
Submitted:
18 November 2024
Posted:
19 November 2024
You are already at the latest version
Abstract
Considering all our previous experience in the design of new cholinesterase inhibitors, especially resveratrol analogs, in this research, the basic stilbene skeleton was used as a structural unit for new carbamates. Inhibitory activity was tested toward the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) by newly prepared carbamates 1–13. In the tested group of compounds, the leading inhibitors were 1 and 7, which achieved excellent selective inhibitory activity for BChE with IC50 values of 0.12 ± 0.09 μM and 0.38 ± 0.01 μM, respectively. Both were much more active than standard inhibitor galantamine against BChE. Molecular docking of the most promising inhibitor candidates, compounds 1 and 7, revealed that stabilizing interactions between the active site residues of BChE and the ligands involve π-stacking, alkyl-π interactions, and, when the carbamate orientation allows, H-bond formation. MD analysis confirmed the stability of the obtained complexes. Some bioactive resveratrol-based carbamates displayed complex-forming capabilities with Fe3+ ions as metal centers. Spectrophotometric investigation indicated that they coordinate one or two metal ions, which is in accordance with their chemical structure, offering two binding sites: an amine and a carboxylic group in the carbamate moiety. Based on all obtained in silico, experimental, and computational results on biological activity in the present work, new carbamates 1 and 7 represent potential selective BChE inhibitors as new therapeutics for neurological disorders.
Keywords:
ADMET
; carbamates
; butyrylcholinesterase inhibition
; biometal complexation
; docking
; molecular dynamics
; synthesis
1. Introduction
Alzheimer's disease is a progressive neurodegenerative disease characterized by the deterioration of synapses and neurons in the cortical and subcortical regions of the brain and impairment of the function of nerve impulse transmitter systems [1,2]. Alzheimer's disease (AD) is a complex multifactorial disease featured by several pathophysiological factors linked to various hypotheses about the origin and causes of AD: the cholinergic hypothesis, the amyloid hypothesis, the tau hypothesis, and the oxidative stress hypothesis. Also, it was observed that in certain parts of the brain affected by AD, there is an increased accumulation of metals, which leads to a change in the homeostasis of biometals. Changes in the biodistribution of metals in presynaptic and postsynaptic neurons cause abnormal neurotransmission, affect the activities of metal-dependent enzymes, and increase the secretion of amyloid β-peptides. Furthermore, metal ions with amyloid β-peptides form stable complexes that are more toxic than Aβ plaques and can generate reactive oxygen species that cause oxidative stress, lipid peroxidation, and protein damage [3,4,5,6,7].
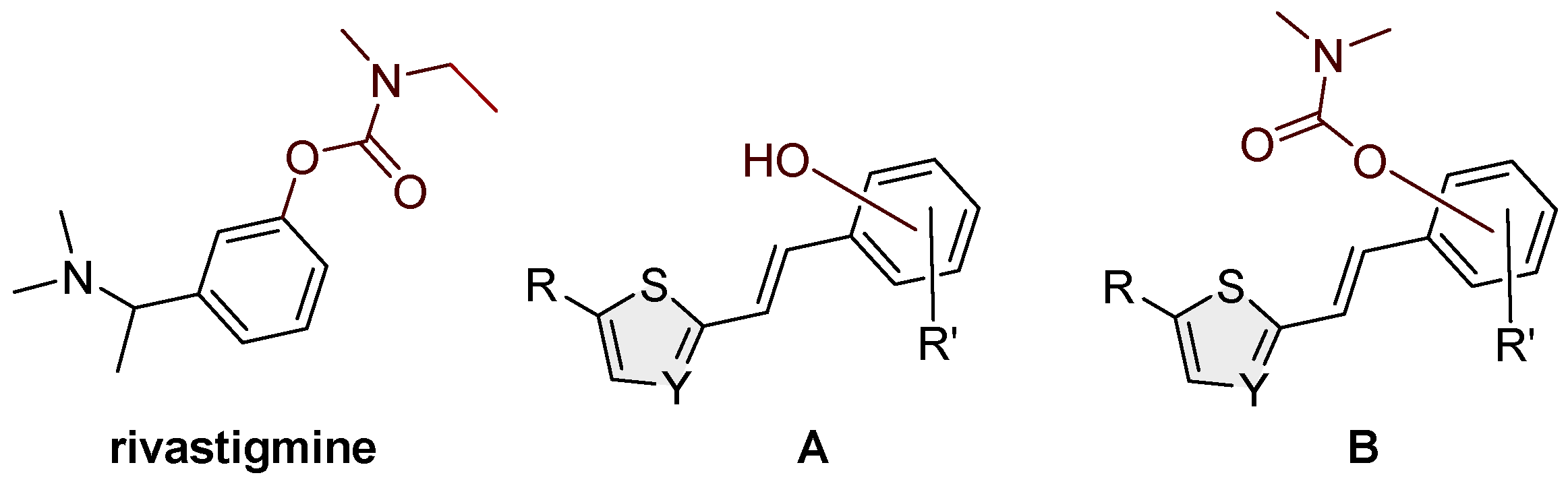
The complex pathophysiology of AD indicates the need to develop new molecules that can simultaneously act on multiple targets responsible for the onset and development of the disease. The investigations have shown that hybrid molecules designed based on existing cholinesterase (ChE) inhibitors (galantamine, donepezil, and tacrine) also have additional activities depending on the fragments introduced into the structure. Today, the treatment of AD is exclusively symptomatic and affects mainly the alleviation of symptoms and not the course, development, and final outcome of the disease [5,8]. It is based mainly on increasing the concentration of acetylcholine (ACh) by inhibiting cholinesterase, and three of the five drugs currently used for the treatment of AD are ChE inhibitors: galantamine, donepezil, and rivastigmine. The carbamate rivastigmine (Figure 1) is a progressive non-selective inhibitor of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) [9,10] that acts in the central nervous system and is particularly active in the cortex and hippocampus.
To date, a number of carbamates of different structures have been synthesized and tested, and they have been shown to inhibit both cholinesterases, AChE, and BChE. The mechanism of action of carbamates on cholinesterase is similar to that of ACh on AChE. However, unlike the extremely fast hydrolysis of acetylcholine, the hydrolysis of carbamates is much slower [11,12,13]. The first known cholinesterase inhibitor is the first carbamate drug, physostigmine, a methylcarbamate ester, also known as eserine, which was initially used as a drug in treating AD [14]. Rivastigmine (Figure 1) is a BChE and AChE inhibitor used in the treatment of mild, moderate, and severe AD, as well as mild to moderate dementia in Parkinson's disease [15,16]. The design of new cholinesterase inhibitors is based on modifying the structure of existing carbamate cholinesterase inhibitors or the synthesis of carbamate derivatives of existing AD drugs to find new, more effective drugs for the treatment of neurodegenerative diseases. To date, a number of carbamates of different structures have been synthesized and tested, which have been shown to inhibit both cholinesterases, AChE and BChE [17,18,19,20,21,22,23]. Since the selective inhibition of BChE stood out as a promising approach to treating AD, one part of the research was directed toward finding a selective BChE inhibitor with a carbamate group in its structure. So far, the carbamate cymserine, a structural analog of physostigmine with an isopropylphenyl instead of a methyl group at N-4, has proven to be an extremely selective as a BChE inhibitor, which is 15 times more selective BChE inhibitor compared to AChE. However, the use of cymserine for the treatment of AD was stopped due to the formation of the toxic metabolite eseroline.
Taking into account all our previous experience in the design of new cholinesterase inhibitors [24,25,26,27,28,29], especially resveratrol analogs [30,31,32] (Figure 1, structure A), in this research, the skeleton of the last ones was used as a structural basis for new carbamates (Figure 1, structure B). Several of those resveratrol analogs exhibited significantly enhanced BChE inhibitory and antioxidant activity compared to established standards such as galantamine or resveratrol (Figure 1, structure A) which was the excellent starting point for this research. Based on all obtained in silico, experimental, and computational results on biological activity in the present work, novel carbamates (Figure 1, structure B) represent potential selective BChE inhibitors as new therapeutics for neurological disorders.
2. Results and Discussion
2.1. Design, Synthesis and Characterization of New Carbamates 1–13
Analogs of resveratrol previously synthesized by our group [30,31,32] were used as the starting material in synthesizing carbamates 1–13 in dichloroethane (Scheme 1). At room temperature in an inert atmosphere, 1.8 eq of triethylamine and a catalytic amount of DMAP were added. After that, the reaction mixture was stirred under argon, followed by adding 2 eq of dimethylcarbamoyl chloride. In the end, the reaction mixture was worked up (see Materials and Methods section).
The resulting products, 1–13, were purified through column chromatography, using petroleum ether and diethyl ether in differing ratios as eluent, and isolated in a wide and different range of yields depending on the nature and position of the substituents (9-87%, Figure 2). It can be noted that the nature of the heterocyclic nucleus, the substituent on it, and the position of the carbamate group do not affect the isolated yields of carbamates and that for most derivatives, these yields are in the range of 40-50%. The highest yield is given by the derivative 6 (87%), in which the methoxy group is in the meta-position in relation to the carbamate group, unlike most derivatives that have substituents in the para-position in relation to the carbamate substituent. The lowest yields are on products 2 (9%) and 7 (31%), which have one methyl group in addition to the carbamate, either on the heterocyclic or the aryl ring.
The structure of all newly synthesized trans-resveratrol-based carbamates 1–13 was confirmed by NMR (1H and 13C) and HRMS analyses (Figure 3, Figures S1–S63). The purity of the newly synthesized carbamates was determined by the HPLC method and ranged from 85 to 99%, depending on the compound.
As in this research, trans-resveratrol-based carbamates 1–13 were designed as selective BChE inhibitors; it is worth briefly referring to BChE-assisted substrate (carbamate) hydrolysis (Scheme 2) that is analogous to the mechanism of AChE-assisted hydrolysis of ACh. The mechanism includes the formation of the Michaelis complex, acylation of the enzyme, and its spontaneous deacylation with water (Scheme 2). In the first step, under the influence of Glu325, His438 acts as a base that attracts a proton from the hydroxyl group of Ser198, thus making it more nucleophilic than other serines in the molecule. The nucleophilic oxygen of Ser198 reacts with the carbonyl carbon atom of the carbamate, resulting in a tetrahedral transition state. After that, serine remains acetylated. Water, which, under the influence of His438, becomes more nucleophilic than other water molecules in the active site of the enzyme, reacts with the carbonyl carbon of the acylated enzyme, which leads to the formation of a new tetrahedral transition state and ultimately to the deacylation of the enzyme [33,34].
2.2. Cholinesterases Inhibitory Activity of Carbamate Derivatives
Previously tested resveratrol analogs [32] with thiophene or thiazole ring on one side and phenol ring on the other side of stilbene moiety were converted into carbamates (Scheme 1, Figure 2). Inhibitory activity was tested toward the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) by newly prepared carbamates 1–13. The assays were carried out using Ellman's method [35] with minor modifications, and the results were expressed as IC50 values and presented in Table 1.
The most dramatic result of translation from resveratrol analogs into carbamate derivatives is the complete loss of activity towards acetylcholinesterase; namely, none of the tested derivatives achieved more than 35% inhibition of this enzyme at the maximum tested concentrations. This is not surprising considering the presence of the carbamate group, which has been shown in various derivatives to be highly selective for BChE [36,37,38,39]. Furthermore, butyrylcholinesterase inhibition is highly successful and has better concentration values than resveratrol analogs. In the tested group of compounds, the leading inhibitors were 1 and 7, which achieved excellent inhibitory activity for BChE with IC50 values of 0.12 ± 0.09 μM and 0.38 ± 0.01 μM, respectively. Both were much more active than galantamine against BChE (Table 2). The common structural feature of these two inhibitors is that they have no substituent on the carbamate side of stilbene, and they differ only in the presence/absence of methyl on thiophene. Changing the position of the carbamate group from ortho (derivative 1) to meta (derivative 12) drastically reduces the activity towards the BChE by two orders of magnitude (IC50 for 12 is 11.37 ± 2.48 μM).
In the group of compounds without the methyl at the thiophene ring (1–6), almost all derivatives achieved excellent inhibition values, better than the standard galantamine. A slight decline in activity was found for derivative 5, the one with methoxy substituent. The effect of substituents on the benzene ring was similar for resveratrol derivatives [32]. Methylation of thiophene ring in derivatives 8–10 reduces the inhibitory effect, but the obtained IC50 values were still in the moderate range. Two thiazole derivatives, 12 and 13, were also tested, and 13 stood out with a very good inhibition value of 4.03 ± 0.5 μM, better than the galantamine. Finding a selective inhibitor of BChE continues to be one of the approaches to discovering new therapeutic treatments for AD. It is shown that the carbamates synthesized and tested here show very strong and utterly selective inhibition of BChE, which makes this structural basis interesting for the development of potent BChE inhibitors.
2.3. In Silico Evaluation of ADME(T) Properties and In Silico Assessment of the Possibility of a Compound Passing Through the Blood-Brain Barrier by Passive Transport
Absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) studies are important in any drug development stage. They give insights into whether the compound exhibits drug-like pharmacokinetic properties and whether it has properties that will cause safety concerns in people [40]. ADMET properties are investigated in silico, in vitro, and in vivo. In early drug discovery, in silico tools are used when molecules are either not yet synthesized in a pure enough form, or the quantities synthesized are insufficient, as the synthetic pathways are still developed and optimized in the early stages. This study used free in silico tools [41,42] to screen the candidate compounds.
It can be seen (Table 2) that the water solubility is very low. As these carbamate derivatives tested positive for selective BChE inhibition, in order to work as potential CNS drugs for Alzheimer's disease, they have to be able to cross the BBB barrier. The in-silico data that will indicate the CNS potential are the ones under distribution in the above table.
The BBB (as log BB = the drug concentration in the brain divided by the concentration in the blood) for both compounds is around 0.3. This result is very promising, as compounds with log BB >0.3 readily cross the BBB. The second indicator is the Log PS (permeability surface area); here, log PS >-2 value signifies molecules that can penetrate the CNS. For compounds 1 and 7, this value is above −0.94, indicating that they are very good candidates. As the CNS is very dependent on the general indicators of distribution in the body, the VDss and the fraction unbound (human) (Fu) were also calculated. VDss is considered the least biased indicator of the extent of distribution. This pharmacokinetic parameter represents the hypothetical volume into which the drug dose would have to be evenly distributed to give rise to the same concentration observed in the blood plasma. When these two parameters are calculated and commented on together, they show how the decreased plasma protein binding leads to an increase in free plasma fraction, causing an increase in the volume of distribution and a shorter elimination half-life. They link distribution and elimination. For a CNS drug, the unbound fraction has a twofold effect, and the interpretation of this indicator based on in-silico preliminary results is complex. Drugs highlighted as potential CNS candidates are very sensitive to the unbound portion of the drug, as a maximal concentration in the plasma is one of the key factors with the CNS candidates. Usually, the bonds to the blood proteins are reversible, but this would require further experimental results for the tested structures. If we consider that the unbound drug is the effective form, then the higher Fu can indicate a better bioavailability, and the drug can better distribute in the central nervous system.
Since these candidates can be regarded as analogs containing an active carbamate moiety, it was practical to compare the aforementioned indicators with well-known drugs that possess the same structural fragment. Bambuterol (Figure 4) was selected due to its structural resemblance to rivastigmine and its similar therapeutic indication (see introductory section).
As these are well-known drugs, the in-silico prediction was made only for the distribution parameters VDss, BBB and PS. The parameter VDss is similar to the one for compounds 1 and 7. Log BB is greater for the known drugs, but as drugs that are above 0.3 signify that they readily cross the BBB barrier, the lead compounds 1 and 7 still meet the criteria.
From all of the parameters that were tested in the range of in-silico ADME(T) studies and from the study of potential genotoxicity, it is safe to promote compounds 1 and 7 into promising leads that could be further developed into drug products.
2.4. Docking Study and Molecular Dynamics
Compounds 1 and 7, identified as the most potent inhibitors, were docked into the active site of BChE. Figure 5 illustrates the molecular structures of the resulting ligand-protein complexes, highlighting key interactions that contribute to their stability.
In the most stable complex of compound 1 with BChE, the carbamate group is oriented toward the esteratic site, where the carbonyl oxygen forms a hydrogen bond with the hydroxyl group of Ser198. Additionally, the dimethylamino group of the carbamate is oriented toward Leu286 in the acyl pocket but also engages in hydrophobic interactions with Phe329. The aromatic thiophene and phenyl rings of the ligand participate in π-stacking interactions with Trp82. In the complex with compound 7, the ligand adopts a different orientation. The carbamate group is positioned toward the peripheral anionic site (PAS); methyl groups of the dimethylamino carbamate fragment engage in alkyl-π interaction with Tyr330 and Trp82. Another alkyl-π attraction is observed between the -CH₃ substituent on the thiophene and His438 in the esteratic site, while the thiophene ring interacts with Trp82 at the anionic site.
The binding free energies estimated from docking for compounds 1 and 7 are −6.85 kcal mol⁻¹ and −7.43 kcal mol⁻¹, respectively (Table S1). For comparison, the binding free energy for the reference ligand galantamine, calculated using the same method, was −7.54 kcal mol⁻¹.
The stability of ligand-enzyme complexes obtained by docking was evaluated through molecular dynamics (MD) simulations. The production simulation lasted 100 ns, and root-mean-square deviation (RMSD) values were calculated to monitor structural changes in the protein-ligand complex over the simulation period. RMSD analysis was conducted on all atoms, excluding hydrogens, for both complexes. Additionally, root-mean-square fluctuation (RMSF) values were computed as average quadratic fluctuations in the positions of α carbons over the trajectory, identifying the most mobile regions of the protein backbone. To estimate protein compactness in the complexes with ligands, the radius of gyration (Rg) was calculated as the RMS average of the distance of all atoms from the protein-ligand complex's center of mass. A significant increase in Rg suggests a more expanded or flexible protein conformation in the presence of the ligand. The MD simulation results for BChE complexes with compounds 1 and 7 are shown in Figure 6, Figure 7, and Figure 8.
In both systems, convergence was achieved after 30 ns. For complex with ligand 1, RMSD values ranged from 0.78 to 2.59 Å, averaging 2.07 Å, with similar values for ligand 7 (0.81 to 2.44 Å, averaging 2.02 Å). The average RMSD for the final 70 ns decreased to 1.89 and 1.90 Å for ligands 1 and 7, respectively. RMSF analysis (Figure 7) revealed that in the BChE-ligand 1 complex (red line), the most mobile protein backbone regions were α carbons 7559 and 5786, corresponding to residues Asn485 and Trp376, with RMSF values of 4.50 and 3.61 Å, respectively. In the BChE-ligand 7 complex (blue line), the highest RMSF (6.59 Å) was observed for Val529 (positioned at a distance of 24 Å from the active site, thus not influencing its structure), with other residues showing significantly less mobility throughout the simulation.
Finally, the radius of gyration for both complexes, shown in Figure 8, indicates that protein compactness was maintained during the simulation, with Rg values within a narrow range between 22.79 and 23.34 Å for complex with ligand 1 and 22.81 and 23.28 Å for BChE-ligand 7, respectively.
2.5. Complex Formation of Biometals with Bioactive Carbamates
Based on our previous studies on other cholinesterase inhibitory active resveratrol derivatives [43], it was reasonable to investigate the interaction of some of these resveratrol-based carbamates with Fe3+, a potential biometal. For this purpose, the following compounds have been selected: 1, 3, 6, and 7. The Fe3+ ions strongly hydrolyze, decreasing the pH of the aqueous system; thus, accordingly, acidic solutions were used to study the possible complex formation by spectrophotometric titrations. Although generally, in these experiments, the concentration of the potential ligand (L) is increased at constant concentration of the metal center (Fe3+ in this case), in our work, also the other type of titration was carried out, i.e., increasing the concentration of the metal center at constant CL. Nevertheless, the previous method provided more useful pieces of information in our case.
First, the absorption spectra of the selected ligands were recorded for spectrophotometric titration. Figure 9 shows the corresponding molar absorptivity spectra, which are very similar to each other from the viewpoint of the band positions. The shorter-wavelength bands (at about 233-238 nm) may be designated as thiophene-centered ones due to a π-π* transition. The longer-wavelength bands (at about 322-331 nm) belong to the whole 2-styryl thiophene moiety due to the conjugation of the two aromatic rings through the vinyl bridge. The substituents hardly influence the band positions; instead, they affect the molar absorptivities.
The molar absorptivities at the longer-wavelength band maxima are given in Table 3.
Figure 10 demonstrates a titration series of spectra, in the case of which the concentration of the ligand (6) was increased up to 3.5×10-5 M. During the titration, the band position did not change, but the absorbance at the band maximum appreciably deviated from the straight line (see inset). This phenomenon unambiguously suggests an interaction between the metal ion and the potential ligand. However, the absorption spectrum of the formed associate or complex is very similar to that of the ligand; only the corresponding absorptivities are lower. Since the deviation from the straight line is quite small, the estimation of the formation constant is very uncertain. Nevertheless, molar absorptivity and the composition of the complex may be suggested based on the absorbance vs. ligand concentration plot (Figure 9 inset). As shown in the inset, which can be seen in a much larger version in Figure S64, the absorbance (at 330 nm) vs. ligand concentration plot indicates an appreciable breaking point, before which the slope is higher than that of the red line representing the theoretical absorbance of the ligand (calculated by using its molar absorptivity at 330 nm). The slope values are the molar absorptivities of the corresponding species at 330 nm. Hence, the molar absorptivity of the complex formed in this system is about 26 700 M-1 cm-1, which is ca. 64% higher than that of the ligand, indicating a considerable interaction between the metal ion and the ligand. The composition of the complex can be estimated using the position of the breaking point. The concentration of the ligand is about half of Fe3+ at the breaking point (0.02 mM vs. 0.038 mM), which suggests a 2:1 (M2L) composition. This result indicates that metal ions may be bound to both coordination sites of this carbamate ligand, i.e., to both the carboxylic and the amine moiety. Nevertheless, the carboxylic group is preferred in this respect. Since the slope of the absorbance vs. ligand concentration plot after the breaking point well agrees with that of the absorptivity of the ligand, i.e., the slope of the red straight line, no further complex formation occurred in this range.
A similar phenomenon was observed for compound (3) (Figure S65). In that case, the absorptivity of the complex (13 200 M-1cm-1) just moderately (by about 26%) exceeds that of the ligand (10 400 M-1cm-1). The position of the breaking point, i.e., the ligand-to-metal concentration ratio, suggests a 1:1 composition. This is in accordance with the lower absorptivity of the complex, indicating a weaker interaction. The absorbance vs. ligand concentration plot does not show any deviation from the straight line for the compounds with no substituent on the benzene ring (apart from the common carbamate group), i.e., for 1 and 7. Since the absorption spectra of the complexes, when they formed, were very similar to those of the ligands, the spectral series of titration with the metal center did not prove to be as informative as those obtained by increasing the ligand concentration.
2.6. In-Silico Genotoxicity Evaluation
Determination of impurities is a vital part of active substance and drug development. Within the scope of impurities of drug substances and drug products, possible mutagenic/cancerogenic impurities take a special role as they are controlled with much lower limits (ICH M7 Guideline). It is common when developing new drug substances and products that the impurities will also be new compounds, and there will be no experimental data available for them in the literature databases. In these cases, the Q(SAR) approach is vital. (Q)SAR models make predictions based on structural components [44]. This approach for evaluation of the mutagenic potential of compounds can also be used to determine the mutagenic potential during the early stages of searching for potentially active drug substances. This is extremely important as compounds with biological activity and mutagenic potential can be eliminated early on. The Lhasa software is the most commonly used tool because two complementary models (one rule base and one statistical-based) are used and their predictions are reviewed one more time by an expert.

Table 3.
The mutagenic potential of carbamates 1–13 by Lhasa M7 evaluation (green square – negative, red square – positive, white square – no data available); grey highlight – negative, orange highlight – positive, white – strongly negative).
Table 3.
The mutagenic potential of carbamates 1–13 by Lhasa M7 evaluation (green square – negative, red square – positive, white square – no data available); grey highlight – negative, orange highlight – positive, white – strongly negative).
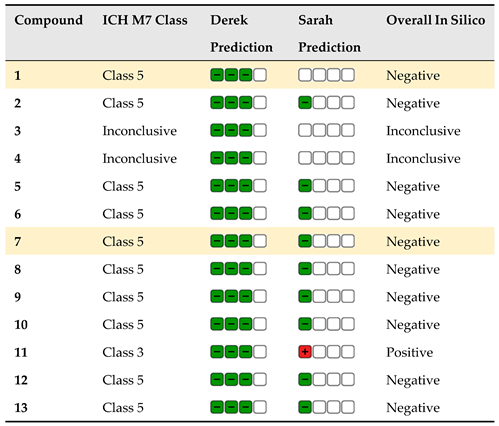 |
In the case of compounds 1–13 that are investigated for their potential biological activity with the emphasis on selective inhibition of enzyme BChE, only one structure is identified by the software to have a potential for mutagenicity. This is the compound 11. Here, the Sarah prediction gives a 9% chance of some of the structural features in this precise structural environment being positive. It is important to note that the most active molecules, 1 and 7, are reviewed as negative and can be good future leads for further early drug product development stages.
3. Materials and Methods
3.1. General Procedure
NMR spectra were recorded using a Bruker AV300 or AV600 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) with a 5-mm probe head at the Ruđer Bošković Institute. The standard 1H and proton-decoupled 13C{1H} NMR spectra were recorded at 300.000 and 600.130 MHz, and 75.432 and 150.903 MHz, respectively. The chemical shifts (δ/ppm) of the 1H and 13C spectra were related to the tetramethylsilane signal (TMS). All spectra were recorded in deuterated chloroform (CDCl3-d) at 25 °C. The signal at 2.36 ppm in the proton spectra belongs to toluene.
Reactions were monitored by thin-layer chromatography performed on silica gel coated plates (0.2 mm, 60/Kieselguhr F254) immersed in 10 mL of the dissolution system. Workup after each synthesis included an extraction between water and DCE, with a single addition of 2.4 M HCl. After separating the organic and aqueous layer, the organic layer was dried over anhydrous magnesium sulfate, MgSO4. Obtained compounds were purified by column chromatography performed in glass columns of different diameters, filled with silica gel (60 Å, technical grade) of different heights. Abbreviations used in the experimental part are as follows: TEA – triethylamine, DMAP – 4-dimethylaminopyridine, DCE – 1,2-dichloroethane, PE – petroleum ether, E – diethyl ether, HCl – hydrochloric acid, NMR – nuclear magnetic resonance, s – singlet, d – doublet, m – multiplet, t – triplet, dd – doublet of doublets. High-resolution mass spectrometry (HRMS) analyses were performed on a MALDI TOF/TOF analyzer mass spectrometer fitted with an Nd:YAG laser at 355 nm (fitting rate of 200 Hz).
3.2. Synthesis of Carbamates 1–13
Analogues of resveratrol previously synthesized by our group [30,31,32] were used as the starting material in the synthesis of carbamates 1–13. In a 25 mL round bottomed flask, depending on the starting resveratrol, 50-100 mg was dissolved in 1 mL DCE. While stirring at room temperature in an inert atmosphere, 1.8 eq of TEA and a catalytic amount of DMAP were added. The reaction flask was then submerged in an oil bath and the mixture stirred at 60 °C for 15 min under argon. Afterwards, 2 eq of dimethylcarbamoyl chloride was added dropwise and stirring continued at 60 °C in an inert atmosphere for 2h. At the end of the reaction, the reaction mixture was extracted three times between DCE and distilled water, with a single addition of 1 mL of 2.4 M HCl. The organic layer was dried over anhydrous magnesium sulfate, MgSO4, filtered, and evaporated to dryness using a rotary evaporator. The resulting products, 1–13, were purified through column chromatography, using petroleum ether and diethyl ether in differing ratios as eluent. NMR analyses indicated carbamates 1–13 were obtained as trans-isomers (with traces of cis-isomer in some samples), with the exception of 12 which was obtained with a prevalence of cis-isomer (trans : cis = 0.3 : 1).
(E)-2-(2-(thiophen-2-yl)vinyl)phenyl
dimethylcarbamate (1): 67 mg (isolated 50%), brown oil; Rf
(40% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.61 (dd, J = 7.6, 1.3 Hz, 1H), 7.25 – 7.11 (m, 5H), 7.08
– 6.97 (m, 3H), 3.20 (s, 3H), 3.04 (s, 3H); 13C NMR (CDCl3,
75 MHz) δ/ppm: 154.6,
148.8, 143.1, 129.7, 128.3, 127.6, 126.4, 125.9, 125.6, 124.5, 123.4, 123.1,
121.9, 36.8, 36.4; HRMS (ESI) (m/z) for C15H15NO2S:
[M + H]+calcd = 273.0823, and [M + H]+measured
= 273.0824.
(E)-4-methyl-2-(2-(thiophen-2-yl)vinyl)phenyl dimethylcarbamate (2): 12 mg (isolated 9%), yellow oil; Rf (40% PE/E) = 0.59; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.33 (br s, 1H), 7.13 (d, J = 8.6 Hz, 1H), 7.11 – 7.08 (m, 1H), 7.01 – 6.89 (m, 5H), 3.12 (s, 3H), 2.96 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 153.8, 145.7, 142.2, 134.0, 128.1, 126.6, 125.3, 125.2, 123.4, 122.1, 121.8, 121.1, 35.8, 35.4, 19.9; HRMS (ESI) (m/z) for C16H17NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0982.
(E)-4-fluoro-2-(2-(thiophen-2-yl)vinyl)phenyl
dimethylcarbamate (3): 99 mg (isolated 75%), yellow oil; Rf (40%
PE/E) = 0.59; 1H NMR (CDCl3, 300 MHz)
δ
/ppm: 7.42 – 7.27 (m, 1H), 7.23 – 6.91 (m, 6H), 3.19 (s, 3H), 3.04 (s,
3H); 13C NMR (CDCl3, 75 MHz)
δ
/ppm: 160.1 (d, JCF = 244.2 Hz), 154.5, 146.7, 144.7
(d, JCF = 3.2 Hz), 142.5, 127.7, 127.0, 126.2 (d, JCF
= 7.7 Hz), 125.0, 124.5 (d, JCF = 9.9 Hz), 122.1, 114.9 (d, JCF
= 24.1 Hz), 111.8 (d, JCF = 24.1 Hz), 36.9, 36.5; HRMS (ESI)
(m/z) for C15H14FNO2S: [M + H]+calcd
= 291.0729, and [M + H]+measured = 291.0735.
(E)-4-chloro-2-(2-(thiophen-2-yl)vinyl)phenyl
dimethylcarbamate (4): 40 mg (isolated 47%), brown oil; Rf (40%
PE/E) = 0.59; 1H NMR (CDCl3, 300 MHz)
δ
/ppm: 7.58 (d, J = 2.2 Hz, 1H), 7.24 – 7.16 (m, 3H), 7.11 – 7.06
(m, 2H), 7.03 – 6.99 (m, 1H), 6.93 (d, J = 16.1 Hz, 1H), 3.19 (s, 3H),
3.04 (s, 3H); 13C NMR (CDCl3,
75 MHz)
δ
/ppm: 154.3, 147.2, 142.5, 131.3, 131.0,
128.0, 127.8, 127.1, 124.6, 124.5, 36.9, 36.5; HRMS (ESI) (m/z) for C15H14ClNO2S:
[M + H]+calcd = 307.0434, and [M + H]+measured
= 307.0439.
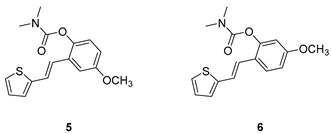
(E)-4-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl
dimethylcarbamate (5): 69 mg (isolated 53%), brown oil; Rf (40%
PE/E) = 0.34; 1H NMR (CDCl3, 300 MHz)
δ
/ppm: 7.23 – 7.13 (m, 2H), 7.09 (d, J = 2.8 Hz, 1H), 7.06 – 7.03
(m, 2H), 7.02 – 6.94 (m, 2H), 6.81 (dd, J = 8.7, 2.8 Hz, 1H), 3.83 (s,
3H), 3.19 (s, 3H), 3.03 (s, 3H); 13C NMR (CDCl3, 75 MHz)
δ
/ppm: 157.0, 155.0, 142.8, 132.7, 130.4, 127.6, 126.5, 124.6, 123.9,
123.6, 121.9, 114.1, 110.3, 55.7, 36.9, 36.5; HRMS (ESI) (m/z) for C16H17ClNO3S:
[M + H]+calcd = 303.0929, and [M + H]+measured
= 303.0936.
(E)-5-methoxy-2-(2-(thiophen-2-yl)vinyl)phenyl dimethylcarbamate (6): 57 mg (isolated 87%), colourless oil; Rf (40% PE/E) = 0.24; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.52 (d, J = 8.6 Hz, 1H), 7.16 (d, J = 4.8 Hz, 1H), 7.09 (d, J = 16.3 Hz, 1H), 7.03 – 6.97 (m, 3H), 6.78 (dd, J = 9.2, 2.1 Hz, 1H), 6.71 (d, J = 2.4 Hz, 1H), 3.81 (s, 3H), 3.20 (s, 3H), 3.05 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 159.8, 154.4, 149.7, 143.4, 127.6, 126.6, 125.6, 123.9, 122.3, 121.9, 121.4, 112.5, 108.3, 55.5, 36.9, 36.5; HRMS (ESI) (m/z) for C16H17ClNO3S: [M + H]+calcd = 303.0929, and [M + H]+measured = 303.0933.
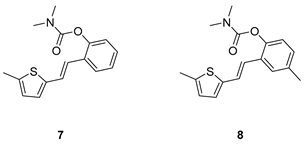
(E)-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl dimethylcarbamate (7): 26 mg (isolated 31%), yellow oil; Rf (40% PE/E) = 0.43; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.58 (dd, J = 7.4, 1.1 Hz, 1H), 7.24 – 7.08 (m, 4H), 6.93 – 6.81 (m, 2H), 6.67 – 6.62 (m, 1H), 3.20 (s, 3H), 3.05 (s, 3H), 2.48 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 154.6, 148.7, 141.0, 139.5, 129.9, 128.0, 126.7, 125.8, 125.8, 125.6, 123.8, 123.1, 120.6, 36.9, 36.5, 15.6; HRMS (ESI) (m/z) for C16H18NO2S: [M + H]+calcd = 287.0980, and [M + H]+measured = 287.0982.
(E)-4-methyl-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl dimethylcarbamate (8): 37mg (isolated 50%), yellow oil; Rf (40% PE/E) = 0.50; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.38 (s, 1H), 7.10 (d, J = 16.1 Hz, 1H), 7.06 – 6.96 (m, 2H), 6.89 – 6.80 (m, 2H), 6.66 – 6.61 (m, 1H), 3.19 (s, 3H), 3.03 (s, 3H), 2.48 (s, 3H), 2.34 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 154.8, 146.6, 141.1, 139.4, 135.0, 129.4, 128.8, 126.6, 126.2, 125.8, 123.5, 122.8, 120.8, 36.9, 36.5, 20.9, 15.6; HRMS (ESI) (m/z) for C17H19NO2S: [M + H]+calcd = 301.1136, and [M + H]+measured = 301.1142.
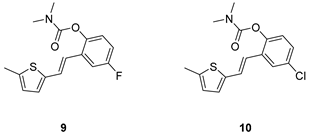
(E)-4-fluoro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl dimethylcarbamate (9): 32 mg (isolated 49%), yellow oil; Rf (40% PE/E) = 0.42; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.22 – 7.15 (m, 1H), 7.06 – 6.97 (m, 2H), 6.88 – 6.83 (m, 1H), 6.80 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 16.0 Hz, 1H), 6.60 – 6.56 (m, 1H), 3.12 (s, 3H), 2.97 (s, 3H), 2.41 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.29 (d, JCF = 243.1 Hz), 157.7, 144.7, 140.5 (d, JCF = 25.4 Hz), 131.7 (d, JCF = 8.7 Hz), 129.2, 127.6, 126.1, 125.0, 124.6 (d, JCF = 8.9 Hz), 119.6, 114.7 (d, JCF = 23.8 Hz), 111.8 (d, JCF = 24.5 Hz), 37.1, 36.6, 15.8; HRMS (ESI) (m/z) for C16H16FNO2S: [M + H]+calcd = 305.0886, and [M + H]+measured = 305.0890.
(E)-4-chloro-2-(2-(5-methylthiophen-2-yl)vinyl)phenyl dimethylcarbamate (10): 26 mg (isolated 41%), yellow oil; Rf (40% PE/E) = 0.41; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.22 – 7.06 (m, 4H), 6.91 – 6.82 (m, 2H), 6.66 – 6.61 (m, 1H), 3.21 (s, 3H), 3.04 (s, 3H), 2.47 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 148.8, 141.0, 139.4, 129.9, 128.1, 126.7, 125.9, 125.8, 125.6, 123.8, 123.0, 120.7, 36.8, 36.4, 15.7 (1 quaternary C is missing); HRMS (ESI) (m/z) for C16H16ClNO2S: [M + H]+calcd = 321.0590, and [M + H]+measured = 321.0594.
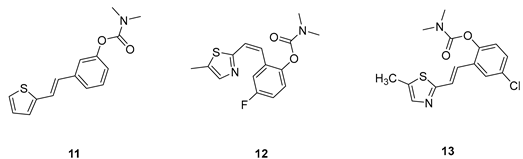
(E)-3-(2-(thiophen-2-yl)vinyl)phenyl dimethylcarbamate (11): 19 mg (isolated 39%), yellow oil; Rf (40% PE/E) = 0.45; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.24 (t, J =7.6, 7.2 Hz, 1H), 7.21 – 7.18 (m, 1H), 7.18 – 7.16 (m, 1H), 7.14 (d, J = 16.1 Hz, 1H), 7.12 (d, J = 5.2 Hz, 1H), 6.99 (d, J = 3.3 Hz, 1H), 6.94 – 6.91 (m, 2H), 6.82 (d, J = 15.9 Hz, 1H), 3.04 (s, 3H), 2.95 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 153.8, 150.9, 141.6, 137.4, 128.4, 126.6, 126.5, 125.3, 123.5, 122.4, 121.5, 119.8, 118.2, 35.7, 35.4; HRMS (ESI) (m/z) for C15H16NO2S: [M + H]+calcd = 273.0824, and [M + H]+measured = 273.0826.
(E)-4-fluoro-2-(2-(5-methylthiazol-2-yl)vinyl)phenyl dimethylcarbamate (12): 30 mg (isolated 4 %), yellow oil; Rf (40% PE/E) = 0.13; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.52 (s, 1H), 7.32 (dd, J = 8.6, 2.5 Hz, 1H), 7.28 (d, J = 2.5 Hz, 1H), 7.14 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 11.8 Hz, 1H), 6.38 (d, J = 11.8 Hz, 1H), 2.95 (s, 3H), 2.91 (s, 3H), 2.58 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 157.8, 146.8, 142.9, 140.7, 132.5, 130.6 129.7, 129.0, 128.0, 127.4, 124.6, 123.3, 122.4, 122.0, 35.7, 35.1, 18.6; MS (ESI) m/z (%, fragment): 304 (100).
(E)-4-chloro-2-(2-(5-methylthiazol-2-yl)vinyl)phenyl dimethylcarbamate (13): 29 mg (isolated 45%), yellow oil; Rf (40% PE/E) = 0.39; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.09 (d, J = 3.0 Hz, 1H), 7.55 (m, 2H), 7.01 – 6.99 (m, 1H), 6.97 (d, J = 16.0 Hz, 1H), 6.81 (dd, J = 8.5, 2.8 Hz, 1H), 3.19 (s, 3H), 3.03 (s, 3H), 2,61 (s, 3H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 157.0, 154.9, 142.9, 142.7, 127.6, 126.5, 124.6, 123.9, 123.6, 121.9, 114.1, 110.3, 55.7, 36.9, 36.5, 20.2; HRMS (ESI) (m/z) for C15H15ClN2O2S: [M + H]+calcd = 322.0543, and [M + H]+measured = 322.0545.
3.2. Cholinesterase Inhibitory Activity
The cholinestrases inhibitory activity of tested compounds were measured using modified Ellman's method. Enzymes acetilcholinsesterase (AChE, E.C. from electric eel) and butyrylcholinesterase (BChE, E.C. from equine serum) were purchased from Sigma Aldrich (St. Louis, MO, USA). Substrates used in this assay, acetylthiocholin iodide (ATChI) and S-butyrylthiocholiniodide (BTChI) as well as trisma base for buffer preparation, were purchased from Sigma Aldrich. Reagent used for colorimetric detection, 5,5´-dithiobis-2-nitrobenzoic acid (DTNB, Ellman's reagnt), was purchased from Zwijndrecht (Belgium). Galantamine was used as a reference standard (Sigma Aldrich). Reaction mixture was composed of 180 µL of Tris buffer (50 mM, pH 8.0), 10 µL of enzyme AChE/BChE (concentration in reaction mixture 0.03 U/mL, prepared in 20 mM Tris buffer, pH 7.5), 10 µL of tested compound dissolved in ethanol (concentration in reaction mixture were in a range of 100 nM – 250 µM, depending on solubility) and 10 µL of DTNB (concentration 0.3 mM in reaction mixture, prepared in Tris buffer). The reaction started by addition of 10 µL of substrate ATChI/BTChI (concentration in reaction mixture 0.5 mM prepared in Tris buffer) and developing yellow color was measured at 405 nm over 6 minutes using 96-well microplate reader (Bio Tek 800TSUV Absorbance Reader, Agilent, Santa Clara, CA, USA). Control measurement was the one without inhibitor which was replaced by 10 µL of buffer. For each measurement, non-enzymatic hydrolysis was measured as a blank, where a 10 µL of buffer replaced the enzyme. Percentage enzyme inhibition was calculated from measured data using equation: Inhibition (%) = [(AC – AT)/AC]·100.
AC represents the enzyme activity in control measurement (without inhibitor), AT is the enzyme activity with the test sample. Inhibition data were used to calculate IC50 value by nonlinear fit of inhibitor concentration vs. response. The inhibitory activity of ethanol was also measured, and its contribution to inhibition was substracted. All experiments were run in triplicate. The IC50 value is represented as a mean value of three measurements ± standard deviation.
3.3. Metal-Chelate or -Associate Formation
The absorption spectra to determine the molar absorptivity and to evaluate the complex formation constant were recorded with a Specord S600 spectrophotometer (Analytic Jena GmbH, Jena, Germany) in the 190-900 nm wavelength range. During the titration the ligand concentration was increased at constant Fe(III) content. Also the iron(III) concentration was increased at constant ligand content, but this method proved to be less informative than the other one.
3.4. Computational Study
The geometries of ligands 1 and 7 were prepared for docking study by geometry optimization using the M062X/6-31+G(d,p) model with the Gaussian16 program package [45], followed by adding Gasteiger charges, detecting rotatable bonds, setting torsional degrees of freedom and identifying aromatic carbons. Crystallographic data for BChE, PDB ID: 3DYI, was obtained from the Protein Data Bank [46] and prepared for docking by removing non-amino acid residues, adding polar hydrogens, and assigning Kollman charges. Molecular docking was performed using Autodock4.0 [47], utilizing the Lamarckian Genetic Algorithm, generating 25 docking poses for each ligand and with enzyme residues rigid throughout the process.
The most stable complexes obtained through docking were used as starting structures for the molecular dynamics (MD) study. The protein-ligand complexes were solvated in a truncated octahedron OPC water box and neutralized with Cl⁻ ions using the Amber16 software package [48], applying the ff14SB force field [49] for the protein and the GAFF force field [50] for the ligands. Ligand partial charges were derived using the RESP procedure. Equilibration of all four systems included energy minimizations and short (20 ps) MD simulations, with harmonic restraints gradually reduced to zero and adjustments made to volume and temperature. Target values for temperature and pressure were set to 300 K and 1 atm, respectively. Production MD simulations were then conducted without constraints for 100 nanoseconds under NPT conditions (300 K and 1 atm).
4. Conclusions
Considering all our previous experience in the design of new cholinesterase inhibitors, especially resveratrol analogs, the basic stilbene skeleton was used as a structural unit for new carbamates in this research. Inhibitory activity was tested toward the enzymes acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) by newly prepared carbamates 1–13. In the tested group of compounds, the leading inhibitors were 1 and 7, which achieved excellent selective inhibitory activity for BChE with IC50 values of 0.12 ± 0.09 μM and 0.38 ± 0.01 μM, respectively. Both were much more active than standard inhibitor galantamine against BChE. Molecular docking of the most promising inhibitor candidates, compounds 1 and 7, revealed that stabilizing interactions between the active site residues of BChE and the ligands involve π-stacking, alkyl-π interactions, and, depending on the orientation of the carbamate group, either an H-bond between the ligand's carbonyl oxygen and the catalytic serine or another alkyl-π attraction, this time between the dimethylamino fragment of carbamate group with Trp82 and Tyr330. MD analysis confirmed the stability of the obtained complexes. Based on all obtained in silico, experimental, and computational results on biological activity in the present work, novel carbamates 1–13 represent potential selective BChE inhibitors as new therapeutics for neurological disorders, and they can be incorporated into chemical libraries used in high-throughput screening and drug development. Complex formation studies indicated that some selected representatives of these compounds proved to bind Fe3+ ions, which confirmed their coordination abilities, due to mainly the carboxylic moieties of the carbamate part.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. 1H and 13C NMR spectra of carbamates 1–13 (Figures S1-S50). Mass spectra and HRMS analyses of carbamates 1–13 (Figures S51-S63). Table S1, free energies of binding obtained by docking. Cartesian coordinates of docked ligands and enzyme. Table S2, RMSD, RMSF, and Rg for protein-ligand complexes from MD simulations. Complex formation of biometals with bioactive carbamates 3 and 6 (Figures S64-S65).
Author Contributions
Conceptualization, I.Š. (Irena Škorić); methodology, I.S. (Ivana Šagud) and L.F.; formal analysis, S.R.; investigation, M.S., M.M., I.O. and D.B.; resources, I.O., D.B., O.H. and I.Š. (Irena Škorić); writing—original draft preparation, M.S., M.M., I.O., O.H., L.F., I.S. (Ivana Šagud), D.B. and I.Š. (Irena Škorić); writing—review and editing, all authors; supervision, I.Š. (Irena Škorić); All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministry for Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the 2021 Thematic Excellence Program funding scheme (grant number TKP2021-NKTA-21).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors. The data are not publicly available due to privacy.
Acknowledgments
This work was supported by grants from the University of Zagreb for short-term scientific support for 2024 under the title Synthesis and Biological Activity of New Heteropolycycle Systems. We thank the University of Zagreb (Croatia) Computing Centre (SRCE) for granting computational time on the Supercomputer Supek. The authors thank Davor Margetić, Ph.D., for kindly providing the initial carbamoyl reagent for the synthesis and Kornelija Lasić, Ph.D. for kindly performing HRMS analyses.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer's disease: causes and treatment. Molecules 2020, 25, e5789. [Google Scholar] [CrossRef]
- Lyness, S.A.; Zarow, C.; Chui, H.C. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol. Aging 2003, 24, 1–23. [Google Scholar] [CrossRef]
- Alzheimer's Association, 2021 Alzheimer's disease facts and figures. Alzheimer. Dement. 2021, 17, 321–387. [CrossRef]
- Szeto, J.Y.Y.; Lewis, S.J.G. Current treatment options for Alzheimer's disease and Parkinson's disease dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef]
- Sharma, M. Preventing Alzheimer's disease. J. Am. Coll. Cardiol. 2019, 74, 1924–1925. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer's disease. Transl. Neurodegener. 2020, 9, e10. [Google Scholar] [CrossRef]
- Dos Santos Picançoa, L.C.; Ozelaa, P.F.; de Brito Brito, M.d.F.; Pinheiroa, A.A.; Padilhab, E.C.; Bragac, F.S.; Tomich de Paula da Silvad, C.D.; Dos Santos, C.B.; Rosad, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer's disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Anand, R.; Veach, J. A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer's disease. Int. J. Geriatr. Psychopharmacol. 1998, 1, 55–65. [Google Scholar]
- Müller, T. Rivastigmine in the treatment of patients with Alzheimer's disease. Neuropsychiatr. Dis. Treat. 2007, 3, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Rotenberg, M.; Almog, S. Evaluation of the decarbamylation process of cholinesterase during assay of enzyme activity. Clin. Chim. Acta 1995, 240, 107–116. [Google Scholar] [CrossRef]
- Hofer, P.; Fringeli, U.P. Acetylcholinesterase kinetics. Biophys. Struct. Mech. 1981, 8, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Reiner, E. Spontaneous reactivation of phosphorylated and carbamaylated cholinesterases. Bull. Wld. Hlth. Org. 1971, 44, 109–112. [Google Scholar]
- Mahase, E. Aducanumab: European agency rejects Alzheimer's drug over efficacy and safety concerns. BMJ 2021, 375, e3127. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Bartolini, M.; Cavalli, A.; Andrisano, V.; Rosini, M.; Minarini, A.; Melchiorre, C. Design, synthesis, and biological evaluation of conformationally restricted rivastigmine analogues. J. Med. Chem. 2004, 47, 5945–5952. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; Toth, G.; Zagyva, A.; Zekany, A.; Finberg, J.; Lavian, G.; Gross, A.; Friedman, R.; Razin, M.; Huang, W.; Krais, B.; Chorev, M.; Youdim, M.B.; Weinstock, M. Novel dual inhibitors of AChE and MAO derived from hydroxy aminoindan and henethylamine as potential treatment for Alzheimer's disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Ucar, G.; Gokhan, N.; Yesilada, A.; Bilgin, A.A. 1-N-Substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: a novel cholinesterase and selective monoamine oxidase B nhibitors for the treatment of Parkinson's and Alzheimer's diseases. Neurosci. Lett. 2005, 38, 327–331. [Google Scholar] [CrossRef]
- Toda, N.; Tago, K.; Marumoto, S.; Takami, K.; Ori, M.; Yamada, N.; Koyama, K.; Naruto, S.; Abe, K.; Yamazaki, R.; Hara, T.; Aoyagi, A.; Abe, Y.; Kaneko, T.; Kogen, H. A conformational restriction approach to the development of dual inhibitors of acetylcholinesterase and serotonin transporter as potential agents for Alzheimer's disease. Bioorg. Med. Chem. 2003, 11, 4389–4415. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Štěpánková, Š.; Vorčáková, K.; Švarcová, M.; Vinšová, J. Novel cholinesterase inhibitors based on O-aromatic N,N-disubstituted carbamates and thiocarbamates. Molecules 2016, 21, e21020191. [Google Scholar] [CrossRef] [PubMed]
- Groner, E.; Ashani, Y.; Schorer-Apelbaum, D.; Sterling, J.; Herzig, Y.; Weinstock, M. The kinetics of inhibition of human acetylcholinesterase and butyrylcholinesterase by two series of novel carbamates. Molec. Pharmacol. 2007, 71, 1610–1617. [Google Scholar] [CrossRef]
- Košak, U.; Strašek, N.; Knez, D.; Jukič, M.; Žakelj, S.; Zahirović, A.; Pišlar, A.; Brazzolotto, X.; Nachon, F.; Kos, J.; Gobec, S. N-alkylpiperidine carbamate as potential anti- Alzheimer`s agensts. Eur. J. Med. Chem. 2020, 197, 112282. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Đurčević, E.; Odak, I.; Barić, D.; Juričević, I.; Šagud, I.; Burčul, F.; Lasić, Z.; Marinić, Ž.; Škorić, I. Thieno-thiazolostilbenes, thienobenzo-thiazoles, and naphtho-oxazoles: Computational study and cholinesterase inhibitory activity. Molecules 2023, 28, 3781. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Faraho, I.; Odak, I.; Kovačević, B.; Raspudić, A.; Šagud, I.; Bosnar, M.; Škorić, I.; Barić, D. Cholinesterase inhibitory and anti-inflammatory activity of the naphtho- and thienobenzo-triazole photoproducts: Experimental and computational study. Int. J. Mol. Sci. 2023, 24, 14676. [Google Scholar] [CrossRef]
- Mlakić, M.; Selec, I.; Ćaleta, I.; Odak, I.; Barić, D.; Ratković, A.; Molčanov, K.; Škorić, I. New thienobenzo/naphtho-triazoles as butyrylcholinesterase inhibitors. Design, synthesis and computational study. Int. J. Mol. Sci. 2023, 24, 5879. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Odak, I.; Faraho, I.; Talić, S.; Bosnar, M.; Lasić, K.; Barić, D.; Škorić, I. New naphtho/thienobenzo-triazoles with interconnected anti-inflammatory and cholinesterase inhibitory activity. Eur. J. Med. Chem. 2022, 241, 114616. [Google Scholar] [CrossRef]
- Mlakić, M.; Faraho, I.; Odak, I.; Talić, S.; Vukovinski, A.; Raspudić, A.; Bosnar, M.; Zadravec, R.; Ratković, A.; Lasić, K.; Marinić, Ž.; Barić, D.; Škorić, I. Synthesis, photochemistry and computational study of novel 1,2,3-triazole heterostilbenes: Expressed biological activity of their electrocyclization photoproducts. Bioorg. Chem. 2022, 121, 105701. [Google Scholar] [CrossRef] [PubMed]
- Modrić, M.; Božičević, M.; Faraho, I.; Bosnar, M.; Škorić, I. Design, synthesis and biological evaluation of new 1,3-thiazole derivatives as potential anti-inflammatory agents. J. Mol. Struct. 2021, 1239, 130526. [Google Scholar] [CrossRef]
- Mlakić, M.; Odak, I.; Barić, D.; Talić, S.; Šagud, I.; Štefanić, Z.; Molčanov, K.; Lasić, Z.; Kovačević, B.; Škorić, I. New resveratrol analogs as improved biologically active structures: design, synthesis and computational modeling. Bioorg. Chem. 2024, 143, 106965. [Google Scholar] [CrossRef]
- Mlakić, M.; Rajić, L.; Ljubić, A.; Vušak, V.; Zelić, B.; Gojun, M.; Odak, I.; Čule, I.; Šagud, I.; Šalić, A.; Škorić, I. Synthesis of new heterocyclic resveratrol analogues in milli- and microreactors: Intensification of the Wittig reaction. J. Flow Chem. 2022, 12, 429–440. [Google Scholar] [CrossRef]
- Mlakić, M.; Talić, S.; Odak, I.; Barić, D.; Šagud, I.; Škorić, I. Cholinesterase Inhibition and Antioxidative Capacity of New Heteroaromatic Resveratrol Analogs: Synthesis and Physico—Chemical Properties. Int. J. Mol. Sci. 2024, 25, 7401. [Google Scholar] [CrossRef]
- E. Giacobini, R. Spiegel, A. Enz, A. E. Veroff i N. R. Cutler, Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer's disease by rivastigmine: correlation with cognitive benefit. J. Neural. Transm. 2002, 109, 1053–1065. [CrossRef] [PubMed]
- Y. Zhou, S. Wang i Y. Zhang, Catalytic reaction mechanism of acetylcholinesterase determined by Born-Oppenheimer ab initio QM/MM molecular dynamics simulations. J. Phys. Chem. B. 2010, 114, 8817–8825. [CrossRef] [PubMed]
- Ellman, G.L.; Courtnex, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Matošević, A.; Knežević, A.; Zandona, A.; Maraković, N.; Kovarik, Z.; Bosak, A. Design, synthesis and biological evaluation of biscarbamates as potential selective butyrylcholinesterase inhibitors for the treatment of Alzheimer's disease. Pharmaceuticals 2022, 15, 1220. [Google Scholar] [CrossRef] [PubMed]
- Matošević, A.; Radman Kastelic, A.; Mikelić, A.; Zandona, A.; Katalinić, M.; Primožič, I.; Bosak, A.; Hrenar, T. Quinuclidine-Based Carbamates as Potential CNS Active Compounds. Pharmaceutics 2021, 13, 420. [Google Scholar] [CrossRef]
- Tunek, A.; Svensson, L.A. Bambuterol, a carbamate ester prodrug of terbutaline, as inhibitor of cholinesterases in human blood. Drug Metab. Dispos. 1988, 16, 759–764. [Google Scholar]
- Kamal, M.A.; Qu, X.; Yu, Q.S.; Tweedie, D.; Halloway, H.W.; Li, Y.; Tan, Y.; Greig, N.H. Tetrahydrofurobenzofuran cymserine, a potent butyrylcholinesterase inhibitor and experimental Alzheimer drug candidate, enzyme kinetic analysis. J. Neural. Transm. 2008, 115, 889–898. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development, 2nd Ed.; Faqi, A.S., Ed.; Academic Press: Cambridge, USA, 2017; pp. 39–67. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Available online: https://ai-druglab.smu.edu/admetresult (accessed on 3 November 2024).
- Mlakić, M.; Fodor, L.; Odak, I.; Horváth, O.; Lovrić, M.J.; Barić, D.; Milašinović, V.; Molčanov, K.; Marinić, Ž.; Lasić, Z.; et al. Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure. Molecules 2022, 27, 6379. [Google Scholar] [CrossRef]
- Hasselgren, C.; Bercu, J.; Cayley, A.; Cross, K.; Glowienke, S.; Kruhlak, N.; Muster, W.; Nicolette, J.; Reddy, M.V.; Saiakhov, R.; Dobo, K. Management of pharmaceutical ICH M7 (Q)SAR predictions – The impact of model updates. Regulatory Toxicology and Pharmacology 2020, 118, 104807. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Carletti, E.; Li, H.; Li, B.; Ekstrom, F.; Nicolet, Y.; Loiodice, M.; Gillon, E.; Froment, M.T.; Lockridge, O.; Schopfer, L.M.; Masson, P.; Nachon, F. Nonaged Form of Human Butyrylcholinesterase Inhibited by Tabun. J. Am. Chem. Soc. 2008, 130, 16011–16020. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDock-823 Tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Case, D. A.; Ben-Shalom, I. Y.; Brozell, S. R.; Cerutti, D. S.; Cheatham, III; T. E.; Cruzeiro, V. W. D.; Darden, T. A.; Duke, R. E.; Ghoreishi, D.; Gilson, M. Ket al. (2016), AMBER 2016, University of California, San Francisco.
- Maier, J. A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K. E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. A.; Case, D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
Scheme 1.
Newly designed and synthesized carbamates 1–13.
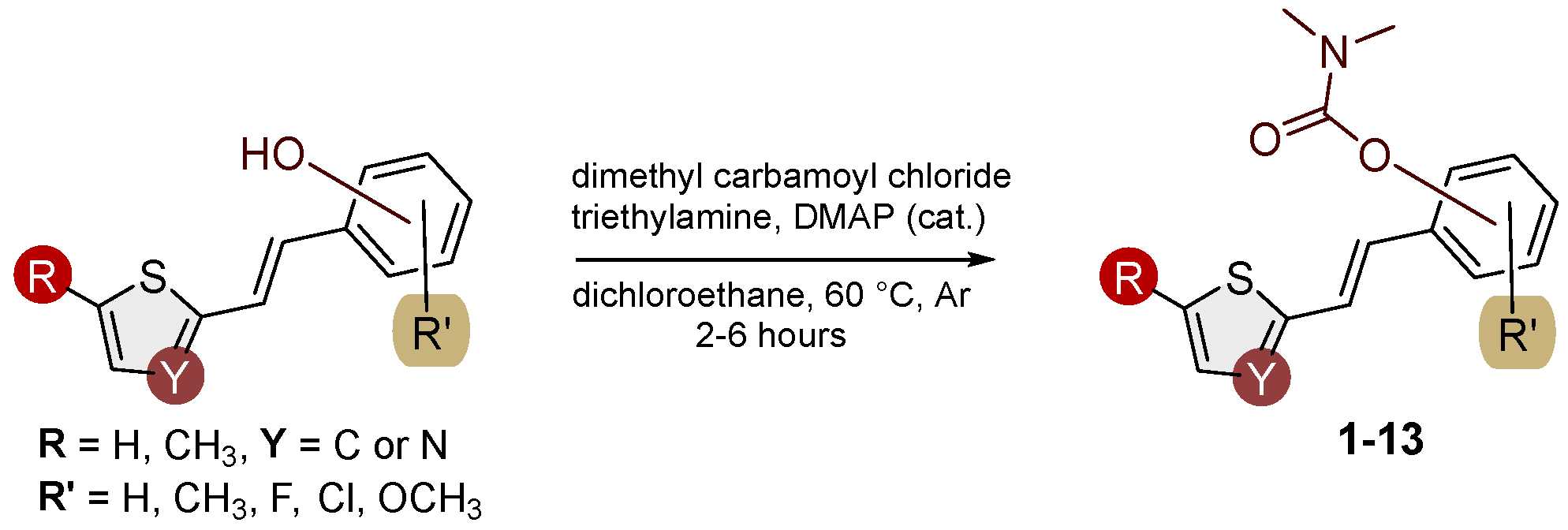
Figure 2.
New synthesized carbamates 1–13 bearing various functionalities (isolated yields for individual compounds are shown in parentheses).
Figure 2.
New synthesized carbamates 1–13 bearing various functionalities (isolated yields for individual compounds are shown in parentheses).
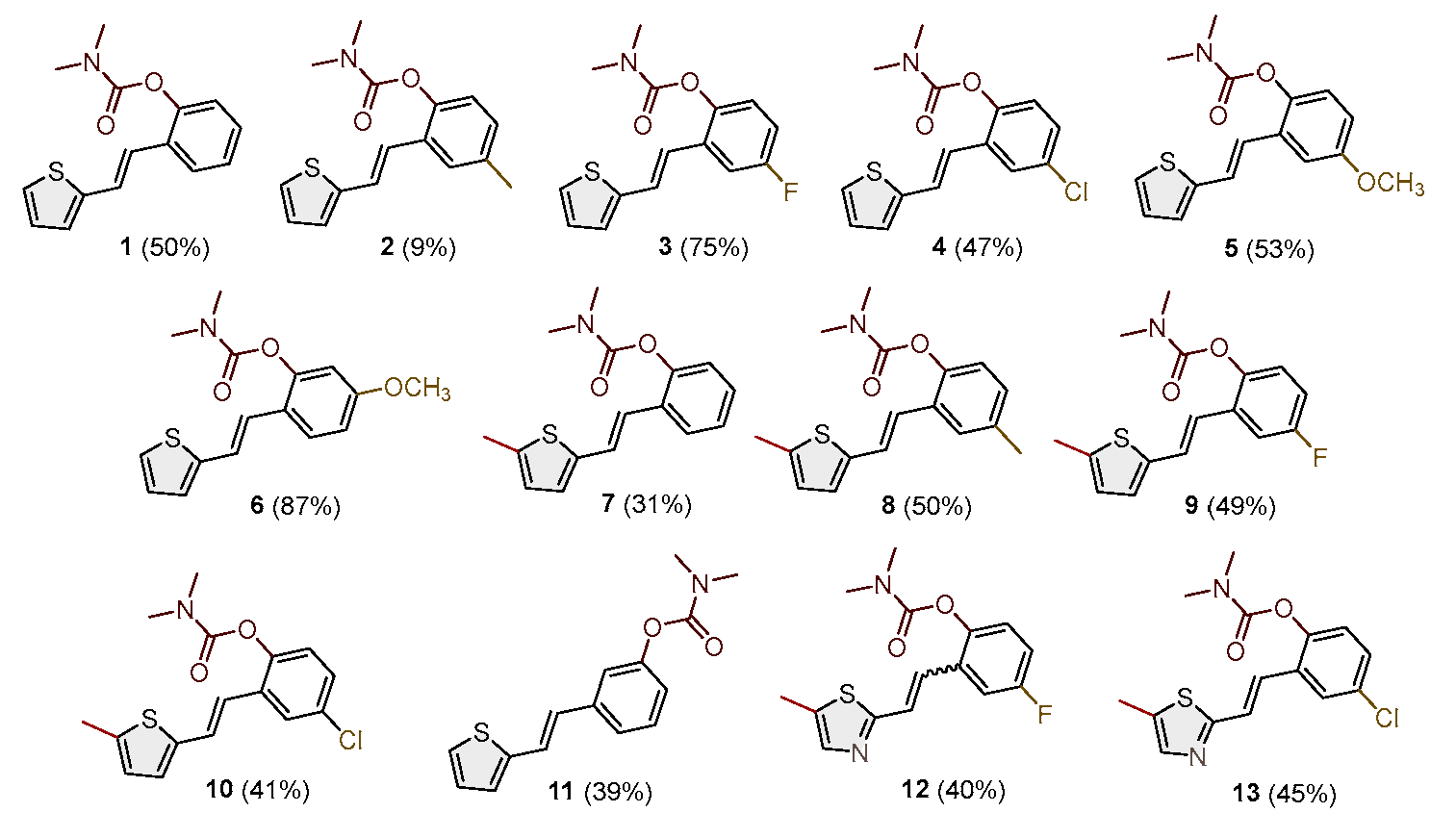
Figure 3.
The parts of the 1H NMR spectra of the most active carbamates as selective BChE inhibitors were compared: (a) 1, (b) 5, and (c) 8.
Figure 3.
The parts of the 1H NMR spectra of the most active carbamates as selective BChE inhibitors were compared: (a) 1, (b) 5, and (c) 8.
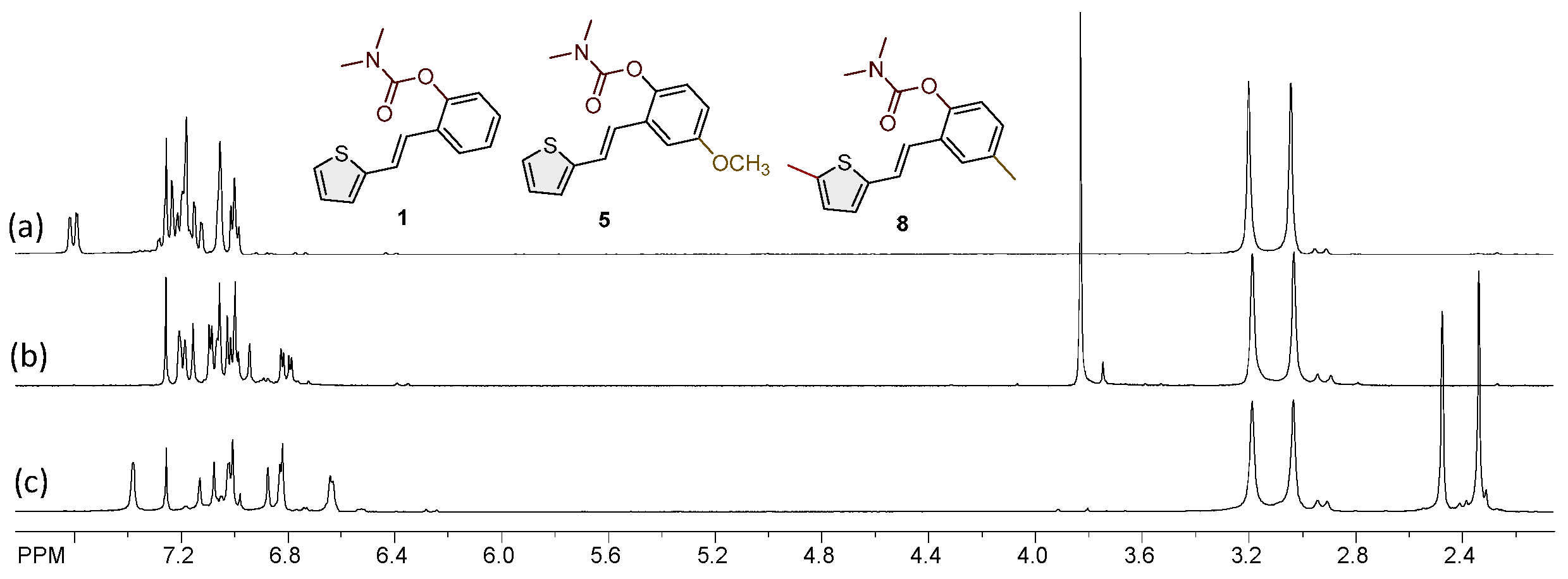
Scheme 2.
Possible mechanism of action of carbamate 1 in the enzyme's active site by analogy with acetylcholine and rivastigmine.
Scheme 2.
Possible mechanism of action of carbamate 1 in the enzyme's active site by analogy with acetylcholine and rivastigmine.
Figure 4.
Structure of bambuterol.
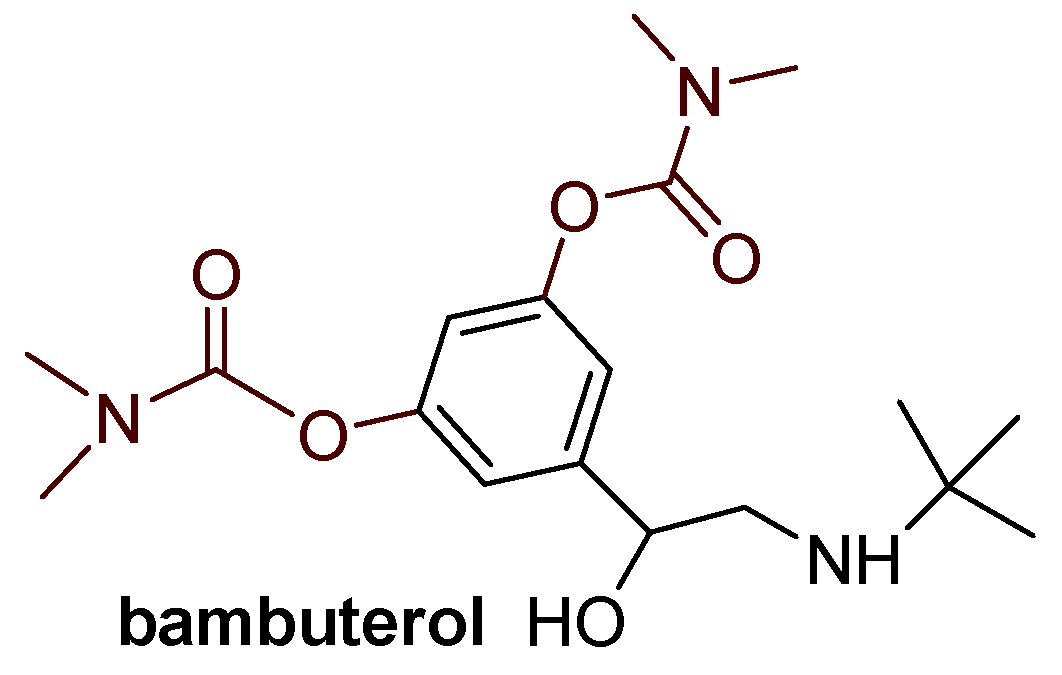
Figure 5.
(a) Molecule 1 docked into the active site of BChE; (b) structure of the active site of BChE docked by molecule 7. All distances in angstroms. Docked molecules are presented using a ball-and-stick model.
Figure 5.
(a) Molecule 1 docked into the active site of BChE; (b) structure of the active site of BChE docked by molecule 7. All distances in angstroms. Docked molecules are presented using a ball-and-stick model.
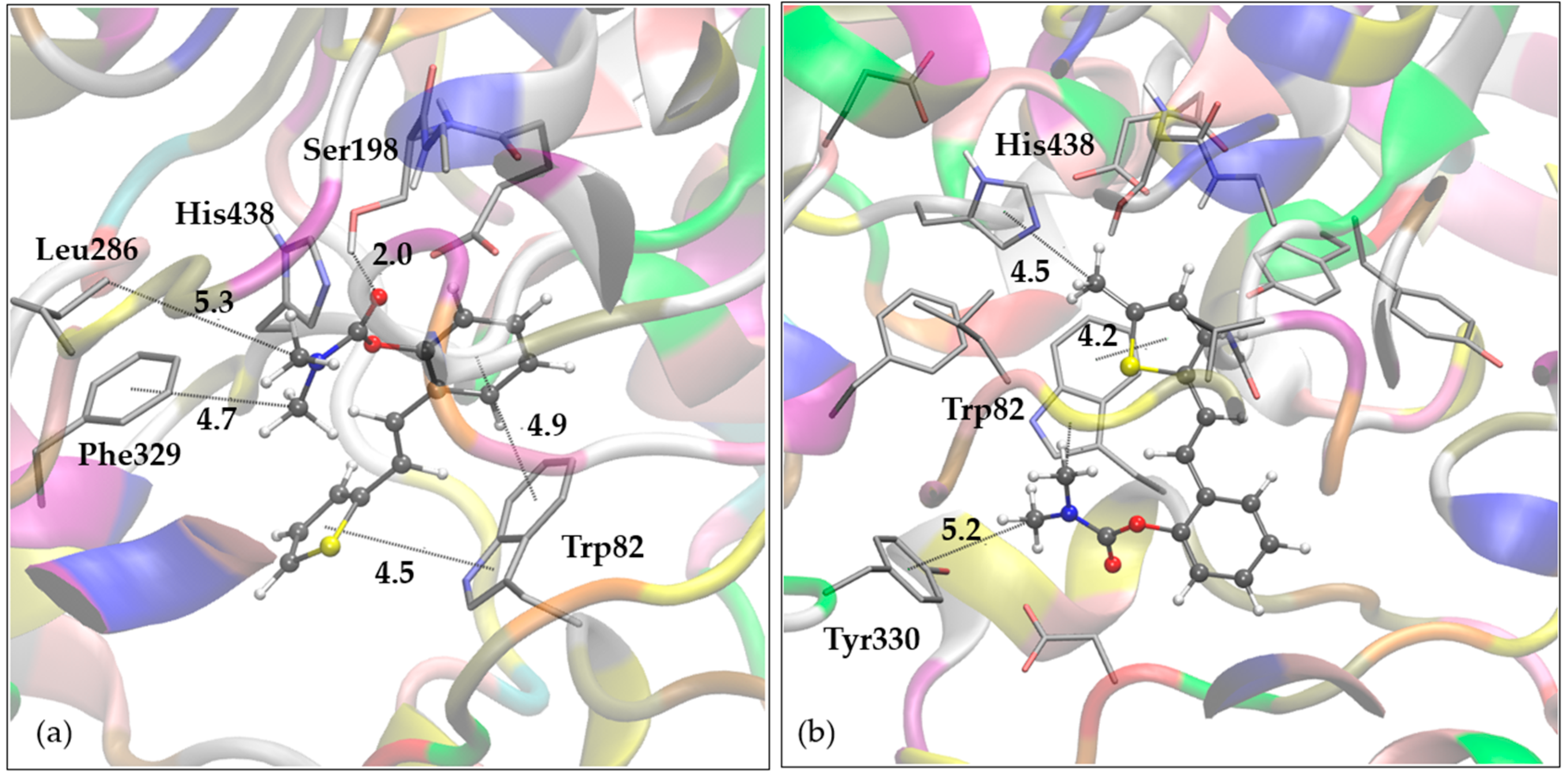
Figure 6.
Root-mean-square deviation values from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 and 7, respectively.
Figure 6.
Root-mean-square deviation values from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 and 7, respectively.
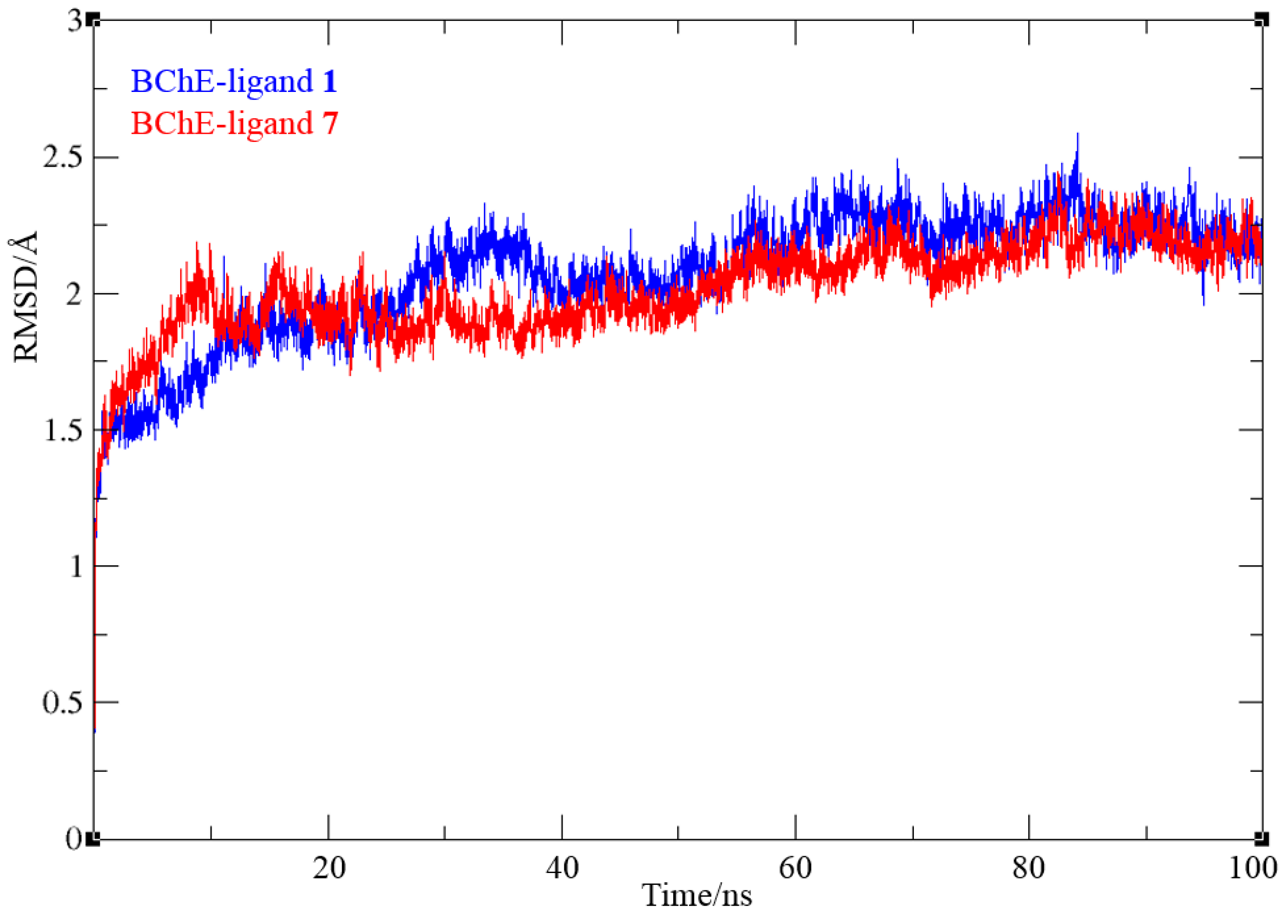
Figure 7.
Root-mean-square fluctuation values from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 (blue) and 7 (red line), respectively.
Figure 7.
Root-mean-square fluctuation values from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 (blue) and 7 (red line), respectively.
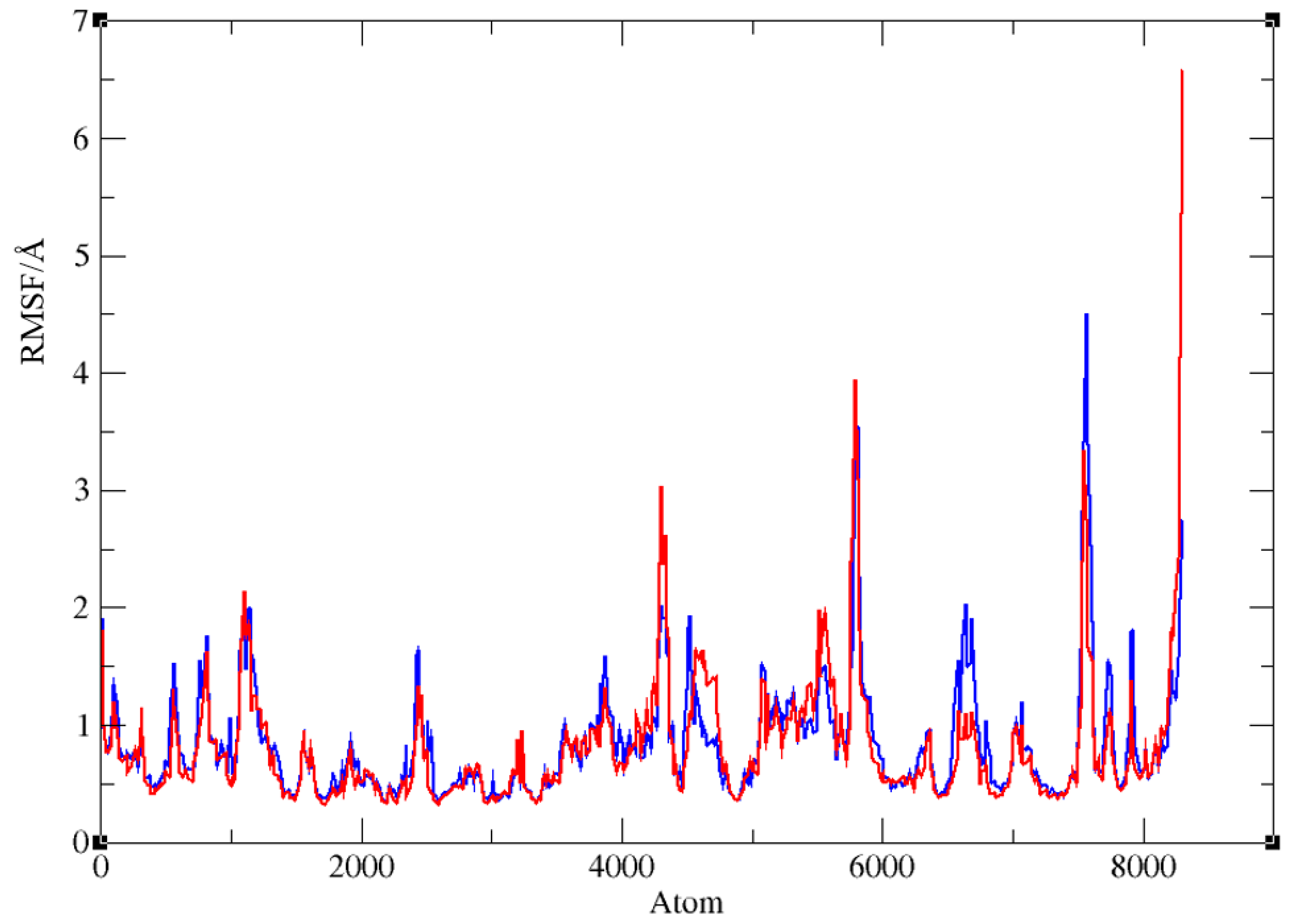
Figure 8.
Values of radius of gyration from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 (blue) and 7 (red line), respectively.
Figure 8.
Values of radius of gyration from molecular dynamics simulation of a protein-ligand complex of BChE with compounds 1 (blue) and 7 (red line), respectively.
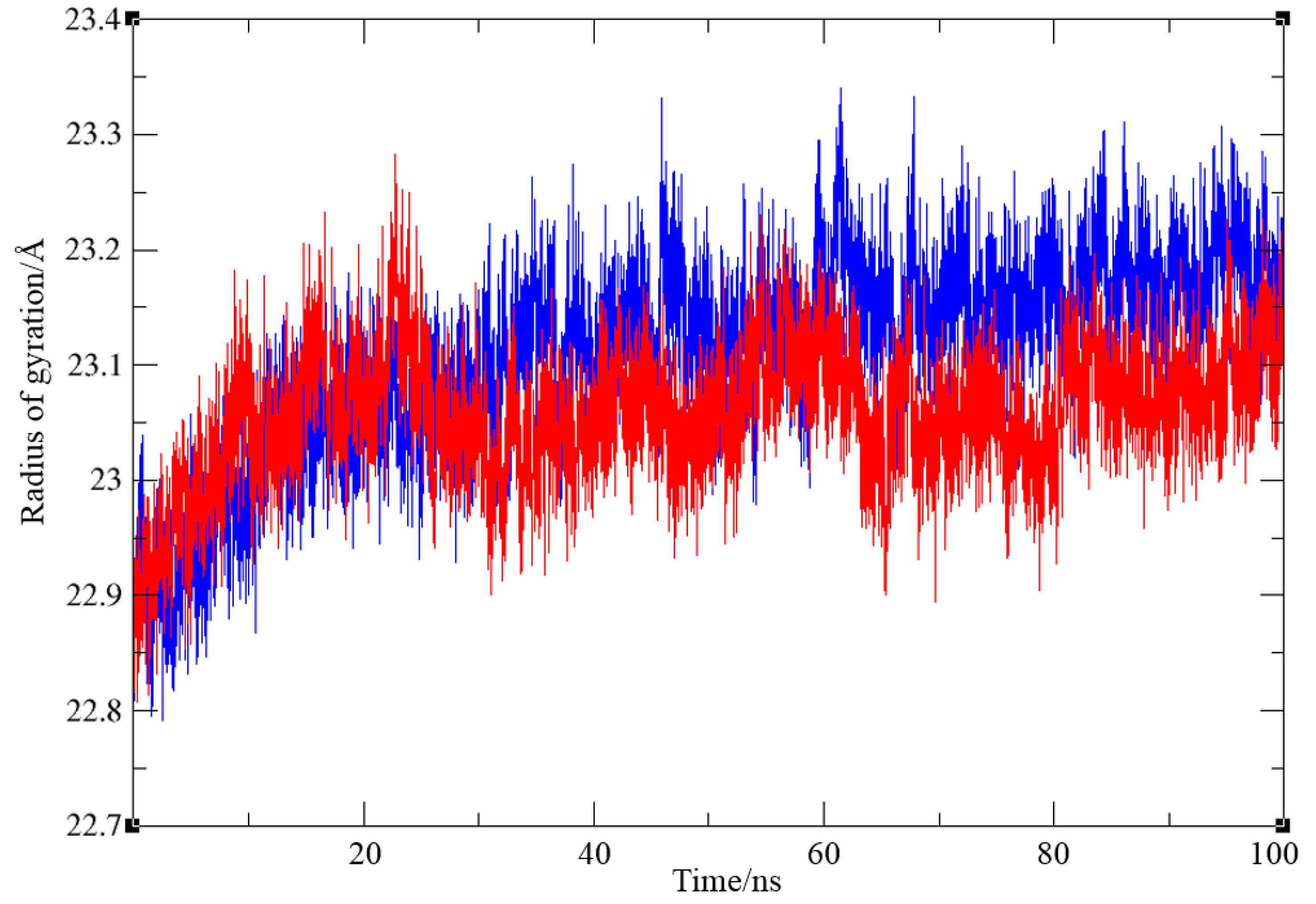
Figure 9.
Molar absorptivity spectra of compound 1 (black), 3 (blue), 6 (green), and 7 (red).
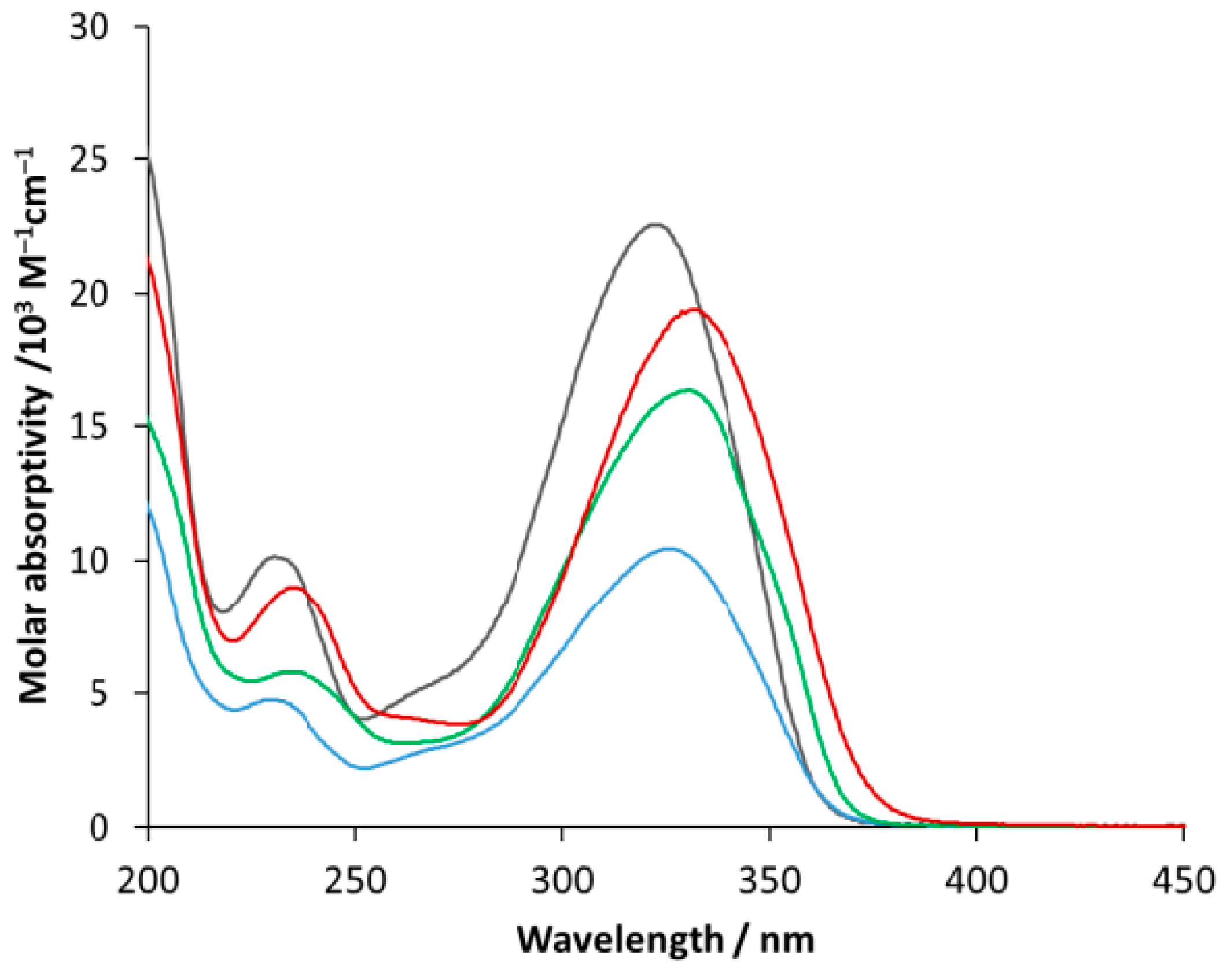
Figure 10.
Difference absorption spectra obtained by titration of 1.93×10–5 M of Fe2(SO4)3 aqueous solution with compound 6 in the 0−0.35 mM concentration range. The orange curve is the spectrum of the starting pure Fe2(SO4)3 solution, while the green spectrum belongs to the pure ligand. The inset shows the changes of absorbance measured at 330 nm (blue) and the theoretical absorbance calculated for the ligand at the same wavelength (red) as a function of the concentration of 6.
Figure 10.
Difference absorption spectra obtained by titration of 1.93×10–5 M of Fe2(SO4)3 aqueous solution with compound 6 in the 0−0.35 mM concentration range. The orange curve is the spectrum of the starting pure Fe2(SO4)3 solution, while the green spectrum belongs to the pure ligand. The inset shows the changes of absorbance measured at 330 nm (blue) and the theoretical absorbance calculated for the ligand at the same wavelength (red) as a function of the concentration of 6.
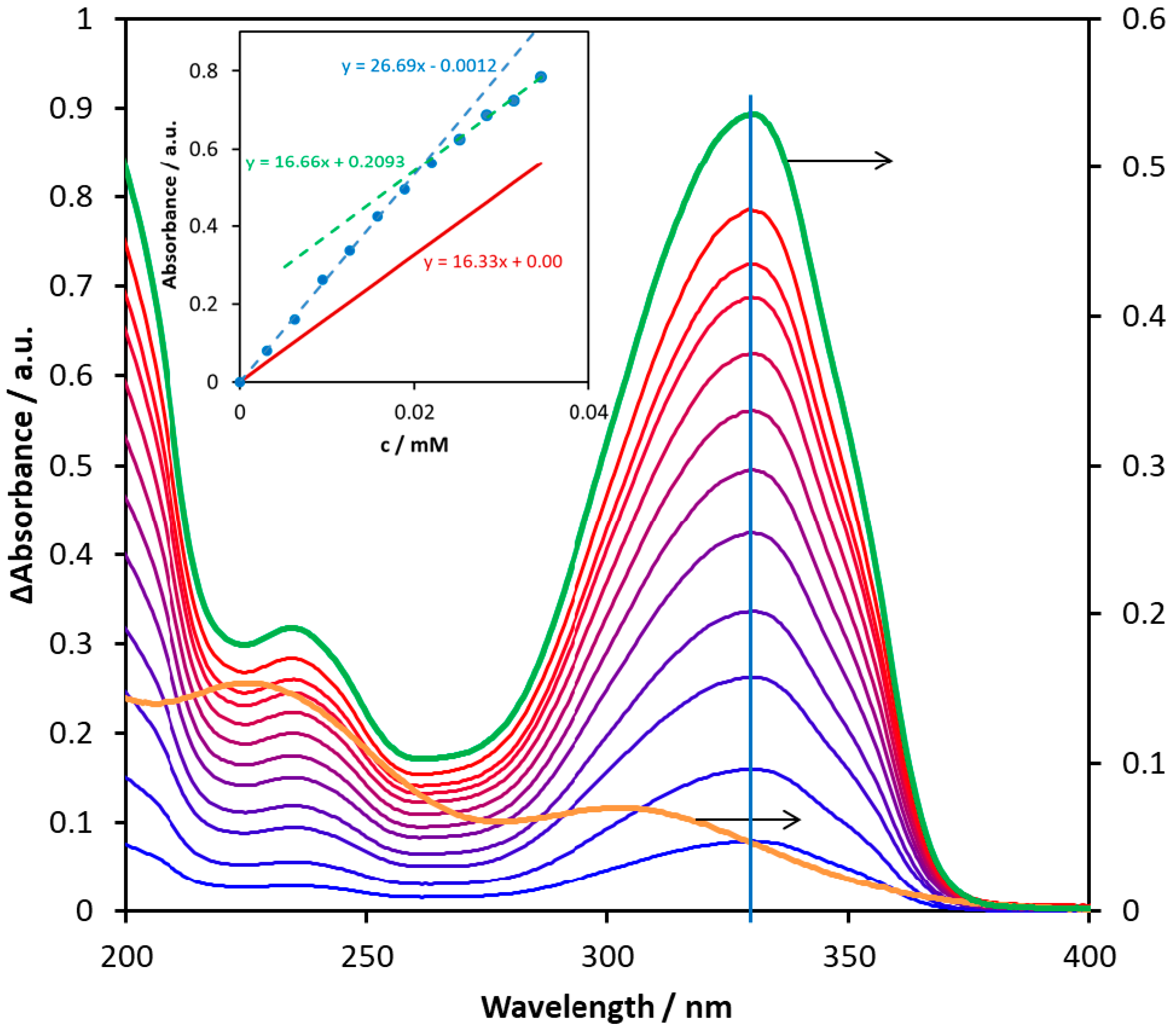
Table 1.
Cholinesterase inhibition (AChE and BChE) for carbamate derivatives 1–13.
| Carbamate derivative | AChE Inhibition* (%) |
AChE IC50 (μM) |
BChE Inhibition* (%) |
BChE IC50 (μM) |
|---|---|---|---|---|
| 1 | 30.0 (100) | - | 85.0 (50) | 0.12 ± 0.09 |
| 2 | 16.7 (100) | - | 85.0 (10) | 1.05 ± 0.09 |
| 3 | 12.6 (100) | - | 85.0 (100) | 1.48 ± 0.36 |
| 4 | 35.0 (100) | - | 85.3 (25) | 2.83 ± 0.03 |
| 5 | 11.0 (100) | - | 84.0 (100) | 11.59 ± 0.18 |
| 6 | 1.0 (100) | - | 84.4 (25) | 0.90 ± 0.14 |
| 7 | 17.7 (100) | - | 86.3 (25) | 0.38 ± 0.01 |
| 8 | 25.2 (100) | - | 72.5 (100) | 25.50 ± 0.21 |
| 9 | 23.6 (100) | - | 85.2 (100) | 15.05 ± 2.32 |
| 10 | 27.0 (100) | - | 85.7 (100) | 23.17 ± 3.78 |
| 11 | 19.0 (100) | - | 83.4 (100) | 11.37 ± 2.48 |
| 12 | 23.9 (100) | - | 81.4 (100) | 23.39 ± 2.43 |
| 13 | 37.5 (100) | - | 84.1 (50) | 4.03 ± 0.50 |
| Galantamine | 90.0 (4.5) | 0.15 ± 0.07 | 90.1 (25) | 7.54 ± 1.64 |
* The Numbers shown in parentheses represent maximal concentrations tested in μM.
Table 2.
In silico study for more indicators of absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) in the human body of carbamates 1 and 7.
Table 2.
In silico study for more indicators of absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) in the human body of carbamates 1 and 7.
| Property | Model Name | 1 | 7 | bambuterol | rivastigmine | Unit |
|---|---|---|---|---|---|---|
| Absorption | Water solubility | -4.1 | -4.43 | log mol/L | ||
| Caco2 permeability | 1.911 | 1.843 | log Papp in 10-6 cm/s | |||
| Intestinal absorption | 90.749 | 91.133 | % Absorbed | |||
| Skin Permeability | -2.687 | -2.689 | log Kp | |||
| P-glycoprotein substrate | No | No | ||||
| P-glycoprotein I inhibitor | No | No | ||||
| P-glycoprotein II inhibitor |
No | No | ||||
| Distribution | VDss (human) | 0.313 | 0.412 | 0.401 | 0.451 | log L/kg |
| Fraction unbound (human) |
0.033 | 0.023 | - | - | Fu | |
| BBB permeability | 0.31 | 0.322 | 0.707 | 0.968 | log BB | |
| CNS permeability | -0.941 | -0.955 | -1.95 | -0.801 | log PS | |
| Metabolism | CYP2D6 substrate | No | No | |||
| CYP3A4 substrate | Yes | Yes | ||||
| CYP1A2 inhibitior | Yes | Yes | ||||
| CYP2C19 inhibitior | Yes | Yes | ||||
| CYP2C9 inhibitior | Yes | No | ||||
| CYP2D6 inhibitior | No | No | ||||
| CYP3A4 inhibitior | No | No | ||||
| Excretion | Total Clearance | 0.167 | 0.111 | log ml/min/kg | ||
| Renal OCT2 substrate | No | No | Yes/No | |||
| Toxicity | AMES toxicity | No | No | Yes/No | ||
| Max. tolerated dose (human) |
0.245 | 0.166 | log mg/kg/day | |||
| hERG I inhibitor | No | No | ||||
| hERG II inhibitor | No | Yes | ||||
| Oral Rat Acute Toxicity (LD50) | 2.579 | 2.618 | mol/kg | |||
| Oral Rat Chronic Toxicity (LOAEL) | 1.874 | 1.855 | log mg/kg_bw/day | |||
| Hepatotoxicity | No | Yes | ||||
| Skin Sensitisation | No | No | ||||
| T.Pyriformis toxicity | 1.785 | 1.927 | log ug/L | |||
| Minnow toxicity | 0.379 | -0.096 | log mM |
Table 3.
Molar absorptivities at the longer-wavelength band maxima of the selected compounds.
| Compound | λ / nm | ε / M–1cm–1 |
|---|---|---|
| 1 | 322 | 22570 |
| 3 | 325 | 10440 |
| 6 | 330 | 16330 |
| 7 | 331 | 19410 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



