Submitted:
28 April 2025
Posted:
29 April 2025
You are already at the latest version
Abstract
The development of large language models (LLMs) has grown exponentially since the release of ChatGPT. Large language models have gained attention for their robust performance across various tasks. The ability of LLMs to understand and produce general-purpose language is achieved by training billions of parameters. These models have emerged as a transformative force in increasing natural language understanding, representing an important step toward general artificial intelligence(AI). LLMs have become powerful tools for various tasks, including natural language processing (NLP), machine translation(MT), vision applications, and question-answering(QA). The expanded reach of LLMs goes beyond the conventional linguistic bounds and includes specialized languages created in different scientific disciplines. The intensification of interest in this new subclass of scientifically oriented LLMs has led to the birth of the scientific LLMs. These scientific LLMs are gradually gaining a foothold as an exciting research area for science study. Theoretically, they share a structure in common with general LLMs. In practice, however, they differ regarding input and usage. This paper undertakes an exhaustive effort to study all the scientific LLMs, the types of structures offered, the datasets, the parameters, and the context of use. Our analysis uses a focused lens that focuses on the biological and chemical domains, which enables an in-depth examination of LLMs for textual knowledge, small molecules, macromolecules, proteins, genomic sequences, and combinations. By providing an overview of the technical advances in the field, this survey is a valuable resource for researchers navigating the complex landscape of scientific LLMs.
Keywords:
large language models
; biological data
; natural language processing
; pre-trained language models
Introduction
The introduction of the Turing test [1] in the 1950s led humans to discover machines’ dominance of linguistic intelligence. Language is a complex communicative system of human utterances governed by grammatical rules called language structures. Language modelling is challenging to develop artificial intelligence (AI) algorithms capable of understanding due to its non-numerical and non-mathematical structure [2]. Language modelling has been studied widely for the last two decades for the understanding, representation and production of language, evolving as one would expect, from statistical language models (SLM)[3] to neural language models. The idea of pre-trained language models (PLMs)[4] emerged from pre-training transformer models on large-scale datasets, making them quite effective in performing various tasks pertaining to natural language processing (NLP). As the researchers find that model scaling increases model capacity, they test the scaling effect further by increasing the parameter scale to an even larger size. Interestingly, when the parameter scale exceeds a certain level, these scaled-up language models achieve significant performance improvements and provide some special capabilities (e.g., in context learning) that are not available in small-scale language models (e.g., BERT). To distinguish language models at different parameter scales, the research community has coined the term LLM for PLMs of significant size (e.g., containing tens or hundreds of billions of parameters). Recently, research on LLM has advanced dramatically in both academia and industry. The technical evolution of LLM has had a significant impact on the entire AI community, revolutionizing existing methods and making the use of AI methods more efficient[2].
Large language models (LLMs) are considered advanced tools in natural language processing and global knowledge gathering. Typically, LLMs refer to transformer-based architectures with hundreds of millions (or even billions) of trainable parameters trained on a large text set [5]. These models have been presented in various data structures and variations; typical examples include GPT-3 [6], PalM [7], Galactica[8], LLaMA [9], ChatGLM [10] and Baichuan2 [11]. In addition to natural languages, a set of scientific languages has been developed to incorporate more specialized scientific knowledge. These language models include text expressions in scientific research domains, mathematical languages for defining mathematical formulas, and chemical languages. These models can also include molecular structures and biological languages that describe proteins or genomes and detail complex structures.
Like in natural language, each concept and term is given a distinct vocabulary. In scientific languages, each concept and term is given a distinct vocabulary. For example, the character "C" in English represents the amino acid Cystine in protein languages [12], while in chemistry, the symbol C represents carbon with atomic number 6 [13]. In addition, experts in specific fields create grammatical rules to organize these terms, allowing for the construction of sentences with more precise semantic functions. The world of general LLMs can be limited to natural languages, which are presented in English, Chinese, etc. Researchers have invented large scientific language models (Sci-LLMs) customized for different scientific fields and disciplines to facilitate the understanding of scientific languages.
Background and Motivation
Language is a prominent human ability to express and communicate, which develops in early childhood and evolves throughout life. However, machines cannot naturally grasp the ability to understand and communicate in human language unless equipped with powerful artificial intelligence (AI) algorithms. Achieving this goal has been a long-standing research challenge that enables machines to read, write, and communicate like humans [1]. Technically, language modelling (LM) is one of the main approaches to advancing machine language intelligence. In general, LM aims to model the probability of generating word sequences to predict the probabilities of future (or missing) tokens. The way text sequences are written is represented by tokens, which can be viewed as a sequence of discrete observations of words or characters. Suppose that they are presented in a text sequence of length. Language objectives Model Estimate the joint probability of the entire sequence [14]:
LM generally aims to draw tokens at a time, which can be expressed as . LM is the basis of various NLP tasks. Early NLP systems were essentially based on hand-written rules, which were time-consuming and laborious and could not cover various linguistic phenomena. In the 1980s, statistical LMs were proposed to assign probabilities to a sequence of N tokens, e.g.
Where denotes the word i in the sequence s, the probability of a sequence of words can be divided by the product of the conditional probability of the next word given its predecessors, commonly called the context history or context. The following forms can be used to calculate this probability:
- Unigram model:
- Bigram model:
- Trigram model:
- N-gram model:
A (-gram model is derived from the Markov order k assumption. This assumption states that the current state depends only on the k previous states, that is[15]:
N-gram LM is a classical, statistical language model based on counting the occurrences of n-grams in a corpus of text. In its simplest form, the probability of a token with a context is estimated as:
where is the number of times the n-gram w appears in the training data D (i.e., a corpus), and n is a predefined meta-parameter [15].
Typically, n-gram LMs are implemented by constructing an n-gram count table from the training data. This table stores all the unique n-grams in the training data, each associated with its count. Such n-gram count tables are large and grow almost exponentially. As a result, previous n-gram LMs are limited to tiny n, typically , and only frequent n-grams. An n-gram LM is defined as follows[15]:
are all the tokens before in the document and
LM research has received widespread attention in the literature, which can be divided into four categories:
- Statistical language models (SLM): SLMs [16,17,18] are developed based on statistical learning methods proposed in the 1990s. The main idea is to build a word prediction model based on the Markov hypothesis. Bigram and trigram language models SLMs have been widely used to improve task performance in information retrieval (IR)[19] and natural language processing (NLP)[20]. These models often suffer from the curse of dimensionality. Also, accurate estimation of language models is difficult due to the many transition probabilities that need to be estimated.
- Neural language models (NLM): NLMs [21,22] characterize the probability of word sequences by neural networks, for example, multilayer perceptron (MLP) and recurrent neural network (RNN). In these models, neural networks try to learn feature selection and representation by gradient. Various approaches such as Word2vec, Glove, Fasttext, and Bert have been proposed for learning distributed word representations, which have been very effective in various NLP tasks.
- Pre-trained language models (PLM): These are neural networks trained on the large-scale unlabeled corpus, from which various downstream tasks can be further tuned. One of the first models presented in this category is ELMo [23]. This model captures context-aware word representations by pre-training a bi-directional LSTM (biLSTM) network (instead of learning fixed word representations) and then fine-tuning the biLSTM network. Other models have been developed based on this idea, the most important of which are GPT-2[24] and BART [25].
- Large language models (LLM):. Researchers use the term LLM for large PLMs. They find that scaling PLMs often leads to improved model capacity on downstream tasks (i.e., following the scaling law [26]). One notable application of LLMs is ChatGPT2, which adapts the GPT series LLMs for conversation, offering the ability to converse with humans.
Search Method
In the process of selecting relevant articles, a search was conducted in reputable databases, including Google Scholar, Scopus, arXiv, and bioRxiv. For each category, Boolean query terms were used (according to Table 1). For example, for the section on Large Language Models for Protein Sequence Representation (LLMs for Protein Sequence Representation), keywords such as "Protein Sequence," "Representation," and "LLM" were searched. In these searches, articles in which the keywords were mentioned in the title, abstract, or keyword section were identified and selected. After the initial search, articles were screened by reading their abstracts, and irrelevant articles were removed. In selecting articles, relevance to the research field (direct relationship with biological, medical, or chemical data), publication date (articles published from 2020 onwards), and scientific importance (a high number of citations or a close alignment with the focus of this review article) were considered as the main selection criteria. Then, the full text of the relevant articles was reviewed to ensure their exact relevance to the research topics. The arXiv and bioRxiv databases were selected due to their open access and the rapid publication of new articles in biology and computational sciences. Additionally, databases such as Scopus and Google Scholar were used to cover high-quality peer-reviewed articles. However, this study did not review some paid articles in the Springer and Elsevier databases due to limited access.
Problem Statement and Research Questions
This review provides an in-depth overview of the application of large language models in the analysis of biological data. It is a nice reference to help researchers understand recent developments regarding large language models. The paper compares different models, their strengths and limitations, and how they work. This review provides strategic insights by discussing limitations in existing models and paves the roadmap to guide future research efforts. It also introduces researchers to the large family of LLMs, their applications in analyzing biological data, and the basic tools that can facilitate choosing the most appropriate model for future research purposes.
This review answers the following key questions:
- What are the capabilities of Large Language Models (LLMs) in biological data analysis?
- Which models perform better in predicting biological traits and behaviors?
- How can LLMs model long-range dependencies and genetic interactions?
- What are the limitations and challenges in applying LLMs to biological data?
- How effective are LLMs in predicting complex biological structures and molecular interactions?
- What are the differences between supervised, unsupervised, and hybrid learning methods in biological models?
0.1. Big Picture of the Literature Review
This study aims to provide a comprehensive review of the LLMs approaches presented in the literature for scientific texts and data. This comprehensive review includes the approaches, data, and scope used. An overview of the study is given in Figure 1. Some of the common libraries used for LLMs training are listed in Appendix A.
Document Outline
The paper is organized as follows: Section 1 elaborates on the Textual Scientific Large Language Models, exploring Medical, Biology, and Chemistry large language models, along with the datasets and benchmarks used in these fields. Molecular Large Language Models are reviewed in Section 2, where applications of large language models are explained for various prediction tasks, including Molecule Property Prediction, Interaction Prediction, Reaction Prediction, and Molecule Generation Designing and Editing. Section 2.5 provides an overview of the related datasets and benchmarks. Section 3 is dedicated to Protein Large Language Models, investigating tasks related to protein sequences, primarily divided into Protein Sequence Representation and Protein Sequence Generation and Designing. As in previous sections, the final part of this section reviews the datasets and benchmarks. Genomic Large Language Models, studied in Section 4, focus on applications of LLMs for Genomic Data Modeling, Function Prediction, Variants and Evolution Prediction, DNA Protein Interaction Prediction, and RNA Prediction. This section concludes by discussing its datasets and benchmarks. Finally, Multimodal Scientific Large Language Models are covered in Section 5, categorized into Molecule and Text, Protein and Text, Protein and Molecule, and Cell and Text. Datasets and benchmarks for each category are reviewed in detail at the end of this section.
1. Textual Scientific Large Language Models
This section aims to review large scientific language models trained using text datasets (i.e., Text-Sci-LLM). This review includes the datasets, language models, the core of the language models, the domain used, and the results obtained by these language models. An overview of this section is shown in Figure 2.
1.1. Medical Large Language Models
This sub section discusses the current LLMs approaches for clinical medicine.Also, the general classification of methods and subcategories of the Medical Large Language Models examined is shown in Figure 3.
Clinical notes contain information about patients, such as laboratory values or medications, beyond unstructured data. However, clinical notes have been less widely used than structured data because they are high-dimensional and sparse. ClinicalBERT [27] aims to develop and evaluate a continuous representation of clinical notes for predicting 30-day hospital readmissions at different time points during admission, including early admission and discharge. The model applies bidirectional encoder representations from a transformer (BERT) to clinical text. Publicly published BERT parameters are trained on standard datasets such as Wikipedia and BookCorpus, which differ from clinical text. Therefore, the authors pre-trained BERT using clinical notes, tuned the network for predicting hospital readmissions, and named it ClinicalBERT. Clinical-BERT reveals high-quality relationships between medical concepts as judged by clinicians. This model outperformed various baselines in predicting 30-day hospital readmission using discharge summaries and first few days in the intensive care unit (ICU) notes across multiple measures, achieving an AUROC of , compared to for BERT. ClinicalBERT’s attentional weight can also be used to interpret the predictions. In [28], the authors first developed a large clinical language model - GatorTron - using over 90 billion texts (including over 82 billion words of unclear clinical text). This language model was systematically applied to five clinical NLP tasks: medical relation extraction, semantic text similarity, natural language inference (NLI), and Medical Question answering (MQA). In this study, to demonstrate how (1) increasing the number of parameters and (2) increasing the size of the training data can be beneficial for NLP tasks, the GatorTron models increased the clinical language model from 110 million to 8.9 billion parameters. The model achieved a maximum of F1 = 0.900 in Clinical concept extraction and F1 = 0.9627 in Medical relation extraction. This model also achieved a Pearson correlation= 0.8903 in Semantic textual similarity, Accuracy= 0.9020 in Natural language inference, and F1 score= 0.9719 in Question answering.
Another example of the GatorTron application was introduced in [29]. This study created a generative clinical LLM, GatorTronGPT, using 277 billion words of text, including 82 billion words of clinical text from 126 clinical departments and approximately 2 million patients at the University of Florida Health System, and 195 billion words of variety. In this study, they trained GatorTronGPT using the GPT-3 architecture with up to 20 billion parameters and evaluated its application for biomedical natural language processing (NLP) and healthcare text generation. The comparison results show that the synthetic text generated by GatorTronGPT in this study contains 40.4 million: 4.82 million unigrams and 416.35 million: 62.51 million bigrams. The synthetic text also has higher entropy than real-world clinical text.
MEDITRON [30] is a set of open-source LLMs with 7B and 70B parameters compatible with the medical domain, improving the accessibility of medical LLMs at a large scale. MEDITRON is built on Llama-2 and extends pre-training on a comprehensive medical dataset, including selected PubMed articles, abstracts and internationally recognized medical guidelines. MEDITRON GAP-REPLAY combines 48.1 billion tokens from four datasets:
- Clinical Guidelines: a new dataset of 46 thousand clinical practice guidelines from various healthcare-related sources.
- Article Abstracts: abstracts available from 16.1 million closed-access PubMed and PubMed Central articles.
- Medical Articles: full-text articles extracted from 5 million publicly available PubMed and PubMed Central articles.
- Replay dataset: public domain data distilled to write 1% of the total corpus.
MEDITRON achieved an absolute performance increase of 6% over the best public baseline in its parameter class and 3% over the most substantial baseline (Llama-2). MEDITRON-70B outperforms GPT-3.5 and Med-PaLM and is within 5% of GPT-4 and 10% of Med-PaLM-2.
While recent advances in commercial large language models (LM) have yielded promising results in medical applications, their closed-source nature raises significant privacy and security concerns, hindering their widespread use in the medical field. Despite efforts to create open-source models, their limited parameters often do not result in sufficient multi-step reasoning capabilities to solve complex medical problems. To address this issue, the authors introduce Meerkat [31] to address these problems. This model is a new family of medical AI systems that range from 7 to 70 billion parameters. The models were trained using a new synthetic dataset consisting of high-quality chain reasoning paths from 18 medical textbooks and a diverse dataset that follows instructions. Meerkat achieved remarkable accuracy on three benchmarks, Mistral-7B, Gemma-7B, and LLaMA-3-8B, outperforming previous best models such as MediTron and BioMistral and GPT-3.5 by a wide margin. In addition, Meerkat-70B correctly identified 21 out of 38 complex clinical cases, which was better than 13.8 for humans and nearly matched GPT-4’s 21.8. Meerkat provided more accurate open-ended answers to clinical questions than existing small models, approaching the performance level of large commercial models.
ClinicalGPT [32] is another language model explicitly designed and optimized for clinical scenarios. ClinicalGPT is better prepared for multiple clinical tasks by incorporating extensive and diverse real-world data, such as medical records, domain-specific knowledge, and multi-step conversational consultations, into the training process. This language model has been trained and evaluated on cMedQA2, cMedQA-KG, MD-EHR, MEDQA-MCMLE, and MedDialog, which are the core of this language model. In medical conversation and examination comparisons, this model has shown better results than LLAMA-7B, ChatGLM-6B, and BLOOM-7B.
The authors in [33] addressed the challenge of integrating LLMs. Integrating large language models (LLMs) in healthcare has excellent potential but faces challenges. Pre-training LLMs from scratch for domains such as medicine is resource-intensive and often impractical. On the other hand, relying solely on Supervised Fine-tuning (SFT) can lead to overly confident predictions. In response, they proposed a multi-stage training method called Qilin-Med. Qilin-Med combines Continued Pre-training (CPT), SFT, and Direct Preference Optimization (DPO). In addition, the Chinese Medicine dataset (ChiMed) was presented in this study, which consists of medical question answers, simple texts, knowledge edge graphs, and dialogues divided into three training stages. Medical LLMs trained with our pipeline, Qilin-Med, show significant performance improvements.
ChatDoctor[34] is a medical chat model fine-tuned on a LLaMA Using Medical Domain Knowledge. The primary goal of ChatDoctor is to address the observed limitations in medical knowledge of standard large language models (LLMs), such as ChatGPT, by creating a highly accurate specialized language model for medical advice. In ChatDoctor [8], the authors adapted and refined the Large Language Model Meta-AI (LLaMA) using a large dataset of 100,000 patient-doctor conversations from a widely used online medical consultation platform. In addition to refining the model, they also incorporated an autonomous information retrieval mechanism that allows the model to access and use real-time information from online sources such as Wikipedia and data from offline medical databases. The model performed better in answering questions than ChatGPT. Another example of LLMs proposed for Chinese is HuaTuo [35]. Large language models (LLMs), such as the LLaMA model, have shown their effectiveness in various general-domain natural language processing (NLP) tasks. However, LLMs have not yet performed well in biomedical tasks due to the need for medical expertise in the response. In response to this challenge, the authors proposed HuaTuo, a model based on LLaMA that is supervised with generated QA (question-answer) examples. Experimental results show that HuaTuo produces responses that have more reliable medical knowledge. This model provided more reliable results than LLaMA, Alpaca, and ChatGLM. HuatuoGPT [36] was also proposed for Chinese, similar to HuaTuo. In HuatuoGPT, the authors proposed a large language model (LLM) for medical consultation. The core premise of HuatuoGPT is to use data distilled from ChatGPT and real-world data from supervised clinicians in the fine-tuning phase. ChatGPT responses are usually detailed, well-presented, and informative, but they cannot act like a clinician in many aspects. To better utilize the strengths of both data, they trained a reward model to align the language model with the merits that both data bring, by the RLAIF (Reinforced Learning from Artificial Intelligence Feedback) model.
Another example of LLMs proposed for Chinese is HuaTuo [35]. Large language models (LLMs), such as the LLaMA model, have shown their effectiveness in various general-domain natural language processing (NLP) tasks. However, LLMs have not yet performed well in biomedical tasks due to the need for medical expertise in the response. In response to this challenge, the authors proposed HuaTuo, a model based on LLaMA that is supervised with generated QA (question-answer) examples. Experimental results show that HuaTuo produces responses that have more reliable medical knowledge. This model provided more reliable results than LLaMA [9], Alpaca[37], and ChatGLM [38]. HuatuoGPT [36] was also proposed for Chinese, similar to HuaTuo. In HuatuoGPT, the authors proposed a large language model (LLM) for medical consultation. The core premise of HuatuoGPT is to use data distilled from ChatGPT and real-world data from supervised clinicians in the fine-tuning phase. ChatGPT responses are usually detailed, well-presented, and informative, but they cannot act like a clinician in many aspects. To better utilize the strengths of both data, they trained a reward model to align the language model with the merits that both data bring, by the RLAIF (Reinforced Learning from Artificial Intelligence Feedback) model.
The Baize language model[39] was extended based on ChatGPT. ChatGPT is accessible through a limited API, which poses obstacles to new research and advancement in various fields. Baize proposes a pipeline that can automatically generate a high-quality multi-round chat set by using ChatGPT to engage in a conversation with itself. The authors use efficient parameter tuning to improve LLaMA, a large open-source language model. The resulting model, named Baize, shows good performance in multi-round conversations with guardrails that minimize potential risks. Furthermore, this research proposes a new technique, named Self-Distill with Feed Back, to improve the performance of Baize models with ChatGPT feedback. This model is mainly presented for research purposes. The model was compared to LLaMA[9], Alpaca [40], Vicuna [41] and ChatGPT [42] evaluated by GPT-4 [43]. It provided comparable results. Given the fact that at least 82% of LLaMA’s pre-training data is from before 2020, Baize may provide outdated answers to certain questions.
There is a performance lag in LMMs in general use cases in some specialized fields, such as Chinese medicine. Existing efforts to integrate Chinese medicine into LLM rely on Supervised Fine-Tuning (SFT) with single-turn and distilled dialogue data. These models lack the ability of physician-like pre-emptive querying and multi-directional understanding and cannot coordinate responses with experts’ intentions. Zhongjing [44] is the first LLaMA-based Chinese medical LLM that provides a complete training pipeline from continuous pre-training, SFT, to Reinforcement Learning from Human Feedback (RLHF). In this study, the authors additionally provide a Chinese multi-turn medical dialogue dataset of 70,000 authentic doctor-patient conversations, CMtMedQA, which significantly enhances the model’s ability to handle complex dialogues and initiate active research. They also define a modified annotation rule and evaluation criteria considering the unique characteristics of the biomedical field. Multi-turn evaluation of this model achieved a 49% win over ChatGPT.
The authors in [45] investigated the process of building a robust open-source language model specifically for medical applications. This model was named PMC-LLaMA. The main goal of this research was to systematically investigate the adaptation process of a general-purpose fundamental language model in the medical domain, which involves data-driven knowledge injection through integrating 4.8 million biomedical academic articles, 30,000 medical textbooks, and comprehensive fine-tuning to align with domain-specific guidelines. We provide a comprehensive and large-scale dataset for guideline tuning. The dataset includes medical question answering (QA), reasoning logic, and conversational dialogues, which total 202 million tokens. The model achieved an average accuracy of 64.43 on three datasets: MedQA, MedMCQA, and PubMedQA, while ChatGPT achieved an average accuracy of 54.97. Clinical Prediction with Large Language Models (CPLLM) [46] presented a method that involves fine-tuning a pre-trained large language model (LLM) for clinical disease and readmission prediction. This model used quantization, and the LLM was trained using commands to predict the diagnosis using historical diagnosis records. The main goal of this study was to provide a model for whether patients will be diagnosed with a target disease during their next visit or in the subsequent diagnosis, leveraging their historical diagnosis records. Their findings show that CPLLM outperformed all the tested models, including ETAIN, Med-BERT, and Logistic Regression, in both PR-AUC and ROC-AUC. This model in Adult respiratory failure achieved and , which is a 0.912% improvement over the previous best model (Logistic Regression).
Med PaLM was the first model to achieve a passing score on the US Medical Licensing Examination (USMLE), scoring 67.2% on the MedQA dataset. However, this and other prior work suggested significant room for improvement, especially when models’ answers were compared to clinicians’ answers. To address these gaps, the authors proposed Med-PaLM 2 [47]. This model uses a combination of improvements to the LLM baseline (PaLM 2), fine-tuning of the medical domain, and stimulus strategies, including a novel set modification approach. To evaluate the model, the authors used a dataset of multiple-choice questions that included the benchmarks: MedQA [48], MedM-CQA [49], PubMedQA [50], and MMLU clinical topics [51], as well as a dataset of long-form questions that included the benchmarks MultiMedQA 140, MultiMedQA 1066, Adversarial (General), and Adversarial (Health equity). Med-PaLM 2 achieved up to 86.5% of the scores on the MedQA dataset, in fact achieving a 19% improvement over Med-PaLM.
Clinical Camel [52] is an open expert-level medical language model with dialogue-based knowledge encoding. This open large language model (LLM) is explicitly designed for clinical research. Clinical Camel improves performance on medical benchmarks among open medical LLMs by fine-tuning LLaMA-2 using QLoRA. Using efficient single GPU training, Clinical Camel outperforms five-shot evaluations on all benchmarks evaluated, including GPT-3.5. The language model uses ShareGPT, Clinical Articles, and MedQA as training datasets. Clinical Camel delivers competitive performance to proprietary LLMs through efficient training, achieving improved results among open medical models and proving its efficiency by outperforming GPT-3.5 on QA benchmarks.
Another example of LLMs proposed for Chinese medical care is DoctorGLM [53]. In this model, the developers first collected a database of Chinese medical conversations using ChatGPT and applied several techniques to train an easy-to-deploy LLM. They set up ChatGLM-6B on an A100 80G in 13 hours. No comparison of the model’s performance on tasks is provided. This language model is open source.
In real-world medical consultations, doctors usually use repeated queries to understand the patient’s condition thoroughly. These queries enable them to provide practical and personalized suggestions, which can be defined as a chain of questioning (CoQ) for LLMs. To demonstrate the CoQ of LLMs, the developers proposed BianQue [54]. BianQue is a ChatGLM-based LLM that is trained on the self-built health dialogue dataset BianQueCorpus[54], which contains multiple rounds of health questions and suggestions processed by ChatGPT. Experimental results show that the proposed BianQue can simultaneously balance the querying capabilities and health suggestions, which helps promote the research and application of LLM in preventive health. On four datasets, MedDialog-CN [55], IMCS-V2 [56], CHIP-MDCFNPC [57], and MedDG [58], this approach provided better results than ChatGLM-6B[59], DoctorGLM [53], and ChatGPT [60].
While these LLMs show competitive performance on automated medical text benchmarks, they are pre-trained and evaluated with a focus on one language (mainly English or Chinese). This has made it difficult for them to apply to other domains (languages). This is especially true for text-to-text models, which typically require large amounts of domain-specific pre-training data that are often not readily available for many languages. Medical mT5 [61] is an open-source multilingual text-to-text LLM for the medical domain. Medical has addressed these shortcomings by assembling the most extensive multilingual collection for the medical domain in four languages: English, French, Italian, and Spanish. This new collection for mT5 Medical is the first open-source, multilingual text-to-text model. In this study, the word sets for English were used from the sources ClinicalTrials, EMEA, and PubMed; for Spanish from the sources EMEA, PubMed, Medical Crawler, SPACC, UFAL, and WikiMed; for French from the sources PubMed, Science Direct, Wikipedia - Médecine, EDP, and Google Patents, and for Italian from the sources: Medical Commoncrawl-I, Drug instructions, Wikipedia - Medicina, E3C Corpus - IT, Medicine descriptions, Medical theses, Medical websites, Medical test simulations, PubMed, Supplement description, Medical notes, Pathologies, and Clinical cases. The results of this model are more acceptable and interpretable compared to the most prominent models in the languages studied.
The researchers in [62] aim to develop foundational medical LLMs by training open-source LLaMA models with domain-specific and large-scale datasets to enhance their performance in various medical text analysis and medical diagnosis tasks. Me-LLaMA is a medical foundation LLMs for comprehensive text analysis and beyond. Me-LLaMA, a new family of medical LLMs that includes the Me-LLaMA 13/70B foundation models and their chat-enhanced versions, is pre-trained and tuned using continuous LLaMA2 instructions using biomedical literature and clinical notes. This language model used the most comprehensive medical dataset, including 129B pre-training tokens and 214K instruction tuning samples from diverse biomedical and clinical data sources. Training the 70B models required significant computational resources, exceeding 100,000 A100 GPU hours. The results of the Me-LLaMA models outperform LLaMA and other open-source medical LLMs in zero-shot and supervised learning settings for most text analysis tasks. This model suffers from reinforcement learning from human feedback (RLHF), as do other models in the literature. Appendix B includes other examples of open-source LLMs.
In [63], the researchers introduced BiMediX, the first bilingual medical fusion of LLM experts. It is designed to interact seamlessly in both English and Arabic. BiMediX facilitates various medical interactions in English and Arabic, including multi-turn chats to inquire about more details such as patient symptoms and medical history, answering multiple-choice and open-ended questions. This study provided a bilingual Arabic-English training set covering 1.3 million diverse medical interactions, resulting in over 632 million healthcare expertise tokens for order generation. The BiMed1.3M dataset provided consists of 250,000 multi-turn doctor-patient chats. This model outperformed the general English-Arabic bilingual LLM, Jais-30B, with an average absolute gain of 10% in the Arabic medical benchmark and 15% in the bilingual assessments across different datasets. The summary of medical Large Language Models(LLMS) is given in Table 2.
1.2. Biology Large Language Models
An overview of the methods and subcategories of Biology Large Language Models. is shown in Figure 4.
Word text embeddings derived from pre-trained language models (LMs) have significantly improved NLP tasks. Pre-trained text embeddings have achieved significant task performance in specific domains, such as biomedical articles. In [64], researchers conducted exploratory experiments to determine what additional information is inherently carried by the pre-trained text embeddings within the domain. To do this, they used pre-trained LMs as fixed feature extractors. They compared the language models BERT[65], ELMo [66], BioBERT [67], and BioELMo, a biomedical version of ELMo trained on 10M PubMed abstracts. These comparisons were examined in two biomedical natural language inference (NLI) and biomedical named entity recognition (NER) tasks. For this purpose, for the NER task, the BC2GM stands for BioCreative II gene mention dataset [68] and the general-domain CoNLL 2003 NER dataset [69] were used. Also, for the NLI task, they use the MedNLI datase[70]. According to the experiments, BioBERT performed better than BioELMo in the biomedical NER and NLI tasks.
BioBERT, a pre-trained biomedical language for biomedical text mining, was introduced in [67]. This language model was trained on the datasets of English Wikipedia, BooksCorpus, PubMed Abstracts, and PMC full-text articles. BioBERT significantly outperformed BERT and previous state-of-the-art models on various biomedical text mining tasks when pre-trained on biomedical datasets. While BERT achieved comparable performance to previous state-of-the-art models, BioBERT significantly outperformed them on three biomedical text mining tasks (biomedical entity name recognition 0.62% F1 score improvement, biomedical relationship extraction 2.80% F1 score improvement, and biomedical question answering 12.24% MRR improvement). The evaluation of BERT and ELMo on ten benchmark datasets was investigated in [71]. The Biomedical Language Understanding Evaluation (BLUE) benchmark was introduced in this study. BLUE consists of five tasks with ten sets that cover a wide range of data quantities and problems. These tasks and data include:
-
Sentence similarity with data:
- MedSTS:sentence pairs
- BIOSSES: sentence pairs
-
Named entity recognition with data:
- BC5CDR-disease: mentions
- BC5CDR-chemical: mentions - ShARe/CLEFE mentions
-
Relation extraction with data:
- DDI: relations
- ChemProt: relations
- i2b2 2010: relations
-
Document classification with data:
- HoC: documents
-
Inference with data:
- MedNLI: pairs
In this study, four pre-trained models were also used, including BERT-Base (PubMed), BERT-Large (PubMed), BERT-Base (PubMed + MIMIC-III), and BERT-Large (PubMed + MIMIC-III). According to the reported results, the BERT model trained on PubMed abstracts and clinical notes has achieved better results than other models in the literature. BioMegatron [72] is another example of a language model trained on a larger domain set of biological data, showing consistent improvements in benchmark scores, which helps us understand the applications of the domain language model. This language model greatly improves Named Entity Recognition, Relation Extraction, and Question Answering. This language model was trained on 6.1 billion words. This model achieved a 0.6 improvement in MRR over the BioMegatron-800m model in QA.
A dominant assumption in language model research is that domain-specific pretraining can benefit domain-general language models. In [73], researchers took this assumption as the basis for their research. Indeed, challenging this issue for domains with abundant unlabeled text, such as biomedicine and pretraining language models from scratch, has led to significant gains over continuous pretraining of general-domain language models. BLURB, a comprehensive benchmark for biomedical NLP, was introduced in this work. The biomedical datasets BC5-chem [74], BC5-disease [74], NCBI-disease [75], BC2GM [68], JNLPBA [76], EBM PICO [77], ChemProt [78], DDI [79], GAD[80], BIOSSES[81], HoC[82], PubMedQA [50], and BioASQ [83] were used to pretrain BLURB. The core of this model is BERT, which was used for tasks such as Token Classification, Sequence Classification, Sequence Regression, and Sequence Classification. The combination of PubMedBERT and BLURB achieved remarkable results in these tasks.
In [84], a case study comparison between Large Biomedical Language and language models presented in this field was conducted. In this study, the authors introduced BioM-Transformers. These models were extended to build large biomedical language models with BERT, ALBERT and ELECTRA. For this purpose, four transformer-based models, namely ELECTRABase, ELECTRALarge, BERTLarge and ALBERTxxlarge, were pre-trained using Tensor Processing Units TPUs on biomedical domain datasets. The pre-trained models were calibrated and evaluated on several downstream biomedical tasks, highlighting the impact of design choices on the performance of biomedical language models.
LinkBERT [85] was a different study that expanded on Document Links. Existing methods such as BERT and other reviewed methods model a single document and do not include dependencies or knowledge that extends across documents. In LinkBERT, they proposed a pre-training LM method that uses links between documents, i.e., hyperlinks. Given a pre-training dataset that can be considered as a graph of documents, they created LM inputs by placing a pair of linked documents in a context (linked), in addition to the existing options of placing a single document (connected) or a pair of random documents (random). They then trained LM with two supervised objectives: masked language modelling (MLM), which predicts masked tokens in the input, and document relation prediction (DRP), which classifies the relation of the two text segments in the input (contiguous, random, or linked). The LinkBERT model achieved an average F1 of 81.0 on HotpotQA, TriviaQA, SearchQA, NaturalQ, NewsQAm, and SQuAD, while BERT achieved an average F1 of 78.5.
In [86], BioGPT, a domain-specific generative language model, was introduced. This model has been pre-trained in the biomedical literature on a large scale. Training only on in-domain data is important for a specific domain from the beginning, so BioGPT only considers in-domain text data and performs training on in-domain texts. In this model, the researchers collected all PubMed items updated before 2021 from the official website using the wget tool containing 15 million items. The GPT-2 architecture was considered the backbone of BioGPT. BioGPT was evaluated on six biomedical NLP tasks, outperforming previous models in most tasks. The model achieved 44.98%, 38.42%, and 40.76% F1 scores in BC5CDR, KD-DTI, and DDI for end-to-end relationship extraction, respectively, and 78.2% accuracy in PubMedQA.
Foundation models (FMs) have demonstrated remarkable performance in a wide range of tasks in many domains, including medical domains. However, general-purpose FMs often face challenges when dealing with domain-specific problems due to their limited access to dedicated training data in a particular domain. Applying these FMs in some domains is impossible due to the lack of sufficient data. In biomedicine, various biological methods such as molecules, proteins, and cells are encoded by the language of life, which has significant differences from human natural language. Researchers developed BioMedGPT [87] to overcome this challenge. BioMedGPT is an open-source multimodal generative pre-trained transformer for biomedicine. This language model is developed with the aim of bridging the gap between biological language and human natural language. BioMedGPT allows users to easily “communicate” with various biological methods through free text. BioMedGPT-LM can serve as a bridge to connect different biomedical methods. The model has the ability to understand and reason about diverse biological methods, including molecules, proteins, transcriptomics, and more, through feature space alignment. The model achieved accuracies of 51.4, 76.1, and 50.4 on the MedMCQA(ID), PubMedQA(ID), and USMLE(OOD) datasets, respectively.
BioinspiredLLM [88] is another domain-specific model developed for structural biological and bio-inspired materials. The model is tuned with over a thousand peer-reviewed articles on structural biological and bio-inspired materials, can help with information recall research tasks, and functions as an engine for creativity. The model has been proven to recall information about biological materials accurately and is enhanced with enhanced reasoning capabilities. It also uses Retrieval-Augmented Generation (RAG) to incorporate new data during generation, which can also help with resource tracking and updating. The autoregressive transformer in decoder-based large language models was used to train the language model. BioinspiredLLM can also act as an engine for scientific creativity. The model can answer open questions about previously unseen topics and suggest new predictions or hypotheses to help researchers.
In [89], BioMistral was introduced, an open source LLM designed for the biomedical domain. The model uses Mistral as the base model and is pre-trained on PubMed Central. The PMC Open Access Subset was considered to adapt the LLM to the medical domain. In this model, Activation-aware Weight Quantization (AWQ) and BitsandBytes (BnB) were considered as Quantization techniques. BioMistral was provided in Multilingual mode. The model achieved an accuracy of 55.4 on 10 tested datasets. A summary of Biology Large Language Models is given in Table 3.
1.3. Chemistry Large Language Models
Access to structured chemical reaction data is of key importance for chemists in performing bench experiments and modern applications such as computer-aided drug design. Human curators have generally collected existing reaction databases through manual abstraction from published texts (e.g., patents and journals). Collecting these data is time-consuming and labour-intensive, especially with the exponential growth of the chemical literature in recent years. In the remainder of this section, we will discuss some research and benchmarks in this area. The general classification of methods and subcategories of the Chemistry Large Language Models examined is shown in Figure 5.
In [91], the authors focused on developing automated deep learning-based methods for extracting reactions from chemical texts. They considered journal publications as the target information source, which is more comprehensive and better reflects the latest developments in chemistry compared to patents. About 194,516 articles were collected from the Journal of the American Chemical Society, The Journal of Organic Chemistry, Organic Letters, Journal of Organic Chemistry, and Organic Process Research and Development. They first devised a chemical reaction scheme to implement the reaction extraction system, mainly consisting of a central product and a set of reaction roles. For this purpose, the Reaction Roles of Product, Catalyst/Reagents, Workup reagents, Solvent, Temperature, Time, Reaction type, and Yield were used. They formulated the task as a structure prediction problem and solved it with a two-step Transformer encoder consisting of product extraction and reaction role labelling. ChemRxnBERT introduced in this research achieved precision= 79.3 in Reaction Role prediction and precision= 84.6 in Product Extraction.
MatSciBERT is another example of a materials-aware language model introduced in [92]. This model uses domain adaptive pretraining for initial training. In this study, they initialized the MatSciBERT weights with a suitable LM and then trained it on MSC. To determine the appropriate initial weights for MatSciBERT, the authors trained on a wordpiece vocabulary without letters based on MSC. Due to the larger overlap with the SciBERT vocabulary, they labelled their set using SciBERT vocabulary and initialized the MatSciBERT weights with the publicly available SciBERT weights. This model achieved accuracy in glass vs. non-glass.
[93] used NLP to extract material property data from polymer literature abstracts automatically. This research proposed MaterialsBERT. A language model trained on 2.4 million materials science abstracts outperformed other baseline models on three out of five named entity recognition datasets. This pipeline resulted in the extraction of 300,000 material property records from 130,000 abstracts, which took 60 hours to extract from this volume of data. The ontology used to annotate Polymer Abstracts included POLYMER, , MONOMER, , , , , , and OTHER. The extracted data for various applications, such as fuel cells, supercapacitors, and polymer solar cells, were analyzed to recover redundant insights and can be used to locate material property data recorded in abstracts. The core of this model is the BERT-base. This model achieved F1-scores of 69.2, 68.6, 86.0, and 71.4 on materials science NER datasets ChemDNER, Inorganic Synthesis recipes, Inorganic Abstracts, and ChemRxnExtractor, respectively. The researchers also extended ChemDFM [94], a language model trained on 34B tokens from chemistry texts and textbooks using 2.7 million instructions. The core of this model is LLaMa. A set of instructions was considered for the initial collection of the dataset. These instructions include MD: Molecule Description, TBMD: Text-Based Molecule Design, MPP: Molecular Property Prediction, RC: Reaction Completion, MNA: Molecular Notation Alignment. This model was able to achieve an accuracy of 81.0 in reaction prediction and retrosynthesis tasks.
ChemLLM [95] is another study that provides LLM in chemistry. This study introduced ChemData, a dataset specifically designed for instructional design, and ChemBench, a robust benchmark covering nine essential tasks. To create ChemData, they used the PubChem[96], ChEMBL[97], ChEBI[98], ZINC[99], USPTO[100], ORDerly[101], ChemXiv[102], LibreTexts Chemistry1, Wikipedia2, and Wikidata3 datasets. They also introduced ChemBench, an innovative benchmark consisting of nine tasks on chemical molecules and reactions to assess a language model’s chemical understanding accurately. These nine tasks are similar to the ChemData tasks, consisting of 4100 multiple-choice questions with one correct answer. The ChemLLM model was trained on InternLM2-Base-7B. The ChemLLM model achieved an accuracy of 96.7% in product prediction.
SMolInstruct was proposed in [103]. A large-scale, comprehensive, and high-quality dataset for setting chemistry instructions. SMolInstruct contains 14 selected chemistry tasks and more than three million examples, which provides a solid foundation for teaching and assessing LLMs for chemistry. SMolInstruct includes the following tasks:
- Name Conversion: IUPAC to Molecular Formula (NC-I2F), IUPAC to SMILES (NC-I2S), SMILES to Molecular Formula (NC-S2F), and SMILES to IUPAC (NC-S2I)
- Molecule Description: Molecule Captioning (MC), and Molecule Generation (MG)
- Property Prediction: ESOL (PP-ESOL), LIPO (PP-LIPO), BBBP (PP-BBBP), ClinTox (PP-ClinTox), HIV (PP-HIV), and SIDER (PP-SIDER)
- Chemical Reaction: Forward Synthesis (FS) and Retrosynthesis (RS)
Using SMolInstruct, the authors set up a set of open-source LLMs called LlaSMol (Large language models on Small Molecules). These models included , , , and , among which they found that Mistral served as the best base model for chemistry tasks. This model achieved accuracies of 99.6 and 74.6 on the two tasks of name conversion (NC) and property prediction (PP), respectively.
Another example of the presented studies is PharmaGPT [104]. This study includes domain-specific large language models for biopharmaceuticals and chemistry. PharmaGPT is a set of domain-specific LLMs with 13 billion and 70 billion parameters, specifically trained on a comprehensive set tailored to the biopharmaceutical and chemical domains. A summary of Chemistry Large Language Models is given in Table 4.
1.4. Datasets and Benchmarks
This subsection lists some of the most important related datasets. These datasets are commonly used for model training and model evaluation. In [105], the eICU database was presented. Philips Healthcare has developed a telehealth system called the eICU program that uses this data to support managing critically ill patients. In this study, the developers presented a multicenter intensive care unit (ICU) database for more than 200,000 ICU admissions monitored by eICU programs across the United States. The database includes 200,859 patient units for 139,367 unique patients admitted between 2014 and 2015. Patients were admitted to one of 335 units in 208 hospitals across the United States. The database is deidentified and includes vital signs, laboratory measurements, medications, APACHE components, care plan information, admission diagnosis, patient history, time-stamped diagnoses, and other freely available information.
cMedQA v2.0 was introduced in [106]. In this study, the problem of Chinese medical question answer selection was investigated. Chinese medical question answer selection is considered a critical subtask in automated question answering and is relatively challenging due to its language and domain characteristics. To answer the questions accurately, they proposed a multi-scale interactive network framework that can extract semantic information at different granularity levels and interactive information between the question and the answer. The data for cMedQA v2.0 was collected from an online Chinese medical question-answering forum (http://www.xywy.com/). In this platform, qualified doctors answer questions from Internet users in the forum. Doctors make diagnoses and make suggestions based on the symptoms described by users. These data, consisting of 108,000 questions and 203,569 answers, were used as training and testing data in cMedQA v2.0. The core of their proposed End-to-End model was bidirectional gated recurrent units networks (GRUs) and multi-scale convolutional neural networks (CNNs). This model achieved an accuracy of 72.1 on the test data.
MedDialog is a large-scale medical conversation dataset introduced in [106]. This dataset includes 1) a Chinese dataset with 3.4 million conversations between patients and doctors, 11.3 million utterances, 660.2 million tokens, covering 172 specialties of diseases, and 2) an English dataset with 0.26 million conversations, 0.51 million utterances, 44.53 million tokens, covering 96 specialties of diseases. This dataset contains the richest information of its kind. GPT achieved Perplexity=8.9 on this dataset. MultiMedQA was proposed in [107] to evaluate large language models. MultiMedQA consists of multiple-choice question-answering datasets. It consists of datasets that require longer answers to questions asked by medical professionals and datasets that require longer answers to questions that non-professionals may ask. These datasets consist of a combination of the MedQA [50], MedMCQA [49], PubMedQA [50], LiveQA[108], MedicationQA [109], and MMLU clinical topics [110]. This dataset was also combined with the HealthSearchQA dataset, which contains health questions. Flan-PaLM was used to evaluate this dataset, achieving 67.6% on MedQA. The authors [48] presented an approach to develop a radiology examination dataset, collecting images and radiologist narrative reports. Initially, 8121 images and 3996 reports were collected for this dataset. Then, through a preprocessing process using Norman MeSH and RadLex codes, 3087 images and 1526 (38%) reports were selected. For preprocessing, 101 MeSH codes and 76 RadLex codes were used to represent the content of the Impressions and Findings sections of 2470 abnormal reports. These codes included Cardiomegaly, Pulmonary atelectasis, Calcified granuloma, Aorta/ tortuous, Lung/hypoinflated, Opacity/lung base, Pleural effusion, Lung/ hyperinflation, Cicatrix/lung, and Calcinosis/lung. The dataset was made publicly available online. MEDQA (Medical Question Answering) [48] introduces a large-scale open-domain question-answering dataset designed for solving complex medical problems. Derived from real-world professional medical exams in the US, Mainland China, and Taiwan, it offers multilingual support with datasets in English, simplified Chinese, and traditional Chinese. Comprising over 60,000 questions, MEDQA requires models to apply advanced domain-specific medical knowledge, integrate prior learning, and perform multi-hop logical reasoning across extensive medical text repositories. Despite advancements in neural and rule-based models, state-of-the-art methods achieve limited accuracy, demonstrating the datasets potential as a challenging benchmark for enhancing OpenQA systems in clinical contexts. MedMCQA [49] introduces a large-scale dataset of multiple-choice questions designed for medical domain question answering, leveraging over 194,000 questions derived from AIIMS and NEET PG exams. Covering 21 subjects and 2,400 healthcare topics, this dataset is uniquely comprehensive, providing not only questions and answers but also detailed explanations. The dataset aims to evaluate models; reasoning abilities across 10+ cognitive domains, challenging existing state-of-the-art models, which achieve only 47% accuracy compared to human candidates 90%. This benchmark serves as a valuable resource for advancing natural language processing in healthcare applications.
Evaluation of large language models (LLMs) on medical question-answering (QA) tasks, particularly in challenging clinical contexts, is explored. Two new datasets, JAMA Clinical Challenge and Medbullets, include high-quality expert-written explanations. These datasets aim to test models capabilities beyond simple medical licensing questions, incorporating complex reasoning and realistic clinical scenarios. Seven LLMs were benchmarked, revealing limitations in accuracy and explainability. The study [111] emphasizes the need for improved metrics and strategies to align model reasoning with medical decision-making. It highlights the datasets as benchmarks for advancing explainable medical QA systems.
The document [112] introduces Huatuo-26M the largest Chinese medical QA dataset, containing 26 million QA pairs. It highlights the dataset construction from various sources like online medical consultations, encyclopedias, and knowledge bases. Huatuo-26M aims to enhance medical QA research and practical applications for doctors and patients. Benchmarks show existing retrieval and generation models struggle with the datasets complexity. The dataset also supports advancements in transfer learning, retrieval-augmented generation, and pre-training for medical NLP tasks. Despite its potential, the dataset faces limitations, including possible inaccuracies and static answers unsuited for diverse medical contexts.
Authors in [51] introduces a new benchmark for evaluating the multitask performance of language models across 57 diverse subjects, ranging from STEM to humanities and professional topics. This benchmark measures text models multitask accuracy using questions of varying difficulty, from elementary to advanced levels. Notable findings include GPT-3’s lopsided performance, where it excels in some areas but struggles with others, particularly procedural tasks like mathematics. The study emphasizes that while GPT-3 represents progress, it remains far from expert-level accuracy and lacks calibration in confidence. The benchmark serves as a tool to analyze the breadth and depth of language models understanding, highlighting key limitations and areas for future improvement.
The article [113] presents C-EVAL, the first comprehensive Chinese evaluation benchmark designed for large language models (LLMs). It includes 13,948 multiple-choice questions across 52 disciplines, covering four difficulty levels: middle school,high school, college, and professional. The benchmark assesses knowledge and reasoning in diverse subjects, emphasizing advanced Chinese cultural and scientific topics. Results reveal that GPT-4 is the top performer, achieving a 66.4% accuracy, while other models, including Chinese-focused LLMs, lag significantly in reasoning-heavy tasks. The study highlights C-EVAL’s role in advancing LLMs for Chinese users and identifies challenges like limited reasoning ability and adaptation to complex scenarios. Authors introduces AGIEval[114], a human-centric benchmark for evaluating large language models (LLMs) on real-world cognitive tasks. Derived from official exams like SAT, LSAT, and Gaokao, AGIEval tests understanding, reasoning, and problem-solving across diverse topics. The benchmark highlights GPT-4’s impressive performance, often surpassing average human results, especially in standardized tests. However, all evaluated models, including ChatGPT and Text-Davinci-003, show limitations in tasks requiring deep domain-specific knowledge or complex reasoning. AGIEval emphasizes the need for more robust LLMs capable of handling intricate human-centric tasks, with future research focusing on enhancing reasoning abilities, multilingual generalization, and incorporating external knowledge sources.
SCIENCEQA [115], a large-scale multimodal dataset for science question answering, featuring 21,208 questions annotated with lectures and explanations. The dataset spans diverse topics across natural, social, and language sciences, with 48.7% of questions including image contexts and 48.2% text contexts. Advanced models like GPT-3 achieve up to 75.17% accuracy, significantly benefiting from chain-of-thought (CoT) reasoning, which improves few-shot learning by 1.20% and fine-tuning by 3.99%. While the human accuracy is 88.40%, current models fall short, particularly in multimodal reasoning. CoT allows models to achieve similar performance with 40% less data, underscoring its efficiency in learning and reasoning.
Xiezhi [116] is a comprehensive, ever-updating benchmark designed to evaluate domain knowledge across 516 disciplines from 13 categories, including science, engineering, medicine, and art, comprising 249,587 questions. It features two subsets: Xiezhi-Specialty for single-domain tasks and Xiezhi-Interdiscipline for multi-domain reasoning, making it ideal for testing advanced large language models (LLMs). Evaluating 47 LLMs revealed that GPT-4 outperformed human practitioners in fields like science, engineering, and medicine, while humans still excelled in economics, jurisprudence, and literature. The benchmark uses a 50-choice multiple-choice format to reduce random guessing, revealing significant performance gaps among LLMs. Smaller, fine-tuned models like DoctorGLM excel in specialized tasks but lose general reasoning capabilities. Observations highlight that fine-tuning combined with pretraining yields the best performance, while many models struggle with few-shot learning, except for GPT-4 and ChatGPT. Xiezhi surpasses other benchmarks like MMLU and C-Eval in identifying LLM disparities, offering a broader, more nuanced evaluation to drive improvements in multi-disciplinary reasoning and domain-specific understanding.
SciEval [117] introduces a multi-level benchmark for evaluating large language models (LLMs) in scientific research. It includes 18,000 questions across biology, chemistry, and physics, systematically assessing LLMs through four dimensions: basic knowledge, knowledge application, scientific calculation, and research ability, aligned with Bloom’s taxonomy. SciEval combines static data, dynamic data (updated regularly to prevent data leakage), and experimental data for subjective assessments. Results show that GPT-4 achieves the highest static accuracy of 73.93% and performs best in experimental reasoning with a score of 93.31, followed by GPT-3.5-turbo and Claude-v1.3. However, all models demonstrate weaknesses in dynamic data, particularly in physics calculations, while Galactica-30B excels in specific computational tasks. The findings highlight significant gaps in calculation and reasoning, underscoring the need for further advancements in LLMs for scientific applications.
BIOINFO-BENCH [118] is a benchmark framework designed to evaluate the bioinformatics skills of large language models (LLMs), assessing their academic knowledge and practical data-mining abilities. The framework focuses on three tasks: knowledge acquisition, sequence verification, and practical data analysis. It consists of 150 multiple-choice questions (BIOINFO-BENCH-qa), 20 sequence verification tasks (BIOINFO-BENCH-seq), and 30 patient data-based analysis tasks (BIOINFO-BENCH-div). The study evaluates models like ChatGPT, Llama-7B, and Galactica-30B, showing that while these models perform well in knowledge retention, with ChatGPT scoring highest (86.6% on multiple-choice and 90% on sequence verification), they struggle with tasks requiring reasoning, such as real-world data analysis. The results highlight a need for further training on domain-specific data and practical applications to improve LLM performance in bioinformatics.
The ARC (AI2 Reasoning Challenge) [119] introduces a benchmark aimed at improving question-answering systems through challenging science questions designed for standardized tests. The dataset contains 7,787 multiple-choice questions, divided into an easy set of 5,197 questions and a challenge set of 2,590 questions. The challenge set includes questions that retrieval-based and co-occurrence models fail to answer, requiring deeper reasoning and commonsense knowledge. ARC also provides a 14-million-sentence science corpus to support QA tasks. Leading models such as DecompAttn, DGEM, and BiDAF were tested, achieving around 25 to 27 percent accuracy on the challenge set, which is comparable to random guessing. On the easy set, these models performed significantly better, scoring between 55 to 65 percent. The results indicate that while current systems excel at surface-level questions, they face substantial difficulties with tasks that involve multi-step reasoning, advanced inference, and integrating commonsense knowledge, highlighting the ARC benchmark as a critical step for advancing AI systems.
2. Molecular Large Language Models
Molecular discovery is a scientific field where LLMs have shown great potential to accelerate discovery. This section provides an overview of molecular language-trained LLMs (Mol-LLMs). An overview of this section is shown in Figure 6. This section provides a brief overview of the datasets, new approaches, and a brief overview of these approaches.
2.1. Large Language Models for Reaction Prediction
The advent of large language models (LLMs) has significantly influenced various fields, and chemistry is no exception. By leveraging the power of Transformer architectures, initially designed for natural language processing, researchers have been able to model complex chemical reactions and retrosynthetic pathways as sequence-to-sequence tasks. These models treat molecules as text-based representations, such as SMILES strings, enabling them to process chemical information in much the same way as human languages. This approach has unlocked new possibilities in reaction prediction, retrosynthesis planning, and the discovery of innovative synthetic routes, offering unparalleled accuracy and scalability. This literature review explores how LLM-inspired techniques have been adapted to the domain of organic chemistry, focusing on Transformer-based models and their extensions. These methods tackle key challenges, including chemical plausibility, grammatical validity, and reaction diversity. By integrating principles from large language models, such as attention mechanisms, positional encoding, and pre-training strategies, these studies demonstrate transformative advancements in retrosynthesis prediction. The following sections outline recent breakthroughs, showcasing how the adoption of LLM techniques is reshaping the landscape of computational chemistry. Also, the general classification of methods and subcategories of the Large Language Models for Reaction Prediction examined is shown in Figure 7.
Paper [120] proposes a Self-Corrected Retrosynthesis Predictor (SCROP) which is a Transformer-based neural network designed for retrosynthesis prediction. The problem is framed as a sequence-to-sequence translation. The model takes a product molecule represented as a SMILES string (optionally prefixed with a reaction type token) as input and outputs SMILES strings of the predicted reactants. Its encoder captures the molecular structure’s local and global features using multi-head self-attention and positional encodings, while the decoder generates reactants step-by-step by attending to both the encoded product and its prior predictions. To improve accuracy, a Transformer-based syntax corrector refines the outputs by fixing invalid SMILES strings, ensuring syntactical and chemical validity. SCROP’s end-to-end design eliminates the need for predefined templates or rules, allowing it to generalize to novel reactions.
[121] Explores the application of Transformer architectures combined with data augmentation for predicting retrosynthesis and direct synthesis reactions. It utilizes SMILES to encode chemical reactions as sequences, enabling the adoption of NLP methods. The authors introduced novel augmentation strategies, including randomizing SMILES sequences for both input and target data on four scenarios: Products only (xN), Products and reactants/reagents (xNF), Products and reactants/reagents with shuffled reactant/reagent order (xNS), and Mixed forward and reverse reactions (xNM). Augmentation significantly reduced model overfitting and improved prediction accuracy. Key results include a top-5 accuracy of 84.8% on the USPTO-50k dataset for retrosynthesis and a remarkable 97% on the USPTO-MIT dataset for direct synthesis predictions. The Beam search was also employed to explore multiple plausible predictions during inference to enhance performance. The paper also incorporates a novel "MaxFrag" metric for retrosynthesis evaluation, focusing on predicting the largest reaction fragment.
Authors in [122] introduce a novel transformer-based model for retrosynthesis in organic chemistry that simultaneously addresses grammatical validity, chemical plausibility, and diversity which are the key challenges in template-free retrosynthesis. Their model employs two tied transformers: one for retrosynthesis (backward prediction) and another for forward reaction prediction, which are interconnected through shared parameters and a cycle consistency check. This ensures that predicted reactants can regenerate the input product, enhancing accuracy and plausibility. To generate diverse pathways, the model utilizes multinomial latent variables with a learned prior by which the model can explore multiple modes in the reaction space. The architecture features a shared encoder-decoder structure with 6 transformer layers, multi-head attention and feed-forward sublayers, leveraging parameter tying to improve data efficiency and generalization. The model demonstrated superior top-k accuracy with 47.1% top-1 and 78.5% top-10 on the USPTO-50K. It also shows minimal grammatical errors (0.1% invalid rate), and a unique molecule rate of 91.4%.
The model in [123] is built on the rxnfp framework, using a BERT-based encoder to process reaction SMILES (a text-based representation of chemical reactions) as input. It combines the encoder with a regression layer to output continuous yield predictions, enabling the estimation of reaction efficiency as a percentage. The model leverages contextualized token representations from the SMILES strings, capturing intricate relationships between reactants, catalysts, and conditions. Trained on high-throughput experimental (HTE) datasets for Buchwald-Hartwig and Suzuki-Miyaura reactions, Yield-BERT achieved high accuracy with an R² score of 0.956 on random splits, surpassing traditional approaches like random forests and descriptor-based models. Despite its success with HTE data, the model struggled with patent data due to inconsistencies and noise, such as varying yield measurement scales.
[124] Introduces a Grammar Ontology-based Prediction of Reaction Outcomes (GO-PRO) framework, leveraging context-free grammars (CFGs) and transformer architectures to predict chemical reaction outcomes. Chemical reactions are seen as a sequence-to-sequence translation task, where reactants and agents as input are translated into product molecules as output sequences. GO-PRO uses a transformer-based encoder-decoder architecture enhanced by multi-head attention mechanisms and positional encoding to model complex molecular transformations efficiently. The input molecules are hierarchically encoded using SMILES-based grammar to ensure syntactic validity and reduce computational complexity. Training employs the Adam optimizer with a warmup-based learning rate schedule, dropout regularization, and sparse categorical cross-entropy loss. The model has only 5 million parameters and achieves 80.1% top-1 accuracy and 99% syntactic validity on a USPTO dataset.
Author in [125] proposed the Graph Enhanced Transformer (GET) for retrosynthesis prediction by integrating sequential and graphical representations of molecules. The model combines SMILES-based Transformer encoding with graph neural network (GNN)-based atom representations through a novel GNN variant called Graph Attention with Edge and Skip-connection (GAES). This encoder-decoder structure leverages both molecular graph topology and SMILES sequences to improve the chemical validity and accuracy of retrosynthesis predictions. GET was evaluated on the USPTO-50K dataset and outperformed other state-of-the-art template-free methods, achieving the top-1 prediction accuracy of 59.1 which is the highest among such models. Graph2SMILES [126] introduces a novel graph-to-sequence model for retrosynthesis and reaction prediction, designed to overcome limitations of SMILES-based methods. Its architecture employs a Directed Message Passing Neural Network (D-MPNN) to encode molecular graphs, capturing local chemical environments, combined with a global attention encoder enriched with graph-aware positional embeddings to incorporate long-range interactions. The transformer-based decoder generates SMILES strings representing molecular transformations. The model’s input is a molecular graph, while its output is a SMILES representation of the target molecule. By ensuring permutation invariance and avoiding input-side data augmentation, Graph2SMILES achieves state-of-the-art accuracy on benchmark datasets, obtaining top-1 accuracy of 52.9% in USPTO_50k dataset (9.8% improvement) for one-step retrosynthesis and 1.9% improvement for reaction outcome prediction, demonstrating its efficiency and scalability across large datasets.
[127] Proposes a retrosynthetic prediction model, RetroTRAE, based on the Transformer architecture using atom environments (AEs) as molecular representations, instead of traditional SMILES strings. The input consists of AEs, which are topological fragments of molecules defined by atom connectivity within a certain radius, and the output is the predicted reactants for a given product molecule. This approach captures chemically meaningful transformations while avoiding common SMILES-based errors. RetroTRAE achieves a top-1 exact match accuracy of 58.3%, improving to 61.6% with highly similar predictions, which outperforms the state-of-the-art methods. It also enhances interpretability by highlighting reaction centers and delivers robust performance across diverse datasets.
A novel approach is proposed in [128] to enhance diversity in single-step retrosynthesis predictions using a modified Transformer-based model. The model incorporates a "cluster token prompt," which adds reaction class information to the input SMILES representation during training. This method enables the model to generate predictions guided by diverse disconnection strategies at inference that address limitations in chemical class diversity observed in baseline models. The input to the model consists of SMILES strings of target molecules with added classification token, and the output includes diverse sets of precursor molecules. Evaluation against public datasets showed a significant increase in diversity (e.g., average class diversity of 5.3 compared to 1.9 in the baseline) while maintaining competitive metrics such as 62% round-trip accuracy. This enhancement facilitates more robust recursive synthesis strategies which can improve chemical pathway exploration and reduce biases in retro synthesis planning.
[129] presents a novel "disconnection-aware" language model designed to enhance retrosynthesis predictions by leveraging prompt-based learning. Using a sequence-to-sequence Transformer architecture, the model accepts SMILES strings of target molecules and optional disconnection site prompts as inputs, generating precursor sets as outputs. This method enables user-guided or automated specification of disconnection sites which improves prediction accuracy and diversity compared to a baseline Molecular Transformer model. Results demonstrated a 39% accuracy improvement and a more than 100% of increase in reaction class diversity showing the model’s ability to address training data biases and generate more innovative retrosynthetic pathways. Summary of LLMs for Reaction Prediction is given in Table 5.
2.2. Datasets and Benchmarks
[130], introduces ZINC15, an updated version of the ZINC database designed to support virtual screening for ligand discovery. It features an expanded collection of over 750 million purchasable compounds curated for drug discovery and lead identification. ZINC15 allows researchers to access ready-to-dock small molecules, categorized by physical properties and annotated with biologically relevant information. The database emphasizes accessibility, providing tools for molecule search, filtering, and download, enabling both expert and novice researchers to perform virtual screening efficiently. The authors highlight advancements in usability and scalability, making the database a versatile resource for drug discovery and cheminformatics research. [99] introduces ZINC20, an extensive and freely accessible chemical database designed for virtual screening and ligand discovery. It aims to support drug discovery efforts by providing a comprehensive collection of commercially available small molecules. ZINC20 is an evolution of earlier ZINC databases, offering improvements in scalability, chemical diversity, and accessibility for large-scale computational chemistry and pharmacology studies. Its design also supports future expansion, making it suitable for large-scale virtual screening projects critical in pharmaceutical research. [131] introduces the ZINC database, a comprehensive resource designed to facilitate virtual drug screening. The database contains over 3.6 million commercially available chemical compounds, each pre-processed into formats suitable for molecular docking studies. ZINC provides these compounds in multiple 3D conformations to enable more accurate modeling of ligand-receptor interactions. They also emphasize its free accessibility, extensive coverage, and compatibility with common docking programs. The goal of ZINC is to streamline the virtual screening process and support drug discovery by offering researchers a practical tool to identify promising compounds efficiently. PubChem[96] is a large, publicly accessible database maintained by the National Institutes of Health (NIH) which stores information about chemical compounds, including their structures, properties, biological activities, and related scientific literature. The 2023 update to PubChem, as detailed by Kim et al., introduces several significant enhancements to this widely-used chemical information resource. Over the past two years, PubChem has incorporated data from more than 120 new sources, expanding its repository to over 870 contributors. Notably, the integration of Google Patents data has substantially increased the coverage of chemical information within the PubChem Patent data collection. Additionally, new data collections for Cell Line and Taxonomy have been established, facilitating quick access to chemical information pertinent to specific cell lines and taxa.
[132] presents the development of OPSIN (Open Parser for Systematic IUPAC Nomenclature), It focuses on developing methods to automatically extract chemical structures and reactions directly from scientific literature, likely utilizing computer algorithms to identify relevant chemical information within text and translate it into structured data like molecular formulas and reaction schemes. The paper[133] introduces the Open Graph Benchmark Large-Scale Challenge (OGB-LSC), designed to push the frontiers of machine learning (ML) on graphs by providing large-scale datasets and standardized evaluation protocols. The challenge aims to address the limitations of existing graph learning benchmarks that focus on small-scale datasets. The paper promotes research in scalable and efficient graph learning algorithms by providing robust, real- world benchmark datasets. Also, it can bridge the gap between research and practical graph-based applications.
[134] presents the winning solution for the KDD Cup 2021 and the OGB Large-Scale Challenge (OGB-LSC) in the graph prediction track. The challenge focused on predicting quantum chemical properties of molecular graphs using the PCQM4M-LSC dataset. The authors address the complexity of large-scale graph machine learning problems by implementing an ensemble of models, leveraging advanced graph representation techniques. Furthermore, the techniques and insights presented in this paper contribute to advancements in graph representation learning and its applications in quantum chemistry predictions.
The paper [135] introduces the GEOM dataset, which comprises energy-annotated molecular conformations to aid in property prediction and molecular generation. The GEOM dataset is designed to aid in training and evaluating machine learning models focused on predicting molecular properties, modeling molecular energy landscapes, and generating novel molecular conformations. It provides energy-annotated 3D conformations of molecules, helping models learn important chemical properties and spatial configurations relevant to computational chemistry tasks. The paper [136] discusses adversarial graph augmentation techniques to enhance graph contrastive learning, particularly in the context of molecular data. The authors present various datasets, including QM9, ZINC12k, Tox21, and others, detailing their characteristics and the tasks associated with them, such as classification and regression. The work emphasizes the importance of improving human hazard characterization of chemicals and explores the limitations of existing molecular machine learning approaches. The authors also provide an overview of the statistics related to the datasets, including the number of molecules, graph labels, and types of data. Additionally, they compare their methods with existing literature and highlight the significance of their contributions to the field of molecular discovery and drug development.
The paper [137] focuses on the ChEMBL database as a foundational drug discovery platform providing bio activity data for various applications, including predictive modeling and machine learning. A Drug Discovery Platform, details the evolution and enhancements of the ChEMBL database, a comprehensive open-access resource for drug discovery. Since its inception in 2009, ChEMBL has grown significantly, now offering over 20.3 million bioactivity measurements and 2.4 million unique compounds. It serves as a valuable tool for medicinal chemistry, chemical biology, and predictive modeling applications. ChEMBL is a critical resource for life sciences research, facilitating data-driven advancements in drug discovery and development. [138] focuses on significant updates to DrugBank 5.0, a comprehensive online resource for drug discovery and pharmacological research, providing molecular information about drugs, interactions, targets, and mechanisms. It is a major update to the DrugBank Database and it presents a comprehensive overhaul of the DrugBank platform, offering significant enhancements in data content, search capabilities, and usability for drug discovery and pharmacological research. Since its first release in 2006, DrugBank has evolved into one of the world’s most widely used drug reference resources. It supports advancements in pharmacogenomics, drug repurposing, precision medicine, and computational drug discovery by providing comprehensive, high-quality datasets and search capabilities. Also, it marks a significant step in enhancing the utility and scope of drug-related data for academic and industry research alike.
The paper [139] describes the creation of the GDB-17 database, a comprehensive resource containing 166.4 billion organic small molecules. These molecules are composed of up to 17 non-metallic atoms, including carbon (C), nitrogen (N), oxygen (O), sulfur (S), and halogens (F, Cl, Br, I). The purpose of this database is to explore and expand the chemical space relevant to drug discovery and materials science. GDB-17 is a valuable resource for virtual screening in drug discovery, as it allows researchers to identify new chemical scaffolds and isomers of existing drugs. It also supports molecular shape analysis, structural diversity studies, and advanced cheminformatics research.
The paper [140] focuses on the creation of ExCAPE-DB, a curated chemogenomics database. It provides data for building predictive models for polypharmacology, off-target effects, and cheminformatics approaches in drug discovery. It introduces ExCAPE-DB, a comprehensive open-access chemogenomics database designed to facilitate large-scale data analysis in drug discovery and cheminformatics. The resource integrates data from PubChem and ChEMBL to provide a standardized and searchable platform for predicting polypharmacology and off-target effects, as well as for benchmarking machine learning algorithms. It provides a vital resource for both academia and industry, promoting advancements in predictive modeling and cheminformatics research.
The paper [141] presents MoleculeNet, a benchmark designed to standardize and advance molecular machine learning by curating datasets, defining evaluation metrics, and providing open-source implementations of algorithms via the DeepChem library. MoleculeNet addresses the challenges in molecular machine learning, such as limited datasets, heterogeneity in molecular structures, and the wide range of molecular properties to predict. The benchmark facilitates reproducibility and comparability across molecular machine learning studies, akin to the role of ImageNet in computer vision.
[142] presents MARCEL (MoleculAR Conformer Ensemble Learning), the first benchmark dedicated to evaluating molecular representation learning (MRL) models using conformer ensembles. MARCEL focuses on capturing molecular flexibility, which arises from thermodynamically- permissible bond rotations and vibrational motions, often overlooked by traditional MRL approaches that treat molecules as static objects. Encoding all conformers is computationally expensive, requiring trade-offs between efficiency and accuracy, so future research should develop more efficient ensemble learning architectures and explore additional datasets and tasks.
The paper [143] presents GuacaMol, a comprehensive benchmarking framework for evaluating generative models used in de novo molecular design. It aims to standardize the assessment of both classical and neural network-based methods, enabling fair comparisons and identifying strengths and weaknesses in molecular generative models. The framework is open-source and provides tools to facilitate reproducibility in molecular design research.
The paper [144] introduces MOSES, a benchmarking platform designed to standardize the evaluation of molecular generative models. It addresses the lack of unified metrics and datasets in the field of molecular design, which hinders meaningful comparisons between different models. The authors aim to foster innovation and fair comparison in generative chemistry research, particularly in drug discovery.
The paper [145] introduces ADMETlab 2.0, a comprehensive web-based platform designed to predict Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties of chemical compounds. This upgraded version builds on its predecessor, addressing limitations such as incomplete endpoints, redundant modules, and a lack of batch processing. ADMETlab 2.0 offers a broader range of predictive capabilities and improved computational efficiency. [146] introduces SPECTRA, a spectral evaluation framework designed to comprehensively assess and understand the generalizability of machine learning models applied to molecular datasets. Generalizability is crucial in biological contexts, as models must perform well on unseen data to be useful in real-world applications like drug discovery, protein engineering, and disease resistance prediction. [147] introduces Molecule3D, a benchmark and dataset for predicting ground-state 3D molecular geometries using machine learning methods. Molecular geometry is crucial for understanding molecular properties and behavior, particularly in fields like drug discovery and quantum chemistry. Traditional methods for obtaining 3D geometries, such as Density Functional Theory (DFT), are computationally expensive and time-consuming.
3. Protein Large Language Models
Large language models have been increasingly used in proteomic research. These models have provided new insights and capabilities in understanding and manipulating proteins. This section presents a comprehensive review of LLMs for proteins (called Prot-LLM). An overview of this section is shown in Figure 8.
Also, the general classification of methods and subcategories of the models examined is shown in Figure 9.
3.1. LLMs for Protein Sequence Representation
Transformer for Protein Language Model is proposed in [148]. This model used a Deep Transformer with Self-Supervised Learning to predict protein structure. The model is trained on 250M protein sequences using self-supervised learning (masked language modeling) to extract representations encoding secondary/tertiary structure, biochemical properties, and homology. It performs better than LSTMs and traditional methods in protein structure prediction, contact prediction, and mutational effect analysis. It also enables state-of-the-art supervised predictions for multiple tasks. The model uses the masked language modeling technique and processes protein sequences through self-attention to build representations of secondary/tertiary structure and biological properties. Structural predictions use linear projections. Regarding the model’s limitation, large datasets are required for pertaining, while large sequence datasets are underfitted due to limited model capacity. The model is computationally expensive, and performance degrades with sparse or highly diverse data.
MSA transformer [149] was developed to predict protein structure using axial attention. MSA Transformer models aligned protein sequences (MSAs) using self-supervised learning to extract structural and functional information from protein sequences. The model outperforms previous models like Potts and ESM-1b with fewer parameters, effectively handles low-depth MSAs, and uses fewer resources. It combines row and column attention to capture sequence alignments efficiently and uses masked language modeling to predict structure from corrupted MSAs. This model requires large MSAs for training, and performance decreases with highly sparse or extensive input sequences. Moreover, it is computationally intensive.MSA Transformer. This model was developed to predict protein structure using axial attention. MSA Transformer models aligned protein sequences (MSAs) using self-supervised learning to extract structural and functional information from protein sequences. The model outperforms previous models like Potts and ESM-1b with fewer parameters, effectively handles low-depth MSAs, and uses fewer resources. It combines row and column attention to capture sequence alignments efficiently and uses masked language modeling to predict structure from corrupted MSAs. This model requires large MSAs for training, and performance decreases with highly sparse or extensive input sequences. Moreover, it is computationally intensive.
HOLOPORT [150] is a protein representation model that was developed using Multi-scale Graph Representation. This model constructs a multi-scale graph combining proteins’ sequence, structure, and surface information. It uses molecular superpixels for efficient memory representation while maintaining accuracy. It outperforms state-of-the-art methods on ligand binding affinity prediction with fewer parameters and improved efficiency and handles surface and structural features simultaneously for robust representation learning. The model combines graph representation of protein structure (nodes as residues, edges based on proximity) with surface representation (nodes as triangulated regions). It propagates information between layers via directed edges and message-passing neural networks. However, the model requires precomputed protein structures and surface manifolds. The preprocessing step is computationally expensive, and performance heavily depends on structural data quality, making it less suited for proteins without available structural data.
ESM-1v [151] was designed for protein function prediction by employing a Transformer-based Protein Language Model. ESM-1v predicts the functional effects of protein mutations using a Transformer trained on a diverse set of 98M protein sequences. It enables zero-shot and few-shot predictions without additional training. Regarding the model’s advantages, as an efficient and scalable model, it performs better than Outperforms prior models like DeepSequence and EVMutation in mutational effect prediction. Additionally, there is no need for task-specific training or MSA generation. The model uses masked language modeling to learn protein representations. For zero-shot inference computes the probability of a mutation occurring in a given context directly from pre-trained weights. Few-shot uses MSA sequences as additional context. This model requires large-scale pretraining datasets. Performance may degrade when applied to proteins or mutations not well-represented in the training data, and it is computationally expensive for large-scale applications.
ProtTrans [152] is a protein representation model based on transformer-based protein language models. This model leverages pre-trained language models (e.g., ProtBERT, ProtT5) on protein sequences to predict structural and functional properties. It uses self-supervised learning with masked language modeling. The model that scales to large protein databases eliminates the need for MSAs and evolutionary information, achieving competitive results with lower computational costs. It processes protein sequences like natural language sentences, learning representations directly from sequences. These embeddings are utilized in downstream tasks such as structure prediction and subcellular localization. Nevertheless, this model is computationally expensive during pre-training, and performance may degrade on proteins not well-represented in the training data. In addition, it requires substantial HPC resources for large-scale datasets.
Pairwise Masked Language Model (PMLM) [153] is a protein representation model developed through the Pairwise Masked Language Model (PMLM). PMLM enhances protein sequence representation by pre-training a transformer with pairwise masked token prediction, capturing co-evolutionary information directly from protein sequences without requiring MSAs. This model outperforms MSA-based methods in contact prediction on the TAPE benchmark and effectively captures inter-residue dependencies, leading to improved protein structure and function predictions. It utilizes token-level masked language modeling (MLM) and pairwise masked modeling (PMLM), builds pairwise labels for residues, and predicts residue pairs with transformer encoders. Nevertheless, it requires extensive computational resources and large-scale pre-training datasets, and its performance may decrease for sequences with sparse or missing evolutionary information.
Joint Representations of Sequence and Structure [154] is another protein representation model designed with Semi-Supervised Learning with SE(3)-Equivariant Transformer. It jointly encodes protein sequence and structure information using semi-supervised learning to create a unified embedding space for downstream tasks like mutation effect prediction and stability estimation. By capturing complex relationships between amino acids in sequential and structural spaces, the model outperforms sequence-only baselines (e.g., ESM-1b) in mutation effect prediction. Additionally, it does not require experimentally validated structures, which makes it more versatile. The model combines sequence embeddings (from ESM-1b) and structural information in a SE(3)-equivariant transformer. In this model, embedding space is used for masked sequence and structure recovery, as well as fine-tuned tasks like thermal stability prediction. It should be noted that the model requires high computational resources for training, and it is dependent on predicted or experimental structural data, which may affect performance for low-quality inputs.
ProteinBERT [155] The model is a protein representation and function prediction model developed through Multi-scale Graph Representation. ProteinBERT introduces a self-supervised approach with a novel pre-training task combining bidirectional language modeling and Gene Ontology (GO) annotation prediction to capture protein sequence and function information. It achieves near state-of-the-art performance on multiple protein tasks while smaller and faster than other models. This model does not require MSAs or evolutionary data, making it efficient and scalable. ProteinBERT combines sequence embeddings with GO annotations to create local and global representations using transformer-like blocks. Embedding space is used for downstream tasks like structure prediction and stability estimation. However, the model requires large-scale protein datasets for training. In addition, it is computationally expensive in the pre-training stage, and performance may degrade with low-quality data. The model does not directly handle pairwise interactions (e.g., contact predictions).
LM-GVP [156] is a protein property representation model built with the application of an End-to-End Deep Learning Framework Combining Protein LM and GNN. The model integrates sequence and structural information using a pre-trained protein language model (Protein LM) with a Graph Neural Network (GNN) to predict protein properties. The framework supports end-to-end training. It achieves state-of-the-art performance in predicting protein properties, including fluorescence, protease stability, and gene ontology functions. The model preserves structural information in LMs and enhances zero-shot mutation prediction. LM-GVP combines amino acid embeddings from Protein LM with structural features represented in a graph (GVP network). The structural features are processed as scalar and vector embeddings, and gradients are propagated for joint optimization. The model requires substantial computational resources for training, and its performance is dependent on the quality of structural data and may degrade with incomplete or low-quality input structures.
Retrieved Sequence Augmentation (RSA) [157] is a protein representation model developed employing the Retrieval-Augmented Language Model. It enhances protein representation by retrieving sequences similar to the input (homology or structural similarity) and augmenting them for downstream predictions without requiring MSAs or alignment. The model outperforms MSA-based methods in tasks like protein structure and property prediction while being 373x faster and exhibiting better generalizability to novel protein domains and efficient on-the-fly retrieval. It uses dense sequence retrieval to find homologous or structurally similar proteins, combines the query and retrieved sequences, and processes them with a transformer encoder for property prediction. However, the model requires high-quality pre-trained embeddings and large protein databases for effective retrieval. It may show decreased performance on proteins lacking homologs in the database.
OntoProtein [158] model used a Knowledge-Enhanced Pre-training with KG and MLM for protein representation and pre-training. OntoProtein integrates external biological knowledge from Gene Ontology (GO) into protein language models using a hybrid encoder and contrastive learning with knowledge-aware negative sampling. The model outperforms existing models such as ProtBert and achieves competitive performance without MSAs. It also enhances protein-protein interaction and function prediction tasks using external structured knowledge. OntoProtein combines sequence embeddings from pre-trained ProtBERT with GO knowledge via a hybrid encoder and optimizes jointly for masked protein modeling (MLM) and knowledge graph embedding (KE) objectives. However, the model requires high computational resources for training and pre-processing. It is limited by the quality and coverage of Gene Ontology data, which may restrict performance on specific protein subsets.
ESMFold [159] another protein structure prediction model is ESMFold, which leverages a Transformer-based Protein Language Model. ESMFold predicts atomic-level protein structures using pre-trained protein language models (ESM-2) without requiring MSAs or templates, providing faster structure predictions. The model provides an order of magnitude faster inference than AlphaFold2, with comparable accuracy for low-perplexity sequences. It also efficiently predicts structures for large metagenomic datasets with low resource requirements. The model utilizes the ESM-2 language model for sequence encoding and predicts structure directly from single sequences. The folding trunk processes the sequence, and the structure module generates atomic-resolution 3D coordinates. The model’s performance degrades for sequences with high perplexity or limited representation in the training data. ESMFold is computationally expensive for training and highly dependent on the quality of pre-trained language models.
PromptProtein [160] is a protein representation and function prediction model developed using a Prompt-Guided Multi-Task Pre-Training Framework. During pre-training, PromptProtein leverages prompt-based learning to integrate multi-level structural information (primary, secondary, tertiary, quaternary). The model learns task-specific representations for diverse protein prediction tasks. It outperforms state-of-the-art models in function prediction and protein engineering tasks. The model is especially effective in low-resource settings, achieving up to 17% improvement in such scenarios and reducing task interference in multi-task learning. This model utilizes three pre-training tasks: Masked Language Modeling (MLM) for primary structure, Alpha-Carbon Coordinate Prediction (CRD) for secondary/tertiary structures, and Protein-Protein Interaction (PPI) for quaternary structures. However, PromptProtein is computationally expensive for training, and its Performance depends heavily on the quality of pre-training data, so some structural information may not transfer effectively to downstream tasks.
KeAP (Knowledge-exploited Auto-encoder for Protein) [161] enhances protein representation by performing token-level exploration of knowledge graphs using QKV attention, enabling granular integration of biological knowledge into primary structure modeling. The model focuses on improving masked amino acid restoration by attending to relational and attribute terms from knowledge graphs. It outperforms OntoProtein and ProtBert on 9 downstream tasks, including contact prediction and protein-protein interaction identification. It uses a simpler optimization strategy (only MLM objective) and achieves higher granularity in encoding knowledge. This model integrates relation and attribute terms into amino acid representations via token-level cross-attention blocks. The extracted knowledge is incorporated into protein sequences through residual learning and MLM-guided training. However, the model is computationally expensive for training. Its performance heavily relies on the quality and comprehensiveness of the underlying knowledge graph. The model lacks focus on local structural details, affecting tasks like secondary structure prediction.
ProtFlash [162] is a lightweight protein language model designed to efficiently extract semantic and structural information from protein sequences. It uses a mixed chunk attention mechanism combining local and global attention for improved performance. This language model achieves equivalent or superior performance to state-of-the-art models while significantly reducing computational complexity and memory usage. Linear complexity makes it more hardware-friendly for long sequences. It uses chunking to divide sequences into smaller segments. Local attention captures nearby interactions and global attention models long-range dependencies. It also employs the UniRef50 dataset for pre-training and fine-tuning tasks. However, the model’s performance can still lag on secondary structure prediction compared to larger models like ESM-1b. Its effectiveness depends on sequence quality and may require additional tuning for specific downstream tasks.
ESM-GearNet [163] is a protein representation and function prediction that employs a Sequence-Structure Hybrid Encoder with Structure-Based Pre-Training. It combines the ESM-1b protein language model with GearNet protein structure encoder, incorporating sequence and structural information through a series fusion approach and pre-training with Multiview Contrast to align sequence-structure representations. The model performs better than existing PLMs and structure-based methods on protein function prediction benchmarks (e.g., EC and GO tasks). It benefits from both sequence-based and structure-based pre-training for enhanced biological correlation. ESM-GearNet processes protein sequences with ESM-1b and encodes residue-level structures with GearNet. Pre-training aligns sequence and structure representations using contrastive learning, and series fusion integrates outputs for final predictions. Regarding the model’s limitation, it should be mentioned that it is computationally expensive for pre-training on large datasets. The model’s performance depends on high-quality structure data and the sequence and structural learning balance. SaProt [164] introduces a novel structure-aware vocabulary that integrates residue and 3D structural information into SA tokens. The model is trained on 40 million protein sequences and structures with a BERT-style MLM objective. It outperforms ESM family models across 10 protein function and structure prediction tasks. This model also achieves superior residue-level and protein-level predictions with consistent improvements over baselines. SaProt combines residue and structure tokens derived from Foldseek into SA tokens. It uses a transformer-based architecture (ESM-2 backbone) and employs masking strategies for sequence and structure recovery during training. However, the model is computationally expensive, and its performance relies heavily on Foldseek’s accuracy and high-quality structure data. It may also struggle with less accurate or incomplete structures. ProteinNPT [165] leverages a non-parametric transformer architecture to jointly represent protein sequences and associated property labels. It uses semi-supervised learning with denoising and target prediction objectives for property prediction and design tasks. The model achieves state-of-the-art performance in protein fitness prediction and iterative redesign while handling multi-task optimization effectively and adapting to label-scarce settings. It also allows conditional sampling of protein sequences for specific properties. This model embeds sequences and labels jointly, applying row and column self-attention to learn interdependencies. ProteinNPT uses masked language modeling for amino acid reconstruction and target prediction and incorporates auxiliary labels for better predictions. The model is computationally expensive due to the use of tri-axial attention and large datasets, and its performance depends on high-quality embeddings and accurate auxiliary labels.
CaLM[166] is a codon language model trained on cDNA sequences using a masked language modeling objective to extract biologically meaningful representations. The model uses codon space, which contains richer biological information than amino acid sequences. This model outperforms state-of-the-art amino acid-based models in tasks such as species recognition, melting point prediction, and solubility estimation despite having fewer parameters and being computationally efficient. It maps codon sequences into a latent embedding space, processes them through a stack of transformer encoder layers, and employs masked language modeling to learn representations. Then, it combines codon embeddings to capture sequence-level information. However, the model is not without limitations. It relies on high-quality cDNA datasets. The performance decreases if codon usage patterns are corrupted or incomplete. The model is limited by preprocessing complexity and the need for large-scale training resources.
ProteinINR[167] integrates protein sequences, structures, and molecular surfaces using Implicit Neural Representations (INRs) to provide comprehensive protein representations for downstream tasks like function prediction and fold classification. It outperforms previous methods by incorporating surface features alongside sequence and structural information and better represents molecular interactions and surface-driven biological processes. The model encodes sequence information using ESM-1b, structure information using GearNet, and surface features with INR-based embeddings. Multi-view contrastive learning aligns these modalities for comprehensive protein representation. ProteinINR relies heavily on high-quality structural and surface data. It is also computationally intensive, requiring large-scale training resources, and performance may degrade for proteins without experimentally validated structures.
ESM-AA (ESM All-Atom)[168] integrates residue and atom scales for unified molecular modeling by pre-training on multi-scale code-switch protein sequences and capturing relationships through multi-scale position encoding. It performs better than previous methods in protein-molecule tasks, capturing both molecular knowledge and protein understanding, enabling versatile applications in protein-molecule interaction, drug discovery, and enzyme engineering. ESM-AA constructs code-switched protein sequences by unzipping residues into atoms and aligning multi-scale relationships using rotary position embeddings for residues and spatial distance matrices for atoms. This model is computationally expensive due to multi-scale pre-training, requiring high-quality structural data and large-scale datasets. The model’s performance may degrade when atom or residue data is incomplete or noisy. KnowRLM [169] integrates domain knowledge using an Amino Acid Knowledge Graph (AAKG) to enhance protein-directed evolution tasks. It employs a knowledge-aware policy network and dynamic sliding windows for efficient protein mutation exploration. The model efficiently identifies high-fitness mutants with fewer experimental rounds. It improves prediction accuracy by integrating amino acid biochemical properties, reducing experimental costs and time. This model constructs an AAKG to model amino acid relationships. A knowledge-aware policy predicts mutation sites and types using preferential random walks on the AAKG, and a fitness predictor provides rewards to optimize the policy iteratively. However, KnowRLM is computationally expensive due to RL iterations and AAKG construction, and its performance depends on the quality of the knowledge graph. It can degrade with limited or noisy biochemical data.
ESM3[170] integrates sequence, structure, and function modalities into a unified latent space using tokenization, enabling multimodal protein representation. The model outperforms prior models in sequence, structure, and function prediction tasks. It is capable of generating novel proteins that simulate evolutionary processes over millions of years. This model uses a bidirectional transformer with tokens for sequence, structure, and function. It was trained with a masked language modeling objective, enabling generation from any combination of modalities. The model is computationally expensive due to large-scale pre-training (98 billion parameters), and its performance relies on high-quality training datasets. In Addition, generative outputs may degrade with incomplete or noisy input data. Summary of LLMs for Protein Sequence Representation is given in Table
3.2. LLMs for Protein Sequence Generation and Designing
The paper [171] introduces ProGen, a powerful AI model with 1.2 billion parameters. It’s trained on a massive dataset of 280 million protein sequences and can design proteins with specific features. ProGen has shown remarkable ability to create proteins that function and look just like natural ones, making it an exciting tool for developing new proteins or enhancing existing ones. ProtGPT2 [172] is another AI model focused on protein design. Think of it as an AI that understands and speaks the language of proteins. It can create protein sequences that closely resemble natural ones and even tackle challenging structures, like membrane proteins. However, while ProtGPT2 shows a lot of promise, it still relies on computer models for validation, so lab testing is essential to confirm its effectiveness. Fold2Seq[173] takes a creative approach by focusing on 3D protein shapes rather than just their linear structures. This model uses advanced AI techniques to generate diverse and functional proteins, even when the input data is incomplete or low quality. That said, there’s still work to do in capturing the finer details of protein structures. ZymCTRL [174] is designed specifically for enzyme generation. Using a huge database of enzyme sequences, it can produce enzymes with user-defined functions tailored to specific chemical reactions. This model has the potential to address complex chemical challenges and offers a fresh way to engineer enzymes. MSA2Prot[175] is a model that generates proteins based on multiple sequence alignments (MSAs). It’s particularly efficient because it doesn’t need to be retrained for every new protein family, making it faster and more flexible than traditional methods. It’s also great at predicting how changes in a protein’s sequence might affect its function. RITA[176] is a family of protein-generating AI models that scales up to an impressive 1.2 billion parameters. These models are designed to predict things like the next amino acid in a sequence or the fitness of a protein. Larger models in the RITA family consistently perform better, positioning RITA as a game-changer in protein research. IgLM[177] focuses on designing antibodies. It creates synthetic libraries of antibodies that mimic natural ones but come with added benefits, like reduced immune responses. This tool could significantly speed up the discovery of new antibody-based therapies. LM-Design[178] combines protein sequence data with structural information to design high-quality proteins. It’s especially good at handling complex structures, outperforming many existing methods. This makes it a flexible tool for designing everything from simple proteins to more complicated ones, like antibodies. MSA Transformer [179] uses a clever masking technique to generate protein sequences. The sequences it creates are similar to natural ones and work particularly well for large protein families. This approach offers a strong alternative to older methods of protein design. [180] a new reinforcement learning framework optimizes protein functionality by tweaking their latent features, which are like hidden characteristics, rather than directly editing their sequences. This technique works well for tasks like improving the brightness of fluorescent proteins or boosting cell fitness. It’s a fresh way of thinking about protein design. ProstT5[181] is an AI model that bridges protein sequences and 3D structures by translating between the two. Using tools like AlphaFold2, ProstT5 can create proteins with specific shapes and functions. This opens up exciting new possibilities for protein engineering. xTrimoPGLM[182] is a next-generation AI model for understanding and creating proteins. With 100 billion parameters, it performs incredibly well across a range of tasks, from predicting protein functions to generating entirely new sequences. However, it’s very computationally demanding and still struggles with unusual data, leaving room for improvement.
Small-Scale Protein Language Model (SS-pLM)[183], a more accessible approach that requires training on merely millions of representative sequences, reducing the number of trainable parameters to 14.8M. This model significantly reduces the computational load, thereby democratizing the use of foundational models in protein studies. The authors have shown that the performance of our model, when tuned to a set of specific sequences for production, is comparable to that of the larger and more computationally intensive PLM.
In [184], the authors introduce and evaluate the pAbT5 model, which demonstrates its efficiency in capturing complex antibody pairing patterns and generating chain pairing sequences with remarkable accuracy. Its performance-targeting properties compared to existing models suggest its utility as a valuable tool in advancing antibody research and therapeutic discovery. They show that our model respects conservation in framework regions and diversity in hypervariable domains, as demonstrated by the agreement with sequence alignments and variable-length CDR loops. In [185], the results show that it is possible to continue scaling the model size (parameters) and see substantial improvements in fitting the distribution of natural sequences. Large protein language models can generate libraries of living sequences that span the sequence and structural space of natural proteins. The test-max50 results and the broad fitness perspective suggest that scaling may particularly show benefits for out-of-distribution, difficult, or tail-end distribution problems. However, our other zero-shot fitness prediction results suggest that the authors needed a better fit to the distribution of the data and the ability to predict desirable performance. Collecting or reducing redundancy through clustering of sequence alignments may not be sufficient.
ProteinRL [186] is a flexible, data-driven approach to de novo design of protein sequences optimized for specific properties. For two different protein design tasks, including single- and multi-target designs, applied to different protein families, ProteinRL has shown high success in generating sequences optimized for the desired scenario. In both cases, few sequences with similar levels of the authors desired properties were found in natural sequences or sequences generated from PLM without fine- tuning ProteinRL. Although the cases highlighted in this study represent real goals in protein design, ProteinRL offers great flexibility in the engineering tasks to which it can be applied. Any property that can be calculated from the sequence or structure of proteins can be used in a reward function for feature-based sequence design. The authors believe that ProteinRL is a promising method for designing novel engineered proteins suitable for use in therapeutic, industrial, and biotechnological applications.
PoET [187] is a transformer-based autoregressive generative model of complete protein families. By framing family generation as a sequence generation problem, tens of millions of protein sequence clusters can be trained to encode the fundamental rules of protein evolution in the PoET model. This enables the model to generalize to protein families not seen during training and to generalize from a small number of conditioned sequences. The sequence generative framework allows PoET to be used as a retrieval-boosted language model, generating new sequences conditional on a set of sequences that represent the family or other features of interest. The authors show that PoET outperforms other protein language models and evolutionary sequence models for predicting fitness on a wide range of deep mutational scan datasets. PoET also enables efficient sequence generation, and the distribution of the product can be controlled through conditioning. Mutase sequences such as lysozyme and chorismate phage were sampled. PoET is novel and is predicted to fold with high confidence. PoET can be supported by other sequence databases and naturally improves as the databases grow without the need for retraining. We predict that PoET will become an essential component of ML-enabled protein design in the future.
[188] proposed Smooth Discrete Sampling (SDS), a new paradigm for modeling discrete distributions that uses Langevin Markov-Chain Monte Carlo to sample smooth data distributions. They introduced the Discrete Walk-Jump Sampling (dWJS) algorithm and evaluated it on antibody discovery and design problems, demonstrating the ability of their method to generate novel, diverse, and functional antibodies that are characterized by the distribution of synthetic biophysical properties, similarity metrics, and measurements. Laboratory experiments The robust regularization provided by fitting the energy function to noisy data completely avoids overfitting and training instability, resulting in fast and efficient training and sampling with low computational requirements. dWJS eliminates many of the common techniques for improving EBM training with Langevin MCMC (replay buffers, standard penalty, simulated annealing, rejection sampling, etc.) and reduces the engineering complexity of training EBMs and diffusion-based models to a single hyper parameter choice: the noise level, . Overall, our results suggest a simpler, more general, and more robust framework for training and sampling energy- and score-based models with applications in the design of therapeutic molecules. Summary of LLMs for Protein Sequence Generation and Designing is given in Table 6
4. Genomic Large Language Models
Genomic language models refer to models that are trained on DNA sequences. These models have the potential to significantly advance our understanding of genomes and how DNA elements interact at different scales to produce complex functions. This section reviews the work done in the LLM for genome sequencing. An overview of the approaches reviewed in this section is given in Figure 10 and Figure 11.
4.1. LLMs for Genomic Data Modeling
Decoding the non-coding DNA language is a fundamental problem in genomic research. The gene regulatory code is extremely complex due to polysemantics and distant semantic relationships, which previous informatics methods often fail to capture, especially in data-poor scenarios. To address this challenge, the authors [189] developed a novel pre-trained bidirectional encoder representation, DNABERT, to obtain a global and transferable understanding of genomic DNA sequences based on upstream and downstream nucleotide contexts. They compared DNABERT with the most widely used programs for genome-wide regulatory element prediction and demonstrated its ease of use, accuracy, and efficiency. They showed that the pre-trained single transformer model can simultaneously achieve improved performance in predicting promoters, binding sites, and transcription factor binding sites, after easy fine-tuning using small, task-specific labeled data. Furthermore, DNABERT enables direct visualization of nucleotide-level significance and semantic relationship within input sequences for better interpretability and accurate identification of conserved sequence motifs and functional genetic variant candidates. In [190], the authors present a new self-supervised MoDNA framework. Instead of directly changing the NLP paradigm to DNA language, they have performed motif patterns in the pre-training stage. By learning from the replaced tokens from the generator, it overcomes the data mismatch limitation in BERT. Meanwhile, the MoDNA discriminator is used to learn and discriminate from all input tokens, which also helps the learning efficiency. They have developed self-supervised motif prediction tasks by incorporating the domain knowledge pattern into the MoDNA pre-training. They have performed motif prediction in the generator and motif occurrence in the checker, respectively. With the help of prior domain knowledge, MoDNA can learn a rich semantic DNA representation. By fine-tuning MoDNA, they have achieved advanced performance in the promoter prediction and transcription factor binding sites prediction of downstream tasks, which proves the power of MoDNA.
The authors in [191] train a masked language model on over 800 species spanning over 500 million years of evolution and show that explicit species modeling is useful in capturing conserved yet evolving regulatory elements and in controlling oligomeric biases. They extract embeddings for the untranslated region of Saccharomyces cerevisiae and Schizosaccharomyces pombe and use them to obtain mRNA half-life predictions that are better than or on par with the state-of-the-art, demonstrating the utility of the approach for surveillance. Genomics Furthermore, they show that the probability of each base being reconstructed in their model significantly predicts protein-binding sites directly bound to RNA. Collectively, their work establishes a self-supervising framework to utilize large genome assemblies of evolutionary distant species for regulatory genomics and to aid alignment-free comparative genomics. In [192], the authors developed a set of DNA LMs based on the transformer, NT, which learned generic nucleotide sequence representations from 6 kb of unannotated genomic data. Inspired by the NLP trend, where larger training datasets and model sizes show better performance, transformer models with different parameter sizes and datasets were built:
- A 500 million parameter model trained on sequences extracted from the human reference genome.
- A 2.5 billion parameter model, both trained on 3202 genetically diverse human genomes
- A 2.5 billion parameter model, including 850 species from different phyla including 11 model organisms.
To assess the performance of these models in predicting diverse molecular phenotypes, they selected 18 genomic datasets from publicly available sources, including binding site prediction tasks, promoter and histone modification tasks, and enhancer tasks, each designed to be of reasonable size to allow for rapid and accurate convergence. Validation Procedures While larger datasets are available for supervised models, this set of 18 selected genomic datasets provides a diverse and robust set of data to rigorously examine the models’ performance across tasks in a statistically rigorous manner and for comparison with other DNA-supervised fundamental models. The datasets were processed in a standardized format to facilitate testing and ensure reproducibility in evaluating the performance of large LM. They evaluated their transformer models through two different techniques: probing and fine-tuning. Exploration refers to the use of LM embeddings learned from DNA sequences as input features for simpler models to predict genomic tags.
HyenaDNA [193] is a genomic foundation model that processes sequences of up to 1M nucleotides at single-nucleotide resolution, pre-trained on the human genome using Implicit Convolution & Long Context Handling. The model increases 500 times more in sequence context length compared to previous models, and it has a faster training time (160x faster than transformers). In addition, it achieves state-of-the-art genomic benchmarks. HyenaDNA uses a decoder-only Hyena operator with single-nucleotide tokens and soft prompting and incorporates a sequence warm-up schedule for stability and a token-based adaptation. However, the model requires significant computational resources, and it relies on high-quality training data.
GENA-LM [194] is a transformer-based foundational model designed to handle long DNA sequences containing up to 36,000 base pairs by employing Recurrent Memory Mechanisms to capture long-range dependencies. The model outperforms existing models like DNABERT, enabling extended sequence handling, enhanced performance for genomic tasks, and fine-tuning across various taxa. It also provides state-of-the-art genomic task benchmarks and web service integration. This model uses Byte-Pair Encoding tokenization for diverse sequence lengths, sparse memory attention mechanisms for extended contexts, and fine-tuning functional genomic elements like promoters, splicing, and chromatin profiles. However, the model is computationally intensive due to memory and sparse attention mechanisms. It relies heavily on high-quality datasets for training and task-specific fine-tuning, and its performance decreases with evolutionarily distant genomes.
Geneformer[195] is a transformer-based model pre-trained on 30 million single-cell transcriptomes to encode gene regulatory networks. The model facilitates fine- tuning for specific tasks in network biology, even with limited data. Regarding the models advantages, it enables context-aware gene expression and regulation predictions. Geneformer performs state-of-the-art in modeling gene networks, predicting therapeutic targets, and functional genomics tasks. The model employs masked self-supervised learning to pre-train on rank-based transcriptomic data, capturing dependencies between genes. It applies transformers to model gene regulatory networks in specific biological contexts during fine-tuning. However, the model is computationally intensive due to large-scale pre-training, requiring high-quality and diverse datasets to generalize across biological contexts. Its Performance may degrade for cell states or conditions not well-represented in the training data.
EpiGePT [196] is a genomic language model developed using a pre-trained transformer with multi-task learning—the model is designed for cross-cell-type prediction of epigenomic signals and chromatin interactions. EpiGePT surpasses baseline models like Enformer in epigenomic predictions. It also integrates 3D chromatin interaction data for enhanced predictions and enables cross-context predictions for regulatory elements. The model employs sequence and transcription factor (TF) modules to encode genomic and contextual features, multi-task learning, and masked training to handle incomplete data. To predict chromatin interactions, an attention mechanism has been developed to identify dependencies and predict interactions. However, it is computationally intensive due to large-scale data and multi-task learning. In addition, its performance relies on high-quality training data and accurate annotation of chromatin interactions. The prediction accuracy will decrease if the chromatin interaction data is incomplete or noisy.
The ENBED (Ensemble Nucleotide Byte-level Encoder-Decoder) model[197] is a genetic sequence analysis model that uses Byte-level tokenization techniques, Encoder-Decoder architecture, and Sub-quadratic Attention mechanism. This model performs Sequence-to-Sequence tasks such as mutation prediction, enhancer identification, and splice site detection. Compared to the previous models, the model predicts genetic sequence noises with higher accuracy (97.6%), demonstrate improved performance in 21 of 25 genetic benchmarks. Using byte-level tokenization increases the accuracy of managing single nucleotide changes and reduces sensitivity to noise. This model uses a combined attention mechanism (Sliding window and Global Attention) to process long sequences. The 2:1 ratio between Encoder and Decoder blocks is designed for balanced learning. Self-supervised Learning and Masked Language Modeling (MLM) algorithms train the model. However, the model contains limitations. Due to the byte-level tokenization and the attention mechanisms complexity, the models computational cost is high. Large and diverse data sets are needed for pre-training and proper performance. In addition, increasing the computational operations (FLOPs) becomes challenging for long sequences. DNAGPT[198] is a generalized pre-training model for DNA analysis that employs multi-task pre-training strategies, such as Next Token Prediction, guanine-cytosine content prediction, and Sequence Order Prediction. This model uses a transformer- based architecture and a masked self-attention mechanism. Tokens include DNA sequences, numeric values, and specialized commands. The model can do various tasks, including predicting genomic signal regions (GSR) and mRNA expression levels and generating synthetic DNA sequences. In GSR tasks, DNAGPT surpasses previous models such as DeepGSR and GSRNET. In mRNA expression prediction, the model outperforms Xpresso. This model performs better than GAN and RBM models in generating synthetic genomic sequences. DNAGPT uses the transformer architecture to pre-train and process sequence and numerical data, and the main tasks include predicting content and sequence order. The models limitations include focusing solely on DNA data and requiring significant computational resources. It is also limited to non-genetic tasks, as the current focus of the model is only on DNA sequences, and support for multimodal data integration remains unimplemented.
Evo [199] is a foundational genomic model with 7 billion parameters designed for prediction and generation tasks, ranging from molecular to genome scale. It processes sequences of 131 thousand nucleotides with single-nucleotide accuracy. The initial training of the model was conducted using the OpenGenome dataset containing more than 300 billion nucleotides. The training data includes bacterial and viral genomes of prokaryotes. The model can process very long sequences, has high performance in predicting the effects of protein mutations, and uses large- scale data. It has shown high performance in predicting the impact of protein mutations and the function of non-coding RNAs (ncRNA) without special training. Additionally, the model can produce complex biological systems such as CRISPR- Cas molecules and genetic transfer systems (Transposable Systems). It uses the StripedHyena architecture, which is a combination of Attention and data-driven filters. It consists of 29 layers of Hyena Layers and 3 Multi-Head Attention layers. However, the model has limitations; for example, it is trained only on prokaryotic data and is limited in predicting the effects of human protein mutations. In addition, the computational cost of the model is high, and challenges with uniformity and precision arise when generating long sequences.
GenomicLLM [200] is a pioneering model that integrates natural language and genomic data. It analyzes DNA sequences and textual descriptions to perform classification, regression, and sequence generation tasks. The model was trained on three large datasets: GenomicLLM_GRCh38 (human genome sequence and annotation data), Genome Understanding Evaluation (GUE) (promoter and binding site prediction), and GenomicBenchmarks (genomic sequence classification). GenomicLLM performs comparable or superior to DNABERT-2 and HyenaDNA on classification tasks, outperforming these models on 7 out of 13 benchmarks. The model uses a hybrid tokenization method, including BPE tokenization for text and a unique algorithm for genetic data. Classification tasks include Splice Site Prediction, Promoters and Enhancers Detection, and Transcription Factor Binding Sites Prediction. Regression tasks involve predicting guanine-cytosine content. Generation tasks include producing amino acid sequences from gene sequences, complementary and reverse-complementary sequences, and gene descriptions. While GenomicLLM achieves impressive results, it is computationally intensive due to large-scale data and hybrid tokenization. Additionally, the model is limited in its application to unusual genomic tasks.
4.2. LLMs for Function Prediction
Enformer[201] is a genomics and gene expression prediction model that uses the Transformer architecture to extend the Receptive Field and analyze long-range genetic interactions. Enformer is designed to predict gene expression and chromatin states from DNA sequences. The receptive field of the model has been increased from 20 kb to 100 kb, which increases the accuracy of the prediction. This model, capable of understanding and processing long-range regulatory interactions, performs better than the Basenji2 and ExPecto models and has improved accuracy in predicting the effects of gene mutations. The model is constructed by integrating Transformer and Convolutional layers, and Attention Layers connect long-range elements better. Also, the model makes more accurate predictions using multitask learning on human and mouse data. Limitations of the model include the need for extensive data sets, computationally intensive processing, and limitations in generalizing the model to new cell types or experiments that were not in the training data.
iEnhancer-BERT [202] is a genomic analysis model using transfer learning with BERT and CNN architecture. It predicts DNA enhancers and their strength using a BERT-based DNA language model pre-trained on the human genome, fine-tuned with transfer learning for enhancer tasks. This model outperforms existing models (e.g., BERT-2D, SENIES) on benchmark datasets, achieving state-of-the-art performance in enhancer identification and strength differentiation tasks. The model uses DNABERT for sequence representation and employs a CNN module for feature extraction. All outputs from 12 transformer encoder layers are concatenated for enhanced feature extraction and classification. However, it requires significant computational resources. The model is limited by fixed-length input sequences (200 bp) and struggles with cell-type-specific enhancer prediction and variable- length sequences. iDNA-ABF[203] is a DNA Methylation Prediction model developed using a Multi-scale Biological Language Model (with k-mers and BERT architecture). It predicts DNA methylation sites across species and methylation types (4mC, 5hmC, 6mA). It uses multi-scale tokenization (3-mer and 6-mer) to enhance feature representation. This model surpasses state-of-the-art predictors on 15 out of 17 benchmark datasets, offering interpretable predictions and robust performance across species. It uses multi-scale tokenization to represent DNA sequences as "biological words," employs BERT encoders for contextual embedding, and integrates adversarial training to enhance robustness. iDNA-ABF utilizes a fusion module for classification that combines multi-scale embeddings. The model requires considerable computational resources for training. The generalization of new methylation types and cell types may be limited, and computational costs increase with sequence length.
iEnhancer-ELM [204] is a genomic analysis model that leverages a BERT-based Enhancer Language Model with multi-scale k-mers tokenization. The model is optimized to identify enhancers by tokenizing DNA sequences into multi-scale k-mers and using contextual embeddings through a pre-trained BERT model. It outperforms state-of-the-art methods in enhancer identification and provides interpretable results, including motif discovery. It tokenizes sequences into k-mers of varying lengths. The model utilizes BERT-based embeddings to capture contextual relations, fine-tunes pre-trained weights on enhancer datasets, and applies an MLP classifier for prediction. However, the model is limited, requiring significant computational resources for training and fine-tuning. In addition, its generalization faces challenges in generalizing to datasets with highly variable enhancer sequences.
[205] presents miProBERT, a new model for predicting microRNA (miRNA) promoters using a pre-trained BERT model, DNABERT. Despite older methods that depend on biological signals like CpG islands or histone marks, miProBERT works directly with gene sequences to make predictions. The model’s accuracy is improved by creating a strong negative dataset with a random substitution technique, which helped the model find deeper patterns and reduced false positives to 0.0421. When tested on 33 experimentally validated miRNA promoters, miProBERT performed better than other existing tools, achieving 78.13% precision and 75.76% recall. The results were further confirmed by analyzing biological signals like conservation, CpG content, and histone marks, proving the reliability of the identified promoters. It also provides accurate predictions using only sequence information, without needing extra data from other sources.
PLPMpro [206] presents a new model that enhances the prediction of promoter sequences using a combination of prompt learning and a pre-trained language model (PLM). Unlike traditional models, in order to maximize the potential of the PLM, the model exploits soft templates and verbalizers, achieving better performance in identifying promoter regions. Extensive experiments are conducted to fine-tune the prompt settings and evaluate the effect of various template configurations and their results show that soft modules significantly boost prediction accuracy. Also, the study highlights how the model learns biological patterns, such as recognizing motifs like the TATA-box in promoter sequences, which proves its ability to capture meaningful biological semantics.
A novel metagenomic pre-trained model designed named FGBERT for impring the understanding of gene functions in metagenomic data is presented in [207]. The model uses a protein-based gene representation as a tokenizer to ensure context-aware and biologically meaningful encoding of sequences. FGBERT takes the advantage of Masked Gene Modeling (MGM) to uncover inter-gene relationships and Triplet Enhanced Metagenomic Contrastive Learning (TMC) to connect gene sequences with their functions. Over 100 million sequences are used for training and the results exhibit exceptional performance across various tasks, such as predicting gene structures, antimicrobial resistance, and nitrogen cycles. The proposed model addresses key challenges in metagenomics, such as the One-to-Many (OTM) and Many-to-One (MTO) relationships, through advanced contextual analysis and contrastive learning.
4.3. LLMs for Variants and Evolution Prediction
Authors in [208] introduced the Genomic Pre-trained Network (GPN), a model designed to predict the effects of genetic variations across an entire genome using only DNA sequences. Inspired by natural language models, GPN learns patterns and structures in DNA, like gene regions and motifs, without requiring labeled data or functional genomics information. It is trained it on genomic data from Arabidopsis thaliana and related species, and the results indicate that it performs better than existing methods like phyloP and phastCons. A special capability of GPN is capturing the context of DNA sequences, making it highly adaptable to other species and useful for tasks like understanding genetic traits or rare diseases. GenSLMs which is presented in [209] is a powerful genome-scale language model for studying the evolution of SARS-CoV-2. GenSLMs uses a unique architecture that combines two advanced techniques: GPT for understanding short-range patterns in the genome and a diffusion-based model for capturing long-range relationships across the entire sequence. By training on over 110 million gene sequences from bacteria and fine-tuning on 1.5 million SARS-CoV-2 genomes, it can quickly and accurately identify concerning variants. The model addresses the challenges of working with long and highly similar genome sequences by learning meaningful biological patterns. AI hardware like the Cerebras CS-2 and GPU supercomputers are used to handle the complexity of such large-scale data. This made the model capable of processing extremely long sequences and being trained efficiently.
[210] introduces GPN-MSA, a powerful new DNA language model created to predict the effects of genetic variants across the entire genome. What makes this model special is its ability to learn from the aligned genomes of 100 vertebrate species, combining biological insights about evolutionary conservation with the advanced capabilities of the Transformer architecture. GPN-MSA works by analyzing small, 128-base pair windows of DNA, masking certain positions, and predicting what’s likely to be there based on the surrounding context and the patterns seen in other species. The model performs exceptionally well, outperforming widely used tools like CADD and SpliceAI in tests involving real-world datasets such as ClinVar and gnomAD only after training in less than five hours on just four GPUs.
4.4. LLMs for DNA-Protein Interaction Prediction
TFBert [211] is a new model for predicting DNA–protein binding sites by leveraging a pre-trained BERT architecture adapted specifically for biological sequences. TFBert treats DNA sequences as natural language sentences and employs the k-mer method to tokenize sequences, where k-mers serve as words. The model’s architecture includes a 12-layer transformer encoder with multi-head attention mechanisms, allowing it to capture the bidirectional context of DNA sequences effectively. Pre-training was conducted on 690 large-scale, unlabeled ChIP-seq datasets using a masked language model strategy, which enhances the model’s ability to generalize, especially for small datasets. TFBert significantly outperformed existing methods and achieved an average AUC of 94.7% in predicting DNA–protein interactions. Authors in [212] present a novel Genomic Language Model (gLM) designed to explore the intricate relationships between genes and their genomic neighbourhoods. Built on a transformer architecture, gLM learns from millions of metagenomic sequences, combining insights from protein language models (pLMs) and the broader genomic context. By masking parts of the input and predicting them based on surrounding genes, the model uncovers how genes work together (forming operons or co-regulated modules). With its attention mechanisms, gLM is very powerful at capturing functional and regulatory patterns in ways that traditional models cannot.
4.5. LLMs for RNA Prediction
In the paper [213] SpliceBERT is proposed to improve our understanding of RNA splicing which is a crucial process in gene expression. The researchers trained SpliceBERT on over 2 million pre-mRNA sequences from 72 vertebrates, using a method that hides parts of the sequence and teaches the model to predict them. This approach helped SpliceBERT learn the relationships between nucleotides and capture evolutionary information embedded in these sequences. There are six Transformer layers and over 19 million parameters in SpliceBERT which cause the model to excel in tasks like predicting splice sites and branchpoints, even outperforming other models trained only on human genome data and by including data from multiple species, SpliceBERT can even demonstrates better generalization.
RNA-MSM, presented in [214] is a language model designed to understand RNA structures by analyzing homologous sequences. Its design is based on transformer architecture where it uses advanced attention mechanisms to focus on the relationships between nucleotides, allowing it to capture structural details like base-pairing and solvent accessibility. Using data from RNAcmap3, which generates rich sequence alignments, the model can predict RNA properties more accurately than existing methods such as SPOT-RNA2. One standout feature of RNA-MSM is its ability to generalize well to RNA families it has not encountered before which is perfect for uncovering the mysteries of RNA structure and function.
An innovative tool for predicting multiple types of RNA modifications, including m6A, m5C, m1A, and pseudouridine, across species like Mus musculus, Arabidopsis thaliana, and Saccharomyces cerevisiae is presented in [215] named MRM-BERT, which is inspired by how humans process language and treats RNA sequences as a biological language using the BERT architecture to understand patterns in these sequences. MRM-BERT avoids the repetitive training required by traditional methods and focuses on extracting meaningful features with its powerful self-attention mechanism. It combines an embedding module, a BERT encoder, and a classification module to deliver precise predictions. MRM-BERT can handle multiple tasks with one model, which makes it easier to explore RNA modifications across different species.
Authors in [216] introduced an innovative deep learning model, Uni-RNA, inspired by the BERT architecture, which is designed to explore the mysteries of RNA structure and function. This model incorporates advanced techniques like rotary embeddings and flash attention to efficiently handle large and complex datasets. Uni-RNA was trained on a dataset of one billion RNA sequences, enabling it to uncover hidden patterns and evolutionary information within RNA molecules. After fine-tuning, the model demonstrated great accuracy in tasks like predicting RNA structures and functions, outperforming previous methods. RiNALMo [217] introduces the largest-to-date RNA language model with 650M parameters, built using a BERT-style Transformer encoder architecture. RiNALMo incorporates advanced features such as rotary positional embeddings (RoPE) for encoding relative and absolute token positions, SwiGLU activation for efficient learning, and FlashAttention-2 for memory-efficient exact attention. The model consists of 33 Transformer blocks, each including multi-head attention with 20 heads and feed-forward networks interconnected through residual connections with layer normalization. It was pre-trained on a curated dataset of 36 million non-coding RNA sequences using masked language modeling (MLM), where 15% of tokens were masked to learn sequence reconstruction. RiNALMo generates 1280-dimensional embeddings that capture structural and evolutionary features of RNA sequences. These embeddings significantly outperform existing models in downstream tasks, including intra- and inter-family secondary structure prediction, splice-site detection, and mean ribosome loading (MRL) regression. It demonstrated exceptional generalization on unseen RNA families, addressing limitations of prior methods. Authors in [218] proposed RNAErnie, a transformer-based model designed for versatile RNA sequence analysis. Its architecture integrates motif-aware pretraining with multilevel masking (base, subsequence, and motif levels) to encode RNA-specific features and a type-guided fine-tuning strategy to adapt to diverse tasks. The model is trained on 23 million RNA sequences from RNAcentral and incorporates biological priors like RNA motifs for enhanced representations. RNAErnie predicts RNA types during fine-tuning, appending them to sequences for downstream tasks, including classification, RNA–RNA interaction prediction, and secondary structure prediction. Experimental results demonstrate significant performance gains, with up to 1.8% higher accuracy in classification, 2.2% better accuracy in interaction prediction, and a 3.3% F1 score improvement in structure prediction, showcasing RNAErnie’s robustness and adaptability.
4.6. Datasets and Benchmarks
Recent breakthroughs in bioinformatics have been driven by the potential of machine learning, particularly through the use of transformer architectures and large language models (LLMs). Originally developed for processing human language, these models have been adapted to analyze biological sequences, such as RNA and DNA, with unprecedented accuracy. Transformers excel at capturing long-range dependencies and contextual relationships, making them ideally suited for modeling the complexities of genetic data. By applying the principles behind LLMs to bioinformatics, researchers are uncovering novel insights into molecular biology, from structural prediction to functional annotation. Papers in this section highlight the growing role of LLMs in bioinformatics, showcasing their ability to generate rich, context-aware embeddings for biological sequences and perform complex tasks like RNA structure prediction and protein interaction analysis. The models, trained on massive, curated datasets, incorporate innovations like multilevel masking and biological priors to enhance their predictive power. The Mammalian Gene Collection (MGC) explained in [219] is an NIH initiative to create a comprehensive resource of full-length complementary DNA (cDNA) sequences and clones for human and other mammalian genes. Aiming to sequence 5,000 to 7,000 full-length cDNAs with insert sizes up to 3-4 kb in its first year, the project plans to expand its pipeline to produce at least 20,000 highly accurate sequences annually, adhering to 99.99% accuracy standards. At the time of writing, only 6,000 full-length sequences were available out of an estimated 80,000 to 100,000 human genes, highlighting the project’s importance. Backed by an initial budget of $10 million, the MGC employs advanced cDNA library technologies to ensure more than 50% of clones contain full open reading frames, with early outputs, such as 5’ and 3’ EST sequences, immediately deposited in GenBank. This publicly accessible resource is set to support genetic and biomedical research by addressing challenges like rare and long transcript identification and cataloging. The paper [220] elaborates on the improvements and impacts of the GRCh38 human reference genome compared to its predecessor, GRCh37, on high-throughput sequencing data analysis. GRCh38 incorporates significant enhancements, including reduced gaps, corrected misassembled regions, added centromere sequences, and increased genetic diversity with alternate loci. It demonstrates improved alignment accuracy, reduced false positives in structural variant detection, and better exome mapping, attributed to a larger and more accurately defined exome size. The findings underline GRCh38’s superior genomic representation and reliability for bioinformatics analysis, marking a substantial advancement in sequencing-based research applications. Authors in [221] investigate convolutional neural network (CNN) architectures to predict DNA–protein binding by exploring various configurations in depth, width, and pooling strategies. The input to their models is a one-hot encoded representation of DNA sequences, and the output is a binary classification indicating binding affinity. Their architecture starts with convolutional layers acting as motif scanners, followed by global max-pooling, a fully connected layer with dropout to prevent overfitting, and a final layer for classification. They demonstrate that increasing the number of convolutional kernels improves performance, particularly for tasks like motif discovery and occupancy. However, deeper architectures and local pooling show limited or negative effects in simpler tasks like motif discovery. Their optimized CNNs outperform existing methods like DeepBind and gkm-SVM, achieving high classification accuracy (AUC close to 0.8) in more complex binding prediction tasks while emphasizing the importance of sufficient training data for complex models.
[222] introduces DeepSEA, a deep learning-based framework for predicting the functional effects of noncoding genomic variants directly from DNA sequences with single-nucleotide sensitivity. DeepSEA uses a convolutional neural network (CNN) architecture comprising three convolutional layers with 320, 480, and 960 kernels, respectively, followed by pooling layers to extract hierarchical sequence features across spatial scales, and a fully connected layer that integrates information from a 1,000-bp sequence. The final sigmoid output layer predicts probabilities for 919 chromatin features, including transcription factor (TF) binding, DNase I hypersensitivity, and histone modifications. Input DNA sequences are represented as 1,000 × 4 binary matrices encoding nucleotide positions. The model employs multitask learning, enabling shared feature representations across chromatin features, which improves computational efficiency and prediction accuracy. Trained on data from ENCODE and Roadmap Epigenomics, DeepSEA achieved high prediction performance, with area under the curve (AUC) values of 0.958 for TF binding. Additionally, it can prioritize functional variants, such as eQTLs and disease-associated SNPs, outperforming other methods. DeepSEA also supports in silico saturated mutagenesis to analyze the impact of base substitutions on chromatin features which offers a powerful tool for interpreting noncoding genomic variants. 1000 Genomes Project dataset [223] is a comprehensive resource of human genetic variation, encompassing the genomes of 2,504 individuals from 26 populations across Africa, East Asia, Europe, South Asia, and the Americas. This dataset includes over 88 million genetic variants, such as single nucleotide polymorphisms (SNPs), insertions/deletions (indels), and structural variants, all organized into high-quality haplotypes. The dataset provides extensive representation of common and rare genetic variants, achieving over 99% detection for variants with frequencies above 1%. This resource facilitates genetic studies by enabling genotype imputation, variant cataloging, and the filtering of neutral variants, and serves as a reference for exploring the genetic diversity and evolutionary history of human populations.
EPDnew [224] is an updated version of the Eukaryotic Promoter Database (EPD) which provides comprehensive, high-quality collections of experimentally defined promoters for key eukaryotic organisms, including humans, mice, and Drosophila. The initial versions of EPDnew include 9,716 promoters for humans, 9,773 for mice, and 11,389 for Drosophila, representing significant expansions compared to the older database, with 40% gene coverage for humans and mice and 70% for Drosophila. EPDnew integrates transcription start site (TSS) data from next-generation sequencing technologies, including CAGE and oligo-capped datasets, and uses ChIP-Seq data for H3K4me3 modifications to enhance precision. Approximately 1,274 promoters with low mRNA 5’-end tag coverage were rescued using chromatin signatures, resulting in sharper TSS peaks and enriched motif occurrences such as TATA-boxes. The dataset is built using an automated pipeline supported by over 2,000 ChIP-Seq datasets, ensuring broad coverage, high resolution, and suitability for genome-wide promoter analysis.
[225] introduces PanglaoDB, that is a user-friendly online database of mouse and human single-cell RNA sequencing (scRNA-seq) data. It aggregates and standardizes data from 1054 single-cell experiments, comprising over 4 million cells from a wide array of tissues and organs, collected from both mouse (845 samples) and human (209 samples). PanglaoDB incorporates pre-processed and pre-computed analyses, enabling researchers to explore cell types, genetic pathways, and regulatory networks without requiring extensive computational resources. It includes a manually curated cell-type marker compendium with 6631 gene markers mapped to 155 cell types, further grouped into 26 organs and 3 germ layers, supporting automated cell-type annotation. The dataset spans major scRNA-seq platforms, including 10X Chromium, Drop-seq, and SMART-seq2, and maintains quality by including only cells with at least 1000 uniquely mapped reads and clusters containing a minimum of 10 cells. As a database with the size of 22 GB, PanglaoDB serves as an evolving and accessible resource for scRNA-seq data exploration and analysis. ExPecto [226] is a deep learning framework designed to predict tissue-specific gene expression levels ab initio from DNA sequences, with a focus on promoter-proximal regions extending 40 kb around transcription start sites. The architecture integrates three components: a deep convolutional neural network to extract epigenomic features, a spatial feature transformation module to summarize these features, and L2-regularized linear models for tissue-specific expression prediction. Inputs are DNA sequences, and outputs are predicted expression levels in log(RPKM) across 218 tissues. ExPecto demonstrated median Spearman correlation of 0.819 in recapitulating gene expression and successfully identified causal variants for immune-related diseases such as Crohn’s disease and hepatitis B. The framework also enabled in silico mutagenesis of over 140 million variants, highlighting evolutionary constraints and advancing the interpretation of non-coding genomic variations. The dataset introduced in [227], the UCSC Genome Browser Database, is a resource that integrates genome sequence data with extensive annotations, facilitating genomic analysis. It includes positional data such as genome start-stop coordinates and non-positional data such as mRNA details, all stored in an optimized MySQL relational database. The dataset supports multiple genomes, such as human, mouse, and others, with continuous updates. Annotations encompass gene predictions, mRNA alignments, SNPs, cross-species homologies, and high-level genomic maps. The database is accessible through a web-based graphical interface, enabling interactive visualization, and provides tools for data downloading, querying, and custom annotation uploads.
The Bacterial and Viral Bioinformatics Resource Center (BV-BRC)[228] is a comprehensive database formed by merging PATRIC, IRD, and ViPR, designed to support research on bacterial and viral pathogens. The BV-BRC hosts over 600,000 bacterial genomes, 11,000 archaeal genomes, and more than 8.5 million viral genomes, including over 6 million SARS-CoV-2 genomes, along with 22,000 bacteriophage genomes and 10 eukaryotic host genomes. Metadata includes laboratory-derived antimicrobial susceptibility test (AST) results for 90,829 bacterial genomes and curated attributes such as host, geographic, and clinical data. The resource integrates annotations for antimicrobial resistance genes, virulence factors, and metabolic pathways, consistently applying tools like RASTtk for bacteria and VIGOR4 for viruses. It also hosts 170 million predicted protein domains and motifs, 300,000 immune epitopes, and 77,000 protein structures, along with tools for comparative analyses. The dataset is enriched with non-genomic data and is continuously updated with new genomes from sources like NCBI GenBank and the Sequence Read Archive, ensuring it remains a vital tool for pathogen research and analysis.
The Arabidopsis Information Resource (TAIR)[229] is a comprehensive genome database for Arabidopsis thaliana, a model organism in plant biology. TAIR provides extensive genetic and genomic data, including 27,416 protein-coding genes, 3,903 transposable element genes, 924 pseudogenes, and various RNA genes such as 689 tRNA, 177 miRNA, and 394 other RNA genes in its TAIR10 release. The database integrates information through manual curation, computational pipelines, and community submissions, supporting tools like genome browsers and advanced search systems. TAIR10 introduced 126 new genes, 2,099 new splice variants, and updated 707 coding sequences. The dataset usage is over 45,000 and 1.8 million users monthly and yearly, respectively, making it an invaluable resource for plant biology research. RNAcmap[230] is an automated method for predicting RNA contact maps using evolutionary coupling analysis. The architecture integrates a two-stage homology search: an initial sequence-based BLAST search, followed by a secondary structure-informed covariance model search using Infernal. Secondary structure prediction is performed with RNAfold or the deep-learning-based SPOT-RNA, enhancing homologous sequence identification. The model outputs base-pair and distance-based contact maps. Evaluation shows that RNAcmap achieves accuracy comparable to Rfam’s manually curated alignments for sequences within Rfam and robust performance for non-Rfam sequences. RNAcmap demonstrates potential in RNA structure prediction by providing reliable structural restraints. [231] introduces the Encyclopedia of DNA Elements (ENCODE) dataset, a comprehensive resource aimed at annotating all functional elements in the human genome. The dataset includes 1,640 experiments across 147 cell types, categorized into three tiers, and reveals that 80.4% of the genome participates in at least one biochemical event, with 95% of the genome within 8 kilobases (kb) of a regulatory element. Key features include 399,124 enhancer-like regions, 70,292 promoter-like regions, and 636,336 DNA-binding sites covering 231 megabases (8.1%) of the genome. The dataset also maps 2.89 million DNase I hypersensitive sites across 125 cell types and 8.4 million DNase I footprints in 41 cell types, while providing transcription data for 20,687 protein-coding genes, 9,640 long non-coding RNA loci, and 62,403 transcription start sites. Additionally, it profiles 1.2 million CpG sites for methylation across 82 cell lines and identifies 127,417 promoter-centered chromatin interactions. This dataset offers an expansive resource for understanding genome regulation and its implications for human biology and disease. The Viral Genome DataBase (VGDB) introduced in [232] is a mySQL-based system designed for storing and analyzing genes and proteins from large viral genomes (>100 kb), encompassing 15 fully sequenced viral genomes and 2847 genes. The database includes comprehensive information such as DNA and protein sequences, molecular weight, isoelectric point, amino acid content, nucleotide frequency, and codon usage. It is accessible via a user-friendly JAVA GUI and supports advanced querying, sorting, and data export. VGDB enables efficient analysis of large datasets, including detecting gene prediction errors and studying genome composition.
[233] presents the Evolutionary Action (EA) method, a computational framework used to predict the functional impact of genetic variants by calculating a mutation’s fitness effect. The method uses sequence homology data and calculates fitness as a product of sensitivity to residue changes and the magnitude of substitution, normalized to percentile ranks. EA was applied to multiple challenges in the Critical Assessment of Genome Interpretation (CAGI5) experiment, addressing problems such as protein function prediction, stability, and clinical association of variants. The EA model demonstrated robust performance across diverse tasks, showing particular reliability in tasks like clinical interpretation of BRCA variants and predictions of protein stability. [234]systematically evaluates 37 machine learning methods for predicting RNA-binding protein (RBP) interactions, focusing on deep learning models. It addresses the challenge posed by heterogeneous datasets and protocols, using a uniform preprocessing strategy and benchmarking 11 representative methods across 313 unique CLIP-seq datasets from three repositories. The study evaluates models based on architecture, input modalities, and sampling strategies for generating negative samples, highlighting that deep learning methods outperform traditional machine learning approaches like SVMs. It explores the impact of input features such as RNA secondary structure, genomic context, and conservation scores on model performance. Based on the findings of authors, multi-task learning models like DeepRiPe and Multi-resBind showed higher resilience to CLIP-seq biases, outperforming binary classifiers. The study emphasizes that auxiliary inputs like conservation scores significantly enhance model accuracy, and it underscores the importance of long input sequences for capturing broader binding contexts. This work provides a better insight for selection and development of computational methods for RBP-binding prediction.
Author Contributions
Ramin Mousa, Ali Sarabadani, Amir Ali Bengari, Omid Eslamifar, and Mohammad Alijanpour Shalmani contributed to the design and implementation of the study, data collection, and initial drafting of the manuscript. Tania Taami performed the statistical analysis and contributed to the interpretation of the data. All authors read and approved the final manuscript.
Funding
No funding.
Informed Consent Statement
Not applicable.
Acknowledgments
In this section you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no competing interests.
Appendix A
Some of the common libraries used for LLM training are listed below:
Transformers: Transformers [235] provides thousands of pre-trained models for tasks in different modes, such as text, vision, and audio. It is also used for training, fine-tuning, inference, and custom models. It provides text models for tasks such as text classification, information extraction, question answering, summarization, translation, and text generation in over 100 languages. It provides image classification, object recognition, and segmentation in the image category, as well as speech recognition and audio classification in the audio category.
DeepSpeed: DeepSpeed[236] provides access to the world’s most powerful language models, such as MT-530B and BLOOM. It is an easy-to-use deep learning optimization software suite providing unprecedented scale and speed for training and inference. It also provides dense or sparse train/inference models with billions or trillions of parameters. It achieves excellent system throughput and efficient scaling to thousands of GPUs. It also enables extreme compression for unparalleled inference latency and model size reduction at low cost. The library is capable of training the following large-scale models:
Megatron-LM: Megatron-LM[238] is one of the first frameworks for LLM, introduced in 2019. Many of the most popular LLM development frameworks are inspired by and built on the open-source Megatron-LM library. Some of the most popular LLM frameworks built on Megatron-LM include Colossal-AI, HuggingFace Accelerate, and NVIDIA NeMo Framework. This library provides GPU-optimized techniques for LLM. Here is a list of projects that have directly used Megatron:
- Training Multi-Billion Parameter Language Models Using Model Parallelism (Megatron-LM)[238].
- Larger Biomedical Domain Language Model(BioMegatron)[72].
- End-to-End Training of Neural Retrievers for Open-Domain Question Answering[239]
- Large Scale Multi-Actor Generative Dialog Modeling[240].
- Local Knowledge Powered Conversational Agents[241]
- MEGATRON-CNTRL[242]
- InstructRetro[243]
JAX: A Python library for high-performance numerical computing and scalable machine learning. JAX[244] can differentiate native Python and NumPy functions and run them on GPUs.
Colossal-AI: Colossal-AI[245] provides a set of parallel components. The tool also provides the ability to create distributed deep-learning models. This tool includes different models including: Baichuan-7B, Baichuan-13B-Base, Baichuan2-7B-Base,Baichuan2-13B-Base, ChatGLM-6B, ChatGLM2-6B, InternLM-7B, Qwen-7B,Llama-2-7B, Linly-AI/Chinese-LLaMA-2-7B-hf, wenge-research/yayi-7b-llama2, ziqingyang/chinese-llama-2-7b, TigerResearch/tigerbot-7b-base, LinkSoul/Chinese-Llama-2-7b, FlagAlpha/Atom-7B, IDEA-CCNL/Ziya-LLaMA-13B-v1.1, Colossal-LLaMA-2-7b-base, and Colossal-LLaMA-2-13b-base.
BMTrain: BMTrain13 is an efficient large model training toolbox that can be used to train large models with tens of billions of parameters.
LLMBox: LLMBox[246] is a comprehensive library for implementing LLMs. It includes an integrated training pipeline and comprehensive model evaluation. This library provides a one-stop solution for training and using LLMs. It also provides access to large language models such as: LLaMA [9], Mistral[247], Qwen [248], Baichuan [11], BLOOM[249], Vicuna [250], CodeGen [251], StarCoder[252], Llemma[253], and DeepSeekMath[254].
Volcano Engine Reinforcement Learning for LLM(veRL): veRL[255] is a flexible, efficient RL training framework for large language models (LLM).
Torchtune: torchtune[257] is a PyTorch library for easy writing, fine-tuning, and experimenting with LLMs, enabling popular LLMs such as Llama, Gemma, Mistral, Phi, and Qwen.
Torchtitan: Torchtitan[256] is a tool for large-scale LLM training that uses PyTorch. The overall goal of this library is to train LLM with minimal changes to the model code when applying multidimensional parallelism and to speed up its deployment.
Appendix B
-
Meta
- Llama 3.2-1|3|11|90B: https://llama.meta.com/
- Llama 3.1-8|70|405B: https://llama.meta.com/
- Llama 3-8|70B: https://llama.meta.com/llama3/
- Llama 2-7|13|70B: https://llama.meta.com/llama2/
- Llama 1-7|13|33|65B: https://ai.facebook.com/blog/large-language-model-llama-meta-ai/
- OPT-1.3|6.7|13|30|66B: https://arxiv.org/abs/2205.01068
-
Mistral AI
- Codestral-7|22B https://mistral.ai/news/codestral/
- Mistral-7B: https://mistral.ai/news/announcing-mistral-7b/
- Mixtral-8x7B: https://mistral.ai/news/mixtral-of-experts/
- Mixtral-8x22B: https://mistral.ai/news/mixtral-8x22b/
-
Google
- RecurrentGemma-2B: https://github.com/google-deepmind/recurrentgemma
-
Apple
- OpenELM-1.1|3B: https://huggingface.co/apple/OpenELM
-
Microsoft
- Phi1-1.3B: https://huggingface.co/microsoft/phi-1
- Phi2-2.7B: https://huggingface.co/microsoft/phi-2
- Phi3-3.8|7|14B: https://huggingface.co/microsoft/Phi-3-mini-4k-instruct
-
AllenAI
-
xAI
- Grok-1-314B-MoE https://x.ai/blog/grok-os
-
Cohere
- Command R-35B: https://huggingface.co/CohereForAI/c4ai-command-r-v01
-
DeepSeek
- DeepSeek-Math-7B: https://huggingface.co/collections/deepseek-ai/
- DeepSeek-Coder-1.3|6.7|7|33B: https://huggingface.co/collections/deepseek-ai/
- DeepSeek-VL-1.3|7Bhttps://huggingface.co/collections/deepseek-ai/
- DeepSeek-MoE-16B: https://huggingface.co/collections/deepseek-ai/
- DeepSeek-v2-236B-MoE: https://arxiv.org/abs/2405.04434
- DeepSeek-Coder-v2-16|236B-MOE: https://github.com/deepseek-ai/DeepSeek-Coder-V2
-
Alibaba
- Qwen-1.8B|7B|14B|72B: https://huggingface.co/collections/Qwen/
- Qwen1.5-0.5B|1.8B|4B|7B|14B|32B|72B|110B|MoE-A2.7B: https://qwenlm.github.io/blog/qwen1.5/
- Qwen2-0.5B|1.5B|7B|57B-A14B-MoE|72B: https://qwenlm.github.io/blog/qwen2
- Qwen2.5-0.5B|1.5B|3B|7B|14B|32B|72B: https://qwenlm.github.io/blog/qwen2.5/
- CodeQwen1.5-7B: https://qwenlm.github.io/blog/codeqwen1.5/
- Qwen2.5-Coder-1.5B|7B|32B: https://qwenlm.github.io/blog/qwen2.5-coder/
- Qwen2-Math-1.5B|7B|72B: https://qwenlm.github.io/blog/qwen2-math/
- Qwen2.5-Math-1.5B|7B|72B: https://qwenlm.github.io/blog/qwen2.5-math/
- Qwen-VL-7B: https://huggingface.co/Qwen/Qwen-VL
- Qwen2-VL-2B|7B|72B: https://qwenlm.github.io/blog/qwen2-vl/
- Qwen2-Audio-7B: https://qwenlm.github.io/blog/qwen2-audio/
-
01-ai
- Yi1.5-6|9|34B: https://huggingface.co/collections/01-ai/
- Yi-VL-6B|34B: https://huggingface.co/collections/01-ai/
-
Baichuan
- Baichuan-7|13B: https://huggingface.co/baichuan-inc
- Baichuan2-7|13B: https://huggingface.co/baichuan-inc
-
Nvidia
- Nemotron-4-340B: https://huggingface.co/nvidia/Nemotron-4-340B-Instruct
-
BLOOM
-
Zhipu AI
- GLM-2|6|10|13|70B: https://huggingface.co/THUDM
- CogVLM2-19B: https://huggingface.co/collections/THUDM/
-
OpenBMB
- MiniCPM-2B: https://huggingface.co/collections/openbmb/
- OmniLLM-12B: https://huggingface.co/openbmb/OmniLMM-12B
- VisCPM-10B: https://huggingface.co/openbmb/VisCPM-Chat
- CPM-Bee-1|2|5|10B: https://huggingface.co/collections/openbmb/
-
RWKV Foundation
- RWKV-v4|5|6: https://huggingface.co/RWKV
-
ElutherAI
- Pythia-1|1.4|2.8|6.9|12B: https://github.com/EleutherAI/pythia
-
Stability AI
- StableLM-3B: https://huggingface.co/collections/stabilityai/
- StableLM-v2-1.6|12B: https://huggingface.co/collections/stabilityai/
- StableCode-3B: https://huggingface.co/collections/stabilityai/
-
BigCode
- StarCoder-1|3|7B: https://huggingface.co/collections/bigcode/
- StarCoder2-3|7|15B: https://huggingface.co/collections/bigcode/
-
DataBricks
- DBRX-132B-MoE: https://www.databricks.com/
-
Shanghai AI Laboratory
- InternLM2-1.8|7|20B: https://huggingface.co/collections/internlm/
- InternLM-Math-7B|20B: https://huggingface.co/collections/internlm/
- InternLM-XComposer2-1.8|7B: https://huggingface.co/collections/internlm/
- InternVL-2|6|14|26: https://huggingface.co/collections/OpenGVLab/
References
- Turing, A. Computing Machinery and Intelligence. Mind. Vol. LIX,?. 236. In Proceedings of the Computers & Thought, 1950.
- Zhao, W.X.; Zhou, K.; Li, J.; Tang, T.; Wang, X.; Hou, Y.; Min, Y.; Zhang, B.; Zhang, J.; Dong, Z.; et al. A survey of large language models. arXiv 2023, arXiv:2303.18223 2023. [Google Scholar]
- Bellegarda, J.R. Statistical language model adaptation: review and perspectives. Speech communication 2004, 42, 93–108. [Google Scholar] [CrossRef]
- Hu, L.; Liu, Z.; Zhao, Z.; Hou, L.; Nie, L.; Li, J. A survey of knowledge enhanced pre-trained language models. IEEE Transactions on Knowledge and Data Engineering 2023. [Google Scholar] [CrossRef]
- Dauphin, Y.N.; Fan, A.; Auli, M.; Grangier, D. Language modeling with gated convolutional networks. In Proceedings of the International conference on machine learning. PMLR; 2017; pp. 933–941. [Google Scholar]
- Hoffmann, J.; Borgeaud, S.; Mensch, A.; Buchatskaya, E.; Cai, T.; Rutherford, E.; Casas, D.d.L.; Hendricks, L.A.; Welbl, J.; Clark, A.; et al. Training compute-optimal large language models. arXiv 2022, arXiv:2203.15556 2022. [Google Scholar]
- Chowdhery, A.; Narang, S.; Devlin, J.; Bosma, M.; Mishra, G.; Roberts, A.; Barham, P.; Chung, H.W.; Sutton, C.; Gehrmann, S.; et al. Palm: Scaling language modeling with pathways. Journal of Machine Learning Research 2023, 24, 1–113. [Google Scholar]
- Taylor, R.; Kardas, M.; Cucurull, G.; Scialom, T.; Hartshorn, A.; Saravia, E.; Poulton, A.; Kerkez, V.; Stojnic, R. Galactica: A large language model for science. arXiv 2022, arXiv:2211.09085 2022. [Google Scholar]
- Touvron, H.; Martin, L.; Stone, K.; Albert, P.; Almahairi, A.; Babaei, Y.; Bashlykov, N.; Batra, S.; Bhargava, P.; Bhosale, S.; et al. Llama 2: Open foundation and fine-tuned chat models. arXiv 2023, arXiv:2307.09288 2023. [Google Scholar]
- GLM, T.; Zeng, A.; Xu, B.; Wang, B.; Zhang, C.; Yin, D.; Zhang, D.; Rojas, D.; Feng, G.; Zhao, H.; et al. Chatglm: A family of large language models from glm-130b to glm-4 all tools. arXiv 2024, arXiv:2406.12793 2024. [Google Scholar]
- Yang, A.; Xiao, B.; Wang, B.; Zhang, B.; Bian, C.; Yin, C.; Lv, C.; Pan, D.; Wang, D.; Yan, D.; et al. Baichuan 2: Open large-scale language models. arXiv 2023, arXiv:2309.10305 2023. [Google Scholar]
- GDR, H.B.; Sharon, N.; Australia, E. Nomenclature and symbolism for amino acids and peptides. Pure and Applied Chemistry 1984, 56, 595–624. [Google Scholar]
- Wang, Y.; Ivison, H.; Dasigi, P.; Hessel, J.; Khot, T.; Chandu, K.; Wadden, D.; MacMillan, K.; Smith, N.A.; Beltagy, I.; et al. How far can camels go? exploring the state of instruction tuning on open resources. Advances in Neural Information Processing Systems 2023, 36, 74764–74786. [Google Scholar]
- Neubig, G. Neural machine translation and sequence-to-sequence models: A tutorial. arXiv 2017, arXiv:1703.01619 2017. [Google Scholar]
- Liu, J.; Min, S.; Zettlemoyer, L.; Choi, Y.; Hajishirzi, H. Infini-gram: Scaling unbounded n-gram language models to a trillion tokens. arXiv 2024, arXiv:2401.17377 2024. [Google Scholar]
- Jelinek, F. Statistical methods for speech recognition; MIT press, 1998.
- Gao, J.; Lin, C.Y. Introduction to the special issue on statistical language modeling, 2004.
- Rosenfeld, R. Two decades of statistical language modeling: Where do we go from here? Proceedings of the IEEE 2000, 88, 1270–1278. [Google Scholar] [CrossRef]
- Liu, X.; Croft, W.B. Statistical language modeling for information retrieval. Annu. Rev. Inf. Sci. Technol. 2005, 39, 1–31. [Google Scholar] [CrossRef]
- Thede, S.M.; Harper, M. A second-order hidden Markov model for part-of-speech tagging. In Proceedings of the 37th annual meeting of the Association for Computational Linguistics; 1999; pp. 175–182. [Google Scholar]
- Bengio, Y.; Ducharme, R.; Vincent, P. A neural probabilistic language model. Advances in neural information processing systems 2000, 13. [Google Scholar]
- Mikolov, T.; Karafiát, M.; Burget, L.; Cernockỳ, J.; Khudanpur, S. Recurrent neural network based language model. In Proceedings of the Interspeech. Makuhari; 2010; Vol. 2, pp. 1045–1048. [Google Scholar]
- Zettlemoyer, L. Deep contextualized word representations. In Proceedings of the Proceedings of the; 2018. [Google Scholar]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I.; et al. Language models are unsupervised multitask learners. OpenAI blog 2019, 1, 9. [Google Scholar]
- Lewis, M. Bart: Denoising sequence-to-sequence pre-training for natural language generation, translation, and comprehension. arXiv 2019, arXiv:1910.13461 2019. [Google Scholar]
- Kaplan, J.; McCandlish, S.; Henighan, T.; Brown, T.B.; Chess, B.; Child, R.; Gray, S.; Radford, A.; Wu, J.; Amodei, D. Scaling laws for neural language models. arXiv 2020, arXiv:2001.08361 2020. [Google Scholar]
- Huang, K.; Altosaar, J.; Ranganath, R. Clinicalbert: Modeling clinical notes and predicting hospital readmission. arXiv 2019, arXiv:1904.05342 2019. [Google Scholar]
- Yang, X.; Chen, A.; PourNejatian, N.; Shin, H.C.; Smith, K.E.; Parisien, C.; Compas, C.; Martin, C.; Flores, M.G.; Zhang, Y.; et al. Gatortron: A large clinical language model to unlock patient information from unstructured electronic health records. arXiv 2022, arXiv:2203.03540 2022. [Google Scholar]
- Peng, C.; Yang, X.; Chen, A.; Smith, K.E.; PourNejatian, N.; Costa, A.B.; Martin, C.; Flores, M.G.; Zhang, Y.; Magoc, T.; et al. A study of generative large language model for medical research and healthcare. NPJ digital medicine 2023, 6, 210. [Google Scholar] [CrossRef]
- Chen, Z.; Cano, A.H.; Romanou, A.; Bonnet, A.; Matoba, K.; Salvi, F.; Pagliardini, M.; Fan, S.; Köpf, A.; Mohtashami, A.; et al. Meditron-70b: Scaling medical pretraining for large language models. arXiv 2023, arXiv:2311.16079 2023. [Google Scholar]
- Kim, H.; Hwang, H.; Lee, J.; Park, S.; Kim, D.; Lee, T.; Yoon, C.; Sohn, J.; Choi, D.; Kang, J. Small language models learn enhanced reasoning skills from medical textbooks. arXiv 2024, arXiv:2404.00376 2024. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, G.; Du, Z.; Fan, L.; Li, X. ClinicalGPT: large language models finetuned with diverse medical data and comprehensive evaluation. arXiv 2023, arXiv:2306.09968 2023. [Google Scholar]
- Ye, Q.; Liu, J.; Chong, D.; Zhou, P.; Hua, Y.; Liu, A. Qilin-med: Multi-stage knowledge injection advanced medical large language model. arXiv 2023, arXiv:2310.09089 2023. [Google Scholar]
- Li, Y.; Li, Z.; Zhang, K.; Dan, R.; Jiang, S.; Zhang, Y. Chatdoctor: A medical chat model fine-tuned on a large language model meta-ai (llama) using medical domain knowledge. Cureus 2023, 15. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Xi, N.; Qiang, Z.; Zhao, S.; Qin, B.; Liu, T. Huatuo: Tuning llama model with chinese medical knowledge. arXiv 2023, arXiv:2304.06975 2023. [Google Scholar]
- Zhang, H.; Chen, J.; Jiang, F.; Yu, F.; Chen, Z.; Li, J.; Chen, G.; Wu, X.; Zhang, Z.; Xiao, Q.; et al. Huatuogpt, towards taming language model to be a doctor. arXiv 2023, arXiv:2305.15075 2023. [Google Scholar]
- Taori, R.; Gulrajani, I.; Zhang, T.; Dubois, Y.; Li, X.; Guestrin, C.; Liang, P.; Hashimoto, T.B. Stanford alpaca: An instruction-following llama model, 2023.
- Zeng, A.; Liu, X.; Du, Z.; Wang, Z.; Lai, H.; Ding, M.; Yang, Z.; Xu, Y.; Zheng, W.; Xia, X.; et al. Glm-130b: An open bilingual pre-trained model. arXiv 2022, arXiv:2210.02414 2022. [Google Scholar]
- Xu, C.; Guo, D.; Duan, N.; McAuley, J. Baize: An open-source chat model with parameter-efficient tuning on self-chat data. arXiv 2023, arXiv:2304.01196 2023. [Google Scholar]
- Taori, R.R. Ingredients for Accessible and Sustainable Language Models. PhD thesis, Stanford University, 2024.
- Zheng, L.; Chiang, W.L.; Sheng, Y.; Li, T.; Zhuang, S.; Wu, Z.; Zhuang, Y.; Li, Z.; Lin, Z.; Xing, E.P.; et al. Lmsys-chat-1m: A large-scale real-world llm conversation dataset. arXiv 2023, arXiv:2309.11998 2023. [Google Scholar]
- Roumeliotis, K.I.; Tselikas, N.D. Chatgpt and open-ai models: A preliminary review. Future Internet 2023, 15, 192. [Google Scholar] [CrossRef]
- Sanderson, K. GPT-4 is here: what scientists think. Nature 2023, 615, 773. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, H.; Zhu, S.; Zhou, G.; Xu, H.; Jia, Y.; Zan, H. Zhongjing: Enhancing the chinese medical capabilities of large language model through expert feedback and real-world multi-turn dialogue. In Proceedings of the AAAI Conference on Artificial Intelligence; 2024; Vol. 38, pp. 19368–19376. [Google Scholar]
- Wu, C.; Lin, W.; Zhang, X.; Zhang, Y.; Xie, W.; Wang, Y. PMC-LLaMA: toward building open-source language models for medicine. Journal of the American Medical Informatics Association 2024, ocae045. [Google Scholar] [CrossRef] [PubMed]
- Shoham, O.B.; Rappoport, N. Cpllm: Clinical prediction with large language models. arXiv 2023, arXiv:2309.11295 2023. [Google Scholar]
- Singhal, K.; Tu, T.; Gottweis, J.; Sayres, R.; Wulczyn, E.; Hou, L.; Clark, K.; Pfohl, S.; Cole-Lewis, H.; Neal, D.; et al. Towards expert-level medical question answering with large language models. arXiv 2023, arXiv:2305.09617 2023. [Google Scholar] [CrossRef]
- Jin, D.; Pan, E.; Oufattole, N.; Weng, W.H.; Fang, H.; Szolovits, P. What disease does this patient have? a large-scale open domain question answering dataset from medical exams. Applied Sciences 2021, 11, 6421. [Google Scholar] [CrossRef]
- Pal, A.; Umapathi, L.K.; Sankarasubbu, M. Medmcqa: A large-scale multi-subject multi-choice dataset for medical domain question answering. In Proceedings of the Conference on health, inference, and learning; PMLR, 2022; pp. 248–260. [Google Scholar]
- Jin, Q.; Dhingra, B.; Liu, Z.; Cohen, W.W.; Lu, X. Pubmedqa: A dataset for biomedical research question answering. arXiv 2019, arXiv:1909.06146 2019. [Google Scholar]
- Hendrycks, D.; Burns, C.; Basart, S.; Zou, A.; Mazeika, M.; Song, D.; Steinhardt, J. Measuring massive multitask language understanding. arXiv 2020, arXiv:2009.03300 2020. [Google Scholar]
- Toma, A.; Lawler, P.R.; Ba, J.; Krishnan, R.G.; Rubin, B.B.; Wang, B. Clinical camel: An open expert-level medical language model with dialogue-based knowledge encoding. arXiv 2023, arXiv:2305.12031 2023. [Google Scholar]
- Xiong, H.; Wang, S.; Zhu, Y.; Zhao, Z.; Liu, Y.; Huang, L.; Wang, Q.; Shen, D. Doctorglm: Fine-tuning your chinese doctor is not a herculean task. arXiv 2023, arXiv:2304.01097 2023. [Google Scholar]
- Chen, Y.; Wang, Z.; Xing, X.; Xu, Z.; Fang, K.; Wang, J.; Li, S.; Wu, J.; Liu, Q.; Xu, X.; et al. Bianque: Balancing the questioning and suggestion ability of health llms with multi-turn health conversations polished by chatgpt. arXiv 2023, arXiv:2310.15896 2023. [Google Scholar]
- Liu, X.; Segonne, V.; Mannion, A.; Schwab, D.; Goeuriot, L.; Portet, F. MedDialog-FR: a French Version of the MedDialog Corpus for Multi-label Classification and Response Generation related to Women’s Intimate Health. In Proceedings of the First Workshop on Patient-Oriented Language Processing (CL4Health)@ LREC-COLING 2024; 2024; pp. 173–183. [Google Scholar]
- Liao, Y.; Jiang, S.; Wang, Y.; Wang, Y. MING-MOE: Enhancing Medical Multi-Task Learning in Large Language Models with Sparse Mixture of Low-Rank Adapter Experts. arXiv 2024, arXiv:2404.09027 2024. [Google Scholar]
- Dinghao, P.; Zhihao, Y.; Hongfei, L.; Jian, W. Dialogue Symptom Inference Based on Structured Self-Attention Network. Journal of Computer Engineering & Applications 2024, 60. [Google Scholar]
- Liu, W.; Tang, J.; Qin, J.; Xu, L.; Li, Z.; Liang, X. Meddg: A large-scale medical consultation dataset for building medical dialogue system. Europe PMC 2020. [Google Scholar]
- Zhang, X.; Zhang, X.; Yu, Y. ChatGLM-6B Fine-Tuning for Cultural and Creative Products Advertising Words. In Proceedings of the 2023 International Conference on Culture-Oriented Science and Technology (CoST). IEEE; 2023; pp. 291–295. [Google Scholar]
- An, J.; Ding, W.; Lin, C. ChatGPT. tackle the growing carbon footprint of generative AI 2023, 615, 586. [Google Scholar]
- García-Ferrero, I.; Agerri, R.; Salazar, A.A.; Cabrio, E.; de la Iglesia, I.; Lavelli, A.; Magnini, B.; Molinet, B.; Ramirez-Romero, J.; Rigau, G.; et al. MedMT5: An Open-Source Multilingual Text-to-Text LLM for the Medical Domain. In Proceedings of the 2024 Joint International Conference on Computational Linguistics, Language Resources and Evaluation (LREC-COLING 2024), 2024, pp. 11165–11177.
- Xie, Q.; Chen, Q.; Chen, A.; Peng, C.; Hu, Y.; Lin, F.; Peng, X.; Huang, J.; Zhang, J.; Keloth, V.; et al. Me llama: Foundation large language models for medical applications. arXiv 2024, arXiv:2402.12749 2024. [Google Scholar]
- Pieri, S.; Mullappilly, S.S.; Khan, F.S.; Anwer, R.M.; Khan, S.; Baldwin, T.; Cholakkal, H. Bimedix: Bilingual medical mixture of experts llm. arXiv preprint arXiv:2402.13253, arXiv:2402.13253 2024.
- Jin, Q.; Dhingra, B.; Cohen, W.W.; Lu, X. Probing biomedical embeddings from language models. arXiv 2019, arXiv:1904.02181 2019. [Google Scholar]
- Kenton, J.D.M.W.C.; Toutanova, L.K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the Proceedings of naacL-HLT. Minneapolis, Minnesota, 2019, Vol. 1, p. 2.
- Peters, M.E.; Ruder, S.; Smith, N.A. To tune or not to tune? adapting pretrained representations to diverse tasks. arXiv 2019, arXiv:1903.05987 2019. [Google Scholar]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020, 36, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Tanabe, L.K.; Ando, R.J.n.; Kuo, C.J.; Chung, I.F.; Hsu, C.N.; Lin, Y.S.; Klinger, R.; Friedrich, C.M.; Ganchev, K.; et al. Overview of BioCreative II gene mention recognition. Genome biology 2008, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sang, E.F.; De Meulder, F. Introduction to the CoNLL-2003 shared task: Language-independent named entity recognition. arXiv 2003, arXiv:cs/0306050. [Google Scholar]
- Romanov, A.; Shivade, C. Lessons from natural language inference in the clinical domain. arXiv 2018, arXiv:1808.06752 2018. [Google Scholar]
- Peng, Y.; Yan, S.; Lu, Z. Transfer learning in biomedical natural language processing: an evaluation of BERT and ELMo on ten benchmarking datasets. arXiv 2019, arXiv:1906.05474 2019. [Google Scholar]
- Shin, H.C.; Zhang, Y.; Bakhturina, E.; Puri, R.; Patwary, M.; Shoeybi, M.; Mani, R. BioMegatron: larger biomedical domain language model. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing (EMNLP); 2020; pp. 4700–4706. [Google Scholar]
- Gu, Y.; Tinn, R.; Cheng, H.; Lucas, M.; Usuyama, N.; Liu, X.; Naumann, T.; Gao, J.; Poon, H. Domain-specific language model pretraining for biomedical natural language processing. ACM Transactions on Computing for Healthcare (HEALTH) 2021, 3, 1–23. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Johnson, R.J.; Sciaky, D.; Wei, C.H.; Leaman, R.; Davis, A.P.; Mattingly, C.J.; Wiegers, T.C.; Lu, Z. BioCreative V CDR task corpus: a resource for chemical disease relation extraction. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Doğan, R.I.; Leaman, R.; Lu, Z. NCBI disease corpus: a resource for disease name recognition and concept normalization. Journal of biomedical informatics 2014, 47, 1–10. [Google Scholar] [CrossRef]
- Collier, N.; Ohta, T.; Tsuruoka, Y.; Tateisi, Y.; Kim, J.D. Introduction to the bio-entity recognition task at JNLPBA. In Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and its Applications (NLPBA/BioNLP); 2004; pp. 73–78. [Google Scholar]
- Nye, B.; Li, J.J.; Patel, R.; Yang, Y.; Marshall, I.J.; Nenkova, A.; Wallace, B.C. A corpus with multi-level annotations of patients, interventions and outcomes to support language processing for medical literature. In Proceedings of the conference. Association for Computational Linguistics. Meeting. NIH Public Access; 2018; Vol. 2018, p. 197. [Google Scholar]
- Krallinger, M.; Rabal, O.; Akhondi, S.A.; Pérez, M.P.; Santamaría, J.; Rodríguez, G.P.; Tsatsaronis, G.; Intxaurrondo, A.; López, J.A.; Nandal, U.; et al. Overview of the BioCreative VI chemical-protein interaction Track. In Proceedings of the sixth BioCreative challenge evaluation workshop; 2017; Vol. 1, pp. 141–146. [Google Scholar]
- Herrero-Zazo, M.; Segura-Bedmar, I.; Martínez, P.; Declerck, T. The DDI corpus: An annotated corpus with pharmacological substances and drug–drug interactions. Journal of biomedical informatics 2013, 46, 914–920. [Google Scholar] [CrossRef]
- Bravo, À.; Piñero, J.; Queralt-Rosinach, N.; Rautschka, M.; Furlong, L.I. Extraction of relations between genes and diseases from text and large-scale data analysis: implications for translational research. BMC bioinformatics 2015, 16, 1–17. [Google Scholar] [CrossRef]
- Soğancıoğlu, G.; Öztürk, H.; Özgür, A. BIOSSES: a semantic sentence similarity estimation system for the biomedical domain. Bioinformatics 2017, 33, i49–i58. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Nentidis, A.; Bougiatiotis, K.; Krithara, A.; Paliouras, G. Results of the seventh edition of the bioasq challenge. In Proceedings of the Machine Learning and Knowledge Discovery in Databases: International Workshops of ECML PKDD 2019, Proceedings, Part II. Springer. Würzburg, Germany, 16–20 September 2019; 2020; pp. 553–568. [Google Scholar]
- Alrowili, S.; Vijay-Shanker, K. BioM-transformers: building large biomedical language models with BERT, ALBERT and ELECTRA. In Proceedings of the 20th workshop on biomedical language processing; 2021; pp. 221–227. [Google Scholar]
- Yasunaga, M.; Leskovec, J.; Liang, P. Linkbert: Pretraining language models with document links. arXiv 2022, arXiv:2203.15827 2022. [Google Scholar]
- Luo, R.; Sun, L.; Xia, Y.; Qin, T.; Zhang, S.; Poon, H.; Liu, T.Y. BioGPT: generative pre-trained transformer for biomedical text generation and mining. Briefings in bioinformatics 2022, 23, bbac409. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Fan, S.; Yang, K.; Wu, Y.; Qiao, M.; Nie, Z. Biomedgpt: Open multimodal generative pre-trained transformer for biomedicine. arXiv 2023, arXiv:2308.09442 2023. [Google Scholar]
- Luu, R.K.; Buehler, M.J. BioinspiredLLM: Conversational Large Language Model for the Mechanics of Biological and Bio-Inspired Materials. Advanced Science 2024, 11, 2306724. [Google Scholar] [CrossRef]
- Labrak, Y.; Bazoge, A.; Morin, E.; Gourraud, P.A.; Rouvier, M.; Dufour, R. Biomistral: A collection of open-source pretrained large language models for medical domains. arXiv 2024, arXiv:2402.10373 2024. [Google Scholar]
- Peng, Y.; Chen, Q.; Lu, Z. An empirical study of multi-task learning on BERT for biomedical text mining. arXiv 2020, arXiv:2005.02799 2020. [Google Scholar]
- Guo, J.; Ibanez-Lopez, A.S.; Gao, H.; Quach, V.; Coley, C.W.; Jensen, K.F.; Barzilay, R. Automated chemical reaction extraction from scientific literature. Journal of chemical information and modeling 2021, 62, 2035–2045. [Google Scholar] [CrossRef]
- Gupta, T.; Zaki, M.; Krishnan, N.A.; Mausam. MatSciBERT: A materials domain language model for text mining and information extraction. npj Computational Materials 2022, 8, 102. [Google Scholar] [CrossRef]
- Shetty, P.; Rajan, A.C.; Kuenneth, C.; Gupta, S.; Panchumarti, L.P.; Holm, L.; Zhang, C.; Ramprasad, R. A general-purpose material property data extraction pipeline from large polymer corpora using natural language processing. npj Computational Materials 2023, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ma, D.; Chen, L.; Sun, L.; Li, Z.; Xu, H.; Zhu, Z.; Zhu, S.; Fan, S.; Shen, G.; et al. Chemdfm: Dialogue foundation model for chemistry. arXiv 2024, arXiv:2401.14818 2024. [Google Scholar]
- Zhang, D.; Liu, W.; Tan, Q.; Chen, J.; Yan, H.; Yan, Y.; Li, J.; Huang, W.; Yue, X.; Zhou, D.; et al. Chemllm: A chemical large language model. arXiv 2024, arXiv:2402.06852 2024. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic acids research 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: towards direct deposition of bioassay data. Nucleic acids research 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic acids research 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—a free ultralarge-scale chemical database for ligand discovery. Journal of chemical information and modeling 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Marco, A.C.; Myers, A.; Graham, S.J.; D’Agostino, P.; Apple, K. The USPTO patent assignment dataset: Descriptions and analysis. SSRN 2015. [Google Scholar] [CrossRef]
- Wigh, D.S.; Arrowsmith, J.; Pomberger, A.; Felton, K.C.; Lapkin, A.A. ORDerly: Data Sets and Benchmarks for Chemical Reaction Data. Journal of Chemical Information and Modeling 2024, 64, 3790–3798. [Google Scholar] [CrossRef]
- Lawlor, B. Preprints and Scholarly Communication in Chemistry: A look at ChemRxiv. Chemistry International 2018, 40, 18–21. [Google Scholar] [CrossRef]
- Yu, B.; Baker, F.N.; Chen, Z.; Ning, X.; Sun, H. Llasmol: Advancing large language models for chemistry with a large-scale, comprehensive, high-quality instruction tuning dataset. arXiv 2024, arXiv:2402.09391 2024. [Google Scholar]
- Chen, L.; Wang, W.; Bai, Z.; Xu, P.; Fang, Y.; Fang, J.; Wu, W.; Zhou, L.; Zhang, R.; Xia, Y.; et al. PharmGPT: Domain-Specific Large Language Models for Bio-Pharmaceutical and Chemistry. arXiv 2024, arXiv:2406.18045 2024. [Google Scholar]
- Pollard, T.J.; Johnson, A.E.; Raffa, J.D.; Celi, L.A.; Mark, R.G.; Badawi, O. The eICU Collaborative Research Database, a freely available multi-center database for critical care research. Scientific data 2018, 5, 1–13. [Google Scholar] [CrossRef]
- Zeng, G.; Yang, W.; Ju, Z.; Yang, Y.; Wang, S.; Zhang, R.; Zhou, M.; Zeng, J.; Dong, X.; Zhang, R.; et al. MedDialog: Large-scale medical dialogue datasets. In Proceedings of the 2020 conference on empirical methods in natural language processing (EMNLP); 2020; pp. 9241–9250. [Google Scholar]
- Singhal, K.; Azizi, S.; Tu, T.; Mahdavi, S.S.; Wei, J.; Chung, H.W.; Scales, N.; Tanwani, A.; Cole-Lewis, H.; Pfohl, S.; et al. Large language models encode clinical knowledge. Nature 2023, 620, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Abacha, A.B.; Agichtein, E.; Pinter, Y.; Demner-Fushman, D. Overview of the medical question answering task at TREC 2017 LiveQA. In Proceedings of the TREC; 2017; pp. 1–12. [Google Scholar]
- Abacha, A.B.; Mrabet, Y.; Sharp, M.; Goodwin, T.R.; Shooshan, S.E.; Demner-Fushman, D. Bridging the gap between consumers’ medication questions and trusted answers. In MEDINFO 2019: Health and Wellbeing e-Networks for All; IOS Press, 2019; pp. 25–29.
- Singh, S.; Romanou, A.; Fourrier, C.; Adelani, D.I.; Ngui, J.G.; Vila-Suero, D.; Limkonchotiwat, P.; Marchisio, K.; Leong, W.Q.; Susanto, Y.; et al. Global mmlu: Understanding and addressing cultural and linguistic biases in multilingual evaluation. arXiv 2024, arXiv:2412.03304 2024. [Google Scholar]
- Chen, H.; Fang, Z.; Singla, Y.; Dredze, M. Benchmarking Large Language Models on Answering and Explaining Challenging Medical Questions. arXiv 2024, arXiv:2402.18060 2024. [Google Scholar]
- Li, J.; Wang, X.; Wu, X.; Zhang, Z.; Xu, X.; Fu, J.; Tiwari, P.; Wan, X.; Wang, B. Huatuo-26m, a large-scale chinese medical qa dataset. arXiv 2023, arXiv:2305.01526 2023. [Google Scholar]
- Huang, Y.; Bai, Y.; Zhu, Z.; Zhang, J.; Zhang, J.; Su, T.; Liu, J.; Lv, C.; Zhang, Y.; Fu, Y.; et al. C-eval: A multi-level multi-discipline chinese evaluation suite for foundation models. Advances in Neural Information Processing Systems 2024, 36. [Google Scholar]
- Zhong, W.; Cui, R.; Guo, Y.; Liang, Y.; Lu, S.; Wang, Y.; Saied, A.; Chen, W.; Duan, N. Agieval: A human-centric benchmark for evaluating foundation models. arXiv 2023, arXiv:2304.06364 2023. [Google Scholar]
- Lu, P.; Mishra, S.; Xia, T.; Qiu, L.; Chang, K.W.; Zhu, S.C.; Tafjord, O.; Clark, P.; Kalyan, A. Learn to explain: Multimodal reasoning via thought chains for science question answering. Advances in Neural Information Processing Systems 2022, 35, 2507–2521. [Google Scholar]
- Gu, Z.; Zhu, X.; Ye, H.; Zhang, L.; Wang, J.; Zhu, Y.; Jiang, S.; Xiong, Z.; Li, Z.; Wu, W.; et al. Xiezhi: An ever-updating benchmark for holistic domain knowledge evaluation. In Proceedings of the AAAI Conference on Artificial Intelligence; 2024; Vol. 38, pp. 18099–18107. [Google Scholar]
- Sun, L.; Han, Y.; Zhao, Z.; Ma, D.; Shen, Z.; Chen, B.; Chen, L.; Yu, K. Scieval: A multi-level large language model evaluation benchmark for scientific research. In Proceedings of the AAAI Conference on Artificial Intelligence; 2024; Vol. 38, pp. 19053–19061. [Google Scholar]
- Chen, Q.; Deng, C. Bioinfo-Bench: A Simple Benchmark Framework for LLM Bioinformatics Skills Evaluation. bioRxiv, 2023; 2023–10. [Google Scholar]
- Clark, P.; Cowhey, I.; Etzioni, O.; Khot, T.; Sabharwal, A.; Schoenick, C.; Tafjord, O. Think you have solved question answering? try arc, the ai2 reasoning challenge. arXiv 2018, arXiv:1803.05457 2018. [Google Scholar]
- Zheng, S.; Rao, J.; Zhang, Z.; Xu, J.; Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. Journal of chemical information and modeling 2019, 60, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nature communications 2020, 11, 5575. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, D.; Kwon, Y.; Park, M.S.; Choi, Y.S. Valid, plausible, and diverse retrosynthesis using tied two-way transformers with latent variables. Journal of Chemical Information and Modeling 2021, 61, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, P.; Vaucher, A.C.; Laino, T.; Reymond, J.L. Prediction of chemical reaction yields using deep learning. Machine learning: science and technology 2021, 2, 015016. [Google Scholar] [CrossRef]
- Mann, V.; Venkatasubramanian, V. Predicting chemical reaction outcomes: A grammar ontology-based transformer framework. AIChE Journal 2021, 67, e17190. [Google Scholar] [CrossRef]
- Mao, K.; Xiao, X.; Xu, T.; Rong, Y.; Huang, J.; Zhao, P. Molecular graph enhanced transformer for retrosynthesis prediction. Neurocomputing 2021, 457, 193–202. [Google Scholar] [CrossRef]
- Tu, Z.; Coley, C.W. Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction. Journal of chemical information and modeling 2022, 62, 3503–3513. [Google Scholar] [CrossRef]
- Ucak, U.V.; Ashyrmamatov, I.; Ko, J.; Lee, J. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nature communications 2022, 13, 1186. [Google Scholar] [CrossRef]
- Toniato, A.; Vaucher, A.C.; Schwaller, P.; Laino, T. Enhancing diversity in language based models for single-step retrosynthesis. Digital Discovery 2023, 2, 489–501. [Google Scholar] [CrossRef]
- Thakkar, A.; Vaucher, A.C.; Byekwaso, A.; Schwaller, P.; Toniato, A.; Laino, T. Unbiasing retrosynthesis language models with disconnection prompts. ACS Central Science 2023, 9, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. Journal of chemical information and modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC- a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Lowe, D.M. Extraction of chemical structures and reactions from the literature. PhD thesis, 2012.
- Hu, W.; Fey, M.; Ren, H.; Nakata, M.; Dong, Y.; Leskovec, J. Ogb-lsc: A large-scale challenge for machine learning on graphs. arXiv 2021, arXiv:2103.09430 2021. [Google Scholar]
- Ying, C.; Yang, M.; Zheng, S.; Ke, G.; Luo, S.; Cai, T.; Wu, C.; Wang, Y.; Shen, Y.; He, D. First place solution of kdd cup 2021 & ogb large-scale challenge graph prediction track. arXiv 2021, arXiv:2106.08279 2021. [Google Scholar]
- Axelrod, S.; Gomez-Bombarelli, R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Scientific Data 2022, 9, 185. [Google Scholar] [CrossRef]
- Beaini, D.; Huang, S.; Cunha, J.A.; Li, Z.; Moisescu-Pareja, G.; Dymov, O.; Maddrell-Mander, S.; McLean, C.; Wenkel, F.; Müller, L.; et al. Towards foundational models for molecular learning on large-scale multi-task datasets. arXiv 2023, arXiv:2310.04292 2023. [Google Scholar]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic acids research 2024, 52, D1180–D1192. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic acids research 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. Journal of chemical information and modeling 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: an integrated large scale dataset facilitating Big Data analysis in chemogenomics. Journal of cheminformatics 2017, 9, 1–9. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: a benchmark for molecular machine learning. Chemical science 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Zhu, Y.; Hwang, J.; Adams, K.; Liu, Z.; Nan, B.; Stenfors, B.; Du, Y.; Chauhan, J.; Wiest, O.; Isayev, O.; et al. Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks. In Proceedings of the The Twelfth International Conference on Learning Representations; 2023. [Google Scholar]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: benchmarking models for de novo molecular design. Journal of chemical information and modeling 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular sets (MOSES): a benchmarking platform for molecular generation models. Frontiers in pharmacology 2020, 11, 565644. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic acids research 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Ektefaie, Y.; Shen, A.; Bykova, D.; Marin, M.; Zitnik, M.; Farhat, M. Evaluating generalizability of artificial intelligence models for molecular datasets. bioRxiv 2024. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, Y.; Zhang, X.; Xu, X.; Xie, Y.; Liu, M.; Dickerson, K.; Deng, C.; Nakata, M.; Ji, S. Molecule3d: A benchmark for predicting 3d geometries from molecular graphs. arXiv 2021, arXiv:2110.01717 2021. [Google Scholar]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Rao, R.M.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.; Abbeel, P.; Sercu, T.; Rives, A. MSA transformer. In Proceedings of the International Conference on Machine Learning. PMLR; 2021; pp. 8844–8856. [Google Scholar]
- Somnath, V.R.; Bunne, C.; Krause, A. Multi-scale representation learning on proteins. Advances in Neural Information Processing Systems 2021, 34, 25244–25255. [Google Scholar]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language models enable zero-shot prediction of the effects of mutations on protein function. Advances in neural information processing systems 2021, 34, 29287–29303. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE transactions on pattern analysis and machine intelligence 2021, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, S.; Wu, L.; Xia, H.; Ju, F.; Zhang, H.; Liu, S.; Xia, Y.; Zhu, J.; Deng, P.; et al. Pre-training co-evolutionary protein representation via a pairwise masked language model. arXiv 2021, arXiv:2110.15527 2021. [Google Scholar]
- Mansoor, S.; Baek, M.; Madan, U.; Horvitz, E. Toward more general embeddings for protein design: Harnessing joint representations of sequence and structure. bioRxiv, 2021. [Google Scholar]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Wang, Z.; Combs, S.A.; Brand, R.; Calvo, M.R.; Xu, P.; Price, G.; Golovach, N.; Salawu, E.O.; Wise, C.J.; Ponnapalli, S.P.; et al. Lm-gvp: an extensible sequence and structure informed deep learning framework for protein property prediction. Scientific reports 2022, 12, 6832. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, H.; Zheng, L.; Xin, J.; Li, Q.; Wu, L.; Deng, Z.; Lu, Y.; Liu, Q.; Kong, L. Retrieved sequence augmentation for protein representation learning. bioRxiv, 2023. [Google Scholar]
- Zhang, N.; Bi, Z.; Liang, X.; Cheng, S.; Hong, H.; Deng, S.; Lian, J.; Zhang, Q.; Chen, H. Ontoprotein: Protein pretraining with gene ontology embedding. arXiv 2022, arXiv:2201.11147 2022. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv 2022, 2022, 500902. [Google Scholar]
- Wang, Z.; Zhang, Q.; Shuang-Wei, H.; Yu, H.; Jin, X.; Gong, Z.; Chen, H. Multi-level protein structure pre-training via prompt learning. In Proceedings of the The Eleventh International Conference on Learning Representations; 2022. [Google Scholar]
- Zhou, H.Y.; Fu, Y.; Zhang, Z.; Bian, C.; Yu, Y. Protein representation learning via knowledge enhanced primary structure modeling. arXiv 2023, arXiv:2301.13154 2023. [Google Scholar]
- Wang, L.; Zhang, H.; Xu, W.; Xue, Z.; Wang, Y. Deciphering the protein landscape with ProtFlash, a lightweight language model. Cell Reports Physical Science 2023, 4. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, M.; Lozano, A.; Chenthamarakshan, V.; Das, P.; Tang, J. Enhancing protein language model with structure-based encoder and pre-training. In Proceedings of the ICLR 2023-Machine Learning for Drug Discovery workshop; 2023. [Google Scholar]
- Su, J.; Han, C.; Zhou, Y.; Shan, J.; Zhou, X.; Yuan, F. Saprot: Protein language modeling with structure-aware vocabulary. bioRxiv, 2023. [Google Scholar]
- Notin, P.; Weitzman, R.; Marks, D.; Gal, Y. Proteinnpt: Improving protein property prediction and design with non-parametric transformers. Advances in Neural Information Processing Systems 2023, 36, 33529–33563. [Google Scholar]
- Outeiral, C.; Deane, C.M. Codon language embeddings provide strong signals for use in protein engineering. Nature Machine Intelligence 2024, 6, 170–179. [Google Scholar] [CrossRef]
- Lee, Y.; Yu, H.; Lee, J.; Kim, J. Pre-training Sequence, Structure, and Surface Features for Comprehensive Protein Representation Learning. In Proceedings of the The Twelfth International Conference on Learning Representations; 2023. [Google Scholar]
- Zheng, K.; Long, S.; Lu, T.; Yang, J.; Dai, X.; Zhang, M.; Nie, Z.; Ma, W.Y.; Zhou, H. ESM All-Atom: Multi-Scale Protein Language Model for Unified Molecular Modeling. In Proceedings of the Forty-first International Conference on Machine Learning.
- Wang, Y.; Zhang, Q.; Qin, M.; Zhuang, X.; Li, X.; Gong, Z.; Wang, Z.; Zhao, Y.; Yao, J.; Ding, K.; et al. Knowledge-aware Reinforced Language Models for Protein Directed Evolution. In Proceedings of the Forty-first International Conference on Machine Learning.
- Hayes, T.; Rao, R.; Akin, H.; Sofroniew, N.J.; Oktay, D.; Lin, Z.; Verkuil, R.; Tran, V.Q.; Deaton, J.; Wiggert, M.; et al. Simulating 500 million years of evolution with a language model. bioRxiv, 2024. [Google Scholar]
- Madani, A.; McCann, B.; Naik, N.; Keskar, N.S.; Anand, N.; Eguchi, R.R.; Huang, P.S.; Socher, R. Progen: Language modeling for protein generation. arXiv 2020, arXiv:2004.03497 2020. [Google Scholar]
- Ferruz, N.; Schmidt, S.; Höcker, B. A deep unsupervised language model for protein design. BioRxiv, 2022. [Google Scholar]
- Cao, Y.; Das, P.; Chenthamarakshan, V.; Chen, P.Y.; Melnyk, I.; Shen, Y. Fold2Seq: A joint sequence (1D)-Fold (3D) embedding-based generative model for protein design. In Proceedings of the International Conference on Machine Learning. PMLR; 2021; pp. 1261–1271. [Google Scholar]
- Munsamy, G.; Lindner, S.; Lorenz, P.; Ferruz, N. ZymCTRL: a conditional language model for the controllable generation of artificial enzymes. In Proceedings of the NeurIPS Machine Learning in Structural Biology Workshop; 2022. [Google Scholar]
- Ram, S.; Bepler, T. Few Shot Protein Generation. arXiv 2022, arXiv:2204.01168 2022. [Google Scholar]
- Hesslow, D.; Zanichelli, N.; Notin, P.; Poli, I.; Marks, D. Rita: a study on scaling up generative protein sequence models. arXiv 2022, arXiv:2205.05789 2022. [Google Scholar]
- Shuai, R.W.; Ruffolo, J.A.; Gray, J.J. Generative language modeling for antibody design. BioRxiv, 2021. [Google Scholar]
- Zheng, Z.; Deng, Y.; Xue, D.; Zhou, Y.; Ye, F.; Gu, Q. Structure-informed language models are protein designers. In Proceedings of the International conference on machine learning. PMLR; 2023; pp. 42317–42338. [Google Scholar]
- Sgarbossa, D.; Lupo, U.; Bitbol, A.F. Generative power of a protein language model trained on multiple sequence alignments. Elife 2023, 12, e79854. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Vecchietti, L.F.; Jung, H.; Ro, H.; Cha, M.; Kim, H.M. Protein sequence design in a latent space via model-based reinforcement learning. OpenReview 2023. [Google Scholar]
- Heinzinger, M.; Weissenow, K.; Sanchez, J.G.; Henkel, A.; Steinegger, M.; Rost, B. Prostt5: Bilingual language model for protein sequence and structure. bioRxiv. bioRxiv 2023. [Google Scholar]
- Chen, B.; Cheng, X.; Li, P.; Geng, Y.a.; Gong, J.; Li, S.; Bei, Z.; Tan, X.; Wang, B.; Zeng, X.; et al. xTrimoPGLM: unified 100B-scale pre-trained transformer for deciphering the language of protein. arXiv 2024, arXiv:2401.06199 2024. [Google Scholar]
- Serrano, Y.; Roda, S.; Guallar, V.; Molina, A. Efficient and accurate sequence generation with small-scale protein language models. bioRxiv, 2023. [Google Scholar]
- Chu, S.K.; Wei, K.Y. Generative Antibody Design for Complementary Chain Pairing Sequences through Encoder-Decoder Language Model. arXiv 2023, arXiv:2301.02748 2023. [Google Scholar]
- Nijkamp, E.; Ruffolo, J.A.; Weinstein, E.N.; Naik, N.; Madani, A. Progen2: exploring the boundaries of protein language models. Cell systems 2023, 14, 968–978. [Google Scholar] [CrossRef]
- Sternke, M.; Karpiak, J. ProteinRL: Reinforcement learning with generative protein language models for property-directed sequence design. In Proceedings of the NeurIPS 2023 Generative AI and Biology (GenBio) Workshop; 2023. [Google Scholar]
- Truong Jr, T.; Bepler, T. Poet: A generative model of protein families as sequences-of-sequences. Advances in Neural Information Processing Systems 2023, 36, 77379–77415. [Google Scholar]
- Frey, N.C.; Berenberg, D.; Zadorozhny, K.; Kleinhenz, J.; Lafrance-Vanasse, J.; Hotzel, I.; Wu, Y.; Ra, S.; Bonneau, R.; Cho, K.; et al. Protein discovery with discrete walk-jump sampling. arXiv 2023, arXiv:2306.12360 2023. [Google Scholar]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- An, W.; Guo, Y.; Bian, Y.; Ma, H.; Yang, J.; Li, C.; Huang, J. MoDNA: motif-oriented pre-training for DNA language model. In Proceedings of the 13th ACM international conference on bioinformatics, computational biology and health informatics, 2022, pp. 1–5.
- Gankin, D.; Karollus, A.; Grosshauser, M.; Klemon, K.; Hingerl, J.; Gagneur, J. Species-aware DNA language modeling. bioRxiv, 2023. [Google Scholar]
- Dalla-Torre, H.; Gonzalez, L.; Mendoza-Revilla, J.; Carranza, N.L.; Grzywaczewski, A.H.; Oteri, F.; Dallago, C.; Trop, E.; de Almeida, B.P.; Sirelkhatim, H.; et al. The nucleotide transformer: Building and evaluating robust foundation models for human genomics. BioRxiv, 2023. [Google Scholar]
- Nguyen, E.; Poli, M.; Faizi, M.; Thomas, A.; Wornow, M.; Birch-Sykes, C.; Massaroli, S.; Patel, A.; Rabideau, C.; Bengio, Y.; et al. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. Advances in neural information processing systems 2024, 36. [Google Scholar]
- Fishman, V.; Kuratov, Y.; Petrov, M.; Shmelev, A.; Shepelin, D.; Chekanov, N.; Kardymon, O.; Burtsev, M. Gena-lm: A family of open-source foundational models for long dna sequences. bioRxiv. bioRxiv 2023. [Google Scholar]
- Theodoris, C.V.; Xiao, L.; Chopra, A.; Chaffin, M.D.; Al Sayed, Z.R.; Hill, M.C.; Mantineo, H.; Brydon, E.M.; Zeng, Z.; Liu, X.S.; et al. Transfer learning enables predictions in network biology. Nature 2023, 618, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, Q.; Zeng, W.; Jiang, R.; Wong, W.H. EpiGePT: a Pretrained Transformer model for epigenomics. bioRxiv 2023. [Google Scholar]
- Malusare, A.; Kothandaraman, H.; Tamboli, D.; Lanman, N.A.; Aggarwal, V. Understanding the natural language of DNA using encoder–decoder foundation models with byte-level precision. Bioinformatics Advances 2024, 4, vbae117. [Google Scholar] [CrossRef]
- Zhang, D.; et al. DNAGPT: A generalized pre-trained tool for versatile DNA sequence analysis tasks. Preprint at https://doi. org/10.48550/arXiv 2023, 2307. [Google Scholar]
- Nguyen, E.; Poli, M.; Durrant, M.G.; Thomas, A.W.; Kang, B.; Sullivan, J.; Ng, M.Y.; Lewis, A.; Patel, A.; Lou, A.; et al. Sequence modeling and design from molecular to genome scale with Evo. BioRxiv, 2024. [Google Scholar]
- Liu, H.; Zhou, S.; Chen, P.; Liu, J.; Huo, K.G.; Han, L. Exploring Genomic Large Language Models: Bridging the Gap between Natural Language and Gene Sequences. bioRxiv, 2024. [Google Scholar]
- Avsec, Ž.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective gene expression prediction from sequence by integrating long-range interactions. Nature methods 2021, 18, 1196–1203. [Google Scholar] [CrossRef]
- Luo, H.; Chen, C.; Shan, W.; Ding, P.; Luo, L. iEnhancer-BERT: a novel transfer learning architecture based on DNA-language model for identifying enhancers and their strength. In Proceedings of the International Conference on Intelligent Computing. Springer; 2022; pp. 153–165. [Google Scholar]
- Jin, J.; Yu, Y.; Wang, R.; Zeng, X.; Pang, C.; Jiang, Y.; Li, Z.; Dai, Y.; Su, R.; Zou, Q.; et al. iDNA-ABF: multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome biology 2022, 23, 219. [Google Scholar] [CrossRef]
- Li, J.; Wu, Z.; Lin, W.; Luo, J.; Zhang, J.; Chen, Q.; Chen, J. iEnhancer-ELM: improve enhancer identification by extracting position-related multiscale contextual information based on enhancer language models. Bioinformatics Advances 2023, 3, vbad043. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, X.; Wang, G.; Li, D. miProBERT: identification of microRNA promoters based on the pre-trained model BERT. Briefings in bioinformatics 2023, 24, bbad093. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, J.; Long, W.; Wei, L. PLPMpro: Enhancing promoter sequence prediction with prompt-learning based pre-trained language model. Computers in Biology and Medicine 2023, 164, 107260. [Google Scholar] [CrossRef]
- Duan, C.; Zang, Z.; Xu, Y.; He, H.; Liu, Z.; Song, Z.; Zheng, J.S.; Li, S.Z. FGBERT: Function-Driven Pre-trained Gene Language Model for Metagenomics. arXiv 2024, arXiv:2402.16901 2024. [Google Scholar]
- Benegas, G.; Batra, S.S.; Song, Y.S. DNA language models are powerful predictors of genome-wide variant effects. Proceedings of the National Academy of Sciences 2023, 120, e2311219120. [Google Scholar] [CrossRef]
- Zvyagin, M.; Brace, A.; Hippe, K.; Deng, Y.; Zhang, B.; Bohorquez, C.O.; Clyde, A.; Kale, B.; Perez-Rivera, D.; Ma, H.; et al. GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics. The International Journal of High Performance Computing Applications 2023, 37, 683–705. [Google Scholar] [CrossRef]
- Benegas, G.; Albors, C.; Aw, A.J.; Ye, C.; Song, Y.S. GPN-MSA: an alignment-based DNA language model for genome-wide variant effect prediction. bioRxiv 2023. [Google Scholar]
- Luo, H.; Shan, W.; Chen, C.; Ding, P.; Luo, L. Improving language model of human genome for DNA–protein binding prediction based on task-specific pre-training. Interdisciplinary Sciences: Computational Life Sciences 2023, 15, 32–43. [Google Scholar] [CrossRef]
- Hwang, Y.; Cornman, A.L.; Kellogg, E.H.; Ovchinnikov, S.; Girguis, P.R. Genomic language model predicts protein co-regulation and function. Nature communications 2024, 15, 2880. [Google Scholar] [CrossRef]
- Chen, K.; Zhou, Y.; Ding, M.; Wang, Y.; Ren, Z.; Yang, Y. Self-supervised learning on millions of pre-mRNA sequences improves sequence-based RNA splicing prediction. bioRxiv 2023, 2023–01. [Google Scholar]
- Zhang, Y.; Lang, M.; Jiang, J.; Gao, Z.; Xu, F.; Litfin, T.; Chen, K.; Singh, J.; Huang, X.; Song, G.; et al. Multiple sequence alignment-based RNA language model and its application to structural inference. Nucleic Acids Research 2024, 52, e3–e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ge, F.; Li, F.; Yang, X.; Song, J.; Yu, D.J. Prediction of multiple types of RNA modifications via biological language model. IEEE/ACM transactions on computational biology and bioinformatics 2023, 20, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, R.; Chen, Z.; Li, Y.; Ji, X.; Ke, G.; Wen, H. UNI-RNA: universal pre-trained models revolutionize RNA research. bioRxiv, 2023. [Google Scholar]
- Penić, R.J.; Vlašić, T.; Huber, R.G.; Wan, Y.; Šikić, M. Rinalmo: General-purpose rna language models can generalize well on structure prediction tasks. arXiv 2024, arXiv:2403.00043 2024. [Google Scholar]
- Wang, N.; Bian, J.; Li, Y.; Li, X.; Mumtaz, S.; Kong, L.; Xiong, H. Multi-purpose RNA language modelling with motif-aware pretraining and type-guided fine-tuning. Nature Machine Intelligence 2024, 1–10. [Google Scholar] [CrossRef]
- Strausberg, R.L.; Feingold, E.A.; Klausner, R.D.; Collins, F.S. The mammalian gene collection. Science 1999, 286, 455–457. [Google Scholar] [CrossRef]
- Guo, Y.; Dai, Y.; Yu, H.; Zhao, S.; Samuels, D.C.; Shyr, Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017, 109, 83–90. [Google Scholar] [CrossRef]
- Zeng, H.; Edwards, M.D.; Liu, G.; Gifford, D.K. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nature methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Consortium, G.P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef]
- Dreos, R.; Ambrosini, G.; Cavin Périer, R.; Bucher, P. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic acids research 2013, 41, D157–D164. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.M.; Björkegren, J.L. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Rusk, N. Sequence-based prediction of variants’ effects. Nature Methods 2018, 15, 571–571. [Google Scholar] [CrossRef]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic acids research 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the bacterial and viral bioinformatics resource center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic acids research 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic acids research 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Zhang, T.; Singh, J.; Litfin, T.; Zhan, J.; Paliwal, K.; Zhou, Y. RNAcmap: a fully automatic pipeline for predicting contact maps of RNAs by evolutionary coupling analysis. Bioinformatics 2021, 37, 3494–3500. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57. [Google Scholar] [CrossRef]
- Hiscock, D.; Upton, C. Viral Genome DataBase: storing and analyzing genes and proteins from complete viral genomes. Bioinformatics 2000, 16, 484–485. [Google Scholar] [CrossRef]
- Katsonis, P.; Lichtarge, O. CAGI5: Objective performance assessments of predictions based on the Evolutionary Action equation. Human mutation 2019, 40, 1436–1454. [Google Scholar] [CrossRef]
- Horlacher, M.; Cantini, G.; Hesse, J.; Schinke, P.; Goedert, N.; Londhe, S.; Moyon, L.; Marsico, A. A systematic benchmark of machine learning methods for protein–RNA interaction prediction. Briefings in Bioinformatics 2023, 24, bbad307. [Google Scholar] [CrossRef]
- Wolf, T. Transformers: State-of-the-Art Natural Language Processing. arXiv 2020, arXiv:1910.03771 2020. [Google Scholar]
- Rasley, J.; Rajbhandari, S.; Ruwase, O.; He, Y. Deepspeed: System optimizations enable training deep learning models with over 100 billion parameters. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining; 2020; pp. 3505–3506. [Google Scholar]
- Bajaj, P.; Xiong, C.; Ke, G.; Liu, X.; He, D.; Tiwary, S.; Liu, T.Y.; Bennett, P.; Song, X.; Gao, J. Metro: Efficient denoising pretraining of large scale autoencoding language models with model generated signals. arXiv 2022, arXiv:2204.06644 2022. [Google Scholar]
- Shoeybi, M.; Patwary, M.; Puri, R.; LeGresley, P.; Casper, J.; Catanzaro, B. Megatron-lm: Training multi-billion parameter language models using model parallelism. arXiv 2019, arXiv:1909.08053 2019. [Google Scholar]
- Sachan, D.S.; Patwary, M.; Shoeybi, M.; Kant, N.; Ping, W.; Hamilton, W.L.; Catanzaro, B. End-to-end training of neural retrievers for open-domain question answering. arXiv 2021, arXiv:2101.00408 2021. [Google Scholar]
- Boyd, A.; Puri, R.; Shoeybi, M.; Patwary, M.; Catanzaro, B. Large scale multi-actor generative dialog modeling. arXiv 2020, arXiv:2005.06114 2020. [Google Scholar]
- Santhanam, S.; Ping, W.; Puri, R.; Shoeybi, M.; Patwary, M.; Catanzaro, B. Local knowledge powered conversational agents. arXiv 2020, arXiv:2010.10150 2020. [Google Scholar]
- Xu, P.; Patwary, M.; Shoeybi, M.; Puri, R.; Fung, P.; Anandkumar, A.; Catanzaro, B. MEGATRON-CNTRL: Controllable story generation with external knowledge using large-scale language models. arXiv 2020, arXiv:2010.00840 2020. [Google Scholar]
- Wang, B.; Ping, W.; McAfee, L.; Xu, P.; Li, B.; Shoeybi, M.; Catanzaro, B. Instructretro: Instruction tuning post retrieval-augmented pretraining. arXiv 2023, arXiv:2310.07713 2023. [Google Scholar]
- Bradbury, J.; Frostig, R.; Hawkins, P.; Johnson, M.J.; Leary, C.; Maclaurin, D.; Zhang, Q. JAX: Composable Transformations of Python+ NumPy Programs. https://github.com/google/jax, 2018. Accessed: 2024-02-07.
- Li, S.; Liu, H.; Bian, Z.; Fang, J.; Huang, H.; Liu, Y.; Wang, B.; You, Y. Colossal-ai: A unified deep learning system for large-scale parallel training. In Proceedings of the 52nd International Conference on Parallel Processing, 2023, pp. 766–775.
- Tang, T.; Hu, Y.; Li, B.; Luo, W.; Qin, Z.; Sun, H.; Wang, J.; Xu, S.; Cheng, X.; Guo, G.; et al. LLMBox: A Comprehensive Library for Large Language Models. arXiv 2024, arXiv:2407.05563 2024. [Google Scholar]
- Jiang, A.Q.; Sablayrolles, A.; Mensch, A.; Bamford, C.; Chaplot, D.S.; Casas, D.d.l.; Bressand, F.; Lengyel, G.; Lample, G.; Saulnier, L.; et al. Mistral 7B. arXiv 2023, arXiv:2310.06825 2023. [Google Scholar]
- Bai, J.; Bai, S.; Chu, Y.; Cui, Z.; Dang, K.; Deng, X.; Fan, Y.; Ge, W.; Han, Y.; Huang, F.; et al. Qwen technical report. arXiv 2023, arXiv:2309.16609 2023. [Google Scholar]
- Workshop, B.; Scao, T.L.; Fan, A.; Akiki, C.; Pavlick, E.; Ilić, S.; Hesslow, D.; Castagné, R.; Luccioni, A.S.; Yvon, F.; et al. Bloom: A 176b-parameter open-access multilingual language model. arXiv 2022, arXiv:2211.05100 2022. [Google Scholar]
- Ghosal, D.; Chia, Y.K.; Majumder, N.; Poria, S. Flacuna: Unleashing the problem solving power of vicuna using flan fine-tuning. arXiv 2023, arXiv:2307.02053 2023. [Google Scholar]
- Nijkamp, E.; Pang, B.; Hayashi, H.; Tu, L.; Wang, H.; Zhou, Y.; Savarese, S.; Xiong, C. Codegen: An open large language model for code with multi-turn program synthesis. arXiv 2022, arXiv:2203.13474 2022. [Google Scholar]
- Li, R.; Allal, L.B.; Zi, Y.; Muennighoff, N.; Kocetkov, D.; Mou, C.; Marone, M.; Akiki, C.; Li, J.; Chim, J.; et al. Starcoder: may the source be with you! arXiv 2023, arXiv:2305.06161 2023. [Google Scholar]
- Azerbayev, Z.; Schoelkopf, H.; Paster, K.; Santos, M.D.; McAleer, S.; Jiang, A.Q.; Deng, J.; Biderman, S.; Welleck, S. Llemma: An open language model for mathematics. arXiv 2023, arXiv:2310.10631 2023. [Google Scholar]
- Shao, Z.; Wang, P.; Zhu, Q.; Xu, R.; Song, J.; Bi, X.; Zhang, H.; Zhang, M.; Li, Y.; Wu, Y.; et al. Deepseekmath: Pushing the limits of mathematical reasoning in open language models. arXiv 2024, arXiv:2402.03300 2024. [Google Scholar]
- Sheng, G.; Zhang, C.; Ye, Z.; Wu, X.; Zhang, W.; Zhang, R.; Peng, Y.; Lin, H.; Wu, C. Hybridflow: A flexible and efficient rlhf framework. arXiv 2024, arXiv:2409.19256 2024. [Google Scholar]
- Liang, W.; Liu, T.; Wright, L.; Constable, W.; Gu, A.; Huang, C.C.; Zhang, I.; Feng, W.; Huang, H.; Wang, J.; et al. TorchTitan: One-stop PyTorch native solution for production ready LLM pre-training. arXiv 2024, arXiv:cs.CL/2410.06511]. [Google Scholar]
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 |
Figure 1.
Textual Scientific Large Language Models.
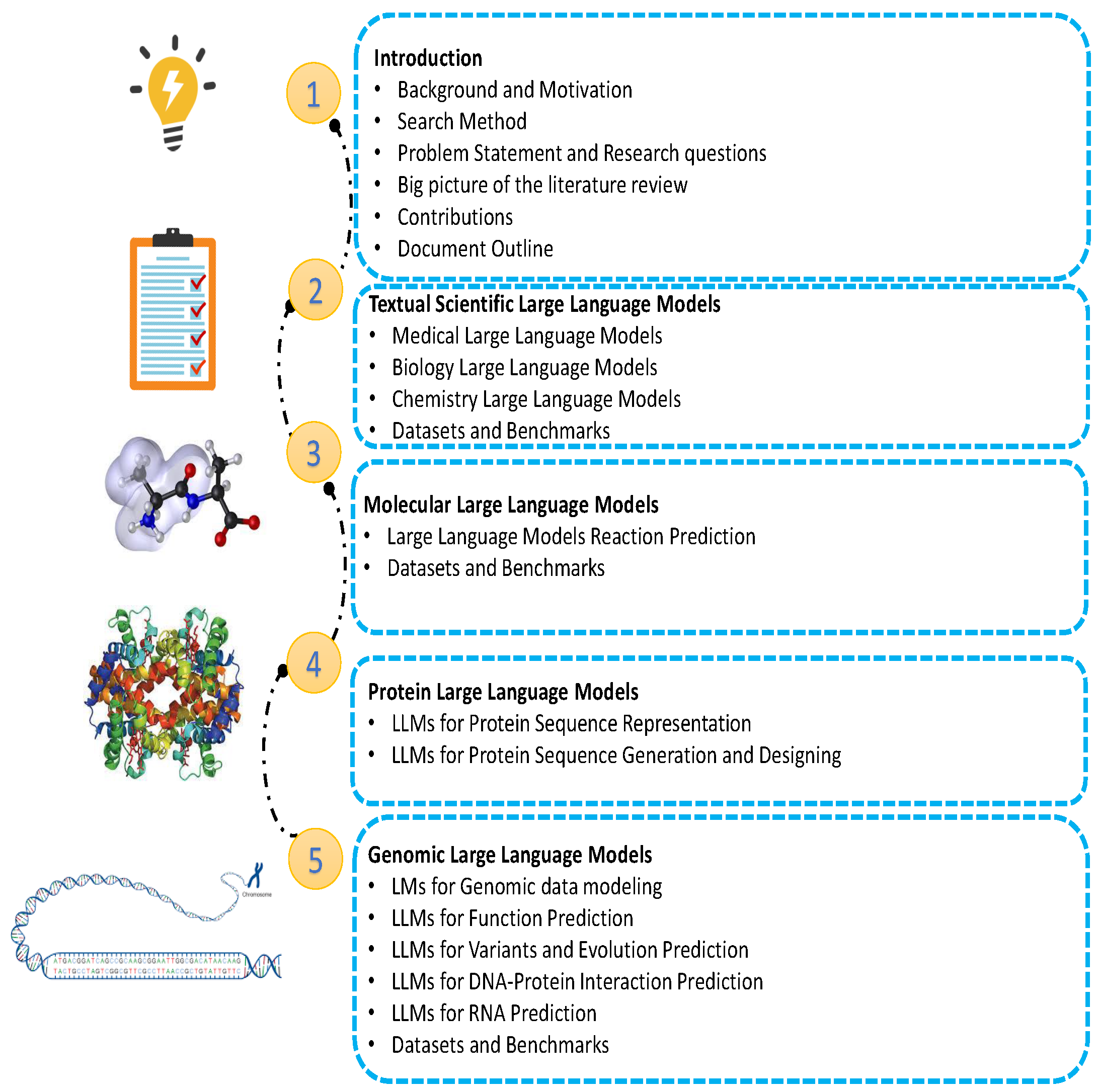
Figure 2.
Textual Scientific Large Language Models.
Figure 3.
Overview of the methods and subcategories of Medical Large Language Models.
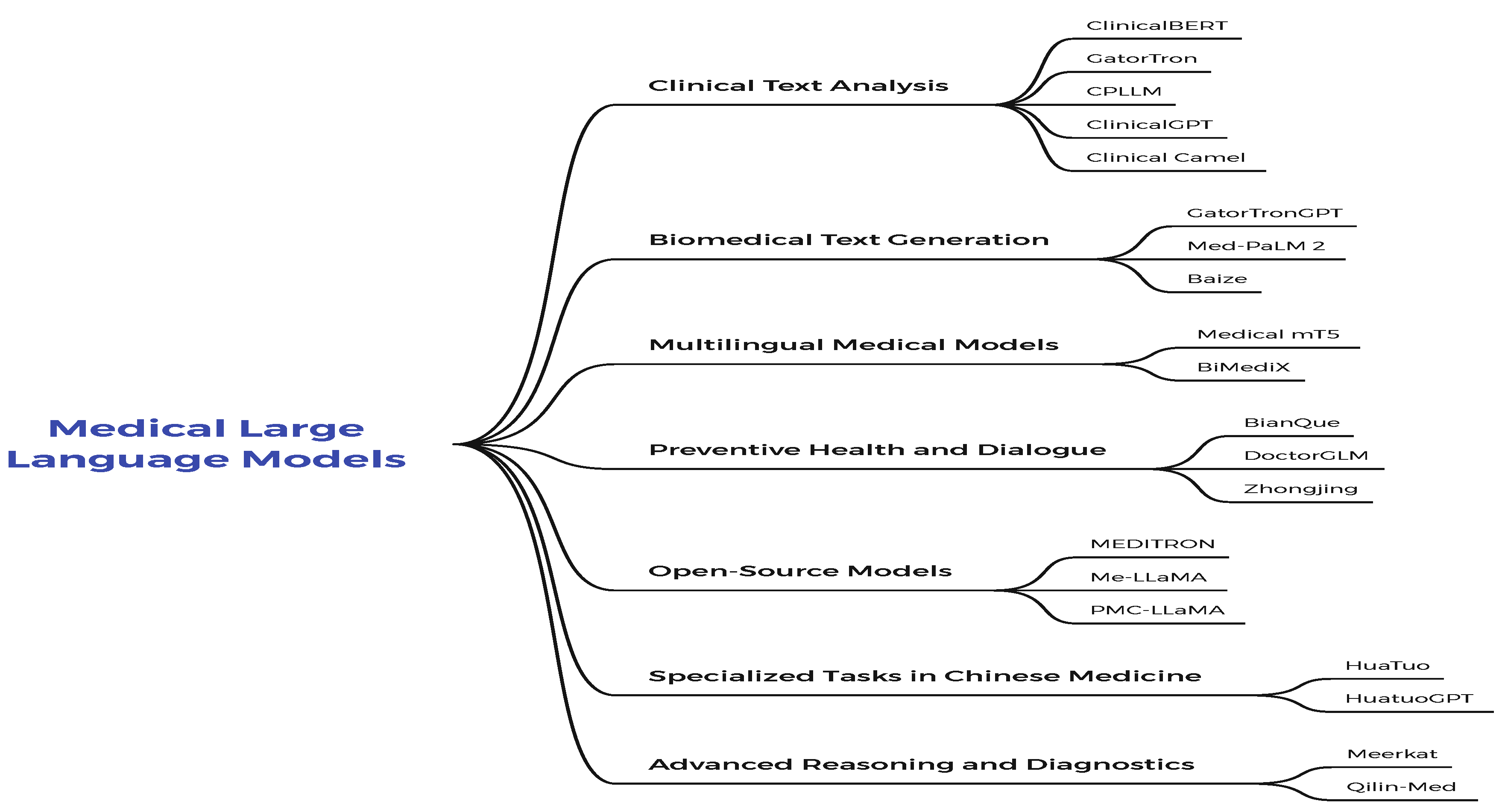
Figure 4.
Overview of the methods and subcategories of Biology Large Language Models.
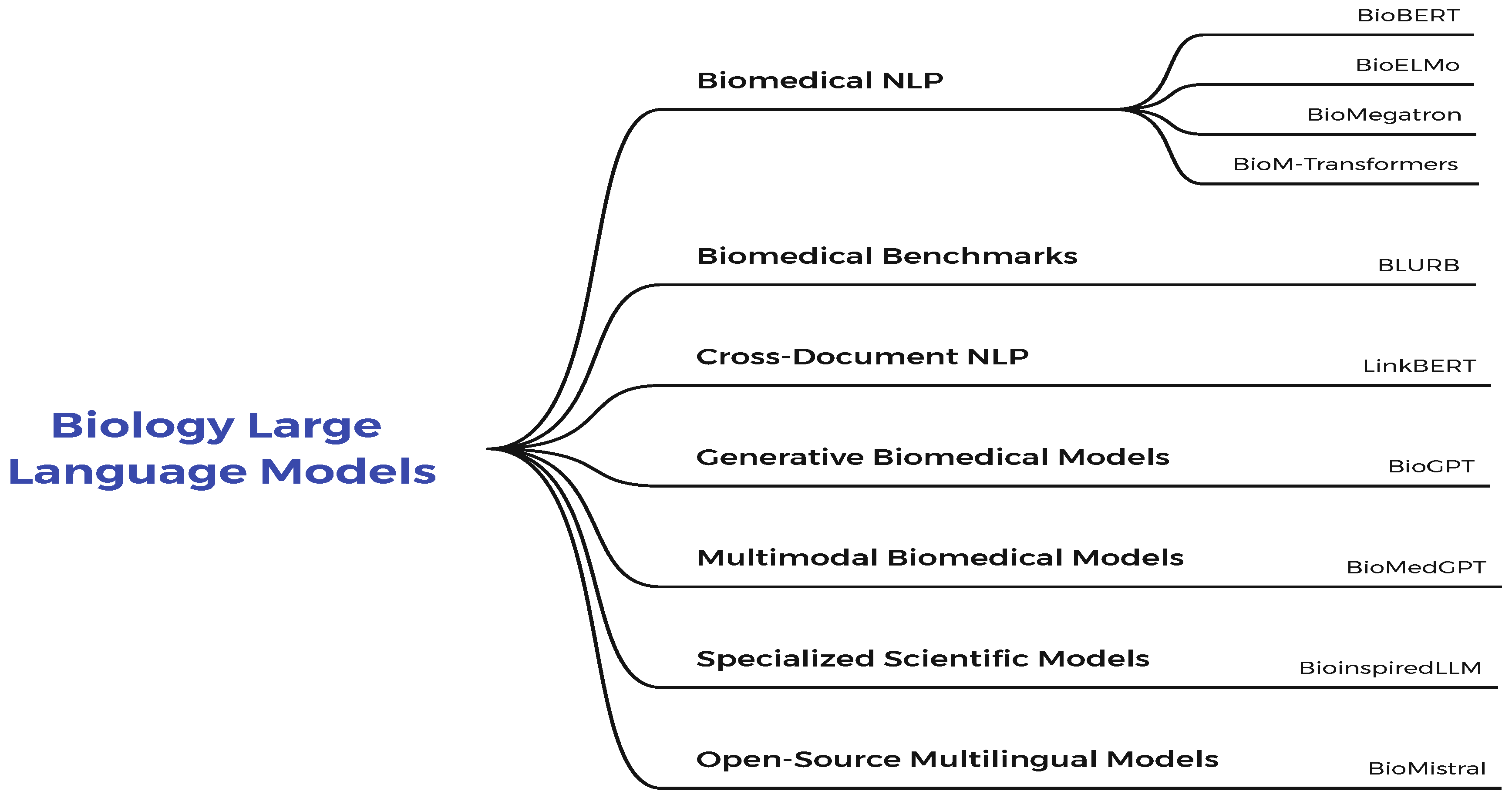
Figure 5.
Overview of the methods and subcategories of Chemistry Large Language Models.
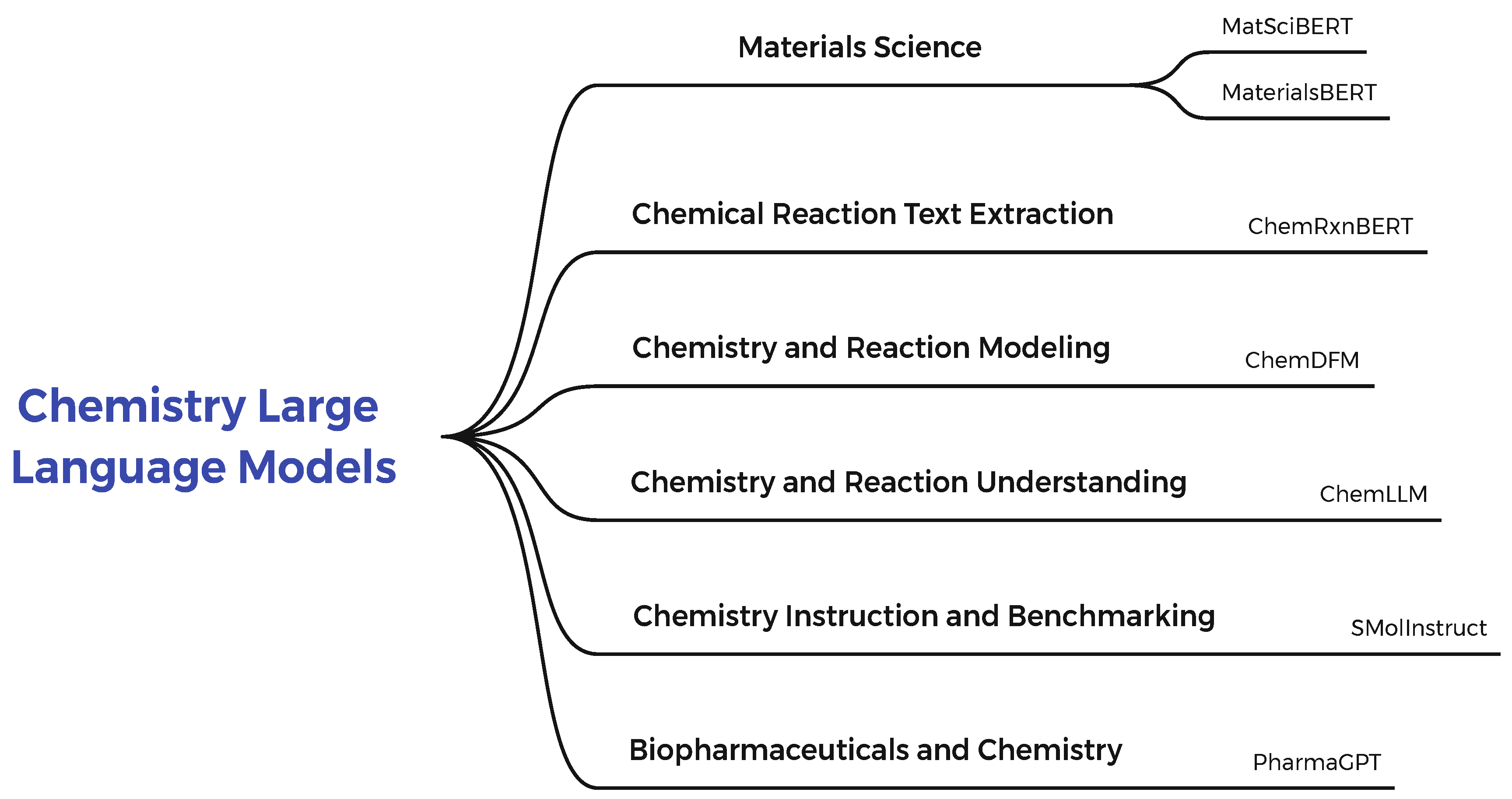
Figure 6.
Molecular Large Language Models.
Figure 7.
Overview of the methods and subcategories of Large Language Models for Reaction Prediction.
Figure 7.
Overview of the methods and subcategories of Large Language Models for Reaction Prediction.
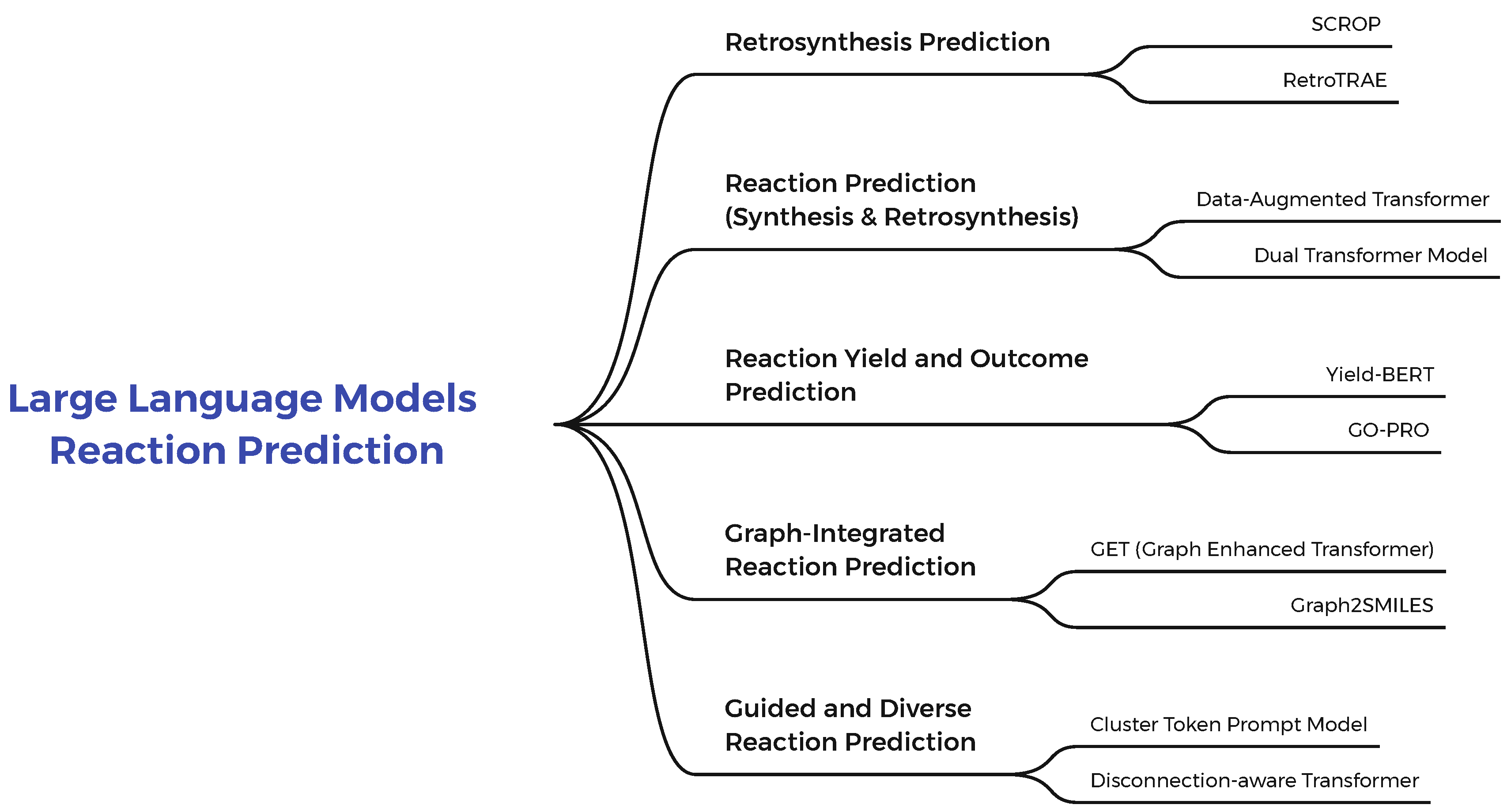
Figure 8.
Protein Large Language Models.
Figure 9.
Overview of the methods and subcategories of Protein Large Language Models.
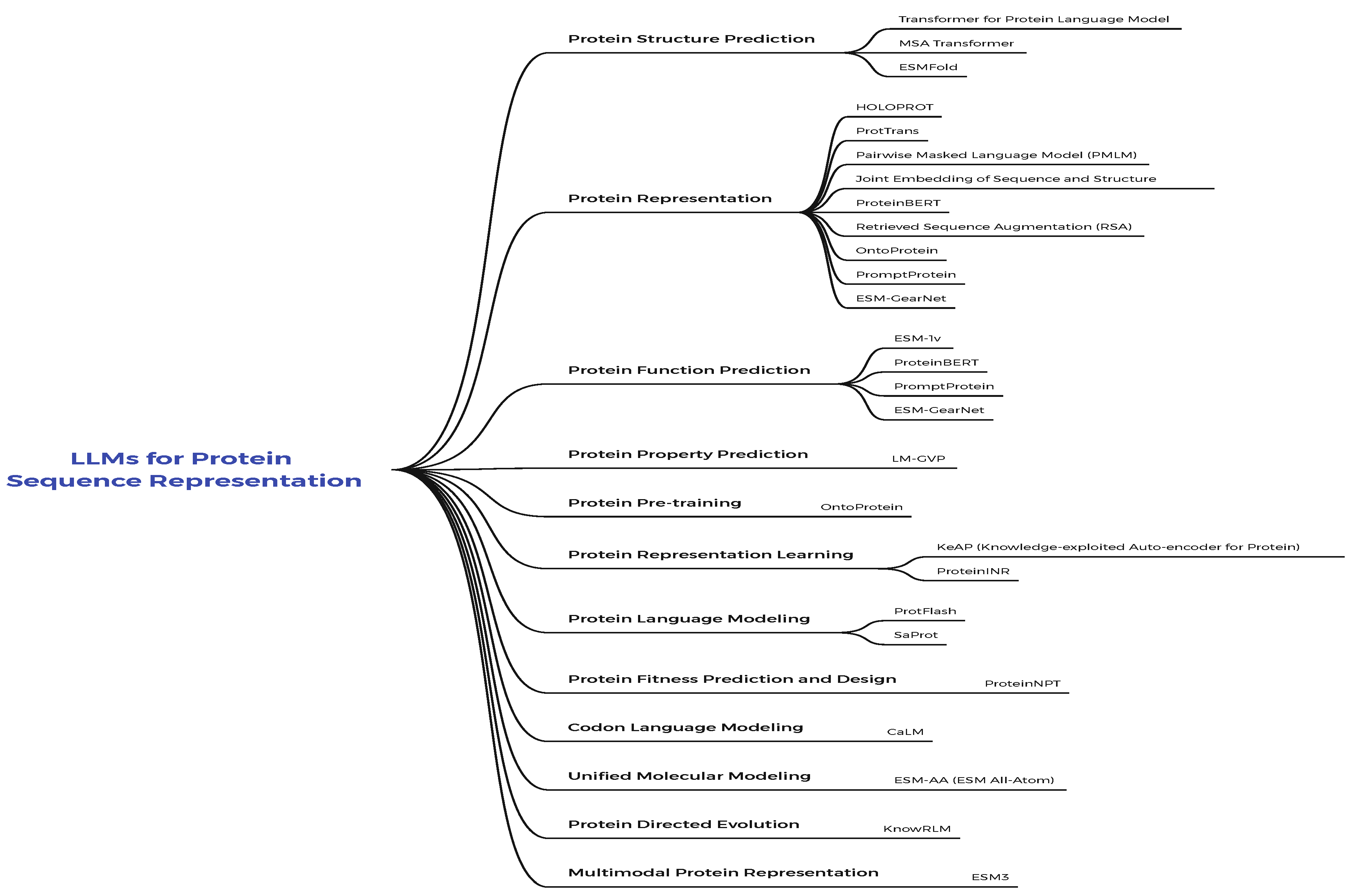
Figure 10.
Genomic Large Language Models.
Figure 11.
Overview of the methods and subcategories of Genomic Large Language Models.
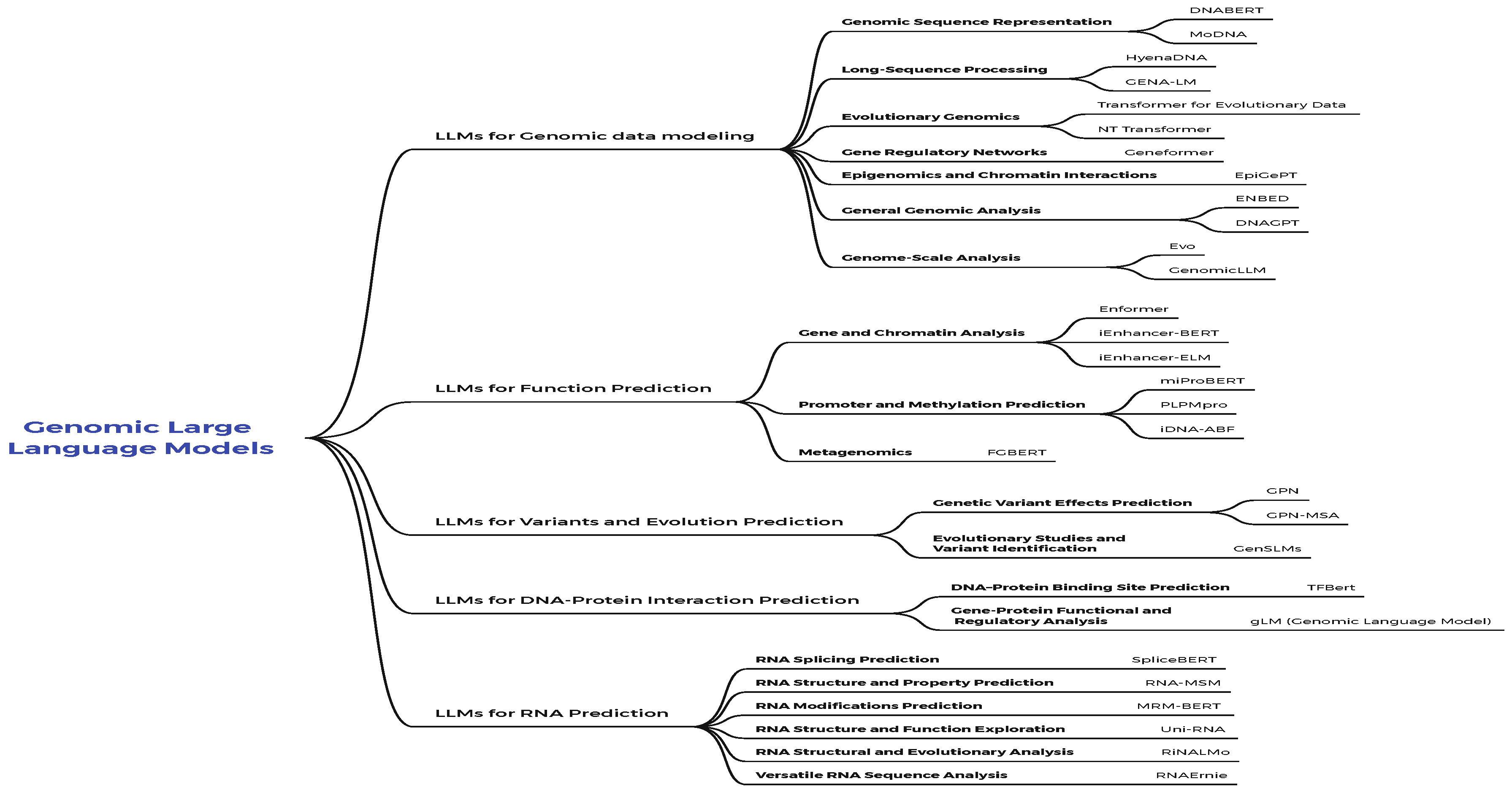

Table 1.
Query for searching articles.
| Row | Topic | Query for Searching Articles |
|---|---|---|
| 1 | Medical Large Language Models | (‘LLM’ OR ‘Large Language Model’ OR ‘Generative AI’ OR ‘Transformer Models’) AND (‘Medical Data’ OR ‘Electronic Health Records’ OR ‘EHR’) |
| 2 | Biology Large Language Models | (‘LLM’ OR ‘Large Language Model’) AND (‘Biology Data’ OR ‘Genomics’ OR ‘Protein Data’) AND (‘Sequence Representation’ OR ‘Function Prediction’ OR ‘Evolution Modeling’) |
| 3 | Chemistry Large Language Models | (‘LLM’ OR ‘Transformer’ OR ‘BERT’) AND (‘Molecular Data’ OR ‘Chemical Structures’) AND (‘Molecule Property Prediction’ OR ‘Reaction Prediction’ OR ‘Molecule Design’) |
| 4 | Datasets and Benchmarks (General) | (‘Datasets’ OR ‘Benchmarks’) AND (‘LLM Evaluation’ OR ‘Scientific LLMs’ OR ‘Biological Data’ OR ‘Chemical Data’ OR ‘Medical Data’) |
| 5 | LLMs for Molecule Property Prediction | (‘LLM’ OR ‘Large Language Model’) AND (‘Molecular Property’ OR ‘Chemical Properties’) AND (‘Prediction’ OR ‘Transformer-based Analysis’) |
| 6 | LLMs for Interaction Prediction | (‘LLM’ OR ‘Large Language Model’) AND (‘Protein-Ligand Interaction’ OR ‘DNA-Protein Binding’ OR ‘RNA Interaction’) |
| 7 | LLMs for Molecule Generation | (‘LLM’ OR ‘Generative Models’ OR ‘Molecule Generation’) AND (‘Drug Design’ OR ‘Protein Engineering’ OR ‘DNA Synthesis’) |
| 8 | LLMs for Reaction Prediction | (‘LLM’ OR ‘Reaction Prediction’) AND (‘Transformer Models’ OR ‘Molecular Modeling’) AND (‘Chemical Reactions’ OR ‘Organic Chemistry’) |
| 9 | Protein Sequence Representation | (‘LLM’ OR ‘Large Language Model’) AND (‘Protein Sequence’ OR ‘Sequence Representation’) AND (‘Transformer’ OR ‘Masked Language Modeling’) |
| 10 | Protein Sequence Generation | (‘LLM’ OR ‘Protein Design’) AND (‘Sequence Generation’ OR ‘Generative Modeling’) |
| 11 | Genomic Data Modeling | (‘LLM’ OR ‘Large Language Model’) AND (‘Genomic Data’ OR ‘DNA Modeling’) AND (‘Sequence-to-Function’ OR ‘Gene Prediction’) |
| 12 | Function Prediction | (‘LLM’ OR ‘Transformer Model’) AND (‘Gene Function Prediction’ OR ‘Protein Function Prediction’) |
| 13 | Variants and Evolution Prediction | (‘LLM’ OR ‘Evolution Modeling’) AND (‘Variants’ OR ‘Mutations’) |
| 14 | DNA-Protein Interaction | (‘LLM’ OR ‘Transformer’) AND (‘DNA-Protein Interaction’ OR ‘Binding Affinity’) |
| 15 | RNA Prediction | (‘LLM’ OR ‘RNA Modeling’) AND (‘Sequence Prediction’ OR ‘RNA Binding Proteins’) |
Table 2.
Summary of Medical Large Language Models (LLMS).
| Model | # Parameter | Evaluation Metrics | Core Model | Pretraining Datasets | Language | Evaluation Tasks | Open-source | Model Link |
|---|---|---|---|---|---|---|---|---|
| ClinicalBERT | 110M | AUROC AUPRC RP80 | BERT | MIMIC-III | English | predicts 30-day readmission using discharge summaries | ✓ | https://github.com/kexinhuang12345/clinicalBERT |
| Gatortron | 8.9B | Pearson , Accuracy, F1 score ,Exact Match | BERT | MIMIC-III, PubMed, etc. | English | Semantic textual similarity and Question answering | ✓ | https://github.com/uf-hobi-informatics-lab/GatorTron |
| GatorTronGPT | 5B, 20B | Precision, Recall, and F1 | GPT | Pubmed, and Custom Data | English | Clinical concept extraction, and Question answering | ✓ | https://github.com/uf-hobi-informatics-lab/GatorTronGPT |
| MEDITRON-70B | 7B, 70B | Accuracy | LLAMA2 | Pubmed, PMC, etc. | English | Question answering | ✓ |
https://github.com/epfLLM/meditron, https://huggingface.co/epfl-llm/ |
| Meerkat-7B | 7B | Accuracy | Mistral | Custom Data | English | Multiple-choice QA | ✓ | https://huggingface.co/dmis-lab/meerkat-7b-v1.0 |
| CLINICALGPT | 7B | Win, Lose, and Tie | BLOOM | cMedQA2, MedDialog, etc. | Chinese | Medical question answering | ✗ | - |
| Qilin-Med | 7B | Accuracy, BLEU, and ROUGE | Baichuan | ChiMed | Chinese | medical exam and practice question | ✓ | https://github.com/williamliujl/Qilin-Med |
| ChatDoctor | 7B | Precision, Recall, and F1 | LLAMA | HealthCareMagic | English | question answering | ✓ | https://github.com/Kent0n-Li/ChatDoctor |
| HuaTuo | 7B,69B | Safety, Usability, and Smoothness | LLaMA | Custom Data | Chinese | question answering | ✓ | https://github.com/SCIR-HI/Huatuo-Llama-Med-Chinese |
| HuatuoGPT | 7B | BLEU, GLEU, ROUGE, and Distinct | BLOOM | Custom Data | Chinese | Efficacy,Medical Expenses, and Consequences Description | ✓ | https://github.com/FreedomIntelligence/HuatuoGPT |
| Baize | 7B | ARC | LLAMA | MedQuAD | English | question answering | ✓ | https://github.com/project-baize/baize-chatbot |
| Zhongjing | 13B | Safety, Prof, Fluency, and Length | LLAMA | Medical Books, Wiki, etc. | Chinese | single-turn and multi-turn dialogue capabilities of the Chine | ✓ | https://github.com/SupritYoung/Zhongjing |
| PMC-LLaMA | 13B | Accuracy | LLAMA | PMC, Medical books, etc | English | Medical question answering | ✓ | https://github.com/chaoyi-wu/PMC-LLaMA |
| CPLLM | 2.7B, 13B | PR-AUC ROC-AUC | LLAMA-2 | eICU-CRD, MIMIC-IV | English | disease prediction | ✓ | https://github.com/nadavlab/CPLLM |
| Med-PaLM 2 | 340B | Accuracy | PaLM2 | MultiMedQA | English | Long-form question | ✗ | - |
| Clinical Camel | 13B, 70B | Zero-shot five-shot |
LLAMA-2 | PubMed | English | Long-form question | ✗ | - |
| DoctorGLM | 6.2B | - | ChatGLM | CMD., HealthCareMagic, etc. | Chinese | Long-form question | ✓ | https://github.com/xionghonglin/DoctorGLM |
| BianQue | 6.2B | BLEU | ChatGLM | BianQueCorpus | Chinese | chain of questioning (CoQ) | ✓ | https://github.com/scutcyr/BianQue |
| Medical mT5 | 738M, 3B | Zero-shot F1 scores | T5 | Custom Data | English, French, Italian and Spanish | Named Entity Recognition, Argument Mining, and Question Answering | ✓ | https://huggingface.co/DHEIVER/Medical-mT5-large |
| Me-LLaMA | 13B, 70B | Accuracy, F1-score | LLAMA | Custom Data | English | Question Answering | ✓ | https://github.com/BIDS-Xu-Lab/ |
| BiMediX | 7B | Accuracy | Mixtral | BiMed1.3M | English and Arabic | Multiple-Choice Question Answering (MCQA)and Question Answering (QA) | ✓ | https://github.com/mbzuai-oryx/BiMediX |
Table 3.
Summary of Biology Large Language Models.
| Model | Reference | #Parameters | Base Model | Pretraining dataset | Open source | Link |
|---|---|---|---|---|---|---|
| BioELMo | [64] | - | ELMo | PubMed | ✓ | https://github.com/mbzuai-oryx/BiMediX |
| BioBERT | [67] | 117M | BERT | PubMed, PMC | ✓ | https://github.com/Andy-jqa/bioelmo |
| BlueBERT | [90] | 117M | BERT | PubMed | ✓ | https://github.com/ncbi-nlp/bluebert |
| BioMegatron | [72] | 345M-1.2B | BERT | PubMed, PMC | ✓ | https://github.com/NVIDIA/NeMo |
| PubMedBERT | [73] | 117M | BERT | PubMed | ✗ | - |
| BioM-BERT | [84] | 235M | BERT | PubMed, PMC | ✓ | https://github.com/BioMedBERT/biomedbert |
| BioLinkBERT | [85] | 110M, 340M | BERT | ioL PubMed | ✓ | https://github.com/michiyasunaga/LinkBERT |
| BioGPT | [86] | 347M | GPT | PubMed | ✓ | https://github.com/microsoft/BioGPT |
| BioMedGPT-LM | [87] | 7B | LLaMA | PMC, arXiv, WIPO | ✓ | https://github.com/PharMolix/OpenBioMed |
| BioinspiredLLM | [88] | 13B | Llama-2 | Biological article | ✓ | https://huggingface.co/lamm-mit/BioinspiredLLM |
| BioMistral | [89] | 7B | Mistral | PMC | ✓ | https://github.com/BioMistral/BioMistral |
Table 4.
Summary of Chemistry Large Language Models.
| Model | Reference | #Parameters | Base Model | Pretraining dataset | Open source | Link |
|---|---|---|---|---|---|---|
| ChemBERT | [91,105] | 120M | BERT | Chemical journals | ✓ | https://github.com/jiangfeng1124/ChemRxnExtractor |
| MatSciBERT | [92] | 117M | BERT | Elsevier journals | ✓ | https://github.com/M3RG-IITD/MatSciBERT |
| MaterialsBERT | [293] [93] | ✗ | BERT | Material journals | ✓ | https://huggingface.co/pranav-s/MaterialsBERT |
| PharmGPT | [104] | 13B, 70B | LLaMA | Paper, report, book, etc. | ✗ | - |
Table 5.
Summary of LLMs for Reaction Prediction.
| Model | Ref | param | Eval met | Dataset | O-source | Model link |
|---|---|---|---|---|---|---|
| SCROP | [120] | N/A | Accuracy | USPTO-50k | ✓ | https://github.com/sysu-yanglab/Self-Corrected-Retrosynthetic-Reaction-Predictor. |
| Tetko et. al | [121] | N/A | Accuracy | USPTO-50k | ✓ | https://github.com/bigchem/synthesis |
| Two-way Transformers | [122] | 34.8 M | Accuracy | USPTO | ✓ | http://github.com/ejklike/tied-twoway-transformer/ |
| Transformer+Regressor | [123] | N/A | USPTO | ✓ | https://rxn4chemistry.github.io/rxn_yields/ | |
| GO-PRO | 98 | 5M | Top-1,2,3 accuracies, BLEU, Syntactic validity, and Character based similarity | Jin’s USPTO and Human Chemists | Upon Request | Upon Request |
| Graph Enhanced Transformer | [125] | N/A | Accuracy | USPTO-50k | ✓ | https://github.com/papercodekl/MolecularGET |
| Graph2SMILES | [126] | N/A | Accuracy | USPTO | ✓ | https://github.com/coleygroup/Graph2SMILES |
| RetroTRAE | [127] | N/A | Accuracy, Tanimoto Coefficient (Tc), and Sørensen–Dice Coefficient (S) | USPTO | ✓ | https://github.com/knu-lcbc/RetroTRAE |
| Toniato et. Al | [128] | 12 M | Accuracy, Round-trip Accuracy, Coverage, and Class diversity | Pistachio Dataset, USPTO-50k |
✓ | https://github.com/rxn4chemistry/rxn_cluster_token_prompt |
| Thakkar et. al | [129] | N/A | Accuracy, Round Trip Accuracy, Disconnection Accuracy, and Reaction Class Diversity | Pistachio Dataset, USPTO, ECReact |
✓ | https://github.com/rxn4chemistry/disconnection_aware_retrosynthesis |
Table 6.
Summary of LLMs for Protein Sequence Generation and Designing.
| Model | Reference | #Parameters | Base Model | Pretraining dataset | Open source | Link |
|---|---|---|---|---|---|---|
| ProGen | [171] | 1.2B | GPT | Uniparc SWISS-Prot | ✓ | https://github.com/salesforce/progen |
| ProtGPT2 | [172] | 738M | GPT | Uniref50 | ✓ | https://huggingface.co/nferruz/ProtGPT2 |
| ZymCTRL | [174] | 738M | GPT | BRENDA | ✓ | https://huggingface.co/AI4PD/ZymCTRL |
| RITA | [176] | 1.2B | GPT | UniRef100 | ✗ | |
| IgLM | [177] | 13M | GPT | - | https://github.com/Graylab/IgLM | |
| ProGen2 | [185] | 151M - 6.4B | GPT | Uniref90, BFD30, PDB | ✓ | https://github.com/salesforce/progen |
| ProteinRL | [186] | 764M | GPT | - | ✗ | |
| PoET | [187] | 201M | GPT | - | ✗ | |
| C. Frey et al. | [188] | 9.87M-1.03M | GPT | hu4D5 antibody mutant | ✗ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



