understanding of their therapeutic potential, and they may ultimately serve as a critical component of future treatments aimed at slowing the progression of neurodegenerative diseases.
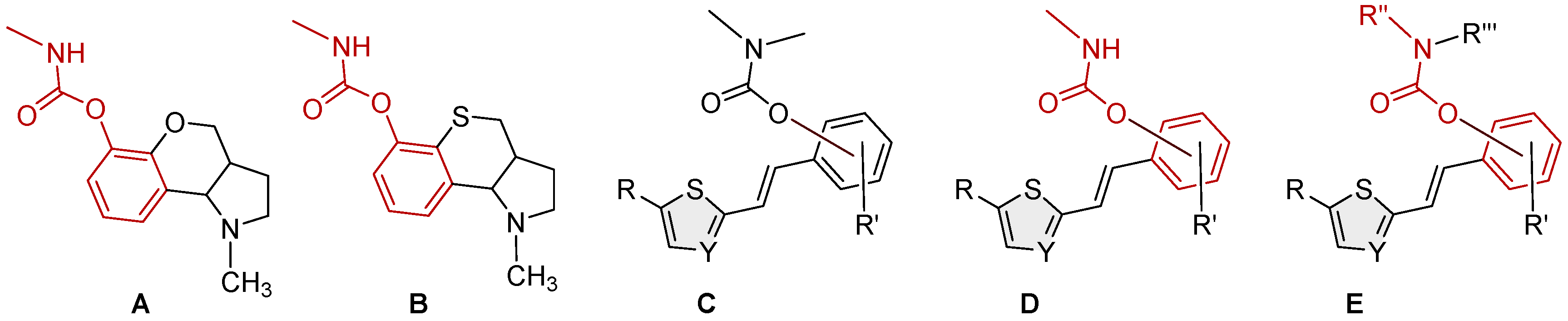
The structures of various other carbamates as cholinesterase inhibitors are well known in the literature [10,11,12,13,14,15,16,17,18] (Figure 1, structures A and B [16]). Considering our experience in the design of new cholinesterase inhibitors, especially resveratrol analogs, the basic stilbene skeleton was used as a structural unit for carbamates in the previous research (Figure 1, structure C) [19]. Inhibitory activity was tested toward AChE and BChE enzymes. In the tested group of compounds, two leading inhibitors achieved excellent selective inhibitory activity for BChE, with IC50 values of 0.12 ± 0.09 μM and 0.38 ± 0.01 μM. Both were much more active than the galantamine, the standard inhibitor against BChE. Molecular docking of the most promising inhibitor candidates revealed that stabilizing interactions between the active site residues of BChE and the ligands involve π-stacking and alkyl-π interactions depending on the orientation of the carbamate group.
Based on in silico, experimental, and computational results on biological activity, the previous heterostilbene carbamates represented potential selective resveratrol-like BChE inhibitors as new therapeutics for neurological disorders. These compounds were an excellent initial basis for the design and synthesis of new heterostilbene carbamates (Figure 1, structures D and E) to create a larger chemical library for high-throughput screening and drug development.
2. Materials and Methods
2.1. General Remarks
NMR spectra were recorded using a Bruker AV300 or AV600 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) equipped with a 5 mm probe head at the Ruđer Bošković Institute. The standard 1H and proton-decoupled 13C{1H} NMR spectra were recorded at frequencies of 300.000 and 600.130 MHz for 1H, and 75.432 and 150.903 MHz for 13C, respectively. Chemical shifts (δ/ppm) for both 1H and 13C NMR spectra were referenced to the tetramethylsilane (TMS) signal. All measurements were recorded in deuterated chloroform (CDCl3) at 25 °C. Reactions were monitored by thin-layer chromatography (TLC) using silica gel-coated plates (0.2 mm, 60/Kieselguhr F254) immersed in 10 mL of the solvent system. Post-synthesis workup involved extraction between water and DCM, with a single addition of 2.4 M HCl in the second step. After separating the organic and aqueous phases, the organic layer was dried over anhydrous magnesium sulfate (MgSO4). The products were purified via column chromatography using glass columns of varying diameters, filled with silica gel (60 Å, technical grade) to different heights. The following abbreviations were used in the experimental section: TEA – triethylamine, DMAP – 4-dimethylaminopyridine, DCE – 1,2-dichloroethane, PE – petroleum ether, E – diethyl ether, HCl – hydrochloric acid, NMR – nuclear magnetic resonance, s – singlet, d – doublet, m – multiplet, t – triplet, dd – doublet of doublets, dt – doublet of triplets, dq – doublet of quartets. High-resolution mass spectrometry (HRMS) analyses were performed on a MALDI TOF/TOF analyzer, using an Nd:YAG laser at 355 nm with a fitting rate of 200 Hz.
2.2. Synthesis of Heterostilbene Carbamates 1–19
Initially synthesized trans-resveratrol analogues [20,21,22,23] were used as preliminary compounds in synthesizing heterostilbene carbamates 1–19. Accordingly, 50–100 mg of the appropriate resveratrol analogue was dissolved in 1.2 mL of DCE in a round-bottomed flask. Then, 1.8 eq of TEA was added to the reaction flask and 0.09 eq of DMAP. The reaction mixture was stirred at room temperature for 15 minutes, after which it was purged with argon for up to 1 minute. Following this, 1.2 eq of the corresponding carbamoyl chloride was added dropwise, and the mixture was stirred at 60 °C under inert conditions for 4 hours. Upon completion, the reaction mixture was extracted with 10 mL DCM and 10 mL distilled water, followed by adding 1 mL of 2.4 M HCl in the second extraction step. The organic phase was dried over anhydrous MgSO4, filtered, and evaporated to dryness using a rotary evaporator. The resulting carbamates 1–19 were purified by column chromatography, using various ratios of petroleum ether and diethyl ether as eluents. NMR analysis revealed that carbamates 1–19 were obtained as trans-isomers (with traces of cis-isomer in some cases), except for thiazole carbamate 8, which showed a higher proportion of the corresponding cis-isomer.
(E)-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (1): 37 mg (isolated 31%), white powder; m.p. 117-119 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.61 (d, J = 7.4 Hz, 1H), 7.26 (m, 3H), 7.20 (d, J = 5.3 Hz, 2H), 7.13 (d, J = 17.5 Hz, 1H), 7.12–7.10 (m, 1H), 7.05–7.00 (m, 1H), 5.10 (br s, 1H), 2.92 (d, J = 4.8 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 155.0 , 154.0, 148.3, 142.9, 128.3, 127.6, 126.4, 126.1, 125.8, 124.6, 123.6, 123.0, 121.8, 27.9; HRMS (ESI) (m/z) for C14H13NO2S: [M + H]+calcd = 259.0667, and [M + H]+measured = 259.0666.
(E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (2): 39 mg (isolated 33%), white powder; m.p. 110-111 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.28 (d, J = 9.7, 1H), 7.22 (d, J = 4.5 Hz, 1H), 7.18 (d, J = 18.4 Hz, 1H), 7.09 –7.02 (m, 2H), 7.01 (t, J = 2.7 Hz, 1H), 7.00–6.9 (m, 2H), 5.10 (br s, 1H), 2.92 (d, J = 4.2, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 160.2 (d, JCF = 243.3 Hz), 155.1, 144.3, 142.0, 130.6 (d, JCF = 7.7 Hz), 127.7, 125.1, 124.6, 124.0 (d, JCF = 8.7 Hz), 120.7, 114.9, 111.9, 27.6; HRMS (ESI) (m/z) for C14H12FNO2S: [M + H]+calcd = 277.0573, and [M + H]+measured = 277.0572.
(E)-4-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (3): 27 mg (isolated 23%), white powder; m.p. 123-124 °C; Rf (50% PE/E) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.33–7.21 (m, 2H), 7.20 (d, J = 3.1 Hz, 1H), 7.10 (d, J = 3.1 Hz, 1H), 7.03 (d, J = 8.9 Hz, 1H), 7.00 (t, J = 3.9 Hz, 1H), 6.91 (d, J = 16.3 Hz, 1H), 6.82 (dd, J = 2.7, 9.6 Hz, 1H), 5.00 (br s, 1H), 3.81 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 157.1, 155.4, 142.7, 142.2, 129.9, 130.7, 127.6, 126.5, 124.7, 123.8, 121.8, 114.1, 110.4, 55.7, 29.7; HRMS (ESI) (m/z) for C15H15NO3S: [M + H]+calcd = 289.0772, and [M + H]+measured = 289.0773.
(E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl methylcarbamate (4): 22 mg (isolated 18%), white powder; m.p. 109-111 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.41 (s, 1H), 7.22–7.17 (m, 2H), 7.09–7.04 (m, 2H), 7.03–6.96 (m, 3H), 5.04 (br s, 1H), 2.92 (d, J = 4.8 Hz, 3H), 2.35 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 155.2, 146.2, 143.0, 135.3, 129.4, 129.1, 127.6, 126.4, 126.3, 124.5, 123.3, 122.7, 121.9, 27.9, 21.0; HRMS (ESI) (m/z) for C15H15NO2S: [M + H]+calcd = 273.0824, and [M + H]+measured = 273.0826.
(E)-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (5): 18 mg (isolated 43%), white powder; m.p. 114-116 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.66–7.55 (m, 1H), 7.24 – 7.16 (m, 2H), 7.15–7. 09 (m, 2H), 6.89 (t, J = 16.7 Hz, 1H), 6.85 (d, J = 3.8 Hz, 1H), 6.60 (d, J = 3.5 Hz, 1H), 5.07 (br s, 1H), 2.93 (d, J = 4.6 Hz, 3H), 2.48 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 155.0, 148.2, 140.9, 139.5, 130.0, 128.0, 126.8, 125.9, 125.8, 125.7, 124.0, 123.0, 120.5, 29.7, 15.6; HRMS (ESI) (m/z) for C15H15NO2S: [M + H]+calcd = 273.0824, and [M + H]+measured = 273.0825.
(E)-4-fluoro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (6): 19 mg (isolated 42%), white powder; m.p. 105-107 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.29–7.23 (m, 2H), 7.09 (d, J = 16.4 Hz, 2H), 6.87 (d, J = 3.4 Hz, 1H), 6.80 (d, J = 16.4 Hz, 1H), 6.66–6.64 (m, 1H), 5.06 (br s), 2.93 (d, J = 4.9 Hz, 3H), 2.48 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 160.3 (JCF = 243.1 Hz), 155.0, 144.0, 140.3, 140.2, 131.9 (d, JCF = 8.3 Hz), 127.5, 125.9, 125.0, 124.4 (d, JCF = 8.3 Hz) , 119.4, 114.7 (d, JCF = 24.1 Hz), 111.8 (d, JCF = 24.1 Hz), 29.5, 15.7; HRMS (ESI) (m/z) for C15H14FNO2S: [M + H]+calcd = 291.0729, and [M + H]+measured = 291.0731.
(E)-4-chloro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl methylcarbamate (7): 12 mg (isolated 25%), white powder; m.p. 122-123 °C; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.55 (d, J = 2.5 Hz, 1H), 7.13–7.09 (m, 2H), 7.06 (d, J = 8.9 Hz, 1H), 6.87 (d, J = 3.4, 1H), 6.80–6.76 (m, 1H), 6.66–6.64 (m, 1H), 5.07 (br s), 2.93 (d, J = 4.9 Hz, 3H), 2.47 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 154.7, 147.7, 146.5, 140.3, 131.8, 131.2, 127.7, 127.5, 125.9, 125.5, 125.2, 125.1, 124.3, 29.7, 15.7; HRMS (ESI) (m/z) for C15H14ClNO2S: [M + H]+calcd = 307.0434, and [M + H]+measured = 307.0433.
Mixture of (E) and (Z)-4-chloro-2-(2-(5-methylthiazol-2-yl)vinyl)phenyl methylcarbamate (8); for (E)-izomer the data are the following: 8 mg (isolated 17%), Rf (50% PE/E) = 0.30; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.52 (s, 1H), 7.26 (s, 1H), 7.12 (d, J = 16.6 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 16.3 Hz, 1H), 2.58 (d, J = 5.0 Hz, 3H), 2.57 (s, 3H); MS (ESI) (m/z) for C17H19NO3S: [M + H]+ 308 (100).
(E)-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (9): 40 mg (isolated 75%), colourless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.63–7.58 (m, 1H), 7.38–7.31 (m, 1H), 7.27–7.23 (m, 1H), 7.22–7.17 (m, 3H), 7.13 (t, J = 7.15 Hz, 1H), 7.05 (d, J = 3.3 Hz, 1H), 7.00–6.98 (m, 1H), 3.50 (dq, J = 7.3, 80.6 Hz, 2H), 3.10 (d, J = 87.6 Hz, 3H), 1.28–1.22 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 154.3, 148.8, 143.1, 130.4, 129.7, 128.8, 128.3, 127.6, 126.3, 125.6, 124.5, 123.4, 122.8, 44.1, 21.0, 13.5; HRMS (ESI) (m/z) for C16H17NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0985.
(E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (10): 28 mg (isolated 53%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.33–7.27 (m, 1H), 7.22 (d, J = 3.89 Hz, 1H), 7.17–7.04 (m, 3H), 7.03–6.86 (m, 3H), 3.49 (dq, J = 7.3, 39.1 Hz, 2H), 3.09 (d, J = 43.3 Hz, 3H), 1.25–1.20. (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1 (d, JCF = 242.6 Hz), 158.5, 154.1, 144.7, 142.5, 127.7, 127.1, 126.5, 126.3, 125.0 (d, JCF = 8.7 Hz), 120.8 (d, JCF = 8.7 Hz), 115.1 (d, JCF = 24.1 Hz), 111.8 (d, JCF =24.1 Hz), 44.1, 34.5, 14.2.
(E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (11): 85 mg (isolated 91%), colourless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.41 (s, 1H), 7.21–7.15 (m, 2H), 7.10–6.94 (m, 5H), 3.49 (dq, J = 7.0, 79.1 Hz, 2H), 3.08 (d, J = 86.4 Hz, 3H), 2.34 (s, 3H), 1.33–1.28 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: too small quantity for recording; HRMS (ESI) (m/z) for C17H19NO2S: [M + H]+calcd = 301.1136, and [M + H]+measured = 301.1141.
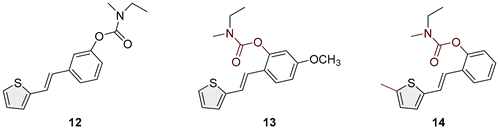
(E)-3-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (12): 63 mg (isolated 87%), colourless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.31 (t, J = 7.7 Hz, 1H), 7.28–7.22 (m, 3H), 7.20 (d, J = 4.5 Hz, 1H), 7.06 (d, J = 1.8 Hz, 1H), 7.03–6.97 (m, 2H), 6.90 (d, J = 15.8 Hz, 1H), 3.46 (dq, J = 7.2, 39.8 Hz, 2H), 3.04 (d, J = 45.6 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 154.5, 151.9, 142.6, 138.4, 129.4, 128.3, 127.6, 126.3, 124.5, 123.3, 122.5, 120.9, 119.2, 44.1, 34.3, 13.2; HRMS (ESI) (m/z) for C16H17NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0982.
(E)-5-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (13): 18 mg (isolated 53%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.52 (d, J = 8.9 Hz, 1H), 7.16–7.03 (m, 2H), 7.02–6.84 (m, 3H), 6.80–6.66 (m, 2H), 3.81 (s, 3H), 3.50 (dq, J = 7.2, 37.7 Hz, 2H), 3.09 (d, J = 42.4 Hz, 3H), 1.26–1.20 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1, 159.8, 149.6, 143.4, 127.6, 126.4, 125.7, 123.8, 122.4, 121.7, 121.3, 112.4, 108.3, 55.5, 44.2, 29.7, 13.5; MS (ESI) (m/z) for C17H19NO3S: [M + H]+ 318 (100).
(E)-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (14): 25 mg (isolated 23%), colourless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.58–7.50 (m, 1H), 7.16–7.11 (m, 1H), 7.05 (dt, J = 8.5; 2.1 Hz, 1H), 7.02–6.93 (m, 2H), 6.75 (d, J = 15.0 Hz, 1H), 6.39 (dd, J = 15.0; 1.8 Hz, 1H), 3.49 (dq, J = 7.9, 72.5 Hz, 2H), 3.05–2.84 (m, 3H), 2.60–2.32 (m, 3H), 1.24–1.18 (m, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: too small quantity for recording; MS (ESI) (m/z) for C17H19NO2S: [M + H]+ 302 (100).
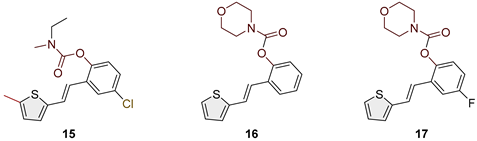
(E)-4-chloro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl ethyl(methyl)carbamate (15): 8 mg (isolated 13%), colourless oil; Rf (50% PE/E) = 0.80; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.20–7.14 (m, 2H), 7.07–6.96 (m, 5H), 3.50 (dq, J = 7.0, 79.1 Hz, 2H), 3.09 (d, J = 86.4 Hz, 3H), 2.35 (s, 3H), 1.31–1.27 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: too small quantity for recording; HRMS (ESI) (m/z) for C17H18ClNO2S: [M + H]+calcd = 335.0747, and [M + H]+measured = 335.0744.
(E)-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (16): 75 mg (isolated 90%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.62 (dd, J = 1.7, 7.5 Hz, 1H) 7.21–7.09 (m, 4H), 7.03–6.84 (m, 4H), 3.88–3. 44 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.4, 148.5, 142.9, 139.6, 130.4, 128.4, 127.7, 126.7, 125.9, 124.6, 123.1, 121.6, 66.8, 66.6 (one quaternary C is missing); HRMS (ESI) (m/z) for C17H17NO3S: [M + H]+calcd = 315.0929, and [M + H]+measured = 315.0932.
(E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (17): 40 mg (isolated 71%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.33–7.20 (m, 3H), 7.16–6.92 (m, 4H), 6.88 (d, J = 15.5 Hz, 1H), 3.88–3.43 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.3 (d, J = 244.3 Hz), 153.3, 144.3, 142.3, 131.4 (d, J = 8.5 Hz), 127.8, 127.3, 126.5 (d, J = 8.5 Hz), 125.1, 124.8, 120.5, 115.0 (d, J = 23.9 Hz), 111.9 (d, J = 23.9 Hz), 66.7, 66.6; HRMS (ESI) (m/z) for C17H16FNO3S: [M + H]+calcd = 333.0835, and [M + H]+measured = 333.0838.
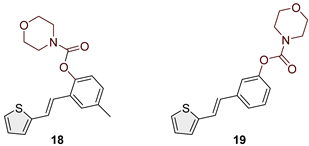
(E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (18): 78 mg (isolated 88%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.41 (s, 1H), 7.26–7.14 (m, 2H), 7.11–7.00 (m, 4H), 6.93 (d, J = 16.3 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.6, 146.4, 143.0, 135.4, 129.2, 127.7, 126.5, 126.4, 124.5, 123.4, 122.7, 121.7, 66.7, 66.5, 21.0 (one quaternary C is missing); HRMS (ESI) (m/z) for C18H19NO3S: [M + H]+calcd = 329.1086, and [M + H]+measured = 329.1091.
(E)-3-(2-(thiophen-2-yl)vinyl)phenyl morpholine-4-carboxylate (19): 87 mg (isolated 93%), colourless oil; Rf (50% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.38 – 7.28 (m, 2H), 7.25–7.18 (m, 3H), 7.14–6.95 (m, 3H), 6.89 (d, J = 16.0 Hz, 1H), 3.80 – 3.50 (m, 8H) 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.6, 151.6, 142.6, 138.6, 129.5, 127.6, 127.4, 126.4, 124.6, 120.7, 119.1, 66.6, 66.5 (two quaternary C are missing); HRMS (ESI) (m/z) for C22H23NO3S: [M + H]+calcd = 315.0929, and [M + H]+measured = 315.0934.
2.3. X-Ray Crystallography
Single crystal measurements were performed on an XtaLAB Synergy diffractometer, using micro-focus sealed X-ray tube CuKα (1.54184 Å) radiation at 100 K for compound 1 and room temperature [293(2) K] for compound 4. The CrysAlisPRO package [24] was used for data collection and reduction. The crystal structures were solved and refined within the OLEX2 program [25] using SHELXT [26] and SHELXL [26]. The model was refined using the full matrix least squares refinement; all non-hydrogen atoms were refined anisotropically. Hydrogen atoms were modelled as riding entities using the AFIX command. Molecular geometry calculations were performed by PLATON [27], and the molecular graphics were prepared using CCDC-Mercury [28]. Experimental details on crystal data, data collection, and structure refinement are summarized in Table 1.
Supplementary crystallographic data for this paper can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk). CCDC-2443931 and CCDC-2441980 contain the supplementary crystallographic data for this paper.
2.4. In Vitro ChE Activity Assay
The inhibitory activity of test compounds against acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) was evaluated using a modified Ellman’s spectrophotometric assay [29]. AChE (EC 3.1.1.7, Electrophorus electricus, Type V-S), BChE (EC 3.1.1.8, equine serum), and Trisma base (2-amino-2-(hydroxymethyl)-1,3-propanediol) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Substrates acetylthiocholine iodide (ATChI) and S-butyrylthiocholine iodide (BTChI) were also purchased from Sigma-Aldrich, while Ellman's reagent 5,5-dithiobis-(2-nitrobenzoic acid) (DTNB), was sourced from Zwijndrecht (Antwerpen, Belgium). Tested compounds were dissolved in ethanol, and 10 µL of each solution was added to 180 µL of Tris buffer (50 mM, pH 8.0) and 10 µL of enzyme solution (final concentration 0.03 U/mL). After adding 10 µL of DTNB (final concentration 0.3 mM), the enzymatic reaction was initiated by adding 10 µL of ATChI/BTChI (final concentration of 0.5 mM). To account for non-enzymatic hydrolysis, blank samples were prepared by replacing the enzyme with an equal amount (10 µL) of buffer solution. The absorbance of the reaction mixture was monitored at 405 nm over 5 minutes at room temperature using a 96-well microplate reader (Bio Tek 800TSUV Absorbance Reader, Agilent). Enzymatic activity in the absence of inhibitors was considered 100%, and all measurements were performed in triplicate. The percentage of inhibition was calculated using the formula: Inhibition (%) = [(AC – AT)/AC]×100, where AC is the enzyme activity without the test sample, and AT is the enzyme activity in the presence of the test compound, calculated as mean values ± standard deviation. IC50 values were obtained through nonlinear regression analysis of log-transformed inhibitor concentrations versus enzyme activity.
2.5. Anti-Inflammatory Activity
The effect of compounds on lipopolysaccharide (LPS)-stimulated tumour necrosis factor alpha (TNFα) production was evaluated as described previously [30]. In short, peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats obtained from healthy adult volunteers and resuspended in RPMI1640 medium (Capricorn Scientific) supplemented with 10% heat-inactivated FBS (Biowest), 1% GlutaMAX (Gibco), and 1% Antibiotic-Antimycotic (Gibco). 2×105 PBMCs were seeded per well of a 96-well plate. Test compounds were dissolved in dimethyl sulfoxide (DMSO, Sigma), and three-fold serial dilutions in DMSO were prepared and added to cells, with a starting concentration of 100 µM. After 1 h pre-incubation with test compounds, cells were stimulated with 1 ng/mL LPS from E. coli 0111:B4 (Sigma). Cells were incubated for 24 h at 37°C, 5% CO2, followed by the collection of supernatants for the measurement of TNF-α and cell viability assessment.
To evaluate cell viability, CellTiter-Glo reagent (Promega) was used according to the manufacturer's instructions, and signals obtained in compound-treated wells were compared with signals in LPS-stimulated vehicle-treated samples.
TNFα concentration in supernatants was measured by ELISA using antibodies and recombinant human TNFα protein (standard) from R&D Systems. Lumitrac 600 384-well plates (Greiner Bio-One) were coated with 1 µg/mL of TNFα capture antibody diluted in phosphate-buffered saline (PBS; Gibco) overnight at 4°C. The following day, plates were blocked with 5% sucrose (Kemika) in assay diluent (1% bovine serum albumin (BSA; Sigma) in PBS) for 4 h at RT. After the blocking step, samples and standards were added to the plates and incubated overnight at 4°C. Afterwards, 250 ng/mL of TNFα detection antibody was added to wells, followed by a 2 h incubation at RT. Finally, after the plates were incubated with streptavidin-HRP (Invitrogen), chemiluminescence ELISA Substrate (Roche) was added to wells and luminescence was measured using EnVision 2105 multilabel reader (Revvity). Measured luminescence was used to calculate concentrations of TNFα in the supernatants by interpolation from the standard curve. Percentages of inhibition were calculated from obtained cytokine concentrations, and IC50 values were determined using GraphPad Prism v9 software using nonlinear regression curve fit (four parameters with variable slope).
2.6. Computational Details
The preparation of ligands for docking study included optimization of their geometries at the M06-2X/6-31+G(d,p) level of theory, using the Gaussian16 program [31]. The molecular docking was conducted using the Autodock program package [32], with crystal structures of BChE taken from the Protein Data Bank (PDB code 7AIY) [33]. Molecular docking was performed using the Lamarckian Genetic Algorithm, with 25 genetic algorithm dockings generating 25 binding poses for ligands, while the residues of the enzyme were kept rigid. Complexes obtained by docking were used as starting structures for molecular dynamics simulations. A truncated octahedron of the OPC water box was used for solvation of the enzyme-ligand complexes. Neutralization with Cl- ions using the Amber16 program package was performed [34], with the ff14SB force field [35] for the protein part of the enzyme, and the General Amber Force Field (GAFF) [36] for ligands. Partial charges for ligands were derived using the RESP procedure. Equilibrations of all four systems consisted of energy minimizations and short MD simulations of 20 ns, with systematic decreases of the harmonic restraints to zero and relaxation of the volume and temperature, with target values of the temperature and pressure set to 300 K and 1 atm, respectively. Productive MD simulation with no constraints was performed for 40 nanoseconds under NPT conditions, i.e., 300 K and 1 atm.
3. Results and Discussion
3.1. Synthesis and Characterization of New Heterostilbene Carbamates 1–19
As the starting material for the synthesis of carbamates 1–19, analogs of resveratrol were used, synthesized by Wittig reaction (Scheme 1) [21,22,23]. New heterostilbene carbamates 1–19 were prepared at room temperature in dichloroethane in an inert atmosphere, with 1.8 eq of triethylamine and a catalytic amount of DMAP. After that, the reaction mixture was stirred under argon, followed by adding 1.2 eq of the corresponding carbamoyl chlorides. In the end, the reaction mixture was worked up (see section Materials and Methods).
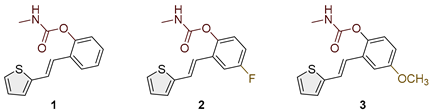
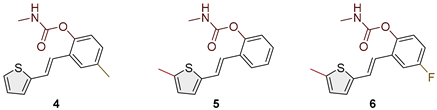
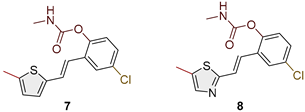
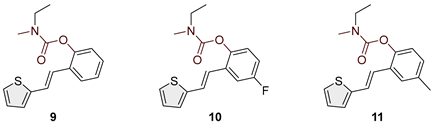
Heterostilbene carbamates 1–19 were purified by column chromatography, using petroleum ether and diethyl ether in different ratios as the eluent, isolated in a broad and different range of yields, primarily depending on the carbamoyl chlorides used and, on the nature and position of the substituents (13–93%, Figure 2 and Figure 3). When comparing the isolated yields on methyl carbamates 1–8 (Figure 2) and ethyl-methyl carbamates 9–19 (Figure 3), it is obvious that the isolated yields for some derivatives in the second group are very high in some cases (compounds 11, 16, and 19). It can be noticed that within certain groups of carbamates (derivatives 1–8 or 9–19), the nature of the heterocyclic nuclei, the substituent on it, and the carbamate group's position affect the carbamates' isolated yields.
For carbamates 9–19, the highest yields were achieved with heterostilbene carbamates 11, 12, 16, 18, and 19 (Figure 3, 87-93%), which, besides the carbamoyl group, either lack additional substituents on the aromatic nucleus, or contain only a methyl group, with the thiophene ring remaining unsubstituted in all cases. It should also be emphasized that all four derivatives with a morpholine carbamate group (16–19) give high yields. The presence of a methyl substituent on the thiophene ring is not favorable for synthesis regarding the isolated yields of the carbamate products (compounds 14 and 15).
The newly synthesized heterostilbene carbamates 1–19 were spectroscopically characterized, and their structures and purity were confirmed via NMR (1H and 13C) and HRMS analyses (Figures S16–S68). As in the previous research, new heterostilbene carbamates 1–19 were designed as selective BChE inhibitors, so it is worth referring again to BChE-assisted substrate (carbamate) hydrolysis [20], which is analogous to the mechanism of the AChE-assisted hydrolysis of ACh. The mechanism includes the formation of the Michaelis complex, acylation of the enzyme, and its spontaneous deacylation with water.
3.2. Crystal Structure of Carbamates 1 and 4
The structures of 1 and 4 were confirmed by single-crystal X-ray diffraction analysis (Figure 4). In both cases, the crystal packing is primarily governed by intermolecular N–H···O hydrogen bonds and π–π stacking interactions (Figure 5), which organize the molecules into one-dimensional chains extending along the [100]-crystallographic axis. These chains are further stabilized by weak van der Waals forces acting between adjacent layers. In the crystal structure of carbamate 1, two symmetry-independent intermolecular hydrogen bonds, N1–H1A···O2 and N2–H2A···O4, link adjacent molecules (Table 2). The presence of multiple aromatic and heteroaromatic rings facilitates π–π stacking interactions, with interplanar distances ranging from 2.87 Å to 3.75 Å (Table 3), contributing significantly to the layered packing arrangement. In carbamate 4, the key intermolecular interaction is the N1–H1A···O2 hydrogen bond, with geometric parameters of d(N–H) = 0.86 Å, d(H···O) = 2.14 Å, d(N···O) = 2.892(3) Å, with an N–H···O angle of 145.4°, and a symmetry operation of –1 + x, y, z. In addition to hydrogen bonding and π–π stacking, C–H···π interactions are observed in compound 4, providing further stabilization. A representative interaction, C15–H15A···C7–C12, exhibits a C···Cg distance of 3.63 Å and occurs via the symmetry operation 1 + x, y, z (Table 4).
3.3. Cholinesterase Inhibition of Heterostilbene Carbamates 1–19
Previously tested carbamate analogues of resveratrol [20] (Figure 1, structure C) showed an interesting structural basis for developing completely selective and powerful inhibitors of BChE. Therefore, using Ellman's modified method, a new portion of variously substituted carbamate derivatives was assayed for their inhibitory effect on AChE and BChE [29]. Results expressed as IC50 values of carbamate derivatives and standard galantamine are presented in Table 5 and Figure 6.
Studied compounds can be divided into three groups according to the type of carbamate part: secondary carbamates 1–8, tertiary carbamates 9–14, and tertiary morpholine carbamates 16–19. Inside each group, variations were made in terms of substituent present on the thiophene or benzene ring, position of the carbamate group, and even the type of heterocycle. Only one derivative tested had a thiazole ring (derivative 8), while all others were thiophenes. Derivative 8 was the only one to show some inhibition of AChE, although with a weak IC50 value of about 275.5 μM. Thiophene derivatives can be considered fully selective BChE inhibitors. Within the first group, the most successful inhibitor is structure 1 with an IC50 value of 0.5 μM, which is in complete agreement with previous results regarding structural features, namely that resveratrol analogues benefit from the absence of a substituent on the carbamate side of the stilbene. Slightly reduced, but still excellent inhibition, is shown by the derivative with a methyl substituent on the thiophene 5 (IC50 0.913 μM), and with a methyl in the meta position on the carbamate side 4 (IC50 1.102 μM). The inhibitory activity decreases with the introduction of the remaining substituents, all with lone electron pairs –F, -Cl, -OCH3 (Table 5). A similar finding was observed in the group of secondary carbamates. Again, the leading inhibitor is the one without a substituent on the carbamate side of stilbene, derivative 9 with an IC50 value of 0.583 μM, and there is almost no difference between 9 and the secondary analogue 1. Introducing a methyl group on the thiophene in derivative 14 significantly reduces the inhibition to IC50 85.130 μM. In contrast, the introduction of methyl on the benzene part in structure 11 preserved excellent inhibitory activity with an IC50 of 1.541 μM.
Changing the position of the carbamate group from ortho to meta in derivative 12 reduces the activity threefold. However, the inhibition achieved is still excellent with an IC50 value of 1.503 μM. The presence of the methoxy group in the para position maintains the activity in an excellent range of concentrations, and derivative 13 has an IC50 of 1.883 μM. The most successful inhibitors are morpholine carbamates 16–19, among which structure 16 stands out as the most potent inhibitor in this group and the entire series of tested compounds. An excellent inhibition value of 26.5 nM is achieved by the derivative without additional substituents on the benzene or thiophene. The same structural feature favors inhibition as in the previous two groups. Introducing fluorine and methyl reduces the activity, but derivatives 17 and 18 are still in the excellent range of concentrations (Table 5). When the carbamate group is in the meta position of the stilbene part in derivative 19, the activity is reduced one hundred times; however, it still ranks among the highly active inhibitors. This group of compounds, with morpholine on the carbamate nitrogen, proved to be the most effective in inhibiting BChE, highlighting its potential for further investigation through derivative studies.
3.4. Anti-Inflammatory activity of heterostilbene carbamates 1–19
The potential anti-inflammatory activity of compounds was evaluated in vitro in an assay where PBMCs were stimulated with LPS to induce TNFα production. Of all the tested compounds, only carbamate 16 inhibited LPS-stimulated TNFα production (Figure 7). The compound was active at the two highest concentrations tested, with an average IC50 of 31.6 µM in PBMCs from two donors. Dexamethasone, a corticosteroid widely used to treat inflammatory conditions, was used in this assay as a reference compound. As expected, it was highly potent in inhibiting LPS-stimulated TNFα production with an IC50 value of 3.5 nM, which aligns with results obtained previously in this assay [30].
3.5. ADME(T) properties and genotoxicity - ICH (M7) Q(SAR)
The first step in any investigation of compounds with biological activity is the study of toxicity. This will be done before the ADMET (see below) to see which compounds should be tested further and which have structural alerts for genotoxicity. (Q)SAR models are used to predict biological activity based on structural components [37]. (Q)SAR models are especially vital during the early stages of searching for potentially active drug substances. The elimination of all compounds that have mutagenic potential saves money and time. The most commonly used tool is the Lhasa software package because it uses two complementary models, and their predictions are then reviewed one more time by an expert. The results for compounds 1–19 are shown in Table 6.
From the compounds that have shown biological activity (1, 9, 16, and 18), only compound 1 will not be a probable candidate for further testing, as it has shown a potential for mutagenicity. If this compound was of particular interest, further in vitro AMES and even in vivo qualification studies would have to be done to show that it can be used as an active drug. Studies related to bioavailability, that is, Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADME(T)), are a cornerstone of any drug development stage. They are here to give insights into the compound's drug-like pharmacokinetic properties and whether there are any safety concerns in people [38]. These properties can be investigated in silico, in vitro, and in vivo. In the early phases of drug discovery, in silico tools provide a practical advantage when target compounds are either insufficiently pure or available only in minimal quantities. This study used free in silico tools [39,40] to screen the candidate compounds. Results are presented in Table 7.
The data presented in Table 2 show that water solubility is very low. The in silico data indicating CNS potential are the ones under distribution in Table 7.
The BBB (as log BB = the drug concentration in the brain divided by the concentration in the blood) for compounds 9 and 16 is approximately. 0.3. This result is promising, as compounds with log BB >0.3 readily cross the BBB. The second indicator is the Log PS (permeability surface area); a log PS > −2 signifies molecules that can penetrate the CNS. This value is above −0.98 for compounds tested, so they are considered good candidates. If we consider that the unbound drug is the effective form, then the higher Fu can indicate better bioavailability and the drug can better distribute in the central nervous system. Since these candidates are analogs containing an active carbamate moiety, the abovementioned indicators were compared with drugs containing a carbamate group. Bambuterol and rivastigmine (Figure 8) were selected due to structural resemblance and similar therapeutic indications as tested compounds.
As these are well-known drugs, the in silico prediction was made only for VDss, BBB, and PS distribution parameters. The parameter VDss is similar to the one for compounds 6, 9, and 18. Log BB is bigger for the known drugs, but as drugs above 0.3 signify that they readily cross the BBB barrier, the lead compounds 6 and 9 meet the criteria. Based on the range of in silico ADME(T) parameters and genotoxicity assessments, all three compounds appear to be promising leads for further drug development.
3.6. Computational Study
The measurements of inhibitory activity toward cholinesterases (Table 5) show that compound 16, one of the carbamates with a morpholine substituent, stands out as the most promising BChE inhibitor. Among the methyl and ethyl carbamates, methyl carbamate 1 exhibited the highest inhibitory activity. We performed molecular docking studies for these two compounds to gain insight into the enzyme-ligand structures and to characterize stabilizing interactions between the ligands and residues within the enzyme's active site.
Docking of molecule 1 resulted in two relevant complexes, as presented in Figure 9. The first one, labeled BChE-1(I), contains the ligand in a conformation that results in the most stable complex structure. The second structure is obtained with the representative ligand pose belonging to the most populated cluster (Table S1). In these two complexes, the ligand orientations are similar, with the thiophene and the double CC bond engaging in parallel π–π stacking interactions with Trp82 in the anionic subdomain of the BChE active site, while the phenyl ring of the ligand interacts with Tyr128. However, in the BChE-1(I) complex, a hydrogen bond is observed between the amide group of the carbamate and the nitrogen atom of one of the glycine residues in the oxyanionic hole (Gly115). In the BChE-1(II) structure, this interaction disappears due to the rotation of the carbamate group. This conformational change allows the carbamate to approach the esteratic subdomain, where an alkyl-π interaction occurs between the ligand’s methyl group and His438, and the carbonyl carbon of the ligand comes within 3.9 Å of the oxygen atom of Ser198. However, this orientation of the carbamate group is not favorable, as the sp² oxygen of the ligand’s carbonyl is positioned too close to the serine hydroxyl group.
Analogously to methyl carbamate 1, molecular docking of molecule 16 resulted in two pertinent enzyme-ligand complexes, both shown in Figure 10. BChE-16(I) represents the most stable complex, while BChE-16(II) corresponds to the complex containing the representative ligand pose from the most populated docking cluster.
In complex BChE-16(I), the hydrophobic region of the morpholine fragment engages in alkyl-π interactions with Trp82. At the same time, the oxygen of the morpholine ring forms a hydrogen bond with the polar hydrogen of the pyrrole moiety of Trp82. The thiophene ring of the ligand participates in T-shaped π–π stacking with Phe329, while the phenyl ring and double CC bond engage in π–π stacking with His438. The ligand orientation in complex BChE-16(II) differs, with the thiophene and morpholine interacting with Trp82, while the phenyl ring of the ligand interacts with Phe329. Most notably, the orientation of the carbamate group in this structure allows favorable placement of the carbonyl carbon in proximity to the catalytic serine oxygen.
To evaluate the structural stability of enzyme-ligand complexes presented above, we performed molecular dynamics (MD) simulations, with a duration of a production simulation of 40 ns. The analysis of MD trajectories included calculating the root-mean-square deviation values (RMSD) for all atoms, except for hydrogens. RMSD values reflect structural changes during the simulation; all four complexes are presented in Figure 11. For both complexes with ligand 1, the convergence was achieved after 10-15 ns, with average RMSD values of 2.02 (ranging from 0.78 to 2.37) and 2.05 (from 0.80 to 2.45 Å), for BChE-1(I) and BChE-1(II), respectively (Table S2). Complexes with morpholine carbamate 16, I and II, behaved similarly, with average RMSD values of 1.93 and 2.17 Å, respectively. For complex BChE-16(I), the convergence was achieved after 10 ns; however, for complex BChE-16(II), the stabilization of RMSD occurred in the second half of the simulation.
Further analysis of the molecular dynamics trajectories involved calculating root mean square fluctuation (RMSF) values. RMSF quantifies the average positional deviations of the α-carbon atoms from their mean positions throughout the simulation, effectively reflecting the extent of α-carbon mobility over time. Thus, they offer insight into the dynamic behavior of the protein backbone and help identify its most mobile protein regions. The results, depicted in Figure 12 and Table S2, show that the average RMSF for all four complexes is between 0.8 and 0.9 Å, indicating low overall backbone mobility. The highest RMSF value is observed in complex BChE-1(II), reaching 4.65 Å and corresponding to residue Val529. The second highest value refers to Asn485 in complex BChE-16(I), with an RMSF of 3.89 Å. Both these residues are located far away from the active site (> 25 Å), and thus their mobility does not affect the structural stability.
Finally, the compactness of the protein upon ligand binding was evaluated by calculating the radius of gyration (Rg). The Rg value reflects the root-mean-square distance of all atoms from the complex’s center of mass and indicates protein flexibility in the presence of a ligand. A narrow Rg range suggests that ligand binding does not substantially disrupt the enzyme’s structural compactness. The radius of gyration values for all four complexes are summarized in Figure 13, with detailed data provided in Table S2.
For both complexes with ligand 1, Rg values ranged from 22.86 to 23.27 Å, while for complexes with ligand 16, they ranged from 22.81 to 23.34 Å. These findings indicate that changes in the radius of gyration were minimal across all complexes, supporting the conclusion that the enzyme retained its compact structure upon ligand binding.
3. Conclusions
In this study, a new series of heterostilbene carbamates was successfully synthesized and characterized as selective butyrylcholinesterase (BChE) inhibitors. Through systematic modification of the stilbene scaffold and carefully selecting substituents, 19 novel compounds were obtained, displaying defined structures confirmed by NMR, HRMS, and X-ray crystallography. The biological evaluation demonstrated that most compounds selectively inhibited BChE over acetylcholinesterase (AChE), with several derivatives, particularly compound 16, showing outstanding potency (IC₅₀ = 26.5 nM).
The selective inhibition profile aligns with the therapeutic strategy to preserve cholinergic function while minimizing side effects associated with dual cholinesterase inhibition. In addition to BChE inhibition, compound 16 exhibited moderate anti-inflammatory activity by reducing lipopolysaccharide (LPS)-induced TNF-α production, further supporting its potential as a multifunctional therapeutic candidate for neurodegenerative diseases. Computational studies, including molecular docking and molecular dynamics simulations, confirmed favorable binding modes and stable interactions between the carbamate derivatives and the active site of BChE.
Furthermore, in silico ADME(T) profiling indicated good predicted absorption, distribution, and low mutagenic potential for the leading compounds, strengthening their suitability for future pharmacological development. Overall, these findings highlight heterostilbene carbamates as promising scaffolds for the design of new therapeutic agents targeting cholinergic dysfunction and neuroinflammation in Alzheimer’s disease and other neurodegenerative conditions. Future work will focus on the optimization of lead compounds through further structural modifications and exploration of additional biological activities relevant to neuroprotection.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, Dose-response curves for the inhibition of BChE by 2–8, 10–14, and 17 – 19. Mass spectra and HRMS analyses of carbamates 1–19. 1H and 13C NMR spectra of carbamates 1–19. Cartesian coordinates of docked ligands. Free energies of binding, the number of conformational clusters, and the distribution of conformations obtained by molecular docking. Analysis of MD trajectories (RMSD, RMS fluctuations, and radius of gyration) for enzyme-ligand complexes.
Author Contributions
Conceptualization, I.S. and D.B.; methodology, I.O., M.B., D.B., V.M. and I.Š.; investigation, A.R., K.K., K.B., M.M., P.P., D.S. and A.J.; resources, I.S., D.B., I.O.; writing—original draft preparation, I.S., D.B., I.O., I.Š., V.M., M.B., and A.J.; writing—review and editing, I.S. and D.B.; supervision, I.S.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available from the authors on request.
Acknowledgments
This work was supported by grants from the University of Zagreb for short-term scientific support for 2024 under the title Synthesis and Biological Activity of New Heteropolycycle Systems. We thank the University of Zagreb (Croatia) Computing Centre (SRCE) for granting computational time on the Supercomputer Supek. The authors thank Zlata Lasić for kindly performing the HRMS analyses. We also acknowledge Sunčica Roca from the NMR Centre at Ruđer Bošković Institute for recording all the NMR spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. A Structural Model of Butyrylcholinesterase. Proc. Natl. Acad. Sci. USA 1997, 94, 3398–3403. [Google Scholar]
- Santos, A.L.; Carvalho, M.M.; Silva, D.F.; Amado, F.; Oliveira, C.R.; Pereira, C.M. Dual Inhibition of Acetylcholinesterase and Butyrylcholinesterase by Phenserine Improves Cognitive Function in Alzheimer's Disease. J. Alzheimers Dis. 2009, 16, 767–774. [Google Scholar] [CrossRef]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Hashimoto, Y. The Role of Butyrylcholinesterase in Alzheimer's Disease and Its Potential as a Therapeutic Target. Neurobiol. Aging 2001, 22, 471–478. [Google Scholar]
- Caccamo, A.; Oddo, S.; Sugarman, M.C.; LaFerla, F.M. Molecular Targets for Alzheimer's Disease Therapy: Amyloid-Beta Aggregation and Cholinergic Dysfunction. Neurobiol. Aging 2010, 31, 987–999. [Google Scholar] [CrossRef]
- Vila, J.L.; Medina, M.; Correa, J.; Romero, L. Butyrylcholinesterase Inhibitors as New Drugs for Alzheimer's Disease. J. Med. Chem. 2001, 44, 2022–2027. [Google Scholar]
- Pereira, R.M.; Marques, L.A.; Silva, D.F.; Oliveira, C.R.; Pereira, C.M. Phenserine: A Selective Butyrylcholinesterase Inhibitor. Curr. Alzheimer Res. 2002, 4, 1–11. [Google Scholar]
- Röcken, C.; Bartels, M.; Jessberger, S.; Stühmer, W.; Alzheimer, C. The Pharmacological Profile of Rivastigmine as a Selective Inhibitor of Acetylcholinesterase and Butyrylcholinesterase. Neurochem. Int. 2003, 43, 315–323. [Google Scholar]
- Giacobini, E. Cholinesterase Inhibitors: From the Bench to the Bedside. Neurochem. Int. 2004, 45, 1–10. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Jakobsen, J.; Høgh-Rasmussen, E.; Holm, S.; Paulson, O.B. The Role of Acetylcholine and Acetylcholinesterase Inhibitors in Parkinson’s Disease. J. Clin. Neurosci. 2007, 14, 610–615. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A Review on Cholinesterase Inhibitors. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, Synthesis, and Biological Evaluation of Conformationally Restricted Rivastigmine Analogues. J. Med. Chem. 2004, 47, 5945–5952. [Google Scholar] [CrossRef]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; Toth, G.; Zagyva, A.; Zekany, A.; Finberg, J.; Lavian, G.; Gross, A.; Friedman, R.; Razin, M.; Huang, W.; Krais, B.; Chorev, M.; Youdim, M.B.; Weinstock, M. Novel Dual Inhibitors of AChE and MAO Derived from Hydroxy Aminoindan and Phenethylamine as Potential Treatment for Alzheimer's Disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef] [PubMed]
- Ucar, G.; Gokhan, N.; Yesilada, A.; Bilgin, A.A. 1-N-Substituted Thiocarbamoyl-3-Phenyl-5-Thienyl-2-Pyrazolines: Novel Cholinesterase and Selective Monoamine Oxidase B Inhibitors for the Treatment of Parkinson’s and Alzheimer’s Diseases. Neurosci. Lett. 2005, 38, 327–331. [Google Scholar] [CrossRef]
- Toda, N.; Tago, K.; Marumoto, S.; Takami, K.; Ori, M.; Yamada, N.; Koyama, K.; Naruto, S.; Abe, K.; Yamazaki, R.; Hara, T.; Aoyagi, A.; Abe, Y.; Kaneko, T.; Kogen, H. A Conformational Restriction Approach to the Development of Dual Inhibitors of Acetylcholinesterase and Serotonin Transporter. Bioorg. Med. Chem. 2003, 11, 4389–4415. [Google Scholar] [CrossRef]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Švarcová, M.; Vinšová, J. Novel Cholinesterase Inhibitors Based on O-Aromatic N,N-Disubstituted Carbamates and Thiocarbamates. Molecules 2016, 21, 191. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Wang, Y.; Li, X.; Wang, S.; Wang, Z. Recent Advances on Carbamate-Based Cholinesterase Inhibitors as Potential Multifunctional Agents Against Alzheimer's Disease. Eur. J. Med. Chem. 2022, 240, 114606. [Google Scholar] [CrossRef] [PubMed]
- Yanovsky, I.; Finkin-Groner, E.; Zaikin, A.; Lerman, L.; Shalom, H.; Zeeli, S.; Weill, T.; Ginsburg, I.; Nudelman, A.; Weinstock, M. Carbamate Derivatives of Indolines as Cholinesterase Inhibitors and Antioxidants for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2012, 55, 10700–10715. [Google Scholar] [CrossRef]
- Sviben, M.; Odak, I.; Barić, D.; Mlakić, M.; Horváth, O.; Fodor, L.; Roca, S.; Šagud, I.; Škorić, I. Resveratrol-Based Carbamates as Selective Butyrylcholinesterase Inhibitors: Design, Synthesis, Computational Study and Biometal Complexation Capability. Molecules 2025, 30, 316. [Google Scholar] [CrossRef]
- Ljubić, A.; Vušak, V.; Cingesar, I.K.; Vrsaljko, D.; Šalić, A.; Škorić, I. Application of natural deep eutectic solvents in the continuous process for synthesis of resveratrol analogues by the Wittig reaction. J. Flow Chem. 2025. [Google Scholar] [CrossRef]
- Mlakić, M.; Odak, I.; Barić, D.; Talić, S.; Šagud, I.; Štefanić, Z.; Molčanov, K.; Lasić, Z.; Kovačević, B.; Škorić, I. New Resveratrol Analogs as Improved Biologically Active Structures: Design, Synthesis and Computational Modeling. Bioorg. Chem. 2024, 143, 106965. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Rajić, L.; Ljubić, A.; Vušak, V.; Zelić, B.; Gojun, M.; Odak, I.; Čule, I.; Šagud, I.; Šalić, A.; Škorić, I. Synthesis of New Heterocyclic Resveratrol Analogues in Milli- and Microreactors: Intensification of The Wittig Reaction. J. Flow Chem. 2022, 12, 429–440. [Google Scholar] [CrossRef]
- Mlakić, M.; Talić, S.; Odak, I.; Barić, D.; Šagud, I.; Škorić, I. Cholinesterase Inhibition and Antioxidative Capacity of New Heteroaromatic Resveratrol Analogs: Synthesis and Physico–Chemical Properties. Int. J. Mol. Sci. 2024, 25, 7401. [Google Scholar] [CrossRef] [PubMed]
- Rigaku OD. CrysAlis PRO. Rigaku Corporation, Wrocław, Poland, 2024.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Mlakić, M.; Faraho, I.; Odak, I.; Kovačević, B.; Raspudić, A.; Šagud, I.; Bosnar, M.; Škorić, I.; Barić, D. Cholinesterase Inhibitory and Anti-Inflammatory Activity of the Naphtho- and Thienobenzo-Triazole Photoproducts: Experimental and Computational Study. Int. J. Mol. Sci. 2023, 24, 14676. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Coquelle, N.; Colletier, J.P. Crystal Structure of Human Butyrylcholinesterase in Complex with 2-{1-[4-(12-Amino-3-chloro-6,7,10,11-tetrahydro-7,11-methanocycloocta[b]quinolin-9-yl)butyl]-1H-1,2,3-triazol-4-yl}-N-[4-hydroxy-3-methoxybenzyl]acetamide. PDB 7AIY. Available online: https://doi.org/10.2210/pdb7aiy/pdb.
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E. III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2016, University of California, San Francisco, 2016.
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hasselgren, C.; Bercu, J.; Cayley, A.; Cross, K.; Glowienke, S.; Kruhlak, N.; Muster, W.; Nicolette, J.; Vijayaraj Reddy, M.; Saiakhov, R.; Dobo, K. Management of Pharmaceutical ICH M7 (Q)SAR Predictions – The Impact of Model Updates. Regul. Toxicol. Pharmacol. 2020, 118, 104807. [Google Scholar] [CrossRef] [PubMed]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development, 2nd ed.; Faqi, A.S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 39–67. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- AI-DrugLab, Singapore Management University. Available online: https://ai-druglab.smu.edu/admetresult (accessed April/May 2025).
Figure 1.
Structures of some carbamates as cholinesterase inhibitors (A and B) [16], resveratrol-based bioactive carbamates (C), and structures investigated in the current research (D and E).
Figure 1.
Structures of some carbamates as cholinesterase inhibitors (A and B) [16], resveratrol-based bioactive carbamates (C), and structures investigated in the current research (D and E).
Scheme 1.
Synthetic pathway to new heterostilbene carbamates 1–19.
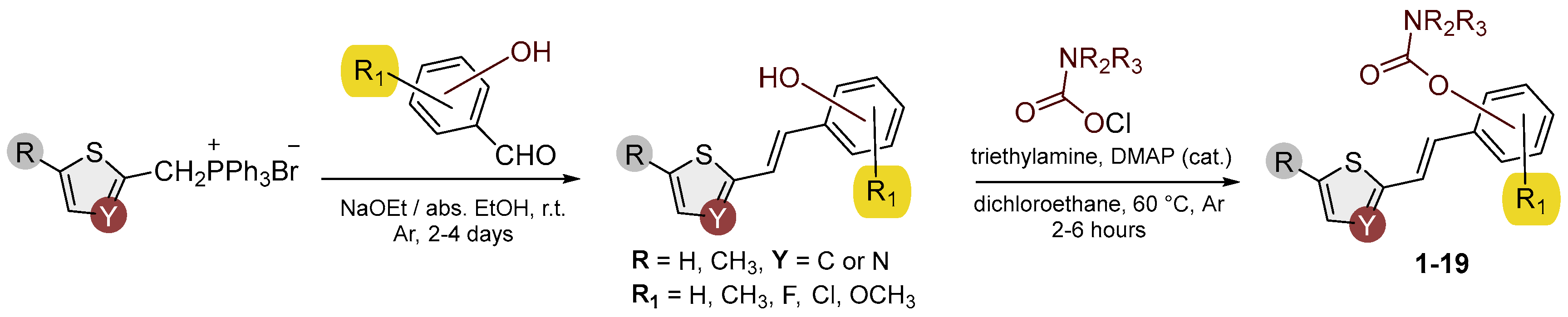
Figure 2.
Structures and isolated yields of new methyl carbamates 1–8.
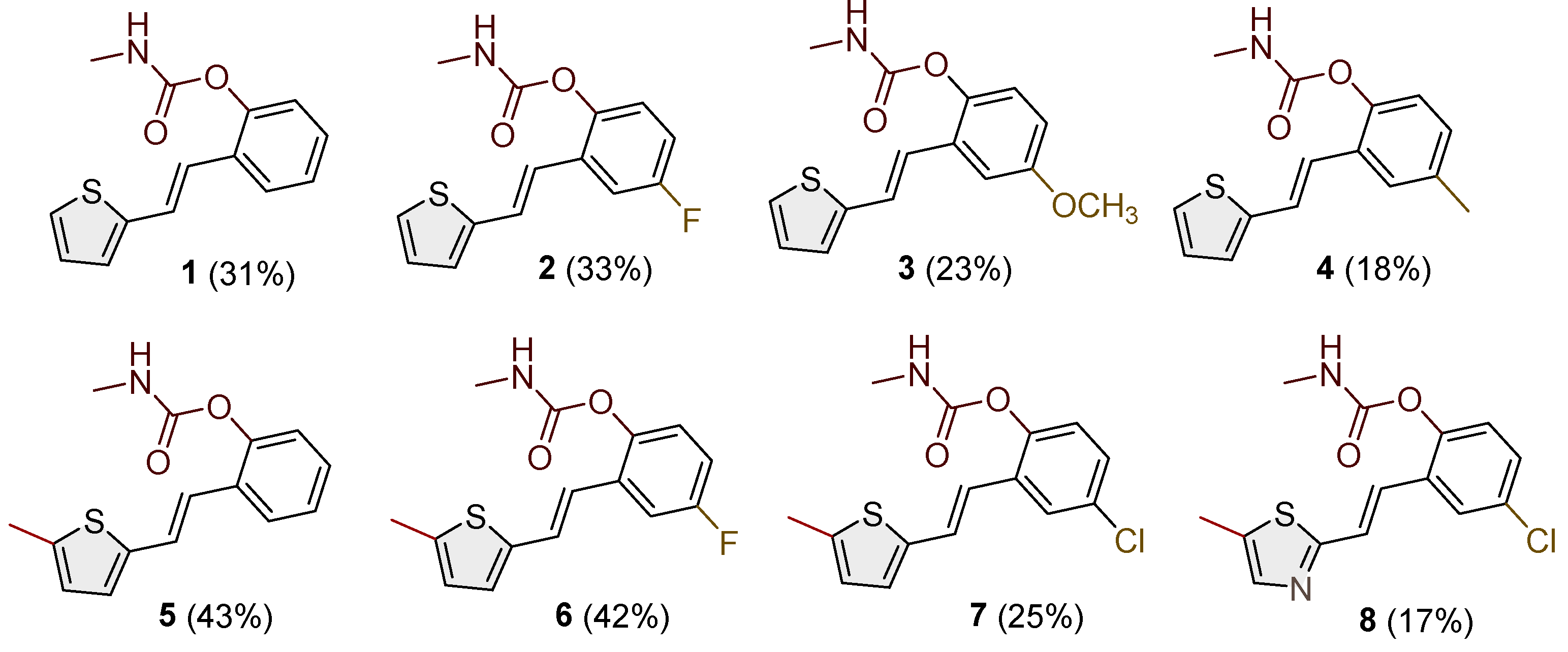
Figure 3.
Structures and isolated yields of new ethylmethyl carbamates 9–15 and morpholine carbamates 16–19.
Figure 3.
Structures and isolated yields of new ethylmethyl carbamates 9–15 and morpholine carbamates 16–19.
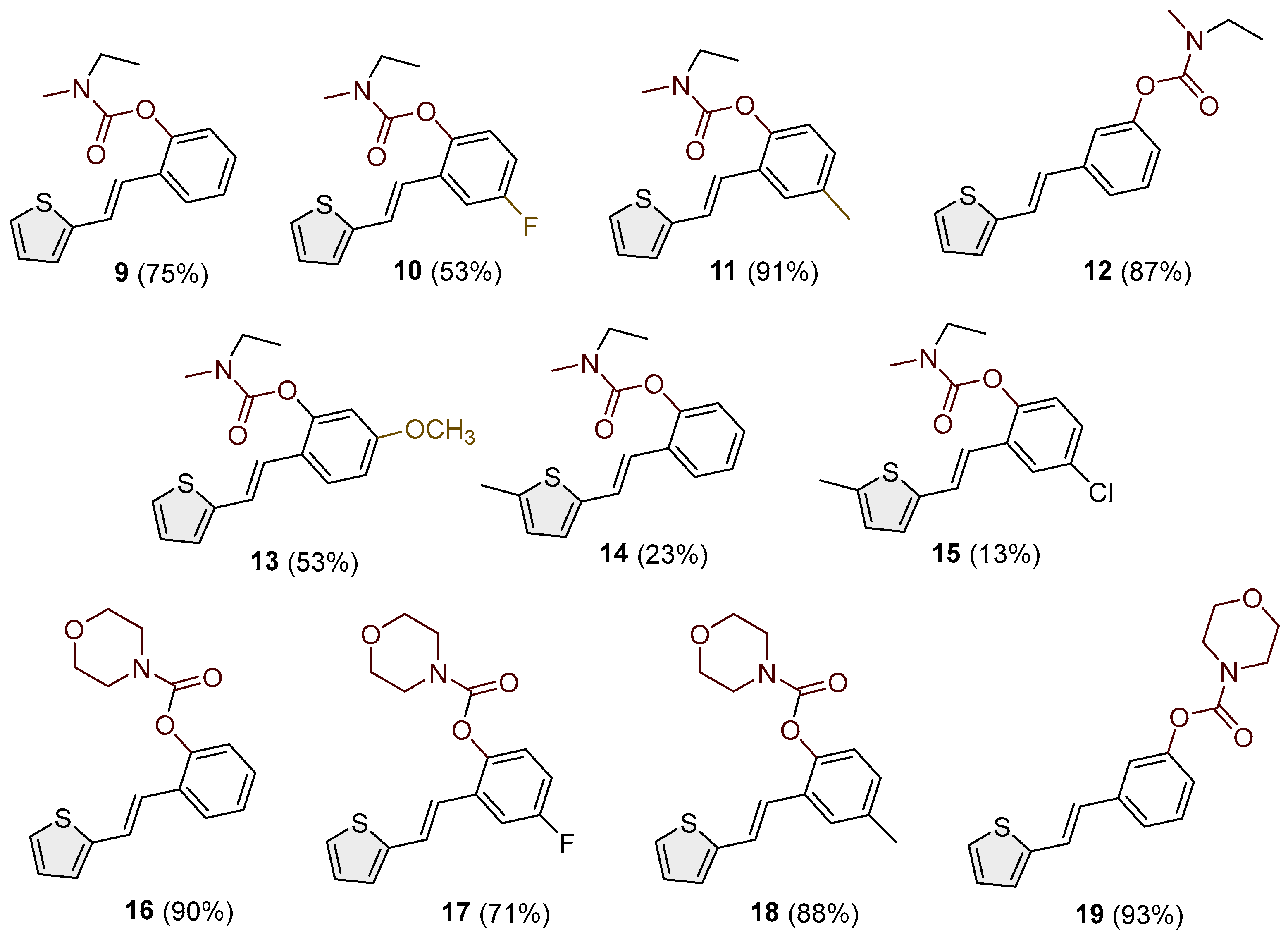
Figure 4.
Molecular structure of compounds 1 (left) and 4 (right). Displacement ellipsoids are drawn for the probability of 30 %, and hydrogen atoms are shown as spheres of arbitrary radii.
Figure 4.
Molecular structure of compounds 1 (left) and 4 (right). Displacement ellipsoids are drawn for the probability of 30 %, and hydrogen atoms are shown as spheres of arbitrary radii.
Figure 5.
Hydrogen-bonding and stacking interaction motifs in the crystal structures of 1 (left) and 4 (right). Hydrogen-bonded chains extend along the [100]-crystallographic direction. N–H···O hydrogen bonds are shown as dashed lines.
Figure 5.
Hydrogen-bonding and stacking interaction motifs in the crystal structures of 1 (left) and 4 (right). Hydrogen-bonded chains extend along the [100]-crystallographic direction. N–H···O hydrogen bonds are shown as dashed lines.
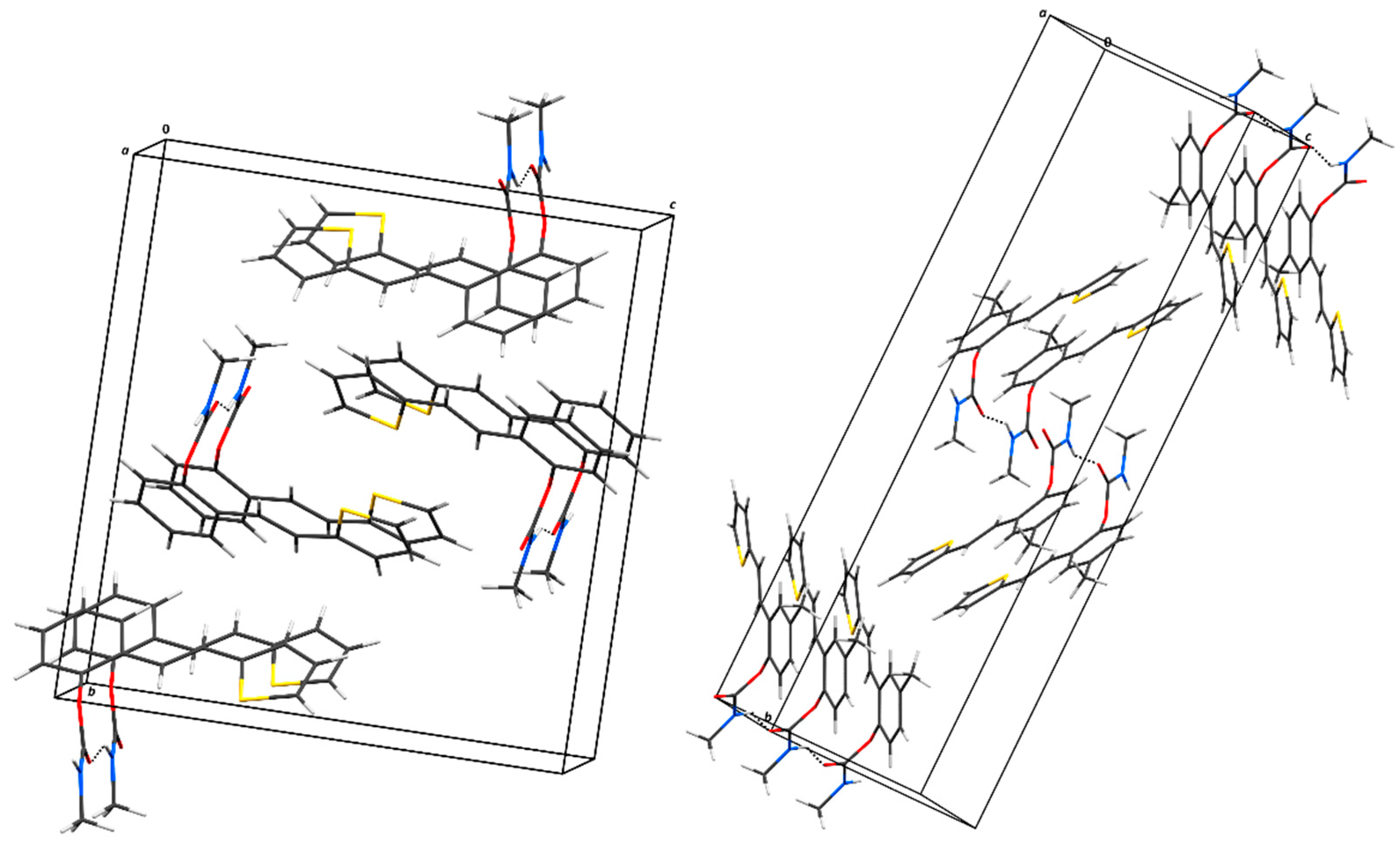
Figure 6.
Dose-response curve for the inhibition of BChE by 1 (a), 9 (b), and 16 (c).
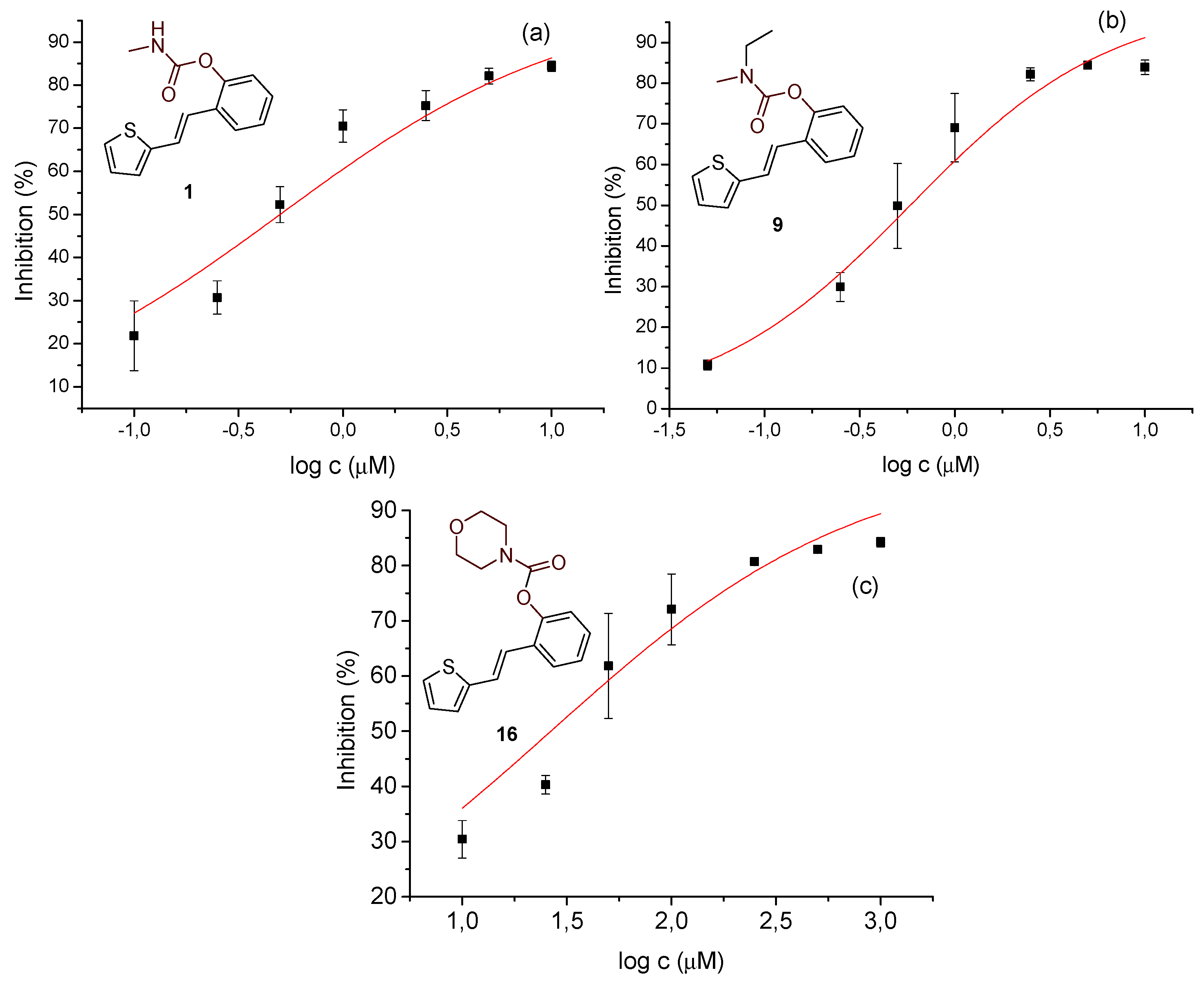
Figure 7.
Inhibition of LPS-stimulated TNFα production in PBMCs from 2 donors.
Figure 8.
Structure of bambuterol (left) and rivastigmine (right).
Figure 9.
Molecule 1 docked into the active site of BChE. (a) The structure of the most stable complex, BChE-1(I), with the hydrogen bond length indicated in green. (b) Structure BChE-1(II) with ligand pose from the most populated cluster; distance to serine hydroxyl oxygen marked red. Docked molecules are presented using a ball-and-stick model, distances given in angstroms.
Figure 9.
Molecule 1 docked into the active site of BChE. (a) The structure of the most stable complex, BChE-1(I), with the hydrogen bond length indicated in green. (b) Structure BChE-1(II) with ligand pose from the most populated cluster; distance to serine hydroxyl oxygen marked red. Docked molecules are presented using a ball-and-stick model, distances given in angstroms.
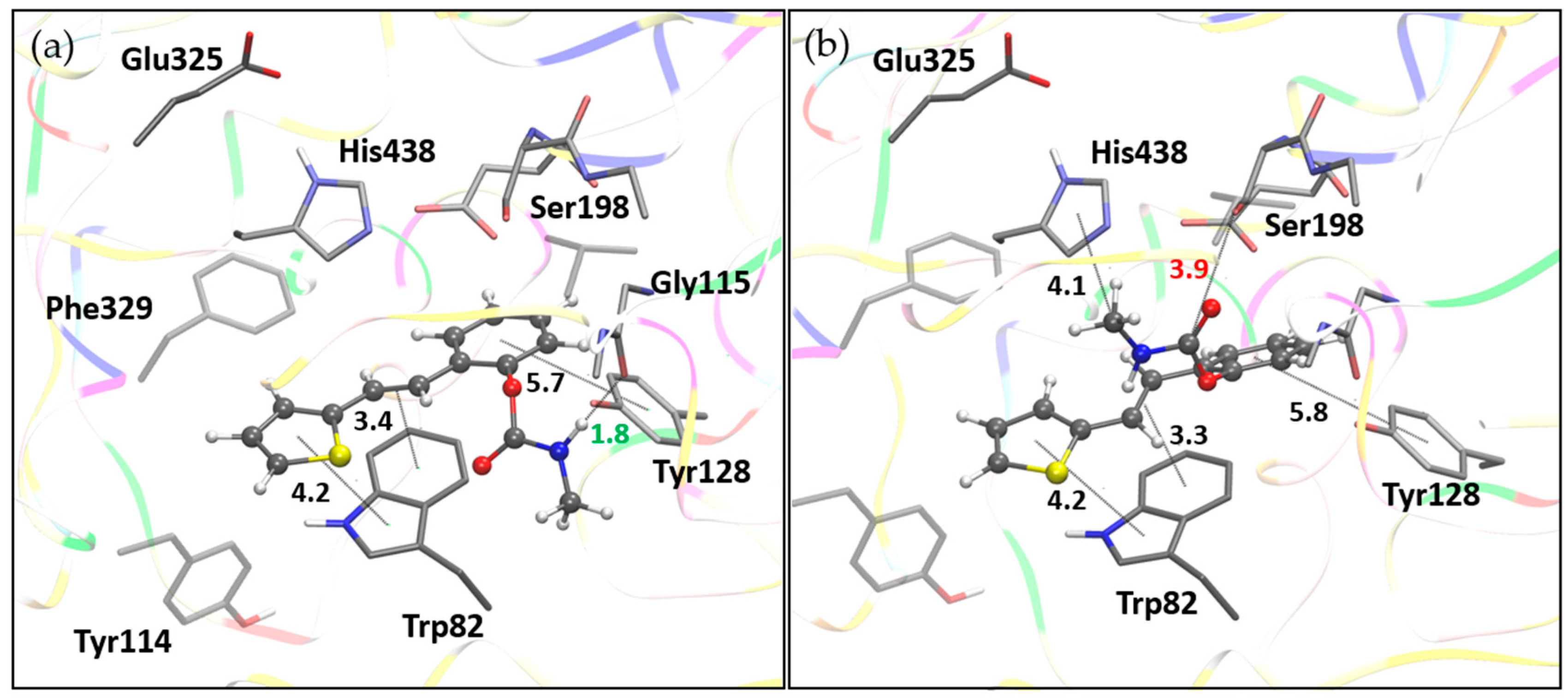
Figure 10.
Molecule 16 docked into the active site of BChE. (a) The structure of the most stable complex, BChE-16(I), with the hydrogen bond length marked in cyan. (b) Structure BChE-1(II) with ligand pose from the most populated cluster; distance to serine hydroxyl oxygen marked in magenta. Docked molecules are presented using a ball-and-stick model. Distances given in angstroms.
Figure 10.
Molecule 16 docked into the active site of BChE. (a) The structure of the most stable complex, BChE-16(I), with the hydrogen bond length marked in cyan. (b) Structure BChE-1(II) with ligand pose from the most populated cluster; distance to serine hydroxyl oxygen marked in magenta. Docked molecules are presented using a ball-and-stick model. Distances given in angstroms.
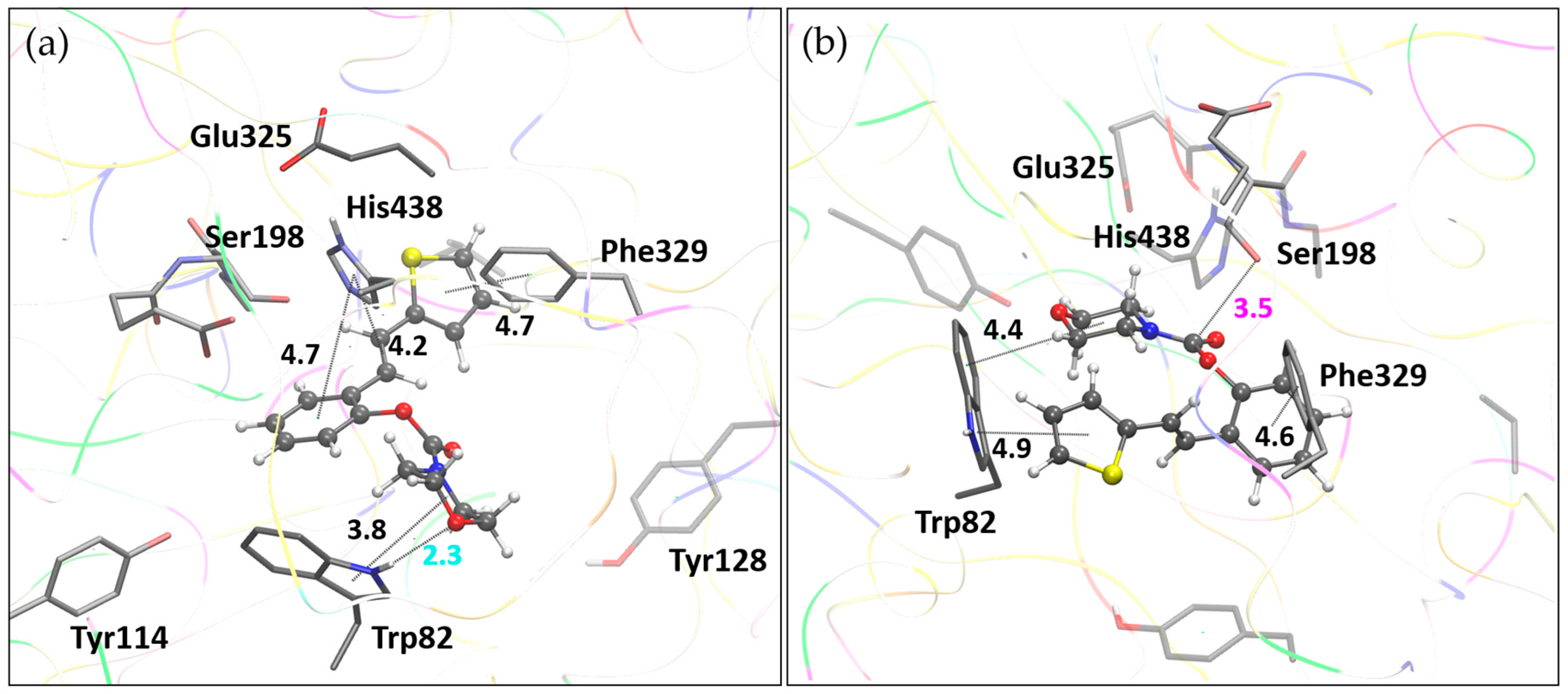
Figure 11.
Root-mean-square deviation values from molecular dynamics simulations of an enzyme-ligand complex of BChE with compound 1 (left) and compound 16 (right).
Figure 11.
Root-mean-square deviation values from molecular dynamics simulations of an enzyme-ligand complex of BChE with compound 1 (left) and compound 16 (right).
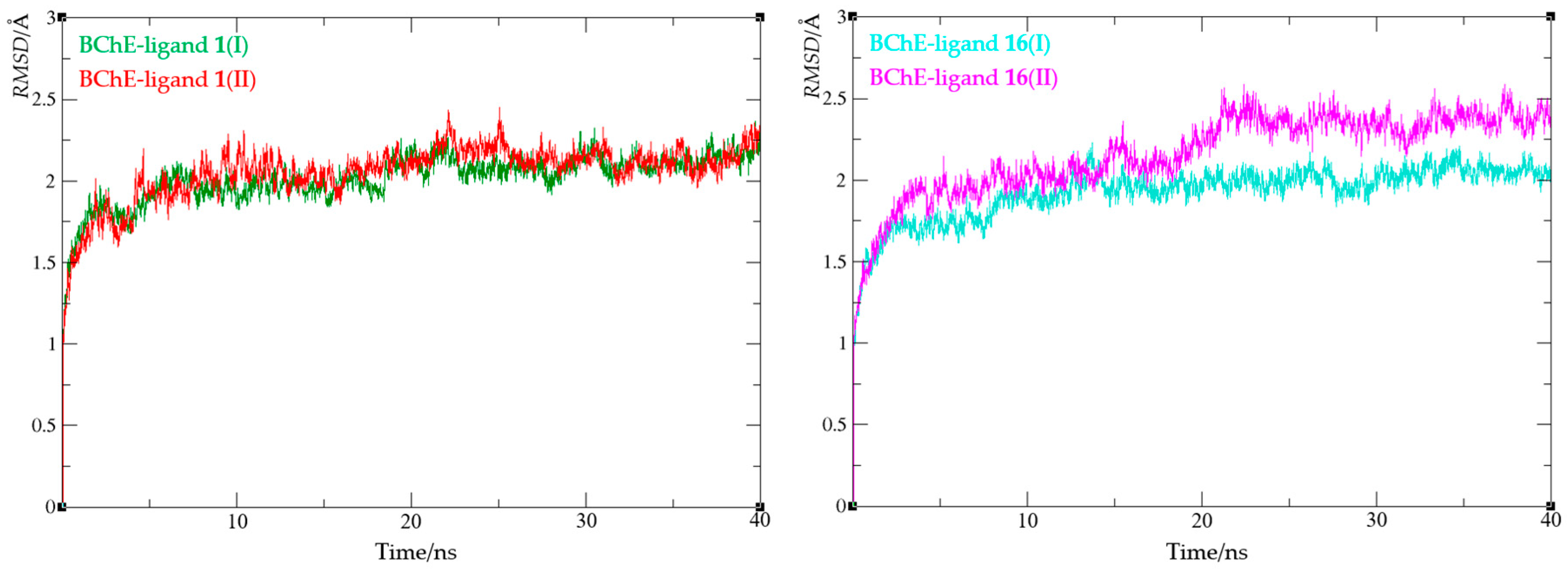
Figure 12.
Root-mean-square fluctuation values of the enzyme-ligand complexes of BChE with compound 1 (left) and compound 16 (right).
Figure 12.
Root-mean-square fluctuation values of the enzyme-ligand complexes of BChE with compound 1 (left) and compound 16 (right).
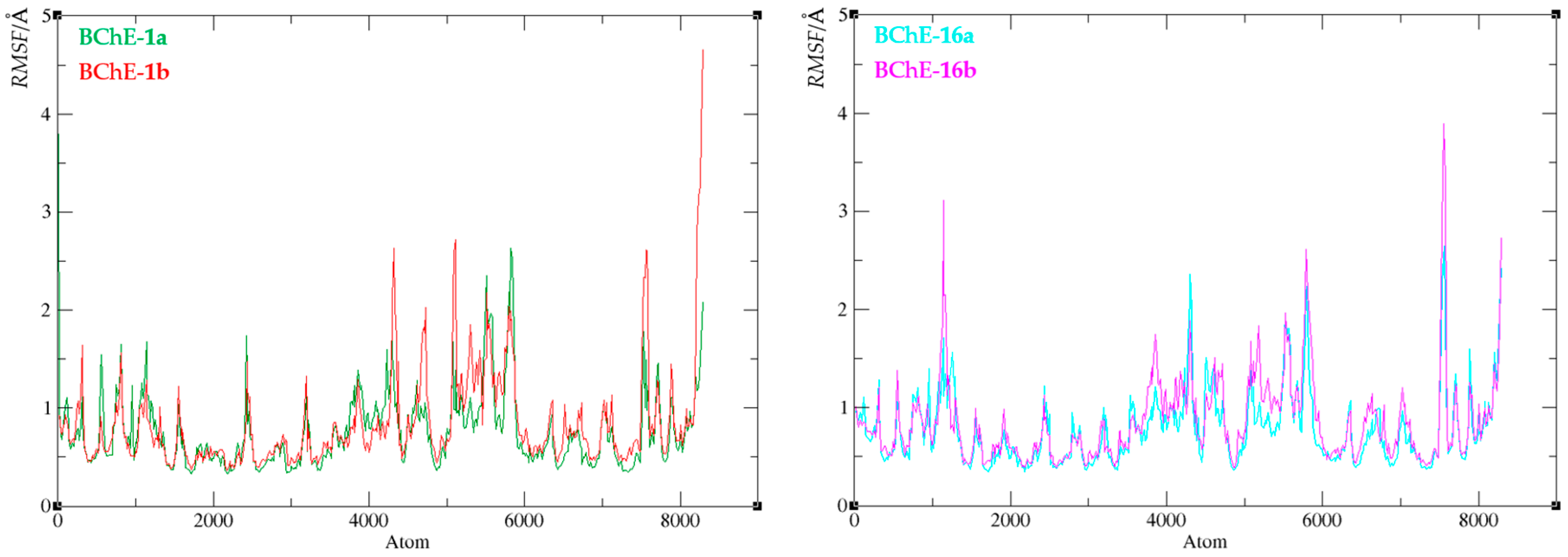
Figure 13.
Values of the radius of gyration from molecular dynamics simulation of an enzyme-ligand complexes of BChE with compounds 1 (left) and 16 (right).
Figure 13.
Values of the radius of gyration from molecular dynamics simulation of an enzyme-ligand complexes of BChE with compounds 1 (left) and 16 (right).
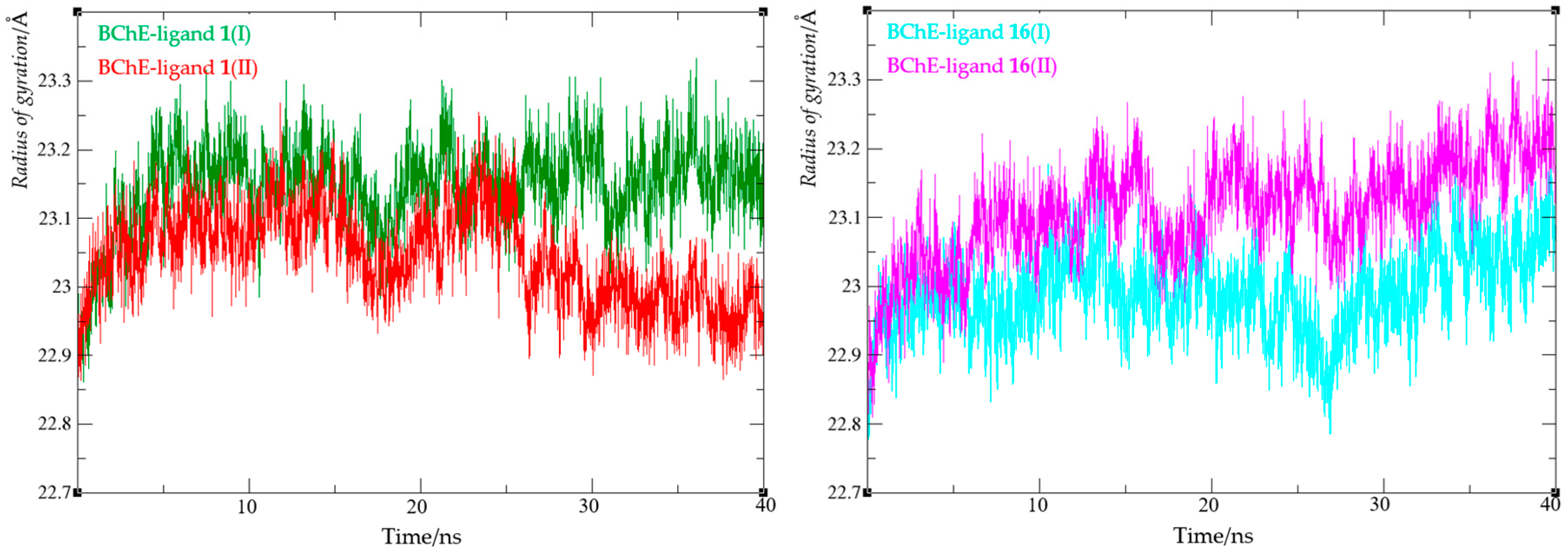

Table 1.
Crystallographic data and structure refinement details for carbamates 1 and 4.
| Compound | 1 | 4 |
| Empirical formula | C14H13NO2S | C15H15NO2S |
| Formula wt. / g mol-1 | 259.31 | 273.34 |
| Crystal dimensions / mm | 0.3 x 0.03 x 0.02 | 0.1 × 0.05 × 0.02 |
| Space group | P21 | P21/c |
| a / Å | 5.07910(10) | 5.12806(17) |
| b / Å | 16.6438(2) | 30.1746(7) |
| c / Å | 15.4486(2) | 9.2923(4) |
| α / ° | 90 | 90 |
| β / ° | 98.0250(10) | 105.137(4) |
| γ / ° | 90 | 90 |
| Z | 4 | 4 |
| V / Å3 | 1293.17(3) | 1387.97(8) |
| Dcalc / g cm-3 | 1.332 | 1.308 |
| μ / mm-1 | 2.171 | 2.049 |
| Θ range / ° | 3.93 – 80.03 | 5.12 – 75.85 |
| T(K) | 104(6) | 293(2) |
| Radiation wavelength | 1.54184 (CuKα) | 1.54184 (CuKα) |
| Diffractometer type | XtaLAB Synergy, Dualflex, HyPix |
XtaLAB Synergy, Dualflex, HyPix |
| Range of h, k, l | –6 > h > 6; –21 > k > 21; –19 > l > 19 |
–6 > h > 6; –26 > k > 37; –11 > l > 11 |
| Reflections collected | 17951 | 25937 |
| Independent reflections | 5485 | 2990 |
| Observed reflections (I ≥ 2σ) |
5146 | 2337 |
| Rint | 0.0776 | 0.0899 |
| R (F) | 0.0508 | 0.0743 |
| Rw (F2) | 0.1369 | 0.2130 |
| No. of parameters, restraints | 335, 1 | 181, 0 |
| Goodness of fit | 1.039 | 1.054 |
| Δρmax, Δρmin (eÅ–3) | 0.48; –0.64 | 0.66; –0.51 |
Table 2.
Geometric parameters of hydrogen bonds determined from geometric analysis.
| D–H / Å | H···A / Å | D···A / Å | D–H···A / º | Symm. op. on A | |
| Compound 1 | |||||
| N1–H1A∙∙∙O2 | 0.84 | 2.11 | 2.824(4) | 143 | –1+x, y, z |
| N2–H2A∙∙∙O4 | 0.93 | 2.10 | 2.821(4) | 134 | 1+x, y, z |
| C6–H6∙∙∙S1 | 0.95 | 2.73 | 3.156(5) | 108 | x, y, z |
| C6–H6∙∙∙O1 | 0.95 | 2.46 | 2.828(5) | 103 | x, y, z |
| C10–H10∙∙∙O4 | 0.95 | 2.57 | 3.477(5) | 160 | 1+x, y, 1+z |
| C16–H16∙∙∙O2 | 0.95 | 2.60 | 3.397(5) | 142 | x, y, z |
| C20–H20∙∙∙S2 | 0.95 | 2.73 | 3.158(3) | 108 | x, y, z |
| C20–H20∙∙∙O3 | 0.95 | 2.46 | 2.821(4) | 103 | x, y, z |
| Compound 4 | |||||
| N1–H1A∙∙∙O2 | 0.86 | 2.14 | 2.892(3) | 145.4 | –1+x, y, z |
| C6–H6∙∙∙S1 | 0.93 | 2.83 | 3.210(4) | 106 | x, y, z |
| C6–H6∙∙∙O1 | 0.93 | 2.45 | 2.800(4) | 103 | x, y, z |
Table 3.
Geometric parameters of π interactions.
| π···π | Cga···Cg / Å |
αb / º | βc / º | Cg···plane(Cg2) / Å | Offset/ Å |
Symm. |
| Compound 1 | ||||||
| S1→C4∙∙∙S1→C4 | 5.079(3) | 0.0(3) | 50.0 | 3.264(2) | 3.891 | –1+x, y, z |
| S1→C4∙∙∙S2→C18 | 4.246(3) | 6.1(2) | 33.7 | 3.751(2) | 2.355 | x, y, z |
| C7→C12∙∙∙C7→C12 | 5.079(2) | 0.0(17) | 50.4 | 3.2388(15) | 3.912 | –1+x, y, z |
| C7→C12∙∙∙C21→C26 | 5.521(2) | 0.78(17) | 58.4 | 2.8750(15) | 4.703 | 1+x, y, z |
| S2→C18∙∙∙S1→C4 | 4.246(3) | 6.1(2) | 27.9 | 3.5336(19) | 1.989 | x, y, z |
| S2→C18∙∙∙S2→C18 | 5.079(3) | 0.0(2) | 52.6 | 3.0846(19) | 4.035 | –1+x, y, z |
| C21→C26∙∙∙C7→C12 | 5.521(2) | 0.78(17) | 58.6 | 2.8916(15) | 4.713 | –1+x, y, –1+z |
| C21→C26∙∙∙C21→C26 | 5.079(2) | 0.03(17) | 49.6 | 3.2914(15) | 3.868 | –1+x, y, z |
| Compound 4 | ||||||
| S1→C4∙∙∙S1→C4 | 5.128(2) | 0.00(19) | 54.6 | 2.9734(16) | 4.178 | –1+x, y, z |
| S1→C4∙∙∙C7→C12 | 5.269(2) | 8.71(18) | 41.0 | 3.4207(16) | 3.458 | –1+x, y, –1+z |
| C7→C12∙∙∙C7→C12 | 5.128(2) | 0.00(16) | 46.1 | 3.5526(14) | 3.698 | –1+x, y, z |
a Cg = centre of gravity of the ring; b α = angle between planes of two interacting rings; c β = angle between Cg···Cg line and normal to the plane of the first interacting ring.
Table 4.
Geometric parameters of the C–H···π interactions in 4.
| C–H···π | H···Cg / Å |
γa / º | C–H···Cg / Å | C···Cg / Å | Symm. Operation on Cg |
| C15–H15A∙∙∙C7→C12 | 2.68 | 11.24 | 171 | 3.627(4) | 1+x, y, z |
a γ = angle defined by a line connecting the centre of gravity of the aromatic ring with the H atom and the normal to the aromatic ring.
Table 5.
Inhibition of BChE and calculated IC50 values by compounds 1–14 and 16–19.
| Compound | BChE | |
|---|---|---|
| % Inhibition* | IC50/μM | |
| 1 | 84.37±1.18 (10) | 0.500 |
| 2 | 84.66±3.20 (100) | 3.740 |
| 3 | 82.15±2.00 (500) | 149.7 |
| 4 | 85.38±2.29 (10) | 0.913 |
| 5 | 74.79±2.36 (5) | 1.102 |
| 6 | 74.00±3.20 (50) | 14.053 |
| 7 | 69.80±1.15 (100) | 30.403 |
| 8 | 87.21±0.47 (500) | 59.108 |
| 9 | 83.91±1.77 (10) | 0.583 |
| 10 | 83.48±1.49 (27) | 3.081 |
| 11 | 82.75±1.57 (10) | 1.541 |
| 12 | 83.19±0.84 (50) | 1.067 |
| 13 | 84.41±2.29 (25) | 1.883 |
| 14 | 70.00±0.80 (250) | 90.001 |
| 16 | 84.25±0.82 (1) | 0.0265 |
| 17 | 82.28±2.44 (25) | 0.645 |
| 18 | 87.48±1.31 (25) | 0.430 |
| 19 | 79.67±2.51 (50) | 2.045 |
| Galantamine | 90.10±3.40 (4.5) | 7.90 |
* Numbers given in parentheses represent maximal concentrations tested in μM.
Table 6.
The mutagenic potential of carbamates 1–19 through Lhasa M7 evaluation (green square—negative, red square—positive, white square—no data available); grey highlight—negative, orange highlight—positive, white—strongly negative.
Table 6.
The mutagenic potential of carbamates 1–19 through Lhasa M7 evaluation (green square—negative, red square—positive, white square—no data available); grey highlight—negative, orange highlight—positive, white—strongly negative.
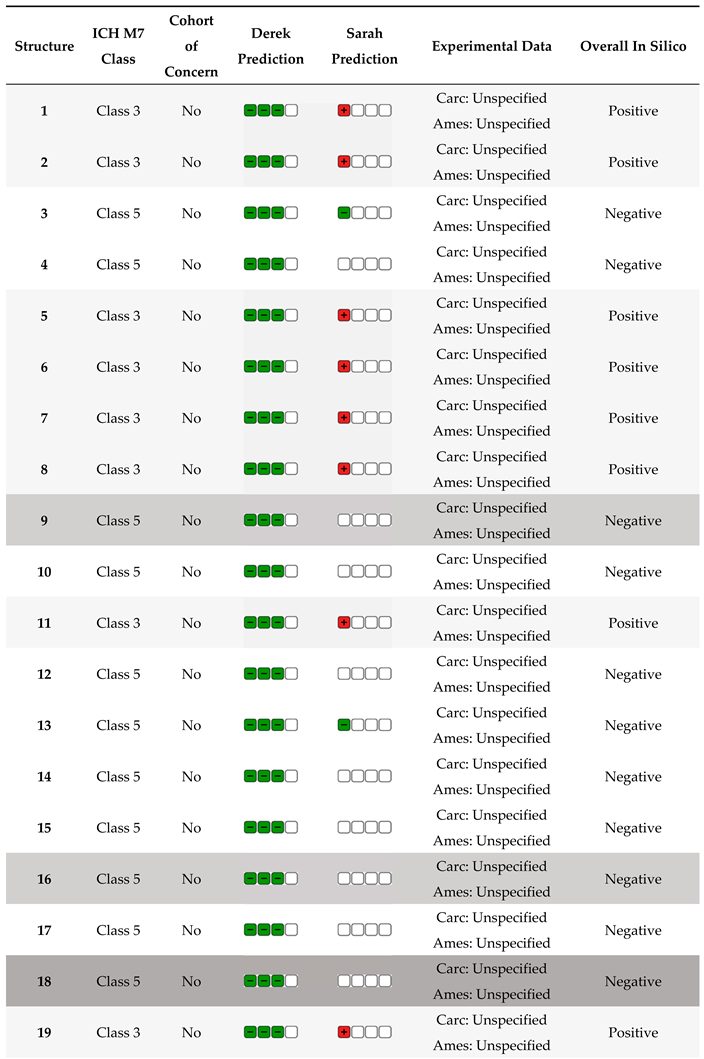
Table 7.
In silico analysis of additional ADME(T) indicators—absorption, distribution, metabolism, excretion, and toxicity—in the human body for carbamates 9, 16, and 18.
Table 7.
In silico analysis of additional ADME(T) indicators—absorption, distribution, metabolism, excretion, and toxicity—in the human body for carbamates 9, 16, and 18.
| Property | Model Name | 9 | 16 | 18 | Bambuterol | Rivastigmine | Unit |
| Absorption | Water solubility | -4.358 | -4.272 | -4.547 | log mol/L | ||
| Caco2 | 1.907 | 2.117 | 1.962 | log Papp in 10−6 cm/s | |||
| Intestinal absorption | 90.33 | 91.775 | 92.085 | % Absorbed | |||
| Skin permeability | -2.693 | -2.743 | -2.757 | log Kp | |||
| P−glycoprotein substrate | No | No | Yes | ||||
| P−glycoprotein I inhibitor | No | Yes | Yes | ||||
| P−glycoprotein II inhibitor | No | No | No | ||||
| Distribution | VDss (human) | 0.4 | 0.298 | 0.324 | 0.401 | 0.451 | log L/kg |
| Fraction unbound | 0.022 | 0.033 | 0.025 | − | − | Fu | |
| BBB permeability | 0.343 | 0.315 | 0.261 | 0.707 | 0.968 | log BB | |
| CNS permeability | -0.979 | -1.069 | -1.063 | −1.95 | −0.801 | log PS | |
| Metabolism | CYP2D6 substrate | No | No | No | |||
| CYP3A4 substrate | Yes | Yes | Yes | ||||
| CYP1A2 inhibitor | Yes | Yes | Yes | ||||
| CYP2C19 inhibitor | Yes | Yes | Yes | ||||
| CYP2C9 inhibitor | Yes | No | Yes | ||||
| CYP2D6 inhibitor | No | No | No | ||||
| CYP3A4 inhibitor | No | No | No | ||||
| Excretion | Total clearance | 0.138 | 0.373 | 0.262 | log ml/min/kg | ||
| Renal OCT2 substrate | Yes | No | No | Yes/No | |||
| Toxicity | AMES toxicity | No | No | No | Yes/No | ||
| Max. tolerated dose | 0.292 | -0.157 | -0.249 | log mg/kg/day | |||
| hERG I inhibitor | No | No | No | ||||
| hERG II inhibitor | Yes | Yes | Yes | ||||
| Oral rat acute toxicity (LD50) | 2.648 | 2.618 | 2.656 | mol/kg | |||
| Oral rat chronic toxicity (LOAEL) | 1.896 | 1.281 | 1.34 | log mg/kg_bw/day | |||
| Hepatotoxicity | Yes | No | No | ||||
| Skin sensitization | No | No | No | ||||
| T. pyriformis toxicity | 1.812 | 1.465 | 1.542 | log ug/L | |||
| Minnow toxicity | 0.214 | -0.487 | -0.62 | log mM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



